phylogeny from whole genome comparison
TRANSCRIPT

Filogenia de Proteomas
Graziela Santos de Araujo
Dissertacao de Mestrado
Orientacao: Prof. Dr. Nalvo Franco de Almeida Junior
Dissertacao apresentada como requisito para o obtencao do tıtulo de mestreem Ciencia da Computacao.
Departamento de Computacao e EstatısticaCentro de Ciencias Exatas e Tecnologia
Universidade Federal de Mato Grosso do Sul22 de maio de 2003

Dedicatoria
Para meus pais e meu irmao.
ii

Agradecimentos
Tenho a feliz oportunidade de agradecer a todos aqueles que me ajudaramde alguma forma para a conclusao de mais essa etapa na minha vida, e soumuito grata por essa oportunidade.
Agradeco aos meus pais e ao meu irmao, por entenderem e apoiarem a decisaode continuar estudando e deixa-los um pouco de lado; ao Marcio Medina pelaimensa paciencia, apoio, compreensao e carinho; e aos meus novos e velhosamigos que sempre me deram apoio para que o estımulo nao acabasse.
Ao professor Marcelo Henriques de Carvalho pelo constante esforco em me-lhorar o Mestrado em Ciencia da Computacao na Universidade Federal deMato Grosso do Sul, sempre nos ajudando e dando forca e incentivo emnossas pesquisas.
Ao professor Edson Norberto de Caceres que nunca mede esforcos para me-lhorar a Ciencia da Computacao nesta universidade.
Um agradecimento mais que especial ao meu orientador, professor Nalvo,pela sua atencao, amizade, confianca em meu trabalho e principalmente pelapaciencia. Obrigada pelas conversas, dicas e compreensao. Vou sentir faltada “ordem” da tarde para fazer o terere.
Agradeco aos membros da banca examinadora, pela boa vontade em aceitaro convite para participar da minha defesa.
Aos meus amigos de Sao Paulo, Leonardo e Said, Valguima e Ana Lucia, quemesmo a distancia contribuıram para que hoje este trabalho fosse concluıdo,gastando o pouco tempo que tinham procurando artigos para mim. Naodeixando de esquecer, o amigo e professor Fabio Viduani que me salvou dodesespero em alguns pontos da pesquisa, mostrando caminhos um pouco maisfaceis a seguir.
Ao restante da minha turma do mestrado, que assim como eu, lutou e estalutando para vencer essa etapa. Valeu pelo apoio e torcida. Em particular,a amiga Edna, o mestrado nao teria sido o mesmo sem o seu apoio e ajuda;
iii

dct-ufms
e tambem a Luciana, pelas conversas, alegrias e desesperos compartilhados.
Enfim, agradeco a TODOS que de alguma forma me ajudaram a concluiresse trabalho. Muito Obrigado.
iv

Resumo
A explicacao da historia evolutiva das especies e os seus possıveis relacio-namentos sao preocupacoes centrais na Biologia. Esses aspectos podem serverificados pela construcao de arvores filogeneticas, tambem conhecidas comofilogenias, que sao arvores onde as folhas representam as especies e os nosinternos representam possıveis ancestrais. Com a descoberta de tecnologiaspara sequenciamento de DNA, e consequente disponibilizacao de genomascompletos, podemos inferir filogenias utilizando dados relativos a ordem dosgenes de cada especie. Esses dados podem ser distancias ou caracterısticas.As distancias representam uma estimativa da distancia evolutiva entre os pa-res de organismos. As caracterısticas dizem respeito, por exemplo, a presencade genes especıficos em alguns genomas e ausencia em outros. Nosso objetivoe o de propor uma metodologia para a construcao de arvores filogeneticas,que consiste em extrair informacoes de comparacoes entre conjuntos de genesde especies. Estas informacoes podem ser: genes encontrados em ambos osgenomas e regioes em que houve a conservacao da ordem dos genes. Alemdisso, tambem propomos a construcao de filogenias utilizando caracterısticasenvolvendo genes e regioes, obtidas tambem dos genomas das especies. Pro-pomos ainda uma medida de distancia entre arvores, com o objetivo de avaliara qualidade das mesmas.
v

Abstract
The explanation of evolutionary history of extant species and their possiblerelationships are major concerns in Biology. These aspects can be verifiedthrough the construction of phylogenetic trees, also known as phylogenies,which are trees where leaves represent species and internal nodes representcommon ancestors of those species. With the advances of DNA sequencingtechniques, and so the availability of whole genomes, we can infer phylogeniesby using data from gene order of each species. These data can be distances orcharacters. The distances represent an estimate of evolutive distance betweenpairs of organisms. Characters are, for example, presence of specific genes insome genomes and absence on others. Our goal is to propose a methodologyfor constructing phylogenetic trees, that consists in getting information fromcomparisons of set of genes. Such information can be: genes found in bothgenomes and regions with gene order conservation. Besides, we also proposephylogenies by using characters envolving genes and regions, gotten of thegenomes. We propose also a measure to evaluate the quality of the trees.
vi

Conteudo
Dedicatoria ii
Agradecimentos iii
Resumo v
Abstract vi
Conteudo xi
1 Introducao 1
1.1 Justificativa . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Conceitos basicos . . . . . . . . . . . . . . . . . . . . . . . . . 3
1.3 Sumario de resultados . . . . . . . . . . . . . . . . . . . . . . 3
1.4 Organizacao do texto . . . . . . . . . . . . . . . . . . . . . . . 4
2 Fundamentos de Biologia Molecular Computacional 5
2.1 Sıntese de proteınas . . . . . . . . . . . . . . . . . . . . . . . . 7
2.2 Visao computacional do genoma . . . . . . . . . . . . . . . . . 8
2.3 Filogenia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
3 Filogenia baseada em distancias 14
3.1 Arvores ultrametricas . . . . . . . . . . . . . . . . . . . . . . . 15
3.2 Arvores aditivas . . . . . . . . . . . . . . . . . . . . . . . . . . 21
vii

Conteudo dct-ufms
3.3 Heurıstica para construcao de filogenias baseadas em distancias 27
4 Filogenia baseada em caracterısticas 32
4.1 Aspectos da filogenia baseada em caracterısticas . . . . . . . . 33
4.2 Algoritmo para numero fixo de estados . . . . . . . . . . . . . 36
4.3 Parsimonia e compatibilidade . . . . . . . . . . . . . . . . . . 39
4.4 Mix . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
5 Comparacao de proteomas e filogenia 47
5.1 Metodologia . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
5.2 Filogenia baseada em distancias . . . . . . . . . . . . . . . . . 55
5.2.1 Resultados . . . . . . . . . . . . . . . . . . . . . . . . . 56
5.3 Filogenia baseada em caracterısticas . . . . . . . . . . . . . . . 58
5.3.1 Resultados . . . . . . . . . . . . . . . . . . . . . . . . . 61
6 Consideracoes finais 63
Apendice A 65
Referencias Bibliograficas 69
viii

Lista de Figuras
1.1 Representacao de uma arvore filogenetica para alguns primatas. 2
2.1 Cada gi e um gene pertencente ao proteoma de G. . . . . . . . 9
2.2 Exemplo de grafo com cinco vertices. . . . . . . . . . . . . . . 10
2.3 Exemplo de grafo bipartido. A particao X contem os verticesX = {1, 3, 5} e a particao Y contem os vertices Y = {2, 4}. . . 10
2.4 (a) Exemplo de subgrafo do grafo da Figura 2.2. (b) Exemplode um subgrafo gerador do grafo da Figura 2.2. . . . . . . . . 11
2.5 Exemplo de grafo que nao e conexo. . . . . . . . . . . . . . . . 11
2.6 Exemplo de grafo que e uma arvore. . . . . . . . . . . . . . . . 11
2.7 Exemplo de uma subarvore. . . . . . . . . . . . . . . . . . . . 12
2.8 Exemplo de arvore geradora. . . . . . . . . . . . . . . . . . . . 12
2.9 (a) Exemplo de grafo com peso associados as suas arestas. (b)Exemplo de arvore geradora de custo mınimo para o grafo (a). 12
3.1 a) Exemplo de uma matriz simetrica M . b) Arvore ul-trametrica para a matriz M . . . . . . . . . . . . . . . . . . . . 16
3.2 Exemplo de subarvore generica contendo as folhas i, j e k. . . 17
3.3 a) Duas linhas de uma matriz simetrica M . A linha do objetoa e usada para obter o caminho ate a folha a, que e mos-trada na Figura b). Os numeros nos nos ao longo do caminhoparticionam os demais objetos. . . . . . . . . . . . . . . . . . . 17
3.4 Exemplo de matrizes de entrada M l e Mh. . . . . . . . . . . . 19
3.5 Grafo Gh e sua arvore geradora mınima T . . . . . . . . . . . . 19
ix

Lista de Figuras dct-ufms
3.6 Arvore ultrametrica para as matrizes da Figura 3.4. Os rotulosnas arestas sao distancias entre os nos. . . . . . . . . . . . . . 19
3.7 Exemplo das possıveis subarvores conectando quatro objetosquaisquer i, j, k e l. . . . . . . . . . . . . . . . . . . . . . . . . 22
3.8 Exemplo de matriz aditiva. . . . . . . . . . . . . . . . . . . . . 23
3.9 Exemplo de execucao do algoritmo para a matriz da Figura 3.8. 25
3.10 (a) Exemplo de matriz aditiva M . A maior entrada tem valor9 e esta na linha do objeto a. (b) Matriz ultrametrica M ′.(c) Arvore ultrametrica T ′ ja com os pesos nas arestas. (d)Arvore resultante T apos subtrairmos ma −Mai das arestasfolhas. A arvore original e obtida apos contrair a aresta depeso zero onde esta a folha a. . . . . . . . . . . . . . . . . . . 26
3.11 (a) Arvore estrela com nenhuma estrutura hierarquica e (b)Arvore em que as especies 1 e 2 estao agrupadas. . . . . . . . 28
3.12 Matriz de distancias para arvores da Figura 3.13. . . . . . . . 29
3.13 Exemplo da aplicacao do metodo Neighbor-Joining para oitoespecies. Os numeros sobre as arestas sao os tamanhos dasarestas. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
4.1 Exemplo de matriz de estados. . . . . . . . . . . . . . . . . . . 33
4.2 Filogenia com exemplos de reversao e evolucao paralela. . . . . 34
4.3 Exemplo de filogenia que nao e uma filogenia perfeita. . . . . . 35
4.4 Exemplo de matriz de estados que admite filogenia perfeita. . 35
4.5 Filogenia correspondente a matriz da Figura 4.4. . . . . . . . . 36
4.6 Matriz de estados da Figura 4.4 com ordenacao das colunas. . 38
4.7 Matriz auxiliar L, calculada para verificar se a matriz de es-tados da Figura 4.4 e aditiva. . . . . . . . . . . . . . . . . . . 39
4.8 Exemplo de execucao para o metodo de Wagner. . . . . . . . . 43
4.9 Exemplo de arvore que sofrera rearranjo local. A aresta pon-tilhada e um segmento interno. . . . . . . . . . . . . . . . . . 45
4.10 Arvore apos rearranjo. . . . . . . . . . . . . . . . . . . . . . . 45
5.1 Exemplo de run entre os proteomas de Xylella fastidiosa eXanthomonas citri. . . . . . . . . . . . . . . . . . . . . . . . . 48
x

Lista de Figuras dct-ufms
5.2 Exemplo de um run. Note que pode haver a participacao deproteınas preditas em fitas distintas. . . . . . . . . . . . . . . 49
5.3 Exemplo de buracos encontrados entre os runs. . . . . . . . . . 49
5.4 Exemplo de regiao ortologa que podemos encontrar entre osgenomas de Xylella fastidiosa e Xanthomonas campestris. . . . 50
5.5 Matriz de distancias para sequencias 16S rRNA de bacterias. . 51
5.6 Arvore filogenetica obtida a partir da matriz 5.5. . . . . . . . . 51
5.7 Arvore filogenetica obtida apos bootstrap. . . . . . . . . . . . 52
5.8 Arvore T1, cuja matriz de distancias e baseada no numero dematches encontrados entre os pares de genomas. . . . . . . . . 57
5.9 Arvore T2, cuja matriz de distancias e baseada no numero deBBHs encontrados entre os pares de genomas. . . . . . . . . . 57
5.10 Filogenia obtida a partir da caracterıstica presenca/ausenciade genes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
5.11 Filogenia obtida a partir da caracterıstica presenca/ausenciade regioes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
7.1 Matriz de probabilidade de substituicao entre bases. . . . . . . 66
7.2 Matriz de probabilidade de substituicao para o modelo de Ki-mura. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
7.3 Matriz de probabilidade de substituicao. . . . . . . . . . . . . 68
7.4 Matriz de probabilidade de substituicao. . . . . . . . . . . . . 68
xi

Capıtulo 1
Introducao
Todas as especies de organismos existentes na Terra passaram por um pro-cesso de transformacao ao longo dos tempos. Tal processo e chamado deevolucao. Um dos problemas centrais da Biologia, conhecido como pro-blema da filogenia, e explicar a historia evolutiva das especies hoje existentes,bem como verificar relacionamentos entre essas especies, a fim de determinarpossıveis ancestrais comuns entre elas.
Para tentar explicar fatos como os mencionados acima, frequentemente utili-zamos arvores. Em geral, uma arvore filogenetica, ou simplesmente filogenia,e uma arvore onde as folhas representam os organismos e os nos internosrepresentam supostos ancestrais. As arestas da arvore denotam as relacoesevolutivas.
Podemos construir arvores filogeneticas para populacoes, especies, generos ououtros grupos de indivıduos, inclusive sequencias de proteınas ou de acidosnucleicos. Na Figura 1.1, temos um exemplo de filogenia1 construıda paraverificacao do relacionamento entre especies de macacos e a especie humana,onde podemos ver que o homem e o chimpanze sao geneticamente maisproximos que os outros pares presentes na arvore.
Hoje em dia, ja e possıvel usar sequencias moleculares como fonte de dados,o que proporciona potencialmente uma maior fidelidade, visto que a evolucaoocorre nas moleculas. Alem disso, ha muitos projetos genomas concluıdos eoutros em andamento, o que nos fornece uma grande quantidade de dados.Para se ter uma ideia, 113 genomas de procariotos e 17 de eucariotos ja foramsequenciados [7, 31].
1Esta filogenia foi transcrita do capıtulo 6 do livro de Setubal e Meidanis [40].
1

1.1. Justificativa dct-ufms
Siamang Gibão Orangotango Gorila Humano Chimpanzé
Figura 1.1: Representacao de uma arvore filogenetica para alguns primatas.
Um projeto genoma consiste de tres grandes etapas: o sequenciamento, aanotacao e a analise. O sequenciamento consiste na descoberta da sequenciaexata de acido desoxirribonucleico (DNA) de cada cromossomo do organismo;a anotacao consiste na descoberta da posicao exata de cada gene do genoma,incluindo a determinacao de sua funcao; a analise compreende a obtencao deuma visao geral do organismo, visando obter uma caracterizacao funcional,baseada na anotacao e em outras informacoes, tais como a comparacao degenomas. As filogenias construıdas neste trabalho sao originadas da com-paracao de genomas.
A construcao de filogenias e um componente essencial em pesquisas modernasnas areas da medicina e da biologia, para descobrir novas drogas, entenderrapidamente as mutacoes de patogenos, a dispersao de especies, a evolucaodos genomas, dentre outras aplicacoes.
1.1 Justificativa
Com os avancos da tecnologia, varios projetos genoma foram concluıdos,assim como outros iniciaram. Com isso, a quantidade de informacoes resul-tantes desses projetos aumenta em grandes proporcoes. No entanto, muitasinformacoes relevantes ainda precisam ser interpretadas e manipuladas. Eessa descoberta de informacoes, que pode ser feita por meio da comparacaode genomas, pode trazer sinais de relacionamentos entre eles.
Logo, podemos utilizar ferramentas de comparacao de genomas para extrairinformacoes que possam ser utilizadas como entrada no processo de cons-trucao de filogenias. Essas informacoes podem ser relacionadas a organizacao
2

1.2. Conceitos basicos dct-ufms
dos genes nos diversos genomas.
1.2 Conceitos basicos
Pressupoe-se que o leitor tenha um conhecimento basico de Biologia Molecu-lar e Ciencia da Computacao, especificamente Estrutura de Dados e Desen-volvimento e Analise de Algoritmos.
Para entender os conceitos basicos em Biologia Molecular Computacional,recomendamos os livros de Setubal e Meidanis [40] e Gusfield [24].
1.3 Sumario de resultados
Em nosso estudo detalhamos como podemos comparar os proteomas dos ge-nomas e obtermos informacoes que podem ser utilizadas na construcao defilogenias. Em particular, descrevemos medidas de distancia entre genomasobtidas a partir da comparacao dos proteomas, e tambem propostas de ca-racterısticas dos proteomas que podem ser usadas na inferencia filogeneticade especies. Propomos uma metodologia para construcao de arvores filo-geneticas, que consiste na aplicacao de algoritmos baseados em distanciase baseados em caracterısticas, sobre os dados obtidos pelas comparacoes dedois genomas completos. Com isso, obtemos arvores que precisam ser vali-dadas, ou seja, precisamos verificar a qualidade dessas arvores; se elas real-mente refletem a verdadeira historia evolutiva dos genomas envolvidos. Pararealizar essa validacao, criamos uma medida de distancia entre arvores. Apartir dessa medida podemos identificar melhor quais medidas de distanciasao bons instrumentos para serem utilizadas no processo de inferencia filo-genetica, alem de tambem avaliar as arvores obtidas a partir das propostasde caracterısticas baseadas em genes e regioes.
Em particular, podemos observar que matches, BBHs e regioes ortologasencontrados na comparacao dos proteomas das especies, sao de fato bonsinstrumentos para serem utilizados no processo de inferencia filogenetica.Matches, BBHs e regioes ortologas sao estruturas resultantes da comparacaode dois genomas, que serao definidos ao longo do texto.
Resultados preliminares deste trabalho, que compreendem a construcao de fi-logenias utilizando medidas de distancias entre genomas, ja foram publicadosem [4].
3

1.4. Organizacao do texto dct-ufms
1.4 Organizacao do texto
O texto e organizado da seguinte maneira: no Capıtulo 2 apresentamos fun-damentos teoricos em biologia, uma visao computacional dos genomas e umabreve descricao do processo de construcao de filogenias. No Capıtulo 3, des-crevemos o problema da construcao de filogenias baseadas em distancias.Em particular, descrevemos a construcao de arvores aditivas e ultrametricas.Apresentamos tambem a construcao de arvores aditivas por meio de arvoresultrametricas. E finalmente, descrevemos uma heurıstica para construir filo-genias baseadas em distancias, que foi escolhida para ser utilizada em nossotrabalho. No Capıtulo 4, descrevemos o problema da filogenia perfeita, quee o problema da construcao de filogenias baseada em caracterısticas, e comoresolve-lo. Apresentamos, em particular, um conhecido algoritmo para resol-ver o problema quando o numero de estados das caracterısticas e igual a dois;uma abordagem para construir filogenias baseadas em caracterısticas quandonao podemos construir filogenias perfeitas, conhecido como criterio de par-simonia; e descrevemos um algoritmo de parsimonia que e utilizado paraconstruir as arvores baseadas em caracterısticas em nosso trabalho. Pro-pomos uma metodologia para construcao de filogenias no Capıtulo 5, ondedescrevemos o programa utilizado para comparar os proteomas das especies,bem como descrevemos seis medidas baseadas em distancias e duas baseadasem caracterısticas que podem ser obtidas a partir da comparacao dos prote-omas das especies. Apresentamos ainda, os resultados obtidos nessas duasabordagens de construcao. Descrevemos tambem uma medida de distanciaentre arvores que e utilizada para verificar a qualidade das arvores obtidas apartir das medidas de distancias e das caracterısticas. No Capıtulo 6 apre-sentamos as consideracoes finais.
4

Capıtulo 2
Fundamentos de BiologiaMolecular Computacional
Assim como encontramos no planeta uma grande diversidade de microor-ganismos, plantas e animais, encontramos tambem uma diversidade biomo-lecular enorme, ja que cada organismo contem de milhares a milhoes detipos de biomoleculas diferentes, dependendo da especie. As principais bio-moleculas sao os acidos nucleicos, as proteınas, os carboidratos (ou acucares)e os lipıdios (ou gorduras).
Os tipos de acidos nucleicos mais conhecidos sao o acido desoxirribonucleico,abreviado na lıngua inglesa como DNA, e o acido ribonucleico, tambem abre-viado por RNA. O DNA e uma longa molecula filamentosa formada por duascadeias (ou fitas) que se “torcem” uma sobre a outra formando uma dupla-helice, como uma escada em caracol.
Cada cadeia da molecula de DNA e um polımero formado pela uniao e com-binacao de quatro tipos diferentes de unidades basicas que sao chamadasnucleotıdeos. Os nucleotıdeos sao formados por um acucar, a desoxirri-
bose, um grupo fosfato e uma molecula chamada base nitrogenada. O quedistingue os quatro diferentes nucleotıdeos entre si e a base nitrogenada. Asbases nitrogenadas presentes no DNA sao: adenina, citosina, timina e gua-nina. Os quatro diferentes nucleotıdeos sao convencionalmente representadospelas iniciais dos nomes das bases nitrogenadas: A (adenina), C (citosina),T (timina) e G (guanina).
O tamanho de uma molecula de DNA e dado em pares de bases (pb), umareferencia aos pares de nucleotıdeos descritos acima. Assim, se uma moleculade DNA tem 1000 pares de nucleotıdeos, diz-se que o tamanho dela e de 1000
5

dct-ufms
pares de bases (1000 pb).
O RNA e formado apenas por uma cadeia de nucleotıdeos. Porem, possuiduas diferencas em relacao ao DNA: o acucar presente e a ribose, e o RNAnao possui a base timina (T) e sim a uracila (U). Ele e sintetizado poruma enzima que toma uma das fitas do DNA como molde para fazer umacadeia complementar dos nucleotıdeos de RNA. Neste processo chamado detranscricao, os nucleotıdeos A, C, G e T do DNA sao transcritos a U, G, Ce A na molecula de RNA, respectivamente.
Ao longo do DNA se encontram os genes. Os genes sao pedacos de DNA,ou seja, sequencias de nucleotıdeos representados por letras [42]. A maio-ria dos genes tem uma sequencia especıfica que carrega um codigo para aproducao de uma proteına. Ha inumeras proteınas com as mais variadasfuncoes biologicas. Por exemplo, os anticorpos participam da defesa imu-nologica, a hemoglobina transporta oxigenio e as enzimas sao catalisadoresque aceleram as reacoes bioquımicas. Embora um gene possa codificar maisde uma proteına, e apesar de haver genes que nao codificam proteına, vamos,quando for conveniente, usar os termos gene e proteına indistintamente.
Todas essas informacoes apresentadas acima sao encontradas no genoma deum ser vivo. O genoma e o conjunto de todo o material genetico que defineum ser vivo [42]. O material genetico contem todas as informacoes para gerarum organismo vivo e determinar suas caracterısticas. As biomoleculas querepresentam o material genetico sao o DNA e, mais raramente, o RNA. Ja oconjunto de proteınas produzidas em um genoma e chamado de proteoma.
Toda a experiencia e conhecimento obtidos no campo da biologia molecularlevaram a comunidade cientıfica a almejar um novo e grande desafio: des-vendar a sequencia completa do genoma humano. Este seria o grande passopara responder a perguntas do tipo: Por que somos do jeito que somos? Oque faz com que algumas pessoas possuam cabelos escuros enquanto outrassao loiras? Sera que nosso comportamento e tendencias psicologicas estaonos genes? Os genes podem responder a algumas destas perguntas, mas naopodemos desprezar fatores ambientais e culturais na determinacao das nossascaracterısticas.
Assim como o genoma humano, muitos outros genomas foram e estao sendosequenciados, principalmente de organismos procariotos. De posse dessasinformacoes, o nosso principal foco de estudo em um genoma sao os genesnele contido, pois sao possuidores das informacoes para produzir proteınas.Alem disso, em nosso trabalho, estamos interessados em encontrar geneshomologos. Ou seja, genes que evoluıram a partir de um gene ances-
6

2.1. Sıntese de proteınas dct-ufms
tral comum. Mais especificamente, estamos interessados em encontrar genesortologos. Dois genes g e h sao ortologos se sao descendentes de um mesmogene ancestral e pertencem a especies distintas. Dizemos, neste caso, que(g, h) e um par de ortologos.
Na Secao 2.1 descrevemos o processo de sıntese de proteınas. Ja na Secao 2.2apresentamos o genoma do ponto de vista computacional e na Secao 2.3apresentamos brevemente como as informacoes para construcao de filogeniaspodem ser obtidas a partir dos proteomas.
2.1 Sıntese de proteınas
Como sabemos, as proteınas tem um papel muito importante em nosso or-ganismo. As proteınas sao biomoleculas formadas pela uniao de unidadesbasicas chamadas aminoacidos. Cada proteına possui uma sequencia es-pecıfica de aminoacidos em sua estrutura, a fim de que possa realizar seu pa-pel biologico. Existem 20 diferentes aminoacidos mais comuns que compoema estrutura das proteınas. A sequencia correta de aminoacidos que deter-minam uma proteına e obtida a partir da sequencia de DNA dos genes. Osgenes possuem na sua sequencia de nucleotıdeos a informacao basica para aordenacao dos aminoacidos da proteına. A celula possui um mecanismo que“traduz” a sequencia de nucleotıdeos dos genes na sequencia de aminoacidos.Este processo chama-se traducao [42]. O RNA, neste processo, assume opapel de fazer uma especie de “elo” entre os genes no DNA e as proteınas.
Existem tres tipos principais de RNA: o RNA mensageiro (mRNA), o RNAribossomal (rRNA) e o RNA transportador (tRNA). Todos sao transcritos apartir da sequencia de um gene, mas somente o mRNA possui as informacoespara a sıntese de proteınas. Esta informacao esta codificada na sequenciade tres nucleotıdeos consecutivos de uma molecula de mRNA. Este grupode tres nucleotıdeos e chamado de codon. Cada codon corresponde a umaminoacido. Cada um dos 20 diferentes aminoacidos e codificado por um oumais codons especıficos. Chama-se codigo genetico a relacao dos codonscom os diferentes aminoacidos. Tendo-se o codigo genetico em maos, pode-sedecifrar a sequencia de aminoacidos de uma proteına a partir da sequenciado mRNA ou do DNA a partir do qual ele foi transcrito [42].
Ja o rRNA se associa a algumas proteınas e faz parte da composicao de umaestrutura celular chamada ribossomo. Sao nos ribossomos que ocorrem osprocessos de sıntese de proteınas.
Para serem levados ate os ribossomos onde serao montados na estrutura
7

2.2. Visao computacional do genoma dct-ufms
das proteınas, os aminoacidos se associam a moleculas de tRNA. Existemdiferentes tRNA ligados a cada um dos 20 aminoacidos. O que os difere ea presenca de uma sequencia de 3 nucleotıdeos chamada anticodon. Estasequencia e complementar a sequencia de um codon especıfico na sequenciado mRNA [42].
A sıntese da cadeia de proteınas se inicia quando os mRNA comecam a ser“lidos” pelo ribossomo, que se movimenta de codon em codon. O primeirocodon a ser lido chama-se codon de iniciacao e e ele que determina a fasede 3 em 3 em que sera lido o mRNA. A cada movimento do ribossomo, umcodon e exposto e um tRNA unido ao seu aminoacido se liga ao mRNA pelainteracao codon-anticodon. Neste momento, o aminoacido trazido pelo tRNAse liga ao ultimo aminoacido incorporado na cadeia de proteına que esta seformando. Completada a adicao do aminoacido, o tRNA sem aminoacido eexpulso e o ribossomo se desloca para o proximo codon iniciando um novociclo. O processo de traducao prossegue ate o ribossomo encontrar um codonespecial chamado codon de terminacao que e um sinal de que a proteına estacompleta e pode ser liberada do ribossomo.
2.2 Visao computacional do genoma
Como mencionado neste capıtulo, o genoma contem informacoes necessariaspara produzir proteınas. Para o proposito deste trabalho, estamos interessa-dos no conjunto de genes que codificam proteınas, nas duas fitas originais doDNA de uma especie (chamadas de fita ‘+’ e fita ‘-’). Assim, vamos traba-lhar com o proteoma de uma especie, onde cada gene codificado possui umaposicao de acordo com a ordem em que aparece no genoma. Essa ordem elevada em consideracao no processo de inferencia de filogenias.
Suponha que as bases do genoma sejam numeradas a partir do numero 1 (um).A ordem dos genes do proteoma e definida da seguinte forma. Seja Ii aposicao da primeira base de gi, caso gi tenha sido codificado na fita ‘+’;ou a posicao da ultima base do penultimo codon (ultimo antes do codonde terminacao), caso gi tenha sido codificado na fita ‘-’. A ordem dos ge-nes g1, g2, . . . , gn e tal que I1 ≤ I2 ≤ . . . ≤ In .
A Figura 2.1 mostra representacoes graficas simplificadas do proteoma de umgenoma G. Na primeira representacao, os genes com uma seta orientada paraa direita indicam os genes pertencentes a fita ‘+’ do genoma, enquanto que osgenes com seta orientada para a esquerda representam os genes pertencentesa fita ‘-’. A segunda representacao e a visao mais adequada que devemos
8

2.3. Filogenia dct-ufms
ter do proteoma de um genoma, onde levamos em consideracao a ordem dosgenes, baseada na descricao feita acima.
G g
1 g
2 g
3 g
4 g 5 g
6 g
7 g
8 g
9 g
10 g
11
G g 1
g 2
g 3
g 4
g 5
g 6
g 7 g
8
g 9
g 10
g 11
Figura 2.1: Cada gi e um gene pertencente ao proteoma de G.
Uma vez que temos um breve conhecimento dos elementos de um proteoma,veremos na secao seguinte, como podemos obter informacoes dos proteomase como utiliza-las no processo de inferencia de filogenias.
2.3 Filogenia
Neste trabalho, as filogenias foram construıdas utilizando como fonte de in-formacoes os proteomas das especies.
Na construcao de filogenias, podemos utilizar duas categorias de dados:distancias e caracterısticas. As medidas de distancias sao estimativas dasdistancias evolutivas entre especies. As caracterısticas sao dados relativos aofenotipo ou mesmo a presenca de certas proteınas. Cada caracterıstica podeassumir varios estados, podendo esses serem discretos ou contınuos. Ambasas categorias de dados podem ser obtidas dos proteomas das especies.
Para obtermos as distancias, realizamos comparacoes entre proteomas depares de especies. Ao compararmos dois proteomas, estamos interessados emencontrar pares de genes ortologos, identificar as regioes com conservacao naordem dos genes, identificar regioes especıficas de um proteoma em relacao aoutro, entre outros interesses. A comparacao nao e uma tarefa facil, uma vezque genes similares ou identicos podem ser encontrados em posicoes distintasde genomas diferentes.
Antes de descrevermos como e realizada a comparacao dos proteomas de doisgenomas, vamos definir alguns conceitos da teoria dos grafos, transcritos dasnotas de Carvalho e Almeida [10]. Um grafo G e uma tripla ordenada(V (G), E(G), ψ(G)) consistindo de um conjunto nao vazio V (G) de vertices,um conjunto E(G) (disjunto de V (G)) de arestas, e uma funcao de incidencia
ψ(G) que associa a cada aresta um par nao ordenado (e nao necessariamentedistinto) de vertices de G. Se e e uma aresta e u e v sao vertices tais que
9

2.3. Filogenia dct-ufms
ψ(e) = (u, v) entao dizemos que e liga u a v. Os vertices u e v sao os extremosde e. Na Figura 2.2 apresentamos um grafo composto de n = 5 vertices. Oconjunto de vertices do grafo e V = {1, 2, 3, 4, 5} e o conjunto de arestas edado por E = {e1, e2, e3, e4, e5}.
2
3
e1
1
4
5
e2
e3e4
e5
Figura 2.2: Exemplo de grafo com cinco vertices.
Um grafo e bipartido se o seu conjunto de vertices pode ser particionado emdois subconjuntos X e Y , tais que cada aresta do grafo possui um extremoem X e outro em Y . Um grafo bipartido com cinco vertices e mostrado naFigura 2.3.
Y
X1 3 5
2 4
Figura 2.3: Exemplo de grafo bipartido. A particao X contem os verticesX = {1, 3, 5} e a particao Y contem os vertices Y = {2, 4}.
Um subgrafo H = (V (H), E(H)) de um grafo G = (V (G), E(G)) e umgrafo tal que V (H) ⊆ V (G) e E(H) ⊆ E(G). Um subgrafo geradorH ′(V (H ′), E(H ′)) de G e um subgrafo de G tal que V (H ′) = V (G). NaFigura 2.4(a) temos o exemplo de um subgrafo H do grafo apresentado naFigura 2.2. Na Figura 2.4(b) temos o exemplo de um subgrafo gerador dografo apresentado na Figura 2.2.
Um caminho entre dois vertices u e v em um grafo e uma sequencia finitade vertices e arestas alternadamente, sendo que nao ha repeticao de vertices.Por exemplo, no grafo da Figura 2.2, um caminho entre os vertices 1 e 4e dado pela sequencia W = 1e35e42e24. Um grafo e conexo se existe umcaminho entre todo par de vertices no grafo. Os grafos apresentados nasFiguras 2.2 e 2.3 sao grafos conexos, ja o grafo da Figura 2.5 nao e conexo,
10

2.3. Filogenia dct-ufms
5
e3
2
e1e5
2
3
1
5
4
1
3
(a) (b)
e3
e4
Figura 2.4: (a) Exemplo de subgrafo do grafo da Figura 2.2. (b) Exemplode um subgrafo gerador do grafo da Figura 2.2.
pois nao existe um caminho entre todo par de vertices, por exemplo, entreos vertices 1 e 4 ou entre os vertices 3 e 5.
2
3
e1
1
4
5
e2
e3
Figura 2.5: Exemplo de grafo que nao e conexo.
Um ciclo em um grafo e um caminho cujos vertices de inıcio e termino docaminho sao iguais. No grafo da Figura 2.2 temos um ciclo C dado porC = 2e24e53e12. Um grafo e acıclico se nao contem ciclo. Uma arvore eum grafo acıclico e conexo. Na Figura 2.6 temos um exemplo de um grafocom seis vertices que e uma arvore. Uma subarvore de uma arvore T e umsubgrafo conexo de T . Na figura 2.7 temos o exemplo de uma subarvore daarvore mostrada na Figura 2.6.
Figura 2.6: Exemplo de grafo que e uma arvore.
11

2.3. Filogenia dct-ufms
Figura 2.7: Exemplo de uma subarvore.
Quando o subgrafo gerador de um grafo G e uma arvore, ele recebe o nome dearvore geradora. Na Figura 2.8 temos uma arvore geradora para o grafo daFigura 2.2. Todo grafo pode possuir pesos associados as suas arestas, comomostrado no grafo da Figura 2.9(a). A partir desse grafo, podemos construiruma arvore geradora de custo mınimo, ou seja, uma arvore geradoracuja soma dos pesos das arestas da arvore e mınima. Uma arvore geradorade custo mınimo para o grafo da Figura 2.9(a) e mostrada na Figura 2.9(b).
2
3
e1
1
4
5
e3e4
e5
Figura 2.8: Exemplo de arvore geradora.
1
2 2
2
63 4
5 52
2
1
3
(a) (b)
Figura 2.9: (a) Exemplo de grafo com peso associados as suas arestas. (b)Exemplo de arvore geradora de custo mınimo para o grafo (a).
A comparacao dos proteomas de dois genomas G1 e G2 e realizada da seguinteforma:
• Fase 1: comparacao dos genes do genoma G1 com todos os genes deG2, e vice-versa;
12

2.3. Filogenia dct-ufms
• Fase 2: construcao de um grafo bipartido, considerando os resultadosda fase 1. Neste grafo, os vertices sao os genes e as arestas representamrelacoes entre os genes ortologos.
• Fase 3: determinacao de estruturas organizacionais presentes no grafo,como regioes de conservacao de ordem.
Entre as estruturas encontradas na fase 3, estao os pares de genes ortologose as regioes ja mencionadas. A partir destas estruturas, geramos distanciaspara cada par de especies e as utilizamos como entrada em um algoritmopara construcao de filogenias baseadas em distancias. Em particular, cadaestrutura encontrada pode ser utilizada na determinacao de uma medida parainferir filogenia.
O objetivo e construir filogenias cuja distancia observada na arvore entreduas especies seja exatamente igual a distancia armazenada numa matrizde distancias entre os pares de genomas. Nem sempre isto e possıvel, de-vido as distancias nao possuırem determinada propriedade, que sera apre-sentada posteriormente. Com isso, para construirmos as filogenias recorre-mos a heurısticas polinomiais, muito conhecidas por sua rapidez e exatidaotopologica.
Para inferirmos filogenias a partir de caracterısticas utilizamos duas abor-dagens. Na primeira, consideramos como caracterıstica a presenca/ausenciados genes nos proteomas das especies. Ou seja, uma especie possuira o es-tado 1 para a caracterıstica se possuir o gene e o estado 0 se nao possuir. Nasegunda, a caracterıstica utilizada e a presenca/ausencia de regioes. Essasregioes sao as mesmas mencionadas nesta secao.
Uma vez que obtemos essas informacoes, construımos uma matriz binariapara cada uma das duas abordagens acima. As linhas nestas matrizes repre-sentam as especies e as colunas representam as caracterısticas. Essas matrizessao utilizadas como entrada em um algoritmo que infere filogenias a partirde matrizes de caracterısticas.
Apos termos construıdo essas filogenias, fazemos uma analise das mesmaspara verificar a proximidade com a arvore filogenetica considerada verdadeira.
No Capıtulo 5, apresentamos em mais detalhes o que foi descrito acima,ou seja, como filogenias podem ser construıdas a partir das comparacoes deproteomas.
13

Capıtulo 3
Filogenia baseada em distancias
A construcao de arvores filogeneticas baseia-se tambem em dados numericosresultantes de comparacoes entre n objetos. A entrada e uma matriz qua-drada M de ordem n, cujo elemento Mij e um numero real nao-negativo,chamado de distancia entre os objetos i e j.
As matrizes de distancias entre objetos podem ser utilizadas para inferirarvores ultrametricas e arvores aditivas, definidas nas Secoes 3.1 e 3.2, res-pectivamente.
Um dos nossos principais objetivos e construir arvores ultrametricas, poiselas demonstram o tempo evolutivo decorrido de uma especie para outra;alem de mostrar quais especies sao mais proximas ou qual especie surgiuprimeiro. Um no interno nessa arvore representa um evento divergente, ouseja, um ponto no tempo quando as historias evolutivas de pelo menos duasespecies divergiram. Segundo Gusfield [24], geralmente e mais natural se con-centrar em arvores ultrametricas, pois os dados biologicos reais normalmenteaproximam o tempo evolutivo desde um evento de divergencia.
Os dados utilizados para construir arvores ultrametricas sao baseados emmutacoes aceitas ocorridas numa proteına, ou seja, mutacoes ocorridas nasequencia de aminoacidos que codificam a proteına (e no DNA) mas quenao alteram a funcao da proteına. Isto se deve ao fato de que o numero demutacoes aceitas em qualquer intervalo de tempo e proporcional ao tamanhodaquele intervalo [24].
Os primeiros metodos para estimar o numero de mutacoes aceitas entreduas especies eram realizados em laboratorio e utilizavam reacoes quımicase fısicas, obtendo dados como a temperatura de fusao das hibridizacoesde DNA, entre outros [24]. Metodos mais recentes estimam o numero de
14

3.1. Arvores ultrametricas dct-ufms
mutacoes aceitas baseando-se diretamente nas sequencias de DNA ou nassequencias de aminoacidos. Para duas especies, o numero de mutacoes acei-tas entre elas e calculado examinando as diferencas nas sequencias de DNAou nas sequencias de aminoacidos codificadas para proteınas. Esta estimativae chamada de distancia de edicao.
As matrizes de distancias utilizadas para inferir arvores ultrametricas saochamadas matrizes ultrametricas e sao definidas na Secao 3.1. Entretanto,nem sempre e possıvel construir uma arvore ultrametrica, conforme descritoacima. Isso acontece porque os dados reais nao sao ultrametricos e mesmoquando sao, nao necessariamente refletem o tempo decorrido desde a di-vergencia verdadeiramente [24]. Alem disso, os dados podem conter errospequenos ou problemas maiores no modelo evolutivo. Um problema maior eque a evolucao (talvez de plantas e certamente de bacterias) nao e sempredivergente (isto e, como arvore). O material genetico pode mergir, fazendocom que as historias evolutivas sofram mais o efeito de mergir do que divergir.
Ja que nao podemos construir arvores ultrametricas sempre, podemos ten-tar inferir arvores aditivas, que nao indicam relacoes de ancestralidade oudirecao de evolucao das especies, mas mostram a proximidade evolutiva en-tre elas. Com isso, essas arvores fornecem menos informacoes que uma arvoreultrametrica. As matrizes de distancias utilizadas para inferir arvores aditi-vas sao chamadas matrizes aditivas e sao definidas na Secao 3.2.
Veremos ainda neste capıtulo, que a condicao para que uma matriz dedistancias admita uma arvore aditiva e mais fraca que a condicao para queuma matriz seja ultrametrica.
O objetivo deste capıtulo e o de descrever o problema e os algoritmos para aconstrucao de arvores ultrametricas e aditivas. Na Secao 3.1 apresentamos adefinicao de arvore ultrametrica, exemplos e algoritmos para sua construcao.Na Secao 3.2 apresentamos a definicao de arvore aditiva, exemplos e umaalgoritmo para construcao da mesma. Ja na Secao 3.3, descrevemos umaheurıstica para construcao de filogenias baseadas em distancias.
3.1 Arvores ultrametricas
Nesta secao apresentamos o conceito de arvore ultrametrica, condicoes paraque uma matriz de distancias possibilite a construcao de uma arvore ul-trametrica e descrevemos algoritmos para tal construcao.
Dada uma matriz simetrica M para n objetos, uma arvore ultrametrica
15

3.1. Arvores ultrametricas dct-ufms
para M , segundo Gusfield [24], e uma arvore enraizada, com n folhas, sendocada folha correspondente a uma linha da matriz M . Um no interno daarvore e rotulado com uma entrada da matriz M e tem pelo menos doisfilhos. Os rotulos dos nos internos sao estritamente decrescentes ao longo dequalquer caminho da raiz ate uma folha. E para quaisquer duas folhas i e jna arvore, Mij e o rotulo do ancestral comum mais proximo entre i e j.
Os conceitos apresentados acima, que definem uma arvore ultrametrica, po-dem ser visualizados na arvore da Figura 3.1.
A B A B C
C
D
D
E
E 0
0 0
0 0
8 8 8 8 8 8
5
5 3
3
A E
D B C 3
3 5
8 a) b)
Figura 3.1: a) Exemplo de uma matriz simetrica M . b) Arvore ultrametricapara a matriz M .
Uma matriz simetrica M de numeros reais define uma distancia ul-trametrica se, e somente se, para quaisquer tres ındices i, j e k, o maximoentre Mij, Mik e Mjk nao e unico.
Quando M define uma distancia ultrametrica, dizemos que M e uma matrizultrametrica. O resultado abaixo caracteriza uma arvore ultrametrica.
Teorema 3.1 Uma matriz simetrica M tem uma arvore ultrametrica se, e
somente se, M e uma matriz ultrametrica.
Prova. Suponha que M tem uma arvore ultrametrica. A Figura 3.2 mostrauma subarvore contendo as folhas i, j e k quaisquer. A subarvore originalpode conter outros nos. Como a arvore mostrada e uma arvore ultrametrica,entao o numero escrito em u deve ser estritamente maior que o numero emv. Por definicao, Mij e o numero escrito em v e Mik = Mjk. Os tres valoressatisfazem a condicao de que o maximo nao e unico. Portanto, se M temuma arvore ultrametrica entao M e uma matriz ultrametrica.
Vamos provar agora que, se M e uma matriz ultrametrica, entao existe umaarvore ultrametrica para M . Vamos construir uma arvore ultrametrica T apartir de M , nos concentrando inicialmente num unico no, por exemplo, a
16

3.1. Arvores ultrametricas dct-ufms
folha i. Se ha d entradas distintas na linha i de M , entao qualquer arvoreultrametrica T para M contem um caminho da raiz a folha i com exatamented nos, incluindo a raiz e a folha i. Cada no neste caminho e rotulado por umadas d entradas distintas na linha i, e estes rotulos devem aparecer em ordemdecrescente no caminho. Podemos visualizar estes conceitos na Figura 3.3,considerando i como sendo o objeto a.
Qualquer no interno v neste caminho, rotulado Mij, e o ancestral comummais proximo da folha i e da folha j. Isto fixa onde a folha j deve aparecerem T , em relacao ao caminho a folha i.
Desta forma, o caminho a folha i particiona as n − 1 folhas remanescentesem d − 1 classes. Chamamos esta particao de D. Duas folhas j e k estaojuntas na mesma classe de D se, e somente se, Mij = Mik. Cada classe emD e definida por um no distinto no caminho a i. O no que define a classecontendo j, por exemplo, e o no rotulado com Mij.
Dada a particao D definida pelo caminho a i, basta resolver o problema daarvore ultrametrica recursivamente em cada uma das d − 1 classes em D eentao conectar estas arvores para formar a arvore ultrametrica para a matrizM . �
v
i j k
u
Figura 3.2: Exemplo de subarvore generica contendo as folhas i, j e k.
a b c d e
f g
h
a b c d e f g h 0 4 3 4 5 4 3 4
0 4 2 5 1 4 4
5
4
3
a c,g
b,d,f,h
e a) b)
Figura 3.3: a) Duas linhas de uma matriz simetrica M . A linha do objetoa e usada para obter o caminho ate a folha a, que e mostrada na Figura b).Os numeros nos nos ao longo do caminho particionam os demais objetos.
17

3.1. Arvores ultrametricas dct-ufms
A prova do Teorema 3.1 nos fornece um algoritmo para construir uma arvoreultrametrica. Este algoritmo foi proposto por Gusfield [24]. Alem disso, deacordo com Gusfield [24], se D e uma matriz ultrametrica, entao uma arvoreultrametrica para D pode ser construıda em tempo O(n2).
Como ja dissemos, os dados nem sempre possibilitam a construcao de arvoresultrametricas. Com isso, podemos tentar relaxar as medidas de distancias,impondo limites superior e inferior para as mesmas. Esta abordagem e apre-sentada por Setubal e Meidanis [40] e sera descrita a seguir.
O Algoritmo de Farach, Kannan e Warnow
Na construcao de filogenias, as distancias que utilizamos, as vezes, podem serincertas. Entao, podemos quantificar tal incerteza expressando as medidasem forma de intervalos. O intervalo define um limite inferior e superiorpara a distancia verdadeira. Obtemos entao, as matrizes M l e Mh, quecontem, respectivamente, limites inferiores e superiores para a distancia entreos respectivos pares de objetos.
Assim, temos que construir uma arvore evolutiva cujas distancias medidasna arvore se “encaixem” entre as duas matrizes, ou seja, se dij e a medidada distancia na arvore entre dois objetos i e j, a seguinte desigualdade deveocorrer:
M lij ≤ dij ≤Mh
ij.
Se impusermos o requisito adicional de que a arvore seja ultrametrica, pode-mos resolver o problema eficientemente. Vamos agora descrever um algoritmoeficiente para a construcao de uma arvore ultrametrica.
Inicialmente, interpretamos a matriz de distancias como um grafo nao-orientado com peso nas arestas. Uma matriz de distancias Mn×n pode ser in-terpretada como um grafo completo de n vertices, onde o peso da aresta (i, j)e dado por Mij. Assumimos que Mij e definido para todo par i,j, tal queo grafo correspondente e sempre conexo. Assumimos tambem que Mii = 0,para todo i. Com isto, obtemos os grafos Gl e Gh, correspondentes, respecti-vamente, a matriz M l e Mh. Referenciamos os pesos nas arestas pela funcaoW , como em W (e) ou W (a, b), onde e = (a, b) e uma aresta.
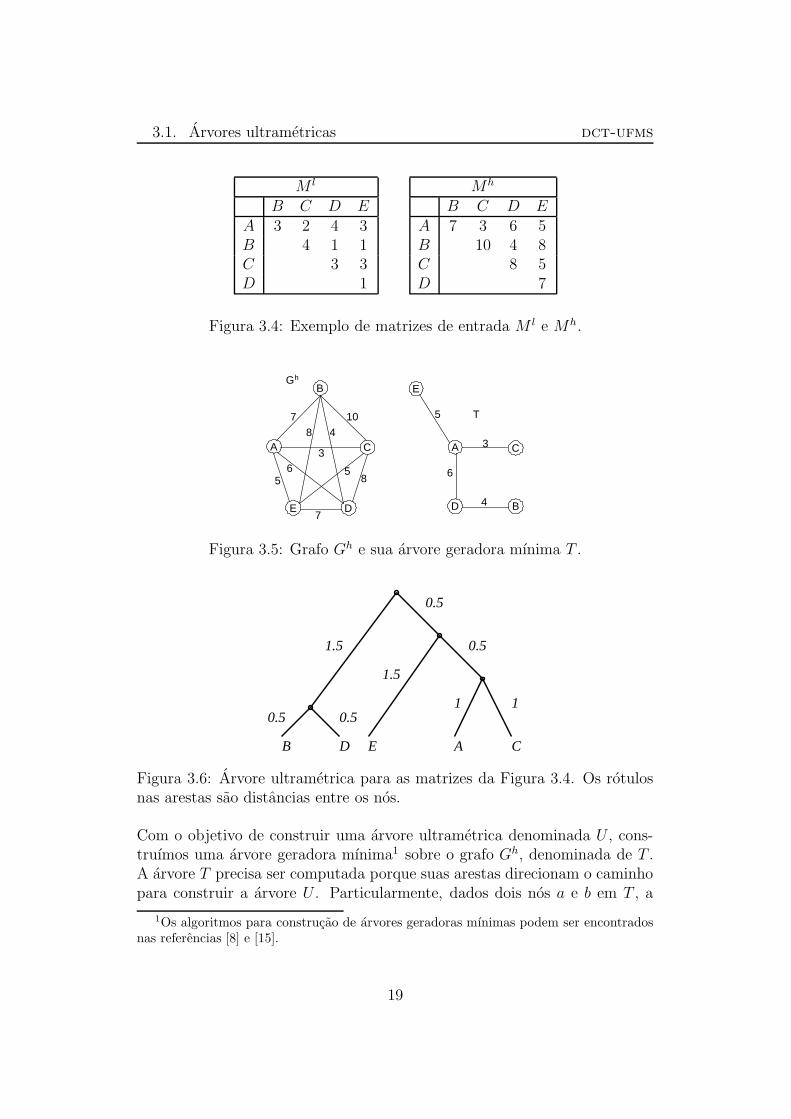
Na Figura 3.4, mostramos um exemplo de matrizes M l e Mh. Na Figura 3.5apresentamos o grafo Gh correspondente a matriz Mh e sua arvore geradoramınima T . Ja na Figura 3.6 temos a arvore filogenetica obtida no metodo aser apresentado para as matrizes da Figura 3.4.
18
3.1. Arvores ultrametricas dct-ufms
M l
B C D E
A 3 2 4 3B 4 1 1C 3 3D 1
Mh
B C D E
A 7 3 6 5B 10 4 8C 8 5D 7
Figura 3.4: Exemplo de matrizes de entrada M l e Mh.
A
B
C
D E
G h
A
E
C
B D
T 5
3
6
4
4
5 5
3 6
7
7 10
8
8
Figura 3.5: Grafo Gh e sua arvore geradora mınima T .
CAEDB
0.5
0.5
1 1
1.5
1.5
0.5 0.5
Figura 3.6: Arvore ultrametrica para as matrizes da Figura 3.4. Os rotulosnas arestas sao distancias entre os nos.
Com o objetivo de construir uma arvore ultrametrica denominada U , cons-truımos uma arvore geradora mınima1 sobre o grafo Gh, denominada de T .A arvore T precisa ser computada porque suas arestas direcionam o caminhopara construir a arvore U . Particularmente, dados dois nos a e b em T , a
1Os algoritmos para construcao de arvores geradoras mınimas podem ser encontradosnas referencias [8] e [15].
19

3.1. Arvores ultrametricas dct-ufms
aresta de maior peso no unico caminho de a a b em T e chamado de link
de a e b e denotado por (a, b)max.
A partir destas definicoes, apresentamos o seguinte teorema:
Teorema 3.2 Uma arvore ultrametrica U para as matrizes M l e Mh existe
se, e somente se, para todo par de objetos a e b e verdade que M lab ≤
W ((a, b)max).
Prova. Esta prova pode ser encontrada em [24].
O link tambem e usado na determinacao da seguinte funcao sobre as arestasde T . Para uma aresta e de uma arvore geradora mınima de Gh, considere afuncao:
CW (e) = max{M la,b|e = (a, b)max}.
Dada a arvore geradora mınima de Gh, denotada por T , cada aresta e = (a, b)que e um link de pelo menos um par de vertices de T tambem pode ser olink de mais pares. Se tomarmos todos esses pares de vertices e escolhermosa maior entrada da matriz M l entre estes pares, este e o valor da funcaoCW (e).
Para construir a arvore ultrametrica, precisamos inicialmente computar aarvore geradora mınima T de Gh e, para cada aresta e ∈ T computar CW (e).A seguir, apresentamos um algoritmo resumido para construcao da arvoreultrametrica U .
Entrada: Arvore Geradora Minima T,
o valor de CW(e) para todas as arestas de T
Saida: Arvore Ultrametrica U
Inicio
Para cada objeto i faca
Crie um conjunto S_i := i
Crie um no para i
Altura[i] := 0
FimPara
Ordene as arestas de T em ordem nao-decrescente de valores CW(e)
Para cada aresta e=(x_i, x_j) em T nesta ordem faca
Seja S_i o conjunto que contem x_i
Seja S_j o conjunto que contem x_j
20

3.2. Arvores aditivas dct-ufms
Se S_i != S_j entao
u_i := no que contem x_i
u_j := no que contem x_j
Crie uma arvore U
Atribua u_i como filho esquerdo de U
Atribua u_j como filho direito de U
Altura[U] := CW(e)/2
W(u_i, U) := Altura[U] - Altura[u_i]
W(u_j, U) := Altura[U] - Altura[u_j]
Una os conjuntos S_i e S_j
FimSe
FimPara
Fim
O algoritmo acima para construcao de arvores ultrametricas foi proposto por Fa-rach, Kannan e Warnow em 1995 [17], e seu tempo de execucao e O(n2).
Como pudemos ver, uma arvore ultrametrica mostra informacoes importantes,uma vez que podemos identificar melhor quem e ancestral de quem, qual no veioantes, em que ponto ocorreu uma divergencia e fez com que um ancestral produzisseduas especies diferentes. Porem, as distancias utilizadas na pratica nao atendemao criterio ultrametrico e com isso temos que recorrer a outra abordagem paraconstrucao de filogenias, que e a construcao de arvores aditivas. Na verdade, arvoreaditiva e uma condicao mais fraca de arvore ultrametrica, e sera apresentada nasecao seguinte.
3.2 Arvores aditivas
Nesta secao apresentamos o conceito de arvore aditiva, exemplos, a condicao paraque uma matriz de distancias admita a construcao de uma arvore aditiva e des-crevemos um algoritmo para a construcao da mesma. No processo de construcaode arvores filogeneticas, quando trabalhamos com distancias, o conceito de espacometrico e necessario. Isto e importante para verificar a qualidade dos dados emelhorar a confiabilidade da analise filogenetica, segundo Dress et al [16]. Aspropriedades abaixo, portanto, devem ser obedecidas.
Um espaco metrico e um conjunto de objetos O tal que para todo par i, j ∈ Oassociamos um numero real nao-negativo dij com as seguintes propriedades:
21

3.2. Arvores aditivas dct-ufms
dij > 0 para i 6= j, (I)dij = 0 para i = j, (II)dij = dji para todo i e j, (III)dij ≤ dik + dkj para todo i, j e k (desigualdade triangular). (IV)
Queremos que nossa matriz de entrada M seja tal que os objetos formem umespaco metrico. Logo, da propriedade (III), vemos que a matriz sera simetrica. Aarvore construıda com base em M tem n folhas, pois a matriz e de ordem n.
O peso do caminho entre quaisquer dois nos i e j, deve ser igual a Mij . Se talarvore T puder ser construıda, dizemos que M e T sao aditivas. O Lema 3.3,a seguir, chamado de “a condicao dos quatro pontos”, caracteriza uma matrizaditiva.
Lema 3.3 (Condicao dos quatro pontos) Uma matriz de estados M e aditiva se, esomente se, dados quaisquer quatro objetos i, j, k e l de M, vale uma das seguintespropriedades:
(i) dik + djl = dil + djk
(ii) dil + dkj = dij + dkl
(iii) dik + djl = dij + dkl.
Prova. Assuma que a matriz M e aditiva. Logo, podemos construir uma arvoreaditiva T para M . Escolha quaisquer quatro objetos i, j, k e l em T e considerea subarvore conectando esses quatro objetos. Esta subarvore corresponde a pelomenos uma das arvores apresentadas na Figura 3.72 . Em todos os tres casos temosque
dm1m2+ dm3m4
= dm1m3+ dm2m4
,
onde < m1,m2,m3,m4 > e alguma permutacao de < i, j, k, l >.
i j
k l
k
l
i
j
l i
j k (a) (b) (c)
Figura 3.7: Exemplo das possıveis subarvores conectando quatro objetosquaisquer i, j, k e l.
2Esta figura foi transcrita de Patrinos e Hakimi [37].
22

3.2. Arvores aditivas dct-ufms
Para mostrarmos que a matriz M e aditiva, caso uma das condicoes (i), (ii), (iii)seja verdadeira, basta notarmos que essas condicoes equivalem, repectivamente, asarvores das Figuras 3.7(a), 3.7(b) e 3.7(c). Por exemplo, se (i) for verdadeira,entao basta construirmos uma arvore que tenha a topologia da Figura 3.7(a), emrelacao aos nos i, j, k e l. �
A matriz da Figura 3.8 e aditiva, ja que o Lema 3.3 e verdadeiro.
A B C D E
A 0 2 7 4 7B 0 7 4 7C 0 7 6D 0 7E 0
Figura 3.8: Exemplo de matriz aditiva.
Em 1977, Waterman et al [46] propuseram um algoritmo cujo tempo de execucaoe O(n2) para construcao de arvores aditivas. O algoritmo e apresentado a seguir.
Algoritmo
• Verifique a condicao do Lema 3.3 para a matriz de entrada M .
• Caso M nao seja aditiva, entao pare. Caso contrario, prossiga.
• Escolha um par de objetos i e j e construa a primeira aresta da arvore, cujopeso e dado por Mij.
• Escolha um terceiro objeto k. Divida a unica aresta da arvore, criando umno interno, que chamaremos de c. Crie uma nova aresta partindo de c, onde ksera colocado como folha. Devemos descobrir onde exatamente c dividira aaresta que liga i a j. As distancias das arestas sao dadas por:
dic =Mij + Mik − Mjk
2(3.1)
djc =Mij + Mjk − Mik
2(3.2)
dkc =Mik + Mjk − Mij
2. (3.3)
23

3.2. Arvores aditivas dct-ufms
• Para adicionar o objeto k + 1, escolha um par de folhas ja adicionadas aarvore e aplique as Equacoes (3.1), (3.2) e (3.3). Com isso, computamosa posicao do novo no interno. Se este no nao coincide com qualquer outrono ja existente na arvore, encontramos a aresta que deve ser dividida paraadicionar o objeto k + 1. Caso contrario, a posicao de divisao cai em umno ja existente, por exemplo u. Como u e um no interno, sabemos que hauma subarvore suspensa a partir dele. Escolha qualquer objeto pertencentea esta subarvore, digamos r, e aplique as Equacoes (3.1), (3.2) e (3.3) sobreos objetos i (ou j), r e o objeto k + 1. Repita este processo ate a posicao dedivisao correta ser encontrada.
Note que a inclusao de um novo no interno, a cada passo do algoritmo, so e possıvelporque sempre vale uma das propriedades (i), (ii) ou (iii). Se alguma delas naovaler em algum passo, logo a matriz nao e aditiva. No entanto, para testarmosa aditividade de M , nao precisamos do algoritmo. Basta, para isso, testarmos avalidade da condicao dos quatro pontos para M .
No algoritmo, para todo objeto que adicionamos a arvore, podemos ter que verificartodos os outros objetos ja colocados, gastando tempo constante por verificacao.Isto significa que no pior caso, o algoritmo executa em tempo O(n2).
Vamos construir uma arvore aditiva para os objetos mostrados na Figura 3.8,usando o algoritmo acima. Inicialmente criamos a aresta (A,B) de tamanho 2,como mostrado na Figura 3.9(a). Em seguida, adicionamos o objeto C. Aplicandoas Equacoes (3.1), (3.2) e (3.3), encontramos que um novo no x1 deveria ser criado auma distancia 1 de A e 1 de B. Alem disso, adicionamos uma nova aresta (x1, C)de tamanho 6, conforme visto na Figura 3.9(b). Considerando agora o no D,ao aplicarmos as equacoes usando A e B, que ja estao na arvore, encontramosque D deveria ficar na mesma subarvore, como C. Ou seja, o novo no internocoincidiu com o no ja existente x1. Entao aplicamos as equacoes novamente agoraconsiderando B, C e D. Com isso, encontramos a posicao correta do no x2, queesta a uma distancia 1 de x1 e 5 de C. Prosseguindo com estes passos, obtemos aarvore final mostrada na Figura 3.9(d).
Note que, atraves de uma arvore aditiva nao ha predicao de ancestralidade, poisa arvore construıda nao possui raiz. Com isso, nao conseguimos ver que no e oancestral de todos ou que no vem antes de outro no.
A seguir veremos que e possıvel construir uma arvore aditiva a partir de umaarvore ultrametrica.
Arvore aditiva vista como um problema ultrametrico
Vamos agora mostrar como reduzir o problema da arvore aditiva ao problema daarvore ultrametrica. Vamos reduzir o problema criando uma matriz M ′, que e
24

3.2. Arvores aditivas dct-ufms
A B 2 1
1 6
A
B C
1
1
1
5
2
A
B
C
D
1
2
A
B
C
D
2 3
3
1
1
E
(b) (a)
(c) (d)
Figura 3.9: Exemplo de execucao do algoritmo para a matriz da Figura 3.8.
ultrametrica se, e somente se, a matriz M e aditiva.
Segundo Gusfield [24], podemos reduzir M a M ′ sem precisar construir a matrizaditiva T ou a matriz ultrametrica T ′, a partir do seguinte resultado:
Lema 3.4 Sem conhecer T e T ′ explicitamente, podemos deduzir que M ′ij = mv +
(Mij − Mvi − Mvj)/2.
No Lema 3.4, v e a linha de D que contem o maior valor e mv e o maior valor.Entao, dado Lema 3.4, temos
Teorema 3.5 Se M e uma matriz aditiva, entao M ′ e uma matriz ultrametrica,onde M ′
ij = mv + (Mij − Mvi − Mvj)/2.
Prova. Esta prova pode ser encontrada em [24].
Entao, se dado uma matriz M queremos estabelecer se M e aditiva, podemos criaruma matriz M ′ e testar se M ′ e ultrametrica. Caso nao seja, entao M nao eaditiva.
Teorema 3.6 Se a matriz M ′ e ultrametrica entao a matriz M e aditiva.
Prova. Esta prova pode ser encontrada em [24].
Em suma, uma matriz M e aditiva se, e somente se, M ′, conforme construcaoacima, e ultrametrica. Alem disso, se M e aditiva entao a arvore aditiva T podeser criada com os seguintes passos:
25

3.2. Arvores aditivas dct-ufms
• Crie uma matriz M ′ a partir de M e construa uma arvore ultrametrica T ′
de M ′;
• Atribua para cada aresta de T ′ uma distancia igual a diferenca absoluta entreos rotulos dos nos extremos, sendo que as folhas possuem rotulos iguais azero;
• Para cada folha i, subtraia mv − Mvi da distancia na aresta da folha i;
• A arvore resultante T e uma arvore aditiva para a matriz M .
A Figura 3.10 mostra todos os passos citados acima.
3 9 7
8 6
6
(a)
M
a
b
c
b c d
9 9 9
7 7
4
(b)
M´
a
b
c
T´ T
9
97
7
2
4
4
3
0
4 2
(c) (d)
c d c d
13
2
4b
a
b
a
b c d
Figura 3.10: (a) Exemplo de matriz aditiva M . A maior entrada tem valor9 e esta na linha do objeto a. (b) Matriz ultrametrica M ′. (c) Arvoreultrametrica T ′ ja com os pesos nas arestas. (d) Arvore resultante T apossubtrairmos ma −Mai das arestas folhas. A arvore original e obtida aposcontrair a aresta de peso zero onde esta a folha a.
As matrizes de distancias usadas na pratica raramente sao aditivas, devido, porexemplo, a erros nas medidas das distancias. Ja em sequencias biologicas, podemocorrer varias mudancas em um mesmo local, ou seja, o registro de mudancasanteriores e destruıdo pelas posteriores. Alem disso, podemos ter ocorrencia deconvergencia paralela. Essas possibilidades podem resultar em distancias entrepares de objetos que nao sao aditivas. E de acordo com Setubal e Meidanis [40],ao tentarmos minimizar a ausencia de aditividade, o problema se torna NP-difıcil.
Como vimos neste capıtulo, podemos construir arvores aditivas ou ultrametricas,desde que nossas matrizes de distancias sejam aditivas ou ultrametricas. Podemos
26

3.3. Heurıstica para construcao de filogenias baseadas em distanciasdct-ufms
ate resolver o problema da arvore aditiva usando de algoritmos para construirarvores ultrametricas. No entanto, geralmente nao podemos assegurar que nossasmatrizes sejam aditivas ou que sejam ultrametricas. Apresentamos entao umaheurıstica polinomial para construir filogenias: o algoritmo de Neighbor-Joining,proposto por Nei e Saitou [36], que sera abordado na secao seguinte.
3.3 Heurıstica para construcao de filogenias
baseadas em distancias
Um dos metodos mais justificados estatisticamente para aproximar uma matrizde distancias e a abordagem least squares, segundo Shamir [41]. Na Equacao 3.4,para cada par de especies, a distancia medida Mij entre elas, e o peso w(i, j),intuitivamente quantificam a precisao desta medida. A quantidade w e o numerode vezes que cada distancia foi replicada. Em casos simples, este valor e usadocomo um, mas o usuario pode, como uma opcao, especificar o grau de replicacaopara cada distancia.
Nosso objetivo e encontrar uma arvore T cujas folhas sao as n especies dadas,e que prediga as distancias dij entre as especies, tal que a expressao seguinte eminimizada.
SSQ(T ) =∑
i6=j
wij(Mij − dij)2. (3.4)
SSQ e uma medida de discrepancia entre as distancias observadas Mij e asdistancias dij preditas por T . Os pesos wij sao normalmente todos 1, ou wij = 1
M2
ij
,
de acordo com Shamir [41].
O metodo Neighbor-Joining (NJ) e um dos metodos que utiliza a abordagem acima.A ideia do metodo e unir duas especies que, alem de estarem proximas umas dasoutras, tambem estejam juntas, distantes do restante das especies.
A distancia de uma especie i para o restante da arvore, denotada por ui, e estimadapela formula
ui =∑
k 6=i
Mik
(n − 1)(3.5)
De acordo com Nei e Saitou [36], a arvore inicial e uma arvore estrela, conformemostrado na Figura 3.11(a), produzida sobre a hipotese de que ainda nao hanenhum par de especies agrupadas. A Figura 3.11(b), mostra o primeiro parde especies escolhido para serem unidas. Porem, qualquer par de especies poderiaocupar as posicoes de 1 e 2 na arvore, e ha n(n−1)
2 formas de escolhe-las, onde n eo numero de especies.
27

3.3. Heurıstica para construcao de filogenias baseadas em distanciasdct-ufms
Figura 3.11: (a) Arvore estrela com nenhuma estrutura hierarquica e (b)Arvore em que as especies 1 e 2 estao agrupadas.
Esse metodo trabalha objetivando minimizar a soma dos tamanhos de todas asarestas da arvore, tambem conhecido como o criterio de evolucao mınima (Mini-mum Evolution - ME). Entao, escolhemos o par (i,j) cujo valor Mij − ui − uj e omenor.
Uma vez escolhido o par de especies a ser agrupado como e o caso do par (1-2)na Figura 3.11(b), calculamos os tamanhos das novas arestas pelo metodo Fitch-Margoliash, de acordo com Nei e Saitou [36], cujas equacoes sao apresentadas naFormula 3.6.
Se as especies 1 e 2 sao designadas como vizinhas e sao ligadas, uma nova especiecombinada e criada, representada por (1-2) e as especies 1 e 2 isoladas sao retiradasda matriz. A distancia entre uma nova especie combinada, por exemplo (1-2), eoutra especie j e calculada pela Formula 3.7.
O numero de especies e reduzido de um e o procedimento e novamente aplicadopara encontrar novos vizinhos. O ciclo se repete ate o numero de especies tornar-seigual a dois.
Se a arvore e aditiva, o metodo da o tamanho correto das arestas para todas asarestas, conforme mostrado por Nei e Saitou [36].
A seguir, apresentamos o algoritmo Neighbor-Joining, transcrito do artigo de Sha-mir [41].
Algoritmo Neighbor-Joining
• Inicializacao
– Crie n nos com as dadas especies, uma especie por no.
• Iteracao
– para cada especie, compute ui =∑
k 6=iMik
(n−1) .
– escolha i e j para os quais Mij − ui − uj e mınimo.
– una as especies i e j numa nova especie - (ij), com um no correspon-dente em T . Calcule o tamanho das arestas de i e j para o novo no,
28

3.3. Heurıstica para construcao de filogenias baseadas em distanciasdct-ufms
da seguinte forma:
di(ij) =1
2Mij +
1
2(ui − uj), dj(ij) =
1
2Mij +
1
2(uj − ui) (3.6)
– compute as distancias entre a nova especie e cada outra especie:
M(ij)k =Mik + Mjk − Mij
2(3.7)
– exclua as especies i e j da tabela e as substitua por (ij).
– se restam mais que dois nos (especies), volte ao primeiro item do passoda iteracao. Caso contrario, conecte os dois nos remanescentes poruma aresta de tamanho Mij .
Dada uma matriz de entrada M , como a matriz mostrada abaixo, podemos cons-truir uma arvore filogenetica para as especies conforme descrito nesta secao. NaFigura 3.13, apresentamos um exemplo de execucao do algoritmo para uma matriz3
de entrada com oito objetos.
1 2 3 4 5 6 72 73 8 54 11 8 55 13 10 7 86 16 13 10 11 57 13 10 7 8 6 98 17 14 11 12 10 13 8
Figura 3.12: Matriz de distancias para arvores da Figura 3.13.
Conforme mostrado na figura, o par de especies 1 e 2 e o primeiro a ser escolhido,pois o valor M12 − u1 −u2 foi o menor entre todos. Em seguida, calculamos os ta-manhos de arestas entre os nos pelas Formulas 3.6. Combinamos 1 e 2 e formamosa nova especie combinada (1-2), sendo as distancias medias (M(1−2,j); j = 3, . . . , 8)computadas pela Equacao 3.7. No proximo ciclo, o par de especies escolhido sao 5e 6. Novamente, calculamos os tamanhos das arestas para os nos envolvidos. Nociclo 3, o par (1-2, 3) e o par de especies escolhido. No ciclo 4, as especies es-colhidas sao (1-2-3) e 4. E no ciclo 5, [1-2-3-4, 5-6] e identificado como o par aser unido, conforme mostrado na Figura 3.13 (f). No ciclo seguinte, unem-se asespecies [1-2-3-4-5-6] e 7. E finalmente, como restaram apenas duas especies, quesao [1-2-3-4-5-6-7] e 8, fazemos a uniao das mesmas por uma aresta de tamanho
3Esta matriz foi obtida de um exemplo de Nei e Saitou [36].
29

3.3. Heurıstica para construcao de filogenias baseadas em distanciasdct-ufms
X Y
3 4
5
6
7
8
5
2
1
2
1
23
45
68 7
1
4
Y X
12
34 5
7
8
1
2
X Y
12
3
45 6
7
8
1
3
12
3
4
5
6 8
7
2
2
X YX Y
12
3
4
5
6
7
8
1
2
6
X
(a) (b) (c)
(d) (e) (f)
6
Figura 3.13: Exemplo da aplicacao do metodo Neighbor-Joining para oitoespecies. Os numeros sobre as arestas sao os tamanhos das arestas.
M(1−2−3−4−5−6−7)8 . As duas ultimas ligacoes e a topologia da arvore encontradasao mostradas na Figura 3.13 (f).
O algoritmo Neighbor-Joining possui tempo de execucao O(n2).
Por que usar Neighbor-Joining
O algoritmo Neighbor-Joining e bastante utilizado nos mais diversos aspectos con-siderados para construcao de filogenias. Existem varias frentes de pesquisas noestudo de filogenias que utilizam o algoritmo Neighbor-Joining por possuir algu-mas caracterısticas que o destacam dos demais algoritmos.
Wang et al [45] fizeram um dos primeiros estudos de metodos para construcaorapida de filogenias para dados relacionados a ordem de genes, usando abordagensbaseadas em distancias e baseadas em parsimonia. Neste trabalho, o metodo NJfoi utilizado para a construcao das arvores baseadas em breakpoints e inversoes. Ometodo NJ executa muito mais rapido que todos os outros algoritmos utilizados naexperiencia. Em relacao a exatidao topologica das arvores produzidas, o metodoNJ foi o melhor metodo baseado em distancia, obtendo precisao igual ou superioraos outros.
Nakhleh et al [34] estudaram a precisao, taxa de convergencia e velocidade devarios metodos de construcao de filogenias rapidos, entre eles o metodo Neighbor-Joining. Estes metodos foram submetidos a um numeroso conjunto de sequencias
30

3.3. Heurıstica para construcao de filogenias baseadas em distanciasdct-ufms
longas. Embora a precisao do metodo NJ tenha sido afetada significantementenesses conjuntos maiores, sua velocidade ainda foi melhor que todos os outrosmetodos avaliados. Em estudos realizados pelos mesmos, eles mostraram que NJpode recuperar a arvore verdadeira com alta probabilidade quando as sequenciasdadas sao de tamanho limitado por uma funcao que cresce exponencialmente emn, onde n e o numero de especies.
Cosner et al [12] apresentaram uma nova heurıstica para construir arvores evo-lutivas a partir de dados da ordem de genes. Eles apresentaram e discutiram osresultados dos experimentos realizados com dados artificiais (sinteticos) e dadosreais, sobre tres metodos, entre eles Neighbor-Joining.
Quando as taxas de evolucao sao suficientemente baixas, todos os metodos recu-peram boas estimativas da arvore verdadeira. Enquanto NJ executa em tempopolinomial, os outros metodos nao o fazem. Tambem notaram que o metodo NJatua tao bem quanto sua nova heurıstica em termos de precisao topologica.
Tateno et al [44] estudaram, por meio de simulacao de computador, a eficiencia re-lativa de alguns metodos entre eles Maximum Likelihood (ML) e Neighbor-Joining.Foram levados em consideracao se a topologia produzida e correta e a estimacaodo tamanho das arestas para o caso de quatro sequencias de DNA de 1000 nu-cleotıdeos.
O metodo NJ possui uma eficiencia maior em obter a arvore correta em relacaoa outros modelos, mesmo quando estes produzem arvores consistentes. E alemdisso, o metodo NJ pode dar uma topologia correta mesmo quando as medidas dedistancia usadas nao sao estimadoras imparciais de substituicao de nucleotıdeos.
Alguns autores sao preocupados com o fato que o metodo NJ gera somente umaarvore final e que esta arvore pode nao ser a melhor em termos do criterio deevolucao mınima (Minimum Evolution - ME). Atualmente, simulacoes de compu-tador tem mostrado que na maioria dos casos a arvore NJ tem a mesma topologiaque a arvore real ME, a menos que o numero de sequencias usadas seja muitogrande [38, 39].
Atteson et al [5] analisaram a performance de NJ, determinando que este metodofaz o melhor possıvel para determinar a topologia da arvore entre todos os metodosbaseados em distancia.
Como descrito nesta secao, o algoritmo NJ apresenta uma eficiencia confiavel,tanto em relacao a topologia construıda como em sua performance, para dadosbaseados em distancia. Por este motivo, nos o utilizamos para testar as medidaspropostas na secao 5.2.
31

Capıtulo 4
Filogenia baseada emcaracterısticas
As filogenias sao construıdas com base nas comparacoes entre as especies. Vamosnos referir as especies e outras taxonomias como objetos. De acordo com Setubale Meidanis [40], podemos construir filogenias baseadas nas seguintes categorias dedados de entrada:
• Caracterısticas. Os dados utilizados sao caracterısticas como: a forma dobico, numero de dedos na pata, presenca ou ausencia de certas proteınas,habitos alimentares, ciclo de vida e outras. Cada caracterıstica pode ou naoter um numero finito de estados. Essas caracterısticas podem ser agrupadasem uma matriz, de modo que cada linha da matriz representa um objetoe cada coluna representa uma caracterıstica. Chamamos esta matriz dematriz de estados.
• Dados comparativos numericos, que chamamos de distancias entre os obje-tos. Essas distancias sao uma estimativa da distancia evolutiva entre os ob-jetos. A matriz resultante destes dados e chamada matriz de distancias,sendo que a mesma e uma matriz triangular, pois temos uma distancia paracada par de objetos e essas distancias sao simetricas.
Neste capıtulo tratamos do problema da filogenia baseada em caracterısticas. Fi-logenia baseada em distancias e descrita no Capıtulo 3. O capıtulo e organizado daseguinte forma: na Secao 4.1 apresentamos alguns aspectos relacionados a filogeniabaseada em caracterısticas; na Secao 4.2 apresentamos as condicoes para determi-nar se uma matriz binaria admite uma filogenia perfeita, descrita logo a seguir;na Secao 4.3 apresentamos a abordagem de parsimonia para construir filogeniase na Secao 4.4 apresentamos um programa de parsimonia utilizado na construcaode filogenia baseada em caracterısticas de estado binario.
32

4.1. Aspectos da filogenia baseada em caracterısticas dct-ufms
4.1 Aspectos da filogenia baseada em carac-
terısticas
Na construcao de filogenias baseadas em caracterısticas, os seguintes aspectos saoconsiderados:
• as caracterısticas podem ser herdadas independentemente umas das outras;
• todos os estados de uma caracterıstica devem evoluir de um estado originaldo ancestral comum mais proximo dos objetos em estudo;
• os nos internos da arvore representam especies ancestrais hipoteticas;
• a distancia entre um no interno e uma folha pode ser interpretada como umaestimativa do tempo que um no (no interno) levou para evoluir para outrono (no caso, a folha).
Podemos entao definir uma matriz de estados como sendo uma matriz M comn linhas (objetos) e m colunas (caracterısticas), onde Mij denota o estado que oobjeto i tem para a caracterıstica j.
Objeto/Caracterıstica c1 c2 c3 c4 c5A 1 1 0 0 0B 0 0 1 0 1C 1 1 0 0 1D 0 1 1 1 0E 1 1 0 0 1
Figura 4.1: Exemplo de matriz de estados.
A construcao de uma filogenia a partir de uma matriz de estados, segundo Setubale Meidanis [40], depara-se com algumas dificuldades, descritas a seguir:
• Convergencia ou evolucao paralela. Os metodos para reconstrucao da arvorefilogenetica se baseiam no fato de que objetos que compartilham o mesmo es-tado para uma dada caracterıstica sao geneticamente mais relacionados queaqueles que nao compartilham. Entretanto, existe a possibilidade que doisobjetos compartilhem um estado mas nao sejam geneticamente proximos.Tal fenomeno e chamado de convergencia ou evolucao paralela.
• Reversao de estados. Tal dificuldade diz respeito a relacao entre os estadosde cada caracterıstica. Considerando a matriz da Figura 4.1, por exemplo,suponha que A e B evoluıram de um objeto ancestral X. Que estado de-verıamos atribuir a X em relacao a caracterıstica c1? Podemos observar pela
33

4.1. Aspectos da filogenia baseada em caracterısticas dct-ufms
matriz que c1 = 1 para A e c1 = 0 para B. Se fizermos c1 = 1 para X, ealgum ancestral de X possuir o estado 0 para c1, entao o objeto B apresentauma reversao de estados para a caracterıstica c1.
Na Figura 4.2, temos uma filogenia na qual sao representados os eventos deevolucao paralela e reversao. Ao lado de cada no temos dois numeros, que saoestados de duas caracterısticas. Analisando o segundo numero ao lado de cada no,verificamos que os nos B e F apresentam o evento chamado evolucao paralela. Jao evento de reversao pode ser notado observando o primeiro numero dos nos, e emparticular o no E apresenta um reversao de estado em relacao a esta caracterısticaque o primeiro numero representa.
Figura 4.2: Filogenia com exemplos de reversao e evolucao paralela.
No exemplo da Figura 4.1, temos caracterısticas de estado binario. De acordo como relacionamento entre os estados, as caracterısticas podem ser classificadas comoordenadas e nao-ordenadas. Para uma caracterıstica nao-ordenada, nao assumimosnada quanto a mudanca de estados, ou seja, qualquer estado pode mudar paraqualquer outro estado. No caso em que as caracterısticas sao ordenadas, temosuma informacao a mais. Por exemplo, uma dada caracterıstica com 4 estados podeter a seguinte ordem linear: 3 ↔ 1 ↔ 4 ↔ 2, isto quer dizer que, se ha um no comestado 3 para uma dada caracterıstica, nao deve haver nenhuma aresta ligando-o a um no com estado 4, o no com estado 1 deve ser sempre um intermediario.No caso de caracterısticas de estado binario, se as caracterısticas sao ordenadas,entao conhecemos o estado ancestral, por exemplo 0, e o estado derivado, nocaso 1. Enfim, para caracterısticas ordenadas, conhecemos a sequencia direta detransicoes para cada caracterıstica quando observamos a arvore filogenetica da raizate as folhas.
Para evitarmos eventos de convergencia e reversao de estados, o projeto de umaarvore T deve possuir a seguinte propriedade: o conjunto de todos os nos (ob-jetos) que possuem o mesmo estado para uma determinada caracterıstica deveformar uma subarvore de T . Uma filogenia com esta propriedade e uma filogenia
34

4.1. Aspectos da filogenia baseada em caracterısticas dct-ufms
perfeita. Na Figura 4.3, apresentamos uma filogenia correspondente a matrizda Figura 4.1, onde podemos observar que a filogenia nao atende a propriedadedescrita acima. Por exemplo, os objetos B, C e E possuem o estado 1 para a carac-terıstica c5 e no entanto nao estao na mesma subarvore. Na verdade, esta filogeniaapresenta o problema da evolucao paralela, logo nao e uma filogenia perfeita.
C, E A D B
Figura 4.3: Exemplo de filogenia que nao e uma filogenia perfeita.
O problema central de reconstrucao da filogenia baseado em matrizes de estados econhecido como problema da filogenia perfeita. O problema consiste em, dados umconjunto O com n objetos, um conjunto C de m caracterısticas, cada caracterısticatendo no maximo r estados, determinar se existe uma filogenia perfeita para O.
Para a matriz da Figura 4.4, a filogenia perfeita e mostrada na Figura 4.5. Umacaracterıstica rotulando uma aresta indica que a transicao de um estado 0 para 1ocorre ao longo desta aresta, tal que a subarvore abaixo da aresta contem todosos objetos que tem estado 1 para aquela caracterıstica.
Objeto/Caracterıstica c1 c2 c3 c4 c5 c6A 0 0 0 1 1 0B 1 1 0 0 0 0C 0 0 0 1 1 1D 1 0 1 0 0 0E 0 0 0 1 0 0
Figura 4.4: Exemplo de matriz de estados que admite filogenia perfeita.
Sempre que um conjunto de objetos definidos por uma matriz de estados admiteuma filogenia perfeita dizemos que as caracterısticas sao compatıveis.
35

4.2. Algoritmo para numero fixo de estados dct-ufms
B D E A C
c 1
c 2 c 3
c 4
c 5
c 6
Figura 4.5: Filogenia correspondente a matriz da Figura 4.4.
4.2 Algoritmo para numero fixo de estados
Nesta secao apresentamos como identificar se uma matriz de estados binarios ad-mite uma filogenia perfeita.
Como veremos no Capıtulo 5, estaremos interessados em construir filogenia deproteomas com base na informacao de que eles contem ou nao determinados genesou regioes. Assim, nao trataremos neste trabalho do problema da filogenia perfeitaquando os estados nao forem binarios. Neste contexto, as especies sao descritaspor caracterısticas cujos estados tem valor 0 ou 1, significando que a especie temou nao uma determinada caracterıstica.
O problema da filogenia perfeita pode ser resolvido polinomialmente para carac-terısticas com estados binarios. Um algoritmo para esse problema foi propostopor Gusfield em 1991 [23], cujo tempo de execucao e O(nm), onde n representa onumero de objetos e m o numero de caracterısticas. Este algoritmo possui duasfases: na primeira ele verifica se a matriz de entrada M admite uma filogeniaperfeita. Se admitir, entao a segunda fase consiste na construcao de uma possıvelfilogenia.
Conforme Gusfield [24], a principal propriedade de uma arvore T com raiz, que euma filogenia perfeita para a matriz M , e: toda caracterıstica em M correspondea uma aresta em T , sendo que esta aresta marca a transicao do estado 0 para oestado 1 para aquela caracterıstica. A raiz sempre tem estado 0 para todas ascaracterısticas. Logo, quando percorremos um caminho ligando um objeto i emuma folha a raiz, as arestas encontradas ao longo do caminho correspondem ascaracterısticas para as quais o objeto i tem estado 1.
Na matriz M , cada coluna j e uma caracterıstica, sendo que os termos colunae caracterıstica sao usados sem distincao, assim como os termos objeto e linha.Entao, para cada coluna j de M , denotaremos por 1j o conjunto dos objetos cujoestado e 1 e denotaremos por 0j o conjunto dos objetos cujo estado e 0 para a
36

4.2. Algoritmo para numero fixo de estados dct-ufms
coluna j.
A condicao necessaria e suficiente para determinarmos se uma matriz M admitefilogenia perfeita e dada a seguir.
Lema 4.1 Uma matriz binaria M admite uma filogenia perfeita se, e somente se,para cada par de caracterısticas i e j, ou os conjuntos 1i e 1j sao disjuntos, ou umdeles contem o outro.
Prova. Suponha que M admite uma filogenia perfeita. Como M e binaria, paracada caracterıstica i podemos associar uma unica aresta (u, v) na arvore. Alemdisso, a subarvore com raiz em v (assumindo que u e ancestral de v) contem todosos nos com estado 1 para a caracterıstica i, e qualquer no que possui estado 0para a caracterıstica i nao pertence a esta subarvore. Suponha agora que ha tresobjetos A, B e C, tal que A,B ∈ 1i, C /∈ 1i e B,C ∈ 1j , A /∈ 1j . Ou seja, conformea caracterıstica i, A e B pertencem a mesma subarvore enquanto C nao pertence.No entanto, conforme caracterıstica j, B e C pertencem a mesma subarvore, o quee uma contradicao.Suponha agora que todos os pares de colunas satisfazem a condicao declarada nolema. Vamos mostrar indutivamente como construir uma filogenia perfeita. Para ocaso base, assuma somente uma caracterıstica, por exemplo, c1. Essa caracterısticaparticiona os objetos em dois conjuntos, A = 1c1 e B = 0c1 . Crie um no raiz. Crieum no a para o conjunto A e o ligue a raiz por uma aresta rotulada com c1. Crieum no b para o conjunto B e o ligue a raiz por uma aresta sem rotulo. Finalmente,divida cada filho da raiz em folhas, sendo uma folha para cada objeto pertencenteao conjunto. A arvore resultante e uma filogenia perfeita.Assuma agora, que temos uma arvore T para k caracterısticas, onde o ultimo passonao foi executado (isto e, nao ha folhas; os nos contem conjuntos de objetos).Vamos processar a caracterıstica k + 1. Esta caracterıstica tambem induz umaparticao no conjunto de objetos, e se nos a usassemos seriamos capazes de obteruma arvore T ′ a partir de T . Faremos isto enquanto a particao induzida pelacaracterıstica k + 1 separar os objetos pertencentes ao mesmo no. Se este naofor o caso, seremos forcados a rotular duas arestas com ck+1 e a arvore resultantenao sera uma filogenia perfeita. Mas tal situacao nao pode acontecer. Suponhaentao que ck+1 separe os objetos pertencentes aos nos a e b. Como estao em nosdiferentes, deve haver uma caracterıstica i que fez com que eles ficassem em nosdistintos em primeiro lugar. Neste caso, temos que 1i ∩ 1k+1 6= ∅, pois os objetosem a ou aqueles em b pertencem a 1i. Mas ocorre tambem que 1i nao contem nemesta contido em Ok+1 porque os objetos em a ou aqueles em b nao estao em 1i, oque contradiz nossa hipotese de inducao. �
O Lema 4.1 nos da um algoritmo para a fase de decisao, que e verificar se cadacoluna e compatıvel com todas as outras. Cada verificacao custa O(n), pois cadacoluna tem n linhas (objetos). E como temos que fazer O(m2) verificacoes, o
37

4.2. Algoritmo para numero fixo de estados dct-ufms
algoritmo resultante gasta tempo O(nm2). A partir do mesmo lema tambempodemos obter um algoritmo para a fase de construcao da filogenia perfeita comos mesmos limites de tempo. Mas podemos construir um algoritmo com tempomelhor, um tempo de execucao O(nm).
A fase de decidir se uma matriz admite ou nao uma filogenia perfeita gastatempo O(nm), se utilizarmos a seguinte ideia: ordenamos as colunas da ma-triz M em ordem nao-crescente pelo numero de 1’s da coluna. Tal ordenacao efeita utilizando o algoritmo radix sort, segundo Setubal e Meidanis [40], que gastatempo de execucao O(nm). Em seguida, calcula-se uma matriz auxiliar Ln×m,que a princıpio e totalmente inicializada com zero. A celula Lij recebera o valor kse Mij = 1, onde k e a coluna mais a direita da esquerda de j, tal que Mik = 1.Se nenhuma coluna existe, k = −1. Este passo tambem gasta O(nm). O passofinal consiste em verificar para cada coluna, se existem duas celulas com valoresdiferentes de zero e diferentes entre si. Se tal situacao acontecer em alguma co-luna, a matriz nao admite filogenia perfeita. Logo, a fase de decisao gasta tempode execucao O(nm).
Vamos aplicar o procedimento descrito sobre a matriz da Figura 4.4. A Figura 4.6mostra a matriz da Figura 4.4 ja ordenada de acordo com as colunas binarias.Em seguida, na Figura 4.7 mostramos a matriz L calculada conforme mencionado.Analisando as colunas dessa matriz, podemos dizer se a matriz admite ou naouma filogenia. Para dizer que a matriz nao admite filogenia perfeita, temos queencontrar duas celulas em uma coluna com valores diferentes de zero e diferentesentre si. Todas as nossas colunas nao contem celulas nestas condicoes. Logo, amatriz da Figura 4.4 admite uma filogenia perfeita.
M 1 2 3 4 5 6A 1 1 0 0 0 0B 0 0 1 1 0 0C 1 1 0 0 1 0D 0 0 1 0 0 1E 1 0 0 0 0 0
Figura 4.6: Matriz de estados da Figura 4.4 com ordenacao das colunas.
Na fase de construcao da filogenia perfeita, comecamos a arvore com um unicono: a raiz. Para cada objeto na matriz, observamos as caracterısticas para asquais seu estado e 1. Enquanto nao existir uma aresta rotulada com uma talcaracterıstica, criamos um novo no e o ligamos ao no corrente (inicialmente a raiz)por uma aresta rotulada por essa caracterıstica. O novo no torna-se o no corrente.Se ja existe uma aresta rotulada com essa caracterıstica, nos passamos para aproxima caracterıstica, deixando que o no corrente seja o ponto final dessa aresta.O algoritmo gasta O(nm) pois olhamos para cada objeto de M exatamente uma
38

4.3. Parsimonia e compatibilidade dct-ufms
L 1 2 3 4 5 6A -1 1 0 0 0 0B 0 0 -1 3 0 0C -1 1 0 0 2 0D 0 0 -1 0 0 3E -1 0 0 0 0 0
Figura 4.7: Matriz auxiliar L, calculada para verificar se a matriz de estadosda Figura 4.4 e aditiva.
vez e gastamos tempo O(m) por elemento.
Pelo Lema 4.1, e facil ver que a matriz da Figura 4.1 nao admite uma filogenia per-feita: as colunas c1 e c5 nao satisfazem a condicao do lema pois 1c1
⋂1c5 6= ∅ e 1c1
nao esta contido em 1c5 nem vice-versa. Dizemos entao que estas caracterısticasnao sao compatıveis.
Como nossas matrizes nem sempre admitem uma filogenia perfeita, na secao se-guinte apresentamos abordagens que podem ser utilizadas para construir filogeniasbaseadas em quaisquer matrizes de caracterısticas binarias.
4.3 Parsimonia e compatibilidade
Na secao anterior, vimos que e possıvel verificar polinomialmente se um conjuntode caracterısticas binarias admite ou nao filogenia perfeita. Contudo, para carac-terısticas nao-ordenadas o problema e NP -completo, conforme Setubal e Meida-nis [40]. Alem disso, as matrizes de estados sao improvaveis de admitir filogeniaperfeita, pois os dados com que se trabalham podem conter erros e os requisitos,como por exemplo, nao ocorrer reversao de estados nem evolucao paralela, as ve-zes sao violados. Por isso, precisamos de outras abordagens para reconstruir afilogenia.
De acordo com Setubal e Meidanis [40] e Gusfield [24], uma das abordagens sugerea minimizacao dos eventos de reversao de estados e evolucao paralela, conhecidacomo criterio de parsimonia. Outra abordagem, segundo Setubal e Meidanis [40],e conhecida como criterio de compatibilidade, que propoe evitar tais eventos, ex-cluindo as caracterısticas que causam tais problemas. Ou seja, no criterio decompatibilidade tentamos encontrar um conjunto maximo de caracterısticas com-patıveis entre si. Ambos os criterios resultam em problemas NP -difıceis paracaracterısticas ordenadas.
Day e Sankoff [14] apresentaram em 1986 uma prova de que o criterio de compa-tibilidade e NP -completo. Para o criterio de parsimonia temos uma prova de que
39

4.3. Parsimonia e compatibilidade dct-ufms
este e NP -completo, apresentada por Day et al [13], em 1986.
A filogenia construıda com o metodo da parsimonia explica a evolucao com o menornumero de mudancas evolutivas. De acordo com Swofford et al [43], em termino-logia matematica, podemos definir o problema da parsimonia como o seguinte: apartir do conjunto de todas as arvore possıveis, encontre aquelas que minimizam
B∑
k=1
N∑
j=1
wj.diff(xk′j, xk′′j), (4.1)
onde B e o numero de arestas, N e o numero de caracterısticas, k ′ e k′′ sao osdois nos incidentes para cada aresta k, xk′j e xk′′j representam ou os elementos damatriz de entrada ou atribuicoes de estados de caracterıstica otimas feitas aos nosinternos, diff(y, z) e uma funcao que especifica o custo de uma transformacao doestado y para o estado z ao longo de qualquer aresta. O coeficiente wj atribui umpeso para cada caracterıstica, mas e tipicamente adotado como 1 (um).
O problema de encontrar uma filogenia baseada em caracterısticas pode ser divi-dido em dois subproblemas: o primeiro e como avaliar quao boa e a arvore obtida;o segundo e como procurar uma arvore no conjunto de todas as arvores possıveis,eficientemente.
O primeiro subproblema e conhecido como o problema da parsimonia pequeno e osegundo como o problema da parsimomia grande.
Problema da parsimonia pequeno
O problema da parsimonia pequeno consiste em determinar o custo de umadada filogenia [21]. O custo de uma filogenia refere-se ao numero de mudancasrequeridas para explicar os dados. Existem algoritmos polinomiais para resolvereste problema, que sao variacoes da parsimonia. Entre eles temos a parsimoniade Fitch e a parsimonia de Wagner, onde a entrada e a topologia de uma arvorefilogenetica com raiz, sendo que as especies se encontram nas folhas. SegundoShamir [41], o objetivo e encontrar uma rotulacao para os nos internos da arvore(em termos de caracterısticas), implicando em um numero mınimo de mudancasde estados das caracterısticas ao longo das arestas da arvore.
O metodo de parsimonia de Wagner, formalizado por Kluge e Farris (1969) e Far-ris (1970), e apropriado para caracterısticas de estados binarios e caracterısticas demultiplos estados. Nesse metodo as mudancas de estados em uma ou outra direcaosao igualmente provaveis de acontecer, conforme Olsen et al [43]. Um estado podemudar para outro e vice-versa, ou seja, o estado ancestral e desconhecido. Porexemplo, no caso de caracterısticas binarias, o estado 0 pode mudar para o es-tado 1, assim como o estado 1 pode mudar para o estado 0. Outras hipoteses
40

4.3. Parsimonia e compatibilidade dct-ufms
deste metodo, conforme Felsenstein [19], sao: caracterısticas diferentes evoluemindependentemente, linhagens diferentes evoluem independentemente.
Para determinar o custo mınimo requerido por uma dada caracterıstica c′ sobre ocriterio de Wagner, somente um passo e necessario, progredir das folhas em direcaoa raiz. Alem disso, Olsen et al [43], recomenda que a arvore seja enraizada emuma das especies, denotada por r.
O algoritmo para determinar o custo de uma arvore com raiz sobre o criterio deparsimonia de Wagner, transcrito de Olsen et al [43], e descrito a seguir:
1. Para cada no folha i (incluindo a raiz), atribua um conjunto de estados Si
contendo o estado da caracterıstica atribuıdo para a especie correspondentena matriz de entrada (Mij). Inicialize o custo da arvore com zero.
2. Visite um no interno k para o qual um conjunto de estados Sk nao tenhasido definido, mas para o qual os conjuntos de estados dos dois descendentesimediatos de k tenham sido definidos. Sejam i e j os representantes dos doisdescendentes imediatos. Atribua a k um conjunto de estados Sk de acordocom as seguintes regras:
(a) Se a intersecao dos conjuntos de estados atribuıdos aos nos i e j evazia (Si ∩ Sj 6= ∅), deixe o conjunto de estados de k igual a estaintersecao (isto e, Sk = Si ∩ Sj). A intersecao pode ser representadacomo um intervalo fechado [ak, bk].
(b) Caso contrario (Si ∩Sj = ∅), deixe o conjunto de estados de k igual aomenor intervalo fechado [ak, bk] contendo um elemento de cada um dosconjuntos de estados atribuıdos a i e j. Adicione ao custo da arvore ovalor bk − ak.
3. Se o no k e o descendente imediato do no folha colocado como raiz, o passoinicial terminou; prossiga para o passo 4. Caso contrario, volte ao passo 2.
4. Se o estado atribuıdo ao no raiz da arvore (xr) nao esta contido no conjuntode estados ja atribuıdo ao no descendente imediato da raiz da arvore (Sk),adicione ao custo da arvore o valor da distancia de xr a Sk. (Esta distanciae igual a ak − xr se xr < ak ou xr − ak se xr > bk.)
Um exemplo de execucao do algoritmo e apresentado na Figura 4.8. Computamoso custo da arvore sem raiz da Figura 4.8(a). (Embora a situacao mais usual paradados moleculares seria envolver mais caracterısticas binarias que caracterısticade multiplos estados, tratamos o caso multiplos estados para demonstrar a gene-ralidade do algoritmo.) Inicialmente, enraizamos a arvore no no A (poderıamoster escolhido qualquer no), produzindo a arvore mostrada na Figura 4.8(b). NaFigura 4.8(b) mostramos tambem os conjuntos de estados atribuıdos aos nos fo-lhas de acordo com o passo 1 do algoritmo. Visitando o no interno X na primeira
41

4.3. Parsimonia e compatibilidade dct-ufms
chamada do passo 2, observamos que SB ∩SC = {0}∩{2} = ∅, e entao atribuımoso intervalo [0, 2] a SX , adicionando 2 − 0 = 2 ao custo da arvore. Similarmente,deixamos SY = [1, 3] na segunda chamada do passo 2, e adicionamos 3 − 1 = 2ao custo, que e agora 4. Na terceira e ultima chamada do passo 2, observamosque a intersecao SX ∩ SY = [0, 2] ∩ [1, 3] nao e vazia, e entao atribuımos o inter-valo [1, 2] a SZ . A situacao que chegamos no passo 4 e a mostrada na Figura 4.8(c).Desde que xr = 0 nao e um elemento de SZ = [1, 2], adicionamos 1−0 = 1 ao custo.Entao, a evolucao desta caracterıstica requer um numero mınimo de 5 mudancasde estado em nossa arvore dada.
O procedimento descrito acima e suficiente para obter o custo mınimo requeridopara qualquer caracterıstica numa dada arvore, mas nao atribui estados aos nosinternos. Para obter tal construcao, podemos realizar um segundo passo sobre aarvore, agora partindo da raiz em direcao as folhas, como abaixo:
1. Visite um no interno k para o qual uma atribuicao de estado otima nao tenhasido feito ainda, mas que tal atribuicao tenha sido feita para o ancestralimediato de k, denotado por m. (Note que a primeira vez que este passo eexecutado, k corresponde ao no que e descendente imediato da raiz e m = r,a especie na raiz da arvore.)
2. Atribua a k o estado a partir do conjunto de estados computado no primeiropasso, Sk(= [ak, bk]), que e mais proximo a Mmc′ . Especificamente, se Mmc′
esta contido em Sk, deixamos Mkc′ = Mmc′ . Caso contrario, permaneceMkc′ = ak se Mmc′ < ak ou Mkc′ = bk se Mmc′ > bk.
3. Se todos os nos internos foram visitados, pare. Caso contrario, volte aopasso 1.
Aplicando os passos descritos acima, ao exemplo da Figura 4.8, inicialmente atri-buiremos o estado 1 (o mais proximo de 0 em [1, 2]) ao no Z. Em seguida,atribuımos o estado 1 (o mais proximo de 1 em [0, 2]) para X; da mesma formaatribuımos o estado 1 (o mais proximo a 1 em [1, 3]) para o no Y . A arvore resul-tante e mostrada na Figura 4.8(d), que confirma que o valor 5 e o custo mınimoda arvore para esta caracterıstica.
Problema da parsimonia grande
Uma vez que temos como determinar o custo de uma arvore, consideramos entao oproblema de encontrar a melhor arvore que explique os dados. No problema daparsimonia grande procuramos a melhor arvore no espaco de todas as arvorepossıveis. Neste problema, a entrada e uma matriz de estados e o objetivo eencontrar uma arvore de parsimonia com o menor custo.
42

4.4. Mix dct-ufms
A (0) B (0)
C (2)
D (1)
E (3)
X Y
Z
{0} A
B {0}
C {2}
D {1}
E {3}
X [0,2]
Y [1,3]
Z [1,2]
{0} A
B {0}
C {2}
D {1}
E {3}
X 1
Y 1
Z 1
0 A
B 0
C 2
D 1
E 3
(a)
(b) (c) (d)
1 1 2
1
Figura 4.8: Exemplo de execucao para o metodo de Wagner.
Considere uma arvore com duas folhas. Ha somente uma topologia para estaarvore. Para produzir uma arvore com 3 folhas, temos tres possibilidades deramificacoes para que possamos adicionar uma nova folha. Uma vez obtida aarvore com 3 folhas ha 5 ramificacoes onde podemos adicionar a quarta folha.Desta forma, pode-se mostrar que ha 1 ∗ 3 ∗ 5 ∗ ... ∗ (2n − 3) = (2n − 3)! arvorescom especies nas folhas e nos internos nao rotulados. Como este numero cresceexponencialmente com o numero de folhas n, o problema da parsimonia grande eNP-difıcil [41].
4.4 Mix
Nesta secao vamos apresentar um programa utilizado para construcao de arvoresde parsimonia a partir de caracterısticas discretas.
Mix e um dos programas contidos no pacote de inferencia filogenetica Phylip [20].E um programa de parsimonia geral que realiza os metodos de parsimonia deCamin-Sokal e de Wagner, para caracterısticas discretas. O metodo de parsimoniade Camin-Sokal explica os dados assumindo que mudancas de estado 0 → 1 saopermitidas, mas mudancas 1 → 0 nao. A parsimonia de Wagner permite ambosos tipos de mudancas. (Isto sobre a hipotese que 0 (zero) e o estado ancestral).
43

4.4. Mix dct-ufms
O programa MIX, assim como a maioria dos outros programas que fazem parte dopacote Phylip, utilizam na construcao das arvores filogeneticas, uma abordagemenvolvendo adicoes e rearranjos [19]. Eles tentam minimizar ou maximizar algumaquantidade sobre o espaco de todas as arvores evolutivas possıveis. Cada programacontem uma parte que, dado uma topologia de arvore, avalia a quantidade que estasendo minimizada ou maximizada. No caso do MIX, queremos o numero mınimode mudancas de estados das caracterısticas, seguindo o criterio de Camin-Sokalou o criterio de Wagner. A abordagem direta seria avaliar todas as topologias dearvores possıveis, uma apos a outra, e escolher uma que, de acordo com o criteriosendo usado, seja a melhor. Isto nao seria possıvel para mais que um certo numerode especies, visto que o numero de topologias de arvores possıveis e enorme.
Como nao podemos procurar por todas as topologias, estes programas nao garan-tem encontrar a melhor topologia sempre. Eles empregam a seguinte estrategia:as especies sao tomadas de acordo com a ordem em que aparecem no arquivo deentrada. Tome as duas primeiras e construa uma arvore para elas. Ha somenteuma topologia para esta arvore. Entao, tome a terceira especie e vamos consideraronde ela deve ser adicionada na arvore. Se a arvore inicial e uma arvore enrai-zada com duas especies e queremos que a arvore resultante com tres especies sejauma arvore binaria, ha somente tres lugares onde podemos adicionar a terceiraespecie. Tentamos cada uma destas, sendo que em cada vez avaliamos a arvoreresultante de acordo com o criterio. A melhor arvore e escolhida para ser a arvorebase para mais operacoes. Agora considere a adicao da quarta especie, novamenteem cada um dos cinco lugares possıveis de adicao resultaria numa arvore binaria.Novamente, a melhor destas e aceita.
Rearranjos locais
O processo descrito acima e realizado continuamente com uma excecao. Apos cadaespecie ser adicionada e, antes de adicionarmos a proxima, tentamos um numerode rearranjos da arvore, no proposito de melhora-la. Os algoritmos se movempela arvore, fazendo todos os possıvies rearranjos. Um rearranjo local envolveum segmento interno da arvore. Cada segmento interno e da forma mostrada naFigura 4.9 (onde T1, T2 e T3 sao subarvores - partes da arvore que podem contermais bifurcacoes e folhas):
Um rearranjo local consiste em trocar as subarvores T1 e T3 ou T2 e T3, tal queobtemos uma das seguintes arvores:
Cada vez que um rearranjo tem sucesso e encontra uma arvore melhor, o novorearranjo e aceito. A fase dos rearranjos locais nao termina ate que o programaatravesse a arvore inteira, tentando rearranjos locais, sem encontrar qualquer me-lhora na arvore.
Esta estrategia de adicionar especies e fazer rearranjos verifica cerca de p topologias
44

4.4. Mix dct-ufms
T 1
T 2 T 3
Figura 4.9: Exemplo de arvore que sofrera rearranjo local. A aresta ponti-lhada e um segmento interno.
T 1
T 3 T 2
T 2
T 3 T 1
Figura 4.10: Arvore apos rearranjo.
diferentes, onde p = (n− 1)× (2n− 3), embora esse numero possa ser maior se osrearranjos tem frequentemente sucesso.
Embora nao haja garantia que encontramos a melhor topologia de arvore, temos agarantia que nenhuma topologia proxima (isto e, nenhuma acessıvel por um unicorearranjo local) e a melhor. Neste sentido, alcancamos um otimo local do nossocriterio. Note que o processo inteiro e dependente da ordem que as especies apa-recem no arquivo de entrada. Podemos entao reordenar as especies no arquivo deentrada e executar o programa novamente. Se nenhuma destas tentativas encon-trar uma solucao melhor, teremos alguma indicacao que podemos ter encontradoa melhor topologia, mesmo podendo nunca ter certeza disto.
Rearranjos globais
Uma caracterıstica da maioria dos programas do pacote Phylip, como e o casodo Mix, e a otimizacao global da arvore. Em alguns dos programas do pacote,esta e uma opcao, enquanto que nos outros ela e aplicada automaticamente, comoacontece em Mix. Quando esta presente, ha um estagio adicional na busca pelamelhor arvore. Cada subarvore possıvel e removida da arvore e adicionada de voltaem todos os lugares possıveis. Este processo continua ate que todas as subarvorespossam ser removidas e adicionadas novamente sem qualquer melhora na arvore.
45

4.4. Mix dct-ufms
O objetivo deste rearranjo extra e tornar menos provavel que uma ou mais especiesfiquem “presas” numa regiao subotima do espaco de todas as arvores possıveis.
De acordo com Felsenstein [20], o que Phylip chama de rearranjos globais saomais apropriadamente chamados SPR (subtree pruning and regrafting) por Swof-ford et al [43].
46

Capıtulo 5
Comparacao de proteomas efilogenia
A analise comparativa de genomas tem se mostrado uma ferramenta importantepara estudar a evolucao das especies. Existem varios estudos realizados nessa area.E essa analise e muito poderosa quando utilizamos dados que denotam conservacaodo conteudo genico e da ordem dos genes nos genomas.
Em nosso trabalho, tambem realizamos uma analise comparativa de genomas,comparando os seus proteomas e inferindo uma historia evolutiva para as especiescorrespondentes aos genomas por meio de uma arvore filogenetica.
Os dados que utilizamos para construir as filogenias foram obtidos por meio daferramenta EGG (Extended Genome-Genome Comparison), desenvolvida por Al-meida [1]. Essa ferramenta encontra todos os pares de genes ortologos, descritoslogo a seguir, resolvendo o seguinte problema: dada uma sequencia de entrada g1
(gene) de G, listar os genes de H que sao similares a g1, onde g1 e o gene de en-trada; G e H sao genomas de especies diferentes. Cada um desses genes retornadosrecebe o nome de hit.
Para realizar essa tarefa, EGG utiliza o programa BLASTP, que e um dos pro-gramas do pacote BLAST (Basic Local Alignment Search Tool) [2, 3], um dosmais populares programas de busca em base de dados de biomoleculas. Trata-sede uma heurıstica, cujo sucesso se deve principalmente a sua velocidade e pelo fatode devolver uma lista de possıveis hits, acompanhados de alinhamentos e tambemde uma estimativa de significancia, denominada e-value. O e-value e uma medidaproporcional a probabilidade de que um hit com aquele valor de alinhamento sejaencontrado em um conjunto de sequencias aleatorias. Ou seja, quanto menor oe-value, mais significante e o hit. O BLAST tambem retorna, para cada hit en-contrado, um alinhamento entre a sequencia de entrada e a sequencia hit. Cadaalinhamento possui um valor chamado score. Apesar do BLAST usar como base
47

dct-ufms
os dados das bases de sequencias do GenBank, NCBI e outros, e possıvel criarbases locais de sequencias, comparando apenas o proteoma de um genoma com ode outro genoma, por exemplo.
Com isso, a entrada para EGG sao os conjuntos das proteınas preditas1 de dois ge-nomas, G e H, por exemplo. Para cada genoma, existe um arquivo de extensao .pttque contem informacoes sobre cada uma das proteınas preditas, como fita, nome,produto, identificador no GenBank (gi); e um arquivo de extensao .faa que traz asequencia de todas as proteınas preditas do genomas, no formato FASTA. Os ar-quivos .ptt e .faa sao obtidos a partir do Genbank [6], disponıvel no site do NCBI
(http://www.ncbi.nih.gov).
O programa EGG faz uma comparacao das proteınas preditas, todas-contra-todas,usando BLAST. Mais especificamente, EGG cria um grafo bipartido, onde osvertices sao genes, particionados pelos seus genomas. Seja gi um gene do genomaG e hj um gene do genoma H. Uma aresta (gi, hj), chamada de match, existe se,e somente se, hj e hit de gi e vice-versa; o e-value da comparacao de gi contra ogene de hj e o e-value da comparacao de hj contra gi sao ambos menores ou iguaisa 10−5; e o alinhamento retornado cobre pelo menos de 60% de gi e 60% de hj. Opeso de um match e definido como sendo a media dos scores dos pares (pi,qj)e (qj ,pi), denotado por peso(p, q). Alem disso, caso o par (pi, qj) forme um matche, qj tenha sido o melhor hit de pi e vice-versa, temos o que chamamos de BBH(Bidirectional Best Hit). Para efeitos do EGG, dizemos que os genes g e h saoortologos, se e somente se, g e um match de h e vice-versa.
Apos a construcao desse grafo, o algoritmo de Almeida [1] encontra alguns tipos deestruturas. Uma delas, chamada de run, consiste em uma sequencia contıgua dematches. Esses runs serao utilizados para construir outra estrutura a ser descrita.Alem disso, a existencia de muitos runs e um bom motivo para acreditarmos queos genomas estao relacionados e que, portanto, sao proximos filogeneticamente.Na Figura 5.1 apresentamos um exemplo de run.
Figura 5.1: Exemplo de run entre os proteomas de Xylella fastidiosa eXanthomonas citri.
1Daqui em diante, usaremos indistintamente os termos gene e proteına predita.
48

dct-ufms
Na Figura 5.2, mostramos outro exemplo de estrutura que podemos encontrarentre os genomas de Xylella fastidiosa e Xanthomonas citri.
Figura 5.2: Exemplo de um run. Note que pode haver a participacao deproteınas preditas em fitas distintas.
Outra estrutura determinada pelo algoritmo e chamada de regiao ortologa, econsiste na uniao de runs consecutivos e proximos. Para criar tais regioes, ousuario informa como parametro de entrada para o programa EGG, o numeromınimo de matches que uma regiao deve conter para ser formada. Outros doisparametros sao fornecidos ao EGG . O primeiro e o numero mınimo de genes quepodemos ter entre dois runs, para que eles sejam unidos e formem uma regiao.O segundo parametro e o numero maximo de genes que podemos ter entre doisruns que serao unidos. Em nosso trabalho, utilizamos os seguintes valores paracada um dos parametros citados acima, respectivamente: 4, 2 e 5. Os buracos quemencionamos sao formados por genes que nao formaram matches com nenhumoutro gene na regiao. Na Figura 5.3, mostramos um exemplo de regiao ortologacontendo buracos.
Figura 5.3: Exemplo de buracos encontrados entre os runs.
Tais regioes sao uteis na descoberta de genes falsos e tambem da funcionalidadede genes hipoteticos, no sentido de que pode haver um par de genes entre doisruns consecutivos, cuja similaridade e baixa (por isso a aresta nao foi gerada), masque, pelo fato de estarem “sanduichados” por dois runs, podem ainda sim seremrelacionados evolutivamente. Novamente, podemos inferir que, quanto maior o
49

5.1. Metodologia dct-ufms
numero de regioes ortologas, mais os genomas sao relacionados evolutivamente.Na Figura 5.4 apresentamos um exemplo de regiao ortologa.
Figura 5.4: Exemplo de regiao ortologa que podemos encontrar entre osgenomas de Xylella fastidiosa e Xanthomonas campestris.
Essas regioes, bem como os matches e BBHs, sao utilizados como medidas parainferir filogenia, como descreveremos nas proximas secoes. Na Secao 5.1 propo-mos uma metodologia para construcao de filogenias. Na Secao 5.2 descrevemosas medidas utilizadas na construcao de filogenias baseadas em distancias e os re-sultados obtidos a partir dessas construcoes. Ja na Secao 5.3 descrevemos ascaracterısticas utilizadas na construcao de filogenias, como foram determinadas eapresentamos os resultados obtidos a partir das construcoes. Os resultados apre-sentados na Secao 5.2 foram publicados em [4].
5.1 Metodologia
Considerando nosso objetivo, que e o de propor metodologias para inferencia defilogenia de proteomas, precisamos de medidas de qualidade das arvores obtidas.Para tanto, precisamos de uma arvore que possa ser considerada verdadeira, paracada conjunto de organismos. Alem disso, precisamos de uma medida de distanciaentre arvores, para sabermos qualifica-las em comparacao com a arvore verdadeira.
Como arvore verdadeira, vamos usar o modelo que propoe filogenia baseada emsequencias 16S rRNA. Os genomas de procariotos, em geral, possuem sequenciasdenominadas Small Subunit Ribosomal RNA (SSU rRNA). A sequencia 16S rRNAe uma delas. Essas sequencias sao encontradas em praticamente todos os genomasde procariotos disponıveis atualmente. Embora existam outras sequencias destetipo, o numero de sequencias 16S rRNA completamente disponıveis e muito maiorque os outros tipos de sequencias. Alem disso, tratam-se de sequencias muitoconservadas nas especies. Estes sao alguns dos motivos por que as utilizamospara construir a arvore filogenetica considerada como verdadeira. Alem disso, elascompartilham similaridades suficientes para serem reconhecidas e usadas comopotenciais marcadores para inferir filogenia [32]
50

5.1. Metodologia dct-ufms
Como exemplo, a Figura 5.5 mostra a matriz de distancias baseada em sequencias16S rRNA dos genomas de Escherichia coli K-12 MG1655 (EC), Pseudomonasaeruginosa PA01 (PA), Staphylococcus aureus MU50 (SM), Staphylococcus aureusN315 (SN), Salmonella typhi (ST) e Salmonella typhimorium LT12 (SL) e a Fi-gura 5.6 mostra a arvore obtida a partir da matriz.
As sequencias 16S rRNA de cada genoma foram obtidas a partir do Gen-bank [6], disponıvel no site do NCBI. Em seguida, criamos uma matriz dedistancias 16S rRNA, utilizando o programa Dnadist [18], disponıvel no pacotePhylip, que calcula uma matriz de distancias a partir de sequencias de nucleotıdeos.A partir dessa matriz, apresentada na Figura 5.5, construımos a arvore filogeneticacorrespondente utilizando o algoritmo Neighbor-Joining.
SM SN EC PA SL STSM 0.0000 0.0019 4.7443 5.9752 5.2363 4.9180SN 0.0000 4.7443 5.9752 5.2363 4.9180EC 0.0000 4.1879 0.0314 0.0287PA 0.0000 4.0768 4.2328SL 0.0000 0.0066ST 0.0000
Figura 5.5: Matriz de distancias para sequencias 16S rRNA de bacterias.
ST
PA
SL
EC
SM
SN
Figura 5.6: Arvore filogenetica obtida a partir da matriz 5.5.
Para confirmarmos a veracidade das ramificacoes obtidas na arvore da Figura 5.6,realizamos o teste de bootstrap sobre as sequencias 16S rRNA. O bootstrap e umatecnica estatıstica de computacao intensiva que testa se realmente um conjunto deespecies formam uma determinada ramificacao na arvore. Para realizar tal tarefa,utilizamos o programa MEGA [30], que baseado em sequencias de DNA ou emsequencias de aminoacidos calcula o alinhamento entre elas e verifica as colunasdo alinhamento que sao significativas para a inferencia filogenetica. As colunasconsideradas significativas sao aquelas que possuem diferencas entre os caracteres
51

5.1. Metodologia dct-ufms
das sequencias. O processo de bootstrap entao remove algumas dessas colunas eduplica outras, de forma que obtemos o mesmo numero de colunas original. Umaarvore filogenetica e construıda, e o processo se repete quantas vezes o usuariodesejar, sendo que a cada vez as colunas a serem removidas sao alternadas. Aofinal do processo, cada ramificacao da arvore possui uma porcentagem, que indicaa porcentagem de vezes que aquela ramificacao se conservou nos demais testes [9].Dizemos que uma arvore suporta os grupos de genomas se o valor das porcentagenspara as ramificacoes for maior que 95%, segundo Brown [9]. Dentre os algoritmosoferecidos pelo programa MEGA para a construcao de filogenias, utilizamos oalgoritmo Neighbor-Joining.
Ao executarmos o bootstrap, a partir do programa MEGA, sobre as sequencias16S rRNA dos seis genomas, obtivemos uma arvore cujas ramificacoes tem porcen-tagem acima de 99%. Logo, nossa arvore 16S rRNA pode ser uma representacaosignificativa da historia evolutiva dessas especies. A arvore e mostrada na Fi-gura 5.7.
SL
ST
EC
PA
SM
SN
99
100
100
Figura 5.7: Arvore filogenetica obtida apos bootstrap.
Para compararmos as arvores que apresentaremos nas proximas secoes com aarvore 16S rRNA, vamos propor agora uma medida de distancia δ(Ti, Tj) entreduas arvores filogeneticas Ti e Tj , envolvendo os mesmos organismos, conformesegue:
δ(Ti, Tj) =∑
a,b folhasa6=b
|∆i(a, b) − ∆j(a, b)|, (5.1)
onde ∆i(a, b) e o numero de arestas no unico caminho entre a e b, na arvore Ti.Vamos considerar duas arvores Ti e Tj isomorfas, e vamos denotar como Ti
∼= Tj ,quando ∆i(a, b) = ∆j(a, b) ∀ a, b.
A medida δ acima satisfaz as propriedades abaixo:
1. δ(Ti, Tj) = 0 ⇔ Ti∼= Tj,
52

5.1. Metodologia dct-ufms
2. δ(Ti, Tj) ≥ 0 ∀ Ti, Tj,
3. δ(Ti, Tj) = δ(Tj , Ti) ∀ Ti, Tj ,
4. δ(Ti, Tj) + δ(Tj , Tk) ≥ δ(Ti, Tk) ∀ Ti, Tj e Tk.
Ou seja, a medida δ proposta e uma metrica. A prova de que a distancia apresen-tada e uma metrica e mostrada abaixo:
1. Dadas duas arvores Ti e Tj , temos que
δ(Ti, Tj) = 0 ⇔∑
a,b folhas |∆i(a, b) − ∆j(a, b)| = 0
⇔ |∆i(a, b) − ∆j(a, b)| = 0.
Logo, ∆i(a, b) = ∆j(a, b). Ou seja, Ti∼= Tj .
2. Por definicao, o modulo de um termo e sempre maior ou igual a zero. Comonossa distancia e constituıda de uma soma de valores em modulo, temos queδ(Ti, Tj) ≥ 0 ∀ Ti, Tj.
3. Dadas duas arvores Ti e Tj , sabemos que
δ(Ti, Tj) =∑
a,b folhas |∆i(a, b) − ∆j(a, b)|
=∑
a,b folhas |∆j(a, b) − ∆i(a, b)|
= δ(Tj , Ti).
4. Vamos provar a validade da propriedade 4 por inducao em n, o numero defolhas das arvores. Note que podemos supor que todas as arvores contem omesmo conjunto de folhas. Vamos considerar tambem n ≥ 2.
Para n = 2, temos apenas uma topologia possıvel. Assim, para quaisquerarvores Ti, Tj e Tk, temos δ(Ti, Tj) = δ(Ti, Tk) = δ(Tj , Tk) = 0. Ou seja,δ(Ti, Tk) ≤ δ(Ti, Tj) + δ(Tj , Tk).
Para n = 3, temos tres topologias possıveis de arvores, onde observamosbasicamente um par de objetos ligados por um no interno w, e w por sua vezesta ligado ao no interno z, que esta ligado ao terceiro objeto por uma aresta.Ou seja, considerando tres objetos A, B e C, por exemplo, em T1 teremos(AB,C), em T2 (AC,B) e em T3 (BC,A). Assim, para quaisquer arvoresTi, Tj e Tk, com n = 3 folhas, temos δ(Ti, Tj) = δ(Ti, Tk) = δ(Tj , Tk) = 2.Ou seja, δ(Ti, Tk) ≤ δ(Ti, Tj) + δ(Tj , Tk).
Considere agora arvores Ti, Tj e Tk, todas com n ≥ 4 folhas. Sejam a e bfolhas tais que
∆i(a, b) ≤ ∆j(a, b) ≤ ∆k(a, b). (5.2)
(Note que sempre e possıvel encontrar tais folhas).
Por hipotese de inducao, vale a desigualdade triangular para as novas arvoresobtidas com a retirada das folhas a e b. Ou seja,
53

5.1. Metodologia dct-ufms
∑
a6=ba,b 6=a,b
|∆i(a, b) − ∆j(a, b)| +∑
a6=ba,b6=a,b
|∆j(a, b) − ∆k(a, b)| ≥
∑
a6=ba,b 6=a,b
|∆i(a, b) − ∆k(a, b)|.
Acrescentando convenientemente os valores ∆i(a, b), ∆j(a, b) e ∆k(a, b) nainequacao acima, obtemos
∑
a6=ba,b 6=a,b
|∆i(a, b) − ∆j(a, b)| + ∆j(a, b) − ∆i(a, b)+
∑
a6=ba,b 6=a,b
|∆j(a, b) − ∆k(a, b)| + ∆k(a, b) − ∆j(a, b) ≥
∑
a6=ba,b 6=a,b
|∆i(a, b) − ∆k(a, b)| + ∆k(a, b) − ∆i(a, b).
Por outro lado, pelas inequacoes 5.2, sabemos que
∆j(a, b) − ∆i(a, b) = |∆i(a, b) − ∆j(a, b)|,∆k(a, b) − ∆j(a, b) = |∆j(a, b) − ∆k(a, b)| e
∆k(a, b) − ∆i(a, b) = |∆i(a, b) − ∆k(a, b)|.
Assim, ∑
a6=ba,b 6=a,b
|∆i(a, b) − ∆j(a, b)| + |∆i(a, b) − ∆j(a, b)|+
∑
a6=ba,b 6=a,b
|∆j(a, b) − ∆k(a, b)| + |∆j(a, b) − ∆k(a, b)| ≥
∑
a6=ba,b 6=a,b
|∆i(a, b) − ∆k(a, b)| + |∆i(a, b) − ∆k(a, b)|,
que conclui a demonstracao. �
54

5.2. Filogenia baseada em distancias dct-ufms
Dessa forma, utilizando a medida de distancia δ, realizamos a comparacao daarvore verdadeira com as arvores obtidas a partir das matrizes baseadas emdistancias e com as arvores baseadas em matrizes de caracterısticas, ambas apre-sentadas nas secoes seguintes.
5.2 Filogenia baseada em distancias
Podemos realizar a construcao de filogenias baseada em dois aspectos da orga-nizacao dos genomas. O primeiro diz respeito a presenca ou ausencia de cadagene individualmente. Podemos fazer isto, procurando pares de genes ortologosentre eles. Para tanto, podemos utilizar os matches e BBHs, apresentados anteri-ormente. O segundo diz respeito a grupos de pares de genes ortologos que foramconservados durante a evolucao. Para esse caso, podemos usar a ideia de regioesortologas.
Como tais estruturas e elementos, descritos no inıcio deste capıtulo, parecem sersignificativos na determinacao da similaridade entre os genomas, nos propomos autilizacao das mesmas como instrumentos para a obtencao de medidas de distanciasentre os genomas e construımos arvores filogeneticas para verificar a eficiencia detais medidas. As medidas que propomos e que foram construıdas a partir de BBHs,matches e regioes ortologas sao descritas a seguir.
Considere dois genomas G e H com m e n genes, respectivamente. Seja M oconjunto de matches, B o conjunto de BBHs e R o conjunto das regioes ortologasentre os genomas G e H. A primeira das medidas de distancias (D1) e baseada nonumero de matches entre os genomas, ou seja, a razao entre |M | e m.n (o numeromaximo de possıveis matches). A segunda medida, D2, e baseada na razao entreB e min{m,n} (o numero maximo de BBHs). Note que quanto maior o numerode matches e BBHs, a distancia filogenetica entre os genomas tende a ser menor.Por isso, precisamos inverter as estimativas, como mostrado a seguir.
D1 = 1|M|m.n
e D2 = 1|B|
min{m,n}
.
As normalizacoes feitas em D1 e D2 servem para nao permitir que pares de genomasmuito grandes sejam favorecidos, em detrimento dos menores.
As outras duas medidas sao baseadas nos pesos dos matches e BBHs, respectiva-mente.
D3 = 1∑
(g,h)∈M
s(g, h)e D4 = 1∑
(g,h)∈B
s(g, h).
55

5.2. Filogenia baseada em distancias dct-ufms
Para as medidas baseadas nas regioes ortologas dos genomas, considere como mr
e br, respectivamente, o numero de matches e BBHs de uma regiao r encontradaentre G e H. Entao,
D5 = 1∑
r∈R
mr
e D6 = 1∑
r∈R
br
.
Essas seis medidas foram utilizadas para inferir filogenias e os resultados sao apre-sentados na secao seguinte.
Alguns trabalhos utilizando o conteudo genico dos genomas para inferir filogeniasja foram realizados. Por exemplo, Korbel et al [29] construıram uma ferramenta,denominada SHOT, que constroi filogenias de genomas baseadas em distancias.As distancias sao calculadas a partir do conteudo genico e da conservacao da or-dem dos genes no genoma. Eles comparam os genomas das especies e selecionamos pares de genes ortologos. Para filogenias baseadas no conteudo genico dos ge-nomas, a similaridade entre dois genomas e definida como o raio do numero degenes ortologos compartilhados e um valor normalizado que reflete a variacao dostamanhos dos genomas. O valor normalizado e dominado pelo numero de genesno menor dos dois genomas comparados, porque esse numero determina o maiornumero possıvel de genes entre os dois genomas que podem ser compartilhados.Alem disso, e possıvel construir as filogenias utilizando qualquer das seguintespossıveis definicoes de tamanho: numero de genes que codificam proteınas anota-das, numero de genes com pelo menos um homologo nos outros genomas, numerode genes com pelo menos um ortologo nos outros genomas.
Ja a construcao baseada na conservacao da ordem dos genes calcula a similaridadeentre dois genomas a partir do numero de pares de genes ortologos conservados.Um par de genes conservados e definido como um par de genes ortologos queformam nos dois genomas um par de genes adjacentes com as mesmas direcoesde transcricao conservadas. Os genes considerados para definicao de pares degenes sao: genes em geral e genes compartilhados entre ambos os genomas. Nessaabordagem, os numeros de pares de genes conservados sao normalizados de acordocom o tamanho do menor genoma (o maximo numero possıvel de pares de genesconservados). O tamanho do genoma pode ser escolhido entre: o numero de genes;o numero de genes com pelo menos um homologo nos outros genomas; o numerode genes com pelo menos um ortologo nos outros genomas; o numero de ortologoscompartilhados entre dois genomas.
5.2.1 Resultados
Nesta secao vamos mostrar as arvores T1, T2, . . ., T6, obtidas a partir das medidasde distancias propostas na secao anterior, D1, D2, . . ., D6, respectivamente. Uti-lizando o mesmo conjunto de especies usado para construir a arvore baseada em
56

5.2. Filogenia baseada em distancias dct-ufms
sequencias 16S rRNA, no inıcio deste capıtulo, construımos matrizes de distanciasentre as especies para cada uma das medidas e inferimos as filogenias utilizando oalgoritmo de Neighbor-Joining, apresentado no Capıtulo 3, Secao 3.3.
E importante ressaltar que, embora o tamanho da aresta represente uma estimativapara a matriz de distancias, acreditamos que o principal fator na avaliacao dasmedidas e a capacidade de topologicamente unir as especies mais proximas. Assim,estamos mais interessados na topologia da arvore do que no tamanho das arestas,ja que uma boa medida de distancia deveria, pelo menos, levar a uma arvore cujatopologia concorda com a arvore verdadeira.
A figura 5.9 mostra a arvore T2. As arvores T3, T4 e T5 tem a mesma topologia,que concorda com a topologia da arvore 16S rRNA. A arvore T1 e mostrada nafigura 5.8. A mesma topologia foi obtida para T6. Embora as arvores T1 e T6
tenham unido os pares de especies proximos, a topologia encontrada e ligeiramentediferente da topologia obtida na arvore 16S rRNA (Figura 5.6).
ST
PASL
EC
SM
SN
Figura 5.8: Arvore T1, cuja matriz de distancias e baseada no numero dematches encontrados entre os pares de genomas.
ST
PA
SL
EC
SM
SN
Figura 5.9: Arvore T2, cuja matriz de distancias e baseada no numero deBBHs encontrados entre os pares de genomas.
O valor da medida de distancia entre duas arvores Ti e Tj proposta na Secao 5.1nos da uma estimativa da qualidade das arvores obtidas. Ao realizarmos o calculodessa medida para as arvore obtidas, verificamos que δ(Ti, Tj) = 0 para Ti = T16S e
57

5.3. Filogenia baseada em caracterısticas dct-ufms
Tj , onde 2 ≤ j ≤ 5. Ou seja, as arvores obtidas sao realmente mais verdadeiras. Naverdade, elas sao isomorfas. Ja para j = 1 e j = 6, o valor obtido foi δ(Ti, Tj) = 8,que embora sendo um valor diferente de zero, sao valores baixos, e ainda nos revelaque as duas medidas D1 e D6 sao boas estimativas de distancias entre os genomas.
A afirmacao acima, de que D1 e D6 podem ser consideradas verdadeiras, pode serconfirmada pelo fato de que, ao construirmos 100 arvores aleatorias com as mesmasespecies, obtivemos uma distancia media de 20.15 para a arvore 16S rRNA. Noteque a maior distancia esta abaixo da media menos tres desvios-padrao, dado queo desvio padrao e de 4.15.
Como pudemos observar, tres medidas de distancias baseadas no conteudo genicodos genomas foram capazes de projetar uma topologia de arvore desejada, o quenos da bons indıcios sobre a inferencia filogenetica pela comparacao de proteo-mas baseada em conteudo genico. Outra medida de distancia, baseada em regioesortologas tambem obteve sucesso. Essa medida e baseada em numero de matchespor regiao, e a arvore obtida a partir dela tambem e identica a arvore 16S rRNA.Ja a medida baseada no numero de BBHs por regiao obteve topologia ligeiramentediferente da arvore 16S rRNA. Isto faz sentido, ja que podemos detectar, especial-mente entre genomas relativamente proximos, um numero expressivo de pequenasregioes com um grande numero de matches, mas com um pequeno numero de BBHsdentro delas. Alem disso, muitos dos BBHs isolados (fora das regioes ortologas)foram detectados e esses BBHs nao foram utilizados nesse tipo de medida.
Embora a arvore baseada no numero de BBHs por regiao nao tenha sido muito boa,nao podemos simplesmente descartar esse tipo de medida, pois o EGG trabalhade forma que as regioes ortologas sao encontradas apenas pela uniao de matchesproximos, sem qualquer outro criterio. Isso pode fazer com que algumas pequenasregioes sejam encontradas ao acaso. Essa pode ser uma outra razao porque onumero de matches dentro de uma regiao produz boas arvores.
Os resultados apresentados ate aqui ja foram publicados no I Workshop Brasileirode Bioinformatica [4], realizado em Gramado, em outubro de 2002.
5.3 Filogenia baseada em caracterısticas
A inferencia de filogenias tambem pode ser realizada pelas caracterısticas dos ge-nomas das especies envolvidas. Com isso, podemos utilizar dados relacionados adescricao fısica da organizacao dos genomas. Pode-se levar em conta as posicoesrelativas dos genes no genoma, o sentido de transcricao dos genes ou mesmo apresenca ou ausencia dos genes nos genomas.
O proposito desta abordagem e comparar os genomas globalmente ao inves de pa-res. A comparacao nao e baseada na observacao de rearranjos entre duas especies,
58

5.3. Filogenia baseada em caracterısticas dct-ufms
mas numa descricao fısica de todo genoma.
A morfologia dos genomas engloba, entre outras caracterısticas, a forma (linearou circular), o tamanho da molecula de DNA na presenca de estruturas particu-lares. Por estrutura entendemos qualquer parte distinta do genoma como genes,sequencias repetidas, etc. Um genoma e caracterizado pela distribuicao dessasestruturas. Cada uma destas estruturas tambem possui um conjunto de carac-terısticas, que podem ser: sua funcao, seu tamanho, sua sequencia de nucleotıdeos,seu sentido de transcricao, sua posicao no genoma e assim por diante.
Gallut et al [22] utilizaram duas abordagens de posicao de genes como carac-terısticas na inferencia de filogenias. A primeira e chamada de juncao. Umajuncao de gene e o contato de dois genes contıguos. Toda juncao encontradaem uma especie e vista como uma caracterıstica binaria em termos de pre-senca/ausencia. Ja na segunda abordagem, a caracterıstica considerada e a posicaorelativa de cada gene presente em pelo menos uma das especies. A posicao relativade um gene B em um genoma, e caracterizado pelos dois genes ao seu redor. Osgrupos de genes que rodeam o gene B nos genomas envolvidos, constituem os dife-rentes estados da caracterıstica “posicao relativa do gene B”, que e tratada comouma caracterıstica de multiplos estados.
Com o objetivo de realizar uma analise mais adequada, as caracterısticas e es-truturas apresentadas acima podem ser consideradas, desde que as caracterısticasdo genoma sejam postuladas como homologas, conforme apresentado por Gal-lut et al [22].
Com o objetivo de inferir filogenia utilizando caracterısticas sobre a conservacaona ordem dos genes, nos detemos a utilizar somente as caracterısticas: pre-senca/ausencia de genes e presenca/ausencia de regioes. Para tal tarefa, criamosduas matrizes binarias. A primeira e uma matriz de presenca/ausencia de genesnos genomas que queremos inferir uma filogenia e a segunda e uma matriz depresenca/ausencia de regioes nos genomas. Tais regioes foram apresentadas naSecao 5.2. Para explicar tais matrizes, considere G = G1, G2, . . . , Gn o conjuntodos genomas das especies que queremos inferir uma filogenia.
Presenca/Ausencia de GenesA matriz C de presenca/ausencia de genes, foi obtida da seguinte forma: dadosdois genomas Gi e Gj quaisquer, realizamos a comparacao dos dois utilizando oprograma EGG, que gera, entre outros arquivos, um arquivo com extensao .set.Este arquivo possui todos os pares de BBHs entre dois genomas Gi e Gj . Com isso,podemos obter todos os genes e seus respectivos BBHs dos genomas pertencentesa G.
Inicialmente, cada gene individualmente e representante de uma famılia. Dadostres genes distintos ga, gb e gc, dois genes passam a fazer parte da mesma famıliada seguinte forma:
59

5.3. Filogenia baseada em caracterısticas dct-ufms
• se ga e gb formam BBH, entao gb fara parte da famılia de ga;
• se gb faz parte da famılia de ga, e gb forma um BBH com gc, entao gc tambemfara parte da famılia de ga e gb.
Fazemos isso para todos os pares de genes que formam BBHs, obtidos do arquivo.set, para cada par de genomas pertencente a G. Logo, obtemos um conjuntode famılias F =< f1, f2, ..., fk >, tal que |fj| ≥ 1. Cada famılia representa umacoluna da matriz binaria C e cada linha representa um genoma. Seja fj umafamılia e seja gr um gene tal que gr ∈ fj. A matriz C obtida, possui ordem n × ke e preenchida da seguinte forma:
C[i, j] = 1 se gr ∈ fj e gr ∈ Gi para i = 1 . . . npara j = 1 . . . |F |para r = 1 . . . |fj|.
Em outras palavras, C[i, j] = 1 se, e somente se, o genoma Gi contem algum geneda famılia fj. Cada famılia constitui um potencial conjunto de genes ortologos.
Presenca/Ausencia de RegioesA matriz de presenca/ausencia de regioes C ′, foi obtida da seguinte forma: dadosdois genomas Gi e Gj, realizamos a comparacao dos dois utilizando o programaEGG, que gera, entre outros arquivos, um arquivo com extensao .mul. Estearquivo possui informacoes sobre todas as regioes ortologas encontradas entre osdois genomas Gi e Gj .
No arquivo temos inicialmente a quantidade de regioes ortologas encontradaspelo EGG entre os dois genomas comparados. Daı em diante, temos as variasregioes encontradas, cujas informacoes estao dispostas em linhas da seguinte forma:nome da regiao (padronizado), seguido dos intervalos de genes que compoem aregiao no genoma 1 e no genoma 2, respectivamente. Depois temos a quantidadede matches encontrados nesta regiao. Por fim, temos a lista dos matches e BBHsna regiao, formado pelo numero do gene no genoma 1 e pelo numero do gene nogenoma 2 que formam match. Um terceiro numero binario ao lado dos matches,indica se este casamento e um match (0) ou BBH (1).
Toda regiao e identificada por quatro numeros (i1, f1, i2, f2), que representam seuinıcio e fim em Gi(i1,f1) e seu inıcio e fim em Gj(i2, f2). Inicialmente, cadaregiao formada entre dois genomas representa uma famılia. Dadas duas regioesri(i1, f1, i2, f2) e rj(i
′1, f
′1, i
′2, f
′2), dizemos que ri se sobrepoe a rj se, e somente se,
ri e uma regiao formada entre genomas Gx(i1, f1) e Gy(i2, f2) tal que um delestambem forma rj . Suponha que Gx tambem forma rj em (i′1, f
′1), entao i1 ≤ i′1 ≤ f1
ou i′1 ≤ i1 ≤ f ′1. A mesma ideia e aplicada se supusermos que Gy forma rj, entao
temos que i2 ≤ i′1 ≤ f2 ou i′1 ≤ i2 ≤ f ′1.
Alem disso, duas regioes passam a fazer parte da mesma famılia nos seguintescasos:
60

5.3. Filogenia baseada em caracterısticas dct-ufms
• se r1 e r2 se sobrepoem, entao r2 fara parte da famılia de r1; ou
• se r2 faz parte da famılia de r1, e r2 se sobrepoe a r3, entao r3 tambem faraparte da famılia de r1.
As matrizes binarias C e C ′ foram utilizadas como entrada para o programa Mix,incluıdo no pacote Phylip [20], utilizado para construir filogenias baseadas em ma-trizes de caracterısticas. O programa ja foi apresentado no Capıtulo 4, Secao 4.4.
5.3.1 Resultados
Nesta secao vamos mostrar as arvores filogeneticas obtidas a partir da aplicacaodas duas caracterısticas apresentadas na secao anterior.
Para testar essas medidas construımos as matrizes C e C ′ para o conjunto de seisgenomas, apresentado na Secao 5.2 e fornecemos como entrada para o programaMix, que forneceu as filogenias apresentadas nas Figuras 5.10 e 5.11, respectiva-mente.
ST
PA
SL
EC
SM
SN
Figura 5.10: Filogenia obtida a partir da caracterıstica presenca/ausencia degenes.
ST
PA
SL
EC
SM
SN
Figura 5.11: Filogenia obtida a partir da caracterıstica presenca/ausencia deregioes.
61

5.3. Filogenia baseada em caracterısticas dct-ufms
Observe que as arvores obtidas nao tem a mesma topologia da arvore 16S rRNA,mas tambem nao podem ser desprezadas visto que conseguiram identificar os paresde genomas proximos. Alem disso, o valor da medida de distancia entre a arvoreverdadeira e a arvore baseada em genes foi δ = 5. Ja a comparacao com a arvore emfuncao das regioes o valor obtido foi δ = 12. Logo, a arvore baseada em genes foi aque obteve menor distancia, o que nos leva a crer novamente que o conteudo genicodos genomas sao estruturas significativas para determinar a historia evolutiva deespecies. E importante ressaltar que a arvore baseada em regioes tambem e umaboa estimativa de filogenia, mesmo obtendo um valor δ maior que zero, tambemconseguiu uma arvore que unisse as especies proximas.
A afirmacao acima, de que as filogenias obtidas a partir dos genes e das regioes po-dem ser consideradas boas, pode ser confirmada pelo fato de que, ao construirmos100 arvores aleatorias com as mesmas especies, obtivemos uma distancia media de20.15 para a arvore 16S rRNA. Note que a maior distancia esta abaixo da mediamenos dois desvios-padrao, dado que o desvio pardrao e de 4.15.
E evidente que os valores obtidos para δ nao sao os mais desejaveis, pois saomaiores que zero. Entretanto, as arvores produzidas conseguiram unir as especiesmais proximas. Alem disso, um dos valores de δ obtido nessa abordagem e menorque os valores obtidos na abordagem de construcao de filogenias baseadas emdistancias, o que nos permite enfatizar que essas caracterısticas tambem podemser bons instrumentos para inferencia filogenetica.
62

Capıtulo 6
Consideracoes finais
Este trabalho tem como principal resultado uma metodologia para inferir filogeniasde genomas completos, a partir do conteudo genico e tambem da conservacao daordem dos genes.
A metodologia proposta para construcao de filogenias utiliza como principal fer-ramenta o programa EGG. Propomos entao, medidas de distancias para inferirfilogenias utilizando as estruturas geradas pelo EGG, verificando quais delas po-dem ser utilizadas no processo de inferencia e que podem resultar em filogeniassignificativas.
Podemos destacar que no processo de construcao de filogenias baseadas emdistancias, dentre as estruturas geradas por EGG, os matches, BBHs e regioesortologas sao bons instrumentos que podem ser utilizados para inferir a historiaevolutiva das especies. No processo de construcao de filogenias baseadas em carac-terısticas podemos destacar que as caracterısticas baseadas em genes e em regioestambem produzem filogenias de genomas significativas.
Com este trabalho, temos uma forma de inferir relacoes de ancestralidade de umconjunto de especies a partir de seus genomas, permitindo ainda que possamosavaliar as arvore construıdas, visto que nossa metodologia inclui a descricao deuma medida de distancia entre arvores, que foi criada para avaliar a qualidade dasarvores obtidas.
Nosso trabalho resultou em uma publicacao no I Workshop Brasileiro de Bioin-formatica [4].
63

dct-ufms
Trabalhos futuros
Muitas aspectos deste trabalho podem ser melhorados e acrecentados aos resulta-dos obtidos. A seguir, listamos algumas das possibilidades.
Interface Grafica
As filogenias construıdas em nosso trabalho sao baseadas nos resultados fornecidospelo egg, que geralmente estao no formato de arquivo texto. Com base nessesarquivos, construımos filogenias utilizando matches, BBHs e regioes. Podemosentao construir uma interface grafica que possibilite ao usuario a visualizacao defilogenias de genomas, que serao escolhidos por ele, bem como permita a escolhada medida de distancia ou da caracterıstica a ser utilizada na construcao dasfilogenias. Ou seja, criar uma interface semelhante a ferramenta SHOT [29].
Disponibilizacao na Web
Outro importante trabalho e a disponibilizacao da interface mencionada acima pelaWeb, para que outras pessoas possam usufruir dos resultados dessa metodologia.
Outras medidas
Como vimos, alguns dos numeros retornados por egg podem ser utilizados comopossıveis medidas de distancias entre os genomas comparados, como por exem-plo, numero de matches e BBHs, regioes ortologas etc. Entretanto, alem des-sas medidas, podemos envolver tambem medidas que indiquem as funcionalidadescompartilhadas entre os genomas.
Outras possıveis caracterısticas a serem utilizadas sao aquelas que envolvem as ca-tegorias dos genes, COG (Cluster of Orthologous Groups of Proteins), por exemplo.Nesse caso, terıamos uma matriz binaria onde cada linha da matriz representa umgenoma e cada coluna representa uma COG, sendo que um genoma pode possuirou nao cada uma das COGs.
64

Apendice A
DNADIST
O programa DnaDist e um dos programas encontrados no pacote de inferencia fi-logenetica Phylip [20]. DnaDist e utilizado para calcular distancias entre especiesa partir de suas sequencias de nucleotıdeos.
A distancia para cada par de especies estima o tamanho da ramificacao total entreas duas especies e pode ser utilizada para montar uma matriz de distancias, queserve de entrada para programas que constroem filogenias baseado em distancias,como Fitch, Kitsch e Neighbor, encontrados tambem no pacote Phylip.
A distancia e calculada de acordo com as mutacoes que ocorrem em cada posicaodas sequencias dos genomas, que sao notadas quando as comparamos entre si.
As mutacoes envolvem substituicoes de uma base para outra. Uma transicaoe uma substituicao de uma base purina (A ou G) para outra base purina ou deuma base pirimidina (C ou T) para outra base pirimidina. Os outros tipos desubstituicoes sao chamados de transversoes. Em geral, as substituicoes sao maisfrequentes entre bases que sao, de acordo com a bioquımica, mais similares. Dessaforma, as transicoes (A→G, G→A, C→T, T→C) sao mais frequentes que as trans-versoes (C→A, A→T, C→G, G→T, e o reverso), de acordo com Merriwether [33].
Para calcular a matriz de distancias, a partir das sequencias de nucleotıdeos, oprograma dispoe de tres modelos diferentes de substituicao de nucleotıdeos. Os tresmodelos sao: o modelo de Jukes-Cantor [26], o modelo Kimura 2-parameter [27],o modelo de maximum likelihood (ML) que e devido a Kishino e Hasegawa [28], eo modelo de Jin e Nei [25].
Modelo de Jukes-Cantor
Este modelo assume que as mudancas ocorrem independentemente entre todas asposicoes do DNA, com igual probabilidade. Uma mudanca de base e independentede sua identidade, e quando ela muda, ha uma probabilidade igual de finalizar com
65

dct-ufms
cada uma das outras tres bases. A Figura 7.1 mostra a matriz de probabilidadede mudanca entre as bases, conforme Felsenstein [18].
A G C TA 1 − 3a a a a
G a 1 − 3a a a
C a a 1 − 3a a
T a a a 1 − 3a
Figura 7.1: Matriz de probabilidade de substituicao entre bases.
onde a e udt, que e o produto da taxa de substituicao por unidade de tempo (u)e o tamanho (dt) do intervalo de tempo.
Podemos estimar o tamanho da ramificacao u′ entre as especies, pela formulaabaixo:
u′ = −3
4loge(1 −
4
3p),
onde p e a proporcao de nucleotıdeos que sao diferentes entre quaisquer duassequencias.
Modelo de Kimura
O modelo de Kimura 2-parameter atribui pesos diferentes para transicoes e trans-versoes. A seguir, na Figura 7.2, temos uma matriz de probabilidade de substi-tuicao para este modelo, conforme Felsenstein [18].
A G C TA 1 − a− 2b a b b
G a 1 − a− 2b b b
C b b 1 − a− 2b a
T b b a 1 − a− 2b
Figura 7.2: Matriz de probabilidade de substituicao para o modelo de Ki-mura.
Na matriz, a e udt, o produto da taxa de transicao por unidade de tempo e dte o tamanho do intervalo de tempo, e b e vdt, o produto da metade da taxa detransversao e o tamanho dt do intervalo de tempo.
Sejam P e Q respectivamente, as fracoes de locais que mostram diferencas dotipo transicao e transversao entre duas sequencias comparadas. Entao, a distancia
66

dct-ufms
evolutiva para cada par de sequencias, segundo Naylor e Eulenstein [35], e dadapor:
1
2ln(
1
1 − 2P − Q) +
1
4ln(
1
1 − 2Q).
Maximum Likelihood
Neste modelo de substituicao de base, as frequencias esperadas para as quatrobases podem ser diferentes, assim como as frequencias esperadas de transicoes etransversoes podem ser diferentes.
O modelo possui as seguintes hipoteses: 1) cada posicao na sequencia evolui in-dependentemente; 2) diferentes linhagens evoluem independentemente; 3) cadaposicao submete-se a uma substituicao em uma taxa esperada, que e escolhida deuma serie de taxas que especificamos; 4) todas as posicoes relevantes sao inclusasna sequencia, nao apenas aquelas que tem mudancas e que sao filogeneticamenteinformativas.
Uma substituicao consiste de dois tipos de eventos:
• substituicao da base existente (purina ou pirimidina) por uma base extraıdade um conjunto de purinas ou de um conjunto de pirimidinas, respectiva-mente. Isto pode levar a uma transicao ou a nenhuma mudanca.
• substituicao da base existente por uma base extraıda aleatoriamente de umconjunto de bases de frequencias conhecidas, independentemente da identi-dade da base que esta sendo substituıda. Isto pode levar a uma transicao, auma transversao ou a nenhuma mudanca.
Em um evento do tipo I, a base A, por exemplo, do tipo purina e substituıda poroutra base A com probabilidade πA/(πA + πG) e por um G com probabilidadeπG/(πA + πG). O mesmo ocorre para uma base do tipo pirimidina. Um evento dotipo II substitui uma base por uma outra de um conjunto de todas as bases possıveiscom probabilidade identica a sua frequencia. Assim, uma base A, por exemplo, esubstituıda pela base A com probabilidade πA, pela base C com probabilidade πC ,pela base G com probabilidade πG e pela base T com probabilidade πT .
Exemplo 7.1 (Felsenstein e Churchil, 1996) Suponha que as frequencias das ba-ses sejam πA = 0.24, πC = 0.28, πG = 0.27 e πT = 0.21.
Evento I:
• determine se a base a ser substituıda e uma purina ou pirimidina;
• substitua a base a partir do conjunto apropriado:
67

dct-ufms
0.4706 A0.5294 G
0.5714 C0.4286 T
Evento II: substitua a base a partir do conjunto completo.
0.24 A0.28 C0.27 G0.21 T
A matriz de mudancas para este modelo, segundo Kishino e Hasegawa [28] e mos-trada em duas partes, nas Figuras 7.3 e 7.4. Os nucleotıdeos estao na ordem A,G, C, T. O termo k define o relacionamento entre transicoes e transversoes e fi ea frequencia da base i.
A GA −[k/(fA + fG) + 3]bfA [k/(fA + fG) + 1]bfA
G [k/(fA + fG) + 1]bfG −[k/(fA + fG) + 3]bfG
C bfC bfC
T bfT bfT
Figura 7.3: Matriz de probabilidade de substituicao.
C TA bfA bfA
G bfG bfG
C −[k/(fC + fT ) + 3]bfC [k/(fC + fT ) + 1]bfC
T [k/(fC + fT ) + 1]bfT −[k/(fC + fT ) + 3]bfT
Figura 7.4: Matriz de probabilidade de substituicao.
Vamos analisar agora o relacionamento das sequencias sobre este modelo. Comduas sequencias, a arvore e trivial: e uma unica aresta de tamanho s que asune. O objetivo e encontrar o tamanho da aresta. Esta tarefa requer encontrar aprobabilidade de no final de uma distancia evolutiva d, a primeira sequencia estejaconvertida exatamente na segunda sequencia; s e entao o valor de d na qual a
68

dct-ufms
probabilidade e maximizada. Se xij e a identidade do nucleotıdeo j na sequenciai, entao
L(x1j , x2j ; d) = fx1jMx1j,x2j(d) (7.1)
onde L(v, w; d) e a probabilidade de observarmos o nucleotıdeo v na primeirasequencia e w na segunda sequencia, sobre a condicao que as sequencias estaoseparadas por uma distancia d. De acordo com Swofford e Olsen [43], a Equacao 7.1e utilizada para obter a estimativa maximum likelihood das distancias entre ospares de sequencias.
Modelo de Jin/Nei
Esse modelo assume que a taxa de substituicao varia de posicao para posicao deacordo com um coeficiente de variacao que e especificado pelo usuario. Alem disso,ele tambem usa taxas de transicao e transversao. A estimativa de distancia entreos pares de especies e calculada pela equacao abaixo:
0.5a((1 − 2P − Q)−1/a + 0.5(1 − 2Q)−1/a − 3/2),
onde
L = substituicao media = taxa transicao + 2 ∗ taxa transversao
a = (media L)2/(variacao de L)
P = transicoes/npos
Q = transversoes/npos,
e npos e o numero de posicoes pontuadas.
Segundo Carver [11], Jin e Nei [25], sugerem em geral, que a distancia seja calculadacom o valor a = 1. Entretanto, o usuario pode especificar seu proprio valor.
69

Referencias Bibliograficas
[1] N.F. Almeida. Tools for Genome Comparison. PhD thesis, IC-UNICAMP,2002.
[2] S.F. Altschul, W. Gish, W. Miller, E.W. Myers, and D.J. Lipman. A basiclocal alignment search tool. Journal of Molecular Biology, 215:403–410, 1990.
[3] S.F. Altschul, T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller,and D.J. Lipman. Gapped blast and psi-blast: a new generation of proteindatabase search programs. Nucleic Acid Research, 25:3389–3402, 1997.
[4] G.S. Araujo and N.F. Almeida. Phylogeny from whole genome comparison.In Proceedings of the 1st Brazilian Workshop on Bioinformatics, pages 9–15.Gramado-RS, Brazil, October 18, 2002.
[5] K. Atteson. The performance of the Neighbor-Joining method of phylogenyreconstruction. In COCOON, pages 101–110, 1997.
[6] D. Benson, I. Karsch-Mizrachi, D. Lipman, J. Ostell, B. Rapp, and D. Whe-eler. Genbank. Nucleic Acids Res., 28(1):15–18, 2000.
[7] A. Bernal, U. Ear, and N. Kyrpides. Genomes online database (GOLD): amonitor of genome projects world-wide. Nucleic Acids Research, 29(2):126–127, 2001.
[8] J.A. Bondy and U.S.R. Murty. Graph Theory with Applications. North-Holland, 1976.
[9] J.K.M. Brown. Bootstrap hypothesis tests for evolutionary trees and otherdendrograms. In Proceedings of the National Academy of Sciences of theUnited States of America, volume 91, pages 12293–12297, December 1994.
[10] M.H. Carvalho and N.F. Almeida. Introducao a teoria dos grafos. Notas deAula, maio 2001.
[11] T. Carver. Emboss - distmat. texto, March 2001. (web site:http://www.hgmp.mrc.ac.uk/Software/EMBOSS/Apps/distmat.html).
70

Referencias Bibliograficas dct-ufms
[12] M.E. Cosner, R. Jansen, B. Moret, L. Raubeson, L. Wang, and S. Wyman. Anew fast heuristic for computing the breakpoint phylogeny and experimentalanalyses of real and synthetic data. In Proceedings of the 8th InternatinalConference on Intelligent Systems for Molecular Biology, pages 104–115, LaJolla, USA, 2000.
[13] W.E. Day, D.S. Johnson, and D. Sankoff. The computational complexity ofinferring rooted phylogenies by parsimony. Mathematical Biosciences, 81:33–42, 1986.
[14] W.E. Day and D. Sankoff. Computational complexity of inferring phylogeniesby compatibility. Systematic Zoology, 2(35):224–229, 1986.
[15] R. Diestel. Graph Theory. Springer, 1997.
[16] A. Dress, K. T. Huber, and V. Moulton. Metric spaces in pure anda ap-plied mathematics. In Documenta Mathematica, Special Volume ProceedingsQuadratic Forms LSU, pages 121–139, La Jolla, USA, 2001.
[17] M. Farach, S. Kannan, and T. Warnow. A robust model for finding optimalevolutionary trees. Algorithmica, 13:155–179, 1995.
[18] J. Felsenstein. DNADIST, a program to compute dis-tance matrix from nucleotide sequences. (web site:http://evolution.genetics.washington.edu/phylip/doc/dnadist.html).
[19] J. Felsenstein. MIX - Mixed method discrete characters parsimony. (web site:http://evolution.genetics.washington.edu/phylip/doc/mix.html).
[20] J. Felsenstein. Phylip (phylogeny inference package) version 3.5c. Distributedby the author, 1993. Department of Genetics, University of Washington,Seattle.
[21] J. Felsenstein. Phylogeny i: Parsimony and tree space. Lecture Note, 1998.
[22] C. Gallut, V. Barriel, and R. Vignes. Gene order and phylogenetic informa-tion. In D. Sankoff and J.H. Nadeau, editors, Comparative Genomics, pages123–132. Kluwer Academic Publishers, 2000.
[23] D. Gusfield. Efficient algorithms for inferring evolutionary trees. Networks,21:19–28, 1991.
[24] D. Gusfield. Algorithms on Strings, Trees, and Sequences. Computer Scienceand Computational Biology. Cambridge University Press, 1997.
[25] L. Jin and M. Nei. Limitations of the evolutionary parsimony method ofphylogenetic analysis. Molecular Biology and Evolution, 7:82–102, 1990.
71

Referencias Bibliograficas dct-ufms
[26] T.H. Jukes and C.R. Cantor. Evolution of protein molecules. Algorithmica,13:123–132, 1969.
[27] M. Kimura. A simple model for estimating evolutionary rates of base subs-titutions through comparative studies of nucleotide sequences. Journal ofMolecular Evolution, 16:111–120, 1980.
[28] H. Kishino and M. Hasegawa. Evaluation of the maximum likelihood esti-mate of the evolutionary tree topologies from DNA sequence data, and thebranching order in hominoidea. Journal of Molecular Evolution, 29:170–179,1989.
[29] J.O. Korbel, B. Snel, M.A. Huynen, and P. Bork. Shot: a web server forthe construction of genome phylogenies. Trends in Genetics, 18(3):158–162,March 2002.
[30] S. Kumar, K. Tamura, I. B.Jakobsen, and M. Nei. Mega2: Molecular evolu-tionary genetics analysis software. Bioinformatics, 17(12):1244–1245, 2001.
[31] N. Kyrpides. Genomes online database (GOLD): a monitor of complete andongoing genome projects world wide. Bioinformatics, 15(2):773–774, 1999.
[32] W. Ludwig and K. Schleifer. Phylogeny of bacteria beyond the 16S rRNAstandard. ASM News, 1999.
[33] D.A. Merriwether. Phylogenetics: Tree Building. Lecture Notes -University of Washington, October 1996. (web site: http://www-personal.umich.edu/andym/MolPhylo.htm).
[34] L. Nakhleh, B.M. Moret, U. Roshan, K.St. John, J. Sun, and T. Warnow.The accuracy of phylogenetic methods for large datasets. In Proceedings ofFifth Pacific Symposium of Biocomputing (PSB’02), pages 211–222, Hawaii,USA, 2002.
[35] G. Naylor and O. Eulenstein. Computation Phylogenetics. Lectu-re Notes - University of Washington, January 2002. (web site:http://www.cs.iastate.edu/cs550/lectures/Dist lectB&W.pdf).
[36] M. Nei and N. Saitou. The Neighbor-Joining method: A new method forreconstructing phylogenetic trees. Molecular Biology Evolution, 4(4):406–425,1987.
[37] A.N. Patrinos and S.L. Hakimi. The distance matrix of a graph and its treerealization. Quarterly of apllied mathematics, pages 255–269, October 1972.
[38] A. Rzhetsky and M. Nei. A simple method for estimating and testingminimum-evolution trees. Molecular Biology Evolution, 9:945–967, 1992.
72

Referencias Bibliograficas dct-ufms
[39] N. Saitou and T. Imanishi. Relative efficiencies of the fitch-margoliash, maximum-parsimony, maximum likelihood, minimum-evolutionand neighbor-joining methods of phylogenetic tree construction in obtainingthe correct tree. Molecular Biology Evolution, 6:514–525, 1989.
[40] J.C. Setubal and J. Meidanis. Introduction to Computational Molecular Bio-logy. PWS Publishing Co., 1997.
[41] R. Shamir. Algorithms for molecular biology. Lecture Note, 2001.
[42] M.V. Souza, F.A. Torres, C.A. Ricart, W. Fontes, and M.A. Silva. Gestao davida?: Genoma e Pos-genoma. Bluhm - UnB, 2001.
[43] D.L. Swofford and G.L. Olsen. Phylogeny Reconstruction Molecular Systema-tics (Hillis and Moritz eds). Sinauer Associates, Sunderland, Massachusetts,U.S.A., 1990.
[44] Y. Tateno, N. Takezaki, and M. Neiji. Relative efficiencies of the maximum-likelihood, neighbor-joining, and maximum-parsimony methods when subs-titution rate varies with site. Molecular Biology Evolution, 11(2):261–277,1994.
[45] L. Wang, R. Jansen, B. Moret, L. Raubeson, and T. Warnow. Fast phy-logenetic methods for genome rearrangement evolution: an empirical study.In Proceedings of Fifth Pacific Symposium of Biocomputing (PSB’02), pages524–535, Hawaii, USA, 2002.
[46] M.S. Waterman, T.F. Smith, M. Singh, and W.A. Beyer. Additive evolutio-nary trees. Journal of Theoretical Biology, pages 199–213, 1977.
73