kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection: kynurenine...
TRANSCRIPT

UNCORRECTED PROOF
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13 1
Journal of Neurochemistry, 2001, 78, 1±13
Kynurenine pathway metabolism in human astrocytes: a paradox
for neuronal protection
Gilles J. Guillemin,* Stephen J. Kerr,* George A. Smythe,³ Danielle G. Smith,* Vimal Kapoor,§Patricia J. Armati,¶ Juliana Croitoru** and Bruce J. Brew*,²
*Centre for Immunology and ²Departments of Neurology and HIV Medicine, St. Vincent's Hospital, Sydney, Australia
³The Ray Williams Biomedical Mass Spectrometry Facility and §School of Physiology and Pharmacology, Faculty of Medicine,
University of New South Wales, Sydney, Australia
School of Biological Sciences, University of Sydney, Australia
**Commissariat a l'Energie Atomique, Department of Neurovirology, Fontenay-aux-Roses, France
Abstract
There is good evidence that the kynurenine pathway (KP) and
one of its products, quinolinic acid (QUIN), play a role in the
pathogenesis of neurological diseases, in particular AIDS
dementia complex. Although QUIN has been shown to be
produced in neurotoxic concentrations by macrophages and
microglia, the role of astrocytes in QUIN production is
controversial. Using cytokine-stimulated culture of human
astrocytes, we assayed key enzymes and products of the KP.
We found that human astrocytes lack kynurenine hydroxylase
so that large amounts of kynurenine and the QUIN antagonist
kynurenic acid were produced. In contrast, the amounts of
QUIN that were synthesized were subsequently completely
degraded. We then showed that kynurenine in concentrations
comparable with those produced by astrocytes led to
signi®cant production of QUIN by macrophages. These results
suggest that astrocytes alone are neuroprotective by mini-
mizing QUIN production and maximizing synthesis of kynure-
nic acid. However, it is likely that, in the presence of
macrophages and/or microglia, astrocytes become indirectly
neurotoxic by the production of large concentrations of
kynurenine that can be secondarily metabolized by neighbour-
ing or in®ltrating monocytic cells to form the neurotoxin QUIN.
Keywords: human astrocyte, kynurenine pathway, kynure-
nine, quinolinic acid, neurotoxicity.
J. Neurochem. (2001) 78, 1±13.
There is good evidence that the kynurenine pathway (KP) is
involved in the neurocytotoxicity associated with AIDS
dementia complex and probably other in¯ammatory brain
diseases (Heyes 1996). The KP is a major route of
l-tryptophan catabolism, resulting in the production of
nicotinamide adenine dinucleotide and other neuroactive
intermediates (Fig. 1) (Bender and McCreanor 1982; Stone
1993; Botting 1995; Curzon 1996; Moroni 1999). These
include kynurenine (KYN) (Lapin et al. 1982), kynurenic
acid (KYNA), 3-hydroxykynurenine (3-HK) (Eastman 1989;
Moroni 1999), picolinic acid (PIC) (Melillo et al. 1996) and
quinolinic acid (QUIN) (Lapin et al. 1982; Schwarcz et al.
1983; Stone 1993). Of the metabolites, the NMDA receptor
agonist and neurotoxin QUIN is the most important.
Moreover, the KP may play a role in certain physiological
functions (Stone 1993; Curzon 1996) such as behaviour,
sleep, thermoregulation and pregnancy (Munn et al. 1998).
The cellular localization of the KP has been shown to be
primarily in macrophages and microglial cells (Heyes et al.
1996; Espey et al. 1997) and possibly in astrocytes (Moffett
et al. 1993; Saito et al. 1993b; Speciale and Schwarcz 1993;
Received April 19, 2001; revised manuscript received May 18, 2001;
accepted May 27, 2001.
Address correspondence and reprint requests to Gilles Guillemin,
Centre for Immunology, St. Vincent's Hospital, Darlinghurst, 2010
Sydney, NSW, Australia. E-mail: g.guillemin@c®.UNSW.edu.au
Abbreviations used: 3-HAA, 3-hydroxyanthranilic acid; 3-HAO,
3-hydroxyanthranilate dioxygenase; 3-HK, 3-hydroxykynurenine;
GC-MS, gas chromatography±mass spectrometry; GFAP, glial ®brillary
acid protein; GM-CSF, granulocyte2macrophage colony-stimulating
factor; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; IL, inter-
leukin; KAT, kynurenine amino transferase; KP, kynurenine pathway;
KYN, kynurenine; KYNA, kynurenic acid; KYNase, kynureninase;
KYN-OHase, kynurenine hydroxylase; MdM, blood monocyte derived
macrophages; PBS, phosphate-buffered saline; PIC, picolinic acid;
QPRTase, quinolinate phosphoribosyltransferase; QUIN, quinolinic
acid; TNF, tumour necrosis factor.
JNC498 JNC jn6489 CD 12/7/1 12:38 ALDEN

UNCORRECTED PROOF
Heyes et al. 1996, 1997a, b; Espey et al. 1997). In support of
the latter, increased indoleamine 2,3-dioxygenase (IDO)
activity has been found in astrocytes (Moffett et al. 1993;
Speciale and Schwarcz 1993; Heyes et al. 1997a, b), but it is
not clear whether the other enzymes in the KP are present
and functional in man (Schwarcz et al. 1983; Okuno et al.
1987; KoÈhler et al. 1988b; Du et al. 1992; Heyes et al. 1992,
1997; Heyes 1993; Saito et al. 1993a, b; Stone 1993;
Guidetti et al. 1997; Sanni et al. 1998; Tamburin et al.
1999).
To de®ne the KP in human astrocytes, we used human
fetal astrocyte cultures stimulated with some of the
cytokines involved in brain in¯ammation, especially those
associated with AIDS dementia complex, namely, inter-
leukin (IL)-1b, IL-6, granulocyte2macrophage colony
stimulating factor (GM-CSF), interferon (IFN)-g and
tumour necrosis factor (TNF)-a. Although IFN-g and
TNF-a are known to induce the ®rst enzyme of the KP
(IDO) (Heyes et al. 1997a; Pemberton et al. 1997), their role
in stimulating the other KP enzymes is unde®ned. We
therefore designed primer sets for seven of the major KP
enzymes, namely, IDO (EC 1.13.11.17), kynurenine-
pyruvate aminotransferase (KAT-I; EC 2.6.1.64), kynure-
nine-2-oxoglutarate aminotransferase (KAT-II; EC 2.6.1.7),
kynurenine hydroxylase (KYN-OHase; EC 1.14.13.9),
kynureninase (KYNase; EC 3.7.1.3), 3-hydroxyanthranilate
dioxygenase (3-HAO; EC 1.13.11.6) and quinolinate
phosphoribosyltransferase (QPRTase; EC 2.4.2.19). Expres-
sion of these enzymes was then assessed by RT-PCR. In
addition, we measured the concentration of important KP
intermediates KYN and KYNA and end-products QUIN and
picolinic acid.
Materials and methods
Reagents and chemicals
All cell culture media and additives were from Life Technologies
(Gaithersburg, MA, USA) unless otherwise stated. TNF-a, IL-1b,
IL-6, IFN-g and mouse monoclonal antibody anti-MAP2 came
from Boehringer Mannheim (Mannheim, Germany). GM-CSF was
kindly provided by Schering-Plough (Sydney, NSW, Australia).
3-HAA, KYN, PIC, QUIN, polyclonal antibody antigalactocerebro-
side protein and DAPI were obtained from Sigma Chemical Co.
(Sydney, NSW, Australia). Anti-mouse IgG1 antibodies coupled to
magnetic microbeads were obtained from Dynal (Carlton, VIC,
Australia). Mouse monoclonal antibody anti-glial ®brillary acid
protein (GFAP) clone GA-5 was obtained from Novacostra
(Newcastle, UK). Mouse monoclonal antibody anti-CD68 clone
KiM1P was gratefully provided by Dr Parwaresch and was used at
a concentration of 10 mg/mL. Mouse monoclonal antibody anti-
CD14 clone TUK-4 and mAb anti-factor VIII clone F8/86 were
from Dako (Carpintera, CA, USA). Secondary goat anti-mouse
IgG1 and anti-rabbit ¯uorescein isothiocyanate or Texas Red-
conjugated antibodies were purchased from Southern Biotech-
nology Associates (Birmingham, AL, USA). All commercial
Fig. 1 Simpli®ed kynurenine pathway in
astrocytes.
Q1
Q2
JNC498 JNC jn6489 CD 12/7/1 12:38 ALDEN
2 G. J. Guillemin et al.
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13

UNCORRECTED PROOF
antibodies were used at the concentrations recommended by the
manufacturer.
Cell cultures
Human fetal brains were obtained from 14218-week-old aborted
fetuses collected after therapeutic termination following informed
consent. Astrocytes were prepared using a protocol adapted from
previously described methods (Guillemin et al. 1997; Kerr et al.
1997). Brie¯y, cerebral portions were washed thoroughly with
phosphate-buffered saline (PBS), forced through a 100-mm nylon
mesh with the plunger of a plastic syringe. The suspension was
centrifuged at 500 g for 5 min and the cell pellet resuspended in
RPMI-1640 medium containing 10% heat-inactivated fetal calf
serum, 2 mm glutamine, 200 IU/mL penicillin G, 200 mg/mL
streptomycin sulfate and 0.5% glucose, then plated onto 75 cm2
culture ¯asks (Corning, NY, USA). Cultures were kept at 378C at
5% CO2 in a humidi®ed atmosphere. After 3 days, medium was
changed at 5 and 10 days. The cells became con¯uent by 10±12
days. Microglia were removed from the cultures by gently shaking
¯asks for 2 h at 220 r.p.m. at room temperature (228C) and the
medium changed, thereby removing ¯oating cells. A second
puri®cation step was performed to ensure that highly pure astrocyte
cultures were obtained by using immunomagnetic depletion of
monocyte-lineage cells using the monoclonal antibodies anti-CD14
and anti-CD68 coupled to magnetic microbeads. The remaining
cells were rinsed twice with PBS and cultured as above in non-
coated ¯asks with the culture medium and maintained for up to 6
weeks. The medium was changed twice a week. Blood monocyte-
derived macrophages (MdM) were obtained from human volunteers
(Centre for Immunology, Sydney, Australia) using a classic
adherence method (Kerr et al. 1997).
Immunocytochemistry
The method for brain cell characterization has been described
previously (Guillemin 1997). The following three controls were
performed for each labelling experiment: (i) isotypic antibody
controls, (ii) incubation with only the secondary labelled anti-
bodies, and (iii) estimation of auto¯uorescence of unlabelled cells.
Astrocyte culture treatments
Based on standard curve optimizations (data not shown), 1 mm
QUIN or 100 mm of 3-HAA was added to 4-week-old pure
astroglial cultures in the presence or absence of 100 IU/mL IL-1b,
IL-6, TNF-a, GM-CSF or IFN-g. RNA and culture supernatants
were collected after 6, 24, 48 and 72 h. Each experiment was
performed in triplicate using cultures derived from three different
fetal brains.
Macrophage culture treatments
Either 12.5 or 50 mm of KYN was added to 10-day-old MdM
cultures stimulated or not with 100 IU/mL IFN-g. MdM culture
stimulated with 100 IU/mL IFN-g was used as positive controls for
IDO activation and QUIN production. Culture medium AIM-Vw
(serum-free medium) was used for these experiments. Supernatants
were collected after 24 and 48 h incubation, and RNA after 48 h.
Experiments were performed in triplicate on cultures derived from
three different fetal brains.
Mass spectrometry
Culture supernatants were assayed for QUIN as described
previously (Kerr et al. 1997). QUIN levels were calculated using
the formula [Total nm of QUIN detected ± nm of QUIN present in
the 10% fetal calf serum culture medium] or the levels of QUIN
effectively catabolized after 48 h. The levels were calculated by the
following formula: [Total nm of QUIN detected ± 1000 nm of
QUIN added in the medium ± nm present in the 10% fetal calf
serum culture medium]. Following the same derivatization
methodology, samples were prepared for PIC analysis using
d4-picolinic acid as an internal standard. QUIN and PIC samples
were analysed by GC-MS with the spectrometer operating in
electron capture negative ionization mode. Selected ions (m/z 273
for PIC and m/z 277 for d4-PIC) were then monitored (Smythe,
Kerr, Guillemin and Brew, manuscript submitted). Effective
PIC de novo synthesis was calculated using the formula [PIC in
sample ± PIC in culture medium]. All results are expressed as the
mean ^SEM.
HPLC
Kynurenine was measured by reverse-phase HPLC with UV
detection at 365 nm, using an ammonium acetate buffer (0.1 m,
pH 4.65) containing 0.02% acetonitrile as the organic modi®er. The
limit of detection was estimated to be 0.6 mm of KYN [Total nm of
KYN detected ± nm of KYN present in the 10% fetal calf serum
culture medium]. Kynurenic acid was measured by reverse-phase
HPLC with ¯uorescence detection after post column derivatization
with zinc acetate as described previously (Kapoor et al. 1994). The
limit of detection was estimated to be 10 fm of KYNA. Amounts of
KYNA were calculated with the formula [Total nm of KYNA
detected ± nm of KYNA present in the 10% fetal calf serum culture
medium]. HPLC with electrochemical detection was used for the
quantitation of 3-hydroxykynurenine (3-HK) essentially according
to the method of Heyes et al. (1998). The limit of detection was
estimated at 0.1 nm of 3-HK. All results are expressed as the
mean ^SEM.
RT-PCR detection of mRNA expression of KP enzymes
cDNAs were synthesized for 1 h at 458C from 1 mg of RNA
extracted using Trizolw reagent (Life Technologies, Mulgrave,
Australia), in a 100-mL ®nal volume of 10 mm Tris2HCl (pH 8.3),
25 mm KCl, 0.6 mm MgCl2, 1 mm dNTP, 5 U/mL of Avian
Myeloblastosis virus reverse transcriptase (Promega, Madison, WI,
USA), 0.01 mg/mL of oligo(dT)15 (Boehringer), 2 U/mL of RNasin
(Promega). PCR was performed on 1 mg of cDNA in a 50-mL
reaction mixture containing 10 mm Tris2HCl pH 8.3, 50 mm KCl,
1.5 mm MgCl2, 10 mm of dNTPs, 0.25 mm of speci®c primers
(Geneworks, Sydney, Australia), and 1.25 U/mL Taq DNA
polymerase (Boehringer). Individual sets of primers were used to
identify mRNA transcripts for human IDO, KAT-I, KAT-II,
KYNase, KYN-OHase, 3-HAO and QPRTase, and spanned at
least one intron to distinguish ampli®cation of cDNA from genomic
DNA. Primers were designed using the application Primers! for the
Mac v1.0a (q1996, Apple Pi, Ashland, MA, USA). Amplify 1.2
(William Engels, Genetics Department, University of Wisconsin,
Madison, WI, USA) was used to estimate the number and size of
PCR products. KP enzyme gene sequences were obtained from
GenBank, see Table 1. Reporter gene GAPDH primers have been
described previously (Villinger et al. 1993). Previously published
primers for IDO (Koide and Yoshida 1994) were used to con®rm
our IDO results (data not shown), and sequences of PCR products
were veri®ed for all primer sets. PCR conditions and cycling
Q3
Q4
JNC498 JNC jn6489 CD 12/7/1 12:38 ALDEN
Kynurenine pathway in astrocytes 3
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13

UNCORRECTED PROOF
parameters were optimized using primary human fetal astrocytes
stimulated with 100 IU/mL IFN-g (data not shown). Negative
controls were: (i) omission of a target template, (ii) omission of
reverse transcriptase, and (iii) genomic DNA. PCR was performed
in an eppendorf gradient cycler (Hamburg, Germany) according to
the following protocol: 35 cycles at 928C for 1 min, 608C for
1 min, and 728C for 90 s. PCR products were loaded in 2%
agarose/1 mg/mL ethidium bromide gel. Ampli®ed products were
visualized under UV light, and quanti®ed after scanning using nih
image 1.61 (NIH, Bethesda, MA, USA). Experiments were
performed in duplicate on cultures derived from three different
fetal brains. Based on image analysis intensity ratios of KP enzyme
mRNA expressed relative to GAPDH mRNA, the standard error
was between 4 and 5%.
Results
Purity of primary cultures of human fetal astrocytes
Cultures of fetal brain cells immunostained with the
astrocytic marker GFAP were 90±95% astrocytic after 7
days in culture and . 99% pure after 1 month of culture
(Fig. 2a). Polyclonal or monoclonal antibodies tested
against galactocerebroside protein (oligodendrocytes),
MAP2 (neurons), factor VIII (endothelial cells) did not
stain any of the cells at 15 or 30 days after seeding.
Monoclonal antibodies directed against CD14 and CD68
proteins showed that , 5% of cells were microglia (Fig. 2b),
Table 1 Sequences of primer sets used to detect KP enzyme expression
Genes Primers Sequences
hu-IDO Forward 5 0-ACCACAAGTCACAGCGCC-3 0
Reverse 5 0-CCCAGCAGGACGTCAAAG-3 0
hu-KAT-1 Forward 5 0-TCGAACCCTTTTTTGACTGC-3 0
Reverse 5 0-CTGCTCCCGTTCAAAGCTCT-3 0
hu-KAT-2 Forward 5 0-AGCTGGCATGTTTCTATGGA-3 0
Reverse 5 0-AAGTGGTTCAGGGAAATTGC-3 0
hu-KYN-OHase Forward 5 0-GATGAGGAAGATAAGCTGAGGC-3 0
Reverse 5 0-CTTAAGGTTTCTTCCCCCTCTC-3 0
hu-KYNase Forward 5 0-GACTATTCCACCTAAGAACGGAGA-3 0
Reverse 5 0-ACAGGAAGACACAAACTAAGGTCG- 3 0
hu-3HAO Forward 5 0-GAGGGCCTTCTTGGTGTG-3 0
Reverse 5 0-CATTGATTGGTTGTGAGTGG-3 0
hu-QPRTase Forward 5 0-GGTCACAGGAGCAGCAGG-3 0
Reverse 5 0-AAGCCAGAGGAGCTGCAC-3 0
Gene sequences have been obtained from GenBank (Internet) and primers have been calculated using Primers for the Mac v1.0a (q1996, Apple
Pi, Ashland, USA).
Fig. 2 Purity of primary cultures of
human fetal astrocytes. (a) GFAP immu-
nocytochemical staining of puri®ed human
primary astrocyte cultures (�400). (b) CD68
immunocytochemical staining of isolated
human primary microglial cells removed
from astrocyte cultures (�400).
JNC498 JNC jn6489 CD 12/7/1 12:38 ALDEN
4 G. J. Guillemin et al.
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13
UNCORRECTED PROOF
however, shaking, immunodepletion and successive trypsi-
nizations led to their total disappearance so that after 3±5
weeks of culture, only astrocytes were present in the
cultures.
Kynurenine and kynurenic acid, but not
3-hydroxykynurenine, are synthesized by puri®ed
cultures of human fetal astrocytes
Kynurenine was not detectable in the culture medium
(containing 10% fetal calf serum). The concentration of
KYNA in the medium was 85.5 ^ 5.1 nm. Unstimulated
astrocytes did not produce detectable amounts of KYN and
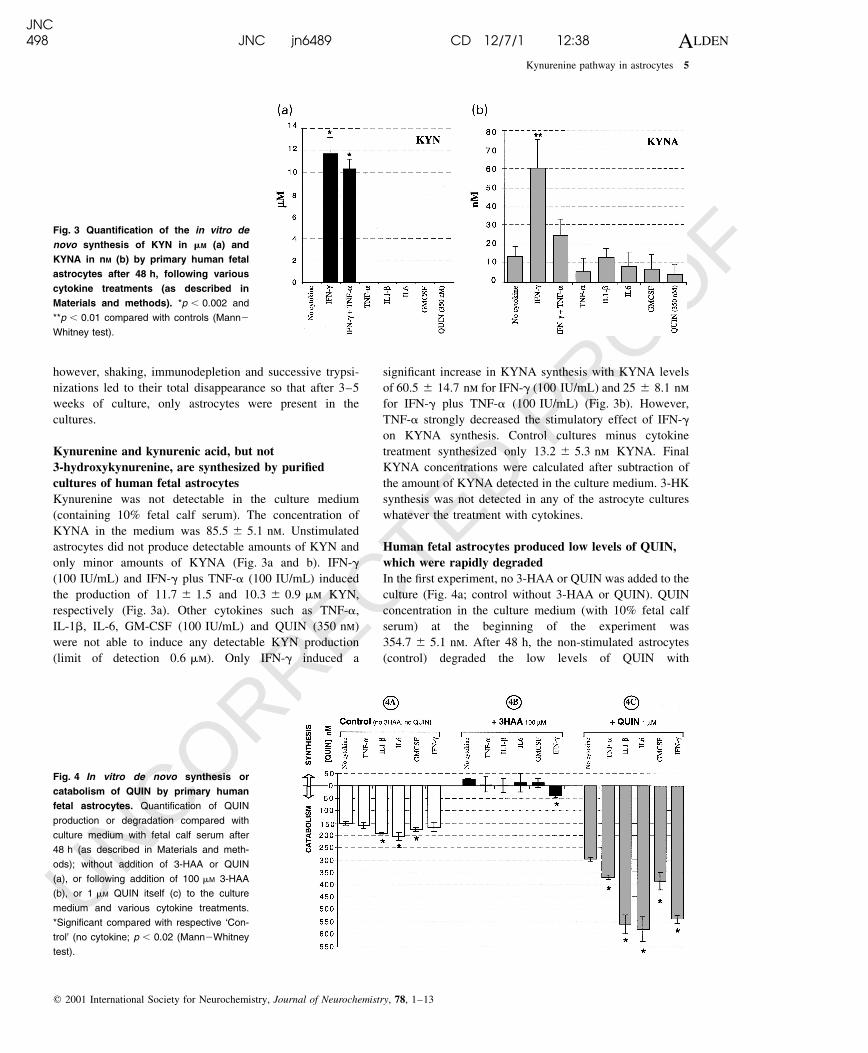
only minor amounts of KYNA (Fig. 3a and b). IFN-g
(100 IU/mL) and IFN-g plus TNF-a (100 IU/mL) induced
the production of 11.7 ^ 1.5 and 10.3 ^ 0.9 mm KYN,
respectively (Fig. 3a). Other cytokines such as TNF-a,
IL-1b, IL-6, GM-CSF (100 IU/mL) and QUIN (350 nm)
were not able to induce any detectable KYN production
(limit of detection 0.6 mm). Only IFN-g induced a
signi®cant increase in KYNA synthesis with KYNA levels
of 60.5 ^ 14.7 nm for IFN-g (100 IU/mL) and 25 ^ 8.1 nm
for IFN-g plus TNF-a (100 IU/mL) (Fig. 3b). However,
TNF-a strongly decreased the stimulatory effect of IFN-g
on KYNA synthesis. Control cultures minus cytokine
treatment synthesized only 13.2 ^ 5.3 nm KYNA. Final
KYNA concentrations were calculated after subtraction of
the amount of KYNA detected in the culture medium. 3-HK
synthesis was not detected in any of the astrocyte cultures
whatever the treatment with cytokines.
Human fetal astrocytes produced low levels of QUIN,
which were rapidly degraded
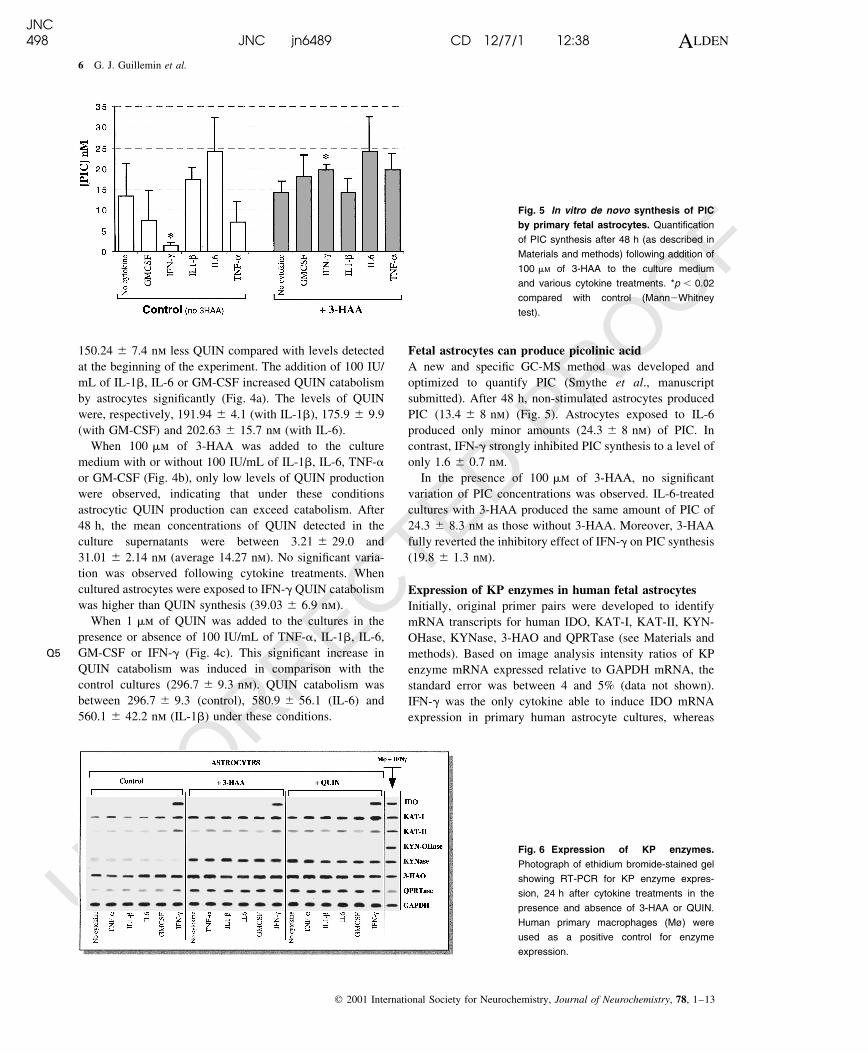
In the ®rst experiment, no 3-HAA or QUIN was added to the
culture (Fig. 4a; control without 3-HAA or QUIN). QUIN
concentration in the culture medium (with 10% fetal calf
serum) at the beginning of the experiment was
354.7 ^ 5.1 nm. After 48 h, the non-stimulated astrocytes
(control) degraded the low levels of QUIN with
Fig. 3 Quanti®cation of the in vitro de
novo synthesis of KYN in mM (a) and
KYNA in nM (b) by primary human fetal
astrocytes after 48 h, following various
cytokine treatments (as described in
Materials and methods). *p , 0.002 and
**p , 0.01 compared with controls (Mann2
Whitney test).
Fig. 4 In vitro de novo synthesis or
catabolism of QUIN by primary human
fetal astrocytes. Quanti®cation of QUIN
production or degradation compared with
culture medium with fetal calf serum after
48 h (as described in Materials and meth-
ods); without addition of 3-HAA or QUIN
(a), or following addition of 100 mM 3-HAA
(b), or 1 mM QUIN itself (c) to the culture
medium and various cytokine treatments.
*Signi®cant compared with respective `Con-
trol' (no cytokine; p , 0.02 (Mann2Whitney
test).
JNC498 JNC jn6489 CD 12/7/1 12:38 ALDEN
Kynurenine pathway in astrocytes 5
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13
UNCORRECTED PROOF
150.24 ^ 7.4 nm less QUIN compared with levels detected
at the beginning of the experiment. The addition of 100 IU/
mL of IL-1b, IL-6 or GM-CSF increased QUIN catabolism
by astrocytes signi®cantly (Fig. 4a). The levels of QUIN
were, respectively, 191.94 ^ 4.1 (with IL-1b), 175.9 ^ 9.9
(with GM-CSF) and 202.63 ^ 15.7 nm (with IL-6).
When 100 mm of 3-HAA was added to the culture
medium with or without 100 IU/mL of IL-1b, IL-6, TNF-a
or GM-CSF (Fig. 4b), only low levels of QUIN production
were observed, indicating that under these conditions
astrocytic QUIN production can exceed catabolism. After
48 h, the mean concentrations of QUIN detected in the
culture supernatants were between 3.21 ^ 29.0 and
31.01 ^ 2.14 nm (average 14.27 nm). No signi®cant varia-
tion was observed following cytokine treatments. When
cultured astrocytes were exposed to IFN-g QUIN catabolism
was higher than QUIN synthesis (39.03 ^ 6.9 nm).
When 1 mm of QUIN was added to the cultures in the
presence or absence of 100 IU/mL of TNF-a, IL-1b, IL-6,
GM-CSF or IFN-g (Fig. 4c). This signi®cant increase in
QUIN catabolism was induced in comparison with the
control cultures (296.7 ^ 9.3 nm). QUIN catabolism was
between 296.7 ^ 9.3 (control), 580.9 ^ 56.1 (IL-6) and
560.1 ^ 42.2 nm (IL-1b) under these conditions.
Fetal astrocytes can produce picolinic acid
A new and speci®c GC-MS method was developed and
optimized to quantify PIC (Smythe et al., manuscript
submitted). After 48 h, non-stimulated astrocytes produced
PIC (13.4 ^ 8 nm) (Fig. 5). Astrocytes exposed to IL-6
produced only minor amounts (24.3 ^ 8 nm) of PIC. In
contrast, IFN-g strongly inhibited PIC synthesis to a level of
only 1.6 ^ 0.7 nm.
In the presence of 100 mm of 3-HAA, no signi®cant
variation of PIC concentrations was observed. IL-6-treated
cultures with 3-HAA produced the same amount of PIC of
24.3 ^ 8.3 nm as those without 3-HAA. Moreover, 3-HAA
fully reverted the inhibitory effect of IFN-g on PIC synthesis
(19.8 ^ 1.3 nm).
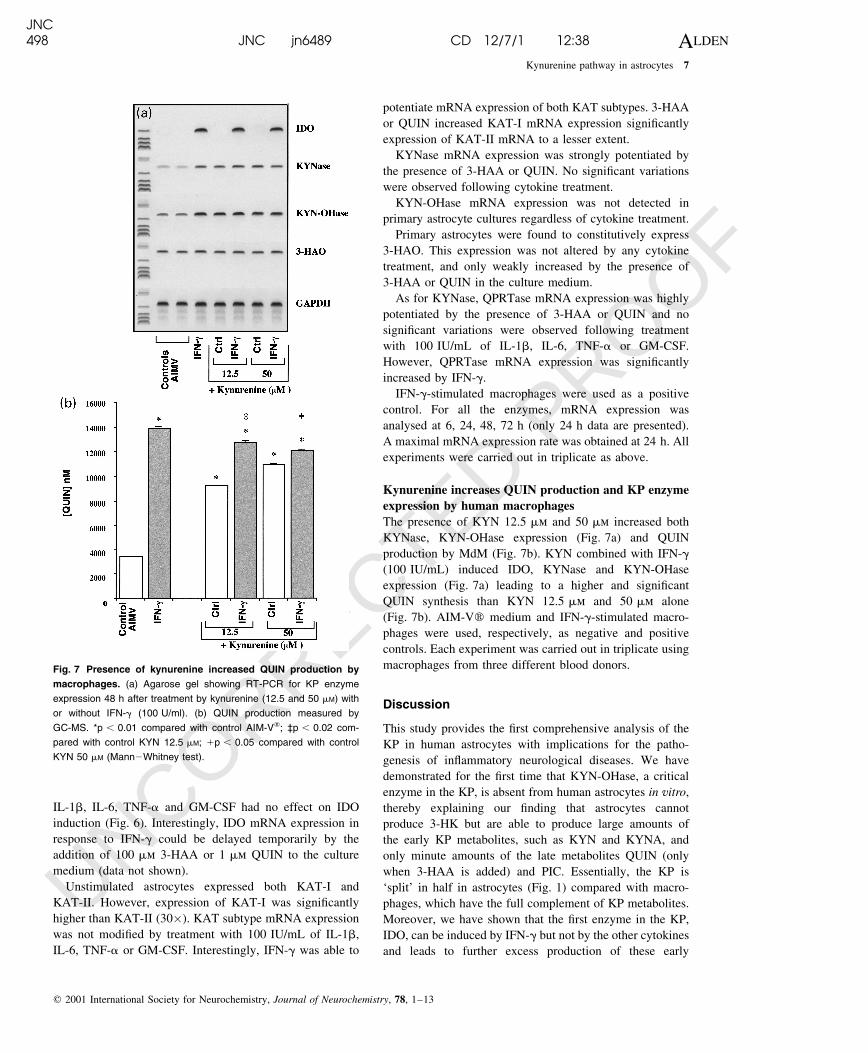
Expression of KP enzymes in human fetal astrocytes
Initially, original primer pairs were developed to identify
mRNA transcripts for human IDO, KAT-I, KAT-II, KYN-
OHase, KYNase, 3-HAO and QPRTase (see Materials and
methods). Based on image analysis intensity ratios of KP
enzyme mRNA expressed relative to GAPDH mRNA, the
standard error was between 4 and 5% (data not shown).
IFN-g was the only cytokine able to induce IDO mRNA
expression in primary human astrocyte cultures, whereas
Fig. 5 In vitro de novo synthesis of PIC
by primary fetal astrocytes. Quanti®cation
of PIC synthesis after 48 h (as described in
Materials and methods) following addition of
100 mM of 3-HAA to the culture medium
and various cytokine treatments. *p , 0.02
compared with control (Mann2Whitney
test).
Fig. 6 Expression of KP enzymes.
Photograph of ethidium bromide-stained gel
showing RT-PCR for KP enzyme expres-
sion, 24 h after cytokine treatments in the
presence and absence of 3-HAA or QUIN.
Human primary macrophages (Mù) were
used as a positive control for enzyme
expression.
Q5
JNC498 JNC jn6489 CD 12/7/1 12:38 ALDEN
6 G. J. Guillemin et al.
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13
UNCORRECTED PROOF
IL-1b, IL-6, TNF-a and GM-CSF had no effect on IDO
induction (Fig. 6). Interestingly, IDO mRNA expression in
response to IFN-g could be delayed temporarily by the
addition of 100 mm 3-HAA or 1 mm QUIN to the culture
medium (data not shown).
Unstimulated astrocytes expressed both KAT-I and
KAT-II. However, expression of KAT-I was signi®cantly
higher than KAT-II (30�). KAT subtype mRNA expression
was not modi®ed by treatment with 100 IU/mL of IL-1b,
IL-6, TNF-a or GM-CSF. Interestingly, IFN-g was able to
potentiate mRNA expression of both KAT subtypes. 3-HAA
or QUIN increased KAT-I mRNA expression signi®cantly
expression of KAT-II mRNA to a lesser extent.
KYNase mRNA expression was strongly potentiated by
the presence of 3-HAA or QUIN. No signi®cant variations
were observed following cytokine treatment.
KYN-OHase mRNA expression was not detected in
primary astrocyte cultures regardless of cytokine treatment.
Primary astrocytes were found to constitutively express
3-HAO. This expression was not altered by any cytokine
treatment, and only weakly increased by the presence of
3-HAA or QUIN in the culture medium.
As for KYNase, QPRTase mRNA expression was highly
potentiated by the presence of 3-HAA or QUIN and no
signi®cant variations were observed following treatment
with 100 IU/mL of IL-1b, IL-6, TNF-a or GM-CSF.
However, QPRTase mRNA expression was signi®cantly
increased by IFN-g.
IFN-g-stimulated macrophages were used as a positive
control. For all the enzymes, mRNA expression was
analysed at 6, 24, 48, 72 h (only 24 h data are presented).
A maximal mRNA expression rate was obtained at 24 h. All
experiments were carried out in triplicate as above.
Kynurenine increases QUIN production and KP enzyme
expression by human macrophages
The presence of KYN 12.5 mm and 50 mm increased both
KYNase, KYN-OHase expression (Fig. 7a) and QUIN
production by MdM (Fig. 7b). KYN combined with IFN-g
(100 IU/mL) induced IDO, KYNase and KYN-OHase
expression (Fig. 7a) leading to a higher and signi®cant
QUIN synthesis than KYN 12.5 mm and 50 mm alone
(Fig. 7b). AIM-Vw medium and IFN-g-stimulated macro-
phages were used, respectively, as negative and positive
controls. Each experiment was carried out in triplicate using
macrophages from three different blood donors.
Discussion
This study provides the ®rst comprehensive analysis of the
KP in human astrocytes with implications for the patho-
genesis of in¯ammatory neurological diseases. We have
demonstrated for the ®rst time that KYN-OHase, a critical
enzyme in the KP, is absent from human astrocytes in vitro,
thereby explaining our ®nding that astrocytes cannot
produce 3-HK but are able to produce large amounts of
the early KP metabolites, such as KYN and KYNA, and
only minute amounts of the late metabolites QUIN (only
when 3-HAA is added) and PIC. Essentially, the KP is
`split' in half in astrocytes (Fig. 1) compared with macro-
phages, which have the full complement of KP metabolites.
Moreover, we have shown that the ®rst enzyme in the KP,
IDO, can be induced by IFN-g but not by the other cytokines
and leads to further excess production of these early
Fig. 7 Presence of kynurenine increased QUIN production by
macrophages. (a) Agarose gel showing RT-PCR for KP enzyme
expression 48 h after treatment by kynurenine (12.5 and 50 mM) with
or without IFN-g (100 U/ml). (b) QUIN production measured by
GC-MS. *p , 0.01 compared with control AIM-Vw; ³p , 0.02 com-
pared with control KYN 12.5 mM; 1p , 0.05 compared with control
KYN 50 mM (Mann2Whitney test).
JNC498 JNC jn6489 CD 12/7/1 12:38 ALDEN
Kynurenine pathway in astrocytes 7
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13

UNCORRECTED PROOF
metabolites. Although there is overproduction of early
metabolites, the amounts of KYNA, an antagonist of
QUIN, are 200-fold less than those of KYN, probably
secondary to a 30-fold lower expression of KAT-II
compared with KAT-I. Our results also demonstrate that
the production of the late metabolites was more complex.
Astrocytes express an ef®cient IFN-g-inducible QPRTase,
which explains our ®nding that astrocytes degrade QUIN.
However, we also showed that the excess of KYN,
synthesized by the cytokine-stimulated astrocytes could be
metabolized by macrophages to produce signi®cant con-
centrations of QUIN. These ®ndings have implications not
only for AIDS dementia complex, but also for other
in¯ammatory conditions of the human CNS.
Until now, the capacity of astrocytes to produce QUIN
and other KP metabolites has been controversial and the KP
enzymes within astrocytes have not been delineated in full.
Of particular interest under our culture conditions with
puri®ed cells, human fetal astrocytes were primarily
associated with QUIN catabolism rather than synthesis. In
contrast, in rat brain, several biochemical (Schwarcz et al.
1988a) and immunocytochemical (KoÈhler et al. 1988a, b)
studies have shown that QUIN production from 3-HAA can
take place within astrocytes (Speciale and Schwarcz 1993)
and injection of l-KYN (Guidetti et al. 1995) or 3-HAA
(Heyes and Markey 1988; Speciale and Schwarcz 1993)
induces an increase in QUIN production. The controversial
issue regarding astrocytic participation in the in¯ammatory/
neurotoxic process arises from con¯icting results from other
studies of rat and human brain cell cultures and gerbil
astrocytes. For example, either human fetal brain cell
cultures enriched in astrocytes (Heyes et al. 1997a) or
activated gerbil astrocytes (Heyes et al. 1997b) have the
capacity to produce l-KYN and QUIN but the levels
produced are very low compared with microglia or
macrophages (Heyes et al. 1996; Espey et al. 1997).
Conversely, other studies in rat brain have shown that
QUIN immunoreactivity is only detectable in activated
macrophages/microglia but not in reactive astrocytes
(Moffett et al. 1997). Also, in rat primary mixed brain cell
cultures composed of 60% astrocytes, no QUIN production
was detected after 3-HAA was added (Heyes et al. 1997a).
Thus, our ®nding that puri®ed primary cultures of human
fetal astrocytes can produce small amounts of QUIN is in
agreement with other reports (KoÈhler et al. 1988b, c;
Schwarcz et al. 1988a; Speciale and Schwarcz 1993; Heyes
et al. 1996, 1997a). The important difference in our system
is that this production occurred only after 3-HAA was added
to the culture medium, a precursor not normally available to
brain tissue (Saito et al. 1993a). These con¯icting results
may well re¯ect species differences but could also relate to
both the relative insensitivity of immunocytochemical
methods and the use of mixed brain cell cultures rather
than puri®ed primary cultured human astrocytes.
Our results for the expression of KP enzymes in human
astrocytes con®rm and extend previous studies (Okuno et al.
1987; KoÈhler et al. 1988a, b; Schwarcz et al. 1988a; Du et al.
1991; Roberts et al. 1992) in that IDO is present in these
astrocytes and is inducible by IFN-g (Werner-Felmayer et al.
1989; Saito et al. 1993b; Alberati-Giani et al. 1996; Heyes
et al. 1996, 1997a). However, in contrast with studies
employing macrophages (Heyes et al. 1997a; Pemberton
et al. 1997), TNF-a did not activate IDO or induce QUIN
production.
Our results concerning KYNase and KYN-OHase are
only partly in accord with published data. We have shown
that KYNase is expressed by isolated human astrocytes and
3-HAA or QUIN potentiate this expression. Biochemical
studies by others, however, have shown that KYNase
activity is very low in astrocytoma cell lines and not
detectable in human fetal brain cell cultures (Heyes et al.
1996, 1997a). KYN-OHase has recently been detected in
adult rat neurons and astrocytes using immunohistochem-
istry (Chiarugi et al. 2001) but KYN-OHase activity is
undetectable in either human astrocytoma or human fetal
brain cell cultures (Heyes et al. 1996, 1997a). However, we
demonstrated that KYN-OHase expression is absent in
human fetal astrocytes in accord with existing literature.
Moreover, we did not detect any expression of KYN-OHase
mRNA in puri®ed and concentrated mitochondrial fraction
from human fetal astrocytes (Almeida and Medina 1998)
stimulated or not with IFN-g (data not shown). These results
were con®rmed by the lack of production of 3-HK, a KP
metabolite able to potentiate QUIN neurotoxicity (Eastman
1989). Thus, we propose that 3-HAA is either not
synthesized by human astrocytes or produced in very low
amounts by an alternative pathway (Baran and Schwarcz
1990) mediated by KYNase or following a potential uptake
of exogenous 3-HK (Eastman et al. 1992) (Fig. 1).
In accordance with previous studies showing that KAT
immunoreactivity and expression have been detected mostly
in astrocytes in the rat brain (Roberts et al. 1992; Guidetti
et al. 1997), we have shown that human astrocytes express
both KAT-I and KAT-II but with a KAT-II expression
30-fold than that of KAT-I. This enzyme expression is well
correlated with the ability of astrocytes to produce KYNA
from KYN (Speciale and Schwarcz 1990; Guidetti et al.
1997). In accordance with the fact that inhibition of KYN-
OHase leads to increase of KYNA production in mouse
brain (Chiarugi et al. 1996) and previous results from
Speciale and Schwarcz (1990), we showed that astrocytes
produce only a limited quantity of KYNA from KYN
(Fig. 3). Indeed, the ratio KYNA/KYN is 0.0051 (i.e. 195-
fold less KYNA than KYN). This poor production yield is
probably associated with the difference of expression of the
KAT subtypes in human astrocytes (Guidetti et al. 1997).
Nevertheless, we observed a very good correlation between
IFN-g induction of increased expression of KAT-I and
Q6
Q7
JNC498 JNC jn6489 CD 12/7/1 12:39 ALDEN
8 G. J. Guillemin et al.
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13

UNCORRECTED PROOF
KAT-II, and increased KYNA synthesis in astrocytes. We
con®rmed the hypothesis that the astroglial KP stops at the
KYN step and that this leads chie¯y to the synthesis of
KYNA mainly by KAT-II, in accordance with Schwarcz
et al. (1998) and Speciale and Schwarcz (1990).
Previous immunocytochemical studies have shown that
most of 3-HAO, but not the majority of QPRTase, is
contained in astrocytes (KoÈhler et al. 1988a, b, c; Roberts
et al. 1994). These observations are in accordance with our
molecular results which show strong constitutive 3-HAO
expression and weak QPRTase expression in our astrocyte
cultures. Because of the lack of KYN-OHase expression and
thus 3-HK production, only in the presence of 3-HAA did
3-HAO activity `overtake' QPRTase activity leading to a
limited production of QUIN by astrocytes (Fig. 4b). In our
in vitro conditions, astrocytes were more capable of
catabolizing QUIN than producing it. Moreover, the facts
that: (i) the addition of QUIN increased QPRTase expres-
sion con®rms that the speci®c enzyme for QUIN catabolism
is functional in astrocytes, and (ii) the addition of 3-HAA
increased QPRTase expression, con®rm both that astrocytes
can produce QUIN from 3-HAA even if it is a low amount.
The presence of QUIN in astrocytes is the result of an
imbalance between the activities of the QUIN synthetic and
catabolic enzymes which apparently can only be modi®ed
by addition of 3-HAA leading to saturation of QPRTase
by QUIN.
The exact mechanism of QUIN catabolism by astrocyte
is still not identi®ed. QUIN is probably taken up within the
cell but no speci®c receptor or membranous system for
QUIN has been identi®ed on the astrocyte yet. Moreover,
the fact that several studies have shown that QPRTase is
mainly detected within astrocytes (KoÈhler et al. 1988a, b, c;
Du et al. 1991, 1992, 1993) and that others failed to
detect QUIN within astrocyte (Moffett et al. 1993, 1997),
make it highly likely that QUIN catabolism is intra-
cellular. A less likely possibility, is that QPRTase may be
secreted by astrocytes, leading to an extracellular catabolism
of QUIN. Nonetheless, entry of QUIN and/or release of
QPRTase require the presence of a transport system across
the plasma membrane, the identity of which is still
unknown.
The integration of our results leads us to hypothesize that
KYNase activity is only just enough to bypass the missing
KYN-OHase step via production of minor amounts of
anthranilic acid, which may then lead to 3-HAA production,
and later to minor amounts of QUIN, which are then
catabolized (Fig. 1). This is supported both by previous
studies showing that anthranilic acid can be converted to late
KP metabolites (Bender and McCreanor 1982; Baran and
Schwarcz 1990, 1991; Fujigaki et al. 1998) and secondarily
by the high constitutive expression of 3-HAO by astrocytes.
However, results from Eastman et al. (1992), which showed
that astrocytes are able to take up 3-HK, lead to the
hypothesis that this intermediate KP metabolite may be used
as a substrate for QUIN synthesis by the astrocytes. But
again, there must be a membranous transport system for
3-HK, which is still unknown.
Although, our data relate to fetal astrocytes cultures, we
think that it is highly likely that they re¯ect what is
happening in adult astrocytes. The fetal astrocytes exhibit all
characteristics of mature cells (GFAP1, vimentin1,
proliferation). Moreover, our more recent preliminary
results obtained with primary human adult astrocytes have
shown that the levels of expression of KP enzymes are very
close and well conserved between fetal and adult cells (data
not shown). However, the interest of our human fetal model
is emphasized by the relevance of the rat model for KP
studies. Indeed, the rat has a limited capacity to induce IDO
in brain and thereby a limited capacity for QUIN production
Fig. 8 Model for kynurenine pathway
interactions between astrocytes and
leukocytes during brain in¯ammation.
Q8
JNC498 JNC jn6489 CD 12/7/1 12:39 ALDEN
Kynurenine pathway in astrocytes 9
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13

UNCORRECTED PROOF
in response to immune activation (Mukhopadhyay et al.
1990; Heyes et al. 1997a, b; Fujigaki et al. 1999).
An interesting aspect of our results is the parallel between
what we have shown and what new drugs have been
developed to modify the KP in preventing or treating brain
damages associated with in¯ammatory diseases, neurode-
generative disorders and stroke. New generations of KYNA
analogues, acting directly on the glutamate receptors, and
the KYN-OHase inhibitors, blocking the synthesis of QUIN
and promoting the formation of KYNA, are both very
promising developments (Stone 2000a, b). The KYN-OHase
inhibitors essentially mimic what the astrocyte does. More-
over, our model may be useful in developing, testing or
validating these new drugs.
Our ®nding of the limited KP expression in human
astrocytes leads to the possibility that astroglial KP may be
involved not only in AIDS dementia complex, but also in the
pathogenesis of a number of other brain diseases (Heyes
et al. 1991, 1992, 1998; Heyes, 1993; Brew et al. 1993,
1996), Huntington's disease (Schwarcz et al. 1988a, b),
Alzheimer's disease (Baran et al. 1999), Down syndrome
(Baran et al. 1996), septicaemia-related encephalopathy
(Heyes and Lackner 1990), toxoplasmosis (Daubener et al.
1993), malaria (Sanni et al. 1998), epilepsy (Du et al. 1993)
and traumatic brain injury (Sinz et al. 1998).
In the absence of macrophages and/or microglia, we
suggest that astrocytes protect neurons by catabolizing any
synthesized QUIN and by producing KYNA, an antagonist
of QUIN (Perkins and Stone 1982). Further, we propose that
during in¯ammation of the brain, activated astrocytes may
`inadvertently' and indirectly contribute to neuronal damage
by producing KYN thus `unwittingly' providing a substrate
for QUIN production by in®ltrating macrophages (Heyes
et al. 1997a) and/or activated resident microglia (Saito et al.
1993a; Heyes et al. 1996) (Fig. 8). In support of this model,
we and colleagues have shown that macrophages and
microglia can metabolize KYN to QUIN (Venkateshan
et al. 1996; Heyes et al. 1997a). Indeed, our results
demonstrate that 12.5 mm of KYN (equivalent to the
concentration of KYN produced by cultured astrocytes
after IFN-g stimulation) is suf®cient to signi®cantly increase
QUIN synthesis by blood macrophages to levels that are
neurotoxic. Moreover, there is evidence that the astrocytic
QPRTase cannot catabolize all the QUIN produced by
in®ltrating macrophages during brain in¯ammation. First,
3-HAO activity is signi®cantly higher than QPRTase within
the brain (Foster et al. 1985, 1986). Secondly, our results
imply that QPRTase levels are easily saturable. For
example, after 48 h, QUIN catabolism in astrocytes
switched to synthesis in the presence of 10 mm (data not
shown) or 100 mm of 3-HAA. Moreover, astrocytes only
degraded < 50% of QUIN added in the culture (Fig. 4). If
this latter catabolic factor is applied to levels of QUIN
produced by macrophages after IFN-g stimulation for 48 h,
the theoretical capacity of astroglial QPRTase to catabolize
QUIN is overcome (Fig. 7). Finally, Nottet et al. (1996)
showed that QUIN production by lipopolysaccharide-activated
macrophages in coculture with primary astrocytes (1 : 1) is
decreased only slightly and the QUIN concentration is still
high enough to be neurotoxic (270 nm).
In conclusion, we have shown that astrocytes lack KYN-
OHase. This could prove neuroprotective in the absence of
in®ltrating macrophages and/or activated resident microglia.
During in¯ammation, however, the in®ltration of activated
macrophages and the activation of microglia could result in
the contribution of astrocytes to neurotoxicity.
Acknowledgements
This work was supported by La Fondation pour la Recherche
Medicale/SIDACTION, Institute LILLY, INSERM, Australian
Brain Foundation, NHMRC (grant 157136), and NSW Health
Department.
References
Alberati-Giani D., Ricciardi-Castagnoli P., Kohler C. and Cesura A. M.
(1996) Regulation of the kynurenine metabolic pathway by
interferon-gamma in murine cloned macrophages and microglial
cells. J. Neurochem. 66, 996±1004.
Almeida A. and Medina J. M. (1998) A rapid method for the isolation of
metabolically active mitochondria from rat neurons and astrocytes
in primary culture. Brain Res. Brain Res. Protoc. 2, 209±214.
Baran H. and Schwarcz R. (1990) Presence of 3-hydroxyanthranilic acid
in rat tissues and evidence for its production from anthranilic acid
in the brain. J. Neurochem. 55, 738±744.
Baran H. and Schwarcz R. (1991) Evidence for the preferential
production of 3-hydroxyanthranilic acid from anthranilic acid in
the rat brain. Adv. Exp. Med. Biol. 294, 485±488.
Baran H., Cairns N., Lubec B. and Lubec G. (1996) Increased kynurenic
acid levels and decreased brain kynurenine aminotransferase I in
patients with Down syndrome. Life Sci. 58, 1891±1899.
Baran H., Jellinger K. and Deecke L. (1999) Kynurenine metabolism in
Alzheimer's disease. J. Neural Transm. 106, 165±181.
Bender D. A. and McCreanor G. M. (1982) The preferred route of
kynurenine metabolism in the rat. Biochim. Biophys. Acta 717,
56±60.
Botting N. P. (1995) Chemistry and neurochemistry of the kynurenine
pathway of tryptophan metabolism. Chem. Soc. Rev. 24, 401±412.
Brew B. J., Pemberton L., Evans L. and Heyes M. (1993) Quinolinic
acid production by macrophages infected with demented and non-
demented isolates of HIV. Clin. Neuropathol. 12, S1.
Brew B. J., Wesselingh S. L., Gonzales M., Heyes M. P. and Price R.
W. (1996) How HIV leads to neurological disease. Med. J. Aust.
164, 233±234.
Chiarugi A., Carpenedo R. and Moroni F. (1996) Kynurenine
disposition in blood and brain of mice: effects of selective
inhibitors of kynurenine hydroxylase and kynureninase.
J. Neurochem. 67, 692±698.
Chiarugi A., Cozzi A., Ballerini C., Massacesi L. and Moroni F. (2001)
Kynurenine 3-mono-oxygenase activity and neurotoxic kynure-
nine metabolites increase in the spinal cord of rats with
experimental allergic encephalomyelitis. Neuroscience 102,
687±695.
Q9
JNC498 JNC jn6489 CD 12/7/1 12:39 ALDEN
10 G. J. Guillemin et al.
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13

UNCORRECTED PROOF
Curzon G. (1996) Brain tryptophan. Normal and disturbed control. Adv.
Exp. Med. Biol. 398, 27±34.
Daubener W., Pilz K., Seghrouchni Zennati S., Bilzer T., Fischer H. G.
and Hadding U. (1993) Induction of toxoplasmostasis in a human
glioblastoma by interferon gamma. J. Neuroimmunol. 43, 31±38.
Du F., Okuno E., Whetsell W. O., KoÈhler C. and Schwarcz R. (1991)
Immunohistochemical localization of quinolinic acid phosphor-
ibosyltransferase in the human neostriatum. Neuroscience 42,
397±406.
Du F., Schmidt W., Okuno E., Kido R., Kohler C. and Schwarcz R.
(1992) Localization of kynurenine aminotransferase immuno-
reactivity in the rat hippocampus. J. Comp. Neurol. 321, 477±487.
Du F., Williamson J., Bertram E., Lothman E., Okuno E. and Schwarcz
R. (1993) Kynurenine pathway enzymes in a rat model of chronic
epilepsy: immunohistochemical study of activated glial cells.
Neuroscience 55, 975±989.
Eastman C. L. (1989) Cytotoxicity of 3-hydroxykynurenine in a
neuronal hybrid cell line. Brain Res. 495, 225±231.
Eastman C. L., Guilarte T. R. and Lever J. R. (1992) Uptake of
3-hydroxykynurenine measured in rat brain slices and in a
neuronal cell line. Brain Res. 584, 110±116.
Espey M. G., Chernyshev O. N., Reinhard J. J., Namboodiri M. A. and
Colton C. A. (1997) Activated human microglia produce the
excitotoxin quinolinic acid. Neuroreport 8, 431±434.
Foster A. C., Whetsell W. O. Jr, Bird E. D. and Schwarcz R. (1985)
Quinolinic acid phosphoribosyltransferase in human and rat brain:
activity in Huntington's disease and in quinolinate-lesioned rat
striatum. Brain Res. 336, 207±214.
Foster A. C., White R. J. and Schwarcz R. (1986) Synthesis of
quinolinic acid by 3-hydroxyanthranilic acid oxygenase in rat
brain tissue in vitro. J. Neurochem. 47, 23±30.
Fujigaki S., Saito K., Takemura M., Fujii H., Wada H., Noma A.
and Seishima M. (1998) Species differences in l-tryptophan±
kynurenine pathway metabolism: quanti®cation of anthranilic acid
and its related enzymes. Arch. Biochem. Biophys. 358, 329±335.
Fujigaki S., Saito K., Fujii H., Wada H. and Seishima M. (1999)
Quanti®cation of anthranilic acid and its related enzyme activity
in several different species. Adv. Exp. Med. Biol. 467, 625±628.
Guidetti P., Okuno E. and Schwarcz R. (1997) Characterization of rat
brain kynurenine aminotransferases I and II. J. Neurosci. Res. 50,
457±465.
Guillemin G., Boussin F., Croitoru J., Le Grand R. and Dormont D.
(1997) Obtention and characterization of astrocytes and microglia
from adult monkey brains. J. Neurosci. Res. 49, 576±591.
Heyes M. P. and Lackner A. (1990) Increased cerebrospinal ¯uid
quinolinic acid, kynurenic acid and l-kynurenine in acute
septicemia. J. Neurochem. 55, 338±341.
Heyes M. P., Brew B. J., Martin A., Price R. W., Salazar A. M., Sidtis
J. J., Yergey J. A., Mouradian M. M., Salder A. E., Keilp J.,
Rubinow D. and Markey S. P. (1991) Quinolinic acid in
cerebrospinal ¯uid and serum in HIV-1 infection: relationship to
clinical and neurological status. Ann. Neurol. 29, 202±209.
Heyes M. P., Saito K., Crowley J. S., Davis L. E., Der Demitrack
M. A. M., Dilling L. A., Elia J., Kruesi M. J. P., Lackner A.,
Larsen S. A., Lee K., Leonard H. L., Markey S. P., Martin A.,
Milstein S., Mouradian M. M., Pranzatelli M. R., Quearry B. J.,
Salazar A., Smith M., Strauss S. E., Sunderland T., Swedo S. W.
and Tourtellotte W. W. (1992) Quinolinic acid and kynurenine
pathway metabolism in in¯ammatory and non-in¯ammatory
neurological disease. Brain 115, 1249±1273.
Heyes M. P. (1993) Metabolism and neuropathologic signi®cance of
quinolinic acid and kynurenic acid. Biochem. Soc. Trans. 21,
83±89.
Heyes M. P. (1996) The kynurenine pathway and neurologic disease.
Therapeutic strategies. Adv. Exp. Med. Biol. 398, 125±129.
Heyes M. P., Achim C. L., Wiley. C. A., Major E. O., Saito K. and
Markey S. P. (1996) Human microglia convert l-tryptophan into
the neurotoxin quinolinic acid. Biochem. J. 320, 595±597.
Heyes M. P., Chen C. Y., Major E. O. and Saito K. (1997a) Different
kynurenine pathway enzymes limit quinolinic acid formation by
various human cell types. Biochem. J. 326, 351±356.
Heyes M. P., Saito K., Chen C. Y., Proescholdt M. G., Nowak T. S.,
Li J., Beagles K. E., Proescholdt M. A., Zito M. A., Kawai K. and
Markey S. P. (1997b) Species heterogeneity between gerbils
and rats: quinolinate production by microglia and astrocytes and
accumulations in response to ischemic brain injury and systemic
immune activation. J. Neurochem. 69, 1519±1529.
Heyes M. P., Saito K., Lackner A., Wiley. C. A., Achim C. L. and
Markey S. P. (1998) Sources of the neurotoxin quinolinic acid in
the brain of HIV-1- infected patients and retrovirus-infected
macaques [In Process Citation]. Faseb J. 12, 881±896.
Kapoor V., Kapoor R. and Chalmers J. (1994) Kynurenic acid, an
endogenous glutamate antagonist, in SHR and WKY rats: possible
role in central blood pressure regulation. Clin. Exp. Pharmacol.
Physiol. 21, 891±896.
Kerr S. J., Armati P. J., Pemberton L. A., Smythe G., Tattam B. and
Brew B. J. (1997) Kynurenine pathway inhibition reduces toxicity
of HIV-infected macrophages. Neurology 49, 1671±1681.
KoÈhler C., Eriksson L. G., Flood P. R., Hardie J. A., Okuno E. and
Schwarcz R. (1988a) Quinolinic acid metabolism in the rat brain.
Immunohistochemical identi®cation of 3-hydroxyanthranilic acid
oxygenase and quinolinic acid phosphoribosyltransferase in the
hippocampal region. J. Neurosci. 8, 975±987.
KoÈhler C., Eriksson L. G., Okuno E. and Schwarcz R. (1988b)
Localization of quinolinic acid metabolising enzymes in the rat
brain using antibodies to 3-hydroxyanthranilic acid oxygenase
and quinolinic acid phosphoribosyltransferase. Neuroscience 27,
49±76.
KoÈhler C., Peterson A., Eriksson L. G., Okuno E. and Schwarcz R.
(1988c) Immunohistochemical identi®cation of quinolinic acid
phosphoribosyltransferase in glial cultures from rat brain.
Neurosci. Lett. 84, 115±119.
Koide Y. and Yoshida A. (1994) The signal transduction mechanism
responsible for gamma-interferon induced indoleamine 2,3-
dioxygenase gene expression. Infect. Immun. 57, 3254±3256.
Lapin I. P., Prakhie I. B. and Kiseleva I. P. (1982) Excitatory effects of
kynurenine and its metabolites, amino acids and convulsants
administered into brain ventricles: differences between rats and
mice. J. Neural Transm. 54, 229±238.
Melillo G., Bosco M. C., Musso T. and Varesio L. (1996)
Immunobiology of picolinic acid. Adv. Exp. Med. Biol. 398,
135±141.
Moffett J. R., Els T., Espey M. G., Walter S. A., Streit W. J. and
Namboodiri M. A. (1997) Quinolinate immunoreactivity in
experimental rat brain tumors is present in macrophages but not
in astrocytes. Exp. Neurol. 144, 287±301.
Moffett J. R., Espey M. G., Gaudet S. J. and Namboodiri M. A. A.
(1993) Antibodies to quinolinic acid reveal localization in select
immune cells rather than neurons or astroglia. Brain Res. 623,
337±340.
Moroni F. (1999) Tryptophan metabolism and brain function: focus on
kynurenine and other indole metabolites [In Process Citation].
Eur. J. Pharmacol. 375, 87±100.
Mukhopadhyay A., Mungre S. M. and Deshmukh D. R. (1990)
Comparison of lysine and tryptophan catabolizing enzymes in
rat and bovine tissues. Experientia 46, 874±876.
Munn D. H., Zhou M., Attwood J. T., Bondarev I., Conway S. J.,
JNC498 JNC jn6489 CD 12/7/1 12:39 ALDEN
Kynurenine pathway in astrocytes 11
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13

UNCORRECTED PROOF
Marshall B., Brown C. and Mellor A. L. (1998) Prevention of
allogeneic fetal rejection by tryptophan catabolism [see com-
ments]. Science 281, 1191±1193.
Nottet H. S., Flanagan E. M., Flanagan C. R., Gelbard H. A.,
Gendelman H. E. and Reinhard J. J. (1996) The regulation of
quinolinic acid in human immunode®ciency virus-infected mono-
cytes. J. Neurovirol. 2, 111±117.
Okuno E., Kohler C. and Schwarcz R. (1987) Rat 3-HAO: puri®cation
from the liver and immunocytochemical localisation in the brain.
J. Neurochem. 49, 771±780.
Pemberton L. A., Kerr S. J., Smythe G. and Brew B. J. (1997)
Quinolinic acid production by macrophages stimulated with
IFN-gamma, TNF-alpha, and IFN-alpha. J. Interferon Cytokine
Res. 17, 589±595.
Perkins M. N. and Stone T. W. (1982) An ionophoretic investigation of
the actions of convulsant kynurenines and their interaction with
the endogenous excitant quinolinic acid. Brain Res. 247, 184±187.
Roberts R. C., Du F., McCarthy K. E., Okuno E. and Schwarcz R.
(1992) Immunocytochemical localization of kynurenine amino-
transferase in the rat striatum: a light and electron microscopic
study. J. Comp. Neurol. 326, 82±90.
Roberts R. C., McCarthy K. E., Du F., Okuno E. and Schwarcz R.
(1994) Immunocytochemical localization of the quinolinic acid
synthesizing enzyme, 3-hydroxyanthranilic acid oxygenase, in the
rat substantia nigra. Brain Res. 650, 229±238.
Saito K., Crowley J. S., Markey S. P. and Heyes M. P. (1993a)
A mechanism for increased quinolinic acid formation follow-
ing acute systemic immune stimulation. J. Biol. Chem. 268,
15496±15503.
Saito K., Nowak T. S., Suyama K., Quearry B. J., Saito M., Crowley
J. S., Markey S. P. and Heyes M. P. (1993b) Kynurenine pathway
enzymes in brain: responses to ischaemic brain injury versus
systemic immune activation. J. Neurochem. 61, 2061±2070.
Sanni L. A., Thomas S. R., Tattam B. N., Moore D. E., Chaudhri G.,
Stocker R. and Hunt N. H. (1998) Dramatic changes in oxidative
tryptophan metabolism along the kynurenine pathway in experi-
mental cerebral and non-cerebral malaria. Am. J. Pathol. 152,
611±619.
Schwarcz R., Okuno E., White R. J., Bird E. D. and Whetsell W. O. Jr
(1988a) 3-Hydroxyanthranilate oxygenase activity is increased in
the brains of Huntington disease victims. Proc. Natl Acad. Sci.
USA 85, 4079±4081.
Schwarcz R., Tamminga C. A., Kurlan R. and Shoulson I. (1988b)
Cerebrospinal ¯uid levels of quinolinic acid in Huntington's
disease and schizophrenia. Ann. Neurol. 24, 580±582.
Schwarcz R., Whetsell W. O. J. and Mangano R. M. (1983) Quinolinic
acid: an endogenous metabolite that produces axon-sparing
lesions in rat brain. Science 219, 316±318.
Sinz E. H., Kochanek P. M., Heyes M. P., Wisniewski S. R., Bell M. J.,
Clark R. S., DeKosky S. T., Blight A. R. and Marion D. W. (1998)
Quinolinic acid is increased in CSF and associated with mortality
after traumatic brain injury in humans. J. Cereb. Blood Flow
Metab. 18, 610±615.
Speciale C. and Schwarcz R. (1990) Uptake of kynurenine into rat brain
slices. J. Neurochem. 54, 156±163.
Speciale C. and Schwarcz R. (1993) On the production and disposition
of quinolinic acid in rat brain and liver slices. J. Neurochem. 60,
212±218.
Stone T. W. (1993) Neuropharmacology of quinolinic and kynurenic
acids. Pharmacol. Rev. 45, 309±379.
Stone T. W. (2000a) Development and therapeutic potential of
kynurenic acid and kynurenine derivatives for neuroprotection.
Trends Pharmacol. Sci. 21, 149±154.
Stone T. W. (2000b) Inhibitors of the kynurenine pathway. Eur. J. Med.
Chem. 35, 179±186.
Tamburin M., Mostardini M. and Benatti L. (1999) Kynurenine
aminotransferase I (KATI) isoform gene expression in the rat
brain: an in situ hybridization study. Neuroreport 10, 61±65.
Venkateshan C. N., Narayanan R., Espey M. G., Moffett J. R., Gajdusek
D. C., Gibbs C. J. Jr and Namboodiri M. A. (1996) Immuno-
cytochemical localization of the endogenous neuroexcitotoxin
quinolinate in human peripheral blood monocytes/macrophages
and the effect of human T-cell lymphotropic virus type I infection.
Proc. Natl Acad. Sci. USA 93, 1636±1641.
Villinger F., Hunt D., Mayne A., Vuchetiv M., Findley H. and Ansari
A. A. (1993) Qualitative and quantitative studies of cytokines
synthetized and secreted by non-human primate peripheral blood
mononuclear cells. Cytokine 5, 169±179.
Werner-Felmayer G., Werner E. R., Fuchs D., Hausen A., Reibnegger
G. and Wachter H. (1989) Characteristics of interferon induced
tryptophan metabolism in human cells in vitro. Biochim. Biophys.
Acta 1012, 140±147.
JNC498 JNC jn6489 CD 12/7/1 12:39 ALDEN
12 G. J. Guillemin et al.
q 2001 International Society for Neurochemistry, Journal of Neurochemistry, 78, 1±13