nadh/nadph oxidase and vascular function
TRANSCRIPT

Thompson CB: 1995. Apoptosis in the patho-genesis and treatment of disease. Science267:1456-1462.
TroyCM, StefanisL, GreeneLA, ShelanskiML:1997. Nedd2 is required for apoptosis aftertrophic factor withdrawal, but not superox-ide dismutase (SOD1) downregulation, insympathetic neurons and PC12 cells. J Neu-rosci 17:1911–1918.
Vaux DL, Aguila HL, Weissman IL: 1992. Bc1-2prevents death of factor-deprived cells butfails to prevent apoptosis in targets of cellmediated killing. Int Immunol 4:821–824.
Vaux DL: 1997. CED-4—thethird horseman ofapoptosis. Cell 90:389–390.
Verheij M, Bose R, Lin XH, et al.: 1996. Re-quirement for ceramide-initiated signalingin stress-induced apoptosis. Nature 380:75–79.
Wang H-G, Rapp UR, Reed JC: 1996. Bc1-2tar-gets theprotein kinaseraf-1 to mitochondria.Cell 87:629+38.
Wang S, Miura M, Jung Y, et a].: 1996. identifi-cation and characterizationof Ich-3, a mem-ber of the interleukin-1~converting enzyme(ICE)/ced-3 family and an upstream regula-tor of ICE. J Biol Chem 271:20,580–20,587.
Wilson DJ, FortnerKA, Lynch DH, et al,: 1996.JNK, but not MAPK, activation is associatedwith FAS-mediated apoptosis in human Tcells. Eur J Immunol 26:989–994.
Wy]]ie AH, Kerr JFR, Currie AR: 1980. Celldeath: the significance of apoptosis. Int RevCytol 68:251-306.
Xia Z, Dickens M, Raingeaud J, Davis RJ,Greenberg ME: 1995. Opposing effects ofERK and JNK-P38 MAP kinases on apopto-sis. Science 270:1326–1331.
Xue D, Shaham S, Horvitz HR: 1996. The Cae-norhabditiselegans cell-death protein CED-3is a cysteineprotease with substratespecific-ities similar to those of the human CPP32protease. Genes Dev 10:1073-1083.
YuanJ, Shaham S, Ledoux S, EllisHM,HorvitzHB: 1993.The C. elegans cell death geneced-3 encodes a protein similar to mamma-lian interleukin-1~-convertingenzyme. Cell75:641–652.
Zha J, Harada H, Yang E, Jockel J, KorsmeyerSJ: 1996. Serine phosphorylation of deathagonist BAD in response to survival factorresults in binding to 14-3-3- not bcl-xL. Cell87:619-628.
Zhang J, AfterN, Reed JC,Borner C, Obeid LM,Hannun YA: 1996. Bc1-2interruptsthe cera-mide-mediated pathway of cell death. ProcNatl Acad Sci USA 93:5325-5328.
Zou H, Henze] WJ, Liu X, et af.: 1997. Apaf-1, ahuman protein homologous to C. elegansCED-4, participates in cytochrome c-depen-dent activation of caspase-3. Cell90:405-$13.
TCM
NV F
Kathy K. Griendling and Masuko Ushio-Fukai
T v a sN A Do xh b s t b t ms oo s u pi t v ew R w h pi n si ni s t ra a ci v ac T e ii n vi b v a ss mm h ya i sf oo i m pe n d o t hr eB ot is ti g ep a ri t p ao h ya a t h e r ot e nt p rr eo sm b i m pd e t eo t c o v d( T rC a rM 1 97 : 30 E S I
Oxidativestresshasbeen the subjectof avast amount of research over the years,initially because of its propensity to in-duce DNA damage. It has now becomeapparent,however, that reactive oxygenspecies may act as intercellular and in-tracellular second messengers in bothnormal and pathophysiologic respon-siveness. In the vascular system, thesourcesof oxidative stresshave not beenclearly established. Recent work hasshown that the traditional sourcesof ox-idative molecules (xanthine oxidase,mi-tochondrial oxidases, arachidonic acid)play a relatively minor role in the pro-duction of reactive oxygen species in thevesselwall and thata nonmitochondrial,membrane-associated NADH/NADPHoxidase is the major source of superox-ide (.02–) in vascular cells (Griendling etal. 1994,Mohazzab-H and Wolin 1994b,Rajagopalan et al. 1996a).
. The NADI-UNADPHOxidaseas a Source of OxidantStressin VascularCells
Vascularcells are exposed to both para-crine and autocrine regulation by reac-
Kathy K. Griendling and Masuko Ushio-Fu-kai are at the Emory University School ofMedicine, Division of Cardiology, Atlanta, GA30322, USA.
tive oxygen species. Endothelial andsmooth muscle cells produce .02– andH202 (Friedl et al. 1989,Pagano et al.1993, Panus et al. 1993, Mohazzab-Hand Wolin 1994b, Rajagopalan et al.1996a),and are exposed to free radicalsreleased by circulating blood cells andinflammatory cells.Cell associated-reac-tive oxygen species appear to derivemainly from an NADH/NADPH oxidase(Mohazzab-H et al. 1994,Rajagopalan etal. 1996a). Acetylcholine-insensitive.02- generation from rabbit aorta wasnoted by Pagano et al. (1993). These in-vestigatorsconcluded that vascular .02–is derived mainly from a nonendothelialsourceand is modulated by endogenoussuperoxide dismutase. Subsequently,a.02--generating NADH oxidase was ob-served in pulmonary arteries by Mo-hazzab-H and Wolin (1994a). This mi-crosomal NADH-dependent productionof .02– is decreased by hypoxia and ap-parently utilizes a cytochrome b~~~elec-tron transport system (see later here).Simultaneously,our laboratory demon-strated (Griendling et al. 1994)that an-giotensin II increases NADH- andNADPH-dependent .02- production incultured vascular smooth muscle cells(VSMC). This activity is membrane as-sociated and is inhibited by diphenyleneiodinium (DPI), an inhibitor of flavin-containing enzymes (Figure la). Paganoet al. (1995) reported an NADPH-depen-
TCM vol. 7, No. 8, 1997 @ 1997,ElsevierScienceInc., 1050-1738/97/$17.00PIIS105O-1738(97)OOO88-1 301
—.-DPI +DPI
T
-DPI
**—
+DPI
+Angll
vector p22phox-antiaanaa
+Angll
——vector p22phox-
antisenaa
1
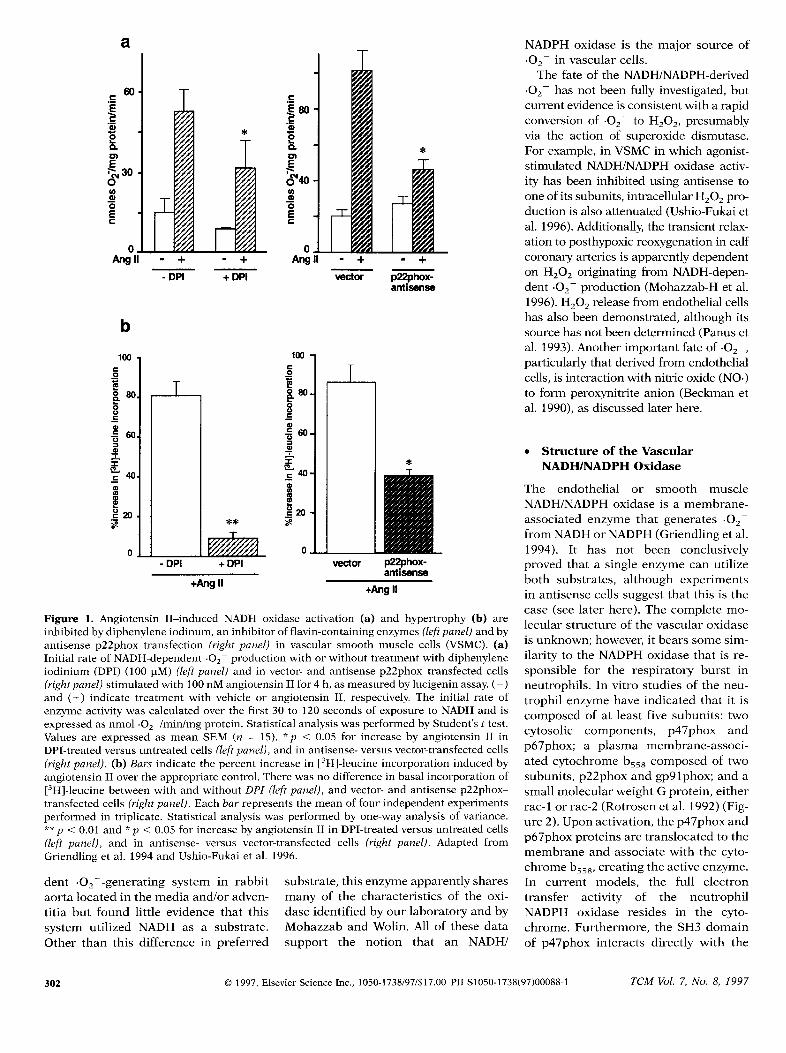
Figure 1. Angiotensin II–induced NADH oxidase activation (a) and hypertrophy (b) areinhibited by diphenylene iodinum, an inhibitor of flavin-containing enzymes (leftpanel) and byantisense p22phox transection (right panel) in vascular smooth muscle cells (VSMC). (a)Initial rate of NADH-dependent .02- production with or without treatment with diphenyleneiodinium (DPI) (100 pM) (left panel) and in vector- and antisense p22phox–transfected cells(rightpanel) stimulated with 100 nM angiotensin II for 4 h, as measured by lucigenin assay. (-)and (+) indicate treatment with vehicle or angiotensin II, respectively. The initial rate ofenzyme activity was calculated over the first 30 to 120 seconds of exposure to NADH and isexpressed as nmol O-/min/mg protein. Statistical analysis was performed by Student’sttest.Values are expressed as mean SEM (n = 15). “ p < 0.05 for increase by angiotensin II inDPI-treated versus untreated cells (leftpanel), and in antisense- versus vector-transfected cells(rightpanel). (b) Bars indicate the percent increase in [3H]-leucine incorporation induced byangiotensin II over the appropriate control. There was no difference in basal incorporation of[3H]-leucine between with and without DPI (left panel), and vector- and antisense p22phox-transfected cells (rightpanel). Each bar represents the mean of four independent experimentsperformed in triplicate. Statistical analysis was performed by one-way analysis of variance.**p <0.01 and *p <0.05 for increase by angiotensin II in DPI-treated versus untreated cells(left panel), and in antisense- versus vector-transfected cells (right panel). Adapted fromGriendling et al. 1994 and Ushio-Fukai et al. 1996.
dent O--generating system in rabbit substrate, this enzyme apparently sharesaorta located in the media andlor adven- many of the characteristics of the oxi-titia but found little evidence that this dase identified by our laboratory and bysystem utilized NADH as a substrate. Mohazzab and Wolin. All of these data
Other than this difference in preferred support the notion that an NADH/
NADPH oxidase is the major source of.02- in vascular cells.
The fate of the NADH/NADPH-derived.02- has not been fully investigated,butcurrentevidenceis consistentwith a rapidconversionof .02– to H202,presumablyvia the action of superoxide dismutase.For example,in VSMC in which agonist-stimulatedNADH/NADPHoxidase activ-ity has been inhibited using antisensetooneof itssubunits,intracellularH202pro-ductionis alsoattenuated(Ushio-Fukaietal. 1996).Additionally,the transientrelax-ationto posthypoxicreoxygenationin calfcoronaryarteriesis apparentlydependenton H202 originating from NADH-depen-dent .02- production(Mohazzab-H et al.1996).H202releasefi-omendothelialcellshas also been demonstrated,althoughitssourcehasnot beendetermined(Panusetal. 1993).Another importantfate of .02 ,particularlythatderived from endothelialcells,is interactionwith nitric oxide (NO.)to form peroxynitnte anion (Beckmanetal. 1990),as discussedlaterhere.
● Structureof the VascularNADWNADPH Oxidase
The endothelial or smooth muscleNADH/NADPH oxidase is a membrane-associated enzyme that generates .02from NADH or NADPH (Griendling et al.1994). It has not been conclusivelyproved that a single enzyme can utilizeboth substrates, although experimentsin antisensecells suggestthat this is thecase (see later here). The complete mo-lecular structure of the vascular oxidaseis unknown; however, it bears some sim-ilarity to the NADPH oxidase that is re-sponsible for the respiratory burst inneutrophils. In vitro studies of the neu-trophil enzyme have indicated that it iscomposed of at least five subunits: twocytosolic components, p47phox andp67phox; a plasma membrane-associ-ated cytochrome b~~~ composed of twosubunits, p22phox and gp91phox; and asmall molecular weight G protein, eitherrac-1 or rac-2 (Rotrosen et al. 1992) (Fig-ure 2). Upon activation, the p47phox andp67phox proteins are translocated to themembrane and associate with the cyto-chrome b~~~,creating the active enzyme.In current models, the full electrontransfer activity of the neutrophilNADPH oxidase resides in the cyto-chrome. Furthermore, the SH3 domainof p47phox interacts directly with the
01997, ElsevierScienceInc., 1050-1738/97/$17.00PIIS105O-1738(97)OOO88-1 TCA4vol. 7, /(0. 8, 1997

cytochrome bS5tl
:*h,pbx
active
Figure2. Molecular structureof the neutrophilNADPHoxidase. The molecular structureof theneutrophiloxidase has been clearlyestablished.The c,ytochromeb~~gresides in the membraneand consists of two subunits, p22phox andgp9tphox. In resting cells, the remaining reW-Iatmy subunits (p47phox, p67phcrx,rac2) [L-side in the cytosol. IJpon stimulation, p47phoxassociates with p22phox via an SH3 domaininteraction, and p67plmx associates withp47phox via a differentSH3 domain, Rac2 alsobecomes part of the active erwymc complex.
prolinc-rich region of p22phox (Leto etal. 1994), whereas the C-terminal SH3domain of p67phox associates with aprolinc-rich region of p47phox (Finan etal. 1994). Thus, the cytochrome mole-cule, and in particular the p22phuxsub-unit, is central to the normal functioningof the oxidase because it is responsiblefor electron transfer and serves a dock-ing function for the cytosolic factors.
Many of the characteristicsof the neu-trophil NADPH oxidase are shared bythe VSMC enzyme, most notably stimu-lation by phosphatidic and arachidonicacids, association with the membrane,and sensitivity to the flavin-containingenzyme inhibitor DPI (Griendling et al.1994).The VSMC oxidase differs, how-ever, from that of neutrophils in manyrespects. First and foremost, the timecourse of stimulation of the oxidase dif-fers dramatically in the two cell types.Superoxidegeneration by the neutrophilNADPH oxidase is massive and beginswithin secondsof exposureto hormones(Watson et al. 1991),whereas accumula-tion of .02- in agonist-stimulatedVSMC(about one-tenth the amount in phago-cytes) occursover a period of hours.Sec-ond, the .02– generated in VSMC ap-pears to be mostly intracellular, withonly a limited amount of .02 being re-leased to the exterior (Griendling et al.1994), as occurs in phagocytes. Third,the VSMC enzyme appears
TCM vol. 7, No. 8, 1997
to utilize
NADH to a greater extent than NADPH,which is exactly the opposite 0[ lhe sit-uation in neutrophils. Finally, the phys-iologic function of the .02– that accu-mulates is quite different from that ofphagocytic cells, perhaps serving a sig-naling function for control of vasculartone or growth (Griendling et al. 1994).
On a molecular level, the vascular oxi-dase may share only limited homologyto the neutrophil enzyme. Antibodyscreening and polymerase chain reac-tion (PCR) experiments have shown thatnot all the components of the neutrophiloxidase are expressed in vascular cells.In endothelial cells, gp91phox, p22phox,p47phox, and p67phox are all detectableby reverse transcriptase (RT)-PCR, butonly p22phoxcan be detected by North-ern analysis and only the cytosolic fac-tors are detectable by Western analysis(Jones ct al. 1996).Using hcme spectro-scopy,cytochrome b55xe x p=not bc detected. Expression of a consti-tutively active mutantof rac-1,however,increases the production of reactive ox-ygen species in endothelial cells (SUl-ciner et al. 1996),suggestingthatat leastthe G protein neutrophil NADPH oxi-dase activating factor is functional in
endothelial cells.In smooth muscle cells, p22phox has
beencloned and is relatively abundantatthe mRNA level (Fukui et al. 1995),bulgp91phoxdoes not appear to be present(unpublished data). Functionally, it ap-pears that p22phox may be part of theagonist-sensitive oxidase, as antisenseinhibition of p22phox expression inVSMC leadsto a diminutionof .02- gen-eration (Ushio-Fukai et al. 1996). Inter-estingly,in these cells the spectrophoto-metric peak occurs not at 558 nm, butrather at 553 nm (Ushio-Fukai et al.1996), suggesting that the cytochromeassociated with the vascular enzymemay be different from the neutrophil cy-tochrome and may even be a c-type cy-tochrome. In theneutrophilNADPH oxi-dase, the large subunit of cytochromebSSS(gpglphox) functions as the sub-strate recognition site (Rotrosen et al.1992),the FAD binding site (Sumimotoet al. 1992),and most likely binds heme(Cross et al. 1995).Thus, the unknownlarge subunit of the vascular NADH/NADPH oxidase may be the componentthat confers on smooth muscle itsunique kinetics, substrate specificity,and activation requirements.
PercentRelaxations
o— .!,s
K
— — a,.,
20 \A(T 1
+ Ang II-EL— ~-” U-SOD
T
t— -...m-EL
%
-9- ShAm-SOD
\*
.
T“
I .\-t
60
80
100-9.0 -8.0 -7:0 -6:0
log M [acetylcholine]
Figure 3. Vascular relaxation to acetylcholinein aortic segmentsfrom sham-operatedand an-giotensin II-infused rats treatedwith liposome-encapsulated superoxide dismutase (SOD, 750U/mL) (Sham-SOD and angiotensinII-SOD,re-spectively)or with empty liposomes (Sham-ELand Ang II-EL,respectively).AngiotensinII wasinfused at a rate of 0.7 mg/kg.day for 5 days,which causes increased vasculal oxidativestress.Vesselsfrom both groups were exposedfor 30q5 min to eithe~JjLor SOfJ,and vasol-e-laxationswere examined aftercontraction withphenylepbrinc. ‘ p <0.05 versus sham for per-cent maximal relaxation; + p =: 0.05 versussham for t31350.Reprinkd with permissionfrom Rajagopalan et al. 1996a,
● Role of ReactiveOxygenSpeciesin Endothelial Function
Even before the identification of endo-thelium-derived relaxing factor as NO.,several groups showed that its integritywas compromised by oxygen-derivedfree radicals (Gryglewski et al. 1986,Rubanyi and Vanhoutte 1986) (Figure3). In fact, if the antioxidantdefense sys-tems are inhibited, endothelium-depen-dent relaxations to acetylcholine are im-paired (Mugge et al. 1991a). This isapparently not due to a decrease in theamount of nitrogen oxides released,butrather to the functional destruction ofNO. by .02- (Nlugge et al. 1991a).Super-oxide reacts rapidly with NO. to formperoxynitrite, which itself can alter en-dothelium-dependent relaxations andmay contribute to lipid peroxidation(Radl et al. 1991),or can be dissociatedto an hydroxyllike radical and nitrogendioxide. Nitrogen dioxide is highly reac-tive and may be involved in nitrosationreactions (Pryor and Squandrito 1995).Thus, the balance between ambient lev-els of .02– and releasedNO. is critical tothe normal functioning of the endothe-lium. As noted later here, elevated .02-production is a characteristic of severalvascular diseases,paired endothelial
01997, ElsevierScienceInc., 1050-1738/97/$17,00PIIS105O-I738(97)OOO88-1
suggesting that ir-responsiveness may
303
be a function of the increase in oxygenfree radicals.
Hemodynamic stressalsohasaneffecton the oxidative state of endothelialcells. In a recent paper, Howard et al.(1997) showed that cyclic strain causedan early (2 h), transient increase inNADH/NADPH oxidase activity and asustained increase in H202 in culturedporcine aortic endothelial cells. This in-crease in oxidative stress was reflectedin an increase in lipid peroxidationproducts released from the cells, sug-gesting that reactive oxygen speciesmayserve to transduce mechanical signalsinto biochemical events.
Another effect of reactive oxygen spe-cies on endothelial function is to shiftthe thrombotic or antithrombotic bal-ance to favor adhesionof leukocytes.Forexample, interleukin-l–induced expres-sion of vascular cell adhesion molecule(VCAM-1) is inhibited by treatmentwiththe intracellular antioxidant pyrro-lidinedithiocarbamate (PDTC) (Marui etal. 1993).The redox-sensitive transcrip-tion of VCAM-1 appears to depend, atleast in part, on tandem NFKB-like ele-ments in the promoter region of thisgene. Induction of monocyte chemotac-tic protein (MCP-1) (Cushinget al. 1990)and heme oxygenase-1 (HO-1) (Nath etal. 1992),among others,is alsomediatedby oxidative stress.
Finally, externally applied oxidantstress leads to an upregulation of theantioxidant defense enzymes, includingsuperoxidedismutase,catalase,and glu-tathioneperoxidase (Lu et al. 1993).Thissuggeststhat factors that influence vas-cular oxidantstressare tightly regulated,a requirement if reactive oxygen speciesserve a second messenger function invascular cells.
● ReactiveOxygenSpeciesand VascularSmooth Muscle
In addition to their influenceon vasculartone via alterations in endothelium-de-pendentrelaxations,reactive oxygen spe-cies also directly affect vascular smoothmuscle.Mohazzab-Handcolleagues(Mo-hazzab-Het al. 1996)haveshownthatthetransient pulmonary vasorelaxation oc-curringduringposthypoxicreoxygenationis a direct result of H202 derived fromNADH-dependent superoxide formation.This effect is apparentlydue to the stimu-lation of guanylate cyclase via catalase-
aBlood
Pressure(nmnlig)
120~1 2 3450
b(mmHg)
100
0
D o Infusion
*
PIIX4SUE
onmd/midmg
120001I
10
5
042- production
moo
4(XIO
o
‘
❑ Sham❑ Angll❑ Angll+l-lydmlazine
*
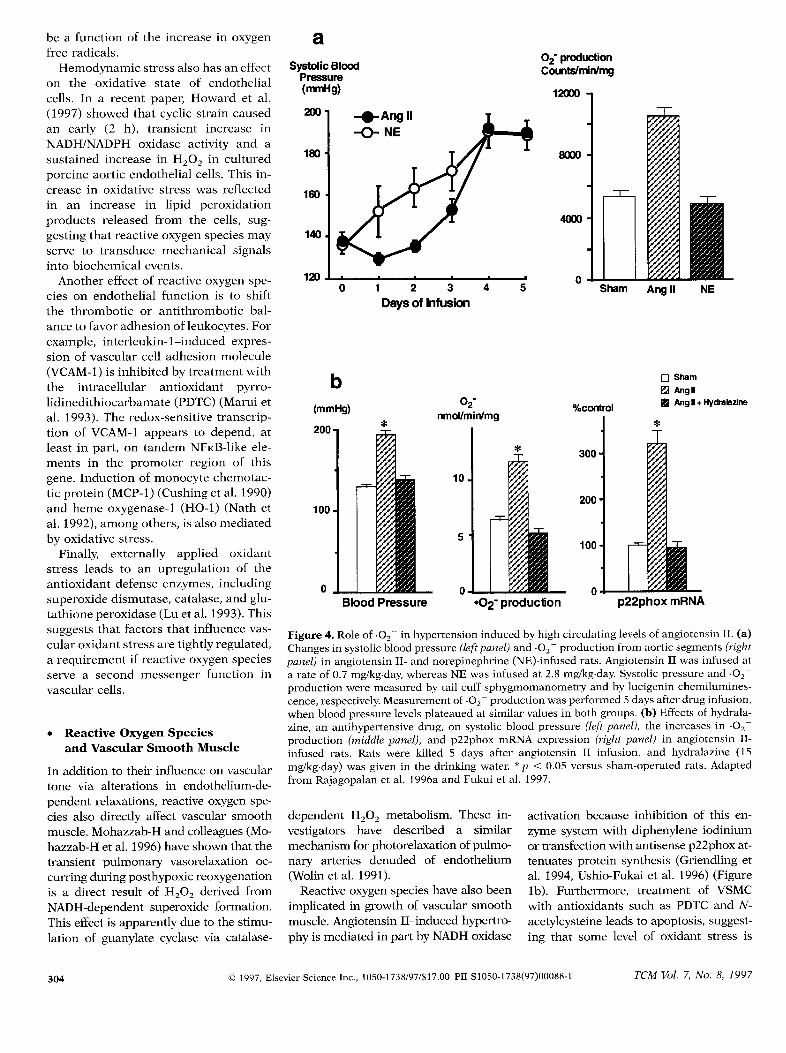
Figure 4. Role of .02- in hypertension induced by high circulating levels of angiotensin II. (a)Changes in systolic blood pressure (Zefipanel)and .02- production from aortic segments (rightpanel) in angiotensin II- and norepinephrine (NE)-infused rats. Angiotensin II was infused ata rate of 0.7 mg/kg.day, whereas NE was infused at 2.8 mg/kg.day. Systolic pressure and .02–production were measured by tail cuff sphygmomanometry and by lucigenin chemilumines-cence, respectively.Measurement of .02– production was performed 5 days after drug infusion,when blood pressure levels plateaued at similar values in both groups. (b) Effects of hydrala-zine, an antihypertensive drug, on systolic blood pressure (left panel), the increases in .02–production (middle panel), and p22phox mRNA expression (right panel) in angiotensin II-infused rats. Rats were killed 5 days after angiotensin II infusion, and hydralazine (15mg/kg.day) was given in the drinking water. ‘<p < 0.05 versus sham-operated rats. Adaptedfrom Rajagopalan et al. 1996a and Fukui et al. 1997.
dependent H202 metabolism. These in-vestigators have described a similarmechanismfor photorelaxationof pulmo-nary arteries denuded of endothelium(Wolin et al. 1991).
Reactive oxygenspecieshavealsobeenimplicated in growth of vascularsmoothmuscle.AngiotensinII–inducedhypertro-phy is mediatedin partby NADH oxidase
activation because inhibition of this en-zyme systemwith diphenylene iodiniumor transection with antisensep22phoxat-tenuatesprotein synthesis(Griendling etal. 1994,Ushio-Fukaiet al. 1996)(Figurelb). Furthermore, treatment of VSMCwith antioxidantssuch as PDTC and N-acetylcysteineleadsto apoptosis,suggest-ing that some level of oxidant stress is
304 @ 1997,ElsevierScienceInc., 1050-1738/97/$17.00PIIS105O-1738(97)OOO88-1 TCM vol. 7, No. 8, 1997

required for normal growth (Tsai et al.1996).Tncfeed,H202and 02- activatem-merous growth-related signaling path-ways in VSMC, including activation of
kinases (BaasandBerk 1995);induc-tion of’ the protooncogencsand c-imyc (Rao and Berk 1992,Rao et al.1993aand b); and activationof the AP-1transcriptionfactor (Puri et al. 1995).Arecent study(Traniet al. 1997)on the roleof supcmxide in mitogenic signaling infibroblasts afso implicates ras, a smallguanosinetrisphosphatebinding proteinknown LObe important in proliferationand differentiation. Fibroblasts h-ans-formedwith a constitutivelyactivemutantof ras produce large amountsof .O1–viaactivationof the NADPH oxidasecompo-nent rac-1,and thesecells have a greatlyincreased DNA turnovm If a similarmechanismholdsin VSMC, thisstudynotonlyprovidessupportfor a role of rac-1 inthe nonphagocyticoxidasc but also sug-gestsa new mitogenic signalingpathwayby which oxidantsexert their effects.
● ReactiveOxygenSpeciesand Microphage Function
Reactive oxygen species are both pro-duced by,and alter,the function of mac-rophages and leukocytes. In leukocytes,.02- and H202 promote CD1lb andCD18 expression and increased adhe-sion to endothelial cells (Fraticelli et al.1996),similar to their effect on endothe-lial cell adhesion molecule expression.Furthermore,microphage-derived foamcells from hypercholesterolemic rabbitsproduce .02– and H202 (Rajagopalan etal. 1996b).These reactive oxygen speciesregulate the activity of smooth muscle-derived matrix metalloproteinasesMMP-2 and MMP-9 (Rajagopalan et al.1996b), suggesting that oxidant stressmay contribute to vascular remodeling.
. The NADI-UNADPHOxidasein Hypertension
Recently,there has been intense interestin the role of .02– in hypertension in-ducedby high circulating levels of angio-tensin II. Rajagopalan et al. (1996a)demonstrated that in rats made hyper-tensive by prolonged infusion of angio-tensin II, vascular NADH/NADPH oxi-dase activity is selectively increased andcontributes to impaired endothelium-dependent vasodilation (Figure 3). This
elevated enzymatic activity is accompa-nied by an increase expression0[p22phoxmRNA (Fukui et al. 1997)(Fig-ure 4), suggesting that the amount ofoxidase present in the vessel wall mightbc upregulated in this model of hyper-tension. Notably, infusion of anothervasoconsLrictol;nolepincphrine, had noeffect on oxidase activity even though itcaused an equivalent in bloodpressure (Rajagopalan et al. 1996a).Both oxidase activity and p22phox ex-pression were blocked by the antihypcr-tensive agent hydralazine (Figure 4) andthe angiotensin ATI receptor blockerlosartan, raising the possibility thatblood pressureitself might contribute tooxidase upregulation in the setting ofelevated angiotensin H (Fukui et al.1997).A possiblealternativeexplanationarises from the recent obsemation thathydralazine has antioxidant proper-ties(Mtinzel et al. 1996). These findingswould thusalsobe consistentwith a rolefor oxidant stressin blood pressurereg-ulation. In fact, administrationof super-oxide dismutasebeginning 3 daysbeforeangiotensin II infusion significantly at-tenuates the subsequent rise in bloodpressure (Bech-Laursen et al. 1997).Consistent with the lack of effect ofnorepinephrine on oxidase activity, su-peroxide dismutasehas no effect on thenorepinephrine-induced increase inblood pressure.
● OxidativeStressin Atherosclerosis
There is accumulatingevidence thatath-erosclerosis is accompanied by, or isconsequentupon,oxidative stresswithinthe vessel wall. Oxygen free radical pro-duction is higher in aortas from rabbitsfed a high-cholesterol diet for severalweeks than in aortas from animals onnormal diets (Harrison et al. 1987,Ohara et al. 1993). Hypercholester-olemic animals exhibit impaired endo-thelial-dependentrelaxation, which canbe restored with antioxidants (Miigge etal. 1991b,Keaney et al. 1995).Further-more, it hasbeen demonstratedthataor-tic atheroscleroticlesion formation is as-sociated with the accumulation of lipidperoxidation products and induction ofinflammatory genesby NFKB-like(redoxsensitive) transcription factors (Liao etal. 1994). In vitro data suggest that ex-pression of endothelial-leukocyteadhe-
sion molecules,an early event in athero-genesis that mediates migration ofmonocytes into the vessel wall, is alsoredox sensitive (Marui et al. 1993,Khanet al. 1995). The increased free radicalproduction may thushave multiple con-sequences, including monocyte recruit-ment into the vessel wall (Alexander1995), inactivation of NO resulting inendothelial dysfunction (Miigge et al.1991b), and increased smooth musclecell growth (Griendling et al. 1994).To-gether,these data suggestthaLincreasedoxidative stress in the arterial wall maybe a fundamental feature of atheroscle-rosis (Afexander 1995).
● The NADH Oxidasein NitrateTolerance
The clinical problem of the developmentof tolerance to nitrates has receivedenormous attention, but the underlyingmechanisms remain elusive. A recentstudy by Miinzel et al. (1996) providesevidence for a role for the vascularNADH oxidase in nitrate tolerance,basedon ananalysisof the drug hydrala-zinc. In rabbits exposed to nitroglycerincontinuously for 3 days, vascular .02production is increased, and this in-crease is completely abolished by hy-dralazine. In vitro studies indicate thatthis drug apparently exerts its effect byinhibiting the NADH oxidase. Impor-tantly, concomitant administration ofhydralazine and nitroglycerin preventsthe development of tolerance to acutenitroglycerin-induced endothelium-de-pendent relaxation, suggesting thatNADH-derived .02- may contribute tothe vascular dysfunction that is pathog-nomonic for nitrate tolerance.
. Future Directions
The recent work summarized here sug-geststhat the proteins and enzymes thatproduce reactive oxygen speciesor serveas the antioxidant defense system areimportant determinantsof the course ofvascular disease. A corollary of thispremise is that the mechanismscontrol-ling activity or expression of these pro-teins or enzymes are also critical. Fur-ther investigation is necessary todetermine the subunit structure of thevascularNADH/NADPHoxidase,the sig-nals that regulate oxidase activity, thefate of the .02- generated, and the func-
TCM vol. 7, No. 8, 1997 Q 1997,ElsevierScienceInc., 1050-1738/97/$17.00PIIS105O-1738(97)OOO88-1 305

tional consequences of increased .02–production in both endothelial andsmooth muscle cells (Rajagopalan et al.1996b). Of particular interest will beidentification of redox-sensitive genesthat play a role in both normal vascularfunction and the pathogenesis of vascu-lar disease.
● Acknowledgments
This work was supported by NationalInstitutes of Health grant HL38206.Wethank Dr. David Harrison for a criticalreading of the manuscript and BarbaraMerchant-Bailey for editorial assistance.
References
Afexander RW: 1995. Hypertension and thepathogenesis of atherosclerosis. Oxidativestress and the mediation of arterial inflam-matory response: a new perspective. Hyper-tension 25:155–161.
Baas AS, Berk BC: 1995. Differentialactivationof mitogen-activatedprotein kinasesby HZ02and 02 in vascular smooth muscle cells.Circ Res 77:29-36.
Bech-Laursen J, Rajagopalan S, Galis Z, et al.:1997. Role of superoxide in angiotensin II-induced but not catecholamine-induced hy-pertension. Circulation 95:588-593.
Beckman JS, Beckman TW, Chen J, MarshallPA, Freeman BA: 1990. Apparent hydroxylradical production by peroxynitrite:implica-tions for endotheliaf injury from nitric oxideand superoxide. Proc Natl Acad Sci USA 87:1620-1624.
Cross AR, Rae J, Curnutte JT: 1995. Cyto-chrome b–245of the neutrophil superoxide-generatingsystem contains two nonidenticalheroes. J Biol Chem 270:17,075-17,077.
Cushing SD, Berliner JA, Valente AJ, et al.:1990. Minimally modified low density lipo-protein induces monocyte chemotactic pro-tein 1 in human endotheliafcells and smoothmuscle cells. Proc Nad Acad Sci USA 87:5134-5138.
Finan P,Shimizu Y, Gout I, et al.: 1994.An SH3domain and proline-rich sequence mediatean interaction between two components ofthe phagocyte NADPH oxidase complex. JBiol Chem 269:13,752-13,755.
Fraticelli A, Serrano CV, Bochner BS, Capo-grossi MC, Zweier JL: 1996.Hydrogen perox-ide and superoxidemodulate leukocyteadhe-sion molecule expression and leukocyteendothelial adhesion. Biochim Biophys Acta1310:251-259.
Friedl HP, Till GO, Ryan US, Ward PA: 1989.Mediator-inducedactivation of xanthine oxi-
dase in endotheliaf cells. FASEB J 3:2512-2518.
FukuiT,Lass&ue B, Kai H, AlexanderRW andGriendling KK: 1995. cDNA cloning andmRNA expression of cytochrome b558.-sub-unit in ratvascularsmooth muscle cells. Bio-chim Biophys Acta 1231:215–219.
FukuiT,IshizakaN, Rajagopalan S, etaL: 1997.p22phox mRNA expressionand NADPHofi-dase activity are increased in aortas fromhypertensiverats. Circ Res 80:45-51.
Griendling KK, Minieri CA, Ollerenshaw JD,Alexander RW: 1994. Angiotensin II stimu-lates NADH and NADPH oxidase activity incultured vascular smooth muscle cells. CircRes 74:1141–1148.
GryglewskiRJ, PafmerRMJ, Moncada S: 1986.Superoxide anion is involved in the break-down of endothelium-derivedrelaxingfactor.Nature 320:454-456.
Harrison DG, Armstrong ML, Freiman PC,Heistad DD: 1987. Restoration of endothe-lium-dependent relaxation by dietary treat-ment of atherosclerosis. J Clin Invest80:1808-1811.
Howard AB, AfexanderRW,Nerem RM, Grien-dling KK, TaylorWR: 1997. Cyclic strain in-duces an oxidativestressin endothelialcells.Am J Physiol 272:C421-C427.
IraniK, Xia Y,ZweierJL,et al.: 1997.Mitogenicsignafing mediated by oxidants in ras-trans-formed fibroblasts. Science 275:1649-1652.
Jones SA, O’DonnellVB, Wood JD, et al.: 1996.Expression of phagocyte NADPH oxidasecomponents in human endotheliafcelfs.Am JPhysiol 271:H1626-H1634.
KeaneyJFJr,Xu A, CunninghamD, etal.: 1995.Dietaryprobucol preservesendotheliaf func-tion in cholesterol-fedrabbits by limitingvas-cular oxidativestressand superoxidegenera-tion. J Clin Invest95:2520–2529.
Khan BV, Parthasarathy SS, Afexander RW,Medford RM: 1995. Modified low density li-poprotein and its constituents augment cy-tokine-activatedvascularcell adhesion mole-cule-l gene expression in human vascularendothelialcells. J Clin Invest95:1262–1270.
Leto TL, Adams AG, DeMendezI: 1994.Assem-bly of the phagocyte NADPH oxidase: bind-ing of Src homology 3 domains to proline-rich targets. Proc Natl Acad Sci USA91:10,650-10,654.
Liao F,AndalibiA, Qiao JH, etal.: 1994.Geneticevidence for a common pathway mediatingoxidative stress, inflammatory gene induc-tion, and aortic fatty streak formation inmice. J Clin Invest94:877–884.
Lu D, Maulik N, Moraru II, KreutzerDL, DasDK: 1993. Molecufar adaptation of vascularendotheliaf cells to oxidative stress. Am JPhysiol 264:C715-C722.
Marui N, OffernmnM, SwerlickR, et al.: 1993.Vascularcell-adhesion molecule-1 (VCAM-1)
gene-transcription and expression are regu-lated through an antioxidantsensitivemech-anism in human vascular endotheliaf cells. JClin Invest92:1866-1874.
Mohazzab-H KM, Wolin MS: 1994a.Propertiesof a superoxide anion-generating microso-mal NADH oxidoreductase, a potential pul-monary arteryP02 sensor.Am J Physiol 267:L823-L831.
Mohazzab-H KM, Wolin MS: 1994b. Sites ofsuperoxide anion production detected by lu-cigenin in caff pulmonary artery smoothmuscle. Am J Physiol 267:L815–L822.
Mohazzab-H KM, Kaminski PM, Wolin MS:1994. NADH oxidoreductase is a majorsource of superoxide anion in bovine coro-nary artery endothelium. Am J Physiol 266:H2568-H2572.
Mohazzab-H KM, Kaminski PM, FayngershRP, Wolin MS: 1996. Oxygen-elicited re-sponses in caff coronary arteries: role ofH202 production via NADH-derivedsuperox-ide. Am J Physiol 27O:H1O44–H1O53.
MuggeA, ElwellJH,PetersonTE, HarrisonDG:1991a.Releaseof intactendothelium-derivedrelaxingfactor depends on endothelialsuper-oxide dismutase activity.Am J Physiol 260:C219-C225.
MtiggeA, ElwellJH, PetersonTE, et al.: 1991b.Chronic treatment with polyethylene-glyco-lated superoxide dismutase partiallyrestoresendothelium-dependentvascular relaxationsin cholesterol-fed rabbits. Circ Res 69:1293–1300.
Mtinzel T, Kurz S, Rajagopalan S, et al.: 1996.Hydralazinepreventsnitroglycerintoleranceby inhibiting activation of a membrane-bound NADH oxidase. J Clin Invest98:1465-1470.
Nath KA, Balla G, VercellottiGM, et al.: 1992.Induction of heme oxygenase is a rapid, pro-tectiveresponse in rhabdomyolysis in therat.J Clin Invest 90:267-270.
Ohara Y, PetersonTE, Harrison DG: 1993. Hy-percholesterolemia increases endothelial su-peroxide anion production. J Clin Invest 91:2546-2551.
Pagano PJ, Tomheim K, Cohen RA: 1993. Su-peroxide anion production by rabbit thoracicaorta:effectof endothelium-derivednitricox-ide. Am J Physiol 265:H707–H712.
Pagano PJ, Ito Y, Tomheim K, et al.: 1995. AnNADPH oxidase superoxide-generating sys-tem in the rabbit aorta. Am J Physiol 268:H2274-H2280.
Panus PC, Radi R, Chumfey PH, Lillard RH,FreemanBA: 1993.Detectionof H202 releasefrom vascularendothelialcells. FreeRad BiolMed 14:217–233.
PryorW, Squandrito G: 1995. The chemistry ofperoxynitrite:a product from the reaction ofnitric oxide with superoxide. Am J Physiol268:L699-L722.
@ 1997,ElsevierScienceInc., 1050-1738/97/$17.00PIIS105O-1738(97)OOO88-1 TCM vol. 7, No. 8, 1997

Puri PL, AvantaggiatiML, Burgio VL, et al.:1995.Reactiveoxygen intermediatesmediateangiotensin II-induced c-jun, c-fos het-erodimer DNA binding activity and prolifer-ative hypertrophic responses in myogeniccells. J Biol Chem 270:22,129–22,134.
Radl R, Beckman JW, Bush KM, Freeman BA:1991. Peroxynitriteinduced membrane lipidperoxidation: thecytotoxic potentiafof super-oxide and nitric oxide. Arch Biochem Bio-phys Res Commun 288:481487.
Rajagopalan S, Kurz S, Miinzel T, et al.: 1996a.Angiotensin II mediated hypertension in therat increasesvascular superoxide productionvia membrane NADWNADPH oxidase acti-vation: contribution to alterationsof vasomo-tor tone. J Clin Invest 97:1916–1923.
Rajagopalan S, Meng XP, Ramasamy S, Hafi-son DG, Galis ZS: 1996b. Reactive oxygenspecies produced by microphage-derivedfoam cells regulate the activity of vascularmatrix metalloproteinasesin vitro. J Clin In-vest 98:2572–2579.
Rao GN, Berk BC: 1992. Active oxygen speciesstimulate vascular smooth muscle cellgrowth and proto-oncogene expression. CircRes 70:593–599.
Rao GN, LassegueB, GriendlingKK, AlexanderRW: 1993a. Hydrogen peroxide stimulatestranscription of c-jun in vascular smoothmuscle cells: role of arachidonic acid. Onco-gene 8:2759-2764.
Rao GN, LassegueB, GriendlingKK, AfexanderRW, Berk BC: 1993b. Hydrogen peroxide-in-duced c-fos expressionis mediatedby arachi-donic acid release: role of protein kinase C.Nucleic Acids Res 21:1259–1263.
Rotrosen D, Yeung CL, Leto TL, Malech HL,Kwong CH: 1992. Cytochrome b~~~:the fla-vin-binding component of the phagocyteNADPH oxidase. Science 256:1459-1462.
Rubanyi GM, VanhouttePM: 1986. Oxygen-de-rived free radicals, endothelium, and the re-sponsivenessof vascular smooth muscle. AmJ Physiol 250:H815-H821.
Sulciner DJ, Irani K, Yu Z, et af.: 1996. Raclregulates a cytokine-stimulated, redox-de-pendentpathwaynecessa~ for NF-tcBactiva-tion and VCAM-1 expression. Circulation 94:1-708.
Sumimoto H, Sakamoto N, Nozaki M, et al.:1992. Cytochrome b,~g, a component of thephagocyte NADPHoxidase, is a ffavoprotein.Biochem Biophys Res Commun 186:1368-1375.
TsaiJ-C,Jain M, Hsieh C-M, et al.: 1996.Induc-tion of apoptosis by pyrrolidinedithiocar-bamate and N-acetylcysteine in vascularsmooth muscle cells. J Biol Chem 271:3667–3670.
Ushio-FukaiM, Zafari AM, FukuiT,IshizakaN,Griendling KK: 1996. p22phox is a criticalcomponent of the superoxide-generating
NADWNADPHoxidase systemand regulates activation of the NADPH oxidase of humanangiotensinII-induced hypertrophyin vascu- neutrophils.J Biol Chem 266:7432–7439.lar smooth muscle cells. J Biol Chem 271: Wolin MS, Omar HA, MortellitiMP,CherryPD:23,317-23,321. 1991. Association of pulmonary artery pho-
Watson F,Robinson J, Edwards SW: 1991. Pro- torelaxationwith H202 metabolism by cata-tein kinase C-dependent and -independent lase. Am J Physiol 261:H1141-H1 147. TCM
K PRT a S G D SJulie Chao and Lee Chao
K a l l if id i sa a h k a l lp it c i r cs hh h ow o p s pt e ii n h i( s eI f a c ol c wt ik a l la i nk a la cS ueh a c c ui r ey i nt k am p ar oi b lp r er e gi n do i i nwt ik a l lI n ti no k ai r a mr e si a r aa t r ar eo b p i ad o s e -m aF u na ni t rm oe x p rr k a l l i kp ra a no h kl i s tr e vt t hm h s i gl b pc o mw c ol i t tA d e nt d o th uk a l lg c c s i gb p rf 4 w ei s p o nh y pr F k aci nv a s o ri i sr a r a r rp e r -p r ei t i sp ek T tf i ns u ga d ir f k a li r eb pa r at p o s sf t d e vo n p ht r e af h y p e( TC aM 1 73 1 0 1E l sS cI
. Discoveryof Kzdlistatinas aTissueKallikrein-BindingProtein
Tissue kallikrein activity was first iden-tified in humanurine in 1909as a resultof its profound but transientblood pres-sure lowering effect on dogs (Abelousand Bardier 1909).In 1928,a tissuekal-
Julie Chao and Lee Chao are in the Depart-ment of Biochemistry and Molecular Biology,Medical Universityof South Carolina, Charle-ston, SC 29425, USA.
likrein-binding protein (KBP), whichrapidly inhibited kallikrein’s activity,was first observed in human plasma(Frey and Kraut 1928), but its identityremained unclear. Geiger et al. (1981)reported that human cxl-antitrypsin (cil-AT) binds to and inhibits human tissuekallikrein. Although al-AT is present in anenormous quantity in the circulation, theformation of the complex between al-ATand kallikrein is a very slow process. Thesignificance of its physiological functionin regulating tissue kallikrein’s function
TCM vol. 7, No. 8, 1997 @ 1997,ElsevierScienceInc., 1050-1738/97/$17.00PIIS105O-1738(97)OOO89-3 307