production and titration of lentiviral vectors
TRANSCRIPT

UNIT 12.10Production and Titration of LentiviralVectors
Patrick Salmon1 and Didier Trono2
1Department of Neuroscience, Faculty of Medicine, University of Geneva, Geneva,Switzerland2School of Life Sciences, Ecole Polytechnique Federale de, Lausanne and “Frontiers inGenetics,” National Center for Competence in Research, Lausanne, Switzerland
ABSTRACT
Lentiviral vectors have emerged over the last decade as powerful, reliable and safe toolsfor stable gene transfer in a wide variety of mammalian cells. Unlike other vectorsderived from oncoretroviruses, they allow for stable gene delivery into most nondividingprimary cells. This is why LVs are becoming useful and promising tools for future geneand cell therapy approaches. Lentivectors (LVs) derived from HIV-1 have graduallyevolved to display many desirable features aimed at increasing both their safety andtheir versatility. These latest designs are reviewed in this unit. This unit also describesprotocols for production and titration of LVs that can be implemented in a researchlaboratory setting, with an emphasis on standardization to improve transposability ofresults between laboratories Curr. Protoc. Hum. Genet. 54:12.10.1-12.10.24. C© 2007 byJohn Wiley & Sons, Inc.
Keywords: Lentiviral vectors � gene therapy � gene delivery
INTRODUCTION
Retroviral vectors have three characteristics of a highly attractive gene delivery system.First, they integrate their genetic cargo into the chromosome of the target cell, a likelyprerequisite for long-term expression. Second, they have a relatively large capacity, closeto 10 kb, allowing the delivery of most cDNAs. Finally, they do not transfer sequencesthat encode for proteins derived from the packaging virus, thus minimizing the riskthat vector-transduced cells will be attacked by virus-specific cytotoxic T lymphocytes.Conventional retroviral vectors (e.g., UNIT 12.5) are derived from oncoretroviruses such asthe mouse leukemia virus (MLV), and, as a consequence, cannot transduce nondividingcells. Many cells, including most in the adult nervous system, heart, and blood vessels,proliferate very little, if at all. In contrast to oncoretroviruses, lentiviruses, such as thehuman immunodeficiency virus (HIV), are a subfamily of retroviruses that can infect bothgrowth-arrested and dividing cells. Accordingly, lentiviral vectors efficiently transducenonreplicating cells both in tissue culture and in vivo.
An infectious retroviral particle comprises an RNA genome that carries cis-acting se-quences necessary for packaging, reverse transcription, and nuclear translocation andintegration, as well as structural proteins encoded by the gag and env genes, and the en-zymatic products of the pol gene. The assembly of these components leads to the buddingof the virion at the plasma membrane of the producer cell. In lentiviruses, the efficientexpression of Gag and Pol requires a virally-encoded post-transcriptional activator calledRev.
The envelope protein (Env) mediates the entry of the vector particle into its target.HIV-1 Env specifically recognizes CD4, a molecule present on the surface of helper
Current Protocols in Human Genetics 12.10.1-12.10.24, July 2007Published online July 2007 in Wiley Interscience (www.interscience.wiley.com).DOI: 10.1002/0471142905.hg1210s54Copyright C© 2007 John Wiley & Sons, Inc.
Vectors for GeneTherapy
12.10.1
Supplement 54

Production andTitration of
Lentiviral Vectors
12.10.2
Supplement 54 Current Protocols in Human Genetics
T cells, macrophages, and some glial cells. Fortunately, as with all retroviruses, theHIV-1 envelope protein can be substituted by the corresponding protein of another virus.This process, which alters the tropism of the virion, is called pseudotyping. Very often,the G protein of vesicular stomatitis virus (VSV-G) is used to pseudotype lentiviralas well as oncoretroviral vector particles because it is highly stable, allowing for theconcentration of the vector by ultracentrifugation, and because its phospholipid receptoris ubiquitously expressed in mammalian cells.
When producing vector stocks, it is mandatory to avoid the emergence of replication-competent recombinants (RCRs). In the retroviral genome, a single RNA molecule thatalso contains critical cis-acting elements carries all the coding sequences. Biosafety fora vector production system is therefore best achieved by distributing the sequences en-coding its various components over as many independent units as possible, to maximizethe number of recombination events that would be required to recreate a replication-competent virus. In the lentiviral vector systems described here, vector particles are gen-erated from three or four separate plasmids. This ensures that only replication-defectiveviruses are produced, because the plasmids would have to undergo multiple and complexrecombination events to regenerate a replication-competent entity.
HIV is a human pathogen. However, its pathogenic potential stems from the presence ofnine genes that all encode for important virulence factors. Fortunately, six of these genescan be deleted from the HIV-derived vector system without altering its gene-transferability. The resulting multiply-attenuated design of HIV vectors ensures that the parentalvirus cannot be reconstituted.
This unit describes the production of lentiviral vectors by transient transfection of 293Tcells and concentration of the particles by centrifugation (see Basic Protocol). Protocolsfor the titration of LV stocks (see Support Protocols 1 to 3) and RCRs in transduced cells(see Support Protocol 4) are also given.
CAUTION: VSV G-pseudotyped lentiviral vectors have a broad tropism, both in vitroand in vivo; biosafety precautions need to take into account the nature of the transgene.A P2 laboratory is required. Procedures using lentiviral vectors must be reviewed andapproved by the local biosafety committee of the institution where they are conducted.
BASICPROTOCOL
PRODUCTION OF HIGH-TITER HIV-1-BASED VECTOR STOCKSBY TRANSIENT TRANSFECTION OF 293T CELLS
The VSV G-pseudotyped vector is the best choice for most gene-delivery experiments,both in vitro and in vivo. This protocol describes production of the VSV G vectors.
Materials
293T/17 cells (ATCC cat. no. SD-3515)Dulbecco’s modified Eagle medium/10% FBS (DMEM-10; APPENDIX 2D)0.05% trypsin/EDTA (e.g., Invitrogen)Plasmids (available from the Trono lab, http://tronolab.epfl.ch):
pMD2G (encoding the VSV G envelope protein)pWPT-GFP (second-generation transfer vector, abbreviated “pWPT”)pRRL-cPPT-PGK-GFP-W-SIN (third-generation transfer vector, abbreviated
“pRRL”)psPAX2 (encoding HIV-1 Gag, Pol, Tat and Rev proteins)pMDLgag/polRRE (encoding the HIV-1 Gag and Pol proteins)pRSVrev (encoding the HIV-1 Rev protein)
TE buffer, pH 8.0 (APPENDIX 2A)Buffered water (see recipe)

Vectors for GeneTherapy
12.10.3
Current Protocols in Human Genetics Supplement 54
0.5 M CaCl2 (see recipe)2× HeBS (see recipe)Phosphate-buffered saline (PBS; APPENDIX 2D), 37◦C75% (v/v) ethanol in spray bottle20% (w/v) sucrose (SigmaUltra from Sigma), filter-sterilized through 0.22-µm
filter (store at 4◦C)CellGro Stem Cell Growth Medium (CellGenix GmbH, http://www.cellgenix.com/;
optional, if subsequent experiments require absence of serum)Phosphate-buffered saline containing Ca2+ and Mg2+ (e.g., Invitrogen; optional, if
subsequent experiments require absence of protein)
10-cm tissue culture dishes15- and 50-ml conical centrifuge tubes, sterile50-ml syringes and 0.45-µm pore size PVDF filters30-ml Beckman Konical ultracentrifuge tubes (Beckman Coulter)Ultracentrifuge with SW 28 rotor (Beckman Coulter) or equivalent
Additional reagents and equipment for tissue culture (APPENDIX 3G) and quantitationof DNA by absorption spectroscopy (APPENDIX 3D)
CAUTION: P2 practices require that open tubes always be handled in the laminar flowhood. Tubes can be taken out of the laminar flow only when they are closed, and that theybe sprayed with 75% ethanol. All solid waste and plasticware must be discarded in a trashbin in the laminar flow hoods and all liquids must be aspirated into a liquid waste bottlecontaining fresh concentrated bleach. Refill the liquid waste bottle with fresh bleachwhen the color of the liquid is no longer yellow. When full, bags are closed inside thelaminar flow hood, then autoclaved. When full, and at least 15 min after neutralizationwith fresh bleach, the liquid waste bottle can be emptied into a regular sink. In case ofa major spill of vector-containing liquid, absorb liquid with paper towels and neutralizewith fresh concentrated bleach prior to disposal. In case there is a leak in the SW 28buckets, remove the tubes in the hood, fill the buckets with 75% ethanol, and invert themseveral times. Leave under the hood for ≥20 min. Discard the 75% ethanol and removethe conical adapters under the hood. Spray the adapters with 75% ethanol and leave themunder the hood for >20 min.
NOTE: All solutions and equipment coming into contact with living cells must be sterile,and proper aseptic technique should be used accordingly.
NOTE: All culture incubations should be performed in a humidified, 37◦C, 5% CO2
incubator unless otherwise specified.
Prepare 293T cells for transfection1. Maintain 293T/17 cells in DMEM-10 medium, in 10-cm tissue culture dishes
(APPENDIX 3G). Split at a ratio of 1:4 to 1:10 using trypsin/EDTA, three times perweek (e.g., every Monday, Wednesday and Friday).
Frequent passages and keeping the 293T/17 cells as individual cells will ensure hightransfection efficiency. Therefore, do not grow cells beyond 80% confluence.
2. The day before the transfection, seed one to ten 10-cm culture dishes at 1–3 × 106
293T/17 cells per dish, such that the cells will be ∼1/4 to 1/3 confluent on the dayof transfection. Incubate overnight.
Cotransfect plasmids encoding vector components3. Adjust the DNA concentration of all plasmids: pMD2G, pWPT, pRRL, psPAX2 (or
pMDLg/pRRE, and pRSVrev) to 1 mg/ml in TE buffer, pH 8.0.
APPENDIX 3D describes quantitation of DNA using absorption spectroscopy.
Production andTitration of
Lentiviral Vectors
12.10.4
Supplement 54 Current Protocols in Human Genetics
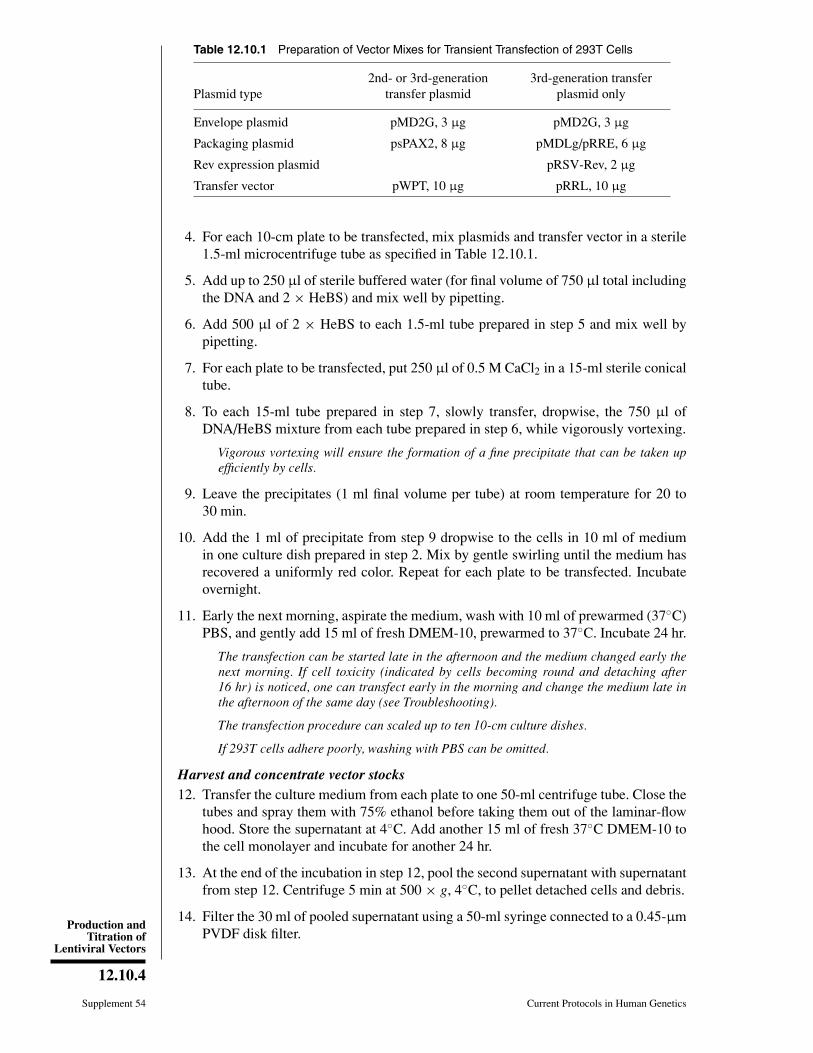
Table 12.10.1 Preparation of Vector Mixes for Transient Transfection of 293T Cells
Plasmid type2nd- or 3rd-generation
transfer plasmid3rd-generation transfer
plasmid only
Envelope plasmid pMD2G, 3 µg pMD2G, 3 µg
Packaging plasmid psPAX2, 8 µg pMDLg/pRRE, 6 µg
Rev expression plasmid pRSV-Rev, 2 µg
Transfer vector pWPT, 10 µg pRRL, 10 µg
4. For each 10-cm plate to be transfected, mix plasmids and transfer vector in a sterile1.5-ml microcentrifuge tube as specified in Table 12.10.1.
5. Add up to 250 µl of sterile buffered water (for final volume of 750 µl total includingthe DNA and 2 × HeBS) and mix well by pipetting.
6. Add 500 µl of 2 × HeBS to each 1.5-ml tube prepared in step 5 and mix well bypipetting.
7. For each plate to be transfected, put 250 µl of 0.5 M CaCl2 in a 15-ml sterile conicaltube.
8. To each 15-ml tube prepared in step 7, slowly transfer, dropwise, the 750 µl ofDNA/HeBS mixture from each tube prepared in step 6, while vigorously vortexing.
Vigorous vortexing will ensure the formation of a fine precipitate that can be taken upefficiently by cells.
9. Leave the precipitates (1 ml final volume per tube) at room temperature for 20 to30 min.
10. Add the 1 ml of precipitate from step 9 dropwise to the cells in 10 ml of mediumin one culture dish prepared in step 2. Mix by gentle swirling until the medium hasrecovered a uniformly red color. Repeat for each plate to be transfected. Incubateovernight.
11. Early the next morning, aspirate the medium, wash with 10 ml of prewarmed (37◦C)PBS, and gently add 15 ml of fresh DMEM-10, prewarmed to 37◦C. Incubate 24 hr.
The transfection can be started late in the afternoon and the medium changed early thenext morning. If cell toxicity (indicated by cells becoming round and detaching after16 hr) is noticed, one can transfect early in the morning and change the medium late inthe afternoon of the same day (see Troubleshooting).
The transfection procedure can scaled up to ten 10-cm culture dishes.
If 293T cells adhere poorly, washing with PBS can be omitted.
Harvest and concentrate vector stocks12. Transfer the culture medium from each plate to one 50-ml centrifuge tube. Close the
tubes and spray them with 75% ethanol before taking them out of the laminar-flowhood. Store the supernatant at 4◦C. Add another 15 ml of fresh 37◦C DMEM-10 tothe cell monolayer and incubate for another 24 hr.
13. At the end of the incubation in step 12, pool the second supernatant with supernatantfrom step 12. Centrifuge 5 min at 500 × g, 4◦C, to pellet detached cells and debris.
14. Filter the 30 ml of pooled supernatant using a 50-ml syringe connected to a 0.45-µmPVDF disk filter.

Vectors for GeneTherapy
12.10.5
Current Protocols in Human Genetics Supplement 54
15. For each pooled supernatant, pipet 4 ml of 20% sucrose into the bottom of a 30-mlBeckman Konical tube. Very slowly pour the supernatant from step 14 onto thesurface of the sucrose cushion until the tube is full (allow a 2-mm dry zone tothe top of the tube). Ultracentrifuge 120 min at 50,000 × g, 16◦C. Gently discardthe supernatant by inversion. Let the tube dry inverted for 5 min.
16. Resuspend the pellet (not always visible) with DMEM-10, serum-free medium,such as CellGro, if subsequent experiments require the absence of serum, or PBScontaining Ca2+ and Mg2+ if subsequent experiments require the absence of protein,by pipetting up and down 50 times.
The vector pellet of one tube can be resuspended in a minimal volume of 30 µl. In thiscase, a 1000-fold concentration will be obtained.
Try to avoid bubbles when resuspending the pellet since their presence will result indecrease of final volume, and hence decrease of yield. The use of protein-containingmedium is preferable over PBS to resuspend the viral pellet for two reasons. First, thepresence of proteins will stabilize the viral particles, and, second, the surfactant effect ofproteins will help redissolve the pellet. Store the final concentrate in aliquots at −80◦C.Try to avoid repeated freeze-thaw cycles, which may result in a drop in titer, althoughthe VSV-G pseudotyped particles are more resistant to this procedure than particlespseudotyped with retrovirus-derived envelopes.
SUPPORTPROTOCOL 1
DETERMINATION OF TOTAL VECTOR CONCENTRATION USINGANTI-p24 IMMUNOASSAY
Titers of viruses in general and lentivectors in particular critically depend on the methodand cells used for titration. The quantification of vector particles capable of achievingevery step from cell binding to expression of the transgene depends on both vector andcell characteristics. First, the cell used as target must be readily permissive to all stepsfrom viral entry to integration of the vector genetic cargo. Second, the expression of theforeign gene must be easily monitored and rapidly reach levels sufficient for reliablequantification. Early vectors had the lacZ bacterial gene as reporter, under the controlof the CMV promoter. Current vectors now have the green fluorescent protein (GFP)gene as a reporter, under the control of promoters that are active in most primary cells.Measured titers can also vary with the conditions used for titration, i.e., volume of sampleduring vector-cell incubation, time of vector-cell incubation, number of cells used, etc.For several years now, numerous laboratories have been using HeLa cells as target cellsfor LVs. These cells are stable, easy to grow, and 100% susceptible to transduction byVSV-G-pseudotyped LVs. Their wide use as reference target cells helps in comparingtiters between laboratories.
Determination of total particle concentration (i.e., physical titer) is important to monitorthe efficiency of vector production and packaging. One must keep in mind, however,that high concentrations of pelletable p24 viral capsid antigen can be measured fromvector particles that do not contain any genomic RNA and/or that are devoid of envelopeprotein. Thus, this assay cannot substitute for a biological transduction assay as describedin Support Protocol 2, which determines the biological titer.
Two p24 antigen capture assay kits are suitable. Since they are very similar, only theprocedure using the Perkin-Elmer kit is described in detail below. The second assay can beobtained through the AIDS Vaccine Program by E-mailing [email protected] minimal p24 concentration detected by this assay is 150 pg/ml. HIV-1- or vector-containing supernatants are lysed by adding Triton X-100 to a final concentration of 1%.Lysed samples can be analyzed immediately or stored at −20◦C.
These assays have different sensitivities but will give similar results. Reliable resultsare obtained only by interpolation of unknown values within the range covered by the

Production andTitration of
Lentiviral Vectors
12.10.6
Supplement 54 Current Protocols in Human Genetics
standard curve. If one sample is below or above the standard values, one must performanother p24 assay with different dilutions. Most unconcentrated vector stocks have a p24concentration between 100 ng/ml and 5 µg/ml.
Materials
Lentiviral vector sample for titration (see Basic Protocol) and positive control(provided with p24 ELISA kit)
Phosphate-buffered saline (PBS; APPENDIX 2D)5% (v/v) Triton X-100 (store at room temperature)p24 ELISA kit (Perkin-Elmer cat. no. NEK05000 1KT) including:
Anti-p24-coated ELISA 96-well plates (or well strips)Adhesive plate coversp24 wash bufferBiotinylated anti-p24 polyclonal antibody for primary reactionStreptavidin–horseradish peroxidase (HRP) for secondary reactionSubstrate: o-phenylenediamine HCl (OPD) tabletsSubstrate diluentStop solution: 4 N sulfuric acid
ELISA plate reader with 492 nm filter
Dilute and lyse vector samples1. In 1.5-ml microcentrifuge tubes, dilute vector samples to 450 µl with PBS so that the
expected final concentration will fall into the range of values covered by the standardcurve.
Serial 10-fold dilutions of concentrated samples (cell culture supernatants) should bedone in DMEM-10.
2. Add 50 µl of 5% Triton X-100 solution. Mix well.
Prepare the standards3. First dilute the positive control (initial concentration = 200 ng/ml) to a final concen-
tration of 200 pg/ml (1000-fold dilution) in 1 ml (final volume) PBS. Prepare 1-mlserial 2-fold dilutions in PBS, ending with a dilution corresponding to 6.25 pg/ml(six dilution points, total).
Refer to the kit manual for further instructions on preparing the control.
4. Lyse the final dilutions by adding 1/10 vol of 5% Triton X-100 solution.
Set up ELISA and perform primary and secondary reactions5. Add 200 µl sample or standard dilution to duplicate wells of the coated strips or plate.
6. Cover with adhesive film and incubate at 37◦C for 2 hr.
7. Remove adhesive film, discard samples by inverting the strips or plate, and washfour times with the p24 wash buffer (provided in kit).
8. Remove remaining liquid by tapping the inverted strips or plate on absorbing paper.
9. Follow steps for primary and secondary reactions as indicated in the manual, withcareful washing and tapping out of the residual liquid as described in steps 7 and 8between reactions.
Perform enzymatic (detection) reaction10. For one plate, dilute one OPD tablet in 11 ml of the substrate diluent, 5 min before use.
11. Add 100 µl of the OPD solution per well.
12. Incubate 30 min in the dark at room temperature.
IMPORTANT NOTE: Do not wash the plate.

Vectors for GeneTherapy
12.10.7
Current Protocols in Human Genetics Supplement 54
13. Stop the reaction by adding 100 µl of the stop solution. Read OD values at 492 nmin an ELISA plate reader.
SUPPORTPROTOCOL 2
BIOLOGICAL TITRATION OF LENTIVECTORS USING FLOWCYTOMETRY
This method can only be used to titer stocks of vectors that carry a transgene which iseasily monitored by flow cytometry (such as GFP, or other living colors, or any membraneprotein that can be detected by flow cytometry), and whose expression is governed by apromoter that is active in HeLa cells (tissue-specific promoter-containing vector must befunctionally assayed in specific cells, and titered by QPCR in HeLa cells; see below). Thisprotocol describes the titration of a PGK-GFP vector (pRRL, also see Basic Protocol).
Materials
HeLa cells (ATCC cat. no. CCL-2)Dulbecco’s modified Eagle medium/10% FBS (DMEM-10; APPENDIX 2D)Lentiviral vector sample for titration, carrying GFP transgene (see Basic Protocol)
and positive control (lentiviral supernatant already titered)Phosphate-buffered saline (PBS), pH 7.4 (APPENDIX 2D)0.05% trypsin/EDTA (e.g., Invitrogen)1% (w/v) formaldehyde: dilute 1 ml of 37% formaldehyde (Sigma) in 36 ml PBS
(APPENDIX 2D); store at 4◦C
6-well tissue culture plates (e.g., BD Biosciences)Fluorescence-activated cell sorter (FACS, Becton Dickinson; with 488 nm excitation
laser and green filter) or equivalent flow cytometer, and appropriate tubes
NOTE: All solutions and equipment coming into contact with living cells must be sterile,and aseptic technique should be used accordingly.
NOTE: All culture incubations should be performed in a humidified, 37◦C, 5% CO2
incubator unless otherwise specified.
Perform assay1. On day 0, seed HeLa cells at 50,000 cells per well in 6-well tissue culture plate in
2 ml DMEM-10. Make sure that the HeLa cells are well separated and uniformlydistributed in the well. Incubate cells.
2. On day 1, put 500, 50, or 5 µl of the vector suspension (either pure from unconcen-trated supernatants or diluted if it comes from a concentrated stock) in three separatewells containing the growing HeLa cells. Continue incubation.
3. On day 2, remove the supernatant and replace with 2 ml fresh DMEM-10.
4. On day 5, wash the cells with 2 ml PBS, detach them by adding 250 µl per well oftrypsin/EDTA and incubating 1 min at 37◦C.
5. Add 250 µl of DMEM-10 and mix well to resuspend the cells.
This step inactivates the trypsin and EDTA.
6. Transfer the cells (500 µl) into a 1.7-ml microcentrifuge tube and microcentrifuge2 min at 2000 rpm, room temperature. Remove the supernatant.
7. Resuspend the pellet in 500 µl of 1% formaldehyde in PBS.
This step will fix the cells and inactivate the vector particles. Samples can thus be takenout of the P2 laboratory at this point.
8. Transfer cells to a FACS tube.
Production andTitration of
Lentiviral Vectors
12.10.8
Supplement 54 Current Protocols in Human Genetics
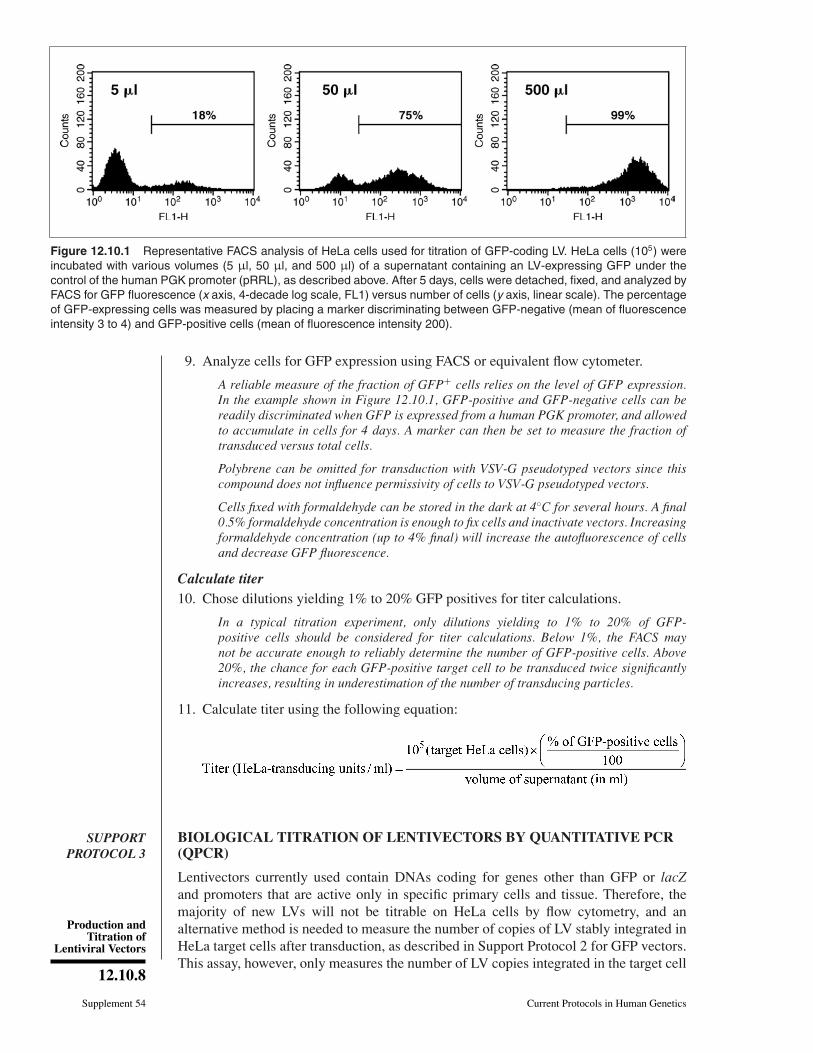
Figure 12.10.1 Representative FACS analysis of HeLa cells used for titration of GFP-coding LV. HeLa cells (105) wereincubated with various volumes (5 µl, 50 µl, and 500 µl) of a supernatant containing an LV-expressing GFP under thecontrol of the human PGK promoter (pRRL), as described above. After 5 days, cells were detached, fixed, and analyzed byFACS for GFP fluorescence (x axis, 4-decade log scale, FL1) versus number of cells (y axis, linear scale). The percentageof GFP-expressing cells was measured by placing a marker discriminating between GFP-negative (mean of fluorescenceintensity 3 to 4) and GFP-positive cells (mean of fluorescence intensity 200).
9. Analyze cells for GFP expression using FACS or equivalent flow cytometer.
A reliable measure of the fraction of GFP+ cells relies on the level of GFP expression.In the example shown in Figure 12.10.1, GFP-positive and GFP-negative cells can bereadily discriminated when GFP is expressed from a human PGK promoter, and allowedto accumulate in cells for 4 days. A marker can then be set to measure the fraction oftransduced versus total cells.
Polybrene can be omitted for transduction with VSV-G pseudotyped vectors since thiscompound does not influence permissivity of cells to VSV-G pseudotyped vectors.
Cells fixed with formaldehyde can be stored in the dark at 4◦C for several hours. A final0.5% formaldehyde concentration is enough to fix cells and inactivate vectors. Increasingformaldehyde concentration (up to 4% final) will increase the autofluorescence of cellsand decrease GFP fluorescence.
Calculate titer10. Chose dilutions yielding 1% to 20% GFP positives for titer calculations.
In a typical titration experiment, only dilutions yielding to 1% to 20% of GFP-positive cells should be considered for titer calculations. Below 1%, the FACS maynot be accurate enough to reliably determine the number of GFP-positive cells. Above20%, the chance for each GFP-positive target cell to be transduced twice significantlyincreases, resulting in underestimation of the number of transducing particles.
11. Calculate titer using the following equation:
SUPPORTPROTOCOL 3
BIOLOGICAL TITRATION OF LENTIVECTORS BY QUANTITATIVE PCR(QPCR)
Lentivectors currently used contain DNAs coding for genes other than GFP or lacZand promoters that are active only in specific primary cells and tissue. Therefore, themajority of new LVs will not be titrable on HeLa cells by flow cytometry, and analternative method is needed to measure the number of copies of LV stably integrated inHeLa target cells after transduction, as described in Support Protocol 2 for GFP vectors.This assay, however, only measures the number of LV copies integrated in the target cell

Vectors for GeneTherapy
12.10.9
Current Protocols in Human Genetics Supplement 54
genome. The overall functionality of the vector must be tested at least once in cells inwhich the promoter is active and/or with appropriate techniques to detect expression ofthe transgene product.
The QPCR assay described here proceeds as follows, using a real-time PCR machine.HeLa cells are transduced as in Support Protocol 2, but instead of being detached bytrypsin, they are lysed directly in the plate and the DNA is extracted using a genomicDNA extraction kit. Finally, a fraction of the total DNA is analyzed for copy number ofHIV sequences using the real-time PCR protocol.
It is advisable to run a dual titration—FACS (Support Protocol 2) plus the TaqMan QPCRdescribed below—using one GFP vector alongside the other vectors—and compare theresults obtained by the two methods.
NOTE: Always use pipet tips containing aerosol-barrier filters when preparing solutions,mixes, samples, and plates for QPCR, to prevent cross-contamination.
Materials
HeLa cells (ATCC cat. no. CCL-2)Lentiviral vector (LV) sample for titration (see Basic Protocol) and DNA from
positive and negative control cells (see annotation to step 1)DNAeasy Genomic DNA Extraction Kit (Qiagen)Kit for preparing QPCR master mix (RT-QP2X-03; Eurogentec), including 2×
reaction buffer10× TaqMan GAG set (see recipe)10× TaqMan HB2 set (see recipe)
MicroAmp 96-well optical reaction plate (Applied Biosystems)Optical caps (Applied Biosystems)Centrifuge with microtiter plate carrierReal-time PCR machine (7700 Sequence Detector, Applied Biosystems)Computer running ABI Prism software (Applied Biosystems) and Microsoft Excel
Additional reagents and equipment for transducing HeLa cells with lentivectors(Support Protocol 2)
Prepare DNA sample1. Transduce HeLa cells with lentiviral vector (LV) of interest carrying GFP transgene
(see Support Protocol 2) in 6-well tissue culture plates. Grow cells to confluence.
DNA typically comes from 2 × 106 HeLa cells (one confluent well of a 6-well plate)extracted and resuspended in 100 µl of Buffer AE from the DNAeasy Tissue Kit.
Standards of cells containing 10, 1, 0.1, and 0.01 copy of LV per cell (for use in thecalculation performed at step 12) can be prepared from HeLa cells transduced with aGFP vector, using serial 10-fold dilutions. A typical set of DNA standards can be preparedas follows. Prepare HeLa cells as in Support Protocol 2 for the FACS titration with serial10-fold dilutions of a GFP vector. Dilute vector so that one dilution will give a value asclose to 10% GFP-positive cells as possible. Before analysis, split cells for simultaneousanalysis with FACS and for DNA extraction. The sample giving a value of 10% of GFP-positive cells will correspond to a value of 0.1 copy per cell. Depending on the initialconcentration of the GFP vector used, the samples corresponding to lower and higherdilutions will provide DNA corresponding to copy numbers ranging from 10 to 0.001copies per cell. These DNAs can then be stored at –20◦C and used as standards for QPCRtitration.
2. Extract target cell DNA from each individual well of the 6-well plate using aDNAeasy kit according to the manufacturer’s recommendations. For the DNA elutionstep, use 100 µl of AE buffer instead of 200 µl. Store DNA at −20◦C until use.
Production andTitration of
Lentiviral Vectors
12.10.10
Supplement 54 Current Protocols in Human Genetics
Figure 12.10.2 Legend at right.

Vectors for GeneTherapy
12.10.11
Current Protocols in Human Genetics Supplement 54
Perform reaction3. Prepare a mix containing everything but the sample DNA for the number of wells
needed for the QPCR analysis, including all samples and standards in duplicates,according to the following recipe (14 µl per well):
7.5 µl 2× reaction buffer from Eurogentec QPCR MasterMix1.5 µl 10× GAG set1.5 µl 10× HB2 set3.5 µl H2O.
The Taq polymerase is part of the 2× reaction buffer.
4. Place 14 µl of this mix into each well of a 96-well optical reaction plate.
5. Add 1 µl sample DNA (from step 1) to each of the appropriate wells. Close platewith optical caps.
6. Centrifuge the plate 1 min at 200 × g, room temperature, to bring all liquid to thebottoms of the wells.
7. Place the 96-well plate in the real-time PCR machine and run the appropriate pro-gram depending on the fluorochromes (e.g., FAM, VIC, Yakima Yellow, TET) andquenchers (e.g., TAMRA, Eclipse Dark Quencher) used in the TaqMan probes (seeReagents and Solutions).
The precise settings of a QPCR protocol depend on the real-time PCR machine used. Thisaspect is beyond the scope of this protocol. If not familiar with QPCR techniques, oneshould seek advice from a local QPCR expert or from the technical support departmentof the supplier of the real-time PCR machine.
Analyze results8. Analyze the amplification reactions using ABI Prism software.
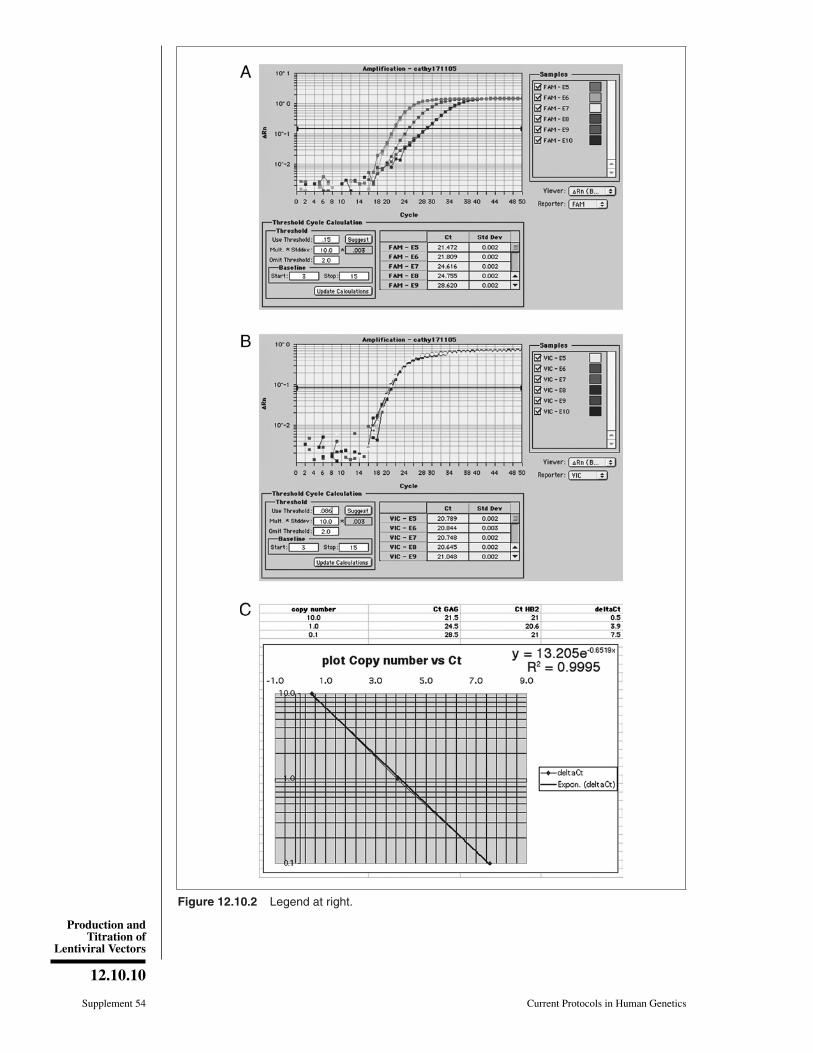
An example of amplification profiles of HIV sequences in human DNA is given inFigure 12.10.2 (as displayed by ABI Prism).
9. Set the threshold values (Ct) where the amplification curve is the steepest, both forthe gene of interest (GAG-FAM, panel A) and for the internal control (HB2-VIC,panel B).
These Ct values are the number of cycles required for the amplification curve to cross theabsorbance threshold values.
10. Export the results as a Microsoft Excel spreadsheet.
11. Using standards of cells containing 10, 1, 0.1, and 0.01 copies of LV per cell, calculatethe �Ct values (CtGAG – CtHB2) using Excel.
Figure 12.10.2 (at left) Representative QPCR analysis used for titration of HIV-1-based LVs. DNAfrom HeLa cells transduced with serial 10-fold dilutions of RRL-GFP vectors was subjected simul-taneously to FACS titration analysis (Support Protocol 2) and QPCR analysis (Support Protocol 3).A sample of each dilution was submitted to QPCR amplification and monitoring using a Perkin-Elmer 7700 (Applied Biosystems) and sets of primers and probes specific for HIV gag sequences(GAG-FAM, panel A) or β-actin sequences (HB2-VIC, panel B). Amplification plots were displayedand cycle threshold values (Ct) were set as described in text. Values of GAG Ct and HB2 Ct wereexported in an Excel worksheet to calculate �Ct values (x axis, linear scale) and plot them againstcopy number values (y axis, log scale); panel C). The sample giving 10% of GFP-positive cellswas set as HeLa DNA containing 0.1 copy of HIV sequences per cell. The regression curvecan then be used to calculate GAG copy numbers (y value) of unknown samples by applyingthe formula to �Ct values (y values) of the sample. For the color version of this figure go tohttp://www.currentprotocols.com.

Production andTitration of
Lentiviral Vectors
12.10.12
Supplement 54 Current Protocols in Human Genetics
12. Using Excel, display an exponential formula giving the copy number as a functionof �Ct.
13. Apply the formula to the unknown samples in order to calculate their correspondingcopy number of HIV sequences.
14. Calculate the titers by applying the following formula:
A prototype Excel worksheet for calculation of QPCR titers can be downloaded athttp://www.medecine.unige.ch/∼salmon/lentilab.
Using standard DNA extraction procedures in a laboratory context where HIV sequencesare often handled, one can expect a level of background contamination with HIV sequencescorresponding to cells containing 1 copy per 1000 genomes. In this case, consider highercopy numbers for calibration. Vector stocks failing to give higher than 0.01 copy pergenome in a QPCR assay have an infectivity index that renders them unusable for mostapplications (see Anticipated Results).
Using careful DNA extraction procedures and standardization as described above, onecan expect reproducibility within a 2-fold range. Investigators should consult a localQPCR expert if a more stringent quantitative PCR procedure is needed.
SUPPORTPROTOCOL 4
RCR ASSAY FOR LVS: REAL-TIME QUANTITATIVE PCR (QPCR)DETECTION OF REPLICATION-COMPETENT RECOMBINANTS
It is essential to ensure the absence of replication-competent recombinants (RCRs) inorder to downgrade the biohazard level of cells that have been transduced by retroviralvectors, including LVs. The test described in this protocol is based on the detection (orabsence of detection)—in the chromosomal DNA of transduced cells—of HIV sequencesthat are absent in the transfer plasmid (vector genome) but that are present in the packagingplasmid and which are essential for HIV (or RCR) replication. The target sequence chosenin this assay is located in the extended packaging signal (vector genome) or in the POLgene. Although the assay described here is performed on a small number of cells, at leastthree weeks after initial transduction it can be scaled up to meet requirements for thedetection of RCR in preclinical vector batches.
NOTE: Always use pipet tips containing aerosol-barrier filters when preparing solutions,mixes, samples, and plates for QPCR, to prevent cross-contamination.
Materials
HeLa cells (ATCC cat. no. CCL-2)Lentiviral vector (LV) sample(s) and standards for titration (see Basic Protocol;
also see last annotation to step 1 below)8E5 cells (ATCC cat. no. CRL-8993)DNAeasy Genomic DNA Extraction Kit (Qiagen)Kit for preparing QPCR master mix (RT-QP2X-03; Eurogentec), including 2×
reaction buffer10× TaqMan GAG set (see recipe) or 10× TaqMan POL set (see recipe)10× TaqMan HB2 set (see recipe)
MicroAmp 96-well optical reaction plate (Applied Biosystems)Optical caps (Applied Biosystems)Centrifuge with microtiter plate carrierReal-time PCR machine (7700 Sequence Detector, Applied Biosystems)Computer running ABI Prism software (Applied Biosystems) and Microsoft Excel

Vectors for GeneTherapy
12.10.13
Current Protocols in Human Genetics Supplement 54
Additional reagents and equipment for transducing HeLa cells with lentivectors(Support Protocol 2)
CAUTION: ATCC recommends that 8E5 cells be handled in a P2 laboratory. Indeed,although they contain a full copy of noninfectious HIV, they can form syncytia withuninfected CD4+ cells.
Prepare sample1. At least 3 weeks prior to assay, transduce HeLa cells with lentiviral vector (LV) of
interest (see Support Protocol 2) and with standards (see below).
CAUTION: Cells being analyzed for the absence of RCR must be confined cells in aculture flask with vented cap until result of RCR analysis. If the result is negative, thebiohazard level of the cells can be downgraded; after spraying the flask with 75% ethanol,it can be transferred outside of the culture laboratory.
The extended growth period allows for dilution of packaging DNA carried over fromvector production steps.
In contrast to Support Protocol 3, in this protocol, two types of standards are needed.One standard corresponds to cells containing vector sequences only (LV standard, targetfor GAG oligo set), and one corresponds to cells containing all HIV sequences (HIVstandard, target for GAG and POL oligo sets). The first is provided by cells transducedwith LV as described in Support Protocol 3. The second is provided by cells having onecopy of full-length HIV genome, such as 8E5 cells.
2. After ≥3 weeks of cell growth, extract DNA from the transduced cells using aDNAeasy kit according to the manufacturer’s instructions. Store DNA at –20◦Cuntil use.
The number of cells and final volume should be such that 1 µl of the final DNA solutioncorresponds to 104 cells.
Perform reaction and analyze results3. For each sample or standard, prepare two independent mixes containing everything
but the sample DNA for the number of wells needed for the QPCR analysis, includingall samples and standards in duplicates, according to the following recipe (14 µl perwell):
7.5 µl 2× reaction buffer1.5 µl 10× GAG set or 10× POL set1.5 µl 10× HB2 set3.5 µl H2O.
The Taq polymerase is part of the 2× reaction buffer.
4. Place 14 µl of the GAG mix or POL mix from step 3 in duplicate independent wellsof a 96-well optical reaction plate.
5. Add 1 µl sample DNA (from step 2) to each of the appropriate wells. Close platewith optical caps.
6. Centrifuge the plate 1 min at 200 × g, room temperature, to bring all liquid to thebottoms of the wells.
7. Place the 96-well plate in the real-time PCR machine and run the appropriate programdepending on the fluorochromes (FAM, VIC, Yakima Yellow, TET) and quenchers(e.g., TAMRA, Eclipse Dark Quencher) used in the TaqMan probes (see Reagentsand Solutions).

Production andTitration of
Lentiviral Vectors
12.10.14
Supplement 54 Current Protocols in Human Genetics
8. Analyze results.
Analysis is performed basically as described in Support Protocol 3. In this case, however,two types of standards are needed. One standard corresponds to cells containing vectorsequences only (LV standard, target for GAG oligo set), and one corresponds to cellscontaining all HIV sequences (HIV standard, target for GAG and POL oligo sets). Thefirst is provided by cells transduced with LV as described in Support Protocol 3. Thesecond is provided by cells having one copy of full-length HIV genome, such as 8E5cells. In the case of 8E5, the DNA will contain 1 copy of HIV per genome. Serial 10-folddilutions of 8E5 DNA into human DNA (up to 10−3 copy per genome) can be performedto provide a HIV DNA standard curve. A negative control both for LV sequences and HIVsequences will be provided by HeLa cells.
Results are expressed as �Ct values for each oligo set, i.e., GAG-HB2 �Ct and POL-HB2�Ct. The sample DNA will be considered negative for POL sequences and hence negativefor RCR if its POL-HB2 �Ct value is similar to the POL-HB2 �Ct value of HeLa cells,with a GAG-HB2 �Ct value above the range corresponding to 1 copy of LV sequence pergenome.
Other RCR tests have been described in the literature. One recent paper describes atrue RCR assay which failed to detect any RCR in vector batches produced from third-generation packaging systems (Escarpe et al., 2003). Other tests have been described,but they detect biological entities that need trans complementation to replicate. Althoughthese assays can measure the level of recombination during the production of lentivectors,they are not suitable to detect genuine RCR that may represent a biological hazard dueto potential dissemination within primary human cells.
REAGENTS AND SOLUTIONSUse deionized, distilled water in all recipes and protocol steps. For common stock solutions, seeAPPENDIX 2D; for suppliers, see SUPPLIERS APPENDIX.
Buffered water
50 ml H2O125 µl 1 M HEPES, pH 7.3 (2.5 mM final)Filter sterilize through 0.22-µm nitrocellulose filterStore up to 6 months at 4◦C
The authors have observed that the appearance and quality of the DNA/CaCl2 precipitate(see Basic Protocol) can vary depending upon the batch of distilled water used to dilute theplasmids. To circumvent this problem, it is advisable to buffer this distilled water. A finalconcentration of 2.5 mM HEPES in the water will help maintainin a proper pH and willnot compete for the final pH with the HeBS pH 7.00 (see recipe) added subsequently (i.e.,the HeBS, pH 7.00, provides for a final concentration of 25 mM HEPES final, whereas thebuffered water, pH 7.3, results in a final concentration of 0.625 mM HEPES).
CaCl2, 0.5 M
36.75 g CaCl2·2H2O (mol. wt., 147; SigmaUltra)500 ml H2OFilter sterilize through 0.22-µm nitrocellulose filterStore up to 2 years at −70◦C in 50-ml aliquots
Once thawed, the CaCl2 solution can be kept at 4◦C for several weeks without significantchange in the transfection efficiency.
HEPES-buffered saline (HeBS), 2×Dissolve the following reagents (all SigmaUltra from Sigma) in 800 ml H2O:16.36 g NaCl (mol. wt., 58.44; 0.28 M final)11.9 g HEPES (mol. wt., 238.3; 0.05 M final)0.213 g anhydrous Na2HPO4 (mol. wt., 142; 1.5 mM final)
continued

Vectors for GeneTherapy
12.10.15
Current Protocols in Human Genetics Supplement 54
Adjust pH to 7.00 with 10 M NaOH (proper pH is critical)Add H2O to 1000 ml and make final pH adjustment to pH 7.00Filter sterilize through a 0.22-µm nitrocellulose filterStore up to 2 years at −70◦C in 50-ml aliquots
Once thawed, the HeBS solution can be kept at 4◦C for several weeks without significantchange in the transfection efficiency.
It is critical that the pH be adjusted accurately; below 6.95, the precipitate will not form;above 7.05, the precipitate will be coarse and the transfection efficiency will be low.
TaqMan GAG set, 10×1 µM GAG-P (probe, antisense): 5′-(FAM)- ACAGCCTTCTGATGTTTCTAAC
AGGCCAGG -(Eclipse)-3′
3 µM GAG-F (forward primer): 5′-GGAGCTAGAACGATTCGCAGTTA-3′
3 µM GAG-R (reverse primer): 5′-GGTTGTAGCTGTCCCAGTATTTGTC-3′
These oligos are used for amplification of HIV-1 derived vector sequences and are specificfor the 5′ end of the gag gene (GAG). This sequence is present in all HIV-1 vectors, as it ispart of the extended packaging signal.
Oligos can be ordered on-line from several companies such as Eurogentec or Sigma. Eclipse(full name, Eclipse Dark Quencher) is a registered trademark of Eurogentec.
Concentrations of probe and primer may vary when using two (or more) oligo sets simul-taneously (multiplex analysis).
Stocks of probes and primers usually come lyophilized and are diluted to 10 µM in water.
TaqMan human β-actin set, 10×1 µM HB2-P (probe, sense): 5′-(YY)- CCTGGCCTCGCTGTCCACCTTCCA -
(Eclipse)-3′
300 nM HB2-F (forward primer): 5′-TCCGTGTGGATCGGCGGCTCCA-3′
300 nM HB2-R (reverse primer): 5′-CTGCTTGCTGATCCACATCTG-3′
These oligos are used to normalize for the amount of genomic DNA and are specific for thehuman β-actin gene.
For multiplex analysis using HB2 and GAG oligo sets, HB2 primers are used at a 10×concentration of 300 nM (30 nM final).
Oligos can be ordered online from several companies such as Eurogentec or Sigma. YakimaYellow (YY) and Eclipse (full name, Eclipse Dark Quencher) are registered trademarks ofEurogentec.
Concentrations of probe and primer may vary when using two (or more) oligo sets simul-taneously (multiplex analysis).
Stocks of probes and primers usually come lyophilized and are diluted to 10 µM in water.
TaqMan POL set, 10×1 µM POL-P (probe, sense): 5′-(FAM)-TGGACAGTACAGCCTATAGTGCTG
CCAGAAA-(TAMRA)-3′
3 µM POL-F (forward primer): 5′-TTCCTTTGGATGGGTTATGAACTC-3′
3 µM POL-R (reverse primer): 5′-GTATGTCATTGACAGTCCAGCTGTC-3′
These oligos are used for amplification of sequences present in RCRs and are specific forthe region of the pol gene coding for the reverse transcriptase (POL). See Support Protocol4 introduction.
Oligos can be ordered online from several companies such as Eurogentec or Sigma. Concen-trations of probe and primer may vary when using two (or more) oligo sets simultaneously(multiplex analysis).
Stocks of probes and primers usually come lyophilized and are diluted to 10 µM in water.

Production andTitration of
Lentiviral Vectors
12.10.16
Supplement 54 Current Protocols in Human Genetics
COMMENTARY
Background InformationBecause lentiviruses can infect both divid-
ing and nondividing cells, vectors were devel-oped from this subgroup of retroviruses withthe hope that they would be able to transducecells that proliferate very little or not at all.The proof of principle of this concept was firstprovided with vectors derived from HIV-1, us-ing the adult rat brain as an in vivo paradigm.Since then, gene-delivery systems based onanimal lentiviruses such as the simian and fe-line immunodeficiency viruses (SIV and FIV)and the equine infectious anemia virus (EIAV)have been described. This unit presents exclu-sively the HIV-based vector system because itis presently the most advanced, and because,in its latest version, it offers a level of biosafetythat matches, if not exceeds, that of the MLV-derived vectors currently used in the clinic.In addition, there is mounting evidence thatintracellular lines of defense limit the suscep-tibility of mammalian cells to retroviral trans-duction. As could be expected, retroviruseshave evolved to surmount this so-called intrin-sic immunity (Bieniasz, 2004; Mangeat andTrono 2005), but have done so in a largelyspecies-specific manner. HIV-derived vectorsthus often prove more efficient than homologsderived from animal lentiviruses when humancells are the targets.
Vector design: Evolution of cis-acting andtrans-acting plasmids
The potential of lentiviral vectors wasfirst revealed in 1996 through the demon-stration that they could transduce neurons invivo (Naldini et al., 1996). Since then, manyimprovements have been brought about toachieve high levels of efficiency and biosafety.The principle, however, remains the same andconsists in building replication-defective re-combinant chimeric lentiviral particles fromthree different components: the genomic RNA,the internal structural and enzymatic proteins,and the envelope glycoprotein. A schematic di-agram of the evolution of HIV-based vectorsis represented in Figure 12.10.3. The genomicRNA contains all the cis-acting sequences andthe packaging plasmids contain the genes thatcode for all the trans-acting proteins necessaryfor adequate transcription, packaging, reversetranscription, and integration.
The first-generation lentiviral vectors weremanufactured using a packaging system thatcomprised all HIV genes but the envelope(Naldini et al., 1996). In a so-called second-
generation system, five of the nine HIV-1genes were eliminated, leaving the gag andpol reading frames, which encode for the struc-tural and enzymatic components of the virion,respectively, and the tat and rev genes, ful-filling transcriptional and post-transcriptionalfunctions (Zufferey et al., 1997). Sensitivetests have so far failed to detect replication-competent-recombinants (RCRs) with thissystem. This good safety record, combinedwith its high efficiency and ease of use, ex-plains why the second-generation lentiviralvector packaging system is utilized for mostexperimental purposes. In a third-generationsystem, geared up towards clinical applica-tions, only gag, pol, and rev genes are stillpresent, using a chimeric 5′ LTR (long terminalrepeat) to ensure transcription in the absenceof Tat (see below).
The genetic information contained in thevector genome is the only one transferredto the target cells. Early genomic vectorswere composed of the following compo-nents: the 5′ LTR, the major splice donor,the packaging signal (encompassing the 5′
part of the gag gene), the Rev-responsive el-ement (RRE), the envelope splice acceptor,the internal expression cassette containing thetransgene, and the 3′ LTR. In the latest gener-ations, several improvements have been intro-duced. The Woodchuck Hepatitis Virus Post-transcriptional Regulatory Element (WPRE)has been added to increase the overall lev-els of transcripts both in producer and targetcells, hence increasing titers and transgene ex-pression (Zufferey et al., 1999). The centralpolypurine tract of HIV has also been addedback in the central portion of the genomeof the transgene RNA (Follenzi et al., 2000;Zennou et al., 2000). This increases titersat least in some targets. The U3 region 3′
LTR is essential for the replication of a wild-type retrovirus, since it contains the viralpromoter in its RNA genome. It is dispens-able for a replication-defective vector and hasbeen deleted to remove all transcriptionallyactive sequences, creating the so-called self-inactivating (SIN) LTR (Zufferey et al., 1998).SIN vectors are thus unable to reconstitutetheir promoter and are safer than their counter-parts with full-length LTRs. Finally, chimeric5′ LTRs have been constructed, in order torender the LV promoter Tat-independent. Thishas been achieved by replacing the U3 re-gion of the 5′ LTR with either the CMV en-hancer (CCL LTR) or the corresponding Rous

Vectors for GeneTherapy
12.10.17
Current Protocols in Human Genetics Supplement 54
Figure 12.10.3 Evolution in the design of HIV-1 based LV vectors. HIV-1-based LV vectors are derived from wild-type HIV-1 by dissociation of the trans-acting components (blue boxes, above HIV genome) coding for structural and accessory pro-teins, and the cis-acting sequences required for packaging and reverse transcription of the genomic RNA (gold boxes, belowHIV genome). Sequences added between two vector versions are in red. Abbreviations: CMV, human cytomegalovirusimmediate-early promoter; RRE, rev-responsive element; RSV, Rous sarcoma promoter; poly(A), polyadenylation site;U3-R-U5, HIV-1 LTR; SD, major splice donor; �, HIV-1 packaging signal; cPPT, central polypurine tract; SA, splice accep-tor; dPPT, distal (3′) polypurine tract; Prom, promoter of the internal expression cassette; TG, transgene of the internalexpression cassette; �U3, self-inactivating deletion of the U3 part of the HIV-1 LTR; WPRE, Woodchuck Hepatitis VirusPosttranscriptional Regulatory Element. For the color version of this figure go to http://www.currentprotocols.com.
sarcoma virus (RSV) U3 sequence (RRL LTR;Dull et al., 1998). Vectors containing such pro-moters can be produced at high titers in the ab-sence of the Tat HIV transactivator. However,the Rev-dependence of these third-generationLVs has been maintained, in order to max-imize the number of recombination eventsthat would be necessary to generate an RCR.
A schematic diagram of the evolution oftransfer plasmids and packaging plasmids isdepicted in Figure 12.10.3.
The latest generation with a chimeric LTRthat can be produced in the absence of Tat (alsocalled third generation) represents the systemof choice for future therapeutics projects. Inthe laboratory, however, this third generationis not mandatory, and the second-generationsystem offers a high level of safety for P2 con-ditions. The second-generation packaging sys-tem also has the advantage of working on bothsecond- and third-generation vectors. Thus, forin vitro and vivo research, it is easier to work

Production andTitration of
Lentiviral Vectors
12.10.18
Supplement 54 Current Protocols in Human Genetics
with one all-purpose packaging plasmid, suchas the psPAX2, which encodes for the HIV-1Gag, Gag/Pol, Tat, and Rev proteins.
Vector design: Loaded LTRsOwing to the replication strategy of retro-
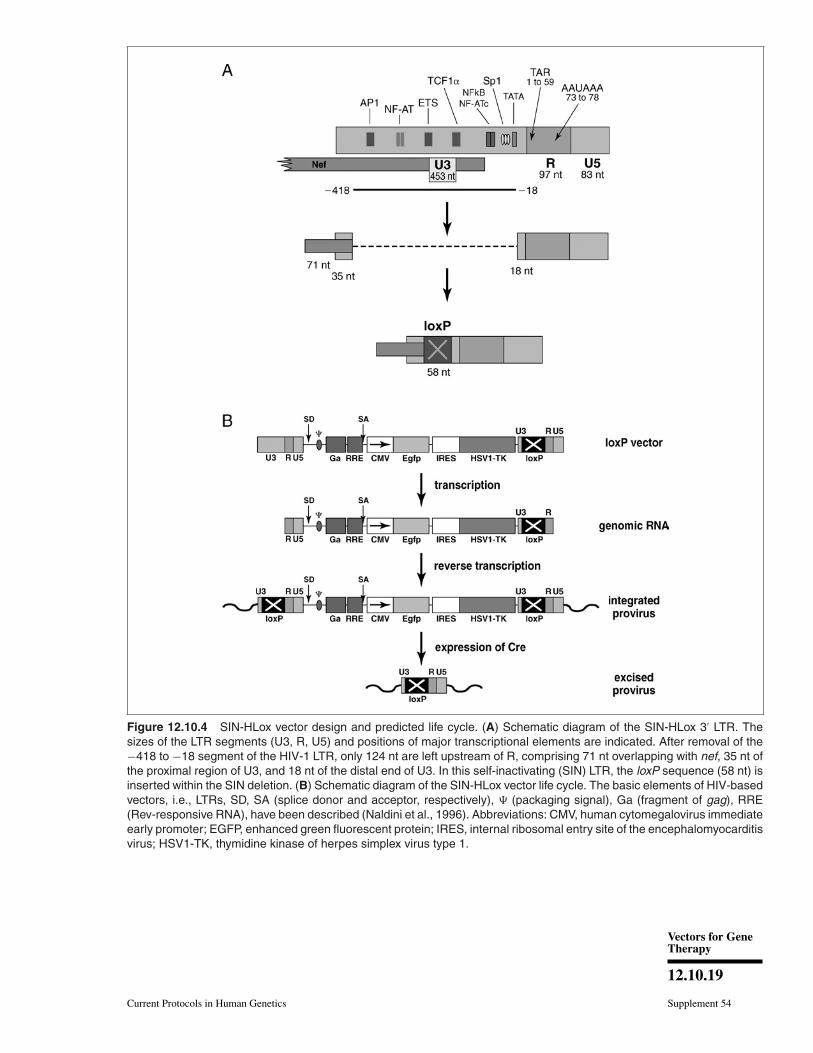
viruses in general and lentiviruses in par-ticular, the U3 part of the 3′ LTR presentin the viral RNA genome becomes dupli-cated once inserted into the host-cell genome.This property has been exploited to inac-tivate the LV promoter (SIN designs, seeabove). It has also been used to duplicatesequences such as loxP sequences to allowfor transgene excision when required (Salmonet al., 2000a). This strategy is illustrated inFigure 12.10.4.
Other recent vectors have been developedto take advantage of the duplication of 3′ LTRU3 sequences (Fig. 12.10.5). One is drug-controlled expression of siRNAs, which hasbeen described in Wiznerowicz and Trono(2003). This allows a final integrated trans-gene with two transcriptional units express-ing small interfering RNAs. Another vectorhas been constructed to flank the integratedLV provirus by two insulator sequences, thusshielding the transgene from influences fromits integration site, i.e., transcriptional activa-tion induced by nearby enhancers, or silenc-ing caused by chromatin remodeling. Theseinsulators may also minimize the influence ofthe transgene transcriptional cassette on genespresent in the vicinity of its integration site.This represents an important biosafety fea-ture against the risk of insertional activation.This sequence represents the shorter fragmentderived from the chicken β-globin gene, de-scribed in the literature, which displays bothactivities.
More detailed information (e.g., maps andsequences) is available at the authors’ in-stitutional Web sites: http://tronolab.epfl.ch/and http://www.medecine.unige.ch/∼salmon/lentilab.
Vector design: Multicistronic vectorsLentiviral vectors can theoretically accom-
modate up to 9 kb of transgenic sequence.Owing to this packaging capacity, a DNA se-quence encoding more than one protein can bedelivered to the target cells. However, express-ing more than one protein from an LV trans-gene requires specific strategies using cellularmechanisms to read several cistrons, either atthe transcriptional or at the translational level.Examples of such vectors using these strate-gies are given in Figure 12.10.6.
In addition to the human PGK promoterdescribed above, several other ubiquitous pro-moters are available. Among these are EF1(EF1α promoter, long version containing itsintron; Salmon et al., 2000b), EFS (EF1α pro-moter, short version after removal of its intron;Kostic et al., 2003), and UBI (ubiquitin genepromoter; Lois et al., 2002). The historicalCMV promoter is seldom used now. Indeed,although it is highly active in some cell linessuch as HeLa, it is not ubiquitously expressedin several primary cells of interest. In the caseof hematopoietic cells, it has been shown tobe active only in B cells and virtually silent inall others (Salmon et al., 2000b; Bovia et al.,2003).
The current rule of thumb is that promotersexpressed in most primary cells and tissuesin vivo display an intermediate transcriptionalactivity. This is the case of the PGK and UBIpromoters.
Bicistronic vectors can carry an internal ri-bosomal entry site (IRES) between the twocoding sequences (Figure 12.10.6B). IRESsare sequence elements which directly re-cruit ribosomes, thus allowing for translationof open reading frames downstream of themRNA cap site.
Vectors containing two different promot-ers can also be used. The two transcriptionalcassettes can be positioned in tandem. In thiscase, the two internal cassettes as well as thegenomic RNA will all use the same polyadeny-lation signal, i.e., from the LV 3′ LTR(Fig. 12.10.6C).
Alternatively, the two transcriptional cas-settes can be positioned back to back. In thiscase, the antisense transcript will require itsown polyadenylation signal, also in antisenseorientation (Fig. 12.10.6D). Note that the useof a bidirectional, or sense poly(A) signal be-fore the 3′ LTR poly(A) signal will result inpremature termination of LV genomes in pro-ducer cells (transcript 1), greatly increasingthe fraction of defective particles. The tan-dem configuration may be a problem for tran-script 2, being too long and unstable in thiscase. The antisense configuration may be re-quired if an intron is important for internalcassette expression. Indeed, in antisense ori-entation, the splice signals are not recognizedand are kept in the LV genome. The inconve-nience will be that, if the antisense promoter isactive in producer cells, it will generate an-tisense transcripts to the LV genome tran-scripts, thus decreasing titers. This canbe avoided by using a tissue-specific pro-moter in the antisense cassette. Note finally
Vectors for GeneTherapy
12.10.19
Current Protocols in Human Genetics Supplement 54
Figure 12.10.4 SIN-HLox vector design and predicted life cycle. (A) Schematic diagram of the SIN-HLox 3′ LTR. Thesizes of the LTR segments (U3, R, U5) and positions of major transcriptional elements are indicated. After removal of the−418 to −18 segment of the HIV-1 LTR, only 124 nt are left upstream of R, comprising 71 nt overlapping with nef, 35 nt ofthe proximal region of U3, and 18 nt of the distal end of U3. In this self-inactivating (SIN) LTR, the loxP sequence (58 nt) isinserted within the SIN deletion. (B) Schematic diagram of the SIN-HLox vector life cycle. The basic elements of HIV-basedvectors, i.e., LTRs, SD, SA (splice donor and acceptor, respectively), � (packaging signal), Ga (fragment of gag), RRE(Rev-responsive RNA), have been described (Naldini et al., 1996). Abbreviations: CMV, human cytomegalovirus immediateearly promoter; EGFP, enhanced green fluorescent protein; IRES, internal ribosomal entry site of the encephalomyocarditisvirus; HSV1-TK, thymidine kinase of herpes simplex virus type 1.

Production andTitration of
Lentiviral Vectors
12.10.20
Supplement 54 Current Protocols in Human Genetics
Figure 12.10.5 Loaded LTRs. (A) Schematic diagram of the LTR present in pLVTHM vectors asdescribed in Wiznerowicz and Trono (2003). This LTR is based on the SIN-Hlox LTR describedabove. �U3 represents the deleted SIN U3, TetO is composed of seven Tet operator sequences,H1 is the polymerase-III H1-RNA gene promoter, and siRNA is where the small interfering RNAsequence is cloned. In the presence of tetracycline (depicted as the light-shaded circle underKRAB at the upper left of panel A), tTR-KRAB does not bind to the TetO sequences. In theabsence of tetracycline, tTR-KRAB binds to the TetO sequences and represses transcription fromthe H1 promoter. (B) Schematic diagram of the LTR present in pRIX vectors. This LTR is based onthe SIN-Hlox LTR described above. �U3 represents the deleted SIN U3. Insulator is composed ofa 242 bp sequence derived from the chicken β-globin gene (Genbank locus number GGU78775,nt 26 to 245), which contains DNA elements with both chromatin-barrier and enhancer-blockingactivities (Recillas-Targa et al., 2002).
that the presence of two promoters in thesame LV may cause transcriptional interfer-ence. However, the authors did not observesuch phenomenon using an insulin promoterback-to-back with and EF1 promoter. Bothpromoters retain their individual activities.
More detailed information on the sequenceof these plasmids is available on the Websites http://tronolab.epfl.ch and http://www.medecine.unige.ch/∼salmon/lentilab. The firstWeb site can also be used to request all theplasmids mentioned in this unit.
Critical Parameters andTroubleshooting
Plasmid preparationPlasmids containing retroviral long termi-
nal repeats (LTRs) are prone to undergo dele-tion in some E. coli strains. The HB101 strainis strongly recommended for propagating theplasmids used in this section. The authors alsorecommend the ElectroTen strain (Stratagene)for difficult clonings with large fragment ofgenomic DNA.
The authors recommend JetStar (GenomedGmbH, http://www.genomed-dna.com/) or Qi-agen kits (Qiagen) to prepare DNAs for trans-fection. The last step of the DNA prep shouldbe an additional precipitation with ethanol andresuspension in TE buffer. Do not treat DNAwith phenol/chloroform, as this may result inchemical alterations. Also, to avoid salt copre-cipitation, do not precipitate DNA below 20◦C.
Troubleshooting vector productionA troubleshooting guide for lentiviral
production and transduction is given inFigure 12.10.7. Transfection efficiency is themost critical parameter affecting vector titer.293T/17 cells are highly transfectable using avariety of protocols. When establishing vec-tor production procedures, it is highly rec-ommended that the transfection protocol beoptimized using a plasmid encoding GFP.Transfection efficiency should not be assessedsolely on the basis of the percentage of GFP-positive cells, but also based on the mean flu-orescence intensity, which reflects the number

Vectors for GeneTherapy
12.10.21
Current Protocols in Human Genetics Supplement 54
Figure 12.10.6 Multicistronic vector LTRs. Schematic diagram of current LV used for expression of multiple cistrons(shown as plasmids in producer cells). (A) Schematic diagram of the pRRL LV. Abbreviations: RSV, Rous Sarcoma VirusLTR U3 sequence; �U3, SIN deletion of the U3 region of LTR; �, packaging signal; cPPT, central HIV polypurine tract;hPGK, human PGK promoter; GFP, enhanced green fluorescent protein. (B) Schematic diagram of the pRRL-PGK-IRES-GFP LV. Abbreviations: GOI, gene of interest; IRES, internal ribosomal entry site of the encephalomyocarditis virus (seeFig. 12.10.4). (C) Schematic diagram of a bicistronic LV with a tissue-specific cassette in line with the ubiquitous PGK-GFP cassette. Abbreviations: Pro, tissue-specific promoter; Intron, intronic sequence containing regulatory elements; Enh,tissue-specific enhancer. (D) Schematic diagram of the pRPA LV. Same as in C but with an inverted cassette for theexpression of the gene of interest, back-to-back with PGK-GFP ubiquitous cassette. Abbreviations: pA, polyadenylationsignal from the herpes simplex 1 virus thymidine kinase gene (Genbank locus number HE1CG, nt 46565 to 46693).Transcription units (mRNAs) are represented as dotted arrows. Transcript 1 is the LV genome. It is synthesized only inproducer cells from the active RSV-RU5 LTR. Transcripts 2 and 3 are synthesized from internal cassettes. They can bepresent in producer cells and in target cells.

Production andTitration of
Lentiviral Vectors
12.10.22
Supplement 54 Current Protocols in Human Genetics
of plasmid copies taken up by the cells. Thismakes FACS analysis of the transfected cellsmandatory. FACS can be done as soon as 15 hrafter the transfection, allowing many variablesto be tested rapidly. The factors most likely toimpact on the transfection efficiency are thepH of the 2× HeBS solution, the quality ofthe batch of fetal bovine serum (FBS) used, thecell density, the total amount of DNA per plate,and the quality of the DNA. A coarse precipi-tate will give poor transfection, whereas a fineprecipitate (barely visible after application oncells) will give good transfection. As a rule ofthumb, the precipitate will become coarser asthe pH of the 2× HeBS increases, as the DNAquantity decreases, and as the temperature orincubation time for precipitate formation in-creases.
In the case of lack of transduction of a spe-cific cell type with a specific lentiviral vector,a synoptic diagram (Fig. 12.10.7) is providedbelow to help addressing most of the problemsthat could account for it.
Figure 12.10.7 Troubleshooting diagram for lentiviral vector production and transduction.
Safety issuesThe system presented here contains nu-
merous safeguards as compared to the first-generation HIV vectors, in which genes encod-ing all HIV-1 proteins, except for Env, werepresent (Naldini et al., 1996). A second gen-eration was characterized by the exclusion offour accessory genes (vif, vpr, vpu, and nef;Zufferey et al., 1997). These deletions im-proved the safety of the vector considerably,because they excluded major determinants ofHIV-1 virulence. In the third-generation sys-tem, described in this unit, Gag, Pol, and Revare the only HIV-1 proteins still present (Dullet al., 1998). Vectors with self-inactivating(SIN) LTR, produced with the third-generationpackaging system, have been tested for RCR.Thus far, no RCR have been detected among atotal of 1.4 × 1010 transducing units (Escarpeet al., 2003).
In general, transduced cells must al-ways be fixed, using formaldehyde as de-scribed in Support Protocol 2, or, alternatively,

Vectors for GeneTherapy
12.10.23
Current Protocols in Human Genetics Supplement 54
paraformaldehyde, before being taken outof the P2 laboratory. If live cells must besorted outside of the P2 laboratory, carefulhandling techniques must be exercised andthe equipment used must be decontaminatedafterwards.
Given the very broad tropism of VSV G–pseudotyped lentiviral vectors both in vitroand in vivo, extra precautions must be takenwhen working with transgenes that are them-selves potential biohazards. For instance, a P3laboratory is recommended for the lentivector-mediated transfer of genes involved in cell pro-liferation.
Anticipated ResultsWhen applied optimally, the procedure
described here yields crude unconcentratedvector titers between 1 × 106 and 1 × 107
transducing units (TU) per ml. After centrifu-gation, a yield of at least 50% is expected. Asimilar 50% yield is also expected after onefreeze/thaw cycle. The cells produce constantamounts of vector during the 48 hr period post-transfection; yield is maximized by harvestingtwice.
Infectivity indexThere are approximately 2000 molecules
of p24 capsid molecules per physical particle(PP) of HIV, which amounts to:2 × 103 × 24 × 103 Da of p24 per PP,
which amounts to:
48 × 106/Avogadro’s no. = 48 × 106/6 ×1023 = 8 × 10−17 g of p24 per PP,
which amounts to:
∼1 PP per 10−16 g of p24,
which amounts to:
104 PP per pg of p24.A reasonably well packaged retrovirus will
have an infectivity index in the range of 1transducing unit (TU) per 300 physical par-ticles (PP) to 1 TU per 10,000 PP. Thus, therange for a lentivector infectivity index is ap-proximately 1 to 30 TU/pg of p24. Lower val-ues indicate that the vector has experiencedproblems during packaging, either because ofa problem during production or because of anintrinsic flaw in the vector design (see CriticalParameters and Troubleshooting).
There is no current procedure for purifica-tion per se of infectious particles. The avail-able methods (e.g., ion exchange, centrifuga-tion) will only concentrate the vector particlesand/or wash away soluble material. One must
keep in mind that all other particulates gener-ated by the producer cells, such as defectivevector particles and exosomes, are also coatedwith VSV-G proteins and will cosediment orcopurify with infectious vector particles. Thisimplies that there is no current way to enhancethe number of infectious particles in a vectorstock displaying a poor infectivity index. Us-ing the abovementioned existing techniques,defective particles will be simultaneously en-riched, causing an increase in cell toxicity.
Time ConsiderationsIf 293T cells are plated for transfection on
day 1, transfected on day 2, and washed on day3, and vector stocks are harvested and concen-trated on days 4 and 5, concentrated vectorstocks will be ready by day 5 and can be usedfor titration. Titration using flow cytometry orQPCR is performed in 5 days. In this way, thewhole procedure takes exactly 10 days fromtransfection to calculation of the titer.
Preparation of cells for titration requires30 min for flow cytometry and 1 hr for QPCRanalysis (DNA extraction). Flow cytometryanalysis can be done in 1 hr. Plate prepara-tion and real-time PCR analysis can be donein 3 to 4 hr.
Literature CitedBieniasz, P.D. 2004. Intrinsic immunity: A front-
line defense against viral attack. Nat. Immunol.5:1109-1115.
Bovia, F., Salmon. P., Matthes, T., Kvell, K.,Nguyen, T.H., Werner-Favre, C., Barnet, M.,Nagy, M., Leuba, F., Arrighi, J.F., Piguet, V.,Trono, D., and Zubler, R.H. 2003. Efficienttransduction of primary human B lymphocytesand nondividing myeloma B cells with HIV-1-derived lentiviral vectors. Blood 101:1727-1733.
Dull, T., Zufferey, R., Kelly, M., Mandel, R.J.,Nguyen, M., Trono, D., and Naldini, L. 1998.A third-generation lentivirus vector with a con-ditional packaging system. J. Virol. 72:8463-8471.
Escarpe, P., Zayek, N., Chin, P., Borellini, F.,Zufferey, R., Veres, G., and Kiermer, V. 2003.Development of a sensitive assay for detection ofreplication-competent recombinant lentivirus inlarge-scale HIV-based vector preparations. Mol.Ther. 8:332-341.
Follenzi, A., Ailles, L.E., Bakovic, S., Geuna, M.,and Naldini, L. 2000. Gene transfer by lentiviralvectors is limited by nuclear translocation andrescued by HIV-1 pol sequences. Nat. Genet.25:217-222.
Kostic, C., Chiodini, F., Salmon, P., Wiznerowicz,M., Deglon, N., Hornfeld, D., Trono, D.,Aebischer, P., Schorderet, D.F., Munier, F.L.,and Arsenijevic, Y. 2003. Activity analysis ofhousekeeping promoters using self-inactivating

Production andTitration of
Lentiviral Vectors
12.10.24
Supplement 54 Current Protocols in Human Genetics
lentiviral vector delivery into the mouse retina.Gene Ther. 10:818-821.
Lois, C., Hong, E.J., Pease, S., Brown, E.J., andBaltimore, D. 2002. Germline transmission andtissue-specific expression of transgenes deliv-ered by lentiviral vectors. Science 295:868-872.
Mangeat, B. and Trono, D. 2005. Lentiviral vectorsand antiretroviral intrinsic immunity. Hum.Gene Ther. 16:913-920.
Naldini, L., Blomer, U., Gallay, P., Ory, D.,Mulligan, R., Gage, F.H., Verma, I.M., andTrono, D. 1996. In vivo gene delivery and stabletransduction of nondividing cells by a lentiviralvector. Science 272:263-267.
Recillas-Targa, F., Pikaart, M.J., Burgess-Beusse,B., Bell, A.C., Litt, M.D., West, A.G., Gaszner,M., and Felsenfeld, G. 2002. Position-effectprotection and enhancer blocking by the chickenbeta-globin insulator are separable activities.Proc. Natl. Acad. Sci. U.S.A. 99:6883-6888.
Salmon, P., Kindler, V., Ducrey, O., Chapuis, B.,Zubler, R.H., and Trono, D. 2000a. High-leveltransgene expression in human hematopoieticprogenitors and differentiated blood lineages af-ter transduction with improved lentiviral vec-tors. Blood 96:3392-3398.
Salmon, P., Oberholzer, J., Occhiodoro, T., Morel,P., Lou, J., and Trono, D. 2000b. Reversibleimmortalization of human primary cells bylentivector-mediated transfer of specific genes.Mol. Ther. 2:404-414.
Wiznerowicz, M. and Trono, D. 2003. Conditionalsuppression of cellular genes: Lentivirus vector-mediated drug-inducible RNA interference. J.Virol. 77:8957-8961.
Zennou, V., Petit, C., Guetard, D., Nerhbass, U.,Montagnier, L., and Charneau, P. 2000. HIV-1genome nuclear import is mediated by a centralDNA flap. Cell 101:173-185.
Zufferey, R., Nagy, D., Mandel, R.J., Naldini,L., and Trono, D. 1997. Multiply attenuatedlentiviral vector achieves efficient gene deliveryin vivo. Nat. Biotechnol. 15:871-875.
Zufferey, R., Dull, T., Mandel, R.J., Bukovsky, A.,Quiroz, D., Naldini, L., and Trono, D. 1998.Self-inactivating lentivirus vector for safe andefficient in vivo gene delivery. J. Virol. 72:9873-9880.
Zufferey, R., Donello, J.E., Trono, D., and Hope,T.J. 1999. Woodchuck hepatitis virus post-transcriptional regulatory element enhances ex-pression of transgenes delivered by retroviralvectors. J. Virol. 73:2886-2892.