mikrokinetik - lehrstuhl für technische chemie ii: home · molekül stabil. bei einer reaktion...
TRANSCRIPT

1
MikrokinetikMikrokinetik
• Begriffe, Definitionen– Reaktionsgeschwindigkeit
• Reaktionskinetik– Differentialgleichungen– Ordnung– Mechanismus
• Parallele Reaktionen• Sequentielle Reaktionen
– Stationärer Zwischenzustand– Geschwindigkeitsbestimmender Schritt
• Theorie des Übergangszustandes– Aktivierungsenergie
DefinitionDefinition
Als Kinetik bezeichnet man die quantitative Erfassung undBeschreibung der dynamischen Parameter chemischerUmsetzungen und ihrer Abhängigkeit von Parametern wieDruck, Temperatur, usw.
Beschreibung der Geschwindigkeit der Annäherung einer chemischen Reaktion an das Gleichgewicht
• Verständnis des Mechanismus über das Studium von
ZielZiel
• Verständnis des Mechanismus über das Studium von Reaktionsraten
Zerlegung der Gesamtreaktion in eine Folge von elementaren Einzelreaktionen
• Auslegung von Reaktionsapparaten

2
ReaktionsmechanismusReaktionsmechanismus
Ein Reaktionsmechanismus ist eine Hypothese (Modell) über den Ablauf einer chemischen Reaktion. Er kann daher nie b i d i t t t d D T tbewiesen, sondern immer nur getestet werden. Das Testen einer Hypothese und das Ermitteln eines Modells läuft in Stufen ab.
1) Bestimmen der Kinetik der Gesamtreaktion und die Aufgliederung dieser Reaktion in Teilschritte.
2) Bestimmung der Reaktionszwischenprodukte und damit der Einzelreaktionen der Gesamtreaktion, sowie des Energieprofils der Reaktion.
3) Bestimmung der Reaktionsdynamik (der Geschwindig-keiten der Einzelschritte).
Energieprofil einer chemischen ReaktionEnergieprofil einer chemischen Reaktion
Was geschieht bei einer Reaktion?Ein Molekül ist eine stabile Anordnung von Atomen (Minimum der potentiellen Energie) relativ zueinander. Bei einer Reaktion werden di A i Si h i bil A d idie Atome umgruppiert. Sie gehen aus einer stabilen Anordnung in eine andere über.
Wie ändert sich die potentielle Energie im Verlauf der Reaktion?Zwischen den Energieminima der Reaktanten und der Produkte muss ein Maximum der potentiellen Energie liegen, das bei einer Reaktion überschritten werden muss. Diesen Zustand größter potentieller
ÜEnergie bezeichnet man als Übergangszustand oder aktiviertenKomplex.

3
Chemische Reaktion Chemische Reaktion
A → B
Übergangszustand • Man denkt sich in der g g
Energiebarriere desElementarschritts
Reaktionskoordinate
Kurve die potentielle Energie dargestellt, d.h., die Energie, die bei 0 K durchlaufen würde.
• Bei allen anderen Temperaturen käme noch thermische Energie dazu
λ
Epot ΔE
Endotherme Reaktion → Zunahme der potentiellen Energie im ReaktionsschrittExotherme Reaktion → Abnahme der potentiellen Energie im Reaktionsschritt
Energie dazu.
Darstellung von Reaktionsabläufen Darstellung von Reaktionsabläufen
Die Abszisse bezeichnet man als Reaktionskoordinate, sie soll die jeweilige Lage aller in den Vorgang involvierten Atomkerne darstellen.
Für eine genauere Darstellung müsste man ein vieldimensionales Koordinatensystem verwenden. Beschreibt man die Lage jedes Atomkerns mit drei Koordinaten, so würden für n Atome zur Beschreibung ihrer Lage 3n Koordinaten gebraucht, also 3n + 1Koordinaten, um auch noch die potentielle Energie darzustellen. Man erhielte dann eine Potentialfläche im vieldimensionalen Raum, eine sogenannte Hyperfläche. Ein Ausschnitt durch diese Hyperfläche ist das dargestellte Energieprofil.

4
cis cis -- trans trans -- Isomerisierung von 2Isomerisierung von 2--ButenButen
Übergangszustand
A
BB
A
0°0
90°π/2
180°π
Epo
t
A
AB
BA
BB
A
Reaktionskoordiante
Die stabile Lage eines Olefins ist die planare, d.h. jene, bei der die Liganden der C - Atome in einer Ebene liegen. Isomerisierung bedeutet hier die Drehung der Doppelbindung um 180°. Dem Energiemaximum entspricht eine Drehung um 90°. Hier ist der Drehwinkel die Reaktionskoordinate.
0
PotentialhyperflächePotentialhyperfläche
H + H2 → H2 + H
Z + XY → YZ + X
In Energieprofilen wird über der roten Linie als Abszisse die potentielle Energie als Ordinate aufgezeichnet Durch den Schnitt
Aaufgezeichnet. Durch den Schnitt durch die Potentialfläche erscheint A als Maximum.

5
Was bedeutet dies für die Stabilität des Moleküls?Was bedeutet dies für die Stabilität des Moleküls?
Es bedeutet, dass das reagierende Molekül in der Konfiguration des aktivierten Komplexes nur in und pgegen die Reaktionsrichtung instabilist, d.h., nur in diesen Richtungen nimmt die Energie ab. In allen anderen Richtungen ist das Molekül stabil.
Bei einer Reaktion kann es mehr als nur ein Potentialmaximum geben. Zwischen Maxima müssen dann notwendigerweise Minima liegen, die wir als Zwischenzustände, Zwischenstufen oder Zwischenprodukte bezeichnen.
Kollisionen zwischen Atomen/MolekülenKollisionen zwischen Atomen/Molekülen
H + H2 → H2 + H

6
Kollisionen zwischen Atomen/MolekülenKollisionen zwischen Atomen/Molekülen
Cl + HJ → HCl + J
Successful collision
Unsuccessful collision
Die AktivierungsenergieDie AktivierungsenergieMechanisches Analogon
Nur jene Moleküle reagieren, die ausreichende kinetische Energie besitzen.
In diesem Gefäß sollen sich Kugeln Die kinetische Energie einer Kugel, di üb h ll
RTE
eΔ
−
gbefinden, zunächst im Ausgangszustand (linke Seite des Gefäßes). Durch Schütteln des Gefäßes auf einem Vibrator (thermische Bewegung) werden die Kugeln in den Endzustand (rechte Seite), gebracht.
die übergehen soll, muss mindestens so groß sein, dass sie den Übergangszustand erreicht. Dabei wird ihre kinetische Energie zumindest teilweise in potentielle Energie umwandelt.

7
Sind alle Reaktionen, die gleiches ΔE haben, gleich schnell?Nein! Manche Übergangszustände sind komplexer als andere (Entropie). Gibt es viele mögliche equivalente Anordnungsmöglichkeiten, so wirkt sich das für die Reaktion günstig aus (hohe Entropie des Übergangszustands).Dies wird im präexponentiellen Faktor zusammengefasst sodass dasDies wird im präexponentiellen Faktor zusammengefasst, sodass das Arrhenius - Gesetz folgt.
RTE
eAkΔ
−⋅=
Die Reaktion erfolgt immer in beiden Richtungen (dynamisches Verhalten). Auch im Endzustand befinden sich Moleküle auf beiden Seiten. Es stellt sich das thermodynamische Gleichgewicht ein.
Hin- und Rückreaktion verlaufen entlang desselben Weges. Dies nennt man das Prinzip der mikroskopischen Reversibilität.
AktivierungsenergieAktivierungsenergie
RTE
eAkA−
= *
dTkdRT
TdkdREA
ln)/1(
ln 2=−=Definition der Aktivierungsenergie

8
Als Reaktionsgeschwindigkeit definiert man die Anzahl der Teilchen, die in der Zeiteinheit einen Prozess durchmachen. Wie schnell dies im Einzelfall erfolgt, ist belanglos.
Definition der ReaktionsgeschwindigkeitDefinition der Reaktionsgeschwindigkeit
dd
ddp
ddc
ddn ξ
===
dtdnr −= n = Anzahl der potentiellen Reaktionsteilnehmer
n kann durch Größen, die proportional Zur Teilchenzahl sind ersetzt werdendtdtdtdt Zur Teilchenzahl sind ersetzt werden.
dtdξξ =&
dtdnr J
J
⋅=ν1
ReaktionsordnungReaktionsordnung
Reaktionsordnung ist der Exponent, bzw. die Summe der Exponenten in der einfachsten Form (Exponentialform) der Geschwindigkeitsgleichung einer Reaktion
....][][][ zyx CBAkr =
0 ≤ x,y,z,... ≤ 2 für Einzelkomponenten0 ≤ x,y,z,... ≤ 4 für Gesamtordnung
meistens
Beispiele
)]([)]([][2
22 gOgNOkdtNOd
=
252
2 )]([][ gONkdtOd
=
2 NO(g) + O2(g) 2 NO2(g)
2 N2O5(g) 4 NO2(g) + O2(g)
Beispiele

9
Reaktion erster OrdnungReaktion erster Ordnung
A B ( + C + ...) Typische ReaktionenRadioaktiver ZerfallOlefin cis-trans-Isomerisierung
nkdtdn
⋅=− Reaktionsrate
Da n, p und c proportional sind ckdtdc
⋅=− pkdtdp
⋅=−dt dt
Reaktionen erster Ordnung zeigen lineare Abhängigkeit zwischen der Reaktionsgeschwindigkeit und der Konzentration.
AA nk
dtdn
⋅=−
dtkdnA
Reaktionsrate bezogen auf A
Separation der VariablendtknA
A ⋅−=
constknA +⋅−= 0ln 0,
⎞⎛
Separation der Variablen
Randbedingungenfür t = 0; nA = nA,0
tknn
A
A ⋅−=⎟⎟⎠
⎞⎜⎜⎝
⎛
0,
ln ktAA enn −⋅= 0,
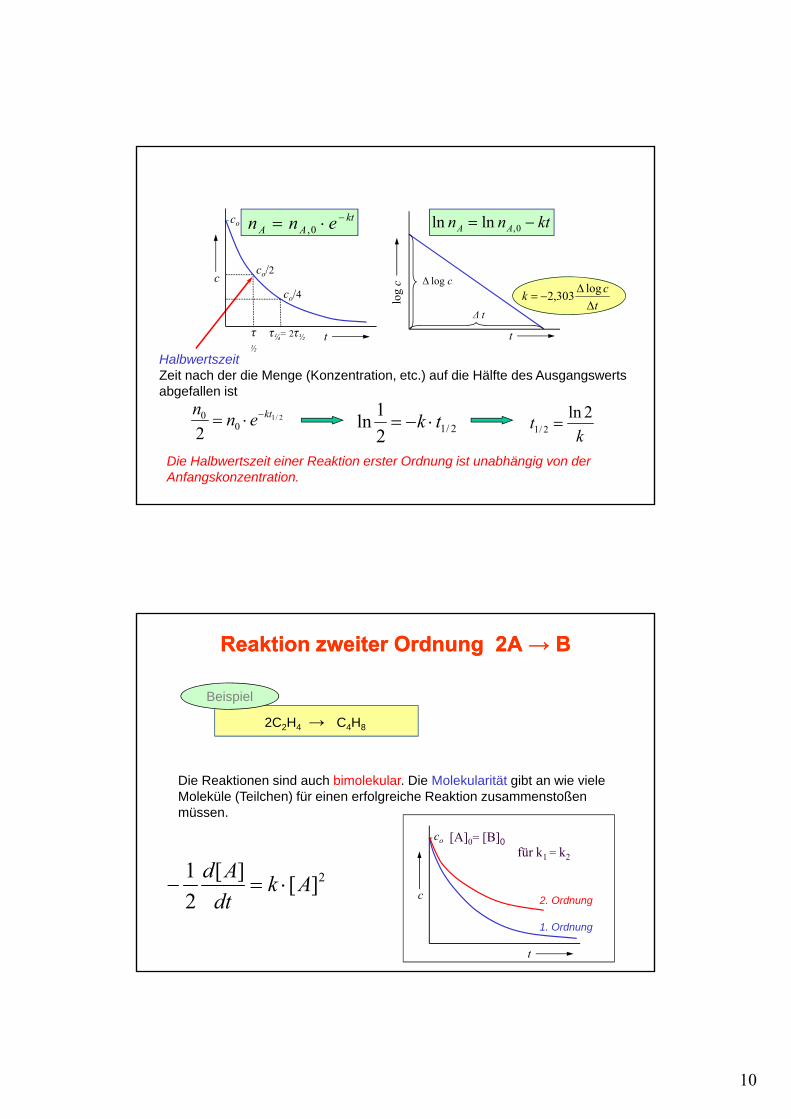
10
ktAA enn −⋅= 0, ktnn AA −= 0,lnln
/
co
tck
ΔΔ
−=log303,2
HalbwertszeitZeit nach der die Menge (Konzentration, etc.) auf die Hälfte des Ausgangswerts
τ½
τ¼= 2τ½
co/2
co/4c
t t
log
c
Δ log c
Δ t
2/10
0
2ktenn −⋅=
abgefallen ist
kt 2ln
2/1 =2/121ln tk ⋅−=
Die Halbwertszeit einer Reaktion erster Ordnung ist unabhängig von der Anfangskonzentration.
Reaktion zweiter Ordnung 2A Reaktion zweiter Ordnung 2A →→ BB
Beispiel
2C2H4 → C4H8
Die Reaktionen sind auch bimolekular. Die Molekularität gibt an wie viele Moleküle (Teilchen) für einen erfolgreiche Reaktion zusammenstoßen müssen.
[A]0= [B]0für k = k
co
2][][21 Ak
dtAd
⋅=−2. Ordnung
1. Ordnung
für k1 = k2
c
t

11
Wie verändert sich A mit der Zeit?Wie verändert sich A mit der Zeit?
2][][21 Ak
dtAdr ⋅=−= dtk
AAd
⋅=− 2][
][2
11
consttkA
+⋅−= 2][
1
Für t = 0 und A=A0 const = 1/A0
tkAA
⋅−=− 2][
1][
1
0tk
AAAA
⋅−=− 2
]][[][][
0
0
0
0
][21][][
AktAA
+=
Halbwertszeit einer Reaktion 2. OrdnungHalbwertszeit einer Reaktion 2. Ordnung
[ A] =[A]0
2für t = t1/ 2
2[ A]0
−1
[A]0
= kt1/ 2
02/1 ][
1Ak
t =
Die Halbwertszeit ist umso größer, je kleiner die Konzentration ist.

12
Vergleich der Reaktionsgeschwindigkeiten Vergleich der Reaktionsgeschwindigkeiten
Identische Konzentrationen von A und B, sowie gleiche Anfangsgeschwindigkeiten
20201 BkAk =
00 BA =
02021 AkBkk ==c
[A]0=[B]0
2. Ordnung
Kon
zent
ratio
n
Die Reaktion 2.Ordnung ist langsamer als die Reaktion 1.Ordnung.
1. Ordnung
K
Zeit
0. Ordnung
Vergleich der ReaktionsgeschwindigkeitenVergleich der ReaktionsgeschwindigkeitenAlle Reaktionen haben gleiche Anfangskonzentrationen und erste Halbwertszeiten
12ln=
2. OrdnungKon
zent
ratio
n
021 Bkk=
2ln1
2ln1
001
2
ABkk
==
2ln021 Bkk =
1. Ordnung0. Ordnung
Zeit

13
Reaktion zweiter Ordnung A + B Reaktion zweiter Ordnung A + B →→ CC
Beispiel
C2H4 + H2O → C2H5-OH
][][][][ BAkdtBd
dtAdr ⋅⋅=−=−=
Eine der beiden Konzentrationen muss als Funktion der anderen ausgedrückt werden
( )][][][][ 00 BBAA −−=ausgedrückt werden.
Wenn [A] = [B] dann gilt:
0
0
][1][][][Akt
ABA+
==
Dimension der Geschwindigkeitskonstante k
0. Ordnung - dc/dt = k0 ko[mol.l-1.s-1]
1 Ordnung - dc/dt = k1c k1[s-1]1. Ordnung dc/dt k1c k1[s ]
2. Ordnung - dc/dt = k2c2 k2[l.mol-1.s-1]
n. Ordnung - dc/dt = kncn kn[ln-1.(moln-1.s)-1]
Halbwertszeit
1 O d t l 2/(k A 0)1. Ordnung t1/2 = ln 2/(k.A00)
2. Ordnung t1/2 = 1/(k.A01)
3. Ordnung t1/2 = 1/(k.A02)
nte Ordnung t1/2 = 1/(k.A0n-1)

14
Methoden zur Bestimmung der Reaktionsordnung Methoden zur Bestimmung der Reaktionsordnung
• Linearisierung der erwarteten Reaktionsgleichungen • Überprüfung der HalbwertszeitenÜberprüfung der Halbwertszeiten• Isolationsmethoden
– Methode der Anfangsgeschwindigkeiten– Überschussmethode
Bestimmung der ReaktionsordnungBestimmung der ReaktionsordnungEinsetzen von erwarteten integrierten Geschwindigkeitsgleichungen
1. Ordnung 2. Ordnung
2][][][][ AkdtAdAk
dtAd
BA ⋅=⋅−=
ktt eAA −⋅= 0][ ][ tk
AA⋅=−
][1
][1
0

15
Bestimmung der ReaktionsordnungBestimmung der ReaktionsordnungLinearisierung für Reaktionen höherer Ordnung
Überprüfen der Halbwertszeiten
kt 2ln
2/1 =][ 0
12/1 Ak
t =
1. Ordnung 2. Ordnung
Bestimmung der ReaktionsordnungBestimmung der ReaktionsordnungMethode der Anfangsgeschwindigkeiten
ba BAkr ][][= Durch den geringen Umsatz
Methode des Überschusses eines Reaktanten
][][
]ln[]ln[lnln BbAakr ++=
]ln['lnln Aakr +=
g goder den Überschuss eines Reaktanten bleibt dessen Konzentration in erster Näherung konstant
➜ Pseudo-Ordnung
Natürlich bleibt bei der Durchführung der Methode des Reaktantüberschusses, die Reaktionsgeschwindigkeit auch abhängig von der Konzentration der Komponente im Überschuss.