blg_undp_82.1_eng.pdf - who | world health organization
TRANSCRIPT


GENERAL TESTS

• WORLD HEALTH ORGANIZATION
ORGANISATION MONDIALE DE LA SANTE
Y.J H , .. ·
BLG/UNDP/82.1
ORIGINAL: ENGLISH
I
MANUAL OF DETAILS OF TESTS REQUIRED ON FINAL VACCINES
USED IN THE WHO EXPANDED PROGRAMME OF Il~NIZATION
The issue of this document does not constitute
formal publication. It should not be reviewed,
abstracted or quoted without the agreement of
the World Health Organization. Authors alone
are responsible for views expressed in signed
articles.
Ce document ne constitue pas une publication. II ne doit faire l'objet d'aucun compte rendu ou
resume ni d'aucune citation sans l'autorisation de
!'Organisation Mondiale de Ia Sante. Les opinions
exprimees dans les articles signes n"engagent que leurs auteurs.

MANUAL OF TESTS FOR BIOLOGICAL SUBSTANCES
BLG/UNDP/8.21 Page 1
This manual has been prepared, in the first instance, to give details of the tests applied to vaccines in the final form. Such tests may be applied by control authorities to vaccines as they are given to children in order to check that they have adequate potency and do not have undue toxicity.
The tests are identified by the vaccine to which they are applicable or as a general test and are classified according to the complexity of the equipment and facilities required for their execution.
Class A: is a test requiring only the laboratory equipment expected to be present in any microbiological laboratory.
Class B: is a test requ~r~ng special items of equipment but does not require the use of animals.
Class C: is a test requiring the use of animals and, thereby, demanding the facilities of an animal holding unit, an animal breeding station or a reliable source of good quality animals.

BLG/UNDP/82.1 Page 1 bis

GENERAL TESTS
BLG/UNDP/82.1 Page 1 ter
BLG/UNDP/82.1 Page 2
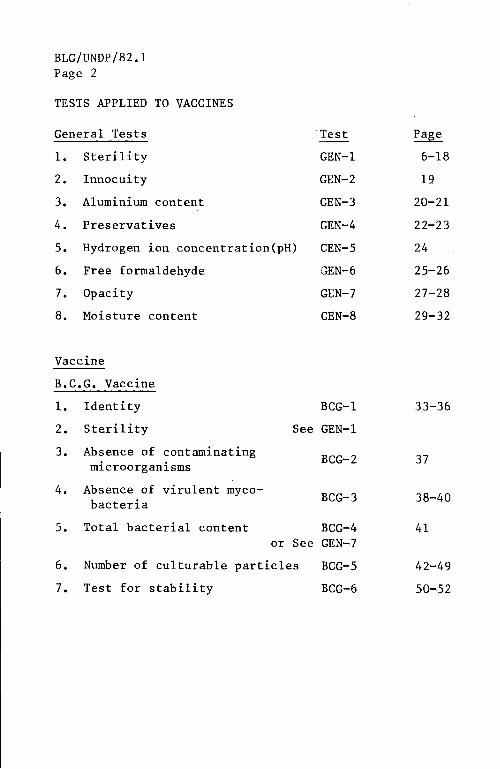
TESTS APPLIED TO VACCINES
General Tests Test Page
1. Sterility GEN-1 6-18
2. Innocuity GEN-2 19
3. Aluminium content GEN-3 20-21
4. Preservatives GEN-4 22-23
5. Hydrogen ion concentration(pH) GEN-5 24
6. Free formaldehyde GEN-6 25-26
7. Opacity GEN-7 27-28
8. Moisture content GEN-8 29-32
Vaccine
B.C.G. Vaccine
1. Identity BCG-1 33-36
2. Sterility See GEN-1
3. Absence of contaminating BCG-2 37 microorganisms
4. Absence of virulent myco-BCG-3 38-40 bacteria
5. Total bacterial content BCG-4 41 or See GEN-7
6. Number of culturable particles BCG-5 42-49
7. Test for stability BCG-6 50-52

BLG/UNDP/82.1 Page 3
Vaccine
Diphtheria Vaccine Test Page
1. Potency DIP-1 53-68
2. Sterility See GEN-1
3. Innocuity " GEN-2
4. Adjuvant content " GEN-3
5. Preservative content " GEN-4
6. Test for pH " GEN-5
7. Identity DIP-2 69-70
See also test for free formaldehyde GEN-6 See also stability and
test reveral
DIP-3 71-73
Pertussis Vaccine
1. Potency PER-l 74-90
2. Sterility See GEN-1
3. Innocuity GEN-2
4. Adjuvant content II GEN-3
5. Preservative content II GEN-4
6. Test for pH II GEN-5
7. Identity PER-3 93
See also test for opacity GEN-7 See also test for toxicity PER-2 91-92

BLG/UNDP/82.1 Page 4
Vaccine
Tetanus Vaccine
1. Potency
2. Sterility
3.
4.
5.
6.
Innocuity
Adjuvant content
Preservative content
Test for pH
7. Identity
See also test for free formaldehyde
See also test for irreversibility and test for specific toxicity
Poliomyelitis Vaccine (oral)
1. Virus titration (potency)
2, Test for bacteria and fungi
3, Innocuity
4. Identity
Measles Vaccine (Live)
l, Virus concentration (potency)
2. Sterility
3. Innocuity
4. Identity
5. Stability
Test
TET-1
See GEN-1
"
"
"
"
GEN-2
GEN-3
GEN-4
GEN-5
TET-2
GEN-6
TET-3
POL-l
GEN-1
GEN-2
POL-2
MEA-l
See GEN-1
" GEN-2
MEA-2
MEA-3
94-107
108-109
110-111
112-116
117
118-121
122
123

BLG/UNDP/82.1 Page 5
SUMMARY PROTOCOLS FOR THE REPORTING OF RESULTS OF TESTS
Protocol
Diphtheria, pertussis, tetanus
Diphtheria toxoid
Pertussis Vaccine
Tetanus toxoid
BCG Vaccine
Poliomyelitis Vaccine (Oral) Sabin strains
Measles Vaccine (Live)
124-131
132-136
137-140
141-144
145-149
150-161
162-167

BLG/UNDP/82.1 Page 6
VACCINE: ALL VACCINES
CLASS OF TEST: A
GEN-1
TEST: Test for Sterility
GENERAL REQUIREMENTS FOR THE STERILITY OF BIOLOGICAL SUBSTANCES
(Requirements for Biological Substances No. 6) (Revised 1973)
Part A Section 5
THE TEST
1. Sampling from final lot
Samples of final containers from each final lot to be tested shall be taken in such a manner as to be representative of the lot to be tested. Appropriate periodic samples shall be taken including samples at the beginning and the end of the filling operation.
If a product lot is filled through several outlets from a single bulk, samples should be taken from each outlet (filling lot) so as to be representative of the filling assembly.
The number of samples taken shall be at least that approved by the national control authority, provided that for final lots containing 500 or more containers, at least 20 samples shall be taken, including samples at the beginning and the end of the filling operation.

BLG/UNDP/82.1 Page 7
For final lots containing fewer than 500 containers, not less than 10 containers from the lot should be taken for a sample, except that for lots of less than 100 containers, only 10% of the lot need be tested.
1 A suggested rule for
calculating the number of samples is to take 0.4 ./'Ff samples, where N is the number of final containers in the lot.
2. Sterility test for bacteria and fungi
2.1 Culture media
The culture media used for sterility tests for bacteria and fungi shall be those approved by the national control authority. Such media shall have been shown to be capable of supporting the growth of a wide variety of micro-organisms, with both aerobic and anaerobic growth characteristics, including the types found in the environment of the manufacturing operations.
More than one culture medium, generally will be needed to fulfil these criteria. The media used in many countries for sterility tests are fluid thioglycolate medium
2and soybean-casein digest.
medium • Any other media that are used, however, should have been demonstrated to have at least equivalent growth supporting properties.
1 2 Wld Hlth Org. techn. Rep. Ser. 1960 No 200, p. 31
The formulae of these culture media are given in Appendix 1

BLG/UNDP/82.1 Page 8
For testing the growth supporting qualities of each culture medium, strains of microorganisms should be used with exacting nutritive and aerobicanaerobic requirements, in an inoculum of only a small number of the organisms (less than 100). The media should be incubated at the temperatures at which they will be used in the sterility test.
Each lot of dehydrated medium or each lot of medium prepared from basic ingredients should be tested for its growth supporting properties, since every lot may not support the growth of microorganisms as well as desirable. It is desirable that lots so tested should be kept separate and reserved for use in sterility testing. This is a safeguard against occasional unsatisfactory components in a particular lot, or destruction of certain components by over-heating or over-sterilization, which may cause differences in growth response.
In some countries an additional test is made to verify the growth supporting properties of the medium, after the completion of a sterility test in which no growth has occurred in the medium, by inoculation with a small number of suitable organisms (less than 100).

BLG/UNDP/82.1 Page 9
2.2 Performance of the test
Prior to conducting a sterility test on any product, it shall have been determined whether or not the material to be tested itself has the property of killing or inhibiting the growth of micro-organisms, or contains preservatives or other substances that have this effect. If such an effect is shown, the sterility test shall be made using a suitable procedure to counteract the effect.
2.3 Inoculum
The inhibitory effect in a preparation to be tested for sterility may be overcome by increasing the volume of culture medium used, so that the inhibitors are rendered ineffective.
The volume of medium required to overcome the inhibitory effect should be determined. Once established, these quantities may be used for subsequent sterility tests unless a change is made in the composition of the product.
For sterility testing of bulk material, at least 5 ml must be used for inoculation into each culture medium and for each temperature of incubation.
For sterility testing of a final lot from each of the final containers that contain more than 20 ml, an amount of at least 1.0 ml shall be inoculated into each culture medium and for each temperature of incubation. If the volume in each final container is less than 1.0 ml the amount to be inoculated shall be the entire content of the container for each culture medium and for each temperature of incubation. If the volume in each final container is greater than 20 ml, but not more than 100 ml, the amount to be inoculated shall be at least 5 ml for each culture medium and for each temperature of incubation,

BLG/UNDP/82.1 Page 10
2.4 Medium
The quantityof medium put into each vessel for conducting the sterility test shall be sufficiently large to ensure that the volume of material inoculated does not impair the growth supporting properties of the medium by dilution.
All vessels of inoculated media shall be clearly identified by labelling that is adequate to identify the product being tested, each medium used, and each temperature of incubation.
2.5 Incubation
All vessels shall be incubated at the appropriate temperatures for the media. The temperatures selected shall be those approved by the national control authority and shall include 20°-25°C and 30°-36°C.
If only one temperature is selected for a particular medium, generally fluid thioglyco6ate medium is incubated at 30 -36°C and casein diRest medium at 20°-25°C.
In some countries additional temperatures are used for psychrophilic and thermophilic organisms.
It is desirable that the incubation apparatus should have a continuous temperature recording device.
All vessels shall be incubated for a period of at least 14 days and shall be examined at regular intervals and on the last day of incubation for evidence of microbial growth.

BLG/UNDP/82.1 Page 11
If the preparation inoculated into the test vessels has clouded the medium to such an extent that it is difficult to recognize whether or not growth has taken place, subcultures of not less than 1.0 ml should be made from the cloudy medium between the third and seventh days after the start of the test. The original and transfer vessels should each be incubated and observed for a total of not less than 14 days.
3. Interpretation of test results
If no evidence of growth is found in any of the vessels inoculated for the test for sterility, the bulk material or final lot, whichever is applicable, meets the requirements for this test. If evidence of growth is found, the preparation tested fails to meet the requirements for the test for sterility, unless it can be demonstrated to the satisfaction of the national control authority, by retests or by other means, that the test was invalid.
The decision between failure of the product to pass the test and invalidity of the test procedure requires competent judgement of a trained individual.
4. Sterility test for mycoplasmas
For viral vaccines made by growing the virus in animal tissues or cell cultures, sterility test for mycoplasmas of virus culture fluid and control fluid specified for the particular product shall be made by a

BLG/UNDP/82.1 Page 12
h d d b h . 1 1 h . 1 a met o approve y t e nat~ona contro aut or~ty.
The samples taken may include material prior to clarification or filtration in the case of live vaccines produced from in vitro living cell cultures or an~mal tissues. In the case of inactivated virus vaccines produced from such cell cultures or tissues, the samples may be taken prior to inactivation, from each virus harvest pool and from control pooled fluid.
Control cultures should be included in each test.
5. Sterility test for viruses
The sterility tests for the detection of extraneous viruses in products where their presence is unacceptable shall be performed as specified in the requirements for the individual products.
6. Tests for specific micro-organisms
The tests to ensure the non-survival of microorganisms in inactivated vaccines and additional tests for the absence of specific extraneous micro-organisms in such products shall be performed as specified in the requirements for the individual products.
7. Records
Records shall be permanent and clearly indicate all steps in the sterility testing of the product, including temperatures and incubation times, as well as the relation-
1 A description of tests for mycoplasmas is given
in Appendix 2

BLG/UNDP/82.1 Page 13
ship of the samples under test to the manufacturing operations followed in the production of the lot under test. The records shall be of a type approved by the national control authority. Written records shall be kept of all tests performed, irrespective of their results. They shall be retained throughout the dating period of the product, and be available for inspection by the national control authority. Records shall be maintained of all cultures kept in the establishment. Such records shall include clear labelling for the identification of the cultures and the purposes for which the cultures are used.
For each sterility test performed a record shall be kept of the name and identifying numbers of the product under test, the quantities inoculated, the batch, type and tests of culture media used, the temperatures of incubation, the dates of inoculation and the results.
8. Additional samples
For each final lot for which sterility tests are made, additional samples shall be retained as reference material throughout the dating period of the product, in a manner that ensures the identity of the lot or batch of the product.
It is desirable, that where possible, manufacturers should retain sufficient additional samples to permit repetition of the sterility tests.

BLG/UNDP/82.1 Page 14
Appendix 1
MEDIA FOR THE DETECTION OF AEROBIC & ANAEROBIC BACTERIA & FUNGI*
1. Fluid thioglycolate medium
L-cystine 0.5 sodium chloride 2.5 glucose (C6Hl206H20) 5.5
agar 0.75 yeast extract, water soluble 5.0 pancreatic digest of casein 15.0 distilled water 1000 thioglycolic acid 1 0.3 (or sodium thioglycolate 0.5 resazurin sodium (0.10% fresh solution) 1.0
Final pH • 7.0- 7.2
Preparation:
g g g
g g g ml ml g) ml
Thoroughly grind the first six ingredients, in the order given above, in a mortar. Stir in some heated water, transfer to a suitable container, add the remainder of the water and complete the solution by_ heating in a boiling water bath, taking special care to ensure complete solution of the L-cystine. Add the thioglycolic compound, then 1 N sodium hydroxide so that the pH of the completed and sterilized medium will be 7.0-7.2. Reheat the solution, but do not boil, filter (if necessary) through a moistened filter paper and add the resazurin solution. Distribute into suitable vessels and sterilize by autoclaving for 18-20 minutes at 121°C.
*
1
The quality of the components should be in accordance with Specifications for Reagents mentioned in the International Pharmacopoeia, Geneva, World Health Organization 1967, unless otherwise stated. Reagent quality sodium thioglycolate is more stable than thioglycolic acid.

BLG/UNDP/82.1 Page 15
0 0 0 Cool promptly to 25 C and store at 20 C-30 C, avoiding excessive light. If the uppermost portion of the medium has changed to a pink colour and this exceeds one-third of the depth of the medium, it is unsuitable for use, but may be restored once by heating in steam. Medium more than 3 weeks old should not be used.
2. Soybean-casein digest medium
pancreatic digest of casein papaic digest of soybean meal sodium chloride dipotassium hydrogen phosphate glucose (C
6H
12o6H20)
dist~lled water
Final pH- 7.1 - 7.5
Preparation:
17.0 3.0 5.0 2.5 2.5
1000
g g g g g
ml
Dissolve the solids in the water, warming slightly, then cool the solution to room temperature. Adjust the reaction with 1 N sodium hydroxide if necessary, so that the pH of the completed and sterilized medium will be 7.1-7.5. Filter, if necessary, to clarify, distribute into suitable vessels and sterilize in an autoclave for 18-20 minutes at 121°C.

BLG/UNDP/82.1 Page 16
Appedix 2
TESTS FOR MYCOPLASMAS
Allvarieties of mycoplasma are not capable of growth in the same medium. Some strains require special culture media. Suitable media should therefore be selected having regard to particular conditions, e.g. the tissue in which a virus is grown or source of animal serum, and the growth requirements of the potential contaminants, e.g., the need for yeast extract. Solid and semi-solid or liquid media coytaining serum fractions rich in certain lipids (sterols and 'phospholipids) are used to provide the nutritive requirements. Control cultures on solid and semi-solid or liquid media of known fastidious strains of mycoplasma, both sterol-requiring and non-sterolrequiring, are included in each test to show that the media are capable of supporting the growth of mycoplasmas.
Samples of the material of not less than 6 ml for a single test (i.e. using one solid and one semi-solid or liquid medium) are stored either between 2° and 8°C for no longer than 24 hours or at -20°C or lower if stored for longer than 24 hours.
Not less than 2.0 ml of each sample are inoculated and evenly distributed over the surface of at least 10 plates of each solid medium used, and not less than 1.0 ml of each sample is inoculated into each of at least 4 tubes of each semi-solid or liquid medium used. Each tube should contain 10 ml of medium. Larger volumes of inoculum, e.g. making a total of 50 ml of material in 400 ml of medium, have been used advantageously. Ultracentrifugation before testing such samples has also been used.
0 0 The plates and tubes are incubated at 36 C + 1.0 C
for not less than 14 days. One half of each set-is
1 Non-sterol-requiring mycoplasmas will grow on media containing sterols.

BLG/UNDP/82.1 Page 17
incubated aerobically in an environment of adequate humidity, and the other half is incubated anaerobically in an environment of 5-10% carbon dioxide in nitrogen, also of adequate humidity. At the end of 3 days and again at 14 days, 0.5 ml from each of 2 tubes being incubated aerobically are combined and inoculated on to not less than 4 plates. The 2 tubes being incubated anaerobically are treated"likewise. · Since some mycoplasmas may not grow on media in Petri dishes, sucf subcultures on solid media may also be made in tubes.
All material seeded originally on solid media is subcultured on the day after the primary inoculation, on the third day and on the sixth day, on other suitable solid and liquid media, and these subcultures are incubated similarly, half aerobically and the other half anaerobically as described, and examined after 10 days, while the original cultures are examined at the end of the 14-day period. All plates (and tubes) after incubation for not less than 14 days are observed for the growth of mycoplasmas by appropriate procedures.
If no evidence is found of the presence of mycoplasmas the material, e.g. virus pool, meets the requirements of these tests. If evidence of growth of mycoplasmas is found, the mycoplasmas should be identified and the source traced.
For detection and identification of mycoplasmas the following procedures are suggested but other methods are available:
1. For detection of mycoplasmas
(a) typical colonies or specific coloration (blue) by Diene's stain of colonies spread on blocks of agar in Petri dishes, and viewed under the microscope;
1 In order to induce mycoplasmas to grow in liquid media, similar subculturing could be done, but three or four passages may be needed.

BLG/UNDP/82.1 Page 18
1. For detection of mycoplasmas Cont ...
(b) phase contrast microscopy of colonies (strips or plaques) for globular bodies, filaments and granules;
(c) examination under the electron microscope of the pellet centrifuged from a semisolid culture;
(d) examination under the light microscope (x 1000) of a film stained by Giemsa.
2. For separation of mycoplasmas into classes
(a) enzymatic effects; fermentation of glucose, breakdown of arginine and reduction of triphenyltetrazole, using decoloration of phenol red as indicator for the first two effects and red colour of the tetrazole derivative for the last;
(b) haemolysis in a 4% suspension of washed guineapig cells, added and incubated for 18-48 hours aerobically at 37°C;
(c) haemadsorption of a 4% suspension of guineapig cells poured over the growth surface or fixation of red blood corpuscles by the colonies after incubation at 37°C for 30 minutes.
3. For specific identification
Growth inhibition by specific anti-mycoplasma serum (prepared in the rabbit); such inhibition might be demonstrated by depositing paper discs impregnated with the serum on 2-4 hour growth cultures and observing them from the third day.
Immunofluorescence techniques for this purpose are being developed.

VACCINE: Diphtheria Toxoid Pertussis vaccine and Tetanus Toxoid
TEST: Test for innocuity
BLG/UNDP/82.1 Page 19
CLASS OF TEST C
GEN-2
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE TETANUS TOXOID AND COMBINED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Addendum 1980
Part A Section 19
THE TEST
Each final lot shall be tested for abnormal toxicity by the injection of one human dose, but not more than 1 ml, into each of 5 mice (weighing 17-22 g) and at least one human dose, but not more than 1 ml, into each of 2 guineapigs (weighing 250-350 g) by the intraperitoneal route. The final product shall be considered as innocuous if the animals survive for at least 7 days without showing significant signs of toxicity.
The animals used in this test shall be from a healthy stock. If there is any doubt about the health of the stock similar groups of animals should be injected with 1 ml of saline and the results compared with those animals given the vaccine. If any animals die within 7 days of being given saline the health of the animals should be investigated.

BLG/UNDP/82.1 Page 20
VACCINES: All Vaccines containing adjuvants
TEST: Test for Aluminium content
CLASS OF TEST A
GEN-3
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE TETANUS TOXOID AND CO}ffiiNED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Section 3.4.3
Procedure
To duplicate 3 ml samples of the product in boiling tubes are added 1 ml of concentrated sulfuric acid and six drops of concentrated nitric acid. The tubes are heated until dense white fumes evolve. Heating is continued, with dropwise addition of nitric acid, until the contents of the tubes are colourless. The tubes are cooled and 10 ml of distilled water is carefully added. If the solution is cloudy the tubes are boiled until clear. Sodium hydroxide (50% w/v) is added to each tube until a pink-yellow endpoint is obtained with methyl orange , indicator. Any precipate forming is dissolved by the addition of dilute sulfuric acid. The contents of the tubes are transferred to separate 250 ml Erlemeyer flasks, the tubes washed out with 25 ml of distilled water and the washings pooled with the contents of the flasks. To each flask are added 25 ml of M/100 disodium ethylenediamine tetra acetate, and 10 ml of acetate buffer (containing 68 g of sodium acetate, 38.5 g of ammonium acetate, 125 ml of glacial acetic acid in a total volume of 500 ml). The flasks are boiled gently for three minutes and 1 ml of pyridylazonaphthol indicator (0.1% solution in 95% ethanol) is added. Each hot

BLG/UNDP/82.1 Page 21
solution is titrated with M/100 copper sulfate solution until a purple-brown endpoint is reached. Simultaneously a blank determination is also carried out using distilled water in the digestion procedure instead of sample. Each ml of M/100 Cuso
4.5H
20 = 0.2698 mg Al+++.
Calculation of the results:
mg of Al per ml of sample = Blank titre-Sample titre Volume of sample (ml)
X 0.2698 X 4.52
Criteria for acceptance
The aluminium content in the final bulk product should not exceed 1.25 mg Al per single human dose.

BLG/UNDP/82.1 Page 22
VACCINE: All Vaccines containing Thiomersal
TEST: Test for Thiomersal
CLASS OF TEST A
GEN-4
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE TETANUS TOXOID AND COMBINED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Section 3.4.2
TEST FOR THIOMERSAL CONTENT
By chemical means
Procedure
The thiomersal content (merthiolate, thimerosal, ethylmercuri-thiosalicylate) is determined spectrophotometrically using diphenylthiocarbazone (dithizone).
Separatory funnels are washed with concentrated nitric acid and rinsed with tap and distilled_ water. Two aliquots of a standard 0.01% acueous solution of thiomersal (0.5 and 1.0 ml) and two samples of the test solution (1 ml) are added to individual separatory funnels and the volume adjusted to 10 ml with a 1% solution of ammonium acetate at pH 6.0. Ten milli-litres of a 1 in 10 dilution of a fresh solution of 0.01% dithizone in chloroform is added to each funnel and the contents shaken vigorously for 45 seconds. The chloroform layer is carefully separated.

BLG/UNDP/82.1 Page 23
The 0 and 100% transmission points on the spectrophotometer are set at 490 nm using the diluted dithizone solution and a scan is taken of the test solutions from 470 to 520 nm. The transmission at 520 nm is plotted against the thiomersal concentration of the standards on semi-logarithmic paper and the concentration of thiomersal in the sample determined.
By microbiological means
Nutrient agar is poured to a depth of 4 mm on a suitable rectangular glass plate set in a metal frame (a large Petri dish may be used but can accommodate only a few samples). It is essential that the plate is absolutely horizontal when the agar is being poured. Circular wells of approximately 8 mm diameter are cut in the agar so that adjacent wells are at leas-t 30 mm apart. The plate is seeded with sufficient Staphylococcus aureus organisms to give a confluent growth at 48 hours. Into each well 100 microlitre samples of 1:2.5, 1:5, 1:10, 1:20, 1:40 and 1:80 dilutions of a standard solution of 1 g/litre sodium thiomersalate are randomly dispersed as well as samples of the test vaccines. The location of each preparation is carefully recorded.
The plates are incubated for 48 hours. The diameter of the zones of inhibition, produced by each of the test vaccines, is measured and compared with the zones produced by the reference dilutions of thiomersal. The effective concentration is expressed in terms of the dilution of reference thiomersal solution to which it most closely approximates.
Criteria for acceptance
The thiomersal content should be between 0.005 and 0.02%.

BLG/UNDP/82.1 Page 24
VACCINES: Diphtheria, Pertussis, Tetanus and Poliomyelitis
CLASS OF TEST A
GEN-5
TEST: Test for Hydrogen Ion concentration pH
TEST FOR pH
Procedure
A pH meter with glass and calomel reference electrodes is standardized with standard buffer solutions at pH 5.0 and 7.0. The electrodes are rinsed with distilled water, dried and the pH of the test solution recorded.
Criteria for acceptance
The pH value of DPT vaccine shall be 6.7-7.0, of Diphtheria, Tetanus and DT vaccines between 6.0-6.7, and for Pertussis vaccine alone 7.0 + 0.3. Poliomyelitis vaccine (ora~) should be 6.8 but should never be more than 7.0. Poliomyelitis vaccine (oral) contains an indicator (phenol red) and the vaccine should be an orange colour. If the colour is bright red or purple the pH is too high and should be checked. Vaccine containing magnesium chloride as a stabilizer but at a high pH will not be stable.

BLG/UNDP/8.21 Page 25
CLASS OF TEST A
GEN-6
VACCINES: Diphtheria, Pertussis and Tetanus
TEST: Test for free Formaldehyde
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE TETANUS TOXOID AND COMBINED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Section 3.5.7
TEST FOR FREE FORMALDEHYDE
Procedure
1 ml of the vaccine or toxoid is diluted with distilled water to 10 ml. 2 ml of the diluted sample is transferred into 50 ml Nessler tube (with about 65 ml capacity) and diluted with distilled water to about 35 ml. 10 ml diluted sulfuric acid (one vol. H2so
4 of sp. gr.
1.84: one vol. water) is added and the Ilu1d is diluted with distilled water to the 50 ml mark. Similarly a series of tubes with known amounts of formaldehyde are prepared to serve as formaldehyde standards for comparison, using a 0.01% dilution of Analar solution of formaldehyde (containing not less than 37% of CH 0) and selecting quantities close to that expected in t5e sample. To the sample tube and to each of the standard tubes 10 ml of fuchsin-sulfurous acid test solution is added. The mixtures are allowed to stand one hour at room temperature and a colorimetric comparison of the sample tube with the standards is made.
Fuchsin-sulfurous acid test solution
0.2 g of basic fuchsin is dissolved in 120 ml of hot

BLG/UNDP/82.1 Page 26
distilled water and the solution allowed to cool. A solution of 2 g of anhydrous sodium sulfite in 20 ml of distilled water is added and then 2 ml of hydrochloric acid. The solution is diluted with distilled' water to 200 ml and allowed to stand at least one hour. This solution must be freshly prepared.
Criteria for acceptance
The vaccine shall be shown to have not more than 0.02% free formaldehyde.
Note: If the B. pertussis organisms in the pertussis vaccine are killed with chemicals other than formaldehyde or by heat then there is no purpose in applying this test to pertussis vaccine not combined with toxoids.

VACCINE: Pertussis and BCG
TEST: Test for opacity
BLG/UNDP/82.1 Page 27
CLASS OF TEST A
GEN-7
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE TETANUS TOXOID AND COMBINED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Section 3.3.2
TEST FOR OPACITY
This test is applied as an "in process" control to the pertussis component before any admixture takes place and before the addition of adjuvant; it is applied also to final BCG vaccine. The test is included in this manual because control laboratories should be aware of the test even though it cannot be applied to the final vaccine.
The opacity of each single harvest shall be determined not later than two weeks after harvesting and before the bacterial suspension has been subjected to a process capable of altering its opacity. The opacity shall be determined in comparison with the International Reference Preparation of Opacity or an equivalent reference preparation approved by the national control authority.
A suspension of pertussis organisms that on visual inspection has an opacity approximately equal to 10 IU of opacity may be considered for practical purposes to

BLG/UNDP/82.1 Page 28
1 have a concentration of 10 000 million organisms per ml .
Procedure
The sample shall be diluted until the opacity is identical with the Fifzh International Reference Preparation of Opacity when compared by EYE. The opacity of such a suspension is 10 Opacity Units.
Taking the dilution factor into account, the opacity of the sample is calculated.
If the final bulk, when diluted with an equal volume of saline has the same opacity as the Fifth International Reference Preparation of Opacity, the bulk has an opacity of 20 Opacity Units.
1
2
The concentration determined by direct count, however, would probably be less than 10 000 million per ml. The question of relationship of the international unit of opacity to concentration of organisms has been considered by the Expert Committee on Biological Standardization (WHO Technical Report Series, No. 96, 1955, page 8, and No. 259, 1963, page 26).
Available from Chief, Biologicals, WHO, Geneva

VACCINE: BCG and Measles
TEST: Test for moisture content
TEST FOR MOISTURE CONTENT
BLG/UNDP/82.1 Page 29
CLASS OF TEST A
GEN-8
The moisture content is determined by measuring the loss of weight of the test sample after drying under detiined conditions.
The acceptance is assessed according to the criterion specified for each vaccine.
Test Procedure
A weighing bottle with a stopper provided with a capillary of 0.20 to 0.25 mm inside diameter is dried and accurately weighed at a relative humidity of no higher than 45%.
A 30-50 mg portion of the test sample is transferred into the weighing bottle and the bottle is accurately weighed to obtain the net weight of the test sample.
The loaded bottle is dried over anhydrous P2o~ at a pressure of no higher than 5 mmHg and at 60°C for 3hours, then placed in a dessicator containing anhydrous P
2o
5 or
silica gel, cooled to room temperature and weighed accurately.

BLG/UNDP/82.1 Page 30
The moisture content is calculated by the following formula:
Moisture content
%
the weight lost of the test sample by drying the weight lost of the test sample before drying
X 100
The moisture shall be less than 3% for BCG vaccine and less than 2% for measles vaccine.

VACCINE: BCG
General Remarks
BLG/UNDP/82.1 Page 31
The system for the control of BCG vaccine is different from that of other vaccines. In view of the special problems in standardizing production and control of this vaccine it is a WHO-centralized system based on the principle that the number of BCG production and control centres should be kept to a minimum.
As far as the quality control is concerned, this is coordinated by the BCG Dept. of the State Serum Institute, Copenhagen. The data from the most important test (estimation of the number of culturable particles - see below) are collected in Copenhagen, punched on cards and calculated in Geneva by computer. In this way at least the vaccines available internationally are standardized in order to ensure their efficacy.
The production and control of BCG vaccine is possibly only by specially trained staff. As a rule such training takes place at the Copenhagen laboratory, where there is an ongoing training programme for this purpose.
Control tests of BCG vaccine are now performed in a number of control laboratories (Budapest, Prague, Bucharest and Madras for vaccines supplied to UNICEF) as well as in other national control laboratories and also production laboratories. The results from most of these laboratories are analysed in the coordination programme and it is clear that this system functions efficiently.
The backgrounds and philosophy of the present situation in the field of BCG vaccine control has been fully described in WHO/TB/Technical Guide/77.8, "WHOsponsored International Quality Control of BCG Va~e". The details of the in-vitro test methods are to be found in

BLG/UNDP/82.1 Page 32
detail in WHO/TB/Technical Guide/77.9, "In vitro Assays of BCG Products".
The most recent WHO Requirements were published in 1979: "Revised Requirements for dried BCG Vaccine", WHO Technical Report Series, No. 630 1979 Annex 2, in which the test methods are found on pages 128-135.

BCG VACCINE

VACCINE: BCG
TEST: Test for Identity
BLG/UNDP/82.1 Page 33
CLASS OF TEST B
BCG-1
REQUIREMENTS FOR DRIED BCG VACCINE (Requirements for Biological Substances No. 11)
(Revised 1978)
Part A Section 5.1
THE TEST
Identity Test
An identity test shall be performed on samples of vaccine from each final lot.
The identity of each filling lot of vaccine shall be verified by the morphological apperance of the bacilli in stained smears and by the characteristic appearance of the colonies grown on solid media.
The medium used is generally the Lowenstein-Jensen or a similar medium. The smears are stained by the Ziehl- Neelsen method.
Lowenstein-Jensen Medium
Lowenstein's medium with 0.75% Glycerin (usually called Lowenstein-Jensen medium) (Zentralblatt fur Bakteriologie, Parasitenkunde u. Infektionskrankheiten I. Abt. Orig. 125, 222, 932 and Advances in the Control of Zoonoses, p. 11 and 35, World Health Organization, Geneva 1953).

BLG/UNDP/82.1 Page 34
Lowenstein-Jensen medium Cont ...
For the culture medium, the following mixture is made up:
Monopotassium phosphate Magnesium sulfate Magnesium citrate 1-Asparagin Glycerine (twice distilled)
0.4% 0.04% 0.1% 0.6% 2.0%
A flask containing 100 ml of this solution is boiled for 2 hours; after cooling, 30g of potato flour are added and the mixture is then boiled in a water-bath with continual and thorough stirring to complete pastiness. The mixture is then cooled to about 56°C before the eggs are added. 20-22 fresh eggs with large yellow yolks are cleaned carefully, broken into a large flask, shaken vigorously and filtered through sterile gauze. One litre of egg and 600 ml of the salt-potato flour solution are mixed; after shaking 20 ml of 2% aqueous malachite-green solution is added. The medium is tubed, slanted, and coagulated at 85°C for 40 minutes (Fig. 2). The following day the tubes are heated to 70°C for 30 minutes. The cotton plugs are then parafined or closed in another way, and stored in a refrigerator at 4°C.
(From: Jensen, Bulletin International Union against Tuberculosis 24: 102-103)
Ziehl-Neelsen Method:
Concentrated Alcoholic Solution of basic Fuchsin
Basic fuchsin (certified) 95 per cent ethyl alcohol
90 g 945 ml
1. Add 300 ml alcohol to the 90 g fuchsin. 2. Heat in water bath to boiling for 2 minutes. 3. Allow to stand 1 minute and decant off; save
the supernatant.

BLG/UNDP/82.1 Page 35
4. Add a second 300 ml portion of the alcohol to the sediment, boil and decant as before.
5. Add the remaining 345 ml alcohol to the sediment, boil as before and add to portions collected previously.
5 per cent Phenol:
Distilled water Phenol crystals
950 ml 50 g
Carbol Fuchsin Solution for Staining Acid-fast Bacilli:
Concentrated alcoholic solution of basic fuchsin
5 per cent phenol
Filter stain just before using to remove
105 ml
895 ml
any acid-fast organisms which may be present.
Acid Alcohol for Decolorizing:
Hydrochloric acid 95 per cent ethyl alcohol Mix thoroughly
Staining Procedure
3 ml 97 ml
It is difficult for stains to penetrate cells of the Mycobacteria because of their high lipoid content. Only after prolonged treatment with carbol fuchsin, one of the most penetrating stains, or the application of heat during the staining process, can these bacteria be coloured. Once stained, however, the waxy material of the cells holds the carbol fuchsin even in the presence of acid alcohol. The steps of the Ziehl-Neelsen method which demonstrate the

BLG/UNDP/82.1 Page 36
property of acid-fastness are:
1. Smear out material (cultures or specimens) dry and fix as usual.
2. Cover smear with carbol fuchsin and steam over a low flame or a water bath for 5 minutes. The stain should never boil nor should it be allowed to dry.
3. Let slide cool a minute. Wash thoroughly in water.
4. Decolorize in acid alcohol (95 ml of 95 per cent ethyl alcohol plus 5 ml concentrated HCl) for 5 minutes or until no more color leaves the smear.
5. Wash in water. 6. Counterstain with methylene blue for 30 seconds. 7. Wash, dry and examine with oil immersion objective.
By this method acid-fast bacteria appear red against a blue background.
(From: Kelly and Rite, Microbiology, ed. AppletonCentury-Crofts, New York)

VACCINE: BCG
TEST: Test for absence of contaminating micro-organisms
BLG/UNDP/82.1 Page 37
CLASS OF TEST B
BCG-2
REQUIREMENTS FOR DRIED BCG VACCINE (Requirements for Biological Substances No. 11)
(Revised 1978)
Part A Section 5.2
THE TEST
Samples from each final lot shall be tested for bacterial and mycotic contamination according to the requirements given in Part A, Section 5, of Requirements for Biological Substances No. 6 (Requirements for the Sterility of Biological Substances) (11, page 4).
For details of this test see GEN-1.

BLG/UNDP/82.1 Page 38
VACCINE: BCG
CLASS OF TEST C
BCG-3
TEST: Test for absence of virulent Mycobacteria
REQUIREMENTS FOR DRIED BCG VACCINE (Requirements for Biological Substances No. 11)
(Revised 1978)
Part a Section 5.3.1
THE TEST
"If the test for the absence of virulent mycobacteria applied to the final bulk, is unsatisfactory, it shall be repeated with a sample of a final lot (see Part A, section 3.4.3).
This test may be applied to the final lot and omitted from the final bulk".
Part A Section 3.4.3
A sample of the final bulk intended for this test shall be stored for not more than 72 hours after harvest at 4°C.
At least 6 guinea-pigs, all of the same sex, each weighing 250-350 g are used.
Some countries prefer to use guinea-pigs that have been shown previously to be tuberculinnegative.

BLG/UNDP/82.1 Page 39
A dose of BCG organisms corresponding to at least 50 human doses of vaccine intended for intradermal injection shall be injected into ealh guinea-pig by the subcutaneous or intramuscular route • The animals shall have been maintained on a diet that is free from added substances, such as antibiotics, that might interfere with the test. The guinea-pigs shall be observed for at least 6 weeks. If, during that time, they remain healthy and gain weight, the final bulk shall be considered to be free from virulent mycobacteria.
At the end of the observation period, the animals shall be killed and examined post mortem for macroscopic evidence of progressive tuberculous disease; similarly, any animals that die before the end of the observation period shall be subjected to post mortem examination.
The vaccine lot shall pass the test if none of the guinea-pigs shows evidence of progressive tuberculous disease and if at least two-thirds of them survive the observation period.
Should more than one-third of the guinea-pigs die during the observation period (and freedom from progressive tuberculous disease is verified), the test shall be repeated with another sample of the final lot on at least 6 more guinea-pigs •
If, on the second occasion, more than one-third of the animals fail to survive the observation period, the vaccine lot shall not pass the test.
1 When a more concentrated vaccine, intended for administration by the percutaneous route, is tested, a dilution factor approved by the national control authority shall be applied, so that the mass of BCG injected corresponds to 50 human doses of intradermal vaccine.

BLG/UNDP/82.1 Page 40
Should a vaccine lot fail to satisfy the requirements of this test because animals die from causes other than tuberculosis, the procedure to be followed by the manufacturer should be determined with the approval of the national control authority.
If evidence of progressive tuberculous disease is seen, the vaccine lot shall be rejected, all subsequent vaccine lots shall be withheld, and all current vaccine stocks shall be held pending further investigation. The manufacture of BCG vaccine shall be discontinued and it shall not be resumed until a thorough investigation has been made and the cause or causes of the failure determined, and then only with the approval of the national control authority.

VACCINE: BCG
TEST: Test for total bacterial content
BLG/UNDP/82.1 Page 41
CLASS OF TEST A
BCG-4
REQUIREMENTS FOR DRIED BCG VACCINES (Requirements for B-iological Substances No. 11)
(Revised 1978)
Part A Section 5.4
THE TEST
The total bacterial content of the reconstituted vaccine shall be estimated for each vaccine lot by a method approved by the national control authority, and shall have a value within a range approved by the national control authority.
The estimation of total bacterial content may be made either directly by determining the dry weight of organisms, or indirectly, by an opacity method that has been calibrated in relation to the dry weight of organisms.
It is desirable that one method of estimation should be adhered to for all the vaccine lots produced by a manufacturer.
An International Reference Preparation of opacity is available for the determination of the opacity of a suspension of BCG organisms. The comparison is made by EYE, and the test is described in GEN-7.

BLG/UNDP/82.1 Page 42
VACCINE: BCG
CLASS OF TEST B
BCG-5
TEST: Test for number of culturable particles
REQUIREMENTS FOR DRIED BCG VACCINES (Requirements for Biological Substances No. 11)
(Revised 1978)
Part A Section 5.5
THE TEST
The number of culturable particles of each final lot shall be determined by an appropriate method approved by the national control authority (See Part A section 3.4.5) The viable count shall have a value within a range approved by the national control authority. By comparison with the results of the test for number of culturable particles carried out on the final bulk, as described in Part A section 3.4.5, the percentage survival on freeze-drying may be calculated and this value should be not less than one approved by the national control authority.
Part A Section 3.4.5
The survival rate usually varies between 75% and 20% although a survival rate as low as 10% is permitted by some national control authorities.
The number of culturable particles of each final bulk shall be determined by an appropriate method approved

by the national control authority.
BLG/UNDP/82.1 Page 43
The medium used in this test should be such that the number of culturable particles may be determined not more than 4 weeks after the medium has been inoculated with dilutions of vaccine.
There are various methods of determining the number of culturable particles in BCG vaccine, and it is desirable that one method should be adhered to for all the vaccine lots produced by a manufacturer (7). It is also desirable that the test should be carried out in parallel with a reference preparation.
In the case of retesting a vaccine, which has been tested before issue by the producer, the test that will give the most essential information, is the Test for Culturable Particles. The total bacterial content, however, is not important, as this will not change. As the test for contamination has been done by the producer, the ampoules should not be contaminated. Such tests will be done, therefore, only if there are special indications for retesting the vaccine.
The importance of checking the number of culturable particles is that it may have changed during transport from the producer, or during the vaccination
e.g. by a failure of the cold chain. Moreover, such information may be useful where vaccines have been stored under correct conditions, but for a long time during which the organisms may have died. This is a good check on the stability of the vaccine.

BLG/UNDP/82.1 Page 44
ADDITIONAL INFO~~TION FOR CARRYING OUT THE TEST
A. In the document TB/77.8 (available from WHO), it is stated that:
The estimation of the number of culturable particles is the most informative viability test, and the method developed is widely used, not only in quality control laboratories but also in most production centres. Traditionally this test was carried out by inoculating on solid medium tenfold different dilutions (of the already highly diluted vaccine), but for the last decade a series of three twofold different dilutions (1:2:4) are chosen (sometimes after a preliminary test) in such a way that estimates are always based on an optimal number of colonies. The use of several containers (tubes, dishes) for each dilution level makes it possible to compute routinely the chi-squares(thePoisson-distribution giving the expected variance). The use of three dilution levels makes it possible to compute "t" values as an indication of the dilution errors, whether systematic or stochastic.
The calculation of these statistics as well as the calculation of the final estimate of the number of culturable particles must be carried out for each sample examined. The computations for each examination, as well as the copy-typing of the report, is therefore a major task that requires utmost accuracy. Since standard procedures are used, however, the data are nowadays easily processed electronically.
Solid media sometimes vary in quality, and this could cause serious errors in comparisons of vaccines. Also, incubation might affect the different containers in an investigation in different ways. The containers of solid media used in ay investigation are therefore randomized and coded. Also in this respect the use of the computer is profitable: the sheets of random permutations according to which the prenumbered containers are selected are easily prepared by computer. Moreover no decoding is needed (which excludes bias): thF

BLG/UNDP/82.1 Page 45
counts for each container number are punched and the computer decodes before it compute.s. and prints the results.
Thus all the painstaking arithmetic, which for many laboratory workers made the determination of the number of culturable particles a relatively "complicated" test, is now done by the computer, which moreover presents the final result directly in printed form.
B. Document TB/77.9 (available from WHO) gives the following data about the materials and design of the test:
Materials
No standard diluent, container, or solid medium is specified here. Tools or recipes that have come to work well in one laboratory (after much trial and error) do not necessarily work if copied elsewhere; it would scarcely be possible, for example, to prescribe a particular medium for viability counts as the standard to be used in all laboratories.
The basic suspension should normally be vaccine ready for use in the standard strength recommended by the manufacturer. Freeze-dried products should be reconstituted as recommended by the manufacturer. The sample to be tested may be an ampoule of liquid or reconstituted vaccine or a pool of two or more ampoules (to even out possible ampoule-to-ampoule differences), or it may be a sample of the stock suspension not yet in ampoules (either in original concentration or diluted down to normal vaccine strength). Further dilution is in terms of volume, i.e. in terms of millilitre of vaccine and not in terms of milligramme semi-dry weight (though a note is made of the nominal strength, as specified by the manufacturer, in terms of mg per ml for the liquid or reconstituted vaccine).

BLG/UNDP/82.1 Page 46
Commonly used media for diluting the reconstituted vaccine are: liquid Sauton medium1 (as used for BCG cultures) diluted 1:4 (i.e. 1 + 3); isotonic saline with or without buffer. A wetting agent (e.g. Tween 80) or bovine albumin is sometimes added to the diluent.
Reference is made to descriptions in t£e litera2ure of the Lowenstein-Jensen medium, theogawa medium3 and the Oleic-acid-albumin-agar medium with blood, which are the most commonly used solid media. A variety of containers are used for the solid medium, such as cotton stoppered glass tubes sealed with paraffin, Legroux flasks, Petri dishes, screw-capped tubes, or flat screw-capped bottles. Major considerations in selecting such a container are the control of ventilation, the avoidance of contamination, and the size of, the surface area. Only solid or semi-solid media should be used, as these allow the number of colonies to be counted. Liquid media, which yield only "growth" or "no growth" for each tube, require the use of a lar.ge number of tubes for reasonably accurate estimates.+ and are therefore not recommended for routine use.
1
2
3
4
Jensen, K.A. Bulletin of the International Union against Tuberculosis, 24: 78-112 (1954)
Obayashi, Y. Dried BCG Vaccine, World Health Organ. Geneva (1955)
Dubos, R.J. & Middlebrook, G.,American review of tuberculosis and pulmonary disease, 56: 334-345(1947)
For statistical handling of such data, see Fisher, R.A. & Yates, F. (1957) Statistical tables for biological, agricultural and medical research 5th ed., Edinburgh, Table VIII2

Design
BLG/UNDP/82.1 Page 47
If the viability is roughly known in advance, a dilution level can be chosen that is likrly to yield a number of colonies optimal for counting. In anticipation of variations in viability, three dilution levels are seeded at the same time, in dilution steps of 1:2:4. The advantage of twofold dilutions (as compared with fourfold or tenfold) is that the validity of the levels will overlap (with a tenfold series, one level may give confluence and the next one too few colonies). Furthermore, two or even all three dilution levels will contribute significantly to the estimate for as long as they show neither confluent nor zero growth. Examples of possible steps in the dilution process are shown in Appendix 1, Sheet 3.
In selecting the dilution levels, it is necessary first to define an optimal number of colonies per container, i.e., a number not too small yet small enough to assure discrete, easily counted colonies. One aims at obtaining this number at the intermediate dilution level, so that for moderate variations (within the range 1:4) one of the three levels will always yield an adequate number of colonies. In the statistical analysis full weight is given to colonies for dilution levels that yield, on the average, the optimum number or less, while reduced weight is given to a dilution level that yields up to twice the optimum, and little or no weight to levels yielding more than twice the optimum. Colonies are nevertheless counted up to 2.5 times the optimum, beyond which counts are reported as "exceeding limit". As will be seen later. tpe
1 If the viability is not known at all, a preliminary viability test must be carried out. The design might be in tenfold steps (1:10:100) or in more than three twofold steps, e.g. 1:2:4:8:16:32:64:128. In the latter case a single container of solid medium per dilution level may suffice, since only a preliminary estimate is aimed at.

BLG/UNDP/82.1 Page 48
statistical interpretation of the latter class is quite different from that of "missing data", e.g. in case of contaminated containers.
For the most concentrated suspension and for the middle suspension the same number of containers is inoculated, e.g. five for each. For the most diluted suspension twice the number of containers is inoculated, in this case 10. Or the respective number of containers may be three, three, and six. It may be noted that in this design the amount of vaccine inoculated at the most concentrated level is the same as the sum of the amounts inoculated at the more dilute levels (1 = 0.5 + 2 x 0.25). Similarly, the amounts inoculated at the two more dilute levels are equal (0.5 = 2 x 0.25). Thus the number of colonies counted will be at least 10 times (or six times respectively) the optimum count for a single container, on the condition that the count for the most concentrated level is not below the optimum and for the most dilute level not above the optimum. Thus the precision will be fairly uniform over this range.
From one ampoule, or pool of ampoules, may be prepared either one dilution series, to be inoculated in 5 + 5 + 10 containers, or two dilution series each to be inoculated in 3 + 3 + 6 containers. The purpose of the latter design is to checf the experimental error inherent in the dilution process.
1 The same design cannot immediately be used for checking the ampoule-to-ampoule variation (by preparing one dilution series from each of two different ampoules) because the estimate of experimental error in this case would confound ampoule-to-ampoule variation with dilution error. A proper check on ampoule-to-ampoule variation requires four dilution series as a basic design, two for each ampoule.

BLG/UNDP/82.1 Page 49
C. Document TB/77.9 (available from WHO), gives a randomization, the reporting and the statistical analysis. If it is planned to perform this test it is essential to study the complete documentrB /77.9, which is available from the Tuberculosis Unit of the WHO, Geneva.

BLG/UNDP/82.1 Page 50
CLASS OF TEST B
BCG-6
VACCINE: BCG
TEST: Test for Stability
REQUIREMENTS FOR DRIED BCG VACCINE (Requirements for Biological Substances No. 11)
(Revised 1978)
Part A Section 5.6
STABILITY TEST
Each final lot shall be tested for its degree of stability by a method 1approved by the national control authority.
The object of this test is to obtain experimental evidence on which to base the statements concerning storage temperature and expiry dates that appear on the label and the leaflet as required in Part A, Section 8.
The test shall involve the determination of the number of culturable particles before and after the samples have been held at appropriate temperatures and for appropriate periods.
An accelerated degradation test shall be carried out by_taking samples of the vaccine and incubating them at
1 Part A, Section 3.4.5 concerns the determination of the number of culturable particles and is discussed in detail in doc. "BCG-5".

BLG/UNDP/82.1 Page 51
-u 37 C for 28 days. The percentage fall in the number of culturable particles is Shen compared with that of the same vaccine stored at 4 C. The number of culturable particles in the vaccine after heating shall be not less than 20% of that in the refrigerated vaccine. The absolute value shall be approved by the national control laboratory. The test shall also be made in parallel with a reference preparation. One method of determining the number of culturable particles shall be adhered to, as suggested in Part A, Section 3.4.5.
The purpose of including a reference preparation is to have a check on the quality of the medium used for the determination of the number of culturable particles. It is not intended to adjust the count of the vaccine by comparison with the reference preparation.
All manufacturers shall keep their product for the permitted storage period and shall determine the number of culturable particles to demonstrate that this has been maintained at an adequate level.
In some countries, the stability test is carried out only after the vaccine has been stored for 3-4 weeks after drying, since it is considered that the degree of stability during the first 3 weeks may not be related to the longterm stability of the product.
As a guide to stability, some manufacturers of dried BCG vaccine determine the residual water content of the final vaccine, since failure to achieve a certain degree of desiccation results in an

BLG/UNDP/82.1 Page 52
unstable product. However, such a test cannot be regarded as an alternative to tests involving the determination of the number of culturable particles

BCG VACCINE

VACCINE: BCG
TEST: Test for Identity
BLG/UNDP/82.1 Page 33
CLASS OF TEST B
BCG-1
REQUIREMENTS FOR DRIED BCG VACCINE (Requirements for Biological Substances No. 11)
(Revised 1978)
Part A Section 5.1
THE TEST
Identity Test
An identity test shall be performed on samples of vaccine from each final lot.
The identity of each filling lot of vaccine shall be verified by the morphological apperance of the bacilli in stained smears and by the characteristic appearance of the colonies grown on solid media.
The medium used is generally the Lowenstein-Jensen or a similar medium. The smears are stained by the Ziehl- Neelsen method.
Lowenstein-Jensen Medium
Lowenstein's medium with 0.75% Glycerin (usually called Lowenstein-Jensen medium) (Zentralblatt fur Bakteriologie, Parasitenkunde u. Infektionskrankheiten I. Abt. Orig. 125, 222, 932 and Advances in the Control of Zoonoses, p. 11 and 35, World Health Organization, Geneva 1953).

BLG/UNDP/82.1 Page 34
Lowenstein-Jensen medium Cont ...
For the culture medium, the following mixture is made up:
Monopotassium phosphate Magnesium sulfate Magnesium citrate 1-Asparagin Glycerine (twice distilled)
0.4% 0.04% 0.1% 0.6% 2.0%
A flask containing 100 ml of this solution is boiled for 2 hours; after cooling, 30g of potato flour are added and the mixture is then boiled in a water-bath with continual and thorough stirring to complete pastiness. The mixture is then cooled to about 56°C before the eggs are added. 20-22 fresh eggs with large yellow yolks are cleaned carefully, broken into a large flask, shaken vigorously and filtered through sterile gauze. One litre of egg and 600 ml of the salt-potato flour solution are mixed; after shaking 20 ml of 2% aqueous malachite-green solution is added. The medium is tubed, slanted, and coagulated at 85°C for 40 minutes (Fig. 2). The following day the tubes are heated to 70°C for 30 minutes. The cotton plugs are then parafined or closed in another way, and stored in a refrigerator at 4°C.
(From: Jensen, Bulletin International Union against Tuberculosis 24: 102-103)
Ziehl-Neelsen Method:
Concentrated Alcoholic Solution of basic Fuchsin
Basic fuchsin (certified) 95 per cent ethyl alcohol
90 g 945 ml
1. Add 300 ml alcohol to the 90 g fuchsin. 2. Heat in water bath to boiling for 2 minutes. 3. Allow to stand 1 minute and decant off; save
the supernatant.

BLG/UNDP/82.1 Page 35
4. Add a second 300 ml portion of the alcohol to the sediment, boil and decant as before.
5. Add the remaining 345 ml alcohol to the sediment, boil as before and add to portions collected previously.
5 per cent Phenol:
Distilled water Phenol crystals
950 ml 50 g
Carbol Fuchsin Solution for Staining Acid-fast Bacilli:
Concentrated alcoholic solution of basic fuchsin
5 per cent phenol
Filter stain just before using to remove
105 ml
895 ml
any acid-fast organisms which may be present.
Acid Alcohol for Decolorizing:
Hydrochloric acid 95 per cent ethyl alcohol Mix thoroughly
Staining Procedure
3 ml 97 ml
It is difficult for stains to penetrate cells of the Mycobacteria because of their high lipoid content. Only after prolonged treatment with carbol fuchsin, one of the most penetrating stains, or the application of heat during the staining process, can these bacteria be coloured. Once stained, however, the waxy material of the cells holds the carbol fuchsin even in the presence of acid alcohol. The steps of the Ziehl-Neelsen method which demonstrate the

BLG/UNDP/82.1 Page 36
property of acid-fastness are:
1. Smear out material (cultures or specimens) dry and fix as usual.
2. Cover smear with carbol fuchsin and steam over a low flame or a water bath for 5 minutes. The stain should never boil nor should it be allowed to dry.
3. Let slide cool a minute. Wash thoroughly in water.
4. Decolorize in acid alcohol (95 ml of 95 per cent ethyl alcohol plus 5 ml concentrated HCl) for 5 minutes or until no more color leaves the smear.
5. Wash in water. 6. Counterstain with methylene blue for 30 seconds. 7. Wash, dry and examine with oil immersion objective.
By this method acid-fast bacteria appear red against a blue background.
(From: Kelly and Rite, Microbiology, ed. AppletonCentury-Crofts, New York)

VACCINE: BCG
TEST: Test for absence of contaminating micro-organisms
BLG/UNDP/82.1 Page 37
CLASS OF TEST B
BCG-2
REQUIREMENTS FOR DRIED BCG VACCINE (Requirements for Biological Substances No. 11)
(Revised 1978)
Part A Section 5.2
THE TEST
Samples from each final lot shall be tested for bacterial and mycotic contamination according to the requirements given in Part A, Section 5, of Requirements for Biological Substances No. 6 (Requirements for the Sterility of Biological Substances) (11, page 4).
For details of this test see GEN-1.

BLG/UNDP/82.1 Page 38
VACCINE: BCG
CLASS OF TEST C
BCG-3
TEST: Test for absence of virulent Mycobacteria
REQUIREMENTS FOR DRIED BCG VACCINE (Requirements for Biological Substances No. 11)
(Revised 1978)
Part a Section 5.3.1
THE TEST
"If the test for the absence of virulent mycobacteria applied to the final bulk, is unsatisfactory, it shall be repeated with a sample of a final lot (see Part A, section 3.4.3).
This test may be applied to the final lot and omitted from the final bulk".
Part A Section 3.4.3
A sample of the final bulk intended for this test shall be stored for not more than 72 hours after harvest at 4°C.
At least 6 guinea-pigs, all of the same sex, each weighing 250-350 g are used.
Some countries prefer to use guinea-pigs that have been shown previously to be tuberculinnegative.

BLG/UNDP/82.1 Page 39
A dose of BCG organisms corresponding to at least 50 human doses of vaccine intended for intradermal injection shall be injected into ealh guinea-pig by the subcutaneous or intramuscular route • The animals shall have been maintained on a diet that is free from added substances, such as antibiotics, that might interfere with the test. The guinea-pigs shall be observed for at least 6 weeks. If, during that time, they remain healthy and gain weight, the final bulk shall be considered to be free from virulent mycobacteria.
At the end of the observation period, the animals shall be killed and examined post mortem for macroscopic evidence of progressive tuberculous disease; similarly, any animals that die before the end of the observation period shall be subjected to post mortem examination.
The vaccine lot shall pass the test if none of the guinea-pigs shows evidence of progressive tuberculous disease and if at least two-thirds of them survive the observation period.
Should more than one-third of the guinea-pigs die during the observation period (and freedom from progressive tuberculous disease is verified), the test shall be repeated with another sample of the final lot on at least 6 more guinea-pigs •
If, on the second occasion, more than one-third of the animals fail to survive the observation period, the vaccine lot shall not pass the test.
1 When a more concentrated vaccine, intended for administration by the percutaneous route, is tested, a dilution factor approved by the national control authority shall be applied, so that the mass of BCG injected corresponds to 50 human doses of intradermal vaccine.

BLG/UNDP/82.1 Page 40
Should a vaccine lot fail to satisfy the requirements of this test because animals die from causes other than tuberculosis, the procedure to be followed by the manufacturer should be determined with the approval of the national control authority.
If evidence of progressive tuberculous disease is seen, the vaccine lot shall be rejected, all subsequent vaccine lots shall be withheld, and all current vaccine stocks shall be held pending further investigation. The manufacture of BCG vaccine shall be discontinued and it shall not be resumed until a thorough investigation has been made and the cause or causes of the failure determined, and then only with the approval of the national control authority.

VACCINE: BCG
TEST: Test for total bacterial content
BLG/UNDP/82.1 Page 41
CLASS OF TEST A
BCG-4
REQUIREMENTS FOR DRIED BCG VACCINES (Requirements for B-iological Substances No. 11)
(Revised 1978)
Part A Section 5.4
THE TEST
The total bacterial content of the reconstituted vaccine shall be estimated for each vaccine lot by a method approved by the national control authority, and shall have a value within a range approved by the national control authority.
The estimation of total bacterial content may be made either directly by determining the dry weight of organisms, or indirectly, by an opacity method that has been calibrated in relation to the dry weight of organisms.
It is desirable that one method of estimation should be adhered to for all the vaccine lots produced by a manufacturer.
An International Reference Preparation of opacity is available for the determination of the opacity of a suspension of BCG organisms. The comparison is made by EYE, and the test is described in GEN-7.

BLG/UNDP/82.1 Page 42
VACCINE: BCG
CLASS OF TEST B
BCG-5
TEST: Test for number of culturable particles
REQUIREMENTS FOR DRIED BCG VACCINES (Requirements for Biological Substances No. 11)
(Revised 1978)
Part A Section 5.5
THE TEST
The number of culturable particles of each final lot shall be determined by an appropriate method approved by the national control authority (See Part A section 3.4.5) The viable count shall have a value within a range approved by the national control authority. By comparison with the results of the test for number of culturable particles carried out on the final bulk, as described in Part A section 3.4.5, the percentage survival on freeze-drying may be calculated and this value should be not less than one approved by the national control authority.
Part A Section 3.4.5
The survival rate usually varies between 75% and 20% although a survival rate as low as 10% is permitted by some national control authorities.
The number of culturable particles of each final bulk shall be determined by an appropriate method approved

by the national control authority.
BLG/UNDP/82.1 Page 43
The medium used in this test should be such that the number of culturable particles may be determined not more than 4 weeks after the medium has been inoculated with dilutions of vaccine.
There are various methods of determining the number of culturable particles in BCG vaccine, and it is desirable that one method should be adhered to for all the vaccine lots produced by a manufacturer (7). It is also desirable that the test should be carried out in parallel with a reference preparation.
In the case of retesting a vaccine, which has been tested before issue by the producer, the test that will give the most essential information, is the Test for Culturable Particles. The total bacterial content, however, is not important, as this will not change. As the test for contamination has been done by the producer, the ampoules should not be contaminated. Such tests will be done, therefore, only if there are special indications for retesting the vaccine.
The importance of checking the number of culturable particles is that it may have changed during transport from the producer, or during the vaccination
e.g. by a failure of the cold chain. Moreover, such information may be useful where vaccines have been stored under correct conditions, but for a long time during which the organisms may have died. This is a good check on the stability of the vaccine.

BLG/UNDP/82.1 Page 44
ADDITIONAL INFO~~TION FOR CARRYING OUT THE TEST
A. In the document TB/77.8 (available from WHO), it is stated that:
The estimation of the number of culturable particles is the most informative viability test, and the method developed is widely used, not only in quality control laboratories but also in most production centres. Traditionally this test was carried out by inoculating on solid medium tenfold different dilutions (of the already highly diluted vaccine), but for the last decade a series of three twofold different dilutions (1:2:4) are chosen (sometimes after a preliminary test) in such a way that estimates are always based on an optimal number of colonies. The use of several containers (tubes, dishes) for each dilution level makes it possible to compute routinely the chi-squares(thePoisson-distribution giving the expected variance). The use of three dilution levels makes it possible to compute "t" values as an indication of the dilution errors, whether systematic or stochastic.
The calculation of these statistics as well as the calculation of the final estimate of the number of culturable particles must be carried out for each sample examined. The computations for each examination, as well as the copy-typing of the report, is therefore a major task that requires utmost accuracy. Since standard procedures are used, however, the data are nowadays easily processed electronically.
Solid media sometimes vary in quality, and this could cause serious errors in comparisons of vaccines. Also, incubation might affect the different containers in an investigation in different ways. The containers of solid media used in ay investigation are therefore randomized and coded. Also in this respect the use of the computer is profitable: the sheets of random permutations according to which the prenumbered containers are selected are easily prepared by computer. Moreover no decoding is needed (which excludes bias): thF

BLG/UNDP/82.1 Page 45
counts for each container number are punched and the computer decodes before it compute.s. and prints the results.
Thus all the painstaking arithmetic, which for many laboratory workers made the determination of the number of culturable particles a relatively "complicated" test, is now done by the computer, which moreover presents the final result directly in printed form.
B. Document TB/77.9 (available from WHO) gives the following data about the materials and design of the test:
Materials
No standard diluent, container, or solid medium is specified here. Tools or recipes that have come to work well in one laboratory (after much trial and error) do not necessarily work if copied elsewhere; it would scarcely be possible, for example, to prescribe a particular medium for viability counts as the standard to be used in all laboratories.
The basic suspension should normally be vaccine ready for use in the standard strength recommended by the manufacturer. Freeze-dried products should be reconstituted as recommended by the manufacturer. The sample to be tested may be an ampoule of liquid or reconstituted vaccine or a pool of two or more ampoules (to even out possible ampoule-to-ampoule differences), or it may be a sample of the stock suspension not yet in ampoules (either in original concentration or diluted down to normal vaccine strength). Further dilution is in terms of volume, i.e. in terms of millilitre of vaccine and not in terms of milligramme semi-dry weight (though a note is made of the nominal strength, as specified by the manufacturer, in terms of mg per ml for the liquid or reconstituted vaccine).

BLG/UNDP/82.1 Page 46
Commonly used media for diluting the reconstituted vaccine are: liquid Sauton medium1 (as used for BCG cultures) diluted 1:4 (i.e. 1 + 3); isotonic saline with or without buffer. A wetting agent (e.g. Tween 80) or bovine albumin is sometimes added to the diluent.
Reference is made to descriptions in t£e litera2ure of the Lowenstein-Jensen medium, theogawa medium3 and the Oleic-acid-albumin-agar medium with blood, which are the most commonly used solid media. A variety of containers are used for the solid medium, such as cotton stoppered glass tubes sealed with paraffin, Legroux flasks, Petri dishes, screw-capped tubes, or flat screw-capped bottles. Major considerations in selecting such a container are the control of ventilation, the avoidance of contamination, and the size of, the surface area. Only solid or semi-solid media should be used, as these allow the number of colonies to be counted. Liquid media, which yield only "growth" or "no growth" for each tube, require the use of a lar.ge number of tubes for reasonably accurate estimates.+ and are therefore not recommended for routine use.
1
2
3
4
Jensen, K.A. Bulletin of the International Union against Tuberculosis, 24: 78-112 (1954)
Obayashi, Y. Dried BCG Vaccine, World Health Organ. Geneva (1955)
Dubos, R.J. & Middlebrook, G.,American review of tuberculosis and pulmonary disease, 56: 334-345(1947)
For statistical handling of such data, see Fisher, R.A. & Yates, F. (1957) Statistical tables for biological, agricultural and medical research 5th ed., Edinburgh, Table VIII2

Design
BLG/UNDP/82.1 Page 47
If the viability is roughly known in advance, a dilution level can be chosen that is likrly to yield a number of colonies optimal for counting. In anticipation of variations in viability, three dilution levels are seeded at the same time, in dilution steps of 1:2:4. The advantage of twofold dilutions (as compared with fourfold or tenfold) is that the validity of the levels will overlap (with a tenfold series, one level may give confluence and the next one too few colonies). Furthermore, two or even all three dilution levels will contribute significantly to the estimate for as long as they show neither confluent nor zero growth. Examples of possible steps in the dilution process are shown in Appendix 1, Sheet 3.
In selecting the dilution levels, it is necessary first to define an optimal number of colonies per container, i.e., a number not too small yet small enough to assure discrete, easily counted colonies. One aims at obtaining this number at the intermediate dilution level, so that for moderate variations (within the range 1:4) one of the three levels will always yield an adequate number of colonies. In the statistical analysis full weight is given to colonies for dilution levels that yield, on the average, the optimum number or less, while reduced weight is given to a dilution level that yields up to twice the optimum, and little or no weight to levels yielding more than twice the optimum. Colonies are nevertheless counted up to 2.5 times the optimum, beyond which counts are reported as "exceeding limit". As will be seen later. tpe
1 If the viability is not known at all, a preliminary viability test must be carried out. The design might be in tenfold steps (1:10:100) or in more than three twofold steps, e.g. 1:2:4:8:16:32:64:128. In the latter case a single container of solid medium per dilution level may suffice, since only a preliminary estimate is aimed at.

BLG/UNDP/82.1 Page 48
statistical interpretation of the latter class is quite different from that of "missing data", e.g. in case of contaminated containers.
For the most concentrated suspension and for the middle suspension the same number of containers is inoculated, e.g. five for each. For the most diluted suspension twice the number of containers is inoculated, in this case 10. Or the respective number of containers may be three, three, and six. It may be noted that in this design the amount of vaccine inoculated at the most concentrated level is the same as the sum of the amounts inoculated at the more dilute levels (1 = 0.5 + 2 x 0.25). Similarly, the amounts inoculated at the two more dilute levels are equal (0.5 = 2 x 0.25). Thus the number of colonies counted will be at least 10 times (or six times respectively) the optimum count for a single container, on the condition that the count for the most concentrated level is not below the optimum and for the most dilute level not above the optimum. Thus the precision will be fairly uniform over this range.
From one ampoule, or pool of ampoules, may be prepared either one dilution series, to be inoculated in 5 + 5 + 10 containers, or two dilution series each to be inoculated in 3 + 3 + 6 containers. The purpose of the latter design is to checf the experimental error inherent in the dilution process.
1 The same design cannot immediately be used for checking the ampoule-to-ampoule variation (by preparing one dilution series from each of two different ampoules) because the estimate of experimental error in this case would confound ampoule-to-ampoule variation with dilution error. A proper check on ampoule-to-ampoule variation requires four dilution series as a basic design, two for each ampoule.

BLG/UNDP/82.1 Page 49
C. Document TB/77.9 (available from WHO), gives a randomization, the reporting and the statistical analysis. If it is planned to perform this test it is essential to study the complete documentrB /77.9, which is available from the Tuberculosis Unit of the WHO, Geneva.

BLG/UNDP/82.1 Page 50
CLASS OF TEST B
BCG-6
VACCINE: BCG
TEST: Test for Stability
REQUIREMENTS FOR DRIED BCG VACCINE (Requirements for Biological Substances No. 11)
(Revised 1978)
Part A Section 5.6
STABILITY TEST
Each final lot shall be tested for its degree of stability by a method 1approved by the national control authority.
The object of this test is to obtain experimental evidence on which to base the statements concerning storage temperature and expiry dates that appear on the label and the leaflet as required in Part A, Section 8.
The test shall involve the determination of the number of culturable particles before and after the samples have been held at appropriate temperatures and for appropriate periods.
An accelerated degradation test shall be carried out by_taking samples of the vaccine and incubating them at
1 Part A, Section 3.4.5 concerns the determination of the number of culturable particles and is discussed in detail in doc. "BCG-5".

BLG/UNDP/82.1 Page 51
-u 37 C for 28 days. The percentage fall in the number of culturable particles is Shen compared with that of the same vaccine stored at 4 C. The number of culturable particles in the vaccine after heating shall be not less than 20% of that in the refrigerated vaccine. The absolute value shall be approved by the national control laboratory. The test shall also be made in parallel with a reference preparation. One method of determining the number of culturable particles shall be adhered to, as suggested in Part A, Section 3.4.5.
The purpose of including a reference preparation is to have a check on the quality of the medium used for the determination of the number of culturable particles. It is not intended to adjust the count of the vaccine by comparison with the reference preparation.
All manufacturers shall keep their product for the permitted storage period and shall determine the number of culturable particles to demonstrate that this has been maintained at an adequate level.
In some countries, the stability test is carried out only after the vaccine has been stored for 3-4 weeks after drying, since it is considered that the degree of stability during the first 3 weeks may not be related to the longterm stability of the product.
As a guide to stability, some manufacturers of dried BCG vaccine determine the residual water content of the final vaccine, since failure to achieve a certain degree of desiccation results in an

BLG/UNDP/82.1 Page 52
unstable product. However, such a test cannot be regarded as an alternative to tests involving the determination of the number of culturable particles

DIPHTHERIA VACCINE

BLG/UNDP/82.1 Page 53
CLASS OF TEST C
DIP-1
VACCINE: Diphtheria Toxoid
TEST: Test for Potency
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE TETANUS TOXOID ANC COMBINED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A, Section 5.3
The control procedures suggested in the manual follow them closely. Although the method of potency control suggested in the manual may not be identical with that used i.n many countries, the quality of the final toxoid will satisfy the requirements of all countries.
Glossary of Terms
Kf
Lf
Lf/mg N
Lf/mg PN
Flocculation time (in minutes) as observed in the flocculation reaction.
Limes flocculationis; the amount of toxin or toxoid which when mixed with 1 International Unit of antitoxin gives a Ramon flocculation in the shortest time.
Flocculation units per milligram of total nitrogen as determined by the Kjeldahl analysis.
Flocculation units per milligram of protein nitrogen (PN) which is usually determined by the precipitation of the proteins with trichloroacetic acid.

BLG/UNDP/82.1 Page 54
I.U
L+
L+/10
Lr
M.L.D.
M.T.V.
A.B.V.
T.C.P.
International Unit is the specific activity of a stated amount of the International Standard as defined by the WHO Expert Committee on Biological Standardization.
The minimum amount of toxin which when combined with 1 I.U. of antitoxin kills an animal of a defined weight in four days (the L+ is dependent upon the animal species used).
The minimum amount of toxin which when combined with 0.1 I.U. of antitoxin kills an animal of a defined weight in four days.
The minimum amount of toxin which when combined with a fixed amount of antitoxin (usually 0.002 I.U. of antitoxin) in a volume of 0.2 ml causes a local skin reaction that is just visible.
The amount of toxin that kills 50% of a group of animals within four days (the LD differs for different animal species).
Minimal lethal dose, the amount of toxin which. kills animals within four days (M.L.D. is different for different animal species). In general the M.L.D. has been replaced by the
LD5o'
Maximum toxin value, the quantity of antitoxin that can bind toxin; this is determined by the neutralization of the toxic effect in animals of toxin-antitoxin mixtures.
Antitoxin binding value, a value which defines the toxin plus toxoid in a mixture (determined in animals).
Total combining power, the reciprocal of the volume of toxoid which when diluted is found to be equivalent to 1 International Unit.
The dose of a vaccine which protects 50% of the immunized animals against a challenge dose of virulent bacteria or toxin.

BLG/UNDP/82.1 Page 55
POTENCY TESTING OF DIPHTHERIA TOXOID
In process testing
A full potency test of diphtheria toxoid requires rather a large number of guinea-pigs and in many countries it will be important to economize as far as laboratory animals are concerned. For the final bulk of a vaccine it is necessary to perform a full test as described below but economy in guinea-pigs is possible by either producing large batches of vaccine or by combining the final potency test for a number of batches.
For the potency tests, required as "in process" tests, a smaller number of guinea-pigs may be used which were just sufficient to give a general indication that the material has at least a reasonable potency. Bulk fluid toxoids could be screened using a few guinea-pigs by a method such as that used in the United States of America: 10 guinea-pigs of 250-300 g are injected with not more than one-sixth of the total human immunizing dose and not more than six weeks later the animals are challenged with 10 lethal challenge doses of diphtheria toxin. At least 80% of the guinea-pigs should survive.
A reasonable acceptance level for an adsorbed diptheria vaccine is that the vaccine should contain about 30 Ill/single human dose. Therefore·, as a rough guide it should be satisfactory to show that a vaccine is not less potent than a reference material of such a potency level. This could be checked by injecting a group of 10 guinea-pigs - as required by WHO - with the vaccine and a similar group of 10 guinea-pigs with the reference material followed four weeks later by the titration of the number of antitoxin units in the blood of both groups of guinea-pigs. If the mean titre (preferably geometric mean) for the vaccine is not lower than the one for the standard, one can expect that the vaccine will be satisfactory in the field. Another possibility would be to inject both groups of guinea-pigs with a suitably chosen dilution of vaccine and standard, followed by toxin challenge after four weeks. The survival percentage of the pigs for the vaccine under test should be not lower than that for the standard vaccine.

BLG/UNDP/82.1 Page 56
It is left to the National Control Authority to decide in which way the potency shall be measured and to define a minimum level of acceptancy.
Potency tests
The final bulk vaccine or the vaccine in its final container must be tested for immunizing potency. Although the WHO Requirements do not specify any particular method it is essential that the potency is measured against a national standard which has been calibrated against the International Standard for Diphtheria Toxoid.
The inclusion of a reference preparation is essential because the antitoxin response of guinea-pigs to a diphtheria toxoid may be affected markedly by diet, conditions of maintenance, strain of guinea-pigs and such variab]Psmake it difficult to identify real differences between different toxoids.
Tests based upon the use of a standard preparation generally involve vaccination of animals with multiple dilutions of the test and reference vaccines, and comparison of the log dose response curves of the preparation. If the response curves are both reasonably linear and do not differ significantly in slope (b value) the difference in log dose required to achieve a given response can be calculated to give an estimate of the relative potency of the test preparation, i.e. the ratio of the volume of the standard preparation required to elicit a given response over the value of the test preparation required to elicit the same response. Since the potency of the national standard will have been calibrated in International Units it is possible, therefore, to assign a potency of the test preparation also in International Units. As a guide it is considered satisfactory if a diphtheria toxoid has a potency of 30 IU/single human dose or more.

BLG/UNDP/82.1 Page 57
Suggested method of assay for adsorbed vaccine
Prepare three dilutions in saline of the national reference preparation of diphtheria vaccine (adsorbed) and three similar dilutions of each of the toxoids or vaccines under test. The choice of dilutions will depend upon the strain of guinea-pigs and the condition of the animals. Serial dilutions of each preparation shouldconform to a two-fold range, e.g. 1/20, 1/40, 1/80. The particular dilutions used are selected in the light of experience of local conditions such that the median dose for each preparation tested may be expected to produce a mean intradermal challenge score of approximately 2 when an intradermal challenge method is used or to protect approximately half the animals when a subcutaneous lethal challenge dose is used.
Inoculate 1 ml of the diluted vaccine subcutaneously into each group of guinea-pigs, which should include not less than eight animals for the intradermal challenge test and not less than 16 for the subcutaneous lethal challenge. Five uninoculated guinea-pigs are used as controls.
It is advisable to distribute the guinea-pigs of each treatment group randomly through several cages since the level of response can be affected by local conditions within different cages. When randomization is carried out it is necessary to identify each animal individually. A scheme for the identification of individual animals by dyemarking is shown at the end of this appendix.
The responsesafthe guinea-pigs to the single dose of vaccine are determined 28 days after inoculation.

BLG/UNDP/82.1 Page 58
1. By the multiple intradermal challenge test
Shave the depiliatethe flayks of all the guinea-pigs. Prepare a dilution of a suitable toxin in borate buffered saline containing 0.0512 Lf units of toxin per 0.2 ml. Prepare from this a further series of three eight-fold dilutions to contain respectively 0.0064, 0.0008 and 0.0001 Lf units in 0.2 ml. Inject four dilutions intradermally into four separate sites on each of the vaccinated guinea-pigs in such a way as to minimize interference between adjacent sites. It is suggested that the injection sites should be placed 2.5 em from the spine and that adjacent sites should be at least 5 em apart in the following pattern.
0.0064 Lf X
0.08 Lf X
s p
I
N
E
Head of guinea-pig
X 0.0001 Lf
X 0.0512 Lf
Inject the unvaccinated control animals with dilutions containing 100, 50,25, 12.5 and 6.25 millionths of an Lf unit of the challenge toxin in borate buffered saline.
1 A suitable toxin contains 0.67-1.33 Lr in 1 Lf unit and 1 Lf unit contains between 25 000 and 50 000 minimal reacting doses for guinea-pig skin. It is preferable for the toxin to be unpurified and to have been stored for a considerable time before use.

BLG/UNDP/82.1 Page 59
Examine all injection sites 48 hours after intradermal injection and record the incidence of specific diphtheria erythema. Record also the number of sites free from such reactions as the intradermal challenge score. Tabulate together the intradermal challenge scores for all the animals receiving each dilution of vaccine and use those data without further transformation to obtain an estimate of the relative potency for each of the test preparations by parallel line quantitative analysis (Finney ).
An assay is acceptable provided that {i) the dose response regression of the test and reference preparation are linear and parallel; (ii) the toxin dilution which contains 50 millionths of an Lf gives a positive erythema in at least 80% of the control guinea-pigs; and (iii) the dilution containing 20 millionths of an Lf gives no reaction in at least 80% of the guinea-pigs. If these criteria are not met a different toxin should be selected.
2. By the subcutaneous challenge test
This test involves the immunization of guinea-pigs in the same way as in the first test, but the challenge is given by subcutaneous inoculation with 1 ml of a diphtheria toxin containing approximately 100 LD
0• The
guinea-pigs are observed twice daily for five dajs, and the number of animals protected by each vaccine dilution over the period are recorded.
Similarly, a control group of unvaccinated animals is included, each of which is given 1 ml of a 1:99 dilution of the toxin solution used to challenge the vaccinated animals (1 Ln
50).
1 Statistical Methods in Biological Assay. London {Charles Griffin and Co.) Chapters 4 and 6

BLG/UNDP/82.1 Page 60
The best method of estimating the relative potencies of the preparations from the proportion of animals surviviyg the challenge is by parallel line assay (Finney).
A small and inexpensive pocket calculator suitable for this analysis is the Texas Instrument 59 electronic calculator which must be equipped with the so called "Applied Statistics Module". In addition a set of programmed magnetic cards are requ~red in which the probit analysis programme has been stored . The operation of the equipment is as follows:-
Probit Analysis of Potency Tests
I Mode of Operation
Insert the Applied Statistics Module
Switch on.
Press: 2nd CP
"Read" first field of card 1 (let pass card hole from right to left, black side down).
1
2
D.J. Finney, Probit Analysis 3rd ed. London(l971) (Cambridge University Press)
These cards may be obtained from Chief, Biologicals, WHO, Geneva.

Press: CLR (clear)
BLG/UNDP/82.1 Page 61
"Read" second field of card 1 (same card, also from right to left, but card reversed, again black side down).
Press: CLR
"Read" card 2 (same way as for the first field of card 1).
3 I I
(Each time a card has passed, the number of the card will be visible. If the display flashes off and on, repeat procedure. Eventually clear the card carefully or the machine reading head or drive roller.)
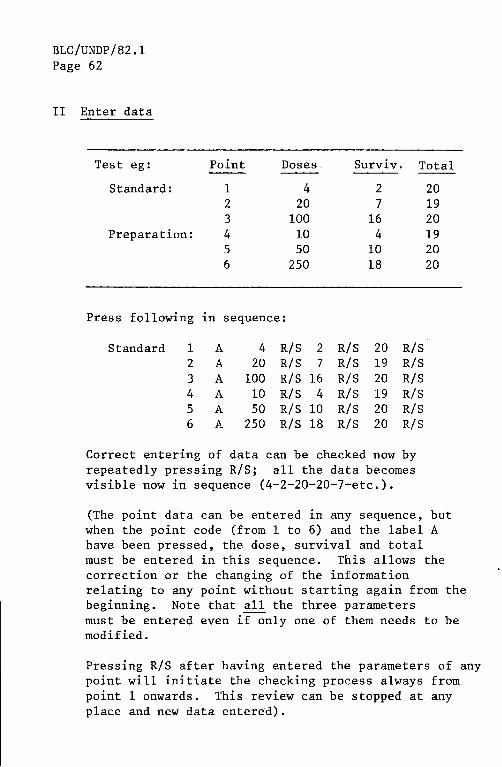
BLG/UNDP/82.1 Page 62
II Enter data
Test eg:
Standard:
Preparation:
Press following
Standard 1 2 3 4 5 6
Correct entering
Point Doses Surviv. Total
1 4 2 20 2 20 7 19 3 100 16 20 4 10 4 19 5 50 10 20 6 250 18 20
in sequence:
A 4 R/S 2 R/S 20 R/S A 20 R/S 7 R/S 19 R/S A 100 R/S 16 R/S 20 R/S A 10 R/S 4 R/S 19 R/S A 50 R/S 10 R/S 20 R/S A 250 R/S 18 R/S 20 R/S
of data can be checked now by repeatedly pressing R/S; all the data becomes visible now in sequence (4-2-20-20-7-etc.).
(The point data can be entered in any sequence, but when the point code (from 1 to 6) and the label A have been pressed, the dose, survival and total must be entered in this sequence. This allows the correction or the changing of the information relating to any point without starting again from the beginning. Note that all the three parameters must be entered even if only one of them needs to be modified.
Pressing R/S after having entered the parameters of any point will initiate the checking process always from point 1 onwards. This review can be stopped at any place and new data entered).

BLG/UNDP/82.1 Page 63
Ill Probit analysis
(a) Computation of successive iterations (Please note that doses are transformed in log.10 values for computation)
(b)
Press B (no display during the calculation period) Wait (+ 5 minutes) until number of iterations appears in display. (In example given, this will be 2). Usually the number of iterations is between 2 and 5.
(The iteration process stops when the difference between two successive estimates of the common slope is equal or less than 5% of the standard error of the last slope estimate. The programme allows for eight successive iterations. If after pressing B the machine stops with 9 in display (the computation may take about ten minutes!), it means that the above condition was not satisfied after eight iterations. The rest of the programme may however still be executed).
Results
Press c Press R/S
Press R/S Press R/S
Press D
Press R/S
(common slope) - sb (S.E. of b)
- ED50 standard - ED50 test
preparation 2
- X lin. (2d.f.,
In example: 1.50 In example: 0.25
In example: 30.1
In example: 40.7
crit. value at 5%.
2 probability level = 6) - X paral. (1 d.f., crit. value at 5%
probability level = 3.84)
(If values found are above crit. value this means significant deviation from linearity and/or parallelism, test statistically invalid. The potency estimate and its confidence limits are neverthe-

BLG/UNDP/82.1 Page 64
less still calculated, but should be interpreted with great caution if not rejected. Note that in case of significant departure from linearity (at 5% probability level) the heterogeneity factor is used in the determination of the 95% confidence interval of the relative potency.)
2 In example given, one finds x lin. 0.497
Press E Press R/S
Press R/S
2 x paral. = 0.014
-Relative potency (in example: 0.74) - Lower 95% confidence limit (in
example: 0.33) - Higher 95% confidence limit (in
example: 1.67)
(Never press B a second time during this procedure. If this is done, one must press CLR and repeat entering of data. C, D and E can be pressed again in any sequence)~
IV Other Tests
If immediately after the calculation another test has to be calculated, simply press CLR and enter the new series of data (proceed from II on).
If a laboratory wishes to copy the programmed magnetic cards, this may be done as follows:
Copying of Magnetic cards TI-59
1. "Read" set of original programmed cards in the normal way (see "Probit analysis of potency tests"). (The principle is to be found in the TI 59 Manual under VII).

2 (a) Press: 1
2nd
Write
BLG/UNDP/82.1 Page 65
Pass blank card side 1 (number 1 appears in display)
(b) Press: 2
2nd
Write
Pass blank card reversed (side 2)
(c) Press: 3
2nd
Write
Pass second blank card (3)
The new cards contain the programme now. Check the new cards.
A test is valid provided that:
(i) some but not all of the control guinea-pigs challenged with 1/100 of the challenge dose of toxin die; ·
(ii) the lowest dilution of vaccine has protected more than half of the animals;
(iii) the highest dilution has protected less than half; and
(iv) the dose response curves(of the test and reference vaccine do not deviate significantly from parallelism and linearity. If no standardization test of parellelism is available a test may be accepted provided that the slope of the steeper curve is not more than 150% of the shallower curve.

BLG/UNDP/82.1 Page 66
3. Method of assay of unadsorbed vaccine
The principles for assaying unadsorbed vaccine are similar to those already described for the assay of adsorbed vaccine. There are, however, three major differences:
(i) vaccines are tested against an unadsorbed standard;
(ii) the dose response curve is less steep as a result of which the interval between doses should be three-fold rather than two-fold;
(iii) the precision of the assay is lower necessitating the use of larger numbers of animals in the experimental groups.
4. Suggested method for individual identification of guinea-pigs in an animal room
The following scheme allows for the individual identification of more than 800 guinea-pigs. The identification consists of dye marks on head, back and legs. For recording purposes head and back colours are designated as a capital letter, thus:
A Red head s Red head, red back B Red back T Green head, green back c Green head w Mauve head, mauve back D Green back X Yellow head E Mauve head z Yellow back F Mauve back AA Yellow head, red back H Red head, green BB Yellow head, green back
back cc Yellow head, mauve back K Red head, mauve
DD Red head, yellow back hack L Green head, EE Green head, yellow back
red back FF Mauve head, yellow back
N Green head, mauve back
p Mauve head, red back
R Mauve head, green back
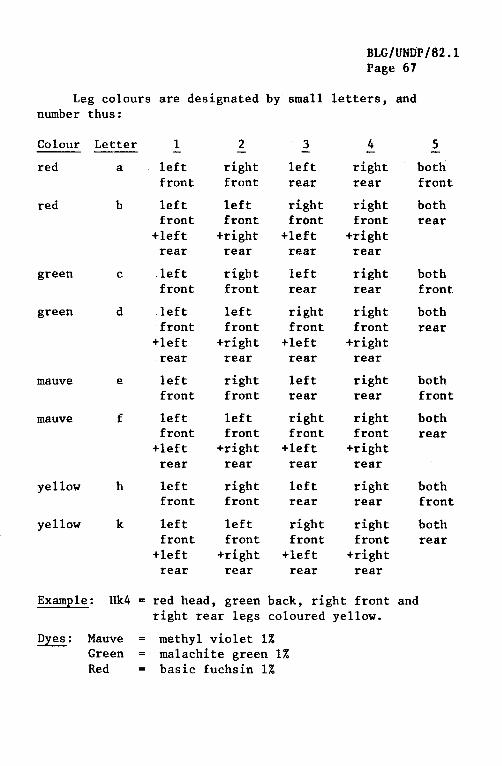
BLG/UNl)P/82.1 Page 67
Leg colours are designated by small letters, and number thus:
Colour Letter 1 2 3 4 5
red a left right left right both front front rear rear front
red b left left right right both front front front front rear
+left +right +left +right rear rear rear rear
green c left right left right both front front rear rear front
green d left left right right both front front front front rear
+left +right +left +right rear rear rear rear
mauve e left right left right both front front rear rear front
mauve f left left right right both front front front front rear
+left +right +left +right rear rear rear rear
yellow h left right left right both front front rear rear front
yellow k left left right right both front front front front rear
+left +right +left +right rear rear rear rear
ExamEle: Hk4 red head, green back, right front and right rear legs coloured yellow.
Dyes: Mauve methyl violet 1% Green malachite green 1% Red basic fuchsin 1%

BLG/UNDP/82.1 Page 68
Boil 1 litre distilled water. Remove from heat, sprinkle 10 g of dye on the surface and stir in.
Yellow picric acid 0.5%
Boil 1 litre distilled water. sprinkle 5 g of dye on the surface.
Remove from heat, Do not stir.
The adhesion of dyes to animal furs is improved by the addition of one drop Lissopol (BDH) to 100 ml of solution.

VACCINE: Diphtheria Toxoid
TEST: Test for Identity
BLG/UNDP/82.1 Page 69
CLASS OF TEST B
DIP-2
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE TETANUS TOXOID AND COMBINED VACCINE
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Section 5.1
IDENTITY TEST
The identity tests on a batch of toxoid can be carried out by both in vitro and in vivo methods. Although it is a simple procedure to establish the identity of fluid toxoid by a flocculation test, it is necessary, in the case of adsorbed toxoids, to dissolve the adsorbent (adjuvant) by the addition of sodium citrate,
For fluid toxoid, a diphtheria antitoxin is diluted, so as to contain 20 IU per ml. Increasing volumes of 0.3, 0.4, 0.5, 0.6, 0.7, 0.8 ml are pipetted into a series of tubes; the volumes are made up with normal saline to 1 ml and 1 ml of fluid toxoid, diluted to approximately 10 Lf/ml is added to each of the tubes. The tubes are placed in a water bath at 50°C and observed continuously. The tube containing 0.5 ml of antitoxin or the tube on either side of it may show flocculation afid such flocculation establishes the identity of the product.
For an adsorbed toxoid about 0.5 g of sodium citrate is added to 10 ml of the adsorbed toxoid. The mixture is incubated at 37°C for one or two days to dissolve. After

BLG/UNDP/82.1 Page 70
the aluminium salt has dissolved the flocculation test similar to the one stated above is used. The flocculation occurs in the same way though it make take a longer time.
In the in vivo test a suitable dose of toxoid is injected into guinea-pigs and the presence of antitoxin ~n the guinea-pig blood demonstrated by carrying out a passive-protection test in guinea-pig skin or by intradermal challenge of the test animal. The guinea-pig used in the test for freedom from abnormal toxicity will have received a suitable dose of toxoid, but the response should not be assessed until 14 days after injection.

VACCINE: Diphtheria Toxoid
TEST: Test for stability and reversal
BLG/UNDP/82.1 Page 71
CLASS OF TEST C
DIP-3
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE TETANUS TOXOID AND COMBINED VACCINE
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Sections 3.5.5 and 3.5.8
STABILITY AND REVERSAL TEST
Although this test is applied to the bulk toxoid and not on the final vaccine, control laboratories should be in a position to ensure that the diphtheria toxoid used in the vaccine is stable and is not subject to reversal to toxin. In countries with a high ambient temperature Test B of this test for diphtheria toxin should be applied to the final vaccine.
There are two tests for detecting reversal of toxoid.
A Intradermal test
A sufficient amount of the bulk toxoid is diluted so as to give 260 ml of a solution containing 80 Lf units/ml. The solution is divided into six lots each of 40 ml, and the residue tested by flocculation. Two of the 40 ml lots are stored at 4°C, two at 20°C and two at 35°C. One sample stored at each temperature is removed after four weeks and another after 12 weeks.

BLG/UNDP/82.1 Page 72
The following tests are applied to each sample:
(a) A volume of 0.2 ml of the sample is injected intradermally into the skin of a shaved depilated guinea-pig. The site of injection is observed 48 hours later. If erythema is seen its specificity is confirmed by injecting a further 0.2 ml of the samples into another normal guinea-pig and injecting 0.2 ml of a mixture prepared by adding 80 units of diphtheria antitoxin to 1 ml of the sample. If erythema is seen at the site of injection of the sample above but not at the site of injection of the mixture of the sample and antitoxin, this is evidence of reversion of toxicity and the bulk toxoid must be returned for further detoxification and resubmitted to the above test procedure.
(b) Each sample is tested for Lf content by the flocculation test. Toxoid is satisfactory provided:
(1)
(2)
(3)
the Lf content of the sample stored for 12 weeks at 37°C is not less than 60% of the Lf content of the initial sample;: the Lf content of the sample stored for 12 weeks at 20°C is not less than 80% of the initial samples; the Lf content of the sample stored for 12 weeks at 4°C is not significantly less than the initial sample.
B Subcutaneous test
A sample of purified toxoid is diluted in saline containing 0.01% thiomersal to give 300 ml of 10 Lf per ml toxoid. Aliguots of 100 ml of the diluted toxoid are held at 4°C, 20 C and 35°C for six weeks. Five millilitres of each sample are injected subcutaneously into each of five guinea-pigs weighing between 280 and 300 g, 2.5 ml on each side of the abdomen.

BLG/UNDP/82.1 Page 73
The pigs are weighed on the first, third and seventh day following injection, then weekly for a further five weeks. The guinea-pigs are examined daily for any evidence of diphtheria intoxication.
Criteria for acceptance
The animals should show no signs of diphtheria intoxication.

BLG/UNDP/82.1 Page 73 bis

PERTUSSIS VACCINE

PERTUSSIS VACCINE
BLG/UNDP/82.1 Page 73 ter

BLG/UNDP/82.1 Page 74
CLASS OF TEST C
PER-l
VACCINE: Pertussis and DPT
TEST: Test for potency of Pertussis
REQUIREMENTS FOR PERTUSSIS VACCINE (Requirements for Biological Substances Nos. 8 and 10)
(Revised 1978)
Part A Section 3.4.6
THE TEST
1. Glossary of terms
I.U.
o.u.
The number of organisms that kills 50% of a group of mice within 14 days when injected by the intracerebral route.
International Unit is the specific activity of a stated amount of the International Standard as defined by the WHO Expert Committee on Biological Standardization.
Opacity is the measure of opacity as determined in comparison with the International Reference Preparation of Opacity.
The dose of vaccine which protects 50% of the immunized mice against a challenge dose of virulent B. Pertussis given by the intracerebral route.

2. Procedure
BLG/UNDP/82.1 Page 75
Immunization: Healthy male or female mice, of the same sex, three to four weeks old and weighing not less than 12 g and not more than 18 g but all within a weight range of 4 g are used. Fivefold serial dilutions of the test vaccine, at 20 opacity units (O.U.), and of a reference pertussis vaccine the potency of which has been calibrated in international units are made in 0.85% saline. Three groups each of 20 mice are injected with the test vaccine and three similar groups with the reference vaccine (if the calculation method of Wilson and Worcester is used, 16 mice are used for each dose of the vaccines; however, to ensure that this number of mice would be available for challenge it is recommended that 18 mice be immunized at each level). Each mouse, in each group, is injected intraperitoneally with 0.5 ml of the appropriate dilution of vaccine. The range of dilution of reference and test vaccine is chosen so as to ensure 70-80%-, 40-50% and 10-20% survival, respectively, at the three levels of immunization. Each group is housed individually and randomly distributed with regard to shelf position and containers. This can be reached either by distributing the mice into the containers by means of latin square randomization tables or - a simpler and also very useful procedure - by putting one mouse injected with each dilution into every single container. If this procedure is adopted the mice must be identified by a system of marking.
At the time of immunization three groups each of 12 mice of the same weight are put into cages. and are injected by the intracerebral route in order to determine the virulence of the challenge suspension on the same day as the immunized mice are challenged. A fourth group of 12 mice is kept and used for checking the challenge suspension itself.

BLG/UNDP/82.1 Page 76
Challenge culture: Bordetella pertussis, strain 183231
, is stored in the lyophilized state. When a challenge is required an ampoule of the challenge strain is reconstituted in 4 ml of casamino acids and 1 ml of the suspen~ion spread on each of four Bordet Gengou (BG) agar plates . The plates are incubated at 35°Cfor three to four
h. 0
days: t 1s stock culture can then be stored at 2-10 C for three weeks. The organisms are scraped from the surface of a plate and seeded on BG slopes containing 1% peptone in 20 x 4 em tubes. The slopes are incubated for 44-48 hours, at 35°C and then subcultured on fresh slopes which are incubated for 20-24 hours at 35°C. After a microscopic check for purity enough culture is scraped into a 1% solution of casamino acids so that 5he suspension has an optical density of 10 O.U. (10 x 10 organisms per ml). The suspension is drawn off and further diluted to give the challenge dose which should contain approximately 100 000 organisms in 0.03 ml. (This is a 1:3000 dilution of 10 O.U) Dilutions of challenge are made also to contain a calculated 2000, 400 and 80 (1:50, 1:250 and 1:1250) organisms per 0.03 ml. These dilutions are made using no greater than serial, 1:10 or 1:5 dilutions changing the pipette between each dilution. The viable count of the challenge dose is determined by seeding the dilution containing 80 organisms per 0.03 ml on a BG plate at the beginning and end of challenge. The count has been found to range from 10 to 50 organisms from test to test, the average being about 25. No more than 2.5 hours should elapse between the time the challenge culture is suspended in the casamino acids solution and the completion of challenge of the last group of mice.
1
2
The challenge strain 18323 may be obtained from the WHO Secretariat (Chief, Biologicals).
See Annex 1.

BLG/UNDP/82.1 Page 77
Challenge: All groups of mice are challenged 14 days after immunization in a random order with 0.03 ml of the challenge suspension; these mice may be anaesthetized with ether. The injections are performed intracerebrally using a 27 gauge, three-eighth inch needle fitted to a 0.25 ml syringe. The mice are kept under observation for 14 days. Animals dying in the first three days are nut counted when computing the results ("surplus" mice are discarded 72 hours after challenge in order to have 16 mice at each immunization level). Deaths are recorded daily up to the fourteenth day. The responses of the mice to the reference and to the test vaccine are compared and analyzed.
Criteria for acceptance of the test (WHO Requirements):
The test is valid if the ED50 of each vaccine is between the largest and the smallest immunizing doses, the limits of one standar1 error of each ED
50 fall within a
range of 64% to 156%, the immune response is graded in relation to the immunizing doses, and the slopes of the dose-response curves of the vaccine under test and the standard vaccine are not significantly different.
The potency of the vaccine will be the ED50 of the reference preparation divided by the En
50 of tne test
preparation and multiplied by the potency of the reference preparation in international units per ml.
The vaccine shall have a potency of not less than four international units (I.U.) per single human immunizing dose.
Calculations of the2results: The results should be analysed by probit analysis.
1
2
This range has been calculated assuming that 16 mice per group are included and that the slope of the log-dose response line, calculated by probit analysis, is 1
D.J. Finney, Probit Analysis 3 rd ed. London 1971 (Cambridge University Press)

BLG/UNDP/82.1 Page 78
A small and inexpensive pocket calculator suitable for this analysis is the Texas Instrument 59 electronic calculator which must be equipped with the so called "Applied Statistics Module". In addition a set of programmed magnetic cards are required in which the probit analysis prograrnrn has been stored. The operation of the equipment is as follows:
PROBIT ANALYSIS OF POTENCY TESTS
I Mode of Operation
Insert the Applied Statistics Module
Switch on
Press: 2nd CP
"Read" fhst field of card 1 (let pass card through hole from right to left, black side down).
Press: CLR (clear).
"Read" second field of card 1 (same card, also from right to left, but card reversed, again black side down).
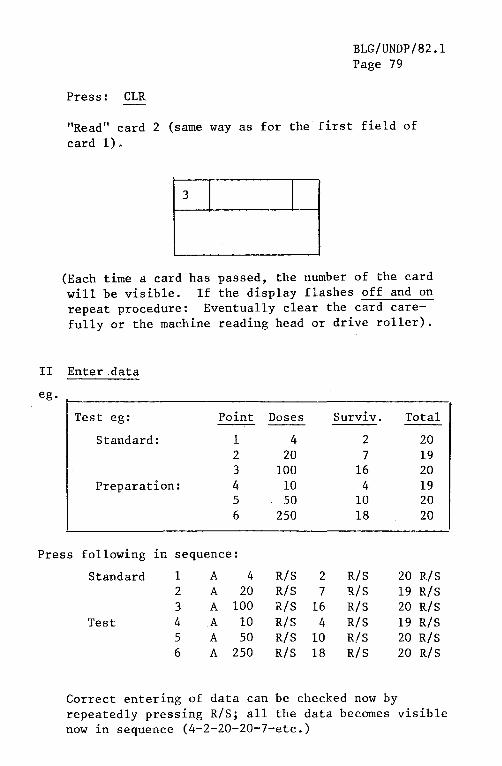
II
eg.
Press: CLR
BLG/UNDP/82.1 Page 79
"Read" card 2 (same way as for the first field of card 1),
I
(Each time a card has passed, the number of the card will be visible. If the display flashes off and on repeat procedure: Eventually clear the card carefully or the machine reading head or drive roller).
Enter.data
Test eg: Point Doses Surviv. Total
Standard: 1 4 2 20 2 20 7 19 3 100 16 20
Preparation: 4 10 4 19 5 50 10 20 6 250 18 20
Press following in sequence:
Standard 1 A 4 R/S 2 R/S 20 R/S 2 A 20 R/S 7 R/S 19 R/S 3 A 100 R/S 16 R/S 20 R/S
Test 4 A 10 R/S 4 R/S 19 R/S 5 A 50 R/S 10 R/S 20 R/S 6 A 250 R/S 18 R/S 20 R/S
Correct entering of data can be checked now by repeatedly pressing R/S; all the data becomes visible now in sequence (4-2-20-20-7-etc.)

BLG/UNDP/82.1 Page 80
(The point data can be entered in any sequence, but when the dose, survival and total must be entered in this sequence. This allows the correction or the changing of the information relating to any point without starting again from the beginning. Note that all the three parameters must be entered even if only one of them needs to be modified.
Pressing R/S after having entered the parameters of any point will initiate the checking process always from point 1 onwards. This review can be stopped at any place and new data entered.)
III Probit analysis
(a) Computation of successive iterations (Please note that doses are transformed in log10 values for computation)
(b)
Press B (no display during the calculation period) Wait (~ 5 minutes) until number of iterations appears in display. (In example given, this will be 2). Usually the number of iterations is between 2 and 5.
(The iteration process stops when the difference between two successive estimates of the common slope is equal or less than 5% of the standard error of the last slope estimate. The programme allows for eight successive iterations. If after pressing B the machine stops with 9 in display (the comp~tation may take about ten minutes!), it means that the above condition was not satisfied after eight iterations. The rest of the programme may however still be executed.)
Results
Press c b (common slope) In example: 1.50 Press R/S s (S.E. of b) In example: 0.25 Press R/S Efl50 standard In example: 30.1

Press R/S
Press D
Press R/S
BLG/UNDP/82.1 Page 81
ED50 test prep. In example: 40.7
2 x lin. (2 d.f., crit. value
at 5% probability level = 6)
2 x paral. (1 d.f., crit. value
at 5% probaility level = 3.84)
(If values found are above crit. value this means significant deviation from linearity and/or parallelism, test statistically invalid. The potency estimate and its confidence limits are nevertheless still calculated, but should be interpreted with great caution if not rejected. Note that in case of significant departure from linearity (at 5% probability level), the heterogeneity factor is used in the determination of the 95% confidence interval of the relative potency.)
In example given, one finds x~ lin. x paral.
0.497 0.014
Press E Relative potency (in example: 0.74) Press R/S Lower 95% confidence limit (in
example: 0.33) Press R/S Higher 95% confidence limit (in
example: 1.67)
(Never press B a second time during this procedure. If this is done, one must press CLR and repeat entering of data. C, D and E can be pressed again in any sequence)~
IV Other tests
If immediately after the calculation another test has to be calculated, simply press CLR and enter new series of

BLG/UNDP/82.1 Page 82
data (proceed from II on).
If a laboratory wishes to copy the programmed magnetic cards this may be done as follows:-
Copying of Magnetic Cards TI-59
1. "Read" set of original programmed cards in the normal way (see "Probit analysis of potency tests"). (The principle is to be found in the TI 59 Manual under VII.)
2(a) Press: 1
2nd
Write
Pass blank card side 1 (number 1 appears in display)
(b) Press: 2
2nd
Write
Pass blank card reversed (side 2)
(c) Press: 3
2nd
Write
Pass second blank card (3)
The new cards contain the programme now. Check the new cards.

ANNEX I
MEDIA
BLG/UNDP/82.1 Page 83
Many media have been described for the growth of B.pertussis but only a few are used regularly today in the production of vaccine. The particular one chosen will depend on experience and the availability of raw materials. Those described are known to yield good vaccine. The formulae and methods of preparation given, however, differ in some respects from those described originally mainly because alternative materials have proved to be equally effective, sometimes even better, or are more readily available. For completeness the reference to the original publication in which the medium is described is given.
1. Bordet-Gengou medium (B~)l
B. pertussis was first isolated on BG medium. The medium is still used as originally described except that sheep. blood may be used in place of rabbit blood and that proteose peptone may be added, a modification introduced by Eldering & Kendrick in 1936. Its main uses for the recovery of B. pertussis from the freeze-dried state, for the initial stages of the propagation of seed cultures, for maintaining the viability of cultures over a period of time and for the growth of the challenge culture used in the mouse-protection test.
Potatoes are washed in cold, distilled water and sliced into a meat-mincer fitted with a coars.e cutter: 500 g of minced potatoes are added to one litre of cold, distilled water containing 40 ml of glycerol. The mixture is boiled until the potatoes are soft, usually about 30 minutes, and filtered through cheesecloth, muslin or other suitable material.
1 Original description: Bordet, J. & Gengou, 0 (1906) Ann. de l'Inst. Pasteur, 20, 731-741

BLG/UNDP/82.1 Page 84
Sodium chloride (11.25 g) is dissolved in 1.5 litres of water to which 65 g of Bacto agar is added. The mixture is placed in a double boiler consisting of an outer, 40-litre vessel with an inner, 20.litre vessel, the water in the outer vessel being heated with a steam coil. When the agar is melted 500 ml of the potato extract is added, the whole heated for one hour, filtered through several layers of suitable material as before, and dispensed in 900 ml amounts in sterile, four-litre bottles. The bottles and contents are then steamed for 15 minutes and autoclaved for 40 minutes at 121°C.
To prepare the final medium, the agar-base is melted cooled, and citrated sheep blood is added to the ratio of two parts of blood to nine parts agar-base. This is achieved by heating one bottle of agar-base (900 rnl) in a boiling-water bath to melt the base: this takes about 2.5 hours. The base is then cooled to about 60°C and 200 ml of blood added and mixed.
A variant of this medium is:
1 Bordet-Gengou medium with added proteose peptone
Ten millilitres of glycerol and 10 g of proteose peptone are added to 30 ml of Bordet-Gengou agar-base (Difco) diluted in one litre of warm, distilled water. The mixture is steamed for 15 minutes and autoclaved for 40 minutes at 121°C. The mixture is then cooled as before and citrated sheep blood added in the ratio of two parts of blood to nine parts of base.
Again, the volume prepared should be calculated to meet needs.
1 Eldering, G. & Kendrick, P.L. (1936), Am. J. Publ. Health, 26 206-511

2. Cohen and wneeler medium
BLG/UNDP/82.1 Page 85
This medium is essentially a modified form of Hornibrook's medium (see following paragraph 3) in which the optimum levels of different components were more carefully assessed.
Cohen and Wheeler medium (1946) Pertussis vaccine grown in fluid medium (American Journal of Public
Health, 36 371-376)
Casarnino acids (Bacto) technical1
NaCl KH2Po
4 MgC1
2.6H
20
Starch, soluble, powdered CaC1
2, 1% solution
Feso4
.7H20, 0.5% solution
Cuso4
.sH2o, 0.05% solution
Cysteine hydroc2loride 1% solution2
Yeast dialysate Distilled water to
10 g 2.5 g 0.5 g
0.4 g 1.5 g 1 ml 2 ml
1 ml
2.5 ml 50 ml
1 kg
1 This preparation is rather expensive. It could be replaced - after sufficient experience has been gained in the vaccine production - by locally prepared, hydrochloric acid - hydrolysed casein.
2 Experience in some laboratories has shown that it may be preferable to add the cysteine hydrochlo~ide and the yeast dialysate under sterile conditions to the medium after sterilization in the autoclave, followed by a mild heating of the whole medium for 15 minutes at 105° - 110°C. Heating to 121°C may be harmful to these ingredients.

BLG/UNDP/82.1 Page 86
The casamino acids, salt, phosphate and magnesium chloride are dissolved in part of the water. The remaining ingredients are added and made up to 1 kg. pH is adjusted to 7.2 to 7.3. The solution is boiled until the starch is dissolved and the medium clear. The fluid is filtered and dispensed in 200 ml amounts in onelitre Roux bottles. This medium is also frequently used for fermenter cultures. The medium is autoclaved at 10 lb pressure for 15 minutes.
Yeast dialysate
Brewers' yeast (Fleischmann, pure dry, type 2019)
Distilled water Distilled water
500 g
800 g 2 000 ml
A paste is made of the yeast with the 800 g of water and transferred to a cellophane tube (500-550 mm long, about 114 mm diameter, 0.089 mm wall thickness). Each end of the tube is tied with heavy twine, immersed in the 2 000 ml of water in such a manner that the liquid cannot spill or seep through the ends of the tube, and its position adjusted to give~appropriate equality of liquid level inside and outside the tuoe. It is heated to 78 - 80°C for seven hours. The tube is removed and the dialysate transferred to a bottle. 5 ml of chloroform is added, the bottle is stoppered with a rubber stopper, and stored in the coldroom. This dialysate is clear, requires no filtration, and remains satisfactory for use even after several months' storage.
This medium is often solidified by the addition of 2-3% agar and 0.4% charcoal (Norit SG), modifications suggested by Powell, Culbertson & Ensminger (1951) Charcoal agar culture medium for preparing Hemophilus pertussis vaccine, Public Health Reports, 66 346-348
1 Could also be produced locally.

3. Hornibrook medium
BLG/UNDP/82.1 Page 87
This was the first, simple, liquid medium described for the growth of B. pertussis. it represented a real advance in that it was ac:i.d:-hydrolysed casein as a carbon and nitrogen source, and for the first time made it possible to eliminate blood from the growth medium. A suggested modification was the replacement of the yeast dialysate by nicotinic acid.
Hornibrook midium (1939) Cultivation of Phase 1 B. pertussis in a semi-synthetic liquid medium
(Public Health Reports, ~ 1S47-1851)
Distilled water 1 000 ml Hydrolysed casein (sulfuric acid
hydrolysed) NaCl KCl CaC1
2 anhydrous
MgC12
6H2o
Na2co
3 anhydrous
KHl04
.H20
7 g
5 g 0.2 g 0.2 g
0.1 g
0.5 g
0.25 g Soluble starch (reagent grade) Yeast extract (opti~nal) Glutathione, 0.2% solution, added
0.25 g
0.5 ml separately to the bulk (see below)
The casein hydrolysate is dissolved in 70 ml of distilled water and the sodium chloride and potassium chloride, dissolved in about 40 ml of water, are added. The magnesium chloride, nicotinic acid, dipotassium phosphate and calcium chloride are then added as the powders. Finally the starch, in solution, is stirred in,
1 The original name Haemophilus pertussis has s1nce been changed to Bordetella pertussis.

BLG/UNDP/82.1 Page 88
the volume is brought to one litre and adjusted to pH 7.0 with sodium carbonate. The starch solution is made by adding the starch to 2 ml of water, forming a paste and then dissolving the paste in 30 ml of boiling water. Where more than 100 litres is required an aliquot of the medium is titrated against a 1% solution of sodium carbonate and the required amount of sodium carbonate calculated and added. The medium is filtered through a fine-mesh nylon cloth or other suitable material ro remove debris.
This medium may be dispensed into flasks. For sterilization the flasks are steamed for 15 minutes and then autoclaved for 40 minutes at 121°C. The containers should be removed from the autoclave as soon as possible and cooled as quickly as possible.
A 0.2% solution of glutathione is prepared separately and sterilized by filtration, preferably through a 0.22 micron membrane. It is added to the flasks, at a concentration of 0.6 ml% prior to inoculation with the organisms.
Casein hydrolysate. In his original paper, Hornibrook used a sulfuric acid hydrolysate of casein, but this can be replaced by more readily available hydrochloric acid hydrolysates (the equivalent of 1% dry weight of hydrolysate should be present in the final medium).
With commercially available hydrolysates, a 10%solution is made. Before use 2.5% powdered, activated Norit A charcoal is added to the hydrolysate, the mixture stirred vigorously and allowed to stand for one hour. The charcoal is removed by filtration through Republic K5, filter-pad pulp (see Appendix P.l8 for equivalent) supported in a Buchner funnel. The chloride content of the final medium should be between 0.5 and 0.6%. Since some commercial hydrolysates may have a high chloride content it may be necessary to make allowances for this in the basal medium which also contains sodium chloride.

BLG/UNDP/82.1 Page 89
Starch.1
Agar contains at least two inhibitors of B. pertussis . These can be inactivated by a number of reagents, e.g. starch, blood, red blood cells, albumin, charcoal. The simplest to include in a medium is starch. The quality of starch, however, is important, and although all starches examined will enable growth of B. pertussis to take place, the antigenicity of the ~arvested cells depends largely on the quality of the starch . For this reason starch should be monitored by comparison with that of a lot known to yield good vaccine. Once identified, sufficient of the good lot should be purchased to secure at least two years' production. It is not necessary to use soluble starch: many millers' starches are highly effective and much cheaper.
Hornibrook medium (1940) Nicotinic acid as a growth factor,
(Proceedings of the Society for Experimental Biology and Medicine, 45, 598-599)
Solution A
NaCl 0.7 g KCl 0.02 Ca c1
2, anhydrous 0.02
Mg Cl2
.6H20 0.01
Na2co3
0.05
g g g g
KH2Po
4.H
20 0.025 g
NH4Cl 0.02 g
Soluble starch 0.1 g Water 50 ml
The ingredients are mixed, autoclaved, cooled and filtered through paper.
1 Cameron, J. & Dunning, J.M. (1970) Tenth International Congress for Microbiology Mexico.

BLG/UNDP/82.1 Page 90
Solution B1
d glutamic 1 tyrosine glycine 1 proline 1 histidinr d arginine
. 1 ac1d 0.18 g
0.02 g 0.0045 g 0.04 g 0.01 g 0.02 g
The above are mixed dry and to them is added 0.001 g of cystine dissolved in a drop or two of lON HCl. Ten millilitres of H
20 is added, and normal NaOH is added
until the acids are in solution.
The addition of 0.5 to 0.001 mg of nicotinic acid per litre allows the organism to grow.
1 This is Hornibrook's original formula. The 1-forms of glutamic acid and arginine would be used today.

VACCINE: Pertussis
TEST: Test for Toxicity
BLG/UNDP/82.1 Page 91
CLASS OF TEST C
PER-2
REQUIREMENTS FOR PERTUSSIS VACCINE (Requirements for Biological Substances No. 8)
(Revised 1978)
Part A Section 3.4.5
THE TEST
There is little information on the relationship between toxicity of vaccines in animals and the occurrence of untoward reactions following vaccination in man. Toxicity tests in animals are required by many national control authorities, but in most tests the results obtained depend to some extent on factors independent of the vaccine, such as the strain of animal used and the conditions under which the animals are kept. One of the tests most widely used is the mouse toxicity test •. This has been of some value in ensuring the production of vaccine which, in general, are satisfactory in that they cause minimal untoward reactions in man.
This test may be performed as follows:
No fewer than 10 healthy mice are used, each weighing 14-16 g. They should have access to food and water for at least 2 hours before injection and continuouslyafter injection for the duration of the test. The total weight of the group of mice is determined immediately before injection. The mice used for testing the vaccine(s) and the control group of mice should be of

BLG/UNDP/82.1 Page 92
the same sex. If both sexes are used, they should be equally distributed in all groups. Each mouse is given an intraperitoneal injection with 0.5 ml, containing a volume of the final bulk under test that is equivalent to not less than half the largest volume recommended as a single human dose. A similar control group of mice is inoculated with 0.5 ml of physiological saline, preferably containing the same concentration of the same preservative as that present in the final bulk vaccine. The total weight of each group of mice is determined at the end of 72 h and at the end of 7 days after injection. The final bulk is considered satisfactory if (a) at the end of 72 h the total weight of the group is not less than that preceding the injection, (b) at the end of 7 days the average weight gain per mouse is not less than 60% of that in the control group of mice, and (c) not more than 5% of the mice die.
Since the toxicity of pertussis vaccine is not well understood, manufacturers and national control laboratories should continue to study tests for toxicity in order to understand it better.

VACCINE: Pertussis
TEST: Test for Identity
BLG/UNDP/82.1 Page 93
CLASS OF TEST A
PER-3
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE, TETANUS TOXOID AND COMBINED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Section 5.1
TEST FOR IDENTITY
These tests should be applied to the final vaccine as well as to the strains of B. pertussis used for making vaccine.
Procedure
The vaccine is diluted to about 50 O.U and three loo~fuls are mixed with one loopful of monospecific antiserum on a microscope slide. After mixing, the slide is rocked gently for a few minutes. The procedure is carried out with sera specific for agglutinogens 1, 2 and 3.
Criteria for acceptance
Rapid, heavy agglutination should be observed with all three specific antisera within about three m1nutes.
1 These typing sera may be obtained by application to Chief, Biologicals, WHO, Geneva.

BLG/UNDP/82.1 Page 93 bis

TETANUS VACCINE

TETANUS VACCINE
BLG/UNDP/82 .1 Page 93 ter

BLG/UNDP/82.1 Page 94
CLASS OF TEST C
TET-1
VACCINE: Tetanus Toxoid
TEST: Test for Potency
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE, TETANUS TOXOID, AND COMBINED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Section 5.3
Glossary of terms
Kf
Lf
Lf/mg N
Lf/ml PN
I.U.
Flocculation time (in minutes) as observed in the flocculation reaction. Limes flocculationis; the amount of toxin or toxoid which when mixed with 1 International Unit of antitoxin gives a Ramon flocculation in the shortest time. Flocculation units per milligram of total nitrogen as determined by the Kjeldahl analysis. Flocculation units per milligram of protein nitrogen (PN) which is usually determined by the precipitation of the proteins with trichloroacetic acid. International Unit is the specific activity of a stated amount of the International Standard as defined by WHO Expert Committee on Biological Standardization.

L+
L+/10
M.L.D.
A.B.V.
T.C.P.
BLG/UNDP/82.1 Page 95
The minimum amount of toxin which when combined with 1 I.U. of antitoxin kills an animal of defined weight in four days (the L+ is dependent upon the animal species used). The minimum amount of toxin which when combined with 0.1 I.U. of antitoxin kills a mouse of a defined weight in four days. The amount of toxin that kills 50% of a group of animals within four days (the 10
50 differs for different
animal species). Minimal lethal dose, the amount of toxin which kills animals within four days
(M.L.D. is different for different animal species). In general the M.L.D. has been replaced by the Ln
50•
Antitoxin binding value, a value which defines the toxin plus toxoid in a mixture (determined in animals). Total combining power, the reciprocal of the volume of toxoid which when diluted is found to be equivalent to 1 International Unit. The dose of a vaccine which protects 50% of the immunized animals against a challenge dose of toxin.
POTENCY TEST
Although the WHO Requirements do not specify any particular method it is essential that potency be measured against that of a national standard which has been calibrated against the International Standard for tetanus toxoid.
The inclusion of a reference preparation is essential because the antitoxin response of mice or guinea-pigs to a tetanus toxoid may be affected markedly by diet, conditions of maintenance, strain of animal used; such variables make

BLG/UNDP/82.1 Page 96
it difficult to identify real differences between toxoids. Furthermore different values are obtained in guinea-pigs and mice.
Tests based upon the use of a standard preparation generally involve vaccination of groups of animals with multiple dilutions of the test and reference vaccine, and comparison of the log dose response curves of the preparations. If the response curves are reasonably linear and do not differ significantly in slope (b value) the difference in log dose required to achieve a given response can be calculated to give an estimate of the relative potency of the test preparation, i.e. the ratio of the volume of the standard preparation required to elicit a given response over the value of the test preparation required to elicit the same response. Since the potency of the national or laboratory standard will have been calibrated in International Units, it is possible to assign a potency to the test preparation also in International Units.
1. Suggested method of assay for adsorbed vaccine
(a) In guinea-pigs
Prepare three dilutions in saline of the pational or laboratory standard preparation of tetanus vaccine (adsorbed) or of the International Standard, and three similar dilutions of each of the toxoids or vaccines under test. The choice of dilutions used will vary depending upon the strain of guinea-pig and the conditions of maintenance, but successive dilution of each preparation should conform to a twofold series (e.g. 100, 200, 400). The particular dilutions used are selected in the light of experience of local conditions such that the median dose for each preparation tested may be expected to protect approximately half the animals against challenge.

BLG/UNDP/82.1 Page 97
Inoculate 1 ml of the diluted vaccine subcutaneously into each group of guinea-pigs which should contain not less than 16 animals for the subcutaneous lethal challenge. Five uninoculated guinea-pigs are used as controls.
It is advisable to distribute the guinea-pigs in each treatment group randomly through several cages since the level of response can be affected by local conditions within different cages. When randomization is carried out it is necessary to identify each animal individually. A scheme for the identification of animals by dye marking is shown later.
The responses of the guinea-pigs to the single dose of vaccine are determined by challenge 28 days after inoculation. The challenge is given by the subcutaneous injection of 1 ml of a dilution of tetanus toxin containing approximately 100 Ln
50• A suitable toxin should contain
at least 10 000 LD50 1 per Lf. The guinea-pigs are observed twice daily for five days, and the number of animals protected by each vaccine dilution over that period recorded.
The control group of unvaccinated animals is divided into three sub-groups each of two animals which is inoculated with 1 ml of a 1:50, 1:100 and 1:200 dilution respectively of the toxin solution used to challenge the vaccinated animals.
1 To determine the Ln50
of a toxin prepare dilutions of the toxin contain~ng 0.0002 Lf, 0.0001 Lf, 0.00005 Lf, 0.000025 Lf per ml, and inject 1 ml doses of each dilution into groups of five guineapigs. Observe the animals for four days and ~ount the proportion of animals which die showing symptoms of tetanus during that period. From the data derive the Ln
50 by the method of Reed & Muench.

BLG/UNDP/82.1 Page 98
The best method of estimating the relative potencies of the preparations from the proportion of animals surviving the chy.llenge is by parallel line probit analysis (Finney).
S small and in expensive pocket calculator suitable for this analysis is the Texas Instrument 59 electronic calculator which must be equipped with the so called "Applied Statistics Module". In addition a set of programmed magnetic cards are required in ~hich the probit analysis programme has been stored. The operation of the equipment is as follows:-
PROBIT ANALYSIS OF POTENCY TESTS
I Mode of Operation
Insert the Applied Statistics Module
Switch on.
Press: 2nd CP
"Read" first field of card 1 (let pass card through hole from right to left, black side down).
Press: CLR (clear)
"Read" second field of card 1 (same card, also from right to left, but card reversed, again black side down).
2
1 & 2 See page 104
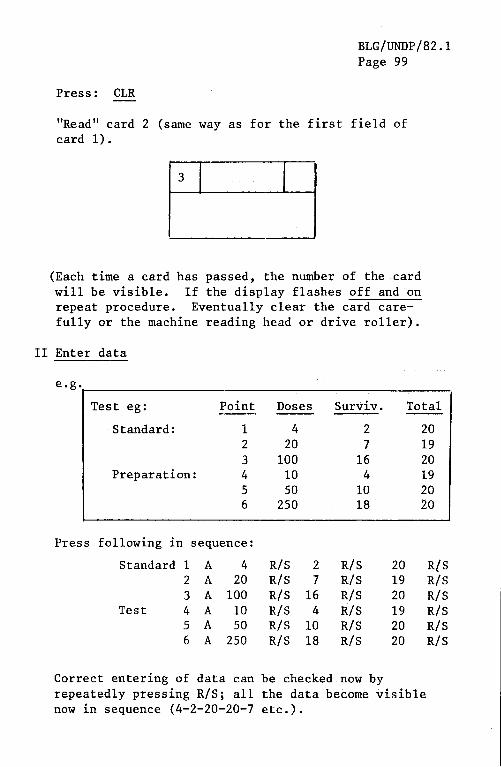
II
BLG/UNDP/82.1 Page 99
Press: CLR
"Read" card 2 (same way as for the first field of card 1).
3 l
(Each time a card has passed, the number of the card will be visible. If the display flashes off and on repeat procedure. Eventually clear the card care-fully or the machine reading head or drive roller).
Enter data
e.g.
Test eg: Point Doses Surviv. Total
Standard: 1 4 2 20 2 20 7 19 3 100 16 20
Preparation: 4 10 4 19 5 50 10 20 6 250 18 20
Press following in sequence:
Standard 1 A 4 R/S 2 R/S 20 R/S 2 A 20 R/S 7 R/S 19 R/S 3 A 100 R/S 16 R/S 20 R/S
Test 4 A 10 R/S 4 R/S 19 R/S 5 A 50 R/S 10 R/S 20 R/S 6 A 250 R/S 18 R/S 20 R/S
Correct entering of data can be checked now by repeatedly pressing R/S; all the data become visible now in sequence (4-2-20-20-7 etc.).

BLG/UNDP/82.1 Page 100
(The point data can be entered in any sequence, but when the point code (from 1 to 6) and the label A have been pressed, the dose, survival and total must be entered in this sequence. This allows the correction or the changing of the information relating to any point without starting again from the beginning. Note that all the three parameters must be entered even if only one of them needs to be modified.
Pressing R/S having entered the parameters of any point will initiate the checking process always from point 1 onwards. This review can be stopped at any place and new data entered).
III Probit analysis
(a) Computation of successive iterations (Please note that doses are transformed in log
10 values for computation)
Press B (no display during the calculation period). Wait (+ 5 minutes) until number of iterations appears in display. (In example given, this will be 2). Usually the number of iterations is between 2 and 5.
(The iteration process stops when the difference between two successive estimates of the common slope is equal or less than 5% of the standard error of the last slope estimate. The programme allows for eight
.successive iterations. If after pressing B the machine stops with 9 indisplay (the computation may take about ten minutes!), it means that the above condition was not satisfied after eight iterations. The test of the programme may however still be executed).
(b) Results
Press C Press R/S
b (common slope) sb (S.E. of b)
In example: In example:
1.50 0.25

Press R/S Press R/S
Press D
Press R/S
ED50 standard ED50 test prep.
BLG/UNDP/82.1 Page 101
In example: 30.1 In example: 40.7
2 x lin. (2 d.f., crit. value at 5%
2 probability level = 6) x paral. (1 d.f., crit. value at 5%
probability level = 3.84)
(If values found are above crit. value this means significant deviation from
linearity and/or parallelism, test statistically invalid. The potency estimate and its confidence limits are nevertheless still calculated, but should be interpreted with great caution if not rejected. Note that in case of significant departure from linearity (at 5% probability level), the heterogeneity factor is used in the determination of the 95% confidence interval of the relative potency).
2 In example given, one finds x2 lin. = 0.497
x paral. = 0.014
Press E Press R/S
Press R/S
Relative potency (in example: 0.74) Lower 95% confidence limit (in
example : G.33) Higher 95% confidence limit (in
example: 1. 6 7)
(Never press ! a second time during this procedure. If this is done, one must press CLR and repeat entering of data. C, D and E can be pressed again in any sequen~e)~ -
IV Other tests
If immediately after the calculation another test has to be calculated, simply press CLR and enter the new series of data (proceed from II on).

BLG/UNDP/82.1 Page 102
If a laboratory wishes to copy the programmed magnetic cards this may be done as follows:-
Copying of Magnetic Cards TI-59
1. "Read" set of original programmed cards in the normal way (see "Probit analysis of potency tests"). (The principle is to be found in the TI 59 Manual under VII).
2(a) Press: 1
2nd
Write
Pass blank card side 1 (number 1 appears in display)
(b) Press: 2
2nd
Write
Pass blank card reversed (side 2).
(c) Press: 3
2nd
Write
Pass second blank card (3).
The new cards contain the programme now. Check the new cards.
A test is valid provided that:
(i) all of the control guinea-pigs challenged with 1 in 50 dilution of the challenge dose of toxin die, and that none of the guinea-pigs inoculated with the 1 in/200 dilution of the challenge dies within the period of observation of the test;

BLG/UNDP/82.1 Page 103
(ii) the lowest dilution of vaccine protects more than half of the animals;
(iii) the highest dilution has protected less than half; and
(iv) that the dose response curves of the test and reference vaccine do not deviate significantly from parallelism and linearity. If no standardized test for parellelism is available a test may be accepted provided that the slope of the steeper curve is not more than 150% that of the shallower curve.
In the l~O Requirements no minimum potency for adsorbed tetanus toxoid has been formulated. As a guideline these vaccines should contain not less than 40 I.U/ single human dose.
In Mice
Prepare three dilutions in saline of the national standard preparation of tetanus vaccine (adsorbed) or of the International Standard, and three similar dilutions of each of the toxoids or vaccines under test. The choice of dilution used will vary depending upon the strain of mice and the conditions of maintenance, but successive dilutions of each preparation should conform to a twofold series (e.g. 30, 60, 120). The particular dilutions used are selected in the light of experience of local conditions such that the median dose for each preparation tested may be expected to protect approximately half of the animals against challenge.
Inoculate 0.5 ml of the diluted vaccine subcutaneously into each group of mice which should contain not less than 16 animals for the subcutaneous lethal challenge. Twelve uninoculated mice are used as controls.
The responses of the mice to the single dose of vaccine are determined by challenge 28 days after inoculation. The challenge is given by the subcutaneous inocula-

BLG/UNDP/82.1 Page 104
tion of 0.5 ml of a dilution of tetanus toxin containing approximately 200 LD
50 per ml.
The mice are observed twice daily for five days, and the number of animals protected by each vaccine dilution over that period recorded.
The control group of unvaccinated animals is divided into three sub-groups each of four animals which are inoculated with 1 ml each of 1:50, 1:100 and 1:200 dilutions respectively of the toxin solution used to challenge the vaccinated animals.
The best method of estimating the relative potencies of the preparations from the proportion of animals survivin~ the challenge is by parallel probit analysis (Finney) .
The use of a computer programme for this analysis is described under the analysis of the data for the test in guinea-pigs.
A test is valid provided that:
1
2
(i) all of the control mice challenged with 1 in 50 dilution of the challenge dose of toxin die, and that none of the mice inoculated with the 1/200 dilution of the challenge dies within the period of observation of the test;
A suitable toxin should contain at least 10 000 1050 per Lf. To determine the 10
50 of a toxin prepare
dilutions of toxin containing 0.0002 Lf, 0.0001 Lf, 0.00005 Lf, 0.000025 Lf per ml, and inject 1 ml doses of each dilution into groups of five mice. Observe the animals for four days and count the proportion of animals which die of tetanus symptoms during that period. From these data derive the 10
50 by the method of Reed
& Muench. D.J. Finney, Probit Analysis 3rd ed. London 1971 (Cambridge University Press)

BLG/UNDP/82.1 Page 105
(ii) the lowest dilution of vaccine protects more than half of the animals;
(iii) the highest dilution has protected less than half; and
(iv) the dose response curves of the test and reference vaccines do not deviate significantly from parallelism and linearity. If no standardized test for parallelism is available a test may be accepted provided that the slope of the steeper curve is not more than 150% that of the shallower curve.
It has been found that the presence of pertussis vaccine has a strong adjuvant effect on the tetanus potency in mice. It will be found, therefore, that DPT vaccines show a higher tetanus potency of about 50% when tested in mice than when tested in guinea-pigs. For this reason in some requirements a higher tetanus potency is required for DPT vaccines, when tested in mice. A reasonable potency level for adsorbed tetanus toxoids can be considered to be 40 I.U./single human dose (60 I.U./ single human dose in DPT vaccines when tested in mice).
Method of assay for unadsorbed
The principles for assaying unadsorbed vaccine are similar to those already described for the assay of adsorbed vaccine. There are however four major differences:
(i) the test must be performed in guinea-pigs; (ii) vaccines are tested against an unadsorbed
standard;

BLG/UNDP/82.1 Page 106
(iii) the dose response curve is less steep as a result of which the interval between doses should be threefold rather than twofold;
(iv) the precision of the assay is lower necessitating the use of larger numbers of animals in the experimental groups.
3. Suggested method for individual identification of guinea-pigs in an animal room
The following scheme allows for the individual identification of more than 800 guinea-pigs. The identification consists of dye marks on head, back and legs. For recording purposes head and back colours are designated as a capital letter, thus:
A Red head B Red back C Green head D Green back E Mauve head F Mauve back H Red head, green back K Red head, mauve back
R Mauve head, green back S Red head, red back T Green head, green back W Mauve head, mauve back X Yellow head Z Yellow back
L Green head, red back N Green head, mauve back P- Mauve head, red back
AA Yellow head, red back BB Yellow head, green back CC Yellow head, mauve back DD Red head, yellow back EE Green head, yellow back FF Mauve head, yellow back
Leg colours are designated by small letters, and number thus:
Colour
red
red
Letter 1
a left front
b left front
+left rear
2
right front
left front
+right rear
3
left rear
right front
+left rear
4
right rear
right front
+right rear
5
both front
both rear

BLG/UNDP/82.1 Page 107
Colour Letter 1 2 3 4 5
green c left right left right both front front rear rear front
green d left left right right both front front front front rear
+left +right +left +right rear rear rear rear
mauve e left right left right both front front rear rear front
mauve f left left right right both front front front front rear
+left +right +left +right rear rear rear rear
yellow h left right left right both front front rear rear front
yellow k left left right right both front front front front rear
Example: Hk4 = Red head, green back, right front and right rear legs coloured yellow
Dyes: Mauve
Green
Red
methyl violet 1%
malachite green 1%
basic fuchsin 1%
Boil 1 litre distilled water. Remove from heat, sprinkle 10 g of dye on the surface and stir in.
Yellow - Picric acid 0.5%
Boil 1 litre distilled water. Remove from heat, sprinkle 5 g of dye on the surface. Do not stir.
The adhesion of dyes to animal furs is improved by the addition of 1 drop Lissopol (BDH) to 100 ml of solution.

BLG/UNDP/82.1 Page 108
VACCINE: Tetanus Toxoid
TEST: Test for Identity
CLASS OF TEST B
TET-2
REQUIREMENT FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE, TETANUS TOXOID, AND COMBINED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Section 5.1
IDENTITY TEST
The identity tests on a batch of toxoid can be carried out by both "in vitro" and "in vivo" methods. Although it is a simple procedure to establish the identity of fluid toxoid by a flocculation test, in the case of adsorbed toxoids it is necessary to dissolve the adsorbed material by the addition of sodium citrate.
For fluid toxoids a tetanus antitoxin is diluted so as to contain 20 IU per ml. Increasing volumes of 0.3, 0.4, 0.5, 0.6, 0.7, 0.8 ml are pipetted into a series of tubes, the volumes are made up with normal saline to 1 ml and 1 ml of fluid toxoid, diluted to approximately 10 Lf/ml, is added to each of0the tubes. The tubes are placed in a water bath at 50 C and observed continuously. The tube containing 0.5 ml of antitoxin or the tube on either side of it may show flocculation and such flocculation establishes the identity of the product.
For an adsorbed toxoid about 0.5 g of sodium citrate is added to 10 ~1 of the adsorbed toxoid. The mixture is

BLG/UNDP/82.1 Page 109
incubated at 37°C for one or two days to dissolve. After the aluminium salt has dissolved the flocculation test similar to one stated above is used. The flocculation occurs in the same way though it may take a longer time.
In the "in vivo" test a suitable dose of toxoid is injected into guinea-pigs and the presence of antitoxin in the guinea-pigs' blood can be demonstrated by the protection test carried out in mice.

BLG/UNDP/82.1 Page 110
VACCINE: Tetanus Toxoid
TEST: Test for irreversibility and test for specific toxicity
CLASS OF TEST C
TET-3
REQUIREMENTS FOR DIPHTHERIA TOXOID, PERTUSSIS VACCINE, TETANUS TOXOID, AND COMBINED VACCINES
(Requirements for Biological Substances Nos. 8 and 10) (Revised 1978)
Part A Section 3.5.5
Although this is applied to the bulk toxoid and not on the final vaccine control laboratories should be in a position to ensure that the tetanus toxoid used in the vaccine has not reverted to toxin. In countries with a high ambient temperature the test for tetanus toxin should be applied to the final vaccine.
Procedure
A sample of purified toxoid is diluted in saline containing 0.01% thiomersal to give 300 ml of 10 Lf per ml toxoid. Aliquots of 100 ml of the diluted toxoid are held at 4°, 20° and 32° for six weeks. 5 ml of each sample is injected subcutaneously into each of five guinea-pigs weighing between 280 and 300 mg, 2.5 ml on each side of the abdomen. The pigs are weighed on the first, third and seventh day following injection, then weekly for a further three weeks. The guinea-pigs are examined daily for any evidence of tetanus toxin poisoning.

Criteria for acceptance
BLG/UNDP/82.1 Page 111
The animals should shown no signs of tetanus intoxication due to lethal toxin.

BLG/UNDP/82.1 Page 111 bis

POLIOMYELITIS VACCINE

POLIOMYELITIS VACCINE
BLG/UNDP/82.1 Page 111 ter

BLG/UNDP/82.1 Page 112
VACCINE: Poliomyelitis (oral)
TEST: Test for virus titration (potency)
CLASS OF TEST B
POL-l
REQUIREMENTS FOR POLIOMYELITIS VACCINE (ORAL) (Requirements for Biological Substances No. 7)
(Revised 1971)
Part A Section 5.3
Addendum 1980
TEST FOR VIRUS CONTENT
There are few tests applied to Poliomyelitis vaccine (oral) in its final form. This is because all tests to demonstrate the presence of extraneous agents have been applied to the appropriate stages of production and the tests for safety (neurovirulence and markers) are applied to the monovalent bulk materials. The vaccines at the monovalent bulk stage are released by national control authorities and the protocols of production of the final vaccine include data on the dilution of each of the monovalent pools incorporated into each mono-, di-or trivalent final vaccine.
It is important that imported vaccines are checked for the virus content of each virus type not only at the time of importation but at regular intervals during their storage. It is particularly important to ensure that all vaccinated subjects are given vaccine containing a virus titre for each virus type known to give immunity.

BLG/UNDP/82.1 Page 113
THE ASSAY METHOD FOR THE DETERMINATION OF THE VIRUS CONTENT OF FINAL BULK SUSPENSIONS
OF ORAL POLIOVIRUS VACCINE
The preparation to be assayed and a reference preparation, the virus titre of which has been determined on several occasions, are diluted in Earle's balanced Basal Medium Eagle (BME) containing 0.13% sodium bicarbonate and 4% fetal calf serum with penicillin (200 pg/ml) and streptomycin (100 pg/ml).
It is convenient to make tenfold dilgtion s!7ps of the virus suspensions initially to about 10 or 10 , but for those dilutions which are to be inoculated into cell cultures the dilutions should be prepared in twofold steps. A preliminary assay may be required to ensure that in the test the dilution range selected encompasses at least 3 dilutions, infecting between 10% and 90% of the cultures inoculated.
Groups of 12 flat-bottomed wells in a microtitre plate are inoculated with 0.1 ml of each of the selected dilutions of virus followed by 0.1 ml of a
3cell
suspension containing approximately 5 x 10 of the Hep-2 (Cincinnati) line. (See below for the technique of subcultivation of the cells). The plates are sealed and incubated at 35-36~ for 7 days.
The cultures are examined for the presence of a specific viral cytopathic effect on days 3-5 and again on day 7. The observations are recorded and the TCID
0 is
calculated on the basis of the observation on day f. The The assay should be repeated on at least two occasions and a mean taken. For the estimated potency of the monovalent lot to be accepted as valid the observed mean for the reference should be within 0.5 log
10TCID
50 of the
established mean for this preparat1on.

BLG/UNDP/82.1 Page ll4
CULTIVATION OF HEP-2 CINCINNATI CELLS
1. Receipt of a cell seed
A 150-ml bottle contains a confluent, or2nearly confluent, sheet of cells approximately 50 em • It is filled with growth medium (i.e. BME to which serum and antibiotics have already been added) to prevent the loss of cells in transit. On arrival, decant all except about 15 ml of the medium and incubate the culture at 37°C till a
0 confluent sheet has been formed. Stored at 4 C, the decanted medium may be used for further subcultivation of cells.
2. Subcultivation of cells
2.1 When the cell sheet is quite confluent pour off medium and rinse twice with 5 ml of trypsin solution (see below for reagents). Pour off the trypsin and allow the bottle to remain at room temperature (or incubate at 37°C) until the cells begin to detach from the glass.
2.2 Add about 5 ml of pre-warmed BME (see below for reagents). Agitate the bottles so that the medium washes back and forth and causes complete release of cells from the bottle wall. When release is complete, aspirate thoroughly with a 5 ml pipette with a bulb in order to effect maximum disaggregation of cells.
2.3 Add a further 55 ml of medium and either use the suspension for a virus assay or distribute one-quarter of the total volume of cell suspension to each of 3 additional bottles having the same surface area as the "parent" bottle and incubate the cultures at 37°C. This is a 1:4 "sElit" with cells at a concentration of approximately 5 x 10 cells per ml.
2.4 Subculture cells (1:4 "split") at intervals of 3-4 days, when the cells growing normally will have formed confluent sheets.

BLG/UNDP/82.1 Page 115
2.5 It is important to keep a record of the number of passages through which the cells have been propagated. The virus sensitivity of the cells may decrease during subcultivation.
Reagents
1. Trypsin: Final concentration 0.25% in Balanced Salt Solution (BS~ (Hanks', Earle's, or
2.
Phosphate Buffered Saline (PBS), pH about 7.8.
Medium: Basal Medium Eagle (BME) in Earle's BSS 95%(by volume) Fetal calf serum 5%(by volume) Antibiotic(s):
Penicillin (100 units/ml) +
Streptomycin (100 pg/ml)
To buffer, add sodium bicarbonate to a final concentration of 0.088%. Pre-warm medium and trypsin solution to 37°C before use.
Titration of Virus Titre in a Multivalent Vaccine
In the determination of virus titre of a particular type in a bivalent or trivalent mixture the types of virus not being assessed must be suppressed by neutralizing antisera. Such antisera must be monospecific and of a high titre. Almost all antisera have the ability to suppress. heterologous virus types to a low titre of antibody and the sera should be of such high titre that non specific suppression can be diluted out.
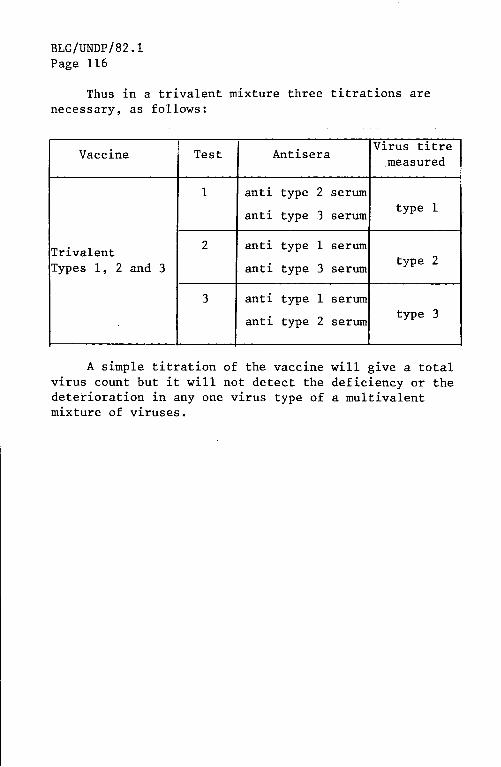
BLG/UNDP/82.1 Page 116
Thus in a trivalent mixture three titrations are necessary, as follows:
Vaccine Test Antisera Virus titre I measured
1 anti type 2 serum
anti type '3 serum type 1
Trivalent 2 anti type 1 serum
Types 1, 2 and 3 anti type 3 serum type 2
3 anti type 1 serum
anti type 2 serum type 3
A simple titration of the vaccine will give a total virus count but it will not detect the deficiency or the deterioration in any one virus type of a multivalent mixture of viruses.

VACCINE: Poliomyelitis (oral)
TEST: Test for Identity
BLG/UNDP/82.1 Page 117
CLASS OF TEST B
POL-2
REQUIREMENTS FOR POLIOMYELITIS VACCINE (ORAL) (Requirements for Biological Substances No. 7)
(Revised 1971)
Part A Section 5.1
TEST FOR IDENTITY
The virus(es) in the vaccine are identified by neutralization with specific serum.
The virus in the vaccine is incubated with a quantity of monospecific antiserum known to neutralize the virus titre of the vaccine. The mixtures are incubated at 35°C for 3 hours and inoculated into cell cultures such as primary monkey kidney or Hep-2 cells. The identity is confirmed by the neutralization of the virus by the monovalent antiserum.
The potency test (POL-l) of polyvalent vaccine may serve as an identity test.

BLG/UNDP/82.1 Page 117 bis

MEASLES VACCINE

MEASLES VACCINE
BLG/UNDP/82.1 Page 117 ter

BLG/UNDP/82.1 Page 118
VACCINE: Measles (live)
CLASS OF TEST B
MEA-l
TEST: Test for Virus Contentration (potency)
REQUIREMENTS FOR MEASLES VACCINE (LIVE) (Requirements for Biological Substances No. 12)
Part A Section 5.3
TEST FOR VIRUS CONTENT
The virus concentration in the vaccine is measured by plaque assay in vera cells.
The virus is diluted in chilled sucrose/phosphate/ glutamate/albumin (SPGA) (see Annex 2) in 0.5 log10 intervals using a separate pipette for each dilut1on. The methods of measuring the virus content of vaccine either by a plaque assay in microtitre plates or by a 50% tissue culture infectious dose (TCID
50) are described.
Propagation of VERO cells and titration of virus
Vera cell cultures
Vera cells are used within ten passages of a cloned, mycoplasma free stock held in liquid nitrogen.
2 Cells are grown as monolayers in 120 em (500 ml)
prescription bottles at 36.5°C in 60 ml volumes of medium. Growth medium consists of 199 medium (Earl's base) supplemented with 5% v/v fetal calf serum, 100 pg/ml penicillin G and 100 pg/ml streptomycin sulphate. Stock

BLG/UNDP/82.1 Page 119
cultures are passaged weekly at a 1:8 split ratio after resuspension of cells by a brief treatment with cation free PBS containing O.OS% w/v trypsin (Difco 1:12SO) and 0:1% w/v EDTA.
Prior to assays, cells from S-8 days confluent cultures are resuspended insgrowth medium and concentration adjusted to S - 10 cells/rnl on the basis of haernacytorneter counts. For each batch of assays, a single homogenous pool of cells is prepared, dispensed into 100 rnl aliquots and held at 4°C for use within three hours. Cell ~ield at this Szage is USUally in the region of 3S X 10 cells per 120 ern culture bottle.
Plaque assay
Virus is diluted in prerneasured volumes of chilled sucrose/phosphate/glutamate/albumen (SPGA) stabilizer to give an estimated SO p.f.u. in SO ul. Where titre is unknown, a half log dilution series is used. When parallel plaque and TCDSO assays are carried out, the diluent for the latter method is used in place of SPGA.
Immediately after preparation, SO ul volumes of test dilutions are dispensed into each of three or more wells in a 24 well plate (Falcon 3008 or Linbro FB 16-24-TC). On completion of each plate, 1 rnl of standard VERO cell suspension is added to each well by pipette or Cornwall syringe. Plates are transferred in groups of three or four to a humidified incubator with S% co
2 in air at 36°C.
After a 3 h incubation, medium is aspirated from the adsorbed cells and replaced with VERO growth medium containing 0.7S% w/v carboxyrnethylcellulose (CMC).
Plates are then incubated at 36°C usually for 10 days but depending on the strain of virus •tsed. Mono layers are fixed with 10% forrnol saline, stained with dilute crystal violet solution, washed and allowed to dry.
Plaques, appearing as clear or granular foci in a uniformly stained cell sheet, are counted and the titre per rnillilitre or per vial is then calculated.

BLG/UNDP/82.1 Page 120
TCID50
Assay
Serial fourfold dilutions of virus are prepared in chilled 199 medium (Earle's base) supplemented with 2% v/v fetal calf serum, 25 mM hepes buffer pH 7.2 and 100 ,ug/ml penicillin and streptomycin. 50 ul aliquots of test dilutions are immediately dispensed into each of 12 wells in a microtitre plate (Falcon, Microtest 11). On completion of each plate, 100 ul of standard Vero cell suspension is added; to each well. Plates are incubated at 36°C for 10 days.
The entire surface of each well is then examined for cytopathic effects on an inverted microscope. Titre i~ calculated by the Spearman/Karber method (Finney, 1952 ). In both plaque and TCD50 assays, all dilutions and dispensing of small volumes is carried out using fixed volume micropipettes with disposable polypropylene tips.
Sucrose/phosphate/glutamate/albumen diluting fluid
Composition:
1
Sucrose
KH2
Po4
K2HP04
Glutamate
Serum albumen (bovine or human)
0.218 Molar
0.0038 Molar
0.0072 Molar
0.0049 Molar
1%
D.J. Finney (1952) Statistical Method in Biological Assay, London, Griffith

Trypsin 0.25%
For 50 litres:
Trypsin GIBCO
NaC1
KC1
Na2HP04 (2H2o)
Phenol red
Phosphate buffered
For 1000 litres:
NaCl
KCl
Na2HP04
(2H20)
KH2Po4
Cat. 840-7072
saline (PBS)
BLG/UNDP/82.1 Page 121
125 g
400 g
15 g
70.5 g
1.0 g
1200 g
200 g
1130.5 g
200 g

BLG/UNDP/82.1 Page 122
VACCINE: Measles
TEST: Test for Identity
CLASS OF TEST B
MEA-2
REQUIREMENTS FOR MEASLES VACCINE (LIVE) (Requirements for Biological Substances No. 12)
Part A Section 5.1
THE TEST
The measles virus in the final container is identified by reconstitution of the vaccine and adding a portion of it to monospecific neutralizing antibody.
The neutralized mixture is added to VERO cell cultures (see Test for Virus Titration MEA-l); two control groups are included - (i) cell cultures from the same batch are inoculated with the non-neutralized measles vaccine, and (ii) uninoculated cell cultures. The cell cultures are incubated at 36°C for 10 days.
The non-neutralized measles virus shows a cytopathic effect typical of measles virus whereas the neutralized virus has no effect on the cell cultures which should have the appearance of cells similar to theuninoculated control cultures.

VACCINE: Measles
TEST: Test for Stability
BLG/UNDP/82.1 Page 123
CLASS OF TEST B
MEA-3
REQUIREMENTS FOR MEASLES VACCINE (LIVE) (Requirements for Biological Substances No. 12)
Addendum 1981
Part A Section 5.7
THE TEST
Samples of the vaccine in the freeze dried final form shall be incubated at 37°C for seven days. At the end of the incubation period the incubated vaccine shall be reconstituted and the virus titre compared with that of samples of vaccine stored at -20°C or below and reconstituted in a similar manner. The content of virus shall be compared by a plaque assay technique (see MEA-l).
For the vaccine to pass the stability test two criteria shall be satisfied:
1. After in3ubation the vaccine shall have retained at least 10 plaque forming units (PFU) per human dose and
2. During incubation the virus titre shall not have been decreased by more than 1.0 Log
10 PFU per human dose.
Both criteria must be satisfied.

BLG/UNDP/82.1 Page 123 bis

PROTOCOLS

PROTOCOLS
BLG/UNDP/82.1 Page 123 ter

BLG/UNDP/82.1 Page 124
SUMMARY PROTOCOL FOR DIPHTHERIA PERTUSSIS AND TETANUS VACCINE:
PRODUCTION AND TESTING
(Based on Requirements for Biological Substances, Nos 8/10 (Requirements for Diphtheria Toxoid, Pertussis Vaccine, Tetanus Toxoid and Combined Vaccines) Revision 1978)
Identification of Final Lot
Name and address of manufacturer
Lot No.
Date of manufacture of final lot
Nature of final product (plain or adsorbed)
Volume of recommended single human dose
No. of containers in final lot for each filling volume
DIPHTHERIA TOXOID
Information on Manufacture
1. Strain
Identity of C. diphtheriae strain used in vaccine

2. Single hanests included in final bulk*
Medium
Period of incubation
Date of earliest harvest included
Conditions of storage
3. Bulk purified toxoid
Results of test for antigenic purity (Lfjmg protein N)
PERTUSSIS VACCINE
Information on Manufacture
I. Strains
Identity of B. pertussis strain used in vaccine
Serological type of strain
Date(s) of reconstitution of ampoule(s) for manufacture
2. Single hanests included in final bulk
Medium
Date of inoculation
Date of harvest
Method of killing
Opacity
Conditions of storage
3. Bulk material
Results of tests for living organisms
BLG/UNDP/82.1 Page 125
• A list of the identification numbers of the single harvests and bulk purified toxoids should be included.

BLG/UNDP/82.1 Page 126
TETANUS TOXOID
Information on Manufacture
1. Strain
Identity of C. tetani strain used in vaccine
2. Single hanests included In final bulk*
Medium
Period of incubation
Date of earliest harvest included
Conditions of storage
3. Bulk toxoid
Nature of bulk toxoid
Results of test for antigenic purity, if applicable (Lf/mg protein N)
Purified/Unpurified
INFORMATION ON BLENDING
Composition of the Final Bulk
1. Identity of diphtheria toxoid component
Lfjml
Volume added
2. Identity of tetanus toxoid component
Lf/ml
Volume added
3. Identity of pertussis vaccine component
Opacity units
Volume added
• A list of the identification numbers of the single harvests and bulk purified toxoids should be included.

4. AdjuYant
Nature and concentration
mg/AI orCa in final bulk
Volume added
5. PreserntiYe
Nature
Concentration in final product (by assay or calculation)
6. Buffer
Concentration
7. Date of completion of final bulk
Volume of final bulk
Tests on Final Bulk
I. Sterility
Date of test and results
Was a repeat test necessary?
2. Specific toxicity
Date of test for diphtheria toxoid and results
Date of test for pertussis vaccine and results
Date of test for tetanus toxoid and results
3. Potency
(i) Diphtheria
Weight of animals
No. of animals per dose of toxoid
BLG/UNDP/82.1 Page 127

BLG/UNDP/82.1 Page 128
Date of immunization and volume of dilutions administered
Date of challenge or bleed
Challenge dose
Date of end of test
(a) Results of lethal challenge test (if done)
Reference toxoid ( IU/ml)
Test toxoid
Dilution No. of survivors
Potency of test toxoid IU per single human dose
95% confidence limits of potency
(b) Results of intradermal challenge test (if done)
Reference toxoid ( IU/ml)
Test toxoid
Potency of test toxoid
Dilution
IU per single human dose
95% confidence limits of potency
ED 5o
ml
ml
Mean score

(ii) Pertussis
Weight and sex of mice
No. of mice per dose of vaccine
Date of immunization
Challenge dose
Date of challenge
Date of end of test
Results of challenge test
Reference vaccine ( IU/ml)
Test vaccine
Dilution No. of survivors
Potency of test vaccine I U per single human dose
95% confidence limits of potency
(iii) Tetanus
Species of animals
Weight of animals
No. of animals per dose of toxoid
Date of immunization and volume of dilutions administered
Date of challenge or bleed
Challenge dose
Date of end of test
BLG/UNDP/82.1 Page 129
ED 5o
ml
ml

BLG/UNDP/82,1 Page 130
Results of challenge test
Reference toxoid ( IUjml)
Test toxoid
Dilution No. of survivors
Potency of test toxoid IU per single human dose
95% confidence limits of potency
TESTS ON FINAL LOT
I. Identity
Test for diphtheria toxoid and results
Test for pertussis vaccine and results
Test for tetanus toxoid and results
2. Sterility
No. of containers examined
Method of test
Date of start of test
Date of end of test
Results
3. Potency
ED so
ml
ml
If this test has not been performed on the final bulk, report these data in the space provided under "Tests on final bulk".

4.
5.
6.
lnnoculty
No. of animals
Route of injection
Volume of injection
Date of start of test
Date of end of test
Results
Preservative
Concentration of preservative
pH
Results of pH test
Signature of head of laboratory
Mice
BLG/UNDP/82.1 Page 131
Guinea-pigs
Certification by person taking overall responsibility for production of the vaccine:
I certify that lot No. of the vaccine satisfies Part A of the WHO Requirements for Diphtheria Toxoid, Pertussis Vaccine, and Tetanus Toxoid.
Signature
Name typed
The protocol must be accompanied by a sample of the label and a copy of the leaflet.

BLG/UNDP/82.1 Page 132
SUMMARY PROTOCOL FOR DIPHTHERIA TOXOID PRODUCTION AND TESTING
Identification of Final Lot
Name and address of manufacturer
Lot No.
Date of manufacture of final lot
Nature of final product (plain or adsorbed)
Volume of recommended single human dose
No. of containers in final lot for each filling volume
Information on Manufacture
I. Strain
Identity of C. diphtheriae strain used in vaccine
2. Single harvests included in final bulk •
Medium
Period of incubation
Date of earliest harvest included
Conditions of storage
3. Bulk purified toxoid
Results of test for antigenic purity (Lfjmg protein N)
4. Final bulk
Date of preparation
Lfper ml
Results of test for residual free formaldehyde
pH
* A list of the identification numbers of the single harvests and bulk purified toxoids should be included.

5. Adjuvant
Nature
mgJAI orCa
6. Presenative
Nature
Concentration in final product {by assay or calculation)
7. Buffer
Concentration
Tests on Final Bulk
1. Sterility
Date of test and results
Was a repeat test necessary?
2. Specific toxicity
No. of animals
Date of injection
Dose of toxoid injected {Lf per animal)
Route of injection
Period of test
Results of test
3. Potency
Weight of animals
No. of animals per dose of toxoid
Date of immunization and volume of dilutions administered
Date of challenge or bleed
Challenge dose
Date of end of test
Bulk purified toxoid
BLG/UNDP/82.1 Page 133
Final bulk or final lot

BLG/UNDP/82.1 Page 134
Results of lethal challenge test (if done)
Reference toxoid ( IUjml)
Test toxoid
Dilution
Potency of test toxoid
No. of survivors
IU per single human dose
95% confidence limits of potency
Results of multiple intradermal test (if done)
ED 5o
ml
ml
Dilution Mean score
Reference toxoid ( IUjml)
Test toxoid
Potency of test toxoid IU per single human dose
95% confidence limits of potency

Tests on Final Lot
I. Identity
Test for diphtheria toxoid and results
Test for pertussis vaccine and results
Test for tetanus toxoid and results
2. Sterility
No. of containers examined
Method of test
Date of start of test
Date of end of test
Results
3. Potency
BLG/UNDP/82.1 Page 135
If this test has not been performed on the final bulk, report these data in the space provided under "Tests on final bulk".
4. lnnocuity Mice Guinea-pigs
No. of animals
Route of injection
Volume of injection
Date of start of test
Date of end of test
Results
s. Preservative
Concentration of preservative
6. pH
Results of pH test
Signature of head of laboratory

BLG/UNDP/82.1 Page 136
Certification by person taking overall responsibility for production of the vaccine:
I certify that lot No. of the vaccine satisfies Part A of the WHO Requirements for Diphtheria Toxoid.
Signature
Name typed
The protocol must be accompanied by a sample of the label and a copy of the leaflet.

BLG/UNDP/82.1 Page 137
SUMMARY PROTOCOL FOR PERTUSSIS VACCINE PRODUCTION AND TESTING
Identification of Final Lot
Name and address of manufacturer
Lot No.
Date of manufacture of final lot
Nature of final product (plain or adsorbed)
Volume of recommended single human dose
No. of containers in final lot for each filling volume
Information on Manufacture
1. Strain
Identity of B. pertussis strain used in vaccine
Serological type of strain
Date(s) of reconstitution of ampoule(s) for manufacture
2. Single harvests Included in final bulk •
Medium
Date of inoculation
Date of harvest
Method of killing
• A list of the identification numbers of the single harvests and bulk suspensions should be included.

BLG/UNDP/82.1 Page 138
Opacity
Conditions of storage
3. Bulk material
Method of killing
Results of tests for living organisms
Date of preparation of final bulk
Volume of final bulk
4. Adjuvant
Nature
mgJAI orCa
Volume added
5. Preservative
Nature
Concentration in final product (by assay or calculation)
6. Buffer
Concentration
Tests on Final Bulk l. Sterility
Date of test and results
Was a repeat test necessary?
2. Toxicity
Species, weight, and sex of animals
No. of animals
Date of injection
Route of injection
Period of test
Results of test

3. Potency
Weight and sex of mice
No. of mice per dose of vaccine
Date of imm.unization
Challenge dose
Date of challenge
Date of end of test
Raults of potncy test
Reference vaccine
IUfml)
Test vaccine
Dilution
Potency of test vaccine
BLG/UNDP/82.1 Page 139
No. of survivors
ml
ml
IU per single human dose
95% confidence limits of potency
Tests 011 Final Lot
I. ldntlty
Test for diphtheria toxoid and results
Test for pertussis vaccine and results
Test for tetanus toxoid and results
1. Sterility
No. of containers examined
Method of test

BLG/UNDP/82.1 Page 140
Date of start of test
Date of end of test
Results
3. Potency
4.
5.
6.
If this test has not been performed on the final bulk, report these data in the space provided under "Tests on final bulk".
lnnoculty Mice Guinea-pigs
No. of animals
Route of injection
Volume of injection
Date of start of test
Date of end of test
Results
Preservative
Concentration of preservative
pH
Results of pH test
Signature of head of laboratory
Certification by person taking overall responsibility for production of the vaccine: I certify that lot No. of the vaccine satisfies Part A of the WHO Requirements for Pertussis Vaccine.
Signature
Name typed
The protocol must be accompanied by a sample of the label and a copy of the leaflet.

BLG/UNDP/82.1 Page 141
SUMMARY PROTOCOL FOR TETANUS TOXOID PRODUCTION AND TESTING
ldelltlflcatloa of Fl•al Lot
Name and address of manufacturer
Lot No.
I >ate oi manufacture of finltllot
Nature of final product (plain or adsorbed)
Volume of recommended single human dose
No. of containers in final lot for each filling volume
lnformatloa on Manufacture
I. Stnln
Identity of C. tetani strain used in vaccine
2. Sl .. le llanelts IKI-*41n final Hlk •
Medium
Period ofincubation
Date of earliest harvest included
Conditions of storage
3. Bulk toxoid
Nature of bulk toxoid
Results of test for antigenic purity, if applicable (Lf/mg protein N)
4. Flnalllulk
Date of preparation
Lfperml
Results of test for residual free formaldehyde
pH
PurifiedfUnpurified
~ A list of th~ identification numbers of the single harvests and bulk purified toxotds should be mcluded.

BLG/UNDP/82,1 Page 142
5. Adjuvant
Nature
mg/AI orCa
6. Preservative
Nature
Concentration in final product (by assay or calculation)
7. Buffer
Concentration
Tests on Final Bulk
I. Sterility
Date of test and results
Was a repeat test necessary?
l. Specific toxicity
No. of animals
Date of injection
Dose of toxoid injected (Lfper animal)
Route of injection
Period of observation
Results of test
3. Potency
Species of animals
Weight and sex of animals
NQ. of animals per dose of toxoid
Daie of immunization and volume of dilutions administered
Date of challenge or bleed
Challenge dose
Date of end of test
Bulk purified toxoid
Final bulk or final lot

Re~~~lts of challenie test
Reference toxoid ( IUjml)
Test toxoid
Dilution No. of survivors
BLG/UNDP/82.1 Page 143
ml
ml
Potency of test toxoid IV per single human dose
95% confidence limits of potency
Tests on Final Lot
I. t•entlty
Test for diphtheria toxoid and results
Test for pertussis vaccine and results
Test for tetanus toxoid and results
1. Sterility
No. of containers examined
Method of test
Date of start of test
Date of end of test
Results
J. Potency
If this test has not been performed on the final bulk, report these data in the space provided under "Tests on final bulk".

BLG/UNDP/82.1 Page 144
4. lnnoculty
No. of animals
Route of injection
Volume of injection
Date of start of test
Date of end of test
Results
5. Presenatlve
Concentration of preservative
6. pH
Results of pH test
Signature of head of laboratory
Mice Guinea-pigs
Certification by person taking overall responsibility for production of the vaccine:
I certify that lot No. of the vaccine satisfies Part A of the WHO Requirements for Tetanus Toxoid.
Signature
Name typed
The protocol must be accompanied by a sample of the label and a copy of the leaflet.

BLG/UNDP/82.1 Page 145
SUMMARY PROTOCOL FOil DillED BCG VACCINE PRODUCfiON AND TESTING
(Based on the Requirements for Biological Substances, No. ItRequirements for Dried BCG Vaccine, WHO Technical Report Series, No. 638, 1979)
IM!Itlficatiell of Flaall...et
Name and address of manufacturer
Lot No. and type of vaccine
Date of manufacture of final lot
I. SeM lot
Identity of seed lot
Origin of seed lot
Date of preparation of seed lot
Date of reconstitution of seed lot a~poule
Date of receipt of seed lot
Intradermal{PercutaneousfOther
1. Te!ib oa leetl lot (If these data on the same seed lot have been submitted before, completion of this paragraph is not necessary.)
(a) Identity test
Identified as BCG yes/no
(b) Test for absence of contaminating microorganisms
Date of start of test

BLG/UNDP/82.1 Page 146
Date of end of test
Results
(c) Safety test
Absence of virulent mycobacteria
Dose injected
No. of guinea-pigs given injection
Weight range and sex of guinea-pigs
Observation period
Health of animals during test
Weight gain
Results (Passed/Failed)
Seed lot approved
Date of approval
First/est
yesfno
Information on Manufacture
3. Single hanest
No. of passages from seed
Medium
No. of containers inoculated
Date of inoculation
Date of harvest
Visual inspection and results
4. Final bulk
Date of preparation
No. of single harvests included
(a) Absence of contamination
Quantity tested
Media
Duration of test
First test
Repeat/est if necessary
Repeat/est if necessary

Results
(b) Absence of virulent mycobacteria
No. of human doses injected
No. of guinea-pigs given injection
Weight range and sex of guinea-pigs
Observation period
Health of animals during test
Weight gain
Results (Passed/Failed)
(c) Substances added to final bulk and concentration
5. Freeze-drying
Type and size of containers
No. of doses per container
First test
BLG/UNDP/82.1 Page 147
Repeat test if necessary
Method used for sealing the containers Vacuum-sealing/Sealing under gas
No. of containers of each size in the final lot
Information on Final Product Controls
Recommended reconstitution fluid
Volume of reconstitution fluid per final container
6. Identity test
Type of test
Results
7. Absence of eontamination
No. of containers tested
Media
First test Repeat test if necessary

BLG/UNDP/82.1 Page 148
Duration of test
Results
8. Safety tests
(a) Absence of virulent mycobacteria (if test not performed on final bulk)
No. of human doses injected
Dilution factor applied if percutaneous vaccine
No. of guinea-pigs given injection
Weight range and sex of guinea-pigs
Observation period
Health of animals during test
Weight gain
Results (Passed/Failed)
(b) Skin reactivity test(ifperformed)
Period of test
(c) Total bacterial content
Method of estimation
Result (content of semi-dry organisms in mgfml)
(d) Culturable particles
Method of determination
First test
Vaccine
Dilution Mean injected diameter
Repeat test if necessary
Reference
Dilution Mean injected diameter

Medium
No. of containers tested
Mean count of culturable particles per ml and standard deviation
Date of start of test
9. Stability test
Date of start of test
Temperature of incubation
Time of incubation
No. of containers tested
Mean percentage of survival
Information on Release
Is the vaccine satisfactory?
Has the lot been released by the national control authority?
If yes, date
Can a certificate be supplied by the national control laboratory?
Which laboratory would supply such a certificate?
When was the vaccine tested in children in your country? (summary of results)
Signature of head of laboratory
yes/no
yes/no
yesfno
BLG/UNDP/82.1 Page 149
Certification by person taking overall responsibility for production of the vaccine:
certify that lot No. of BCG vaccine satisfies Part A of the WHO Requirements for BCG Vaccine.
Signature
Name typed
The protocol must be accompanied by a sample of the label, a copy of the leaflet, and a copy of the national control release certificate, if issued.

BLG/UNDP/82.1 Page 150
SUMMARY PROTOCOL FOR POLIOMYELITIS VACCINE (ORAL) SABIN STRAINS BASED ON REQUIREMENTS FOR POLIOMYELITIS VACCINE (ORAL)
(REQUIREMENTS FOR BIOLOGICAL SUBSTANCES NO. 7)
REVISED 1971 AND ADDENDUM 1980
NAME AND ADDRESS OF MANDF ACTURER
PROPRIETARY NAME -----------------------------------------------------------------
LOT. NO. OF VACCINE TRIVALENT BLEND-----------------------------------------------
FILLING LOT NO. NO. OF FILLED CONTAINERS -----------------
DATE OF INITIATION OF LAST TEST FOR VIRUS CONCENTRATION --------------------------------
EXPIRY DATE-------------------
NATURE AND CONCENTRATION OF STABILIZER ----------------------------------------------
VOLUME OF VACCINE CONTAINER------------------------------------------------------
VOLUME OF HUMAN DOSE (IN DROP AND/OR m1)
PRESCRIBED VIRUS CONCENTRATION PER HUMAN DOSE - TYPE
TYPE
TYPE
CONCENTRATION OF ANTIBIOTICS/HUMAN DOSE ------------------------------------------
MAXIMUM RESIDUAL CONCENTRATION OF SERUM/HUMAN DOSE
PRODUCTION CELL TISSUE:
MONOVALENT BULK SUSPENSIONS BLENDED IN TRIVALENT VACCINE
BULK NO.
DATE OF APPROVAL OF PROTOCOL OF BULK INDICATING COMPLIANCE WITH WHO REQUIREMENTS
TYPE 1 TYPE 2 TYPE 3

HLG/UNDP/82.1 Page 15]
The following sections are for the recording of the results of the tests performed during the production of the vaccine. It is intended that the complete document will provide evidence for consistency of production, thus if any test had to be repeated this must be indicated, Any abnormal result must be detailed on a separate sheet.
If any cell lot or virus harvest intended for production was rejected during the control testing this also should be recorded either in the following sections or on a separate sheet.

BLG/UNDP/82,1 Page 152
PRODUCTION IN HUMAN DIPLOID CELL CULTURES
POLIOVIRUS TYPE ______________________________ __ MONOVALENT BULK NO.
VIRUS SEED LOT USED FOR PRODUCTION --------------------------------------
DATE OF APPROVAL OF PROTOCOL INDICATING COMPLIANCE WITH WHO REQUIREMENTS ________________________________________________ __
CELL STRAIN USED FOR PRODUCTION:
MANUFACTURERS WORKING CELL BANK (MWCB) NO.
POPULATION DOUBLING LEVEL (PDL) OF MWCB
DATE OF APPROVAL OF PROTOCOL INDICATING COMPLIANCE WITH WHO REQUIREMENTS
NATURE AND CONCENTRATION OF ANTIBIOTICS USED IN PRODUCTION CELL CULTURE MAINTENANCE MEDIUM
PRODUCTION DETAILS
PRODUCTION CELL LOT NO.
DATE OF THAWING AMPOULE OF MWCB
TESTS OF CONTROL CELL CULTURES
RATIO OR PROPORTION OF CONTROL TO PRODUCTION CELL CULTURES
PERIOD OF OBSERVATION OF CULTURES -----------
RESULT
TESTS ON POOLED SUPERNATANT FLUIDS
DATE OF SAMPLING FROM PRODUCTION CELL CULTURES
TESTS FOR EXTRANEOUS AGENTS
VOLUME TESTED/CELL CULTURE TYPE
OBSERVATION PERIOD
DATE OF COMPLETION OF TESTS
RESULT
TESTS FOR BACTERIA, FUNGI & MYCOPLASMA
MEDIA USED AND DATE OF TEST
RESULT

BLG/UNDP/82.1 Page 153
PRODUCTION IN HUMAN DIPLOID CELL CULTURES CONT ...
PRODUCTION CELL CULTURES
VIRUS SEED LOT NO.
VIRUS INFECTIVITY/CELL RATIO
POPULATION DOUBLING LEVEL FOR VIRUS GROWTH
NO. OF CULTURES INOCULATED
DATE OF INOCULATION
TEMPERATURE OF INCUBATION
PERIOD OF INCUBATION
NO. OF CULTURES HARVESTED
TESTS ON SINGLE HARVESTS
VOLUME HARVESTED
DATE OF SAMPLING
TESTS FOR BACTERIA, FUNGI & MYCOPLASMA
RESULT
TESTS OF NEUTRALIZED SINGLE HARVESTS IN MONKEY KIDNEY AND HUMAN CELL CULTURES
BATCH NO. OF ANTISERUM USED
VOLUME TESTED
DATE OF INITIATING PRIMARY CELL CULTURE TESTS
PERIOD OF OBSERVATION
DATE OF SAMPLING CELL CULTURE FLUIDS -----
PERIOD OF OBSERVATION
DATE OF COMPLETION OF TEST
RESULT
TESTS IN RABBIT KIDNEY CELL CULTURES
VOLUME TESTED
DATE OF COMPLETION OF TEST
RESULT

BLG/UNDP/82.1 Page 154
PRODUCTION IN HUMAN DIPLOID CELL CULTURES CONT •..
TESTS IN ANIMALS
TESTS IN RABBITS
NO. AND WEIGHT OF ANIMALS
DATE OF INOCULATION
RESULTS OF INJECTION
QUANTITY INJECTED
RESULTS (SURVIVAL NOS. ETC)
TESTS IN ADULT MICE
NO. AND WEIGHT OF ANIMALS
DATE OF INOCULATION
RESULTS OF INJECTION
QUANTITY INJECTED
RESULTS (SURVIVAL NOS. ETC)
TESTS IN SUCKLING MICE
NO. AND WEIGHT OF ANIMALS
DATE OF INOCULATION
RESULTS OF INJECTION
QUANTITY INJECTED
RESULTS (SURVIVAL NOS. ETC)
TESTS IN GUINEA PIGS
NO. AND WEIGHT OF ANIMALS
DATE OF INOCULATION
RESUlTS OF INJECTION
QUANTITY INJECTED
RESULTS (SURVIVAL NOS. ETC)

BLG/UNDP/82.1 Page 155
PRODUCTION IN MONKEY KIDNEY CELL CULTURES
POLIOVIRUS TYPE
VIRUS SEED LOT USED FOR PRODUCTION
DATE OF APPROVAL OF PROTOCOL INDICATING COMPLIANCE WITH WHO REQUIREMENTS
MONKEY SPECIES USED FOR PRODUCTION:
QUARANTINE BATCH NO.
MONOVALENT BULK NO.
PERCENTAGE OF MONKEYS SURVIVING QUARANTINE PERIOD
NATURE AND CONCENTRATION OF ANTIBIOTICS USED IN PRODUCTION CELL CULTURE MAINTENANCE MEDIUM
PRODUCTION DETAILS
PRODUCTION MONKEY NO.
DATE OF TRYPSINISING
NO. OF CULTURES PREPARED
TESTS OF CONTROL CELL CULTURES
RATIO OR PROPORTION OF CONTROL TO PRODUCTION CELL CULTURES
PERIOD OF OBSERVATION OF CULTURES
RATIO OR PROPORTION OF CULTURES DISCARDED FOR NON SPECIFIC REASONS
RESULT
TESTS ON POOLED SUPERNATANT FLUIDS
DATE OF SAMPLING FROM PRODUCTION CELL CULTURES
TESTS FOR EXTRANEOUS AGENTS
VOLUME TESTED/CELL CULTURE TYPE
OBSERVATION PERIOD
DATE OF COMPLETION OF TESTS
RESULT
TESTS FOR BACTERIA, FUNGI & MYCOPLASMA
MEDIA USED AND DATE OF TEST
RESULT

BLG/UNDP/82.1 Page 156
PRODUCTION IN MONKEY KIDNEY CELL CULTURES CONT .•.
PRODUCTION CELL CULTURES
VIRUS SEED LOT NO.
VIRUS INFECTIVITY/CELL RATIO
NO. OF CULTURES INOCULATED
DATE OF INOCULATION
TEMPERATURE OF INCUBATION
PERIOD OF INCUBATION
NO. OF CULTURES HARVESTED
TESTS ON SINGLE HARVESTS
VOLUME HARVESTED
DATE OF SAMPLING
TESTS FOR BACTERIA, FUNGI & MYCOPLASMA
RESULT
TESTS OF NEUTRALIZED SINGLE HARVESTS IN MONREY KIDNEY AND HUMAN CELL CULTURES
BATCH NO. OF ANTISERUM USED
VOLUME TESTED
DATE OF INITIATING PRIMARY CELL CULTURE TESTS
PERIOD OF OBSERVATION
DATE OF SAMPLING CELL CULTURE FLUIDS -----
PERIOD OF OBSERVATION
DATE OF COMPLETION OF TEST
RESULT
TESTS IN RABBIT KIDNEY CELL CULTURES
VOLUME TESTED
DATE OF COMPLETION OF TEST
RESULT

BLG/UNDP/82.1 Page 157
PRODUCTION IN MONKEY KIDNEY CELL CULTURES CONT .•.
TESTS IN ANIMALS
TESTS IN RABBITS
NO. AND WEIGHT OF ANIMALS
DATE OF INOCULATION
RESULTS OF INJECTION
QUANTITY INJECTED
RESULTS (SURVIVAL NOS. ETC)
TESTS IN ADULT MICE
NO. AND WEIGHT OF ANIMALS
DATE OF INOCULATION
RESULTS OF INJECTION
QUANTITY INJECTED
RESULTS (SURVIVAL NOS. ETC)
TESTS IN SUCKLING MICE
NO. AND WEIGHT OF ANIMALS
DATE OF INOCULATION
RESULTS OF INJECTION
QUANTITY INJECTED
RESULTS (SURVIVAL NOS. ETC)
TESTS IN GUINEA PIGS
NO. AND WEIGHT OF ANIMALS
DATE OF INOCULATION
RESULTS OF INJECTION
QUANTITY INJECTED
RESULTS (SURVIVAL NOS. ETC)

BLG/UNDP/82,1 Page 158
TESTS OF BULK SUSPENSION
DATE OF FILTRATION OF BULK
POROSITY OF FILTERS USED
DATE OF SAMPLING
IDENTITY TEST
DATE
METHOD USED
RESULT
VIRUS CONCENTRATION
DATE
METHOD USED
RESULT
TESTS FOR CONSISTENCY OF VIRUS CHARACTERISTICS
NEUROVIRULENCE TEST
SPECIES OF MONKEY INOCULATED
DILUTION OF VACCINE INJECTED
NO. OF VALID MONKEYS INOCULATED WITH TEST SAMPLE ----------------
NO. OF POSITIVE MONKEYS OBSERVED
REFERENCE PREPARATION
DILUTION OF REFERENCE INJECTED
NO. OF VALID MONKEYS INOCULATED WITH REFERENCE
NO. OF POSITIVE MONKEYS OBSERVED
DATE OF TEST
RESULT
MEAN LESION SCORE OF TEST SAMPLE
MEAN LESION SCORE OF REFERENCE
(SEE ALSO ATTACHED FORMS RECORDING DETAILS OF THE HISTOLOGICAL OBSERVATIONS AND ASSESSMENT)

llLG/UNDP/82.1
PagP 1'i9
INTRASPINAL NE.UROVIRULENCE TEST IN MONKEYS HISTOLOGY
All dala !rom dll monkeys must be recorded whrch mdy rt->qurrf' expansron o! !111~ torm Clrnrcal ~rqns ot paralysrs musT be recorded nn a ;.eparate lorm
Test No Type F1nal Bulk No Spec1es -
Post Inoculation T1trg Log~<, TCID~.,/ml Ollulron ----
Date of InoculatiOn ---- Date ol end of test-------
---------- -- ------------ --- --- ----.,---, Hrs!Oiogrcal lesrons of pollomyelltrs
-------- ~- - -- ------------ --- ·-- r·- ·----------~-Lesion
Lumbar sectroos Av Cervrcal sections Av Brain A" 1----~~,-~-· ,- score
+++-t-H-t---t.T ~~Cb P M T Co-J--+-----1
123456789101112 ! 12345678910 1 2~---t
-++-+l i+-+-++++-+-+-t-+---l---1-e---t----:~ L-: -+---+
L R ' I I
"" M = Monkey number Cb V ~ Vahdrty of moculum P L "'Left M A = Rrght T Av -= Average Co Mad = Medulla
I I
Cerebellum Pons Mrdbram Thalamus Cortex
t ·t t-++-+-++-+-+--+-+ - - +
Mean lesion score
Srgnature----- Date--------
Name (typed)----------------

BLG/UNDP/82,1 Page 160
IN VITRO MARKER TESTS
RCT 40 MARKER TEST
DATE OF TEST
REDUCTION OF TITRE OF BULK SAMPLE
REDUCTION OF TITRE OF NEGATIVE REFERENCE
REDUCTION OF TITRE OF POSITIVE REFERENCE
RESULT
ADDITIONAL IN VITRO MARKER TEST
TEST METHOD
DETAILS
DATE OF TEST
RESULT
STABILIZER OR PRESERVATIVE ADDED TO MONOVALENT BULK
NATURE AND QUANTITY OF ANY STABILIZER OR PRESERVATIVE ADDED
PREPARATION OF TRIVALENT BULK
LOT NUMBER OF TRIVALENT BLEND
MONOVALENT BULKS IN BLEND
VOLUME IN BLEND
NATURE AND VOLUME OF STABILIZER
NATURE AND VOLUME OF DILUENT
TOTAL VOLUME OF BLEND
STERILITY TEST
DATE
MEDIA USED
RESULT
TYPE 1 TYPE 2 TYPE 3

FINAL FILLING
LOT NO.
BLENDING DETAILS
VOLUME OF TRIVALENT BLEND
NATURE AND VOLUME OF STABILIZER
NATURE AND VOLUME OF DILUENT
TOTAL VOLUME OF FINAL FILLING
DATE OF FINAL FILLING
NO. OF VIALS FILLED
TESTS ON FINAL FILLING
STERILITY TESTS
DATE
MEDIA USED
RESULT
TEST FOR VIRUS CONCENTRATION
TOTAL VIRUS CONTENT
DATE OF TEST
RESULT: VACCINE
REFERENCE1
TITRE OF INDIVIDUAL VIRUS TYPES
ANTISERUM BATCH NOS. USED IN TEST
DATE OF TEST
RESULT FOR TYPE
2
3
Identity reference preparations
BLG/UNDP/82.1 Page 161
VACCINE REFERENCE

BLG/UNDP/82.1 Page 162
SUMMARY PROTOCOL FOR MEASLES VACCINE (LIVE) BASED ON REQUIREMENTS FOR MEASLES VACCINE (LIVE) (REQUIREMENTS FOR BIOLOGICAL SUBSTANCES NO. 12)
NAME AND ADDRESS OF MANUFACTURER
LOT. NO. OF VACCINE
NO. OF FREEZE-DRYING LOT
DATE OF INITIATION OF LAST
TEST FOR VIRUS CONCENTRATION ---------------------------------------------------
EXPIRY DATE
PROPRIETARY NAME OF VACCINE
NO.. AND VOLUME OF AMPOULES OR VIALS IN THE LOT
SEED VIRUS STRAIN
TISSUE USED FOR PREPARING SEED LOT
DATE(S) OF SATISFACTORY TEST(S) FOR FREEDOM FROM EXTRANEOUS AGENTS
NUMBER OF PASSAGES BETWEEN VACCINE AND SEED
DATE OF NEUROVIRULENCE TEST OF SEED LOT
NUMBER OF MONKEYS INOCULATED
QUANTITY INOCULATED
NUMBER OF MONKEYS SURVIVING WITHOUT SPECIFIC SYMPTOMS
RESULT OF BLOOD SERUM TEST
RESULT OF HISTOPATHOLOGICAL
EXAMINATION (SPECIFY ANY ABNORMAL FINDINGS)
SEED VIRUS

BLG/UNDP/82.1 Page 163
CONTROL OF TISSUE CULTURES
TYPE OF TISSUE CULTURE USED FOR VACCINE PRODUCTION
QUANTITY USED FOR CONTROL
PERIOD OF OBSERVATION OF UNINOCULATED CONTROL CELLS
TEST FOR HAEMADSORPTION VIRUSES
TEST METHOD
RESULTS
TISSUE CULTURE TESTS
SAMPLE SIZE
TYPE OF CELL CULTURES INOCULATED
RESULTS
OTHER TESTS IF DONE
TYPE OF TEST(S)
RESULTS
SINGLE HARVESTS USED IN VIRUS POOL
ANTIBIOTICS USED IN TISSUE CULTURE MEDIUM, IF USED
CONCENTRATION
TESTS ON SINGLE HARVESTS
NO. OF HARVESTS
VIRUS CONCENTRATION
DATE OF STERILITY TEST
RESULT OF STERILITY TEST
DATE OF TEST FOR MYCOPLASMA ----------------------------
RESULT

BLG/UNDP/82.1 Page 164
TESTS ON VIRUS POOL
If any test had to be repeated or any abnormal result was observed, this has to be specified. If necessary a separate sheet of paper can be used.
STERILITY TEST
DATE
MEDIA USED
RESULTS
MYCOPLASMA TEST
DATE
MEDIA USED
RESULTS
TEST FOR M. TUBERCULOSIS
DATE
MEDIA USED
RESULTS
TESTS IN TISSUE CULTURE
VOLUME TESTED
TISSUE CULTURES USED
OBSERVATION PERIOD
RESULTS
TEST IN ADULT MICE
NUMBER AND WEIGHT OF MICE
DATE OF INOCULATION
ROUTE OF INOCULATION
QUANTITY INJECTED
RESULTS (SURVIVAL NUMBERS ETC.)

TEST IN SUCKLING MICE
NUMBER AND WEIGHT OF MICE
DATE OF INOCULATION
ROUTE OF INOCULATION
QUANTITY INJECTED
RESULTS (SURVIVAL NUMBERS ETC.)
TEST IN GUINEA PIGS
NUMBER AND WEIGHT OF GUINEA PIGS
DATE OF INOCULATION
ROUTE OF INJECTION
QUANTITY INJECTED
RESULTS (SURVIVAL NUMBERS ETC.)
TEST IN CHICKEN EMBRYOS
VOLUME OF VIRUS POOL TESTED
NUMBER OF EGGS USED
ROUTE(S) OF INOCULATION
RESULTS
ADDITIONAL TESTS (DEPENDING ON TYPE OF CELL USED)
TEST METHOD USED
RESULTS OF TEST
REST FOR NEUROTROPIC AGENTS IN MONKEYS (IF DONE)
DESCRIPTION OF METHOD
RESULTS
BLG/UNDP/82.1 Page 165

BLG/UNDP/82,1 Page 166
FINAL BULK SUSPENSION
DATE OF PREPARATION
ADDED STABILIZERS, DILUENT OR OTHER SUBSTANCES (WITH CONCENTRATION)
STERILITY TEST
DATE
MEDIA USED
RESULT
IDENTITY TEST
DATE
METHOD USED
RESULT
STERILITY TESTS
DATE
MEDIA USED
RESULT
WERE REPEAT TESTS NECESSARY? (IF SO, SPECIFY DETAILS)
TEST FOR VIRUS CONCENTRATION
DATE
METHOD
REFERENCE PREPARATION USED
NUMBER OF CONTAINERS TESTED
VIRUS CONCENTRATION FOUND IN EACH CONTAINER
MEAN VIRUS CONCENTRATION
FINAL PRODUCT

INNOCUITY lESTS
DATE
NO. OF MICE GIVEN INJECTIONS
VOLUME AND ROUTE
OBSERVATION PERIOD
RESULTS (SPECIFY DETAILS IN CASE OF DEATHS)
NO. OF GUINEA PIGS
VOLUME AND ROUTE
OBSERVATION PERIOD
RESULTS (SPECIFY DETAILS IN CASE OF DEATHS)
RESIDUAL MOISTURE
SIZE OF SAMPLE
MOISTURE CONTENT (%)
SIGNATURE OF HEAD OF LABORATORY:
BLG/UNDP/82.1 Page 167
CERTIFICATION BY PERSON TAKING OVERALL RESPONSIBILITY FOR PRODUCTION OF THE VACCINE:
CERTIFY THAT LOT NO. OF VACCINE SATISFIES PART A OF THE WHO REQUIREMENTS FOR MEASLES VACCINE (LIVE).
SIGNATURE:
NAME TYPED:

BLG/UNDP/82.1 Page 168
APPENDIX I
SUGGESTED SOURCES OF CELL CULTURE MATERIALS
The list shown below is representative of manufacturers of cell culture materials from whom it is possible to obtain certain equipment, chemicals, etc. It should be stressed that in most cases there are other manufacturers from whom similar materials of equal quality may be obtained. This list, therefore, does not express any preference for the companies mentioned.
Becton Dickinson AG (Flacon) Alte Reinacherstrasse 1-3 CH-4142 Munchenstein
Bio-Merieux Marcy-l'Etoile 69260 Charbonnieres-les-Bains France
Difco Laboratories Inc. P.O. Box 1058A Detroit, Michigan 48232 United States of America
Dynatech Produkte AG Oberfeldstrasse 20 CH-8302 Kloten
Flow Laboratories Ltd. P.O. Box 17 Second Avenue Industrial Estate Irvine, Ayrshire, KA12 8NB
Telex: 64784
Telex: 3800972 (Sales Office)
Telex: 23-5683
Telex: 57 575
Scotland Telex: 77231

APPENDIX I Cont •••
Flow Laboratories Ltd. P.O. Box 17 Victoria Park Heatherhouse Road Irvine, Ayrshire KA12 8NB Scotland, U.K.
Gibco-Europe b.v. Starnmeerstraat 5-7 P.O. Box 248 2130 AE Hoofddorp Netherlands
Luckham Ltd. Labro Works Victoria Gardens Burgess Hill Sussex RH15 9QN, U.K.
Nissui Seiyaku Co. Ltd. 5-11 Komagome 2-chome Toshima-ku Tokyo 170 Japan
Sterilin Ltd. 43-45 Broad Street Teddington Middlesex TWll 8QZ, U.K.
Tecnomara AG (Costar) Langgonserstrasse 11 D-6338 - Ruttenberg W. Germany
Hellcome Reagents Ltd. 299-303 Hither Green Lane London SE13 6TL U.K.
BLG/UNDP/82.1 Page 169
Telex: 77231
Telex: 41229
Telex: 87323
Telex:
Telex: 88 1 2910
Telex: 482 993
Telex: 896113