repÚblica bolivariana de venezuela la … · distrofia muscular de duchenne -becker...
TRANSCRIPT

REPÚBLICA BOLIVARIANA DE VENEZUELALA UNIVERSIDAD DEL ZULIA
FACULTAD DE MEDICINAPROGRAMA DE GENETICA CLINICA
Ana BrachoEspecialista en Pediatría
MgSc en Genética Médica
HERENCIA RECESIVA
LIGADA AL X

DISTROFIA MUSCULAR DE DUCHENNE –BECKERRECESIVA LIGADA AL X

DISTROFIA MUSCULAR DE DUCHENNE -BECKER
DEFINICION:
La distrofia muscular, definida como debilidad y pérdida
muscular progresiva, existen al menos una docena de
formas distintas. De ellas la distrofia muscular de Duchenne
es la mas grave y frecuente
INCIDENCIA:
DMD 1:3.500 varones
DMB 1:18.000 varones

DISTROFIA MUSCULAR DE DUCHENNE -BECKER
MECANISMO DE TRANSMISION:
Herencia: recesivo ligado al X. El 30% de los casos son"esporádicos".
DMD: cambios en el marco de lectura
DMB: sin cambios en el marco de lectura, suelen ser el resultadode deleciones.
UBICACIÓN GÉNICA:
Cromosoma X.Proteína: distrofina.

DISTROFIA MUSCULAR DE DUCHENNE -BECKER
MANIFESTACIONES CLÍNICAS:
♠ Comienzo: 2 -5 años
♠ Debilidad muscular: caídas frecuentesdificultad para subir y bajar escaleras
♠ Signo o maniobra de Gower presente
♠ Marcha anadeante
♠ Pseudo-hipertrofia de las pantorrillas

DISTROFIA MUSCULAR DE DUCHENNE -BECKER
EVOLUCIÓN:
DMD: Inicio 2 A 5 AÑOS, 12 años silla de rueda,
generalmente mueren alrededor de los 20 años.
DMB: Inicio 5 años, generalmente no dejan de caminar.

DIAGNÓSTICO:
♠ Electromiografía
♠ Creatin-kinasa
♠ Biopsia muscular
♠ Estudio de la distrofina por inmunohistoquimica
♠ Demostración de la deleción por PCR
DISTROFIA MUSCULAR DE DUCHENNE -BECKER

TRATAMIENTO:
Cuidados generales:
Fisioterapia
Ortopedia
Cirugía
DISTROFIA MUSCULAR DE DUCHENNE -BECKER

DIFERENCIACIÓN SEXUAL

DIFERENCIACIÓN SEXUAL
DIFERENCIACIÓN SEXUAL:
♠ Determinación del sexo cromosómico
♠ Determinación sexual de las gónadas
♠ Diferenciación sexual fenotipica
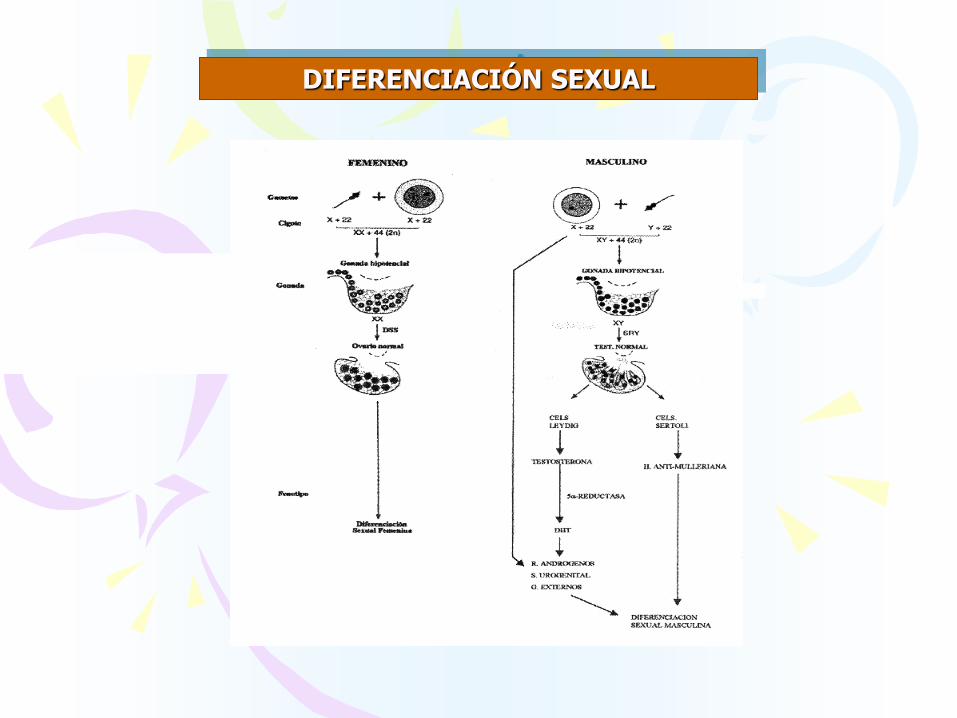
DIFERENCIACIÓN SEXUAL

DIFERENCIACIÓN DE GENITALES INTERNOS

DIFERENCIACIÓN SEXUAL
GENITALES NO DIFERENCIADOS:
Define una variedad de condiciones que afectan la
formación de los genitales del feto durante las primeras
etapas del embarazo
CLASIFICACIÓN DE GENITALES NO
DIFERENCIADOS:
♠ Hermafroditismo verdadero
♠ Pseudohermafroditismo
♠ Disgenesia gonadal

INSENSIBILIDAD ANDROGÉNICA
DEFINICION:
Varones genéticos (XY) con diferenciación de
testículos, que desarrollan genitales externos
femeninos
INCIDENCIA:
1:20.000 nacidos vivos

INSENSIBILIDAD ANDROGÉNICA RECESIVO LIGADO AL X
MECANISMO DE TRANSMISION:
Recesivo ligado al X
UBICACIÓN GÉNICA:
El locus del gen del receptor androgénico (AR) seencuentra ubicado en el cromosoma X

INSENSIBILIDAD ANDROGÉNICA
TIPOS:
♣ Insensibilidad androgénica completa
♣ Insensibilidad androgénica parcial
MANIFESTACIONES CLÍNICAS:
Cariotipo XY
Ausencia de útero, trompas y ovarios
Amenorrea
Genitales externos femeninos
Vagina en fondo de saco

INSENSIBILIDAD ANDROGÉNICA
DIAGNÓSTICO:
Ecograma pélvico
IRM
Laparoscopia
Estimulación con Testosterona
TRATAMIENTO:
Quirúrgico
Hormonal
Psicológico

HEMOFILIA RECESIVA LIGADA AL X

DEFINICION:
La hemofilia se caracteriza por ser un trastornogenético en el cual la sangre no coagula de maneranormal debido a una deficiencia del factor VIII(hemofilia A) y IX (hemofilia B)
INCIDENCIA:
HA: 1/10.000 varones
HB: 15-20/1.000.000
HEMOFILIA

MECANISMO DE TRANSMISION:
Se hereda como un rasgo recesivoligado al X
UBICACIÓN GÉNICA:
HA: Cromosoma Xq28HB: Cromosoma Xq27
HEMOFILIA

MANIFESTACIONES CLÍNICAS:
♠ Tiempo prolongado de sangría
♠ Hemartrosis
♠ Hemorragia intracraneal
TIPOS:
♠ Leve: >5 - <40% del factor VIII normal
♠ Moderada: 1 al 5%
♠ Severa: menos del <1%
HEMOFILIA

HEMOFILIA
EVOLUCIÓN:
♠ Deformidades articulares
♠ Riesgo aumentado para HIV,
hepatitis B
♠ Hemorragia intracraneal

DIAGNÓSTICO:
♠ Clínica
♠ TPT: normal
♠ Tiempo de protrombina: alargado
♠ Tiempo de sangrado: anormal
♠ Niveles normales de fibrinógeno
♠ Actividad baja del factor VIII y IX sérico
HEMOFILIA

DIAGNÓSTICO:
♠ Antecedentes familiares
♠ Análisis de ligamiento (RFLP)
♠ Secuenciación de los genes de los
factores VIII Y IX
HEMOFILIA

TRATAMIENTO:
♠ Reemplazo del factor faltante
♠ Desmopresina (DDAVP)
♠Vacunación contra la hepatitis B
HEMOFILIA

HEMOFILIA

REPÚBLICA BOLIVARIANA DE VENEZUELALA UNIVERSIDAD DEL ZULIA
FACULTAD DE MEDICINAPROGRAMA DE GENETICA CLINICA
Ana BrachoEspecialista en Pediatría
MgSc en Genética Médica
HERENCIA DOMINANTE
LIGADA AL X
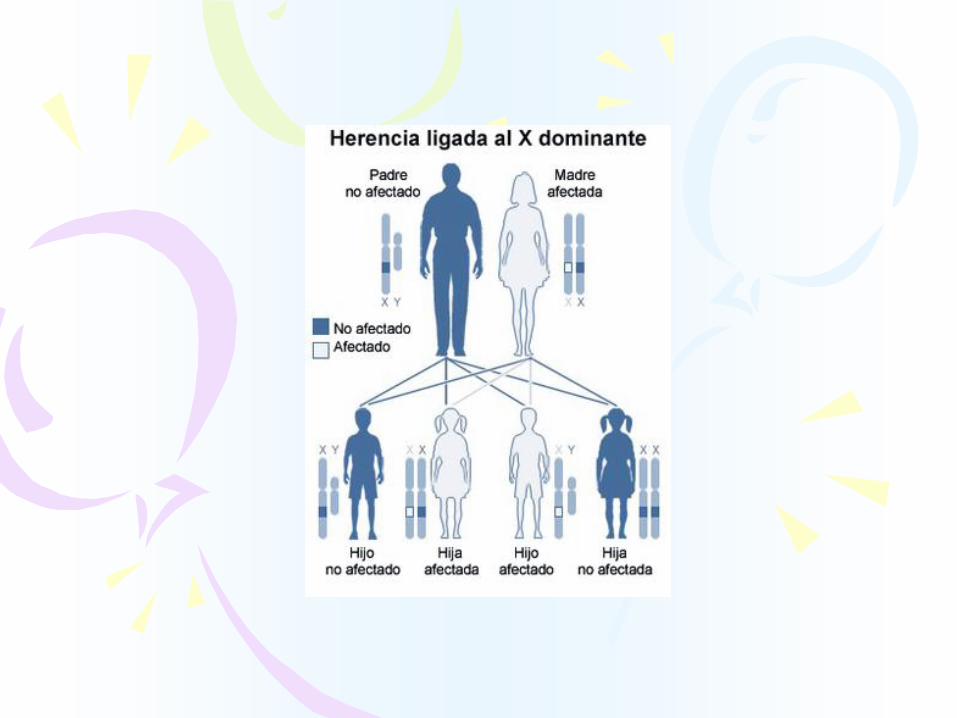


Segunda causa de deficiencia mental hereditaria,
afecta principalmente a varones.
INCIDENCIA
Sexo masculino: 1:4.000 - 6.000.
Sexo femenino: 1:7.000 - 12.000.
Portadoras: 1:250 mujeres.
SINDROME X FRAGIL

MECANISMO DE TRANSMISION
Dominante ligada al X
Penetrancia: incompleta35% mujeres80% hombres
UBICACIÓN GENICA
Xq27.3
Gen Fragile X Mental Retardation (FMR1)Proteína: FMRP

ETIOLOGIA
• Expansión CGG en el gen
• Individuos afectados: expansión > 230, gen metilado.
• Portadores: 55 y 230 repeticiones (premutación) gen no metilado.
• 15-20% SXF: mosaicos, cierta cantidad de FMRP
• <1%: deleciones

CLÍNICA
Retraso mental: moderado en los varones y leve en lasmujeres
Mujeres afectadas, heterocigotas para la mutación se handescrito rasgos faciales similares a los varones, aunque másleves
En varones afectados la clínica varía en relación a la edad yel estadío puberal


CLINICA:
• Retraso mental
• Hiperactividad
• Problemas de atención
• Contacto visual escaso
• Hiperlaxitud articular
• Cara alargada
• mentón prominente
• Orejas prominentes
• Macroorquidismo

DIAGNÓSTICO
Estudio citogenético: sitio frágil en el locus Xq27.3
Técnicas moleculares: Southern blot o PCR.Cuantifica el número de trinucleótidos CGG en elADN
99% de los casos son detectados con estas técnicas
<1% son deleciones o mutaciones puntuales dentrodel gen.
El test molecular también permite determinar elestado de metilación del gen FMR1.


TRATAMIENTO:
No existe tratamiento curativo para el SXF.
Manejo terapeútico:
1) tratamiento farmacológico de los problemasde comportamiento que afectan lainteracción social
2) intervención educativa individualizadadirigida a mejorar la capacidad deaprendizaje, terapia ocupacional, etc


ENFERMEDAD DE FABRY
Panetnica
Incidencia en varones 1: 40.000 - 60.000
Incidencia: 1/117.000 nacidos vivos
Expectativa de vida: 48-53 años

ENFERMEDAD DE FABRY

ENFERMEDAD DE FABRY

ENFERMEDAD DE FABRY
GLA
Ubicación: Xq21.3–q23/24
Gen: αGalactosidasa ( GLA)
Metabolito: Globotriaosilceramida (GL3)
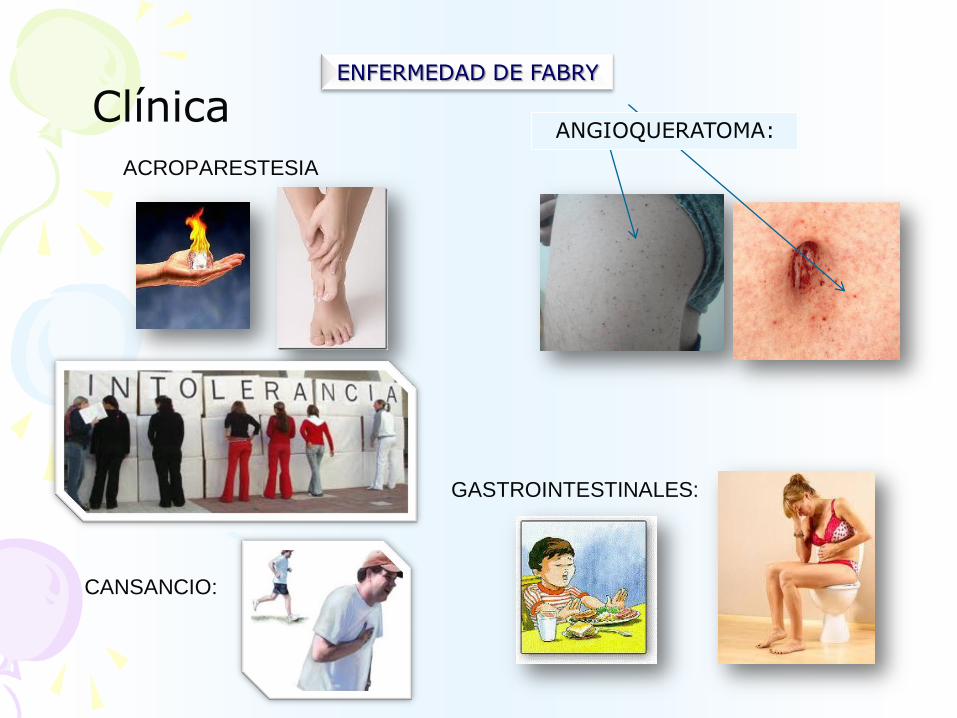
ENFERMEDAD DE FABRY
ANGIOQUERATOMA:Clínica
ACROPARESTESIA
GASTROINTESTINALES:
CANSANCIO:

ENFERMEDAD DE FABRY
RENAL: CORAZON:
SISTEMA NERVIOSO
OJOS:
OIDOS

ENFERMEDAD DE FABRY
Diagnostico
1. Clínico: Historia clínica genética.
2. Bioquímico: actividad α galactosidasa A
3. Molecular: secuenciación del gen completo.

ENFERMEDAD DE FABRY
Equipo multidiciplinario:
Cardiología
Neurología
Oftalmológia
Dermatología
Nefrología
Audiometría

Diagnostico Prenatal:
Actividad de α-GAL-A en:
Líquido amniótico ó
Vellosidades coriónicas.
ENFERMEDAD DE FABRY

ENFERMEDAD DE FABRY
Tratamiento:
Para el dolor: carbamazepina y fenitoína.
Insuficiencia renal: diálisis o trasplante renal.
Terapia de reemplazo enzimático:

ENFERMEDAD DE FABRY
Terapia de reemplazo enzimático:
Agalsidasa Beta
Agalsidasa Alpha
Dosis: 1mg/Kg de peso corporal.
C/15 días infusión EV





DEFINICION:
Síndrome caracterizado por la presencia demúltiples malformaciones orofaciales y digitalesdesde el nacimiento
INCIDENCIA:
1:250.000
SINDROME OROFACIO-DIGITAL tipo I

SINDROME OROFACIO-DIGITAL tipo I
MECANISMO DE TRANSMISION:
DOMINANTE ligada al XLetal para el varón
GEN:
CXORF5
UBICACIÓN GÉNICA:
Xp22.3-p22.2

MANIFESTACIONES CLÍNICAS
Orales
Lengua lobulada-LENGUA Frenillo corto
Suelo de la boca
-ENCIAS Frenillos anormales y supernumerarios.
Hendido-PALADAR Bóveda alta con puentes laterales.
Supernumerarios o ausencia de piezas -DIENTES dentarias.
Malposición dentaria.
-LABIOS Hendidos o muesca del labio superior en sulínea media.

-Ojos HipertelorismoDistopia cantorum
-Nariz Hipoplasia de los cartílagos alaresfiltrum corto
-Mejillas Hipoplasia y aplanamiento de maxilar superior.
MANIFESTACIONES CLÍNICAS
Faciales

PolidactiliaSindactilia
-Dedos CampodactiliaClinodactiliaBraquidactilia
-Óseos Posible osteoporosis.
MANIFESTACIONES CLÍNICAS
Digitales

-Cráneo Frente protuberante
-Piel Seca o seborreicaAlopecia
Retraso mentalHidrocefalia con quistes poroencefálicos
-Neurológico Temblor familiarHidranencefaliaAgenesia del cuerpo calloso
Riñón poliquístico, Hidronefrosis-Órganos internos Poliquistosis hepática
Poliquitosis pancreática
MANIFESTACIONES CLÍNICAS
Otras
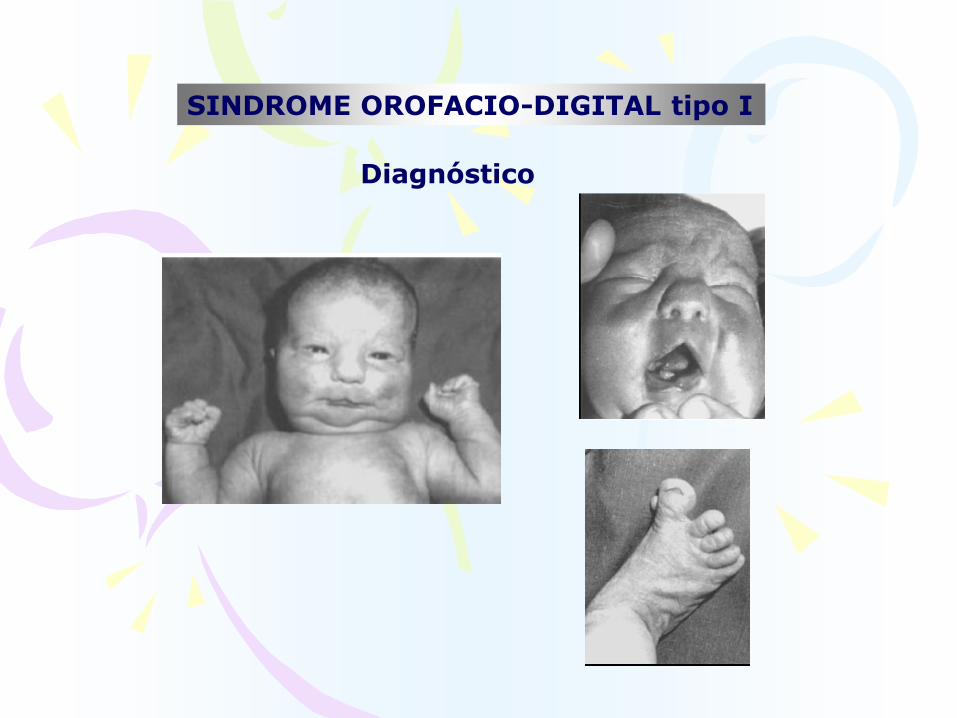
SINDROME OROFACIO-DIGITAL tipo I
Diagnóstico

SINDROME OROFACIO-DIGITAL tipo I
Tratamiento de las anomalías asosiadas al sindrome
Cirugía maxilofacial
Ortodoncia
Terapia del lenguaje
Nefrología
Asesoramiento genético
Tratamiento
