type ii ligands as chemical auxiliaries to favor enzymatic transformations by p450 2e1
TRANSCRIPT

Reprint© Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

� WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
Table of Contents
A. M�nard, C. Fabra, Y. Huang, K. Auclair*
2527 – 2536
Type II Ligands as Chemical AuxiliariesTo Favor Enzymatic Transformationsby P450 2E1
Controlling P450 transformations:Type II ligands contain an aromatic ni-trogen that coordinates to the hemeiron in the active site of cytochromeP450 enzymes. The type II ligand nicoti-nate can serve as a useful chemical aux-iliary for biocatalysis with P450 2E1 bypromoting the predictable oxidation ofsmall hydrocarbon substrates.

DOI: 10.1002/cbic.201200524
Type II Ligands as Chemical Auxiliaries To Favor EnzymaticTransformations by P450 2E1Am�lie M�nard, Camilo Fabra, Yue Huang, and Karine Auclair*[a]
Introduction
Cytochrome P450 enzymes (P450s) are heme monooxygenasesthat are involved in the biosynthesis of secondary metabolitesand play a central role in drug metabolism. Of note is theirremarkable ability to oxidize inactivated C�H bonds with highregio- and stereoselectivities, a feat that is difficult to achievechemically. Despite their tremendous potential to be used asbiocatalysts,[1] our ability to exploit these qualities in vitro hasbeen hampered by the enzyme’s low stability, low activity, andpoor product predictability, as well as the requirement forexpensive cofactors.
Over the years, our lab has been addressing these issueswith a focus on P450 3A4[2] because of its highly promiscuousbehavior, which enhances its potential as a useful and versatilebiocatalyst. Recently, our lab has pioneered a substrate-engi-neering strategy that has proven useful in taming the sub-strate promiscuity of P450 3A4. In this approach, a chemicalauxiliary is covalently linked to the substrate. The role of thechemical auxiliary is to orient the substrate in the P450 activesite in such a way as to promote its oxidation with consistentselectivity. Interestingly, the chemical auxiliary can also serve tofacilitate retrieval of the product when applied to fermentationprocesses.[3] Indeed, product recovery is a major roadblock inwhole-cell biotransformations. In a proof-of-concept study byLarsen et al. , it was found that, when using theobromine asa chemical auxiliary, small hydrocarbon substrates were reliablyhydroxylated at the fourth carbon from the auxiliary with pro-Rfacial selectivity.[2d] This was the first example of hydroxylationat an inactivated secondary C�H bond in which the catalystwas able to predictably discriminate between methylenegroups with similar electronic properties.
Upon ligand binding, P450 enzymes also show interestingbehaviors that can be monitored spectroscopically. In theirresting state, P450s have a water molecule coordinated to theactive-site heme (six-coordinate) iron. Interaction of the
enzyme with ligands usually favors five-coordinate iron with re-lease of the water molecule. Broadly speaking, ligands can leadto three types of UV spectral shifts of the Soret absorptionband: type I, type II, or reverse type I. Type I binders causea shift in the iron spin equilibrium towards the high-spin spe-cies; this results in an increase in absorbance around 390 nmand a decrease around 420 nm. Type II binders generally pos-sess an aromatic nitrogen that coordinates to the heme ironand stabilizes the low-spin state. They cause a decrease inabsorbance around 390 nm and an increase in absorbancearound 425 nm. A reverse type I spectral shift is similar totype II except without a red shift of the Soret peak. It is oftenobserved with ligands containing an oxygen atom that coordi-nates to the heme iron.[4] The type II interaction has oftenbeen called a dead-end complex and is believed to inhibitP450 catalysis in two ways.[4] The first way is by trapping theenzyme in the low-spin state, thus making heme iron reduc-tion by cytochrome P450 reductase (CPR) more difficult. Thesecond way is by competitive exclusion of other potential li-gands from the active site. Moreover, it has been demonstrat-ed that type II ligands have a much higher affinity for P450enzymes than similar type I binders.[5]
These high-affinity inhibitory interactions have been exploit-ed in the design of P450-targeted therapeutic agents by incor-poration of nitrogen-containing aromatic functional groups.This strategy has led to the successful development of inhibi-tors of lanosterol 14-demethylase (P450 51A1) as antifungalagents[6] and of P450 17 for the treatment of prostate cancer.[7]
The remarkable ability of P450 enzymes to oxidize inactivatedC�H bonds and the high substrate promiscuity of many P450isoforms have inspired us and others to investigate their useas biocatalysts. Our lab has pioneered a chemical-auxiliaryapproach to control the promiscuity of P450 3A4 and provideproduct predictability. The recent realization that type II li-gands are sometimes also P450 substrates has prompted thedesign of a new generation of chemical auxiliaries with type IIbinding properties. This approach takes advantage of the high
affinity of type II ligands for the active site of these enzymes.Although type II ligands typically block P450 activity, we reporthere that type II ligation can be harnessed to achieve just theopposite, that is, to favor biocatalysis and afford predictableoxidation of small hydrocarbon substrates with P450 2E1.Moreover, the observed predictability was rationalized by mo-lecular docking. We hope that this approach might find futureuse with other P450 isoforms and yield complimentary prod-ucts.
[a] A. M�nard, C. Fabra, Y. Huang, Prof. K. AuclairDepartment of Chemistry, McGill University801 Sherbrooke St. W., Montreal, Quebec, H3A 0B8 (Canada)E-mail : [email protected]
Supporting information for this article is available on the WWW underhttp ://dx.doi.org/10.1002/cbic.201200524.
ChemBioChem 2012, 13, 2527 – 2536 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 2527
Others have observed that, in some cases, type II binding canalso improve the metabolic stability of drug candidates,[8] animportant criterion in drug development. On the other hand,many drugs containing moieties capable of type II binding areof particular concern to drug developers for their potential tocause inadvertent inhibition of P450 enzymes.[4, 9] In somecases, removal of type II binding character has proven effectivein abolishing unwanted hepatic P450 enzyme inhibition.[10]
Although type II P450 ligands are generally considered to beinhibitors, further scrutiny has revealed that some are also sub-strates.[5a, 11] A recent systematic study of pyridinyl quinoline-4-carboxamide-derived type II ligands of P450 3A4 by Peng et al.showed that type II binding can increase binding affinity by upto 1200 times.[5a] Moreover, whereas the type II ligands studiedare competitive inhibitors of testosterone metabolism by P4503A4, they are also extensively metabolized themselves, insome cases to a greater extent than similar type I binders.These results inspired us to investigate the utility of type IIligands as chemical auxiliaries in order to target substrates tothe active site of P450s. Although type II ligands have beenintroduced into molecules with the goal of generating P450inhibitors, the focus of this report is on the possibility of har-nessing type II ligation to favor biocatalysis and control en-zyme selectivity.
Results and Discussion
Intrigued by the seemingly contradictory properties of P450type II ligands, we wondered if they could be exploited todesign a chemical auxiliary. Having already described theobro-mine as an effective chemical auxiliary to improve the predict-ability of P450 3A4 oxidations,[2d] we turned our attention to-wards another isoform, P450 2E1, in hopes of achieving com-plimentary regioselectivity. We envisaged taking advantage oftype II ligation built into a chemical auxiliary to target sub-strates to the P450 2E1 active site and thus improve turnover.Moreover, the few examples of transformed P450 type II li-gands clearly begged for more studies of these systems.
Selection of an appropriate chemical auxiliary
Although theobromine was highly successful as a chemicalauxiliary to control enzymatic transformations by P450 3A4,[2d]
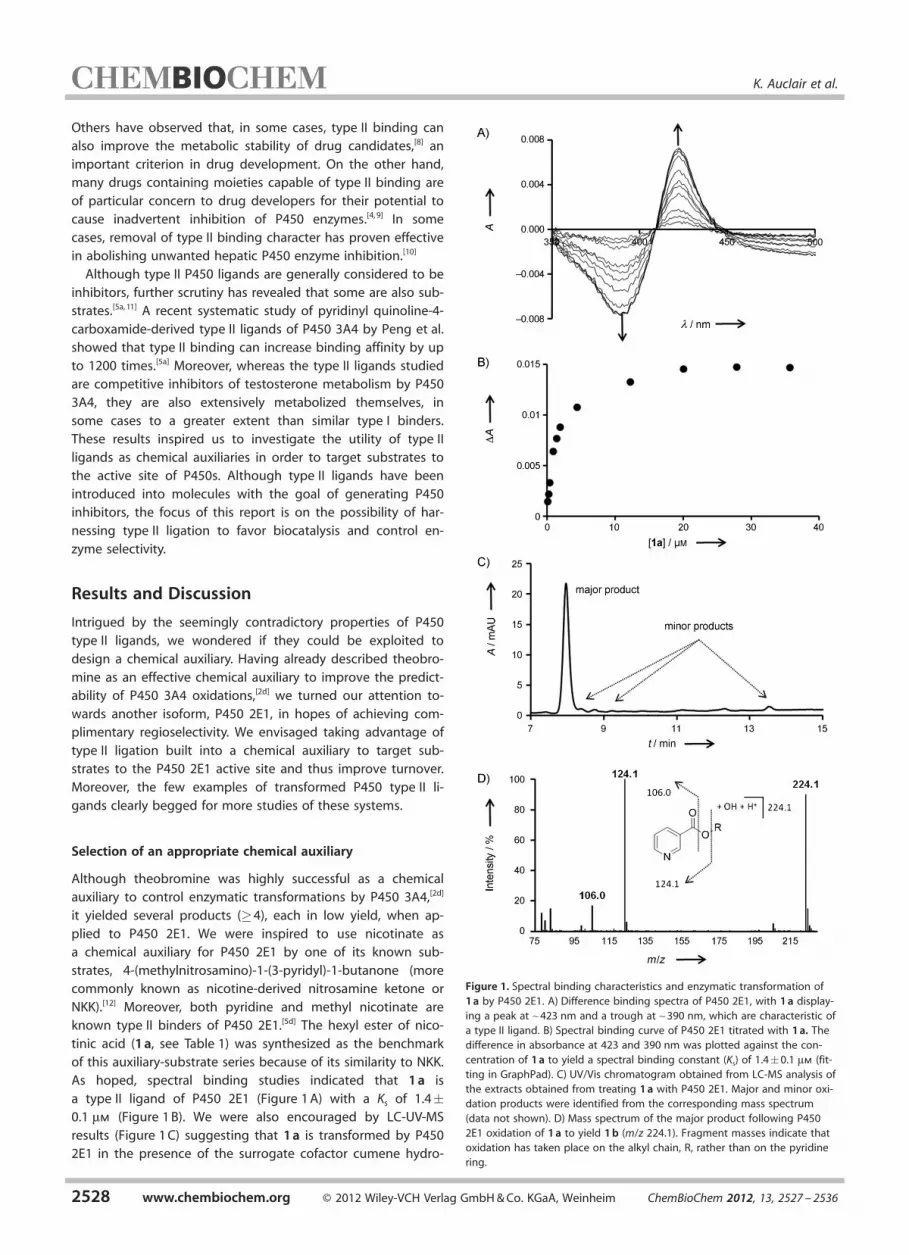
it yielded several products (�4), each in low yield, when ap-plied to P450 2E1. We were inspired to use nicotinate asa chemical auxiliary for P450 2E1 by one of its known sub-strates, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (morecommonly known as nicotine-derived nitrosamine ketone orNKK).[12] Moreover, both pyridine and methyl nicotinate areknown type II binders of P450 2E1.[5d] The hexyl ester of nico-tinic acid (1 a, see Table 1) was synthesized as the benchmarkof this auxiliary-substrate series because of its similarity to NKK.As hoped, spectral binding studies indicated that 1 a is
a type II ligand of P450 2E1 (Figure 1 A) with a Ks of 1.4�0.1 mm (Figure 1 B). We were also encouraged by LC-UV-MSresults (Figure 1 C) suggesting that 1 a is transformed by P4502E1 in the presence of the surrogate cofactor cumene hydro-
Figure 1. Spectral binding characteristics and enzymatic transformation of1 a by P450 2E1. A) Difference binding spectra of P450 2E1, with 1 a display-ing a peak at ~423 nm and a trough at ~390 nm, which are characteristic ofa type II ligand. B) Spectral binding curve of P450 2E1 titrated with 1 a. Thedifference in absorbance at 423 and 390 nm was plotted against the con-centration of 1 a to yield a spectral binding constant (Ks) of 1.4�0.1 mm (fit-ting in GraphPad). C) UV/Vis chromatogram obtained from LC-MS analysis ofthe extracts obtained from treating 1 a with P450 2E1. Major and minor oxi-dation products were identified from the corresponding mass spectrum(data not shown). D) Mass spectrum of the major product following P4502E1 oxidation of 1 a to yield 1 b (m/z 224.1). Fragment masses indicate thatoxidation has taken place on the alkyl chain, R, rather than on the pyridinering.
2528 www.chembiochem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemBioChem 2012, 13, 2527 – 2536
K. Auclair et al.
peroxide (CHP).[2a] MS analysis of the major product (later char-acterized as 1 b) indicates the addition of 16 mass units to thestarting material 1 a (Figure 1 D). Inspection of the fragmenta-tion pattern indicates that oxidation occurred on the alkylchain (the substrate portion) rather than on the aromatic ring(the auxiliary portion).
To study the effect of the chemical auxiliary, several otherauxiliary substrates with a hexyl group as the substrate partwere prepared (Table 1). Each auxiliary substrate was character-ized with respect to its binding mode and spectral dissociationconstant (Ks) in the P450 2E1 complex. The latter was extractedfrom difference-spectra titration curves. Moreover, the ratioand yield of the oxidized products formed by reaction withP450 2E1 was estimated by LC-UV-MS analysis.
The relationships observed between binding mode andspectral binding affinity correlate well with other reports in theliterature.[5] As expected, the Ks values of type I binders were,in most cases, at least an order of magnitude higher thanthose of type II binders, except for compounds 4 a and 6 a. The
presence of an aromatic nitrogen was necessary for type IIbinding to occur; 5 a and 8 a lacked such a nitrogen and exhib-ited type I and reverse type I behavior, respectively. Conversely,not all structures containing an aromatic nitrogen producedtype II binding characteristics, as seen for compounds 2 a, 4 aand 6 a. This has been attributed by others to steric inter-actions restricting access to the heme iron by the nitro-gen.[5a, b, 8, 13] Changing the position of the nitrogen with respectto the ester group in the aromatic ring from meta (1 a) to para(3 a) also decreased the Ks value by an order of magnitude,thus indicating a higher affinity interaction. This is consistentwith observations made by Peng et al. that para-substitutedquinoline-4-carboxamide analogues had a higher affinity to-wards P450 3A4 than their meta-substituted counterparts.[5a]
Also in agreement with the results of this study is our observa-tion that the ortho-substituted auxiliary substrate 2 a exhibitedtype I spectral binding characteristics, and thus was not ableto coordinate to the heme iron. Others have observed similarsteric restrictions to heme iron coordination caused by ortho
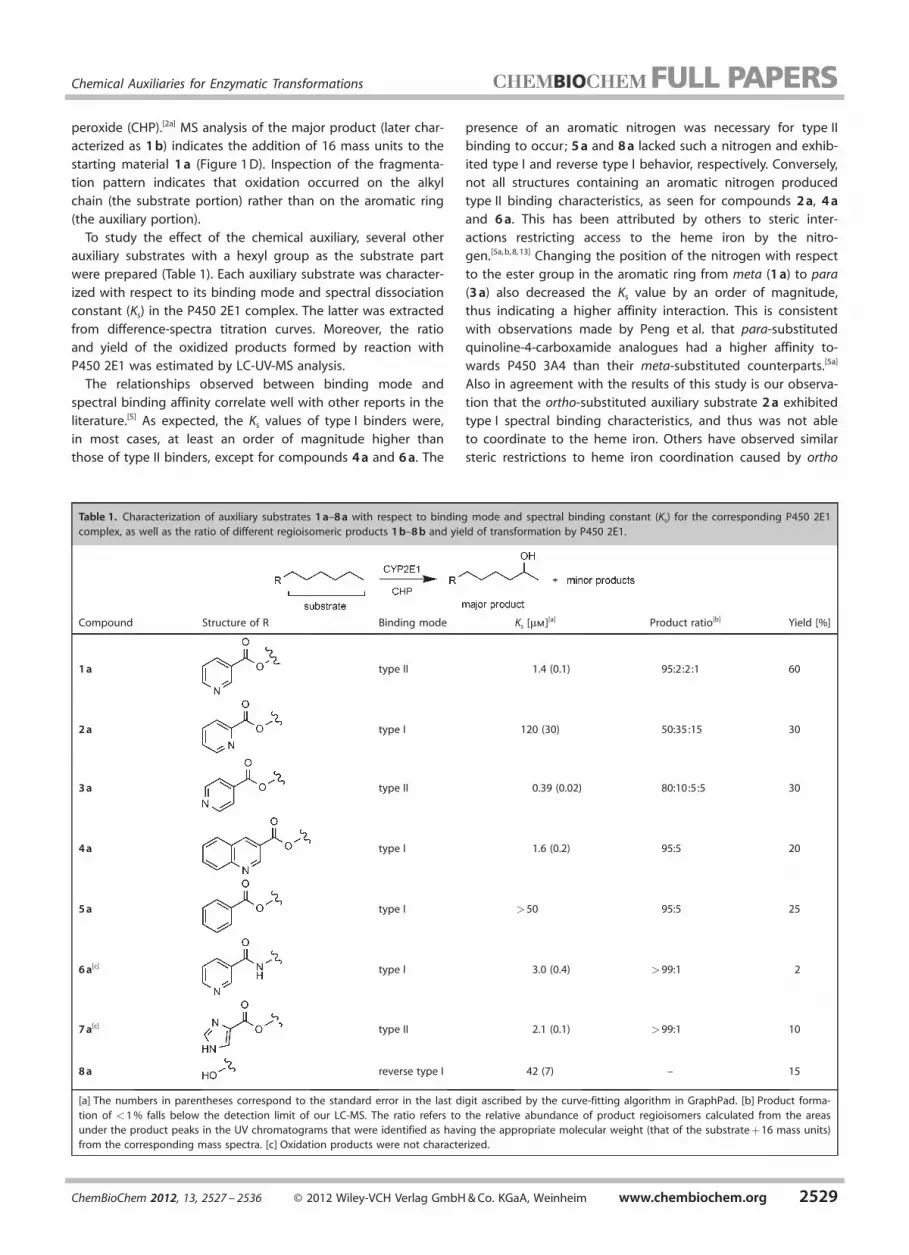
Table 1. Characterization of auxiliary substrates 1 a–8 a with respect to binding mode and spectral binding constant (Ks) for the corresponding P450 2E1complex, as well as the ratio of different regioisomeric products 1 b–8 b and yield of transformation by P450 2E1.
Compound Structure of R Binding mode Ks [mm][a] Product ratio[b] Yield [%]
1 a type II 1.4 (0.1) 95:2:2:1 60
2 a type I 120 (30) 50:35:15 30
3 a type II 0.39 (0.02) 80:10:5:5 30
4 a type I 1.6 (0.2) 95:5 20
5 a type I >50 95:5 25
6 a[c] type I 3.0 (0.4) >99:1 2
7 a[c] type II 2.1 (0.1) >99:1 10
8 a reverse type I 42 (7) – 15
[a] The numbers in parentheses correspond to the standard error in the last digit ascribed by the curve-fitting algorithm in GraphPad. [b] Product forma-tion of <1 % falls below the detection limit of our LC-MS. The ratio refers to the relative abundance of product regioisomers calculated from the areasunder the product peaks in the UV chromatograms that were identified as having the appropriate molecular weight (that of the substrate + 16 mass units)from the corresponding mass spectra. [c] Oxidation products were not characterized.
ChemBioChem 2012, 13, 2527 – 2536 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chembiochem.org 2529
Chemical Auxiliaries for Enzymatic Transformations

substitution; this also explains why compound 4 a is not atype II binder.[5a, b, 8, 13] The low Ks value observed for 4 a despiteits type I binding character is likely due to the additional aro-matic ring. Its presence probably increases the affinity of 4 afor the hydrophobic active site of P450 2E1, which is enclosedon the distal side by five phenylalanine residues.[14]
Interestingly, changing the linking group between the auxili-ary and the substrate from an ester (1 a) to an amide (6 a) wasaccompanied by a change in binding mode (Figure 2). In some
cases, sterically unhindered meta-substituted compounds havebeen reported to be type I binders instead of type II.[5a, b] Reten-tion of binding affinity can be explained, at least in part, bythe fact that amide carbonyl oxygens are better hydrogen-bond acceptors than their ester equivalents. Thus, 6 a can forma stronger bond with the backbone NH of Thr303, which is atan appropriate distance of 2.8 �.
LC-UV-MS analysis revealed that P450 2E1 oxidation yieldedmore than one product for many of the auxiliary substrates,yet in most cases one product was dominant. Only productswith molecular weights corresponding to that of the sub-strate + 16 mass units were observed, thus indicating that hy-droxylation had taken place. The different products detectedwere typically regioisomers, and their relative abundance (re-ported as product ratios) was calculated from the areas underthe product peaks in the UV chromatograms that were identi-fied as having the appropriate molecular weight from the cor-responding mass spectra. This is a reasonable measure of re-gioselectivity in as much as the substrate and the correspond-ing products have similar extinction coefficients. This was infact confirmed to be the case for 1 a, 12 a, and 12 b (see theSupporting Information). As seen in Table 1, there is no obvi-
ous relationship between binding mode and regioselectivity,and values of �95 % can be achieved by both type I andtype II binders. Although transformation of 6 a and 7 a by P4502E1 occurred with excellent regioselectivity (within the detec-tion limits of the MS), product yields were quite poor. The nic-otinate-based auxiliary substrate 1 a stood out as it was oxi-dized in very good yield (60 %) and excellent regioselectivity(95 %).
The major P450 2E1 oxidation products for the reactionswith higher yields (i.e. , reactions of 1 a–5 a) were prepared en-zymatically, then purified and characterized by NMR and high-resolution mass spectrometry. The results revealed that themajor site of oxidation was always at the furthest methylenegroup from the auxiliary (Table 1, top), irrespective of the bind-ing mode of the parent substrate. Comparison to an authenticstandard through GC-MS confirmed this same preferred regio-selectivity for the oxidation of hexan-1-ol (8 a) to producehexane-1,5-diol. Ultimately, nicotinate was retained for subse-quent studies as the most promising auxiliary in terms of yieldand regioselectivity.
Effects of varying the substrate structure
Having selected nicotinate as our chemical auxiliary of choice,we investigated its ability to direct the hydroxylation of variousother hydrocarbon substrates. Thus, a variety of nicotinateesters were synthesized from nicotinic acid and various alco-hols: branching (9 a–11 a), unsaturated straight chain (cis andtrans, 15 a–19 a), and saturated and unsaturated cyclic (20 a–23 a ; Table 2).
All compounds were oxidized to a certain extent by P4502E1, to give again at least two products, with one being pre-dominant in most cases. Spectral binding studies with samplesof each substrate type (10 a, 11 a, 12 a, 14 a, 16 a, 18 a, 20 a)
confirmed their ability to bind P450 2E1 in a type II fashionwith Ks values for their P450 2E1 complex similar to that of 1 a.Unfortunately, in many cases, yields were significantly lowerthan for 1 a. As commercial applications of P450 enzymes arecurrently limited to using whole cells,[15] and because thisenzyme is naturally membrane-bound and less stable in vitro,it is reasonable to expect that yields can be improved withwhole-cell transformations. Regioselectivities were lower withmost substrates than with 1 a, yet very good in most cases.Substrates that were transformed with the best regioselectivi-ties, 10 a–14 a, 18 a, 20 a, and 22 a, were retained for character-ization of the major product. Table 3 lists the correspondingproduct structures 10 b–14 b, 18 b, 20 b, and 22 b. Because ofthe lower stability of 18 b compared to the other products, anauthentic standard of the suspected epoxide product was syn-thesized. Comparison of LC-MS profiles confirmed that thiswas in fact the correct product structure. Taken together, theseresults demonstrate a preference by P450 2E1 for oxidizing thealiphatic or alkenyl secondary C�H bond furthest from the aux-iliary. This is similar to the regioselectivity observed for P4502E1 hydroxylation of some medium- and long-chain fattyacids.[16] These include saturated fatty acids lauric, myristic, pal-
Figure 2. Spectral binding characteristics of 6 a with P450 2E1. A) Differencebinding spectrum of P450 2E1, with 6 a displaying a peak at ~390 nm anda trough at ~423 nm, which are characteristic of a type I ligand. B) Spectralbinding curve of P450 2E1 titrated with 6 a. The difference in absorbance at390 and 423 nm was plotted against concentration of 6 a to yield Ks =
3.0�0.4 mm (fitting in GraphPad).
2530 www.chembiochem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemBioChem 2012, 13, 2527 – 2536
K. Auclair et al.

mitic, and stearic acid, which are hydroxylated at the w�1 po-sition.
Molecular modeling studies
We used molecular modeling to gain insight into the origins ofthe regioselectivity observed with respect to P450 2E1 oxida-tion of these auxiliary substrates. Docking of molecules 1 a–7 aand 9 a–23 a was performed by using Fitted through the web-based platform Forecaster.[17] Preliminary docking with Fitted
in flexible protein mode allowed us to select the most appro-priate X-ray crystal structure. PDB IDs 3E4E (P450 2E1 in com-plex with 4-methylpyrrazole) and 3LC4 (P450 2E1 in complexwith w-imidazolyl-dodecanoic acid, a fatty acid mimic) werecompared for their ability to accommodate 1 a–7 a and 9 a–23 a in the active site. These two structures differ in active site
volume (190 and 440 �, respectively). In 3LC4, Phe298 is rotat-ed toward the B’ helix, thereby opening up the active site toa void above the I helix that is closed off in 3E4E.[18] 3LC4 wasidentified as the better candidate for docking of our com-pounds. This is most likely due to their elongated shape,which somewhat resembles that of a fatty acid. A proteinRMSD value of 0 was obtained for 3LC4 with all moleculesexcept 6 a, thereby indicating that the favored structure fordocking these molecules corresponds exactly to that of 3LC4.(Alternatively, an RMSD value >0 for both PDB files would in-dicate that the favored structure combines features from bothparent input files).
When docked into 3LC4 in rigid-protein mode, each com-pound (1 a–7 a and 9 a–23 a) adopted several binding poses ofsimilar energy. Thus, it was not possible to distinguish betweenthem in order to identify which was favored. However, careful
Table 2. Characterization of auxiliary substrates 9 a–23 a with respect to binding mode and spectral binding constant (Ks) for the corresponding P450 2E1complex, as well as the ratio of different regioisomeric products and yield of transformation by P450 2E1.
Compound Structure of R Binding mode Ks [mm][a] Product ratio[b] Yield [%]
9 a – – 50:50 15
10 a type II 2.6 (0.2) 70:30 15
11 a type II 1.8 (0.1) 85:15 25
12 a type II 5.9 (0.4) 80:20 40
1 a type II 1.4 (0.1) 95:2:2:1 60
13 a – – 80:20 35
14 a type II 2.1 (0.1) 95:5 40
15 a – – 55:30:15 30
16 a type II 1.3 (0.1) 40:40:10:10 70
17 a – – 60:25:10:5 55
18 a type II 12 (1) 90:10 20
19 a – – 55:30:15 50
20 a type II 1.6 (0.2) 70:25:5 20
21 a – – 60:40 20
22 a – – 75:25 15
23 a – – 55:25:10:10 10
[a] The numbers in parentheses correspond to the standard error in the last digit ascribed by the curve-fitting algorithm in GraphPad. [b] Product forma-tion of <1 % falls below the detection limit of our LC-MS. The ratio refers to the relative abundance of product regioisomers calculated from the areasunder the product peaks in the UV chromatograms that were identified as having the appropriate molecular weight (that of the substrate + 16 mass units)from the corresponding mass spectra.
ChemBioChem 2012, 13, 2527 – 2536 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chembiochem.org 2531
Chemical Auxiliaries for Enzymatic Transformations

inspection of the docking results revealed that the compoundsclustered in three separate groupings (clusters A, B, and C; Fig-ure 3 A–C) with respect to the distal face of the heme prosthet-ic group in the active site of P450 2E1. In clusters A and B, theauxiliary portion binds almost directly above the porphyrinring at two discrete distances, with the substrate portion di-rected towards the heme iron. In cluster C, the docked mole-cules were oriented with the auxiliary portion closest to theheme in a manner akin to other known type II binders.
Compounds containing different auxiliaries with the samesubstrate (as in 1 a–7 a) yielded binding poses of similar energythat grouped well into cluster A and/or B, and C. As expected,the binding poses of most type I binders (2 a and 4 a) belong-ing to cluster C have their aromatic nitrogen pointing awayfrom the heme iron. Interestingly, this was not the case for 6 a,which has a cluster C binding pose that suggests type II bind-ing, contrary to the characterization given in Table 1.
The three clusters are especially useful to explain our resultswhen different substrates with the same auxiliary (nicotinate)are compared. Binding poses for 1 a, 9 a, 10 a, 13 a, 14 a, 16 a,17 a, 18 a, 22 a, and 23 a with the auxiliary portion binding fur-ther away (cluster A) from the heme are shown overlapped inFigure 3 A. A similar overlap of molecules 10 a, 12 a, 15 a, 16 aand 18 a–20 a binding closer to the heme (cluster B) is given inFigure 3 B. In general, longer molecules were found in cluster A
and shorter ones in cluster B. Interestingly, some molecules(10 a, 16 a, and 18 a) were found to have binding poses of sim-ilar energy in both clusters (Figure 3 D). This suggests thatshorter auxiliary substrates can navigate between both binding
sites, thus allowing them to beoxidized while residing closer tothe heme (as in cluster B). Givenour observation that type IIbinding auxiliary substratesgenerally gave higher productyields than similar type I bind-ers, it is unlikely that the transi-tion from type II binding to theproductive binding modeoccurs through debinding andrebinding of the auxiliary sub-strate from the enzyme.[5a] It ismore likely that, once bound,the auxiliary substrate reorientsitself within the active sitebefore and/or after activationwith CHP. The productive bind-ing modes observed in clustersA and B are likely stabilized byH-bonding and aromatic inter-actions with nearby active siteresidues (Figure 3 D) throughthe nicotinate moiety. Exactlyhow this transition occurs isbeyond the scope of this work.
For the observed bindingmode predicted to give the
major product (belonging either to cluster A or B), distancesfrom the heme iron to the closest nonaromatic secondary C�Hbond (Fe�H distance) range from 2.70–7.51 � with an averagedistance of 4.50 �. Such distances indicate that these bindingposes could correspond to the productive binding mode forthese compounds within the active site of P450 2E1. In fact,only four such distances (for 5 a, 7 a, 9 a, and 22 a from clus-ter A) fell outside of the 6 � cutoff suggested by Tarcsay et al.to distinguish between productive and nonproductive bindingmodes during computational studies with P450 2C9.[19] Similar-ly, Prasad et al. used a cutoff distance of 4 � from the substrateto the iron-oxo with an Fe�O bond length of 1.9 � (a total of~6 �) in their modeling studies with P450 1A homologues.[20]
As those molecules that fell outside the 6 � range were alsotransformed by P450 2E1, it is likely that, experimentally, theycan adopt a binding mode similar to that of other moleculesin cluster B (even though this was not observed computation-ally). Thus P450 2E1 simply oxidizes the C�H bond with thelowest bond dissociation energy in its vicinity. Greater prefer-ence is given to the oxidation of more electron rich secondaryand tertiary nonaromatic C�H bonds at the subterminal posi-tion rather than the terminal primary position.
Finally, binding poses for nicotinate-derived substrates 1 aand 9 a–22 a in cluster C are shown overlapped in Figure 3 C.The average distance between the heme iron and the aromaticnitrogen on molecules in this cluster was 3.1 �. This is thesame as was observed for the known type II binding indazolewhen cocrystallized with P450 2E1.[14] Other type II binders 3 aand 7 a also adopted a similar pose (data not shown). Taken to-
Table 3. Structures of the major P450 2E1 oxidation products for auxiliary-substrates 10 a–14 a, 18 a, 20 a and22 a.
Substrate R Product R’
10 a 10 b
11 a 11 b
12 a 12 b
13 a 13 b
14 a 14 b
18 a 18 b
20 a 20 b
22 a 22 b
2532 www.chembiochem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemBioChem 2012, 13, 2527 – 2536
K. Auclair et al.

gether, our molecular modeling and experimental results are ingood agreement and help explain how our compounds can beboth type II binders and substrates of P450 2E1 by adoptingmultiple binding modes within the active site.
Necessity of using the chemical auxiliary
In order to confirm the importance of the nicotinate auxiliary,control enzymatic reactions were performed with three sub-strates without the chemical auxiliary part. Thus, hexan-1-ol(8 a ; the substrate part of molecules 1 a–7 a), 3-methylpentan-1-ol (the substrate part of 10 a), and cyclopentylmethanol (thesubstrate part of 20 a) were treated with P450 2E1 under thesame conditions as above. Reaction mixtures were analyzed byGC-MS. As seen in Table 1, 8 a is oxidized by P450 2E1, but isa poorer substrate than compounds 1 a–5 a (15 % yield ob-served for the transformation of 8 a compared to 60 % for thatof 1 a). Conversely, neither 3-methylpentan-1-ol nor cyclopen-tylmethanol was transformed by P450 2E1 at a detectable level
(<1 %). These results support our hypothesis thattype II ligation can be used to favor biocatalysis byP450 enzymes.
Nicotinate as a chemical auxiliary
Ultimately, the type II binder nicotinate, when usedas a chemical auxiliary, was found to improve theyield of hexan-1-ol (1 a vs 8 a, Table 1) oxidation byP450 2E1 without changing the major site of oxida-tion. Moreover, covalent binding of nicotinate madepossible the transformation of some small alcoholsthat are normally not substrates of this enzyme. Notall type II binding auxiliaries had the same effect,and optimization is likely necessary with each P450enzyme. It is possible that some ligands favor theunproductive type II orientation within the P450active site to a greater extent than nicotinate. Thus,1 a exhibits a balance between type II binding,which increases its affinity for P450 2E1, and produc-tive binding, which leads to better oxidation yields.
Interestingly, in all cases, the major product(Tables 1 and 3) is the result of preferential oxidationat the secondary or tertiary aliphatic of alkenyl C�Hfurthest from the auxiliary. This type of regioselectivi-ty is in contrast to what was observed by Larsenet al. with P450 3A4 and the chemical auxiliary theo-bromine.[2d] In this case, the site of oxidation wasalways a set distance from the auxiliary (namely thefourth secondary C�H bond) regardless of substratelength. Nicotinate’s lack of influence on the majorsite of oxidation is likely due to the topology of theP450 2E1 active site and the resemblance of oursubstrate auxiliaries to short fatty acids, in that theycontain groups capable of participating in H-bond-ing (the ester and pyridine nitrogen) concentrated atone extremity and attached to a short alkyl chainyielding a generally oblong shape. Consequently, the
auxiliary can bind at more than one discrete distance from theheme, as was observed with fatty acid analogues of differentlengths cocrystallized with P450 2E1.[18] This leads to an oxida-tion regioselectivity that is controlled by electronics and mobi-lity rather than by the distance of a given C�H bond from thechemical auxiliary (as was observed with P450 3A4 and theo-bromine[2d]). Differences in product ratios can perhaps be ex-plained by differences in the mobility of various auxiliary sub-strates within the active site of P450 2E1. In other words, moremobile auxiliary substrates would expose C�H bonds with sim-ilar bond dissociation energies within a productive range fromthe heme iron in a manner that would result in an increasednumber or proportion of minor oxidation products.
Finally, using nicotinate as a chemical auxiliary has severalpractical advantages. It is achiral, commercially available, andinexpensive. Moreover, it contains one easily derivatizedgroup, the carboxylate, and is therefore readily linked to vari-ous substrates. Importantly, this auxiliary also contains a chro-mophore for easy detection and is easily cleaved off after enzy-
Figure 3. Molecular modeling of compounds 1 a and 9–23 a with P450 2E1 (PDB ID:3LC4). A) Cluster A: overlap of compounds 1 a, 9 a, 10 a, 13 a, 14 a, 16 a, 17 a, 18 a, 22 aand 23 a (purple), with the auxiliary moiety docking closer to the G helix and furtherfrom the heme (black). B) Cluster B: overlap of compounds 2 a, 10 a, 12 a, 15 a, 16 a, 18 a,19 a and 20 a (purple), with the auxiliary moiety binding further from the G helix andcloser to the heme (black). C) Cluster C: overlap of compounds 1 a and 9 a–22 a (purple)docking in a type II mode with their aromatic nitrogen atom ~3.1 � from the heme iron(bright green sphere). D) Overlap of 18 a poses in cluster A (purple) and cluster B(orange). Amino acids within 4 � of either structure are highlighted and labeled.
ChemBioChem 2012, 13, 2527 – 2536 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chembiochem.org 2533
Chemical Auxiliaries for Enzymatic Transformations

matic reactions (see the Supporting Information). Additionally,many nicotinate derivatives are known to cross cell mem-branes; this is an important consideration for whole-cell trans-formations.
Conclusion
In this report, we have shown that linking the type II bindingchemical auxiliary nicotinate to a variety of hydrocarbon sub-strates can successfully promote P450 2E1 biocatalysis withpredictable regioselectivity. P450 2E1’s preferential oxidation ofthe secondary, nonaromatic C�H bond furthest from the auxili-ary was also rationalized with the help of computational dock-ing studies. Fitted was able to predict both the type II and theproductive binding mode. This study not only confirms therecently reported oxidation of type II ligands by yet anotherP450 enzyme, but also demonstrates for the first time thattype II ligands can find a use in targeting various substrates tothe P450 active site for biocatalytic applications, as opposed tothe traditional goal of inhibiting transformation. We expectthat this concept could find use with other P450 isoforms topotentially afford complementary products, after identificationof the optimal type II ligands.
Experimental Section
General: Liquid chromatography mass spectra (LC-MS) were ac-quired on an Agilent 1100 series modular system equipped witha vacuum degasser, an autosampler, a quaternary pump systemand a UV/Vis detector in line with an Agilent 6120 quadrupole MSdetector. Gas chromatography mass spectra (GC-MS) were acquiredon an Agilent 7890A GC system in line with an Agilent 5975C MSdetector.
Addition of a His6 tag to P450 2E1: A C-terminal hexahistidine tagwas generated by insertion mutagenesis with pCW-2E1[21] as thetemplate. This plasmid was kindly donated by Prof. F. Peter Guen-gerich (Vanderbilt University). The following primers were usedalong with the QuikChange II XL mutagenesis kit (Stratagene, Agi-lent): 5’-CATTCC CCGCTC ACATCA CCATCA TCATCA TTGAGTGTGTGG AGGACA CCCTGA ACC-3’ and 5’-ATGTGA GCGGGGAATGAC ACAGAG TTTGTA ACGTGG TGGGAT ACAG-3’. These pri-mers contain non-overlapping regions and were designed accord-ing to Liu and Naismith.[22] Primers designed as per the instructionsprovided with the QuikChange kit did not produce the desired in-sertion.
Expression and purification of P450 2E1: P450 2E1 was expressedas previously described[2e] with a few modifications. During the ex-pression, the growth medium was supplemented with thiami-ne·HCl (10 mm) and trace elements[2a] (188 mL). During purification,P450 2E1 was eluted from Ni·NTA with 500 mm imidazole (insteadof the 200 mm reported by in ref. [2e]). Also, the ion-exchangechromatography step was omitted. The fractions of interest wereidentified by SDS-PAGE. Desired fractions were pooled and dialyzedagainst potassium phosphate (KPi, 0.1 m, pH 7.4) containing glycer-ol (10 %, v/v). Finally, the enzyme was stored at �80 8C. The con-centration of P450 enzyme was calculated from its reduced-COspectrum.[23]
General procedure for synthesis of compounds 1 a–5 a, 9 a–23 a,and 12 b: Nicotinic acid (123 mg, 1 mmol) was dissolved in DMF
(5 mL). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochlo-ride (230 mg, 1.2 mmol), the desired alcohol (1.1 mmol), and DMAP(~5 mg, catalytic) were added, and the reaction mixture was stirredovernight at room temperature. DMF was then evaporated invacuo, and the crude mixture was redissolved in EtOAc (~20 mL)before being washed with NaHCO3 (sat. , 3 � 20 mL) and brine (1 �20 mL). Finally, the organic layer was dried over anhydrous sodiumsulfate and concentrated to afford the desired product. (See theSupporting Information for purification and characterization detailsfor each specific case.)
Small-scale enzymatic transformation of auxiliary-substrates 1a–7a and 9a–23a by P450 2E1 for yield and regioselectivity esti-mates: P450 2E1 (100 pmol, 34.3 mL from a 2.9 mm stock in storagebuffer) was diluted in buffer B (0.1 m KPi at pH 7.4 containing 10 %v/v glycerol, 15.6 mL) and preincubated at 37 8C and 250 rpm for5 min (reaction tubes were placed on their side in an orbitalshaker/incubator). Inclusion of 1,2-dilauroyl-sn-glycero-3-phospho-choline (DLPC, 20 mm) made no significant difference. Reactionswere initiated by sequential addition of the substrate (1.2 mL froma 2 mm stock in acetonitrile) and CHP (400 mm, 1.2 mL from a10 mm stock in buffer containing 10 % MeOH) and incubated for20 min, as above. Then, more P450 2E1 (100 pmol) and CHP (2.1 mLfrom a 10 mm stock) were added, and the reaction mixtures wereincubated for a further 25 min, after which the products were ex-tracted in EtOAc (3 � 500 mL). According to preliminary tests with1a, adding P450 2E1 and CHP in two batches has a positive effecton the reaction yield but no effect on product regioselectivity. Theextracts were combined and evaporated. The residue was redis-solved in acetonitrile (30 mL) and analyzed by LC-MS. Standardswere also prepared by mixing each substrate (1.2 mL from a 2 mm
stock in acetonitrile) with acetonitrile (28.8 mL) before LC-MS analy-sis. Analytical separation and quantification of the products wasachieved by using a SYNERGI 4 mm HydroRP 80 � (4.6 � 250 mm)analytical column (Phenomenex, Torrance, CA) and mobile phasesA (Milli-Q water) and B (acetonitrile) at a flow rate of 0.5 mL min�1
with the UV detector set to monitor at 256 nm. Elution consistedof a linear increase from 50 to 95 % phase B over 20 min followedby an isocratic step at 95 % phase B. Yields were estimated bytaking the ratio of the area under the product peak(s) to the areaunder the substrate peak (injected separately as a “no-reaction”standard) from their respective UV/Vis spectra. Regioselectivity wasestimated from the area under the product peaks from the UV/Visspectra, except for compounds 18a (in this case the SIM spectrumwas used due to poor resolution in the UV/Vis spectrum) and 2a(in this case the NMR spectrum was also used since the major pro-duct 2b could not be separated from one of the minor products).
General procedure for enzymatic synthesis of compounds 1 b–5 b, 9 b, 11 b–14 b, 18 b, 20 b, and 22 b: P450 2E1 (10 mL, froma 7–14 mm stock) was diluted in KPi buffer (57 mL, 0.1 m at pH 7.4)and preincubated at 37 8C and 250 rpm (orbital shaking) for15 min. Reactions were initiated by sequential addition of the sub-strate (1.9 mL, 20 mm stock in acetonitrile) and CHP (6 mL froma 10 mm stock in buffer containing 1 % MeOH) and allowed to con-tinue for 45 min at 37 8C and 250 rpm. The products were extract-ed in EtOAc (3 � 50 mL). The extracts were combined, dried overanhydrous sodium sulfate, and evaporated under reduced pres-sure. The residues were redissolved in CH2Cl2 (~500 mL), and theproducts were purified by preparatory thin-layer chromatographywith hexanes/EtOAc as the mobile phase. Some products requiredfurther purification by reversed-phase semipreparatory HPLC. (Seethe Supporting Information for purification and characterizationdetails for each specific case.)
2534 www.chembiochem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemBioChem 2012, 13, 2527 – 2536
K. Auclair et al.

Enzymatic transformation of hexan-1-ol (8 a), 3-methylpentan-1-ol, and cyclopentylmethanol by P450 2E1: The reaction condi-tions used were as described above for substrates 1 a–7 a and 9 a–23 a, but scaled up three times. At the end of the incubationperiod, NaCl (~10 mg) was added to each mixture before extrac-tion in chloroform (3 � 500 mL). Extracts were combined, evaporat-ed, redissolved in chloroform (50 mL for 8 a and 30 mL for the othertwo alcohols), and analyzed by GC-MS. The reaction yield for 8 awas estimated by comparing the area under the product peak tothat of an authentic standard (hexane-1,5-diol) injected separatelyin the same amount as the substrate initially added to the reaction.The area of the latter was taken as that expected for a reactionwith 100 % yield. Separations were achieved on an Agilent Cyclo-dex-B column (60 m � 0.250 mm � 0.25 mm). The temperature pro-gram consisted of an initial isocratic step at 100 8C for 3 min,followed by a ramp to 150 8C (20 8C min�1) and a final isocratic stepat 150 8C for 15 min. The inlet temperature was set to 250 8C andsplitless injections were performed (1 mL for 8 a and 3 mL for theother two alcohols). The carrier gas (He) flow rate was16.3316 cm s�1 (0.5 mL min�1), and the detector temperature was250 8C.
Removal of the nicotinate auxiliary: KOH (10 mg, 100 mL froma 100 mg mL�1 solution in methanol) was added to compound12 b (1.3 mg), and the mixture was stirred at 37 8C for 1 h. Comple-tion of the reaction was confirmed by TLC (100 % EtOAc).
Spectral binding studies: All UV/Vis spectra of the enzyme–ligandcomplexes were obtained on a Cary 5000 UV/Vis spectrophotome-ter. P450 2E1 (145 pmol, 50 mL from a 2.9 mm stock), KPi buffer(445 mL, 0.1 m KPi at pH 7.4 with 10 % glycerol), and DLPC (20 mm,5 mL from a 2 mm stock) were added to both the reference andsample cuvettes, and a blank was taken. Substrates 1 a–8 a, 10 a–12 a, 14 a, 16 a, 18 a, and 20 a were titrated into the sample cuv-ette from stock solutions in acetonitrile such that the final acetoni-trile concentration never exceeded 3 % (v/v). These acetonitrileconcentrations had no detectable effect on the P450 spectra. Equalvolumes of acetonitrile were added to the reference cuvette, andthe difference spectra were acquired from 350–500 nm. The differ-ence in absorbance at the peak and the trough was plottedagainst substrate concentration to obtain a binding curve. Spectraldissociation constants (Ks) were extracted from the binding curvesby fitting to the following equation:
jDAj ¼ jDAmaxj � ½S�K s þ ½S�
Here DA is the difference in absorbance between the peak and thetrough, DAmax is the maximum reachable value of DA at saturatingsubstrate concentrations, [S] is the substrate concentration, and Ks
is the apparent spectral dissociation constant. Fitting was per-formed by using the GraphPad Software.
Docking studies: Docking was performed by using Fitted throughthe web-based platform Forcaster, both developed by Prof. Nico-las Moitessier and his group.[17] The PDB files (3E4E and 3LC4) wereprepared for docking by using the programs Prepare and Pro-
cess, whereas the ligands were prepared by using Smart. Dockingwas performed by using Fitted in both flexible- and rigid-proteinmode with default settings. All programs are freely available.
Acknowledgements
We would like to thank Prof. Nicolas Moitessier and Dr. Eric Ther-rien for their guidance with the docking studies performed withFitted. This work was funded by the Natural Sciences and Engi-neering Research Council of Canada (NSERC). A.M. was supportedby scholarships from the NSERC, the Centre in Green Chemistryand Catalysis, and the McGill Chemistry Department (Tak-Hang(Bill) and Christina Chan Fellowship in Chemistry). Y.H.’s stipendwas partly covered by an NSERC undergraduate scholarship. Theauthors also thank the Center in Green Chemistry and Catalysisfor its support. The authors declare they have no conflict of inter-est for the work reported here.
Keywords: biocatalysis · C�H activation · chemical auxiliaries ·cytochromes · nicotinate · oxidation
[1] a) E. M. J. Gillam, Arch. Biochem. Biophys. 2007, 464, 176 – 186; b) S.Kumar, Expert Opin. Drug Metab. Toxicol. 2010, 6, 115 – 131; c) E. O’Reilly,V. Kçhler, S. L. Flitsch, N. J. Turner, Chem. Commun. 2011, 47, 2490 –2501.
[2] a) A. Chefson, J. Zhao, K. Auclair, ChemBioChem 2006, 7, 916 – 919; b) A.Chefson, J. Zhao, K. Auclair, J. Biotechnol. 2007, 130, 436 – 440; c) A.Chefson, K. Auclair, ChemBioChem 2007, 8, 1189 – 1197; d) A. T. Larsen,E. M. May, K. Auclair, J. Am. Chem. Soc. 2011, 133, 7853 – 7858; e) A.M�nard, Y. Huang, P. Karam, G. Cosa, K. Auclair, Bioconjugate Chem.2012, 23, 826 – 836.
[3] A. T. Larsen, T. Lai, V. Polic, K. Auclair, Green Chem. 2012, 14, 2206 – 2211.[4] Cytochrome P450 : Structure, Mechanism, and Biochemistry, 3rd ed. (Ed. :
P. R. Ortiz de Montellano), Kluwer Academic/Plenum, New York, 2005.[5] a) C.-C. Peng, J. T. Pearson, D. A. Rock, C. A. Joswig-Jones, J. P. Jones,
Arch. Biochem. Biophys. 2010, 497, 68 – 81; b) C.-C. Peng, J. L. Cape, T.Rushmore, G. J. Crouch, J. P. Jones, J. Med. Chem. 2008, 51, 8000 – 8011;c) C.-C. Peng, T. Rushmore, G. J. Crouch, J. P. Jones, Bioorg. Med. Chem.2008, 16, 4064 – 4074; d) J. P. Jones, C. A. Joswig-Jones, M. Hebner, Y.Chu, D. R. Koop, Chem.-Biol. Interact. 2011, 193, 50 – 56.
[6] a) E. Mercer, Lipids 1991, 26, 584 – 597; b) H. Vanden Bossche, W. Lauw-ers, G. Willemsens, P. Marichal, F. Cornelissen, W. Cool, Pestic. Sci. 1984,15, 188 – 198.
[7] T. S. Vasaitis, R. D. Bruno, V. C. O. Njar, J. Steroid Biochem. Mol. Biol. 2011,125, 23 – 31.
[8] M. Chiba, L. Jin, W. Neway, J. P. Vacca, J. R. Tata, K. Chapman, J. H. Lin,Drug Metab. Dispos. 2001, 29, 1 – 3.
[9] a) W. Zhang, Y. Ramamoorthy, T. Kilicarslan, H. Nolte, R. F. Tyndale, E. M.Sellers, Drug Metab. Dispos. 2002, 30, 314 – 318; b) P. O. Gubbins, ExpertOpin. Drug Metab. Toxicol. 2011, 7, 1411 – 1429; c) Z. Dvorak, Toxicol.Lett. 2011, 202, 129 – 132.
[10] a) M. C. Gerber, G. A. Tejwani, N. Gerber, J. R. Bianchine, Pharmacol. Ther.1985, 27, 353 – 370; b) X. Sun, J. Qiu, S. A. Strong, L. S. Green, J. W. F.Wasley, J. P. Blonder, D. B. Colagiovanni, S. C. Mutka, A. M. Stout, J. P. Ri-chards, G. J. Rosenthal, Bioorg. Med. Chem. Lett. 2011, 21, 5849 – 5853.
[11] a) J. T. Pearson, J. J. Hill, J. Swank, N. Isoherranen, K. L. Kunze, W. M.Atkins, Biochemistry 2006, 45, 6341 – 6353; b) K. L. Kunze, W. L. Nelson,E. D. Kharasch, K. E. Thummel, N. Isoherranen, Drug Metab. Dispos. 2006,34, 583 – 590; c) U. P. Dahal, C. Joswig-Jones, J. P. Jones, J. Med. Chem.2012, 55, 280 – 290; d) K. P. Conner, P. Vennam, C. M. Woods, M. D. Krzya-niak, M. K. Bowman, W. M. Atkins, Biochemistry 2012, 51, 6441 – 6457.
[12] S. Krishnan, D. Wasalathanthri, L. Zhao, J. B. Schenkman, J. F. Rusling, J.Am. Chem. Soc. 2011, 133, 1459 – 1465.
[13] a) M. Chiba, C. Tang, W. E. Neway, T. M. Williams, S. J. Desolms, C. J. Din-smore, J. S. Wai, J. H. Lin, Biochem. Pharmacol. 2001, 62, 773 – 776;b) M. M. Ahlstrçm, I. Zamora, J. Med. Chem. 2008, 51, 1755 – 1763.
[14] P. R. Porubsky, K. M. Meneely, E. E. Scott, J. Biol. Chem. 2008, 283,33698 – 33707.
[15] M. K. Julsing, S. Cornelissen, B. B�hler, A. Schmid, Curr. Opin. Chem. Biol.2008, 12, 177 – 186.
ChemBioChem 2012, 13, 2527 – 2536 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chembiochem.org 2535
Chemical Auxiliaries for Enzymatic Transformations

[16] F. Adas, J. P. Sala�n, F. Berthou, D. Picart, B. Simon, Y. Amet, J. Lipid Res.1999, 40, 1990 – 1997.
[17] E. Therrien, P. Englebienne, A. G. Arrowsmith, R. Mendoza-Sanchez, C. R.Corbeil, N. Weill, V. Campagna-Slater, N. Moitessier, J. Chem. Inf. Model.
2012, 52, 210 – 224.[18] P. R. Porubsky, K. P. Battaile, E. E. Scott, J. Biol. Chem. 2010, 285, 22282 –
22290.
[19] �. Tarcsay, R. Kiss, G. Keseru, J. Comput.-Aided Mol. Des. 2010, 24, 399 –408.
[20] J. C. Prasad, J. V. Goldstone, C. J. Camacho, S. Vajda, J. J. Stegeman, Bio-chemistry 2007, 46, 2640 – 2654.
[21] E. M. J. Gillam, Z. Y. Guo, F. P. Guengerich, Arch. Biochem. Biophys. 1994,312, 59 – 66.
[22] H. Liu, J. H. Naismith, BMC Biotechnol. 2008, 8, 91.[23] T. Omura, R. Sato, J. Biol. Chem. 1962, 237, PC1375 – PC1376.
Received: August 13, 2012Published online on November 5, 2012
2536 www.chembiochem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemBioChem 2012, 13, 2527 – 2536
K. Auclair et al.