abordaje mieloneuropatia
TRANSCRIPT

Diagnostic Approachto Myeloneuropathy
Brent P. Goodman, MD
ABSTRACTDisorders that concomitantly affect the spinal cord and peripheral nerves can becharacterized as myeloneuropathies. Such conditions can be broadly categorized asmetabolic, inflammatory, infectious, or hereditary disorders. Because these disordersmay present with predominantly myelopathic or peripheral neuropathic signs andsymptoms, a careful neurologic examination and a thoughtful diagnostic evaluationare necessary to establish a diagnosis of myeloneuropathy. This article outlines anapproach to the identification, evaluation, and treatment of myeloneuropathy.
Continuum Lifelong Learning Neurol 2011;17(4):744–760.
INTRODUCTIONA thoughtful approach to the evalua-tion of suspected myeloneuropathyrequires an understanding of spinalcord and peripheral nervous systemanatomy and physiology, careful historyand neurologic examination, and ap-propriate utilization and interpretationof diagnostic testing. Recognition of amyeloneuropathy may be challengingfor even the most seasoned cliniciansbecause symptoms can be similar tothose of purely spinal cord or peripheralnerve disease, and examination findingsmay be more prominently myelopathicor (peripheral) neuropathic, making itmore difficult to recognize signs of theother. A diagnostic evaluation usingsome combination of blood work, CSFexamination, and radiographic and elec-trodiagnostic studies is typically neces-sary. A thoughtful, consistent approachto the patient presenting with myelo-neuropathy facilitates an accurate diag-nosis and timely treatment and helps toavoid unnecessary testing.
Conditions known to result in mye-loneuropathy include metabolic, in-flammatory, infectious, toxic, andhereditary disorders (Table 2-1). Rec-
ognition of these disorders is helpedby an understanding of the typicalmode of onset and progression, thepattern of myelopathic and neuro-pathic findings, other potential neuro-logic manifestations (such as opticneuropathy or cognitive impairment),and the laboratory, electrodiagnostic,and radiographic findings seen in theseconditions. For example, a slowly pro-gressive spastic paraparesis associatedwith a demyelinating peripheral neuro-pathy in a patient with a family historyof gait impairment suggests a heredi-tary condition such as adrenomyelo-neuropathy. A rapidly progressivesensory ataxia with cervical MRI studiesdemonstrating T2 hyperintensity in theposterior columns suggests a myelo-neuropathy due to either copper orvitamin B12 deficiency. Of course, it isimportant to remember that morethan one disorder may be responsiblefor a patient’s condition.
CLINICAL RECOGNITION OFMYELONEUROPATHYThe initial and most important com-ponents of an evaluation leading to acorrect diagnosis of myeloneuropathy
Address correspondence toDr Brent P. Goodman, MayoClinic, Department ofNeurology, 13400 E. SheaBoulevard, Scottsdale, AZ 85259,[email protected].
Relationship Disclosure:Dr Goodman reports nodisclosure.
Unlabeled Use ofProducts/InvestigationalUse Disclosure:Dr Goodman discusses theunlabeled or investigationaluse of azathioprine,thalidomide, chloroquine,hydroxychloroquine,intravenous immunoglobulin,plasma exchange,corticosteroids, methotrexate,infliximab, cyclophosphamide,and glyceryl trioleate-glyceryltrierucate.
Copyright B 2011, AmericanAcademy of Neurology. Allrights reserved.
Review Article
744 www.aan.com/continuum August 2011
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

are a careful history and an examina-tion to localize the disorder within theneuraxis. As indicated above, clinical
recognition of a myeloneuropathy maybe difficult. A patient may have beenpreviously diagnosed with a peripheral
TABLE 2-1 Conditions Causing Myeloneuropathy
Disorder Diagnosis Treatment
Metabolic
Vitamin B12 deficiency Serum cobalamin
Serum methylmalonic acid
Serum homocysteine
IM vitamin B12 10002 for 5 days; oncemonthly thereafter or oral/sublingualvitamin B12 10002g daily
Copper deficiency Serum copper, ceruloplasmin
Urinary copper
Discontinue zinc
Copper 8 mg orally dailyfor 1 week; 6 mg daily for1 week; 4 mg daily for 1 week;2 mg daily thereafter
Folate deficiency Serum folate, homocysteine Folate 1 mg orally 3 times a day initially;1 mg daily thereafter
Vitamin E deficiency Serum vitamin E
Ratio of serum vitamin E to thesum of serum cholesteroland triglycerides
Vitamin E dose range 200 mg to200 mg/kg/d oral or IM
Nitrous oxide Serum cobalamin (renderedinactive by nitrous oxide)
Cessation of nitrous oxide exposure
IM vitamin B12
Oral methionine considered
Inflammatory
Sarcoidosis Serum angiotensin-convertingenzyme
Hilar/paratracheal adenopathyon chest imaging
Noncaseating granuloma ontissue biopsy
Corticosteroids
Methotrexate, chloroquine,hydroxychloroquine, cyclophosphamide,infliximab, mycophenolate,azathioprine
Multiple sclerosis MRI studies
EMG studies
CSF studies
Multiple sclerosisdisease-modifying therapy
Corticosteroids
Plasma exchange
Sjogren syndrome SSA, SSB antibodies
Rheumatoid factor;antinuclear antibodies
Schirmer test; rose bengal stain
Salivary gland biopsy
Corticosteroids
Cyclophosphamide, methotrexate
continued on next page
745Continuum Lifelong Learning Neurol 2011;17(4):744–760 www.aan.com/continuum
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

neuropathy or myelopathy in isolation.Myelopathic and neuropathic symptomsare often similar and upper motorneuron signs may predominate, makingit difficult to appreciate lower motorneuron signs. Conversely, significantlower motor neuron signs may masksigns of CNS impairment.
Symptoms that can be attributed tomyelopathy include gait unsteadiness,extremity weakness, bowel and bladderimpairment, and sensory symptoms.Many of these same symptoms are alsopresent in patients with peripheralneuropathy. However, bowel and blad-der impairment is less common in
peripheral neuropathy unless periph-eral autonomic fibers are involved(autonomic neuropathy). Bladder im-pairment associated with a peripheralautonomic neuropathy is typically char-acterized as a high-capacity bladderwith incomplete bladder emptying orurinary retention. Furthermore, sen-sory symptoms associated with periph-eral neuropathy typically involve a‘‘stocking-glove’’ pattern andusually be-gin in the feet. The presence of sensorysymptoms in both the hands and feetshould alert the neurologist to a possi-ble concomitant peripheral neuropathy(myeloneuropathy). As with sensory
Continued
Disorder Diagnosis Treatment
Paraneoplastic Paraneoplastic antibodies Treatment of underlying malignancy
Infectious
HIV HIV serology; CD4+ lymphocytes
Spinal cord MRI
CSF studies
Highly active antiretroviral therapy
Human T-cell lymphotropicvirus type I (HTLV-I)Yassociatedmyelopathy/tropical spasticparaparesis
HTLV-I serum, CSF Supportive
Hereditary
Adrenomyeloneuropathy Serum very long-chain fatty acids
ABCD1 gene testing
Supportive
Glyceryl trioleate-glyceryltrierucate (Lorenzo oil) in clinical trials
Cerebrotendinousxanthomatosis
Normal/low cholesterol
Elevated cholestanol
Chenodeoxycholic acid
Hereditary spastic paraplegia Genetic testing Supportive
Mitochondrial disorders Serum lactate, pyruvate
Muscle biopsy
Blood DNA testing
Muscle DNA testing
Coenzyme Q10, creatine;!-lipoic acid can be considered
Krabbe disease Leukocyte "-galactosidase Supportive
Pelizaeus-Merzbacher disease Proteolipid protein gene testing Supportive
Metachromatic leukodystrophy Leukocyte arylsulfatase assay Supportive
Myeloneuropathy
746 www.aan.com/continuum August 2011
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

symptoms, weakness associated withperipheral neuropathy typically beginsin the distal extremities and is accom-panied by atrophy. The presence of aLhermitte sign with precipitation ofelectrical pain down the spine and intothe extremities with neck flexion wouldsuggest a myelopathy. Signs of myelop-athy include sensory loss, weakness,spasticity, hyperreflexia, and extensorplantar responses. Weakness, atrophy,sensory loss, and hyporeflexia or are-flexia are typical of peripheral neuro-pathy. Foot architecture changes, suchas pes cavus (high arches) or hammertoes, may suggest a long-standing andoften hereditary peripheral neuropathy.As alluded to above, when upper motorsigns predominate, it may be difficultto appreciate more subtle signs of pe-ripheral neuropathy. Absent ankle jerkreflexes and distal lower limb atrophymay be the only definite clinical signs ofperipheral neuropathy in a patient withprominentmyelopathic findings. In someinstances, EMG testing may be necessaryto identify peripheral nerve involvement.
Myelopathic signs may be subtle in apatient with prominent lower motorneuron findings. Deep tendon reflexesmay be reduced or absent and toe ex-tensor paresis may result in no re-sponse with plantar stimulation. Insuch cases, preserved deep tendonreflexes at the knees may be the onlyevidence of myelopathy. Generally,with a peripheral neuropathy of suffi-cient severity to result in lower limbproprioceptive impairment, lower limbareflexia (knee and ankle) is expected.
IDENTIFICATION OFMYELONEUROPATHIC PATTERNSRecognition of certain clinical, radio-graphic, and electrodiagnostic find-ings may suggest particular disorders(Table 2-2). Identification of the post-erolateral syndrome, which manifests asa sensory ataxia with pyramidal signs, is
characteristic of vitamin B12 deficiency,copper deficiency, folate deficiency, hu-man T-cell lymphotropic virus (HTLV),andHIVmyelopathies. Demyelinating fea-tures on EMG are typical of the heredi-tary leukodystrophies but can also beseen with HIV and HTLV and in rarepatients with multiple sclerosis whohave signs of central and peripheralnervous system demyelination. Finally,involvement of peripheral nerve andnerve roots (a polyradiculoneuropathy)can be seenwith neurosarcoidosis, HIV,
KEY POINTS
h Recognition ofmyeloneuropathyrequires a careful historyand neurologicexamination.
h Myelopathic symptomsmay include gaitunsteadiness, extremityweakness, bowel andbladder impairment,and sensory symptoms.
h Bowel and bladderinvolvement is less likelyto occur in peripheralneuropathic disordersunless peripheralautonomic fibers areinvolved.
h Signs of myelopathyinclude sensory loss,weakness, spasticity,hyperreflexia, andextensor plantarresponses.
h Weakness, atrophy,sensory loss, anddiminished or absentreflexes are typicallyassociated with aperipheral neuropathy.
TABLE 2-2 MyeloneuropathyPatterns–PotentialClues to Diagnosis
b Posterolateral Cord Syndrome
Vitamin B12 deficiency
Copper deficiency
Folate deficiency
HIV vacuolar myelopathy
Human T-cell lymphotropicvirus myelopathy
b Demyelination on EMG
Adrenomyeloneuropathy
Cerebrotendinousxanthomatosis
Krabbe disease
Metachromaticleukodystrophy
Pelizaeus-Merzbacher disease
Multiple sclerosis (rare)
HIV (uncommon)
Human T-cell lymphotropic virus
b (Myelo) Polyradiculoneuropathy
Neurosarcoidosis
Multiple sclerosis (rare)
Paraneoplastic disorders
HumanT-cell lymphotropic virus
HIV
747Continuum Lifelong Learning Neurol 2011;17(4):744–760 www.aan.com/continuum
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

HTLV, multiple sclerosis (rare), andparaneoplastic disorders.
METABOLIC DISORDERSVitamin B12 DeficiencyMyeloneuropathyNeurologic signs and symptoms of vi-tamin B12 (cobalamin) deficiency arevaried but are often the initial manifes-tations of myeloneuropathy. Cognitiveand psychiatric impairment, optic neu-ropathy, and orthostatic hypotensionare common. Classically, vitamin B12deficiency results in a myelopathy thathas been termed subacute combineddegeneration.1 This condition resultsfrom impairment in posterior columnand lateral spinothalamic tract functionwith a combination of pyramidal signsand sensory loss (vibration sense, lighttouch, and proprioception) evident onexamination. Paresthesia is the mostcommon initial symptom in patients withvitamin B12 deficiency. The myelopathycan be accompanied by a mild, predom-inantly axonal peripheral neuropathy.2,3
Other constitutional symptoms are alsocommon and include fatigue, weightloss, fever, dyspnea, and gastrointestinalsymptoms. A number of hematologicmanifestations of vitamin B12 deficiencycan occur, including megaloblastic ane-mia, pancytopenia, neutrophil hyperseg-mentation, and macrocytosis.
Vitamin B12 is contained in a num-ber of animal proteins, in fortifiedbreakfast cereals, and in some nutri-tional yeast products.4 Following inges-tion, cobalamin is cleaved from otherproteins by gastric acid and binds tointrinsic factor in the duodenum. Theintrinsic factorYcobalamin complex isabsorbed in the ileum. In addition, 1%of cobalamin is absorbed passively(independent of intrinsic factor) in theterminal ileum. This explains why largeoral doses of vitamin B12 can success-fully treat vitamin B12 deficiency due topernicious anemia.5 Once absorbed,
vitamin B12 binds to transcobalamin II,which is responsible for the delivery ofvitamin B12 to tissues.
The recommended daily intake ofvitamin B12 is 2.4 2g for adults, 2.6 2gfor pregnant women, and 2.8 2g forlactating women.4 Normal total bodystores of vitamin B12 range from 2 to5 mg, with half stored in the liver. Itmay take anywhere from 2 to 5 yearsfor deficiency to develop, even in indi-viduals with severe malabsorption.
Vitamin B12 deficiency has been es-timated to affect between 1.5% and 15%of the general population.6,7 Conditionsknown to cause or be associated withvitamin B12 deficiency include malnutri-tion, atrophic gastritis, pernicious anemia(lack of intrinsic factor), and malabsorp-tion due to gastric bypass surgery orgastrointestinal disorders such as inflam-matory bowel disease. Certain medica-tions, including histamine H2 receptorantagonists8 and metformin, are knownto reduce vitamin B12 absorption.9 Evi-dence is conflicting regarding protonpump inhibitors and vitamin B12 absorp-tion, but it has been suggested that in-dividuals on long-term therapy withthese medications have their vitaminB12 levels monitored.10
Although some patients with vitaminB12 deficiency will have normal serumcobalamin levels, a serum cobalaminmeasurement is the initial test used toinvestigate suspected vitamin B12 defi-ciency. In patients with borderline-lowcobalamin levels, particularly thosestrongly suspected of vitamin B12 defi-ciency, methylmalonic acid and homo-cysteine levels should be checked.Methylmalonic acid and homocysteinelevels are increased in as many as one-third of patients with low-normal serumcobalamin levels and vitamin B12 defi-ciency. A complete blood count maydemonstrate a macrocytic anemia andthrombocytopenia, and a peripheralblood smear may show hypersegmented
KEY POINTS
h The potential neurologicmanifestations ofvitamin B12 deficiencyare varied andmay includemyeloneuropathy,cognitive and psychiatricimpairment, opticneuropathy, andorthostatic hypotension.
h The most commonmanifestation of vitaminB12 deficiency issubacute combineddegeneration, withpyramidal signs andsensory loss indicatingposterolateral spinalcord impairment.
h Some patients withclinical signs of vitaminB12 deficiency mayhave normal serumcobalamin levels. Insuch patients, elevatedmethylmalonic acid andhomocysteine levelsmay suggest vitamin B12deficiency.
Myeloneuropathy
748 www.aan.com/continuum August 2011
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

polymorphonuclear cells, basophilic stip-pling of the erythrocytes, Howell-Jollybodies, anisocytosis, and poikilocytosis.
Spinal cord MRI studies may showT2-weighted signal change in the pos-terior and lateral columns; these areasmay also enhance following the ad-ministrationofgadolinium.11 EMG stud-ies may demonstrate a predominantlyaxonal peripheral neuropathy, andsomatosensory-evoked potentials typi-cally show slowing in central somato-sensory pathways through the spinalcord. Visual-evoked potentials havebeen reported to be abnormal in 60%of patients.12 Autonomic testing maydemonstrate orthostatic hypotensionon tilt-table testing.
With laboratory confirmation of vita-min B12 deficiency, additional evaluationfor possible causes of malabsorption canbe considered. Elevated serum gastrinlevels are common but nonspecific,occurring in up to 30% of the elderlywithout pernicious anemia. Antibodiesto intrinsic factor lack both sensitivityand specificity. Antiparietal cell antibod-ies also lack specificity, as they can bepresent in up to 10% of healthy individ-uals. The Schilling test has traditionallybeen used to identify whether vitaminB12 can be absorbed. This test is nolonger widely used because of concernsover the use of radioisotopes and theinconvenience of the test.
Treatment of neurologic impairmentdue to vitamin B12 deficiency involvesthe administration of high-dose oral,sublingual, or IM cobalamin. For pa-tients with malabsorption, 1000 2g ofcobalamin is administered intramuscu-larly for 5 days and monthly thereafter.Evidence demonstrates that 1000 2gof oral or sublingual cobalamin givendaily is as effective as IM administration.6,7
Lifelong vitamin B12 supplementationtherapy is typically necessary. Completehematologic recovery occurs within thefirst 1 to 2 months. The neurologic con-
dition should stabilize, and improvementmay occur over the first 6 to 12 monthsfollowing the initiation of treatment.Neurologic recovery may be incomplete,however, particularly in those who hadsignificant neurologic deficits prior to theinitiation of therapy.
Nitrous OxideMyeloneuropathyNitrous oxide remains one of the morecommonly used anesthetic agents world-wide and can also be obtained for abusein the form of whipped cream canistersand ‘‘whippets,’’ which are small bulbscontaining nitrous oxide. Surveys ofmedical and dental students in NewZealand and the United States revealedrecreational nitrous oxide use of 12%and 20%, respectively.13,14 Nitrous oxidealters the cobalt core of cobalamin, con-verting it into an inactive, oxidized form.
Most reported cases of nitrous oxidemyeloneuropathy have been associatedwith low or borderline-low vitamin B12
levels. However, several cases withnormal vitamin B12 levels and a clinicalphenotype resembling subacute com-bined degeneration in chronic nitrousoxide abuse have been reported, sug-gesting that chronic exposure may beenough to induce neurologic impair-ment, even in the absence of vitaminB12 deficiency. A single exposure to ni-trous oxide may be enough to precipitateneurologic impairment in an individualwith unsuspected vitamin B12 deficiency.Time to symptom onset ranges from im-mediately to 2 months following nitrousoxide exposure.
Myeloneuropathy, myelopathy, peri-pheral neuropathy, and cognitivechanges have been associated with ni-trous oxide toxicity. Treatment involvescessation of nitrous oxide exposure andIM administration of vitamin B12. Sup-plemental oral methionine has beensuggested as an additional treatmentoption to potentially hasten recovery.15
KEY POINTS
h High-dose vitamin B12supplementation can beadministered orally,sublingually, orintramuscularly.
h Most cases of nitrousoxide myeloneuropathyoccur in individuals withlow or borderline-lowvitamin B12 levels.
749Continuum Lifelong Learning Neurol 2011;17(4):744–760 www.aan.com/continuum
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

A recent literature review of 18 re-ported cases suggested improvementin 15 cases. Dramatic or completeresolution of symptoms without anycorrelation with serum cobalamin levelswas observed in only four cases.16
Sensory and motor impairment per-sisted in most patients.
Copper DeficiencyMyeloneuropathyCopper is an important trace elementfound in a number of metalloenzymesinvolved in nervous system function.Examples include copper-zinc super-oxide dismutase, cytochrome c oxi-dase, and dopamine "-hydroxylase.Copper deficiency in animals was firstrecognized in sheep in 1937, manifest-ing as an enzootic ataxia (also knownas swayback), and was subsequentlynoted to affect other animals similarly.Hematologic abnormalities were thefirst signs of acquired copper defi-ciency recognized in humans, withanemia, neutropenia, and sideroblasticanemia evident in some patients. Theneurologic manifestations of acquiredcopper deficiency have been definedover the past 9 years. Copper defi-ciency in humans results in a myelo-neuropathy that is remarkably similarto the enzootic ataxia described in ani-mals with copper deficiency. Patientswith copper deficiency myeloneurop-athy present with a sensory ataxia thatresembles the subacute combined de-generation of vitamin B12 deficiency.
17
Pyramidal signs, such as brisk deep ten-don reflexes at the knees and extensorplantar responses, are typically present,along with impairment in posteriorcolumn sensory modalities. Neuro-pathic extremity pain may be reportedand distal lower limb weakness andatrophy may develop, suggesting pe-ripheral nerve involvement.
EMG studies demonstrate an axonalsensorimotor peripheral neuropathy,
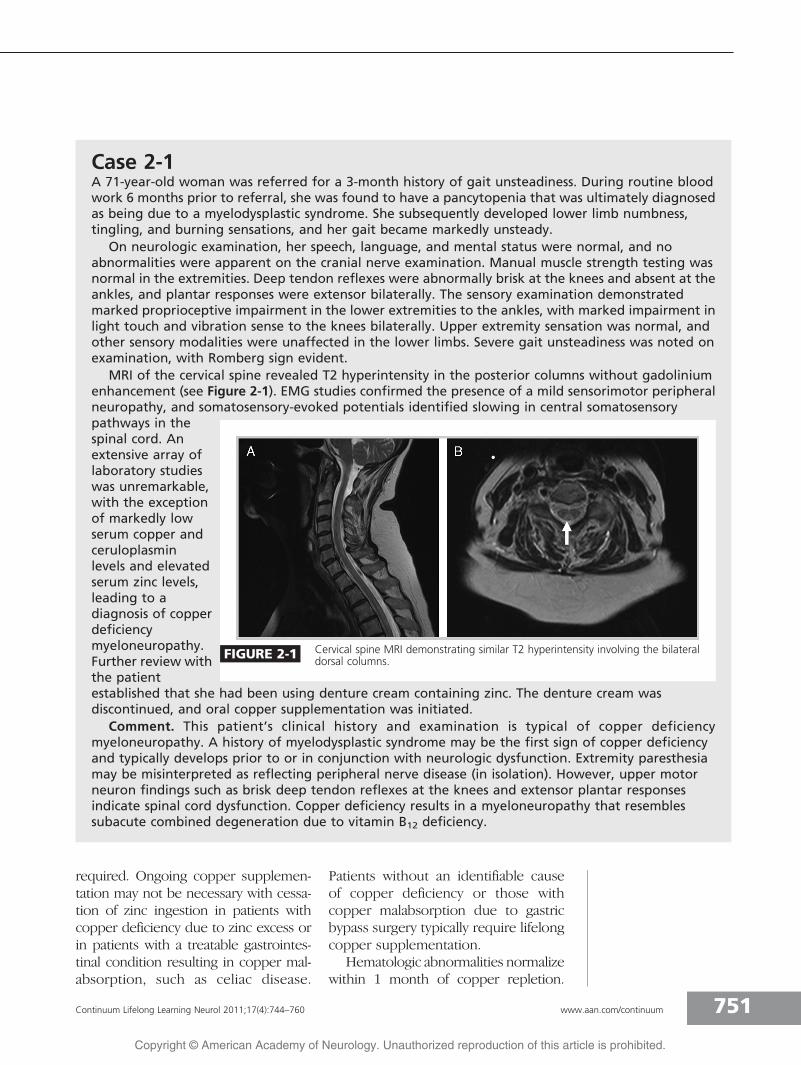
although the sensory loss evident clini-cally is not proportional to the sensorynerve action potential abnormalities onEMG.18 Slowing in central somatosen-sory pathways on somatosensory-evoked potential testing provides fur-ther neurophysiologic proof that theprimary cause of the sensory ataxia isimpairment in posterior column func-tion. Spinal cord MRI studies may showhyperintense lesions on T2-weightedsequences in the posterior columns,particularly in the cervical and thoracicspinal cord (Case 2-1).19 These find-ings resemble those seen with vitaminB12 deficiency and may improve follow-ing copper supplementation.
Low serum copper and ceruloplas-min levels establish a diagnosis ofcopper deficiency. Urinary copper lev-els are also low in copper deficiency butare increased in Wilson disease. Themost common cause of copper defi-ciency is malabsorption following bari-atric surgery, but excess zinc ingestioncan also result in copper deficiency.20
Patients should be queried about theuse of zinc supplements as well asdenture creams, some of which haveexcessive zinc and may induce copperdeficiency.21 Chronic gastrointestinalconditions such as celiac disease mayresult in copper malabsorption andsubsequent copper deficiency myelo-neuropathy and should be consideredin patients with otherwise unexplainedcopper deficiency. Some patients willnot have any identifiable cause forcopper deficiency.20
Treatment involves copper supple-mentation and, in those with excessivezinc consumption, discontinuation ofzinc. Oral copper supplementation istypically adequate. A recommended reg-imen is 8 mg of elemental copper dailyfor 1 week, followed by 6mg daily for thenext week, 4 mg daily during the thirdweek, and 2 mg daily thereafter.22 Occa-sionally IV copper supplementation is
KEY POINTS
h Patients withcopper deficiencymyeloneuropathy presentwith a sensory ataxiathat may resemblevitamin B12 deficiency.
h Low serum and urinecopper levels anddecreasedceruloplasmin levelsestablish the presenceof copper deficiency.
h Treatment ofcopper deficiencymyeloneuropathyrequires coppersupplementation andcessation of zincconsumptionwhen relevant.
Myeloneuropathy
750 www.aan.com/continuum August 2011
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.
required. Ongoing copper supplemen-tation may not be necessary with cessa-tion of zinc ingestion in patients withcopper deficiency due to zinc excess orin patients with a treatable gastrointes-tinal condition resulting in copper mal-absorption, such as celiac disease.
Patients without an identifiable causeof copper deficiency or those withcopper malabsorption due to gastricbypass surgery typically require lifelongcopper supplementation.
Hematologic abnormalities normalizewithin 1 month of copper repletion.
Case 2-1A 71-year-old woman was referred for a 3-month history of gait unsteadiness. During routine bloodwork 6 months prior to referral, she was found to have a pancytopenia that was ultimately diagnosedas being due to a myelodysplastic syndrome. She subsequently developed lower limb numbness,tingling, and burning sensations, and her gait became markedly unsteady.
On neurologic examination, her speech, language, and mental status were normal, and noabnormalities were apparent on the cranial nerve examination. Manual muscle strength testing wasnormal in the extremities. Deep tendon reflexes were abnormally brisk at the knees and absent at theankles, and plantar responses were extensor bilaterally. The sensory examination demonstratedmarked proprioceptive impairment in the lower extremities to the ankles, with marked impairment inlight touch and vibration sense to the knees bilaterally. Upper extremity sensation was normal, andother sensory modalities were unaffected in the lower limbs. Severe gait unsteadiness was noted onexamination, with Romberg sign evident.
MRI of the cervical spine revealed T2 hyperintensity in the posterior columns without gadoliniumenhancement (see Figure 2-1). EMG studies confirmed the presence of a mild sensorimotor peripheralneuropathy, and somatosensory-evoked potentials identified slowing in central somatosensorypathways in thespinal cord. Anextensive array oflaboratory studieswas unremarkable,with the exceptionof markedly lowserum copper andceruloplasminlevels and elevatedserum zinc levels,leading to adiagnosis of copperdeficiencymyeloneuropathy.Further review withthe patientestablished that she had been using denture cream containing zinc. The denture cream wasdiscontinued, and oral copper supplementation was initiated.
Comment. This patient’s clinical history and examination is typical of copper deficiencymyeloneuropathy. A history of myelodysplastic syndrome may be the first sign of copper deficiencyand typically develops prior to or in conjunction with neurologic dysfunction. Extremity paresthesiamay be misinterpreted as reflecting peripheral nerve disease (in isolation). However, upper motorneuron findings such as brisk deep tendon reflexes at the knees and extensor plantar responsesindicate spinal cord dysfunction. Copper deficiency results in a myeloneuropathy that resemblessubacute combined degeneration due to vitamin B12 deficiency.
FIGURE 2-1 Cervical spine MRI demonstrating similar T2 hyperintensity involving the bilateraldorsal columns.
751Continuum Lifelong Learning Neurol 2011;17(4):744–760 www.aan.com/continuum
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

Neurologic deficits are expected to sta-bilize, but there may be little improve-ment in neurologic signs and symptoms,particularly in those with more severeneurologic impairment.
Folate DeficiencyMyeloneuropathyThe active form of folate, tetrahydrofolicacid, is essential in the transfer of one-carbonunits to substrates used in the syn-thesis of purine, thymidine, and aminoacids. Folate is present in animal products,citrus fruits, and green, leafy vegetables.Normal body stores of folate range from5002g to 20,000 2g, and 50 2g to 100 2gare required daily. Folate is absorbed inthe proximal small intestine and ileum.
Folate deficiency does not typicallyoccur in a pure state and is character-istically associated with conditions thatresult in other nutritional deficiencies.22
Alcoholics, premature infants, and ado-lescents are populations at risk. Theprevalence of folate deficiency has de-creased in the United States and Canadasince the mandatory folic acid fortifica-tionprogrambegan in1998.General con-ditions associated with folate deficiencyincludedisorders that result inmalabsorp-tion (eg, celiac disease, bacterial over-growth, inflammatory bowel disease),states of reduced gastric acid secretion(following gastric surgery, atrophic gastri-tis, or with medications that lower gastricacid levels), or use of medications thatinhibit tetrahydrofolate reductase.
Folate deficiency has been associatedwith myelopathy, myeloneuropathy, pe-ripheral neuropathy, and cognitive andpsychiatric disorders. A clinical patternresembling subacute combined degen-eration has been described. An anemiawith macrocytosis and hypersegmentedpolymorphonuclear leukocytes is typi-cal. A low serum folate level, typicallyless than 2.5 ng/mL, can establish a di-agnosis of folate deficiency. However,it has been suggested that a range of
2.5 ng/mL to 5 ng/mL may reflect mildlycompromised folate status. Plasmahomocysteine is elevated in folate defi-ciency and is useful to confirm thepresence of folate deficiency and tomonitor the response to folate supple-mentation. Serum cobalamin levelsshould be assessed with suspectedfolate deficiency, and, if cobalamin lev-els are low, vitamin B12 supplementa-tion should be immediately initiated.Oral administration of folic acid maybe adequate; a typical regimen is 1 mg3 times a day followed by a mainte-nance dose of 1 mg daily.22 Parenteraladministration of folic acid may be con-sidered in patients who are acutely ill.
Vitamin E DeficiencyMyeloneuropathyVitamin E is a fat-soluble vitamin withimportant antioxidant properties thatprotect against oxidative stress and in-hibit the fatty acid peroxidation ofmembrane phospholipids. Foods richin vitamin E include nut oils, sunflowerseeds, whole grains, wheat germ, andspinach. Vitamin E absorption requiresbile salts and pancreatic esterases. Vita-min E is incorporated into chylomi-crons in intestinal enterocytes, and,upon its release into the circulation,lipolysis ensues, resulting in the transferof vitamin E to high-density and otherlipoproteins.22 !-Tocopherol transfer pro-tein in the liver is responsible for theincorporation of vitamin E into very low-density lipoprotein (VLDL), which deliversvitamin E to tissues.
Numerous neurologic manifestationsof vitamin E deficiency have been re-ported, including ophthalmoplegia, retin-opathy, and a spinocerebellar syndromewith an associated peripheral neuropathyresembling Friedreich ataxia. The last-mentioned can be considered a myelo-neuropathy syndrome that clinicallymanifests with signs of a cerebellar ataxia,posterior column sensory loss, pyramidal
KEY POINTS
h Folate deficiencytypically occurs inassociation with othervitamin deficiencies.
h Myeloneuropathy,myelopathy, peripheralneuropathy, andcognitive and psychiatricsymptoms have beenattributed tofolate deficiency.
h Vitamin E deficiencymay result inophthalmoplegia,retinopathy, or aspinocerebellarsyndrome.
Myeloneuropathy
752 www.aan.com/continuum August 2011
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

signs, and signs of peripheral sensoryloss. Somatosensory-evoked potentialstypically indicate central slowing, andEMG studies confirm an axonal, primarilysensory peripheral neuropathy.
Acquired causes of vitamin E defi-ciency include chronic gastrointestinaldisorders that result in malabsorption,liver disease, and pancreatic insufficiency.Hereditary disorders of vitamin E metab-olism should be considered, particularlyin children without evidence of gastro-intestinal disease. These conditions in-clude defects in the tocopherol (alpha)transfer protein gene, the apolipopro-tein B (including Ag(x) antigen) gene,and the microsomal triglyceride transferprotein gene and defects in chylomicronsynthesis and secretion.23
Low serum vitamin E levels establishthe diagnosis. Serum lipids, cholesterol,and VLDL affect serum vitamin E levels.Serum vitamin E levels can be correctedfor these factors by dividing serumvitamin E levels by the sum of serumtriglycerides and cholesterol.22 VitaminE supplementation using doses rang-ing from 200 mg/d to 200 mg/kg/d maybe necessary. Parenteral administrationmay be required with some conditions,particularly those involving severe mal-absorption. Unless the cause of thevitamin E deficiency is reversible, life-long supplementation may be needed.
INFLAMMATORY DISORDERSParaneoplasticMyeloneuropathyA paraneoplastic syndrome results froman immune-mediated response trig-gered by an underlying malignancy.Signs and symptoms of a paraneoplasticdisorder may occur in patients with well-established cancer or those thought tobe in remission; they may also precedethe diagnosis of cancer. A paraneoplasticmyeloneuropathy is a rare manifestationof a paraneoplastic syndrome but shouldbe considered in individuals with symp-
toms that develop subacutely over weeksto months and in individuals with a his-tory ofmalignancy. Other factors, such asa history of smoking or a family history ofmalignancy, may suggest an increasedlikelihood of a paraneoplastic cause.
A myriad of other neurologic mani-festations of paraneoplastic disordershas been reported, including limbic en-cephalitis, ataxia, Lambert-Eaton myas-thenic syndrome, chorea, myelopathy,peripheral neuropathy (often sensorypredominant), and autonomic neuro-pathy. The presence of one or more ofthese other signs in a patient with a mye-loneuropathy may suggest a possibleparaneoplastic etiology. For example, arecent report describes a patient withacute-onset chorea with signs and symp-toms of transverse myelitis and periph-eral neuropathy.24
Elevated paraneoplastic antibodiesmay provide evidence in favor of a para-neoplastic etiology. The most commonantibody reported in association withparaneoplastic myelopathy is antineu-ronal nuclear antibody 1 (anti-Hu), al-though a number of others have beenreported, including ampiphysin, neuro-myelitis optica, antineuronal nuclear anti-body 2 (anti-Ri), voltage-gated potassiumchannel, glutamate decarboxylase 65,and Purkinje cell antibody type 2.25 Para-neoplastic peripheral neuropathy is typi-cally axonal and may involve sensoryand motor fibers (sensorimotor) or maybe isolated to sensory nerves (sensoryneuronopathy). Occasionally the neu-ropathy may be demyelinating, as de-scribed in a recently reported case of asteroid-responsive demyelinating neuro-pathy and myelopathy in a patient withbreast carcinoma.26
When a paraneoplastic disorder issuspected in an individual without anestablished cancer, diagnostic studiesshould be performed to identify an oc-cult malignancy. Immune-modulatingtherapies, such as IV methylprednisolone
KEY POINTS
h A paraneoplasticneurologic disorder mayprecede a clinicaldiagnosis of cancer,occur in those withestablished cancer, oroccur in patientsthought to bein remission.
h When a paraneoplasticdisorder is suspected inan individual without anestablished cancer,diagnostic studiesshould be performed toidentify an occultmalignancy.
753Continuum Lifelong Learning Neurol 2011;17(4):744–760 www.aan.com/continuum
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

and IV immunoglobulin, can be consid-ered in patients without an identifiableunderlying neoplasm.
Peripheral Neuropathy inMultiple SclerosisPeripheral neuropathy is not uncom-mon in multiple sclerosis, althoughclinically significant peripheral nerve dis-ease is rare. In one prospective study of22 patients with multiple sclerosis, 10had abnormalities on EMG testing, withboth axonal and demyelinating featurespresent.27 Sural nerve biopsy specimensin 10 patients with multiple sclerosisshowed a reduction in myelin thicknessand reduced numbers of myelin lamellaecompared with controls.28 Several pa-tients with both central and peripheralnervous system demyelinating diseasewith clinical and diagnostic featuressuggesting concomitant multiple scle-rosis and an acute or chronic demyeli-nating polyradiculoneuropathy havebeen reported. Such patients may pre-sent with a myeloneuropathic pheno-type, and a common pathogenic eventprecipitating spread of T-cell autoim-munity from central to peripheral mye-lin has been suggested.29 Treatmentprotocols for patients with both centraland peripheral nervous system demye-linating disease are not well estab-lished. Some combination of multiplesclerosis disease-modifying therapiesand other immune-modulating thera-pies such as plasma exchange may benecessary.
Sjogren SyndromeSjogren syndrome (SS) is a chronic, sys-temic autoimmune disorder associatedwith exocrine gland impairment thatmanifests as xerophthalmia, xerostomia,and a wide variety of neurologic andsystemic visceral disorders. Peripheralnervous system disease is a major man-ifestation of SS and often precedes theclinical diagnosis. A large cohort of 92
patients with SS classified the forms ofneuropathy as a sensory ataxic neuro-pathy, painful sensory neuropathy, multi-ple mononeuropathy, multiple cranialneuropathy, trigeminal neuropathy, auto-nomic neuropathy, and radiculoneu-ropathy.30 Among the patients with asensory ataxic neuropathy, hyperintensityon T2-weighted cervical spinal cord MRIstudies was seen in 9 of 12 patients.Myelopathy is a considerably less fre-quent manifestation of SS than periph-eral neuropathy. Myelopathy in SS canbe acute, resembling transverse myelitis,or it can be chronic and progressive.
Sural nerve biopsies in patients withSS neuropathy suggest a predominantlyaxonal process and may show findingsof chronic vasculitis of epineurial arte-rioles and mild small vessel perivascularlymphocytic infiltrates.30 Dorsal rootganglion biopsies and necropsy studieshave also demonstrated lymphocyticinfiltration, suggesting a ganglioneu-ronitis may be the cause of the sen-sory ataxic form of neuropathy seen inSS. Similar pathogenic mechanismslikely occur in SS myelopathy. Immune-modulating therapy is considered in SSneuropathy and myelopathy, includingcorticosteroids, plasma exchange, IVimmunoglobulin, methotrexate, inflixi-mab, and cyclophosphamide. Anecdo-tal reports of these medications usedsolely or in combination in patientswith SS have been reported. Systema-tic randomized controlled studies havenot been performed in patients with SSneuropathy and myelopathy. It hasbeen suggested that patients with mul-tiple mononeuropathy or multiple cra-nial neuropathy respond favorably tocorticosteroids, whereas patients withsensory neuropathy and radiculoneur-opathy respond more favorably to IVimmunoglobulin.30 Several anecdotalreports indicate that patients with SSmyelopathy have a favorable responseto corticosteroids, and, in a recently
KEY POINTS
h Paraneoplasticsyndromes respondmost favorably totreatment of theunderlying tumor.
h Peripheral neuropathymay occur in patientswith multiple sclerosisand can rarely manifestas a chronicdemyelinatingpolyradiculoneuropathy.
h Neurologicmanifestationsmay precede thedevelopment of siccasymptoms in patients withSjogren syndrome.
h Peripheral neuropathyis the most commonneurologic manifestationof Sjogren syndrome.
h Myelopathy in Sjogrensyndrome can be acute,resembling transversemyelitis, or chronic andprogressive.
h Immune-modulatingtherapy is considered inSjogren syndromeneuropathy andmyelopathy.
Myeloneuropathy
754 www.aan.com/continuum August 2011
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

reported series of patients with severemyelopathy, 12 of 14 responded favor-ably to a combination of corticosteroidand cyclophosphamide therapy.31,32
The diagnosis of SS can be challeng-ing, particularly when the initial mani-festations are neurologic. Neurologicmanifestations may precede symptomsof exocrine gland failure, and few or noadditional systemic manifestations of SSmay be present. The San Diego andEuropean classification criteria requireobjective evidence of exocrine gland im-pairment, positive autoantibodies, andcharacteristic findings on salivary glandbiopsy. The Schirmer test and rose bengalstaining can be used to demonstratexerophthalmia, and low salivary flow ratesdemonstrated by salivary flow studies,sialography, or salivary scintiscanningestablish the presence of xerostomia.Salivary gland biopsy may demonstratelymphocytic infiltrates with atrophy ofacinar tissue. Rheumatoid factor andantinuclear antibodies are typically ele-vated in SS. Anti-Ro (SSA) and anti-La(SSB) antibodies are elevated in mostpatients with SS.
NeurosarcoidosisSarcoidosis is a granulomatous disorderthat can affect virtually any organ sys-tem and any component of the nervoussystem. Historically, nervous systeminvolvement has been reported in ap-proximately 5% of individuals with sar-coidosis, but recent prospective studieshave suggested a higher frequency ofneurologic involvement. Autopsy stud-ies indicate that only 50% of affectedindividuals are diagnosed with neuro-sarcoidosis prior to death. Sarcoid myel-opathy can be subacute, chronic andprogressive, or relapsing-remitting. Spi-nal cord involvement may be extrame-dullary (either intradural or extradural)or intramedullary. Not surprisingly, amyriad of clinical manifestations is pos-sible, including myelopathic signs and
symptoms and signs of segmental radi-culopathy at affected levels. Polyradiculo-pathy, including cauda equina syndromeand peripheral neuropathy, may accom-pany myelopathic findings (Case 2-2).
The diagnosis of sarcoidosis can bequite challenging, particularly when thesarcoidosis is isolated to the nervoussystem. Serum angiotensin-converting en-zyme levels are not particularly sensitive(they are increased in only two-thirds ofpatients), and chest x-ray is abnormal inonly 30% of patients. Hilar and para-tracheal lymphadenopathy may be betterappreciated on chest CT. Spinal cord MRIstudies may demonstrate leptomeningealenhancement along the surface of thecord and nerve roots. Intramedullary in-volvement may be associated with hyper-intense signal change on T2-weightedimaging, and enhancement may followthe administration of gadolinium. CSFstudies may demonstrate a lymphocyticpleocytosis with depressed glucose andhigh protein levels but can be normal insomepatients.CSFangiotensin-convertingenzyme levels are unreliable.
No standard treatment protocols existfor neurosarcoidosis, but corticosteroidshave traditionally been the initial drugof choice. Other immune-modulatingtherapies are considered when cortico-steroid treatment is ineffective or inpatients who develop adverse steroid-related side effects. Other pharmaco-logic options include methotrexate,cyclophosphamide, azathioprine, chlor-oquine and hydroxychloroquine, thali-domide, and infliximab.33
INFECTIOUS DISORDERS
Myelopathy Associated withHuman T-cell LymphotropicVirus Type I or TropicalSpastic ParaparesisPrior to the descriptions of HTLV-IYassociated tropical spastic paraparesis(TSP) andHTLV-IYassociatedmyelopathy
KEY POINTS
h Nervous systeminvolvement has beenreported inapproximately 5%of individuals withsarcoidosis.
h Spinal cord involvementin sarcoidosis may beextramedullary orintramedullary.
h Sarcoid polyradiculopathyand peripheral neuropathymay accompanymyelopathy or otherCNS disease or may occurin isolation.
h Corticosteroids aretypically the initialtreatment of choice inneurosarcoidosis.
755Continuum Lifelong Learning Neurol 2011;17(4):744–760 www.aan.com/continuum
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.
(HAM) in the mid-1980s, tropical spasticparaparesis was a general term used todescribe a chronic progressive myelop-athy in equatorial countries. It is nowrecognized that at least 80%of individualswho would have previously been diag-nosed with TSP are HTLV-I positive.HTLV-I is a retrovirus that can be trans-mitted sexually, via contaminated nee-dles, and through breastmilk. Breastmilktransmission has been implicated in the
maintenance of infection in high preva-lence areas. HTLV-I is endemic in south-ern Japan, parts of Africa, the Caribbean,and Papua New Guinea. Seropositivityincreases with age and is more commonin females. It has been suggested thatonly 2% of carriers develop HAM/TSP.
HAM/TSP manifests neurologically asa chronic progressive myelopathic disor-der with spasticity, weakness, hyper-reflexia, andurinary symptoms. Peripheral
Case 2-2A 52-year-old female was referred for evaluation of lower limb pain and gait unsteadiness. Shereported a 2-year history of progressive, asymmetric sensory loss affecting the lower extremities andprogressive gait unsteadiness. She experienced severe, burning pain in the lower extremities, and shedeveloped constipation and urinary incontinence. Toward the end of this 2-year period she had begunto fall and had started using a walker.
Neurologic evaluation showed normal cranial nerve and upper extremity strength and sensation.Manual muscle strength testing in the lower limbs revealed right-greater-than-left distal weaknesswith mild spasticity. Deep tendon reflexes were brisk at the knees and absent at the ankles, andextensor plantar responses were noted. The sensory examination showed diminished sensation topinprick and light touch to the knees, with impairment in proprioception in the feet bilaterally. Nodefinite truncal sensory level was identified. Gait was wide based, and a prominent Romberg signwas noted.
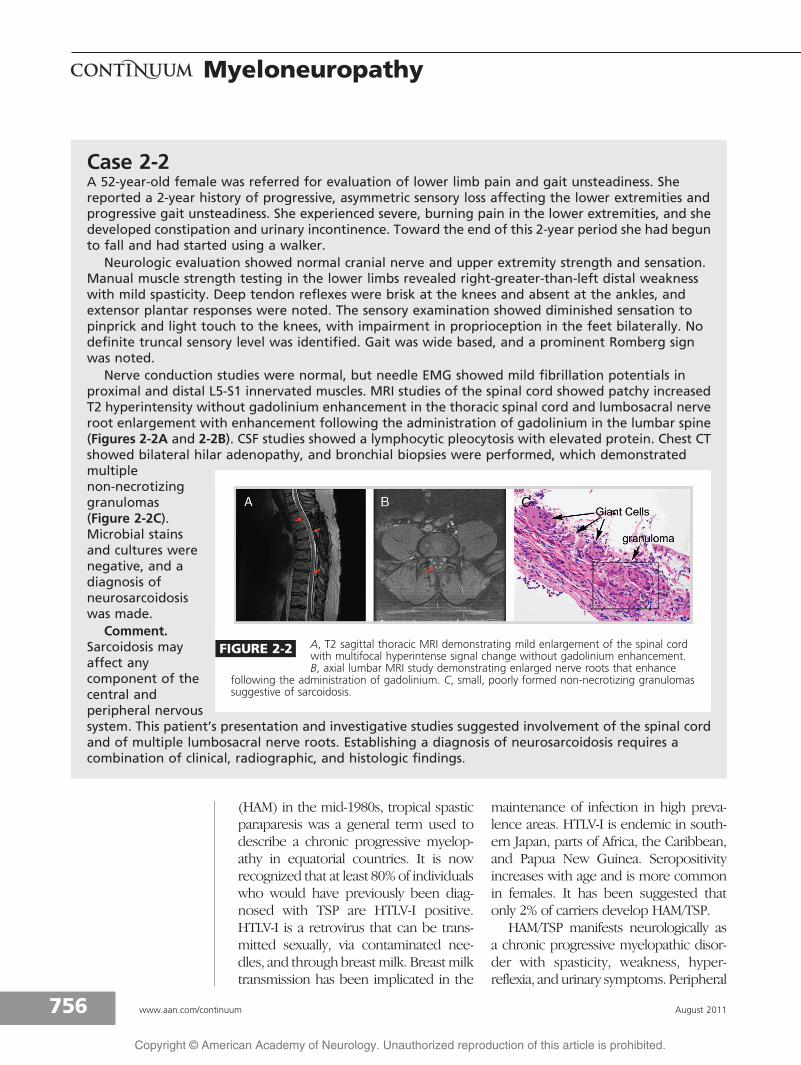
Nerve conduction studies were normal, but needle EMG showed mild fibrillation potentials inproximal and distal L5-S1 innervated muscles. MRI studies of the spinal cord showed patchy increasedT2 hyperintensity without gadolinium enhancement in the thoracic spinal cord and lumbosacral nerveroot enlargement with enhancement following the administration of gadolinium in the lumbar spine(Figures 2-2A and 2-2B). CSF studies showed a lymphocytic pleocytosis with elevated protein. Chest CTshowed bilateral hilar adenopathy, and bronchial biopsies were performed, which demonstratedmultiplenon-necrotizinggranulomas(Figure 2-2C).Microbial stainsand cultures werenegative, and adiagnosis ofneurosarcoidosiswas made.
Comment.Sarcoidosis mayaffect anycomponent of thecentral andperipheral nervoussystem. This patient’s presentation and investigative studies suggested involvement of the spinal cordand of multiple lumbosacral nerve roots. Establishing a diagnosis of neurosarcoidosis requires acombination of clinical, radiographic, and histologic findings.
FIGURE 2-2 A, T2 sagittal thoracic MRI demonstrating mild enlargement of the spinal cordwith multifocal hyperintense signal change without gadolinium enhancement.B, axial lumbar MRI study demonstrating enlarged nerve roots that enhance
following the administration of gadolinium. C, small, poorly formed non-necrotizing granulomassuggestive of sarcoidosis.
Myeloneuropathy
756 www.aan.com/continuum August 2011
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

neuropathy can also occur in patientswho are HTLV-I seropositive, and suralnerve biopsies in affected patients havesuggested the presence of amixed axonaland demyelinating process.34 Optic neu-ropathy, myositis, and cerebellar ataxiahave been attributed to HTLV-I infec-tion. A number of other non-neurologicconditions are associated with HTLV-I,including uveitis, arthritis, pulmonarylymphocytic alveolitis, keratoconjunc-tivitis sicca, dermatitis, and T-cell leu-kemia or lymphoma. HTLV-II has beenassociated with spastic paraparesis andother non-neurologic manifestationssimilar to HTLV-I.
ELISAmay indicateHTLV-I andHTLV-II infection, but Western blot, immuno-fluorescence assay, or PCR must beperformed subsequently to confirmthe diagnosis. Patients diagnosed withHTLV-I or HTLV-II should be screenedfor HIV. Treatment of HTLV infection issupportive at this time.
Tropical ataxic neuropathy is a generalterm used to describe a lower motorneuronYpredominant syndrome associ-atedwith nutritional deficiencies (likely Bvitamins) and cassava consumption.Cassava, an important caloric source intropical countries, contains cyanide,which may be the neurotoxin respon-sible for tropical ataxic neuropathy. Thecyanide content in cassava increasesduring drought, and inadequate process-ing of cassava flour during times ofdrought may additionally increase therisk of cyanide toxicity.
HIVA vacuolar myelopathy may occurduring the late stages of HIV infectionwhenCD4+ lymphocyte counts are low.The vacuolar myelopathy may occur inconjunction with AIDS dementia com-plex and a sensory neuropathy. Sincethe introduction of highly active antire-troviral therapy (HAART), the frequencyof HIV-associated myelopathy has de-
creased from as high as 55% to lessthan 10%. A chronic, slowly progressivespastic paraparesis with a sensory ataxiaand urinary incontinence ensues. A sen-sory neuropathy develops in up to 30%ofindividuals infected with HIV, typicallylate in the course. EMG studies indicatean axonal sensorimotor peripheral neuro-pathy, and autopsy studies have demon-strated particularly severe involvement ofdistal unmyelinated nerves.
CD4+ lymphocyte counts should bechecked, if they are not already known,and vitamin B12 and folate deficiencyshould be excluded. CSF studiesshould be normal in HIV-associatedvacuolar myelopathy and are helpfulin excluding other infectious causes ofmyelopathy, such as cytomegalovirus,herpes simplex virus, varicella-zostervirus, and HTLV-I and HTLV-II infections.MRI studies are indicated to rule outmalignancy and infection. MRI findingsin vacuolar myelopathy are variable butmay show spinal cord atrophy, mostcommonly in the thoracic cord, andnonenhancing, hyperintense signalchange on T2-weighted sequences dif-fusely or primarily in the posterior cord.Anecdotal reports suggest improvementwith HAART.
HEREDITARY DISORDERSAdrenomyeloneuropathyAdrenomyeloneuropathy is an X-linkeddisorder of peroxisomal lipidmetabolismthat leads to abnormal deposition of verylong-chain fatty acids (VLCFAs) in varioustissues, including peripheral and centralmyelin. Neurologic manifestations typi-cally become apparent in the third tofifth decade as a chronic, progressivespastic paraparesis. A peripheral neuro-pathy is present in most patients butmay be difficult to appreciate clinicallybecause of prominent pyramidal signs.EMG studies characteristically suggest amixed axonal and demyelinating pattern,
KEY POINTS
h Most cases of tropicalspastic paraparesisresult from human T-celllymphotropic virustype I infection.
h Tropical spastic paraparesisassociated with humanT-cell lymphotropic virustype I manifestsneurologically as a chronicprogressive myelopathicdisorder with spasticity,weakness, hyperreflexia,and urinary symptoms.
h Tropical ataxic neuropathyis a general term used todescribe a lower motorneuronYpredominantsyndrome associated withnutritional deficienciesand cassava consumption.
h A vacuolar myelopathymay occur during the latestages of HIV infectionwhen CD4+ lymphocytecounts are low.
h Vacuolar myelopathymay occur in conjunctionwith AIDS dementiacomplex and a sensoryneuropathy.
h Neurologic manifestationsof adrenomyeloneuropathytypically becomeapparent in the third tofifth decade as a chronic,progressive spasticparaparesis.
h A peripheral neuropathyis present in mostpatients withadrenomyeloneuropathybut may be difficult toappreciate clinicallybecause of prominentpyramidal signs.
757Continuum Lifelong Learning Neurol 2011;17(4):744–760 www.aan.com/continuum
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

and MRI studies may show spinal cordatrophy (as shown in Case 2-3). Abnor-mal ratios of VLCFAs C:24/C:22 andC:26/C:22 establish a diagnosis andcan be further confirmed through ge-netic analysis of the ATP-binding cas-
sette, sub-family D (ALD), member 1gene, ABCD1. A double-blind, placebo-controlled trial of glyceryl trioleate-glyceryl trierucate (Lorenzo oil) iscurrently recruiting patients with adre-nomyeloneuropathy.35
Case 2-3A 45-year-old man was referred for evaluation of a 10-year history of progressive gait unsteadiness.He reported normal early developmental milestones and played sports competitively throughoutchildhood and in high school. At the age of 35 he recalled becoming increasingly clumsy andultimately stopped running because of clumsiness and frequent tripping. Soon thereafter he becameaware of sensory loss in the feet and distal lower extremities and developed progressive impairmentin bowel and bladder function. Over the past few years he began to notice leg spasms and beganusing a cane while ambulating to prevent falls.
He was not aware of any other family members with neurologic disease, and he did not have anyother significant medical problems. He had no history of cognitive impairment, vision or hearing loss,or seizures.
The neurologicexaminationdemonstrated normalspeech, language, andmental status. Cranialnerve examinationwas normal. Upperextremity strengthwas normal, butmarked distal lowerlimb weakness andatrophy were notedwith moderatebilateral spasticity.Generalizedhyperreflexia waspresent with sustained clonus and extensor plantar responses. Sensation to pinprick, light touch,vibration, and proprioception were impaired in the lower limbs. A spastic gait and Romberg signwere noted.
EMG studies were performed and showed evidence of a diffuse mixed axonal-demyelinatingperipheral neuropathy (Figure 2-3A), and somatosensory-evoked potentials showed slowing in centralproprioceptive pathways, most prominently in thoracic spinal cord segments. Spinal cord MRI studiesdemonstrated diffuse atrophy without signal change (Figure 2-3B). Abnormalities on very long-chainfatty acid testing suggested adrenomyeloneuropathy, which was subsequently confirmed by thepresence of a mutation on the ATP-binding cassette, sub-family D (ALD), member 1 gene, ABCD1.
Comment. This patient has a history and examination consistent with adrenomyeloneuropathy.Upper motor neuron signs and symptoms predominate, with flexor spasms, spasticity, and extensorplantar responses. While the peripheral sensory loss can occur in both myelopathic and neuropathicdisorders, the presence of distal weakness and atrophy most reliably establishes the presence of aperipheral neuropathy in this patient. Adrenomyeloneuropathy is an X-linked disorder that resultsfrom the accumulation of very long-chain fatty acids.
FIGURE 2-3 A, tibial motor nerve conduction study demonstrating slow conduction velocitywithout temporal dispersion or conduction block and normal amplitude. B,sagittal T1 thoracic MRI study demonstrating marked atrophy of the spinal cord.
Myeloneuropathy
758 www.aan.com/continuum August 2011
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

CerebrotendinousXanthomatosisCerebrotendinous xanthomatosis (CTX)is a lipid storage disorder characterized bycataract development in childhood, xan-thomas, and neurologic signs and symp-toms that develop in adulthood. Diarrheain infancy may be the earliest manifesta-tion of cerebrotendinous xanthomatosis.Xanthomas typically occur on the Achillestendon, the extensor tendons of theupper limbs, and in the neck. Neurologicmanifestations include dementia, psychi-atric disease, cerebellar ataxia, myelop-athy, and peripheral neuropathy. Normalor low serum cholesterol and elevatedplasma cholestanol suggest the diagnosis.Genetic testing of the cytochrome P450,family 27, subfamily A, polypeptide 1gene,CYP27A1, is commercially available andcan confirm the diagnosis. Chenodeoxy-cholic acid is the treatment of choice andmay result in clinical improvement.
Hereditary Spastic ParaplegiaHereditary spastic paraplegia (HSP) isassociated with lower limb weakness,spasticity, hyperreflexia, extensor plantarresponses, and sensory loss. HSP is con-sidered ‘‘pure’’ when no other neurol-ogic signs or symptoms are present and‘‘complicated’’ when other neurologicmanifestations are apparent. Other neu-rologic manifestations include peripheralneuropathy, dementia, seizures, amyo-trophy, and extrapyramidal findings. HSPis associated with autosomal-dominant,autosomal-recessive, and X-linked pat-terns of inheritance. Nearly 40 geneticmutations have been identified as beingassociated with HSP, some of which areable to be tested for commercially. MRIfindings are typically normal inHSP.Moreinformation on HSP can be found in thearticle ‘‘Hereditary Myelopathies.’’
CONCLUSIONThe identification of a myeloneuropathyrequires clinical recognition of a combi-
nation of upper and lower motor neu-ron signs and symptoms during theneurologic examination. The pattern ofneurologic impairment may suggestcertain diagnoses and facilitate an accu-rate and timely diagnosis.
REFERENCES1. Russell JSR, Batten FE, Collier J. Subacute
combined degeneration of the spinal cord.Brain 1900;23:39Y110.
2. Healton EB, Savage DG, Brust JC, et al.Neurologic aspects of cobalamin deficiency.Medicine (Baltimore) 1991;70(4):229Y245.
3. Kayser-Gatchalian MC, Neundorfer B.Peripheral neuropathy with vitamin B12deficiency. J Neurol 1977;214(3):183Y193.
4. National Institutes of Health Office ofDietary Supplements. Dietary supplementfact sheet: vitamin B12. ods.od.nih.gov/factsheets/vitaminb12/. Updated May 26,2010. Accessed August 29, 2010.
5. Elia M. Oral or parenteral therapy for B12deficiency. Lancet 1998;352(9142):1721Y1722.
6. Vidal-Alaball J, Butler CC, Cannings-John R,et al. Oral vitamin B12 versus intramuscularvitamin B12 for vitamin B12 deficiency.Cochrane Database Syst Rev 2005;(3):CD004655.
7. Butler CC, Vidal-Alaball J, Cannings-John R,et al. Oral vitamin B12 versus intramuscularvitamin B12 for vitamin B12 deficiency: asystematic review of randomized controlledtrials. Fam Pract 2006;23(3):279Y285.
8. Force RW, Nahata MC. Effect of histamineH2-receptor antagonists on vitamin B12absorption. Ann Pharmacother 1992;26(10):1283Y1286.
9. de Jager J, Kooy A, Lehert P, et al. Long termtreatment with metformin in patients withtype 2 diabetes and risk of vitamin B-12deficiency: randomised placebo controlledtrial [published online ahead of print May20, 2010]. BMJ. doi: 10.1136/bmj.c2181.
10. Natural Medicines Comprehensive Database.www.naturaldatabase.com. AccessedAugust 29, 2010.
11. Ravina B, Loevner LA, Bank W. MR findingsin subacute combined degeneration of thespinal cord: a case of reversible cervicalmyelopathy. AJR Am J Roentgenol 2000;174(3):863Y865.
12. Pandey S, Kalita J, Misra UK. A sequential studyof visual evoked potential in patients withvitamin B12 deficiency neurological syndrome.Clin Neurophysiol 2004;115(4):914Y918.
KEY POINTS
h Neurologicmanifestationsof cerebrotendinousxanthomatosis includedementia, psychiatricdisease, cerebellar ataxia,myelopathy, andperipheral neuropathy.
h Chenodeoxycholic acid isthe treatment of choicefor cerebrotendinousxanthomatosis and mayresult in clinicalimprovement.
759Continuum Lifelong Learning Neurol 2011;17(4):744–760 www.aan.com/continuum
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.

13. Rosenberg H, Orkin FK, Springstead J. Abuseof nitrous oxide. Anesthe Analg 1979;58(2):104Y106.
14. Ng J, O’Grady G, Pettit T, Frith R. Nitrousoxide use in first-year students at AucklandUniversity. Lancet 2003;361(9366):1349Y1350.
15. Alt RS, Morrissey RP, Gang MA, et al.Severe myeloneuropathy from acutehigh-dose Nitrous Oxide (N(2)O) abuse[published online ahead of print June 3,2010]. J Emerg Med. doi: 10.1016/j.jemermed.2010.04.020.
16. Singer MA, Lazaridis C, Nations SP, Wolfe GI.Reversible nitrous oxide-inducedmyeloneuropathy with pernicious anemia:case report and literature review. MuscleNerve 2008;37(1):125Y129.
17. Kumar N, Gross JB, Ahlskog JE. Copperdeficiency myelopathy produces a clinicalpicture like subacute combineddegeneration. Neurology 2004;63(1):33Y39.
18. Goodman BP, Bosch EP, Ross MA, et al.Clinical and electrodiagnostic findings incopper deficiency myeloneuropathy. JNeurol Neurosurg Psychiatry 2009;80(5):524Y527.
19. Kumar N, Ahlskog JE, Klein CJ, Port JD.Imaging features of copper deficiencymyelopathy: a study of 25 cases.Neuroradiology 2006;48(2):78Y83.
20. Kumar N. Copper deficiency myelopathy(human swayback). Mayo Clin Proc 2006;81(10):1371Y1384.
21. Nations SP, Boyer PJ, Love LA, et al.Denture cream: an unusual source of excesszinc, leading to hypocupremia andneurologic disease. Neurology 2008;71(9):639Y643.
22. Kumar N. Neurologic presentations ofnutritional deficiencies. Neurol Clin 2010;28(1):107Y170.
23. Kumar N. Metabolic and toxic myelopathies.Continuum Lifelong Learning Neurol 2008;14(3):91Y115.
24. Kleinig TJ, Thompson PD. Kneebone CS.Chorea, transverse myelitis, neuropathy anda distinctive MRI: paraneoplasticmanifestations of probably small cell lungcancer. J Clin Neurosci 2009;16(7):964Y966.
25. Graber JJ, Nolan CP. Myelopathies in patientswith cancer. Arch Neurol 2010;67(3):298Y304.
26. Rajabally YA, Qaddoura B, Abbott RJ.Steroid-responsive paraneoplasticdemyelinating neuropathy and myelopathyassociated with breast carcinoma. J ClinNeuromuscul Dis 2008;10(2):65Y69.
27. Sarova-Pinhas I, Achiron A, Gilad R, Lampl Y.Peripheral neuropathy in multiple sclerosis:a clinical and electrophysiologic study. ActaNeurol Scand 1995;91(4):234Y238.
28. Pollock M, Calder C, Allpress S. Peripheralnerve abnormality in multiple sclerosis. AnnNeurol 1977;2(1):41Y48.
29. Falcone M, Scalise A, Minisci C, et al.Spreading of autoimmunity from central toperipheral myelin: two cases of clinicalassociation between multiple sclerosis andchronic inflammatory demyelinatingpolyneuropathy. Neurol Sci 2006;27(1):58Y62.
30. Mori K, Iijima M, Koike H, et al. The widespectrum of clinical manifestations inSjogren’s syndrome-associated neuropathy.Brain 2005;128(pt 11):2518Y2534.
31. de Seze J, Delalande S, Fauchais AL, et al.Myelopathies secondary to Sjogren’ssyndrome: treatment with monthlyintravenous cyclophosphamide associatedwith corticosteroids. J Rheumatol 2006;33(4):709Y711.
32. Williams CS, Butler E, RomanGC. Treatment ofmyelopathy in Sjogren syndrome with acombination of prednisone andcyclophosphamide. Arch Neurol 2001;58(5):815Y819.
33. Terushkin V, Stern BJ, Judson MA, et al.Neurosarcoidosis: presentations andmanagement [erratum published inNeurologist 2010;16(2):140]. Neurologist2010;16(1):2Y15.
34. Kiwaki T, Umehara F, Arimura Y, et al.The clinical and pathological features ofperipheral neuropathy accompanied withHTLV-I associated myelopathy.J Neurol Sci 2003;206(1):17Y21.
35. U.S. National Institutes of Health:ClinicalTrials.gov. A phase III trial ofLorenzo’s oil in adrenomyeloneuropathy.clinicaltrials.gov/ct2/show/NCT00545597.Accessed September 2, 2010.
Myeloneuropathy
760 www.aan.com/continuum August 2011
Copyright @ American Academy of Neurology. Unauthorized reproduction of this article is prohibited.