capitolo 68 1507 diabete mellito 11 - doctor33.it · capitolo 68 - diabete mellito e ipoglicemie 11...
TRANSCRIPT

1507
11
© 2
010
ELSE
VIE
R S
.R.L
. Tu
tti
i d
irit
ti r
iser
vati
.
11
CapIToLo 68
Diabete mellito e ipoglicemie
Sara Giunti
Gabriella Gruden
Insulina e omeostasi glucidica
Sintesi e secrezione dell’insulinaL’insulina viene prodotta dalle cellule b delle isole di Lan-gerhans del pancreas in forma di precursore inattivo (proin-sulina). Dal clivaggio della proinsulina si formano il peptide di connessione (C-peptide) e le due catene (a e B) dell’insu-lina. La forma matura dell’insulina, con le due catene unite tramite ponti disolfuro, e il C-peptide sono immagazzinati nelle cellule b e secreti simultaneamente. poiché l’insulina ha un’emivita (5 min) molto più breve del C-peptide (30 min), la secrezione endogena di insulina è più facilmente valutabile misurando i livelli circolanti di C-peptide. La secrezione di insulina è regolata principalmente dai livelli glicemici, sebbene aminoacidi, chetoni, nutrienti, peptidi gastrointestinali e neurotrasmettitori la possano influenzare. Il glucosio, entrato nella cellula b tramite il trasportatore di membrana GLUT-2, viene metabolizzato nella via della glicolisi con formazione di aTp. L’aTp inibisce il canale del K+ sensibile all’aTp con conseguente depolarizzazione della cellula, apertura dei canali del calcio voltaggio-dipendenti, entrata del calcio e rilascio di insulina. Il profilo di secrezione dell’insulina nella giornata mostra una secrezione pulsatile basale continua a cui si sovrappongono picchi secretori (4-5 volte la secrezione basale) della durata di 2-3 ore indotti prin-cipalmente dai pasti. I livelli glicemici regolano strettamente la secrezione non solo di insulina, ma anche di glucagone, rilasciato dalle cellule a delle isole di Langerhans. Se la glice-mia è ≥ 90 mg/dL le cellule b aumentano la loro secrezione di insulina, mentre le cellule a riducono il rilascio di glucagone; se la glicemia scende sotto i 70 mg/dL, diminuisce la secre-zione di insulina, mentre aumenta quella di glucagone. La secrezione dei due ormoni non cessa mai completamente. Un aumento dei livelli di insulina inibisce il rilascio di glu-cagone dalle cellule a; per contro, se il glucagone aumenta, come accade al ridursi dei livelli glicemici, questo stimola le cellule b alla secrezione di insulina, preparandole al suc-cessivo rialzo della glicemia.
Effetti dell’insulinaL’insulina secreta dalle cellule b entra nel circolo portale, dove viene in parte degradata dal fegato (50%). L’insulina che sfugge alla clearance epatica entra nel circolo sistemico e si lega al proprio recettore sito sugli organi insulino-sensibili. Il legame insulina-recettore determina l’autofosforilazione del recettore e l’attivazione di vie di segnale intracellulari responsabili degli effetti metabolici e mitogeni dell’insulina. Gli effetti metabolici dell’insulina sono di tipo anabolizzan-te: essa stimola la sintesi delle proteine e favorisce l’accumulo
di depositi energetici sotto forma di glicogeno e trigliceridi (Tab. 68.1). Nel fegato l’insulina inibisce la gluconeogenesi e stimola la glicogenosintesi. Ciò si traduce in un ridotto rilascio epatico di glucosio e in un accumulo del glucosio in eccesso in forma di glicogeno. a livello periferico, e in parti-colare nel muscolo e nel tessuto adiposo, l’insulina stimola la traslocazione del trasportatore del glucosio GLUT-4 sulla membrana plasmatica. Questo evento è cruciale nel consen-tire la captazione di glucosio da parte delle cellule muscolari e degli adipociti e nel favorire l’utilizzazione del glucosio circolante da parte dei tessuti periferici. poiché l’omeostasi glucidica è il risultato di un equilibrio tra la produzione epatica di glucosio e la sua captazione e utilizzazione da parte dei tessuti periferici, si comprende come l’insulina svolga un ruolo cruciale nel suo controllo. Nel muscolo il glucosio internalizzato viene utilizzato come substrato energetico o, sotto l’influsso dell’insulina che stimola la glicogenosintesi, accumulato in forma di glicogeno. Inoltre, nel muscolo l’insulina stimola la captazione degli aminoa-cidi ed è necessaria per la sintesi proteica. Nell’adipocita il glucosio internalizzato viene convertito in glicerolo fosfato e utilizzato nella sintesi dei trigliceridi insieme agli acidi grassi rilasciati in circolo dalla lipoproteinlipasi, un enzima endoteliale attivato dall’insulina. poiché gli adipociti non hanno l’enzima che converte il glicerolo in glicerolo fosfato, essi dipendono dalla captazione di glucosio dall’esterno e, quindi, dall’insulina per la sintesi di trigliceridi. al contrario, la lipolisi dei trigliceridi che avviene nell’adipocita è sotto il controllo della lipasi ormono-sensibile che è inibita dall’in-sulina e attivata da glucagone e adrenalina. Quindi l’insulina inibisce, mentre il glucagone e l’adrenalina favoriscono la lipolisi dei trigliceridi immagazzinati negli adipociti.
Cenni di omeostasi metabolicaIl bilancio tra insulina e glucagone è cruciale nel mantenere l’omeostasi metabolica e nel consentire il mantenimento di livelli glicemici relativamente costanti, nonostante sia il consumo di glucosio sia il suo apporto con la dieta varino considerevolmente nell’arco della giornata. Ciò è impor-tante perché il cervello usa il glucosio come quasi esclusivo substrato energetico e non è in grado di captare gli acidi grassi e di usarli come fonte energetica alternativa. Il gluca-gone è un ormone ad azione prevalentemente catabolica e ha effetti opposti a quelli dell’insulina. Esso, infatti, induce il rilascio di glucosio dai depositi di glicogeno (glicogeno-lisi) e di acidi grassi dai trigliceridi (lipolisi), e stimola la gluconeogenesi (si veda Tab. 68.1).Dopo i pasti, il glucosio assorbito a livello intestinale in-duce, attraverso un rialzo dei livelli glicemici, il rilascio di

parte 11 - MaLaTTIE DEL RICaMBIo 1508
insulina e l’inibizione del rilascio di glucagone. Il modifi-cato rapporto insulina/glucagone determina l’attivazione della glicogenosintesi, la riduzione della gluconeogenesi e della glicogenolisi e l’aumentata captazione periferica di glucosio. Il fegato, liberamente permeabile al glucosio, se-questra dal circolo splancnico circa il 50% del glucosio e lo accumula in forma di glicogeno che servirà poi a mantene-re costanti i livelli glicemici nelle fasi tra i pasti. La restante parte del glucosio assorbito con il pasto viene internalizzata dal muscolo mediante il GLUT-4 e utilizzata come substra-to energetico o accumulata come glicogeno. Se presente in largo eccesso, il glucosio può essere utilizzato anche a livello del tessuto adiposo per la sintesi dei trigliceridi. Questi processi metabolici consentono un rapido ritorno dei livelli glicemici ai valori basali ed evitano la dispersio-ne del glucosio con le urine. È noto, infatti, che se i livelli glicemici postprandiali superano la soglia renale (180 mg/dL), il glucosio non può essere completamente riassorbito dal tubulo renale e viene disperso con le urine.Nell’intervallo tra i pasti la riduzione dei livelli glicemici causa un ridotto rilascio di insulina e un aumentato rilascio di glucagone. Il risultato metabolico è l’attivazione della glicogenolisi e della gluconeogenesi, con conseguente au-mentato rilascio epatico di glucosio. Ciò consente di man-tenere i livelli glicemici costanti, nonostante il continuo consumo di glucosio da parte del cervello. La gluconeoge-nesi necessita di numerosi substrati tra cui il glicerolo e gli
aminoacidi, forniti rispettivamente da tessuto adiposo e muscolo. I ridotti livelli di insulina diminuiscono la cap-tazione di aminoacidi dal muscolo e la sintesi proteica, rendendo quindi disponibili gli aminoacidi per la glucone-ogenesi epatica. La riduzione dell’insulina e l’aumento del glucagone favoriscono, inoltre, la lipolisi dei trigliceridi nel tessuto adiposo con rilascio di glicerolo e acidi grassi, che vengono utilizzati rispettivamente nella gluconeogenesi e, dai tessuti periferici, come substrato energetico nel meta-bolismo aerobio, con conseguente risparmio di glucosio. L’eccesso di acidi grassi viene convertito dal fegato in corpi chetonici, i cui livelli rimangono bassi in condizioni fisiolo-giche. Durante il digiuno prolungato la marcata riduzione dei livelli insulinemici determina un netto aumento della produzione epatica di corpi chetonici a partire dagli acidi grassi circolanti. I corpi chetonici possono essere utilizzati dal cervello come substrati energetici alternativi al glucoso. In condizioni di digiuno protratto tale meccanismo, che utilizza le riserve di grasso al posto del glucosio, può ga-rantire sino al 50% delle esigenze energetiche del cervello, contribuendo a prolungare la sopravvivenza. Qualora i livelli circolanti di corpi chetonici, che sono acidi deboli, divengano tali da superare le capacità omeostatiche del sistema acido-basico, si instaura uno stato di acidosi meta-bolica. Tuttavia, in condizioni fisiologiche, tale evenienza non si verifica, in quanto i livelli di insulina, che limitano la lipolisi e la chetogenesi, non si azzerano mai.
Definizione
Diabete, alterata glicemia a digiuno e ridotta tolleranza ai carboidratiIl termine “diabete mellito” si applica a un gruppo di di-sordini metabolici caratterizzati da iperglicemia. I valori
glicemici oltre i quali si pone la diagnosi di diabete sono variati nel tempo e sono continuamente oggetto di rivalu-tazione e discussione. In accordo con l’american Diabetes association (aDa) si parla di diabete mellito quando sono presenti i sintomi clinici della malattia (poliuria, poli-dipsia e calo ponderale) e una glicemia casuale misurata
Diabete mellito
Organo Bersaglio Insulina GlucagoneGlicogeno epatico Stimola la glicogenosintesi e quindi
l’immagazzinamento del glucosioStimola la glicogenolisi con rilascio di glucosio
Gluconeogenesi epatica Inibisce la gluconeogenesi con risparmio di aminoacidi
Stimola la gluconeogenesi e il rilascio epatico di glucosio
Captazione di acidi grassi/glucosio e sintesi di trigliceridi
Stimola la captazione di glucosio e ren-de disponibili gli acidi grassi attivando la lipoproteinlipasi endoteliale
Lipolisi dei trigliceridi nell’adipocita
Inibisce la lipolisi riducendo il rilascio di acidi grassi
Attiva la lipolisi aumentando il rilascio di acidi grassi
Captazione di glucosio e sintesi di glicogeno
Stimola la captazione di glucosio e il suo accumulo come glicogeno
Captazione di aminoacidi Stimola la captazione di aminoacidi e il loro utilizzo nella sintesi proteica
Senso di fame Riduce il senso di fame
Tabella 68.1 Effetti metabolici di insulina e glucagone

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1509
in qualsiasi momento del giorno ≥ 200 mg/dL. In assenza di sintomi, si pone diagnosi di diabete se la glicemia a digiuno è ≥ 126 mg/dL o la glicemia 2 ore dopo carico orale con glucosio (oGTT, oral Glucose Tolerance Test) è ≥ 200 mg/dL. In assenza di iperglicemia inequivocabile, è necessario confermare la positività a uno dei criteri in un giorno successivo (Tab. 68.2).alterata glicemia a digiuno (IFG, Impaired Fasting Glu-cose) e ridotta tolleranza ai carboidrati (IGT, Impaired Glucose Tolerance) sono categorie introdotte per indi-care uno stato metabolico intermedio tra la normalità e il diabete. Si parla di IFG quando i livelli di glicemia a digiuno sono compresi tra 100 e 125 mg/dL e di IGT quando la glicemia, determinata 2 ore dopo l’oGTT, è compresa tra 140 e 199 mg/dL (Fig. 68.1). Le condizioni di IFG e IGT non rappresentano patologie di per sé, né si accompagnano ad alcuna sintomatologia clinica, ma sono di interesse medico per l’elevato rischio di diabete e malattie cardiovascolari che le caratterizza. IFG e IGT possono coesistere nello stesso individuo, ma sono spesso presenti in forma isolata.
Classificazione
L’attuale classificazione del diabete mellito è fondata su criteri eziopatogenetici (Tab. 68.3). precedenti classifi-cazioni basate sull’età di insorgenza e sul trattamento terapeutico sono state completamente abbandonate. Il diabete di tipo 1 e quello di tipo 2 rappresentano le forme di più comune riscontro nella pratica clinica.Il diabete di tipo 1 è caratterizzato da distruzione delle cellule b pancreatiche che conduce a deficit insulinico assolu-to con necessità di terapia insulinica sostitutiva al fine di prevenire lo sviluppo di chetoacidosi. La distruzione della cellula b pancreatica avviene di solito su base autoimmune (tipo 1a) e il siero dei pazienti è positivo per la presenza di autoanticorpi. I pazienti che non presentano evidenza di autoimmunità (10%) sono affetti da una forma idiopatica di diabete di tipo 1 (tipo 1B) di più comune riscontro in asia e in africa.Il diabete di tipo 2 comprende un gruppo eterogeneo di di-sordini caratterizzati da variabili gradi di insulino-resisten-za, alterata secrezione insulinica e aumentata produzione epatica di glucosio. Sebbene questi disordini diano luogo a un quadro fenotipico sovrapponibile, i meccanismi
fisiopatologici prevalenti possono essere diversi e richie-dere, quindi, interventi terapeutici diversificati.La categoria altri tipi specifici di diabete comprende un grup-po eterogeneo di forme di diabete secondario a una causa nota. Rientrano in questa categoria: malattie del pancreas esocrino che distruggono oltre l’80% della massa pancrea-tica, affezioni endocrine responsabili di eccessiva secrezione di ormoni controregolatori, farmaci o sostanze chimiche
1 Glicemia a digiuno ≥ 126 mg/dLOppure2 Sintomi classici di iperglicemia (poliuria, polidipsia
e calo ponderale inspiegato) e glicemia casuale (in ogni momento del giorno indipendentemente dall’ultimo pasto) ≥ 200 mg/dL
Oppure3 Glicemia misurata 2 ore dopo la curva da carico
con glucosio (OGTT) ≥ 200 mg/dL
In assenza di iperglicemia inequivocabile, è necessario confermare la diagnosi ripetendo uno dei test in un giorno diverso.
Tabella 68.2 Criteri per la diagnosi di diabete
Figura 68.1Livelli soglia di glicemia per la diagnosi di diabete: IFG e IGT sulla base dei valori glicemici misurati a digiuno (8 ore senza introito calorico) o 2 ore dopo OGTT.
I. Diabete di tipo 1 (distruzione della cellula b che determina di solito un deficit insulinico assoluto)a. Immuno-mediatoB. Idiopatico
II. Diabete di tipo 2 (spazia da predominante insulino-resisten-za con deficit insulinico relativo a un difetto predominante della secrezione insulinica con insulino-resistenza)
III. Altri tipi specifici di diabetea. Difetti genetici della funzione della cellula b (cromoso-
ma 12, HNF-1 (MODY 3), cromosoma 7, glucochinasi (MODY2), cromosoma 20, HNF-4 (MODY 1), cromosoma 13, fattore I del promotore insulinico (IPF-1; MODY 4), cro-mosoma 17, HNF-1b (MODY 5), cromosoma 2, neuroD1 (MODY 6), DNA mitocondriale ecc.)
B. Difetti genetici dell’azione insulinica (insulino-resistenza di tipo A, leprecaunismo, sindrome di Rabson-Mendenhall, lipoatrofia, altri)
c. Malattie del pancreas esocrino (pancreatite, trauma/pancre-asectomia, neoplasia, fibrosi cistica, emocromatosi ecc.)
D. Endocrinopatie (acromegalia, sindrome di Cushing, glucagonoma, feocromocitoma, ipertiroidismo, somatostatinoma ecc.)
e. Diabete indotto da farmaci o chimici (vacor, pentamidina, acido nicotinico, glucocorticoidi, ormone tiroideo, diazossido, agonisti b-adrenergici, tiazidi, dilantina, interferone ecc.)
F. Infezioni (rosolia congenita, cytomegalovirus ecc.)G. Forme rare di diabete immuno-mediato ("stiff-man
syndrome" anticorpi anti-insulina)H. Altre sindromi genetiche talvolta associate al diabete
(sindromi di Down, di Klinefelter, di Turner, di Wolfram, di Laurence-Moon-Biedl e di Prader-Willi, atassia di Friedreich, corea di Huntington, distrofia miotonica, porfiria ecc.)
IV. Diabete mellito gestazionale
Tabella 68.3 Classificazione eziologica del diabete mellito

parte 11 - MaLaTTIE DEL RICaMBIo 1510
iperglicemizzanti, infezioni virali, difetti genetici della fun-zione b-cellulare o dell’azione insulinica. Il MoDY (Maturity onset Diabetes of the Young) è una forma monogenica di diabete a trasmissione autosomica dominante. Sono stati descritti sei difetti genetici diversi che, con meccanismi differenti, determinano un’alterata funzionalità della cellula b (Tab. 68.4). Il MoDY insorge di solito prima dei 25 anni in assenza di marcatori di autoimmunità e il controllo me-tabolico viene mantenuto senza insulina per oltre 2 anni. La trasmissione ereditaria dominante (almeno tre genera-zioni) è ovviamente determinante per l’individuazione di tali pazienti.Il diabete gestazionale è un diabete diagnosticato per la pri-ma volta durante la gravidanza e colpisce circa il 4% delle donne in gravidanza. Lo stato di insulino-resistenza che si instaura nelle fasi avanzate della gravidanza determina un aumento della richiesta di insulina e ne rappresenta il probabile substrato fisiopatologico. La maggior parte delle donne ritorna a uno stato di normale tolleranza glucidica dopo la gravidanza, ma tali soggetti hanno un aumentato rischio di sviluppare il diabete nel corso della vita.
Epidemiologia
Il diabete mellito è una malattia comune e la sua pre-valenza, in particolare quella del diabete di tipo 2, è in drammatico aumento. I soggetti affetti da diabete mellito nel mondo sono oggi 246 milioni (prevalenza 5,9%), di cui il 46% nella fascia di età compresa tra 40 e 59 anni, e si stima che nel 2025 saranno 370 milioni, con una preva-lenza del 7,3%. In Italia, si registra la presenza di 2 milioni e 643 mila persone colpite da diabete, con una prevalenza del 4,5%, ma il dato è probabilmente sottostimato per la presenza di un elevato numero di soggetti con diabete non diagnosticato (prevalenza reale ≈ 6%). Tale “pande-mia” è da attribuirsi principalmente all’aumento, nei paesi sviluppati, dei soggetti sovrappeso e obesi, e al più facile accesso alle fonti alimentari nei paesi in via di sviluppo. particolarmente allarmante è l’aumento dei casi di obesi-tà e sovrappeso tra bambini e adolescenti, con sviluppo di diabete mellito di tipo 2 associato a obesità già in età adolescenziale. Circa il 90% della popolazione diabetica è affetto da diabete di tipo 2, mentre solo una minoranza è affetta da diabete di tipo 1. L’incidenza del diabete di tipo
1 è massima nel Nord Europa (Scandinavia e Regno Unito: 19-37 casi all’anno per 100.000 soggetti nella fascia di età tra 0 e 14 anni), intermedia nel resto dell’Europa e negli Stati Uniti (8-15 casi/100.000/anno) e minima in Cina e Sud america (< 1 caso/100.000/anno). In Italia il diabete di tipo 1 rappresenta dal 3 al 6% di tutti i casi di diabete e l’incidenza è di circa al 10-11 casi/100.000/anno. Fa ecce-zione la Sardegna, che ha un’incidenza e una prevalenza di diabete di tipo 1 tra le più alte del mondo (34 casi/100.000/anno), in Europa seconda soltanto alla Finlandia.
Eziopatogenesi
Diabete di tipo 1Il diabete di tipo 1a è dovuto alla distruzione autoimmu-ne delle cellule b delle isole di Langerhans del pancreas. Questo processo è probabilmente innescato da uno o più fattori ambientali in soggetti il cui sistema immunitario è geneticamente predisposto a sviluppare una reazione autoimmune contro un antigene danneggiato della cellula b (o, meno probabilmente, contro molecole della cellula b simili ad antigeni virali – mimetismo molecolare). La distruzione autoimmune della cellula b è mediata preva-lentemente dai linfociti T attraverso il rilascio di citochine o citotossicità diretta e si realizza nell’arco di mesi o anni durante i quali il soggetto è asintomatico ed euglicemico. Questo lungo periodo di latenza è dovuto al fatto che è necessaria la distruzione di un elevato numero di cellule b (80%) per l’instaurarsi di un deficit funzionale nella produzione di insulina tale da determinare la comparsa di iperglicemia (Fig. 68.2).L’osservazione che il rischio di diabete di tipo 1 è aumen-tato di dieci volte nei parenti e di 50 volte nei gemelli monozigoti dei diabetici di tipo 1 supporta l’ipotesi di una suscettibilità genetica allo sviluppo della malattia. Sono stati identificati numerosi geni le cui varianti polimorfiche si associano allo sviluppo di diabete di tipo 1. Tra questi i più importanti sono i polimorfismi dei geni della regione HLa (Human Leukocyte antigen System) (DQa, DQb, DR) che spiegano il 41% dell’aggregazione familiare. Questi geni codificano per le molecole del complesso maggio-re di istocompatibilità (MHC, Major Histocompatibility Complex) di classe II che sono espresse sulla superficie delle cellule deputate a presentare l’antigene, come i ma-
Tipo Gene Caratteristiche cliniche TrattamentoMODY 1 HNF-4a Diabete; complicanze cardiovascolari; riduzione di trigliceridi nel siero Ipoglicemizzanti orali e insulina
MODY 2 GK Alterazioni glicemiche a digiuno; alterata tolleranza al glucosio; diabete; normale rapporto insulina/proinsulina nel siero
Esercizio fisico e dieta
MODY 3 HNF-1a Diabete; complicanze microvascolari; glicosuria, aumento di sensibilità alle sulfaniluree; aumento del rapporto insulina/proinsulina nel sangue
Ipoglicemizzanti orali e insulina
MODY 4 IPF-1 Diabete Ipoglicemizzanti orali e insulina
MODY 5 HNF-1b Diabete; cisti renali e altre anormalità nello sviluppo del rene; insufficienza renale cronica; anormalità interne nei genitali (donne)
Insulina
MODY 6 NeuroD1 o b2 Diabete Insulina
Tabella 68.4 Classificazione MODY

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1511
crofagi. Queste molecole formano una “nicchia” a cui si lega l’autoantigene responsabile dell’innesco del diabete. Tale legame consente la presentazione dell’antigene ai linfociti T helper e l’avvio della reazione autoimmune. La capacità delle molecole dell’MHC di classe II di presentare l’antigene dipendente in parte dalla loro composizione aminoacidica e la sostituzione di uno o due aminoacidi in posizione critica può aumentare o ridurre il loro legame all’autoantigene e, quindi, influenzare la suscettibilità allo sviluppo del diabete di tipo 1. Il 90% dei pazienti con diabete di tipo 1 è portatore dell’aplotipo DR3-DQ2 o DR4-DQ8 e il 30% li ha entrambi (DR3/4). I geni HLA-DQ sono ancora più precisi marcatori di suscettibilità al diabe-te di tipo 1, perché una particolare variante DQB1*0302 si trova nei pazienti DR4 con diabete di tipo 1, mentre la variante protettiva DQB1*0602 si trova nei soggetti DR4 non affetti da diabete. altri polimorfismi importanti nel conferire suscettibilità/protezione sono quello di un promoter del gene dell’insulina e di una tirosina fosfatasi specifica dei linfociti (pTp) che influenza il signaling del recettore dei linfociti T.anticorpi diretti contro gli antigeni delle cellule b (ICa), come gli anticorpi anti-decarbossilasi dell’acido glutammi-co (GaD), anti-insulina (Iaa) e anti-proteina 2 associata a insulinoma (Ia-2), sono di frequente riscontro nel diabete di tipo 1 di nuova diagnosi. Tuttavia, non è chiaro se que-sti autoanticorpi siano importanti nella patogenesi del diabete o siano meri marcatori di danno della cellula b. La distruzione della cellula b, causata dall’immunità cellulo-mediata, potrebbe infatti causare il rilascio di antigeni che, se non riconosciuti dal sistema immunitario, inducono una reazione autoimmune secondaria che può contribuire alla progressione del danno, senza rappresentarne il fattore causale. a tale proposito, sia studi condotti nell’animale sia un caso di diabete di tipo 1 insorto in un ragazzo con agammaglobulinemia suggeriscono che i linfociti B e la produzione di autoanticorpi non siano necessari per lo sviluppo di diabete e che la distruzione della cellula b sia mediata principalmente dai linfociti T. La possibilità che
uno degli autoantigeni riconosciuti dagli ICa sia respon-sabile dell’innesco della risposta cellulo-mediata sembra improbabile in quanto questi antigeni non sono specifici della cellula b, mentre la distruzione autoimmune della cellula b è estremamente selettiva e risparmia gli altri tipi cellulari delle isole di Langerhans. I fattori ambientali che innescano la risposta autoimmune non sono noti: sono stati associati al diabete di tipo 1 infezioni virali (infezioni da Coxsackie, rosolia, morbillo) e determinati alimenti, come proteine del latte vaccino e cereali contenti glutine, mentre gli acidi grassi 3 e la vitamina D avrebbero un ruo-lo protettivo. I dati non sono, peraltro, univoci e mancano evidenze scientifiche convincenti.
Diabete di tipo 2I fattori genetici svolgono nel diabete di tipo 2 un ruolo ancora più importante che nel diabete di tipo 1. Infatti, la concordanza per il diabete di tipo 2 tra gemelli identici è del 70-90%. Inoltre, soggetti con un genitore affetto da diabete hanno un rischio di sviluppare la malattia durante la vita del 38%, con un aumento al 60% se entrambi i genitori sono diabetici. Nonostante questo chiaro condizionamento genetico, sono raramente identificabili singoli geni causali e, nella maggior parte dei casi, la malattia è poligenica e sensibilmente influenzata da fattori ambientali.In condizioni fisiologiche, la normoglicemia è mantenuta grazie all’equilibrio tra secrezione e azione dell’insulina. La cellula b pancreatica, infatti, è in grado di adattarsi alle variazioni dell’azione dell’insulina: la riduzione di tale azione è compensata da un aumento della secrezione pancreatica di insulina. Il diabete di tipo 2 si sviluppa quando la secrezione insulinica è inadeguatamente bassa per uno specifico grado di attività dell’insulina. pertanto, due sono gli aspetti caratteristici del diabete di tipo 2: la ridotta azione dell’insulina (insulino-resistenza) e l’inade-guata secrezione insulinica. Nelle fasi precoci della ma-lattia la tolleranza glicidica resta normale, nonostante lo stato di insulino-resistenza, perché la cellula b pancreatica compensa la resistenza periferica all’azione dell’insulina
Figura 68.2Fasi dello sviluppo del diabete di tipo 1. In soggetti geneticamente predisposti uno o più fattori ambientali innescano una reazione autoimmune contro un antigene della cellula con conseguente insulite, danno della cellula b e progressiva perdita della massa pancreatica. Un elevato numero di cellule (> 80%) deve essere perso prima che si verifichi un deficit funzionale nella produzione di insulina tale da determinare l’esordio clinico della malattia.

parte 11 - MaLaTTIE DEL RICaMBIo 1512
aumentandone la secrezione con aumento dei livelli insu-linemici. al progredire dello stato di insulino-resistenza, in alcuni individui la cellula b non è più in grado di so-stenere un ulteriore aumento del rilascio di insulina e si sviluppa il diabete.
Insulino-resistenzaSi parla di insulino-resistenza quando gli effetti biologici dell’insulina, consistenti nella rimozione del glucosio circolante da parte del muscolo scheletrico e nella sop-pressione della produzione epatica di glucosio, sono infe-riori a quanto atteso. Nel diabete di tipo 2 vi è un difetto dell’azione dell’insulina sul fegato perché la glicemia a digiuno, determinata principalmente dalla produzione epatica di glucosio, è aumentata, nonostante i livelli di insulina siano elevati. analogamente, dopo il pasto vi è una ridotta rimozione/utilizzo del glucosio circolante da parte del fegato e, soprattutto, del muscolo che rende con-to dell’iperglicemia postprandiale. Il ruolo cruciale del fe-gato nello sviluppo dell’insulino-resistenza è confermato dall’osservazione che l’insulino-resistenza e l’intolleranza glicidica si sviluppano nell’animale da esperimento privo del recettore insulinico a livello epatico e non in quello privo dello stesso recettore nel muscolo o nel tessuto adiposo. Sebbene la causa dell’instaurarsi di uno stato di insulino-resistenza non sia stata ancora chiarita, è noto che l’insulino-resistenza è significativamente associata all’obesità, soprattutto viscerale, e all’inattività fisica. Il tessuto adiposo rilascia ormoni, citochine infiammatorie, adipochine e acidi grassi, sostanze in grado di modulare l’azione dell’insulina in altri tessuti. Un aumento dei trigliceridi nel tessuto adiposo, soprattutto viscerale, in-duce la formazione di adipociti ipertrofici che riducono l’azione dell’insulina mediante il rilascio di adipochine e citochine. Gli adipociti ipertrofici sono essi stessi meno sensibili all’azione antilipolitica dell’insulina e rilasciano, quindi, più acidi grassi, che causano insulino-resistenza. Infine, l’accumulo di trigliceridi non negli adipociti, ma in sedi ectopiche, come nelle cellule muscolari e negli epatociti, rende tali cellule insulino-resistenti. I meccani-smi molecolari attraverso cui gli acidi grassi, le adipochine e le citochine interferiscono con l’azione dell’insulina non sono stati del tutto chiariti. Tuttavia, queste sostanze sembrano agire prevalentemente a livello postrecettoriale, interferendo con le vie intracellulari di segnale attivate dal legame dell’insulina con il suo recettore. Importanti ber-sagli sono il substrato del recettore insulinico (IRS) e la via di segnale della fosfoinositolo-3 chinasi, responsabili della traslocazione del trasportatore GLUT-4 alla membrana e degli effetti metabolici (glicogenosintesi, sintesi proteica) dell’insulina. al contrario, le vie di segnale che mediano gli effetti mitogenici dell’insulina sono preservate e pro-babilmente iperattivate dallo stato iperinsulinemico.
Alterata secrezione insulinicaSebbene i livelli basali di insulina siano aumentati nel sog-getto diabetico e superiori alla norma nella fase postpran-diale, ciò accade in risposta agli aumentati livelli glicemici ed è stato dimostrato che, a parità di livelli glicemici, la secrezione insulinica del soggetto con diabete di tipo 2 è inferiore a quella del soggetto non diabetico. Il difetto b-cellulare è riscontrabile anche prima dello sviluppo di
diabete e riconosce, probabilmente, una componente genetica. Inoltre, l’iperglicemia può determinare di per sé una riduzione della funzione b-cellulare (glucotossicità) aumentando lo stress ossidativo della cellula b. Nel corso della storia naturale del diabete di tipo 2 si assiste, infatti, a un progressivo decadimento della funzionalità pancrea-tica con riduzione dell’efficacia dei farmaci che stimolano la secrezione insulinica e, spesso, con necessità di ricorrere a terapia insulinica sostitutiva come nel diabete di tipo 1. Recenti studi suggeriscono che anche gli elevati livelli di acidi grassi circolanti presenti nel soggetto insulino-resistente/diabetico e l’accumulo ectopico di lipidi nella cellula b possano contribuire al deterioramento funzio-nale della cellula b (lipotossicità).
Manifestazioni cliniche
Diabete di tipo 1Il diabete di tipo 1 esordisce nel 50% dei casi a un’età inferiore ai 20 anni e più spesso durante la pubertà. Non è tuttavia raro un esordio nell’infanzia, anche nel primo anno di vita. L’esor-dio può essere acuto con coma chetoacidosico (si veda oltre, Complicanze acute), spesso in occasione di stress intercorrenti (infezione, intervento chirurgico ecc.), oppure subacuto con sintomi e segni clinici più sfumati e di più difficile riconosci-mento. Quando il diabete esordisce in concomitanza con un evento intercorrente acuto può essere seguito da una fase di regressione spontanea che perdura per alcuni mesi (cosiddetta “luna di miele”). probabilmente l’evento acuto, stimolando un’aumentata produzione di ormoni della controregola-zione, rende manifesto il deficit di insulina e determina il raggiungimento dell’orizzonte clinico. Dopo la risoluzione dell’evento intercorrente, la funzione delle cellule b può di-venire nuovamente adeguata, per poi deteriorarsi nel tempo e divenire insufficiente indipendentemente dalla presenza di eventi intercorrenti scatenanti.I sintomi clinici più comuni all’esordio del diabete sono po-liuria, sete, polidipsia, polifagia, astenia e dimagramento. Il deficit assoluto di insulina e il concomitante aumento dei livelli di glucagone provocano iperglicemia e, quando i livelli glicemici superano la soglia renale per il glucosio (180 mg/dL), compare glicosuria, che, se di entità notevole, provoca diuresi osmotica con conseguente poliuria e perdita di acqua, glucosio ed elettroliti. In genere, un incremento della sete e dell’introduzione di liquidi (polidipsia) bilancia la poliuria senza un rilevante aumento dell’osmolarità plasmatica. Tut-tavia, l’esposizione del cristallino a fluidi iperosmotici può determinare un offuscamento ricorrente della vista e la perdita di liquidi può causare la comparsa di sintomi da ipotensione ortostatica. La perdita di peso, nonostante un’esaltazione dell’appetito e dell’assunzione di alimenti (polifagia), è ini-zialmente dovuta alla perdita di acqua e al depauperamento delle riserve energetiche (glicogeno, trigliceridi) per il venir meno dell’effetto anabolizzante dell’insulina. Successivamen-te, il dimagramento è da attribuirsi alla perdita della massa muscolare. L’insulina, infatti, stimola la sintesi proteica e la sua assenza favorisce la proteolisi. La deplezione di potassio, perduto con le urine, e la riduzione della massa muscolare contribuiscono alla comparsa di astenia e faticabilità. possono, infine, insorgere parestesie dovute a disfunzione transitoria delle fibre nervose sensitive da effetto tossico diretto dell’iper-glicemia (Tabb. 68.5 e 68.6).

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1513
Diabete mellito di tipo 2Il diabete mellito di tipo 2 si manifesta di solito in età adulta e si associa frequentemente alla presenza di obesità. può manifestarsi, analogamente al diabete di tipo 1, con poliuria, sete, polidipsia, astenia, anche se nella maggior parte dei casi il soggetto è asintomatico e la diagnosi viene fatta per occasionale riscontro di iperglicemia in esami
ematochimici di routine. poiché il diabete di tipo 2 può rimanere a lungo asintomatico (in media 7 anni), un’eleva-ta percentuale di soggetti ignara della malattia può, talora, presentare all’esordio manifestazioni cliniche dovute alle complicanze croniche della malattia (neuropatia periferica, macroangiopatia, retinopatia). Le infezioni cutanee croni-che sono frequenti e, nella donna, un prurito generalizzato o lo sviluppo di vulvovaginiti croniche da Candida può di frequente rappresentare la manifestazione d’esordio. Nel soggetto diabetico obeso il tessuto adiposo ha spesso una distribuzione addominale e la compresenza di ipertensione arteriosa è di comune riscontro. I pazienti con diabete di ti-po 2 hanno scarsa tendenza a sviluppare coma chetoacido-sico, anche quando l’alterazione metabolica è molto grave. Tuttavia, i soggetti più anziani, con un meccanismo della sete meno efficiente, possono raggiungere livelli glicemici e di osmolarità plasmatica tali da determinare una grave sin-drome neurologica denominata coma iperosmolare (si veda oltre, Complicanze acute; si vedano Tabb. 68.5 e 68.6).Seppure lo screening di massa per il diabete di tipo 2 sia al momento sconsigliato, i soggetti ad alto rischio (Tab. 68.7) dovrebbero essere sottoposti ogni 2-3 anni a scree-ning mediante misurazione della glicemia a digiuno. poi-ché nel diabete non diagnosticato l’iperglicemia esercita effetti deleteri a livello dei tessuti bersaglio, la diagnosi tempestiva mediante screening può consentire di ridurre il rischio di complicanze croniche. Inoltre, il controllo dell’iperglicemia riduce la glucotossicità e consente di preservare più a lungo la funzionalità b-cellulare.
Diabete di tipo 1 Diabete di tipo 2Poliuria e sete ++ +
Debolezza e faticabilità
++ +
Polifagia con calo ponderale
++ −
Ricorrente offuscamento del visus
+ ++
Vulvovaginiti e prurito
+ ++
Neuropatia periferica
+ ++
Asintomatico − ++
Tabella 68.5 Aspetti clinici del diabete alla diagnosi
Diabete di tipo 1 Diabete di tipo 2Sintomatologia Sempre presente
spesso acutaSpesso modesta o assente
Età all’esordio < 30 anni di solito
> 30 anni di solito
BMI all’esordio < 25 di solito > 25 di solito
Tipo di esordio Generalmente acuto
Subdolo
Tendenza alla chetosi
Sì Generalmente no
Insulina (C-peptide)
Grave deficit o assente
Normale o elevata
Insulinoterapia Necessaria per la sopravvivenza
Talvolta necessaria per il compenso
Comparsa di complicanze croniche
Parecchi anni dopo l’esordio del diabete
Spesso presenti alla diagnosi
Predisposizione genetica
Moderata Elevata
Autoimmunità Sì No
Ambiente Virus? Alimenti? Sedentarietà, obesità
Tabella 68.6 Confronto tra diabete di tipo 1 e di tipo 2
IFG o IGT o pregresso diabete gestazionale
Età ≥ 45 anni, specialmente se con BMI ≥ 25 kg/m2
Età < 45 anni e una o più tra le seguenti condizioni:
d familiarità di primo grado per diabete di tipo 2 (genitori, fratelli)
d appartenenza a gruppo etnico ad alto rischiod ipertensione arteriosa (≥ 140/90 mmHg) o terapia
antipertensiva in attod bassi livelli di colesterolo HDL (≤ 35 mg/dL) e/o elevati
valori di trigliceridi (≥ 250 mg/dL)d nella donna, parto di un neonato di peso > 4 kgd sindrome dell’ovaio policistico o altre condizioni di
insulino-resistenza come l’acanthosis nigricansd evidenza clinica di malattie cardiovascolari
Bambini di età > 10 anni, con BMI > 85° percentile e due tra le seguenti condizioni:d familiarità di primo o secondo grado per diabete di tipo 2d madre con diabete gestazionaled segni di insulino-resistenza o condizioni associate (iperten-
sione, dislipidemia, acanthosis nigricans, ovaio policistico)d appartenenza a gruppo etnico ad alto rischio
Tabella 68.7 Soggetti ad alto rischio di diabete per classi di età

parte 11 - MaLaTTIE DEL RICaMBIo 1514
Diabete autoimmune latente dell’adultoUna proporzione intorno al 10% dei pazienti inizialmente definiti come diabetici tipo 2 è in realtà affetta da una forma di diabete autoimmune a lenta evoluzione verso l’insulino-dipendenza, definito LaDa (Latent autoim-mune Diabetes in adult), che forse non è altro se non il diabete di tipo 1 dell’adulto. Utilizzando il solo criterio clinico, alla diagnosi della malattia i pazienti sono definiti come diabetici di tipo 2 e iniziano il trattamento con dieta o terapia orale. Tuttavia, precocemente si osserva in questi soggetti un deterioramento della funzione b-insulare tale da richiedere la terapia insulinica. Elementi clinici che possono suggerire il sospetto di LaDa sono: età < 50 anni, BMI < 25 kg/m2, familiarità per diabete di tipo 1, anamnesi familiare o personale positiva per malattie autoimmuni; inadeguato compenso glicemico in corso di trattamento con terapia orale a distanza di 6-12 mesi dalla diagnosi.
Esami di laboratorioDiagnosi e screening del diabeteI criteri per porre diagnosi di diabete sono riassunti nella tabella 68.2. La misurazione della glicemia a fini diagno-stici e di screening va effettuata su plasma venoso. Sono sconsigliate le misurazioni su sangue venoso intero o capillare a causa della scarsa possibilità di standardizza-zione. L’esecuzione dell’oGTT o curva da carico orale di glucosio prevede la misurazione della glicemia dopo due ore dall’assunzione per via orale di 75 g di glucosio dissolti in 300 mL di acqua. Il test non va eseguito in presenza di patologie acute intercorrenti e l’assunzione di carboidrati nei tre giorni precedenti l’esame deve essere normale (al-meno 200 g/die). Si ricorre all’utilizzo dell’oGTT sempre meno frequentemente, preferendo la più semplice e stan-dardizzabile misurazione della glicemia a digiuno. Tutta-via, nei soggetti con IFG e obesità addominale o sindrome metabolica può essere utile l’esecuzione di un oGTT per una migliore definizione diagnostica e prognostica.
Definizione del tipo di diabeteSebbene l’anamnesi e le modalità di esordio (si veda Tab. 68.6) siano spesso sufficienti a definire il tipo di diabete, in alcuni casi, per esempio nel sospetto clinico di LaDa, la determinazione dei marcatori di autoimmunità (GaDa, ICa, Ia-2) e i test di valutazione della funzione b-cellulare (misurazione del C-peptide basale o dopo stimolo con glucagone) possono fornire informazioni aggiuntive. In un soggetto a digiuno viene eseguito un prelievo basale 6 minuti dopo la somministrazione e.v. di 1 mg di glu-cagone. Valori di C-peptide basali < 0,2 nmol/L o dopo stimolo < 0,6 nmol/L sono indicativi di un grave deficit di secrezione insulinica con necessità di trattamento so-stitutivo. In presenza di un fondato sospetto clinico di MoDY è necessario rivolgersi a laboratori di riferimento per la caratterizzazione del difetto genetico.
Complicanze acute
Chetoacidosi diabeticaLa chetoacidosi diabetica rappresenta una complicanza metabolica acuta caratteristica del diabete mellito di tipo 1, seppure raramente osservabile anche nel diabete mellito
tipo 2. Tale complicanza si caratterizza per un’incidenza stimata in 4,6-8/1000 soggetti diabetici/anno e per una mortalità inferiore al 5% in centri specializzati. Essa viene definita dalla presenza di iperglicemia (> 250 mg/dL), aci-dosi metabolica (pH < 7,3; HCo3
− < 15 mmol/L) e iperche-tonemia (> 5 mmol/L) o rilievo di chetonuria su campione di urina analizzato mediante striscia reattiva.La chetoacidosi diabetica si instaura in presenza di due condizioni: (1) deficit insulinico, assoluto o relativo; (2) eccesso di glucagone e ormoni della controregolazione (cortisolo, catecolamine e GH). Il deficit insulinico as-soluto può verificarsi in caso di diabete mellito di tipo 1 all’esordio o di non corretto impiego della terapia insu-linica (sospensione o riduzione), mentre quello relativo può instaurarsi in condizioni di stress (infezioni, traumi, eventi cardiovascolari acuti ecc.), in caso di assunzione di farmaci che interferiscono con il metabolismo glucidico (per esempio, steroidi, diuretici tiazidici) o in presenza di endocrinopatie (per esempio, ipertiroidismo, feocro-mocitoma).Come specificato nel paragrafo Insulina e omeostasi glu-cidica, la compresenza di deficit insulinico e di eccesso di glucagone si traduce in riduzione della captazione periferica di glucosio, aumento della glicogenolisi, della gluconeogenesi (anche per l’accresciuta disponibilità di substrati quali aminoacidi e glicerolo) – con incrementato rilascio epatico di glucosio – e della lipolisi dei trigliceridi nel tessuto adiposo, con rilascio di acidi grassi e glicerolo. Tali modifiche del metabolismo provocano lo sviluppo di iperglicemia e la produzione a livello epatico di cor-pi chetonici (acetone, acetoacetato, b-idrossi butirrato). Essendo questi ultimi acidi deboli che, a pH fisiologico, sono dissociati per il 99%, si determina un aumento degli idrogenioni, che entrano nelle cellule scambiandosi con gli ioni K+, che vengono eliminati nelle urine. Da un lato, l’iperglicemia determina, con meccanismo osmotico, il richiamo di acqua dalle cellule – provocandone disidra-tazione – e diuresi osmotica, con deplezione di elettroliti, quando la concentrazione plasmatica di glucosio eccede la soglia renale. Dall’altro, l’aumentata produzione di corpi chetonici, qualora non corrisposta da un loro aumentato metabolismo nei tessuti periferici, comporta lo sviluppo di chetosi con conseguente acidosi metabolica, che a livello polmonare viene compensata mediante iperventilazione. La chetoacidosi determina sviluppo di insulino-resistenza, ipotensione – per effetto vasodilatante a livello periferico e inotropo negativo a livello cardiaco – e depressione ce-rebrale.La chetoacidosi diabetica deve essere sospettata in base ai dati clinici e anamnestici e confermata con alcuni esami chiave.I sintomi sono rappresentati da polidipsia, anoressia, astenia, crampi muscolari, nausea, vomito e addomi-nalgia. Con riferimento a quest’ultima è utile avere presente che, se da un lato i sintomi addominali pos-sono mimare quelli di un addome acuto, dall’altro un addome acuto può costituire il fattore scatenante della chetoacidosi.I segni clinici di chetoacidosi diabetica sono caratterizza-ti da poliuria, calo ponderale, tachicardia, ipotensione, respiro profondo e frequente (Kussmaul), alito acetoni-co, ipotermia, cute secca e calda, grave disidratazione,

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1515
ipotonia muscolare, iporeflessia, alterazione dello stato di coscienza sino al coma, addome trattabile o addome acuto o ileo paralitico.Dal punto di vista anamnestico è opportuno indagare, in caso di diabete noto, sulla terapia (tipo, dose, ultima som-ministrazione) e sulla presenza di condizioni di stress e, in caso di diabete di nuova diagnosi, sulle possibili cause scatenanti, in particolare infezioni, infarto miocardico acuto, embolia polmonare, vasculopatia cerebrale acuta, ischemia mesenterica, pancreatite acuta, traumi, abuso alcolico e impiego di farmaci potenzialmente interferenti con il metabolismo glicidico.La diagnosi di chetoacidosi diabetica si basa sul riscontro di iperglicemia (> 250 mg/dL), acidosi metabolica con gap anionico aumentato (pH < 7,3; HCo3
− < 15 mmol/L) e marcate glicosuria e chetonuria o iperchetonemia (> 5 mmol/L).Gli esami ematochimici possono evidenziare leucocitosi neutrofila (secondaria a disidratazione e/o infezione), aumento della creatininemia (può riflettere disidratazione e/o insufficienza renale prerenale), sodiemia nella norma o ridotta (a causa dello spostamento osmotico di acqua dalle cellule indotto dall’iperglicemia), kaliemia nella norma, lievemente diminuita o aumentata, aumento di CpK, transaminasi, amilasi e lipasi (può essere rilevato un aumento aspecifico in assenza di evidenza clinica di pancreatite) e coagulazione intravascolare disseminata. L’elettrocardiogramma può evidenziare segni di cardio-patia ischemica acuta, aritmie e alterazioni secondarie a variazioni della kaliemia.La terapia della chetoacidosi diabetica consiste nell’idra-tazione, nella somministrazione di insulina e nella corre-zione degli eventuali disordini elettrolitici.La terapia idratante ha l’obiettivo di ristabilire il volu-me plasmatico, mediante infusione preferibilmente di soluzione isotonica. L’utilizzo di soluzione ipotonica è consigliato solo in caso di rilievo di sodiemia > 145 mEq/L all’inizio o > 155 mEq/L durante il trattamento, fino a normalizzazione del parametro. La soluzione ipotonica, infatti, nel causare una riduzione troppo rapida dell’osmo-larità, potrebbe favorire lo sviluppo di edema cerebrale. Le soluzioni colloidali, che sono trattenute esclusivamente a livello intravascolare, possono rendersi necessarie in presenza di shock ipovolemico. L’infusione di liquidi deve essere regolata in base all’osmolarità plasmatica (nor-malmente pari a 280-300 mosm/L) e al deficit idrico del soggetto, calcolati secondo le seguenti formule:
Posm = 2 Na+ (mEq/L) + glicemia (mg/dL)
_______________ 18
Deficit ______ idrico
= 285 − Posm ___________ Posm
× peso ________ corporeo ×
60% (uomo) ______________
o 50% (donna)
Il deficit idrico deve essere corretto in non meno di 24 ore. È utile considerare che il fabbisogno idrico può essere aumentato a causa della diuresi osmotica e dell’eventuale presenza di fattori che favoriscono la deplezione di liquidi (per esempio, febbre, vomito, diarrea ecc.). Nei soggetti a rischio di sviluppare insufficienza ventricolare sinistra (per esempio, cardiopatici, anziani ecc.) è necessario un attento monitoraggio clinico, eventualmente esteso alla valutazione dello stato emodinamico mediante misura-zione della pressione venosa centrale.
La terapia insulinica permette di ridurre la sintesi di corpi chetonici, mediante inibizione della lipolisi e conseguen-te riduzione degli acidi grassi liberi affluenti al fegato, e l’osmolarità plasmatica, mediante inibizione della glu-coneogenesi e aumentato utilizzo tissutale di glucosio. L’insulina regolare viene somministrata mediante in-fusione continua e.v. con pompa automatica alla dose, nell’adulto, di 0,1 U/kg/h. L’infusione può essere prece-duta, una volta esclusa la presenza di ipokaliemia, da una somministrazione – la cui utilità non è peraltro unani-memente riconosciuta – di insulina regolare di 0,15 U/kg in bolo e.v. La velocità di infusione dell’insulina deve esse-re modificata in relazione al variare dei valori glicemici.I valori di kaliemia, che all’esordio possono essere nor-mali, aumentati o ridotti, tendono a diminuire con la progressiva correzione dell’acidosi metabolica. al fine di prevenire l’ipokaliemia è opportuno infondere cloruro di potassio, eventualmente associato a piccole dosi di fosfato di potassio, immediatamente – se la kaliemia iniziale è nor-male o bassa – o eventualmente nel corso della terapia – se la kaliemia iniziale è elevata.L’utilità della somministrazione di bicarbonati nella che-toacidosi diabetica è controversa per i potenziali rischi connessi al loro impiego (alcalosi tardiva da rimbalzo, aumento dell’affinità dell’emoglobina per l’ossigeno, ri-duzione del pH del liquor cerebrospinale con acidosi pa-radossa a livello del sistema nervoso centrale e possibile peggioramento dello stato di coscienza, riduzione della kaliemia e della calcemia) in assenza di provata effica-cia nel ridurre la mortalità nei soggetti chetoacidotici. L’american Diabetes association considera prudente la somministrazione di bicarbonati per valori di pH < 7.poiché la terapia insulinica determina una correzione dei livelli glicemici più rapida rispetto a quella della chetoa-cidosi, quando i valori glicemici raggiungono 250 mg/dL è necessario iniziare l’infusione di glucosio, al fine di prevenire lo sviluppo di ipoglicemia e la repentina ri-duzione dell’osmolarità e di migliorare la chetoacidosi. Si infonde soluzione glucosata a velocità costante, re-golando l’infusione di soluzione fisiologica contenente insulina regolare in base ai valori glicemici, che devono essere mantenuti intorno a 200-250 mg/dL. La terapia infusionale può essere sospesa quando si raggiungano valori glicemici < 200 mg/dL in presenza di pH > 7,3 con bicarbonati plasmatici > 18 mEq/L e il paziente è in grado di alimentarsi. In questo caso, deve essere somministrata terapia insulinica sotto cute 30 min circa prima del pasto e, al fine di garantire adeguati livelli insulinemici, deve essere mantenuta la terapia insulinica e.v. per 1-2 ore dopo la somministrazione sottocute (s.c.). La prosecuzione del trattamento con terapia insulinica s.c. deve essere impo-stata secondo schemi di terapia ottimizzata.La chetosi deve essere monitorata avendo come riferimen-ti il pH e il gap anionico. Non sono, invece, da considerare i corpi chetonici, in quanto le strisce al nitroprussiato, impiegate per la loro determinazione, sono reattive solo all’acetone e all’acetoacetato e non al b-idrossibutirrato. Quest’ultimo, presente nel plasma in concentrazioni molto elevate, viene convertito in acetoacetato duran-te la correzione dell’acidosi. pertanto, nelle ore che se-guono l’inizio della terapia, la positività della reazione può risultare ancora elevata, senza che ciò implichi un

parte 11 - MaLaTTIE DEL RICaMBIo 1516
peggioramento dell’acidosi. Nell’evoluzione del quadro il pH aumenta, il gap anionico si riduce, mentre i bicar-bonati restano bassi per l’ipercloremia da infusione di soluzione fisiologica.
Sindrome iperglicemica iperosmolare non chetoticaLa sindrome iperglicemica iperosmolare non chetotica è una complicanza acuta del diabete mellito definita da concomitante presenza di iperosmolarità (320 mosm/L), iperglicemia (> 600 mg/dL) e significativa disidratazione, per lo più in assenza di acidosi metabolica.Tale complicanza ha incidenza superiore a quella della chetoacidosi diabetica e mortalità stimata del 15%. Essa viene più comunemente riscontrata in soggetti con dia-bete mellito di tipo 2, seppure possa anche riguardare soggetti diabetici di tipo 1 che praticano una terapia in-sulinica inadeguata a mantenere il compenso glicemico, ma sufficiente a prevenire lo sviluppo di chetosi.La sindrome iperglicemica iperosmolare non chetotica è causata da fattori favorenti lo sviluppo di scompenso glicemico e/o disidratazione, quali ridotto apporto di liquidi, stress intercorrenti (diarrea, vomito, infezioni, ictus cerebrale ecc.), endocrinopatie (ipertiroidismo, feo-cromocitoma), terapia con farmaci che possono interferire con il metabolismo glicidico (steroidi, diuretici ecc.). Qua-lora, infatti, lo sviluppo di iperglicemia e la conseguente diuresi osmotica non siano controbilanciati da apporto idrico adeguato a compensare la perdita di liquidi con le urine, si instaura uno stato di grave disidratazione, che provoca la riduzione del filtrato glomerulare e il rilascio di ormoni della controregolazione (glucagone, cortisolo, catecolamine e GH). Questi ultimi determinano l’ulteriore incremento della glicemia, dell’osmolarità e della disidra-tazione sino allo shock.Come sopra anticipato, la sindrome iperglicemica ipero-smolare non chetotica è per lo più caratterizzata da assenza di chetoacidosi. Tale evenienza dipende almeno in parte dalla residua funzione b-cellulare a livello pancreatico, sep-pure la ridotta perfusione tissutale possa indurre lo svilup-po di lieve acidosi da iperchetonemia e iperlattatemia.La sindrome iperosmolare non chetotica deve essere sospet-tata in soggetti diabetici di tipo 2 anziani, poco controllati, che vivono soli o in istituti e si presentano astenici, disidra-tati, tachicardici, ipotesi, con cute secca, mucose asciutte, alterazione dello stato di coscienza fino al coma e disturbi neurologici, quali convulsioni, emiparesi transitoria, iperto-no muscolare e pseudorigor nucale. È utile osservare che i di-sturbi neurologici, la febbre e lo pseudorigor nucale possono erroneamente orientare verso un quadro di meningite. Nelle settimane precedenti lo sviluppo della complicanza possono essere riferiti poliuria, polidipsia e calo ponderale.La diagnosi di sindrome iperosmolare non chetotica si basa sul riscontro di iperglicemia (> 600 mg/dL), marca-ta glicosuria in assenza o presenza di lieve chetonuria, osmolarità > 320 mosm/L ed evidenza all’emogasanalisi di pH > 7,3 e HCo3
− > 15 mmol/L. Gli esami ematochimici possono evidenziare leucocitosi neutrofila, conseguente a disidratazione e/o infezione, aumento della creatininemia a causa della disidratazione, normo/ipersodiemia, normo/ipo/iperkaliemia, aumento di CpK, transaminasi, amilasi e lipasi e coagulazione intravascolare disseminata.
I principi di terapia della sindrome iperosmolare non che-totica sono analoghi a quelli della chetoacidosi diabetica e comprendono l’idratazione, la somministrazione di insulina e la correzione degli eventuali disordini elettro-litici. Tuttavia, poiché tale complicanza si manifesta con maggiore frequenza nei soggetti anziani, con frequen-te comorbilità cardiovascolare e/o renale, è opportuno procedere cautamente con l’idratazione, effettuando il monitoraggio clinico e, se necessario, della pressione ve-nosa centrale. La terapia idratante può di per sé ridurre in misura significativa l’iperglicemia attraverso la correzione dell’ipovolemia, che permette l’aumento del filtrato glo-merulare e dell’escrezione urinaria di glucosio.Il fabbisogno insulinico è di norma inferiore a quello dei soggetti diabetici con chetoacidosi, seppure possano ren-dersi necessarie dosi superiori in presenza di contestuali condizioni suscettibili di determinare insulino-resistenza (infezioni, cardiovasculopatie acute ecc.). L’insulina re-golare viene somministrata mediante infusione continua e.v. con pompa automatica alla dose iniziale, nell’adulto, di 0,1 U/kg/h. L’infusione può essere preceduta, una volta esclusa la presenza di ipokaliemia, da una somministrazio-ne – la cui utilità non è peraltro unanimemente riconosciu-ta – di insulina regolare di 0,15 U/kg in bolo endovena. La velocità di infusione dell’insulina deve essere modificata in relazione al variare dei valori glicemici.È necessario iniziare l’infusione di glucosio quando si raggiungono valori glicemici di 300 mg/dL, al fine di pre-venire episodi ipoglicemici e di evitare repentine ridu-zioni dell’osmolarità plasmatica. Si infonde soluzione glucosata a velocità costante, regolando l’infusione di soluzione fisiologica contenente insulina regolare in base ai valori glicemici, che devono essere mantenuti intorno a 200-250 mg/dL. La terapia infusionale può essere sospesa quando si raggiungano valori glicemici < 250-300 mg/dL, osmolarità < 310 mosm/L e il paziente è in grado di ali-mentarsi. Quindi, deve essere impostata terapia insulinica s.c. secondo schemi di terapia ottimizzata.Le eventuali alterazioni della kaliemia devono essere corret-te avendo presente, da un lato, che nella sindrome ipergli-cemica iperosmolare non chetotica, a causa del più lungo periodo prodromico, il deficit di potassio può essere supe-riore a quello che si verifica nella chetoacidosi diabetica, dall’altro, che nei soggetti con sindrome iperglicemica ipe-rosmolare non chetotica l’insufficienza renale è di comune riscontro ed espone a rischio di sviluppo di iperkaliemia.
IpoglicemiaSi veda oltre, il paragrafo Ipoglicemie.
Complicanze croniche
I pazienti con diabete, sia di tipo 1 sia di tipo 2, hanno una notevole tendenza a presentare complicanze a lun-go termine che, per la loro frequenza, fanno parte della storia naturale della malattia. Le complicanze vascolari del diabete si suddividono in macroangiopatiche e mi-croangiopatiche (retinopatia, nefropatia e neuropatia). Il rischio di svilupparle aumenta con la durata del diabete; tuttavia, poiché il diabete di tipo 2 ha spesso una lunga fase di iperglicemia silente, le complicanze possono essere già presenti al momento della diagnosi. L’iperglicemia

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1517
cronica è un fattore causale cruciale nello sviluppo delle complicanze microangiopatiche, sia nel diabete di tipo 1 sia nel diabete di tipo 2. Studi di intervento hanno infatti dimostrato in modo conclusivo che il miglioramento del compenso glicemico ottenuto con terapia intensiva è in grado di prevenire o ridurre significativamente lo sviluppo di retinopatia, nefropatia e neuropatia. L’osservazione che, a parità di compenso glicemico, soltanto alcuni sog-getti diabetici manifestano microangiopatia suggerisce, peraltro, che altri fattori (predisposizione genetica, iper-tensione arteriosa) possano avere un ruolo nello sviluppo di tali complicanze del diabete. La dimostrazione di un ruolo causale dell’iperglicemia cronica nella macroan-giopatia diabetica è meno convincente. L’associazione tra rischio cardiovascolare e compenso glicemico è stata documentata in numerosi studi epidemiologici, ma gli studi di intervento hanno dimostrato un’efficacia del mi-glioramento del compenso solo nel diabete di tipo 1. altri fattori, quali la dislipidemia e l’ipertensione, svolgono probabilmente un ruolo dominante nello sviluppo della macroangiopatia diabetica nel diabetico di tipo 2.
Meccanismi patogeneticiStudi condotti in vitro e nell’animale da esperimento han-no permesso di chiarire almeno in parte i meccanismi cellulari attraverso cui l’esposizione cronica a elevati li-velli glicemici determina lo sviluppo delle complicanze croniche del diabete. Il glucosio, per poter esercitare i suoi effetti deleteri, deve essere internalizzato dalle cellule dei tessuti bersaglio. È interessante notare come le complican-ze croniche del diabete non colpiscano i tessuti in cui la captazione del glucosio è regolata dall’insulina attraverso il trasportatore di membrana GLUT-4. Infatti, il deficit as-soluto o relativo di insulina, ostacolando l’internalizzazio-ne del glucosio, protegge questi tessuti dagli effetti deleteri degli elevati livelli di glucosio extracellulari. Sono invece colpiti i tessuti in cui l’internalizzazione cellulare del glu-cosio è mediata dal trasportatore di membrana GLUT-1, che è indipendente dall’insulina e la cui espressione è addirittura aumentata in presenza di elevati livelli glice-mici. Sono stati proposti quattro meccanismi attraverso cui gli elevati livelli di glucosio possono determinare una disfunzione o un danno cellulare: formazione di prodotti avanzati di glicosilazione (aGE), attivazione delle vie me-taboliche dei polioli e delle esosamine, attivazione della via di segnale della proteina chinasi C (pKC). Il glucosio intracellulare viene normalmente metabolizzato attraver-so la via della glicolisi, ma, quando presente in eccesso, può seguire anche altre vie metaboliche. Nella via dei polioli il glucosio è convertito a sorbitolo con conseguente alterazione dello stato redox della cellula, formazione di radicali dell’ossigeno e stress ossidativo della cellula. Nella via delle esosamine il glucosio è convertito in glucosio-6-fosfato, un substrato per la glicosilazione delle proteine, e le proteine glicosilate possono presentare alterazioni funzionali. Il glucosio in eccesso può, attraverso l’aumen-tata produzione di diacilglicerolo (DaG), attivare la via di segnale della pKC, che, agendo su fattori trascrizionali, regola l’espressione di numerosi geni potenzialmente deleteri se iperespressi. Infine, l’interazione del glucosio con gli aminogruppi delle proteine (glicosilazione non enzimatica) porta alla formazione di aGE che alterano la
struttura e interferiscono con la funzionalità e il catabo-lismo delle proteine. Recentemente è stata proposta una teoria che unifica questi meccanismi di danno in un uni-co modello. Secondo questa teoria il glucosio, causando stress ossidativo a livello mitocondriale, determinerebbe una riduzione dell’attività della glucosio-6-fosfato deidro-genasi, un enzima cruciale della glicolisi. Il ridotto flusso di glucosio lungo la via della glicolisi favorirebbe il suo utilizzo alternativo nella via dei polioli e delle esosamine e per la sintesi di DaG e aGE.
Macroangiopatia diabeticaLa macroangiopatia diabetica è una tendenza a sviluppare aterosclerosi più precocemente e in misura più significati-va rispetto a quanto osservato nella media della popola-zione. Lo studio di Framingham ha, infatti, dimostrato un marcato aumento di vasculopatia periferica, scompenso cardiaco, coronaropatia, infarto miocardico e ictus in presenza di diabete mellito. Le malattie cardiovascolari sono, inoltre, responsabili di oltre il 50% dei decessi tra i soggetti diabetici. Il diabete è, quindi, un fattore di rischio cardiovascolare maggiore, al pari di ipertensione arteriosa e dislipidemia, e nella valutazione del rischio cardiovasco-lare globale la sua presenza è considerata un equivalente di rischio coronarico. In altre parole, un soggetto diabe-tico ha un rischio di eventi coronarici pari a quello di un soggetto postinfartuato. L’entità del rischio cardiovascola-re associato al diabete è, peraltro, fortemente influenzata dal gruppo etnico. In Italia, per esempio, i diabetici di tipo 2 sembrano esposti a un rischio cardiovascolare inferiore rispetto a quello di diabetici appartenenti a popolazioni nord-europee o statunitensi. Le carte del rischio italiane elaborate dall’Istituto Superiore di Sanità (www.cuore.iss.it) rappresentano uno strumento utile nella valutazione del rischio cardiovascolare dei diabetici italiani. Fattori di rischio cardiovascolare specifici del soggetto diabeti-co includono scarso compenso glicemico, presenza di nefropatia diabetica e di insulino-resistenza. L’approccio diagnostico-terapeutico non differisce sostanzialmente da quello suggerito per i soggetti non diabetici. Tuttavia, occorre tenere conto che i soggetti diabetici hanno più fre-quentemente sintomi anginosi atipici o ischemia/infarto silente, probabilmente per la compresenza di neuropatia autonomica con denervazione del cuore. Inoltre, nel dia-betico la patologia coronarica coinvolge spesso più vasi coronarici e si associa a prognosi più infausta rispetto al soggetto non diabetico. al fine di identificare i soggetti affetti da macroangiopatia, tutti i pazienti con diabete, indipendentemente dal livello di rischio, devono eseguire annualmente un esame dei polsi periferici con ricerca dei soffi vascolari, un ECG basale e una determinazione dell’indice di Winsor. È dibattuta l’utilità di sottoporre a screening con test cardiologico provocativo i soggetti diabetici asintomatici.
Retinopatia diabeticaNegli Stati Uniti la retinopatia diabetica rappresenta la principale causa di cecità nella fascia di età tra i 20 e i 74 anni e i soggetti diabetici hanno un rischio di svi-luppare cecità 25 volte maggiore rispetto ai soggetti non diabetici. Sono stati identificati due stadi di retinopa-tia diabetica: semplice e proliferativa. Nella retinopatia

parte 11 - MaLaTTIE DEL RICaMBIo 1518
semplice la perdita dei periciti e delle cellule endoteliali e l’ispessimento della membrana basale determinano aumento della permeabilità vasale e sviluppo di altera-zioni microvascolari (formazione di microaneurismi, vasi tortuosi e dilatati, shunt arterovenosi e occlusione vasa-le). L’aumentata permeabilità capillare, che può essere evidenziata dalla fuoriuscita di colorante dai capillari all’esame fluoroangiografico, causa il passaggio extrava-sale di materiale plasmatico (lipidi, proteine) con forma-zione di caratteristici essudati definiti “essudati duri”. I microaneurismi, estroflessioni dei capillari retinici dovute almeno in parte a indebolimento della parete vasale per perdita dei periciti, si possono rompere causando piccole emorragie retiniche, che negli strati più profondi della retina hanno un aspetto puntiforme e, negli strati più su-perficiali, assumono, disponendosi lungo le fibre nervose, il cosiddetto aspetto “a fiamma”. L’obliterazione dei vasi con conseguente ischemia, oltre a causare la formazione di aree di microinfarti denominati “essudati cotonosi”, è responsabile del rilascio di fattori vasoproliferativi (VEGF, IGF, eritropoietina), che stimolano la formazione di nuovi vasi nel tentativo di rivascolarizzare il tessuto retinico. Il secondo stadio della retinopatia diabetica, denominato retinopatia proliferativa, è caratterizzato essenzialmente da neoformazione di vasi indotta dall’ischemia retini-ca. Questi nuovi vasi si rompono facilmente, causando emorragie nel vitreo, fibrosi e distacco della retina, con conseguente riduzione del visus sino alla cecità. Un’al-tra importante causa di cecità nel soggetto diabetico è l’edema maculare che si può sviluppare in ogni stadio di retinopatia ed è dovuto ad aumento della permeabilità capillare con edema nella zona della macula e ispessi-mento della retina. La retinopatia semplice colpisce l’80% dei diabetici di tipo 1 con oltre 20 anni di malattia. Non tutti i soggetti con retinopatia semplice sviluppano reti-nopatia proliferativa. Tuttavia, maggiore è la gravità della retinopatia semplice e maggiore è la probabilità di evolu-zione a retinopatia proliferativa entro 5 anni. poiché la retinopatia diabetica diventa sintomatica solo negli stadi molto avanzati di malattia ed esistono trattamenti efficaci, se attuati precocemente, è importante sottoporre tutti i soggetti con diabete a screening annuale con valutazione del fundus oculi in dilatazione per una precoce diagnosi della complicanza. Lo screening va iniziato dopo 3-5 an-ni dall’insorgenza del diabete nel tipo 1 e alla diagnosi in quello di tipo 2. Il controllo intensivo del compenso glicemico e dei livelli di pressione arteriosa sono efficaci nel prevenire e ritardare la progressione della retinopatia diabetica. Inoltre, il trattamento con fotocoa gulazione laser si è dimostrato efficace nel preservare la vista nei pa-zienti con retinopatia diabetica proliferativa o retinopatia semplice in fase avanzata.
Nefropatia diabeticaLa nefropatia diabetica è la causa principale di insuf-ficienza renale cronica nei paesi occidentali e colpisce circa un terzo dei soggetti diabetici. Si ritiene che fattori genetici e la presenza di elevati livelli di pressione ar-teriosa conferiscano suscettibilità allo sviluppo di tale complicanza. La patologia, che inizialmente colpisce soprattutto il glomerulo renale, è caratterizzata da perdita di podociti e accumulo di matrice extracellulare nel me-
sangio, che diventa sclerotico, e nella membrana basale glomerulare, che si ispessisce. Ciò si traduce, dal punto di vista funzionale, in un aumento della permeabilità glomerulare alle proteine, con conseguente aumento della concentrazione delle proteine nelle urine, e in una progressiva perdita della funzionalità renale con caduta del filtrato glomerulare. Un lieve aumento dell’escrezione renale di albumina (aER), denominata microalbuminu-ria, rappresenta lo stadio più precoce della complicanza (nefropatia incipiente) e si accompagna non solo a un elevato rischio di progressione verso la macroalbuminu-ria (nefropatia conclamata), ma anche a un aumentato rischio di patologia cardiovascolare. Nel soggetto con macroalbuminuria si ha un progressivo declino della funzionalità renale e, in assenza di terapia, il 50% dei pazienti necessita entro 7-10 anni di dialisi o trapianto di rene. Diversi interventi terapeutici si sono dimostrati efficaci nel ridurre il rischio e rallentare la progressione della malattia renale. È, quindi, importante sottoporre tutti i diabetici a screening per la presenza di microal-buminuria, iniziando 5 anni dopo l’insorgenza del dia-bete nel tipo 1 e alla diagnosi di diabete nel tipo 2. Lo screen ing prevede la misurazione del rapporto albumina/creatinina (a/C) su un campione di urina (occasionale o del mattino) oppure dell’albuminuria su raccolte tem-porizzate (24 ore/notte) (Fig. 68.3). Valori anormali in almeno due su tre test eseguiti in 6 mesi sono necessari per considerare un soggetto micro/macroalbuminurico. È, inoltre, importante stimare annualmente anche il filtrato glomerulare (eGFR). La terapia ha lo scopo di raggiungere valori glicemici il più possibile vicini alla normoglicemia e di mantenere la pressione arteriosa a livelli inferiori a 130/80 mmHg (< 125/75 mmHg in presenza di protei-nuria > 1 g). oltre al loro effetto antipertensivo i farmaci che interferiscono con il sistema renina-angiotensina II (aCE-inibitori e bloccanti del recettore dell’angiotensina II-aRB) hanno un dimostrato effetto antiproteinurico e protettivo sulla funzionalità renale e devono essere sem-pre usati nel paziente diabetico di tipo 1 (aCE-I) e di tipo 2 (aRB) con micro/macroalbuminuria, incrementando progressivamente la dose sino a ottenere una riduzione della proteinuria. Nei pazienti con nefropatia conclamata l’apporto proteico dovrebbe essere ridotto a 0,8 g/kg/die. Un’ulteriore riduzione (0,6-0,8 g/kg/die) può essere utile nel rallentare il declino del GFR in pazienti in progressio-ne nonostante l’ottimizzazione della terapia.
Neuropatia diabeticaLa neuropatia diabetica, che comprende la neuropatia periferica e la neuropatia autonomica, colpisce circa il 50% dei soggetti con diabete di lunga durata. La forma più comune di neuropatia periferica è la polineuropa-tia simmetrica distale dovuta a un’assonopatia su base metabolica e ischemica (danno dei vasa vasorum delle fibre nervose). Vengono colpite le fibre sensitive sia pic-cole (sensibilità dolorifica, tattile, termica) sia grandi (sensibilità vibratoria e propriocettiva), mentre il coin-volgimento dei motoneuroni si verifica tardivamente e solo nelle forme più severe. poiché il danno è propor-zionale alla lunghezza dell’assone, la patologia dappri-ma interessa piedi e mani (distribuzione calza/guanto) e poi progredisce in senso prossimale. Iniziali disturbi

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1519
irritativi delle piccole fibre (iperestesie, parestesie, dolore urente a riposo prevalentemente notturno) sono seguiti, al progredire del danno neuronale, da anestesia tattile, dolorifica, termica. L’interessamento delle grandi fibre causa perdita della sensibilità vibratoria e propriocettiva con riduzione dei riflessi osteoarticolari e disordini della postura/andatura per anormale senso di posizione. La perdita di sensibilità del piede è responsabile di ripetuti “traumi” silenti che favoriscono lo sviluppo di ulcere plantari o deformità del piede secondarie a fratture mul-tiple silenti e alterata postura (articolazione di Charcot). occorre ricercare la presenza di neuropatia annualmente in tutti i diabetici (dalla diagnosi nel tipo 2 e dopo 5 anni di malattia nel tipo 1) valutando la sensibilità presso-ria mediante monofilamento e la sensibilità vibratoria all’alluce mediante diapason. I pazienti affetti vanno inseriti in un programma di educazione specifico per il piede diabetico, allo scopo di prevenirne le ulcerazioni e le amputazioni, e il loro compenso glicemico deve essere ottimizzato. per alleviare il dolore neuropatico possono essere usati antidepressivi, quali amitriptilina, gabapentina, capsaicina.Il soggetto diabetico può anche sviluppare mononeuro-patie o neuropatie multiple che insorgono improvvisa-mente e in genere regrediscono spontaneamente entro 6-12 settimane. Sono comunemente coinvolti i nervi
cranici, soprattutto il III (diplopia) e il nervo femorale e predominano le alterazioni di tipo motorio.La neuropatia autonomica colpisce il sistema nervoso vegetativo con manifestazioni a carico dei sistemi cardio-vascolare (tachicardia a riposo, ipotensione ortostatica, morte improvvisa), gastrointestinale (atonia gastrica, al-ternanza di stipsi e diarrea per difetti della motilità inte-stinale che favoriscono la colonizzazione batterica e il ma-lassorbimento), genito-urinario (eiaculazione retrograda, impotenza, vescica neurologica), sudoriparo (iperidrosi delle estremità superiori e anidrosi di quelle inferiori). La perdita dei sintomi adrenergici aumenta il rischio di crisi ipoglicemiche gravi.
Il piede diabeticoIl diabete è la principale causa di amputazione non trau-matica degli arti inferiori. Le ulcere e le infezioni del piede rappresentano, inoltre, un’importante causa di morbilità tra i diabetici. La neuropatia e la macroangiopa-tia diabetica concorrono entrambe allo sviluppo dell’ul-cera diabetica. La neuropatia periferica interferisce con i normali meccanismi di protezione ed espone il piede diabetico a danno continuo; essa, inoltre, altera la bio-meccanica del piede favorendo lo sviluppo di alterazioni strutturali. La perdita della sensibilità propriocettiva alte-ra la postura e la deambulazione favorendo lo sviluppo di
Figura 68.3Livelli soglia di albuminuria per la classificazione dei pazienti in normo-, micro- e macroalbuminuria suddivisi in base alle metodiche di raccolta del campione di urina (spot/24 ore/notturna). Nel pannello in basso sono riportate le tappe dello screening.

parte 11 - MaLaTTIE DEL RICaMBIo 1520
calli e ulcerazioni. La neuropatia autonomica, nel causare anidrosi, aumenta la secchezza della cute e favorisce il crearsi di fissurazioni e ulcerazioni. La vasculopatia pe-riferica causa danno ischemico e ostacola i processi di guarigione favorendo le sovrainfezioni. Fondamentale nella prevenzione delle ulcere diabetiche è l’educazio-ne del paziente. Si raccomanda attenzione nella scelta delle calzature, ispezione quotidiana del piede, igiene scrupolosa e di evitare comportamenti a rischio, come camminare a piedi nudi o calzare scarpe strette. L’ulcera neuropatica si sviluppa di solito in sede plantare e può essere accompagnata da infezione locale e osteomielite. La superficie e i tessuti circostanti sono spesso colonizzati da flora polimicrobica e gli esami colturali sono di utilità solo se condotti su materiale (pus, tessuto) prelevato dalla profondità dell’ulcera. La presenza di osteomielite va esclusa mediante esecuzione di esami strumentali (ra-diografia, risonanza magnetica). L’eco-Doppler arterioso e l’arteriografia permettono di valutare la compresenza di insufficienza vascolare. Le procedure di rivascolarizza-zione favoriscono la guarigione e riducono il rischio di amputazione. Riposo, toeletta chirurgica con rimozione del tessuto necrotico, protratta antibioticoterapia a largo spettro ed eventuale rivascolarizzazione rappresentano i principali presidi terapeutici.
Valutazione del compenso metabolico
Nel paziente diabetico i principali strumenti per il con-trollo del compenso metabolico finalizzato a prevenire i sintomi dell’iperglicemia e lo sviluppo di complicanze acute e croniche sono la misurazione della Hba1c, l’auto-monitoraggio domiciliare della glicemia e la valutazione dell’assetto lipidico.
HbA1c e fruttosaminaL’emoglobina glicata è il prodotto della condensazione non enzimatica delle molecole di glucosio con gli ami-no gruppi liberi della globina dell’emoglobina. Maggiori sono i livelli medi di glicemia e più elevato sarà il livello di emoglobina glicata. La forma predominante di glico-emoglobina è la Hba1c (4-6% dell’emoglobina totale). poiché la Hba1c circola con i globuli rossi, che hanno un’emivita di 120 giorni, i suoi livelli riflettono la media della glicemia nei 2-3 mesi precedenti. La frequenza del dosaggio dell’Hba1c dipende dalla situazione clinica; in genere si raccomanda un dosaggio due volte all’anno se il controllo glicemico è ottimale e stabile, e ogni 3 me-si negli altri casi. Il dosaggio della Hba1c rappresenta il principale strumento per valutare l’efficacia della terapia in atto e, insieme con i dati dell’autocontrollo glicemico domiciliare, fornisce informazioni utili all’apporto di mo-difiche terapeutiche. La fruttosamina è il prodotto della glicosilazione non enzimatica delle proteine plasmatiche e in particolare dell’albumina. Condizioni cliniche che riducono i livelli di albumina (sindrome nefrosica, epato-patia) riducono falsamente i livelli di fruttosamina. poiché l’emivita dell’albumina è breve, la fruttosamina riflette la media dei livelli glicemici nelle 2 settimane precedenti il dosaggio. È consigliato il dosaggio della fruttosamina, in sostituzione a quello dell’Hba1c, in pazienti con stati emolitici o varianti dell’emoglobina o quando è necessario
valutare il compenso glicemico a intervalli più ravvicinati, per esempio nella donna in gravidanza.
Automonitoraggio domiciliare della glicemiaConsiste nella misurazione della glicemia su sangue intero capillare da parte del paziente a domicilio. Una goccia di sangue, ottenuta pungendo il dito, viene posta su una striscia reattiva che viene letta da un reflettometro. Le misurazioni possono essere eseguite al mattino a digiuno; prima e 2 ore dopo i pasti; prima, durante e dopo l’eserci-zio fisico e, occasionalmente, durante la notte. La frequen-za del monitoraggio va individualizzata. Nel diabete di tipo 1 la misurazione della glicemia consente al paziente di effettuare pronti aggiustamenti della terapia insulinica e della dieta e di intervenire tempestivamente in caso di ipoglicemia. È, pertanto, utile monitorare la glicemia sempre prima del pasto, periodicamente dopo il pasto, occasionalmente durante la notte. Nel diabete di tipo 2 l’automonitoraggio quotidiano è consigliato solo in caso di terapia insulinica o con farmaci che possono causare ipoglicemia. In tutti i soggetti diabetici l’automonitorag-gio va intensificato in presenza di situazioni cliniche quali patologie intercorrenti, ipoglicemie inavvertite, ipoglice-mie notturne, variazione della terapia ipoglicemizzante. È necessario istruire il paziente all’autocontrollo glicemico, valutare periodicamente la correttezza dell’utilizzo del reflettometro e la capacità di modificare la terapia sulla base dei valori misurati.
Valutazione dell’assetto lipidicoNei soggetti diabetici il controllo del quadro lipidico (colesterolo totale, colesterolo HDL e trigliceridi) deve essere effettuato almeno annualmente e, a intervalli di tempo più ravvicinati, qualora si riscontrino valori alte-rati. Infatti, nel soggetto diabetico si riscontrano spesso alterazioni del profilo lipidico, in quanto il metabolismo lipidico è sensibilmente influenzato dall’insulina. Nel diabetico di tipo 1 scompensato si osserva un modesto aumento del colesterolo LDL e dei trigliceridi, che risulta perlopiù corretto dal controllo della glicemia. Nel paziente con diabete di tipo 2 e obeso si osservano alterazioni del profilo lipidico (dislipidemia diabetica) secondarie alla presenza di uno stato di insulino-resistenza: aumento dei trigliceridi, riduzione del colesterolo HDL e prevalenza di particelle LDL piccole e dense, più suscettibili all’ossida-zione e quindi maggiormente aterogene.
Terapia
Obiettivi terapeuticiper gli obiettivi terapeutici si rimanda alla tabella 68.8.
Obiettivi glicemiciL’obiettivo è quello di normalizzare i livelli glicemici senza provocare ipoglicemie frequenti e gravi. Nei soggetti diabetici i livelli di Hba1c dovrebbero essere inferiori al 7%. L’obiettivo terapeutico può in casi se-lezionati essere ancora più ambizioso (Hba1c < 6,5%), ma ciò comporta un maggior rischio di ipoglicemia.

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1521
Nei pazienti con una ridotta aspettativa di vita, non autonomi, con patologia multisistemica, residenti in case di riposo, affetti da demenza è accettabile un livello di Hba1c compreso tra il 7,5 e l’8,5%. Vengo-no considerati accettabili livelli glicemici compre-si tra 90 e 130 mg/dL al mattino a digiuno e prima del pasto. Elevati livelli di glicemia postprandiale sono stati associati ad aumentato rischio cardiova-scolare indipendentemente dalla glicemia basale. pertanto, nei diabetici con valori ottimali di glicemia preprandiale, ma non di Hba1c, è verosimilmente possibile ottenere un’ulteriore riduzione dell’Hba1c e del rischio cardiovascolare con trattamenti miranti a valori di glicemia postprandiale < 180 mg/dL.
Altri obiettivi terapeuticiÈ opportuno assicurare, oltre al buon compenso glice-mico, anche un adeguato controllo della dislipidemia e dell’ipertensione arteriosa. La mancata riduzione del rischio cardiovascolare con il trattamento glice-mico intensivo che si è osservata negli studi clinici condotti nei pazienti con diabete di tipo 2, riflette verosimilmente la patogenesi multifattoriale della malattia cardiovascolare e sottolinea l’importanza di un approccio terapeutico esteso.
Ipertensione arteriosa
L’ipertensione arteriosa è una comune comorbilità nel diabete e un fattore di rischio maggiore per lo sviluppo di patologia cardiovascolare e complicanze microvascolari (retinopatia e nefropatia). Nel diabete di tipo 1 è spesso la conseguenza di una nefropatia sottostante e nel diabete di tipo 2 può essere presente come parte della sindrome meta-bolica, che si associa ad aumentato rischio di patologia car-diovascolare. Nei pazienti con diabete l’obiettivo terapeutico è quello di ottenere valori di pressione sistolica < 130 mmHg e di pressione diastolica < 80 mmHg. Un obiettivo presso-rio < 125/75 mmHg è raccomandato nei soggetti diabetici con proteinuria > 1 g/die. Infine, nelle donne diabetiche la
pressione sistolica in gravidanza deve essere compresa tra 110-129 mmHg e quella diastolica tra 65-79 mmHg.
Dislipidemia
Il colesterolo LDL è l’obiettivo primario della terapia. Nel paziente senza patologia cardiovascolare manifesta l’obiettivo è il raggiungimento di valori di colesterolo LDL < 100 mg/dL. Nei pazienti con patologia cardiovasco-lare manifesta valori di colesterolo LDL < 70 mg/dL sono un obiettivo terapeutico opzionale. altri obiettivi terapeu-tici sono livelli di trigliceridi < 150 mg/dL e di colesterolo HDL > 40 mg/dL nell’uomo e > 50 mg/dL nella donna.
Terapia del diabete
DietaLa dieta è un aspetto cruciale del trattamento e i pa-zienti diabetici dovrebbero ricevere da un dietista, pre-feribilmente inserito nel team diabetologico, una tera-pia medica nutrizionale individualizzata. Una mode-rata riduzione dell’apporto calorico (300-500 kcal/die), associata ad aumento del dispendio energetico, è raccomandata nei pazienti diabetici sovrappeso o obesi. In questo tipo di pazienti il calo ponderale rappresenta, infatti, il principale obiettivo terapeutico e si associa invariabilmente a un miglioramento del compenso glicemico, soprattutto perché riduce lo sta-to di insulino-resistenza. La composizione della dieta del paziente diabetico non differisce sostanzialmente dalla dieta bilanciata raccomandata a tutti i soggetti. L’obiettivo è quello non solo di evitare rapide escur-sioni dei livelli glicemici, ma anche e soprattutto di prevenire lo sviluppo delle complicanze croniche del diabete controllando il compenso glicemico, i livelli di pressione arteriosa e di colesterolo.L’apporto giornaliero di carboidrati, preferibilmente complessi, dovrebbe essere tale da fornire il 45-50% delle kcal totali giornaliere. Non sono indicate diete a basso contenuto di carboidrati, ma apporti elevati di carboidrati (> 55%) possono favorire l’iperglicemia, l’ipertrigliceridemia e la riduzione del colesterolo HDL e vanno evitati nei pazienti obesi con diabete di tipo 2. Il raggiungimento di un ottimale controllo glicemico può essere facilitato, soprattutto nei pa-zienti in terapia insulinica intensiva, dal controllo di quantità, qualità e orario di assunzione dei car-boidrati. Se il paziente è trattato con dosi costanti di insulina/ipoglicemizzanti è importante che l’in-troduzione dei carboidrati con i pasti sia mantenuta costante nelle quantità e nei tempi. a tale scopo sono utili il conteggio dei carboidrati e le diete a scambio, dove porzioni di cibo inserite nella stessa lista sono intercambiabili (www.eatright.org). Il conteggio dei carboidrati è anche utile al paziente che modifica il dosaggio dell’insulina preprandiale sulla base dei carboidrati che prevede di assumere. Il rapporto in-sulina/carboidrati è di circa 10-15 unità di insulina pronta per ogni 10 g di carboidrati introdotti con la dieta, anche se varia considerevolmente da individuo
Obiettivi glicemiciHbA1c < 7,0%
Glicemia preprandiale 90-130 mg/dL
Glicemia postprandiale < 180 mg/dL
Altri obiettivi
Pressione arteriosa < 130/80 mmHg
Colesterolo LDL < 100 mg/dL
Trigliceridi < 150 mg/dL
Colesterolo HDL > 40 mg/dL
Tabella 68.8 Obiettivi terapeutici

parte 11 - MaLaTTIE DEL RICaMBIo 1522
a individuo. I dolcificanti acalorici (saccarina, aspar-tame, acesulfame K, sucralosio) sono sicuri quando consumati in quantità giornaliere moderate, ma è anche consentito un modesto uso di saccarosio, sem-pre che venga considerato nella conta dei carboidrati. Le proteine dovrebbero fornire il 15-20% dell’energia totale giornaliera introdotta con gli alimenti. Tutta-via, nei soggetti con malattia renale cronica l’apporto proteico non dovrebbe superare gli 0,8 g/kg/die al fine di ridurre il rischio di evoluzione della nefropatia. L’apporto di grassi non deve contribuire per più del 30-35% (≤ 30% se obeso) all’energia totale giornaliera introdotta con gli alimenti (< 7-10% grassi saturi, < 10% grassi poli-insaturi, 10-20% di oli ricchi in acidi grassi monoinsaturi come l’olio d’oliva). L’assunzione di acidi grassi trans deve essere minimizzata (< 1%) e il colesterolo non deve superare i 200 mg/die. Una moderata introduzione di alcol (fino a 10 g/die nella donna e 20 g/die nell’uomo) è accettabile. Viene in-coraggiata l’assunzione di cibi ad alto contenuto di fibre (14 g/1000 Kcal) e di frutta e verdura per il loro ricco contenuto di antiossidanti, microelementi e vitamine; non è, invece, consigliata l’integrazione con antiossidanti, come le vitamine E, C e b-carotene. L’introduzione giornaliera di sale dovrebbe essere, al pari che nel soggetto non diabetico, inferiore a 6 g/die. Infine, è importante sottolineare che non esi-stono evidenze per raccomandare l’uso di alimenti “dietetici” per i soggetti con diabete.
Esercizio fisicoL’esercizio fisico ha plurimi effetti benefici nel pa-ziente diabetico: riduce il rischio cardiovascolare, la pressione arteriosa, il peso corporeo, la massa grassa, la glicemia (durante e dopo l’esercizio) e l’insulino-re-sistenza. Inoltre, l’esercizio fisico può avere favorevoli effetti psicologici. Un programma di esercizio fisico è, pertanto, consigliabile in tutti i soggetti diabetici e rientra nel piano terapeutico del paziente sovrappeso o obeso. Gli individui con diabete di tipo 1 hanno
la tendenza a sviluppare durante l’esercizio fisico iperglicemia-chetoacidosi (se i livelli insulinemici sono bassi) o ipoglicemia (se i livelli insulinemici sono elevati) ed è pertanto necessario controllare la glicemia prima, durante e dopo l’esercizio e ope-rare eventuali aggiustamenti della terapia insulini-ca/introito alimentare. L’esercizio fisico intenso è controindicato nei soggetti con retinopatia prolife-rativa non trattata, perché può favorire il distacco di retina e l’emorragia del vitreo. Test di tolleranza all’esercizio sono consigliati ai pazienti diabetici a elevato rischio cardiovascolare. Un programma sem-plice ed efficace di esercizio consiste nel cammino a passo veloce per 30 min al giorno per 5-7 giorni alla settimana.
Farmaci anti-iperglicemici oraliI farmaci utilizzati nel trattamento farmacologico dell’iperglicemia nel diabete di tipo 2 rientrano in tre categorie: farmaci che stimolano la secrezione insulinica, farmaci che aumentano l’azione periferica dell’insulina e farmaci che interferiscono con l’assor-bimento intestinale di glucosio.1. Farmaci che stimolano la secrezione insulinica. Gli
ipoglicemizzanti orali che stimolano il rilascio di insulina (secretagoghi) dalle cellule b pancreati-che sono le sulfoniluree, le glinidi e le incretine (Tab. 68.9).
Le sulfoniluree sono indicate nel trattamento del diabete di tipo 2 in monoterapia o in associazio-ne con la metformina. Sono efficaci nel miglio-rare il compenso glicemico (riduzione 1-2% della Hba1c), ma causano aumento del peso corporeo e rischio di ipoglicemia. Legandosi a un recetto-re sito sulla cellula b, inducono la chiusura dei canali del potassio con conseguente depolariz-zazione della cellula b, l’entrata di ioni calcio e il rilascio di insulina. Sono indicate solo nei pa-zienti diabetici con residua funzione b-cellulare e hanno ridotta efficacia nel diabete in fase avan-
Nome Formulazioni Dose/die Clearance
Sulfoniluree di seconda generazione
Glimepiride 1-2-4 mg 1-4 mg EpaticaRenale
Glipizide 5-10 mg 2,5-40 mg Epatica
Gliclazide 80 mg 40-320 mg EpaticaRenale
Gliclazide RM 30 mg 30-120 mg EpaticaRenale
Glibenclamide 1,25-2,5-5 mg 1,25-20 mg EpaticaRenale
Glinidi Repaglinide 0,5-1-2 mg 0,5-4 mg Epatica
Tabella 68.9 Caratteristiche dei secretagoghi

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1523
zata, quando viene perduta la funzione b-cellu-lare. La loro efficacia è proporzionale all’affinità con cui si legano al recettore. Le sulfoniluree di prima generazione sono cadute in disuso. Le sul-foniluree di seconda generazione, attualmente preferite, hanno una rapida insorgenza d’azione che consente, quando somministrate 30 min prima del pasto, il controllo dell’iperglicemia postprandiale. poiché riducono non solo la gli-cemia postprandiale, ma anche quella basale, espongono il paziente a rischio di ipoglicemia. per ridurre tale rischio occorre iniziare la terapia a basse dosi e aumentare il dosaggio gradualmente. Età avanzata, pasto ritardato o mancato, esercizio fisico, abuso alcolico, uso di farmaci a lunga durata d’azione (glibenclamide, glimepiride) od a elevato dosaggio espongono a un aumentato rischio di crisi ipoglicemiche.
Le glinidi, repaglinide e nateglinide, si legano a un recettore b-cellulare diverso da quello a cui si legano le sulfoniluree, pur inducendo anch’esse la chiusura dei canali del potassio con conseguente rilascio di insulina. Hanno breve durata d’azione con maggiore effetto sull’iperglicemia postpran-diale e minor rischio di ipoglicemia. La repaglini-de (la nateglinide non è in commercio in Italia) si somministra al dosaggio di 0,5 mg prima di ogni pasto e il dosaggio può essere aumentato a 1 mg se il controllo non è soddisfacente (dose massima 4 mg). poiché è metabolizzata dal fegato ed escreta solo per il 10% dal rene, può essere impiegata nei pazienti con insufficienza renale. Una nuo-va classe di farmaci secretagoghi è rappresentata dalle incretine. Le incretine sono ormoni peptidici, prodotti dal tratto gastroenterico, che regolano l’omeostasi glicidica in risposta al pasto. È stato dimostrato che l’incretina GLp-1, prodotta dalle cellule L dell’intestino tenue, stimola la biosintesi e la secrezione di insulina indotta dal glucosio, ha un effetto trofico sulla cellula b, inibisce la secrezione di glucagone, lo svuotamento gastrico, la secrezione acida e riduce l’introito alimentare. Rispetto agli altri secretagoghi il GLp-1 presenta, quindi, teoricamente un minor rischio di crisi ipoglicemiche e di aumento ponderale; inoltre, per il suo effetto trofico sulla cellula b, potrebbe ritardare la perdita di funzionalità della cellula b. Tuttavia, il GpL-1 non può essere utilizzato a scopo terapeutico perché viene rapidamente degradato dall’enzima dipeptidil-peptidasi IV e ha un’emivita estremamente breve (1-2 min). Sono stati, quindi, prodotti degli analoghi del GLp-1 resistenti all’azione della DDp-IV (exena-tide e liraglutide, somministrabili solo s.c.) e degli agenti che aumentano i livelli di GLp-1 iniben-do la DDp-IV (sitagliptina e vildagliptina a som-ministrazione orale). Seppure gli studi clinici di confronto siano al momento ancora scarsi, tali composti sembrano efficaci in associazione ad altri ipoglicemizzanti orali. Tuttavia, il costo, gli
effetti collaterali gastroenterici, l’efficacia modesta e la via di somministrazione (per gli analoghi del GLp-1) ne limitano, per ora, le indicazioni tera-peutiche.
2. Farmaci che aumentano l’azione periferica dell’in-sulina. Sono la metformina e i glitazoni. La metformina è il farmaco di prima scelta per il trattamento del paziente con diabete di tipo 2, perché è efficace (riduzione Hba1c 1,5%); non induce un aumento del peso corporeo, ma deter-mina al contrario una lieve riduzione ponderale; riduce i trigliceridi e gli acidi grassi circolanti; non causa ipoglicemia ed è di basso costo. Il suo meccanismo d’azione non è totalmente chiarito, ma è noto che inibisce la produzione epatica di glucosio, che riduce l’assorbimento intestinale e aumenta l’utilizzazione periferica (soprattut-to dopo il pasto) del glucosio. può essere usata sia in monoterapia sia in associazione ad altri ipoglicemizzanti o insulina. può dare distur-bi gastrointestinali (sapore metallico in bocca, nausea, diarrea, meteorismo), in particolare se usata ad alte dosi, ma tali disturbi sono spes-so transitori. Si somministra ai pasti, iniziando con basse dosi (per esempio, 500 mg alla sera) che possono essere gradualmente aumentate sino a una dose giornaliera massima di 2000 mg/die in 2 somministrazioni (colazione e cena). Essendo escreta dal rene è controindicata nei pazienti con insufficienza renale cronica (GFR stimato < 60 mL/min) o a rischio di insufficienza renale acuta (per esempio, per esame radiologico che richiede l’infusione di mezzi di contrasto iodati; in questo caso è opportuno sospendere la metformina il giorno prima della procedura e per 48 ore dopo l’esame). Va utilizzata con cautela nei pazienti con aumentata produzione di acido lattico secondaria a grave ipossia tis-sutale (alcolismo, insufficienza epatica, grave insufficienza cardiorespiratoria, shock settico), perché, inibendo la captazione epatica di lattato, può favorire lo sviluppo di acidosi lattica. La fenformina è stata ritirata dalla farmacopea in molti Stati, in quanto espone a un rischio signi-ficativamente più elevato di acidosi lattica fatale. In Italia è ancora disponibile in associazione, ma se ne sconsiglia l’uso.
I glitazoni (rosiglitazone e pioglitazone) agiscono legandosi a una famiglia di recettori nucleari denominati peroxisome proliferator activated Receptors (ppaR)-g, che regolano la trascrizione genica e sono prevalentemente espressi nel tessu-to adiposo, nel muscolo scheletrico e nel fegato. Un importante effetto trascrizionale dei glitazoni è rappresentato dall’aumentata espressione del trasportatore del glucosio GLUT-4, sia a livello del muscolo sia del tessuto adiposo, con conse-guente stimolo all’utilizzazione periferica del glucosio. Nel tessuto adiposo, inoltre, stimolano l’adipogenesi, riducono il rilascio di citochine

parte 11 - MaLaTTIE DEL RICaMBIo 1524
che inducono insulino-resistenza (TNF-a, resi-stina) e aumentano il rilascio di adiponectina, che ha un effetto insulino-sensibilizzante. La loro efficacia nel ridurre lo stato di insulino-resistenza è comprovata dall’osservazione che nei pazienti trattati si ha una diminuzione non solo dei livelli glicemici, ma anche di quelli insu-linemici. Il dosaggio giornaliero è di 4-8 mg/die in 1-2 somministrazioni per il rosiglitazone e di 15-45 mg/die in unica somministrazione per il pioglitazone. La loro efficacia nel ridurre i livelli di Hba1c in monoterapia è simile a quella delle altre terapie orali, ma l’effetto massimo viene raggiunto dopo un tempo più lungo (4-6 set-timane). Il pioglitazone, per la sua capacità di legare anche il recettore ppaR-a, ha un effetto sul profilo lipidico più favorevole del rosigli-tazone. I glitazoni causano un aumento del peso corporeo, specialmente se assunti in as-sociazione con insulina o sulfoniluree, e sono controindicati nei pazienti con insufficienza cardiaca (classi III, IV NYHa), perché tendono a causare ritenzione idrica e a favorire lo sviluppo di insufficienza cardiaca congestizia. Infine, i glitazoni riducono la massa ossea aumentando il rischio di fratture patologiche soprattutto nella donna. L’elevato costo e la presenza di effetti collaterali rilevanti suggerisce il loro utilizzo in monoterapia solo dopo dimostrata intolleranza agli altri farmaci.
3. Farmaci che interferiscono con l’assorbimento inte-stinale di glucosio. Le a-glucosidasi sono enzimi localizzati sulla superficie dei microvilli del tenue e idrolizzano i polisaccaridi e gli oligosaccaridi in monosaccaridi, consentendone l’assorbimento. Gli inibitori delle a-glucosidasi (acarboso, miglitol), inibendo questa funzione enzimatica, causano un rallentamento dell’assorbimento intestinale del
glucosio con riduzione dell’iperglicemia postpran-diale. L’acarboso è disponibile in formulazioni da 50 e 100 mg. Il dosaggio di partenza dell’acarboso è di 50 mg due volte al giorno e può essere gra-dualmente aumentato sino a 100 mg per tre volte al giorno da assumersi all’inizio dei pasti. La sua efficacia in monoterapia è inferiore a quella degli altri ipoglicemizzanti orali. Gli effetti collaterali, flatulenza e diarrea, sono dovuti ai carboidrati indigeriti che raggiungono l’intestino crasso e inducono produzione di gas da parte della flora batterica. Tali effetti collaterali scoraggiano il pa-ziente a eccedere nell’assunzione di carboidrati e ne migliorano, pertanto, la compliance alla die-ta, ma la percentuale di soggetti che sospende il trattamento a causa di tali effetti collaterali è molto elevata. Il miglitol, che ha effetti analoghi a quelli dell’acarboso, viene assorbito ed escreto per via renale ed è, quindi, controindicato nei pazienti con insufficienza renale.
Approccio a gradini nella terapia farmacologica oraleIl farmaco di prima scelta è la metformina. In genere, si inizia la terapia farmacologica orale con metfor-mina quando gli interventi sullo stile di vita non sono più in grado di mantenere il controllo della glicemia (Hba1c ≥ 7%), ma una recente consensus conference raccomanda di iniziare il trattamento far-macologico già al momento della diagnosi di diabete, insieme alle modifiche dello stile di vita. Quando la metformina da sola non riesce a mantenere un buon controllo della glicemia o non è tollerata o è con-troindicata (insufficienza renale) occorre aggiungere/utilizzare un farmaco secretagogo, preferibilmente a breve durata d’azione come la glipizide, oppure un glitazone, preferibilmente il pioglitazone. I secreta-goghi raggiungono più velocemente l’obiettivo, ma possono più rapidamente condurre al fallimento secondario; i glitazoni inducono ritenzione idrica e comportano un aumentato rischio di scompenso cardiaco nei pazienti già a rischio per tale patologia, ma consentono il mantenimento di un buon con-trollo della glicemia per un periodo più prolungato (Fig. 68.4). La repaglinide, sebbene più costosa e di paragonabile efficacia rispetto alle sulfoniluree, può rappresentare una valida alternativa nei pazienti con allergia alle sulfoniluree o con stile di vita variabile, perché ha breve durata d’azione ed espone a un mi-nore rischio di ipoglicemie. La repaglinide diventa il farmaco di prima scelta in monoterapia nei pazienti con insufficienza renale e in quelli intolleranti al-le sulfoniluree. Si ricorre alla terapia tricombinata quando le associazioni metformina-secretagoghi o metformina-glitazoni non sono in grado di mante-nere il controllo della glicemia.
InsulinaL’insulina viene somministrata come terapia sosti-tutiva nel diabete di tipo 1, nel quale esiste carenza
Figura 68.4Il farmaco di
prima scelta è la metformina che, secondo una recente consensus
conference di ADA/EASD,
va iniziata alla diagnosi insieme
ai cambiamenti dello stile di vita.
Se dopo 3 mesi il compenso non è soddisfacente si
aggiunge insulina basale o una
sulfonilurea o un glitazone.
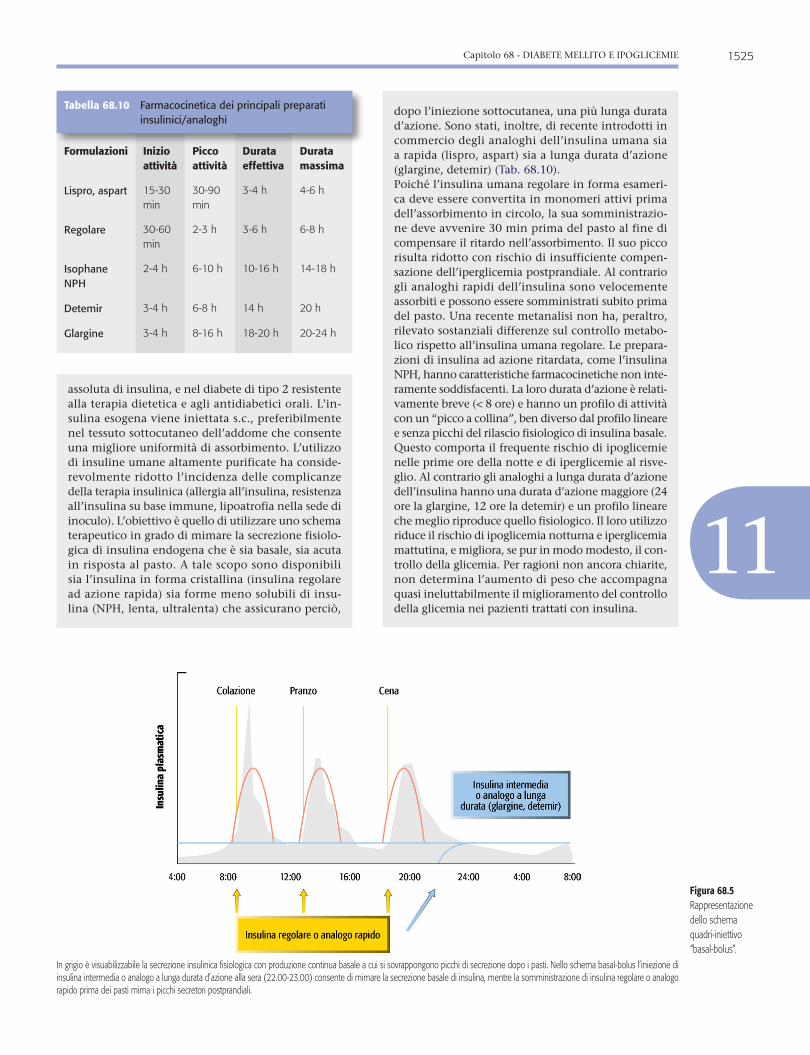
Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1525
assoluta di insulina, e nel diabete di tipo 2 resistente alla terapia dietetica e agli antidiabetici orali. L’in-sulina esogena viene iniettata s.c., preferibilmente nel tessuto sottocutaneo dell’addome che consente una migliore uniformità di assorbimento. L’utilizzo di insuline umane altamente purificate ha conside-revolmente ridotto l’incidenza delle complicanze della terapia insulinica (allergia all’insulina, resistenza all’insulina su base immune, lipoatrofia nella sede di inoculo). L’obiettivo è quello di utilizzare uno schema terapeutico in grado di mimare la secrezione fisiolo-gica di insulina endogena che è sia basale, sia acuta in risposta al pasto. a tale scopo sono disponibili sia l’insulina in forma cristallina (insulina regolare ad azione rapida) sia forme meno solubili di insu-lina (NpH, lenta, ultralenta) che assicurano perciò,
dopo l’iniezione sottocutanea, una più lunga durata d’azione. Sono stati, inoltre, di recente introdotti in commercio degli analoghi dell’insulina umana sia a rapida (lispro, aspart) sia a lunga durata d’azione (glargine, detemir) (Tab. 68.10).poiché l’insulina umana regolare in forma esameri-ca deve essere convertita in monomeri attivi prima dell’assorbimento in circolo, la sua somministrazio-ne deve avvenire 30 min prima del pasto al fine di compensare il ritardo nell’assorbimento. Il suo picco risulta ridotto con rischio di insufficiente compen-sazione dell’iperglicemia postprandiale. al contrario gli analoghi rapidi dell’insulina sono velocemente assorbiti e possono essere somministrati subito prima del pasto. Una recente metanalisi non ha, peraltro, rilevato sostanziali differenze sul controllo metabo-lico rispetto all’insulina umana regolare. Le prepara-zioni di insulina ad azione ritardata, come l’insulina NpH, hanno caratteristiche farmacocinetiche non inte-ramente soddisfacenti. La loro durata d’azione è relati-vamente breve (< 8 ore) e hanno un profilo di attività con un “picco a collina”, ben diverso dal profilo lineare e senza picchi del rilascio fisiologico di insulina basale. Questo comporta il frequente rischio di ipoglicemie nelle prime ore della notte e di iperglicemie al risve-glio. al contrario gli analoghi a lunga durata d’azione dell’insulina hanno una durata d’azione maggiore (24 ore la glargine, 12 ore la detemir) e un profilo lineare che meglio riproduce quello fisiologico. Il loro utilizzo riduce il rischio di ipoglicemia notturna e iperglicemia mattutina, e migliora, se pur in modo modesto, il con-trollo della glicemia. per ragioni non ancora chiarite, non determina l’aumento di peso che accompagna quasi ineluttabilmente il miglioramento del controllo della glicemia nei pazienti trattati con insulina.
Formulazioni Inizio attività
Picco attività
Durata effettiva
Durata massima
Lispro, aspart 15-30 min
30-90 min
3-4 h 4-6 h
Regolare 30-60 min
2-3 h 3-6 h 6-8 h
Isophane NPH
2-4 h 6-10 h 10-16 h 14-18 h
Detemir 3-4 h 6-8 h 14 h 20 h
Glargine 3-4 h 8-16 h 18-20 h 20-24 h
Tabella 68.10 Farmacocinetica dei principali preparati insulinici/analoghi
Figura 68.5Rappresentazione dello schema quadri-iniettivo “basal-bolus”.
In grigio è visuabilizzabile la secrezione insulinica fisiologica con produzione continua basale a cui si sovrappongono picchi di secrezione dopo i pasti. Nello schema basal-bolus l’iniezione di insulina intermedia o analogo a lunga durata d’azione alla sera (22.00-23.00) consente di mimare la secrezione basale di insulina, mentre la somministrazione di insulina regolare o analogo rapido prima dei pasti mima i picchi secretori postprandiali.

parte 11 - MaLaTTIE DEL RICaMBIo 1526
Nel diabete di tipo 1 il trattamento o schema di terapia di prima scelta è il basal-bolus, che prevede tre inie-zioni di insulina o analoghi ad azione rapida prima dei pasti e una somministrazione, prima di coricarsi, di un’insulina ad azione intermedia o di un analogo a lunga durata d’azione (Fig. 68.5). Lo schema con le sole tre iniezioni di insulina regolare prima dei pasti può essere utilizzato in tutti quei pazienti che presentano ancora una quota residua di funzione b-cellulare. La somministrazione di un’unica dose di insulina ad azione ritardata o di due dosi (mattino e sera) di una miscela di insulina regolare e intermedia non è adatta per una terapia ottimale a causa del frequente rischio di ipoglicemia e iperglicemia. La dose di insulina necessaria per ottenere un compenso ottimale è strettamente dipendente dalla sensibilità individuale, dalla quantità di carboidrati del pasto e dal tipo di vita del paziente nelle ore successive all’iniezione (sedentaria o attiva) e va quindi indivi-dualizzata. Di solito il paziente con diabete di tipo 1 necessita di 0,5-1 U/kg/die di cui il 40-50% sommini-strato come insulina basale (NpH, glargine, detemir) e il restante 50-60% suddiviso ai tre pasti (insulina regolare o analoghi rapidi). La dose ai pasti viene
variata utilizzando algoritmi sulla base della glicemia preprandiale e sul quantitativo di carboidrati che si prevede di introdurre con il pasto (Tab. 68.11).Nel paziente con diabete di tipo 2 la terapia insuli-nica viene in genere iniziata quando, nonostante una terapia orale combinata massimale, i valori di Hba1c non sono soddisfacenti; tuttavia una recente consensus conference suggerisce un più precoce ini-zio della terapia insulinica e propone l’associazione metformina-insulina in alternativa alle associazioni metformina-secretagoghi o metformina-glitazoni. Nei pazienti in cui viene iniziata la terapia insulini-ca, la metformina può essere continuata, in quanto, riducendo il rilascio epatico di glucosio, consente di usare dosi minori di insulina. Si può iniziare ag-giungendo un analogo a lunga durata d’azione alla sera (10 unità o 0,2 U/kg) e titolarne la dose (varia-zioni di 2 unità ogni 3 giorni) sino a ottenere una glicemia a digiuno di 70-130 mg/dL. Se i valori di Hba1c restano superiori al 7% si può misurare la glicemia preprandiale (pranzo, cena) e alla sera e, se necessario, introdurre una o più dosi di insulina rapida o analogo rapido (per esempio, se livelli glice-mici prima del pranzo sono > 130 mg/dL si aggiunge insulina rapida o analogo rapido a colazione) sino ad arrivare a uno schema basal-bolus.
MicroinfusoriL’infusione continua di insulina mediante infusori esterni contenenti insulina o analogo ad azione rapi-da offre il vantaggio di riprodurre il modello fisiolo-gico di erogazione insulinica. L’insulina viene anche somministrata come bolo al momento del pasto o per correggere un’iperglicemia intercorrente su comando del paziente. L’infusione basale può essere program-mata in base alle variazioni circadiane del fabbisogno insulinico che si verificano durante la giornata con una riduzione nelle ore notturne e un aumento nelle prime ore del mattino per fronteggiare il fenomeno dell’iperglicemia mattutina. Viene ridotta anche du-rante l’esercizio fisico e nelle ore successive in cui ancora può verificarsi un abbassamento della glice-mia. accanto a questi indiscutibili vantaggi ci sono anche degli inconvenienti che ne limitano l’impiego: il costo elevato, l’impatto psicologico sul paziente e la possibile complicanza cutanea indotta dall’inseri-mento prolungato di un catetere o di un ago sottocu-te. per tale ragione, il microinfusore viene a tutt’oggi riservato a quei casi particolari in cui l’instabilità dell’equilibrio metabolico non viene controllata dalla terapia convenzionale multi-iniettiva.
Trapianto di pancreas e insuleIl trapianto di pancreas può normalizzare la tolleranza al glucosio e rappresenta un’importante opzione tera-peutica nei diabetici di tipo 1. Tuttavia, la severità degli effetti collaterali del trattamento immunosoppressivo limita il suo utilizzo prevalentemente ai pazienti che necessitano di trapianto di rene per nefropatia diabe-tica o sono stati sottoposti ad altro trapianto d’organo
Glicemia Dose di insulina
Altre raccomandazioni
< 50 Ridurre di 2-3 U
Ritardare l’iniezione all’inizio del pasto, includere almeno 10 g di zucchero
50-70 Ridurre di 1-2 U
Ritardare l’iniezione all’inizio del pasto
70-130 Iniettare la dose-base
130-150 Aumentare di 1 U
150-200 Aumentare di 2 U
200-250 Aumentare di 3 U
Ritardare il pasto: 45 min dopo l’iniezione
250-300 Aumentare di 4 U
Ritardare il pasto: 40-60 min dopo l’iniezione
300-350 Aumentare di 5 U
Ritardare il pasto: 40-60 min dopo l’iniezione, controllare la chetonuria
350-400 Aumentare di 6 U
Ritardare il pasto: 40-60 min dopo l’iniezione, controllare la chetonuria
> 400 Aumentare di 7 U
Ritardare il pasto: 50-60 min dopo l’iniezione, controllare la chetonuria
Tabella 68.11 Algoritmo per le variazioni nella somministrazione preprandiale dell’insulina regolare

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1527
Si definisce ipoglicemia una riduzione della glicemia (< 50 mg/dL) tale da determinare l’insorgenza di sintomi che si risolvono prontamente dopo la normalizzazione dei valori glicemici.
Fisiopatologia
Il glucosio rappresenta la principale fonte energetica per il cervello e una sua deprivazione, qualora protratta, può provocare un danno tissutale e conseguenze sul piano funzionale che possono risultare fatali. La vulnerabilità del tessuto cerebrale a seguito della deprivazione di glucosio dipende dal fatto che il cervello non è in grado di produrre o immagazzinare glucosio né, a differenza di altri tessuti, come quelli muscolare ed epatico, di utilizzare gli acidi grassi liberi come substrato energetico alternativo. Esso
può, invece, utilizzare come fonte energetica alternativa i corpi chetonici, la cui sintesi, a partire dagli acidi grassi circolanti, avviene, tuttavia, dopo digiuno prolungato, rendendo questo meccanismo inadeguato a tamponare una deprivazione acuta di glucosio.Diversi meccanismi fisiologici intervengono a preservare il cervello dall’ipoglicemia. Nell’uomo la deprivazione di glucosio induce la riduzione della secrezione di insulina endogena e il rilascio degli ormoni della controregola-zione, in particolare di glucagone, catecolamine e cor-tisolo. Tali modifiche si traducono: (1) nell’induzione a livello epatico della glicogenolisi e della gluconeogenesi, con aumento della disponibilità di glucosio; (2) nel-la riduzione della sintesi proteica e della captazione di aminoacidi dal muscolo, con aumentata disponibilità di aminoacidi per la gluconeogenesi; (3) nell’aumento della
Ipoglicemie
e si trovano, pertanto, già nella necessità di assumere farmaci immunosoppressivi. Rispetto al trapianto di pancreas, quello di insule risulta di più facile esecu-zione oltre che meno traumatico, pur presentando il limite delle difficoltà tecniche dell’isolamento delle insule e, analogamente al trapianto di pancreas, la necessità di un trattamento immunosoppressivo.
Terapia per ridurre il rischio di patologia cardiovascolare
Terapia antiaggreganteLa terapia antiaggregante con acido acetilsalicilico è indicata in prevenzione primaria nei diabetici di età superiore a 40 anni o con almeno un fattore di ri-schio cardiovascolare (ipertensione, fumo di sigaretta, dislipidemia, familiarità per eventi cardiovascolari, microalbuminuria). In prevenzione secondaria è indicata in tutti i soggetti con storia di infarto mio-cardico, by-pass, ictus, TIa, vasculopatia periferica, claudicatio intermittens e angina.
Fumo di sigarettaLa prevalenza di fumatori tra i diabetici è superiore rispet-to ai non diabetici. In prevenzione secondaria l’abolizione del fumo si è dimostrata più efficace della maggior parte degli altri interventi terapeutici. È pertanto importante consigliare a tutti i diabetici la sospensione del fumo di sigaretta. Il counseling alla sospensione del fumo e la terapia sostitutiva nicotinica possono essere considerati componenti terapeutiche della cura del diabete.
Terapia dell’ipertensione arteriosaI pazienti con una pressione sistolica di 130-139 mmHg o una diastolica di 80-89 mmHg devono modificare
lo stile di vita e seguire una terapia comportamentale per un massimo di 3 mesi. Qualora gli obiettivi non vengano raggiunti, è necessario iniziare una terapia farmacologica. I pazienti ipertesi (≥ 140/90 mmHg) devono ricevere subito una terapia farmacologica. L’obiettivo primario è il raggiungimento del target pressorio indipendentemente dalla classe di farmaci utilizzata. Tuttavia, nella scelta della terapia farmaco-logica occorre considerare la presenza di comorbilità che pongano indicazione all’utilizzo/esclusione di una particolare classe di farmaci. per esempio i pazienti con nefropatia diabetica devono essere trattati con aCE-inibitori o aRB. I b-bloccanti sono indicati nel paziente con angina, gli aCE-inibitori e b-bloccanti nei pazienti con pregresso infarto del miocardio, gli aCE-inibitori e i diuretici nei pazienti con scompenso cardiaco. al contrario i b-bloccanti sono controindicati nei pa-zienti con vasculopatia periferica o asma e gli aCE-ini-bitori/aRB nei soggetti con stenosi dell’arteria renale e nelle donne in gravidanza. per raggiungere gli obiettivi pressori è normalmente necessaria una terapia di asso-ciazione con due o più farmaci antipertensivi.
Terapia della dislipidemiaNei diabetici con dislipidemia è fondamentale mo-dificare lo stile di vita (dieta povera di grassi saturi e colesterolo e ricca di fibre, incremento dell’attività fisica ecc.). La terapia farmacologica antidislipidemica è efficace nel ridurre il rischio cardiovascolare nei dia-betici di tipo 2 in prevenzione primaria e secondaria; le statine sono i farmaci di prima scelta. In diabetici di età < 40 anni senza fattori aggiuntivi di rischio cardio-vascolare, la terapia con statine, oltre alle variazioni dello stile di vita, è indicata per valori di colesterolo LDL > 130 mg/dL. Nei diabetici con più di 40 anni o con un alto fattore di rischio cardiovascolare la terapia ipolipemizzante deve essere iniziata indipen-dentemente dal valore di colesterolo LDL.

parte 11 - MaLaTTIE DEL RICaMBIo 1528
lipolisi nel tessuto adiposo, con conseguente rilascio di glicerolo, che viene utilizzato per la gluconeogenesi, e di acidi grassi liberi, che vengono utilizzati come substrato energetico alternativo dai tessuti periferici (rendendo così disponibile il glucosio per il cervello) e convertiti, a livello epatico, in corpi chetonici, utilizzabili come fonte energetica alternativa dal cervello (si veda Cenni di omeostasi metabolica). Il difetto del sistema di regola-zione dell’omeostasi glucidica si traduce nello sviluppo di ipoglicemia.
Classificazione
Le ipoglicemie possono, sotto il profilo clinico, essere distinte in ipoglicemie a digiuno, che si verificano dopo un digiuno più o meno prolungato e sono per lo più as-sociate a condizioni patologiche specifiche, e ipoglicemie postprandiali, che insorgono dopo alcune ore dal pasto e non sottendono necessariamente una malattia organica (Tab. 68.12).
Manifestazioni cliniche
Le manifestazioni cliniche di ipoglicemia dipendono dalla rapidità con cui essa si instaura. La rapida riduzio-ne dei valori glicemici induce una reazione adrenergica da rilascio degli ormoni della controregolazione (gluca-gone, catecolamine, cortisolo), caratterizzata da ansia, irritabilità, cardiopalmo, tremore, senso di fame, tachi-cardia, sudorazione. Questi sintomi adrenergici posso-no non manifestarsi nei soggetti diabetici neuropatici o con diabete di lunga durata, in cui può venire meno la
risposta controregolatoria. Qualora la riduzione dei valori glicemici si verifichi gradualmente o sia protratta e/o la risposta controregolatoria sia inadeguata, insorgono ma-nifestazioni neuroglucopeniche da sofferenza cellulare, caratterizzate da astenia, cefalea, difficoltà di concen-trazione, sonnolenza, parestesie, diplopia, disturbi del comportamento (aggressività, irrequietezza), alterazione dello stato di coscienza sino al coma, convulsioni, segni neurologici focali.
Diagnosi
La diagnosi di ipoglicemia richiede la compresenza di: (1) segni/sintomi clinici compatibili con tale condizione; (2) riscontro di ridotti valori glicemici (< 50 mg/dL nel sog-getto non diabetico e < 70 mg/dL nel soggetto diabetico); (3) risoluzione della sintomatologia dopo normalizzazione della glicemia. Il solo rilievo di ridotti valori glicemici in assenza di sintomi specifici, pur compatibile con la pre-senza di disordine ipoglicemico, deve fare considerare la possibile presenza di problemi preanalitici (per esempio, ritardo nell’analisi di campione prelevato da soggetti con leucemia o severa emolisi, in cui il glucosio viene consu-mato rispettivamente dai globuli bianchi o dai globuli rossi nucleati; assenza di composto antiglicolitico nella provetta utilizzata per il prelievo del campione).Qualora l’ipoglicemia venga riscontrata in un soggetto diabetico, occorre ottenere informazioni relative al tipo di diabete (1 o 2), alla terapia in atto (insulina, sulfoniluree, emivita del farmaco, dose, ultima somministrazione, va-riazioni o errori posologici, sede di iniezione dell’insulina) e all’eventuale presenza di fattori scatenanti (ridotto o ritardato apporto alimentare, aumento dell’attività fisica, altri farmaci, alcol, insufficienza renale e/o epatopatie, alterata controregolazione in caso di presenza di neuro-patia, diabete di lunga durata o terapia con b-bloccanti).Nel caso di episodi ipoglicemici a digiuno in soggetti non diabetici è opportuno indagare altre possibili cause di ipo-glicemia riconducibili a due sottoinsiemi: (1) ipoglicemie non associate ad aumentati livelli insulinemici, riscon-trabili, per esempio, in caso di etilismo, cirrosi epatica, uremia, grave malnutrizione, endocrinopatie diverse dal diabete (per esempio, insufficienza surrenale); (2) ipogli-cemie associate ad aumentati livelli insulinemici, riscon-trabili, per esempio, in caso di insulinoma, assunzione di insulina esogena o sulfoniluree a scopo anticonservativo o simulatorio nei neurolabili (ipoglicemia fittizia), presenza di anticorpi anti-insulina o anti-recettore dell’insulina. Qualora il soggetto giunga all’osservazione medica in corso di un episodio ipoglicemico a digiuno, è necessario effettuare un prelievo ematico per il dosaggio di gluco-sio, C-peptide e insulina. Se il soggetto giunge, invece, all’osservazione medica con normali valori glicemici è opportuno effettuare il test del digiuno, che, nel caso in cui non si verifichino nel frattempo episodi ipoglicemici, deve durare almeno 72 ore e prevedere prelievi ematici per il dosaggio di glicemia, insulina e C-peptide ogni 6 ore o in caso di ipoglicemia. Il rilievo di ipoglicemia, se contestuale a quello di elevati livelli di insulina e C-peptide, deve fare considerare l’ipotesi dell’insulinoma e dell’assunzione a scopo non terapeutico di sulfoniluree, i cui livelli plasmatici devono essere a tal fine verificati.
1. Ipoglicemia a digiunoa. Da ridotta produzione di glucosio
Deficit ormonali (per esempio, deficit di ACTH, cortisolo, GH, catecolamine e glucagone)EpatopatieUremiaIpotermiaFarmaci (per esempio, alcol, salicilati)Difetti enzimatici (per esempio, glucosio-6-fosfatasi, piruvato-carbossilasi)Deficit di substrati (grave malnutrizione)
b. Da aumentata utilizzazione di glucosioCon iperinsulinismo (insulinoma, insulina esogena in eccesso, sulfoniluree, anticorpi anti-insulina o anti-recettore per l’insulina, farmaci, sepsi)Senza iperinsulinismo (tumori extrapancreatici, attività fisica protratta)
2. Ipoglicemia postprandialea. Iperinsulinismo alimentare (per esempio, soggetti
gastroresecati con accelerato transito intestinale)b. Diabete di tipo 2 in fase inizialec. Intolleranza ereditaria al fruttosio, galattosemia,
ipersensibilità alla leucinad. Idiopatica
Tabella 68.12 Classificazione delle ipoglicemie

Capitolo 68 - DIaBETE MELLITo E IpoGLICEMIE
11
1529
aMD, Diabete Italia, SID. Standard italiani per la cura del diabete mellito. Linee Guida e Raccomandazioni 2007. Torino: Ed Infomedica Srl.
american Diabetes association Standards of Medical Care in Diabetes. Diabetes Care 2007; 30:S4–S41.
american Diabetes association Standards of Medical Care in Diabetes 2008. Diabetes Care 2008; 31:S12–S54.
american Diabetes association. Hyperglycemic crises in diabetes. Diab Care 2004;(S1):S94–S101.
Cavallo perin p, Giunti S. Coma metabolici (coma chetoacidosico, coma iperosmolare, coma ipoglicemico, coma lattacidemico). In: Della Corte F, Enrichens F, olliveri F et al., editors. Manuale di medicina d’emergenza. 2a ed. Milano: Mc Graw-Hill; 2007. pp. 439–67.
Comi RJ. approach to acute hypoglycemia. Endocrinol Metab North am 1992;22:247–62.
Cryer pE, Fisher JN, Shamoon H. Hypoglycemia. Diabetes Care 1994;17:734–55.
Cryer pE. Hypoglycaemia. in: Kasper DL, Braunwald E, Fauci a et al., editors. Harrison’s principles of internal medicine. 16th ed. Milano: McGraw-Hill professional; 2005. pp. 2180–84.
De Micheli a. Italian standards for diabetes mellitus 2007. Executive Summary. acta Diabetol 2008;45:107–27.
English p, Williams G. Hyperglycemic crises and lactic acidosis in diabetes mellitus. postgrad Med J 2004;80:253–61.
Goodenow TJ, Malarkey WB. Leukocytosis and artifactual hypoglycaemia. JaMa 1977;237:1961.
Kitabchi aE, Wall BM. Diabetic ketoacidosis. Med Clin North am 1995;79:9–37.
Lorber D. Nonketotic hypertonicity in diabetes mellitus. Med Clin North am 1995;79:39–52.
Macaron CI, Kadri a, Macaron Z. Nucleated red blood cells and artifactual hypoglycemia. Diabetes Care 1981;4:113–5.
Masharani U. Diabetes mellitus & Hypoglycaemia. In: Tierney LM, Mcphee SJ et al., editors. Current medical diagnosis and treatment. New York: Lange Medical Books McGraw-Hill; 2005. pp. 1157–201.
powers aC. Diabetes mellitus. in: Kasper DL, Braunwald E, Fauci a et al., editors. Harrison’s principles of internal medicine. 16th ed. Milano: McGraw-Hill professional; 2005. pp. 2152-79.
Service FJ. Hypoglycemic disorders. New Engl J Med 1995;332: 1144–52.
Siperstein MD. Diabetic ketoacidosis and hyperosmolar coma. Endocrinol Metab Clin North am 1992;21:415–32.
Wallace TM, Matthews DR. Recent advances in the monitoring and management of diabetic ketoacidosis. Q J Med 2004;97: 773–80.
Bibliografia
Il rilievo di ipoglicemia, se contestuale a quello di elevati livelli di insulina e di ridotti valori di C-peptide, deve indurre a considerare l’ipotesi dell’ipoglicemia da auto-somministrazione di insulina esogena, che non possiede il C-peptide e inibisce il rilascio dell’insulina endogena.Nel caso di ipoglicemia postprandiale, è necessario con-siderare la possibilità che si tratti di ipoglicemia reattiva, riscontrabile nella popolazione non diabetica e in alcuni casi di diabete di tipo 2 all’esordio. Il sospetto può essere confermato mediante test del pasto misto o con curva da carico di glucosio orale con 75 g con dosaggio di glicemia, insulinemia e C-peptide ogni ora fino alla quinta.
Terapia
Se il soggetto con ipoglicemia è cosciente è opportu-no somministrare 10-20 g di saccarosio per via ora-le, eventualmente seguiti da carboidrati complessi a lento assorbimento in base ai successivi valori glice-mici e alla terapia ipoglicemizzante in atto in caso di diabete.Se il soggetto non è cosciente ed è disponibile un accesso venoso si somministrano 10-20 g di gluco-sio per via endovenosa in bolo. La dose è ripetibile, dopo alcuni minuti, fino alla ripresa della coscienza.
Successivamente, è opportuno proseguire con infu-sione di glucosio fino a quando il paziente è in grado di alimentarsi.Qualora il soggetto non sia cosciente e l’accesso venoso non disponibile, è opportuno somministrare glucago-ne 1 mg s.c. o i.m. al risveglio, qualora l’accesso venoso non sia disponibile, è opportuno somministrare sac-carosio e carboidrati complessi per via orale, poiché l’effetto del glucagone, che agisce inducendo la rapida metabolizzazione del glicogeno a glucosio, è tempora-neo. L’efficacia del glucagone è scarsa nelle ipoglicemie alcoliche e/o in presenza di epatopatie, poiché essa dipende dalla disponibilità di glicogeno epatico.poiché la risposta alla terapia di un soggetto in coma ipoglicemico è rapida, l’eventuale persistere dello stato di incoscienza o di deficit neurologici può es-sere ricondotto a sequele di ipoglicemia prolungata, edema cerebrale o stroke, poiché l’ipoglicemia può provocare TIa, stroke o crisi comiziale in soggetti con vasculopatia cerebrale. Essa, inoltre, può indurre angina nel coronaropatico.Le forme di ipoglicemia conseguenti a eccesso di te-rapia insulinica possono essere gestite a domicilio, mentre quelle secondarie ad assunzione di sulfonilu-ree o alcol e quelle a digiuno richiedono il ricovero ospedaliero.

This page intentionally left blank