tetanus, botulinum and snake presynaptic neurotoxins
TRANSCRIPT

DOI: 10.1007/s12210-008-0010-zRendiconti Lincei 19, 173 – 188 (2008)
Ornella Rossetto · Laura Morbiato ·Paola Caccin · Michela Rigoni ·Luca Carli · Marco Paoli · MarianaCintra-Francischelli · Cesare Montecucco
Tetanus, Botulinum and Snake Presynaptic Neurotoxins
Received: 18th September 2007 / Accepted: 18th April 2008 – © Springer-Verlag 2008
Abstract Tetanus and botulinum neurotoxins, produced by anaerobic bacte-ria of the genus Clostridium, are the most toxic proteins known and are solelyresponsible for the pathogenesis of tetanus and botulism. They are metallo-proteases that enter nerve terminals and cleave proteins of the neuroexocytosisapparatus causing a persistent, but reversible, inhibition of neurotransmitterrelease. Botulinum neurotoxins are used in the therapy of many human syn-dromes caused by hyperactive nerve terminals. Snake presynaptic PLA2 neuro-toxins block nerve terminals by binding to the nerve membrane and catalyzing
O. RossettoTel. +39 0498276077, Fax: +39 049 8276049, E-mail: [email protected]
L. MorbiatoTel. +39 0498276056, Fax: +39 049 8276049, E-mail: [email protected]
P. CaccinTel. +39 0498276056, Fax: +39 049 8276049, E-mail: [email protected]
M. RigoniTel. +39 0498276056, Fax: +39 049 8276049, E-mail: [email protected]
L. CarliTel. +39 0498276056, Fax: +39 049 8276049, E-mail: [email protected]
M. PaoliTel. +39 0498276069, Fax: +39 049 8276049, E-mail: [email protected]
M. Cintra-FrancischelliTel. +39 0498276056, Fax: +39 049 8276049, E-mail: [email protected]
C. Montecucco (B)Tel.: +39 0498276058, Fax: +39 0498276049, E-mail: [email protected]
Address (for all the authors):Dipartimento di Scienze Biomediche and Istituto CNR di Neuroscienze, Università di Padova,Via G. Colombo 3, 35121 Padova (Italy)

174 O. Rossetto et al.
phospholipid hydrolysis with production of lysophospholipids and fatty acids.These compounds change the membrane conformation causing enhanced fu-sion of synaptic vesicle via hemifusion intermediate with release of neuro-transmitter and, at the same time, inhibition of vesicle fission and recycling. Itis possible to envisage clinical applications of the lysophospholipid/fatty acidmixture to inhibit hyperactive superficial nerve terminals.
Keywords Tetanus and botulinum neurotoxins, snake neurotoxins, neuroexo-cytosis, muscle paralysis, dystonia
Subject code B18006
Abbreviations ACh, acetylcholine; BoNT, botulinum neurotoxin; FA, fattyacid; LPL, lysophospholipid; NMJ, neuromuscular junction; SNAP-25, 25-kDa synaptosomal-associated protein; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; VAMP, vesicle-associated mem-brane protein; SPAN, snake presynaptic PLA2 neurotoxin; SV, synaptic vesi-cles; TeNT, tetanus neurotoxin.
1 Introduction
Toxigenic anaerobic spore-forming bacteria of the genus Clostridium produceseven different botulinum neurotoxins (designated BoNT/A to /G) and onetetanus neurotoxin (TeNT). Ingestion of BoNT-poisoned food causes an intox-ication known as botulism, whose symptoms are the result of a generalizedinhibition of acetylcholine release (ACh) at somatic and autonomic nerve ter-minals. They include diplopia, ptosis, dysphagia and paralysis of facial muscleswhich progressively descends to the trunk eventually involving the respiratoryand visceral muscles. Dysfunctions of the autonomic nervous system includereduced salivation and lacrimation, nausea, vomiting and abdominal pain. Mostpatients survive botulism, but their complete recovery occurs slowly and mayrequire mechanical ventilation. The recovery time is longer after BoNT/A in-toxication than after BoNT/B and BoNT/E intoxications (Tacket and Rogawski1989; Simpson 2000; Schiavo et al. 2000; Rossetto and Montecucco 2004).
The contamination of even minor skin lesions with spores of Clostridiumtetani causes tetanus. The disease develops days/weeks after infection and isdue to TeNT which migrates retroaxonally along motoneurons, is releasedwithin the spinal cord and enters inhibitory interneurons which are blocked.Consequently the neuronal systems, which ensure a balanced contraction ofopposing skeletal muscles, are impaired and this results in the full contractionof the contralateral muscle with the development of a spastic paralysis typicalof tetanus. This usually begins with a characteristic facial trismus (lockjaw

Tetanus, Botulinum and Snake Presynaptic Neurotoxins 175
or risus sardonicus) (Major, 1945, Weinstein, 1973). Subsequently, neck stiff-ness develops and later spreads to involve the muscles of the spinal cord, theabdomen and the legs. The typical tetanic seizure is characterized by a suddenburst of tonic muscle contraction causing opisthotonos, flexion and adductionof the arms and extension of the lower extremities. The patient is consciousduring such episodes and experiences intense pain. Later, autonomic symptomsdevelop with sweating and alterations of blood pressure and cardiac rhythm.Dysphagia may occur and lead to hydrophobia. Glottal and laryngeal spasmmay develop and cause cyanosis and asphyxia if not promptly relieved by med-ical or surgical means. Dysuria or urinary retention may supervene. A milderform of the disease is local tetanus,which may persist for a considerable periodof time without further developments or it may proceed to generalized tetanus.It is due to dysfunction of interneurons which inhibit the alpha motor neuronsof the affected muscles, without further spread through the central nervoussystem.
Following vaccination, tetanus has almost disappeared from the more devel-oped countries, but it still takes hundreds of thousand of lives in those regionsof the world where vaccination is not practised, and, consequently, tetanusneonatorum is frequent (Galazka and Gasse, 1995). This form of the diseasedevelops during or after the cut of the umbilical cord of babies born from nonimmunized mothers. In fact, spore contaminated tools are often employed tosever the cord at birth, and old rags, often soiled with animal faeces, are usedas dressing. Tetanus is often fatal; death follows body exhaustion and usuallyintervenes by respiratory failure or heart failure (Bleck, 1989). Mortality hasdecreased considerably in the more developed parts of the world, thanks tomodern intensive care techniques, but it is still high because usually elder peo-ple are tetanized and their respiration has to be mechanically assisted for longperiods with the risk of developing pulmonary infections.
Presynaptic snake neurotoxins endowed with PLA2 activity (SPANs) aremajor components of the venom of four families of venomous snakes (Crotal-idae, Elapidae, Hydrophiidae and Viperidae). These neurotoxins play a majorrole in envenomation of the prey (Harris, 1997) by causing a persistent block-ade of neurotransmitter release from nerve terminals (Kini, 1997; Schiavo etal., 2000; Montecucco and Rossetto 2000). This process is more complicatedthan botulism as several venom components are biologically active. However,almost invariably, most of the neurological signs and symptoms are due tothe action of SPANs. In fact, independently of the anatomical site of biting,patients are reported to have ptosis, diplopia, ophtalmoplegia, difficulty inswallowing, respiratory paralysis, abdominal pain and autonomic symptoms(Warrell et al., 1983; Theakston et al., 1990; Connolly et al., 1995; Kularatne,2002; Prasarnpun et al., 2005). These symptoms overlap those of botulism.
The progression of paralysis in isolated nerve-muscle preparations exposedto SPANs is rather simple and consists of three phases. A brief initial phase

176 O. Rossetto et al.
of weak inhibition of acetylcholine release (not always present) is followedby a second prolonged phase of facilitated release and then by a third phaseof progressive decline of neurotransmission (Kelly et al., 1976; Howard andGundersen, 1980; Harris, 1997; Montecucco and Rossetto, 2000). Electronmicroscope pictures taken at the third stage show swollen and enlarged axonterminals with depletion of synaptic vesicles (SVs) and several clathrin-coated�-shaped plasma membrane invaginations, also in areas not facing the musclemembrane foldings. At later stages, swollen mitochondria and vacuoles de-velop (Chen and Lee, 1970; Abe et al., 1976; Cull-Candy et al., 1976; Dixonand Harris, 1999; Harris et al., 2000). The kinetics and morphological changesinduced by PLA2 neurotoxins at neuromuscular junctions (NMJs) led to thesuggestion that they may promote fusion of SVs with the presynaptic mem-brane, and, at the same time, inhibit their retrieval (Montecucco and Rossetto,2000). In contrast, TeNT- and BoNT-poisoned nerve terminals have a normalsize and appearance with normal content and shape of synaptic vesicles andmitochondria (Duchen, 1971), but there is an apparent increase in the numberof synaptic vesicles close to the cytosolic face of the presynaptic membrane(Neale et al., 1999). In addition,BoNT/A poisoning of the frog NMJ causes thedisappearance of the small membrane invagination normally detectable closeto the active zones, which are believed to represent SVs fusion sites (Pumplinand Reese, 1977).
2 Toxicity of TeNT, BoNTs and SPANs
Tetanus and botulinum neurotoxins are the most potent toxins known, as fewng/Kg are sufficient to kill most mammals. When injected peripherally, themouse LD50s of TeNT and BoNTs are between 0.4 ng and 1 ng of toxin per kgof body weight (Gill, 1982), but different animal species show a great range ofsensitivity to the different BoNTs. Humans and horses are at least as sensitiveas mice, whereas snakes and amphibians are rather resistant (Payling-Wright,1955; Gill, 1982). The use of BoNT/A as a therapeutic agent for a varietyof dystonias and other diseases, has uncovered significant variations in theresponse of patients to the same dose of BoNT/A, with some individuals beingunresponsive.
ALSO in the case of SPANs, toxicity varies among animal species. Suf-ficient data are presently available only for mice, where their LD50 toxicityvalues range from 1 µg/kg of textilotoxin (present in the venom of the Aus-tralian snake Pseudonaja textilis) to 1300 µg/kg of pseudexin A (present in thevenom of Pseudechis porphyriacus) (Pearson et al. 1993; Kini 1997; Monte-cucco and Rossetto 2000;), when injected intraperitoneally. The time of onsetof paralysis of animals injected with these neurotoxins is variable dependingon the type of toxin, animal species, dose and route of injection. However, a

Tetanus, Botulinum and Snake Presynaptic Neurotoxins 177
lag phase, ranging from several hours up to days, is always present betweenthe time of injection and the appearance of symptoms and this reflects the timeperiods necessary to diffuse and reach the appropriate neuronal target, to bindand enter into cells and to hydrolyze a number of substrate molecules sufficientto cause paralysis.
3 Structure of TeNT, BoNTs and of SPANs
The active form of TeNT and BoNTs consists of two polypeptide chains (L,50 kDa and H, 100 kDa) (Lacy et al., 1998; Swaminathan and Eswaramoor-thy, 2000). The L chain is disulfide linked to the H chain which is essential forbinding and entry into nerves. The binding activity is located in the C-terminalpart of the H chain and is exerted via a specific interaction with a polysialo-ganglioside molecule and a protein molecule (Montecucco, 1986; Montecuccoet al., 2004). The protein receptor has been identified as the luminal domainsof the synaptic vesicle protein synaptotagmin I and II in the case of BoNT/B(Nishiki et al., 1996; Dong et al., 2003) and /G (Rummel et al., 2004) and ofthe synaptic vesicle protein SV2 for BoNT/A (Dong et al., 2006; Mahrhold etal., 2006). TeNT binds two molecules of polysialogangliosides at two differentsites located within the C-terminal part of the H chain (Rummel et al., 2003).
The N-terminal half of the heavy chain features two ∼100 Å-long antiparal-lel α-helices, similar to those of the membrane interacting proteins of bacterialand viral origin (Kielian and Rey, 2006).
The L chains of TeNT and BoNT exert their action inside nerves and cleavespecifically one of the three proteins that form the core of the machinery whichmediates the release of the neurotransmitter from the lumen of synaptic vesiclesinto the intersynaptic space (see below).
At variance from the high structural similarity of BoNTs, SPANs show awide range of structural complexity and of PLA2 activity. The crystallographicstructure of a few of the simpler toxins has been determined (Westerlund et al.,1992; Kwong et al., 1995; Singh et al., 2001). They include a phospholipaseA2 domain with the presence of a calcium atom which stabilizes and activatesthe enzyme. Their typical feature is a hydrophobic channel that accommodatesthe fatty acid chains of the phospholipid molecule and places the ester bondto be cleaved into the active site. The key residues directly involved in catal-ysis are a histidine residue, which hydrogen-binds the water molecule usedfor hydrolysis, followed by an aspartate residue, which positions the Ca2+ ioncoordinating the phosphate and the sn-2 carbonyl groups of the phospholipidsmolecule. In addition, chemical modification studies identified two segmentsof the PLA2 subunit involved in determining neurotoxicity (Yang, 1997). How-ever, this opens the possibility that additional regions and/or subunits may beimplicated, particularly in the case of multi-subunit toxins and this point still

178 O. Rossetto et al.
awaits detailed investigations. In fact, SPANs vary greatly in terms of theirquaternary structures from the single subunit notexin (from the Australiantiger snake Notechis scutatus), to β-bungarotoxin (from Bungarus multicinc-tus) which consists of two covalently linked subunits, the larger of which is aPLA2. Similarly, only one of the three subunits of taipoxin has PLA2 activ-ity. In contrast, textilotoxin (from the Australian elapid Pseudonaja textilis)consists of five subunits, all endowed with PLA2 activity (Kini, 1997).
4 Cellular steps in neuronal intoxication
The actions of TeNTs, BoNTs and SPANs at nerve terminals have severalfeatures in common: a) their binding to the presynaptic membrane becomesirreversible after a few minutes from toxin addition to a bathed NMJ prepa-ration, b) their inhibitory activity is characterized by a lag phase of severalminutes, and c) the rate of nerve terminal inhibition is strongly increased bysynaptic activity (Hughes and Whaler, 1962; Kamenskaya et al., 1974; Kellyand Brown, 1974; Kelly et al. 1976; Howard and Gundersen, 1980; Simpsonet al., 1993). These hallmarks of the activity of the neurotoxins endowed withenzymatic activities must result from their specific mechanism of cell intoxi-cation. In fact, BoNTs paralyze nerve terminals via four sequential steps: (a)rapid and very specific binding to the cholinergic nerve terminal membrane,(b) internalization inside a vesicle endowed with an ATPase proton pump,(c) membrane translocation triggered by acidification of the vesicular lumen,which causes a structural rearrangement with membrane insertion of the toxinand translocation of the L-chain into the cytosol, and (d) expression of theL-chain proteolytic activity against specific substrates (Schiavo et al., 2000;Rossetto and Montecucco, 2004). The same holds for TeNT at the inhibitorysynapses of central nervous tissue (CNS), but it should be recalled that TeNTreaches the CNS after binding, internalizationand retroaxonal migration insidemotorneurons (Lalli et al., 2003).
The cellular steps involved in the action of SPANs at nerve terminals arenot known in similar details, but it appears that they do not need to enter insideneurons via endocytosis, and a low pH-dependent membrane translocation inthe cytosol to display their neurotoxic activity (Simpson et al., 1994). Thekinetics of paralysis of the prey after a snake bite and after SPAN injectionsuggest that, similarly to BoNTs, they find their way rapidly to peripheralnerve terminals to which they bind quickly and specifically (Harris et al., 2000;Prasarnpun et al., 2004, 2005). Since the targets of the enzymatic activity ofthe BoNT L chain are located in the cytosol, at least this part of the moleculehas to translocate the membrane. It has been established that the acidity of thevesicle interior is essential for such a movement (Simpson et al., 1994). Low pHinduces a conformational change of all BoNTs from a water soluble “neutral”

Tetanus, Botulinum and Snake Presynaptic Neurotoxins 179
structure to an “acid” structure characterized by the exposure of hydrophobicpatches on the protein surface, with the consequent insertion of both the H and Lchains into the hydrocarbon core of the lipid bilayer (Montecucco et al., 1988,1989; Puhar et al., 2004). In this process, BoNTs form transmembrane ionchannels of low conductance across a planar lipid bilayer membrane, which isbelieved to conduce the L chain on the other side of the membrane (Koriazovaand Montal, 2003; Fischer and Montal, 2007a). During translocation, as itemerges in the cytosolic neutral pH, the L chain has to re-acquire its three-dimensional enzymatically active structure and to detach from the H chainupon reduction of the interchain disulfide bridge (Fischer and Montal, 2007b)a complex process which is likely to be assisted by cytosolic chaperons of thenerve terminals.
It is clear that the SPAN-induced hydrolysis of phospholipids residing on theneuronal cell surface is sufficient to cause inhibition of neurotransmitter release(Howard and Wu, 1976; Rigoni et al., 2005). However, we cannot exclude that,later on, these neurotoxins enter inside nerve terminals and display their PLA2activity on phospholipids facing the cytosol. Indeed, ammodytoxin (a SPANpresent in the venom of Vipera ammodytes ammodytes) binds to cytosolicand mitochondrial proteins and has been detected inside hippocampal neurons(Sribar et al., 2001, 2003a and b; Petrovic et al., 2004), and taipoxin binds toa calcium-binding protein of 49 kDa located on the ER (Dodds et al., 1997,Kirkpatrick et al., 2000) and has been immuno-detected inside chromaffin cells(Neco et al., 2003).
5 Enzymatic activities of BoNTs and SPANs
The L-chains of the seven BoNTs and of TeNT are remarkably specific pro-teases acting at single, different peptide bonds. TeNT and BoNT/B, /D, /Fand /G cleave only VAMP (vesicle-associated membrane protein) with loss ofmost of its cytosolic domain. BoNT/A and /E cleave only SNAP-25 within itsC-terminus and BoNT/C cleaves both SNAP-25 and syntaxin. SNAP-25 andsyntaxin reside on the plasma membrane facing the nerve cell cytosol. VAMP,SNAP-25 and syntaxin form a trimeric complex (abbreviated as SNARE com-plex) by windingone to another to form a stable, four-helices, coiled-coil struc-ture. Several SNARE complexes then assemble into a rosette that brings theSV membrane close enough to the cytosolic face of the presynaptic membraneto permit their fusion, with subsequent release of the vesicle neurotransmittercontent into the synaptic cleft (Jahn et al., 2003; Montecucco et al., 2005). Pro-teolysis of one SNARE protein prevents the formation of a functional SNAREcomplex and, consequently, the release of neurotransmitter (fig. 1). In mostcases, BoNT cleavage results in the loss of a large part of the cytosolic portionof SNARE proteins, thus preventing the formation of the SNARE complex.
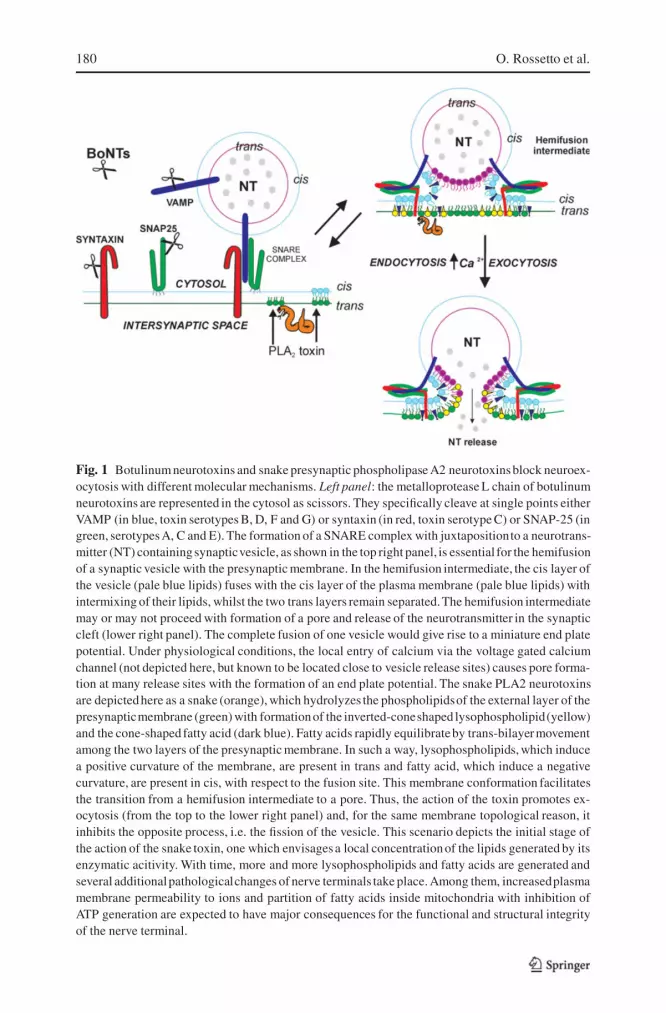
180 O. Rossetto et al.
Fig. 1 Botulinum neurotoxins and snake presynaptic phospholipase A2 neurotoxinsblock neuroex-ocytosis with different molecular mechanisms. Left panel: the metalloprotease L chain of botulinumneurotoxins are represented in the cytosol as scissors. They specifically cleave at single points eitherVAMP (in blue, toxin serotypes B, D, F and G) or syntaxin (in red, toxin serotype C) or SNAP-25 (ingreen, serotypesA, C and E). The formation of a SNARE complex with juxtapositionto a neurotrans-mitter (NT) containingsynapticvesicle, as shown in the top right panel, is essential for the hemifusionof a synaptic vesicle with the presynaptic membrane. In the hemifusion intermediate, the cis layer ofthe vesicle (pale blue lipids) fuses with the cis layer of the plasma membrane (pale blue lipids) withintermixing of their lipids, whilst the two trans layers remain separated.The hemifusion intermediatemay or may not proceed with formation of a pore and release of the neurotransmitter in the synapticcleft (lower right panel). The complete fusion of one vesicle would give rise to a miniature end platepotential. Under physiological conditions, the local entry of calcium via the voltage gated calciumchannel (not depicted here, but known to be located close to vesicle release sites) causes pore forma-tion at many release sites with the formation of an end plate potential. The snake PLA2 neurotoxinsare depictedhere as a snake (orange), which hydrolyzes the phospholipidsof the external layer of thepresynapticmembrane (green) with formationof the inverted-coneshaped lysophospholipid(yellow)and the cone-shaped fatty acid (dark blue). Fatty acids rapidly equilibrateby trans-bilayermovementamong the two layers of the presynaptic membrane. In such a way, lysophospholipids, which inducea positive curvature of the membrane, are present in trans and fatty acid, which induce a negativecurvature, are present in cis, with respect to the fusion site. This membrane conformation facilitatesthe transition from a hemifusion intermediate to a pore. Thus, the action of the toxin promotes ex-ocytosis (from the top to the lower right panel) and, for the same membrane topological reason, itinhibits the opposite process, i.e. the fission of the vesicle. This scenario depicts the initial stage ofthe action of the snake toxin, one which envisagesa local concentrationof the lipids generated by itsenzymatic acitivity. With time, more and more lysophospholipids and fatty acids are generated andseveral additionalpathologicalchanges of nerve terminals take place. Among them, increasedplasmamembrane permeability to ions and partition of fatty acids inside mitochondria with inhibition ofATP generation are expected to have major consequences for the functional and structural integrityof the nerve terminal.

Tetanus, Botulinum and Snake Presynaptic Neurotoxins 181
In the case of BoNT/A and BoNT/C, the truncated SNAP-25 proteins retainmost of their sequences (197 and 198 out of 206 amino acid residues, re-spectively) and are capable of forming stable, though non-functional, SNAREcomplexes. In addition, these defective SNARE complexes appear to havea prolonged life within the nerve terminal, possibly because of their stability.Given the fact that a rosette of SNARE complexes is necessary for the exocyto-sis of one synaptic vesicle, the presence of a single defective SNARE complexis expected to have a dominant negative effect on neurotransmitter release. In-deed, transfection or injection of SNAP-25 truncated at its C-terminus blocksexocytosis (Huang et al., 1998; Criado et al., 1999). Molecular modelling ofthe SNARE complex rosette shows that the SNAP-25 C-terminus, which isremoved by BoNT/A and /C, lies at the site of contact between SNARE com-plexes and therefore is essential for rosette formation (Montecucco et al., 2005).This explains why BoNT/A- and BoNT/C-truncated SNAP-25 are dominantnegatives and also the long duration of the paralytic effect of BoNT/A and /C.Indeed, the long effect of these BoNTs is the combined result of their prote-olytic activity and of the inhibitory activity of C-terminal truncated SNAP-25on the assembly of the rosette of SNARE complexes necessary for exocytosis.
SPANs however do not affect directly proteins involved in neuroexocyto-sis. Instead they act on phospholipids. Indeed, within a few minutes from theiraddition to neurons in culture, phosphatidylcholine is hydrolyzed to lysophos-phatidylcholine (LPC) and fatty acid (FA), with minor production of lysophos-phatidyl-ethanolamine and -serine (Rigoni et al., 2005). This indicates thatSPANs act mainly on the outside layer of the plasma membrane, as PC is themajor phospholipid present on this layer, whilst PE and PS are present onthe cytosolic layer of the plasma membrane and on intracellular membranes,i.e. they are not accessible to SPAN bound to the cell surface (Rigoni et al.,2005). Under physiological conditions, such lipids are present only in minuteamounts, as their molecular shape is not compatible with the bilayer mem-brane structure. LPC is an inverted cone-shaped lipid which forms spheroidalmicelles whereas FAs are cone-shaped. As a consequence of their shapes,these lipids alter the curvature of the membrane (Fuller and Rand, 2001; Cher-nomordik and Kozlov, 2003). Moreover, it should be considered that LPC doesnot flip across the lipid bilayer of the membrane, whereas FAs are capable ofa very rapid trans-bilayer movement (Hamilton, 2003). Consequently, as FAsare produced by SPANs on the external layer of the plasma membrane, theyredistribute among the two membrane layers, whilst LPC remains outside, asschematically depicted in fig. 1. It has been theoretically predicted, and exper-imentally documented in the case of viral membrane fusion, that the presenceof an inverted cone-shaped lipid in trans, and of cone-shaped lipids in cis,with respect to the site of membrane fusion, promotes the process, whilst itinhibits the opposite process of membrane fission (Chernomordik et al., 1997;Chernomordick and Kozlov, 2003). The reason is that such membrane con-

182 O. Rossetto et al.
figuration favours the membrane fusion via pore opening from an hemifusionlipid intermediate which develops by intermixing of the two cis monolayersof the two membranes which have come together, as depicted in fig. 1. Theresults of Rigoni et al. (2005) provide indirect, but compelling, evidence thatthe ready-to-release SVs are indeed already hemifused with the presynapticmembrane and this would well account for the ultra-fast event of neuroexocy-tosis that takes place within a few hundreds of microseconds from the entry ofcalcium (Zimmerberg and Chernomordik, 2005).
SPANs have been shown to induce release of neurotransmitter not followedby recycling of the empty SV (Rigoni et al., 2004; Bonanomi et al. 2005),which accounts for the � figures detected on the intoxicated nerve presynapticmembrane (Cull-Candy et al., 1976). Thus one can envisage the scenario,schematically depicted in fig. 1, which is described in the figure legend.
After this initial stage which envisages a local concentration of the lipidsgenerated by the SPAN enzymatic activity,other molecular events follow. Withtime, more and more phospholipids are hydrolyzed and lysophospholipids areexpected to move laterally on the two-dimensional membrane plane. FAs cando the same but on both membrane layers; in addition, FAs can partition intointernal cell membranes (McArthur et al., 1999). Their transfer into intracel-lular organelles can alter their functionality. FAs are expected to cause a grossalteration of the inner mitochondrial membrane where they are known to act asuncouplers with depression of ATP production. This effect would account forthe change of mitochondrial shape and nerve terminal degeneration detected inlong-term poisoned nerve terminals (Ng and Howard, 1978, 1980; Cull-Candyet al., 1976; Harris et al., 2000). Finally, the scenario presented here does notprovide for the snake toxin inside the cell, but such penetration could well takeplace after the initial stage depicted here, as suggested by several experimentalresults (see above).
6 Comparison of the blockade of nerve terminals induced by BoNTs andSPANs
The final effect of BoNTs and SPANs at nerve terminals is the same: the block-ade of the conversion of the electrical signal into a chemical signal which takesplace at the synapse. However, the mechanism and subcellular sites of actionare different, and this is reflected in the very different morphology of motorneu-ron nerve terminals poisoned by the two classes of enzymatic neurotoxins. Thegross morphological alterations caused by SPANs would suggest that their ac-tion is only partially or not reversible, whilst that of BoNTs would be rapidlyreversed once the toxin and its products are eliminated. The experimental find-ings provide the opposite response. SPAN paralysis is reversed rather rapidly,whereas the duration of action of BoNT is longer or much longer (table 1

Tetanus, Botulinum and Snake Presynaptic Neurotoxins 183
Table 1 Average duration of muscle paralysis produced by a single local injection of distinctBoNTs and SPANs in mice hind limb, as detected with the assay of Aoki, 2001.
Neurotoxin Amount injected Duration of paralysis (days)
BoNT/A 20 pg 35BoNT/B 20 pg 33BoNT/C 15 pg 22BoNT/D 80 pg 30BoNT/E 100 pg 4BoNT/F 50 pg 3TAIPOXIN 100 ng 3NOTEXIN 1000 ng 10TEXTILOTOXIN 100 ng 3β-BUNGAROTOXIN 1000 ng 3
and see Warrell et al., 1983; Theakston et al., 1990; Connolly et al., 1995;Kularatne, 2002; Montecucco and Molgo, 2005; Prasarnpun et al., 2005; Mor-biato et al., unpublished results). In fact, the duration of the inhibitory effectelicited by BoNTs varies with the toxin serotype, the dose, the type of cholin-ergic nerve terminal affected and the animal species (table 1). Their catalyticactivity persists for long periods of time within the intoxicated neurons (Foranet al., 2003). Moreover, in the case of BoNT/A and /C, their truncated SNAP-25 proteins have a dominant negative effect on neurotransmitter release. Inaddition, since the exocytosis apparatus is highly conserved, the BoNTs in-hibitory effect is not limited to the exocytosis of ACh; i.e., they also inhibitthe release of other neurotransmitters and neuropeptides (Schiavo et al., 2000;Simpson, 2000).
Both SPANs and BoNTs do not cause neuronal death in vivo and in vitro,unless very high amounts are used, though they may reach respiratory musclesand autonomic terminals and cause death of the intoxicated animal mainly byrespiratory paralysis. It appears that, with time, the toxins are inactivated, theproducts of their hydrolysis are removed and replaced by de novo synthesistaking place in the motoneuron cell body. Thus, if the intoxicated animalssurvive respiratory paralysis by mechanical ventilation, they appear to recovercompletely, as botulism patients do. Comparable observations aimed at testingthe possibility of long term neurologic sequelae are not available for SPANs.
These neurotoxins combine potency and neurospecificity with reversibilityat the cellular level. These properties are at the basis of the growing thera-peutic use of BoNTs in humans. As far as we know, only one of the SPANs(β-bungarotoxin) has been tested as a muscle weakening agent in experimentalstrabismus in monkeys (Alan Scott, personal communication). It was foundthat it diffused out of the site of injection and that its duration of action wasvery limited, contrary to BoNT/A that does not diffuse significantly and causes

184 O. Rossetto et al.
a paralysis lasting for months (Montecucco and Molgo, 2005). We recently in-jected notexin, β-bugarotoxin, taipoxin and textilotoxin in the posterior legsof mice and recorded complete functional recovery within 3-10 days (table 1)(Morbiato et al., unpublished results). Similar time periods of recovery werefound in rat injected in the hind limb with β-bungarotoxin (Prasarnpun et al.,2005). The possibilityof using SPANs in human therapy deserves attention andappropriate studies. This does not exclude another possibility, that of therapeu-tic indications suggested by the study of the mechanism of action of SPANs. Infact, one could envisage to use the LPL+FA lipid mixture to induce paralysisof superficial nerve terminals that can be reached via transdermal migration.This dermal treatment could be useful in axillary and palmar hyperhydrosis oranal fissures, diseases caused by hyperactive cholinergic nerve terminals thatcould be reached and inhibited by the lipid mixture.
Acknowledgements. The authors studies were supported by research grants from Telethon-Italia GGP06133, from the University of Padova and from FIRB-RBNE01RHZM.
References
1. Abe T, Limbrick AR, Miledi R (1976) Acute muscle denervation induced by beta-bungarotoxin. Proc R Soc Lond B Biol Sci. 194: 545–53
2. Aoki KR (2001) A comparison of the safety margins of botulinum neurotoxin serotypesA, B, and F in mice. Toxicon 39: 1815–1820
3. Bleck TP (1989) Clinical aspects of tetanus. In: Simpson LL (ed.), Botulinum neurotoxinsand tetanus toxin. San Diego: Academic Press: 379–398
4. Bonanomi D, Pennuto M, Rigoni M, Rossetto O, Montecucco C, Valtorta F (2005) Taipoxininduces synaptic vesicle exocytosis and disrupts the interaction of synaptophysin I withVAMP2. Mol. Pharmacol. 67: 1901–1908
5. Chen IL, Lee CY (1970) Ultrastructural changes in the motor nerve terminals caused bybeta-bungarotoxin. Virchows Arch. B. Cell Pathol. 6: 318–325
6. Chernomordik LV, Leikina E, Frolov V, Bronk P, Zimmerberg J (1997) An early stageof membrane fusion mediated by the low pH conformation of influenza hemagglutinindepends upon membrane lipids. J. Cell. Biol. 136: 81–93
7. Chernomordik LV, Kozlov MM (2003) Protein-lipid interplay in fusion and fission ofbiological membranes. Annu. Rev. Biochem. 72: 175
8. Connolly S, Trevett AJ, Nwokolo NC, Lalloo DG, Naraqi S, Mantle D, Schofield IS,Fawcett PR, Harris JB, Warrell DA (1995) Neuromuscular effects of Papuan Taipan snakevenom. Ann. Neurol. 38: 916–920
9. Criado M, Gil A, Viniegra S, Gutierrez LM (1999) A single amino acid near the C terminusof the synaptosome-associated protein of 25 kDa (SNAP-25) is essential for exocytosis inchromaffin cells. Proc. Natl. Acad. Sci. USA 96: 7256–7261
10. Cull-Candy SG, Fohlman J, Gustavsson D, Lullmann-Rauch R, Thesleff S (1976) The ef-fects of taipoxin and notexin on the function and fine structure of the murine neuromuscularjunction. Neuroscience 1: 175–180
11. Dixon RW, Harris JB (1999) Nerve terminal damage by beta-bungarotoxin: its clinicalsignificance. Am J Pathol. 154: 447–455

Tetanus, Botulinum and Snake Presynaptic Neurotoxins 185
12. Dodds DC, Omeis IA, Cushman SJ, Helms JA, Perin MS (1997) Neuronal pentraxin re-ceptor, a novel putative integral membrane pentraxin that interacts with neuronal pentraxin1 and 2 and taipoxin-associated calcium-binding protein 49. J. Biol. Chem. 272: 21488–21494
13. Dong M, Richards DA, Goodnough MC, Tepp WH, Johnson EA, Chapman ER (2003)Synaptotagmins I and II mediate entry of botulinum neurotoxin B into cells. J. Cell. Biol.162: 1293–1303
14. Dong M, Yeh F, Tepp WH, Dean C, Johnson EA, Janz R, Chapman ER (2006) SV2 Is theProtein Receptor for Botulinum Neurotoxin A. Science 312: 592–596
15. Duchen LW (1971) An electron microscopic study of the changes induced by botulinumtoxin in the motor end-plates of slow and fast skeletal muscle fibres of the mouse. J NeurolSci. 14: 47–60
16. Fischer A, Montal M (2007a) Single molecule detection of intermediates during botulinumneurotoxin translocation across membranes. Proc. Natl. Acad. Sci. USA 104: 10447–10452
17. Fischer A, Montal M (2007b) Crucial role of the disulfide bridge between botulinumneurotoxin light and heavy chains in protease translocation across membranes. J BiolChem. In press
18. Foran PG, Mohammed N, Lisk GO, NagwaneyS, Lawrence GW, JohnsonE, Smith L, AokiKR, Dolly JO (2003) Evaluation of the therapeutic usefulness of botulinum neurotoxin B,C1, E, and F compared with the long lasting type A. Basis for distinct durations of inhibitionof exocytosis in central neurons. J. Biol. Chem. 278: 1363–1371
19. Fuller N, Rand RP (2001) The influence of lysolipids on the spontaneous curvature andbending elasticity of phospholipid membranes. Biophys J. 81: 243–254
20. Galazka A, Gasse F (1995) The present status of tetanus and tetanus vaccination. Curr.Top. Microbiol. Immunol. 195: 31–53
21. Gill DM (1982) Bacterial toxins: a table of lethal amounts. Microbiol. Rev. 46: 86–9422. Hamilton JA (2003) Fast flip-flop of cholesterol and fatty acids in membranes: implications
for membrane transport proteins. Curr. Opin. Lipidol. 14: 263–27123. Harris JB (1997) Toxic phospholipases in snake venom: an introductory review. Symp.
zool. Soc. Lond. 70: 235–25024. Harris JB, Grubb BD, Maltin CA, Dixon R. (2000) The neurotoxicity of the venom phos-
pholipases A(2), notexin and taipoxin. Exp. Neurol. 161: 517–52625. Howard BD, Gundersen CB (1980) Effects and mechanisms of polypeptide neurotoxins
that act presynaptically. Annu. Rev. Pharmacol. Toxicol. 20: 307–33626. Howard BD, Wu WC (1976) Evidence that beta-bungarotoxin acts at the exterior of nerve
terminals. Brain Res. 103: 190–19227. Huang X, Wheeler MB, Kang YH, Sheu L, Luckacs GL, Trimble WS, Gaisano HY (1998)
Truncated SNAP-25 (1–197), like botulinum neurotoxin A, can inhibit insulin secretionfrom HIT-T15 insulinoma cells. Mol. Endocrinol. 121060–121070
28. HughesR, Whaler BC (1962) Influence of nerve-endingsactivity and of drugs on the rate ofparalysis of rat diaphragm preparations by Clostridium botulinum type A toxin. J. Physiol.(Lond) 160: 221–233
29. Jahn R, Lang T, Sudhof TC (2003) Membrane fusion. Cell 112: 519–53330. Kamenskaya MA, Thesleff S (1974) The neuromuscular blocking action of an isolated
toxin from the elapid (Oxyuranus scutellactus). Acta Physiol. Scand. 90: 716–72431. Kelly RB, Brown BR (1974) Biochemical and physiological properties of a purified snake
venom neurotoxin which acts presynaptically. J. Neurobiol. 5: 135–15032. Kelly RB, Oberg SG, Strong PN, Wagner GM (1976) beta-Bungarotoxin, a phospholipase
that stimulates transmitter release. Cold Spring Harb. Symp. Quant. Biol. 40: 117–12533. Kielian M, Rey FA (2006) Virus membrane-fusion proteins: more than one way to make a
hairpin. Nat. Rev. Microbiol. 4: 67–76

186 O. Rossetto et al.
34. Kini RM (1997) Venom Phospholipase A2 Enzymes. John Wiley & Sons, Chichester35. Kirkpatrick LL, Matzuk MM, Dodds DC, Perin MS (2000) Biochemical interactions of
the neuronal pentraxins. Neuronal pentraxin (NP) receptor binds to taipoxin and taipoxin-associatedcalcium-binding protein 49 via NP1 and NP2. J. Biol. Chem. 275: 17786–17792
36. Koriazova LK, Montal M (2003) Translocation of botulinum neurotoxin light chain pro-tease through the heavy chain channel. Nat. Struct. Biol. 10: 13–18
37. Kularatne SA (2002) Common krait (Bungarus caeruleus) bite in Anuradhapura, Sri Lanka:a prospective clinical study, 1996-98. Postgrad. Med. J. 78: 276–280
38. Kwong PD, Mcdonald NQ, Sigler PB, Hendrickson WA (1995) Structure of beta 2-bungarotoxin: potassium channel binding by Kunitz modules and targeted phospholipaseaction. Structure 3: 1109–1119
39. Lacy DB, Tepp W, Cohen AC, Dasgupta BR, Stevens RC (1998) Crystal structure ofbotulinum neurotoxin type A and implications for toxicity. Nature Struct. Biol. 5: 898–902
40. Lalli G, Bohnert S, Deinhardt K, Verastegui C, Schiavo G (2003) The journey of tetanusand botulinum neurotoxins in neurons. Trends Microbiol. 11: 431–437
41. Mahrhold S, Rummel A, Bigalke H, Davletov B, Binz T (2006) The synaptic vesicle protein2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 580:2011–2014
42. McArthur M, Atshaves B, Frolov A, Foxworth W, Kier A, Schroeder F (1999) Cellularuptake and intracellular trafficking of long chain fatty acids. J. Lipid Res.: 1371–1383
43. Major RH (1945) Classic descriptions of disease. Springfield, IL: Charles C. Thomas44. Montecucco C (1986) How do tetanus and botulinum toxins bind to neuronal membranes?
Trends Biochem. Sci. 11: 315–31745. Montecucco C, Molgo J (2005) Botulinal neurotoxins: revival of an old killer. Curr Opin
Pharmacol. 5: 274–27946. Montecucco C, Rossetto O (2000) How do presynaptic PLA2 neurotoxins block nerve
terminals? Trends Biochem. Sci. 25: 266–27047. Montecucco C, Rossetto O, Schiavo G (2004) Presynaptic receptor arrays for clostridial
neurotoxins. Trends Microbiol. 195: 221–24148. Montecucco C, Schiavo G, Gao Z, Bauerlein E, Boquet P, Dasgupta BR (1988) Interaction
of botulinum and tetanus toxins with the lipid bilayer surface. Biochem. J. 251: 379–38349. Montecucco C, Schiavo G, Dasgupta BR (1989)EffectofpH on the interaction ofbotulinum
neurotoxins A, B and E with liposomes. Biochem. J. 259: 47–5350. Montecucco C, Schiavo G, Pantano S (2005) SNARE complexes and neuroexocytosis:
how many, how close? Trends Biochem. Sci. 30: 367–37251. Neale EA, Bowers LM, Jia M, Bateman KE, Williamson LC, 1999. Botulinum neurotoxin
A blocks synaptic vesicle exocytosis but not endocytosis at the nerve terminal. J Cell Biol.147: 1249–1260
52. Neco P, Rossetto O, Gil A, Montecucco C, Gutierrez LM (2003) Taipoxin induces F-actin fragmentation and enhances release of catecholamines in bovine chromaffin cells. J.Neurochem. 85: 329–337
53. Ng RH, Howard BD (1978) Degenergization of nerve terminals by beta-bungarotoxin.Biochemistry 17: 4978–4986
54. Ng RH, Howard BD (1980) Mitochondria and sarcoplasmic reticulum as model targets forneurotoxic and myotoxic phospholipases A2. Proc. Nat. Acad. Sci. USA 77: 1346–1350
55. Nishiki T, Tokuyama Y, Kamata Y, Nemoto Y, Yoshida A, Sato K, Sekiguchi M, TakahashiM, Kozaki S (1996) The high-affinity binding of Clostridium botulinum type B neurotoxinto synaptotagmin II associated with gangliosides GT1b/GD1a. FEBS Lett. 378: 253–257
56. Payling-WrightG (1955)The neurotoxins ofClostridium botulinum and Clostridium tetani.Pharmacol. Rev. 7: 413–465

Tetanus, Botulinum and Snake Presynaptic Neurotoxins 187
57. Pearson JA, Tyler MI, Retson KV, Howden ME (1993) Studies on the subunit structureof textilotoxin, a potent presynaptic neurotoxin from the venom of the australian commonbrown snake (pseudonaja textilis). 3. the complete amino-acid sequences of all the subunits.Biochim Biophys Acta, 1161: 223–229
58. Petrovic U, Sribar J, Paris A, Rupnik M, Krzan M, Vardjan N, GubensekF, Zorec R, KrizajI (2004) Ammodytoxin, a neurotoxic secreted phospholipase A(2), can act in the cytosolof the nerve cell. Biochem. Biophys. Res. Commun. 324: 981–985
59. Prasarnpun S, Walsh J, Harris JB (2004) Beta-bungarotoxin-induced depletion of synapticvesicles at the mammalian neuromuscular junction. Neuropharmacology 47: 304–314
60. Prasarnpun S, Walsh J, Awad SS, Harris JB (2005) Envenoming bites by kraits: the bio-logical basis of treatment-resistant neuromuscular paralysis. Brain., 128: 2987–2996
61. Puhar A, Johnson EA, Rossetto O, Montecucco C (2004) Comparison of the pH-inducedconformational rearrangement of different clostridial neurotoxins.Biochem. Biophys.Res.Commun. 319: 66–71
62. Pumplin DW, Reese TS (1977) Action of brown widow spider venom and botulinum toxinon the frog neuromuscular junction examined with the freeze-fracture technique. J. Physiol.273: 443–457
63. Rigoni M, Caccin P, Gschmeissner S, Koster G, Postle Ad, Rossetto O, Schiavo G, Mon-tecucco C (2005) Equivalent Effects of Snake PLA2 Neurotoxins and Lysophospholipid-Fatty Acid Mixtures. Science 310: 1678–1680
64. Rigoni M, Schiavo G, Weston AE, Caccin P,Allegrini F, Pennuto M, Valtorta F, MontecuccoC, Rossetto O (2004) Snake presynaptic neurotoxins with phospholipase A2 activity inducepuntate swellings of neurites and exocytosis of synaptic vesicles. J. Cell Sci. 117: 3561–1570
65. Rossetto O, Montecucco C (2004) Clostridial neurotoxins. In: Proft T (ed.), MicrobialToxins. Molecular and Cellular Biology. Horizon Scientific Press, Norfolk, UK: 149–178
66. Rummel A, Bade S, Alves J, Bigalke H, Binz T (2003) Two carbohydrate binding sitesin the H(CC)-domain of tetanus neurotoxin are required for toxicity. J. Mol. Biol. 326:835–847
67. Rummel A, Karnath T, Henke T, Bigalke H, Binz T (2004) Synaptotagmin I and II act asnerve cell receptors for botulinum neurotoxin G. J. Biol. Chem. 279: 30865–30870
68. Schiavo G, Matteoli M, Montecucco C (2000) Neurotoxins Affecting Neuroexocytosis.Physiol. Rev. 80: 717–766
69. Simpson LL (2000) Identification of the characteristics that underlie botulinum toxin po-tency implications for designing novel drugs. Biochimie 82: 943–953
70. Simpson LL, Coffield JA, Bakry N (1994). Inhibition of vacuolar adenosine triphosphataseantagonizes the effects of clostridial neurotoxins but not phospholipase A2 neurotoxins. J.Pharmacol. Exp. Ther. 269: 256–262
71. Simpson LL, Lautenslager GT, Kaiser II, Middlebrook JL (1993) Identification of the siteat which phospholipase A2 neurotoxins localise to produce their neuromuscular blockingeffects. Toxicon, 31: 13–26
72. Singh G, Gourinath S, Sharma S, Paramasivam M, Srinivasan A, Singh TP (2001) Se-quence and crystal structure determination of a basic phospholipase A2 from commonkrait (Bungarus caeruleus) at 2.4 A resolution: identification and characterization of itspharmacological sites. J. Mol. Biol. 307: 1049–1059
73. Sribar J, Copic A, Paris A, Sherman NE, Gubensek F, Fox JW, Krizaj I (2001) A highaffinity acceptor for phospholipase A2 with neurotoxic activity is a calmodulin. J. Biol.Chem. 276: 12493–12496
74. Sribar J, Copic A, Poljsak-Prijatelj M, Kuret J, Logonder U, Gubensek F, Krizaj I (2003a)R25 is an intracellular membrane receptor for a snakevenom secretori phospholipaseA(2).FEBS Lett. 553: 309–314

188 O. Rossetto et al.
75. Sribar J, Sherman NE, Prijatelj P, Faure G, Gubensek F, Fox JW, Aitken A, Punger-car J, Krizaj I (2003b) The neurotoxic phospholipase A2 associates, through a non-phosphorylatedbinding motif, with 14-3-3 protein gamma and epsilon isoforms. Biochem.Biophys. Res. Commun. 302: 691–696
76. Swaminathan S, Eswaramoorthy S (2000) Structural analysis of the catalytic and bindingsites of Clostridium botulinum neurotoxin B. Nat.Struct. Biol. 7: 693–699
77. Tacket CO, Rogawski MA (1989) Botulism. In: Simpson LL (ed) Botulinum Neurotoxinsand Tetanus Toxin, Academic Press, San Diego: 351–378
78. Theakston RD, Phillips RE, Warrell DA, Galagedera Y, Abeysekera DT, Dissanayaka P,De Silva A, Aloysius DJ (1990) Envenoming by the common krait (Bungarus caeruleus)and Sri Lankan cobra (Naja naja naja): efficacy and complications of therapy with Haffkineantivenom. Trans. R. Soc. Trop. Med. Hyg. 84: 301–308
79. Warrell DA, Looareesuwan S, White NJ, Theakston RD, Warrell MJ, Kosakarn W, Reid HA(1983) Severe neurotoxic envenoming by the Malayan krait Bungarus candidus (Linnaeus):response to antivenom and anticholinesterase. Br. Med. J. 286: 678–680
80. Weinstein L (1973) Tetanus. N. Engl. J. Med. 289: 1293–129681. Westerlund B, Nordlund P, Uhlin U, Eaker D, Eklund H (1992) The three-dimensional
structure of notexin, a presynaptic neurotoxic phospholipase A2 at 2.0 A resolution. FEBSLett. 301: 159–164
82. Yang CC (1997) Chemical modification and functional sites of phospholipasesA2, In: KiniRM (ed), Venom Phospholipase A2 Enzymes. John Wiley & Sons, Chichester: 185–204
83. Zimmerberg J, Chernomordik LV (2005) Neuroscience. Synaptic membranes bend to thewill of a neurotoxin. Science 310: 1626–1627