the endocannabinoid anandamide inhibits potassium conductance in rat cortical astrocytes
TRANSCRIPT

The Endocannabinoid Anandamide Inhibits PotassiumConductance in Rat Cortical Astrocytes
M. VIGNALI,1 V. BENFENATI,1 M. CAPRINI,1 M. ANDEROVA,2 M. NOBILE,3 AND S. FERRONI1*1Department of Human and General Physiology, University of Bologna, 40127 Bologna, Italy2Department of Cellular Neurophysiology, Institute of Experimental Medicine, Videnska 1083, Prague 4, 14220 Czech Republic3Institute of Biophysics, National Research Council, 16100 Genoa, Italy
KEY WORDScortical astroglia; potassium conductance; endocannabi-noids; NG2 glia; brain homeostasis
ABSTRACTEndocannabinoids are a family of endogenous signalingmolecules that modulate neuronal excitability in the centralnervous system (CNS) by interacting with cannabinoid(CB) receptors. In spite of the evidence that astroglial cellsalso possess CB receptors, there is no information on therole of endocannabinoids in regulating CNS functionthrough the modulation of ion channel-mediated homeo-static mechanisms in astroglial cells. We provide electro-physiological evidence that the two brain endocannabinoidsanandamide (AEA) and 2-arachidonylglycerol (2-AG) mark-edly depress outward conductance mediated by delayed out-ward rectifier potassium current (IKDR) in primary culturedrat cortical astrocytes. Pharmacological experiments sug-gest that the effect of AEA does not result from the activa-tion of known CB receptors. Moreover, neither the produc-tion of AEA metabolites nor variations in free cytosolic cal-cium are involved in the negative modulation of IKDR. Weshow that the action of AEA is mediated by its interactionwith the extracellular leaflet of the plasma membrane. Sim-ilar experiments performed in situ in cortical slices indicatethat AEA downregulates IKDR in complex and passiveastroglial cells. Moreover, IKDR is also inhibited by AEA inNG2 glia. Collectively, these results support the notion thatendocannabinoids may exert their modulation of CNS func-tion via the regulation of homeostatic function of the astro-glial syncytium mediated by ion channel activity. VVC 2008
Wiley-Liss, Inc.
INTRODUCTION
Astroglial cells, the most numerous cell population ofthe central nervous system (CNS), play a pivotal role inthe regulation of the neuronal network not only by pro-viding energy substrates, but also by maintaining waterand ion homeostasis and contributing to regulate neuro-transmitter dynamics in the perineuronal space (forreviews, see Simard and Nedergaard, 2004; Volterra andMeldolesi, 2005). Although astrocytes are nonexcitablecells, they are equipped with a variety of voltage- andligand-gated ion channels both in situ and in vitro(Verkhratsky and Steinhauser, 2000). Astroglial cells insitu are endowed with a large K1 conductance owing tothe presence of several types of voltage-gated and ohmic
channels that are open over a large range of transmem-brane voltages (Bordey and Sontheimer, 2000). Analysisof the relative contribution of each K1 channel to totalK1 conductance has permitted the identification of atleast two subtypes of astroglial cells, termed passive andcomplex astrocytes (Wallraff et al., 2004). Specifically,whereas passive astrocytes display a background K1
conductance with no time or voltage dependencies, com-plex cells possess several voltage-gated K1 channelsincluding outward transient and delayed rectifier cur-rents (IKA and IKDR, respectively) and inward rectifierK1 conductance (IKir). The functional relevance of thesedifferences in K1 channel expression is, however, stillunclear (for review, see Walz, 2000). A dynamic regula-tion of certain subtypes of K1 channels has beendescribed in relation with ontogenic period (Bordey andSontheimer, 1997; Kressin et al., 1995) and in responseto various models of traumatic brain injuries (Anderovaet al., 2004; Bordey et al., 2001; D’Ambrosio et al., 1999).Moreover, alterations in the repertoire of K1 channelshave been reported in astroglial cells of epileptic brain,reinforcing the view that the dysregulation of astrocyticK1 permeability might have a pathogenetic role in severeneurological diseases (for review, see Seifert et al., 2006).The cellular mechanisms underlying the dynamicchanges in K1 permeability in astroglial cells are, how-ever, still elusive, and there is very little informationwhether modifications in extracellular signaling mole-cules contribute substantially to such regulation.
Endocannabinoids are a family of endogenous mole-cules that act on at least two G-protein-coupled metabo-tropic receptors, which were initially identified as bind-ing sites of the psychoactive agent D9tetrahydrocanna-binol of the Cannabis sativa and thus were calledcannabinoid receptors (CB) (for review, see Pertwee andRoss, 2002). In the CNS, the two endogenous moleculesanandamide (AEA) and 2-arachidonylglycerol (2-AG)
Grant sponsor: Mariani Foundation of Milan; Grant number: R-07-66; Grantsponsor: Academy of Sciences of the Czech Republic; Grant number:AVOZ50390512; Grant sponsor: Grant Agency of the Czech Republic; Grant num-bers: 305/06/1316 and 305/06/1464; Grant sponsor: Ministry of Education, Youthand Sports of the Czech Republic; Grant number: LC554; Grant sponsor: ItalianMinistry of University and Research.
*Correspondence to: Stefano Ferroni, Department of Human and General Physiol-ogy, via San Donato 19/2, 40127 Bologna (BO), Italy. E-mail: [email protected]
Received 16 May 2008; Accepted 6 October 2008
DOI 10.1002/glia.20807
Published online 20 November 2008 in Wiley InterScience (www.interscience.wiley.com).
GLIA 57:791–806 (2009)
VVC 2008 Wiley-Liss, Inc.

bind to two CB receptors called CB1 and CB2 that dis-play distinct pharmacological profiles (Felder et al.,1993; Vogel et al., 1993). In vitro, these two types ofreceptors are differentially expressed in glial cells(Bouaboula et al., 1995; Carrier et al., 2004; Facchinettiet al., 2003; Molina-Holgado et al., 2002a,b).
It is well recognized that the acute effects of endocan-nabinoids are associated with their action on neuronalcells (for review, see Chevaleyre et al., 2006). In neu-rons, the short-term activation of CB1 receptors modu-lates synaptic transmission by regulating presynapticion channels to depress neurotransmitter release (forreview, see Schlicker and Kathmann, 2001). More sus-tained increase in the production of endocannabinoidsand prolonged activation of CB receptors have beenassociated with neuroprotection following brain injuries(Hansen et al., 2001; Marsicano et al., 2003; Mechoulamand Shohami, 2007), an effect in which glial cells mightalso be involved (Sheng et al., 2005; Stella, 2004).
Astroglial cells in situ express the CB1 receptor (Mol-drich and Wenger, 2000; Rodriguez et al., 2001; Salio etal., 2002). Accumulating evidence indicate that thesecells are deeply involved in the physiology of the endo-cannabinoid system, because they can synthesize,release, reuptake, and inactivate endocannabinoids,thereby potentially contributing to regulate endocanna-binoid levels in the CNS (Beltramo et al., 1997; DiMarzo et al., 1994; Romero et al., 2002; Walter andStella, 2003; Walter et al., 2002, 2004). The mechanisticprocesses whereby endocannabinoids regulate the astro-glial activities are beginning to be unraveled. AEA wasreported to inhibit calcium (Ca21) wave propagationwithin the astroglial syncytium of the striatum throughblockage of gap junctional channels (Venance et al.,1995). In vitro stimulation of the CB1 receptor preservesthe viability of astroglia subjected to apoptotic stimuli(Gomez Del Pulgar et al., 2002), and inhibits nitric oxiderelease in a model of astrogliosis (Molina-Holgado et al.,2002a). Finally, AEA was reported to depress the so-dium-dependent uptake of glutamate in cultured astro-cytes (Shivachar, 2007) and to stimulate glycine trans-port in glioma cells (Pearlman et al., 2003). However,whether endocannabinoids can also regulate astroglialfunctions through the modulation of ion channelsinvolved in homeostatic mechanisms is still unknown.
In this study, we addressed the question whether AEAcan alter K1 conductance of cortical astrocytes in vitro,a condition that mimics several features of in situ astro-gliosis, and in situ by using cortical slices. We show thatin primary cultured rat cortical astrocytes, low micromo-lar concentrations of AEA and 2-AG promote a strongreduction of delayed rectifier outward K1 current. Phar-macological manipulations indicate that AEA action isindependent of CB1 stimulation. Importantly, AEA alsodepressed outward K1 currents in different subtypes ofastrocytes and in NG2 glial cells in situ. The observationthat endocannabinoids can regulate astroglial ion chan-nels might provide a mechanistic clue to explain some ofthe non-neuronal effects of the endocannabinoid systemin vivo.
MATERIALS AND METHODSAstrocyte Culture
The experiments were performed according to Italianand international laws on the protection of laboratoryanimals, with the approval of the local bioethical com-mittee and under the supervision of the veterinary com-mission for animal care and comfort of the University ofBologna. Every effort was made to minimize the numberof animals used and their suffering. Primary cultures ofpure cortical rat astrocytes were prepared as previouslydescribed (Ferroni et al., 1995) from neonatal rats(Sprague–Dawley, Charles River, Italy). Briefly, neonatalcerebral occipital cortices devoid of meninges weregently triturated, filtered with a 70-lm cell strainer(Falcon, BD Bioscience, Bedford, MA) and placed in cellculture flasks containing Dulbecco’s Modified Eagle-glu-tamax medium with 15% fetal bovine serum (FBS) andpenicillin/streptomycin (100 U/mL and 100 lg/mL,respectively). All products were purchased from Gibco-Invitrogen, Milan, Italy. Culture flasks were maintainedfor 2–4 weeks in an incubator with 5% CO2 and ahumidified atmosphere. Before electrophysiologicalexperiments, astroglial cells were enzymatically dis-persed (trypsin-EDTA 0.05%, Gibco-Invitrogen) in Petridishes (35-mm diameter) at a density of 1 3 104 cellsper dish and maintained in 15% FBS-containing culturemedium. Electrophysiological experiments were carriedout 3–6 days later.
Preparation of Brain Slices
All procedures involving the use of animals wereapproved by the local ethical review committee. Rats(P10-12, Wistar) were anesthetized with isoflurane anddecapitated. Brains were quickly dissected and placed incold (4�C) artificial cerebrospinal fluid (ACSF) and gluedwith 3 M Vetbound tissue adhesive (World PrecisionInstruments, FL) to a metal plate. Coronal, 200-lmthick slices for patch-clamp recordings were preparedusing a HM 650 V microtome with a vibrating blade(MICROM International GmbH, Waldorf, Germany). Sli-ces were kept in ACSF at room temperature (22–25�C)for up to 6 h. The solution was continuously gassed witha mixture of 95% O2 and 5% CO2 to maintain a final pHof 7.4.
Electrophysiological RecordingsIn Vitro and In Situ
Petri dishes containing the cultured astrocytes wereplaced on the stage of an inverted microscope (NikonDiaphot, Nikon Italy, Firenze, Italy). Plasma membranecurrents were measured at room temperature (22–25�C)with the whole-cell configuration of the patch clamptechnique (Hamill et al., 1981). Patch pipettes were pre-pared from thin-walled borosilicate glass capillaries tohave a tip resistance of 2–4 MX when filled with the
792 VIGNALI ET AL.
GLIA

standard internal solution. Membrane currents wereamplified (EPC-7, List Electronic Darmstadt, Germany),low-pass filtered at 2 kHz (23 dB), and acquired at asample rate of 5 kHz on a computer for off-line analysis(pClamp 6, Axon Instrument, Foster City, CA and Origin6.0, MicroCal, Northampton, MA). During recording,access resistance was continuously monitored and maxi-mally corrected up to 70% with the analog circuit of theamplifier.
The membrane properties of glial cells in situ werestudied in neocortical slices of 10- to 12-day-old (P10-P12) rats with the whole-cell patch-clamp technique aspreviously described (Anderova et al., 2004). The sliceswere placed in a chamber mounted on the stage of a flu-orescent microscope (Axioskope FX, Carl Zeiss, Germany)and fixed using a U-shaped platinum wire with a grid ofnylon threads. The slices were continuously perfusedwith ACSF at room temperature. Cells were approachedby the patch electrode using INFRAPATCH system(Luigs & Neumann, Ratingen, Germany). To allow post-experiment identification of the recorded cells, the elec-trodes were filled with the internal solution supple-mented with 0.1 mg/mL Alexa Fluor-hydrazide-488 or594 (Molecular Probes, Eugene, OR). Current signalswere amplified with an EPC-10 amplifier (HEKA Elektro-nik, Lambrecht/Pfalz, Germany), low-pass-filtered at 3kHz, and sampled at 5 kHz by an interface connected toan AT-compatible computer system, which also served asstimulus generator. Data acquisition, storage, and analy-sis were performed with PatchMaster/Fitmaster (HEKAElektronik, Lambrecht/Pfalz, Germany). Current tracesobtained in vitro and in situ were not leak subtracted.
Solutions and Chemicals
Salts and other chemicals were of the highest puritygrade (Sigma-Aldrich, St. Louis, MO). For the electro-physiological experiments performed in vitro, the perfus-ing solution contained (in mM): 140 NaCl, 4 KCl, 2CaCl2, 2 MgCl2, 5 HEPES, and 5 glucose. The pH wasadjusted to 7.4 with NaOH and the osmolarity adjustedto �315 mOsm with mannitol. The intracellular (pipette)solution was composed of (in mM): 144 KCl, 2 MgCl2, 10TES, and 5 EGTA, with the pH adjusted to 7.3 withKOH and the osmolarity to �290 mOsm. The differentexternal solutions containing pharmacologic agents wereapplied with a gravity-driven local perfusion system at aflow rate of �200 lL/min positioned within �100 lm ofthe recorded cell. During electrophysiological recordingscarried out in situ, the slices were perfused with ACSFcomposed of (in mM): 122 NaCl, 3 KCl, 1.5 CaCl2, 1.3MgCl2, 1.25 Na2HPO4, 28 NaHCO3,10 glucose; the pHwas adjusted to 7.4 by gassing with a mixture of 95% O2
and 5% CO2. The pipette solution contained (in mM):130 KCl, 0.5 CaCl2, 2 MgCl2, 5 EGTA, and 10 HEPES.The pH was adjusted to 7.2 with KOH. Pharmacologicalagents were dissolved in ACSF and applied to therecorded cell using a pressurized perfusion system(Automate Scientific, Berkley, CA).
Aliquots of AEA were prepared in pure ethanol andstored at 280�C for no longer than 1 month. Perfusingsolutions containing AEA (or methanandamide, arachi-donic acid (AA), 2-arachidonyl glycerol) were used for upto 2 h. Dimethyl sulfoxide (DMSO) was used to preparealiquots of stock solutions of WIN55.212-2 (10 mM,Sigma-Aldrich, Milan, Italy), AA (10 mM, Sigma-Aldrich), methyl-arachidonyl-fluore-phosphonate (5 mM,Sigma), AM251 (10 mM, Tocris, Bristol, United King-dom), and 2-arachidonyl glycerol (10 mM, Tocris). Ali-quots of methanandamide (10 mM, Sigma-Aldrich) wereprepared in pure ethanol. Stock solution (1 mM) of rimo-nabant (SR141716A) (a kind gift of Prof. Santi Spampi-nato, Dept. of Pharmacology, University of Bologna) wasprepared in DMSO. Ethanol and DMSO concentrationsused to dissolve the various agents were tested in con-trol experiments and had no effects on evoked currents.Solutions containing pertussis toxin, methyl-b-cyclodex-trin, and bovine serum albumin (BSA) (all from Sigma-Aldrich) were prepared by dissolving the final amountsdirectly in culture medium or saline solutions.
Immunohistochemistry
For immunohistochemical analysis of the rat cortex,animals were anesthetized with isoflurane and decapi-tated. Brains were dissected out and postfixed in parafor-maldehyde solution (4% paraformaldehyde in 0.2 M phos-phate buffer pH 7.4, PB) overnight, then placed in 30%sucrose in 0.01 M phosphate-buffered saline (PBS) andallowed to sink for cryoprotection. Coronal slices, 30-lmthick, were prepared using a microtome (HM 400, MicromInt. GmbH, Waldorf, Germany). For immunohistochemicalidentification of recorded cells, coronal slices, 200-lmthick, were fixed in paraformaldehyde solution for 1–2 hand then transferred to PBS. To identify cells as astro-cytes, antibodies against glial fibrillary acidic protein(GFAP) and the b-subunit of calcium binding protein(S100b) were used. The slices were incubated overnight at4�C with a mouse monoclonal antibody directed againstS100b (Sigma-Aldrich), diluted 1:200 in PBS containing5% ChemiBLOCKER (Chemicon, Temicula, CA) and 0.5%Triton X-100, followed by incubation with the secondaryantibody, goat anti-mouse IgG conjugated with eitherAlexa-Fluor 488 or 594, depending on the type of AlexaFluor hydrazide that was used for cell identification dur-ing patch-clamp experiments (Molecular Probes). The sli-ces were incubated for 1 h at room temperature with amouse monoclonal antibody directed against GFAP conju-gated with Cy3 (Sigma-Aldrich), diluted 1:200 in PBScontaining 5% ChemiBLOCKER and 0.5% Triton X-100.To identify NG2 glia, we used an antibody directedagainst NG2-proteoglycan (Chemicon, Temecula, CA),diluted 1:400 in PBS containing 5% ChemiBLOCKERand 0.5% Triton X-100. Overnight incubation with theprimary antibody at 4�C was followed by incubation withthe secondary antibody, goat anti-rabbit IgG conjugatedwith Alexa-Fluor 660 (Molecular Probes). After immuno-staining, the slices were mounted using Vectashield
793EAE MODULATION OF ASTROCYTE K+ CHANNELS
GLIA

mounting medium (Vector Laboratories, Burlingame, CA)and examined using a LEICA TCS SP system spectralconfocal microscope equipped with an Ar/HeNe laser.
Protein Immunoblotting
Protein analysis was carried out by western immuno-blotting. Cultured astrocytes, microglial and neuronal cellswere collected and lysed as previously described (Benfenatiet al., 2007). The protein concentration was determined inthe supernatant (Bio-Rad Laboratories, Hercules, CA).Soluble materials (15 lg) from the total protein contentwere separated on a 12% sodium-dodecyl sulfate-polyacryl-amide gel and electrotransferred onto a nitrocellulosemembrane (Bio-Rad laboratories). The nitrocellulose wasblocked in 5% fat-free milk in PBS containing 0.05%Tween 20 (PBST) and probed overnight with primary anti-bodies (both diluted 1:500, Chemicon, Temecula, CA)directed against the CB1 and CB2 receptors. The mem-branes were subsequently washed with PBST and incu-bated with horseradish peroxidase-conjugated anti-rabbitIgG secondary antibody (Sigma-Aldrich) and developedwith the enhancing chemiluminescence detection system(ECL-Plus, Amersham Biosciences, Milan, Italy).
Data Analysis and Statistics
Only those astrocytes in which access resistance didnot change of more than 20% during the experimentwere considered for analysis. Histograms represent thequantity of charge (Q) movement through IKDR afterAEA application (QAEA) normalized to the charge quan-tity under control conditions (QCTRL). Charge quantitywas determined from the integral of the current tracesevoked by a 100-mV voltage step of 300-ms durationfrom a holding potential of 260 mV to 140 mV. The con-centration–response relationship for the effect of AEAwas derived from fitting the data points to the Hillequation: (% inhibition)/(% inhibition)max 5 [C]b/([C]b 1EC50
b), where (% inhibition)/(% inhibition)max is the rateof current inhibition after exposure to a given concentra-tion (C) of AEA, IC50 is the half-maximal inhibitory con-centration, and b is the apparent Hill coefficient.
Data are expressed as mean 6 standard deviation(SD) of several cells (n) under the various conditions.Statistics were performed using one-way analysis of var-iance followed by Bonferroni posthoc test when appropri-ate. A P value <0.05 was considered as the level ofsignificance.
RESULTSAEA Inhibits Delayed Rectifier Outward K1
Current in Cultured Rat Cortical Astrocytes
In previous studies, we showed that in rat corticalastrocytes primary cultured for 3–4 weeks the majorityof voltage-gated K1 conductance is carried by delayed
rectifier K1 current (IKDR) (Ferroni et al., 1995, 2003).Astrocytes were stimulated from a holding potential of260 mV with a protocol (inset in Fig. 1A) consisting of arepetitive hyperpolarizing step to 2120 mV for 500 msfollowed by a slowly depolarizing ramp (180 mV/500 ms)to 160 mV. In physiological extracellular solution rampcurrents were stable over a 20-min recording period.Membrane resistance decreased at potentials more posi-tive than 240 mV owing to the activation of IKDR (Fig.1A). Extracellular application of 1 lM AEA induced arobust decrease of ramp currents at potentials morepositive than 240 mV, but did not affect membraneresistance at more negative potentials. Ramp currentsmeasured at 140 displayed a mean current depressionof 75% 6 4% (mean 6 SD, n 5 28). The onset of currentblockage at 140 mV occurred within 30 s of starting theperfusion and developed slowly, reaching maximal inhi-bition after about 10 min (Fig. 1B; time of half-inhibi-tion, 219 6 20 s). Washout with standard physiologicalsolution for up to 5–10 min did not produce any signifi-cant current recovery. AEA inhibition did not alter sig-nificantly the astrocyte resting membrane potential(Vrest) measured in current clamp mode as upon controlconditions Vrest was 238 6 5 mV, (n 5 56) and follow-ing maximal AEA inhibition Vrest was 239 6 6 mV,(n 5 44).
It was recently demonstrated that membrane lipids,including AEA, can convert some noninactivating sub-types of voltage-gated K1 channels (Kv) that underlieIKDR into rapidly inactivating channels (Oliver et al.,2004). To investigate whether the AEA-induced depres-sion of the ramp current could be partially ascribed tosuch a mechanism, we explored the AEA action byapplying a depolarizing voltage step protocol (Fig. 1C,inset). The comparison of K1 currents evoked prior toAEA application (ctrl), and the residual currents aftermaximal blockage by AEA indicates that AEA did notchange the current kinetics (Fig. 1C,D). The inhibitoryaction of AEA was voltage independent as the rate ofcurrent depression was not altered by the amplitude ofthe voltage stimulation (percentage of blockage at 0 mV5 78% 6 4%; at 160 mV 5 75% 6 3%, n 5 18). Fur-thermore, the time period required to reach the maximaldiminution was not modified by lowering the stimulationfrequency, suggesting that AEA-mediated blockage ofIKDR does not need channel opening to occur (Fig. 1E).To address the potency of AEA, experiments were car-ried out by applying AEA for 10 min at various concen-trations and constructing the dose–response curve ofcurrent inhibition at 140 mV. The best fit of the datapoints with Hill equation indicates that half-maximalinhibition (IC50) of IKDR occurred at �300 nM and theHill coefficient was 0.7 (Fig. 1F).
AEA-Induced Inhibition of IKDR Does Not Involvethe CB1 Receptor
Because there is some evidence that cultured astro-cytes express CB receptors that could mediate the
794 VIGNALI ET AL.
GLIA
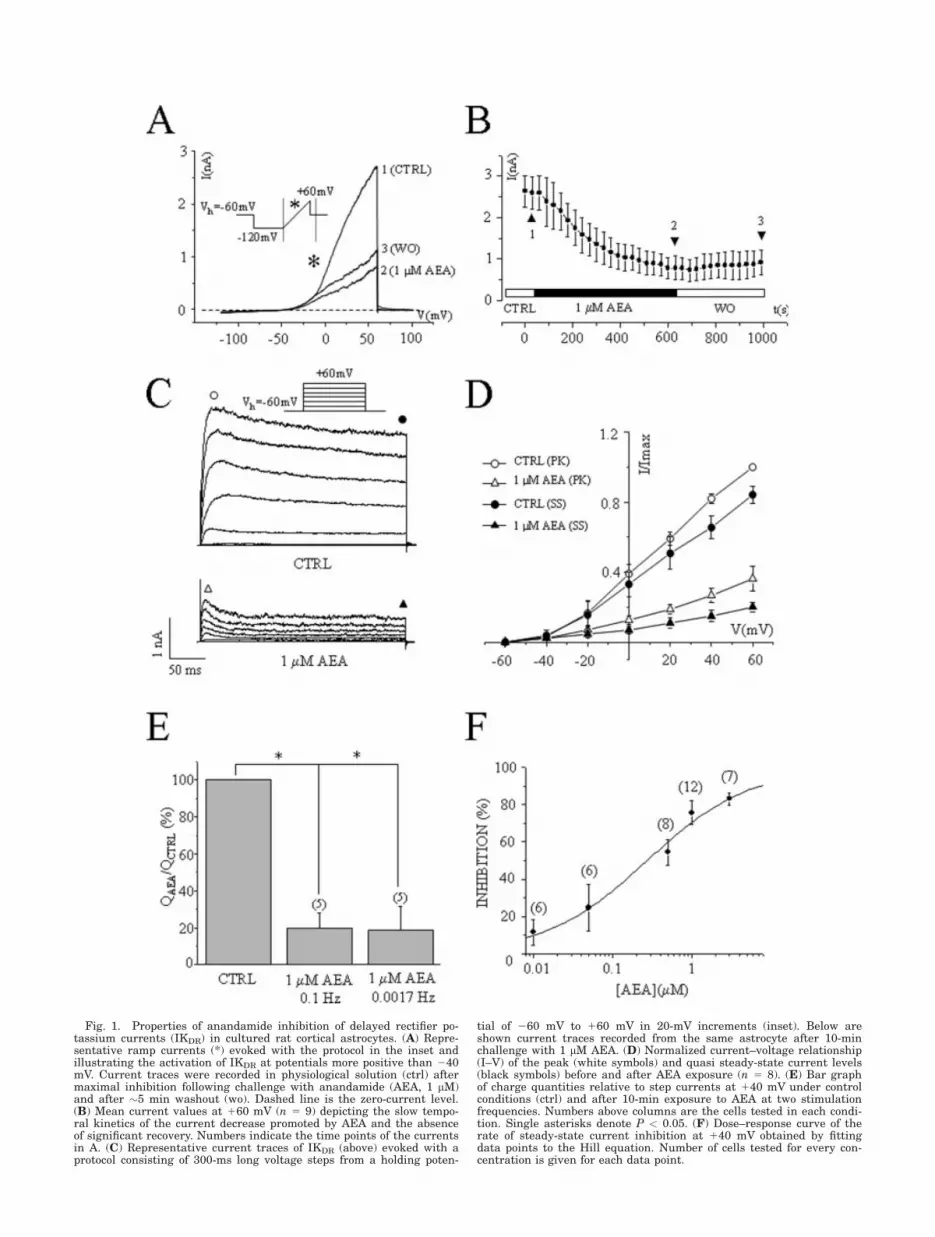
Fig. 1. Properties of anandamide inhibition of delayed rectifier po-tassium currents (IKDR) in cultured rat cortical astrocytes. (A) Repre-sentative ramp currents (*) evoked with the protocol in the inset andillustrating the activation of IKDR at potentials more positive than 240mV. Current traces were recorded in physiological solution (ctrl) aftermaximal inhibition following challenge with anandamide (AEA, 1 lM)and after �5 min washout (wo). Dashed line is the zero-current level.(B) Mean current values at 160 mV (n 5 9) depicting the slow tempo-ral kinetics of the current decrease promoted by AEA and the absenceof significant recovery. Numbers indicate the time points of the currentsin A. (C) Representative current traces of IKDR (above) evoked with aprotocol consisting of 300-ms long voltage steps from a holding poten-
tial of 260 mV to 160 mV in 20-mV increments (inset). Below areshown current traces recorded from the same astrocyte after 10-minchallenge with 1 lM AEA. (D) Normalized current–voltage relationship(I–V) of the peak (white symbols) and quasi steady-state current levels(black symbols) before and after AEA exposure (n 5 8). (E) Bar graphof charge quantities relative to step currents at 140 mV under controlconditions (ctrl) and after 10-min exposure to AEA at two stimulationfrequencies. Numbers above columns are the cells tested in each condi-tion. Single asterisks denote P < 0.05. (F) Dose–response curve of therate of steady-state current inhibition at 140 mV obtained by fittingdata points to the Hill equation. Number of cells tested for every con-centration is given for each data point.

described AEA action, we next sought to evaluate theexpression level of CB1 and CB2 receptor proteins.Whole-cell extracts from pure astroglial (astro) andmixed microglial/astroglial cultures (micro/astro) wereanalyzed by immunoblotting with antibodies directedagainst the CB1 and CB2 receptors. As positive controlfor CB1 receptor, primary cultured hippocampal neurons(neuro) were also probed (Irving et al., 2000). Whereasanti-CB1 antibody recognized two bands at �30 and�60 kDa in all extracts (Fig. 2A), positive immunoreac-tivity for the CB2 receptor (�27 kDa) was detected onlyin mixed glial cultures (Fig. 2B). This latter result is inline with previous work (Carlisle et al., 2002), showingno match between the CB2 band and the molecularweight of �40 kDa extrapolated from the predicted ratCB2 coding sequence. Taken together, these findingsindicate that under our experimental conditions ratcortical astrocytes in vitro express CB1, but not CB2
receptors.We next addressed the question whether the effect of
AEA could be mediated by stimulation of the CB1
receptor. The application of the CB1 synthetic agonistWIN55,212-2 (1 lM) caused a diminution of IKDR, whichwas less than that promoted by AEA (Fig. 2C). Anotherendogenous endocannabinoid, 2-arachidonylglycerol (2-AG), which is structurally related to AEA and acts as afull agonist of the CB1 receptor (Pertwee and Ross,2002), was also tested. Exposure to 2-AG (1 lM) inhib-ited IKDR, even though with lower efficacy comparedwith AEA (Fig. 2D). Next, we examined whether astro-cyte pretreatment with the selective CB1 antagonistAM251 (1 h at room temperature) followed by its co-application with AEA could prevent the current reduc-tion. The data show that the pharmacological inhibitionof CB1 did not avoid or diminish the AEA effect (Fig.2E). A similar result was obtained by using anotherselective antagonist of CB1 receptor, rimonabant(SR141716A), at a concentration (100 nM) able to fullyblock CB1 receptor activity (Jung et al., 1997; Lay et al.,2000) (data not shown).
Because it is well known that the CB1 receptor is aGi/o-protein-coupled receptor, the action of AEA wasinvestigated in astrocytes challenged with the selec-tive Gi/o protein inhibitor pertussis toxin (PTX). Pre-treatment with PTX (500 ng/mL, 16 h at 37�C) nei-ther changed the magnitude of IKDR nor attenuatedthe AEA-mediated current inhibition (Fig. 2F). Thecontrol current density at 140 mV was 54 6 15 pA/pF (n 5 10), whereas following PTX incubation it was51 6 14 pA/pF (n 5 12). This observation also ruledout the possible involvement of Gi/o-coupled receptors,distinct from CB1 and CB2, called non-CB1/-CB2 recep-tors previously described in brain and in culturedastrocytes. These receptors, in fact, mediate theeffects of AEA in a CB1-antagonist-insensitive man-ner, but they are PTX-sensitive (Breivogel et al.,2001; Di Marzo et al., 2000; Sagan et al., 1999; Ven-ance et al., 1995). Taken together, these findings indi-cate that AEA action on IKDR is independent of acti-vation of known CB receptors.
Inhibition of IKDR Is Independent of AEAMetabolism and Ca21 Signaling
There is some evidence that astrocytes express theenzyme fatty acid amide hydrolase (FAAH; Romero et al.,2002), which catalyzes the AEA hydrolysis to the polyun-saturated fatty acid AA and ethanolamine (Shivachar etal., 1996). To explore the possibility that K1 current inhi-bition was due to the formation of by-products of AEAmetabolism, experiments were carried out by pharmaco-logically interfering with AEA hydrolysis using a selec-tive and irreversible inhibitor of FAAH, methyl arachido-nil fluore phosphonate (MAFP). The pretreatment ofastrocytes with MAFP (10 nM, for 1 h at room tempera-ture), followed by its co-application with AEA did not al-ter the efficacy of AEA to depress IKDR (rate of currentinhibition at 140 mV after MAFP: 79% 6 8%, n 5 9),thus strongly suggesting that AEA metabolism is notinvolved. This result was corroborated by the observationthat the nonhydrolyzable AEA analog, methanandamide(methAEA, 1 lM), caused a diminution of IKDR similar tothat exerted by AEA (Fig. 3A). Finally, whereas long ex-posure (15 min) to AEA caused only strong inhibition ofIKDR, astrocyte challenge with one of its metabolic prod-uct, AA, induced a rapid and marked current depressionfollowed by an increase in membrane conductance (Fig.3B), initiating less than 10 min after AA application, pro-duced by activation of AA-sensitive, two-p-domain K1
channels (Ferroni et al., 2003). Collectively, these comple-mentary experiments strongly suggest that AEA actionon IKDR does not need its metabolic degradation.
It is widely recognized that AEA can raise cytoplasmic-free calcium concentration ([Ca21]i) not only by promot-ing Ca21 entry through the TRPV1 (Van der Stelt et al.,2005) and TRPV4 ionotropic receptors (Watanabe et al.,2003), but also by depleting intracellular Ca21 stores(Mombouli et al., 1999; Venance et al., 1997). We did notfind any evidence of the presence of TRPV1 in culturedastrocytes (our unpublished observation), but we recentlydescribed the expression of functional TRPV4 by astrogliain vitro (Benfenati et al., 2007). To explore whether intra-cellular [Ca21]i elevation through Ca21 influx via TRPV4receptors was involved in AEA-mediated effect, experi-ments were performed in the absence of extracellularCa21. Incubation of astrocytes in 0 Ca21 solution (30 minat room temperature) followed by its superfusion in thepresence of AEA did not modify the rate of IKDR depres-sion (Fig. 3C). Furthermore, experiments performedreducing by 10-fold the concentration of the Ca21 chelatorEGTA in the pipette solution (0.5 mM EGTA) to decreaseCa21 buffer capacity, did not significantly modify theAEA ability to inhibit IKDR (Fig. 3D).
AEA Action on IKDR Is Mediated by ItsInteraction with the Extracellular Leaflet
of the Plasma Membrane
We next addressed the question whether AEA exertedits modulatory action by interacting with the intracellular
796 VIGNALI ET AL.
GLIA

or the extracellular leaflet of the plasma membrane. Itis well known, in fact, that AEA can passively cross theplasma membrane (Hillard and Jarrahian, 2003). There
is also evidence that astrocytes can actively reuptakeAEA (Beltramo et al., 1997). Importantly, it wasreported that AEA modulates the activity of different
Fig. 2. Anandamide action is not mediated by cloned CB receptors.(A) Western blotting of cell lysates from pure cortical astrocytes in pri-mary culture (astro), mixed microglia/astroglia culture (micro/astro)and primary cultured hippocampal neurons (neuro) probed with anti-body directed against the CB1 receptor. (B) The same samples as in Aprobed with anti-CB2 antibody. Note that in cultured astrocytes onlythe CB1 signal is clearly detectable. (C) I–V plot of current tracesevoked as in Fig. 1C in the absence (1) and after maximal depression(2) of IKDR following the application of the CB1 agonist WIN55,212-2 (1lM, n 5 6). In the inset are shown representative current traces at140 mV. (D) I–V graph of control currents (1) and after maximal inhi-
bition (2) by the brain endocannabinoid, 2-arachidonyl glycerol (2-AG; n5 7). Representative current traces at 140 mV are shown in the inset.(E) I–V plots of control currents (1) and those evoked after exposure toAEA (2) but following preincubation with the selective CB1 antagonistAM521 (1 lM) 1 h before measurement and during voltage stimulation(n 5 5). Representative currents at 140 mV are shown in the inset. (F)Bar graph of variations in charge quantities at 140 mV following AEA(0.3 lM) challenge in control condition and after preincubation withpertussis toxin (PTX; 16 h). Numbers above columns are the cells testedin each condition Single asterisks denote P < 0.05.
797EAE MODULATION OF ASTROCYTE K+ CHANNELS
GLIA

ion channels by acting from the cytoplasmatic side(Chemin et al., 2001; De Petrocellis et al., 2001; Evanset al., 2004). Hence, experiments were carried out byadding AEA (5 lM) in the patch electrode. Under theseexperimental conditions, ramp currents evoked within 1min of accessing the cell were not significantly differentfrom those elicited after 10–15 min of intracellular dialy-sis, which is a time sufficiently long to achieve equilib-rium of small molecules (Pusch and Neher, 1988). More-over, the extracellular application of AEA after 10 minof its internal dialysis resulted in a reduction of IKDR
that did not differ significantly from that obtained with-out AEA in the recording pipette (Fig. 4A). Because it iswell known that BSA has a high capacity to scavengeAEA (Bojesen and Hansen, 2003) and thus can preventintracellular accumulation of free AEA, further experi-ments were carried out by adding BSA (2.5 mg/mL) intothe recording pipette. The intracellular dialysis of BSAfor 10 min did not significantly change control currentdensity and did not abolish the reduction of ramp cur-rents promoted by the extracellular application of AEA.Since there is some evidence that AEA can intercalate
into the external leaflet of the plasma membrane (Tianet al., 2005), complementary experiments were per-formed to analyze the recovery of IKDR in the presenceof BSA (10 mg/mL) applied extracellularly. Compared towashout with standard solution, BSA exposure gener-ated a rapid and marked increase in K1 conductance(Fig. 4B). BSA challenge did not modify the evoked cur-rents as the temporal kinetics were virtually identical tocontrol traces (Fig. 4C). In the presence of BSA, themaximal recovery was of �80% (n 5 7). These resultsare consistent with the view that BSA effect on restora-tion of K1 conductance is due to its ability to strip offAEA interacting with the external leaflet of the plasmamembrane. It was recently reported that lipid rafts andcaveolae microdomains could have a role in endocanna-binoid signaling (Dainese et al., 2007), and are possiblesites of localization of outwardly rectifying K1 channels(Martens et al., 2004). These observations prompted usto explore whether the interaction of AEA with lipidrafts and/or caveolae could be associated with K1 cur-rent blockage. Experiments were performed by applyingAEA on astrocytes pretreated (1 h at room temperature)
Fig. 3. Anandamide-mediated blockage of IKDR is independent of itsintracellular metabolism and intracellular Ca21 signaling. (A) I–V plotof IKDR in the absence (1) and in the presence (2) of the nonmetaboliz-able analog of AEA methanandamide (n 5 9). In inset representativecurrents at 140 mV are shown. (B) Representative ramp currents (*) ofthe effect of one of the metabolic product of AEA, the polyunsaturatedlipid arachidonic acid (AA), on IKDR. Note that in addition to inhibitingIKDR, AA also causes a delayed increase in K1 conductance owing to
activation of open rectifier K1 channels. (C) Bar graph of maximal var-iations in charge quantities at 140 mV promoted by AEA after removalof extracellular Ca21 (and addition of 0.5 mM EGTA). (D) Histogram ofthe charge quantities at 140 mV under control conditions (ctrl) and fol-lowing the maximal AEA-induced depression of IKDR by using standardinternal solution containing 10-fold different concentrations of the Ca21
chelator EGTA. Numbers above columns are the cells tested in eachcondition. Single asterisks denote P < 0.05.
798 VIGNALI ET AL.
GLIA

with the cholesterol-extracting agent methyl-b cyclodex-trine (MCD, 5 mM), which disrupts the structure andthe functional integrity of lipid rafts and caveolae micro-domains (Brown and London, 2000; Fielding and Field-ing, 2003). The results demonstrate that current densityof IKDR recorded following MCD exposure was not differ-ent from that elicited in control astrocytes and illustratethat MCD pretreatment did not prevent the ability ofAEA to inhibit K1 conductance (rate of blockage withoutMCD at 140 mV: 74% 6 7%, n 5 6; after MCD: 71% 68%, n 5 9).
AEA Reduces IKDR in Glial Cells In Situ
We next explored whether astroglial K1 conductancein situ could also be affected by endocannabinoids. Dur-ing postexperiment analysis, the recorded cells were
identified by their fluorescent signals from Alexa Fluorhydrazide 488 or 594 dialyzed from the recording pipetteand characterized immunohistologically. Based onGFAP-, S100b-, and NG2-immunoreactivity, three typesof glial cells were identified in brain slices of the P10-P12 rat cortex: NG2-proteoglycan expressing cells (NG2-glia) (Nishiyama et al., 1999) and GFAP/S100b- orS100b-positive astroglia. Since S100b was expressed ina larger population of astrocytes than GFAP, co-immuno-staining of S100b and NG2 was used for cell identifica-tion after each patch-clamp recording. When performingelectrophysiological measurements, glial cells were iden-tified by the lack of action potential generation and verynegative (>280 mV) resting membrane potentials. Inthe initial set of experiments, AEA was applied at thesame concentration as AEA used in vitro (1 lM). Underthese conditions, AEA applied for 10–20 min (n 5 10)had no effect (data not shown). The lack of AEA activity
Fig. 4. AEA action on IKDR occurs through its interaction with anextracellular binding site of the plasma membrane. (A) Histogram ofvariations in charge quantities at 140 mV after 10-min intracellular di-alysis with AEA (5 lM) added to the pipette solution, after maximal in-hibition of IKDR by extracellular AEA in the presence of intracellularAEA, following 10-min intracellular dialysis with bovine serum albumin(BSA) and after maximal current depression by extracellular AEA byusing BSA-containing pipette. Numbers above columns are the cells
tested in each condition. Single asterisks denote P < 0.05. (B) Timecourse of current recovery from AEA inhibition at 160 mV duringwashout with control solution (ctrl) and by adding BSA to extracellularsaline (n 5 6 for ctrl, and n 5 5 for BSA). Lines are spline curvesthrough the data points. (C) Representative step currents at 160 mVupon control condition (1), following maximal inhibition with AEA (2),and after largest recovery during BSA administration (3).
799EAE MODULATION OF ASTROCYTE K+ CHANNELS
GLIA

could be due to the poor penetration of AEA into theslice and/or by its segregation in other cells forming thecortical tissue. Both conditions would promote a reduc-tion of AEA concentration close to the recorded cell. Tocircumvent this limitation, AEA was thereafter appliedat a 10-fold higher concentration.
To characterize the effect of 10 lM AEA, inward andoutward currents were activated by a voltage step proto-col (inset in Fig. 5B). Glial cells immunopositive forS100b (Figs. 5A and 6A) displayed a large K1 conduct-ance over an extended range of membrane potentials(insets in Figs. 5A and 6A). The temporal and voltage-dependent kinetics of the evoked currents were different
and were used to separate passive and complex astro-cytes (Anderova et al., 2004; Bordey and Sontheimer,2000; Neprasova et al., 2007). The third type of glialcells, NG2-positive cells (Fig. 7A), expressed a pattern ofK1 currents (inset in Fig. 7A) that was comparable tothat of complex astroglia (Jabs et al., 2005; Matthias etal., 2003).
Passive astrocytes, which represented the majority ofrecorded astrocytes (n 5 14; 39% of recorded cells), hadvery negative resting membrane potentials (Vrest: 283 69 mV) and displayed large time- and voltage-independ-ent outward and inward K1 currents (Fig. 5B). Theapplication of 10-lM AEA for 10 min produced a signifi-
Fig. 5. Anandamide attenuates voltage- and time-independent po-tassium currents in passive astroglia in situ. (A) Representative passiveastroglial cell identified in a cortical slice by immunostaining a cellloaded with Alexa Fluor hydrazide-488 (left panel) with the specificastrocytic marker S100b (middle panel). Overlay (right panel) illus-trates the co-localization of S100 b with the fluorescently labeled cell,whose current pattern is shown in the inset. (B) Representative currentfamily elicited in a passive astrocyte upon voltage stimulation with the
voltage step protocol shown in the inset. (C) In the same astrocyteshown in panel B, a 10-min challenge with 10 lM AEA caused a time-independent depression of outward currents. (D) Current tracesobtained by digital subtraction of currents in B and C (B-C) depictingAEA-sensitive current component. (E) The normalized I–V plot (n 5 7)shows that AEA-induced reduction occurred only in the positive rangeof membrane potentials. [Color figure can be viewed in the online issue,which is available at www.interscience.wiley.com.]
800 VIGNALI ET AL.
GLIA

cant (�40%) time- and voltage-independent reduction ofoutward currents (Fig. 5C,D). Of note, AEA induced thedepression of K1 currents activated at membrane poten-tials more positive than 270 mV (Fig. 5E) without alter-ing significantly the cell resting membrane potential(control Vrest 281 6 9 mV; AEA Vrest: 274 6 2 mV; P >0.05). The second type of astroglial cells, called complexastrocytes, also had a very negative resting membranepotential (Vrest: 291 6 4 mV; n 5 8; 22% of recordedcells). As previously reported (Anderova et al., 2004),these cells exhibited time- and voltage-dependent K1
currents mediated by A-type (KA), delayed rectifier(KDR) and inwardly rectifying (Kir) channels (Fig. 6B).Also in these cells AEA applied for 10 min diminishedoutward currents in a time-independent manner (Fig.
6C,D), but did not affect the resting membrane potential(control Vrest:291 6 4 mV ; AEA Vrest: 289 6 6 mV; P >0.05). The steady-state I–V curve denotes that blockage(�50%) was pronounced at potentials more positive than240 mV, where IKDR channels were mostly activated(Fig. 6E), and was not observed at more negative poten-tials. Finally, we explored the effect of AEA on NG2 glia.These cells had a very negative resting membranepotential (Vrest: 292 6 2 mV; n 5 14; 39% of recordedcells). Moreover, at variance with complex astrocytes theIKA and IKir current components were higher andlower, respectively (Fig. 7B). AEA challenge promoted atime-independent reduction of steady-state currents(Fig. 7C,D) at potentials more positive than 240 mV(�50%), where IKDR is predominant (Fig. 7E), and did
Fig. 6. Anandamide inhibits voltage-gated outward potassium cur-rents in complex astrocytes in situ. (A) A representative complex astro-cyte identified in a cortical slice by immunostaining a cell loaded withAlexa Fluor hydrazide-594 (left panel) with the specific astrocyticmarker S100b (middle panel). Overlay (right panel) illustrates the co-localization of S100 b with the fluorescently labeled cell; the currentpattern of this cell is shown in the inset. (B) Representative current
family from a complex astrocyte stimulated as in Fig. 5B. (C) Currentselicited in the same astrocyte as in B after administration of AEA. (D)Current traces obtained by digital subtraction of currents in B and C(B-C) depicting AEA-sensitive current component. (E) The normalizedI–V plot (n 5 6) shows that AEA-induced inhibition increased at largerdepolarizing potentials. [Color figure can be viewed in the online issue,which is available at www.interscience.wiley.com.]
801EAE MODULATION OF ASTROCYTE K+ CHANNELS
GLIA

not modify the resting membrane potential (control Vrest:292 6 2 mV; AEA Vrest: 291 6 3 mV; P > 0.05).
DISCUSSION
The major finding of this study is the demonstrationthat the endocannabinoid molecule AEA is able to regu-late astroglial activity by modulation of K1 conductance.The effect of AEA was observed in primary cultures of ratcortical astrocytes and in astroglial cells in cortical slices.It appears to be mediated by the regulation of outwardrectifier currents, IKDR, the majority of the voltage-gatedK1 conductance in cultured astroglia and a large compo-nent of K1 permeability in situ (Barres et al., 1990; Bor-dey and Sontheimer, 1997; Ferroni et al., 1995; Nowak et
al., 1987). The contribution of other K1 channels, such aslarge-conductance, Ca21-dependent K1 channels (BKchannel) identified in vitro (Gebremedhin et al., 2003;Nowak et al., 1987; Quandt and MacVicar, 1986) isunlikely because the magnitude and the kinetics of volt-age-activated currents were not significantly affected byremoval of extracellular Ca21 (our unpublished observa-tion) or by changing the capacity of intracellular Ca21
buffering. Moreover, the activation probability of rapidlyinactivating (IA) conductance, which was previouslydescribed in vitro and in situ (Anderova et al., 2004;Bekar et al., 2005; Bordey and Sontheimer, 1999; Nowaket al., 1987) was minimized by low holding potential and/or by using slow-ramp depolarizing protocol.
The pharmacological profile of AEA action is incompati-ble with the stimulation of the two cloned CB receptors
Fig. 7. Anandamide depresses outward potassium conductance incortical NG2 glia in situ. (A) Representative NG2 glial cell loaded withAlexa Fluor-hydrazide-594 and identified by its immunopositive stainingfor proteoglycan NG2. Inset in the left panel shows the current patternof this cell. (B) Representative current family recorded in a NG2 cellstimulated as in Fig. 5B. (C) A 10-min exposure to 10 lM AEA caused a
time-independent depression of outward currents. (D) Current tracesobtained by digital subtraction of currents in B and C (B–C) depictingAEA-sensitive current component. (E) The normalized I–V plot (n 5 5)denotes that AEA was effective at membrane potentials more positivethan 240 mV. [Color figure can be viewed in the online issue, which isavailable at www.interscience.wiley.com.]
802 VIGNALI ET AL.
GLIA

so far identified, for several reasons: (1) Specific antago-nists of the CB1 receptor, the only CB receptor subtypeexpressed in our primary cultures, did not modify theaction of AEA on IKDR. (2) Pharmacological inhibition ofCB receptors by pertussis toxin did not alter the AEAability to diminish IKDR. (3) Experimental manipulationsable to modify [Ca21]i levels did not change AEA effecton IKDR. (4) The synthetic cannabionoid receptor agonistWIN55,212-2, which is more potent at activating CB1
than AEA, was less effective than AEA in depressingIKDR.
Our results also demonstrate that it is unlikely thatthe products of AEA metabolism are mediators of AEAaction, because pharmacological inhibition of the specificenzyme involved in AEA degradation, the FAAH, did notprevent the effects of AEA. Furthermore, the FAAH-re-sistant AEA analog methanandamide also reduced IKDR.Finally, at variance with the present data, the AEAbyproduct AA caused a diminution of IKDR, which, how-ever, was followed by the activation of the open rectifierK1 current likely mediated by TREK-2 (Ferroni et al.,2003).
The observation that capsaicin, an agonist of TRPV1that is also stimulated by AEA (Smart et al., 2000), didnot significantly affect IKDR, together with the lack ofevidence of TRPV1 expression in cultured astroglia,rules out the involvement of TRPV1 in the action ofAEA (our unpublished observation). A role for Ca21
influx through TRPV4, which is another Ca21-permeablecation channel activated by AEA (Watanabe et al., 2003)and expressed by cultured astrocytes (Benfenati et al.,2007), could be excluded because IKDR blockage by AEAwas unaffected by the removal of extracellular Ca21.
Although activation of the CB1 receptor accounts formost of the behavioral effects mediated by endocannabi-noid receptors, several findings obtained in CB1-deficient(knockout) mice have indicated the presence of CB1-re-ceptor-independent actions of endocannabinoids in theCNS (Di Marzo et al., 2000). Of note, the existence ofnovel CB receptors expressed by astroglial cells hasbeen postulated (for review, see Stella, 2004). It wasrecently reported that the orphan receptor GPR55,which is also found in brain (Sawzdargo et al., 1999), isa novel cannabinoid receptor (Ryberg et al., 2007). Inrecombinant expression system (Ryberg et al., 2007),this receptor was shown to be activated by AEA and 2-AG but not by the synthetic agonist WIN55,212-2. Itwas recently demonstrated that in neurons, GPR55stimulation promotes Ca21 mobilization from intracellu-lar stores and inhibits the K1 conductance mediated byM currents (Lauckner et al., 2008). Our results clearlyshow that in vitro IKDR was depressed by AEA with ahigher efficacy compared with the synthetic agonist and2-AG. In addition, the IC50 of AEA effect on K1 conduct-ance in astrocytes in vitro was about 10-fold higher thanthat described for stimulation of GPR55. Finally, single-cell microfluorometry analysis showed that AEA did notcause any significant [Ca21]i elevation in cultured corti-cal astrocytes (our unpublished observation). From thesefindings, we can conclude that it is unlikely that GPR55
is involved in the AEA-induced inhibition of astroglialIKDR. Recently, it was reported that endocannabinoidspromote [Ca21]i increases through CB1 activation in situ(Navarrete and Araque, 2008). The reason of this dis-crepancy remains to be ascertained. It has to be pointedout, however, that from our immunoblot analysis wecannot conclude that the positive signal for CB1 in cul-tured astrocytes is paralleled by its functional signalingactivity.
It is now well recognized that endogenous and syn-thetic cannabinoids can modulate brain cell activities ina CB-independent manner, directly affecting targetedion channels (for review, see Oz, 2006). Such a mecha-nism was shown to underlie the inhibition of voltage-gated Na1 and Ca21 channels by AEA in neuronal cells(Chemin et al., 2001; Fisyunov et al., 2006; Kim et al.,2005). Moreover, neuronal currents through N-methyl-D-aspartate-, serotonin- and glycine-gated receptors werealso reported to be directly modulated by AEA (Fan,1995; Hampson et al., 1998; Lozovaya et al., 2005).Finally, an apparent CB-receptor-independent inhibitionof IKDR and ATP-sensitive K1 currents by AEA wasdescribed in smooth muscle cells and Xenopus laevisoocytes (Oz et al., 2007; Van den Bossche and Vanheel,2000). Similar results have been reported for recombi-nant ion channels expressed in heterologous systems.The current through low-voltage-activated (T) Ca21
channels was markedly depressed by a low concentra-tion of AEA when expressed in various cell lines(Chemin et al., 2001). Recombinant Shaker-related(Kv1.2) and two-pore-domains (TASK-1) K1 channelswere also markedly suppressed by AEA (Maingret et al.,2001; Poling et al., 1996). Both negative and positivedirect modulations by AEA were reported for nicotinicacetylcholine and glycine receptors expressed in Xeno-pus oocytes, respectively (Hejazi et al., 2006; Oz et al.,2003). Our results suggest that AEA must interact withthe extracellular leaflet of the plasma membrane inorder to exert its inhibitory action because: (1) Intracel-lular dialysis of AEA did not cause significant inhibitionof IKDR and did not alter the ability of extracellularAEA to depress the K1 conductance. (2) Whereas intra-cellular dialysis with the lipid scavenger BSA did notchange the efficacy of extracellular AEA, its extracellu-lar application was the only procedure to achieve a rapidand large recovery of IKDR upon AEA washout. The find-ing that the effect of AEA was not use-dependent andwas complete even under experimental conditions inwhich IKDR channels were closed could suggest thatAEA stabilizes the closed state of the channel by bindingto hydrophobic determinants of the protein complex.Whether AEA modifies the phospholipid environment inwhich IKDR channel is embedded, or interacts directlywith the proteins forming the channel or with auxiliarysubunits remains to be established. Interestingly, molec-ular dynamic studies have postulated that the interac-tion of AEA with the transmembrane CB1 receptoroccurs through its binding with the lipid-facing aminoacid motif of the receptor (Lynch and Reggio, 2005). Mu-tagenesis studies of the channel protein(s) underlying
803EAE MODULATION OF ASTROCYTE K+ CHANNELS
GLIA

astroglial IKDR might be useful to address this issue.However, owing to the lack of pharmacological tools thatspecifically inhibit the various subtypes of Kv channels,it is still not known whether IKDR is mediated by a sin-gle channel population or by different channel types.Whereas in cultured cortical astrocytes, Kv1.6 wasreported to be a major component of IKDR (Smart et al.,1997), in cultured spinal cord astroglia Kv1.5 was shownto mediate most of the IKDR (MacFarlane and Son-theimer, 2000a; Roy et al., 1996). This lack of informa-tion also precluded a detailed analysis of AEA influenceon the voltage dependence of activation and the steady-state of inactivation. The possible contribution of dif-ferent channel subtypes to IKDR might also explain theobservation that the synthetic CB receptor agonistWIN55,212-2 produced a significant current depressiononly at potentials positive to 0 mV. In light of this pos-sibility, in fact, it could be envisaged that the inhibi-tory action of WIN55,212-2 is mediated by anothermechanism that would differentially affect the variousK1 channel subtypes. Such divergence of mechanisticprocesses between AEA and other endocannabinoidswas recently described for inhibition of voltage-gatedCa21 channels (Guo and Ikeda, 2004). Further re-search is needed to address this issue, as the maingoal of this work was to characterize AEA action. Adifferential sensitivity of various subtypes of K1 chan-nels to endocannabinoids might also account for thesubstantial difference in efficacy of AEA in vitro and insitu. It must be emphasized, however, that poor pene-tration of AEA in the brain tissue might also contrib-ute to this discrepancy.
Accumulating evidence indicate that brain endocanna-binoids are upregulated as a result of traumatic braininjuries (Hansen et al., 2001; Mechoulam and Shohami,2007). Our results illustrate that the AEA concentrationable to significantly depress IKDR was in the submicro-molar range, which is well below the AEA levels foundin the brain under acute pathological conditions(Franklin et al., 2003; Hansen et al., 1999; Marsicanoet al., 2003). Synthetic cannabinoid agonists were shownto have a neuroprotective effect in vivo and in vitrothrough mechanisms partially CB-receptor insensitive(Marsicano et al., 2002; Nagayama et al., 1999; Van derStelt et al., 2001). Whether the here-described action ofAEA on astroglial cells partially accounts for such a neu-roprotective process remains an open question. Whereasthe role of modulation of IKir in the regulation of astro-glial-mediated homeostasis of CNS is well established(for review, see Butt and Kalsi, 2006), the functionalrelevance of IKDR both in vitro and in situ is stillunclear. Immature astroglia express predominantlyIKDR (Bordey and Sontheimer, 1997; Kressin et al.,1995). In Muller glial cells, IKDR was proposed to oper-ate as shunt current to set the resting membrane poten-tial in the absence of functional inward rectifier K1
channels as it occurs in several pathophysiological condi-tions (Pannicke et al., 2000). From this, it could behypothesized that its blockage by AEA affects cellularprocesses regulated by the level of membrane potential
polarization. However, our study shows that AEA, atleast at the concentrations used, did not alter substan-tially the astroglial resting membrane potential.
It has been postulated that an upregulation of IKDR
may be relevant to astroglial proliferation and scarrepair in an in vitro model of reactive gliosis (MacFar-lane and Sontheimer, 1997). Noteworthy, in corticalastroglia in situ stab wound and freeze lesion injuriescaused an increase in IKDR and augmented astrocyteproliferation in the area surrounding the sites of lesion(Anderova et al., 2004; Bordey et al., 2001). Biochemicaldownregulation of Kv1.5, the Kv channel mediating themajority of IKDR in spinal cord astrocytes, was reportedto markedly depress astroglial cell proliferation in vitro(MacFarlane and Sontheimer, 2000b). In glioma cells,AEA was demonstrated to inhibit proliferation through acomplex mechanism involving CB1 and TRPV1 receptors(Jacobsson et al., 2001). Further studies will clarifywhether the AEA-induced reduction of IKDR plays a rolein the regulation of astroglial proliferation.
In conclusion, this study provides the first evidencethat the endocannabinoid system is able to regulateastroglial activity through modulation of K1 conduct-ance. The existence of a novel astroglial molecular tar-get for endocannabinoids might be useful in formulat-ing new hypotheses for defining a broader role ofthe endocannabinoid system in the regulation of CNSexcitability.
ACKNOWLEDGMENTS
The authors thank Alessia Minardi for preparation andmaintenance of primary cultures of cortical astrocytes.
REFERENCES
Anderova M, Antonova T, Petr�ık D, Neprasova H, Chvatal A, Sykova E.2004. Voltage-dependent potassium currents in hypertrophied ratastrocytes after a cortical stab wound. Glia 48:311–326.
Barres BA, Koroshetz WJ, Chun LL, Corey DP. 1990. Ion channelexpression by white matter glia: The type-1 astrocyte. Neuron 5:527–544.
Bekar LK, Loewen ME, Cao K, Sun X, Leis J, Wang R, Forsyth GW,Walz W. 2005. Complex expression and localization of inactivating Kvchannels in cultured hippocampal astrocytes. J Neurophysiol93:1699–1709.
Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D.1997. Functional role of high-affinity anandamide transport, asrevealed by selective inhibition. Science 277:1094–1097.
Benfenati V, Amiry-Moghaddam M, Caprini M, Mylonakou M, Rapi-sarda C, Ottersen P, Ferroni S. 2007. Expression and functional char-acterization of transient receptor potential vanilloid-related channel4 (TRPV4) in rat cortical astrocytes. Neuroscience 148:876–892.
Bojesen IN, Hansen HS. 2003. Binding of anandamide to bovine serumalbumin. J Lipid Res 44:1790–1794.
Bordey A, Lyonos SA, Hablitz JJ, Sontheimer H. 2001. Electrophysio-logical characteristics of reactive astrocytes in experimental corticaldysplasia. J Neurophysiol 85:1719–1731.
Bordey A, Sontheimer H. 1997. Postnatal development of ionic currentsin rat hippocampal astrocytes in situ. J Neurophysiol 78:461–477.
Bordey A, Sontheimer H. 1999. Differential inhibition of glial K1 cur-rents by 4-AP. J Neurophysiol 82:3476–3487.
Bordey A, Sontheimer H. 2000. Ion channel expression by astrocytes insitu: Comparison of different CNS regions. Glia 30:27–38.
804 VIGNALI ET AL.
GLIA

Bouaboula M, Bourrie B, Rinaldi-Carmona M, Shire D, Le Fur G, Case-llas P. 1995. Stimulation of cannabionoid receptor CB1 induces krox-24 expression in human astrocytoma cells. J Biol Chem 270:13973–13980.
Breivogel CS, Griffin G, Di Marzo V, Martin BR. 2001. Evidence for anew G protein-coupled cannabinoid receptor in mouse brain. MolPharmacol 60:155–163.
Brown DA, London E. 2000. Structure and function of sphingolipid-and cholesterol-rich membrane rafts. J Biol Chem 275:17221–17224.
Butt AM, Kalsi A. 2006. Inwardly rectifying potassium channels (Kir)in central nervous system glia: A special role for Kir4.1 in glial func-tions. J Cell Mol Med 10:33–44.
Carlisle SJ, Marciano-Cabral F, Staab A, Ludwick C, Cabral GA. 2002.Differential expression of the CB2 cannabinoid receptor by rodentmacrophages and macrophage-like cells in relation to cell activation.Int Immunopharmacol 2:69–82.
Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yanh W, Nithipati-kom K, Pfister SL, Campbell WB, Hillard CJ. 2004. Cultured ratmicroglial cells synthesize the endocannabinoid 2-arachidonylgly-cerol, which increases proliferation via a CB2 receptor-dependentmechanism. Mol Pharmacol 65:999–1007.
Chemin J, Monteil A, Perez-Reyes E, Nargeot J, Lory P. 2001. Directinhibition of T-type calcium channels by the endogenous cannabinoidanandamide. EMBO J 20:7033–7040.
Chevaleyre V, Takahashi KA, Castillo PE. 2006. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci 29:37–76.
Dainese E, Oddi S, Bari M, Maccarrone M. 2007. Modulation of theendocannabinoid system by lipid rafts. Curr Med Chem 14:2702–2715.
D’Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. 1999.Impaired K1 homeostasis and altered electrophysiological propertiesof post-traumatic hippocampal glia. J Neurosci 19:8152–8162.
De Petrocellis L, Bisogno T, Maccarrone M, Davis JB, Finazzi-Agro A,Di Marzo V. 2001. The activity of anandamide at vanilloid VR1 recep-tors requires facilitated transport across the cell membrane and islimited by intracellular metabolism. J Biol Chem 276:12856–12863.
Di Marzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, ZimmerAM, Zimmer A, Martin BR. 2000. Level, metabolism and pharmaco-logical activity of anandamide in CB1 cannabinoid receptor knockoutmice: Evidence for non-CB1, non-CB2 receptor-mediated actions ofanandamide in mouse brain. J Neurochem 75:2434–2444.
Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC,Piomelli D. 1994. Formation and inactivation of endogenous cannabi-noid anandamide in central neurons. Nature 372:686–691.
Evans RM, Scott RH, Ross RA. 2004. Multiple actions of anandamideon neonatal rat cultured sensory neurones. Br J Pharmacol141:1223–1233.
Facchinetti F, Del Giuduce E, Furegato S, Passarotto M, Leon A. 2003.Cannabinoids ablate release of TNFa in rat microglial cells stimu-lated with lypopolysaccharide. Glia 41:161–168.
Fan P. 1995. Cannabinoid agonists inhibit the activation of 5-HT3receptors in rat nodose ganglion neurons. J Neurophysiol 73:907–910.
Felder CC, Briley EM, Axelrod J, Simpson JT, Mackie K, Devane WA.1993. Anandamide, an endogenous cannabimimetic eicosanoid, bindsto the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc Natl Acad Sci USA 90:7656–7660.
Ferroni S, Marchini C, Schubert P, Rapisarda C. 1995. Two distinctinwardly rectifying conductances are expressed in long-term dibu-tyryl-cyclic-AMP treated rat cultured cortical astrocytes. FEBS Lett367:319–325.
Ferroni S, Valente P, Caprini M, Nobile M, Schubert P, Rapisarda C.2003. Arachidonic acid activates an open rectifier potassium channelin cultured rat cortical astrocytes. J Neurosci Res 72:363–372.
Fielding CJ, Fielding PE. 2003. Relationship between cholesterol traf-ficking and signaling in rafts and caveolae. Biochim Biophys Acta1610:219–228.
Fisyunov A, Tsintsadze V, Min R, Burnashev N, Lozovaya N. 2006.Cannabinoids modulate the P-type high-voltage-activated calciumcurrents in purkinje neurons. J Neurophysiol 96:1267–1277.
Franklin A, Parmentier-Batteur S, Walter L, Greenberg DA, StellaN. 2003. Palmitoylethanolamide increases after focal cerebral is-chemia and potentiates microglial cell motility. J Neurosci 23:7767–7775.
Gebremedhin D, Yamamura K, Zhang C, Bylund J, Koehler RC, HarderDR. 2003. Matabotropic glutamate receptor activation enhances theactivities of two types of Ca21-activated K1 channels in rat hippo-campal astrocytes. J Neurosci 23:1678–1687.
Gomez Del Pulgar T, De Ceballos ML, Guzman M, Velasco G. 2002.Cannabinoids protect astrocytes from ceramide-induced apoptosisthrough the phosphatidylinositol 3-kinase/protein kinase B pathway.J Biol Chem 277:36527–36533.
Guo J, Ikeda SR. 2004. Endocannabinoids modulate N-type calciumchannels and G-protein-coupled inwardly rectifying potassium chan-nels via CB1 cannabinoid receptors heterologously expressed in mam-malian neurons. Mol Pharmacol 65:665–674.
Hamill OP, Marty A, Neher E, Sackmann B, Sigworth FJ. 1981.Improved patch-clamp techniques for high resolution current record-ing from cells and cell free membrane patches. Pflugers Arch 391:85–100.
Hampson AJ, Bornheim LM, Scanziani M, Yost CS, Gray AT, HansenBM, Leonoudakis DJ, Bickler PE. 1998. Dual effects of anandamideon NMDA receptor-mediated responses and neurotransmission.J Neurochem 70:671–676.
Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F,Manzanares J, Ikonomidou C, Schmid HH, Fernandez-Ruiz JJ,Hansen HS. 2001. Anandamide, but not 2-arachidonoylglycerol,accumulates during in vivo neurodegeneration. J Neurochem 78:1415–1427.
Hansen HS, Moesgaard B, Hansen HH, Schousboe A, Petersen G. 1999.Formation of N-acyl-phosphatidylethanolamine and N-acylethanol-amine (including anandamide) during glutamate-induced neurotoxic-ity. Lipids 34:327–330.
Hejazi N, Zhou C, Oz M, Sun H, Ye JH, Zhang L. 2006. D9-tetrahydro-cannabinol and endogenous cannabinoid anandamide directly potenti-ate the function of glycine receptors. Mol Pharmacol 69:991–997.
Hillard CJ, Jarrahian A. 2003. Cellular accumulation of anandamide:Consensus and controversy. Br J Pharmacol 140:802–808.
Irving AJ, Coutts AA, Harvey J, Rae MG, Mackie K, Bewick GS,Pertwee RG. 2000. Functional expression of cell surface cannabinoidCB(1) receptors on presynaptic inhibitory terminals in cultured rathippocampal neurons. Neuroscience 98:253–262.
Jabs R, Pivneva T, Huttmann K, Wyczynski A, Nolte C, Kettenmann H,Steinhauser C. 2005. Synaptic transmission onto hippocampal glialcells with hGFAP promoter activity. J Cell Sci 118:3791–3803.
Jacobsson SOP, Wallin T, Fowler CJ. 2001. Inhibition of rat C6 gliomacell proliferation by endogenous and synthetic cannabinoids. Relativeinvolvement of cannabinoid and vanilloid receptors. J Pharmacol ExpTher 299:951–959.
Jung M, Calassi R, Rinaldi-Carmona M, Cardenot P, Le Fur G, SoubrieP, Oury-Donat F. 1997. Characterization of CB1 receptors on rat neu-ronal cell cultures: Binding and functional studies using the selectivereceptor antagonist SR141716A. J Neurochem 68:402–409.
Kim HI, Kim TH, Shin YK, Lee CS, Park M, Song J. 2005. Anandamidesuppression of Na1 currents in rat dorsal root ganglion neurons.Brain Res 1062:39–47.
Kressin K, Kuprijanova E, Jabs R, Seifert G, Steinhauser C. 1995.Developmental regulation of Na1 and K1 conductances in glialcells of mouse hippocampal brain slices. Glia 15:173–187.
Lauckner JE, Jensen JB, Chen HY, Lu HC, Mackie K. 2008. GPR55 isa cannabinoid receptor that increases calcium and inhibits M cur-rent. Proc Natl Acad Sci USA 105:2699–2704.
Lay L, Angus JS, Wright CE. 2000. Pharmacological characterisation ofcannabinoid CB(1) receptors in the rat and in mouse. Eur J Pharma-col 391:151–161.
Lozovaya N, Yatsenko N, Beketov A, Tsintsadze T, Burnashev N. 2005.Glycine receptors in CNS neurons as a target for nonretrogradeaction of cannabinoids. J Neurosci 25:7499–7506.
Lynch DL, Reggio PH. 2005. Molecular dynamics simulations of theendocannabinoid N-arachidonoylethanolamine (anandamide) in aphospholipid bilayer: Probing structure and dynamics. Med Chem48:4824–4833.
MacFarlane SN, Sontheimer H. 1997. Electrophysiological changes thataccompany reactive gliosis in vitro. J Neurosci 17:7316–7329.
MacFarlane SN, Sontheimer H. 2000a. Modulation of Kv1.5 currentsby Src tyrosine phosphorylation: Potential role in the differentiationof astrocytes. J Neurosci 20:5245–5253.
MacFarlane SN, Sontheimer H. 2000b. Changes in ion channel expres-sion accompany cell cycle progression of spinal cord astrocytes. Glia30:39–48.
Maingret F, Patel AJ, Lazdunski M, Honore E. 2001. The endocannabi-noid anandamide is a direct and selective blocker of the backgroundK1 channel TASK-1. EMBO J 20:47–54.
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, CannichA, Azad SC, Cascio MG, Ortega Guti�errez S, Van der Stelt M, L�opez-Rodr�ıguez ML, Casanova E, Sch€utz G, Zieglg€ansberger W, Di MarzoV, Behl C, Lutz B. 2003. CB1 cannabinoid receptors and on-demanddefense against excitotoxicity. Science 302:84–88.
Marsicano G, Moosman B, Herman H, Lutz B, Behl C. 2002. Neuropro-tective properties of cannabinoids against oxidative stress: Role ofthe cannabinoid receptor CB1. J Neurochem 80:448–456.
Martens JR, O’Connell K, Tamkun M. 2004. Targeting of ion channelsto membrane microdomains: Localization of Kv channels to lipidrafts. Trends Pharmacol Sci 25:16–21.
805EAE MODULATION OF ASTROCYTE K+ CHANNELS
GLIA

Matthias K, Kirchhoff F, Seifert G, Huttmann K, Matyash M, Ketten-mann H, Steinhauser C. 2003. Segregated expression of AMPA-typeglutamate receptors and glutamate transporters defines distinctastrocyte populations in the mouse hippocampus. J Neurosci 23:1750–1758.
Mechoulam R, Shohami E. 2007. Endocannabinoids and traumaticbrain injury. Mol Neurobiol 36:68–74.
Moldrich G, Wenger T. 2000. Localization of the CB1 cannabinoid re-ceptor in the rat brain. An immunohistochemical study. Peptides21:1735–1742.
Molina-Holgado F, Molina-Holgado E, Guaza C, Rothwell NJ. 2002a.Role of CB1 and CB2 receptors in the inhibitory effects of cannabi-noids on lipopolysaccharide-induced nitric oxide release in astrocytecultures. J Neurosci Res 67:829–836.
Molina-Holgado E, Vela JM, Arevelo-Martin A, Almazan G, Molina-Hol-gado F, Borrel J, Guaza C. 2002b. Cannabinoids promote oligodentro-cyte progenitor survival: Involvement of cannabinoid receptor andphosphatidylinositol-3 kinase/Akt signaling. J Neurosci 22:9742–9753.
Mombouli JV, Schaeffe G, Holzmann S, Kostner GM, Graier WF. 1999.Anandamide-induced mobilization of cytosolic Ca21 in endothelialcells. Br J Pharmacol 126:1593–1600.
Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Green-berg DA. 1999. Cannabinoids and neuroprotection in global and focalcerebral ischemia and in neuronal cultures. J Neurosci 19:2987–2995.
Navarrete M, Araque A. 2008. Endocannabinoids mediate neuron-astro-cyte communication. Neuron 57:883–893.
Neprasova H, Anderova A, Petrik D, Vargova L, Kubinova S, ChvatalA, Sykova E. 2007. High extracellular K1 evokes changes in voltage-dependent K1 and Na1 currents and volume regulation in astrocytes.Pflugers Arch 453:839–849.
Nishiyama A, Chang A, Trapp BD. 1999. NG21 glial cells: A novel glialcell population in the adult brain. J Neuropathol Exp Neurol 58:1113–1124.
Nowak L, Ascher P, Berwald-Netter Y. 1987. Ionic channels in mouseastrocytes in culture. J Neurosci 7:101–109.
Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. 2004.Functional conversion between A-type and delayed rectifier K1 chan-nels by membrane lipids. Science 304:265–270.
Oz M. 2006. Receptor-independent effects of endocannabinoids on ionchannels. Curr Pharm Des 12:227–239.
Oz M, Ravindran A, Diaz-Ruiz O, Zhang L, Morales M. 2003. The en-dogenous cannabinoid anandamide inhibits a7 nicotinic acetylcholinereceptor-mediated responses in Xenopus oocytes. J Pharmacol ExpTher 306:1003–1010.
Oz M, Yang KH, Dinc M, Shippenberg TS. 2007. The endogenous canna-binoid anandamide inhibits cromakalim-activated K1 currents in fol-licle-enclosed Xenopus oocytes. J Pharmacol Exp Ther 323:547–554.
Pannicke T, Faude F, Reichenbach A, Reichelt W. 2000. A function ofdelayed rectifier potassium channels in glial cells: Maintenance of anauxiliary membrane potential under pathological conditions. BrainRes 862:187–193.
Pearlman RJ, Aubrey KR, Vandenberg RJ. 2003. Arachidonic acid andanandamide have opposite modulatory actions at the glycine trans-porter, GLYT1a. J Neurochem 84:592–601.
Pertwee RG, Ross RA. 2002. Cannabinoid receptors and their ligands.Prostaglandins Leukot Essent Fatty Acids 66:101–121.
Poling JS, Rogawski MA, Salem NJ, Vicini S. 1996. Anandamide, anendogenous cannabinoid, inhibits Shaker-related voltage-gated K1
channels. Neuropharmacology 35:983–991.Pusch M, Neher H. 1988. Rates of diffusional exchange between small
cells and a measuring patch pipette. Pflugers Arch 411:204–211.Quandt FN, MacVicar BA. 1986. Calcium-activated potassium channels
in cultured astrocytes. Neuroscience 19:29–41.Rodriguez JJ, Mackie K, Pickel VM. 2001. Ultrastructural localization
of the CB1 cannabinoid receptor in mu-opioid receptor patches of therat caudate putamen nucleus. J Neurosci 21:823–833.
Romero J, Hillard CJ, Calero M, R�abano A. 2002. Fatty acid amide hy-drolase localization in the human central nervous system: An immu-nohistochemical study. Mol Brain Res 100:85–93.
Roy ML, Saal D, Perney T, Sontheimer H, Waxman SG, Kaczmarec LK.1996. Manipulation of the delayed rectifier Kv1.5 potassium channelin glial cells by antisense oligodeoxynucleotides. Glia 18:177–184.
Ryberg E, Larsson N, Sj€ogren S, Hjorth S, Hermansson NO, Leonova J,Elebring T, Nilsson K, Drmota T, Greasley PJ. 2007. The orphanreceptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152:1092–1101.
Sagan S, Venance L, Torrens Y, Cordier J, Glowinski J, Giaume C.1999. Anandamide and WIN55212–2 inhibit cyclic AMP formationthrough G-protein-coupled receptors distinct from CB1 cannabinoidreceptors in cultured astrocytes. Eur J Neurosci 11:691–699.
Salio C, Doly S, Fischer J, Franzoni MF, Conrath M. 2002. Neuronaland astrocytic localization of the cannabinoid receptor-1 in the dorsalhorn of the rat spinal cord. Neurosci Lett 329:13–16.
Sawzdargo M, Nguyen T, Lee DK, Lynch KR, Cheng R, Heng HH,George SR, O’Dowd BF. 1999. Identification and cloning of threenovel human G protein-coupled receptor genes GPR52, PsiGPR53and GPR55: GPR55 is extensively expressed in human brain. BrainRes Mol Brain Res 64:193–198.
Schlicker E, Kathmann M. 2001. Modulation of transmitter release viapresynaptic cannabinoid receptors. Trends Pharmacol Sci 22:565–572.
Seifert G, Schilling K, Steinhauser C. 2006. Astrocyte dysfunction inneurological disorders: A molecular perspective. Nat Rev Neurosci7:194–206.
Sheng WS, Hu S, Min X, Cabral GA, Lokensgard JR, Peterson PK.2005. Synthetic cannabinoid WIN55,212–2 inhibits generation ofinflammatory mediators by IL-1b-stimulated human astrocytes. Glia49:211–219.
Shivachar AC. 2007. Cannabinoids inhibit sodium-dependent, highaffinity excitatory amino acid transport in cultured rat corticalastrocytes. Biochem Pharmacol 73:2004–2011.
Shivachar AC, Martin BR, Ellis EF. 1996. Anandamide- and D9-tetrahy-drocannabinol-evoked arachidonic acid mobilization and blockade bySR141716A [N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichloro-phe-nyl)-4methyl-1H-Pyrazole-3-carboximide hydrochloride]. BiochemPharmacol 51:669–676.
Simard M, Nedergaard M. 2004. The neurobiology of glia in the contextof water and ion homeostasis. Neuroscience 129:877–896.
Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, Cham-bers JK, Randall AD, Davis JB. 2000. The endogenous lipid ananda-mide is a full agonist at the human vanilloid receptor (hVR1). Br JPharmacol 129:227–230.
Smart SL, Bosma SL, Tempel BL. 1997. Identification of the delayedrectifier potassium channel, Kv16, in cultured astrocytes. Glia20:127–134.
Stella N. 2004. Cannabinoid signaling in glial cells. Glia 48:267–277.Tian X, Guo J, Yao F, Yang DP, Makriyannis A. 2005. The conforma-
tion, location, and dynamic properties of the endocannabinoid ligandanandamide in a membrane bilayer. J Biol Chem 280:29788–29795.
Van den Bossche I, Vanheel B. 2000. Influence of cannabinoids on thedelayed rectifier in freshly dissociated smooth muscle cells of the rataorta. Br J Pharmacol 131:85–93.
Van der Stelt M, Trevisani M, Vellani V, De Petrocellis L, Moriello AS,Campi B, McNaughton P, Geppetti P, Di Marzo V. 2005. Anandamideacts as an intracellular messenger amplifying Ca21 influx via TRPV1channels. EMBO J 24:3026–3037.
Van der Stelt M, Veldhuis WB, van Haaften GW, Fezza F, Bisog no T,Bar PR, Veldink GA, Vliegenthart JF, Di Marzo V, Nicolay K. 2001.Exogenous anandamide protects rat brain against acute neuronalinjury in vivo. J Neurosci 21:8765–8771.
Venance L, Piomelli D, Glowinski J, Giaume C. 1995. Inhibition byanandamide of gap junctions and intercellular calcium signalling instriatal astrocytes. Nature 376:590–594.
Venance L, Sagan S, Giaume C. 1997. (R)-methanandamide inhibits re-ceptor-induced calcium responses by depleting internal calcium storesin cultured astrocytes. Pflugers Arch 434:147–149.
Verkhratsky A, Steinhauser C. 2000. Ion channels in glial cells. BrainRes Brain Res Rev 32:380–412.
Vogel Z, Barg J, Levi R, Saya D, Heldman E, Mechoulam R. 1993.Anandamide, a brain endogenous compound, interacts specificallywith cannabinoid receptors and inhibits adenylate cyclase. J Neuro-chem 61:352–355.
Volterra A, Meldolesi J. 2005. Astrocytes, from brain glue to communi-cation elements: The revolution continues. Nat Rev Neurosci 6:626–640.
Wallraff A, Odermatt B, Willecke K, Steinhauser C. 2004. Distincttypes of astroglial cells in the hippocampus differ in gap junction cou-pling. Glia 48:36–43.
Walter L, Dinh T, Stella N. 2004. ATP induces a rapid and pronouncedincrease in 2-arachidonoylglycerol production by astrocytes, aresponse limited by monoacylglycerol lipase. J Neurosci 24:8068–8074.
Walter L, Franklin A, Witting A, Moller T, Stella N. 2002. Astrocytes inculture produce anandamide and other acylethanolamides. J BiolChem 277:20869–20876.
Walter L, Stella N. 2003. Endothelin-1 increases 2-arachidonoyl glycerol(2-AG) production in astrocytes. Glia 44:85–90.
Walz W. 2000. Controversy surrounding the existence of discrete func-tional classes of astrocytes in adult gray matter. Glia 31:95–103.
Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B.2003. Anandamide and arachidonic acid use epoxyeicosatrienoic acidsto activate TRPV4 channels. Nature 424:434–438.
806 VIGNALI ET AL.
GLIA