Download - Sesion de inmunodeficiencias

Catalina BoverMargarida Salvà

Casos clínicos

Antecedentes personales y familiaresCaso 1 Caso 2
• Lactante de 7 semanas• Embarazo y periodo perinatal
inmediato sin incidencias• Lactancia mixta• Calendario vacunal no iniciado• No alergias medicamentosas
conocidas• Barrera idiomática
• Padres y 2 hermanos sanos. Niegan consanguinidad
• Lactante de 22 meses• Embarazo y periodo perinatal
sin incidencias• Lactancia artificial. Beikost
normal• Desarrollo psicomotor normal• Calendario vacunal al día
(incluida antineumocócica)• No alergias medicamentosas
conocidas
• No antecedentes familiares de interés. No consanguinidad

Antecedentes patológicosCaso 1 Caso 2
• Dermatitis desde los primeros días de vida
• Fallo de medro
• Ingreso a los 2 meses por neumonía bilateral que requirió ingreso en UCIP, intubación y ventilación mecánica.
• Otitis y bronquitis de repetición.

Enfermedad actualCaso 1 Caso 2
• Sepsis-meningitis con evolución tórpida
• Dermatitis descamativa severa
• Hepatomegalia
• Aftas orofaríngeas
• Exantema cutáneo

Exploración físicaCaso 1 Caso 2
• Fotos • Fotos

Orientación diagnóstica
Inmunodeficiencia primaria

Pruebas complementariasCaso 1 Caso 2
• Déficit de gammaglobulinas
• Ausencia de linfocitos B en periferia
• Elevación de IgE
• Estudio genético:▫ Mutación en los genes
RAG 1 y RAG 2
• Linfopenia de la subpoblación T CD4+
• Resto de poblaciones linfocitarias normales
• Ig’s y complemento normales
• Estudio genético:▫ Pendiente
Síndrome de Ommen

Evolución (1)Caso 1 Caso 2
• Dermatitis descamativa severa
• Dermatitis del pañal con sobreinfección por C. Parapsilosis
• Esofagitis eosinofílica, hipereosinofília e hiper IgE
• Sospecha APLV no IgE mediada
• Reingreso en UCIP en 2 ocasiones por deshidratación hipernatrémica
• Reingreso en UCIP tras episodio de atragantamiento
• Candidiasis mucocutánea
• Ingreso en UCIP por epiglotitis
• Neumonía basal derecha
• Primoinfección por CMV

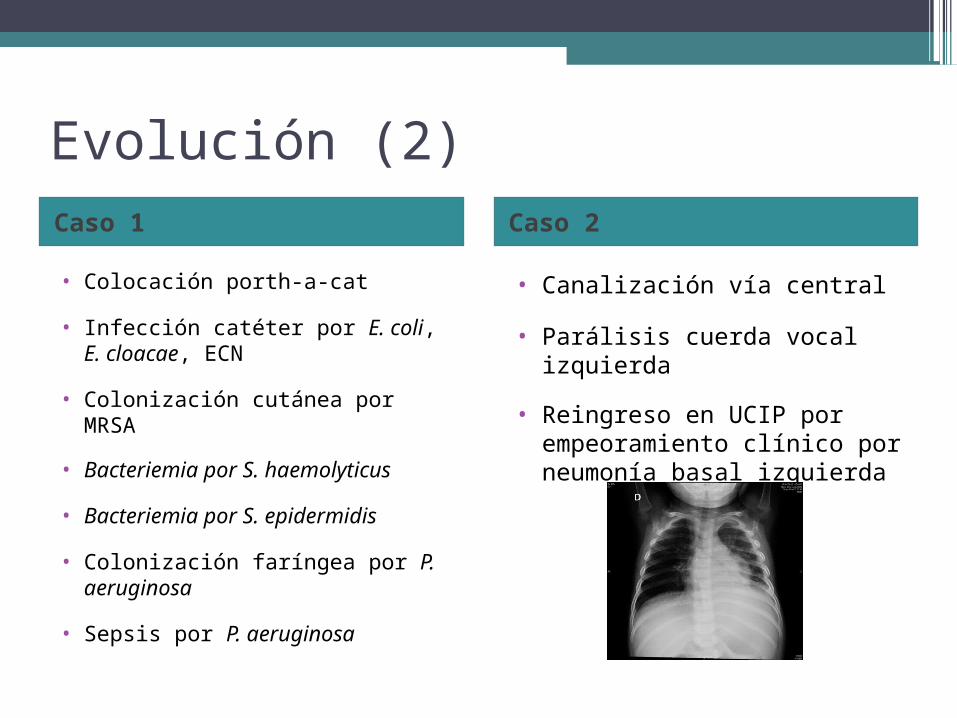
Evolución (2)Caso 1 Caso 2
• Colocación porth-a-cat
• Infección catéter por E. coli, E. cloacae, ECN
• Colonización cutánea por MRSA
• Bacteriemia por S. haemolyticus
• Bacteriemia por S. epidermidis
• Colonización faríngea por P. aeruginosa
• Sepsis por P. aeruginosa
• Canalización vía central
• Parálisis cuerda vocal izquierda
• Reingreso en UCIP por empeoramiento clínico por neumonía basal izquierda

Tratamiento
Caso 1 Caso 2
• Inicial:▫ Inmunoglobulina
humana
• Definitivo:▫ Trasplante de
progenitores hematopoyéticos
• Prueba terapéutica:▫ Inmunoglobulina
humana

Inmunodeficiencias primarias

INTRODUCCIÓNINMUNODEFICIENCIAS
PRIMARIAS (IDP)
Conjunto de enfermedades de origen genético
Alteración cuantitativo i/o funcional del sistema inmune
Mayor predisposición a infecciones, procesos
autoinmunes, alergia y cáncer

INTRODUCCIÓN
• Las inmunodeficiencias primarias (IDP) representan un conjunto de enfermedades basadas en la alteración cuantitativa y/o en la función del sistema inmune.
• Forman parte de las enfermedades “raras”, pero en los últimos años ha habido un aumento en la prevalencia (hasta 1/1200, excluyendo el déficit de IgA).
• Requieren un alto índice de sospecha clínica.
• El desconocimiento por parte del médico conlleva a infradiagnósticos o retrasos en el diagnóstico.

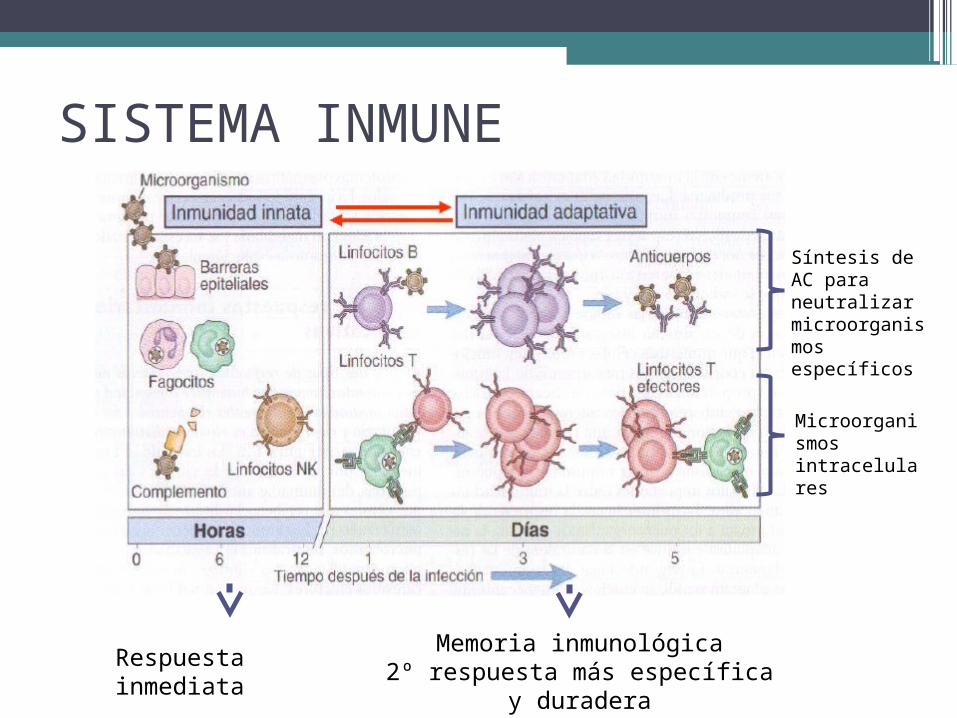
SISTEMA INMUNE
Respuesta inmediata Memoria inmunológica2º respuesta más específica y duradera
Microorganismos intracelulares
Síntesis de AC para neutralizar microorganismos específicos

CLASIFICACIÓN

CLASIFICACIÓN

CLASIFICACIÓN
Hipogammaglobulinemia transitoria de la infancia
Deficiencia selectiva de IgA
ID variable común
Agammaglobulinemia ligada al X (síndrome de Bruton)
1) Defectos predominantes de anticuerpos (ID humoral)
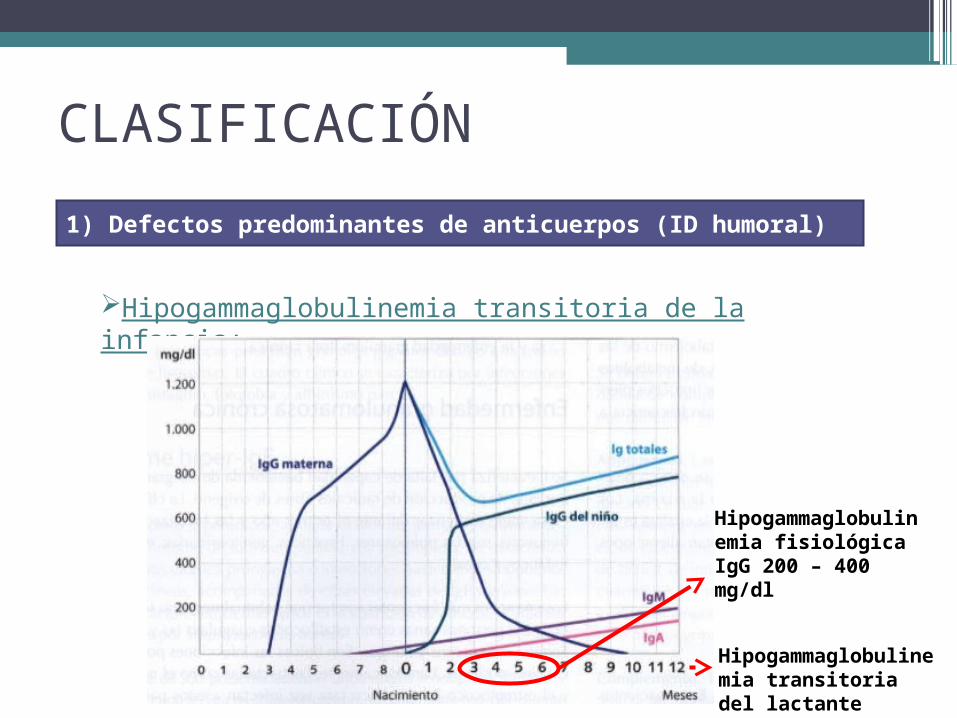
CLASIFICACIÓN
Hipogammaglobulinemia transitoria de la infancia:
1) Defectos predominantes de anticuerpos (ID humoral)
Hipogammaglobulinemia fisiológicaIgG 200 – 400 mg/dl
Hipogammaglobulinemia transitoria del lactante

CLASIFICACIÓN
Deficiencia selectiva de IgA:
1) Defectos predominantes de anticuerpos (ID humoral)
REACCIONES GRAVES ante la administración de hemoderivados
SHOCK ANAFILÁCTICO ante la administración de gammaglobulina
REACCIONES GRAVES ante la administración de hemoderivados
SHOCK ANAFILÁCTICO ante la administración de gammaglobulina
• La más frecuente.• Asintomáticos.• Mayor incidencia de trastornos AI (diabetes tipo 1), alergia y
enfermedad celíaca.• Estudiar si existe deficiencia de subclases de IgG: valores
inferiores de una o más subclases, con cantidad normal de IgG total.

CLASIFICACIÓN
ID variable común:
1) Defectos predominantes de anticuerpos (ID humoral)

CLASIFICACIÓN
Agammaglobulinemia ligada al X (síndrome de Bruton):
1) Defectos predominantes de anticuerpos (ID humoral)
- Sexo masculino (delecciones gen tirosin –cinasa en el cromosoma X)
-Inicio entre los 6 meses y año de vida
-Ausencia de LB circulantes y células plasmáticas
-IgG < 200 mg/dl, IgA y IgM indetectables
-LT normales

URGENCIA
PEDIÁT
RICA
CLASIFICACIÓN
En ausencia de tratamiento correctivo, fallecimiento durante la infancia
2) IDP combinadas
- Aparición temprana de infecciones graves repetidas
- Anorexia o retraso ponderoestatural
- Candidiasis oral resistente al tratamiento tópico
- Neumonías intersticiales- Historia familiar de
fallecimientos tempranos

CLASIFICACIÓN
ID combinada severaSíndrome de Omenn (RAG)Deficiencia de ADADeficiencia HLA 2Síndrome de hiper IgMDisgenesia reticular
Ante la sospecha, como la tríada:
Candidiasis
Diarreas
Neumonías
Evitar vacunación con gérmenes vivos
2) IDP combinadas

CLASIFICACIÓN
ID combinada severa:
Diagnóstico: Tratamiento:
Hipogammaglobulinemia intensaLinfopeniaAusencia de linfocitos funcionantes
Hipogammaglobulinemia intensaLinfopeniaAusencia de linfocitos funcionantes
Trasplante de médula ósea
2) IDP combinadas
- Ausencia de inmunidad mediada por células T y alteración respuesta humoral
- Agenesia/hipogenesia de timo en la rx de tórax

CUANDO SOSPECHARLO: signos de alarma
Signo 1 4 o más otitis en 1 año
Signo 2 2 o más sinusitis graves en 1 año
Signo 3 Dos o más meses tomando ATB con poca mejoría
Signo 4 Dos o más infecciones respiratorias (neumonías) en 1 año o una por año consecutivo.
Signo 5 Retraso ponderoestatural
Signo 6 Abscesos cutáneos o profundos recurrentes
Signo 7 Candidiasis mucocutánea persistente después del año de vida
Signo 8 Necesidad de ATB ev para curar una infección
Signo 9 2 o más infecciones profundas graves, como la septicemia.
Signo 10 Historia familiar de IDP

ORIENTACIÓN DIAGNÓSTICA
Historia familiar
Exploración física
Edad inicio clínica
Pruebas complementarias
ConsanguinidadFamiliar fallecido en la
infanciaInfecciones de repetición
Fenotipo clínico

ORIENTACIÓN DIAGNÓSTICA
Exploración física
Eczema, petequias Wiskott-Aldrich
Telangiectasias (conjuntiva, pabellón auricular)
Ataxia-telangiectasia
Gingivitis, enfermedad periodontal Alteración de la fagocitosis
Microcefalia, “bird face”, frente pequeña, retrognatia, filtrum largo
Sd Nijmegen
Eccema desde el nacimiento (palmas y plantas)
Sd Omenn
Albinismo, nistagmus, fotofobia Sd Chediak-Higashi
Abscesos cutáneos, retraso caída dientes Sd hiper IgE
Fenotipo peculiar (implantación baja pabellones auriculares, úvula bífida) y cardiopatía congénita
Sd Di George

ORIENTACIÓN DIAGNÓSTICAExploración
física
Síndrome Omenn

ORIENTACIÓN DIAGNÓSTICAExploración
física
Ataxia telangiectasia

ORIENTACIÓN DIAGNÓSTICAExploración
física
Síndrome de Hiper IgE

ORIENTACIÓN DIAGNÓSTICAFenotipo clínico
Síndrome de DiGeorge

ORIENTACIÓN DIAGNÓSTICAEdad inicio
clínica

ORIENTACIÓN DIAGNÓSTICA
Prioridad: descartar
formas graves
Prioridad: descartar
formas graves
edad
Citometría de flujo (Ac monoclonales)
Células T maduras (periféricas)
CD3+
T helper CD3+, CD4+
T citotóxicas CD3+, CD8+
Células NKCD3-, CD16+ o CD 56+
T activadas CD3+, HLA-DR+
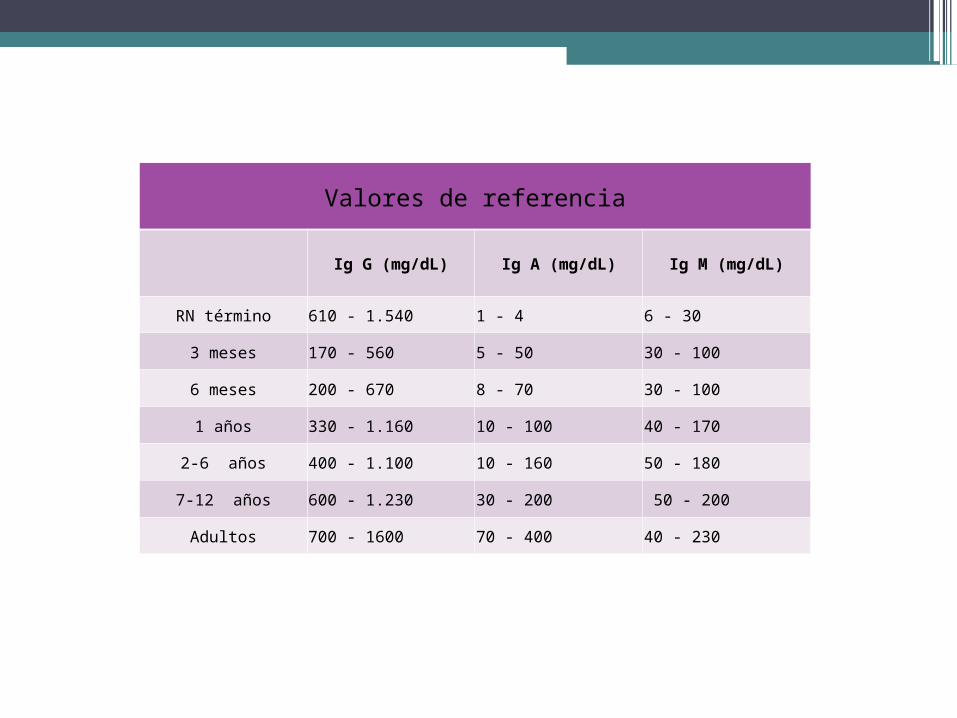
Valores de referencia
Ig G (mg/dL) Ig A (mg/dL) Ig M (mg/dL)
RN término 610 - 1.540 1 - 4 6 ‐ 30
3 meses 170 ‐ 560 5 ‐ 50 30 ‐ 100
6 meses 200 ‐ 670 8 ‐ 70 30 ‐ 100
1 años 330 ‐ 1.160 10 ‐ 100 40 ‐ 170
2‐6 años 400 ‐ 1.100 10 ‐ 160 50 ‐ 180
7‐12 años 600 ‐ 1.230 30 ‐ 200 50 ‐ 200
Adultos 700 ‐ 1600 70 ‐ 400 40 ‐ 230

Valores de referencia
Linfocitos totales
Linfocitos T (CD3+)
T CD4+ (CD3+/CD4+)
T CD8+ (CD 3+/CD8+)
Linfocitos B (CD 19+)
Células NK(CD3+/
CD56+CD16+)
RN término4.100
(2.200‐6.900)2.500
(1.100‐4.200)1.800
(800‐2.900)800
(300‐1800)500
(200‐1000)800
(200‐1800)
1-2 meses6.700
(4000‐11.300)4.600
(2.800‐6.500)3.500
(2.100‐4.900)1000
(500‐1.600)1000
(300‐1700)500
(300‐800)
2-5 meses5.900
(3.900‐8.600)3.600
(2.400‐5.600)2.500
(1.600‐4.200)1000
(700‐1.500)1.300
(800‐2.600)300
(200‐900)
2-5 meses6000
(4000‐9000)3.800
(2.700‐6.100)2.800
(1.700‐4.100)1.100
(700‐1.800)1.300
(800‐2.200)300
(200‐800)
9-15 meses5.500
(3.100‐8.700)3.400
(1.800‐5.900)2.300
(1.300‐4.100)1.100
(500‐1.600)1.400
(700‐2.400)400
(200‐900)
15-24 meses5600
(3.400‐8.900)3.500
(2.200‐5.500)2.200
(1.100‐3.600)1.200
(500‐1.800)1.300
(900‐2.500)400
(100‐1.100)
2-5 años3.300
(2.300-5.600)2.300
(1.400‐3.600)1.300
(700‐2000)800
(500‐1.400)800
(400‐1.500)400
(100‐700)
5-10 años2.800
(1.600‐4.300)1.900
(1.100‐2.800)1000
(500‐1.800)800
(400‐1.200)500
(300‐700)300
(100‐600)
10-16 años2.200
(1.300‐3000)1.500
(1.000‐2.000)800
(500‐1.300)400
(300‐800)300
(200‐500)300
(100‐700)
Adultos1.800
(1.100‐2.5001.200
(700‐1.900)700
(400‐1.300)400
(200‐700)200
(100‐400)300
(100‐400)
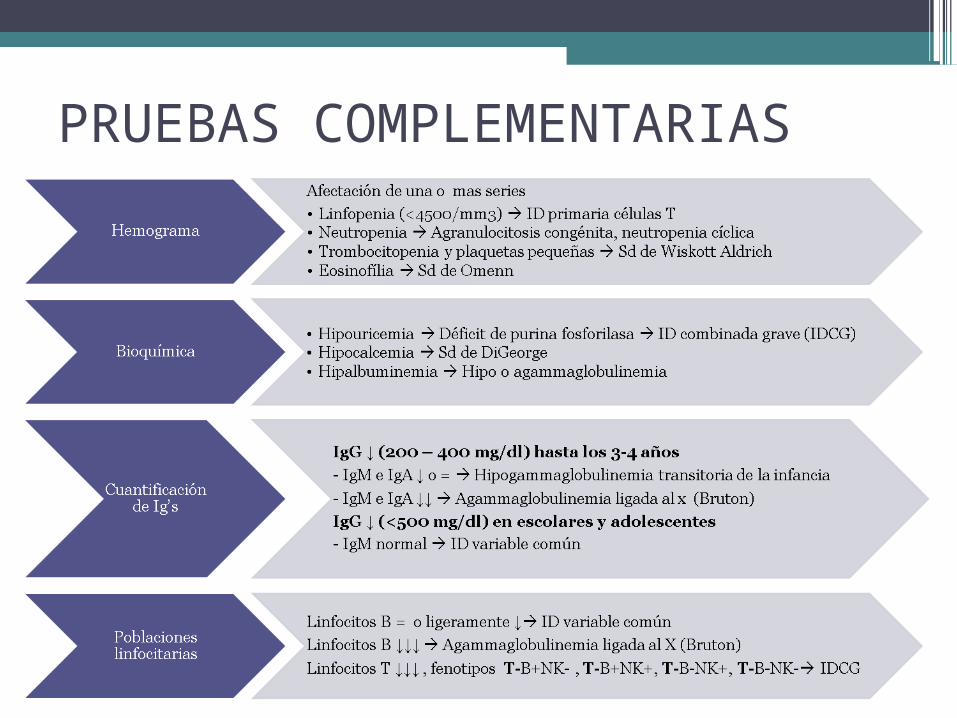
PRUEBAS COMPLEMENTARIAS
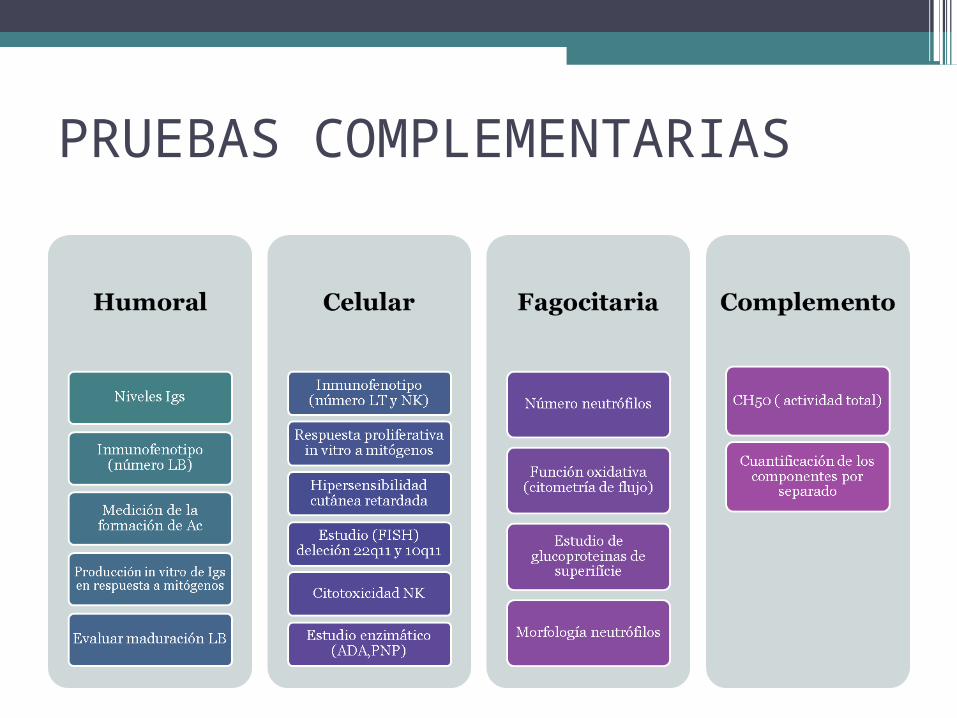
PRUEBAS COMPLEMENTARIAS

CONCLUSIONES• La sospecha de ID primaria empieza con una correcta historia clínica.
• La principal manifestación es la susceptibilidad aumentada a las infecciones.
• Sospechar en < 6 meses si: retraso pondoestatural, muguet persistente, diarrea crónica e infecciones invasivas graves.
• Sospechar en > 6 meses si: infecciones de repetición, infecciones invasivas repetidas y graves, enfermedades alérgicas, AI o neoplasias.
• Infecciones invasivas por Neisseria sugieren defectos del complemento.
• Abscesos de repetición sugieren neutropenia o defecto de la fagocitosis.
• Escrutinio inicial: hemograma, bioquímica, inmunoglobulinas y poblaciones linfocitarias.
GRÀCIES







