genetic features of the x chromosome affect pubertal development and testicular degeneration in...
TRANSCRIPT

Clinical Endocrinology (2006)
65
, 92–97 doi: 10.1111/j.1365-2265.2006.02554.x
O R I G I N A L A R T I C L E
© 2006 The Authors
92
Journal compilation © 2006 Blackwell Publishing Ltd
Blackwell Publishing Ltd
Genetic features of the X chromosome affect pubertal development and testicular degeneration in adolescent boys with Klinefelter syndrome
Anne M. Wikström*, Jodie N. Painter†, Taneli Raivio*, Kristiina Aittomäki‡ and Leo Dunkel§
*
Hospital for Children and Adolescents, Helsinki University Central Hospital, University of Helsinki, Helsinki, Finland,
†
Folkhälsan Institute of Genetics, University of Helsinki, Finland,
‡
Department of Clinical Genetics, Helsinki University Central Hospital, University of Helsinki, Finland and
§
Department of Paediatrics, Kuopio University Hospital, University of Kuopio, Kuopio, Finland
Summary
Objective
To investigate how genetic features of the X chromosome
influence growth, pubertal development and testicular degeneration
in adolescent boys with Klinefelter syndrome (KS). Previous studies
have suggested that genetic features of the X chromosome may
contribute to the wide phenotypic variation in KS.
Design
A prospective clinical study.
Patients
Fourteen nonmosaic 47,XXY boys, aged 10–13·9 years.
Measurements
The relationship of genetic features of the X chro-
mosome, including parental origin of X chromosomes, the CAG
repeat length of the androgen receptor (
AR
) gene, and X inactivation
with progression of pubertal development, growth and testicular
function in KS boys.
Results
Paternal (47,X
m
X
p
Y,
n
= 3) as compared to maternal
(47,X
m
X
m
Y,
n
= 11) origin of the supernumerary X chromosome
was associated with a later onset of puberty. In 47,X
m
X
p
Y patients,
serum LH concentrations increased above 1·0 IU/l at 12·5
±
0·6 years
(mean
±
SD), Tanner stage P2 occurred at 12·5
±
0·7 years, and pubertal
acceleration of growth was noted at 13·9
±
1·4 years and peak velocity
at 14·5
±
0·8 years. All of these occurred 1·3–1·9 years later than in
47,X
m
X
m
Y patients (
P =
0·01–0·09). In 47,X
m
X
m
Y subjects, CAG repeat
length (range 17–26) correlated with age at which serum LH level
first exceeded 1·0 IU/l (
r
s
= 0·63,
P
= 0·06,
n
= 10) and testosterone
1·0 nmol/l (28·8 ng/dl) (
r
s
= 0·78,
P
= 0·02,
n
= 10).
Conclusions
Paternal origin of the supernumerary X chromosome
is associated with later onset of puberty and longer CAG repeats of
the
AR
with later pubertal reactivation of the pituitary–testicular axis
in KS boys. Identifying genetic factors that affect the phenotype may
lead to a better understanding of the pathogenesis of KS.
(Received 9 March 2006; returned for revision 7 April 2006; finally
revised 10 April 2006; accepted 11 April 2006)
Introduction
The classical phenotype of Klinefelter syndrome (KS) with gynae-
comastia, small firm testes, dysgenetic seminiferous tubules with
aspermatogenesis, high levels of gonadotrophins and low normal
testosterone levels is well recognized.
1
The possibility of behavioural
and neurological problems in KS is also acknowledged. There is,
however, a wide variation in the KS phenotype, ranging from indi-
viduals with severe hypogonadism and/or behavioural problems in
childhood to those with infertility as the only presenting symptom as
adults. Only 40% of all individuals with KS are diagnosed; less than
10% are diagnosed prenatally and another 10% during childhood.
2
Several genetic features of the X chromosome have been proposed
to affect the phenotype, but to date this issue has not been thoroughly
investigated. The parental origin of the supernumerary X chromosome
results in different doses of paternally and maternally derived genes.
Furthermore, due to imprinting, the paternal and maternal alleles
could be differentially expressed.
3
As a dosage compensation mechanism
in subjects with two X chromosomes, one of the X chromosomes is
randomly inactivated.
3,4
It has, however, been shown that over 15%
of X chromosomal genes escape inactivation
5
and, consequently, a
widely accepted hypothesis is that these genes are responsible for
many of the features of KS.
6
As in normal females, skewed X chro-
mosome inactivation, defined as > 80% preferential inactivation of
one of the X chromosomes, can also occur in KS.
3,4
This leads to
predominant expression of the genes on one of the X chromosomes,
and as Iitsuka
et al
.
3
suggested, in 47,X
m
X
p
Y subjects skewed inacti-
vation of the X
m
could result in a situation where only paternally
derived genes are expressed. In the case of X
p
inactivation the situation
equals that of a normal male with perhaps a less severe phenotype.
In the maternally derived cases the extra X chromosome is due to
nondisjunction during the first (M-I) or second (M-II) meiotic
division, or in postzygotic mitotic divisions.
3,6,7
In cases of errors in M-
II or in mitosis, the 47,XXY subject has two identical X chromosomes.
7
Anne M. Wikström and Jodie N. Painter contributed equally to the study.Correspondence: Anne Wikström, HUCH, Hospital for Children and Adolescents, PO Box 281, 00029 Helsinki, Finland. Tel.: + 358 9 47175293; Fax: + 358 9471 75888; E-mail: [email protected]

Genetic factors in Klinefelter syndrome
93
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd,
Clinical Endocrinology
,
65
, 92–97
This isodisomy could, as in cases of skewed X inactivation, lead to
expression of recessive mutations of X-linked genes, and accordingly
to a more adverse phenotype.
Androgen-related genes might play a particular role in modulating
the differences in the KS phenotype. The androgen receptor (
AR
)
gene is located on the X chromosome. The N-terminal domain of
exon 1 of the
AR
gene contains a highly polymorphic CAG repeat,
the length of which is inversely associated with the activity of the
receptor.
8
It is possible that a subtle modulation of AR function could
contribute to the variability in KS phenotypes, especially because
most of these patients have low normal or low androgen levels.
The aim of our study was to investigate the impact of genetic features
of the X chromosome on growth during childhood and adolescence,
and on onset and progression of puberty in boys with KS. In an earlier
study
9
we noted wide differences in the testicular degeneration in
adolescent boys with KS, hence we also wanted to evaluate how
genetic factors influence this process.
Subjects and methods
Subjects
Fourteen nonmosaic 47,XXY boys were followed-up prospectively
for 4–25 months (median 18). In addition, data from routine clinical
visits prior to and after the systematic surveillance period were collected
from patient records. None of the subjects was or had previously
been on androgen therapy. At the start of the systematic prospective
follow-up, their median age was 11·5 years (range 10·0–13·9). Some
of the clinical and hormonal data have already been published.
9
The boys visited the Hospital for Children and Adolescents, Helsinki
University Central Hospital every fourth to sixth month. The visits
included physical examination with assessment of puberty according
to Tanner.
10
Body mass index (BMI) was calculated as weight (kg)
divided by the height squared (m
2
). Width and length of the testes
were measured with a ruler to the nearest millimetre; testicular volume
(ml) was calculated by the formula 0·52
×
length
×
width
2
and was
expressed as the mean volume of the left and right testes (Tvol).
11
Serum hormone levels were determined by methods described
previously.
9
Bone age was assessed annually according to the method
of Greulich and Pyle.
12
Reported heights of the parents were recorded.
The parents of each boy gave their informed consent for their son’s and
their own participation in this study, which was approved by the research
ethics committee of the Hospital District of Helsinki and Uusimaa.
Genetic studies
For this study, blood samples were collected from all 14 boys and 27
parents. Genomic DNA was extracted from whole ethylenediamine-
tetraacetic acid (EDTA)-blood with the PUREGENE
®
DNA isolation
kit (Gentra Systems, Minneapolis, MN, USA). Parental origin of the
X chromosomes was determined by genotyping each boy and both
parents (with the exception of one father) at 10 microsatellite loci:
DXS6807, DXS989, DXS1068, DXS1003, DXS6800, DXS6797,
DXS1001, DXS984, DXS1193 and DXS1073. The length of the CAGn
repeat in exon 1 of the
AR
gene and the degree of X chromosome
inactivation were determined essentially as outlined in Suzuki
et al
.
13
The CAG alleles were sized initially by genotyping, and the lengths
of the repeats subsequently confirmed by sequencing all homozygous
samples. The degree of skewing of X inactivation was estimated for
all heterozygous samples according to the equations outlined in
Iitsuka
et al
.
3
using the peak area values for each allele. All genotyping
and sequence reactions were electrophoresed in an ABI 3700
(Applied Biosystems, Foster City, CA, USA) and analysed with Gen-
eMapper v3·7 (Applied Biosystems) and Sequencher v4·5 (Gene
Codes Corporation, Ann Arbor, MI, USA), respectively.
Calculations and statistical analyses
Midparental target height (SD) for each KS boy was calculated by
subtracting 171 from the arithmetic mean of the parents’ heights and
dividing this difference by 10. For predicting adult height for the KS
subjects, the method of Bayley and Pinneau was used.
14
For converting
height in centimetres to SD scores, age-specific growth norms for
normal Finnish boys were used. The X-weighted biallelic mean CAG
repeat length was calculated by a method described previously:
15
each
allele in a genetic pair was multiplied by its percentage of expression
(100%
−
% inactivity) and added together.
Descriptive data are reported as median and range or as mean
±
SD.
Mann–Whitney
U
-tests were used to compare differences between boys
with paternal (X
p
) and maternal (X
m
) origin of the supernumerary
X chromosome. Spearman’s rank correlations were calculated for
associations between continuous parameters and X-weighted biallelic
mean CAG repeat length. Significance was set at
P
< 0·05.
Results
We investigated the impact of all genetic features of the X chromosome
listed in Table 1 on the phenotypes in Table 2. For biochemical markers
of onset of puberty, LH and testosterone levels clearly exceeding
prepubertal levels were used.
Parental origin of the supernumerary X chromosome
The origin of the X chromosomes was unambiguously assigned for all
cases (Table 1). Although the sample from the father was not available
for patient 13, the supernumerary X chromosome was assigned as
maternal because all marker alleles of the proband were maternal.
The extra X chromosome was paternal in three (21%) and maternal
in 11 (79%) cases.
Parental origin of the supernumerary X chromosome did not
influence the growth during childhood (ages 2–9 years). Relative
heights in this period were 0·63
±
1·03 SD and
−
0·19
±
0·54 SD for
the 47,X
m
X
m
Y and 47,X
m
X
p
Y boys, respectively (
P =
ns). Predicted
adult heights were 1·47
±
1·05 SD and 1·34
±
0·79 SD (
P =
ns) for
subjects with 47,X
m
X
m
Y and 47,X
m
X
p
Y, respectively. Furthermore,
there was no difference in body composition between these two
groups during puberty (BMI after age 10 years: 47,X
m
X
m
Y, 19·6
±
2·6
and 47,X
m
X
p
Y, 17·5
±
0·8;
P
= ns).
The onset and progression of puberty was delayed in the 47,X
m
X
p
Y
boys compared to the 47,X
m
X
m
Y boys as indicated by clinical markers
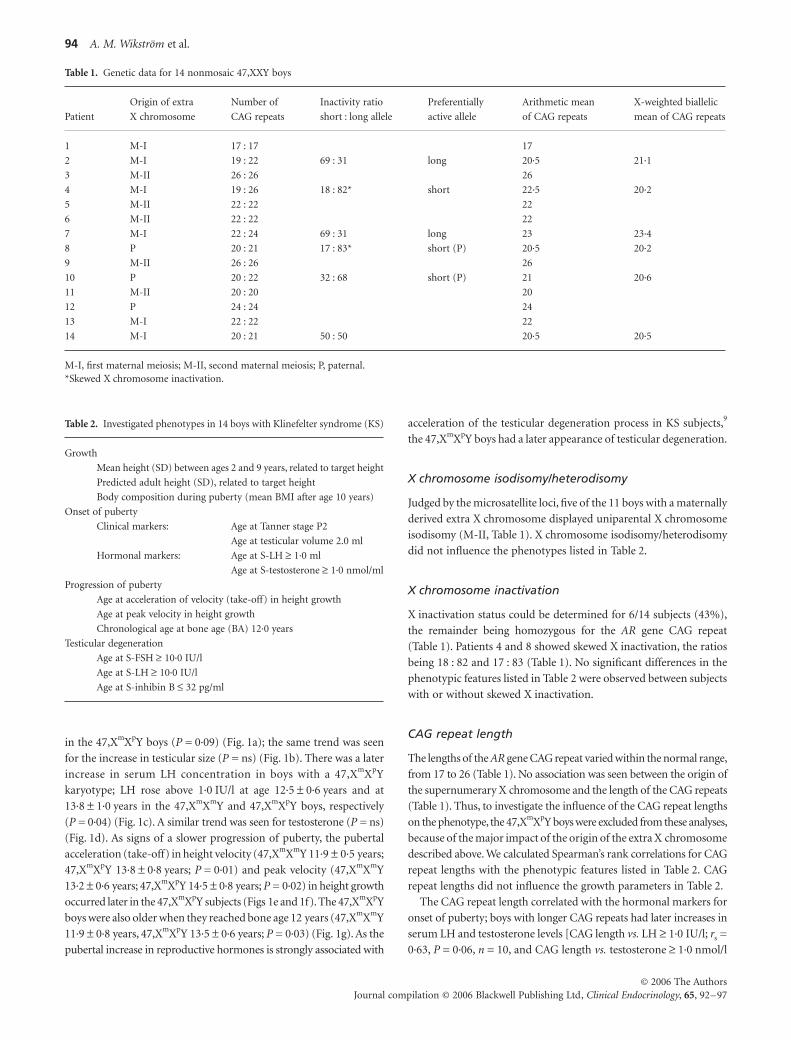
and serum hormone measurements (Fig. 1). Tanner stage P2 was noted
at age 12·5
±
0·7 years in the 47,X
m
X
m
Y boys, and at 13·9
±
1·4 years
94
A. M. Wikström
et al.
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd,
Clinical Endocrinology
,
65
, 92–97
in the 47,X
m
X
p
Y boys (
P =
0·09) (Fig. 1a); the same trend was seen
for the increase in testicular size (
P =
ns) (Fig. 1b). There was a later
increase in serum LH concentration in boys with a 47,X
m
X
p
Y
karyotype; LH rose above 1·0 IU/l at age 12·5
±
0·6 years and at
13·8
±
1·0 years in the 47,X
m
X
m
Y and 47,X
m
X
p
Y boys, respectively
(
P =
0·04) (Fig. 1c). A similar trend was seen for testosterone (
P =
ns)
(Fig. 1d). As signs of a slower progression of puberty, the pubertal
acceleration (take-off) in height velocity (47,X
m
X
m
Y 11·9
±
0·5 years;
47,X
m
X
p
Y 13·8
±
0·8 years;
P
= 0·01) and peak velocity (47,X
m
X
m
Y
13·2
±
0·6 years; 47,XmXpY 14·5 ± 0·8 years; P = 0·02) in height growth
occurred later in the 47,XmXpY subjects (Figs 1e and 1f). The 47,XmXpY
boys were also older when they reached bone age 12 years (47,XmXmY
11·9 ± 0·8 years, 47,XmXpY 13·5 ± 0·6 years; P = 0·03) (Fig. 1g). As the
pubertal increase in reproductive hormones is strongly associated with
acceleration of the testicular degeneration process in KS subjects,9
the 47,XmXpY boys had a later appearance of testicular degeneration.
X chromosome isodisomy/heterodisomy
Judged by the microsatellite loci, five of the 11 boys with a maternally
derived extra X chromosome displayed uniparental X chromosome
isodisomy (M-II, Table 1). X chromosome isodisomy/heterodisomy
did not influence the phenotypes listed in Table 2.
X chromosome inactivation
X inactivation status could be determined for 6/14 subjects (43%),
the remainder being homozygous for the AR gene CAG repeat
(Table 1). Patients 4 and 8 showed skewed X inactivation, the ratios
being 18 : 82 and 17 : 83 (Table 1). No significant differences in the
phenotypic features listed in Table 2 were observed between subjects
with or without skewed X inactivation.
CAG repeat length
The lengths of the AR gene CAG repeat varied within the normal range,
from 17 to 26 (Table 1). No association was seen between the origin of
the supernumerary X chromosome and the length of the CAG repeats
(Table 1). Thus, to investigate the influence of the CAG repeat lengths
on the phenotype, the 47,XmXpY boys were excluded from these analyses,
because of the major impact of the origin of the extra X chromosome
described above. We calculated Spearman’s rank correlations for CAG
repeat lengths with the phenotypic features listed in Table 2. CAG
repeat lengths did not influence the growth parameters in Table 2.
The CAG repeat length correlated with the hormonal markers for
onset of puberty; boys with longer CAG repeats had later increases in
serum LH and testosterone levels [CAG length vs. LH ≥ 1·0 IU/l; rs =
0·63, P = 0·06, n = 10, and CAG length vs. testosterone ≥ 1·0 nmol/l
Table 1. Genetic data for 14 nonmosaic 47,XXY boys
Patient
Origin of extra
X chromosome
Number of
CAG repeats
Inactivity ratio
short : long allele
Preferentially
active allele
Arithmetic mean
of CAG repeats
X-weighted biallelic
mean of CAG repeats
1 M-I 17 : 17 17
2 M-I 19 : 22 69 : 31 long 20·5 21·1
3 M-II 26 : 26 26
4 M-I 19 : 26 18 : 82* short 22·5 20·2
5 M-II 22 : 22 22
6 M-II 22 : 22 22
7 M-I 22 : 24 69 : 31 long 23 23·4
8 P 20 : 21 17 : 83* short (P) 20·5 20·2
9 M-II 26 : 26 26
10 P 20 : 22 32 : 68 short (P) 21 20·6
11 M-II 20 : 20 20
12 P 24 : 24 24
13 M-I 22 : 22 22
14 M-I 20 : 21 50 : 50 20·5 20·5
M-I, first maternal meiosis; M-II, second maternal meiosis; P, paternal.*Skewed X chromosome inactivation.
Table 2. Investigated phenotypes in 14 boys with Klinefelter syndrome (KS)
Growth
Mean height (SD) between ages 2 and 9 years, related to target height
Predicted adult height (SD), related to target height
Body composition during puberty (mean BMI after age 10 years)
Onset of puberty
Clinical markers: Age at Tanner stage P2
Age at testicular volume 2.0 ml
Hormonal markers: Age at S-LH ≥ 1·0 ml
Age at S-testosterone ≥ 1·0 nmol/ml
Progression of puberty
Age at acceleration of velocity (take-off) in height growth
Age at peak velocity in height growth
Chronological age at bone age (BA) 12·0 years
Testicular degeneration
Age at S-FSH ≥ 10·0 IU/l
Age at S-LH ≥ 10·0 IU/l
Age at S-inhibin B ≤ 32 pg/ml
Genetic factors in Klinefelter syndrome 95
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Clinical Endocrinology, 65, 92–97
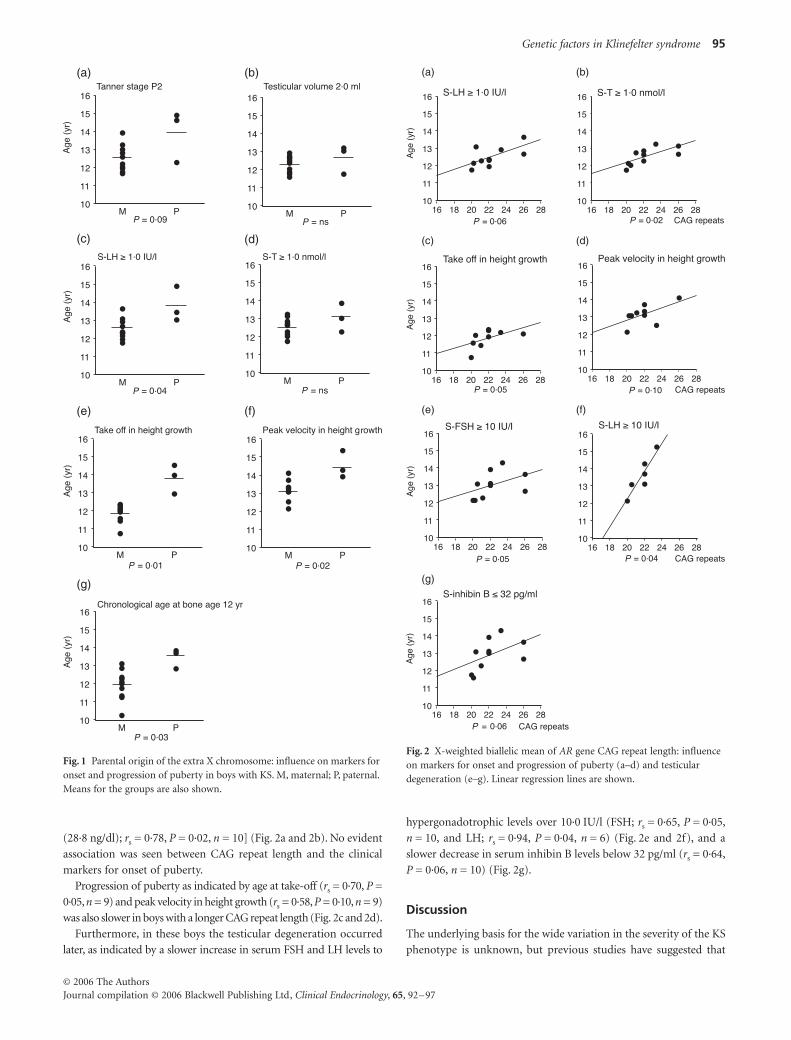
(28·8 ng/dl); rs = 0·78, P = 0·02, n = 10] (Fig. 2a and 2b). No evident
association was seen between CAG repeat length and the clinical
markers for onset of puberty.
Progression of puberty as indicated by age at take-off (rs = 0·70, P =
0·05, n = 9) and peak velocity in height growth (rs = 0·58, P = 0·10, n = 9)
was also slower in boys with a longer CAG repeat length (Fig. 2c and 2d).
Furthermore, in these boys the testicular degeneration occurred
later, as indicated by a slower increase in serum FSH and LH levels to
hypergonadotrophic levels over 10·0 IU/l (FSH; rs = 0·65, P = 0·05,
n = 10, and LH; rs = 0·94, P = 0·04, n = 6) (Fig. 2e and 2f), and a
slower decrease in serum inhibin B levels below 32 pg/ml (rs = 0·64,
P = 0·06, n = 10) (Fig. 2g).
Discussion
The underlying basis for the wide variation in the severity of the KS
phenotype is unknown, but previous studies have suggested that
Fig. 1 Parental origin of the extra X chromosome: influence on markers for onset and progression of puberty in boys with KS. M, maternal; P, paternal. Means for the groups are also shown.
Fig. 2 X-weighted biallelic mean of AR gene CAG repeat length: influence on markers for onset and progression of puberty (a–d) and testicular degeneration (e–g). Linear regression lines are shown.

96 A. M. Wikström et al.
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Clinical Endocrinology, 65, 92–97
genetic features of the X chromosome might play a role. The results
of the present study suggest such genetic effects on the onset and pro-
gression of puberty, and the development of testicular degeneration.
The parental origin of the extra X chromosome could influence the
KS phenotype through altered dosage of paternally and maternally
derived genes, and imprinting. In our study the three subjects with
a paternal additional X chromosome showed later onset and slower
progression of puberty. Jacobs et al.16 suggested that parental origin of
the extra X chromosome has no evident effect on the phenotypes of
KS males. This view was based on the finding of a similar proportion
of maternally and paternally derived cases among subjects diagnosed
prenatally and as newborns, or in adulthood because of signs of
hypogonadism.16 In this study, however, the phenotypes were not
studied.16 Zinn et al.17 found that the parental origin of the super-
numerary X chromosome had no impact on anthropometric and
physical findings, which is in accordance with our results. They also
measured FSH, LH, testosterone and oestradiol concentrations in
their subjects aged 0·1–39 years, and found no differences in testicular
function between the maternally and paternally derived cases.17 The
study by Zinn et al.17 was, however, cross-sectional, while our study was
longitudinal, and in our series the phenotypic differences became
apparent during follow-up.
A predominant hypothesis is that the altered dosage of some X-
linked genes may affect the KS phenotype.18 In KS the supernumerary
X chromosome is probably inactivated in the same manner as in
normal females. Approximately 15% of X-linked genes escape inacti-
vation, 20% show a variable inactivation pattern, and around 65%
are always inactivated.5 Depending on whether the maternally or
paternally derived X chromosome is preferentially inactivated, the
dosage of maternally and paternally derived genes varies. Therefore, it
has been suggested that X chromosome inactivation patterns, especially
skewed X inactivation, influence KS phenotypes.3 To date, no study
has thoroughly evaluated the impact of skewed X inactivation. The
study by Zinn et al. had two subjects with skewed X inactivation, and
no association with phenotypic features was seen.17 This aspect was not
evaluated in the study by Zitzmann et al.,19 who had five individuals
with skewed X inactivation in their cohort of 77 adult KS males.
Similarly, no differences were seen between the two boys with skewed
X inactivation and the other 12 boys in our study. Conclusions
should, however, be drawn with caution because of the small number
of patients in these studies.
In 47,XmXmY subjects the X chromosomes can be either identical
(isodisomy) or different (heterodisomy). Isodisomy could lead to
double dosage of some harmful genes, which escape X inactivation.
However, our study and the earlier study by Zinn et al.17 did not find
an impact on the KS phenotype.
Androgen-related genes located on the X chromosome might play
a particular role in the differences in the KS phenotype. Two recent
studies have shown that KS infants have a physiological increase in
serum testosterone during the first months of life, but that the levels
are lower than in controls.20,21 During puberty, the serum testosterone
levels remain within the low normal range,9,22,23 but in adulthood,
over half of the KS males have serum testosterone levels below
normal.7,24 The length of the CAG stretch in exon 1 of the AR gene
is inversely related to the activity of the receptor, and may modulate its
response to androgens.8 Zitzmann et al.19 found a positive correlation
between CAG repeat length and body height and presence of
gynecomastia, while there was an inverse association with bone
density, social status and testicular volume, and even to response
to androgen substitution. In the study by Zinn et al., the only inves-
tigated parameter that was associated with CAG repeat length was
penile length; the correlation was inverse.17
In our study, the KS boys with a longer CAG repeat showed a later
onset and slower progression of puberty and a slower testicular
degeneration process. These findings are in agreement with diminished
AR response to androgens when the AR gene has a longer CAG
repeat. We have previously reported that the testicular degeneration
process accelerates at the onset of puberty.9 After an increase in
serum testosterone to levels above 2·5 nmol/ l (72·1 ng/dl), there is a
rapid decline in the serum levels of the Sertoli cell-specific markers
inhibin B and anti-Müllerian hormone. This suggests that androgens
may play a role in initiating this degeneration process. The present
finding of lower AR activity with a slower testicular degeneration
process supports this hypothesis.
In our cohort only one boy was diagnosed by an amniocentesis
while the other 13 boys initially presented between the ages of 5 and
10·5 years with speech, learning and/or behavioural problems. Thus,
our patient series may contain ascertainment bias and the phenotypic
features may be different in totally unselected cohorts of patients
diagnosed prenatally or in patients diagnosed in adulthood because
of infertility. Earlier studies have shown that the supernumerary
X chromosome is paternal in 50–60% and maternal in 40–50% of
KS cases,3 while in our study the percentages were 21% and 79%,
respectively. Further studies including a larger number of subjects
are therefore needed to confirm our preliminary results.
In conclusion, genetic features of the X chromosome appear to
play a part in modulating KS phenotypes. In the present study we
have shown that parental origin of the supernumerary X chromosome
and the length of the CAG repeat of the AR gene influence pubertal
development. Furthermore, androgens may play a role in the patho-
genesis of the testicular degeneration in KS, the mechanisms of
which is unknown. Identifying genetic factors that contribute to the
substantial variation in the KS phenotype will lead to a better under-
standing of the pathogenesis of KS, and may through more targeted
therapeutic measures offer better prognosis and improvement in
quality of life for the patients.
Acknowledgements
This work was supported by grants from the Medical Society of
Finland (Finska Läkaresällskapet), the Finnish Medical Foundation
and the Hospital District of Helsinki and Uusimaa.
References
1 Klinefelter, H.F., Reifenstein, E.C. & Albright, F. (1942) Syndromecharacterized by gynecomastia, aspermatogenesis without a-Leydigism,and increased excretion of follicle-stimulating hormone. Journal ofClinical Endocrinology, 2, 615–627.
2 Bojesen, A., Juul, S. & Gravholt, C.H. (2003) Prenatal and postnatalprevalence of Klinefelter syndrome: a national registry study. Journalof Clinical Endocrinology and Metabolism, 88, 622–626.

Genetic factors in Klinefelter syndrome 97
© 2006 The AuthorsJournal compilation © 2006 Blackwell Publishing Ltd, Clinical Endocrinology, 65, 92–97
3 Iitsuka, Y., Bock, A., Nguyen, D.D., Samango-Sprouse, C.A.,Simpson, J.L. & Bischoff, F.Z. (2001) Evidence of skewed X-chromosome inactivation in 47,XXY and 48,XXYY Klinefelterpatients. American Journal of Medical Genetics, 98, 25–31.
4 Willard, H.F. (2001) The sex chromosomes and X chromosomeinactivation. In: C.R. Scriver, A.L. Beaudet, W.S. Sly, D. Valle eds. TheMetabolic and Molecular Bases of Inherited Disease, 8th edn. McGraw-Hill, New York, 1191–1211.
5 Carrel, L. & Willard, H.F. (2005) X-inactivation profile revealsextensive variability in X-linked gene expression in females. Nature,434, 400–404.
6 Simpson, J.L., de la Cruz, F., Swerdloff, R.S., Samango-Sprouse, C.,Skakkebaek, N.E., Graham, J.M. Jr, Hassold, T., Aylstock, M.,Meyer-Bahlburg, H.F., Willard, H.F., Hall, J.G., Salameh, W., Boone, K.,Staessen, C., Geschwind, D., Giedd, J., Dobs, A.S., Rogol, A., Brinton, B.& Paulsen, C.A. (2003) Klinefelter syndrome: expanding the pheno-type and identifying new research directions. Genetics in Medicine,5, 460–468.
7 Lanfranco, F., Kamischke, A., Zitzmann, M. & Nieschlag, E. (2004)Klinefelter’s syndrome. Lancet, 364, 273–283.
8 Zitzmann, M. & Nieschlag, E. (2003) The CAG repeat polymorphismwithin the androgen receptor gene and maleness. International Journalof Andrology, 26, 76–83.
9 Wikstrom, A.M., Raivio, T., Hadziselimovic, F., Wikstrom, S., Tuuri, T.& Dunkel, L. (2004) Klinefelter syndrome in adolescence: onset ofpuberty is associated with accelerated germ cell depletion. Journal ofClinical Endocrinology and Metabolism, 89, 2263–2270.
10 Tanner, J.M. (1962) Growth at Adolescence, 2nd edn. Blackwell,Oxford, UK.
11 Hansen, P. & With, T.K. (1952) Clinical measurements of testes. ActaMedica Scandinavica, 206, 457–465.
12 Greulich, W.W. & Pyle, S.L. (1959) Atlas of Skeletal Development ofthe Hand and Wrist, 2nd edn. Stanford University Press, Stanford,CA.
13 Suzuki, Y., Sasagawa, I., Tateno, T., Ashida, J., Nakada, T., Muroya, K.& Ogata, T. (2001) Mutation screening and CAG repeat lengthanalysis of the androgen receptor gene in Klinefelter’s syndromepatients with and without spermatogenesis. Human Reproduction,16, 1653–1656.
14 Bayley, N. & Pinneau, S.R. (1952) Tables for predicting adult heightfrom skeletal age: revised for use with the Greulich–Pyle hand standards.Journal of Pediatrics, 40, 423–441.
15 Hickey, T., Chandy, A. & Norman, R.J. (2002) The androgen receptorCAG repeat polymorphism and X-chromosome inactivation inAustralian Caucasian women with infertility related to polycysticovary syndrome. Journal of Clinical Endocrinology and Metabolism,87, 161–165.
16 Jacobs, P.A., Bacino, C., Hassold, T., Morton, N.E., Keston, M. &Lee, M. (1988) A cytogenetic study of 47,XXY males of known originand their parents. Annals of Human Genetics, 52, 319–325.
17 Zinn, A.R., Ramos, P., Elder, F.F., Kowal, K., Samango-Sprouse, C.& Ross, J.L. (2005) Androgen receptor CAGn repeat length influencesphenotype of 47,XXY (Klinefelter) syndrome. Journal of ClinicalEndocrinology and Metabolism, 90, 5041–5046.
18 Aksglaede, L., Wikstrom, A.M., Rajpert-De Meyts, E., Dunkel, L.,Skakkebaek, N.E. & Juul, A. (2006) Natural history of seminiferoustubule degeneration in Klinefelter syndrome. Human ReproductionUpdate, 12, 39–48.
19 Zitzmann, M., Depenbusch, M., Gromoll, J. & Nieschlag, E. (2004)X-chromosome inactivation patterns and androgen receptorfunctionality influence phenotype and social characteristics as wellas pharmacogenetics of testosterone therapy in Klinefelter patients.Journal of Clinical Endocrinology and Metabolism, 89, 6208–6217.
20 Lahlou, N., Fennoy, I., Carel, J.C. & Roger, M. (2004) Inhibin B andanti-Mullerian hormone, but not testosterone levels, are normal ininfants with nonmosaic Klinefelter syndrome. Journal of ClinicalEndocrinology and Metabolism, 89, 1864–1868.
21 Ross, J.L., Samango-Sprouse, C., Lahlou, N., Kowal, K., Elder, F.F. &Zinn, A. (2005) Early androgen deficiency in infants and young boyswith 47,XXY Klinefelter syndrome. Hormone Research, 64, 39–45.
22 Salbenblatt, J.A., Bender, B.G., Puck, M.H., Robinson, A., Faiman, C.& Winter, J.S. (1985) Pituitary–gonadal function in Klinefeltersyndrome before and during puberty. Pediatric Research, 19, 82–86.
23 Topper, E., Dickerman, Z., Prager-Lewin, R., Kaufman, H., Maimon, Z.& Laron, Z. (1982) Puberty in 24 patients with Klinefelter syndrome.European Journal of Pediatrics, 139, 8–12.
24 Smyth, C.M. & Bremner, W.J. (1998) Klinefelter syndrome. Archivesof Internal Medicine, 158, 1309–1314.