tepzz¥z_¥957b_t - ep 3 013 957 b1
TRANSCRIPT

Note: Within nine months of the publication of the mention of the grant of the European patent in the European PatentBulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with theImplementing Regulations. Notice of opposition shall not be deemed to have been filed until the opposition fee has beenpaid. (Art. 99(1) European Patent Convention).
Printed by Jouve, 75001 PARIS (FR)
(19)E
P3
013
957
B1
TEPZZ¥Z_¥957B_T(11) EP 3 013 957 B1
(12) EUROPEAN PATENT SPECIFICATION
(45) Date of publication and mention of the grant of the patent: 26.09.2018 Bulletin 2018/39
(21) Application number: 14817610.0
(22) Date of filing: 26.06.2014
(51) Int Cl.:C12N 15/10 (2006.01) C12Q 1/68 (2018.01)
G01N 33/566 (2006.01) G06F 19/20 (2011.01)
(86) International application number: PCT/US2014/044398
(87) International publication number: WO 2014/210353 (31.12.2014 Gazette 2014/53)
(54) COMPOSITIONS AND METHODS FOR SAMPLE PROCESSING
ZUSAMMENSETZUNGEN UND VERFAHREN ZUR PROBENVERARBEITUNG
COMPOSITIONS ET PROCÉDÉS DE TRAITEMENT D’ÉCHANTILLON
(84) Designated Contracting States: AL AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL PT RO RS SE SI SK SM TR
(30) Priority: 27.06.2013 US 201361840403 P10.07.2013 US 201361844804 P13.08.2013 US 20131396615013.08.2013 PCT/US2013/05479726.10.2013 US 201361896060 P27.11.2013 US 201361909974 P07.02.2014 US 201461937344 P14.02.2014 US 201461940318 P09.05.2014 US 201461991018 P
(43) Date of publication of application: 04.05.2016 Bulletin 2016/18
(60) Divisional application: 18189200.1
(73) Proprietor: 10X Genomics, Inc.Pleasanton, CA 94566 (US)
(72) Inventors: • HINDSON, Benjamin
Pleasanton, CA 94566 (US)• HINDSON, Christopher
Livermore, CA 94551 (US)• SCHNALL-LEVIN, Michael
Palo Alto, CA94306 (US)
• NESS, KevinBoulder, CO 80304 (US)
• JAROSZ, MirnaMountain View, CA 94041 (US)
• MASQUELIER, DonaldTracy, CA 95376 (US)
• SAXONOV, SergeOakland, CA 94619 (US)
• MERRILL, LandonPleasanton, CA 94566 (US)
• PRICE, AndrewPleasanton, CA 94566 (US)
• HARDENBOL, PaulSan Francisco, CA 94110 (US)
• LI, YuanDublin, CA 94568 (US)
(74) Representative: Forrest, Graham Robert et alMewburn Ellis LLPCity Tower40 Basinghall StreetLondon EC2V 5DE (GB)
(56) References cited: WO-A1-2010/115154 WO-A1-2012/048341US-A1- 2008 268 431 US-A1- 2012 220 494US-A1- 2012 309 002 US-A1- 2012 316 074US-A1- 2013 225 418 US-A1- 2014 378 322US-A1- 2014 378 345 US-A1- 2015 298 091

EP 3 013 957 B1
2
5
10
15
20
25
30
35
40
45
50
55
Description
BACKGROUND
[0001] Genomic sequencing can be used to obtain in-formation in a wide variety of biomedical contexts, includ-ing diagnostics, prognostics, biotechnology, and forensicbiology. Sequencing may involve basic methods includ-ing Maxam-Gilbert sequencing and chain-terminationmethods, or de novo sequencing methods including shot-gun sequencing and bridge PCR, or next-generationmethods including polony sequencing, 454 pyrose-quencing, Illumina sequencing, SOLiD sequencing, IonTorrent semiconductor sequencing, HeliScope singlemolecule sequencing, SMRT® sequencing, and others.For most sequencing applications, a sample such as anucleic acid sample is processed prior to introduction toa sequencing machine. A sample may be processed, forexample, by amplification or by attaching a unique iden-tifier. Often unique identifiers are used to identify the or-igin of a particular sample.[0002] WO 2010/115154 describes a multi-primer am-plification method for barcoding of target nucleic acids.WO 2012/048341 describes high-throughput methodsfor barcoding analytes derived from single cells, US2012/0316074 describes methods for barcoding sam-ples by bringing together partitions containing barcodemolecules with partitions comprising samples to be la-belled.
SUMMARY
[0003] The present disclosure generally relates tomethods, compositions, devices, and kits for the gener-ation of beads with covalently attached polynucleotides.Such beads may be used for any suitable application.[0004] The invention provides a method of barcodingsample materials, comprising:
providing a bead comprising a plurality of nucleic ac-id barcode molecules releasably associated there-with, the plurality of nucleic acid barcode moleculescomprising the same nucleic acid barcode se-quence;co-partitioning the bead with components of a sam-ple material into a partition;releasing the barcode molecules from the bead intothe partition; andattaching the barcode molecules to one or more ofthe components of the sample material or fragmentsthereof within the partition.
In some cases, the bead may comprise at least 1,000barcode molecules, at least 10,000 barcode molecules,at least 100,000 barcode molecules, or at least 1,000,000barcode molecules associated therewith having thesame barcode sequence. In some cases, the barcodemolecules may be releasably coupled to the bead. The
bead may, for example, be a gel bead. In some cases,the barcode molecules may be encapsulated within ordiffused throughout the bead. Moreover, the partitionmay comprise a droplet and/or may comprise no morethan one bead.[0005] In some cases, the co-partitioning may com-prise combining a first aqueous fluid comprising beadswith a second aqueous fluid comprising the sample com-ponents in a droplet within an immiscible fluid. The bar-code molecules may be released in the partition by de-grading the bead and/or cleaving a chemical linkage be-tween the barcode molecules and the bead. In some cas-es, at least one of crosslinking of the bead and a linkagebetween the bead and the barcode molecules may com-prise a disulfide linkage. In such cases, the barcode mol-ecules may be released from the bead by exposing thebead to a reducing agent (e.g., dithiothreitol (DTT) ortris(2-carboxyethyl)phosphine (TCEP)).[0006] The sample materials may comprise one ormore template nucleic acid molecules and the barcodemolecules may be attached to one or more fragments ofthe template nucleic acid molecules. In some cases, thebarcode molecules may comprise a primer sequencecomplementary to at least a portion of the template nu-cleic acid molecules and the barcode molecules may beattached to the template nucleic acid molecule or frag-ments thereof by extending the barcode molecules toreplicate at least a portion of the template nucleic acidmolecules. Moreover, the sample materials may com-prise the contents of a single cell, such as, for example,a cancer cell or a bacterial cell (e.g., a bacterial cell iso-lated from a human microbiome sample).[0007] Furthermore, a plurality of beads comprising aplurality of different nucleic acid barcode sequences maybe provided. Each of the beads can include a plurality ofat least 1000 nucleic acid barcode molecules having thesame nucleic acid barcode sequence associated there-with. The beads may be co-partitioned with componentsof the sample material into a plurality of partitions. Thenucleic acid barcode molecules may then be releasedinto the partitions. The released nucleic acid barcodemolecules can then be attached to the components ofthe sample material or fragments thereof within the par-titions. In some cases, the plurality of different nucleicacid barcode sequences may comprise at least about1,000 different barcode sequences, at least about 10,000different barcode sequences, at least about 100,000 dif-ferent barcode sequences, or at least about 500,000 dif-ferent barcode sequences. Additionally, in some exam-ples, a subset of the partitions may comprise the samenucleic acid barcode sequence. For example, at leastabout 1%, at least about 2%, or at least about 5% of thepartitions may comprise the same nucleic acid barcodesequence. In addition, in some cases, at least 50% ofthe partitions, at least 70% of the partitions, or at least90% of the partitions may contain no more than one firstpartition. In some cases, at least 50% of the partitions,at least 70% of the partitions, or at least 90% of the par-
1 2

EP 3 013 957 B1
3
5
10
15
20
25
30
35
40
45
50
55
titions may contain exactly one bead.[0008] Fragments of the components of the samplematerial may include one or more fragments of one ormore template nucleic acid sequences. The fragmentsof the template nucleic acid sequences may be se-quenced and characterized based at least in part upona nucleic acid barcode sequence attached thereto. Insome cases, the fragments of the template nucleic acidsequences may be characterized by mapping a fragmentof an individual template nucleic acid sequence of thetemplate nucleic acid sequences to an individual tem-plate nucleic acid sequence of the template nucleic acidsequences or a genome from which the individual tem-plate nucleic acid sequence was derived. In some cases,the fragments of the template nucleic acid sequence maybe characterized by at least identifying an individual nu-cleic acid barcode sequence of the different nucleic acidbarcode sequences and identifying a sequence of an in-dividual fragment of the fragments of the template nucleicacid sequences attached to the individual nucleic acidbarcode sequence.
BRIEF DESCRIPTION OF THE DRAWINGS
[0009]
Fig 1A is a flow diagram for making barcoded beads.Fig 1B is a flow diagram for processing a sample forsequencing.Fig 2 is a flow diagram for making beads.Fig 3A is a flow diagram for adding barcodes tobeads by limiting dilution.Fig 3B is a flow diagram for adding additional se-quences to oligonucleotides attached to beads.Figs 4A-N are diagrams for attaching sequences tobeads. "g/w" means gel-in-water; "g/w/o" means gel-in-water-in-oil;Fig 5 provides an illustration of a gel bead attachedto an oligonucleotide 5A, an image of a microfluidicchip used to make Gel Beads in Emulsions (GEM)5B, as well as images of GEMs 5C, D, E.Fig 6 provides bright-field (A, C, E) and fluorescent(B, D, F) images of beads with attached oligonucle-otides.Figs 7A-C provide fluorescent images of beads at-tached to DNA.Figs 8A-F provide images of barcode-enriched pop-ulations of beads.Figs 9A-D provide images of the dissolution of beadsby heating.Fig 10A provides a schematic of a functionalizedbead. Figs 10B-G provide images of beads dis-solved with a reducing agent.Fig 11A provides a schematic of a functionalizedbead. Figs 11B-D provide graphic depictions of thepresence of barcode oligonucleotides and primer-dimer pairs when beads are prepared using differentconditions.
Fig 12 is a graphic depiction of content attached tobeads.Fig 13A is a flow diagram illustrating the addition ofbarcodes to beads using partitions.Fig 13B is a flow diagram illustrating the addition ofadditional sequences to beads.Fig 13C is a diagram illustrating the use of a combi-natorial approach in microwell plates to make bar-coded beads.Figs 14A-C are diagrams of oligonucleotides con-taining universal sequences (R1, P5) and uracil con-taining nucleotides.Figs 15A-G are diagrams of steps used in the partialhairpin amplification for sequencing (PHASE) proc-ess.Fig 16A is a graphic depiction of including uracil con-taining nucleotides in the universal portion of theprimer.Fig 16B is a graphic depiction of controlling amplifi-cation product length by including acyNTPs in thereaction mixture.Fig 17 is a graphic depiction of reducing start sitebias by adding a blocker oligonucleotide.Fig 18 is a flow diagram of a digital processor andits related components.Fig 19 is a table providing example sequences forIllumina sequencers.Fig 20 is a table providing a list of example capturemoiety concentrations used to label beads.Fig 21 is a table providing a list of sequencing metricsobtained using primers comprising thymine contain-ing nucleotides.Fig 22 is a table providing a list of sequencing metricsobtained using primers comprising uracil containingnucleotides.Figs 23A-D are schematics illustrating the use of anexample ligation-based combinatorial approach tomake barcoded beads.Figs 24A-B are schematics illustrating an exampleuse of spacer bases in a ligation-based combinato-rial approach to make barcoded beads.Figs 25A-C are schematics illustrating the use of anexample ligation-based combinatorial approach tomake barcoded beads.Fig 26 is a schematic illustrating example nucleicacids used in an example ligation-based combina-torial approach to make barcoded beads.Fig 27 is a schematic illustrating an example ligation-based combinatorial approach to make barcodedbeads.Figs 28A-B are schematic representations of exam-ple targeted barcode constructs suitable for strand-specific amplification.Figs 29A-C are structural depictions of examplemonomers and cross-linkers that can be polymer-ized to generate beads.Figs 30A-C are structural depictions of an examplemethod that can be used to generate beads.
3 4

EP 3 013 957 B1
4
5
10
15
20
25
30
35
40
45
50
55
Fig 31 is a schematic depiction of example beadscomprising functional groups that can be used to at-tach species to the beads.Fig 32 provides structural depictions of example in-itiators that may be used during a polymerization re-action.Fig 33A is a schematic depiction of barcode primers.Figs 33B-E are graphic depictions of data corre-sponding to example amplification reaction experi-ments described in Example 16.Figs 34A-C are schematics of example hairpin con-structs.Figs 35A-B are schematics of example methods forfunctionalizing beads.Fig 36 is a photograph of a gel obtained during a gelelectrophoresis experiment described in Example17.Fig 37A is a schematic depiction of oligonucleotidesdescribed in Example 18. Fig 37B is a photographof a gel obtained during a gel electrophoresis exper-iment described in Example 18. Fig 37C is a micro-graph of beads obtained during a fluorescence mi-croscopy experiment described in Example 18.Fig 38 provides a schematic illustration of an exem-plary nucleic acid barcoding and amplification proc-ess.Fig 39 provides a schematic illustration of an exem-plary application of the methods described herein tonucleic acid sequencing and assembly.Fig 40 presents examples of alternative processingsteps following barcoding and amplification of nucle-ic acids, as described herein.
DETAILED DESCRIPTION
I. General Overview
[0010] This disclosure relates to methods, systemsand compositions useful in the processing of sample ma-terials through the controlled delivery of reagents to sub-sets of sample components, followed by analysis of thosesample components employing, in part, the delivered re-agents. In many cases, the methods and compositionsare employed for sample processing, particularly for nu-cleic acid analysis applications, generally, and nucleicacid sequencing applications, in particular. Included with-in this disclosure are bead compositions that include di-verse sets of reagents, such as diverse libraries of beadsattached to large numbers of oligonucleotides containingbarcode sequences, and methods of making and usingthe same.[0011] Methods of making beads can generally in-clude, e.g. combining bead precursors (such as mono-mers or polymers), primers, and cross-linkers in an aque-ous solution, combining said aqueous solution with anoil phase, sometimes using a microfluidic device or drop-let generator, and causing water-in-oil droplets to form.In some cases, a catalyst, such as an accelerator and/or
an initiator, may be added before or after droplet forma-tion. In some cases, initiation may be achieved by theaddition of energy, such, as for example via the additionof heat or light (e.g., UV light). A polymerization reactionin the droplet can occur to generate a bead, in somecases covalently linked to one or more copies of an oli-gonucleotide (e.g., primer). Additional sequences can beattached to the functionalized beads using a variety ofmethods. In some cases, the functionalized beads arecombined with a template oligonucleotide (e.g., contain-ing a barcode) and partitioned such that on average oneor fewer template oligonucleotides occupy the same par-tition as a functionalized bead. While the partitions maybe any of a variety of different types of partitions, e.g.,wells, microwells, tubes, vials, microcapsules, etc., inpreferred aspects, the partitions may be droplets (e.g.,aqueous droplets) within an emulsion. The oligonucle-otide (e.g., barcode) sequences can be attached to thebeads within the partition by a reaction such as a primerextension reaction, ligation reaction, or other methods.For example, in some cases, beads functionalized withprimers are combined with template barcode oligonucle-otides that comprise a binding site for the primer, enablingthe primer to be extended on the bead. After multiplerounds of amplification, copies of the single barcode se-quence are attached to the multiple primers attached tothe bead. After attachment of the barcode sequences tothe beads, the emulsion can be broken and the barcodedbeads (or beads linked to another type of amplified prod-uct) can be separated from beads without amplified bar-codes. Additional sequences, such as a random se-quence (e.g., a random N-mer) or a targeted sequence,can then be added to the bead-bound barcode sequenc-es, using, for example, primer extension methods or oth-er amplification reactions. This process can generate alarge and diverse library of barcoded beads.[0012] Fig 1A illustrates an example method for gen-erating a barcoded bead. First, gel precursors (e.g., linearpolymers and/or monomers), cross-linkers, and primersmay be combined in an aqueous solution, 101. Next, ina microfluidic device, the aqueous solution can then becombined with an oil phase, 102. Combining the oil phaseand aqueous solution can cause water-in-oil droplets toform, 103. Within water-in-oil droplets, polymerization ofthe gel precursors occurs to form beads comprising mul-tiple copies of a primer, 104. Following generation of aprimer-containing bead, the emulsion may be broken,105 and the beads recovered. The recovered beads maybe separated from unreacted components, via, for ex-ample, washing and introduced to any suitable solvent(e.g., an aqueous solvent, a non-aqueous solvent). Insome cases, the primer-containing beads may then becombined (e.g., via limiting dilution methods) with tem-plate barcode sequences in droplets of another emulsion,such that each droplet comprises on average at least onebead and on average one or less molecules of a templatebarcode sequence. The template barcode sequencemay be clonally amplified, using the primer attached to
5 6

EP 3 013 957 B1
5
5
10
15
20
25
30
35
40
45
50
55
the bead, resulting in attachment to the bead of multiplecopies of a barcode sequence complementary to the tem-plate, 106. The barcoded beads may then be pooled intoa population of beads either containing barcodes or notcontaining barcodes, 107. The barcoded beads may thenbe isolated by, for example, an enrichment step. The bar-code molecules may also be provided with additionalfunctional sequence components for exploitation in sub-sequent processing. For example, primer sequencesmay be incorporated into the same oligonucleotides thatinclude the barcode sequence segments, to enable theuse of the barcode containing oligonucleotides to func-tion as extension primers for duplicating sample nucleicacids, or as priming sites for subsequent sequencing oramplification reactions. In one example, random N-mersequences may then be added to the barcoded beads,108, via primer extension or other amplification reactionand a diverse library of barcoded beads, 110, may there-by be obtained, where such random n-mer sequencescan provide a universal primer sequence. Likewise, func-tional sequences may include immobilization sequencesfor immobilizing barcode containing sequences onto sur-faces, e.g., for sequencing applications. For ease of dis-cussion, a number of specific functional sequences aredescribed below, such as P5, P7, R1, R2, sample index-es, random Nmers, etc., and partial sequences for these,as well as complements of any of the foregoing. However,it will be appreciated that these descriptions are for pur-poses of discussion, and any of the various functionalsequences included within the barcode containing oligo-nucleotides may be substituted for these specific se-quences, including without limitation, different attach-ment sequences, different sequencing primer regions,different n-mer regions (targeted and random), as wellas sequences having different functions, e.g., secondarystructure forming, e.g., hairpins or other structures, probesequences, e.g., to allow interrogation of the presenceor absence of the oligonucleotides or to allow pull downof resulting amplicons, or any of a variety of other func-tional sequences.[0013] Also included within this disclosure are methodsof sample preparation for nucleic acid analysis, and par-ticularly for sequencing applications. Sample preparationcan generally include, e.g. obtaining a sample comprisingsample nucleic acid from a source, optionally furtherprocessing the sample, combining the sample nucleicacid with barcoded beads, and forming emulsions con-taining fluidic droplets comprising the sample nucleic acidand the barcoded beads. Droplets may be generated, forexample, with the aid of a microfluidic device and/or viaany suitable emulsification method. The fluidic dropletscan also comprise agents capable of dissolving, degrad-ing, or otherwise disrupting the barcoded beads, and/ordisrupting the linkage to attached sequences, therebyreleasing the attached barcode sequences from thebead. The barcode sequences may be released eitherby degrading the bead, detaching the oligonucleotidesfrom the bead such as by a cleavage reaction, or a com-
bination of both. By amplifying (e.g., via amplificationmethods described herein) the sample nucleic acid in thefluidic droplets, for example, the free barcode sequencescan be attached to the sample nucleic acid. The emulsioncomprising the fluidic droplets can then be broken and,if desired, additional sequences (e.g., sequences that aidin particular sequencing methods, additional barcode se-quences, etc.) can then be added to the barcoded samplenucleic acid using, for example, additional amplificationmethods. Sequencing can then be performed on the bar-coded, amplified sample nucleic acid and one or moresequencing algorithms applied to interpret the sequenc-ing data. As used herein, the sample nucleic acids mayinclude any of a wide variety of nucleic acids, including,e.g., DNA and RNA, and specifically including for exam-ple, genomic DNA, cDNA, mRNA total RNA, and cDNAcreated from a mRNA or total RNA transcript.[0014] Fig 1B illustrates an example method for bar-coding and subsequently sequencing a sample nucleicacid. First, a sample comprising nucleic acid may be ob-tained from a source, 111, and a set of barcoded beadsmay be obtained, e.g., as described herein, 112. Thebeads are preferably linked to oligonucleotides contain-ing one or more barcode sequences, as well as a primer,such as a random N-mer or other primer. The barcodesequences are releasable from the barcoded beads, e.g.,through cleavage of a linkage between the barcode andthe bead or through degradation of the underlying beadto release the barcode, or a combination of the two. Forexample, in certain preferred aspects, the barcodedbeads can be degraded or dissolved by an agent, suchas a reducing agent to release the barcode sequences.In this example, the sample comprising nucleic acid, 113,barcoded beads, 114, and e.g., a reducing agent, 116,are combined and subject to partitioning. By way of ex-ample, such partitioning may involve introducing thecomponents to a droplet generation system, such as amicrofluidic device, 115. With the aid of the micro fluidicdevice 115, a water-in-oil emulsion 117 may be formed,wherein the emulsion contains aqueous droplets thatcontain sample nucleic acid, reducing agent, and bar-coded beads, 117. The reducing agent may dissolve ordegrade the barcoded beads, thereby releasing the oli-gonucleotides with the barcodes and random N-mersfrom the beads within the droplets, 118. The random N-mers may then prime different regions of the sample nu-cleic acid, resulting in amplified copies of the sample afteramplification, wherein each copy is tagged with a bar-code sequence, 119. Preferably, each droplet containsa set of oligonucleotides that contain identical barcodesequences and different random N-mer sequences. Sub-sequently, the emulsion is broken, 120 and additionalsequences (e.g., sequences that aid in particular se-quencing methods, additional barcodes, etc.) may beadded, 122, via, for example, amplification methods (e.g.,PCR). Sequencing may then be performed, 123, and analgorithm applied to interpret the sequencing data, 124.Sequencing algorithms are generally capable, for exam-
7 8

EP 3 013 957 B1
6
5
10
15
20
25
30
35
40
45
50
55
ple, of performing analysis of barcodes to align sequenc-ing reads and/or identify the sample from which a partic-ular sequence read belongs.[0015] The methods and compositions of this disclo-sure may be used with any suitable digital processor.The digital processor may be programmed, for example,to operate any component of a device and/or executemethods described herein. In some embodiments, beadformation may be executed with the aid of a digital proc-essor in communication with a droplet generator. Thedigital processor may control the speed at which dropletsare formed or control the total number of droplets thatare generated. In some embodiments, attaching barcodesequences to sample nucleic acid may be completed withthe aid of a microfluidic device and a digital processor incommunication with the microfluidic device. In some cas-es, the digital processor may control the amount of sam-ple and/or beads provided to the channels of the micro-fluidic device, the flow rates of materials within the chan-nels, and the rate at which droplets comprising barcodesequences and sample nucleic acid are generated.[0016] The methods and compositions of this disclo-sure may be useful for a variety of different molecularbiology applications including, but not limited to, nucleicacid sequencing, protein sequencing, nucleic acid quan-tification, sequencing optimization, detecting gene ex-pression, quantifying gene expression, epigenetic appli-cations, and single-cell analysis of genomic or expressedmarkers. Moreover, the methods and compositions ofthis disclosure have numerous medical applications in-cluding identification, detection, diagnosis, treatment,staging of, or risk prediction of various genetic and non-genetic diseases and disorders including cancer.
II. Beads or Particles
[0017] The methods, compositions, devices, and kitsof this disclosure may be used with any suitable bead orparticle, including gel beads and other types of beads.Beads may serve as a carrier for reagents that are to bedelivered in accordance with the methods describedherein. In particular, these beads may provide a surfaceto which reagents are releasably attached, or a volumein which reagents are entrained or otherwise releasablypartitioned. These reagents may then be delivered in ac-cordance with a desired method, for example, in the con-trolled delivery of reagents into discrete partitions. A widevariety of different reagents or reagent types may be as-sociated with the beads, where one may desire to deliversuch reagents to a partition. Non-limiting examples ofsuch reagents include, e.g., enzymes, polypeptides, an-tibodies or antibody fragments, labeling reagents, e.g.,dyes, fluorophores, chromophores, etc., nucleic acids,polynucleotides, oligonucleotides, and any combinationof two or more of the foregoing. In some cases, the beadsmay provide a surface upon which to synthesize or attacholigonucleotide sequences. Various entities including ol-igonucleotides, barcode sequences, primers, crosslink-
ers and the like may be associated with the outer surfaceof a bead. In the case of porous beads, an entity may beassociated with both the outer and inner surfaces of abead. The entities may be attached directly to the surfaceof a bead (e.g., via a covalent bond, ionic bond, van derWaals interactions, etc.), may be attached to other oli-gonucleotide sequences attached to the surface of abead (e.g. adaptor or primers), may be diffused through-out the interior of a bead and/or may be combined witha bead in a partition (e.g. fluidic droplet). In preferredembodiments, the oligonucleotides are covalently at-tached to sites within the polymeric matrix of the beadand are therefore present within the interior and exteriorof the bead. In some cases, an entity such as a cell ornucleic acid is encapsulated within a bead. Other entitiesincluding amplification reagents (e.g., PCR reagents,primers) may also be diffused throughout the bead orchemically-linked within the interior (e.g., via pores, cov-alent attachment to polymeric matrix) of a bead.[0018] Beads may serve to localize entities or samples.In some embodiments, entities (e.g. oligonucleotides,barcode sequences, primers, crosslinkers, adaptors andthe like) may be associated with the outer and/or an innersurface of the bead. In some cases, entities may be lo-cated throughout the bead. In some cases, the entitiesmay be associated with the entire surface of a bead orwith at least half the surface of the bead.[0019] Beads may serve as a support on which to syn-thesize oligonucleotide sequences. In some embodi-ments, synthesis of an oligonucleotide may comprise aligation step. In some cases, synthesis of an oligonucle-otide may comprise ligating two smaller oligonucleotidestogether. In some cases, a primer extension or other am-plification reaction may be used to synthesize an oligo-nucleotide on a bead via a primer attached to the bead.In such cases, a primer attached to the bead may hybrid-ize to a primer binding site of an oligonucleotide that alsocontains a template nucleotide sequence. The primer canthen be extended by an primer extension reaction or otheramplification reaction, and an oligonucleotide comple-mentary to the template oligonucleotide can thereby beattached to the bead. In some cases, a set of identicaloligonucleotides associated with a bead may be ligatedto a set of diverse oligonucleotides, such that each iden-tical oligonucleotide is attached to a different member ofthe diverse set of oligonucleotides. In other cases, a setof diverse oligonucleotides associated with a bead maybe ligated to a set of identical oligonucleotides.
Bead Characteristics
[0020] The methods, compositions, devices, and kitsof this disclosure may be used with any suitable bead.In some embodiments, a bead may be porous, non-po-rous, solid, semi-solid, semi-fluidic, or fluidic. In someembodiments, a bead may be dissolvable, disruptable,or degradable. In some cases, a bead may not be de-gradable. In some embodiments, the bead may be a gel
9 10

EP 3 013 957 B1
7
5
10
15
20
25
30
35
40
45
50
55
bead. A gel bead may be a hydrogel bead. A gel beadmay be formed from molecular precursors, such as apolymeric or monomeric species. A semi-solid bead maybe a liposomal bead. Solid beads may comprise metalsincluding iron oxide, gold, and silver. In some cases, thebeads are silica beads. In some cases, the beads arerigid. In some cases, the beads may be flexible.[0021] In some embodiments, the bead may containmolecular precursors (e.g., monomers or polymers),which may form a polymer network via polymerization ofthe precursors. In some cases, a precursor may be analready polymerized species capable of undergoing fur-ther polymerization via, for example, a chemical cross-linkage. In some cases, a precursor comprises one ormore of an acrylamide or a methacrylamide monomer,oligomer, or polymer. In some cases, the bead may com-prise prepolymers, which are oligomers capable of fur-ther polymerization. For example, polyurethane beadsmay be prepared using prepolymers. In some cases, thebead may contain individual polymers that may be furtherpolymerized together. In some cases, beads may be gen-erated via polymerization of different precursors, suchthat they comprise mixed polymers, co-polymers, and/orblock co-polymers.[0022] A bead may comprise natural and/or syntheticmaterials, including natural and synthetic polymers. Ex-amples of natural polymers include proteins and sugarssuch as deoxyribonucleic acid, rubber, cellulose, starch(e.g. amylose, amylopectin), proteins, enzymes,polysaccharides, silks, polyhydroxyalkanoates, chi-tosan, dextran, collagen, carrageenan, ispaghula, aca-cia, agar, gelatin, shellac, sterculia gum, xanthan gum,Corn sugar gum, guar gum, gum karaya, agarose, alginicacid, alginate, or natural polymers thereof. Examples ofsynthetic polymers include acrylics, nylons, silicones,spandex, viscose rayon, polycarboxylic acids, polyvinylacetate, polyacrylamide, polyacrylate, polyethylene gly-col, polyurethanes, polylactic acid, silica, polystyrene,polyacrylonitrile, polybutadiene, polycarbonate, polyeth-ylene, polyethylene terephthalate, poly(chlorotrifluor-oethylene), poly(ethylene oxide), poly(ethylene tereph-thalate), polyethylene, polyisobutylene, poly(methylmethacrylate), poly(oxymethylene), polyformaldehyde,polypropylene, polystyrene, poly(tetrafluoroethylene),poly(vinyl acetate), poly(vinyl alcohol), poly(vinyl chlo-ride), poly(vinylidene dichloride), poly(vinylidene difluo-ride), poly(vinyl fluoride) and combinations (e.g., co-pol-ymers) thereof. Beads may also be formed from materialsother than polymers, including lipids, micelles, ceramics,glass-ceramics, material composites, metals, other inor-ganic materials, and others.[0023] In some cases, a chemical cross-linker may bea precursor used to cross-link monomers during polym-erization of the monomers and/or may be used to func-tionalize a bead with a species. In some cases, polymersmay be further polymerized with a cross-linker speciesor other type of monomer to generate a further polymericnetwork. Non-limiting examples of chemical cross-linkers
(also referred to as a "crosslinker" or a "crosslinker agent"herein) include cystamine, gluteraldehyde, dimethylsuberimidate, N-Hydroxysuccinimide crosslinker BS3,formaldehyde, carbodiimide (EDC), SMCC, Sulfo-SM-CC, vinylsilance, N,N’diallyltartardiamide (DATD), N,N’-Bis(acryloyl)cystamine (BAC), or homologs thereof. Insome cases, the crosslinker used in the present disclo-sure contains cystamine.[0024] Crosslinking may be permanent or reversible,depending upon the particular crosslinker used. Revers-ible crosslinking may allow for the polymer to linearize ordissociate under appropriate conditions. In some cases,reversible cross-linking may also allow for reversible at-tachment of a material bound to the surface of a bead.In some cases, a cross-linker may form disulfide linkag-es. In some cases, the chemical cross-linker forming di-sulfide linkages may be cystamine or a modifiedcystamine. In some embodiments, disulfide linkages maybe formed between molecular precursor units (e.g. mon-omers, oligomers, or linear polymers). In some embodi-ments, disulfide linkages may be may be formed betweenmolecular precursor units (e.g. monomers, oligomers, orlinear polymers) or precursors incorporated into a beadand oligonucleotides.[0025] Cystamine (including modified cystamines), forexample, is an organic agent comprising a disulfide bondthat may be used as a crosslinker agent between indi-vidual monomeric or polymeric precursors of a bead.Polyacrylamide may be polymerized in the presence ofcystamine or a species comprising cystamine (e.g., amodified cystamine) to generate polyacrylamide gelbeads comprising disulfide linkages (e.g., chemically de-gradable beads comprising chemically-reducible cross-linkers). The disulfide linkages may permit the bead tobe degraded (or dissolved) upon exposure of the beadto a reducing agent.[0026] In at least one alternative example, chitosan, alinear polysaccharide polymer, may be crosslinked withglutaraldehyde via hydrophilic chains to form a bead.Crosslinking of chitosan polymers may be achieved bychemical reactions that are initiated by heat, pressure,change in pH, and/or radiation.[0027] In some embodiments, the bead may comprisecovalent or ionic bonds between polymeric precursors(e.g. monomers, oligomers, linear polymers), oligonucle-otides, primers, and other entities. In some cases, thecovalent bonds comprise carbon-carbon bonds orthioether bonds.[0028] In some cases, a bead may comprise an acry-dite moiety, which in certain aspects may be used to at-tach one or more species (e.g., barcode sequence, prim-er, other oligonucleotide) to the bead. In some cases, anacrydite moiety can refer to an acrydite analogue gener-ated from the reaction of acrydite with one or more spe-cies, such as, for example, the reaction of acrydite withother monomers and cross-linkers during a polymeriza-tion reaction. Acrydite moieties may be modified to formchemical bonds with a species to be attached, such as
11 12

EP 3 013 957 B1
8
5
10
15
20
25
30
35
40
45
50
55
an oligonucleotide (e.g., barcode sequence, primer, oth-er oligonucleotide). For example, acrydite moieties maybe modified with thiol groups capable of forming a, di-sulfide bond or may be modified with groups already com-prising a disulfide bond. The thiol or disulfide (via disulfideexchange) may be used as an anchor point for a speciesto be attached or another part of the acrydite moiety maybe used for attachment. In some cases, attachment isreversible, such that when the disulfide bond is broken(e.g., in the presence of a reducing agent), the agent isreleased from the bead. In other cases, an acrydite moi-ety comprises a reactive hydroxyl group that may be usedfor attachment.[0029] Functionalization of beads for attachment ofother species, e.g., nucleic acids, may be achievedthrough a wide range of different approaches, includingactivation of chemical groups within a polymer, incorpo-ration of active or activatable functional groups in thepolymer structure, or attachment at the prepolymer ormonomer stage in bead production.[0030] For example, in some examples, precursors(e.g., monomers, cross-linkers) that are polymerized toform a bead may comprise acrydite moieties, such thatwhen a bead is generated, the bead also comprises acry-dite moieties. Often, the acrydite moieties are attachedto an oligonucleotide sequence, such as a primer (e.g.,a primer for one or more of amplifying target nucleic acidsand/or sequencing target nucleic acids barcode se-quence, binding sequence, or the like)) that is desired tobe incorporated into the bead. In some cases, the primercomprises a P5 sequence. For example, acrylamide pre-cursors (e.g., cross-linkers, monomers) may compriseacrydite moieties such that when they are polymerizedto form a bead, the bead also comprises acrydite moie-ties.[0031] In some cases, precursors such as monomersand cross-linkers may comprise, for example, a singleoligonucleotide (e.g., such as a primer or other se-quence) or other species. Fig 29A depicts an examplemonomer comprising an acrydite moiety and single P5sequence linked to the acrydite moiety via a disulfidebond. In some cases, precursors such as monomers andcross-linkers may comprise multiple oligonucleotides,other sequences, or other species. Fig 29B depicts anexample monomer comprising multiple acrydite moietieseach linked to a P5 primer via a disulfide bond. Moreover,Fig 29C depicts an example cross-linker comprising mul-tiple acrydite moieties each linked to a P5 species via adisulfide bond. The inclusion of multiple acrydite moietiesor other linker species in each precursor may improveloading of a linked species (e.g., an oligonucleotide) intobeads generated from the precursors because each pre-cursor can comprise multiple copies of a species to beloaded.[0032] In some cases, precursors comprising a func-tional group that is reactive or capable of being activatedsuch that it becomes reactive can be polymerized withother precursors to generate gel beads comprising the
activated or activatable functional group. The functionalgroup may then be used to attach additional species (e.g.,disulfide linkers, primers, other oligonucleotides, etc.) tothe gel beads. For example, some precursors comprisinga carboxylic acid (COOH) group can co-polymerize withother precursors to form a gel bead that also comprisesa COOH functional group, as shown in Fig 31. In somecases, acrylic acid (a species comprising free COOHgroups), acrylamide, and bis(acryloyl)cystamine can beco-polymerized together to generate a gel bead compris-ing free COOH groups. The COOH groups of the gelbead can be activated (e.g., via 1-Ethyl-3-(3-dimethyl-aminopropyl)carbodiimide (EDC) and N-Hydroxysuccin-imide (NHS) or 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) as shown in Fig31) such that they are reactive (e.g., reactive to aminefunctional groups where EDC/NHS or DMTMM are usedfor activation). The activated COOH groups can then re-act with an appropriate species (e.g., a species compris-ing an amine functional group where the carboxylic acidgroups are activated to be reactive with an amine func-tional group) comprising a moiety to be linked to the bead.[0033] An example species comprising an aminegroup linked to a P5 primer via a disulfide bond (e.g.,H2N-C6-S-S-C6-P5) is shown in Fig 31. COOH functionalgroups of a gel bead can be activated with EDC/NHS orDMTMM to generate an amine reactive species at oneor more of the COOH sites. The amine group of the spe-cies H2N-C6-S-S-C6-P5 moiety can then react with theactivated carboxylic acid such that the moiety and at-tached P5 oligonucleotide becomes covalently linked tothe bead as shown in Fig 31. Unreacted COOH speciescan be converted to other species such that they areblocked.[0034] Beads comprising disulfide linkages in their pol-ymeric network may be functionalized with additionalspecies via reduction of some of the disulfide linkages tofree thiols. The disulfide linkages may be reduced via,for example, the action of a reducing agent (e.g., DTT,TCEP, etc.) to generate free thiol groups, without disso-lution of the bead. Free thiols of the beads can then reactwith free thiols of a species or a species comprising an-other disulfide bond (e.g., via thiol-disulfide exchange))such that the species can be linked to the beads (e.g.,via a generated disulfide bond). In some cases, though,free thiols of the beads may react with any other suitablegroup. For example, free thiols of the beads may reactwith species comprising an acrydite moiety. The free thiolgroups of the beads can react with the acrydite viaMichael addition chemistry, such that the species com-prising the acrydite is linked to the bead. In some cases,uncontrolled reactions can be prevented by inclusion ofa thiol capping agent such as, for example, N-ethyl-malieamide or iodoacetate.[0035] Activation of disulfide linkages within a bead canbe controlled such that only a small number of disulfidelinkages are activated. Control may be exerted, for ex-ample, by controlling the concentration of a reducing
13 14

EP 3 013 957 B1
9
5
10
15
20
25
30
35
40
45
50
55
agent used to generate free thiol groups and/or concen-tration of reagents used to form disulfide bonds in beadpolymerization. In some cases, a low concentration (e.g.,molecules of reducing agent:gel bead ratios of less thanabout 10000, 100000, 1000000, 10000000, 100000000,1000000000, 10000000000, or 100000000000) of re-ducing agent may be used for reduction. Controlling thenumber of disulfide linkages that are reduced to free thiolsmay be useful in ensuring bead structural integrity duringfunctionalization. In some cases, optically-active agents,such as fluorescent dyes may be may be coupled tobeads via free thiol groups of the beads and used to quan-tify the number of free thiols present in a bead and/ortrack a bead.[0036] An example scheme for functionalizing gelbeads comprising disulfide linkages is shown in Fig 35A.As shown, beads 3501 (e.g., gel beads) comprising di-sulfide linkages can be generated using, for example,any of the methods described herein. Upon action of areducing agent 3502 (e.g., DTT, TCEP, or any other re-ducing agent described herein) at a concentration notsuitable for bead degradation, some of the gel bead 3501disulfide linkages can be reduced to free thiols to gener-ate beads 3503 comprising free thiol groups. Upon re-moval of the reducing agent (e.g., via washing) 3504,beads 3503 can be reacted with an acrydite-S-S-speciesmoiety 3505 comprising a species to be loaded (e.g., P5oligonucleotide shown, but the species may be anothertype of polynucleotide such as, for, example, an oligonu-cleotide comprising P5, a barcode sequence, R1, and arandom N-mer) linked to the acrydite via a disulfide bond.Moiety 3505 can couple with the gel beads 3503 viaMichael addition chemistry to generate beads 3506 com-prising moiety 3505. The generated beads 3506 can thenbe purified (e.g., via washing) by removing unwanted(e.g., non-attached) species.[0037] Another example scheme for functionalizing gelbeads comprising disulfide linkages is shown in Fig 35B.As shown, beads 3501 (e.g., gel beads) comprising di-sulfide linkages can be generated using, for example,any of the methods described herein. Upon action of areducing agent 3502 (e.g., DTT, TCEP, or any other re-ducing agent described herein) at a concentration notsuitable for bead degradation, some of the gel beads3501 disulfide linkages can be reduced to free thiols togenerate beads 3503 comprising free thiol groups. Uponremoval of the reducing agent (e.g., via washing) 3504,beads 3503 can be reacted with 2,2’-Dithiopyridine 3507to generate gel beads 3509 linked to a pyridine moietyvia a disulfide bond. As an alternative to 2,2’-Dithiopyri-dine, other similar species, such as 4,4’-Dithiopyridine or5,5’-dithiobis-(2-nitrobenzoic acid) (e.g., DTNB or Ell-man’s Reagent) may be used. 2,2’-Dithiopyridine 3507can couple with the gel beads 3503 via disulfide ex-change to generate beads 3509 comprising a pyridinemoiety linked to the beads 3509 via a disulfide bond. Gelbeads 3509 can then be separated from unreacted spe-cies (e.g., via washing).
[0038] The purified gel beads 3509 can then be reactedwith a moiety 3508 comprising a species of interest (e.g.,a P5 oligonucleotide as shown) to be coupled to the gelbeads and a free thiol group. In some cases, moiety 3508may be generated from another species comprising adisulfide bond, such that when the disulfide bond is re-duced (e.g., via the action of a reducing agent such asDTT, TCEP, etc.), moiety 3508 with a free thiol group isobtained. Moiety 3508 can participate in thiol-disulfideexchange with the pyridine group of beads 3509 to gen-erate gel beads 3510 comprising moiety 3508. The py-ridine group is generally a good leaving group, which canpermit effective thiol-disulfide exchange with the free thiolof moiety 3508. The generated beads 3510 can then bepurified (e.g., via washing) by removing unwanted spe-cies.[0039] In some cases, addition of moieties to a gel beadafter gel bead formation may be advantageous. For ex-ample, addition of a species after gel bead formation mayavoid loss of the species during chain transfer terminationthat can occur during polymerization. Moreover, smallerprecursors (e.g., monomers or cross linkers that do notcomprise side chain groups and linked moieties) may beused for polymerization and can be minimally hinderedfrom growing chain ends due to viscous effects. In somecases, functionalization after gel bead synthesis can min-imize exposure of species (e.g., oligonucleotides) to beloaded with potentially damaging agents (e.g., free rad-icals) and/or chemical environments. In some cases, thegenerated gel may possess an upper critical solutiontemperature (UCST) that can permit temperature drivenswelling and collapse of a bead. Such functionality mayaid in species (e.g., a primer, a P5 primer) infiltration intothe bead during subsequent functionalization of the beadwith the species. Post-production functionalization mayalso be useful in controlling loading ratios of species inbeads, such that, for example, the variability in loadingratio is minimized. Also, species loading may be per-formed in a batch process such that a plurality of beadscan be functionalized with the species in a single batch.[0040] In some cases, acrydite moieties linked to pre-cursors, another species linked to a precursor, or a pre-cursor itself comprise a labile bond, such as, for example,chemically, thermally, or photo-sensitive bonds e.g., di-sulfide bonds, UV sensitive bonds, or the like. Once acry-dite moieties or other moieties comprising a labile bondare incorporated into a bead, the bead may also comprisethe labile bond. The labile bond may be, for example,useful in reversibly linking (e.g., covalently linking) spe-cies (e.g., barcodes, primers, etc.) to a bead. In somecases, a thermally labile bond may include a nucleic acidhybridization based attachment, e.g., where an oligonu-cleotide is hybridized to a complementary sequence thatis attached to the bead, such that thermal melting of thehybrid releases the oligonucleotide, e.g., a barcode con-taining sequence, from the bead or microcapsule. More-over, the addition of multiple types of labile bonds to agel bead may result in the generation of a bead capable
15 16

EP 3 013 957 B1
10
5
10
15
20
25
30
35
40
45
50
55
of responding to varied stimuli. Each type of labile bondmay be sensitive to an associated stimulus (e.g., chem-ical stimulus, light, temperature, etc.) such that releaseof species attached to a bead via each labile bond maybe controlled by the application of the appropriate stim-ulus. Such functionality may be useful in controlled re-lease of species from a gel bead. In some cases, anotherspecies comprising a labile bond may be linked to a gelbead after gel bead formation via, for example, an acti-vated functional group of the gel bead as describedabove. As will be appreciated, barcodes that are releas-ably, cleavably or reversibly attached to the beads de-scribed herein include barcodes that are released or re-leasable through cleavage of a linkage between the bar-code molecule and the bead, or that are released throughdegradation of the underlying bead itself, allowing thebarcodes to be accessed or accessible by other rea-gents, or both. In general, the barcodes that are releas-able as described herein, may generally be referred toas being activatable, in that they are available for reactiononce released. Thus, for example, an activatable bar-code may be activated by releasing the barcode from abead (or other suitable type of partition described herein).As will be appreciated, other activatable configurationsare also envisioned in the context of the described meth-ods and systems. In particular, reagents may be providedreleasably attached to beads, or otherwise disposed inpartitions, with associated activatable groups, such thatonce delivered to the desired set of reagents, e.g.,through co-partitioning, the activatable group may be re-acted with the desired reagents. Such activatable groupsinclude caging groups, removable blocking or protectinggroups, e.g., photolabile groups, heat labile groups, orchemically removable groups.[0041] In addition to thermally cleavable bonds, di-sulfide bonds and UV sensitive bonds, other non-limitingexamples of labile bonds that may be coupled to a pre-cursor or bead include an ester linkage (e.g., cleavablewith an acid, a base, or hydroxylamine), a vicinal diollinkage (e.g., cleavable via sodium periodate), a Diels-Alder linkage (e.g., cleavable via heat), a sulfone linkage(e.g., cleavable via a base), a silyl ether linkage (e.g.,cleavable via an acid), a glycosidic linkage (e.g., cleav-able via an amylase), a peptide linkage (e.g., cleavablevia a protease), or a phosphodiester linkage (e.g., cleav-able via a nuclease (e.g., DNAase)).[0042] A bead may be linked to a varied number ofacrydite moieties. For example, a bead may compriseabout 1, 10, 100, 1000, 10000, 100000, 1000000,10000000, 100000000, 1000000000, or 10000000000acrydite moieties linked to the beads. In other examples,a bead may comprise at least 1, 10, 100, 1000, 10000,100000, 1000000, 10000000, 100000000, 1000000000,or 10000000000 acrydite moieties linked to the beads.For example, a bead may comprise about 1, 10, 100,1000, 10000, 100000, 1000000, 10000000, 100000000,1000000000, or 10000000000 oligonucleotides cova-lently linked to the beads, such as via an acrydite moiety.
In other examples, a bead may comprise at least 1, 10,100, 1000, 10000, 100000, 1000000, 10000000,100000000, 1000000000, or 10000000000 oligonucle-otides covalently linked to the beads, such as via an acry-dite moiety.[0043] Species that do not participate in polymerizationmay also be encapsulated in beads during bead gener-ation (e.g., during polymerization of precursors). Suchspecies may be entered into polymerization reaction mix-tures such that generated beads comprise the speciesupon bead formation. In some cases, such species maybe added to the gel beads after formation. Such speciesmay include, for example, oligonucleotides, species nec-essary for a nucleic acid amplification reaction (e.g., prim-ers, polymerases, dNTPs, co-factors (e.g., ionic co-fac-tors)) including those described herein, species neces-sary for enzymatic reactions (e.g., enzymes, co-factors,substrates), or species necessary for a nucleic acid mod-ification reaction such as polymerization, ligation, or di-gestion. Trapping of such species may be controlled bythe polymer network density generated during polymer-ization of precursors, control of ionic charge within thegel bead (e.g., via ionic species linked to polymerizedspecies), or by the release of other species. Encapsulat-ed species may be released from a bead upon bead deg-radation and/or by application of a stimulus capable ofreleasing the species from the bead.[0044] Beads may be of uniform size or heterogeneoussize. In some cases, the diameter of a bead may be about1mm, 5mm, 10mm, 20mm, 30mm, 40mm, 45mm, 50mm,60mm, 65mm, 70mm, 75mm, 80mm, 90mm, 100mm,250mm, 500mm, or 1mm. In some cases, a bead mayhave a diameter of at least about 1mm, 5mm, 10mm,20mm, 30mm, 40mm, 45mm, 50mm, 60mm, 65mm, 70mm,75mm, 80mm, 90mm, 100mm, 250mm, 500mm, 1mm, ormore. In some cases, a bead may have a diameter ofless than about 1mm, 5mm, 10mm, 20mm, 30mm, 40mm,45mm, 50mm, 60mm, 65mm, 70mm, 75mm, 80mm, 90mm,100mm, 250mm, 500mm, or 1mm. In some cases, a beadmay have a diameter in the range of about 40-75mm,30-75mm, 20-75mm, 40-85mm, 40-95mm, 20-100mm,10-100mm, 1-100mm, 20-250mm, or 20-500mm.[0045] In certain preferred aspects, the beads are pro-vided as a population of beads having a relatively mon-odisperse size distribution. As will be appreciated, insome applications, where it is desirable to provide rela-tively consistent amounts of reagents within partitions,maintaining relatively consistent bead characteristics,such as size, contributes to that overall consistency. Inparticular, the beads described herein may have size dis-tributions that have a coefficient of variation in their cross-sectional dimensions of less than 50%, less than 40%,less than 30%, less than 20%, and in some cases lessthan 15%,less than 10%, or even less than 5%.[0046] Beads may be of a regular shape or an irregularshape. Examples of bead shapes include spherical, non-spherical, oval, oblong, amorphous, circular, cylindrical,and homologs thereof.
17 18

EP 3 013 957 B1
11
5
10
15
20
25
30
35
40
45
50
55
Degradable Beads
[0047] In addition to, or as an alternative to the cleav-able linkages between the beads and the associated mol-ecules, e.g., barcode containing oligonucleotides, de-scribed above, the beads may be degradable, disrupta-ble, or dissolvable spontaneously or upon exposure toone or more stimuli (e.g., temperature changes, pHchanges, exposure to particular chemical species orphase, exposure to light, reducing agent, etc.). In somecases, a bead may be dissolvable, such that materialcomponents of the beads are solubilized when exposedto a particular chemical species or an environmentalchanges, such as, for example, temperature, or pH. Forexample, a gel bead may be degraded or dissolved atelevated temperature and/or in basic conditions. In somecases, a bead may be thermally degradable such thatwhen the bead is exposed to an appropriate change intemperature (e.g., heat), the bead degrades. Degrada-tion or dissolution of a bead bound to a species (e.g., anucleic acid species) may result in release of the speciesfrom the bead.[0048] A degradable bead may comprise one or morespecies with a labile bond such that when the bead/spe-cies is exposed to the appropriate stimuli, the bond isbroken and the bead degrades. The labile bond may bea chemical bond (e.g., covalent bond, ionic bond) or maybe another type of physical interaction (e.g., van derWaals interactions, dipole-dipole interactions, etc.). Insome cases, a crosslinker used to generate a bead maycomprise a labile bond. Upon exposure to the appropriateconditions, the labile bond is broken and the bead is de-graded. For example, a polyacrylamide gel bead maycomprise cystamine crosslinkers. Upon exposure of thebead to a reducing agent, the disulfide bonds of thecystamine are broken and the bead is degraded.[0049] A degradable bead may be useful in more quick-ly releasing an attached species (e.g., an oligonucleotide,a barcode sequence) from the bead when the appropriatestimulus is applied to the bead. For example, for a speciesbound to an inner surface of a porous bead or in the caseof an encapsulated species, the species may have great-er mobility and accessibility to other species in solutionupon degradation of the bead. In some cases, a speciesmay also be attached to a degradable bead via a degra-dable linker (e.g., disulfide linker). The degradable linkermay respond to the same stimuli as the degradable beador the two degradable species may respond to differentstimuli. For example, a barcode sequence may be at-tached, via a disulfide bond, to a polyacrylamide beadcomprising cystamine. Upon exposure of the barcoded-bead to a reducing agent, the bead degrades and thebarcode sequence is released upon breakage of boththe disulfide linkage between the barcode sequence andthe bead and the disulfide linkages of the cystamine inthe bead.[0050] A degradable bead may be introduced into apartition, such as a droplet of an emulsion or a well, such
that the bead degrades within the partition and any as-sociated species are released within the droplet whenthe appropriate stimulus is applied. The free species mayinteract with other species. For example, a polyacryla-mide bead comprising cystamine and linked, via a di-sulfide bond, to a barcode sequence, may be combinedwith a reducing agent within a droplet of a water-in-oilemulsion. Within the droplet, the reducing agent breaksthe various disulfide bonds resulting in bead degradationand release of the barcode sequence into the aqueous,inner environment of the droplet. In another example,heating of a droplet comprising a bead-bound barcodesequence in basic solution may also result in bead deg-radation and release of the attached barcode sequenceinto the aqueous, inner environment of the droplet.[0051] As will be appreciated from the above disclo-sure, while referred to as degradation of a bead, in manyinstances as noted above, that degradation may refer tothe disassociation of a bound or entrained species froma bead, both with and without structurally degrading thephysical bead itself. For example, entrained species maybe released from beads through osmotic pressure differ-ences due to, for example, changing chemical environ-ments. By way of example, alteration of bead pore sizesdue to osmotic pressure differences can generally occurwithout structural degradation of the bead itself. In somecases, an increase in pore size due to osmotic swellingof a bead can permit the release of entrained specieswithin the bead. In other cases, osmotic shrinking of abead may cause a bead to better retain an entrainedspecies due to pore size contraction.[0052] As will be appreciated, where degradablebeads are provided, it may be desirable to avoid exposingsuch beads to the stimulus or stimuli that cause suchdegradation prior to the desired time, in order to avoidpremature bead degradation and issues that arise fromsuch degradation, including for example poor flow char-acteristics, clumping and aggregation. By way of exam-ple, where beads comprise reducible cross-linkinggroups, such as disulfide groups, it will be desirable toavoid contacting such beads with reducing agents, e.g.,DTT or other disulfide cleaving reagents. In such cases,treatments to the beads described herein will, in somecases be provided to be free of reducing agents, suchas DTT. Because reducing agents are often provided incommercial enzyme preparations, it is often desirable toprovide reducing agent free (or DTT free) enzyme prep-arations in treating the beads described herein. Exam-ples of such enzymes include, e.g., polymerase enzymepreparations, ligase enzyme preparations, as well asmany other enzyme preparations that may be used totreat the beads described herein. By "reducing agentfree" or "DTT free" preparations means that the prepa-ration will have less than 1/10th, less than 1/50th, andeven less than 1/100th of the lower ranges for such ma-terials used in degrading the beads. For example, forDTT, the reducing agent free preparation will typicallyhave less than 0.01 mM, 0.005 mM, 0.001 mM DTT,
19 20

EP 3 013 957 B1
12
5
10
15
20
25
30
35
40
45
50
55
0.0005 mM DTT, or even less than 0.0001 mM DTT orless. In many cases, the amount of DTT will be undetec-table.
Methods for Degrading Beads
[0053] In some cases, a stimulus may be used to trig-ger degrading of the bead, which may result in the releaseof contents from the bead. Generally, a stimulus maycause degradation of the bead structure, such as degra-dation of the covalent bonds or other types of physicalinteraction. These stimuli may be useful in inducing abead to degrade and/or to release its contents. Examplesof stimuli that may be used include chemical stimuli, ther-mal stimuli, light stimuli and any combination thereof, asdescribed more fully below.[0054] Numerous chemical triggers may be used totrigger the degradation of beads. Examples of thesechemical changes may include, but are not limited to pH-mediated changes to the integrity of a component withinthe bead, degradation of a component of a bead viacleavage of cross-linked bonds, and depolymerization ofa component of a bead.[0055] In some embodiments, a bead may be formedfrom materials that comprise degradable chemicalcrosslinkers, such as BAC or cystamine. Degradation ofsuch degradable crosslinkers may be accomplishedthrough a number of mechanisms. In some examples, abead may be contacted with a chemical degrading agentthat may induce oxidation, reduction or other chemicalchanges. For example, a chemical degrading agent maybe a reducing agent, such as dithiothreitol (DTT). Addi-tional examples of reducing agents may include β-mer-captoethanol, (2S)-2-amino-1,4-dimercaptobutane(dithiobutylamine or DTBA), tris(2-carboxyethyl) phos-phine (TCEP), or combinations thereof. A reducing agentmay degrade the disulfide bonds formed between gelprecursors forming the bead, and thus, degrade thebead. In other cases, a change in pH of a solution, suchas an increase in pH, may trigger degradation of a bead.In other cases, exposure to an aqueous solution, suchas water, may trigger hydrolytic degradation, and thusdegrading the bead.[0056] Beads may also be induced to release their con-tents upon the application of a thermal stimulus. A changein temperature can cause a variety of changes to a bead.For example, heat can cause a solid bead to liquefy. Achange in heat may cause melting of a bead such that aportion of the bead degrades. In other cases, heat mayincrease the internal pressure of the bead componentssuch that the bead ruptures or explodes. Heat may alsoact upon heat-sensitive polymers used as materials toconstruct beads.[0057] The methods, compositions, devices, and kitsof this disclosure may be used with any suitable agentto degrade beads. In some embodiments, changes intemperature or pH may be used to degrade thermo-sen-sitive or pH-sensitive bonds within beads. In some em-
bodiments, chemical degrading agents may be used todegrade chemical bonds within beads by oxidation, re-duction or other chemical changes. For example, a chem-ical degrading agent may be a reducing agent, such asDTT, wherein DTT may degrade the disulfide bondsformed between a crosslinker and gel precursors, thusdegrading the bead. In some embodiments, a reducingagent may be added to degrade the bead, which may ormay not cause the bead to release its contents. Examplesof reducing agents may include dithiothreitol (DTT), β-mercaptoethanol, (2S)-2-amino-1,4-dimercaptobutane(dithiobutylamine or DTBA), tris(2-carboxyethyl) phos-phine (TCEP), or combinations thereof. The reducingagent may be present at 0.1mM, 0.5mM, 1mM, 5mM, or10mM. The reducing agent may be present at more than0.1mM, 0.5mM, 1mM, 5mM, 10mM, or more. The reduc-ing agent may be present at less than 0.1mM, 0.5mM,1mM, 5mM, or 10mM.
Timing of Degrading Step
[0058] Beads may be degraded to release contentsattached to and contained within the bead. This degrad-ing step may occur simultaneously as the sample is com-bined with the bead. This degrading step may occur si-multaneously when the sample is combined with thebead within a fluidic droplet that may be formed in a mi-crofluidic device. This degrading step may occur afterthe sample is combined with the bead within a fluidicdroplet that may be formed in a microfluidic device. Aswill be appreciated, in many applications, the degradingstep may not occur.[0059] The reducing agent may be combined with thesample and then with the bead. In some cases, the re-ducing agent may be introduced to a microfluidic deviceas the same time as the sample. In some cases, thereducing agent may be introduced to a microfluidic deviceafter the sample is introduced. In some cases, the samplemay be mixed with the reducing agent in a microfluidicdevice and then contacted with the gel bead in the mi-crofluidic device. In some embodiments, the sample maybe pre-mixed with the reducing agent and then added tothe device and contacted with the gel bead.[0060] A degradable bead may degrade instantane-ously upon application of the appropriate stimuli. In othercases, degradation of the bead may occur over time. Forexample, a bead may degrade upon application of anappropriate stimulus instantaneously or within about 0,0.01, 0.1, 0.5, 1, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 5.5,6.0, 6.5, 7.0, 7.5, 8.0, 8.5, 9.0, 9.5, 10.0, 11, 12, 13, 14,15 or 20 minutes. In other examples, a bead may degradeupon application of a proper stimulus instantaneously orwithin at most about 0, 0.01, 0.1, 0.5, 1, 1.5, 2.0, 2.5, 3.0,3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, 8.5, 9.0, 9.5,10.0, 11, 12, 13, 14, 15 or 20 minutes.[0061] Beads may also be degraded at different times,relative to combining with a sample. For example, thebead may be combined with the sample and subsequent-
21 22

EP 3 013 957 B1
13
5
10
15
20
25
30
35
40
45
50
55
ly degraded at a point later in time. The time betweencombining the sample with the bead and subsequentlydegrading the bead may be about 0.0001, 0.001, 0.01,1, 10, 30, 60, 300, 600, 1800, 3600, 18000, 36000,86400, 172800, 432000, or 864000 seconds. The timebetween combining the sample with the bead and sub-sequently degrading the bead may be more than about0.0001, 0.001, 0.01, 1, 10, 30, 60, 300, 600, 1800, 3600,18000, 36000, 86400, 172800, 432000, 864000 secondsor more. The time between combining the sample withthe bead and subsequently degrading the bead may beless than about 0.0001, 0.001, 0.01, 1, 10, 30, 60, 300,600, 1800, 3600, 18000, 36000, 86400, 172800, 432000,or 864000 seconds.
Preparing Beads Pre-functionalized with Oligonucle-otides
[0062] The beads described herein may be producedusing a variety of methods. In some cases, beads maybe formed from a liquid containing molecular precursors(e.g. linear polymers, monomers, cross-linkers). The liq-uid is then subjected to a polymerization reaction, andthereby hardens or gels into a bead (or gel bead). Theliquid may also contain entities such as oligonucleotidesthat become incorporated into the bead during polymer-ization. This incorporation may be via covalent or non-covalent association with the bead. For example, in somecases, the oligonucleotides may be entrained within abead during formation. Alternatively, they may be cou-pled to the bead or the bead framework either duringformation or following formation. Often, the oligonucle-otides are connected to an acrydite moiety that becomescross-linked to the bead during the polymerization proc-ess. In some cases, the oligonucleotides are attached tothe acrydite moiety by a disulfide linkage. As a result, acomposition comprising a bead-acrydite-S-S-oligonucle-otide linkage is formed. Fig 4A is an exemplary diagramof a bead functionalized with an acrydite-linked primer.[0063] In one exemplary process, functionalized beadsmay be generated by mixing a plurality of polymers and/ormonomers with one or more oligonucleotides, such as,for example, one or more oligonucleotides that comprisesa primer (e.g., a universal primer, a sequencing primer).The polymers and/or monomers may comprise acryla-mide and may be crosslinked such that disulfide bondsform between the polymers and/or monomers, resultingin the formation of hardened beads. The oligonucleotidesmay be covalently linked to the plurality of polymersand/or monomers during the formation of the hardenedbeads (e.g., contemporaneously) or may be covalentlylinked to the plurality of polymers and/or monomers afterthe formation of the hardened beads (e.g., sequentially).In some cases, the oligonucleotides may be linked to thebeads via an acrydite moiety.[0064] In most cases, a population of beads is pre-functionalized with the identical oligonucleotide such asa universal primer or primer binding site. In some cases,
the beads in a population of beads are pre-functionalizedwith multiple different oligonucleotides. These oligonu-cleotides may optionally include any of a variety of dif-ferent functional sequences, e.g., for use in subsequentprocessing or application of the beads. Functional se-quences may include, e.g., primer sequences, such astargeted primer sequences, universal primer sequences,e.g., primer sequences that are sufficiently short to beable to hybridize to and prime extension from large num-bers of different locations on a sample nucleic acid, orrandom primer sequences, attachment or immobilizationsequences, ligation sequences, hairpin sequences, tag-ging sequences, e.g., barcodes or sample index se-quences, or any of a variety of other nucleotide sequenc-es.[0065] By way of example, in some cases, the univer-sal primer (e.g., P5 or other suitable primer) may be usedas a primer on each bead, to attach additional content(e.g., barcodes, random N-mers, other functional se-quences) to the bead. In some cases, the universal prim-er (e.g., P5) may also be compatible with a sequencingdevice, and may later enable attachment of a desiredstrand to a flow cell within the sequencing device. Forexample, such attachment or immobilization sequencesmay provide a complementary sequence to oligonucle-otides that are tethered to the surface of a flow cell in asequencing device, to allow immobilization of the se-quences to that surface for sequencing. Alternatively,such attachments sequences may additionally be pro-vided within, or added to the oligonucleotide sequencesattached to the beads. In some cases, the beads andtheir attached species may be provided to be compatiblewith subsequent analytical process, such as sequencingdevices or systems. In some cases, more than one primermay be attached to a bead and more than one primermay contain a universal sequence, in order to, for exam-ple, allow for differential processing of the oligonucleotideas well as any additional sequences that are coupled tothat sequence, in different sequential or parallel process-ing steps, e.g., a first primer for amplification of a targetsequence, with a second primer for sequencing the am-plified product. For example, in some cases, the oligo-nucleotides attached to the beads will comprise a firstprimer sequence for conducting a first amplification orreplication process, e.g., extending the primer along atarget nucleic acid sequence, in order to generate anamplified barcoded target sequence(s). By also includinga sequencing primer within the oligonucleotides, the re-sulting amplified target sequences will include such prim-ers, and be readily transferred to a sequencing system.For example, in some cases, e.g., where one wishes tosequence the amplified targets using, e.g., an Illuminasequencing system, an R1 primer or primer binding sitemay also be attached to the bead.[0066] Entities incorporated into the beads may in-clude oligonucleotides having any of a variety of func-tional sequences as described above. For example,these oligonucleotides may include any one or more of
23 24

EP 3 013 957 B1
14
5
10
15
20
25
30
35
40
45
50
55
P5, R1, and R2 sequences, non cleavable 5’acrydite-P5,a cleavable 5’ acrydite-SS-P5, R1c, sequencing primer,read primer, universal primer, P5_U, a universal readprimer, and/or binding sites for any of these primers. Insome cases, a primer may contain one or more modifiednucleotides nucleotide analogues, or nucleotide mimics.For example, in some cases, the oligonucleotides mayinclude peptide nucleic acids (PNAs), locked nucleic acid(LNA) nucleotides, or the like. In some cases, these oli-gonucleotides may additionally or alternatively includenucleotides or analogues that may be processed differ-ently, in order to allow differential processing at differentsteps of their application. For example, in some casesone or more of the functional sequences may include anucleotide or analogue that is not processed by a partic-ular polymerase enzyme, thus being uncopied in a proc-ess step utilizing that enzyme. For example, e.g., in somecases, one or more of the functional sequence compo-nents of the oligonucleotides will include, e.g., a uracilcontaining nucleotide, a nucleotide containing a non-na-tive base, a blocker oligonucleotide, a blocked 3’ end,3’ddCTP. Fig 19 provides additional examples. As willbe appreciated, sequences of any of these entities mayfunction as primers or primer binding sites depending onthe particular application.[0067] Polymerization may occur spontaneously. Insome cases, polymerization may be initiated by an initi-ator and/or an accelerator, by electromagnetic radiation,by temperature changes (e.g., addition or removal ofheat), by pH changes, by other methods, and combina-tions thereof. An initiator may refer to a species capableof initiating a polymerization reaction by activating (e.g.,via the generation of free radicals) one or more precur-sors used in the polymerization reaction. An acceleratormay refer to a species capable of accelerating the rateat which a polymerization reaction occurs. In some cas-es, an accelerator may speed up the activation of an in-itiator (e.g., via the generation of free radicals) used tothen activate monomers (e.g., via the generation of freeradicals) and, thus, initiate a polymerization reaction. Insome cases, faster activation of an initiator can give riseto faster polymerization rates. In some cases, though,acceleration may also be achieved via non-chemicalmeans such as thermal (e.g., addition and removal ofheat) means, various types of radiative means (e.g., vis-ible light, UV light, etc.), or any other suitable means. Tocreate droplets containing molecular precursors, whichmay then polymerize to form hardened beads, an emul-sion technique may be employed. For example, molec-ular precursors may be added to an aqueous solution.The aqueous solution may then be emulsified with an oil(e.g., by agitation, microfluidic droplet generator, or othermethod). The molecular precursors may then be polym-erized in the emulsified droplets to form the beads.[0068] An emulsion may be prepared, for example, byany suitable method, including methods known in the art,such as bulk shaking, bulk agitation, flow focusing, andmicrosieve (See e.g., Weizmann et al., Nature Methods,
2006, 3(7):545-550; Weitz et al. U.S. Pub. No.2012/0211084). In some cases, an emulsion may be pre-pared using a microfluidic device. In some cases, water-in-oil emulsions may be used. These emulsions may in-corporate fluorosurfactants such as Krytox FSH with aPEG-containing compound such as bis krytox peg (BKP).In some cases, oil-in-water emulsions may be used. Insome cases, polydisperse emulsions may be formed. Insome cases, monodisperse emulsions may be formed.In some cases, monodisperse emulsions may be formedin a microfluidic flow focusing device. (Gartecki et al.,Applied Physics Letters, 2004, 85(13):2649-2651).[0069] In at least one example, a microfluidic devicefor making the beads may contain channel segments thatintersect at a single cross intersection that combines twoor more streams of immiscible fluids, such as an aqueoussolution containing molecular precursors and an oil.Combining two immiscible fluids at a single cross inter-section may cause fluidic droplets to form. The size ofthe fluidic droplets formed may depend upon the flowrate of the fluid streams entering the fluidic cross, theproperties of the two fluids, and the size of the microfluidicchannels. Initiating polymerization after formation of flu-idic droplets exiting the fluidic cross may cause hardenedbeads to form from the fluidic droplets. Examples of mi-crofluidic devices, channel networks and systems forgenerating droplets, both for bead formation and for par-titioning beads into discrete droplets as discussed else-where herein, are described for example in U.S. Provi-sional Patent Application No. 61/977,804, filed April 4,2014.[0070] To manipulate when individual molecular pre-cursors, oligomers, or polymers begin to polymerize toform a hardened bead, an initiator and/or accelerator maybe added at different points in the bead formation proc-ess. An accelerator may be an agent which may initiatethe polymerization process (e.g., in some cases, via ac-tivation of a polymerization initiator) and thus may reducethe time for a bead to harden. In some cases, a singleaccelerator or a plurality of accelerators may be used forpolymerization. Careful tuning of acceleration can be im-portant in achieving suitable polymerization reactions.For example, if acceleration is too fast, weight and ex-cessive chain transfer events may cause poor gel struc-ture and low loading of any desired species. If accelera-tion is too slow, high molecular weight polymers can gen-erate trapped activation sites (e.g., free radicals) due topolymer entanglement and high viscosities. High viscos-ities can impede diffusion of species intended for beadloading, resulting in low to no loading of the species. Tun-ing of accelerator action can be achieved, for example,by selecting an appropriate accelerator, an appropriatecombination of accelerators, or by selecting the appro-priate accelerator(s) and any stimulus (e.g., heat, elec-tromagnetic radiation (e.g., light, UV light), another chem-ical species, etc.) capable of modulating accelerator ac-tion. Tuning of initiator action may also be achieved inanalogous fashion.
25 26

EP 3 013 957 B1
15
5
10
15
20
25
30
35
40
45
50
55
[0071] An accelerator may be water-soluble, oil-solu-ble, or may be both water-soluble and oil-soluble. Forexample, an accelerator may be tetramethylethylenedi-amine (TMEDA or TEMED), dimethylethylenediamine,N,N, N,’N’- tetramethylmethanediamine, N,N’ - dimor-pholinomethane, or N,N,N’,N’-Tetrakis(2-Hydroxypro-pyl)ethylenediamine. For example, an initiator may beammonium persulfate (APS), calcium ions, or any of thecompounds (I-IX) shown in Fig 32. The compounds (I-IX) shown in Fig 32 can function as water-soluble azo-based initiators. Azo-based initiators may be used in theabsence of TEMED and APS and can function as thermalbased initiators. A thermal based initiator can activatespecies (e.g., via the generation of free radicals) ther-mally and, thus, the rate of initiator action can be tunedby temperature and/or the concentration of the initiator.A polymerization accelerator or initiator may include func-tional groups including phosphonate, sulfonate, carbox-ylate, hydroxyl, albumin binding moieties, N-vinyl groups,and phospholipids. A polymerization accelerator or initi-ator may be a low molecular weight monomeric-com-pound. An accelerator or initiator may be a) added to theoil prior to droplet generation, b) added in the line afterdroplet generation, c) added to the outlet reservoir afterdroplet generation, or d) combinations thereof.[0072] Polymerization may also be initiated by electro-magnetic radiation. Certain types of monomers, oligom-ers, or polymers may contain light-sensitive properties.Thus, polymerization may be initiated by exposing suchmonomers, oligomers, or polymers to UV light, visiblelight, UV light combined with a sensitizer, visible lightcombined with a sensitizer, or combinations thereof. Anexample of a sensitizer may be riboflavin.[0073] The time for a bead to completely polymerizeor harden may vary depending on the size of the bead,whether an accelerator may be added, when an accel-erator may be added, the type of initiator, when electro-magnetic radiation may be applied, the temperature ofsolution, the polymer composition, the polymer concen-tration, and other relevant parameters. For example, po-lymerization may be complete after about 5, 6, 7, 8, 9,10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 minutes.Polymerization may be complete after more than about5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20minutes or more. Polymerization may be complete in lessthan about 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17,18, 19, or 20 minutes.[0074] Beads may be recovered from emulsions (e.g.gel-water-oil) by continuous phase exchange. Excessaqueous fluid may be added to the emulsion (e.g. gel-water-oil) and the hardened beads may be subjected tosedimentation, wherein the beads may be aggregatedand the supernatant containing excess oil may be re-moved. This process of adding excess aqueous fluid fol-lowed by sedimentation and removal of excess oil maybe repeated until beads are suspended in a given purityof aqueous buffer, with respect to the continuous phaseoil. The purity of aqueous buffer may be about 80%, 90%,
95%, 96%, 97%, 98%, or 99% (v/v). The purity of aque-ous buffer may be more than about 80%, 90%, 95%,96%, 97%, 98%, 99% or more (v/v). The purity of aque-ous buffer may be less than about 80%, 90%, 95%, 96%,97%, 98%, or 99% (v/v). The sedimentation step may berepeated about 2, 3, 4, or 5 times. The sedimentationstep may be repeated more than about 2, 3, 4, 5 timesor more. The sedimentation step may be repeated lessthan about 2, 3, 4, or 5 times. In some cases, sedimen-tation and removal of the supernatant may also removeun-reacted starting materials.[0075] Examples of droplet generators may includesingle flow focuser, parallel flow focuser, and microsievemembrane, such as those used by Nanomi B.V., andothers. Preferably, a microfluidic device is used to gen-erate the droplets.[0076] An example emulsion based scheme for gen-erating gel beads pre-functionalized with an acrydite moi-ety linked to a P5 primer via a disulfide bond is depictedin Fig 30. As shown in Fig 30A, acrylamide, bis(acry-loyl)cystamine, acrydite-S-S-P5 moieties, and ammoni-um persulfate are combined into a droplets of an emul-sion. TEMED can be added to the emulsion oil phaseand can diffuse into the droplets to initiate the polymeri-zation reaction. As shown in Fig 30A, TEMED action onammonium persulfate results in the generation of SO4
-
free radicals that can then activate the carbon-carbondouble bond of the acrylamide via generation of a freeradical at one of the carbons of the carbon-carbon doublebond.[0077] As shown in Fig 30B, activated acrylamide canreact with non-activated acrylamide (again, at its carbon-carbon double bond) to begin polymerization. Each prod-uct generated can again be activated via the formationof a free radical resulting in polymer propagation. More-over, both the bis(acryloyl)cystamine cross-linker andacrydite-S-S-P5 moieties comprise carbon-carbon dou-ble bonds that can react with activated species and theproducts themselves can then become activated. Theinclusion of the bis(acryloyl)cystamine cross-linker intothe polymerization reaction can result in cross-linking ofpolymer chains that are generated as shown in Fig 30C.Thus, a hydrogel polymer network comprising acrydite-S-S-P5 moieties linked to polymer backbones can begenerated, as depicted in Fig 30C. The polymerizationreaction can continue until it terminates. Upon reactiontermination, continuous phase exchange or other suita-ble method can be used to break the emulsion and obtaingel beads comprising a cross-linked hydrogel (shownschematically in Fig 30A) coupled to the acrydite-S-S-P5 moieties.
Barcode and Random N-mers (introduction)
[0078] Certain applications, for example polynucle-otide sequencing, may rely on unique identifiers ("bar-codes") to identify a sequence and, for example, to as-semble a larger sequence from sequenced fragments.
27 28

EP 3 013 957 B1
16
5
10
15
20
25
30
35
40
45
50
55
Therefore, it may be desirable to add barcodes to poly-nucleotide fragments before sequencing. In the case ofnucleic acid applications, such barcodes are typicallycomprised of a relatively short sequence of nucleotidesattached to a sample sequence, where the barcode se-quence is either known, or identifiable by its location orsequence elements. In some cases, a unique identifiermay be useful for sample indexing. In some cases,though, barcodes may also be useful in other contexts.For example, a barcode may serve to track samplesthroughout processing (e.g., location of sample in a lab,location of sample in plurality of reaction vessels, etc.);provide manufacturing information; track barcode per-formance over time (e.g., from barcode manufacturingto use) and in the field; track barcode lot performanceover time in the field; provide product information duringsequencing and perhaps trigger automated protocols(e.g., automated protocols initiated and executed withthe aid of a computer) when a barcode associated withthe product is read during sequencing; track and trouble-shoot problematic barcode sequences or product lots;serve as a molecular trigger in a reaction involving thebarcode, and combinations thereof. In particularly pre-ferred aspects, and as alluded to above, barcode se-quence segments as described herein, can be used toprovide linkage information as between two discrete de-termined nucleic acid sequences. This linkage informa-tion may include, for example, linkage to a common sam-ple, a common reaction vessel, e.g., a well or partition,or even a common starting nucleic acid molecule. In par-ticular, by attaching common barcodes to a specific sam-ple component, or subset of sample components withina given reaction volume, one can attribute the resultingsequences bearing that barcode to that reaction volume.In turn, where the sample is allocated to that reactionvolume based upon its sample of origin, the processingsteps to which it is subsequently exposed, or on an indi-vidual molecule basis, one can better identify the result-ing sequences as having originated from that reactionvolume.[0079] Barcodes may be generated from a variety ofdifferent formats, including bulk synthesized polynucle-otide barcodes, randomly synthesized barcode sequenc-es, microarray based barcode synthesis, native nucle-otides, partial complement with N-mer, random N-mer,pseudo random N-mer, or combinations thereof. Synthe-sis of barcodes is described herein, as well as in, forexample, in U.S. Patent Application No. 14/175,973, filedFebruary 7, 2014.[0080] As described above, oligonucleotides incorpo-rating barcode sequence segments, which function as aunique identifier, may also include additional sequencesegments. Such additional sequence segments may in-clude functional sequences, such as primer sequences,primer annealing site sequences, immobilization se-quences, or other recognition or binding sequences use-ful for subsequent processing, e.g., a sequencing primeror primer binding site for use in sequencing of samples
to which the barcode containing oligonucleotide is at-tached. Further, as used herein, the reference to specificfunctional sequences as being included within the bar-code containing sequences also envisioned the inclusionof the complements to any such sequences, such thatupon complementary replication will yield the specific de-scribed sequence.[0081] In some examples, barcodes or partial bar-codes may be generated from oligonucleotides obtainedfrom or suitable for use in an oligonucleotide array, suchas a microarray or bead array. In such cases, oligonu-cleotides of a microarray may be cleaved, (e.g., usingcleavable linkages or moieties that anchor the oligonu-cleotides to the array (such as photoclevable, chemicallycleavable, or otherwise cleavable linkages)) such thatthe free oligonucleotides are capable of serving as bar-codes or partial barcodes. In some cases, barcodes orpartial barcodes are obtained from arrays are of knownsequence. The use of known sequences, including thoseobtained from an array, for example, may be beneficialin avoiding sequencing errors associated with barcodesof unknown sequence. A microarray may provide at leastabout 10,000,000, at least about 1,000,000, at leastabout 900,000, at least about 800,000, at least about700,000, at least about 600,000, at least about 500,000,at least about 400,000, at least about 300,000, at leastabout 200,000, at least about 100,000, at least about50,000, at least about 10,000, at least about 1,000, atleast about 100, or at least about 10 different sequencesthat may be used as barcodes or partial barcodes.[0082] The beads provided herein may be attached tooligonucleotide sequences that may behave as uniqueidentifiers (e.g., barcodes). Often, a population of beadsprovided herein contains a diverse library of barcodes,wherein each bead is attached to multiple copies of asingle barcode sequence. In some cases, the barcodesequences are pre-synthesized and/or designed withknown sequences. In some cases, each bead within thelibrary is attached to a unique barcode sequence. In somecases, a plurality of beads will have the same barcodesequence attached to them. For example, in some casesabout 1%, 2%, 3%, 4%, 5%, 10%, 20%, 25%, 30%, 50%,75%, 80%, 90%, 95%, or 100% of the beads in a libraryare attached to a barcode sequence that is identical to abarcode sequence attached to a different bead in thelibrary. Sometimes, about 1%, 2%, 3%, 4%, 5%, 10%,20%, 25%, or 30% of the beads are attached to the samebarcode sequence.[0083] The length of a barcode sequence may be anysuitable length, depending on the application. In somecases, a barcode sequence may be about 2 to about 500nucleotides in length, about 2 to about 100 nucleotidesin length, about 2 to about 50 nucleotides in length, about2 to about 20 nucleotides in length, about 6 to about 20nucleotides in length, or about 4 to 16 nucleotides inlength. In some cases, a barcode sequence is about 2,3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19,20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 85, 90, 95, 100,
29 30

EP 3 013 957 B1
17
5
10
15
20
25
30
35
40
45
50
55
150, 200, 250, 300, 400, or 500 nucleotides in length. Insome cases, a barcode sequence is greater than about2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,19, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 85, 90, 95,100, 150, 200, 250, 300, 400, 500, 750, 1000, 5000, or10000 nucleotides in length. In some cases, a barcodesequence is less than about 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50,55, 60, 65, 70, 85, 90, 95, 100, 150, 200, 250, 300, 400,500, 750, or 1000 nucleotides in length.[0084] The barcodes may be loaded into beads so thatone or more barcodes are introduced into a particularbead. In some cases, each bead may contain the sameset of barcodes. In other cases, each bead may containdifferent sets of barcodes. In other cases, each bead maycomprise a set of identical barcodes.[0085] The beads provided herein may be attached tooligonucleotide sequences that are random, pseudo-ran-dom, or targeted N-mers capable of priming a sample(e.g., genomic sample) in a downstream process. Insome cases, the same n-mer sequences will be presenton the oligonucleotides attached to a single bead or beadpopulation. This may be the case for targeted primingmethods, e.g., where primers are selected to target cer-tain sequence segments within a larger target sequence.In other cases, each bead within a population of beadsherein is attached to a large and diverse number of N-mer sequences to, among other things, diversify the sam-pling of these primers against template molecules, assuch random n-mer sequences will randomly primeagainst different portions of the sample nucleic acids.[0086] The length of an N-mer may vary. In some cas-es, an N-mer (e.g., a random N-mer, a pseudo-randomN-mer, or a targeted N-mer) may be between about 2and about 100 nucleotides in length, between about 2and about 50 nucleotides in length, between about 2 andabout 20 nucleotides in length, between about 5 andabout 25 nucleotides in length, or between about 5 andabout 15 nucleotides in length. In some cases, an N-mer(e.g., a random N-mer, a pseudo-random N-mer, or atargeted N-mer) may be about 2, 3, 4, 5, 6, 7, 8, 9, 10,11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45,50, 55, 60, 65, 70, 85, 90, 95, 100, 150, 200, 250, 300,400, or 500 nucleotides in length. In some cases, an N-mer (e.g., a random N-mer, a pseudo-random N-mer, ortargeted a N-mer) may be greater than about 2, 3, 4, 5,6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25,30, 35, 40, 45, 50, 55, 60, 65, 70, 85, 90, 95, 100, 150,200, 250, 300, 400, 500, 750, 1000, 5000, or 10000 nu-cleotides in length. In some cases, an N-mer (e.g., a ran-dom N-mer, a pseudo-random N-mer, or a targeted N-mer) may be less than about 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 35, 40, 45, 50,55, 60, 65, 70, 85, 90, 95, 100, 150, 200, 250, 300, 400,500, 750, or 1000 nucleotides in length.[0087] N-mers (including random N-mers) can be en-gineered for priming a specific sample type. For example,N-mers of different lengths may be generated for different
types of sample nucleic acids or different regions of asample nucleic acid, such that each N-mer length corre-sponds to each different type of sample nucleic acid oreach different region of a sample nucleic acid. For ex-ample, an N-mer of one length may be generated forsample nucleic acid originating from the genome of onespecies (e.g., for example, a human genome) and an N-mer of another length may be generated for a samplenucleic acid originating from another species (e.g., forexample, a yeast genome). In another example, an N-mer of one length may be generated for sample nucleicacid comprising a particular sequence region of a ge-nome and an N-mer of another length may be generatedfor a sample nucleic acid comprising another sequenceregion of the genome. Moreover, in addition or as analternative to N-mer length, the base composition of theN-mer (e.g., GC content of the N-mer) may also be en-gineered to correspond to a particular type or region ofa sample nucleic acid. Base content may vary in a par-ticular type of sample nucleic acid or in a particular regionof a sample nucleic acid, for example, and, thus, N-mersof different base content may be useful for priming dif-ferent sample types of nucleic acid or different regionsof a sample nucleic acid.[0088] Populations of beads described elsewhereherein can be generated with an N-mer engineered fora particular sample type or particular sample sequenceregion. In some cases, a mixed population of beads (e.g.,a mixture of beads comprising an N-mer engineered forone sample type or sequence region and beads compris-ing another N-mer engineered for another sample typeor sequence region) with respect to N-mer length andcontent may be generated. In some cases, a populationof beads may be generated, where one or more of thebeads can comprise a mixed population of N-mers engi-neered for a plurality of sample types or sequence re-gions.[0089] As noted previously, in some cases, the N-mers, whether random or targeted, may comprise nucle-otide analogues, mimics, or non-native nucleotides, inorder to provide primers that have improved performancein subsequent processing steps. For example, in somecases, it may be desirable to provide N-mer primers thathave different melting/annealing profiles when subjectedto thermal cycling, e.g., during amplification, in order toenhance the relative priming efficiency of the n-mer se-quence. In some cases, nucleotide analogues or non-native nucleotides may be incorporated into the N-merprimer sequences in order to alter the melting tempera-ture profile of the primer sequence as compared to acorresponding primer that includes native nucleotides.In certain cases, the primer sequences, such as the N-mer sequences described herein, may include modifiednucleotides or nucleotide analogues, e.g., LNA bases,at one or more positions within the sequence, in order toprovide elevated temperature stability for the primerswhen hybridized to a template sequence, as well as pro-vide generally enhanced duplex stability. In some cases,
31 32

EP 3 013 957 B1
18
5
10
15
20
25
30
35
40
45
50
55
LNA nucleotides are used in place of the A or T bases inprimer synthesis to replace those weaker binding baseswith tighter binding LNA analogues. By providing en-hanced hybridizing primer sequences, one may generatehigher efficiency amplification processes using suchprimers, as well as be able to operate within differenttemperature regimes.[0090] Other modifications may also be provided to theoligonucleotides described above. For example, in somecases, the oligonucleotides may be provided with pro-tected termini or other regions, in order to prevent or re-duce any degradation of the oligonucleotides, e.g.,through any present exonuclease activity. In one exam-ple, the oligonucleotides may be provided with one ormore phosphorothioate nucleotide analogue at one ormore positions within the oligonucleotide sequence, e.g.,adjacent or proximal to the 3’ and/or 5’ terminal position.These phosphorothioate nucleotides typically provide asulfur group in place of the non-linking oxygen in an in-ternucleotide linkage within the oligonucleotide to reduceor eliminate nuclease activity on the oligonucleotides, in-cluding, e.g., 3’-5’ and/or 5’-3’ exonucleases. In general,phosphorothioate analogues are useful in imparting exoand/or endonuclease resistance to oligonucleotides thatinclude them, including providing protection against, e.g.,3’-5’ and/or 5’-3’ exonuclease digestion of the oligonu-cleotides. Accordingly, in some aspects, these one ormore phosphorothioate linkages will be in one or moreof the last 5 to 10 internucleotide linkages at either the3’ or the 5’ terminus of the oligonucleotides, and prefer-ably include one or more of the last 3’ or 5’ terminal in-ternucleotide linkage and second to last 5’ terminal inter-nucleotide linkage, in order to provide protection against3’-5’ or 5’-3’ exonuclease activity. Other positions withinthe oligonucleotides may also be provided with phospho-rothiate linkages as well. In addition to providing suchprotection on the oligonucleotides that comprise the bar-code sequences (and any associated functional se-quences), the above described modifications are alsouseful in the context of the blocker sequences describedherein, e.g., incorporating phosphorothioate analogueswithin the blocker sequences, e.g., adjacent or proximalto the 3’ and/or 5’ terminal position as well as potentiallyother positions within the oligonucleotides.
Attaching Content to Pre-functionalized Beads
[0091] A variety of content may be attached to thebeads described herein, including beads functionalizedwith oligonucleotides. Often, oligonucleotides are at-tached, particularly oligonucleotides with desired se-quences (e.g., barcodes, random N-mers). In many ofthe methods provided herein, the oligonucleotides areattached to the beads through a primer extension reac-tion. Beads pre-functionalized with primer can be con-tacted with oligonucleotide template. Amplification reac-tions may then be performed so that the primer is extend-ed such that a copy of the complement of the oligonucle-
otide template is attached to the primer. Other methodsof attachment are also possible such as ligation reac-tions.[0092] In some cases, oligonucleotides with differentsequences (or the same sequences) are attached to thebeads in separate steps. For example, in some cases,barcodes with unique sequences are attached to beadssuch that each bead has multiple copies of a first barcodesequence on it. In a second step, the beads can be furtherfunctionalized with a second sequence. The combinationof first and second sequences may serve as a uniquebarcode, or unique identifier, attached to a bead. Theprocess may be continued to add additional sequencesthat behave as barcode sequences (in some cases,greater than 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 barcode se-quences are sequentially added to each bead). Thebeads may also be further functionalized random N-mersthat can, for example, act as a random primer for down-stream whole genome amplification reactions.[0093] In some cases, after functionalization with a cer-tain oligonucleotide sequence (e.g., barcode sequence),the beads may be pooled and then contacted with a largepopulation of random Nmers that are then attached tothe beads. In some cases, particularly when the beadsare pooled prior to the attachment of the random Nmers,each bead has one barcode sequence attached to it, (of-ten as multiple copies), but many different random Nmersequences attached to it. Fig 4 provides a step-by-stepdepiction of one example method, an example limitingdilution method, for attaching oligonucleotides, such asbarcodes and Nmers, to beads.[0094] Limiting dilution may be used to attach oligonu-cleotides to beads, such that the beads, on average, areattached to no more than one unique oligonucleotide se-quence such as a barcode. Often, the beads in this proc-ess are already functionalized with a certain oligonucle-otide, such as primers. For example, beads functional-ized with primers (e.g., such as universal primers) and aplurality of template oligonucleotides may be combined,often at a high ratio of beads: template oligonucleotides,to generate a mixture of beads and template oligonucle-otides. The mixture may then be partitioned into a pluralityof partitions (e.g., aqueous droplets within a water-in-oilemulsion), such as by a bulk emulsification process,emulsions within plates, or by a microfluidic device, suchas, for example, a microfluidic droplet generator. In somecases, the mixture can be partitioned into a plurality ofpartitions such that, on average, each partition comprisesno more than one template oligonucleotide.[0095] Moreover, the template oligonucleotides can beamplified (e.g., via primer extension reactions) within thepartitions via the primers attached to the beads. Amplifi-cation can result in the generation of beads comprisingamplified template oligonucleotides. Following amplifica-tion, the contents of the partitions may be pooled into acommon vessel (e.g., a tube, a well, etc.). The beadscomprising the amplified template oligonucleotides maythen be separated from the other contents of the parti-
33 34

EP 3 013 957 B1
19
5
10
15
20
25
30
35
40
45
50
55
tions (including beads that do not comprise amplifiedtemplate oligonucleotides) by any suitable method in-cluding, for example, centrifugation and magnetic sepa-ration, with or without the aid of a capture moiety as de-scribed elsewhere herein.[0096] Beads comprising amplified template oligonu-cleotides may be combined with additional template oli-gonucleotides to generate a bulk mixture comprising thebeads and the additional template oligonucleotides. Theadditional template oligonucleotides may comprise a se-quence that is at least partially complementary to theamplified template oligonucleotides on the beads, suchthat the additional template oligonucleotide hybridizes tothe amplified template oligonucleotides. The amplifiedtemplate oligonucleotides can then be extended via thehybridized additional template oligonucleotides in an am-plification reaction, such that the complements of the ad-ditional template oligonucleotides are attached to the am-plified template oligonucleotides. The cycle of bindingadditional template oligonucleotides to amplified oligo-nucleotides, followed by extension of the amplified oligo-nucleotides in an amplification reaction, can be repeatedfor any desired number of additional oligonucleotides thatare to be added to the bead.[0097] The oligonucleotides attached to the amplifiedtemplate oligonucleotides may comprise, for example,one or more of a random N-mer sequence, a pseudorandom N-mer sequence, or a primer binding site (e.g..,a universal sequence portion, such as a universal se-quence portion that is compatible with a sequencing de-vice). Any of these sequences or any other sequenceattached to a bead may comprise at least a subsectionof uracil containing nucleotides, as described elsewhereherein.[0098] An example of a limiting dilution method for at-taching a barcode sequence and a random N-mer tobeads is shown in Fig 4. As shown in Fig 4A, beads 401,(e.g., disulfide cross-linked polyacrylamide gel beads)are pre-functionalized with a first primer 403. The firstprimer 403 may be, for example, coupled to the beadsvia a disulfide linkage 402 with an acrydite moiety boundto the surface of the beads 401. In some cases, though,first primer 403 may be coupled to a bead via an acryditemoiety, without a disulfide linkage 402. The first primer403 may be a universal primer for priming template se-quences of oligonucleotides to be attached to the beadsand/or may be a primer binding site (e.g., P5) for use insequencing an oligonucleotide that comprises first primer403.[0099] The first primer 403 functionalized beads 401can then be mixed in an aqueous solution with templateoligonucleotides (e.g., oligonucleotides comprising a firstprimer binding site 404 (e.g., P5c), a template barcodesequence 405, and a template primer binding site 407(e.g., R1c)) and reagents necessary for nucleic acid am-plification (e.g., dNTPs, polymerase, co-factors, etc.) asshown in Fig 4B. The aqueous mixture may also com-prise a capture primer 406 (e.g., sometimes referred to
as a read primer) linked to a capture moiety (e.g., biotin),identical in sequence to the template primer binding site407 of the template oligonucleotide.[0100] The aqueous mixture is then emulsified in a wa-ter/oil emulsion to generate aqueous droplets (e.g., thedroplets comprising one or more beads 401, a templateoligonucleotide, reagents necessary for nucleic amplifi-cation, and, if desired, any capture primers 406) in a con-tinuous oil phase. In general, the droplets comprise, onaverage, at most one template oligonucleotide per drop-let. As shown in Fig 4B and 4C, a first round of thermo-cycling of the droplets results in priming of the templateoligonucleotides at primer binding site 404 by first primer403 and extension of first primer 403 such that oligonu-cleotides complementary to the template oligonucleotidesequences are attached to the gel beads at first primer403. The complementary oligonucleotides comprisesfirst primer 403, a barcode sequence 408 (e.g., comple-mentary to template barcode sequence 405), and a cap-ture primer binding site 415 complementary to both tem-plate primer binding site 407 and capture primer 406.Capture primer binding site 415 may also be used as aread primer binding site (e.g., R1) during sequencing ofthe complementary oligonucleotide.[0101] As shown in Fig 4D, capture primer 406 canbind to capture primer binding site 415 during the nextround of thermocycling. Capture primer 406, comprisinga capture moiety (e.g., biotin) at its 5’ end, can then beextended to generate additional template oligonucle-otides (e.g., comprising sequences 404, 405, and 406),as shown in Fig 4E. Thermocyling may continue for adesired number of cycles (e.g., at least about 1, 5, 10,15, 20, 25, 30, 35, 40, 45, 50 or more cycles) up until allfirst primer 403 sites of beads 401 are linked to a barcodesequence 408 and a capture primer binding site 415.Because each droplet generally comprises one or zerotemplate oligonucleotides to start, each droplet will gen-erally comprise beads attached to multiple copies of asequence complementary to the template oligonucle-otide or no copies of a sequence complementary to thetemplate oligonucleotide. At the conclusion of thermocy-cling, the oligonucleotide products attached to the beadsare hybridized to template oligonucleotides also compris-ing the capture moiety (e.g., biotin), as shown in Fig 4E.[0102] The emulsion may then be broken via any suit-able means and the released beads can be pooled intoa common vessel. Using a capture bead (or other device,including capture devices described herein) 409 linkedto a moiety (e.g., streptavidin) capable of binding withthe capture moiety of capture primer 406, positive beads(e.g., beads comprising sequences 403, 408, and 415)may be enriched from negative beads (e.g., beads notcomprising sequences 403, 408, and 415) by interactionof the capture bead with the capture moiety, as shownin Fig 4F and Fig 4G. In cases where capture beads areused, the beads may be magnetic, such that a magnetmay be used for enrichment. As an alternative, centrifu-gation may be used for enrichment. Upon enrichment of
35 36

EP 3 013 957 B1
20
5
10
15
20
25
30
35
40
45
50
55
the positive beads, the hybridized template oligonucle-otides comprising the capture moiety and linked to thecapture bead may be denatured from the bead-boundoligonucleotide via heat or chemical means, includingchemical means described herein, as shown in Fig 4H.Denatured oligonucleotides (e.g., oligonucleotides com-prising sequences 404, 405 and 406) may then be sep-arated from the positive beads via the capture beads at-tached to the denatured oligonucleotides. As shown inFig 4H, beads comprising sequences 403, 408, and 415are obtained. As an alternative to capture beads, positivebeads may also be sorted from positive beads via flowcytometry by including, for example, an optically activedye in partitions capable of binding to beads or speciescoupled to beads.[0103] In bulk aqueous fluid, the beads comprising se-quences 403, 408, and 415 can then be combined withtemplate random sequences (e.g., random N-mers) 413each linked to a sequence 412 complementary to captureprimer binding site 415, as shown in Fig 4I. As shown inFig 4J, capture primer binding site 415 can prime oligo-nucleotides comprising template random sequences 413at sequence 412 upon heating. Following priming, cap-ture primer binding site 415 can be extended (e.g., viapolymerase) to link capture primer binding site 415 witha random sequence 414 that is complementary to tem-plate random sequence 413. Oligonucleotides compris-ing template random sequences 413 and sequence 412can be denatured from the bead using heat or chemicalmeans, including chemical means described herein.Centrifugation and washing of the beads, for example,may be used to separate the beads from denatured oli-gonucleotides. Following removal of the denatured oli-gonucleotides, beads comprising a barcode sequence408 and a random sequence 414 are obtained, as shownin Fig 4K, 4L, and 4M. Because the attachment of ran-dom sequence 414 was done in bulk, each bead thatcomprises multiple copies of a unique barcode sequence408, also comprises various random sequences 414.[0104] To release bead-bound oligonucleotides fromthe beads, stimuli described elsewhere herein, such as,for example, a reducing agent, may be used. As shownin Fig 4N, contact of a bead comprising disulfide bondsand linkages to oligonucleotides via disulfide bonds witha reducing agent degrades both the bead and the di-sulfide linkages freeing the oligonucleotide from thebead. Contact with a reducing agent may be completed,for example, in another partition (e.g., a droplet of anotheremulsion), such that, upon oligonucleotide release fromthe bead, each droplet generally comprises free oligonu-cleotides all comprising the same barcode sequence408, yet various random sequences 414. Via random se-quence 414 acting as a random primer, free oligonucle-otides may be used to barcode different regions of a sam-ple nucleic acid also in the partition. Amplification or li-gation schemes, including those described herein, maybe used to complete attachment of barcodes to the sam-ple nucleic acid.
[0105] With limiting dilution, the partitions (e.g., drop-lets) may contain on average at most one oligonucleotidesequence per partition. This frequency of distribution ata given sequence-bead dilution follows Poisson distribu-tion. Thus, in some cases, about 6%, 10%, 18%, 20%,30%, 36%, 40%, or 50% of the droplets or partitions maycomprise one or fewer oligonucleotide sequences. Insome cases, more than about 6%, 10%, 18%, 20%, 30%,36%, 40%, 50%, 60%, 70%, 75%, 80%, 85%, 90%, 95%,96%, 97%, 98%, 99%, 99.1%, 99.2%, 99.3%,99.4%,99.5%, 99.6%, 99.7%, 99.8%, 99.9%, or more of thedroplets may comprise one or fewer oligonucleotide se-quences. In other cases, less than about 6%, 10%, 18%,20%, 30%, 36%, 40%, or 50% of the droplets may com-prise one or fewer oligonucleotide sequences.[0106] In some cases, limiting dilution steps may berepeated, prior to the addition of a random N-mer se-quence in order to increase the number of positive beadswith copies of barcodes. For example, a limiting dilutioncould be prepared such that a desired fraction (e.g., 1/10to 1/3) of emulsion droplets comprises a template for am-plification. Positive beads could be generated via ampli-fication of the template (as depicted in Fig 4) such thatpositives generally comprise no more primer for amplifi-cation (e.g., all P5 primer sites have been extended). Theemulsion droplets can then be broken, and subsequentlyre-emulsified with fresh template at limiting dilution for asecond round of amplification. Positive beads generatedin the first round of amplification generally would not par-ticipate in further amplification because their priming siteswould already be occupied. The process of amplificationfollowed by re-emulsification can be repeated for a suit-able number of steps, until the desired fraction of positivebeads is obtained.[0107] In some cases, negative beads obtained duringsorting after a limiting dilution functionalization may berecovered and further processed to generate additionalpositive beads. For example, negative beads may be dis-pensed into wells of a plate (e.g., a 384 well plate) afterrecovery such that each well generally comprises 1 bead.In some cases, dispensing may be achieved with the aidof flow cytometry (e.g., a flow cytometer directs each neg-ative bead into a well during sorting - an example flowcytometer being a BD FACS Jazz) or via a dispensingdevice, such as for example, a robotic dispensing device.Each well can also comprise a template barcode se-quence and the process depicted in Fig 4 repeated, ex-cept that each well partitions each bead, rather than afluidic droplet. Because each well comprises templateand a bead, each well can produce a positive bead. Thebeads can then be pooled from each well and additionalsequences (e.g., a random N-mer sequence) can be add-ed in bulk as described elsewhere herein.[0108] The barcodes may be loaded into the beads atan expected or predicted ratio of barcodes per bead tobe barcoded. In some cases, the barcodes are loadedsuch that a ratio of about 0.0001, 0.001, 0.1, 1, 2, 3, 4,5, 6, 7, 8, 9, 10, 20, 50, 100, 500, 1000, 5000, 10000,
37 38

EP 3 013 957 B1
21
5
10
15
20
25
30
35
40
45
50
55
20000, 50000, 100000, 500000, 1000000,5000000,10000000,50000000,100000000,500000000,1000000000,5000000000,10000000000,50000000000, or 100000000000 barcodes are loadedper bead. In some cases, the barcodes are loaded suchthat a ratio of more than 0.0001, 0.001, 0.1, 1, 2, 3, 4, 5,6, 7, 8, 9, 10, 20, 50, 100, 500, 1000, 5000, 10000, 20000,50000, 100000, 200000, 300000, 400000, 500000,600000, 700000, 800000, 900000, 1000000, 2000000,3000000, 4000000, 5000000, 6000000, 7000000,8000000, 9000000, 10000000, 20000000, 30000000,40000000, 50000000, 60000000, 70000000, 80000000,90000000, 100000000, 200000000, 300000000,400000000, 500000000, 600000000, 700000000,800000000, 900000000, 1000000000, 2000000000,3000000000, 4000000000, 5000000000, 6000000000,7000000000, 8000000000, 9000000000, 10000000000,20000000000, 30000000000, 40000000000,50000000000, 60000000000, 70000000000,80000000000, 90000000000, 100000000000 or morebarcodes are loaded per bead. In some cases, the bar-codes are loaded such that a ratio of less than about0.0001, 0.0002, 0.0003, 0.0004, 0.0005, 0.0006, 0.0007,0.0008, 0.0009, 0.001, 0.002, 0.003, 0.004, 0.005, 0.006,0.007, 0.008, 0.009, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8,0.9, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 50, 100, 500, 1000,5000, 10000, 20000, 50000, 100000, 500000, 1000000,5000000, 10000000, 50000000, 100000000,500000000, 1000000000, 5000000000, 10000000000,50000000000, or 100000000000 barcodes are loadedper bead.[0109] Beads, including those described herein (e.g.,substantially dissolvable beads, in some cases, substan-tially dissolvable by a reducing agent), may be covalentlyor non-covalently linked to a plurality of oligonucleotides,wherein at least a subset of the oligonucleotides com-prises a constant region or domain (e.g., a barcode se-quence, a barcode domain, a common barcode domain,or other sequence that is constant among the oligonu-cleotides of the subset) and a variable region or domain(e.g., a random sequence, a random N-mer, or other se-quence that is variable among the oligonucleotides of thesubset). In some cases, the oligonucleotides may be re-leasably coupled to a bead, as described elsewhereherein. Oligonucleotides may be covalently or non-cov-alently linked to a bead via any suitable linkage, includingtypes of covalent and non-covalent linkages describedelsewhere herein. In some cases, an oligonucleotide maybe covalently linked to a bead via a cleavable linkagesuch as, for example, a chemically cleavable linkage(e.g., a disulfide linkage), a photocleavable linkage, or athermally cleavable linkage. Beads may comprise morethan about or at least about 1, 10, 50, 100, 500, 1000,5000, 10000, 50000, 100000, 500000, 1000000,5000000, 10000000, 50000000, 100000000,500000000, 1000000000, 5000000000, 10000000000,50000000000, 100000000000, 500000000000, or1000000000000 oligonucleotides comprising a constant
region or domain and a variable region or domain.[0110] In some cases, the oligonucleotides may eachcomprise an identical constant region or domain (e.g.,an identical barcode sequence, identical barcode do-main, a common domain, etc.). In some cases, the oli-gonucleotides may each comprise a variable domain witha different sequence. In some cases, the percentage ofthe oligonucleotides that comprise an identical constantregion (or common domain) may be at least about 0.01%,0.1%, 1%, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%,45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%,95%, or 100%. In some cases, the percentage of theoligonucleotides that comprise a variable region with adifferent sequence may be at least about 0.01%, 0.1%,1%, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%,50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%,or 100%. In some cases, the percentage of beads in aplurality of beads that comprise oligonucleotides with dif-ferent nucleotide sequences (including those comprisinga variable and constant region or domain) is at least about0.01%, 0.1%, 1%, 5%, 10%, 15%, 20%, 25%, 30%, 35%,40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%,90%, 95%, or 100%. In some cases, the oligonucleotidesmay also comprise one or more additional sequences,such as, for example a primer binding site (e.g., a se-quencing primer binding site), a universal primer se-quence (e.g., a primer sequence that would be expectedto hybridize to and prime one or more loci on any nucleicacid fragment of a particular length, based upon the prob-ability of such loci being present within a sequence ofsuch length) or any other desired sequence includingtypes of additional sequences described elsewhere here-in.[0111] As described elsewhere herein, a plurality ofbeads may be generated to form, for example, a beadlibrary (e.g., a barcoded bead library). In some cases,the sequence of a common domain (e.g., a common bar-code domain) or region may vary between at least a sub-set of individual beads of the plurality. For example, thesequence of a common domain or region between indi-vidual beads of a plurality of beads may be different be-tween 2 or more, 10 or more, 50 or more, 100 or more,500 or more, 1000 or more, 5000 or more, 10000 or more,50000 or more, 100000 or more, 500000 or more,1000000 or more, 5000000 or more, 10000000 or more,50000000 or more, 100000000 or more, 500000000 ormore, 1000000000 or more, 5000000000 or more,10000000000 or more, 50000000000 or more, or100000000000 or more beads of the plurality. In somecases, each bead of a plurality of beads may comprisea different common domain or region. In some cases,the percentage of individual beads of a plurality of beadsthat comprise a different common domain or region maybe at least about 0.01%, 0.1%, 1%, 5%, 10%, 15%, 20%,25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%,75%, 80%, 85%, 90%, 95%, or 100%. In some cases, aplurality of beads may comprise at least about 2, 10, 50,100, 500, 1000, 5000, 10000, 50000, 100000, 500000,
39 40

EP 3 013 957 B1
22
5
10
15
20
25
30
35
40
45
50
55
1000000, 5000000, 10000000, 50000000, 100000000,500000000, or more different common domains coupledto different beads in the plurality.[0112] As an alternative to limiting dilution (e.g., viadroplets of an emulsion), other partitioning methods maybe used to attach oligonucleotides to beads. As shownin Fig 13A, the wells of a plate may be used. Beads com-prising a primer (e.g., P5, primer linked to the bead viaacrydite and, optionally, a disulfide bond) may be com-bined with a template oligonucleotide (e.g., a templateoligonucleotide comprising a barcode sequence) andamplification reagents in the wells of a plate. Each wellcan comprise one or more copies of a unique templatebarcode sequence and one or more beads. Thermal cy-cling of the plate extends the primer, via hybridization ofthe template oligonucleotide to the primer, such that thebead comprises an oligonucleotide with a sequence com-plementary to the oligonucleotide template. Thermal cy-cling may continue for a desired number of cycles (e.g.,at least about 1, 2, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50or more cycles) up until all primers have been extended.[0113] Upon completion of thermal cycling, the beadsmay be pooled into a common vessel, washed (e.g., viacentrifugation, magnetic separation, etc.), complemen-tary strands denatured, washed again, and then subjectto additional rounds of bulk processing if desired. Forexample, a random N-mer sequence may be added tothe bead-bound oligonucleotides using the primer exten-sion method described above for limiting dilution and asshown in Fig 13B and Fig 4I-M.[0114] As another alternative approach to limiting di-lution, a combinatorial process involving partitioning inmultiwell plates can be used to generate beads with oli-gonucleotide sequences as shown in Fig 13C. In suchmethods, the wells may contain pre-synthesized oligo-nucleotides such as oligonucleotide templates. Thebeads (e.g., beads with preincorporated oligonucleotidessuch as primers) may be divided into the individual wellsof the multiwell plate. For example, a mixture of beadscontaining P5 oligonucleotides may be divided into indi-vidual wells of a multiwell plate (e.g., 384 wells), whereineach well contains a unique oligonucleotide template(e.g., an oligonucleotide including a first partial barcodetemplate or barcode template). A primer extension reac-tion may be performed within the individual wells using,for example, the oligonucleotides templates as the tem-plate and the primer attached to the beads as primers.Subsequently, all wells may be pooled together and theunreacted products may be removed.[0115] The mixture of beads attached to the amplifiedproduct may be re-divided into wells of a second multiwellplate (e.g., 384-well plate), wherein each well of the sec-ond multiwell plate contains another oligonucleotide se-quence (e.g., including a second partial barcode se-quence and/or a random N-mer). In some cases, the ol-igonucleotide sequence may be attached (e.g., via hy-bridization) to a blocker oligonucleotide. Within the wellsof the second multiwell plate, a reaction such as a single-
stranded ligation reaction may be performed to add ad-ditional sequences to each bead (e.g., via ligation of theprimer extension products attached to the beads as inthe first step with the oligonucleotide in the wells of thesecond step). In some cases, a partial barcode sequencelinked to the bead in the first step is ligated to a secondpartial barcode sequence in the second step, to generatebeads comprising full barcode sequences. In some cas-es, the beads comprising full barcode sequences alsocomprise random sequences (e.g., random N-mers)and/or blocking oligonucleotides. In some cases, a PCRreaction or primer extension reaction is performed to at-tach the additional sequence to the beads. Beads fromthe wells may be pooled together, and the unreactedproducts may be removed. In some cases, the processis repeated with additional multi-well plates. The processmay be repeated over 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20,50, 100, 500, 1000, 5000, or 10000 times.[0116] In some combinatorial approaches, ligationmethods may be used to assemble oligonucleotide se-quences comprising barcode sequences on beads (e.g.,degradable beads as described elsewhere herein). Forexample, separate populations of beads may be providedto which barcode containing oligonucleotides are to beattached. These populations may include anchor com-ponents (or linkage) for attaching nucleotides, such asactivatable chemical groups (phosphoramidites, acryditemoieties, or other thermally, optically or chemically acti-vatable groups), cleavable linkages, previously attachedoligonucleotide molecules to which the barcode contain-ing oligonucleotides may be ligated, hybridized, or oth-erwise attached, DNA binding proteins, charged groupsfor electrostatic attachment, or any of a variety of otherattachment mechanisms.[0117] A first oligonucleotide or oligonucleotide seg-ment that includes a first barcode sequence segment, isattached to the separate populations, where differentpopulations include different barcode sequence seg-ments attached thereto. Each bead in each of the sepa-rate populations may be attached to at least 2, 10, 50,100, 500, 1000, 5000, 10000, 50000, 100000, 500000,1000000, 5000000, 10000000, 50000000, 100000000,500000000, 1000000000, or more first oligonucleotidemolecules or oligonucleotide segment molecules. Thefirst oligonucleotide or oligonucleotide segment may bereleasably attached to the separate populations. In somecases, the first oligonucleotide or oligonucleotide seg-ments may be attached directly to respective beads inthe separate populations or may be indirectly attached(e.g., via an anchor component coupled to the beads, asdescribed above) to respective beads in the separatepopulations.[0118] In some cases, the first oligonucleotide may beattached to the separate populations with the aid of asplint (an example of a splint is shown as 2306 in Fig.23A). A splint, as used herein, generally refers to a dou-ble-stranded nucleic acid, where one strand of the nucleicacid comprises an oligonucleotide to-be-attached to one
41 42

EP 3 013 957 B1
23
5
10
15
20
25
30
35
40
45
50
55
or more receiving oligonucleotides and where the otherstrand of the nucleic acid comprises an oligonucleotidewith a sequence that is in part complementary to at leasta portion of the oligonucleotide to-be-attached and in partcomplementary to at least a portion of the one or morereceiving oligonucleotides. In some cases, an oligonu-cleotide may be in part complementary to at least a por-tion of a receiving oligonucleotide via an overhang se-quence as shown in Fig 23A). An overhang sequencecan be of any suitable length, as described elsewhereherein.[0119] For example, a splint may be configured suchthat it comprises the first oligonucleotide or oligonucle-otide segment hybridized to an oligonucleotide that com-prises a sequence that is in part complementary to atleast a portion of the first oligonucleotide or oligonucle-otide segment and a sequence (e.g., an overhang se-quence) that is in part complementary to at least a portionof an oligonucleotide attached to the separate popula-tions. The splint can hybridize to the oligonucleotide at-tached to the separate populations via its complementarysequence. Once hybridized, the first oligonucleotide oroligonucleotide segment of the splint can then be at-tached to the oligonucleotide attached to the separatepopulations via any suitable attachment mechanism,such as, for example, a ligation reaction.[0120] Following attachment of the first oligonucleotideor oligonucleotide segment to the separate populations,the separate populations are then pooled to create amixed pooled population, which is then separated into aplurality of separate populations of the mixed, pooledpopulation. A second oligonucleotide or segment includ-ing a second barcode sequence segment is then at-tached to the first oligonucleotides on the beads in eachseparate mixed, pooled population, such that differentmixed pooled bead populations have a different secondbarcode sequence segment attached to it. Each bead inthe separate populations of the mixed, pooled populationmay be attached to at least 2, 10, 50, 100, 500, 1000,5000, 10000, 50000, 100000, 500000, 1000000,5000000, 10000000, 50000000, 100000000,500000000, 1000000000, or more second oligonucle-otide molecules or oligonucleotide segment molecules.[0121] In some cases, the second oligonucleotide maybe attached to the first oligonucleotide with the aid of asplint. For example, the splint used to attach the first ol-igonucleotide or oligonucleotide segment to the separatepopulations prior to generating the mixed pooled popu-lation may also comprise a sequence (e.g., an overhangsequence) that is in part complementary to at least aportion of the second oligonucleotide. The splint can hy-bridize to the second oligonucleotide via the complemen-tary sequence. Once hybridized, the second oligonucle-otide can then be attached to the first oligonucleotide viaany suitable attachment mechanism, such as, for exam-ple, a ligation reaction. The splint strand complementaryto both the first and second oligonucleotides can then bethen denatured (or removed) with further processing. Al-
ternatively, a separate splint comprising the second oli-gonucleotide may be provided to attach the second oli-gonucleotide to the first oligonucleotide in analogousfashion as described above for attaching the first oligo-nucleotide to an oligonucleotide attached to the separatepopulations with the aid of splint. Also, in some cases,the first barcode segment of the first oligonucleotide andsecond barcode segment of the second oligonucleotidemay be joined via a linking sequence as described else-where herein.[0122] The separate populations of the mixed, pooledpopulation can then be pooled and the resulting pooledbead population then includes a diverse population ofbarcode sequences, or barcode library that is represent-ed by the product of the number of different first barcodesequences and the number of different second barcodesequences. For example, where the first and second ol-igonucleotides include, e.g., all 256 4-mer barcode se-quence segments, a complete barcode library may in-clude 65,536 diverse 8 base barcode sequences.[0123] The barcode sequence segments may be inde-pendently selected from a set of barcode sequence seg-ments or the first and second barcode sequence seg-ments may each be selected from separate sets of bar-code sequence segments. Moreover, the barcode se-quence segments may individually and independentlycomprise from 2 to 20 nucleotides in length, preferablyfrom about 4 to about 20 nucleotides in length, more pref-erably from about 4 to about 16 nucleotides in length orfrom about 4 to about 10 nucleotides in length. In somecases, the barcode sequence segments may individuallyand independently comprise at least 2, 3, 4, 5, 6, 7, 8, 9,10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or more nucle-otides in length. In particular, the barcode sequence seg-ments may comprise 2-mers, 3-mers, 4-mers, 5-mers,6-mers, 7-mers, 8-mers, 9-mers, 10-mers, 11-mers, 12-mers, 13-mers, 14-mers, 15-mers, 16-mers, 17-mers,18-mers, 19-mers, 20-mers, or longer sequence seg-ments.[0124] Furthermore, the barcode sequence segmentsincluded within the first and second oligonucleotide se-quences or sequence segments will typically representat least 10 different barcode sequence segments, at least50 different barcode sequence segments, at least 100different barcode sequence segments, at least 500 dif-ferent barcode sequence segments, at least 1,000 dif-ferent barcode sequence segments, at least about 2,000different barcode sequence segments, at least about4,000 different barcode sequence segments, at leastabout 5,000 different barcode sequence segments, atleast about 10,000 different barcode sequence seg-ments, at least 50,000 different barcode sequence seg-ments, at least 100,000 barcode sequence segments, atleast 500,000 barcode sequence segments, at least1,000,000 barcode sequence segments, or more. In ac-cordance with the processes described above, these dif-ferent oligonucleotides may be allocated amongst a sim-ilar or the same number of separate bead populations in
43 44

EP 3 013 957 B1
24
5
10
15
20
25
30
35
40
45
50
55
either the first or second oligonucleotide addition step,e.g., at least 10, 100, 500, 1000, 2000, 4000, 5000,10000, 50000, 100000, 500000, 1000000, etc., differentbarcode sequence segments being separately added toat least 10, 100, 500, 1000, 2000, 4000, 5000, 10000,50000, 100000, 500000, 1000000, etc., separate beadpopulations.[0125] As a result, resulting barcode libraries mayrange in diversity of from at least about 100 different bar-code sequence segments to at least about 1,000,000,2,000,000, 5,000,000, 10,000,000 100,000,000 or moredifferent barcode sequence segments as described else-where herein, being represented within the library.[0126] As noted previously, either or both of the firstand second oligonucleotide sequences or sequence seg-ments, or subsequently added oligonucleotides (e.g., ad-dition of a third oligonucleotide to the second oligonucle-otide, addition of a fourth oligonucleotide to an addedthird oligonucleotide, etc.), may include additional se-quences, e.g., complete or partial functional sequences(e.g., a primer sequence (e.g., a universal primer se-quence, a targeted primer sequence, a random primersequence), a primer annealing sequence, an attachmentsequence, a sequencing primer sequence, a random N-mer, etc.), for use in subsequent processing. These se-quences will, in many cases, be common among beadsin the separate populations, subsets of populations,and/or common among all beads in the overall popula-tion. In some cases, the functional sequences may bevariable as between different bead subpopulations, dif-ferent beads, or even different molecules attached to asingle bead. Moreover, either or both of the first and sec-ond oligonucleotide sequences or sequence segmentsmay comprise a sequence segment that includes one ormore of a uracil containing nucleotide and a non-nativenucleotide, as described elsewhere herein. In addition,although described as oligonucleotides comprising bar-code sequences, it will be appreciated that such refer-ences includes oligonucleotides that are comprised oftwo, three or more discrete barcode sequence segmentsthat are separated by one or more bases within the oli-gonucleotide, e.g., a first barcode segment separatedfrom a second barcode segment by 1, 2, 3, 4, 5, 6, or 10or more bases in the oligonucleotide in which they arecontained. Preferably, barcode sequence segments willbe located adjacent to each other or within 6 bases, 4bases, 3 bases or two bases of each other in the oligo-nucleotide sequence in which they are contained. To-gether, whether contiguous within an oligonucleotide se-quence, or separated by one or more bases, such col-lective barcode sequence segments within a given oli-gonucleotide are referred to herein as a barcode se-quence, barcode sequence segment, or barcode do-main.[0127] An example combinatorial method for generat-ing beads with sequences comprising barcode sequenc-es as well as specific types of functional sequences isshown in Fig 23. Although described in terms of certain
specific sequence segments for purposes of illustration,it will be appreciated that a variety of different configura-tions may be incorporated into the barcode containingoligonucleotides attached to the beads described herein,including a variety of different functional sequence types,primer types, e.g., specific for different sequencing sys-tems, and the like. As shown in Fig 23A, beads 2301may be generated and covalently linked (e.g., via an acry-dite moiety or other species) to a first oligonucleotidecomponent to be used as an anchoring componentand/or functional sequence or partial functional se-quence, e.g., partial P5 sequence 2302. In each well ofa plate (e.g., a 384-well plate) an oligonucleotide 2303,comprising the remaining P5 sequence and a unique firstpartial barcode sequence (indicated by bases"DDDDDD" in oligonucleotide 2303), can be hybridizedto an oligonucleotide 2304 that comprises the comple-ment of oligonucleotide 2303 and additional bases thatoverhang each end of oligonucleotide 2303. Hybridizedproduct (a "splint") 2306 can thus be generated. Eachoverhang of the splint can be blocked (indicated with an"X" in Fig 23A) with a blocking moiety to prevent sideproduct formation. Non-limiting examples of blockingmoieties include 3’ Inverted dT, dideoxycytidine (ddC),and 3’C3 Spacer. Accordingly, in the example described,different splints can be generated, each with a uniquefirst partial barcode sequence or its complement, e.g.,384 different splints, as described.[0128] As shown in Fig 23B, beads 2301 can be addedto each well of the plate and the splint 2306 in each wellcan hybridize with the corresponding anchor sequence,e.g., partial P5 sequence 2302, of beads 2301, via oneof the overhangs of oligonucleotide 2304. Limited stabilityof the overhang of oligonucleotide 2304 in hybridizingpartial P5 sequence 2302 can permit dynamic samplingof splint 2306, which can aid in ensuring that subsequentligation of oligonucleotide 2303 to partial P5 sequence2302 is efficient. A ligation enzyme (e.g., a ligase) canligate partial P5 sequence 2302 to oligonucleotide 2303.An example of a ligase would be T4 DNA ligase. Follow-ing ligation, the products can be pooled and the beadswashed to remove unligated oligonucleotides.[0129] As shown in Fig 23C, the washed products canthen be redistributed into wells of another plate (e.g., a384-well plate), with each well of the plate comprising anoligonucleotide 2305 that has a unique second partialbarcode sequence (indicated by "DDDDDD" in oligonu-cleotide 2305) and an adjacent short sequence (e.g.,"CC" adjacent to the second partial barcode sequenceand at the terminus of oligonucleotide 2305) complemen-tary to the remaining overhang of oligonucleotide 2304.Oligonucleotide 2305 can also comprise additional se-quences, such as R1 sequences and a random N-mer(indicated by "NNNNNNNNNN" in oligonucleotide 2305).In some cases, oligonucleotide 2305 may comprise auracil containing nucleotide. In some cases, any of thethymine containing nucleotides of oligonucleotide 2305may be substituted with uracil containing nucleotides. In
45 46

EP 3 013 957 B1
25
5
10
15
20
25
30
35
40
45
50
55
some cases, in order to improve the efficiency of ligationof the oligonucleotide comprising the second partial bar-code sequence, e.g., sequence 2305, to the first partialbarcode sequence, e.g., sequence 2303, a duplexstrand, e.g., that is complementary to all or a portion ofoligonucleotide 2305, may be provided hybridized tosome portion or all of oligonucleotide 2305, while leavingthe overhang bases available for hybridization to splint2304. As noted previously, splint 2304 and/or the duplexstrand, may be provided blocked at one or both of their3’ and 5’ ends to prevent formation of side products fromor between one or both of the splint and the duplex strand.In preferred aspects, the duplex strand may be comple-mentary to all or a portion of oligonucleotide 2305. Forexample, where oligonucleotide 2305 includes a randomn-mer, the duplex strand may be provided that does nothybridize to that portion of the oligonucleotide.[0130] Via the adjacent short sequence, oligonucle-otide 2305 can be hybridized with oligonucleotide 2304,as shown in Fig 23C. Again, the limited stability of theoverhang in hybridizing the short complementary se-quence of oligonucleotide 2305 can permit dynamic sam-pling of oligonucleotide 2305, which can aid in ensuringthat subsequent ligation of oligonucleotide 2305 to oligo-nucleotide 2303 is efficient. A ligation enzyme (e.g., aligase) can then ligate oligonucleotide 2305 to oligonu-cleotide 2303. Ligation of oligonucleotide 2305 to oligo-nucleotide 2303 can result in the generation of a full bar-code sequence, via the joining of the first partial barcodesequence of oligonucleotide 2305 and the second partialbarcode sequence of oligonucleotide 2303. As shown inFig 23D, the products can then be pooled, the oligonu-cleotide 2304 can be denatured from the products, andunbound oligonucleotides can then be washed away.Following washing, a diverse library of barcoded beadscan be obtained, with each bead bound to, for example,an oligonucleotide comprising a P5 sequence, a full bar-code sequence, an R1 sequence, and a random N-mer.In this example, 147, 456 unique barcode sequences canbe obtained (e.g., 384 unique first partial barcode se-quence x 384 unique second partial barcode sequences).[0131] In some cases, the inclusion of overhang basesthat aid in ligation of oligonucleotides as described abovecan result in products that all have the same base at agiven position, including in between portions of a barcodesequence as shown in Fig 24A. Limited or no base di-versity at a given sequence position across sequencingreads may result in failed sequencing runs, dependingupon the particular sequencing method utilized. Accord-ingly, in a number of aspects, the overhang bases maybe provided with some variability as between differentsplints, either in terms of base identity or position withinthe overall sequenced portion of the oligonucleotide. Forexample, in a first example, one or more spacer bases2401 (e.g., "1" "2" in Fig 24B at 2401) can be added tosome oligonucleotides used to synthesize larger oligo-nucleotides on beads, such that oligonucleotide productsdiffer slightly in length from one another, and thus position
the overhang bases at different locations in different se-quences. Complementary spacer bases may also beadded to splints necessary for sequence component li-gations. A slight difference in oligonucleotide length be-tween products can result in base diversity at a givenread position, as shown in Fig 24B.[0132] In another example shown in Fig 25, splintscomprising a random base overhang may be used tointroduce base diversity at read positions complementaryto splint overhangs. For example, a double-strandedsplint 2501 may comprise a random base (e.g., "NN" inFig 25A) overhang 2503 and a determined base (e.g.,"CTCT" in Fig 25A) overhang 2506 on one strand and afirst partial barcode sequence (e.g., "DDDDDD" in Fig25A) on the other strand. Using an analogous ligationscheme as described above for the Example depicted inFig 23, the determined overhang 2506 may be used tocapture sequence 2502 (which may be attached to abead as shown in Fig 23) via hybridization for subsequentligation with the upper strand (as shown in Fig 25A) ofsplint 2501. Although overhang 2506 is illustrated as afour base determined sequence overhang, it will be ap-preciated that this sequence may be longer in order toimprove the efficiency of hybridization and ligation in thefirst ligation step. As such determined base overhang2506 may include 4, 6, 8, 10 or more bases in length thatare complementary to partial P5 sequence 2502. More-over, the random base overhang 2503 may be used tocapture the remaining component (e.g., sequence 2504)of the final desired sequence. Sequence 2504 may com-prise a second partial barcode sequence ("DDDDDD" insequence 2504 of Fig 25C), the complement 2505 (e.g.,"NN" at 2505 in Fig 25C) of the random base overhang2503 at one end and a random N-mer 2507 at its otherend (e.g., "NNNNNNNNNN" in sequence 2504 of Fig25C).[0133] Due to the randomness of the bases in randombase overhang 2503, bases incorporated into the ligationproduct at complement 2505 can vary, such that productscomprise a variety of bases at the read positions of com-plement 2505. As will be appreciated, in preferred as-pects, the second partial barcode sequence portion tobe ligated to the first partial barcode sequence will typi-cally include a population of such second partial barcodesequences that includes all of the complements to therandom overhang sequences, e.g., a given partial bar-code sequence will be present with, e.g., 16 differentoverhang portions, in order to add the same second par-tial barcode sequence to each bead in a given well wheremultiple overhang sequences are represented. While on-ly two bases are shown for random overhang 2503 andcomplement 2505 in Fig 25, the example is not meantto be limiting. Any suitable number of random bases inan overhang may be used. Further, while described asrandom overhang sequences, in some cases, theseoverhang sequences may be selected from a subset ofoverhang sequences. For example, in some cases, theoverhangs will be selected from subsets of overhang se-
47 48

EP 3 013 957 B1
26
5
10
15
20
25
30
35
40
45
50
55
quences that include fewer than all possible overhangsequences of the length of the overhang, which may bemore than one overhang sequence, and in some cases,more than 2, more than 4, more than 10, more than 20,more than 50, or even more overhang sequences.[0134] In another example, a set of splints, each witha defined overhang selected from a set of overhang se-quences of a given length, e.g., a set of at least 2, 4, 10,20 or more overhang sequences may be used to intro-duce base diversity at read positions complementary tosplint overhangs. Again, because these overhangs areused to ligate a second partial barcode sequence to thefirst barcode sequence, it will be desirable to have allpossible overhang complements represented in the pop-ulation of second partial barcode sequences. As such,in many cases, it will be preferred to keep the numbersof different overhang sequences lower, e.g., less than50, less than 20, or in some cases, less than 10 or lessthan 5 different overhang sequences. In many cases, thenumber of different linking sequences in a barcode librarywill be between 2 and 4096 different linking sequences,with preferred libraries having between about 2 and about50 different linking sequences. Likewise it will typicallybe desirable to keep these overhang sequences of a rel-atively short length, in order to avoid introducing non-relevant bases to the ultimate sequence reads. As such,these overhang sequences will typically be designed tointroduce no more than 10, no more than 9, no more than8, no more than 7, no more than 6, no more than 5, nomore than 4, and in some cases, 3 or fewer nucleotidesto the overall oligonucleotide construct. In some cases,the length of an overhang sequence may be from about1 to about 10 nucleotides in length, from about 2 to about8 nucleotides in length, from about 2 to about 6 nucle-otides in length, or from about 2 to about 4 nucleotidesin length. In general, each splint in the set can comprisean overhang with a different sequence from other splintsin the set, such that the base at each position of the over-hang is different from the base in the same base positionin the other splints in the set. An example set of splintsis depicted in Fig 26. The set comprises splint 2601 (com-prising an overhang of "AC" 2602), splint 2603 (compris-ing an overhang of "CT" 2604), splint 2605 (comprisingan overhang of "GA" 2606), and splint 2607 (comprisingan overhang of "TG" 2608). Each splint can also comprisean overhang 2609 (e.g., "CTCT" in each splint) and firstpartial barcode sequence ("DDDDDD"). As shown in Fig26, each splint can comprise a different base in eachposition of its unique overhang (e.g., overhang 2602 insplint 2601, overhang 2604 in splint 2603, overhang 2606in splint 2605, and overhang 2608 in splint 2607) suchthat no splint overhang comprises the same base in thesame base position. Because each splint comprises adifferent base in each position of its unique overhang,products generated from each splint can also have a dif-ferent base in each complementary position when com-pared to products generated from one of the other splints.Thus, base diversity at these positions can be achieved.
[0135] Such products can be generated by hybridizingthe first component of the desired sequence (e.g., se-quence 2502 in Fig 25 comprising a first partial barcodesequence; the first component may also be attached toa bead) with the overhang common to each splint (e.g.,overhang 2609 in Fig 26); ligating the first component ofthe sequence to the splint; hybridizing the second partof the desired sequence (e.g., a sequence similar to se-quence 2504 in Fig 25 comprising a second partial bar-code sequence, except that the sequence comprisesbases complementary to the unique overhang sequenceat positions 2505 instead of random bases) to the uniqueoverhang of the splint; and ligating the second compo-nent of the desired sequence to the splint. The unligatedportion of the splint (e.g., bottom sequence comprisingthe overhangs as shown in Fig 26) can then be dena-tured, the products washed, etc. as described previouslyto obtain final products. As will be appreciated, and asnoted previously, these overhang sequences may pro-vide 1, 2, 3, 4, 5 or 6 or more bases between differentpartial barcode sequences (or barcode sequence seg-ments), such that they provide a linking sequence be-tween barcode sequence segments, with the character-istics described above. Such a linking sequence may beof varied length, such as for example, from about 2 toabout 10 nucleotides in length, from about 2 to about 8nucleotides in length, from about 2 to about 6 nucleotidesin length, from about 2 to about 5 nucleotides in length,or from about 2 to about 4 nucleotides in length.[0136] An example workflow using the set of splintsdepicted in Fig 26 is shown in Fig 27. For each splint inthe set, the splint strand comprising the unique overhangsequence (e.g., the bottom strand of splints shown in Fig26) can be provided in each well of one or more plates.In Fig 27, two 96-well plates of splint strands comprisinga unique overhang sequence are provided for each ofthe four splint types, for a total of eight plates. Of the eightplates, two plates (2601a, 2601b) correspond to the bot-tom strand of splint 2601 comprising a unique overhangsequence ("AC") in Fig 26, two plates (2603a, 2603b)correspond to the bottom strand of splint 2603 in Fig 26comprising a unique overhang sequence ("CT"), twoplates (2605a, 2605b) correspond to the bottom strandof splint 2605 in Fig 26 comprising a unique overhangsequence ("GA"), and two plates (2607a, 2607b) corre-spond to the bottom strand of splint 2607 in Fig 26 com-prising a unique overhang sequence ("TG"). The oligo-nucleotides in each 96-well plate (2601a, 2601b, 2603a,2603b, 2605a, 2605b, 2607a, and 2607b) can be trans-ferred to another set of 96-well plates 2702, with eachplate transferred to its own separate plate (again, for atotal of eight plates), and each well of each plate trans-ferred to its corresponding well in the next plate.[0137] The splint strand comprising a unique first par-tial barcode sequence (e.g., the upper strand of splintsshown in Fig 26) and a first partial P5 sequence can beprovided in one or more plates. In Fig 27, such splintstrands are provided in two 96-well plates 2708a and
49 50

EP 3 013 957 B1
27
5
10
15
20
25
30
35
40
45
50
55
2708b, with each well of the two plates comprising anoligonucleotide with a unique first partial barcode se-quence, for a total of 192 unique first partial barcode se-quences across the two plates. Each well of plate 2708acan be added to its corresponding well in four of the plates2702 and each well of plate 2708b can be added to itscorresponding well in the other four of the plates 2702.Thus, the two splint strands in each well can hybridize togenerate a complete splint. After splint generation, eachwell of two of the 96-well plates 2702 in Fig 27 comprisesa splint configured as splint 2601, splint 2603, splint 2605,or splint 2607 in Fig 26 and a unique first partial barcodesequence, for a total of 192 unique first partial barcodesequences.[0138] To each of the wells of the plates 2702, beads2709 comprising a second partial P5 sequence (e.g., sim-ilar or equivalent to sequence 2502 in Fig 25) can thenbe added. The splints in each well can hybridize with thesecond partial P5 sequence via the common overhangsequence 2609 of each splint. A ligation enzyme (e.g., aligase) can then ligate the second partial P5 sequenceto the splint strand comprising the remaining first partialP5 sequence and the first partial barcode sequence. Firstproducts are, thus, generated comprising beads linkedto a sequence comprising a P5 sequence and a first par-tial barcode sequence, still hybridized with the splintstrand comprising the overhang sequences. Followingligation, first products from the wells of each plate canbe separately pooled to generate plate pools 2703. Theplate pools 2703 corresponding to each two-plate set(e.g., each set corresponding to a particular splint con-figuration) can also be separately pooled to generate firstproduct pools 2704, such that each first product pool2704 comprises products generated from splints com-prising only one unique overhang sequence. In Fig 27,four first product pools 2704 are generated, each corre-sponding to one of the four splint types used in the ex-ample. The products in each plate pool 2703 may bewashed to remove unbound oligonucleotides, the prod-ucts in each first product pool 2704 may be washed toremove unbound oligonucleotides, or washing may occurat both pooling steps. In some cases, plate pooling 2703may be bypassed with the contents of each two-plate setentered directly into a first product pool 2704.[0139] Next, each first product pool 2704 can be aliq-uoted into each well of two 96-well plates 2705, as de-picted in Fig 27, for a total of eight plates (e.g., two platesper product pool 2704). Separately, oligonucleotides thatcomprise a unique second partial barcode sequence, aterminal sequence complementary to one of the fourunique overhang sequences, and any other sequence tobe added (e.g., additional sequencing primer sites, ran-dom N-mers, etc.) can be provided in 96-well plates 2706.Such oligonucleotides may, for example, comprise a se-quence similar to sequence 2504 in Fig 25, except thatthe sequence comprises bases complementary to aunique overhang sequence at position 2505 instead ofrandom bases. For example, for splint 2601 shown in Fig
26, the bases in position 2505 would be "TG", comple-mentary to the unique overhang 2602 ("AC") of splint2601. Of the plates 2706, sets of two plates can eachcomprise oligonucleotides comprising sequences com-plementary to one of the four unique overhang sequenc-es, for a total of eight plates and four plate sets as shownin Fig 27. Plates 2706 can be configured such that eachwell comprises a unique second partial barcode se-quence, for a total of 768 unique second partial barcodesequences across the eight plates.[0140] Each plate of plates 2706 can be paired with acorresponding plate of plates 2705, based on the appro-priate unique overhang sequence of first products en-tered into the plate of plates 2705, as shown in Fig 27.Oligonucleotides in each well of the plate from plates2706 can be added to its corresponding well in its corre-sponding plate from plates 2705, such that each wellcomprises an aliquot of first products from the appropri-ate first product pool 2704 and oligonucleotides compris-ing a unique second barcode sequence and any othersequence (e.g., random N-mers) from plates 2706. Ineach well of the plates 2705, the unique overhang se-quence of each first product can hybridize with an oligo-nucleotide comprising the second partial barcode se-quence, via the oligonucleotide’s bases complementaryto the unique overhang sequence. A ligation enzyme(e.g., a ligase) can then ligate the oligonucleotides to thefirst products. Upon ligation, second products comprisingcomplete barcode sequences are generated via joiningof the first partial barcode sequence of the first productswith the second partial barcode sequence of the secondproducts. The second products obtained from plates2705 can be removed and deposited into a common sec-ond product pool 2707. The splint strands comprising theoverhangs (as shown in Fig 26) can then be denaturedin product pool 2707, and the products washed to obtainfinal products. A total of 147,456 unique barcode se-quences can be obtained (e.g., 192 first partial barcodesequences x 768 second partial barcode sequences)with base diversity in base positions complementary tounique overhang sequences used during ligations.[0141] The above example with respect to splint setsis not meant to be limiting, nor is the number and type(s) of plates used for combinatorial synthesis. A set ofsplints can comprise any suitable number of splints.Moreover, each set of splints may be designed with theappropriate first partial barcode sequence diversity de-pending upon, for example, the number of unique bar-code sequences desired, the number of bases used togenerate a barcode sequence, etc.[0142] Using a combinatorial plate method, libraries ofbarcoded beads with high-diversity can be generated.For example, if two 384-well plates are used, each witholigonucleotides comprising partial barcode sequencespre-deposited in each well, it is possible that 384 x 384or 147,456 unique barcode sequences can be generated.The combinatorial examples shown herein are not meantto be limiting as any suitable combination of plates may
51 52

EP 3 013 957 B1
28
5
10
15
20
25
30
35
40
45
50
55
be used. For example, while in some cases, the barcodesequence segments added in each combinatorial stepmay be selected from the same sets of barcode sequencesegments. However, in many cases, the barcode se-quence segments added in each combinatorial step maybe selected from partially or completely different sets ofoligonucleotide sequences. For example, in some cases,a first oligonucleotide segment may include a barcodesequence from a first set of barcode sequences, e.g., 4-mer sequences, while the second oligonucleotide se-quence may include barcode sequences from a partiallyor completely different set of barcode sequence seg-ments, e.g., 4-mer sequences, 6-mer sequences, 8-mersequences, etc., or even sequences of mixed lengths,e.g., where the second oligonucleotide segment is se-lected form a set of oligonucleotides having barcode se-quences having varied lengths and sequences, to gen-erate multiparameter variability in the generated bar-codes, e.g., sequence and length.,[0143] With reference to the example above, for exam-ple, the number and type of plates (and barcodes) usedfor each step in a combinatorial method does not haveto be the same. For example, a 384 well plate may beused for a first step and a 96 well plate may be used fora second step for a total of 36,864 unique barcode se-quences generated. Furthermore, the number of basesof a full barcode sequence added in each combinatorialstep does not need to be the same. For example, in afirst combinatorial step, 4 bases of a 12 base barcodesequence may be added, with the remaining 8 basesadded in a second combinatorial step. Moreover, thenumber of combinatorial steps used to generate a fullbarcode sequence may also vary. In some cases, about2, 3, 4, 5, 6, 7, 8, 9, or 10 combinatorial steps are used.[0144] The primer extension reactions and ligation re-actions can be conducted with standard techniques andreagents in the multiwell plates. For example, the poly-mer, poly-ethylene glycol (PEG), may be present duringthe single-stranded ligation reaction at a concentrationof about 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%,55%, 60%, 65%, 70%, or 75%. In some cases, the PEGmay be present during the ligation reaction at a concen-tration of more than about 6%, 10%, 18%, 20%, 30%,36%, 40%, 50% or more. In some cases, the PEG maybe present during the ligation reaction in the second plateat a concentration of less than about 6%, 10%, 18%,20%, 30%, 36%, 40%, or 50%.[0145] The methods provided herein may reduce nu-cleotide bias in ligation reactions. Better results may oc-cur when the first extension in the first well plate may berun to completion. For the single-strand ligation step inthe second well plate, no competition may be presentwhen only one type of oligonucleotide sequence is used.The partitioning in wells method for attaching content tobeads may avoid misformed adaptors with 8N ends, par-ticularly when the first extension in the first well plate isrun to completion.[0146] Potential modifications to the partitioning in
wells process may include replacing the single-strandligation step with PCR by providing the second oligonu-cleotide sequence with degenerate bases, modifying thefirst oligonucleotide sequence to be longer than the sec-ond oligonucleotide sequence, and/or adding a randomN-mer sequence in a separate bulk reaction after thesingle-strand ligation step, as this may save synthesiscosts and may reduce N-mer sequence bias.[0147] In some cases, the following sequence of proc-esses may be used to attach a barcode sequence to abead. The barcode sequence may be mixed with suitablePCR reagents and a plurality of beads in aqueous fluid.The aqueous fluid may be emulsified within an immisciblefluid, such as an oil, to form an emulsion. The emulsionmay generate individual fluidic droplets containing thebarcode sequence, the bead, and PCR reagents. Indi-vidual fluidic droplets may be exposed to thermocyclingconditions, in which the multiple rounds of temperaturecycling permits priming and extension of barcode se-quences. The emulsion containing the fluidic dropletsmay be broken by continuous phase exchange, de-scribed elsewhere in this disclosure. Resulting barcodedbeads suspended in aqueous solution may be sorted bymagnetic separation or other sorting methods to obtaina collection of purified barcoded beads in aqueous fluid.[0148] In some cases, the following sequence of proc-esses may be used to attach an N-mer sequence to abead. The N-mer sequence may be mixed with suitablePCR reagents and a plurality of pooled barcoded beadsin aqueous fluid. The aqueous fluid may be heated topermit hybridization and extension of the N-mer se-quence. Additional heating may permit removal of thecomplement strand.[0149] The PCR reagents may include any suitablePCR reagents. In some cases, dUTPs may be substitut-ed for dTTPs during the primer extension or other ampli-fication reactions, such that oligonucleotide productscomprise uracil containing nucleotides rather than thym-ine containing nucleotides. This uracil-containing sectionof the universal sequence may later be used togetherwith a polymerase that will not accept or process uracil-containing templates to mitigate undesired amplificationproducts.[0150] Amplification reagents may include a universalprimer, universal primer binding site, sequencing primer,sequencing primer binding site, universal read primer,universal read binding site, or other primers compatiblewith a sequencing device, e.g., an Illumina sequencer,Ion Torrent sequencer, etc. The amplification reagentsmay include P5, non cleavable 5’acrydite-P5, a cleavable5’ acrydite-SS-P5, R1c, Biotin R1c, sequencing primer,read primer, P5_Universal, P5_U, 52-BioR1-rc, a ran-dom N-mer sequence, a universal read primer, etc. Insome cases, a primer may contain a modified nucleotide,a locked nucleic acid (LNA), an LNA nucleotide, a uracilcontaining nucleotide, a nucleotide containing a non-na-tive base, a blocker oligonucleotide, a blocked 3’ end,3’ddCTP. Fig 19 provides additional examples.
53 54

EP 3 013 957 B1
29
5
10
15
20
25
30
35
40
45
50
55
[0151] As described herein, in some cases oligonucle-otides comprising barcodes are partitioned such thateach bead is partitioned with, on average, less than oneunique oligonucleotide sequence, less than two uniqueoligonucleotide sequences, less than three unique oligo-nucleotide sequences, less than four unique oligonucle-otide sequences, less than five unique oligonucleotidesequences, or less than ten unique oligonucleotide se-quences. Therefore, in some cases, a fraction of thebeads does not contain an oligonucleotide template andtherefore cannot contain an amplified oligonucleotide.Thus, it may be desirable to separate beads comprisingoligonucleotides from beads not comprising oligonucle-otides. In some cases, this may be done using a capturemoiety.[0152] In some embodiments, a capture moiety maybe used with isolation methods such as magnetic sepa-ration to separate beads containing barcodes frombeads, which may not contain barcodes. As such, insome cases, the amplification reagents may include cap-ture moieties attached to a primer or probe. Capture moi-eties may allow for sorting of labeled beads from non-labeled beads to confirm attachment of primers anddownstream amplification products to a bead. Exemplarycapture moieties include biotin, streptavidin, glutathione-S-transferase (GST), cMyc, HA, etc. The capture moie-ties may be, or include, a fluorescent label or magneticlabel. The capture moiety may comprise multiple mole-cules of a capture moiety, e.g., multiple molecules of bi-otin, streptavidin, etc. In some cases, an amplificationreaction may make use of capture primers attached to acapture moiety (as described elsewhere herein), suchthat the primer hybridizes with amplification products andthe capture moiety is integrated into additional amplifiedoligonucleotides during additional cycles of the amplifi-cation reaction. In other cases, a probe comprising a cap-ture moiety may be hybridized to amplified oligonucle-otides following the completion of an amplification reac-tion such that the capture moiety is associated with theamplified oligonucleotides.[0153] A capture moiety may be a member of bindingpair, such that the capture moiety can be bound with itsbinding pair during separation. For example, beads maybe generated that comprise oligonucleotides that com-prise a capture moiety that is a member of a binding pair(e.g., biotin). The beads may be mixed with capturebeads that comprise the other member of the binding pair(e.g., streptavidin), such that the two binding pair mem-bers bind in the resulting mixture. The bead-capture beadcomplexes may then be separated from other compo-nents of the mixture using any suitable means, including,for example centrifugation and magnetic separation(e.g., including cases where the capture bead is a mag-netic bead).[0154] In many cases as described, individual beadswill generally have oligonucleotides attached thereto,that have a common overall barcode sequence segment.As described herein, where a bead includes oligonucle-
otides having a common barcode sequence, it is gener-ally meant that of the oligonucleotides coupled to a givenbead, a significant percentage, e.g., greater than 70%,greater than 80%, greater than 90%, greater than 95%or even greater than 99% of the oligonucleotides of orgreater than a given length, e.g., including the full ex-pected length or lengths of final oligonucleotides and ex-cluding unreacted anchor sequences or partial barcodesequences, include the same or identical barcode se-quence segments. This barcode sequence segment ordomain (again, which may be comprised of two or moresequence segments separated by one or more bases)may be included among other common or variable se-quences or domains within a single bead. Also as de-scribed, the overall population of beads will include beadshaving large numbers of different barcode sequence seg-ments. In many cases, however, a number of separatebeads within a given bead population may include thesame barcode sequence segment. In particular, a bar-code sequence library having 1000, 10,000, 1,000,000,10,000,000 or more different sequences, may be repre-sented in bead populations of greater than 100,000,1,000,000, 10,000,000, 100,000,000, 1 billion, 10 billion,100 billion or more discrete beads, such that the samebarcode sequence is represented multiple times withina given bead population or subpopulation. For example,the same barcode sequence may be present on two ormore beads within a given analysis, 10 or more beads,100 or more beads, etc..[0155] A capture device, such as a magnetic bead, witha corresponding linkage, such as streptavidin, may beadded to bind the capture moiety, for example, biotin.The attached magnetic bead may then enable isolationof the barcoded beads by, for example, magnetic sorting.Magnetic beads may also be coated with other linkingentities besides streptavidin, including nickel-IMAC toenable the separation of His-tagged fusion proteins, coat-ed with titanium dioxide to enable the separation of phos-phorylated peptides, or coated with amine-reactive NHS-ester groups to immobilize protein or other ligands forseparation.[0156] In some embodiments, the capture moiety maybe attached to a primer, to an internal sequence, to aspecific sequence within the amplified product, to a bar-code sequence, to a universal sequence, or to a com-plementary sequence. Capture moieties may be at-tached by PCR amplification or ligation. Capture moietiesmay include a universal tag such as biotin attached to aspecific target such as a primer before added to the beadpopulation. In other cases, capture moieties may includea specific tag that recognizes a specific sequence or pro-tein or antibody that may be added to the bead populationindependently. In some embodiments, the capture moi-eties may be pre-linked to a sorting bead, such as a mag-netic bead. In some cases, the capture moiety may be afluorescent label, which may enable sorting via fluores-cence-activated cell sorting (FACS).[0157] In some cases, a nucleic acid label (e.g., fluo-
55 56

EP 3 013 957 B1
30
5
10
15
20
25
30
35
40
45
50
55
rescent label) may be used to identify fluidic droplets,emulsions, or beads that contain oligonucleotides. Sort-ing (e.g., via flow cytometry) of the labeled droplets orbeads may then be performed in order to isolate beadsattached to amplified oligonucleotides. Exemplary stainsinclude intercalating dyes, minor-groove binders, majorgroove binders, external binders, and bis-intercalators.Specific examples of such dyes include SYBR green,SYBR blue, DAPI, propidium iodide, SYBR gold, ethid-ium bromide, propidium iodide, imidazoles (e.g., Hoechst33258, Hoechst 33342, Hoechst 34580, and DAPI), 7-AAD, SYTOX Blue, SYTOX Green, SYTOX Orange,POPO-1, POPO-3, YOYO-1, YOYO-3, TOTO-1, TOTO-3, JOJO-1, LOLO-1, BOBO-1, BOBO-3, PO-PRO-1, PO-PRO-3, BO-PRO-1, BO-PRO-3, TO-PRO-1, TO-PRO-3,TO-PRO-5, JO-PRO-1, LO-PRO-1, YO-PRO-1, YO-PRO-3, PicoGreen, OliGreen, RiboGreen, EvaGreen,SYBR Green, SYBR Green II, SYBR DX, SYTO-40, -41,-42, -43, -44, -45 (blue), SYTO-13, -16, -24, -21, -23, -12,-11, -20, -22, -15, - 14, -25 (green), SYTO-81, -80, -82,-83, -84, -85 (orange), SYTO-64, -17, -59, -61, -62, -60,and -63 (red).
Multi-Functional Beads
[0158] Beads may be linked to a variety of species (in-cluding non-nucleic acid species) such that they are mul-ti-functional. For example, a bead may be linked to mul-tiple types of oligonucleotides comprising a barcode se-quence and an N-mer (e.g., a random N-mer or a targetedN-mer as described below). Each type of oligonucleotidemay differ in its barcode sequence, its N-mer, or any othersequence of the oligonucleotide. Moreover, each beadmay be linked to oligonucleotides comprising a barcodesequence and an N-mer and may also be linked to ablocker oligonucleotide capable of blocking the oligonu-cleotides comprising a barcode sequence and an N-mer.Loading of the oligonucleotide blocker and oligonucle-otide comprising a barcode sequence and an N-mer maybe completed at distinct ratios in order to obtain desiredstoichiometries of oligonucleotide blocker to oligonucle-otide comprising a barcode sequence and an N-mer. Ingeneral, a plurality of species may be loaded to beadsat distinct ratios in order to obtain desired stoichiometriesof the species on the beads.[0159] Moreover, a bead may also be linked to one ormore different types of multi-functional oligonucleotides.For example, a multi-functional oligonucleotide may becapable of functioning as two or more of the following: aprimer, a tool for ligation, an oligonucleotide blocker, anoligonucleotide capable of hybridization detection, a re-porter oligonucleotide, an oligonucleotide probe, a func-tional oligonucleotide, an enrichment primer, a targetedprimer, a non-specific primer, and a fluorescent probe.Oligonucleotides that function as fluorescent probes maybe used, for example, for bead detection or characteri-zation (e.g., quantification of number of beads, quantifi-cation of species (e.g., primers, linkers, etc.) attached to
beads, determination of bead size/topology, determina-tion of bead porosity, etc.).[0160] Other non-limiting examples of species thatmay also be attached or coupled to beads include wholecells, chromosomes, polynucleotides, organic mole-cules, proteins, polypeptides, carbohydrates, saccha-rides, sugars, lipids, enzymes, restriction enzymes, ligas-es, polymerases, barcodes, adapters, small molecules,antibodies, antibody fragments, fluorophores, deoxynu-cleotide triphosphates (dNTPs), dideoxynucleotide tri-phosphates (ddNTPs), buffers, acidic solutions, basic so-lutions, temperature-sensitive enzymes, pH-sensitiveenzymes, light-sensitive enzymes, metals, metal ions,magnesium chloride, sodium chloride, manganese,aqueous buffer, mild buffer, ionic buffer, inhibitors, sac-charides, oils, salts, ions, detergents, ionic detergents,non-ionic detergents, oligonucleotides, nucleotides,DNA, RNA, peptide polynucleotides, complementaryDNA (cDNA), double stranded DNA (dsDNA), singlestranded DNA (ssDNA), plasmid DNA, cosmid DNA,chromosomal DNA, genomic DNA, viral DNA, bacterialDNA, mtDNA (mitochondrial DNA), mRNA, rRNA, tRNA,nRNA, siRNA, snRNA, snoRNA, scaRNA, microRNA,dsRNA, ribozyme, riboswitch and viral RNA, a lockednucleic acid (LNA) in whole or part, locked nucleic acidnucleotides, any other type of nucleic acid analogue, pro-teases, nucleases, protease inhibitors, nuclease inhibi-tors, chelating agents, reducing agents, oxidizing agents,probes, chromophores, dyes, organics, emulsifiers, sur-factants, stabilizers, polymers, water, small molecules,pharmaceuticals, radioactive molecules, preservatives,antibiotics, aptamers, and combinations thereof. Bothadditional oligonucleotide species and other types of spe-cies may be coupled to beads by any suitable methodincluding covalent and non-covalent means (e.g., ionicbonds, van der Waals interactions, hydrophobic interac-tions, encapsulation, diffusion of the species into thebead, etc.). In some cases, an additional species maybe a reactant used for a reaction comprising another typeof species on the bead. For example, an additional spe-cies coupled to a bead may be a reactant suitable for usein an amplification reaction comprising an oligonucle-otide species also attached to the bead.[0161] In some cases, a bead may comprise one ormore capture ligands each capable of capturing a par-ticular type of sample component, including componentsthat may comprise nucleic acid. For example, a beadmay comprise a capture ligand capable of capturing acell from a sample. The capture ligand may be, for ex-ample, an antibody, antibody fragment, receptor, protein,peptide, small molecule or any other species targetedtoward a species unique to and/or over-expressed on thesurface of a particular cell. Via interactions with the celltarget, the particular cell type can be captured from asample such that it remains bound to the bead. A beadbound to a cell can be entered into a partition as describedelsewhere herein to barcode nucleic acids obtained fromthe cell. In some cases, capture of a cell from a sample
57 58

EP 3 013 957 B1
31
5
10
15
20
25
30
35
40
45
50
55
may occur in a partition. Lysis agents, for example, canbe included in the partition such in order to release thenucleic acid from the cell. The released nucleic acid canbe barcoded and processed using any of the methodsdescribed herein.
III. Barcode Libraries
[0162] Beads may contain one or more attached bar-code sequences. In some cases, each bead may be at-tached to about 1, 5, 10, 50, 100, 500, 1000, 5000, 10000,20000, 50000, 100000, 500000, 1000000, 5000000,10000000, 50000000, 100000000, 500000000,1000000000, 5000000000, 10000000000,50000000000, or 100000000000 identical barcode se-quences. In some cases, each bead may be attached toat least about 1, 5, 10, 50, 100, 500, 1000, 5000, 10000,20000, 50000, 100000, 200000, 300000, 400000,500000, 600000, 700000, 800000, 900000, 1000000,2000000, 3000000, 4000000, 5000000, 6000000,7000000, 8000000, 9000000, 10000000, 20000000,30000000, 40000000, 50000000, 60000000, 70000000,80000000, 90000000, 100000000, 200000000,300000000, 400000000, 500000000, 600000000,700000000, 800000000, 900000000, 1000000000,2000000000, 3000000000, 4000000000, 5000000000,6000000000, 7000000000, 8000000000, 9000000000,10000000000, 20000000000, 30000000000,40000000000, 50000000000, 60000000000,70000000000, 80000000000, 90000000000,100000000000 or more identical barcode sequences. Insome cases, each bead may be attached to less thanabout 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70,80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900,1000, 2000, 3000, 4000, 5000, 6000, 7000, 8000, 9000,10000, 20000, 30000, 40000, 50000, 60000, 70000,80000, 90000, 100000, 500000, 1000000, 5000000,10000000, 50000000, 1000000000, 5000000000,10000000000, 50000000000, or 100000000000 identi-cal barcode sequences.[0163] An individual barcode library may comprise oneor more barcoded beads. In some cases, an individualbarcode library may comprise about 1, 5, 10, 50, 100,500, 1000, 5000, 10000, 20000, 50000, 100000, 500000,1000000, 5000000, 10000000, 50000000, 100000000,500000000, 1000000000, 5000000000, 10000000000,50000000000, or 100000000000 individual barcodedbeads. In some cases, each library may comprise at leastabout 1, 5, 10, 50, 100, 500, 1000, 5000, 10000, 20000,50000, 100000, 200000, 300000, 400000, 500000,600000, 700000, 800000, 900000, 1000000, 2000000,3000000, 4000000, 5000000, 6000000, 7000000,8000000, 9000000, 10000000, 20000000, 30000000,40000000, 50000000, 60000000, 70000000, 80000000,90000000, 100000000, 200000000, 300000000,400000000, 500000000, 600000000, 700000000,800000000, 900000000, 1000000000, 2000000000,3000000000, 4000000000,5000000000, 6000000000,
7000000000, 8000000000, 9000000000, 10000000000,20000000000, 30000000000, 40000000000,50000000000, 60000000000, 70000000000,80000000000, 90000000000, 100000000000 or more in-dividual barcoded beads. In some cases, each librarymay comprise less than about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10,20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500,600, 700, 800, 900, 1000, 2000, 3000, 4000, 5000, 6000,7000, 8000, 9000, 10000, 20000, 30000, 40000, 50000,60000, 70000, 80000, 90000, 100000, 500000,1000000, 5000000, 10000000, 50000000, 1000000000,5000000000, 10000000000, 50000000000, or100000000000 individual barcoded beads. The barcod-ed beads within the library may have the same sequenc-es or different sequences.[0164] In some embodiments, each bead may have aunique barcode sequence. However, the number ofbeads with unique barcode sequences within a barcodelibrary may be limited by combinatorial limits. For exam-ple, using four different nucleotides, if a barcode is 12nucleotides in length, than the number of unique con-structs may be limited to 412 = 16777216 unique con-structs. Since barcode libraries may comprise manymore beads than 1677216, there may be some librarieswith multiple copies of the same barcode. In some em-bodiments, the percentage of multiple copies of the samebarcode within a given library may be 1%, 2%, 3%, 4%,5%, 6%, 7%, 8%, 9% ,10%, 15%, 20%, 25%, 30%, 40%,or 50%. In some cases, the percentage of multiple copiesof the same barcode within a given library may be morethan 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9% ,10%, 15%,20%, 25%, 30%, 40%, 50% or more. In some cases, thepercentage of multiple copies of the same barcode withina given library may be less than 1%, 2%, 3%, 4%, 5%,6%, 7%, 8%, 9% ,10%, 11%, 12%, 13%, 14%, 15%, 16%,17%, 18%, 19%, 20%, 25%, 30%, 40%, or 50%.[0165] In some embodiments, each bead may com-prise one unique barcode sequence but multiple differentrandom N-mers. In some cases, each bead may haveone or more different random N-mers. Again, the numberof beads with different random N-mers within a barcodelibrary may be limited by combinatorial limits. For exam-ple, using four different nucleotides, if an N-mer se-quence is 12 nucleotides in length, than the number ofdifferent constructs may be limited to 412 = 16777216different constructs. Since barcode libraries may com-prise many more beads than 16777216, there may besome libraries with multiple copies of the same N-mersequence. In some embodiments, the percentage of mul-tiple copies of the same N-mer sequence within a givenlibrary may be 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%,9% ,10%, 15%, 20%, 25%, 30%, 40%, or 50%. In somecases, the percentage of multiple copies of the same N-mer sequence within a given library may be more than1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9% ,10%, 15%, 20%,25%, 30%, 40%, 50% or more. In some cases, the per-centage of multiple copies of the same N-mer sequencewithin a given library may be less than 1%, 2%, 3%, 4%,
59 60

EP 3 013 957 B1
32
5
10
15
20
25
30
35
40
45
50
55
5%, 6%, 7%, 8%, 9% ,10%, 11%, 12%, 13%, 14%, 15%,16%, 17%, 18%, 19%, 20%, 25%, 30%, 40%, or 50%.[0166] In some embodiments, the unique identifier se-quence within the barcode may be different for each prim-er within each bead. In some cases, the unique identifiersequence within the barcode sequence may be the samefor each primer within each bead.
IV. Combining Barcoded Beads with Sample
Types of Samples
[0167] The methods, compositions, devices, and kitsof this disclosure may be used with any suitable sampleor species. A sample (e.g., sample material, componentof a sample material, fragment of a sample material, etc.)or species can be, for example, any substance used insample processing, such as a reagent or an analyte. Ex-emplary samples can include one or more of whole cells,chromosomes, polynucleotides, organic molecules, pro-teins, nucleic acids, polypeptides, carbohydrates, sac-charides, sugars, lipids, enzymes, restriction enzymes,ligases, polymerases, barcodes (e.g., including barcodesequences, nucleic acid barcode sequences, barcodemolecules), adaptors, small molecules, antibodies, fluor-ophores, deoxynucleotide triphosphate (dNTPs), dide-oxynucleotide triphosphates (ddNTPs), buffers, acidicsolutions, basic solutions, temperature-sensitive en-zymes, pH-sensitive enzymes, light-sensitive enzymes,metals, metal ions, magnesium chloride, sodium chlo-ride, manganese, aqueous buffer, mild buffer, ionic buff-er, inhibitors, oils, salts, ions, detergents, ionic deter-gents, non-ionic detergents, oligonucleotides, templatenucleic acid molecules (e.g., template oligonucleotides,template nucleic acid sequences), nucleic acid frag-ments, template nucleic acid fragments (e.g., fragmentsof a template nucleic acid generated from fragmenting atemplate nucleic acid during fragmentation, fragments ofa template nucleic acid generated from a nucleic acidamplification reaction), nucleotides, DNA, RNA, peptidepolynucleotides, complementary DNA (cDNA), doublestranded DNA (dsDNA), single stranded DNA (ssDNA),plasmid DNA, cosmid DNA, chromosomal DNA, genomicDNA (gDNA), viral DNA, bacterial DNA, mtDNA (mito-chondrial DNA), mRNA, rRNA, tRNA, nRNA, siRNA, sn-RNA, snoRNA, scaRNA, microRNA, dsRNA, ribozyme,riboswitch and viral RNA, proteases, locked nucleic acidsin whole or part, locked nucleic acid nucleotides, nucle-ases, protease inhibitors, nuclease inhibitors, chelatingagents, reducing agents, oxidizing agents, probes,chromophores, dyes, organics, emulsifiers, surfactants,stabilizers, polymers, water, pharmaceuticals, radioac-tive molecules, preservatives, antibiotics, aptamers, andthe like. In summary, the samples that are used will varydepending on the particular processing needs.[0168] Samples may be derived from human and non-human sources. In some cases, samples are derivedfrom mammals, non-human mammals, rodents, amphib-
ians, reptiles, dogs, cats, cows, horses, goats, sheep,hens, birds, mice, rabbits, insects, slugs, microbes, bac-teria, parasites, or fish. Samples may be derived from avariety of cells, including but not limited to: eukaryoticcells, prokaryotic cells, fungi cells, heart cells, lung cells,kidney cells, liver cells, pancreas cells, reproductive cells,stem cells, induced pluripotent stem cells, gastrointesti-nal cells, blood cells, cancer cells, bacterial cells, bacte-rial cells isolated from a human microbiome sample, etc.In some cases, a sample may comprise the contents ofa cell, such as, for example, the contents of a single cellor the contents of multiple cells. Samples may also becell-free, such as circulating nucleic acids (e.g., DNA,RNA).[0169] A sample may be naturally-occurring or synthet-ic. A sample may be obtained from any suitable location,including from organisms, whole cells, cell preparationsand cell-free compositions from any organism, tissue,cell, or environment. A sample may be obtained fromenvironmental biopsies, aspirates, formalin fixed embed-ded tissues, air, agricultural samples, soil samples, pe-troleum samples, water samples, or dust samples. Insome instances, a sample may be obtained from bodilyfluids, which may include blood, urine, feces, serum,lymph, saliva, mucosal secretions, perspiration, centralnervous system fluid, vaginal fluid, or semen. Samplesmay also be obtained from manufactured products, suchas cosmetics, foods, personal care products, and the like.Samples may be the products of experimental manipu-lation including recombinant cloning, polynucleotide am-plification, polymerase chain reaction (PCR) amplifica-tion, purification methods (such as purification of genom-ic DNA or RNA), and synthesis reactions.
Methods of Attaching Barcodes to Samples
[0170] Barcodes (or other oligonucleotides, e.g. ran-dom N-mers) may be attached to a sample by joining thetwo nucleic acid segments together through the actionof an enzyme. This may be accomplished by primer ex-tension, polymerase chain reaction (PCR), another typeof reaction using a polymerase, or by ligation using aligase. When the ligation method is used to attach a sam-ple to a barcode, the samples may or may not be frag-mented prior to the ligation step. In some cases, the ol-igonucleotides (e.g., barcodes, random N-mers) are at-tached to a sample while the oligonucleotides are stillattached to the beads. In some cases, the oligonucle-otides (e.g., barcodes, random N-mers) are attached toa sample after the oligonucleotides are released from thebeads, e.g., by cleavage of the oligonucleotides compris-ing the barcodes from the beads and/or through degra-dation of the beads.[0171] The oligonucleotides may include one or morerandom N-mer sequences. A collection of unique randomN-mer sequences may prime random portions of a DNAsegment, thereby amplifying a sample (e.g., a whole ge-nome). The resulting product may be a collection of bar-
61 62

EP 3 013 957 B1
33
5
10
15
20
25
30
35
40
45
50
55
coded fragments representative of the entire sample(e.g., genome).[0172] The samples may or may not be fragmentedbefore ligation to barcoded beads. DNA fragmentationmay involve separating or disrupting DNA strands intosmall pieces or segments. A variety of methods may beemployed to fragment DNA including restriction digest orvarious methods of generating shear forces. Restrictiondigest may utilize restriction enzymes to make intentionalcuts in a DNA sequence by blunt cleavage to both strandsor by uneven cleavage to generate sticky ends. Exam-ples of shear-force mediated DNA strand disruption mayinclude sonication, acoustic shearing, needle shearing,pipetting, or nebulization. Sonication, is a type of hydro-dynamic shearing, exposing DNA sequences to short pe-riods of shear forces, which may result in about 700 bpfragment sizes. Acoustic shearing applies high-frequen-cy acoustic energy to the DNA sample within a bowl-shaped transducer. Needle shearing generates shearforces by passing DNA through a small diameter needleto physically tear DNA into smaller segments. Nebuliza-tion forces may be generated by sending DNA througha small hole of an aerosol unit in which resulting DNAfragments are collected from the fine mist exiting the unit.[0173] In some cases, a ligation reaction is used toligate oligonucleotides to sample. The ligation may in-volve joining together two nucleic acid segments, suchas a barcode sequence and a sample, by catalyzing theformation of a phosphodiester bond. The ligation reactionmay include a DNA ligase, such as an E.coli DNA ligase,a T4 DNA ligase, a mammalian ligase such as DNA ligaseI, DNA ligase III, DNA ligase IV, thermostable ligases, orthe like. The T4 DNA ligase may ligate segments con-taining DNA, oligonucleotides, RNA, and RNA-DNA hy-brids. The ligation reaction may not include a DNA ligase,utilizing an alternative such as a topoisomerase. To ligatea sample to a barcode sequence, utilizing a high DNAligase concentration and including PEG may achieve rap-id ligation. The optimal temperature for DNA ligase, whichmay be 37 °C, and the melting temperature of the DNAto be ligated, which may vary, may be considered to se-lect for a favorable temperature for the ligation reaction.The sample and barcoded beads may be suspended ina buffer to minimize ionic effects that may affect ligation.[0174] Although described in terms of ligation or directattachment of a barcode sequence to a sample nucleicacid component, above, the attachment of a barcode toa sample nucleic acid, as used herein, also encompassesthe attachment of a barcode sequence to a complementof a sample, or a copy or complement of that complement,e.g., when the barcode is associated with a primer se-quence that is used to replicate the sample nucleic acid,as is described in greater detail elsewhere herein. In par-ticular, where a barcode containing primer sequence isused in a primer extension reaction using the samplenucleic acid (or a replicate of the sample nucleic acid) asa template, the resulting extension product, whether acomplement of the sample nucleic acid or a duplicate of
the sample nucleic acid, will be referred to as having thebarcode sequence attached to it.[0175] In some cases, sample is combined with thebarcoded beads (either manually or with the aid of a mi-crofluidic device) and the combined sample and beadsare partitioned, such as in a microfluidic device. The par-titions may be aqueous droplets within a water-in-oilemulsion. When samples are combined with barcodedbeads, on average less than two target analytes may bepresent in each fluidic droplet. In some embodiments, onaverage, less than three target analytes may appear perfluidic droplet. In some cases, on average, more than twotarget analytes may appear per fluidic droplet. In othercases, on average, more than three target analytes mayappear per fluidic droplet. In some cases, one or morestrands of the same target analyte may appear in thesame fluidic droplet. In some cases, less than 1, 2, 3, 4,5, 6, 7, 8, 9, 10, 50, 100, 1000, 5000, 10000, or 100000target analytes are present within a fluidic droplet. Insome cases, greater than 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 50,100, 1000, 5000, 10000, or 100000 target analytes arepresent within a fluidic droplet. The partitions describedherein are often characterized by having extremely smallvolumes. For example, in the case of droplet based par-titions, the droplets may have overall volumes that areless than 1000 pL, less than 900 pL, less than 800 pL,less than 700 pL, less than 600 pL, less than 500 pL, lessthan 400pL, less than 300 pL, less than 200 pL, less than100pL, less than 50 pL, less than 20 pL, less than 10 pL,or even less than 1 pL. Where co-partitioned with beads,it will be appreciated that the sample fluid volume withinthe partitions may be less than 90% of the above de-scribed volumes, less than 80%, less than 70%, less than60%, less than 50%, less than 40%, less than 30%, lessthan 20%, or even less than 10% the above describedvolumes.[0176] When samples are combined with barcodedbeads, on average less than one bead may be presentin each fluidic droplet. In some embodiments, on aver-age, less than two beads may be present in each fluidicdroplet. In some embodiments, on average, less thanthree beads may be present per fluidic droplet. In somecases, on average, more than one bead may be presentin each fluidic droplet. In other cases, on average, morethan two beads may appear be present in each fluidicdroplet. In other cases, on average, more than threebeads may be present per fluidic droplet. In some em-bodiments, a ratio of on average less than one barcodedbead per fluidic droplet may be achieved using limitingdilution technique. Here, barcoded beads may be dilutedprior to mixing with the sample, diluted during mixing withthe sample, or diluted after mixing with the sample.[0177] The number of different barcodes or differentsets of barcodes (e.g., different sets of barcodes, eachdifferent set coupled to a different bead) that are parti-tioned may vary depending upon, for example, the par-ticular barcodes to be partitioned and/or the application.Different sets of barcodes may be, for example, sets of
63 64

EP 3 013 957 B1
34
5
10
15
20
25
30
35
40
45
50
55
identical barcodes where the identical barcodes differ be-tween each set. Or different sets of barcodes may be,for example, sets of different barcodes, where each setdiffers in its included barcodes. In some cases, differentbarcodes are partitioned by attaching different barcodesto different beads (e.g., gel beads). In some cases, dif-ferent sets of barcodes are partitioned by disposing eachdifferent set in a different partition. In some cases, thougha partition may comprise one or more different barcodesets. For example, each different set of barcodes maybe coupled to a different bead (e.g., a gel bead). Eachdifferent bead may be partitioned into a fluidic droplet,such that each different set of barcodes is partitioned intoa different fluidic droplet. For example, about 1, 5, 10,50, 100, 1000, 10000, 20,000, 30,000, 40,000, 50,000,60,000, 70,000, 80,000, 90,000, 100000, 200,000,300,000, 400,000, 500,000, 600,000, 700,000, 800,000,900,000, 1000000, 2000000, 3000000, 4000000,5000000, 6000000, 7000000, 8000000, 9000000,10000000, 20000000, 50000000, 100000000, or moredifferent barcodes or different sets of barcodes may bepartitioned. In some examples, at least about 1, 5, 10,50, 100, 1000, 10000, 20000, 30000, 40000, 50000,60000, 70000, 80000, 90000, 100000, 200,000,300,000, 400,000, 500,000, 600,000, 700,000, 800,000,900,000, 1000000, 2000000, 3000000, 4000000,5000000, 6000000, 7000000, 8000000, 9000000,10000000, 20000000, 50000000, 100000000, or moredifferent barcodes or different sets of barcodes may bepartitioned. In some examples, less than about 1, 5, 10,50, 100, 1000, 10000, 20000, 30000, 40000, 50000,60000, 70000, 80000, 90000, 100000, 200,000,300,000, 400,000, 500,000, 600,000, 700,000, 800,000,900,000, 1000000, 2000000, 3000000, 4000000,5000000, 6000000, 7000000, 8000000, 9000000,10000000, 20000000, 50000000, or 100000000 differentbarcodes or different sets of barcodes may be parti-tioned. In some examples, about 1-5, 5-10, 10-50,50-100, 100-1000, 1000-10000, 10000-100000,100000-1000000, 10000-1000000, 10000-10000000, or10000-100000000 different barcodes or different sets ofbarcodes may be partitioned.[0178] Barcodes may be partitioned at a particular den-sity. For example, barcodes may be partitioned so thateach partition contains about 1, 5, 10, 50, 100, 1000,10000, 20,000, 30,000, 40,000, 50,000, 60,000, 70,000,80,000, 90,000, 100000, 200,000, 300,000, 400,000,500,000, 600,000, 700,000, 800,000, 900,000, 1000000,2000000, 3000000, 4000000, 5000000, 6000000,7000000, 8000000, 9000000, 10000000, 20000000,50000000, or 100000000 barcodes per partition. Bar-codes may be partitioned so that each partition containsat least about 1, 5, 10, 50, 100, 1000, 10000, 20000,30000, 40000, 50000, 60000, 70000, 80000, 90000,100000, 200,000, 300,000, 400,000, 500,000, 600,000,700,000, 800,000, 900,000, 1000000, 2000000,3000000, 4000000, 5000000, 6000000, 7000000,8000000, 9000000, 10000000, 20000000, 50000000,
100000000, or more barcodes per partition. Barcodesmay be partitioned so that each partition contains lessthan about 1, 5, 10, 50, 100, 1000, 10000, 20000, 30000,40000, 50000, 60000, 70000, 80000, 90000, 100000,200,000, 300,000, 400,000, 500,000, 600,000, 700,000,800,000, 900,000, 1000000, 2000000, 3000000,4000000, 5000000, 6000000, 7000000, 8000000,9000000, 10000000, 20000000, 50000000, or100000000 barcodes per partition. Barcodes may be par-titioned such that each partition contains about 1-5, 5-10,10-50, 50-100, 100-1000, 1000-10000, 10000-100000,100000-1000000, 10000-1000000, 10000-10000000, or10000-100000000 barcodes per partition. In some cas-es, partitioned barcodes may be coupled to one or morebeads, such as, for example, a gel bead. In some cases,the partitions are fluidic droplets.[0179] Barcodes may be partitioned such that identicalbarcodes are partitioned at a particular density. For ex-ample, identical barcodes may be partitioned so thateach partition contains about 1, 5, 10, 50, 100, 1000,10000, 20,000, 30,000, 40,000, 50,000, 60,000, 70,000,80,000, 90,000, 100000, 200,000, 300,000, 400,000,500,000, 600,000, 700,000, 800,000, 900,000, 1000000,2000000, 3000000, 4000000, 5000000, 6000000,7000000, 8000000, 9000000, 10000000, 20000000,50000000, or 100000000 identical barcodes per parti-tion. Barcodes may be partitioned so that each partitioncontains at least about 1, 5, 10, 50, 100, 1000, 10000,20000, 30000, 40000, 50000, 60000, 70000, 80000,90000, 100000, 200,000, 300,000, 400,000, 500,000,600,000, 700,000, 800,000, 900,000, 1000000,2000000, 3000000, 4000000, 5000000, 6000000,7000000, 8000000, 9000000, 10000000, 20000000,50000000, 100000000, or more identical barcodes perpartition. Barcodes may be partitioned so that each par-tition contains less than about 1, 5, 10, 50, 100, 1000,10000, 20000, 30000, 40000, 50000, 60000, 70000,80000, 90000, 100000, 200,000, 300,000, 400,000,500,000, 600,000, 700,000, 800,000, 900,000, 1000000,2000000, 3000000, 4000000, 5000000, 6000000,7000000, 8000000, 9000000, 10000000, 20000000,50000000, or 100000000 identical barcodes per parti-tion. Barcodes may be partitioned such that each partitioncontains about 1-5, 5-10, 10-50, 50-100, 100-1000,1000-10000, 10000-100000, 100000-1000000,10000-1000000, 10000-10000000, or10000-100000000 identical barcodes per partition. Insome cases, partitioned identical barcodes may be cou-pled to a bead, such as, for example, a gel bead. In somecases, the partitions are fluidic droplets.[0180] Barcodes may be partitioned such that differentbarcodes are partitioned at a particular density. For ex-ample, different barcodes may be partitioned so that eachpartition contains about 1, 5, 10, 50, 100, 1000, 10000,20,000, 30,000, 40,000, 50,000, 60,000, 70,000, 80,000,90,000, 100000, 200,000, 300,000, 400,000, 500,000,600,000, 700,000, 800,000, 900,000, 1000000,2000000, 3000000, 4000000, 5000000, 6000000,
65 66

EP 3 013 957 B1
35
5
10
15
20
25
30
35
40
45
50
55
7000000, 8000000, 9000000, 10000000, 20000000,50000000, or 100000000 different barcodes per parti-tion. Barcodes may be partitioned so that each partitioncontains at least about 1, 5, 10, 50, 100, 1000, 10000,20000, 30000, 40000, 50000, 60000, 70000, 80000,90000, 100000, 200,000, 300,000, 400,000, 500,000,600,000, 700,000, 800,000, 900,000, 1000000,2000000, 3000000, 4000000, 5000000, 6000000,7000000, 8000000, 9000000, 10000000, 20000000,50000000, 100000000, or more different barcodes perpartition. Barcodes may be partitioned so that each par-tition contains less than about 1, 5, 10, 50, 100, 1000,10000, 20000, 30000, 40000, 50000, 60000, 70000,80000, 90000, 100000, 200,000, 300,000, 400,000,500,000, 600,000, 700,000, 800,000, 900,000, 1000000,2000000, 3000000, 4000000, 5000000, 6000000,7000000, 8000000, 9000000, 10000000, 20000000,50000000, or 100000000 different barcodes per parti-tion. Barcodes may be partitioned such that each partitioncontains about 1-5, 5-10, 10-50, 50-100, 100-1000,1000-10000, 10000-100000, 100000-1000000,10000-1000000, 10000-10000000, or10000-100000000 different barcodes per partition. Insome cases, partitioned different barcodes may be cou-pled to a bead, such as, for example, a gel bead. In somecases, the partitions are fluidic droplets.[0181] The number of partitions employed to partitionbarcodes or different sets of barcodes may vary, for ex-ample, depending on the application and/or the numberof different barcodes or different sets of barcodes to bepartitioned. For example, the number of partitions em-ployed to partition barcodes or different sets of barcodesmay be about 5, 10, 50, 100, 250, 500, 750, 1000, 1500,2000, 2500, 5000, 7500, or 10,000, 20000, 30000,40000, 50000, 60000, 70000, 80000, 90000, 100,000,200000, 300000, 400000, 500000, 600000, 700000,800000, 900000, 1,000,000, 2000000, 3000000,4000000, 5000000, 10000000, 20000000 or more. Thenumber of partitions employed to partition barcodes ordifferent sets of barcodes may be at least about 5, 10,50, 100, 250, 500, 750, 1000, 1500, 2000, 2500, 5000,7500, 10,000, 20000, 30000, 40000, 50000, 60000,70000, 80000, 90000, 100000, 200000, 300000,400000, 500000, 600000, 700000, 800000, 900000,1000000, 2000000, 3000000, 4000000, 5000000,10000000, 20000000 or more. The number of partitionsemployed to partition barcodes or different sets of bar-codes may be less than about 5, 10, 50, 100, 250, 500,750, 1000, 1500, 2000, 2500, 5000, 7500, 10,000,20000, 30000, 40000, 50000, 60000, 70000, 80000,90000, 100000, 200000, 300000, 400000, 500000,600000, 700000, 800000, 900000, 1000000, 2000000,3000000, 4000000, 5000000, 10000000, or 20000000.The number of partitions employed to partition barcodesmay be about 5-10000000, 5-5000000, 5-1,000,000,10-10,000, 10-5,000, 10-1,000, 1,000-6,000,1,000-5,000, 1,000-4,000, 1,000-3,000, or 1,000-2,000.In some cases, the partitions may be fluidic droplets.
[0182] As described above, different barcodes or dif-ferent sets of barcodes (e.g., each set comprising a plu-rality of identical barcodes or different barcodes) may bepartitioned such that each partition generally comprisesa different barcode or different barcode set. In some cas-es, each partition may comprise a different set of identicalbarcodes, such as an identical set of barcodes coupledto a bead (e.g., a gel bead). Where different sets of iden-tical barcodes are partitioned, the number of identicalbarcodes per partition may vary. For example, about100,000 or more different sets of identical barcodes (e.g.,a set of identical barcodes attached to a bead) may bepartitioned across about 100,000 or more different par-titions, such that each partition comprises a different setof identical barcodes (e.g., each partition comprises abead coupled to a different set of identical barcodes). Ineach partition, the number of identical barcodes per setof barcodes may be about 1,000,000 or more identicalbarcodes (e.g., each partition comprises 1,000,000 ormore identical barcodes coupled to one or more beads).In some cases, the number of different sets of barcodesmay be equal to or substantially equal to the number ofpartitions or may be less than the number of partitions.Any suitable number of different barcodes or differentbarcode sets, number of barcodes per partition, andnumber of partitions may be combined. Thus, as will beappreciated, any of the above-described different num-bers of barcodes may be provided with any of the above-described barcode densities per partition, and in any ofthe above-described numbers of partitions.
Microfluidic Devices and Droplets
[0183] In some cases, this disclosure provides devicesfor making beads and for combining beads (or other typesof partitions) with samples, e.g., for co-partitioning sam-ple components and beads. Such a device may be amicrofluidic device (e.g., a droplet generator). The devicemay be formed from any suitable material. In some ex-amples, a device may be formed from a material selectedfrom the group consisting of fused silica, soda lime glass,borosilicate glass, poly(methyl methacrylate) PMMA,PDMS, sapphire, silicon, germanium, cyclic olefin copol-ymer, polyethylene, polypropylene, polyacrylate, poly-carbonate, plastic, thermosets, hydrogels, thermoplas-tics, paper, elastomers, and combinations thereof.[0184] A device may be formed in a manner that it com-prises channels for the flow of fluids. Any suitable chan-nels may be used. In some cases, a device comprisesone or more fluidic input channels (e.g., inlet channels)and one or more fluidic outlet channels. In some embod-iments, the inner diameter of a fluidic channel may beabout 10mm, 20mm, 30mm, 40mm, 50mm, 60mm, 65mm,70mm, 75mm, 80mm, 85mm, 90mm, 100mm, 125mm, or150mm. In some cases, the inner diameter of a fluidicchannel may be more than 10mm, 20mm, 30mm, 40mm,50mm, 60mm, 65mm, 70mm, 75mm, 80mm, 85mm, 90mm,100mm, 125mm, 150mm or more. In some embodiments,
67 68

EP 3 013 957 B1
36
5
10
15
20
25
30
35
40
45
50
55
the inner diameter of a fluidic channel may be less thanabout 10mm, 20mm, 30mm, 40mm, 50mm, 60mm, 65mm,70mm, 75mm, 80mm, 85mm, 90mm, 100mm, 125mm, or150mm. Volumetric flow rates within a fluidic channel maybe any flow rate known in the art.[0185] As described elsewhere herein, the microfluidicdevice may be utilized to form beads by forming a fluidicdroplet comprising one or more gel precursors, one ormore crosslinkers, optionally an initiator, and optionallyan aqueous surfactant. The fluidic droplet may be sur-rounded by an immiscible continuous fluid, such as anoil, which may further comprise a surfactant and/or anaccelerator.[0186] In some embodiments, the microfluidic devicemay be used to combine beads (e.g., barcoded beadsor other type of first partition, including any suitable typeof partition described herein) with sample (e.g., a sampleof nucleic acids) by forming a fluidic droplet (or other typeof second partition, including any suitable type of partitiondescribed herein) comprising both the beads and thesample. The fluidic droplet may have an aqueous coresurrounded by an oil phase, such as, for example, aque-ous droplets within a water-in-oil emulsion. The fluidicdroplet may contain one or more barcoded beads, a sam-ple, amplification reagents, and a reducing agent. Insome cases, the fluidic droplet may include one or moreof water, nuclease-free water, acetonitrile, beads, gelbeads, polymer precursors, polymer monomers, polyacr-ylamide monomers, acrylamide monomers, degradablecrosslinkers, non-degradable crosslinkers, disulfide link-ages, acrydite moieties, PCR reagents, primers,polymerases, barcodes, polynucleotides, oligonucle-otides, nucleotides, DNA, RNA, peptide polynucleotides,complementary DNA (cDNA), double stranded DNA(dsDNA), single stranded DNA (ssDNA), plasmid DNA,cosmid DNA, chromosomal DNA, genomic DNA, viralDNA, bacterial DNA, mtDNA (mitochondrial DNA), mR-NA, rRNA, tRNA, nRNA, siRNA, snRNA, snoRNA, scaR-NA, microRNA, dsRNA, probes, dyes, organics, emulsi-fiers, surfactants, stabilizers, polymers, aptamers, reduc-ing agents, initiators, biotin labels, fluorophores, buffers,acidic solutions, basic solutions, light-sensitive enzymes,pH-sensitive enzymes, aqueous buffer, oils, salts, deter-gents, ionic detergents, non-ionic detergents, and thelike. In summary, the composition of the fluidic dropletwill vary depending on the particular processing needs.[0187] The fluidic droplets may be of uniform size orheterogeneous size. In some cases, the diameter of afluidic droplet may be about 1mm, 5mm, 10mm, 20mm,30mm, 40mm, 45mm, 50mm, 60mm, 65mm, 70mm, 75mm,80mm, 90mm, 100mm, 250mm, 500mm, or 1mm. In somecases, a fluidic droplet may have a diameter of at leastabout 1mm, 5mm, 10mm, 20mm, 30mm, 40mm, 45mm,50mm, 60mm, 65mm, 70mm, 75mm, 80mm, 90mm,100mm, 250mm, 500mm, 1mm or more. In some cases,a fluidic droplet may have a diameter of less than about1mm, 5mm, 10mm, 20mm, 30mm, 40mm, 45mm, 50mm,60mm, 65mm, 70mm, 75mm, 80mm, 90mm, 100mm,
250mm, 500mm, or 1mm. In some cases, fluidic dropletmay have a diameter in the range of about 40-75mm,30-75mm, 20-75mm, 40-85mm, 40-95mm, 20-100mm,10-100mm, 1-100mm, 20-250mm, or 20-500mm.[0188] In some embodiments, the device may com-prise one or more intersections of two or more fluid inputchannels. For example, the intersection may be a fluidiccross. The fluidic cross may comprise two or more fluidicinput channels and one or more fluidic outlet channels.In some cases, the fluidic cross may comprise two fluidicinput channels and two fluidic outlet channels. In othercases, the fluidic cross may comprise three fluidic inputchannels and one fluidic outlet channel. In some cases,the fluidic cross may form a substantially perpendicularangle between two or more of the fluidic channels formingthe cross.[0189] In some cases, a microfluidic device may com-prise a first and a second input channel that meet at ajunction that is fluidly connected to an output channel. Insome cases, the output channel may be, for example,fluidly connected to a third input channel at a junction. Insome cases, a fourth input channel may be included andmay intersect the third input channel and outlet channelat a junction. In some cases, a microfluidic device maycomprise first, second, and third input channels, whereinthe third input channel intersects the first input channel,the second input channel, or a junction of the first inputchannel and the second input channel.[0190] As described elsewhere herein, the microfluidicdevice may be used to generate gel beads from a liquid.For example, in some embodiments, an aqueous fluidcomprising one or more gel precursors, one or morecrosslinkers and optionally an initiator, optionally anaqueous surfactant, and optionally an alcohol within afluidic input channel may enter a fluidic cross. Within asecond fluidic input channel, an oil with optionally a sur-factant and an accelerator may enter the same fluidiccross. Both aqueous and oil components may be mixedat the fluidic cross causing aqueous fluidic droplets toform within the continuous oil phase. Gel precursors with-in fluidic droplets exiting the fluidic cross may polymerizeforming beads.[0191] As described elsewhere herein, the microfluidicdevice (e.g., a droplet generator) may be used to combinesample with beads (e.g., a library of barcoded beads) aswell as an agent capable of degrading the beads (e.g.,reducing agent if the beads are linked with disulfidebonds), if desired. In some embodiments, a sample (e.g.,a sample of nucleic acids) may be provided to a first fluidicinput channel that is fluidly connected to a first fluidiccross (e.g., a first fluidic junction). Pre-formed beads(e.g., barcoded beads, degradable barcoded beads) maybe provided to a second fluidic input channel that is alsofluidly connected to the first fluidic cross, where the firstfluidic input channel and second fluidic input channelmeet. The sample and beads may be mixed at the firstfluidic cross to form a mixture (e.g., an aqueous mixture).In some cases, a reducing agent may be provided to a
69 70

EP 3 013 957 B1
37
5
10
15
20
25
30
35
40
45
50
55
third fluidic input channel that is also fluidly connected tothe first fluidic cross and meets the first and second fluidicinput channel at the first fluidic cross. The reducing agentcan then be mixed with the beads and sample in the firstfluidic cross. In other cases, the reducing agent may bepremixed with the sample and/or the beads before en-tering the microfluidic device such that it is provided tothe microfluidic device through the first fluidic input chan-nel with the sample and/or through the second fluidicinput channel with the beads. In other cases, no reducingagent may be added.[0192] In some embodiments, the sample and beadmixture may exit the first fluidic cross through a first outletchannel that is fluidly connected to the first fluidic cross(and, thus, any fluidic channels forming the first fluidiccross). The mixture may be provided to a second fluidiccross (e.g., a second fluidic junction) that is fluidly con-nected to the first outlet channel. In some cases, an oil(or other suitable immiscible) fluid may enter the secondfluidic cross from one or more separate fluidic input chan-nels that are fluidly connected to the second fluidic cross(and, thus, any fluidic channels forming the cross) andthat meet the first outlet channel at the second fluidiccross. In some cases, the oil (or other suitable immisciblefluid) may be provided in one or two separate fluidic inputchannels fluidly connected to the second fluidic cross(and, thus, the first outlet channel) that meet the first out-let channel and each other at the second fluidic cross.Both components, the oil and the sample and bead mix-ture, may be mixed at the second fluidic cross. This mix-ing partitions the sample and bead mixture into a pluralityof fluidic droplets (e.g., aqueous droplets within a water-in-oil emulsion), in which at least a subset of the dropletsthat form encapsulate a barcoded bead (e.g., a gel bead).The fluidic droplets that form may be carried within theoil through a second fluidic outlet channel exiting fromthe second fluidic cross. In some cases, fluidic dropletsexiting the second outlet channel from the second fluidiccross may be partitioned into wells for further processing(e.g., thermocycling).[0193] In many cases, it will be desirable to control theoccupancy rate of resulting droplets (or second parti-tions) with respect to beads (or first partitions). Such con-trol is described in, for example, U.S. Provisional patentapplication No. 61/977,804, filed April 4, 2014. In general,the droplets (or second partitions) will be formed suchthat at least 50%, 60%, 70%, 80%, 90% or more droplets(or second partitions) contain no more than one bead (orfirst partition). Additionally, or alternatively, the droplets(or second partitions) will be formed such that at least50%, 60%, 70%, 80%, 90% or more droplets (or secondpartitions) include exactly one bead (or first partition). Insome cases, the resulting droplets (or second partitions)may each comprise, on average, at most about one, two,three, four, five, six, seven, eight, nine, ten, eleven,twelve, thirteen, fourteen, fifteen, sixteen, seventeen,eighteen, nineteen, or twenty beads (or first partitions).In some cases, the resulting droplets (or second parti-
tions) may each comprise, on average, at least aboutone, two, three, four, five, six, seven, eight, nine, ten,eleven, twelve, thirteen, fourteen, fifteen, sixteen, sev-enteen, eighteen, nineteen, twenty, or more beads (orfirst partitions).[0194] In some embodiments, samples may be pre-mixed with beads (e.g., degradable beads) comprisingbarcodes and any other reagent (e.g., reagents neces-sary for sample amplification, a reducing agent, etc.) priorto entry of the mixture into a microfluidic device to gen-erate an aqueous reaction mixture. Upon entry of theaqueous mixture to a fluidic device, the mixture may flowfrom a first fluidic input channel and into a fluidic cross.In some cases, an oil phase may enter the fluidic crossfrom a second fluidic input channel (e.g., a fluidic channelperpendicular to or substantially perpendicular to the firstfluidic input channel) also fluidly connected to the fluidiccross. The aqueous mixture and oil may be mixed at thefluidic cross, such that an emulsion (e.g. a gel-water-oilemulsion) forms. The emulsion can comprise a pluralityof fluidic droplets (e.g., droplets comprising the aqueousreaction mixture) in the continuous oil phase. In somecases, each fluidic droplet may comprise a single bead(e.g., a gel bead attached to a set of identical barcodes),an aliquot of sample, and an aliquot of any other reagents(e.g., reducing agents, reagents necessary for amplifica-tion of the sample, etc.). In some cases, though, a fluidicdroplet may comprise a plurality of beads. Upon dropletformation, the droplet may be carried via the oil continu-ous phase through a fluidic outlet channel exiting fromthe fluidic cross. Fluidic droplets exiting the outlet channelmay be partitioned into wells for further processing (e.g.,thermocycling).[0195] In cases where a reducing agent may be addedto the sample prior to entering the microfluidic device ormay be added at the first fluidic cross, the fluidic dropletsformed at the second fluidic cross may contain the re-ducing agent. In this case, the reducing agent may de-grade or dissolve the beads contained within the fluidicdroplet as the droplet travels through the outlet channelleaving the second fluidic cross.[0196] In some embodiments, a microfluidic devicemay contain three discrete fluidic crosses in parallel. Flu-idic droplets may be formed at any one of the three fluidiccrosses. Sample and beads may be combined within anyone of the three fluidic crosses. A reducing agent maybe added at any one of the three fluidic crosses. An oilmay be added at any one of the three fluidic crosses.[0197] The methods, compositions, devices, and kitsof this disclosure may be used with any suitable oil. Insome embodiments, an oil may be used to generate anemulsion. The oil may comprise fluorinated oil, silicon oil,mineral oil, vegetable oil, and combinations thereof.[0198] In some embodiments, the aqueous fluid withinthe microfluidic device may also contain an alcohol. Forexample, an alcohol may be glycerol, ethanol, methanol,isopropyl alcohol, pentanol, ethane, propane, butane,pentane, hexane, and combinations thereof. The alcohol
71 72

EP 3 013 957 B1
38
5
10
15
20
25
30
35
40
45
50
55
may be present within the aqueous fluid at about 5%,6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%,17%, 18%, 19%, or 20% (v/v). In some cases, the alcoholmay be present within the aqueous fluid at least about5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%,16%, 17%, 18%, 19%, 20% or more (v/v). In some cases,the alcohol may be present within the aqueous fluid forless than about 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%,13%, 14%, 15%, 16%, 17%, 18%, 19%, or 20% (v/v).[0199] In some embodiments, the oil may also containa surfactant to stabilize the emulsion. For example, asurfactant may be a fluorosurfactant, Krytox lubricant,Krytox FSH, an engineered fluid, HFE-7500, a siliconecompound, a silicon compound containing PEG, such asbis krytox peg (BKP). The surfactant may be present atabout 0.1%, 0.5%, 1%, 1.1%, 1.2%, 1.3%, 1.4%, 1.5%,1.6%, 1.7%, 1.8%, 1.9%, 2%, 5%, or 10% (w/w). In somecases, the surfactant may be present at least about 0.1%,0.5%, 1%, 1.1%, 1.2%, 1.3%, 1.4%, 1.5%, 1.6%, 1.7%,1.8%, 1.9%, 2%, 5%, 10% (w/w) or more. In some cases,the surfactant may be present for less than about 0.1%,0.5%, 1%, 1.1%, 1.2%, 1.3%, 1.4%, 1.5%, 1.6%, 1.7%,1.8%, 1.9%, 2%, 5%, or 10% (w/w).[0200] In some embodiments, an accelerator and/orinitiator may be added to the oil. For example, an accel-erator may be Tetramethylethylenediamine (TMEDA orTEMED). In some cases, an initiator may be ammoniumpersulfate or calcium ions. The accelerator may bepresent at about 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%,0.7%, 0.8%, 0.9%, 1%, 1.1%, 1.2%, 1.3%, 1.4%, 1.5%,1.6%, 1.7%, 1.8%, 1.9%, or 2% (v/v). In some cases, theaccelerator may be present at least about 0.1%, 0.2%,0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 1.1%,1.2%, 1.3%, 1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, or 2%(v/v) or more. In some cases, the accelerator may bepresent for less than about 0.1%, 0.2%, 0.3%, 0.4%,0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 1.1%, 1.2%, 1.3%,1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, or 2% (v/v).
V. Amplification
[0201] DNA amplification is a method for creating mul-tiple copies of small or long segments of DNA. The meth-ods, compositions, devices, and kits of this disclosuremay use DNA amplification to attach one or more desiredoligonucleotide sequences to individual beads, such asa barcode sequence or random N-mer sequence. DNAamplification may also be used to prime and extend alonga sample of interest, such as genomic DNA, utilizing arandom N-mer sequence, in order to produce a fragmentof the sample sequence and couple the barcode associ-ated with the primer to that fragment.[0202] For example, a nucleic acid sequence may beamplified by co-partitioning a template nucleic acid se-quence and a bead comprising a plurality of attachedoligonucleotides (e.g., releasably attached oligonucle-otides) into a partition (e.g., a droplet of an emulsion, amicrocapsule, or any other suitable type of partition, in-
cluding a suitable type of partition described elsewhereherein). The attached oligonucleotides can comprise aprimer sequence (e.g., a variable primer sequence suchas, for example, a random N-mer, or a targeted primersequence such as, for example, a targeted N-mer) thatis complementary to one or more regions of the templatenucleic acid sequence and, in addition, may also com-prise a common sequence (e.g., such as a barcode se-quence). The primer sequence can be annealed to thetemplate nucleic acid sequence and extended (e.g., in aprimer extension reaction or any other suitable nucleicacid amplification reaction) to produce one or more firstcopies of at least a portion of the template nucleic acid,such that the one or more first copies comprises the prim-er sequence and the common sequence. In cases wherethe oligonucleotides comprising the primer sequence arereleasably attached to the bead, the oligonucleotidesmay be released from the bead prior to annealing theprimer sequence to the template nucleic acid sequence.Moreover, in general, the primer sequence may be ex-tended via a polymerase enzyme (e.g., a strand displac-ing polymerase enzyme as described elsewhere herein,an exonuclease deficient polymerase enzyme as de-scribed elsewhere herein, or any other type of suitablepolymerase, including a type of polymerase describedelsewhere herein) that is also provided in the partition.Furthermore, the oligonucleotides releasably attached tothe bead may be exonuclease resistant and, thus, maycomprise one or more phosphorothioate linkages as de-scribed elsewhere herein. In some cases, the one ormore phosphorothioate linkages may comprise a phos-phorothioate linkage at a terminal internucleotide linkagein the oligonucleotides.[0203] In some cases, after the generation of the oneor more first copies, the primer sequence can be an-nealed to one or more of the first copies and the primersequence again extended to produce one or more sec-ond copies. The one or more second copies can comprisethe primer sequence, the common sequence, and mayalso comprise a sequence complementary to at least aportion of an individual copy of the one or more first cop-ies, and/or a sequence complementary to the variableprimer sequence. The aforementioned steps may be re-peated for a desired number of cycles to produce ampli-fied nucleic acids.[0204] The oligonucleotides described above maycomprise a sequence segment that is not copied duringan extension reaction (such as an extension reaction thatproduces the one or more first or second copies de-scribed above). As described elsewhere herein, such asequence segment may comprise one or more uracil con-taining nucleotides and may also result in the generationof amplicons that form a hairpin (or partial hairpin) mol-ecule under annealing conditions.[0205] In another example, a plurality of different nu-cleic acids can be amplified by partitioning the differentnucleic acids into separate first partitions (e.g., dropletsin an emulsion) that each comprise a second partition
73 74

EP 3 013 957 B1
39
5
10
15
20
25
30
35
40
45
50
55
(e.g., beads, including a type of bead described else-where herein). The second partition may be releasablyassociated with a plurality of oligonucleotides. The sec-ond partition may comprise any suitable number of oli-gonucleotides (e.g., more than 1,000 oligonucleotides,more than 10,000 oligonucleotides, more than 100,000oligonucleotides, more than 1,000,000 oligonucleotides,more than 10,000,000 oligonucleotides, or any othernumber of oligonucleotides per partition described here-in). Moreover, the second partitions may comprise anysuitable number of different barcode sequences (e.g., atleast 1,000 different barcode sequences, at least 10,000different barcode sequences, at least 100,000 differentbarcode sequences, at least 1,000,000 different barcodesequences, at least 10,000,000 different barcode se-quence, or any other number of different barcode se-quences described elsewhere herein).[0206] Furthermore, the plurality of oligonucleotidesassociated with a given second partition may comprisea primer sequence (e.g., a variable primer sequence, atargeted primer sequence) and a common sequence(e.g., a barcode sequence). Moreover, the plurality ofoligonucleotides associated with different second parti-tions may comprise different barcode sequences. Oligo-nucleotides associated with the plurality of second par-titions may be released into the first partitions. Followingrelease, the primer sequences within the first partitionscan be annealed to the nucleic acids within the first par-titions and the primer sequences can then be extendedto produce one or more copies of at least a portion of thenucleic acids with the first partitions. In general, the oneor more copies may comprise the barcode sequencesreleased into the first partitions.
Amplification within Droplets and Sample Indexing
[0207] Nucleic acid (e.g., DNA) amplification may beperformed on contents within fluidic droplets. As de-scribed herein, fluidic droplets may contain oligonucle-otides attached to beads. Fluidic droplets may furthercomprise a sample. Fluidic droplets may also comprisereagents suitable for amplification reactions which mayinclude Kapa HiFi Uracil Plus, modified nucleotides, na-tive nucleotides, uracil containing nucleotides, dTTPs,dUTPs, dCTPs, dGTPs, dATPs, DNA polymerase, Taqpolymerase, mutant proof reading polymerase, 9 de-grees North, modified (NEB), exo (-), exo (-) Pfu, DeepVent exo (-), Vent exo (-), and acyclonucleotides (acyN-TPS).[0208] Oligonucleotides attached to beads within a flu-idic droplet may be used to amplify a sample nucleic acidsuch that the oligonucleotides become attached to thesample nucleic acid. The sample nucleic acids may com-prise virtually any nucleic acid sought to be analyzed,including, for example, whole genomes, exomes, ampli-cons, targeted genome segments e.g., genes or genefamilies, cellular nucleic acids, circulating nucleic acids,and the like, and, as noted above, may include DNA (in-
cluding gDNA, cDNA, mtDNA, etc.) RNA (e.g., mRNA,rRNA, total RNA, etc.). Preparation of such nucleic acidsfor barcoding may generally be accomplished by meth-ods that are readily available, e.g., enrichment or pull-down methods, isolation methods, amplification methodsetc. In order to amplify a desired sample, such as gDNA,the random N-mer sequence of an oligonucleotide withinthe fluidic droplet may be used to prime the desired targetsequence and be extended as a complement of the targetsequence. In some cases, the oligonucleotide may bereleased from the bead in the droplet, as described else-where herein, prior to priming. For these priming and ex-tension processes, any suitable method of DNA amplifi-cation may be utilized, including polymerase chain reac-tion (PCR), digital PCR, reverse-transcription PCR, mul-tiplex PCR, nested PCR, overlap-extension PCR, quan-titative PCR, multiple displacement amplification (MDA),or ligase chain reaction (LCR). In some cases, amplifi-cation within fluidic droplets may be performed until acertain amount of sample nucleic acid comprising bar-code may be produced. In some cases, amplification maybe performed for about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12,13, 14, 15, 16, 17, 18, 19, or 20 cycles. In some cases,amplification may be performed for more than about 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18,19, 20 cycles, or more. In some cases, amplification maybe performed for less than about 2, 3, 4, 5, 6, 7, 8, 9, 10,11, 12, 13, 14, 15, 16, 17, 18, 19, or 20 cycles.[0209] An exemplary amplification and barcoding proc-ess as described herein, is schematically illustrated inFig 38. As shown, oligonucleotides that include a bar-code sequence are co-partitioned in, e.g., a droplet 3802in an emulsion, along with a sample nucleic acid 3804.As noted elsewhere herein, the oligonucleotides 3808may be provided on a bead 3806 that is co-partitionedwith the sample nucleic acid 3804, which oligonucle-otides are preferably releasable from the bead 3806, asshown in panel A. The oligonucleotides 3808 include abarcode sequence 3812, in addition to one or more func-tional sequences, e.g., sequences 3810, 3814 and 3816.For example, oligonucleotide 3808 is shown as compris-ing barcode sequence 3812, as well as sequence 3810that may function as an attachment or immobilization se-quence for a given sequencing system, e.g., a P5 se-quence used for attachment in flow cells of an IlluminaHiseq or Miseq system. As shown, the oligonucleotidesalso include a primer sequence 3816, which may includea random or targeted N-mer for priming replication ofportions of the sample nucleic acid 3804. Also includedwithin oligonucleotide 3808 is a sequence 3814 whichmay provide a sequencing priming region, such as a"read1" or R1 priming region, that is used to primepolymerase mediated, template directed sequencing bysynthesis reactions in sequencing systems. In many cas-es, the barcode sequence 3812, immobilization se-quence 3810 and R1 sequence 3814 will be common toall of the oligonucleotides attached to a given bead. Theprimer sequence 3816 may vary for random N-mer prim-
75 76

EP 3 013 957 B1
40
5
10
15
20
25
30
35
40
45
50
55
ers, or may be common to the oligonucleotides on a givenbead for certain targeted applications.[0210] Based upon the presence of primer sequence3816, the oligonucleotides are able to prime the samplenucleic acid as shown in panel B, which allows for exten-sion of the oligonucleotides 3808 and 3808a usingpolymerase enzymes and other extension reagents alsoco-partitioned with the bead 3806 and sample nucleicacid 3804. As described elsewhere herein, thesepolymerase enzymes may include thermostablepolymerases, e.g., where initial denaturation of doublestranded sample nucleic acids within the partitions is de-sired. Alternatively, denaturation of sample nucleic acidsmay precede partitioning, such that single stranded tar-get nucleic acids are deposited into the partitions, allow-ing the use of non-thermostable polymerase enzymes,e.g., Klenow, phi29, Pol 1, and the like, where desirable.As shown in panel C, following extension of the oligonu-cleotides that, for random N-mer primers, would annealto multiple different regions of the sample nucleic acid3804; multiple overlapping complements or fragments ofthe nucleic acid are created, e.g., fragments 3818 and3820. Although including sequence portions that arecomplementary to portions of sample nucleic acid, e.g.,sequences 3822 and 3824, these constructs are gener-ally referred to herein as comprising fragments of thesample nucleic acid 3804, having the attached barcodesequences. In some cases, it may be desirable to artifi-cially limit the size of the replicate fragments that areproduced in order to maintain manageable fragment siz-es from the first amplification steps. In some cases, thismay be accomplished by mechanical means, as de-scribed above, e.g., using fragmentation systems like aCovaris system, or it may be accomplished by incorpo-rating random extension terminators, e.g., at low concen-trations, to prevent the formation of excessively long frag-ments.[0211] These fragments may then be subjected to se-quence analysis, or they may be further amplified in theprocess, as shown in panel D. For example, additionaloligonucleotides, e.g., oligonucleotide 3808b, also re-leased from bead 3806, may prime the fragments 3818and 3820. This shown in for fragment 3818. In particular,again, based upon the presence of the random N-merprimer 3816b in oligonucleotide 3808b (which in manycases will be different from other random N-mers in agiven partition, e.g., primer sequence 3816), the oligo-nucleotide anneals with the fragment 3818, and is ex-tended to create a complement 3826 to at least a portionof fragment 3818 which includes sequence 3828, thatcomprises a duplicate of a portion of the sample nucleicacid sequence. Extension of the oligonucleotide 3808bcontinues until it has replicated through the oligonucle-otide portion 3808 of fragment 3818. As noted elsewhereherein, and as illustrated in panel D, the oligonucleotidesmay be configured to prompt a stop in the replication bythe polymerase at a desired point, e.g., after replicatingthrough sequences 3816 and 3814 of oligonucleotide
3808 that is included within fragment 3818. As describedherein, this may be accomplished by different methods,including, for example, the incorporation of different nu-cleotides and/or nucleotide analogues that are not capa-ble of being processed by the polymerase enzyme used.For example, this may include the inclusion of uracil con-taining nucleotides within the sequence region 3812 tocause a non-uracil tolerant polymerase to cease replica-tion of that region. As a result, a fragment 3826 is createdthat includes the full-length oligonucleotide 3808b at oneend, including the barcode sequence 3812, the attach-ment sequence 3810, the R1 primer region 3814, andthe random n-mer sequence 3816b. At the other end ofthe sequence will be included the complement 3816’ tothe random n-mer of the first oligonucleotide 3808, aswell as a complement to all or a portion of the R1 se-quence, shown as sequence 3814’. The R1 sequence3814 and its complement 3814’ are then able to hybridizetogether to form a partial hairpin structure 3828. As willbe appreciated because the random-n-mers differamong different oligonucleotides, these sequences andtheir complements would not be expected to participatein hairpin formation, e.g., sequence 3816’, which is thecomplement to random N-mer 3816, would not be ex-pected to be complementary to random n-mer sequence3816b. This would not be the case for other applications,e.g., targeted primers, where the N-mers may be com-mon among oligonucleotides within a given partition.[0212] By forming these partial hairpin structures, it al-lows for the removal of first level duplicates of the samplesequence from further replication, e.g., preventing itera-tive copying of copies. The partial hairpin structure alsoprovides a useful structure for subsequent processing ofthe created fragments, e.g. ,fragment 3826.[0213] Following attachment of the barcode to the sam-ple, additional amplification steps (e.g. PCR) may be per-formed to amplify the barcoded fragments prior to se-quencing, as well as to optionally add additional function-al sequences to those barcoded fragments, e.g., addi-tional primer binding sites (e.g. Read2 sequence primer,Index primer) that is compatible with a sequencing device(e.g. Illumina MiSeq) and optionally, one or more addi-tional barcode sequences (e.g., see Fig 14C), as well asother functional sequences, e.g., additional immobiliza-tion sequences or their complements, e.g., P7 sequenc-es. In some cases, an additional barcode sequence mayserve as a sample index, with the original barcode andsample index permitting multiplexed sequencing (e.g.,simultaneous molecular tagging and sample identifica-tion). The original barcode can be used during sequenc-ing to align a sequence read corresponding to the nucleicacid molecule associated with the barcode (e.g., identi-fied via the barcode). A different sample index can beincluded in sequencer-ready products generated fromeach different sample. Thus, the sample index can beused during sequencing for identifying the sample towhich a particular sequence read belongs and multiplex-ing can be achieved.
77 78

EP 3 013 957 B1
41
5
10
15
20
25
30
35
40
45
50
55
[0214] In some cases, a sample index can be addedto a sample nucleic acid after the addition of the originalbarcode to the sample nucleic acid, with or without theuse of partitions or the generation of additional partitions.In some cases, the sample index is added in bulk. Insome cases, the addition of a sample index to a samplenucleic acid may occur prior to the addition of a barcodeto the sample nucleic acid. In some cases, the additionof a sample index to a sample nucleic acid may occursimultaneous to or in parallel to the addition of a sampleindex to the sample nucleic acid.[0215] In some cases, a sample index may be addedto a sample nucleic acid after addition of a barcode se-quence to the sample nucleic acid. For example, as de-scribed elsewhere herein, amplification methods may beused to attach a barcode sequence and other sequences(e.g., P5, R1, etc.) to a sample nucleic acid. In somecases, a random amplification scheme, such as PartialHairpin Amplification for Sequencing (PHASE - as de-scribed elsewhere herein), for example, may aid in at-taching a barcode sequence and other sequences to asample nucleic acid. In one example, a plurality of prim-ers, each comprising a different random N-mer, a se-quencer attachment or immobilization site (e.g., P5), abarcode sequence (e.g., an identical barcode sequence),and a sequencing primer binding site (e.g., R1) are usedto randomly prime and amplify a sample nucleic acid.Any of the sequencer primer binding site, the barcodesequence, and/or sequencing primer binding site maycomprise uracil containing nucleotides. The primer mayalso include an oligonucleotide blocker hybridized to theprimer at one or more sequences of the primer to ensurethat priming of the sample nucleic acid occurs only viathe random N-mer. A schematic representation of an ex-ample primer is as follows (oligonucleotide blocker notshown):P5-Barcode-R1-RandomNMer[0216] Random priming of the sample nucleic acid andmultiple rounds of amplification can generate ampliconscomprising a portion of the sample nucleic acid linked atone end to the sequencer attachment or immobilizationsite (e.g., P5), the barcode, the sequencing primer bind-ing site (e.g., R1), and the random N-mer. At its otherend, the portion of the sample nucleic acid can be linkedto a region (e.g., R1c, or R1c partial) that is complemen-tary or partially complementary to the sequencing primerbinding site. A schematic representation of an examplesequence (in a linear configuration) is as follows:
P5-Barcode-R1-RandomNmer-Insert-R1c,partial
where "Insert" corresponds to the portion of the sam-ple nucleic acid copied during amplification. The se-quencing primer binding site (e.g., R1) and its partialcomplement (e.g., R1c, partial) at the opposite endof the portion of the copied sample nucleic acid (In-sert) can intramolecularly hybridize to form a partialhairpin structure as described elsewhere herein.
[0217] Following creation of the barcoded fragmentsof the sample nucleic acid, and as noted above, it maybe desirable to further amplify those fragments, as wellas attach additional functional sequences to the ampli-fied, barcoded fragments. This amplification may be car-ried out using any suitable amplification process, includ-ing, e.g., PCR, LCR, linear amplification, or the like. Typ-ically, this amplification may be initiated using targetedprimers that prime against the known terminal sequencesin the created fragments, e.g., priming against one orboth of the attachment sequence 3810, in Fig 38, andsequence 3814’. Further by incorporating additionalfunctional sequences within these primers, e.g., addition-al attachment sequences such as P7, additional se-quencing primers, e.g., a read 2 or R2 priming sequence,as well as optional sample indexing sequences, one canfurther configure the amplified barcoded fragments.[0218] By way of example, following generation of par-tial hairpin amplicons, intramolecular hybridization of thepartial hairpin amplicons can be disrupted by contactingthe partial hairpin amplicons with a primer that is com-plementary to the duplex portion of the hairpin, e.g., se-quence 3814’, in order to disrupt the hairpin and primeextension along the hairpin structure. In many cases, itwill be desirable to provide these primers with a strongerhybridization affinity than the hairpin structure in order topreferentially disrupt that hairpin. As such, in at least oneexample, the primer comprises a locked nucleic acid(LNAs) or locked nucleic acid nucleotides. LNAs includenucleotides where the ribonucleic acid base comprisesa molecular bridge connecting the 2’-oxygen and 4’-car-bon of the nucleotide’s ribose moiety. LNAs generallyhave higher melting temperatures and lower hybridiza-tion energies. Accordingly, LNAs can favorably competewith intramolecular hybridization of the partial hairpin am-plicons by binding to any of the hybridized sequences ofa partial hairpin amplicon. Subsequent amplification ofthe disrupted amplicons via primers comprising LNAsand other primers can generate linear products compris-ing any additional sequences (including a sample index)to be added to the sequence.[0219] For the example partial hairpin P5-Barcode-R1-RandomNmer-Insert-R1c,partial configuration de-scribed above, the partial hairpin can be contacted witha primer comprising LNAs and a sequence complemen-tary to R1c,partial (e.g., see Fig 14C). The primer mayalso comprise the complement of any additional se-quence to be added to the construct. For example, theadditional sequence (e.g., R2partial) may be a sequencethat, when coupled to Rlc,partial, generates an additionalsequencing primer binding site (e.g., R2). Hybridizationof the primer with the partial hairpin can disrupt the partialhairpin’s intramolecular hybridization and linearize theconstruct. Hybridization may occur, for example, suchthat the primer hybridizes with R1c,partial via its comple-mentary sequence (e.g., see Fig 14C). Extension of theprimer can generate a construct comprising the primerlinked to a sequence complementary to the linearized
79 80

EP 3 013 957 B1
42
5
10
15
20
25
30
35
40
45
50
55
partial hairpin amplicon. A schematic of an example con-struct is as follows:
P5c-Barcode,c-R1c-RandomNmer,c-Insert,c-R1,partial-R2partial,c
where P5c corresponds to the complement of P5,Barcode,c corresponds to the complement of thebarcode, RandomNmer,c corresponds to the com-plement of the random N-mer, Insert,c correspondsto the complement of the portion of the Insert, andR1,partial-R2partial,c corresponds to the comple-ment of R2.
[0220] Upon a further round of amplification with a sec-ond primer (e.g., P5, hybridizing at P5c), a linear con-struct comprising the partial hairpin amplicon sequenceand a sequence complementary to the primer can begenerated. A schematic representation of an exampleconfiguration is as follows:
P5-Barcode-R1-RandomNmer-Insert-R1c,partial-R2partial or
P5-Barcode-R1-RandomNmer-Insert-R2
where the combined sequence of R1c,partial andR2partial can correspond to an additional sequenc-ing primer binding site (e.g., R2).
[0221] Additional sequences can be added to the con-struct using additional rounds of such amplification, forhowever many additional sequences/rounds of amplifi-cation are desired. For the example P5-Barcode-R1-RandomNmer-Insert-R2 construct described above, aprimer comprising a sequence complementary to R2(e.g., R2c), the complement of a sample index sequence(e.g., SIc, SampleBarcode), and the complement of anadditional sequencer primer binding site sequence (e.g.,P7c) can be hybridized to the construct at R2, via R2c ofthe primer (e.g., see Fig 14C). Extension of the primercan generate a construct comprising the primer linked toa sequence complementary to the construct. A schematicrepresentation of an example configuration is as follows:P5c-Barcode,c-R1c-RandomNmer,c-Insert,c-R2,c-SIc-P7c[0222] Upon a further round of amplification with a sec-ond primer (e.g., P5, hybridizing at P5c), a sequencer-ready construct comprising the construct sequence anda sequence complementary to the primer can be gener-ated. A schematic representation of an example config-uration of such a sequencer-ready construct is as follows:[0223] P5-Barcode-R1-RandomNmer-Insert-R2-SampleIndex-P7As an alternative, the starting primermay comprise a barcode sequence, P7, and R2 (insteadof P5 and R1). A schematic representation of an exampleprimer is as follows:P7-Barcode-R2-RandomNmer
[0224] Using an analogous amplification scheme asdescribed above (e.g., amplification with primers com-prising LNAs, additional rounds of amplification, etc.), aninsert comprising a portion of a sample nucleic acid tobe sequenced, P5, R1, and a sample index can be addedto the primer to generate a sequencer-ready product. Aschematic representation of an example product is asfollows:P7-Barcode-R2-RandomNmer-Insert-R1-SampleIndex-P5[0225] In other cases, a sample index may be addedto a sample nucleic acid concurrently with the additionof a barcode sequence to the sample nucleic acid. Forexample, a primer used to generate a barcoded samplenucleic acid may comprise both a barcode sequence anda sample index, such that when the barcode is coupledto the sample nucleic acid, the sample index is coupledsimultaneously. The sample index may be positioned an-ywhere in the primer sequence. In some cases, the prim-er may be a primer capable of generating barcoded sam-ple nucleic acids via random amplification, such asPHASE amplification. Schematic representations of ex-amples of such primers include:
P5-Barcode-R1-SampleIndex-RandomNmer
P5-Barcode-SampleIndex-R1-RandomNmer
P5-SampleIndex-Barcode-R1-RandomNmer
[0226] Upon random priming of a sample nucleic acidwith a respective primer and amplification of the samplenucleic acid in the partition, partial hairpin ampliconscomprising a barcode sequence and a sample index se-quence can be generated. Schematic representations(shown in linear form) of examples of such partial hairpinamplicons generated from the above primers include, re-spectively:
P5-Barcode-R1-SampleIndex-RandomNmer-In-sert-R1c,partial
P5-Barcode-SampleIndex-R1-RandomNmer-In-sert-R1c,partial
P5-SampleIndex-Barcode-R1-RandomNmer-In-sert-R1c,partial
R1c, partial can intramolecularly hybridize with itscomplementary sequence in R1 to form a partial hair-pin amplicon.
[0227] By way of example, in some cases, followingthe generation of partial hairpin amplicons, additional se-quences (e.g., functional sequences like R2 and P7 se-quences) can be added to the partial hairpin amplicons,such as, for example, in bulk. In analogous fashion toamplification methods described elsewhere herein, prim-
81 82

EP 3 013 957 B1
43
5
10
15
20
25
30
35
40
45
50
55
ers that include these additional functional sequencesmay be used to prime the replication of the partial hairpinmolecule, e.g., by priming against the 5’ end of the partialhairpin, e.g., the R1c sequence, described above. Inmany cases, it will be desirable to provide a higher affinityprimer sequence, e.g., to outcompete rehybridization ofthe hairpin structure, in order to provide greater primingand replication. In such cases, tighter binding primer se-quences, e.g., that include in their sequence one or morehigher affinity nucleotide analogues, like LNAs or the like,may be used to disrupt partial hairpin amplicons and addadditional sequences to the amplicons. For example, withreference to the example described above, a primer maycomprise LNAs, a sequence complementary to R1c,par-tial and a sequence comprising the complement toR2partial, such that when the primer is extended and theresulting product further amplified via a P5 primer,R1c,partial and R2partial are joined to generate R2.Schematic representations of examples of such con-structs generated from the above primers include, re-spectively:
P5-Barcode-R1-SampleIndex-RandomNmer-In-sert-R2
P5-Barcode-SampleIndex-R1-RandomNmer-In-sert-R2
P5-SampleIndex-Barcode-R1-RandomNmer-In-sert-R2
[0228] As noted above, additional rounds of amplifica-tion cycles may be used to add additional sequences tothe constructs. For example, a primer may comprise asequence complementary to R2 and a sequence com-prising the complement to P7, such that when the primeris extended and the resulting product further amplifiedvia a P5 primer, P7 is linked to R2 and a sequencer-readyconstruct is generated. Schematic representations of ex-amples of such sequencer-ready constructs generatedfrom the above primers include, respectively:
P5-Barcode-R1-SampleIndex-RandomNmer-In-sert-R2-P7
P5-Barcode-SampleIndex-R1-RandomNmer-In-sert-R2-P7
P5-SampleIndex-Barcode-R1-RandomNmer-In-sert-R2-P7
[0229] Combining a barcode and a sample index intoa primer capable of amplifying regions of a sample nu-cleic acid (e.g., via PHASE amplification) may enableparallelization of sample indexing. Sets of primers maybe used to index nucleic acids from different samples.Each set of primers may be associated with nucleic acidmolecules obtained from a particular sample and com-
prise primers comprising a diversity of barcode sequenc-es and a common sample index sequence.[0230] In some cases, it may be desirable to attachadditional sequence segments to the 5’ end of the partialhairpin molecules described herein, not only to provideadditional functionality to the amplified fragment of thesample nucleic acid as described above, but also to en-sure more efficient subsequent processing , e.g., ampli-fication and/or sequencing, of those molecules. For ex-ample, where a partial hairpin molecule is subjected toextension reaction conditions, it may be susceptible tofilling in of the partial hairpin structure, by priming its own’filling in’ reaction through extension at the 5’ terminus.As a result, a complete hairpin structure may be createdthat is more difficult to amplify, by virtue of the greaterstability of its duplex portion. In such cases, it may bedesirable to preferentially attach additional sequencesegment(s) that is not complementary to the opposingend sequence, in order to prevent the formation of a com-plete hairpin structure. In one exemplary process, theLNA primers described above for the amplification of thepartial hairpin structures, may be provided with additionaloverhanging sequence, including, e.g., the R2 comple-mentary sequence described above, as well as poten-tially complementary sequences to other functional se-quence components, e.g., attachment sequences likeP7, sample index sequences, and the like. Subjectingthe partial hairpin and primer to the extension reactiondescribed above for amplification of that partial hairpin,will also result in extension of the partial hairpin alongthe overhanging sequence on the LNA primer. The ex-tended sequence may comprise simply a non-comple-mentary sequence, or it may comprise additional func-tional sequences, or their complements as noted above,such that the extension reaction results in attachment ofthose functional sequences to the 5’ terminus of the par-tial hairpin structure.[0231] In alternative aspects, additional sequence seg-ments may be ligated to the 5’ end of the partial hairpinstructure where such sequence segments are not com-plementary to the non-overlapped portion of the hairpinstructure. The foregoing are schematically illustrated inFig 40. As shown in path A, a partial hairpin structure,when subjected to primer extension conditions, may actas its own primer and have its 5’ sequence extended, asshown by the dashed arrow, until it forms a complete ornearly complete hairpin structure, e.g., with little or nooverhang sequence. This full hairpin structure will pos-sess far greater duplex stability, thereby potentially neg-atively impacting the ability to disrupt the hairpin structureto prime its replication, even when employing higher af-finity primers, e.g., LNA containing primers/probes.[0232] In order to minimize this possibility, as shownin both paths B and C, a separate sequence segment4006 is added to the 5’end of the hairpin structure, toprovide a partial hairpin with non-complementary tail se-quences 4008, in order to prevent the generation of thecomplete or nearly complete hairpin structure. As shown,
83 84

EP 3 013 957 B1
44
5
10
15
20
25
30
35
40
45
50
55
this may be accomplished in a number of different ways.For example, in a first process shown in path B, an in-vading probe 4010 may be used to disrupt the partialhairpin structure and hybridize to sequence segment4012. Such invading probes may be provided with higheraffinity binding than the inherent partial hairpin structure,e.g., through use of higher affinity nucleotide analoguessuch as LNAs or the like. In particular, that portion of theinvader sequence 4010 that hybridizes to sequence seg-ment 4012 may comprise LNAs within its sequence inthe same fashion described herein for use with LNA prim-er sequences used in subsequent amplification.[0233] Extension of the 5’ portion of the partial hairpin(and sequence segment 4012) as shown by the dashedarrow in path B, then appends the sequence 4006 to the5’ terminus of the partial hairpin structure to provide struc-ture 4008. Alternatively, sequence 4006 may be ligatedto the 5’ end of the partial hairpin structure 4002 (or se-quence segment 4012). As shown in path C, thisachieved through the use of a splint sequence 4014 thatis partially complementary to sequence 4006 and partial-ly complementary to sequence 4012, in order to hold se-quence 4006 adjacent to sequence segment 4012 forligation. As will be appreciated, the splint sequence 4014may again utilize a higher affinity invading probe, likeprobe 4010, to disrupt the hairpin structure and hybridizeto sequence segment 4012. In particular, again, that por-tion of splint sequence 4014 that is intended to hybridizeto sequence segment 4012 may be provided with one ormore LNA nucleotide analogues within its sequence, inorder to preferentially disrupt the partial hairpin structure4002, and allow ligation of sequence 4006 to its 5’ end.[0234] In some cases, a microfluidic device (e.g., a mi-crofluidic chip) may be useful in parallelizing sample in-dexing. Such a device may comprise parallel moduleseach capable of adding a barcode sequence and a sam-ple index to nucleic acid molecules of a sample via prim-ers comprising both the barcode sequence and the sam-ple index. Each parallel module may comprise a primerset comprising a different sample index, such that thesample processed in each module is associated with adifferent sample index and set of barcodes. For example,a microfluidic device with 8 modules may be capable ofsample indexing 8 different samples. Following barcod-ing and sample indexing via attachment of the sequencesto a sample nucleic acid, bulk addition of additional se-quences (e.g., R2, P7, other barcode sequences) via, forexample, serial amplification can be used to generatesequencer-ready products as described elsewhere here-in.[0235] In some cases, sample indexing may beachieved during barcoding without the inclusion of a sep-arate sample index sequence in a primer used to attacha barcode to a sample nucleic acid. In such cases, abarcode sequence, for example, may also serve as asample index. An example configuration of a sequencer-ready construct with a sequence functioning as both abarcode sequence and a sample index is as follows:
P5-BSI-R1- RandomNmer-Insert-R2-P7
where "BSI" is the sequence functioning as both abarcode sequence and a sample index.
A sequencer-ready product may comprise a barcode se-quence that can be used to align sequence reads andprovide a sequence for a sample nucleic acid. The se-quencer-ready product may be generated, for example,using PHASE amplification and subsequent bulk ampli-fication as described elsewhere herein. Moreover, thebarcode sequence may belong to a particular set ofknown barcode sequences. The set of barcode sequenc-es may be associated with a particular sample, such thatidentification of the sample from which a particular se-quencing read originates can be achieved via the readbarcode sequence. Each sample can be associated witha set of known barcode sequences, with each barcodesequence set comprising barcode sequences that do notoverlap with barcode sequence in other barcode sets as-sociated with other samples. Thus, the uniqueness of abarcode sequence and its uniqueness amongst differentsets of barcode sequences may be used for multiplexing.[0236] For example, a sequencing read may comprisethe barcode sequence "GAGCCG". Barcode sequence"GAGCCG" may be a barcode sequence in a set of knownbarcode sequences associated with Sample A. The se-quence is not found in a set of known barcode sequencesassociated with another sample. Upon reading the se-quence "GAGCCG", it can be determined that the se-quence read is associated with Sample A because thesequence "GAGCCG" is unique to the set of barcodesequences associated with Sample A. Moreover, anoth-er sequencing read may comprise the barcode sequence"AGCAGA". Barcode sequence "AGCAGA" may be abarcode sequence in a set of known barcode sequencesassociated with Sample B. The sequence is not found ina set of known barcode sequences associated with an-other sample. Upon reading the sequence "AGCAGA",it can be determined that the sequence read is associatedwith Sample B because "AGCAGA" is unique to the setof barcode sequences associated with Sample B.[0237] In another example, a sample index sequencemay be embedded in a random sequence of a primerused in one or more amplification reactions to attach abarcode to a sample nucleic acid. For example, a primermay comprise a barcode sequence and a random se-quence that can be used to randomly prime a samplenucleic acid and attach the barcode sequence to the sam-ple nucleic acid. In some cases, the random sequencemay be a pseudo-random sequence such that particularbases of the random sequence are conserved betweenall primers. The pattern of the conserved bases may beused as a sample index, such that all sequencer-readyproducts obtained from a particular sample all comprisethe conserved pattern of bases in the random sequenceregion. Each sample can be associated with a differentpattern of conserved bases and, thus, multiplexing can
85 86

EP 3 013 957 B1
45
5
10
15
20
25
30
35
40
45
50
55
be achieved. In some cases, the pattern is a contiguoussequence region of a pseudo-random sequence (e.g.,"NNNATACNNN") or in other cases, the pattern is a non-contiguous sequence region of a pseudo-random se-quence (e.g., "NCNGNNAANN"), where "N" correspondsto a random base. Moreover, any suitable number of bas-es may be conserved in a pseudo-random sequence inany pattern and the examples described herein are notmeant to be limiting. An example configuration of a se-quencer-ready construct with a sequence functioning asboth a barcode sequence and a sample index is as fol-lows:
P5-Barcode-R1-NQNQNNQQNN-Insert-R2-P7
where "Q" is a conserved base in the random region
[0238] For example, a sequencer-ready product maycomprise a 10-mer pseudo-random sequence "NCNGN-NAANN", where the second base ("C"), fourth base ("G"),seventh base ("A"), and eighth base ("A") of the pseudo-random sequence are conserved for all sequencer-readyproducts generated from Sample A. A sequencing readmay comprise such a pattern of conserved bases in therandom sequence region. Upon reading the conservedbase pattern, it can be determined that the sequenceread is associated with Sample A because the "NCNGN-NAANN" conserved pattern of bases is associated withSample A. Moreover, a sequencer-ready product maycomprise a 10-mer pseudo-random sequence "NNGC-NGNGNN", where the third base ("G"), fourth base ("C"),sixth base ("G"), and eighth base ("G") of the pseudo-random sequence are conserved for all sequencer-readyproducts generated from Sample B. A sequencing readmay comprise such a pattern of conserved bases in therandom sequence region. Upon reading the conservedbase pattern, it can be determined that the sequenceread is associated with Sample B because the "NNGC-NGNGNN" conserved pattern of bases is associated withSample B.[0239] In other cases, a sample index may be addedto a sample nucleic acid prior to the addition of a barcodesequence to the sample nucleic acid. For example, asample nucleic acid may be pre-amplified in bulk suchthat resulting amplicons are attached to a sample indexsequence prior to barcoding. For example, sample maybe amplified with a primer comprising a sample indexsequence such that the sample index sequence can beattached to the sample nucleic acid. In some cases, theprimer may be a random primer (e.g., comprising a ran-dom N-mer) and amplification may be random. Producedamplicons that comprise the sample index can then bebarcoded using any suitable method, including barcodingmethods described herein.[0240] Sample nucleic acid molecules can be com-bined into partitions (e.g., droplets of an emulsion) withthe primers described above. In some cases, each par-tition can comprise a plurality of sample nucleic acid mol-
ecules (e.g., smaller pieces of a larger nucleic acid). Insome cases, no more than one copy of a unique samplenucleic acid molecule is present per partition. In somecases, each partition can generally comprise primerscomprising an identical barcode sequence and a samplepriming sequence (e.g., a variable random-Nmer, a tar-geted N-mer), with the barcode sequence generally dif-fering between partitions. In such cases, each partition(and, thus, sample nucleic acid in the partition) can beassociated with a unique barcode sequence and theunique barcode sequence can be used to determine asequence for the barcoded sample nucleic acid gener-ated in the partition.[0241] In some cases, upon generation of barcodedsample nucleic acids, the barcoded sample nucleic acidscan be released from their individual partitions, pooled,and subject to bulk amplification schemes to add addi-tional sequences (e.g., additional sequencing primerbinding sites, additional sequencer primer binding sites,additional barcode sequences, sample index sequenc-es) common to all downstream sequencer-ready prod-ucts. In cases where the partitions are droplets of anemulsion, the emulsion may be broken and the barcodedsample nucleic acids pooled. A sample index can be add-ed in bulk to the released, barcoded sample nucleic acids,for example, using the serial amplification methods de-scribed herein. Where a sample index is added in bulk,each sequencer-ready product generated from the samesample will comprise the same sample index that can beused to identify the sample from which the read for thesequencer-ready product was generated. Where a sam-ple index is added during barcoding, each primer usedfor barcoding may comprise an identical sample indexsequence, such that each sequencer-ready product gen-erated from the same sample will comprise the samesample index sequence.[0242] Partitioning of sample nucleic acids to generatebarcoded (or barcoded and sample indexed) sample nu-cleic acids and subsequent addition of additional se-quences (e.g., including a sample index) to the barcodedsample nucleic acids can be repeated for each sample,using a different sample index for each sample. In somecases, a microfluidic droplet generator may be used topartition sample nucleic acids. In some cases, a micro-fluidic chip may comprise multiple droplet generators,such that a different sample can be processed at eachdroplet generator, permitting parallel sample indexing.Via each different sample index, multiplexing during se-quencing can be achieved.[0243] Upon the generation of sequencer-ready oligo-nucleotides, the sequencer-ready oligonucleotides canthen be provided to a sequencing device for sequencing.Thus, for example, the entire sequence provided to thesequencing device may comprise one or more adaptorscompatible with the sequencing device (e.g. P5, P7), oneor more barcode sequences, one or more primer bindingsites (e.g. Read1 (R1) sequence primer, Read2 (R2) se-quencing primer, Index primer), an N-mer sequence, a
87 88

EP 3 013 957 B1
46
5
10
15
20
25
30
35
40
45
50
55
universal sequence, the sequence of interest, and com-binations thereof. The barcode sequence may be locatedat either end of the sequence. In some cases, the barcodesequence may be located between P5 and Read1 se-quence primer binding site. In other cases, the barcodesequence may be located between P7 and Read 2 se-quence primer binding site. In some cases, a secondbarcode sequence may be located between P7 and Read2 sequence primer binding site. The index sequenceprimer binding site may be utilized in the sequencing de-vice to determine the barcode sequence.[0244] The configuration of the various components(e.g., adaptors, barcode sequences, sample index se-quences, sample sequence, primer binding sites, etc.)of a sequence to be provided to a sequencer device mayvary depending on, for example the particular configura-tion desired and/or the order in which the various com-ponents of the sequence is added. Any suitable config-uration for sequencing may be used and any sequencescan be added to oligonucleotides in any suitable order.Additional sequences may be added to a sample nucleicacid prior to, during, and after barcoding of the samplenucleic acid. For example, a P5 sequence can be addedto a sample nucleic acid during barcoding and P7 can beadded in bulk amplification following barcoding of thesample nucleic acid. Alternatively, a P7 sequence canbe added to a sample nucleic acid during barcoding anda P5 sequence can be added in bulk amplification follow-ing barcoding of the sample nucleic acid. Example con-figurations displayed as examples herein are not intend-ed to be limiting. Moreover, the addition of sequencecomponents to an oligonucleotide via amplification is alsonot meant to be limiting. Other methods, such as, forexample, ligation may also be used. Furthermore, adap-tors, barcode sequences, sample index sequences,primer binding sites, sequencer-ready products, etc. de-scribed herein are not meant to be limiting. Any type ofoligonucleotide described herein, including sequencer-ready products, may be generated for any suitable typeof sequencing platform (e.g., Illumina sequencing, LifeTechnologies Ion Torrent, Pacific Biosciences SMRT,Roche 454 sequencing, Life Technologies SOLiD se-quencing, etc.) using methods described herein.[0245] Sequencer-ready oligonucleotides can be gen-erated with any adaptor sequence suitable for a particularsequencing platform using methods described herein.For example, sequencer-ready oligonucleotides com-prising one or more barcode sequences and P1 and Aadaptor sequences useful in Life Technologies Ion Tor-rent sequencing may be generated using methods de-scribed herein. In one example, beads (e.g., gel beads)comprising an acrydite moiety linked to a P1 sequencevia a disulfide bond may be generated. A barcode con-struct may be generated that comprises a P1sequence,a barcode sequence, and a random N-mer sequence.The barcode construct may enter an amplification reac-tion (e.g., in a partition, such as a fluidic droplet) to bar-code sample nucleic acid. Barcoded amplicons may then
be subject to further amplification in bulk to add the Asequence and any other sequence desired, such as asample index. Alternatively, P1 and A sequences can beinterchanged such that A is added during sample bar-coding and P1 is added in bulk. The complete sequencecan then be entered into an Ion Torrent sequencer. Otheradaptor sequences (e.g., P1 adaptor sequence for LifeTechnologies SOLiD sequencing, A and B adaptor se-quences for Roche 454, etc.) for other sequencing plat-forms can be added in analogous fashion.[0246] Although described herein as generating partialhairpin molecules, and in some cases, preventing forma-tion of complete hairpins, in some cases, it may be de-sirable to provide complete hairpin fragments that includethe barcode sequences described herein. In particular,such complete hairpin molecules may be further subject-ed to conventional sample preparation steps by treatingthe 3’ and 5’ end of the single hairpin molecule as oneend of a double stranded duplex molecule in a conven-tional sequencing workflow. In particular, using conven-tional ligation steps, one could readily attach the appro-priate adapter sequences to both the 3’ and 5’ end of thehairpin molecule in the same fashion as those are at-tached to the 3’ and 5’ termini of a duplex molecule. Forexample, in case of an Illumina based sequencing proc-ess, one could attach a standard Y adapter that includesthe P5 and P7 adapters and R1 and R2 primer sequenc-es, to one end of the hairpin as if it were one end of aduplex molecule, using standard Illumina protocols.
Methods for Reducing Undesired Amplification Products (Partial Hairpin Amplification for Sequenc-ing (PHASE))
[0247] A random N-mer sequence may be used to ran-domly prime a sample, such as genomic DNA (gDNA).In some embodiments, the random N-mer may comprisea primer. In some cases, the random N-mer may primea sample. In some cases, the random N-mer may primegenomic DNA. In some cases, the random N-mer mayprime DNA fragments.[0248] Additionally, a random N-mer sequence may al-so be attached to another oligonucleotide. This oligonu-cleotide may be a universal sequence and/or may containone or more primer read sequences that may be com-patible with a sequencing device (e.g. Read 1 primer site,Read 2 primer site, Index primer site), one or more bar-code sequences, and one or more adaptor segments thatmay be compatible with a sequencing device (e.g. P5,P7). Alternatively, the oligonucleotide may comprisenone of these and may include another sequence.[0249] Via subsequent amplification methods, primingof a sample nucleic acid with a random N-mer may beused to attach an oligonucleotide sequence (e.g., an ol-igonucleotide sequence comprising a barcode se-quence) linked to a random N-mer to the sample nucleicacid, including a sample nucleic acid to be sequenced.Utilizing random primers to prime a sample may intro-
89 90

EP 3 013 957 B1
47
5
10
15
20
25
30
35
40
45
50
55
duce significant sequence read errors, due to, for exam-ple, the production of undesired amplification products.[0250] To mitigate undesired amplification products, atleast a subsection of an oligonucleotide sequence maybe substituted with dUTPs or uracil containing nucle-otides in place of dTTPs or thymine containing nucle-otides, respectively. In some cases, substitution may becomplete (e.g., all thymine containing nucleotides aresubstituted with uracil containing nucleotides), or may bepartial such that a portion of an oligonucleotide’s thyminecontaining nucleotides are substituted with uracil con-taining nucleotides. In some cases, thymine containingnucleotides in all but the last about 10 to about 20, lastabout 10 to 30, last about 10 to 40, or last about 5 to 40nucleotides of an oligonucleotide sequence adjacent toa random N-mer sequence are substituted with dUTPsor uracil containing nucleotides. In addition, a polymer-ase that does not accept or process uracil-containingtemplates may be used for amplification of the samplenucleic acid. In this case, the non-uracil containing por-tion of about 10 to about 20 nucleotides may be amplifiedand the remaining portion containing the dUTPs or uracilcontaining nucleotides may not be amplified. In somecases, the portion of an oligonucleotide sequence com-prising dUTPs or uracil containing nucleotides may beadjacent to the N-mer sequence. In some cases, the por-tion of an oligonucleotide sequence comprising dUTPsor uracil containing nucleotides may be adjacent to thebarcode sequence. Any portion of an oligonucleotide se-quence, including an adaptor segment, barcode, or readprimer sequence may comprise dUTPs or uracil contain-ing nucleotides (e.g., substituted for thymine containingnucleotides), depending upon the configuration of the ol-igonucleotide sequence.[0251] Moreover, the number and positioning of uracilcontaining nucleotide-for-thymine containing nucleotidesubstitutions in an oligonucleotide may be used, for ex-ample, to tune the size of partial hairpin products obtainedwith amplification methods described below and/or totune the binding of the polymerase enzyme with a uracilcontaining primer sequence. Additionally, free uracil con-taining nucleotides, e.g., UTP or an analogue thereof,may also be provided within the reaction mixture, e.g.,within the partition, at a desired concentration to mediatepolymerase/uracil-primer binding kinetics. In some cas-es, smaller partial hairpin products may give rise to moreaccurate sequencing results. Accordingly, an oligonucle-otide may comprise at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10,11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25,26, 27, 28, 29, 30, or more uracil containing nucleotide-for-thymine containing nucleotide substitutions depend-ing upon, for example, the desired length of partial hairpinproducts generated from the oligonucleotide.[0252] Upon random priming of a sample nucleic acidwith a random N-mer linked to an oligonucleotide se-quence (e.g., an oligonucleotide sequence comprisinguracil containing nucleotides described above) Fig 15A,a first round of amplification (e.g., using a polymerase
that does not accept or process a uracil containing nu-cleotide as a template) may result in the attachment ofthe oligonucleotide sequence to a complement of thesample nucleic acid, Fig 15B and Fig 15C. Upon priming(via the random N-mer) and further amplification of theamplification product with another copy of the oligonu-cleotide sequence comprising the random N-mer (Fig15D), an amplification product comprising the oligonu-cleotide sequence, a portion of the sample nucleic acidsequence, and a partial complementary oligonucleotidesequence (e.g., complementary to the portion of the oli-gonucleotide sequence not comprising uracil containingnucleotides) at an end of the amplification product oppo-site the oligonucleotide sequence, can be generated. Thepartial complementary oligonucleotide sequence and theoligonucleotide sequence can hybridize to form a partialhairpin that, in some cases, can no longer participate innucleic acid amplification. A partial hairpin can be gen-erated because a portion of the original oligonucleotidesequence comprising uracil containing nucleotides wasnot copied. Amplification can continue for a desirednumber of cycles (e.g., 5, 10, 15, 20, 25, 30, 35, 40, 45,or 50 cycles), up until all oligonucleotide sequences com-prising random N-mers have been exhausted (Fig 15E-G).[0253] In some embodiments, to ensure priming ofsample nucleic acid (e.g., genomic DNA (gDNA)) withonly a random N-mer and not portions of an attachedoligonucleotide sequence, the oligonucleotide sequencemay be blocked via hybridization of a blocker oligonucle-otide (e.g., black dumbbell in Fig 15). A blocker oligonu-cleotide (also referred to as an oligonucleotide blockerelsewhere herein) may be hybridized to any portion ofan oligonucleotide sequence, including a barcode se-quence, read primer site sequence, all or a portion of auracil containing portion of the oligonucleotides, or all orany other portion of the oligonucleotides, or other se-quence therein. A blocker oligonucleotide may be DNAor RNA. In some cases, a blocker oligonucleotide maycomprise uracil containing nucleotide-for-thymine con-taining nucleotide substitutions. In some cases, all of thethymine containing nucleotides of a blocker oligonucle-otide may be substituted with uracil containing nucle-otides. In some cases, a portion of the thymine containingnucleotides of a blocker oligonucleotide may be substi-tuted with uracil containing nucleotides. In some cases,a blocker oligonucleotide may comprise locked nucleicacid (LNA), an LNA nucleotide, bridged nucleic acid(BNA), and/or a BNA nucleotide. Moreover a blocker ol-igonucleotide may be of any suitable length necessaryfor blocker functionality. A blocker oligonucleotide maybe of length suitable to block a portion of an oligonucle-otide or may be of the same or of substantially the samelength of an oligonucleotide it is designed to block. Theblocker oligonucleotide may ensure that only random N-mers bind to the sample nucleic acid (e.g., genomic DNA)and not other portions of the oligonucleotide sequence.[0254] The stoichiometric ratio of a blocker oligonucle-
91 92

EP 3 013 957 B1
48
5
10
15
20
25
30
35
40
45
50
55
otide to oligonucleotide (e.g., blocker oligonucleotide:ol-igonucleotide) may vary. For example, the blocker oligo-nucleotide:oligonucleotide stoichiometric ratio may beabout 0.01, 0.05, 0.10, 0.15, 0.20, 0.25, 0.30, 0.35, 0.40,0.45, 0.50, 0.55, 0.60, 0.65, 0.70, 0.75, 0.80, 0.85, 0.90,0.95, 1.00, 1.05, 1.10, 1.15, 1.20, 1.25, 1.30, 1.35, 1.40,1.45, 1.50, 1.55, 1.60, 1.65, 1.70, 1.75, 1.80, 1.85, 1.90,1.95, 2.00, 2.10, 2.20, 2.30, 2.40, 2.50, 2.60, 2.70, 2.80,2.90, 3.00, 3.50, 4.00, 4.50, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5,8.0, 8.5, 9.0, 10.0, 20, 30, 40, 50, 100 or more. In somecases, the blocker oligonucleotide:oligonucleotide stoi-chiometric ratio may be at least about 0.01, 0.05, 0.10,0.15, 0.20, 0.25, 0.30, 0.35, 0.40, 0.45, 0.50, 0.55, 0.60,0.65, 0.70, 0.75, 0.80, 0.85, 0.90, 0.95, 1.00, 1.05, 1.10,1.15, 1.20, 1.25, 1.30, 1.35, 1.40, 1.45, 1.50, 1.55, 1.60,1.65, 1.70, 1.75, 1.80, 1.85, 1.90, 1.95, 2.00, 2.10, 2.20,2.30, 2.40, 2.50, 2.60, 2.70, 2.80, 2.90, 3.00, 3.50, 4.00,4.50, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, 8.5, 9.0, 10.0, 20,30, 40, 50, 100 or more. In some cases, the blocker oli-gonucleotide:oligonucleotide stoichiometric ratio may beat most about 0.01, 0.05, 0.10, 0.15, 0.20, 0.25, 0.30,0.35, 0.40, 0.45, 0.50, 0.55, 0.60, 0.65, 0.70, 0.75, 0.80,0.85, 0.90, 0.95, 1.00, 1.05, 1.10, 1.15, 1.20, 1.25, 1.30,1.35, 1.40, 1.45, 1.50, 1.55, 1.60, 1.65, 1.70, 1.75, 1.80,1.85, 1.90, 1.95, 2.00, 2.10, 2.20, 2.30, 2.40, 2.50, 2.60,2.70, 2.80, 2.90, 3.00, 3.50, 4.00, 4.50, 5.0, 5.5, 6.0, 6.5,7.0, 7.5, 8.0, 8.5, 9.0, 10.0, 20, 30, 40, 50, or 100.[0255] Moreover, incorporation of a blocker moiety(e.g., via a dideoxynucleotide (ddNTP), ddCTP, ddATP,ddGTP, ddTTP, etc. at the 3’ or 5’ end of the blockeroligonucleotide) to a blocker oligonucleotide and/or theinclusion of uracil containing nucleotides (e.g., substitut-ed for all or a portion of thymine containing nucleotides)in a blocker oligonucleotide may prevent preferentialbinding of blocked portions of the blocked oligonucleotidesequence to the sample nucleic acid. Additional exam-ples of blocker moieties include 3’ phosphate, a blocked3’ end, 3’ddCTP, C3 Spacer (/3SpC3/), Dideoxy-C(/3ddC/). Blocker oligonucleotides may be cleaved froman oligonucleotide sequence by RNAse, RNAseH, an an-tisense DNA oligonucleotide, and/or alkaline phos-phatase.[0256] In some cases, an oligonucleotide sequencemay be blocked with a blocker oligonucleotide such thatthe oligonucleotide sequence comprises a blocked 5’end, comprises a blocked 3’ end, may be entirely blocked(e.g., may be entirely blocked, except for its random N-mer sequence), or may be blocked at another location(e.g., a partial sequence of the oligonucleotide, differentfrom an oligonucleotide sequence’s random N-mer). Insome cases, an oligonucleotide sequence may comprisea plurality of blockers, such that multiple sites of the oli-gonucleotide are blocked. In some cases, an oligonucle-otide sequence may comprise both a blocked 3’ end anduracil containing nucleotides. In some cases, an oligo-nucleotide sequence comprising uracil containing nucle-otides and a blocked 3’ end may be adjacent to the N-mer sequence. In some cases, an oligonucleotide se-
quence may comprise a blocked 3’ end. In some cases,an oligonucleotide sequence may comprise uracil con-taining nucleotides. In some cases, an oligonucleotidesequence may comprise both a blocked 5’ end and uracilcontaining nucleotides.[0257] In some cases, the oligonucleotide sequencecomprising uracil containing nucleotides and a blocked3’ end may be adjacent to the N-mer sequence. In somecases, the oligonucleotide sequence comprising uracilcontaining nucleotides and a blocked 3’ end may be ad-jacent to the barcode sequence. In some cases, the oli-gonucleotide sequence may comprise a blocked 3’ end.In some cases, the oligonucleotide sequence may com-prise uracil containing nucleotides. In some cases, theoligonucleotide sequence may comprise both theblocked 3’ end and uracil containing nucleotides. Additionof a blocker oligonucleotide may prevent preferentialbinding to portions of the universal sequence, which maynot be desired to be amplified.[0258] In some cases, an oligonucleotide suitable forpriming a sample nucleic acid via its random N-mer mayalso comprise a blocking sequence that can function inthe same role as a blocker oligonucleotide. For example,an oligonucleotide may be arranged in a hairpin config-uration with a blocking sequence that can function in thesame role as a blocker oligonucleotide. An example oli-gonucleotide comprising a random N-mer, an R1c se-quence, a P5 sequence, a barcode sequence, and anR1 sequence may be configured as follows:5’-RandomNmer-R1c-P5-Barcode-R1-3’The R1 sequence and R1c sequence of the oligonucle-otide may hybridize to generate a hairpin with a hairpinloop comprising the P5 and Barcode sequences. TheR1c sequence can function in the same role as a blockeroligonucleotide such that priming of sample nucleic acidwith the oligonucleotide occurs via only the oligonucle-otide’s random N-mer. In some cases, one or more cleav-age sites (e.g., a restriction site, a cleavage site, an aba-sic site, etc.) may be included in an oligonucleotide ar-ranged as a hairpin with a blocking sequence, includingan oligonucleotide’s hairpin loop, to separate sequencecomponents of the oligonucleotide downstream, if de-sired. Separation may occur, for example, via an enzy-matic reaction, oxidation-reduction, radiation (e.g., UV-light), the addition of heat, or other suitable means.[0259] An example uracil containing nucleotide-substi-tuted oligonucleotide sequence linked to a random N-mer is depicted in Fig 14B. Specifically, a random primer(e.g., a random N-mer), of about 8N-12N in length, 1404,may be linked with an oligonucleotide sequence. The ran-dom N-mer may be used to randomly prime and extendfrom a sample nucleic acid, such as, genomic DNA (gD-NA). The oligonucleotide sequence comprises: (1) se-quences for compatibility with a sequencing device, suchas, a flow cell (e.g. Illumina’s P5, 1401, and Read 1 Primersites, 1402) and (2) a barcode (BC), 1403, (e.g., 6-12base sequences). Furthermore, the Read 1 Primer site1402 of the oligonucleotide sequence may be hybridized
93 94

EP 3 013 957 B1
49
5
10
15
20
25
30
35
40
45
50
55
with a blocking oligonucleotide comprising uracil contain-ing nucleotides and a blocker moiety at its 3’ end (e.g.3’ddCTP, indicated by an "X"). The blocking oligonucle-otide can be used to promote priming of a sample nucleicacid with only the random N-mer sequence and preventpreferential binding of the oligonucleotide sequence toportions of the sample nucleic acid that are complemen-tary to the Read 1 Primer site, 1402. Optionally, to furtherlimit product lengths, a small percentage of terminatingnucleotides (e.g., 0.1-2% acyclonucleotides (acyNTPs))(Fig 16B) may be included in oligonucleotide sequencesto reduce undesired amplification products.[0260] An example of partial hairpin amplification forattaching a uracil containing nucleotide-substituted oli-gonucleotide sequence comprising a random N-mer toa sample nucleic acid (e.g., genomic DNA (gDNA)) isdepicted in Fig 15. First, initial denaturation of the samplenucleic acid may be achieved at a denaturation temper-ature (e.g., 98°C, for 2 minutes) followed by priming of arandom portion of the sample nucleic acid with the ran-dom N-mer sequence at a priming temperature (e.g., 30seconds at 4°C), Fig 15A. The oligonucleotide sequenceis hybridized with a blocking oligonucleotide (black dumb-bell in Fig 15), to ensure that only the random N-merprimes the sample nucleic acid and not another portionof the oligonucleotide sequence. Subsequently, se-quence extension (e.g., via polymerase that does not ac-cept or process a uracil containing nucleotide as a tem-plate) may follow as the temperature ramps to highertemperature (e.g., at 0.1°C/second to 45°C (held for 1second)) (Fig 15A). Extension may then continue at el-evated temperatures (e.g., 20 seconds at 70°C), contin-uing to displace upstream strands and create a first phaseof redundancy (Fig 15B). Denaturation of the amplifica-tion product may then occur at a denaturing temperature(e.g., 98°C for 30 seconds) to release the sample nucleicacid and amplification product for additional priming.[0261] After the first cycle, amplification products havea single 5’ tag (Fig 15C) comprising the oligonucleotidesequence. These aforementioned steps are repeated toprime the amplification product and sample nucleic acidwith the oligonucleotide sequence via its random N-mer.The black sequence indicates portions of the added 5’tags (added in cycle 1) that comprise uracil containingnucleotides and thus, will not be copied upon priming andamplification of the amplification product (Fig 15D). Fol-lowing a second round of amplification, both 5’ taggedproducts and 3’ & 5’ tagged products may be generated(Fig 15E). The 3’ & 5’ tagged products comprise a fulloligonucleotide sequence at one end, the sample nucleicacid sequence, and a sequence partially complementaryto the oligonucleotide sequence (e.g., complementary toregions of the oligonucleotide sequence not comprisinguracil containing nucleotides) at the other end of the ol-igonucleotide. The oligonucleotide sequence may hy-bridize with its partially complementary sequence to gen-erate a partial hairpin structure (Fig 15F. Amplificationcan continue repeatedly for a desired number of cycles
(e.g., up to 20 times), up until all oligonucleotide sequenc-es have been exhausted (Fig 15G).[0262] Partial hairpin formation may prevent generat-ing a copy of a copy and may instead encourage onlycopies of the original template to be produced, thus re-ducing potential amplification bias, and other artifacts.Partial hairpin formation may encourage segregation ofthe desired product and may reduce production of copies.[0263] Desirable properties for the uracil-non-readingpolymerase to form the partial hairpin may include anexonuclease deficient polymerase (e.g., having low ex-onuclease activity, having substantially no exonucleaseactivity, having no exonuclease activity), strand displac-ing capabilities (e.g., a thermostable strand displacingpolymerase enzyme), residual activity at temperatures <50 °C, and discrimination against uracil containing nu-cleotides v thymine containing nucleotides. Examples ofsuch polymerases may include 9 degrees North, modi-fied (NEB), exo minus Pfu, Deep Vent exo minus, Ventexo minus, and homologs thereof. Moreover, a polymer-ase with low exonuclease activity may be a polymerasewith less than 90%, less than 80%, less than 70%, lessthan 60%, less than 50%, less than 40%, less than 30%,less than 20%, less than 10%, less than 5%, or 0% ex-onuclease activity of a thermally stable polymerase withnormal exonuclease activity (e.g., Taq polymerase). Insome cases, a polymerase used for partial hairpin am-plification may be capable of strand-displacement. Insome cases, limiting the length of the amplified sequencemay reduce undesired amplification products, whereinlonger length products may include undesired upstreamportions such as a barcode sequence. The amplifiedproduct length may be limited by inclusion of terminatingnucleotides. An example of a terminating nucleotide mayinclude an acyclonucleotide (acyNTPs). Terminating nu-cleotides may be present at about 0%, 0.1%, 0.2%, 0.3%,0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 1.1%, 1.2%,1.3%, 1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, 2%, 2.1%,2.2%, 2.3%, 2.4%, or 2.5% of the amplified productlength. In some cases, terminating nucleotides may bepresent at more than about 0%, 0.1%, 0.2%, 0.3%, 0.4%,0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 1.1%, 1.2%, 1.3%,1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, 2%, 2.1%, 2.2%,2.3%, 2.4%, 2.5%, or more of the amplified productlength. In some cases, terminating nucleotides may bepresent at less than about 0%, 0.1%, 0.2%, 0.3%, 0.4%,0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 1.1%, 1.2%, 1.3%,1.4%, 1.5%, 1.6%, 1.7%, 1.8%, 1.9%, 2%, 2.1%, 2.2%,2.3%, 2.4%, or 2.5% of the amplified product length.[0264] Amplification product length may also be con-trolled by pre-amplification of sample nucleic acid priorto initiation of PHASE amplification. For example, a ran-dom N-mer may be used for pre-amplification of the sam-ple nucleic acid. A random N-mer may be used to primea sample nucleic acid followed by extension of the primerusing suitable thermal cycling conditions. Product lengthcan be controlled by thermal cycling conditions (e.g.,number of thermal cycles, temperatures utilized, cycle
95 96

EP 3 013 957 B1
50
5
10
15
20
25
30
35
40
45
50
55
time, total run time, etc.) in addition to the random primingof the sample nucleic acid. In some cases, pre-amplifi-cation products smaller than the original sample nucleicacid can be obtained. Amplification products generatedduring pre-amplification may then be entered into aPHASE amplification and barcoded as described above.[0265] As shown in Fig 17, addition of a blocking oli-gonucleotide may reduce start site bias by 50%. Incor-poration of uracil containing nucleotides instead of thym-ine containing nucleotides into the universal sequenceand using a polymerase that does not accept or processuracil-containing templates, may significantly reduce se-quencing errors, as reported in Fig 21 and Fig 22. Forexample, Q40 error may be reduced from about 0.002to about 0.001, unmapped fraction ends may be reducedfrom about 0.996 to about 0.03, median insert size maybe reduced from about 399 to about 310, IQR insert sizemay be reduced from about 413 to about 209, and zerocoverage fraction may be reduced from about 0.9242 toabout 0.0093.[0266] Amplification schemes that do not involve thesubstitution of thymine containing nucleotides with uracilcontaining nucleotides are also envisioned for generatingpartial hairpin species. In some cases, other species un-able to be recognized or be copied by a polymerase (e.g.,methylated bases, abasic sites, bases linked to bulkyside groups, etc.) may be used in place of uracil contain-ing nucleotides to generate partial hairpin amplicons. Insome cases, full hairpin amplicons may be generatedand processed post-synthesis to generate partial hairpinspecies. In some cases, full hairpin amplicons may begenerated and portions subsequently removed to gen-erate partial hairpin species. For example, as shown inFig 34A, full hairpin amplicons 3401 can be generatedvia the amplification scheme depicted in Fig 15 whenoligonucleotide primers comprising random N-mers donot comprise uracil containing nucleotides and/or apolymerase capable of accepting or processing a uracilcontaining template is used for amplification. Upon gen-eration of the full hairpin amplicons 3401, the full hairpinamplicons can be enzymatically (e.g., via a restrictionenzyme or other site specific enzyme such as a nickase)or chemically nicked 3403 at one or more appropriatesites to generate partial hairpin species 3402.[0267] In some cases, full hairpin amplicons may begenerated and portions added to the full hairpin ampli-cons to generate partial hairpin species. For example, aprimer comprising a sequencing primer binding site (e.g.,R1) coupled to a random N-mer and not comprising uracilcontaining nucleotides may be used to amplify samplenucleic acid and generate full hairpin amplicons (e.g., afull hairpin comprising the sequencing primer binding site(e.g., R1), the copied sample nucleic acid, and the com-plement to the sequencing primer binding site hybridizedwith the sequencing primer binding site (e.g., R1c) - 3404in Fig 34B) via the amplification scheme depicted in Fig15. Upon generation of the full hairpin amplicons 3404,the full hairpin amplicons can have additional sequences
(e.g., a sequence comprising a P5 sequence and a bar-code sequence) 3405 added, for example, via ligation3406.[0268] In some cases, primers (e.g., oligonucleotidescomprising a random N-mer) used to generate full hairpinamplicons may be covalently modified to comprise anadditional sequence via, for example, a linker (e.g., alinker not comprising nucleic acid or a linker comprisingnucleic acid that does not participate in amplification). Insome cases, the linker may be polyethylene glycol or acarbon-based linker. Full hairpin amplicons generatedfrom the primers (e.g., via an amplification scheme de-picted in Fig 15), thus, can also be covalently linked tothe additional sequence via the linker. The attached se-quence can then be ligated to the full hairpin ampliconto generate a partial hairpin species. An example of a fullhairpin amplicon 3409 comprising an additional se-quence 3408 via a linker 3407 is shown in Fig 34C. Fol-lowing full hairpin generation, the additional sequence3408 can be ligated to the full hairpin amplicon 3409 suchthat a partial hairpin species (3410) comprising the ad-ditional sequence 3408 can be generated.
Targeted N-mers and Targeted Amplification
[0269] In addition to random amplification schemes,barcode constructs (e.g., oligonucleotides comprising abarcode sequence and an N-mer for priming a samplenucleic acid) comprising targeted priming sequences(e.g., a targeted N-mer) and targeted amplificationschemes are also envisioned. Targeted amplificationschemes may be useful, for example, in detecting a par-ticular gene or sequence of interest via sequencing meth-ods, may be useful in detecting a particular type of nucleicacid, may be useful in detecting the a particular strandof nucleic acid comprising a sequence, and combinationsthereof. In general, targeted amplification schemes relyon targeted primers to complete amplification of a par-ticular nucleic acid sequence. In some examples, PCRmethods may be used for targeted amplification, via theuse of primers targeted toward a particular gene se-quence of interest or a particular sequence upstream ofa particular gene sequence of interest, such that the par-ticular gene sequence of interest is amplified during PCR.[0270] The PHASE amplification reaction describedabove may also be modified such that target amplificationof sample nucleic acid is achieved. Barcode constructscomprising a targeted priming sequence (e.g., a targetedN-mer), rather than a random sequence (e.g., a randomN-mer), as described above, may be used to prime aspecific sequence during PHASE amplification. The spe-cific sequence, for example, may be a particular genesequence of interest such that generation of ampliconsis indicative of the sequence’s presence. Or, the specificsequence may be a sequence known to be upstreamfrom a particular gene sequence of interest. Such con-structs may be generated, and, if desired, coupled tobeads, using any of the methods described herein, in-
97 98

EP 3 013 957 B1
51
5
10
15
20
25
30
35
40
45
50
55
cluding limiting dilution schemes depicted in Fig 4 andthe combinatorial plate schemes described elsewhereherein.[0271] For example, as described previously with re-spect to Fig 4, a construct comprising a primer 403 (e.g.,P5), a barcode sequence 408, and a read primer bindingsite (e.g., R1) 415 can be generated (see Fig 4A-4H).As shown in Fig 4I, an additional sequence 413 can beadded (optionally in bulk) to the construct via primer com-prising a sequence 412 complementary to read primerbinding site 415. Sequence 413 may serve as a targetedsequence (e.g., a targeted N-mer) such that the targetedsequence corresponds to a particular target sequenceof interest. The construct may also comprise an oligonu-cleotide blocker, as described elsewhere herein, in orderto ensure that only the targeted sequence, and not othersequence portions of the construct, primes the samplenucleic acid. Upon entry of the completed construct intoa PHASE reaction with sample nucleic acid, for example,the targeted construct may prime the sample nucleic acid(e.g., at the desired sequence site) and the amplificationreaction can be initiated to generate partial hairpins fromthe sample nucleic acid as described above. In somecases, a combination of targeted N-mer primers and ran-dom N-mer primers are used to generate partial hairpinamplicons. In some cases, targeted amplification may beuseful in controlling the size (e.g., sequence length) ofpartial hairpin amplicons that are generated during am-plification for a particular target.[0272] In some cases, a plurality of constructs com-prising a barcode sequence and a targeted N-mer maybe coupled to a bead (e.g., a gel bead). In some cases,the plurality of constructs may comprise an identical bar-code sequence and/or an identical targeted N-mer se-quence. In some cases, the targeted N-mer sequencemay vary amongst individual constructs of the pluralitysuch that a plurality of target sequences on a samplenucleic acid may be primed via the various targeted N-mers. As described above, the beads may be partitioned(e.g., in fluidic droplets) with sample nucleic acid, thebead(s) in each partition degraded to release the coupledconstructs into the partition, and the sample nucleic acidamplified via the targeted N-mer of the constructs. Postprocessing (e.g., addition of additional sequences (e.g.,P7, R2), addition of a sample index, etc.) of the generatedamplicons may be achieved with any method describedherein, including bulk amplification methods (e.g., bulkPCR) and bulk ligation.[0273] In a partition, constructs comprising a barcodesequence and a targeted N-mer may be coupled to abead, may be free in solution (e.g., free in the aqueousinterior of a fluidic droplet), or both. Moreover, a partitionmay comprise both targeted constructs (e.g., constructscomprising a targeted N-mer sequence) and non-target-ed constructs (e.g., constructs comprising a random N-mer sequence). Each of the targeted and non-targetedconstructs may be coupled to a bead, one of the two maybe coupled to a bead, and either construct may also be
in solution within a partition.[0274] Where each type of construct is present in apartition, both targeted and non-targeted amplification ofsample nucleic acids may take place. For example, withrespect to a PHASE amplification reaction, a targetedbarcode construct may be used to initially prime and ex-tend a sample nucleic acid. In general, these steps cor-respond to the first cycle of PHASE amplification de-scribed above with respect to Figs. 15A-C, except thatthe targeted construct is used for initial priming. The ex-tension products can then be primed with a barcode con-struct comprising a random N-mer such that a partial hair-pin is generated, these steps corresponding to the sec-ond cycle of PHASE described above with respect toFigs. 15D-F. Amplification can continue for additionalrounds (e.g., Fig 15G) until the desired number of roundsare complete. Post processing (e.g., addition of addition-al sequences (e.g., P7, R2), addition of a sample index,etc.) of the generated partial hairpin amplicons may beachieved with any method described herein, includingbulk amplification methods (e.g., bulk PCR) and bulk li-gation.[0275] Moreover, targeted barcode constructs may begenerated such that the construct’s targeted N-mer isdirected toward nucleic acid species other than DNA,such as, for example, an RNA species. In some cases,the targeted barcode construct’s targeted N-mer may bedirected toward a particular RNA sequence, such as, forexample, a sequence corresponding to transcribed geneor other sequence on a messenger RNA (mRNA) tran-script. In some cases, sequencing of barcoded productsgenerated from RNA (e.g., an mRNA) may aid in deter-mining the expression level of a gene transcribed by theRNA. In some cases, the targeted N-mer may be a poly-thymine (e.g., poly-T sequence) sequence capable of hy-bridizing with a poly-adenine (poly-A sequence) that can,for example, be found at the 3’ end of an mRNA transcript.Upon priming of an mRNA with a targeted barcode con-struct comprising a poly-T sequence via hybridization ofthe barcode construct’s poly-T sequence with the mR-NA’s poly-A sequence, the targeted barcode constructcan be extended via a reverse transcription reaction togenerate a complementary DNA (cDNA) product com-prising the barcode construct. In some cases, a targetedbarcode construct comprising a poly-T targeted N-mermay also comprise an oligonucleotide blocker as de-scribed elsewhere herein, such that only the poly-T se-quence hybridizes with RNA.[0276] Targeted barcode constructs to RNA speciesmay also be useful in generating partial hairpin ampliconsvia, for example, a PHASE amplification reaction. Forexample, a targeted barcode construct comprising a po-ly-T sequence can hybridize with an mRNA via its poly-A sequence. The targeted barcode construct can be ex-tended via a reverse transcription reaction (e.g., via theaction of a reverse transcriptase) such that a cDNA com-prising the barcode construct is generated. These stepscan correspond to the first cycle of PHASE amplification
99 100

EP 3 013 957 B1
52
5
10
15
20
25
30
35
40
45
50
55
described above with respect to Figs. 15A-C, except thatreverse transcription is used to generate the extensionproduct. Following reverse transcription (e.g., a firstPHASE cycle), a barcode construct comprising a randomN-mer may prime the extension products such that a par-tial hairpin is generated as described above with respectto Figs. 15D-F. Amplification can continue for additionalrounds (e.g., Fig 15G) until the desired number of roundsare complete.[0277] In some cases, a plurality of targeted constructscomprising a barcode sequence and a targeted N-mercomprising a poly-T sequence may be coupled to a bead(e.g., a gel bead). In some cases, the plurality of con-structs may comprise an identical barcode sequence.The beads may be partitioned (e.g., in fluidic droplets)with sample nucleic acid comprising RNA, the bead(s) ineach partition degraded to release the coupled con-structs into the partition, and the sample RNA capturedvia the targeted N-mer of the constructs. Partitions mayalso comprise barcode constructs (e.g., with barcode se-quences identical to the targeted constructs) that com-prise a random N-mer. In a first amplification cycle, ex-tension of the targeted constructs can occur via reversetranscription within each partition, to generate extensionproducts comprising the targeted construct. The exten-sion products in each partition can then be primed withthe barcode constructs comprising the random N-mer togenerate partial hairpin amplicons as described abovewith respect to Figs. 15-A-G. Post processing (e.g., ad-dition of additional sequences (e.g., P7, R2), addition ofa sample index, etc.) of the generated amplicons may beachieved with any method described herein, includingbulk amplification methods (e.g., bulk PCR) and bulk li-gation.[0278] In some cases, reverse transcription of RNA ina sample may also be used without the use of a targetedbarcode construct. For example, sample nucleic acidcomprising RNA may be first subject to a reverse tran-scription reaction with other types of reverse transcriptionprimers such that cDNA is generated from the RNA. ThecDNA that is generated may then undergo targeted ornon-targeted amplification as described herein. For ex-ample, sample nucleic acid comprising RNA may be sub-ject to a reverse transcription reaction such that cDNA isgenerated from the RNA. The cDNA may then enter aPHASE amplification reaction, using a barcode constructwith a random N-mer as described above with respectto Figs 15A-G, to generate partial hairpin ampliconscomprising the construct’s barcode sequence. Postprocessing (e.g., addition of additional sequences (e.g.,P7, R2), addition of a sample index, etc.) of the generatedpartial hairpin amplicons may be achieved with any meth-od described herein, including bulk amplification meth-ods (e.g., bulk PCR) and bulk ligation.[0279] Targeted barcode constructs may also be gen-erated toward specific sequences (e.g., gene sequenc-es) on specific strands of a nucleic acid such that strand-edness information is retained for sequencer-ready prod-
ucts generated for each strand. For example, a samplenucleic may comprise double stranded nucleic acid (e.g.,double-stranded DNA), such that each strand of nucleicacid comprises one or more different target gene se-quences. Complementary DNA strands can comprise dif-ferent gene sequences due to the opposite 5’ to 3’ direc-tionalities and/or base composition of each strand. Tar-geted barcode constructs can be generated for eachstrand (based on 5’ to 3’ directionality of the strand) basedon the targeted N-mer and configuration of the barcodeconstruct. Example sets of targeted barcode constructsdirected to forward and reverse strands of a double-stranded sample nucleic acid are shown in Fig 28A.[0280] Example sets 2801 and 2802 of targeted bar-code constructs each targeted to either of a forward(2801) strand and reverse (2802) strand of a double-stranded sample nucleic acid are shown in Fig 28A. Set2801 comprises targeted barcode constructs 2803 and2804 comprising a P5 sequence, a barcode sequence,and a targeted N-mer to either of a first target sequence(2803) or a second target sequence (2804). Set 2802comprises targeted barcode constructs 2805 and 2806comprising a P5 sequence, a barcode sequence, and atargeted N-mer to either of the first target sequence(2805) and the second target sequence (2806). Eachconstruct can also comprise any additional sequencesbetween the barcode and the targeted N-mer (indicatedby an arrow in each construct shown in Fig 28A).[0281] The barcode constructs in set 2801 are config-ured to prime their respective target sequences on theforward strand of the double-stranded sample nucleic ac-id. The barcode constructs of set 2802 are configured toprime their respective target sequences on the reversestrand of the double-stranded sample nucleic acid. Asshown, the targeted barcode constructs in each set areconfigured in opposite directionality corresponding to theopposite directionality of forward and reverse strands ofthe double-stranded sample nucleic acid. Each barcodeconstruct can prime its respective target sequence on itsrespective strand of sample nucleic acid to generate bar-coded amplicons via an amplification reaction, such asany amplification reaction described herein.[0282] Additional sequences can be added to barcod-ed amplicons using amplification methods describedherein, including bulk amplification, bulk ligation, or acombination thereof. Example sets of primers that maybe used to add a sample index and P7 sequence to am-plicons generated from the targeted barcode constructsin Fig 28A are shown in Fig 28B. Primer set 2808 cor-responds to targeted barcode construct set 2801 (e.g.,targeted barcode construct 2803 corresponds to primer2811, targeted barcode construct 2804 corresponds toprimer 2812) and primer set 2808 corresponds to target-ed barcode construct set 2801 (e.g., targeted barcodeconstruct 2505 corresponds to primer 2809, targeted bar-code construct 2806 corresponds to primer 2810). Eachprimer can prime its respective target sequence on itsrespective strand and bulk amplification (e.g., bulk PCR)
101 102

EP 3 013 957 B1
53
5
10
15
20
25
30
35
40
45
50
55
initiated to generate sequencer-ready constructs that in-clude the P7 and sample index sequences in analogousfashion to bulk amplification methods described else-where herein. Based on the configuration and direction-ality of the various components of each sequencer-readyconstruct (e.g., P5, barcode, targeted N-mer, sample in-sert, etc.), the strand from which the sequencer-readyproduct is generated can be determined/is retained.[0283] Libraries of barcode constructs (e.g., targetedbarcode constructs) may be generated for both forwardand reverse strands of a double stranded nucleic acid.For example, two libraries of beads (e.g., gel beads) com-prising targeted barcode constructs may be generatedusing methods described herein, such that one librarycomprises targeted barcode constructs for forwardstrands of sample nucleic acids and the other library com-prises targeted barcode constructs for reverse strandsof sample nucleic acids. In some cases, each library maycomprise beads each comprising an identical targetedN-mer. In some cases, each library may comprise two ormore sets of beads, with each bead in a set comprisingan identical targeted N-mer (e.g., a targeted N-mer tar-geted toward a particular gene) and different sets com-prising different targeted N-mers. In some cases, the twolibraries may be combined such that a library of forwardstrand and reverse strand beads is generated.[0284] For example, a library can comprise two typesof forward strand beads and two types of reverse strandbeads, for a total of four types of beads. Each bead inthe library may comprise a unique barcode sequence.One type of the forward strand beads and one type ofthe reverse strand beads may comprise targeted N-merscorresponding to a target sequence (e.g., a target genesequence). For example, one type of forward strandbeads may comprise a targeted barcode construct asshown in 2803 in Fig 28A and one type of reverse strandbeads may comprise a targeted barcode construct asshown in 2805 in Fig 28A. Analogously, the second typeof forward strand beads may comprise a targeted bar-code construct as shown in 2804 in Fig 28A and onetype of reverse strand beads may comprise a targetedbarcode construct as shown in 2806 in Fig 28A.[0285] A barcode library comprising forward strandand reverse strand beads (e.g., gel beads), with eachbead comprising a unique barcode sequence may bepartitioned to barcode sample nucleic acids as describedelsewhere herein. For example, the mixed library of twotypes of forward strand and two types of reverse strandbeads described above may be partitioned with a samplenucleic acid (e.g., genomic DNA) and any other desiredreagents (e.g., reagents necessary for amplification ofthe sample nucleic acid, a reducing agent). The partitionsmay be, for example, fluidic droplets such as droplets ofan emulsion. In general, each partition may comprise abead (e.g., a forward strand bead or a reverse strandbead) coupled to a targeted barcode construct compris-ing a unique barcode sequence and a targeted N-mer.In some cases, though, one or more of the partitions may
comprise multiple beads of the same type or of differenttypes. The targeted barcode constructs may be releasedfrom the bead (e.g., via degradation of the bead - forexample, via a reducing agent in cases where the beadis a gel bead comprising disulfide bonds) in the partitionand allowed to prime their target sequence on their re-spective strand (e.g., forward strand or reverse strand)of sample nucleic acid.[0286] A first product strand synthesis may take placein each partition via extension of the hybridized targetedbarcode construct, via, for example, linear amplificationof the sample nucleic acid. Additional rounds of linearamplification of the sample nucleic acid with the targetedbarcode construct, for example, may be used to generateadditional copies of the first product strand. First productstrands may then be removed from the partitions (e.g.,in cases where the partitions are droplets of an emulsion,the emulsion may be broken to release first products)and pooled. The first products may be washed to removetargeted barcode constructs and any other waste prod-ucts. In some cases, an optional double-stranded diges-tion may be completed to digest sample nucleic acid andremove it from the first product strands.[0287] Next, the first product strands may be subjectto bulk amplification to add additional sequences (e.g.,P7, a sample index, etc.) to the first product strands, re-sulting in the generation of second product strands. Thebulk amplification reaction mixture may comprise a plu-rality of primers, with each primer in the plurality corre-sponding to one of the bead types (and, thus, type oftargeted barcode construct) used to generate the firstproducts strands. For the example library comprising twotypes of forward strand beads and two types of reversestrand beads described above, primers shown as 2809,2810, 2811, and 2812 in Fig 28B may be used to addadditional sample index and P7 sequences to first prod-uct strands generated from targeted barcode constructs2803, 2804, 2805, and 2806 respectively via bulk ampli-fication. Second product strands may then be washed toremove primers from the reaction mixture. Fresh primers(e.g., primers comprising P5 and P7 for the example de-scribed above) may then be added one or more additionalrounds of amplification (e.g., via PCR) to generate final,sequencer-ready products. Thus, final products cancomprise the original targeted barcode construct, thestrand of sample nucleic acid amplified, and the addition-al sequences (e.g., P7, sample index) added to first prod-uct strands.[0288] Methods described herein may be useful inwhole genome amplification. In some embodiments ofwhole genome amplification, a random primer (e.g., arandom N-mer sequence) can be hybridized to a genomicnucleic acid. The random primer can be a component ofa larger oligonucleotide that may also include a universalnucleic acid sequence (including any type of universalnucleic acid sequence described herein) and a nucleicacid barcode sequence. In some cases, the universalnucleic acid sequence may comprise one or more uracil
103 104

EP 3 013 957 B1
54
5
10
15
20
25
30
35
40
45
50
55
containing nucleotides. Moreover, in some cases, theuniversal nucleic acid sequence may comprise a seg-ment of at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14,15, 16, 17, 18, 19, 20, or more nucleotides that do notcomprise uracil. The random primer can be extended(e.g., in a primer extension reaction or any other suitabletype of nucleic acid amplification reaction) to form an am-plified product.[0289] As described elsewhere herein, the amplifiedproduct may undergo an intramolecular hybridization re-action to form a hairpin molecule such as, for example,a partial hairpin molecule. In some cases, whole genomeamplification may occur in the presence of an oligonu-cleotide blocker (also referred to as a blocker oligonucle-otide elsewhere herein) that may or may not comprise ablocker moiety (e.g., C3 spacer (/3SpC3/), Dideoxy-C(/3ddC/), 3’ phosphate, or any other type of blocker moi-ety described elsewhere herein). Furthermore, the oligo-nucleotide blocker may be capable of hybridizing to atleast a portion of the universal nucleic acid sequence orany other part of an oligonucleotide comprising the ran-dom primer.[0290] In some embodiments of whole genome ampli-fication, a genomic component (e.g., a chromosome, ge-nomic nucleic acid such as genomic DNA, a whole ge-nome of an organism, or any other type of genomic com-ponent described herein) may be fragmented in a plural-ity of first fragments. The first fragments can be co-par-titioned into a plurality of partitions with a plurality of oli-gonucleotides. The oligonucleotides in each of the par-titions may comprise a primer sequence (including a typeof primer sequence described elsewhere herein) and acommon sequence (e.g., a barcode sequence). Primersequences in each partition can then be annealed to aplurality of different regions of the first fragments withineach partition. The primer sequences can then be ex-tended along the first fragments to produce amplified firstfragments within each partition of the plurality of parti-tions. The amplified first fragments within the partitionsmay comprise any suitable coverage (as described else-where herein) of the genomic component. In some cases,the amplified first fragments within the partitions maycomprise at least 1X coverage, at least 2Xcoverage, atleast 5X coverage, at least 10X coverage, at least 20Xcoverage, at least 40X coverage, or greater coverage ofthe genomic component.
VII. Digital Processor
[0291] The methods, compositions, devices, and kitsof this disclosure may be used with any suitable proces-sor, digital processor or computer. The digital processormay be programmed, for example, to operate any com-ponent of a device and/or execute methods describedherein. The digital processor may be capable of trans-mitting or receiving electronic signals through a computernetwork, such as for example, the Internet and/or com-municating with a remote computer. One or more periph-
eral devices such as screen display, printer, memory,data storage, and/or electronic display adaptors may bein communication with the digital processor. One or moreinput devices such as keyboard, mouse, or joystick maybe in communication with the digital processor. The dig-ital processor may also communicate with detector suchthat the detector performs measurements at desired orotherwise predetermined time points or at time pointsdetermined from feedback received from pre-processingunit or other devices.[0292] A conceptual schematic for an example controlassembly is shown in Fig 18. A computer, serves as thecentral hub for control assembly. The computer is in com-munication with a display, one or more input devices(e.g., a mouse, keyboard, camera, etc.), and optionallya printer. The control assembly, via its computer, is incommunication with one or more devices: optionally asample pre-processing unit, one or more sampleprocessing units (such as a sequence, thermocycler, ormicrofluidic device), and optionally a detector. The con-trol assembly may be networked, for example, via anEthernet connection. A user may provide inputs (e.g., theparameters necessary for a desired set of nucleic acidamplification reactions or flow rates for a microfluidic de-vice) into the computer, using an input device. The inputsare interpreted by the computer, to generate instructions.The computer communicates such instructions to the op-tional sample pre-processing unit, the one or more sam-ple processing units, and/or the optional detector for ex-ecution.[0293] Moreover, during operation of the optional sam-ple pre-processing unit, one or more sample processingunits, and/or the optional detector, each device may com-municate signals back to computer. Such signals maybe interpreted and used by computer to determine if anyof the devices require further instruction. The computermay also modulate the sample pre-processing unit suchthat the components of a sample are mixed appropriatelyand fed, at a desired or otherwise predetermined rate,into the sample processing unit (such as the microfluidicdevice).[0294] The computer may also communicate with a de-tector such that the detector performs measurements atdesired or otherwise predetermined time points or at timepoints determined from feedback received from pre-processing unit or sample processing unit. The detectormay also communicate raw data obtained during meas-urements back to the computer for further analysis andinterpretation.[0295] Analysis may be summarized in formats usefulto an end user via a display and/or printouts generatedby a printer. Instructions or programs used to control thesample pre-processing unit, the sample processing unit,and/or the detector; data acquired by executing any ofthe methods described herein; or data analyzed and/orinterpreted may be transmitted to or received from oneor more remote computers, via a network, which, for ex-ample, could be the Internet.
105 106

EP 3 013 957 B1
55
5
10
15
20
25
30
35
40
45
50
55
[0296] In some embodiments, the method of bead for-mation may be executed with the aid of a digital processorin communication with a droplet generator. The digitalprocessor may control the speed at which droplets areformed or control the total number of droplets that aregenerated. In some embodiments, the method of attach-ing samples to barcoded beads may be executed withthe aid of a digital processor in communication with themicrofluidic device. Specifically, the digital processormay control the volumetric amount of sample and/orbeads injected into the input channels and may also con-trol the flow rates within the channels. In some embodi-ments, the method of attaching oligonucleotides, prim-ers, and the like may be executed with the aid of a digitalprocessor in communication with a thermocycler or otherprogrammable heating element. Specifically, the digitalprocessor may control the time and temperature of cyclesduring ligation or amplification. In some embodiments,the method of sequencing a sample may be executedwith the aid of a digital processor in communication witha sequencing device.
VIII. Kits
[0297] In some cases, this disclosure provides a kitcomprising a microfluidic device, a plurality of barcodedbeads, and instructions for utilizing the microfluidic de-vice and combining barcoded beads with customer sam-ple to create fluidic droplets containing both. As specifiedthroughout this disclosure, any suitable sample may beincorporated into the fluidic droplets. As describedthroughout this disclosure, a bead may be designed tobe degradable or non-degradable. In this case, the kitmay or may not include a reducing agent for bead deg-radation.[0298] In some cases, this disclosure provides a kitcomprising a plurality of barcoded beads, suitable am-plification reagents, e.g., optionally including one or moreof polymerase enzymes, nucleoside triphosphates ortheir analogues, primer sequences, buffers, and the like,and instructions for combining barcoded beads with cus-tomer sample. As specified throughout this disclosure,any suitable sample may be used. As specified through-out this disclosure, the amplification reagents may in-clude a polymerase that will not accept or process uracil-containing templates. A kit of this disclosure may alsoprovide agents to form an emulsion, including an oil andsurfactant.
IX. Applications
Barcoding Sample Materials
[0299] The methods, compositions and systems de-scribed herein are particularly useful for attaching bar-code nucleic acid sequences, to sample materials andcomponents of those sample materials. In general, thisis accomplished by partitioning sample material compo-
nents into separate partitions or reaction volumes inwhich are co-partitioned a plurality of barcodes, whichare then attached to sample components within the samepartition.[0300] In an exemplary process, a bead is providedthat includes a plurality of nucleic acid barcode moleculesthat each comprise a common nucleic acid barcode se-quence. The bead may comprise any suitable type ofbead (e.g., a degradable bead, a gel bead), to which thenucleic acid barcode molecules are releasably attached,releasably coupled, or are releasably associated. More-over, any suitable number of nucleic acid barcode mol-ecules may be included, including numbers of nucleicacid barcode molecules per bead described elsewhereherein. For example, the nucleic acid barcode moleculesmay be releasably attached to, releasably coupled to, orreleasably associated with the bead via a cleavable link-age such as, for example, a chemically cleavable linkage(e.g., a disulfide linkage, or any other type of chemicallycleavable linkage described herein), a photocleavablelinkage, and/or a thermally cleavable linkage. In somecases, the bead may be a degradable bead (e.g., a pho-todegradable bead, a chemically degradable bead, athermally degradable bead, or any other type of degra-dable bead described elsewhere herein). Moreover, thebead may comprise chemically-cleavable cross-linking(e.g., disulfide cross-linking) as described elsewhereherein.[0301] The bead is then co-partitioned into a partition,with a sample material, sample material component, frag-ment of a sample material, or a fragment of a samplematerial component. The sample material (or componentor fragment thereof) may be any appropriate sampletype, including the example sample types described else-where herein. In cases where a sample material or com-ponent of a sample material comprises one or more nu-cleic acid fragments, the one or more nucleic acid frag-ments may be of any suitable length, including, for ex-ample, nucleic acid fragment lengths described else-where herein. The partition may include any of a varietyof partitions, including for example, wells, microwells, na-nowells, tubes or containers, or in preferred cases drop-lets (e.g., aqueous droplets in an emulsion) or microcap-sules in which the bead may be co-partitioned. In somecases, the bead may be provided in a first aqueous fluidand the sample material, sample material component, orfragment of a sample material component may be pro-vided in a second aqueous fluid. During co-partitioning,the first aqueous fluid and second aqueous fluid may becombined within a droplet within an immiscible fluid. Insome cases, the partition may comprise no more thanone bead. In other cases, the partition may comprise nomore than one, two, three, four, five, six, seven, eight,nine, or ten beads. In other cases, the partition may com-prise at least one, two, three, four, five, six, seven, eight,nine, ten, or more beads.[0302] Once co-partitioned, the nucleic acid barcodemolecules may be released from the bead (e.g., via deg-
107 108

EP 3 013 957 B1
56
5
10
15
20
25
30
35
40
45
50
55
radation of the bead, cleaving a chemical linkgage be-tween the nucleic acid barcode molecules and the bead,or any other suitable type of release, including types ofrelease described elsewhere herein) into the partition,and attached to the sample components co-partitionedtherewith. In some cases, the bead may be crosslinkedand the crosslinking of the bead may comprise a disulfidelinkage. In addition, or as an alternative, the nucleic acidbarcode molecules may be linked to the bead via a di-sulfide linkage. In either case, the nucleic acid barcodemolecules may be released from the bead by exposingthe first partition to a reducing agent (e.g., DTT, TCEP,or any other exemplary reducing agent described else-where herein).[0303] As noted elsewhere herein, attachment of thebarcodes to sample components includes the direct at-tachment of the barcode oligonucleotides to sample ma-terials, e.g. through ligation, hybridization, or other asso-ciations. Additionally, in many cases, for example, in bar-coding of nucleic acid sample materials (e.g., templatenucleic acid sequences, template nucleic acid mole-cules), components or fragments thereof, such attach-ment may additionally comprise use of the barcode con-taining oligonucleotides that also comprise as primingsequences. The priming sequence can be complemen-tary to at least a portion of a nucleic acid sample materialand can be extended along the nucleic acid sample ma-terials to create complements to such sample materials,as well as at least partial amplification products of thosesequences or their complements.[0304] In another exemplary process, a plurality ofbeads can be provided that comprise a plurality of differ-ent nucleic acid barcode sequences. Each of the beadscan comprise a plurality of nucleic acid barcode mole-cules having the same nucleic acid barcode sequenceassociated therewith. Any suitable number of nucleic acidbarcode molecules may be associated with each of thebeads, including numbers of nucleic acid barcode mole-cules per partition described elsewhere herein. Thebeads may comprise any suitable number of differentnucleic acid barcode sequences, including, for example,at least about 2, 10, 100, 500, 1000, 5000, 10000, 50000,100000, 500000, 1000000, 5000000, 10000000,50000000, or 1000000000, or more different nucleic acidbarcode sequences.[0305] In some cases, the plurality of beads may com-prise a plurality of different beads where each of the dif-ferent beads comprises a plurality of releasably attached,releasably coupled, or releasably associated oligonucle-otides comprising a common barcode sequence, with theoligonucleotides associated with each different beadscomprising a different barcode sequence. The numberof different beads may be, for example, at least about 2,10, 100, 500, 1000, 5000, 10000, 50000, 100000,500000, 1000000, 5000000, 10000000, 50000000, or1000000000, or more different beads.[0306] The beads may be co-partitioned with samplematerials, fragments of a sample material, components
of a sample material, or fragments of a component(s) ofa sample material into a plurality of partitions. In somecases, a subset of the partitions may comprise the samenucleic acid barcode sequence. For example, at leastabout 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 15%,20%, 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%,70%, 75%, 80%, 85%, 90%, 95%, or more of the parti-tions may comprise the same nucleic acid barcode se-quence. Moreover, the distribution of beads per partitionmay also vary according to, for example, occupancy ratesdescribed elsewhere herein. In cases where the pluralityof beads comprises a plurality of different beads, eachdifferent bead may be disposed within a separate parti-tion.[0307] Following co-partitioning, the nucleic acid bar-code molecules associated with the beads can be re-leased into the plurality of partitions. The released nucleicacid barcode molecules can then be attached to the sam-ple materials, sample material components, fragmentsof a sample material, or fragments of sample materialcomponents, within the partitions. In the case of barcod-ed nucleic acid species (e.g., barcoded sample nucleicacid, barcoded template nucleic acid, barcoded frag-ments of one or more template nucleic acid sequences,etc.), the barcoded nucleic acid species may be se-quenced as described elsewhere herein.[0308] In another exemplary process, a sample of nu-cleic acids may be combined with a library of barcodedbeads (including types of beads described elsewhereherein) to form a mixture. In some cases, the barcodesof the beads may, in addition to a barcode sequence,each comprise one or more additional sequences suchas, for example, a universal sequence and/or a functionalsequence (e.g., a random N-mer or a targeted N-mer, asdescribed elsewhere herein). The mixture may be parti-tioned into a plurality of partitions, with at least a subsetof the partitions comprising at most one barcoded bead.Within the partitions, the barcodes may be released fromthe beads, using any suitable route, including types ofrelease described herein. A library of barcoded beadsmay be generated via any suitable route, including theuse of methods and compositions described elsewhereherein. In some cases, the sample of nucleic acids maybe combined with the library of barcoded beads and/orthe resulting mixture partitioned with the aid of a micro-fluidic device, as described elsewhere herein. In caseswhere the released barcodes also comprise a primer se-quence (e.g., such as a targeted N-mer or a random N-mer as described elsewhere herein), the primer sequenc-es of the barcodes may be hybridize with the samplenucleic acids and, if desired, an amplification reactioncan be completed in the partitions.
Polynucleotide Sequencing
[0309] Generally, the methods and compositions de-scribed herein are useful for preparation of oligonucle-otide fragments for downstream applications such as se-
109 110

EP 3 013 957 B1
57
5
10
15
20
25
30
35
40
45
50
55
quencing. In particular, these methods, compositionsand systems are useful in the preparation of sequencinglibraries. Sequencing may be performed by any availabletechnique. For example, sequencing may be performedby the classic Sanger sequencing method. Sequencingmethods may also include: high-throughput sequencing,pyrosequencing, sequencing-by-ligation, sequencing bysynthesis, sequencing-by-hybridization, RNA-Seq (Illu-mina), Digital Gene Expression (Helicos), next genera-tion sequencing, single molecule sequencing by synthe-sis (SMSS) (Helicos), massively-parallel sequencing,clonal single molecule Array (Solexa), shotgun sequenc-ing, Maxim-Gilbert sequencing, primer walking, and anyother sequencing methods known in the art.[0310] For example, a plurality of target nucleic acidsequences may be sequenced by providing a plurality oftarget nucleic sequences and separating the target nu-cleic acid sequences into a plurality of separate parti-tions. Each of the separate partitions can comprise oneor more target nucleic acid sequences and a plurality ofoligonucleotides. The separate partitions may compriseany suitable number of different barcode sequences(e.g., at least 1,000 different barcode sequences, at least10,000 different barcode sequences, at least 100,000different barcode sequences, at least 1,000,000 differentbarcode sequences, at least 10,000,000 different bar-code sequences, or any other number of different bar-code sequences as described elsewhere herein). More-over, the oligonucleotides in a given partition can com-prise a common barcode sequence. The oligonucle-otides and associated common barcode sequence in agiven partition can be attached to fragments of the oneor more target nucleic acids or to copies of portions ofthe target nucleic acid sequences within the given parti-tion. Following attachment, the separate partitions canthen be pooled. The fragments of the target nucleic acidsor the copies of the portions of the target nucleic acidsand attached barcode sequences can then be se-quenced.[0311] In another example, a plurality of target nucleicacid sequences may be sequenced by providing the tar-get nucleic acid sequences and separating them into aplurality of separate partitions. Each partition of the plu-rality of separate partitions can include one or more ofthe target nucleic acid sequences and a bead having aplurality of attached oligonucleotides. The oligonucle-otides attached to a given bead may comprise a commonbarcode sequence. The oligonucleotides associated witha bead can be attached to fragments of the target nucleicacid sequences or to copies of portions of the target nu-cleic acid sequences within a given partition, such thatthe fragments or copies of the given partition are alsoattached to the common barcode sequence associatedwith the bead. Following attachment of the oligonucle-otides to the fragments of the target nucleic acid sequenc-es or the copies of the portions of the target nucleic acidsequences, the separate partitions can then be pooled.The fragments of the target nucleic acid sequences or
the copies of the portions of the target nucleic acid se-quences and any attached barcode sequences can thenbe sequenced (e.g., using any suitable sequencing meth-od, including those described elsewhere herein) to pro-vide barcoded fragment sequences or barcoded copysequences. The barcoded fragment sequences or bar-coded copy sequences can be assembled into one ormore contiguous nucleic acid sequence based, in part,upon a barcode portion of the barcoded fragment se-quences or barcoded copy sequences.[0312] In some cases, varying numbers of barcoded-oligonucleotides are sequenced. For example, in somecases about 30%-90% of the barcoded-oligonucleotidesare sequenced. In some cases, about 35%-85%, 40%-80%, 45%-75%, 55%-65%, or 50%-60% of the barcoded-oligonucleotides s are sequenced. In some cases, atleast about 30%, 40%, 50%, 60%, 70%, 80%, or 90% ofbarcoded-oligonucleotides are sequenced. In some cas-es, less than about 30%, 40%, 50%, 60%, 70%, 80%, or90% of the barcoded-oligonucleotides are sequenced.[0313] In some cases, sequences from fragments areassembled to provide sequence information for a contig-uous region of the original target polynucleotide that maybe longer than the individual sequence reads. Individualsequence reads may be about 10-50, 50-100, 100-200,200-300, 300-400, or more nucleotides in length.[0314] The identities of the barcodes may serve to or-der the sequence reads from individual fragments as wellas to differentiate between haplotypes. For example,when combining individual sample fragments and bar-coded beads within fluidic droplets, parental polynucle-otide fragments may be separated into different droplets.With an increase in the number of fluidic droplets andbeads within a droplet, the likelihood of a fragment fromboth a maternal and paternal haplotype contained withinthe same fluidic droplet associated with the same beadmay become negligibly small. Thus, sequence readsfrom fragments in the same fluidic droplet and associatedwith the same bead may be assembled and ordered.[0315] In at least one example, the present disclosureprovides nucleic acid sequencing methods, systemscompositions, and combinations of these that are usefulin providing myriad benefits in both sequence assemblyand read-length equivalent, but do so with very highthroughput and reduced sample preparation time andcost.[0316] In general, the sequencing methods describedherein provide for the localized tagging or barcoding offragments of genetic sequences. By tagging fragmentsthat derive from the same location within a larger geneticsequence, one can utilize the presence of the tag or bar-code to inform the assembly process as alluded to above.In addition, the methods described herein can be usedto generate and barcode shorter fragments from a single,long nucleic acid molecule. Sequencing and assemblyof these shorter fragments provides a long read equiva-lent sequence, but without the need for low throughputlonger read-length sequencing technologies.
111 112

EP 3 013 957 B1
58
5
10
15
20
25
30
35
40
45
50
55
[0317] Fig 39 provides a schematic illustration of anexample sequencing method. As shown, a first geneticcomponent 3902 that may comprise, for example, a chro-mosome or other large nucleic acid molecule, is frag-mented into a set of large first nucleic acid fragments,e.g., including fragments 3904 and 3906. The fragmentsof the large genetic component may be non-overlappingor overlapping, and in some cases, may include multifoldoverlapping fragments, in order to provide for high con-fidence assembly of the sequence of the larger compo-nent. In some cases, the fragments of the larger geneticcomponent provide 1X, 2X, 5X, 10X, 20X, 40X or greatercoverage of the larger component.[0318] One or more of the first fragments 3904 is thenprocessed to separately provide overlapping set of sec-ond fragments of the first fragment(s), e.g., second frag-ment sets 3908 and 3910. This processing also providesthe second fragments with a barcode sequence that isthe same for each of the second fragments derived froma particular first fragment. As shown, the barcode se-quence for second fragment set 3908 is denoted by "1"while the barcode sequence for fragment set 3910 is de-noted by "2". A diverse library of barcodes may be usedto differentially barcode large numbers of different frag-ment sets. However, it is not necessary for every secondfragment set from a different first fragment to be barcodedwith different barcode sequences. In fact, in many cases,multiple different first fragments may be processed con-currently to include the same barcode sequence. Diversebarcode libraries are described in detail elsewhere here-in.[0319] The barcoded fragments, e.g., from fragmentsets 3908 and 3910, may then be pooled for sequencing.Once sequenced, the sequence reads 3912 can be at-tributed to their respective fragment set, e.g., as shownin aggregated reads 3914 and 3916, at least in part basedupon the included barcodes, and optionally, and prefer-ably, in part based upon the sequence of the fragmentitself. The attributed sequence reads for each fragmentset are then assembled to provide the assembled se-quence for the first fragments, e.g., fragment sequences3918 and 3920, which in turn, may be assembled intothe sequence 3922 of the larger genetic component.[0320] In accordance with the foregoing, a large ge-netic component, such as a long nucleic acid fragment,e.g., 1, 10, 20, 40, 50, 75, 100, 1000 or more kb in length,a chromosomal fragment or whole chromosome, or partof or an entire genome (e.g., genomic DNA) is fragment-ed into smaller first fragments. Typically, these fragmentsmay be anywhere from about 1000 to about 100000 bas-es in length. In certain preferred aspects, the fragmentswill be between about 1 kb and about 100 kb, or betweenabout 5 kb and about 50 kb, or from about 10kb to about30kb, and in some cases, between about 15 kb and about25 kb. Fragmentation of these larger genetic componentsmay be carried out by any of a variety of convenient avail-able processes, including commercially available shearbased fragmenting systems, e.g., Covaris fragmentation
systems, size targeted fragmentation systems, e.g., BluePippin (Sage Sciences), enzymatic fragmentation proc-esses, e.g., using restriction endonucleases, or the like.As noted above, the first fragments of the larger geneticcomponent may comprise overlapping or non-overlap-ping first fragments. Although described here as beingfragmented prior to partitioning, it will be appreciated thatfragmentation may optionally and/or additionally be per-formed later in the process, e.g., following one or moreamplification steps, to yield fragments of a desired sizefor sequencing applications.[0321] In preferred aspects, the first fragments aregenerated from multiple copies of the larger genetic com-ponent or portions thereof, so that overlapping first frag-ments are produced. In preferred aspects, the overlap-ping fragments will constitute greater than 1X coverage,greater than 2X coverage, greater than 5X coverage,greater than 10X coverage, greater than 20X coverage,greater than 40 X coverage, or even greater coverage ofthe underlying larger genetic component or portion there-of. The first fragments are then segregated to differentreaction volumes. In some cases, the first fragments maybe separated so that reaction volumes contain one orfewer first fragments. This is typically accomplished byproviding the fragments in a limiting dilution in solution,such that allocation of the solution to different reactionvolumes results in a very low probability of more thanone fragment being deposited into a given reaction vol-ume. However, in most cases, a given reaction volumemay include multiple different first fragments, and caneven have 2, 5, 10, 100, 100 or even up to 10,000 ormore different first fragments in a given reaction volume.Again, achieving a desired range of fragment numberswithin individual reaction volumes is typically accom-plished through the appropriate dilution of the solutionfrom which the first fragments originate, based upon anunderstanding of the concentration of nucleic acids inthat starting material.[0322] The reaction volumes may include any of varietyof different types of vessels or partitions. For example,the reaction volumes may include conventional reactionvessels, such as test tubes, reaction wells, microwells,nanowells, or they may include less conventional reac-tion volumes, such as droplets within a stabilized emul-sion, e.g., a water in oil emulsion system. In preferredaspects, droplets are preferred as the reaction volumesfor their extremely high multiplex capability, e.g., allowingthe use of hundreds of thousands, millions, tens of mil-lions or even more discrete droplet/reaction volumeswithin a single container. Within each reaction volume,the fragments that are contained therein are then sub-jected to processing that both derives sets of overlappingsecond fragments of each of the first fragments, and alsoprovides these second fragments with attached barcodesequences. As will be appreciated, in preferred aspects,the first fragments are partitioned into droplets that alsocontain one or more microcapsules or beads that includethe members of the barcode library used to generate and
113 114

EP 3 013 957 B1
59
5
10
15
20
25
30
35
40
45
50
55
barcode the second fragments.[0323] In preferred aspects, the generation of thesesecond fragments is carried out through the introductionof primer sequences that include the barcode sequencesand that are capable of hybridizing to portions of the firstfragment and be extended along the first fragment to pro-vide a second fragment including the barcode sequence.These primers may comprise targeted primer sequenc-es, e.g., to derive fragments that overlap specific portionsof the first fragment, or they may comprise universal prim-ing sequences, e.g., random primers, that will prime mul-tiple different regions of the first fragments to create largeand diverse sets of second fragments that span the firstfragment and provide multifold overlapping coverage.These extended primer sequences may be used as thesecond fragments, or they may be further replicated oramplified. For example, iterative priming against the ex-tended sequences, e.g., using the same primer contain-ing barcoded oligonucleotides. In certain preferred as-pects, the generation of the second sets of fragmentsgenerates the partial hairpin replicates of portions of thefirst fragment, as described elsewhere herein that eachinclude barcode sequences, e.g., for PHASE amplifica-tion as described herein. As noted elsewhere herein, theformation of the partial hairpin is generally desired to pre-vent repriming of the replicated strand, e.g., making acopy of a copy. As such, the partial hairpin is typicallypreferentially formed from the amplification product dur-ing annealing as compared to a primer annealing to theamplification product, e.g., the hairpin will have a higherTm than the primer product pair.[0324] The second fragments are generally selectedto be of a length that is suitable for subsequent sequenc-ing. For short read sequencing technologies, such frag-ments will typically be from about 50 bases to about 1000bases in sequenceable length, from about 50 bases toabout 900 bases in sequenceable length, from about 50bases to about 800 bases in sequenceable length, fromabout 50 bases to about 700 bases in sequenceablelength, from about 50 bases to about 600 bases in se-quenceable length, from about 50 bases to about 500bases in sequenceable length, from about 50 bases toabout 400 bases in sequenceable length, from about 50bases to about 300 bases in sequenceable length, fromabout 50 bases to about 250 bases in sequenceablelength, from about 50 bases to about 200 bases in se-quenceable length, or from about 50 bases to about 100bases in sequenceable length, including the barcode se-quence segments, and functional sequences that aresubjected to the sequencing process.[0325] Once the overlapping, barcoded second frag-ment sets are generated, they may be pooled for subse-quent processing and ultimately, sequencing. For exam-ple, in some cases, the barcoded fragments may be sub-sequently subjected to additional amplification, e.g., PCRamplification, as described elsewhere herein. Likewise,these fragments may additionally, or concurrently, beprovided with sample index sequences to identify the
sample from which collections of barcoded fragmentshave derived, as well as providing additional functionalsequences for use in sequencing processes.[0326] In addition, clean up steps may also optionallybe performed, e.g., to purify nucleic acid componentsfrom other impurities, to size select fragment sets for se-quencing, or the like. Such clean up steps may includepurification and/or size selection upon SPRI beads (suchas Ampure® beads, available from Beckman Coulter,Inc.). In some cases, multiple process steps may be car-ried out in an integrated process while the fragments areassociated with SPRI beads, e.g., as described in Fisheret al., Genome Biol. 2011:12(1):R1 (E-pub Jan 4, 2011).[0327] As noted previously, in many cases, short readsequencing technologies are used to provide the se-quence information for the second fragment sets. Ac-cordingly, in preferred aspects, second fragment sets willtypically comprise fragments that, when including thebarcode sequences, will be within the read length of thesequencing system used. For example, for IlluminaHiSeq® sequencing, such fragments may be betweengenerally range from about 100 bases to about 200 basesin length, when carrying out paired end sequencing. Insome cases, longer second fragments may be se-quenced when accessing only the terminal portions ofthe fragments by the sequencing process.[0328] As noted above with reference to Fig 39, thesequence reads for the various second fragments arethen attributed to their respective starting nucleic acidsegment based in part upon the presence of a particularbarcode sequence, and in some cases, based in part onthe actual sequence of the fragment, i.e., a non-barcodeportion of the fragment sequence. As will be appreciated,despite being based upon short sequence data, one caninfer that two sequences sharing the same barcode likelyoriginated from the same longer first fragment sequence,especially where such sequences are otherwise assem-ble-able into a contiguous sequence segment, e.g., usingother overlapping sequences bearing the common bar-code. Once the first fragments are assembled, they maybe assembled into larger sequence segments, e.g., thefull length genetic component.[0329] In one exemplary process, one or more frag-ments of one or more template nucleic acid sequencesmay be barcoded using a method described herein. Afragment of the one or more fragments may be charac-terized based at least in part upon a nucleic acid barcodesequence attached thereto. Characterization of the frag-ment may also include mapping the fragment to its re-spective template nucleic acid sequence or a genomefrom which the template nucleic acid sequence was de-rived. Moreover, characterization may also include iden-tifying an individual nucleic acid barcode sequence anda sequence of a fragment of a template nucleic acid se-quence attached thereto.[0330] In some cases, sequencing methods describedherein may be useful in characterizing a nucleic acid seg-ment or target nucleic acid. In some example methods,
115 116

EP 3 013 957 B1
60
5
10
15
20
25
30
35
40
45
50
55
a nucleic acid segment may be characterized by co-par-titioning the nucleic acid segment and a bead (e.g., in-cluding any suitable type of bead described herein) com-prising a plurality of oligonucleotides that include a com-mon nucleic acid barcode sequence, into a partition (in-cluding any suitable type of partition described herein,such as, for example, a droplet). The oligonucleotidesmay be releasably attached to the bead (e.g., releasablefrom the bead upon application of a stimulus to the bead,such as, for example, a thermal stimulus, a photo stim-ulus, and a chemical stimulus) as described elsewhereherein, and/or may comprise one or more functional se-quences (e.g., a primer sequence, a primer annealingsequence, an immobilization sequence, any other suita-ble functional sequence described elsewhere herein,etc.) and/or one or more sequencing primer sequencesas described elsewhere herein. Moreover, any suitablenumber of oligonucleotides may be attached to the bead,including numbers of oligonucleotides attached to beadsdescribed elsewhere herein.[0331] Within the partition, the oligonucleotides maybe attached to fragments of the nucleic segment or tocopies of portions of the nucleic acid segment, such thatthe fragmentsor copies are also attached to the commonnucleic barcode sequence. The fragments may be over-lapping fragments of the nucleic acid segment and may,for example, provide greater than 2X coverage, greaterthan 5X coverage, greater than 10X coverage, greaterthan 20X coverage, greater than 40X coverage, or evengreater coverage of the nucleic acid segment. In somecases, the oligonucleotides may comprise a primer se-quence capable of annealing with a portion of the nucleicacid segment or a complement thereof. In some cases,the oligonucleotides may be attached by extending theprimer sequences of the oligonucleotides to replicate atleast a portion of the nucleic acid segment or complementthereof, to produce a copy of at least a portion of thenucleic acid segment comprising the oligonucleotide,and, thus, the common nucleic acid barcode sequence.[0332] Following attachment of the oligonucleotides tothe fragments of the nucleic acid segment or to the copiesof the portions of the nucleic acid segment, the fragmentsof the nucleic acid segment or the copies of the portionsof the nucleic acid segment and the attached oligonucle-otides (including the oligonucleotide’s barcode se-quence) may be sequenced via any suitable sequencingmethod, including any type of sequencing method de-scribed herein, to provide a plurality of barcoded frag-ment sequences or barcoded copy sequences. Followingsequencing, the fragments of the nucleic acid segmentor the copies of the portions of the nucleic acid segmentcan be characterized as being linked within the nucleicacid segment at least in part, upon their attachment tothe common nucleic acid barcode sequence. As will beappreciated, such characterization may include se-quences that are characterized as being linked and con-tiguous, as well as sequences that may be linked withinthe same fragment, but not as contiguous sequences.
Moreover, the barcoded fragment sequences or barcod-ed copy sequences generated during sequencing can beassembled into one or more contiguous nucleic acid se-quences based at least in part on the common nucleicacid barcode sequence and/or a non-barcode portion ofthe barcoded fragment sequences or barcoded copy se-quences.[0333] In some cases, a plurality of nucleic acid seg-ments (e.g., fragments of at least a portion of a genome,as described elsewhere herein) may be co-partitionedwith a plurality of different beads in a plurality of separatepartitions, such that each partition of a plurality of differentpartitions of the separate partitions contains a singlebead. The plurality of different beads may comprise aplurality of different barcode sequences (e.g., at least1,000 different barcode sequences, at least 10,000 dif-ferent barcode sequences, at least 100,000 different bar-code sequences, at least 1,000,000 different barcodessequences, or any other number of different barcode se-quences as described elsewhere herein). In some cases,two or more, three or more, four or more, five or more,six or more, seven or more of the plurality of separatepartitions may comprise beads that comprise the samebarcode sequence. In some cases, at least 0.01%, 0.1%,1%, 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%,50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%,or 99% of the separate partitions may comprise beadshaving the same barcode sequence. Moreover, eachbead may comprise a plurality of attached oligonucle-otides that include a common nucleic acid barcode se-quence.[0334] Following co-partitioning, barcode sequencescan be attached to fragments of the nucleic acid seg-ments or to copies of portions of the nucleic acid seg-ments in each partition. The fragments of the nucleic acidsegments or the copies of the portions of the nucleic acidsegments can then be pooled from the separate parti-tions. After pooling, the fragments of the nucleic acid seg-ments or copies of the portions of the nucleic acid seg-ments and any associated barcode sequences can besequenced (e.g., using any suitable sequencing method,including those described herein) to provide sequencedfragment or sequenced copies. The sequenced frag-ments or sequenced copies can be characterized as de-riving from a common nucleic acid segment, based atleast in part upon the sequenced fragments or se-quenced copies comprising a common barcode se-quence. Moreover, sequences obtained from the se-quenced fragments or sequenced copies may be assem-bled to provide a contiguous sequence of a sequence(e.g., at least a portion of a genome) from which the se-quenced fragments or sequenced copies originated. Se-quence assembly from the sequenced fragments or se-quenced copies may be completed based, at least in part,upon each of a nucleotide sequence of the sequencedfragments and a common barcode sequence of the se-quenced fragments.[0335] In another example method, a target nucleic ac-
117 118

EP 3 013 957 B1
61
5
10
15
20
25
30
35
40
45
50
55
id may be characterized by partitioning fragments of thetarget nucleic acid into a plurality of droplets. Each dropletcan comprise a bead attached to a plurality of oligonu-cleotides comprising a common barcode sequence. Thecommon barcode sequence can be attached to frag-ments of the fragments of the target nucleic acid in thedroplets. The droplets can then be pooled and the frag-ments and associated barcode sequences of the pooleddroplets sequenced using any suitable sequencing meth-od, including sequencing methods described herein. Fol-lowing sequencing, the fragments of the fragments of thetarget nucleic acid may be mapped to the fragments ofthe target nucleic acid based, at least in part, upon thefragments of the fragments of the target nucleic acid com-prising a common barcode sequence.[0336] The application of the methods, compositionsand systems described herein in sequencing may gen-erally be applicable to any of a variety of different se-quencing technologies, including NGS sequencing tech-nologies such as Illumina MiSeq, HiSeq and X10 Se-quencing systems, as well as sequencing systems avail-able from Life Technologies, Inc., such as the Ion Torrentline of sequencing systems. While discussed in terms ofbarcode sequences, it will be appreciated that the se-quenced barcode sequences may not include the entirebarcode sequence that is included, e.g., accounting forsequencing errors. As such, when referring to character-ization of two barcode sequences as being the same bar-code sequence, it will be appreciated that this may bebased upon recognition of a substantial portion of a bar-code sequence, e.g., varying by fewer than 5, 4, 3, 2 oreven a single base.
Sequencing from Small Numbers of Cells
[0337] Methods provided herein may also be used toprepare polynucleotides contained within cells in a man-ner that enables cell-specific information to be obtained.The methods enable detection of genetic variations fromvery small samples, such as from samples comprisingabout 10-100 cells. In some cases, about 1, 5, 10, 20,30, 40, 50, 60, 70, 80, 90 or 100 cells may be used in themethods described herein. In some cases, at least about1, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90 or 100 cells maybe used in the methods described herein. In other cases,at most about 5, 10, 20, 30, 40, 50, 60, 70, 80, 90 or 100cells may be used in the methods described herein.[0338] In an example, a method may comprise parti-tioning a cellular sample (or crude cell extract) such thatat most one cell (or extract of one cell) is present withina partition, e.g., fluidic droplet, and is co-partitioned withthe barcode oligonucleotides, e.g., as described above.Processing then involves lysing the cells, fragmentingthe polynucleotides contained within the cells, attachingthe fragmented polynucleotides to barcoded beads, pool-ing the barcoded beads, and sequencing the resultingbarcoded nucleic acid fragments.[0339] As described elsewhere herein, the barcodes
and other reagents are associated with (e.g. encapsulat-ed within, coated on, associated with, or dispersed within)a bead (e.g. gel bead). The bead may be loaded into afluidic droplet contemporaneously with loading of a sam-ple (e.g. a cell), such that each cell is contacted with adifferent bead. This technique may be used to attach aunique barcode to oligonucleotides obtained from eachcell. The resulting tagged oligonucleotides may then bepooled and sequenced, and the barcodes may be usedto trace the origin of the oligonucleotides. For example,oligonucleotides with identical barcodes may be deter-mined to originate from the same cell, while oligonucle-otides with different barcodes may be determined to orig-inate from different cells.[0340] The methods described herein may be used todetect a specific gene mutation that may indicate thepresence of a disease, such as cancer. For example,detecting the presence of a V600 mutation in the BRAFgene of a colon tissue sample may indicate the presenceof colon cancer. In other cases, prognostic applicationsmay include the detection of a mutation in a specific geneor genes that may serve as increased risk factors fordeveloping a specific disease. For example, detectingthe presence of a BRCA1 mutation in a mammary tissuesample may indicate a higher level of risk to developingbreast cancer than a person without this mutation. Insome examples, this disclosure provides methods ofidentifying mutations in two different oncogenes (e.g.,KRAS and EGRF). If the same cell comprises genes withboth mutations, this may indicate a more aggressive formof cancer. In contrast, if the mutations are located in twodifferent cells, this may indicate that the cancer may bemore benign, or less advanced.
Analysis of Gene Expression
[0341] Methods of the disclosure may be applicable toprocessing samples for the detection of changes in geneexpression. A sample may comprise a cell, mRNA, orcDNA reverse transcribed from mRNA. The sample maybe a pooled sample, comprising extracts from severaldifferent cells or tissues, or a sample comprising extractsfrom a single cell or tissue.[0342] Cells may be placed directly into a fluidic dropletand lysed. After lysis, the methods of the disclosure maybe used to fragment and barcode the oligonucleotides ofthe cell for sequencing. Oligonucleotides may also beextracted from cells prior to introducing them into a fluidicdroplet used in a method of the disclosure. Reverse tran-scription of mRNA may be performed in a fluidic dropletdescribed herein, or outside of such a fluidic droplet. Se-quencing cDNA may provide an indication of the abun-dance of a particular transcript in a particular cell overtime, or after exposure to a particular condition.
Partitioning Polynucleotides from Cells or Proteins
[0343] In one example the compositions, methods, de-
119 120

EP 3 013 957 B1
62
5
10
15
20
25
30
35
40
45
50
55
vices, and kits provided in this disclosure may be usedto encapsulate cells or proteins within the fluidic droplets.In one example, a single cell or a plurality of cells (e.g.,2, 10, 50, 100, 1000, 10000, 25000, 50000, 10000,50000, 1000000, or more cells) maybe loaded onto, into,or within a bead along with a lysis buffer within a fluidicdroplet and incubated for a specified period of time. Thebead may be porous, to allow washing of the contents ofthe bead, and introduction of reagents into the bead,while maintaining the polynucleotides of the one or morecells (e.g. chromosomes) within the fluidic droplets. Theencapsulated polynucleotides of the one or more cells(e.g. chromosomes) may then be processed accordingto any of the methods provided in this disclosure, orknown in the art. This method can also be applied to anyother cellular component, such as proteins.
Epigenetic Applications
[0344] Compositions, methods, devices, and kits ofthis disclosure may be useful in epigenetic applications.For example, DNA methylation can be in indicator of ep-igenetic inheritance, including single nucleotide polymor-phisms (SNPs). Accordingly, samples comprising nucle-ic acid may be treated in order to determine bases thatare methylated during sequencing. In some cases, asample comprising nucleic acid to be barcoded may besplit into two aliquots. One aliquot of the sample may betreated with bisulfite in order to convert unmethylated cy-tosine containing nucleotides to uracil containing nucle-otides. In some cases, bisulfite treatment can occur priorto sample partitioning or may occur after sample parti-tioning. Each aliquot may then be partitioned (if not al-ready partitioned), barcoded in the partitions, and addi-tional sequences added in bulk as described herein togenerate sequencer-ready products. Comparison of se-quencing data obtained for each aliquot (e.g., bisulfite-treated sample vs. untreated sample) can be used todetermine which bases in the sample nucleic acid aremethylated.[0345] In some cases, one aliquot of a split samplemay be treated with methylation-sensitive restriction en-zymes (MSREs). Methylation specific enzymes can proc-ess sample nucleic acid such that the sample nucleicacid is cleaved as methylation sites. Treatment of thesample aliquot can occur prior to sample partitioning ormay occur after sample partitioning and each aliquot maybe partitioned used to generate barcoded, sequencer-ready products. Comparison of sequencing data ob-tained for each aliquot (e.g., MSRE-treated sample vs.untreated sample) can be used to determine which basesin the sample nucleic acid are methylated.
Low Input DNA Applications
[0346] Compositions and methods described hereinmay be useful in the analysis and sequencing of low poly-nucleotide input applications. Methods described herein,
such as PHASE, may aid in obtaining good data qualityin low polynucleotide input applications and/or aid in fil-tering out amplification errors. These low input DNA ap-plications include the analysis of samples to sequenceand identify a particular nucleic acid sequence of interestin a mixture of irrelevant or less relevant nucleic acids inwhich the sequence of interest is only a minority compo-nent, to be able to individually sequence and identify mul-tiple different nucleic acids that are present in an aggre-gation of different nucleic acids, as well as analyses inwhich the sheer amount of input DNA is extremely low.Specific examples include the sequencing and identifi-cation of somatic mutations from tissue samples, or fromcirculating cells, where the vast majority of the samplewill be contributed by normal healthy cells, while a smallminority may derive from tumor or other cancer cells.Other examples include the characterization of multipleindividual population components, e.g., in microbiomeanalysis applications, where the contributions of individ-ual population members may not otherwise be readilyidentified amidst a large and diverse population of micro-bial elements. In a further example, being able to individ-ually sequence and identify different strands of the sameregion from different chromosomes, e.g., maternal andpaternal chromosomes, allows for the identification ofunique variants on each chromosome.[0347] The advantages of the methods and systemsdescribed herein are clearer upon a discussion of theproblems confronted in the present state of the art. Inanalyzing the genetic makeup of sample materials, e.g.,cell or tissue samples, most sequencing technologies re-ly upon the broad amplification of target nucleic acids ina sample in order to create enough material for the se-quencing process. Unfortunately, during these amplifica-tion processes, majority present materials will preferen-tially overwhelm portions of the samples that are presentat lower levels. For example, where a genetic materialfrom a sample is comprised of 95% normal tissue DNA,and 5% of DNA from tumor cells, typical amplificationprocesses, e.g., PCR based amplification, will quicklyamplify the majority present material to the exclusion ofthe minority present material. Furthermore, becausethese amplification reactions are typically carried out ina pooled context, the origin of an amplified sequence, interms of the specific chromosome, polynucleotide or or-ganism will typically not be preserved during the process.[0348] In contrast, the methods and systems describedherein partition individual or small numbers of nucleicacids into separate reaction volumes, e.g., in droplets, inwhich those nucleic acid components may be initially am-plified. During this initial amplification, a unique identifiermay be coupled to the components to the componentsthat are in those separate reaction volumes. Separate,partitioned amplification of the different components, aswell as application of a unique identifier, e.g., a barcodesequence, allows for the preservation of the contributionsof each sample component, as well as attribution of itsorigin, through the sequencing process, including sub-
121 122

EP 3 013 957 B1
63
5
10
15
20
25
30
35
40
45
50
55
sequent amplification processes, e.g., PCR amplifica-tion.[0349] The term "about," as used herein and through-out the disclosure, generally refers to a range that maybe 15% greater than or 15% less than the stated numer-ical value within the context of the particular usage. Forexample, "about 10" would include a range from 8.5 to11.5.[0350] As will be appreciated, the instant disclosureprovides for the use of any of the compositions, libraries,methods, devices, and kits described herein for a partic-ular use or purpose, including the various applications,uses, and purposes described herein. For example, thedisclosure provides for the use of the compositions,methods, libraries, devices, and kits described herein inpartitioning species, in partitioning oligonucleotides, instimulus-selective release of species from partitions, inperforming reactions (e.g., ligation and amplification re-actions) in partitions, in performing nucleic acid synthesisreactions, in barcoding nucleic acid, in preparing polynu-cleotides for sequencing, in sequencing polynucleotides,in polynucleotide phasing, in sequencing polynucleotidesfrom small numbers of cells, in analyzing gene expres-sion, in partitioning polynucleotides from cells, in muta-tion detection, in neurologic disorder diagnostics, in dia-betes diagnostics, in fetal aneuploidy diagnostics, in can-cer mutation detection and forensics, in disease detec-tion, in medical diagnostics, in low input nucleic acid ap-plications, such as circulating tumor cell (CTC) sequenc-ing, in a combination thereof, and in any other application,method, process or use described herein.[0351] Any concentration values provided herein areprovided as admixture concentration values, without re-gard to any in situ conversion, modification, reaction, se-questration or the like. Moreover, where appropriate, thesensitivity and/or specificity of methods (e.g., sequencingmethods, barcoding methods, amplification methods,targeted amplification methods, methods of analyzingbarcoded samples, etc.) described herein may vary. Forexample, a method described herein may have specificityof greater than 50%, 70%, 75%, 80%, 85%, 86%, 87%,88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%,98%, 99%, or 99.5% and/or a sensitivity of greater than50%, 70%, 75%, 80%, 85%, 86%, 87%, 88%, 89%, 90%,91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or99.5%.
X. Examples
Example 1: Creation of Gel Beads Functionalized with Acrydite Primer
[0352] Gel beads are produced according to the meth-od illustrated in Fig 2. In nuclease free water, 1 mL stocksolutions are prepared at the following concentrations:an acrylamide precursor (Compound A) = 40% (v/v) stocksolution, a crosslinker (Bis-acryloyl cystamine - Com-pound B) = 3.19mg/mL in 50:50 mix of acetonitrile:water,
an initiator (Compound C) = 20 mg/mL, and di-sulfideacrydite primer (Compound D) = ImM. From these stocksolutions, 1 mL of an aqueous Gel Bead (GB) workingsolution is prepared by mixing the following volumes: nu-clease free water = 648 mL, Compound A = 150 mL, Com-pound B = 100 mL, Compound C = 100 mL, and Com-pound D = 2 mL. Stock solutions of Compound A and Band GB working solutions are prepared daily.[0353] The Gel Bead (GB) working solution, 201, is anaqueous fluid that contains the crosslinker, BAC, and apolymer precursor solution with di-sulfide-modified acry-dite oligonucleotides at a concentration of between about0.1 and about 100mm. The second fluid, 202, is a fluori-nated oil containing the surfactant, Krytox FSH 1.8% w/wHFE 7500. The accelerator, tetramethylethylenediamine(TEMED) is added a) to the oil prior to droplet generation,203, b) in the line after droplet generation, 205, and/orc) to the outlet reservoir after droplet generation, 206 togive a final concentration of 1% (v/v). TEMED is madefresh daily. Gel beads are generated by sending theaqueous and oil phase fluids to a droplet generator, 204.Polymerization is initiated immediately after droplet gen-eration and continues to the outlet well. Gelation is con-sidered complete after 15-20 minutes. After gelation,generated gel beads are subjected to continuous phaseexchange by washing in HFE 7500, 207, to remove ex-cess oil, and re-suspending the beads in aqueous solu-tion. In some cases, the resulting beads may be presentin an agglomeration. The agglomeration of gel beads areseparated into individual gel beads with vortexing. Gelbeads are visualized under a microscope.
Example 2: Creation of Barcoded Gel Beads by Lim-iting Dilution
[0354] Functionalized gel beads are produced by lim-iting dilution according to the method illustrated in Fig3A and Fig 4. Gel beads with acrydite oligonucleotides(with or without a di-sulfide modification), 301, 401, aremixed with barcode-containing template sequences,302, at a limiting dilution. PCR reagents, 303, includinga biotin labeled read primer, 406, are mixed with the gelbeads and template sequences, 304. The beads, bar-code template, and PCR reagents are emulsified into agel-water-oil emulsion by shaking/agitation, flow focus-ing, or microsieve, 305, preferably such that at most onebarcode template is present in a partition (e.g., droplet)within the emulsion. The emulsion is exposed to one ormore thermal cycles, 306. The first thermocycle incorpo-rates the complement barcode sequence, 408, and im-mobilizes it onto the gel bead.[0355] Continued thermal cycling is performed to clon-ally amplify the barcode throughout the gel bead and toincorporate the 5’ biotin labeled primer into the comple-mentary strand for downstream sorting of beads whichcontain barcode sequences from those that do not. Theemulsion is broken, 307, by adding perfluorodecanol, re-moving the oil, washing with HFE-7500, adding aqueous
123 124

EP 3 013 957 B1
64
5
10
15
20
25
30
35
40
45
50
55
buffer, centrifuging, removing supernatant, removing un-desired products (e.g. primer dimers, starting materials,deoxynucleotide triphosphates (dNTPs), enzymes, etc.)and recovering degradable gel beads into an aqueoussuspension. The functionalized gel beads are re-sus-pended in high salt buffer, 308. Streptavidin-labeled mag-netic beads are added to the re-suspension, which isthen incubated to allow binding to gel beads attached tobiotinylated barcodes 308, 410. A magnetic device isthen used to separate positive barcoded gel beads frombeads that are not attached to barcode, 308. Denatura-tion conditions, 309, (e.g. heat or chemical denaturant)are applied to the gel beads in order to separate the bi-otinylated complementary strand from the barcodedbeads. The magnetic beads are subsequently removedfrom the solution; and the resulting solution of partially-functionalized barcoded beads is pooled for furtherprocessing.
Example 3: Further Functionalization of Barcoded beads
[0356] As shown in Fig 3B, the barcoded gel beads,311, from Example 2, are further functionalized as fol-lows. The beads are combined with an additional tem-plate oligonucleotide, 310, (such as an oligonucleotidecontaining a random N-mer sequence, 413, as shown inFig 4), and PCR reagents, 312, 313, and subjected toconditions to enable hybridization of the template oligo-nucleotide with the read primer attached to the gel bead.An extension reaction is performed so that the barcodestrands are extended, 314, thereby incorporating thecomplementary sequence of the template oligonucle-otide. Resulting functionalized gel beads are re-suspend-ed in aqueous buffer, 315, and exposed to heating con-ditions to remove complement strands, 316, and placedinto aqueous storage buffer, 317.
Example 4: Step-by-Step Description of Bead Func-tionalization
[0357] Fig 4 provides a step-by-step description of anexample process of functionalizing the gel beads withbarcodes and random N-mers. As shown in Fig 4A, theprocess begins with gel beads, 401, that are attached toa universal primer, such as a P5 primer (or its comple-ment), 403. The beads may be linked to the primer via adi-sulfide bond, 402. The gel beads are provided in anaqueous solution (g/w). Using a limiting dilution and par-titioning, unique barcode sequence templates, 405, arecombined with the beads such that at most one uniquebarcode sequence occupies the same partition as a gelbead. Generally, the partitions are aqueous droplets with-in a gel/water/oil (g/w/o) emulsion. As shown in Fig 4B,the barcode sequence template, 405, is contained withina larger nucleotide strand that contains a sequence, 404,that is complementary to the universal primer 403, aswell as a sequence, 407, that is identical in sequence to
a biotin labeled read primer, 406.[0358] As shown in Fig 4C, an amplification reactionis then conducted to incorporate the complement, 408,of the barcode template, 405, onto the strand that is at-tached to the bead. The amplification reaction also re-sults in incorporation of a sequence, 415, that is comple-mentary to sequence, 407. Additional amplification cy-cles result in hybridization of the biotin labeled read prim-er, 406, to sequence, 415 (Fig 4D), and the biotin labeledread primer is then extended (Fig 4E). The emulsion maythen be broken, and the gel beads may then be pooledinto a gel/water common solution.[0359] In the gel/water solution, magnetic capturebeads, 409, are then used to capture the biotinylatednucleic acids attached to the gel beads, which are thenisolated from beads that only contain the original primer(Fig 4F and Fig 4G). The biotinylated strand is then re-moved from the strand attached to the gel bead (Fig 4H).Random N-mer sequences, 414, may then be attachedto the strands attached to the gel bead. For each gelbead, an identical barcode sequence, 408, is attachedto each primer throughout the gel bead; each barcodesequence is then functionalized with a random N-mersequence, 414, such that multiple different random N-mer sequences are attached to each bead. For this proc-ess, a random N-mer template sequence, 413, linked toa sequence, 412, complementary to sequence, 415, isintroduced to the solution containing the pooled beads(Fig 4I). The solution is subjected to conditions to enablehybridization of the template to the strand attached to thebead and sequence 415 is extended to include the ran-dom N-mer, 414. (Fig 4J). The fully functionalized beads(Fig 4K) are then combined with a sample nucleic acidand a reducing agent (e.g., dithiothreitol (DTT) at a con-centration of ImM) and partitioned within droplets of agel/water/oil emulsion (Fig 4L). This combining step maybe conducted with a microfluidic device (Fig 5A). The gelbeads are then degraded within each partition (e.g., drop-let) such as by the action of a reducing agent, and thebarcoded sequence is released from the droplet (Fig 4Mand Fig 4N). The random N-mer within the barcoded se-quence may serve as a primer for amplification of thesample nucleic acid.
Example 5: Use of a Microfluidic Chip to Combine the Gel-Beads-in Emulsions (GEMs) with Sample
[0360] The functionalized gel beads may be combinedwith sample using a double-cross microfluidic device il-lustrated in Fig 5. Degradable gel beads are introducedto the fluidic input, 501, in a fluid stream, which containsabout 7% glycerol. The experimental sample of interestis introduced to the fluidic input, 502, in a fluid stream,which is aqueous phase. The reducing agent, dithiothre-itol (DTT) at a concentration of about ImM is introducedto the fluidic input, 503, in a fluid stream, which containsabout 7% glycerol. Fluidic inputs 501, 502, and 503 mixat a microfluidic cross junction, 504, and enter a second
125 126

EP 3 013 957 B1
65
5
10
15
20
25
30
35
40
45
50
55
microfluidic cross junction, 506. The second microfluidiccross junction can be used to produce emulsified (w/o)droplets containing the el beads. Fluidic input, 505, isused to introduce oil with 2% (w/w) bis krytox peg (BKP).Individual droplets exiting from the second microfluidiccross junction, 507, are added into microplate wells, Fig5C, for further downstream applications. Fig 5D is animage of droplets generated in the absence of DTT (andtherefore containing gel beads). Fig 5E is an image ofdroplets generated with DTT that caused the internal gelbeads to degrade.
Example 6: Fluorescent Identification of Positive Gel Beads
[0361] Fig 6 depicts images of gel beads containingamplified nucleic acids that have been labeled with a flu-orescent label. Functionalization of the gel beads is firstperformed using a limiting dilution so that only a portionof the gel beads are functionalized with barcodes. Gelbeads suspended in a bis krytox peg (BKP) emulsion areimaged at 4X magnification following PCR thermocyclingbut before washing. The bright field image, Fig 6A, showsall emulsion-generated droplets, and the fluorescent im-age, Fig 6B, shows only positive functionalized gelbeads. Many non-fluorescent droplets are generated in-dicating empty droplets, which do not contain either gelbead and/or oligonucleotide. Empty droplets are washedaway by multiple re-suspensions and washing in HFE-7500. Fig 6C and Fig 6D show positive gel bead enrich-ment following emulsion breaking and further washsteps. The bright field images (4X), Fig 6C, and (10X)Fig 6E, show all gel beads. The fluorescent images (4X),Fig 6D, and (10X), Fig 6F, show 30% positive beadsfrom SYBR staining. The 30% positive bead result match-es predicted value from gDNA input.[0362] Fig 7 shows images of gel beads containingsingle stranded (ss) DNA, double-stranded (ds) DNA,and denatured, ssDNA. Gel beads stained with IX Eva-Green are brighter in the presence of dsDNA as evidentfrom the fluorescent images taken at step 1: Make (ssD-NA), Fig 7A, step 2: Extension (dsDNA), Fig 7B, andstep 3: Denature (ssDNA), Fig 7C. Fluorescent imagesshow that beads become brighter after extension andbecome dimmer after denaturation.
Example 7: Enrichment of Positive Gel Beads Using Streptavidin-Coated Magnetic Beads
[0363] Enrichment of positive gel beads using strepta-vidin-coated magnetic beads is depicted in Fig 8. Fig 8A(bright field) and Fig B (fluorescent) provides images ofSYBR-stained gel beads 24 hours following the additionof magnetic beads. Magnetic coated positive gel beadsare brighter due to SYBR staining. Bright field imagesbefore, Fig 8C, and after sorting, Fig 8D, at a magneticbead concentration of 40mg/mL, show positive gel beadenrichment, where coated beads are optically brighter.
Bright field images before, Fig 8E, and after sorting, Fig8F, at a magnetic bead concentration of 60 mg/mL, showpositive gel bead enrichment, where coated beads areoptically brighter. At each magnetic bead working con-centration, a single gel bead is coated by about 100-1000magnetic beads.
Example 8: Dissolution of Gel Beads
[0364] Heating gel beads in basic solution degradesthe gel beads as evident in Fig 9. Gel beads are heatedin basic solution at 95°C and monitored at 5 minute heat-ing intervals: t = 0min, Fig 9A, t = 5min, Fig 9B, t = 10min,Fig 9C, t = 15min, Fig 9D. Following 15 minutes, gelbeads are completely degraded. Gel beads more thandouble in size while they are degrading. Fig 10 depictsdissolution of the gel beads using tris(2-carboxye-thyl)phosphine (TCEP), which is an effective and irre-versible di-sulfide bond reducing agent. Functionalizedgel beads, Fig 10A, are placed into basic solution, pH =8, with ImM TCEP and monitored at 2 minute intervals:t = 0min, Fig 10B, t = 2min, Fig 10C, t = 4min, Fig 10D,t = 6min, Fig 10E, t = 8min, Fig 10F, t = 10min, Fig 10G.Between about 6 and about 10 minutes, the functional-ized gel beads are completely degraded.
Example 9: Analysis of Content After Dissolution Gel Beads (GB)
[0365] An analysis of content attached to gel beads isprovided in Fig 11, and Fig 12. Gel beads are function-alized, 1101, with barcode or barcode complement(N12C) and a random N-mer (8mer) that is 8 nucleotidesin length, 1102. The random N-mer is attached by per-forming a primer extension reaction using a template con-struct containing R1C and a random N-mer 1102. Thelength of the entire oligonucleotide strand (including thebar code and random N-mer) is 82 bp, 1101. The strandlength of the random N-mer and the R1C is 42 base pairs(bp), 1102. The extension reaction is performed using aKAPA HIFI RM Master Mix under high primer concentra-tion (10mm at 65°C for one hour. Increasing the numberof wash steps before the step of degrading the gel beadsresults in a reduction in the amount of primer dimers with-in the sample. When no washes are performed, 1103,both 42 bp products, 1106, and 80 bp products, 1107,can be observed. After three washes, the level of primerdimer, 1104, is reduced relative to the no-wash experi-ment. After six washes, 1105, 80 bp products, 1107, areobserved, but no primer dimers are observed.[0366] The six-wash experiment can also be per-formed using six different temperatures (65°C, 67°C,69°C, 71°C, 73°C, 75°C, Fig 11C) for the extension step.In this specific example, a high primer concentration(10mm) is used and the extension step lasts one hour. Itappears that 67°C is the optimal temperature for bothoptimizing the level of 80bp products and minimizing thenumber of 42 bp products, 1109.
127 128

EP 3 013 957 B1
66
5
10
15
20
25
30
35
40
45
50
55
[0367] The temperature, 67°C, is chosen for subse-quent denaturation studies. Heat denaturation of thecomplementary strand, wherein the sample is heated to95°C six times and washed to remove complementarystrand, results in an 84 bp peak, 1202, before denatura-tion, and shows a reduced peak, 1201, following dena-turation. The control value measured from step 1 isshown at 1203.
Example 10: Creation of Barcoded Gel Beads by Par-titioning in Wells
[0368] Functionalized beads are produced by partition-ing in wells according to the method illustrated in Fig 13Aand 13B. The first functionalization step is outlined in Fig13A, the second functionalization step is outlined in Fig13B. An example multiplex adaptor creation scheme isoutlined in Fig 13C and described in Example 11. Asshown in Fig 13A, functionalized beads, 1301 (e.g.,beads with acrydite oligos and primer (e.g., 5’-AAU-GAUACGGCGACCACCGAGA-3’), the template withbarcode sequence, 1302 (e.g., 5’-XXXXXXTCTCGGT-GGTCGCCGTATCATT-3’), and appropriate PCR rea-gents, 1303, are mixed together, 1304/1305 and dividedinto 384 wells of a multi-well plate. Each well comprisesmultiple copies of a unique barcode sequence and mul-tiple beads. Thermocycling, 1306, with an extension re-action is performed in each individual well to form beadswith attached barcodes. All wells are pooled together andcleaned up in bulk, 1307/1308.[0369] To add a random N-mer, the partially function-alized beads, 1310, the template random N-mer oligo-nucleotides, 1309, and the appropriate PCR reagents,1311, are mixed together, 1312, and the functionalizedbeads 1310 subjected to extension reactions 1313 to adda random N-mer sequence complementary to the ran-dom N-mer template, to the beads. Following thermalcycling, the beads are cleaned up in bulk, 1314-1316.
Example 11: Combinatorial Plate Technique
[0370] As shown in Fig 13C, beads 1317 attached toprimers (e.g., P5 oligomers, 5’-AAUGAUACGGCGAC-CACCGAGA-3’) 1318 are partitioned into wells of a multi-well plate (such as a 5X-1 384-well plate 1319) with mul-tiple copies of a template 1321 comprising a unique tem-plate partial barcode sequence (e.g., 5’-XXXXXXTCTCGGTGGTCGCCGTATCATT-3). Exten-sion reactions (e.g., extension of primer 1318 via tem-plate 1321) are performed to generate Bead-P5-[5X-1],1320 comprising an extension product (e.g., an oligonu-cleotide comprising primer 1318 and a partial barcodesequence complementary to the template partial barcodesequence) in each well. The beads are removed fromthe wells are pooled together and a clean-up step is per-formed in bulk.[0371] The pooled mixture is then re-divided into wellsof a second multiwell plate such as a 384-well plate with
5X-2, 1322, with each well also comprising an oligonu-cleotide comprising a second unique partial barcode se-quence and a random N-mer (e.g., 5’P-YYYYYYCGCACACUCUUUCCCUACACGACGCUC-UUCCGAUCUNNNNNNNN-BLOCK). The oligonucle-otide may have a blocker oligonucleotide attached (e.g.,via hybridization) (e.g., "BLOCK"). Single-stranded liga-tion reactions 1324 are performed between the extensionproduct bound to the bead and the oligonucleotide com-prising the second partial barcode sequence and randomN-mer. Following the ligation reaction, beads comprisinga full barcode sequence (e.g., XXXXXXYYYYYY) and arandom N-mer are generated, 1323 (e.g., Bead-P5-[5X-1][5X-2]R1[8N-Blocker]). The beads also comprise theblocker oligonucleotide. All wells are then pooled togeth-er, the blocking groups are cleaved, and the bead prod-ucts are cleaned up in bulk. Beads comprising a largediversity of barcode sequences are obtained.
Example 12: Partial Hairpin Amplification for Se-quencing (PHASE) Reaction
[0372] Partial Hairpin Amplification for Sequencing(PHASE) reaction is a technique that can be used to mit-igate undesirable amplification products according to themethod outlined in Fig 14 and Fig 15 by forming partialhairpin structures. Specifically, random primers, of about8N-12N in length, 1404, tagged with a universal se-quence portion, 1401/1402/1403, may be used to ran-domly prime and extend from a nucleic acid, such as,genomic DNA (gDNA). The universal sequence compris-es: (1) sequences for compatibility with a sequencing de-vice, such as, a flow cell (e.g. Illumina’s P5, 1401, andRead 1 Primer sites, 1402) and (2) a barcode (BC), 1403,(e.g., 6 base sequences). In order to mitigate undesirableconsequences of such a long universal sequence por-tion, uracil containing nucleotides are substituted forthymine containing nucleotides for all but the last 10-20nucleotides of the universal sequence portion, and apolymerase that will not accept or process uracil-contain-ing templates is used for amplification of the nucleic acid,resulting in significant improvement of key sequencingmetrics, Fig 16A, Fig 21, and Fig 22. Furthermore, ablocking oligonucleotide comprising uracil containing nu-cleotides and a blocked 3’ end (e.g. 3’ddCTP) are usedto promote priming of the nucleic acid by the random N-mer sequence and prevent preferential binding to por-tions of the nucleic acid that are complementary to theRead 1 Primer site, 1402. Additionally, product lengthsare further limited by inclusion of a small percentage ofterminating nucleotides (e.g., 0.1-2% acyclonucleotides(acyNTPs)) (Fig 16B) to reduce undesired amplificationproducts.[0373] An example of partial hairpin formation to pre-vent amplification of undesired products is provided here.First, initial denaturation is achieved at 98°C for 2 minutesfollowed by priming a random portion of the genomic DNAsequence by the random N-mer sequence acting as a
129 130

EP 3 013 957 B1
67
5
10
15
20
25
30
35
40
45
50
55
primer for 30 seconds at 4°C (Fig 15A). Subsequently,sequence extension follows as the temperature rampsat 0.1°C/second to 45°C (held for 1 second) (Fig 15A).Extension continues at elevated temperatures (20 sec-onds at 70°C), continuing to displace upstream strandsand creating a first phase of redundancy (Fig 15B). De-naturation occurs at 98°C for 30 seconds to release ge-nomic DNA for additional priming. After the first cycle,amplification products have a single 5’ tag (Fig 15C).These aforementioned steps are repeated up to 20 times,for example by beginning cycle 2 at 4°C and using therandom N-mer sequence to again prime the genomicDNA where the black sequence indicates portions of theadded 5’ tags (added in cycle 1) that cannot be copied(Fig 15D). Denaturation occurs at 98°C to again releasegenomic DNA and the amplification product from the firstcycle for additional priming. After a second round of ther-mocycling, both 5’ tagged products and 3’ & 5’ taggedproducts exist (Fig 15E). Partial hairpin structures formfrom the 3’ & 5’ tagged products preventing amplificationof undesired products (Fig 15F). A new random primingof the genomic DNA sequence begins again at 4°C (Fig15G).
Example 13: Adding Additional Sequences by Am-plification
[0374] For the completion of sequencer-ready librar-ies, an additional amplification (e.g., polymerase chainreaction (PCR) step) is completed to add additional se-quences, Fig 14C. In order to out-compete hairpin for-mation, a primer containing locked nucleic acid (LNAs)or locked nucleic acid nucleotides, is used. Furthermore,in cases where the inclusion of uracil containing nucle-otides is used in a previous step, a polymerase that doesnot discriminate against template uracil containing nu-cleotides is used for this step. The results presented inFig 17 show that a blocking oligonucleotide reduces startsite bias, as measured by sequencing on an IlluminaMiSeq sequencer. The nucleic acid template in this caseis yeast gDNA.
Example 14: Digital Processor
[0375] A conceptual schematic for an example controlassembly, 1801, is shown in Fig 18. A computer, 1802,serves as the central hub for control assembly, 1801.Computer, 1802, is in communication with a display,1803, one or more input devices (e.g., a mouse, key-board, camera, etc.) 1804, and optionally a printer, 1805.Control assembly, 1801, via its computer, 1802, is in com-munication with one or more devices: optionally a samplepre-processing unit, 1806, one or more sample process-ing units (such as a sequence, thermocycler, or micro-fluidic device) 1807, and optionally a detector, 1808. Thecontrol assembly may be networked, for example, via anEthernet connection. A user may provide inputs (e.g., theparameters necessary for a desired set of nucleic acid
amplification reactions or flow rates for a microfluidic de-vice) into computer, 1802, using an input device, 1804.The inputs are interpreted by computer, 1802, to gener-ate instructions. The computer, 1802, communicatessuch instructions to the optional sample pre-processingunit, 1806, the one or more sample processing units,1807, and/or the optional detector, 1808, for execution.Moreover, during operation of the optional sample pre-processing unit, 1806, one or more sample processingunits, 1807, and/or the optional detector, 1808, each de-vice may communicate signals back to computer, 1802.Such signals may be interpreted and used by computer,1802, to determine if any of the devices require furtherinstruction. Computer, 1802, may also modulate samplepre-processing unit, 1806, such that the components ofa sample are mixed appropriately and fed, at a desiredor otherwise predetermined rate, into the sampleprocessing unit (such as the microfluidic device), 1807.Computer, 1802, may also communicate with detector,1808, such that the detector performs measurements atdesired or otherwise predetermined time points or at timepoints determined from feedback received from pre-processing unit, 1806, or sample processing unit, 1807.Detector, 1808, may also communicate raw data ob-tained during measurements back to computer, 1802, forfurther analysis and interpretation. Analysis may be sum-marized in formats useful to an end user via display,1803, and/or printouts generated by printer, 1805. In-structions or programs used to control the sample pre-processing unit, 1806, the sample processing unit, 1807,and/or detector, 1808; data acquired by executing anyof the methods described herein; or data analyzed and/orinterpreted may be transmitted to or received from oneor more remote computers, 1809, via a network, 1810,which, for example, could be the Internet.
Example 15: Combinatorial Technique via Ligation
[0376] As shown in Fig 23A, beads 2301 are generat-ed and covalently linked (e.g., via an acrydite moiety) toa partial P5 sequence 2302. Separately, in 50 mL of eachwell of 4 96 well plates, an oligonucleotide 2303, com-prising the remaining P5 sequence and a unique partialbarcode sequence (indicated by bases "DDDDDD" in ol-igonucleotide 2303), is hybridized to an oligonucleotide2304 that comprises the reverse complement to oligonu-cleotide 2303 and additional bases that overhang eachend of oligonucleotide 2303. Splint 2306 is generated.Each overhang is blocked (indicated with an "X" in Fig23) with 3’ C3 Spacer, 3’ Inverted dT, or dideoxy-C (ddC)to prevent side product formation.[0377] As shown in Fig 23B, splints 2306 are eachadded to 4 96 deep well plates, with each well comprising2 mL beads 2301 and a splint comprising a unique partialbarcode sequence. In each well, the splint 2306 hybrid-izes with the partial P5 sequence 2302 of beads 2301,via the corresponding overhang of oligonucleotide 2304.Following hybridization, partial P5 sequence 2302 is
131 132

EP 3 013 957 B1
68
5
10
15
20
25
30
35
40
45
50
55
ligated to oligonucleotide 2303 (which will typically havebeen 5’ phosphorylated) via the action of a ligase, e.g.,a T4 ligase, at 16°C for 1 hour. Following ligation, theproducts are pooled and the beads washed to removeunligated oligonucleotides.[0378] As shown in Fig 23C, the washed products arethen redistributed into wells of 4 new 96 well plates, witheach well of the plate comprising 2 mL of beads 2301and an oligonucleotide 2305 that has a unique partialbarcode sequence (indicated by "DDDDDD" in oligonu-cleotide 2305) and an adjacent short sequence (e.g.,"CC" adjacent to the partial barcode sequence and at theterminus of oligonucleotide 2305) complementary to theremaining overhang of oligonucleotide 2304. Oligonucle-otide 2305 also comprises a random N-mer (indicatedby "NNNNNNNNNN" in oligonucleotide 2305). Via theadjacent short sequence, oligonucleotide 2305 is hybrid-ized with oligonucleotide 2304 via the remaining over-hang of oligonucleotide 2304. Oligonucleotide 2305 isthen ligated to oligonucleotide 2303 via the action of aligase at 16°C for 1 hour. Ligation of oligonucleotide 2305to oligonucleotide 2303 results in the generation of a fullbarcode sequence. As shown in Fig 23D, the productsare then pooled, the oligonucleotide 2304 is denaturedfrom the products, and the unbound oligonucleotides arethen washed away. Following washing, a diverse libraryof barcoded beads is obtained, with each bead bound toan oligonucleotide comprising a P5 sequence, a full bar-code sequence, and a random N-mer. The generatedlibrary comprises approximately 147,000 different bar-code sequences.
Example 16: Substitution of Uracil Containing Nucle-otides for Thymine Containing Nucleotides in Bar-code Primers
[0379] As shown in Fig 33A, two barcode primers 3301and 3302 suitable for PHASE amplification were used toamplify sample nucleic acid obtained from a yeast ge-nome. Following PHASE amplification, additional se-quences were added (e.g., via bulk PCR) to generatesequencer-ready products. Barcode primers 3301 (alsoshown as U.2 in Fig 33A) and 3302 (also shown at U.1in Fig 33A) comprised an identical sequence except thatbarcode primer 3301 comprised an additional uracil con-taining nucleotide-for-thymine containing nucleotidesubstitution at position 3306. Sets of amplification exper-iments were run for each barcode primer, with each setcorresponding to a particular blocker oligonucleotidemixed with the respective barcode primer at various sto-ichiometries. For barcode primer 3302, sets of amplifica-tion experiments corresponding to a standard blocker ol-igonucleotide 3303, a full blocker oligonucleotide com-prising bridged nucleic acid (BNAs) 3304 (also shown asBNA blocker in Fig 33A), or a full blocker oligonucleotide3305 were conducted. Blocker oligonucleotides 3303and 3305 comprised uracil containing nucleotide-for-thymine containing nucleotide substitutions at all thymine
containing nucleotide positions and a ddC blocked end.In each set, the blocker oligonucleotide:barcode primerstoichiometry was either 0, 0.4, 0.8, or 1.2. For barcodeprimer 3301, each type of blocker oligonucleotide 3303,3304, and 3305 was tested at a 0.8 blocker oligonucle-otide:barcode primer stoichiometry.[0380] The size results of PHASE amplification prod-ucts are depicted in Fig 33B. As shown, barcode primer3302 (e.g., comprising the extra uracil containing nucle-otide-for-thymine containing nucleotide substitution)coupled to blocker oligonucleotide 3303 generally pro-duced the smallest amplification products across the sto-ichiometries tested. Results for barcode primer 3302 withrespect to blocker oligonucleotides 3304 and 3305 var-ied, with sizes generally larger than results for blockeroligonucleotide 3303. For barcode primer 3301, amplifi-cation product sizes were also generally larger than thoseobtained for barcode primer 3301 coupled to blocker ol-igonucleotide 3303 across the blocker oligonucleotidestested. The size results of sequencer-ready products aredepicted in Fig 33C.[0381] Key sequencing metrics obtained from the am-plification products are depicted in Fig 33D. As shown,the fraction of unmapped reads (panel I in Fig 33D) wasgenerally lower for sequencing runs for amplificationproducts generated from barcode primer 3302. For ex-ample, the fraction of unmapped reads for amplificationproducts generated from barcode primer 3302 and block-er oligonucleotide 3303 at 0.8 blocker oligonucle-otide:barcode primer stoichiometry was approximately7-8%, whereas results obtained using barcode primer3301 at the same conditions was approximately 17-18%.Moreover, Q40 error rates (panel II in Fig 33D) were alsolower for barcode primer 3302. For example, Q40 errorrate for amplification products generated from barcodeprimer 3302 and blocker oligonucleotide 3303 at 0.8blocker oligonucleotide:barcode primer stoichiometrywas approximately 0.105%, whereas results obtainedusing barcode primer 3301 at the same conditions wasapproximately 0.142%. Read 1start site (panel III) andRead 2 start site (panel IV) relative entropies determinedduring sequencing are shown in Fig 33E.
Example 17: Post-Synthesis Functionalization of Gel Beads via Disulfide Exchange
[0382] Gel beads comprising disulfide bonds weregenerated according to one or more methods describedherein. The gel beads were then reacted with TCEP atratios of molecules of TCEP to gel beads (TCEP:GB).The tested ratios were 0, 2.5 billion, and 10.0 billion. TheTCEP functions as a reducing agent to generate freethiols within the gel beads. Following reduction, the gelbeads were washed once to remove the TCEP. Next, thegenerated free thiols of the gel beads were reacted withan acrydite-S-S-P5 species (e.g., 3505 in Fig 35A) tolink the acrydite-S-S-P5 to the gel beads via Michael ad-dition chemistry as shown in Fig 35A. Different ratios of
133 134

EP 3 013 957 B1
69
5
10
15
20
25
30
35
40
45
50
55
acrydite-S-S-P5 to each type (e.g., ratio of TCEP:GBused to generate free thiols on the gel beads) of the ac-tivated gel beads were tested. The tested ratios of acry-dite-S-S-P5 species to activated gel beads (P5:GB) were50 million, 500 million, and 5 billion.[0383] Following syntheses, the gel beads from eachreaction were washed and treated with DTT in a reactionmixture to degrade the gel beads and release any boundacrydite-S-S-P5 species. An aliquot of each reaction mix-ture was entered into a lane of a gel and free oligonucle-otides subject to gel electrophoresis as shown in Fig 36(e.g., lanes 3-11 in Fig 36). A 50 picomole acrydite-S-S-P5 standard was also run (e.g., lane 1 in Fig 36) alongwith a 25 base pair ladder (e.g., lane 2 in Fig 36). Bandscorresponding to loaded acrydite-S-S-P5 were generat-ed in lanes 5 and 8 (indicated by arrows in Fig 36). Lane5 corresponds to gel beads treated at a TCEP:GB ratioof 2.5 billion and the TCEP treated gel beads reactedwith acrydite-S-S-P5 at a P5:GB ratio of 5 billion. Lane8 corresponds to gel beads treated at a TCEP:GB ratioof 10.0 billion and the TCEP treated gel beads reactedwith acrydite-S-S-P5 at a P5:GB ratio of 5 billion.
Example 18: Post-Synthesis Functionalization of Gel Beads via Disulfide Exchange
[0384] Gel beads comprising disulfide bonds weregenerated according to one or more methods describedherein. The gel beads were then reacted with TCEP in0.1M phosphate buffer at a concentration of 4 mgTCEP/100,000 gel beads. The TCEP can function as areducing agent to generate gel beads with free thiolgroups. Following reduction, the gel beads were washedonce to separate the gel beads from the TCEP. Next, thefree thiols of the gel beads were reacted with 2,2’-dithi-opyridine (e.g., 3507 in Fig 35B) in a saturated solution(∼0.2 mM) of 2,2’-dithiopyridine to link pyridine groups tothe gel beads via disulfide exchange chemistry as shownin Fig 35B. Following synthesis, the gel beads werewashed three times to remove excess 2,2’-dithiopyridine.[0385] The washed gel beads were then reacted withan oligonucleotide 3702 comprising a full construct bar-code (FCBC - e.g., an oligonucleotide comprising P5, abarcode sequence, R1, and a random N-mer) sequenceat one end and a free thiol group at its other end. Tworeactions were completed at two different ratios of mol-ecules of FCBC to gel beads (e.g., FCBC:GB) and thereactions were allowed to proceed overnight. The testedFCBC:GB ratios were 400 million and 1.6 billion. Oligo-nucleotide 3702 was initially supplied with its free thiolgroup protected in a disulfide bond, shown as 3701 inFig 37A. To generate the free thiol as in oligonucleotide3702, oligonucleotide 3701 was treated with 0.1 M DTTin IX Tris-EDTA buffer (TE) buffer for 30 minutes. Saltexchange on a Sephadex (NAP-5) column was used toremove DTT after reduction and purify oligonucleotide3702. For each reaction, purified oligonucleotides 3702were then reacted with the dithio-pyridine species of the
gel beads via thiol-disulfide exchange (e.g., see Fig 35B)to generate gel beads comprising oligonucleotide 3702.Following the reaction, the gel beads were purified bywashing the beads three times.[0386] For comparison purposes, gel beads compris-ing disulfide bonds and the FCBC sequence were alsogenerated via polymerization of monomers as describedelsewhere herein. The FCBC was linked to a monomercomprising an acrydite species that was capable of par-ticipating in a polymerization with acrylamide andbis(acryloyl)cystamine to generate the gel beads. TheFCBC sequence was linked to the gel beads via the acry-dite moiety.[0387] Following syntheses, the gel beads from eachreaction were washed and treated with DTT in a reactionmixture to degrade the gel beads and release any boundoligonucleotide 3702. Gel beads comprising the FCBCsequence that were synthesized via polymerization werealso treated with DTT in a reaction mixture. An aliquot ofeach reaction mixture was entered into a lane of a geland free oligonucleotides subject to gel electrophoresisas shown in Fig 37B. As shown in the gel photographdepicted in Fig 37B, lane 1 corresponds to a 50 basepair ladder; lane 2 corresponds to gel beads functional-ized via disulfide exchange chemistry at an FCBC:GBratio of 400 million; lane 3 corresponds to gel beads func-tionalized via disulfide exchange chemistry at anFCBC:GB ratio of 1.6 billion; and lane 4 corresponds tofunctionalized gel beads generated via polymerization ofacrydite species. Bands corresponding to loaded oligo-nucleotides were generated for functionalized gel beadsgenerated at both FCBC:GB ratios and were at a similarposition to the band generated for functionalized gelbeads generated via polymerization of acrydite species.[0388] Following syntheses, gel beads from each re-action were also washed and stained with SYBR Goldfluorescent stain. Gel beads comprising the FCBC se-quence that were synthesized via polymerization werealso stained with SYBR Gold. SYBR Gold can stain func-tionalized beads by intercalating any bound oligonucle-otides. Following staining, the beads were pooled andimaged using fluorescence microscopy, as shown in themicrograph depicted in Fig 37C. Brighter beads (3704)in Fig 37C correspond to beads functionalized duringpolymerization of the beads and dim beads (still showingSYBR gold signal) (3705) correspond to beads function-alized with disulfide exchange chemistry after gel beadgeneration. Loading of oligonucleotides via disulfide-ex-change was approximately 30% of that achieved withfunctionalization of beads during gel bead polymeriza-tion.
Claims
1. A method of barcoding sample materials, compris-ing:
135 136

EP 3 013 957 B1
70
5
10
15
20
25
30
35
40
45
50
55
providing a bead comprising a plurality of nucleicacid barcode molecules releasably associatedtherewith, the plurality of nucleic acid barcodemolecules comprising the same nucleic acidbarcode sequence;co-partitioning the bead with components of asample material into a partition;releasing the barcode molecules from the beadinto the partition; andattaching the barcode molecules to one or moreof the components of the sample material orfragments thereof within the partition.
2. The method of claim 1, wherein the bead comprises:
(i) at least 1,000 barcode molecules releasablyassociated therewith, the at least 1,000 barcodemolecules having the same barcode sequence;(ii) comprises at least 10,000 barcode moleculesreleasably associated therewith, the at least10,000 barcode molecules having the same bar-code sequence;(iii) at least 100,000 barcode molecules releas-ably associated therewith, the at least 100,000barcode molecules having the same barcodesequence; or(iv) at least 1,000,000 barcode molecules re-leasably associated therewith, the at least1,000,000 barcode molecules having the samebarcode sequence..
3. The method of claim 1, wherein the partition is a drop-let.
4. The method of claim 1, wherein the bead comprisesa gel bead, and wherein the barcode molecules arereleasably coupled to the bead.
5. The method of claim 1, wherein the barcode mole-cules are encapsulated within or diffused throughoutthe bead.
6. The method of claim 1, wherein the releasing stepcomprises degrading the bead.
7. The method of claim 4, wherein:
(i) the co-partitioning comprises combining afirst aqueous fluid comprising beads with a sec-ond aqueous fluid comprising the sample com-ponents in a droplet within an immiscible fluid;(ii) the releasing comprises degrading the beadto release the barcode molecules into the sec-ond partition;(iii) the releasing comprises cleaving a chemicallinkage between the barcode molecules and thebead; or(iv) at least one of crosslinking of the bead and
a linkage between the bead and the barcodemolecules comprises a disulfide linkage, and thereleasing comprises exposing the bead to a re-ducing agent, e.g. wherein the reducing agentis DTT or TCEP.
8. The method of claim 1, wherein the sample materialscomprise one or more template nucleic acid mole-cules, and:
(i) the attaching comprises attaching the bar-code molecules to one or more fragments of thetemplate nucleic acid molecules; or(ii) the barcode molecules further comprise aprimer sequence complementary to at least aportion of the template nucleic acid molecules,and the attaching comprises extending the bar-code molecules to replicate at least a portion ofthe template nucleic acid molecules.
9. The method of claim 1, wherein the sample materialscomprise contents of a single cell, and wherein op-tionally the single cell comprises:
(i) a cancer cell; or(ii) a bacterial cell, eg. a bacterial cell isolatedfrom a human microbiome sample.
10. The method of claim 1, wherein the partition com-prises no more than one bead.
11. The method of claim 1, wherein:
the providing comprises providing a plurality ofbeads comprising a plurality of different nucleicacid barcode sequences, wherein each of theplurality of beads comprises a plurality of at least1000 nucleic acid barcode molecules having thesame nucleic acid barcode sequence associat-ed therewith;the co-partitioning comprises co partitioning theplurality of beads with the components of thesample material into a plurality of partitions;the releasing comprises releasing the nucleicacid barcode molecules from the plurality ofbeads into the plurality of partitions; andthe attaching comprises attaching the nucleicacid barcode molecules to the components ofthe sample material or fragments thereof withinthe plurality of partitions.
12. The method of claim 11, wherein the plurality of dif-ferent nucleic acid barcode sequences comprises atleast about 1000 different barcode sequences, atleast about 10,000 different barcode sequences, atleast about 100,000 different barcode sequences, orat least about 500,000 different barcode sequences.
137 138

EP 3 013 957 B1
71
5
10
15
20
25
30
35
40
45
50
55
13. The method of claim 11, wherein a subset of theplurality of partitions comprise the same nucleic acidbarcode sequence.
14. The method of claim 13, wherein at least about 1%,at least about 2%, or at least about 5% of the pluralityof partitions comprise the same nucleic acid barcodesequence.
15. The method of claim 14, wherein the fragments ofthe components of the sample material comprise oneor more fragments of one or more template nucleicacid sequences and the method further comprisessequencing the one or more fragments of the one ormore template nucleic acid sequences and charac-terizing the one or more fragments of the templatenucleic acid sequences based at least in part upona nucleic acid barcode sequence attached thereto.
16. The method of claim 15, wherein the characterizingthe one or more fragments of the one or more tem-plate nucleic acid sequences comprises:
(i) mapping a fragment of an individual templatenucleic acid sequence of the one or more tem-plate nucleic acid sequences to an individualtemplate nucleic acid sequence of the one ormore template nucleic acid sequences or a ge-nome from which the individual template nucleicacid sequence was derived; or(ii) at least identifying an individual nucleic acidbarcode sequence of said plurality of differentnucleic acid barcode sequences, and identifyinga sequence of an individual fragment of the oneor more fragments of the one or more templatenucleic acid sequences attached to the individ-ual nucleic acid barcode sequence.
17. The method of claim 11, wherein:
(i) at least 50%, at least 70%, or at least 90% ofthe plurality of partitions contain no more thanone bead; or(ii) at least 50%, at least 70% or at least 90% ofthe plurality of partitions comprises exactly onebead.
Patentansprüche
1. Verfahren zum Versehen von Probematerialien miteinem Barcode, das Folgendes umfasst:
das Bereitstellen eines Kügelchens, das eineVielzahl von Nucleinsäure-Barcodemolekülenumfasst, die freisetzbar damit assoziiert sind,wobei die Vielzahl von Nucleinsäure-Barcode-molekülen dieselbe Nucleinsäure-Barcodese-
quenz umfasst;das Abtrennen des Kügelchens mit Komponen-ten eines Probematerials in eine Teileinheit;das Freisetzen der Barcodemoleküle von demKügelchen in die Teileinheit; unddas Anbringen der Barcodemoleküle an eineroder mehreren der Komponenten des Probema-terials oder Fragmenten davon innerhalb derTeileinheit.
2. Verfahren nach Anspruch 1, wobei das KügelchenFolgendes umfasst:
(i) zumindest 1000 Barcodemoleküle, die frei-setzbar damit assoziiert sind, wobei die zumin-dest 1000 Barcodemoleküle dieselbe Barcode-sequenz aufweisen;(ii) zumindest 10.000 Barcodemoleküle, die frei-setzbar damit assoziiert sind, wobei die zumin-dest 10.000 Barcodemoleküle dieselbe Bar-codesequenz aufweisen;(iii) zumindest 100.000 Barcodemoleküle, diefreisetzbar damit assoziiert sind, wobei die zu-mindest 100.000 Barcodemoleküle dieselbeBarcodesequenz aufwiesen;(iv) zumindest 1.000.000 Barcodemoleküle, diefreisetzbar damit assoziiert sind, wobei die zu-mindest 1.000.000 Barcodemoleküle dieselbeBarcodesequenz aufweisen.
3. Verfahren nach Anspruch 1, wobei die Teileinheitein Tröpfchen ist.
4. Verfahren nach Anspruch 1, wobei das Kügelchenein Gelkügelchen umfasst und wobei die Barcode-moleküle freisetzbar an das Kügelchen gekuppeltsind.
5. Verfahren nach Anspruch 1, wobei die Barcodemo-leküle innerhalb des Kügelchens verkapselt oder imganzen Kügelchen verteilt sind.
6. Verfahren nach Anspruch 1, wobei der Freiset-zungsschritt das Abbauen des Kügelchens umfasst.
7. Verfahren nach Anspruch 4, wobei:
(i) das Abtrennen das Kombinieren einer erstenwässrigen Flüssigkeit, die Kügelchen umfasst,mit einer zweiten wässrigen Flüssigkeit, welchedie Probekomponenten umfasst, in einemTröpfchen innerhalb einer nichtmischbarenFlüssigkeit umfasst;(ii) das Freisetzen das Abbauen des Kügel-chens umfasst, um die Barcodemoleküle in diezweite Teileinheit freizusetzen;(iii) das Freisetzen das Spalten einer chemi-schen Bindung zwischen den Barcodemoleküle
139 140

EP 3 013 957 B1
72
5
10
15
20
25
30
35
40
45
50
55
und dem Kügelchen umfasst; oder(iv) eine Vernetzung des Kügelchens und/odereine Bindung zwischen dem Kügelchen und denBarcodemolekülen eine Disulfidbindung um-fasst und das Freisetzen das Aussetzen des Kü-gelchens gegenüber einem Reduktionsmittelumfasst, wobei das Reduktionsmittel z.B. DTToder TCEP ist.
8. Verfahren nach Anspruch 1, wobei die Probemate-rialien ein oder mehrere Matrizennucleinsäuremole-küle umfassen und:
(i) das Anbringen das Anbringen der Barcode-moleküle an einem oder mehreren Fragmentender Matrizennucleinsäuremoleküle umfasst;oder(ii) die Barcodemoleküle weiters eine Primerse-quenz umfassen, die komplementär zu zumin-dest einem Abschnitt der Matrizennucleinsäu-remoleküle ist, und das Anbringen das Verlän-gern der Barcodemoleküle umfasst, um zumin-dest einen Abschnitt der Matrizennucleinsäure-moleküle zu replizieren.
9. Verfahren nach Anspruch 1, wobei die Probemate-rialien Inhalte einer Einzelzelle umfassen und wobeidie Einzelzelle gegebenenfalls Folgendes umfasst:
(i) eine Krebszelle; oder(ii) eine Bakterienzelle, z.B. eine aus einermenschlichen Mikrobiomprobe isolierte Bakte-rienzelle.
10. Verfahren nach Anspruch 1, wobei die Teileinheitnicht mehr als ein Kügelchen umfasst.
11. Verfahren nach Anspruch 1, wobei:
das Bereitstellen das Bereitstellen einer Vielzahlvon Kügelchen umfasst, die eine Vielzahl ver-schiedener Nucleinsäure-Barcodesequenzeumfassen, wobei jedes der Vielzahl von Kügel-chen eine Vielzahl von zumindest 1000 Nucle-insäure-Barcodemolekülen umfasst, die diesel-be damit assoziierte Nucleinsäure-Barcodese-quenz aufweisen;das Abtrennen das Trennen der Vielzahl von Kü-gelchen mit den Komponenten des Probemate-rials in eine Vielzahl von Teileinheiten umfasst;das Freisetzen das Freisetzen der Nucleinsäu-re-Barcodemoleküle von der Vielzahl von Kü-gelchen in die Vielzahl von Teileinheiten um-fasst; unddas Anbringen das Anbringen der Nucleinsäure-Barcodemoleküle an den Komponenten desProbematerials oder Fragmenten davon inner-halb der Vielzahl von Teileinheiten umfasst.
12. Verfahren nach Anspruch 11, wobei die Vielzahl vonverschiedenen Nucleinsäure-Barcodesequenzenzumindest etwa 1000 verschiedene Barcodese-quenzen, zumindest etwa 10.000 verschiedene Bar-codesequenzen, zumindest etwa 100.000 verschie-dene Barcodesequenzen oder zumindest etwa500.000 verschiedene Barcodesequenzen umfasst.
13. Verfahren nach Anspruch 11, wobei eine Untergrup-pe der Vielzahl von Teileinheiten dieselbe Nuclein-säure-Barcodesequenz umfasst.
14. Verfahren nach Anspruch 13, wobei zumindest etwa1 %, zumindest etwa 2 % oder zumindest etwa 5 %der Vielzahl von Teileinheiten dieselbe Nucleinsäu-re-Barcodesequenz umfasst/umfassen.
15. Verfahren nach Anspruch 14, wobei die Fragmenteder Komponenten des Probematerials ein oder meh-rere Fragmente einer oder mehrerer Matrizennucle-insäuresequenzen umfassen und das Verfahrenweiters das Sequenzieren des einen oder der meh-reren Fragmente der einen oder mehreren Matrizen-nucleinsäuresequenzen und das Charakterisierendes einen oder der mehreren Fragmente der Matri-zennucleinsäuresequenzen, zumindest teilweisebasierend auf einer daran gebundenen Nucleinsäu-re-Barcodesequenz umfasst.
16. Verfahren nach Anspruch 15, wobei das Charakte-risieren des einen oder der mehreren Fragmente dereinen oder mehreren Matrizennucleinsäuresequen-zen Folgendes umfasst:
(i) das Kartieren eines Fragments einer einzel-nen Matrizennucleinsäuresequenz der einenoder mehreren Matrizennucleinsäuresequen-zen bis zu einer einzelnen Matrizennucleinsäu-resequenz der einen oder mehreren Matrizen-nucleinsäuresequenzen oder eines Genoms,von dem die einzelne Matrizennucleinsäurese-quenz abgeleitet wurde; oder(ii) zumindest das Identifizieren einer einzelnenNucleinsäure-Barcodesequenz der Vielzahl vonverschiedenen Nucleinsäure-Barcodesequen-zen und das Identifizieren einer Sequenz eineseinzelnen Fragments des einen oder der meh-reren Fragmente der einen oder mehreren Ma-trizennucleinsäuresequenzen, die an die einzel-ne Nucleinsäure-Barcodesequenz gebundensind.
17. Verfahren nach Anspruch 11, wobei:
(i) zumindest 50 %, zumindest 70 % oder zu-mindest 90 % der Vielzahl von Teileinheitennicht mehr als ein Kügelchen enthalten; oder(ii) zumindest 50 %, zumindest 70 % oder zu-
141 142

EP 3 013 957 B1
73
5
10
15
20
25
30
35
40
45
50
55
mindest 90 % der Vielzahl von Teileinheiten ge-nau ein Kügelchen umfassen.
Revendications
1. Procédé pour attribuer un code-barres à du matérield’échantillon, comprenant :
la mise à disposition d’une bille comprenant unepluralité de molécules à code-barres d’acide nu-cléique associées à celle-ci de manière libéra-ble, la pluralité de molécules à code-barresd’acide nucléique comprenant la même séquen-ce de code-barres d’acide nucléique ;le co-partitionnement de la bille avec des com-posants d’un matériel d’échantillon en unepartition ;la libération des molécules à code-barres à partirde la bille dans la partition ; etl’attachement des molécules à code-barres à unou plusieurs des composants du matérield’échantillon ou à des fragments de celui-ci ausein de la partition.
2. Procédé selon la revendication 1, dans lequel la billecomprend :
(i) au moins 1 000 molécules à code-barres as-sociées à celle-ci de manière libérable, les aumoins 1 000 molécules à code-barres possé-dant la même séquence de code-barres ;(ii) comprend au moins 10 000 molécules à co-de-barres associées à celle-ci de manière libé-rable, les au moins 10 000 molécules à code-barres possédant la même séquence de code-barres ;(iii) au moins 100 000 molécules à code-barresassociées à celle-ci de manière libérable, les aumoins 100 000 molécules à code-barres possé-dant la même séquence de code-barres ; ou(iv) au moins 1 000 000 molécules à code-barresassociées à celle-ci de manière libérable, les aumoins 1 000 000 molécules à code-barres pos-sédant la même séquence de code-barres.
3. Procédé selon la revendication 1, dans lequel la par-tition est une gouttelette.
4. Procédé selon la revendication 1, dans lequel la billecomprend une bille de gel, et dans lequel les molé-cules à code-barres sont couplées de manière libé-rable à la bille.
5. Procédé selon la revendication 1, dans lequel lesmolécules à code-barres sont encapsulées au seinde ou diffusées dans la bille.
6. Procédé selon la revendication 1, dans lequel l’étapede libération comprend la dégradation de la bille.
7. Procédé selon la revendication 4, dans lequel :
(i) le co-partitionnement comprend la combinai-son d’un premier liquide aqueux comprenantdes billes avec un second liquide aqueux com-prenant les composants d’échantillon dans unegouttelette au sein d’un liquide non miscible ;(ii) la libération comprend la dégradation de labille afin de libérer les molécules à code-barresdans la seconde partition ;(iii) la libération comprend un clivage d’uneliaison chimique entre les molécules à code-bar-res et la bille ; ou(iv) au moins l’une d’une réticulation de la billeet d’une liaison entre la bille et les molécules àcode-barres comprend une liaison disulfure, etla libération comprend l’exposition de la bille àun agent réducteur, par exemple où l’agent ré-ducteur est le DTT ou la TCEP.
8. Procédé selon la revendication 1, dans lequel le ma-tériel d’échantillon comprend une ou plusieurs mo-lécules d’acide nucléique matrice, et :
(i) l’attachement comprend l’attachement desmolécules à code-barres à un ou plusieurs frag-ments des molécules d’acide nucléiquematrice ; ou(ii) les molécules à code-barres comprennenten outre une séquence d’amorce complémen-taire à au moins une partie des molécules d’aci-de nucléique matrice, et l’attachement com-prend l’extension des molécules à code-barresafin de répliquer au moins une partie des molé-cules d’acide nucléique matrice.
9. Procédé selon la revendication 1, dans lequel le ma-tériel d’échantillon comprend le contenu d’une cel-lule individuelle, et dans lequel facultativement la cel-lule individuelle comprend :
(i) une cellule cancéreuse ; ou(ii) une cellule bactérienne, par exemple une cel-lule bactérienne isolée à partir d’un échantillonde microbiome humain.
10. Procédé selon la revendication 1, dans lequel la par-tition comprend une seule bille au maximum.
11. Procédé selon la revendication 1, dans lequel :
la mise à disposition comprend la mise à dispo-sition d’une pluralité de billes comprenant unepluralité de séquences de code-barres d’acidenucléique différentes, où chacune de la pluralité
143 144

EP 3 013 957 B1
74
5
10
15
20
25
30
35
40
45
50
55
de billes comprend une pluralité d’au moins 1000 molécules à code-barres d’acide nucléiquepossédant la même séquence de code-barresd’acide nucléique associée à celles-ci ;le co-partitionnement comprend le co-partition-nement de la pluralité de billes avec les compo-sants du matériel d’échantillon en une pluralitéde partitions ;la libération comprend la libération des molécu-les à code-barres d’acide nucléique à partir dela pluralité de billes dans la pluralité departitions ; etl’attachement comprend l’attachement des mo-lécules à code-barres d’acide nucléique auxcomposants du matériel d’échantillon ou à desfragments de celui-ci au sein de la pluralité departitions.
12. Procédé selon la revendication 11, dans lequel lapluralité de séquences de code-barres d’acide nu-cléique différentes comprend au moins environ 1 000séquences de code-barres différentes, au moins en-viron 10 000 séquences de code-barres différentes,au moins environ 100 000 séquences de code-bar-res différentes, ou au moins environ 500 000 sé-quences de code-barres différentes.
13. Procédé selon la revendication 11, dans lequel unsous-ensemble de la pluralité de partitions com-prend la même séquence de code-barres d’acidenucléique.
14. Procédé selon la revendication 13, dans lequel aumoins environ 1 %, au moins environ 2 %, ou aumoins environ 5 % de la pluralité de partitions com-prend la même séquence de code-barres d’acidenucléique.
15. Procédé selon la revendication 14, dans lequel lesfragments des composants du matériel d’échantilloncomprennent un ou plusieurs fragments d’une ouplusieurs séquences d’acide nucléique matrice et leprocédé comprend en outre le séquençage du un ouplusieurs fragments de la une ou plusieurs séquen-ces d’acide nucléique matrice et la caractérisationdu un ou plusieurs fragments des séquences d’acidenucléique matrice en se basant au moins en partiesur une séquence de code-barres d’acide nucléiqueattachée à ceux-ci.
16. Procédé selon la revendication 15, dans lequel lacaractérisation du un ou plusieurs fragments de laune ou plusieurs séquences d’acide nucléique ma-trice comprend :
(i) la cartographie d’un fragment d’une séquen-ce d’acide nucléique matrice individuelle de laune ou plusieurs séquences d’acide nucléique
matrice à une séquence d’acide nucléique ma-trice individuelle de la une ou plusieurs séquen-ces d’acide nucléique matrice ou d’un génomeà partir duquel la séquence d’acide nucléiquematrice individuelle a été dérivée ; ou(ii) au moins l’identification d’une séquence decode-barres d’acide nucléique individuelle de la-dite pluralité de séquences de code-barresd’acide nucléique différentes, et l’identificationd’une séquence d’un fragment individuel du unou plusieurs fragments de la une ou plusieursséquences d’acide nucléique matrice attachéeà la séquence de code-barres d’acide nucléiqueindividuelle.
17. Procédé selon la revendication 1, dans lequel :
(i) au moins 50 %, au moins 70 %, ou au moins90 % de la pluralité de partitions contient uneseule bille au maximum ; ou(ii) au moins 50 %, au moins 70 %, ou au moins90 % de la pluralité de partitions comprend exac-tement une seule bille.
145 146
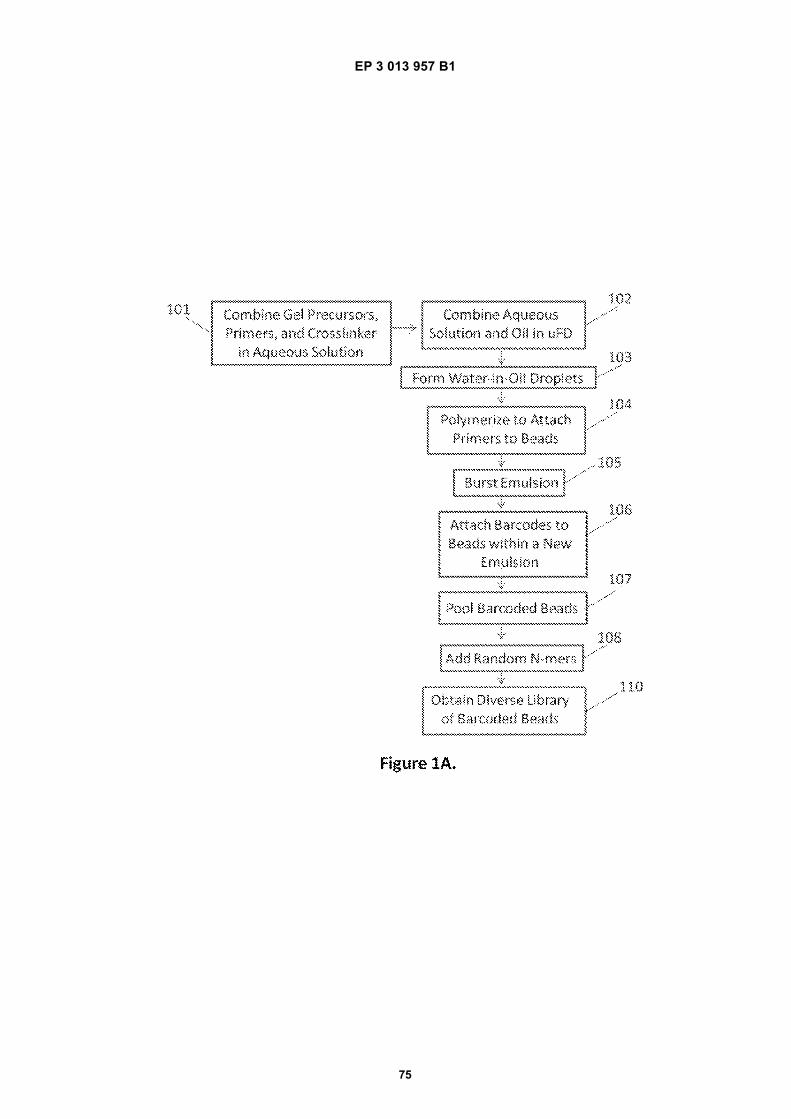
EP 3 013 957 B1
75
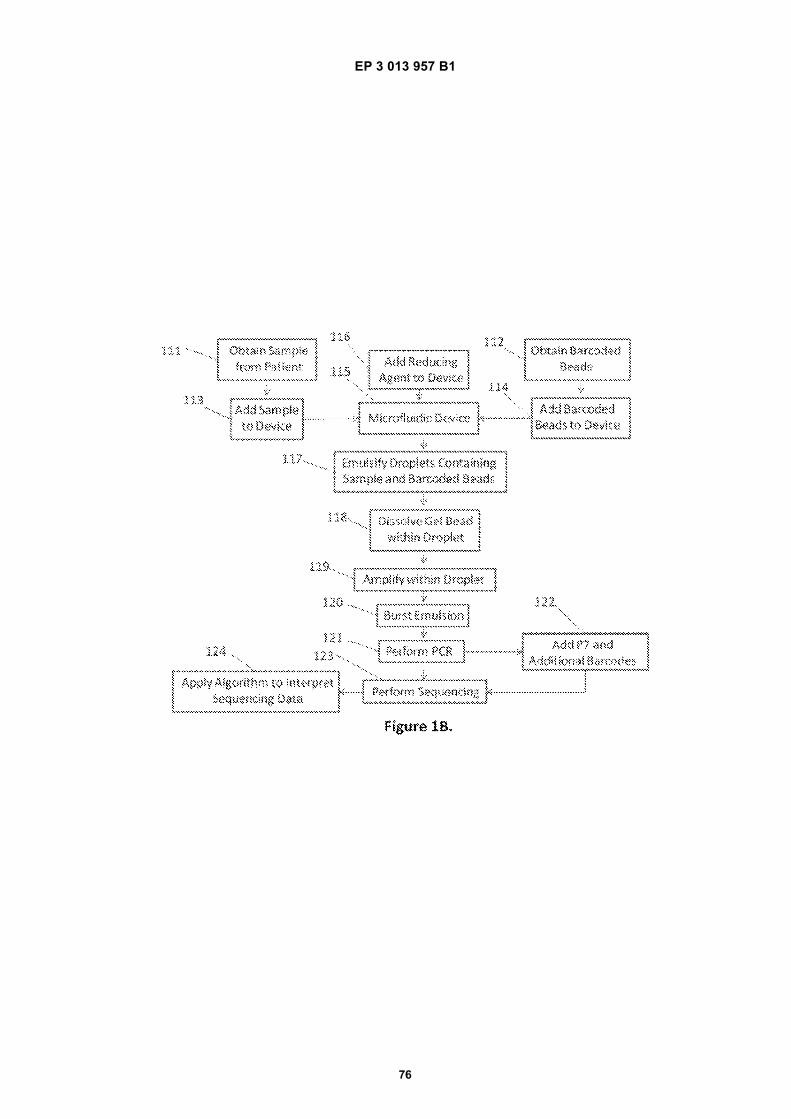
EP 3 013 957 B1
76
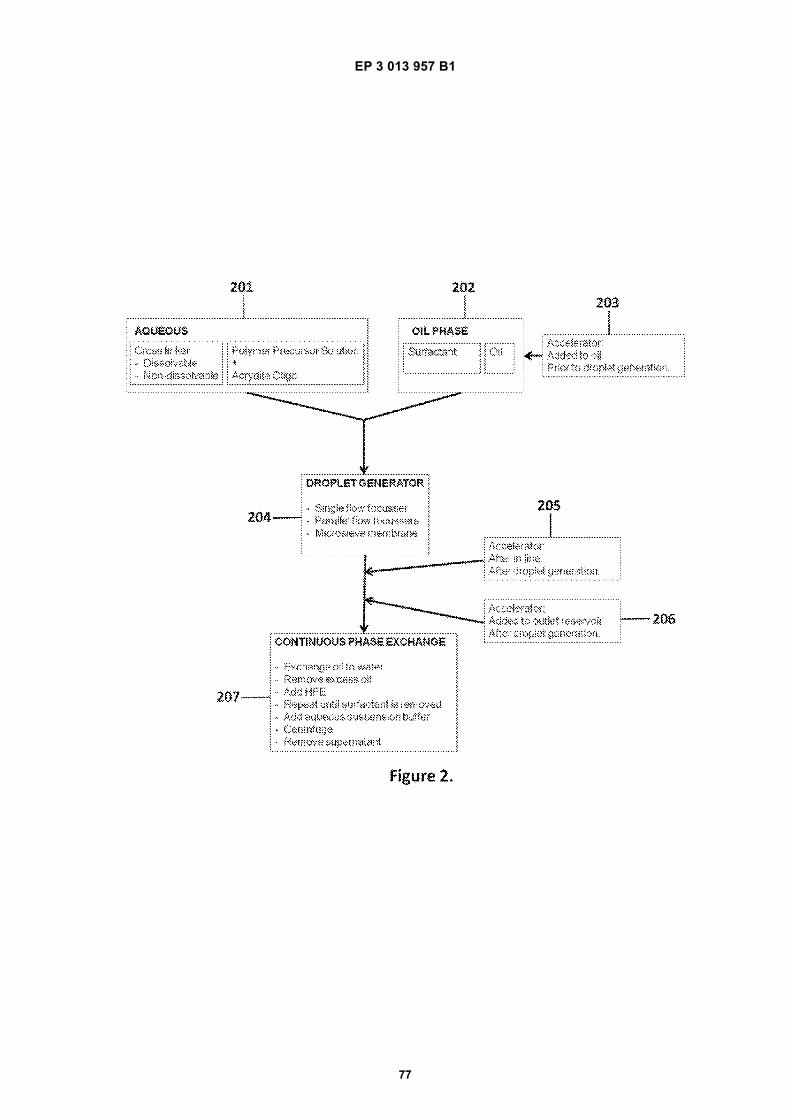
EP 3 013 957 B1
77

EP 3 013 957 B1
78

EP 3 013 957 B1
79
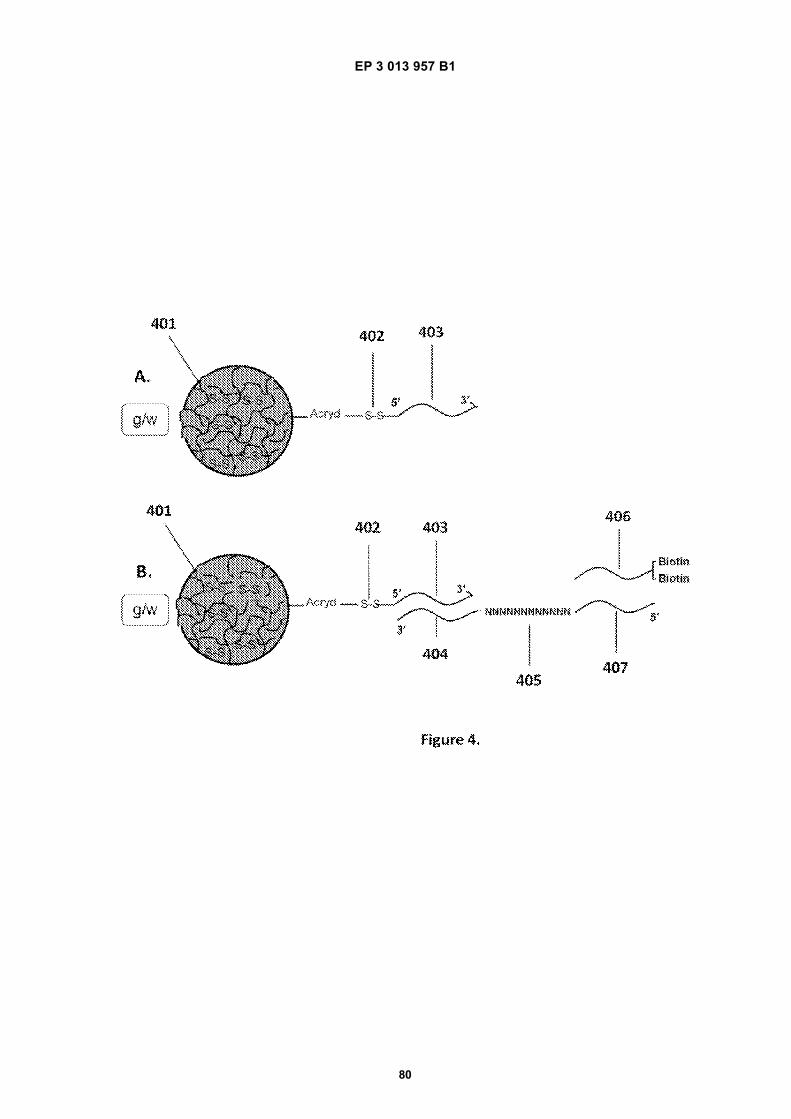
EP 3 013 957 B1
80
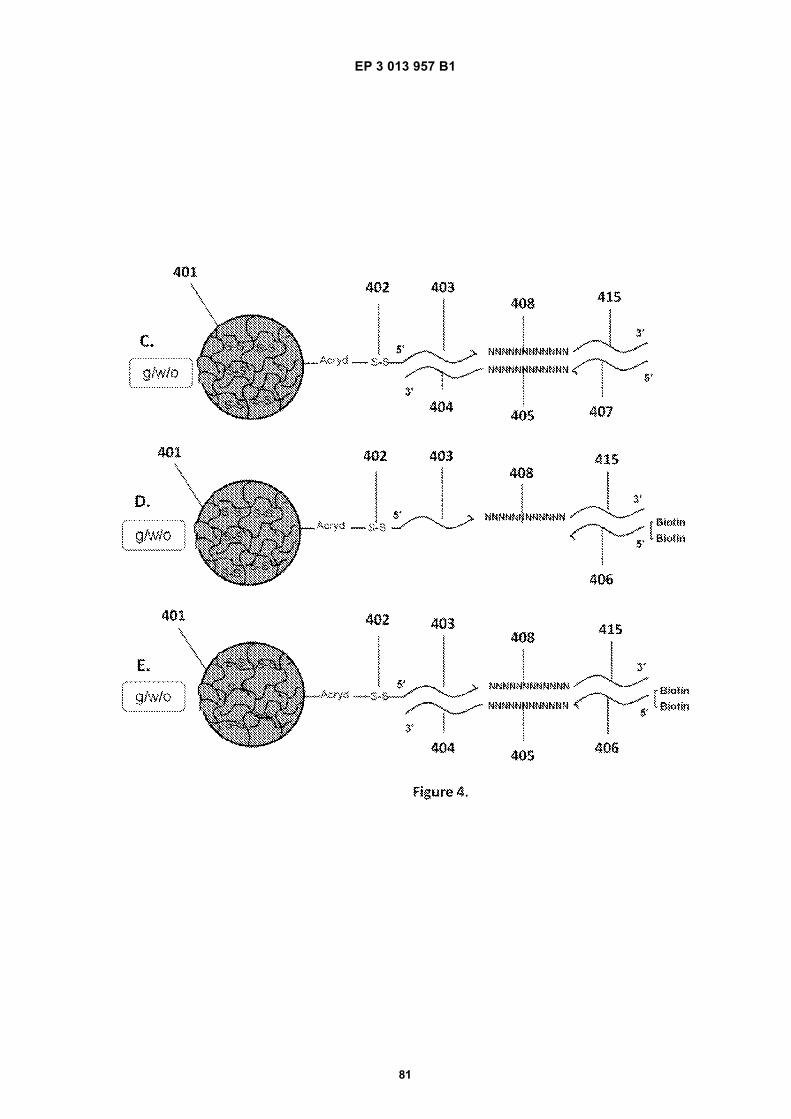
EP 3 013 957 B1
81

EP 3 013 957 B1
82
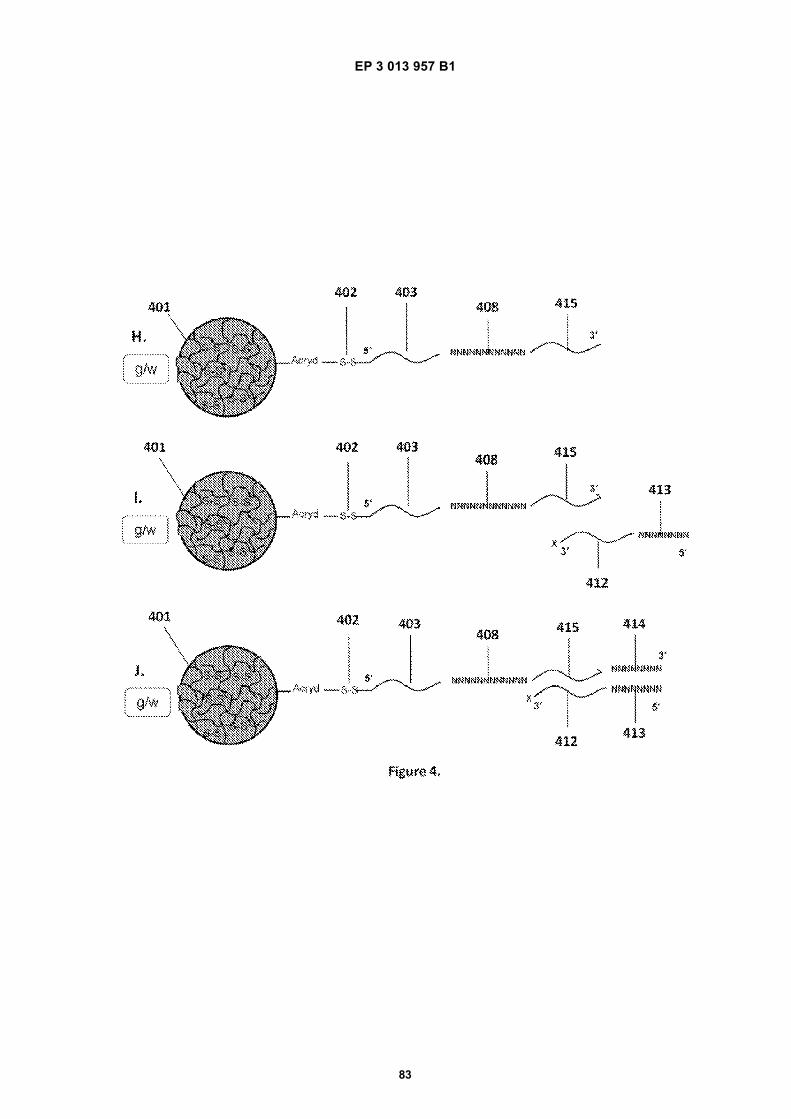
EP 3 013 957 B1
83

EP 3 013 957 B1
84
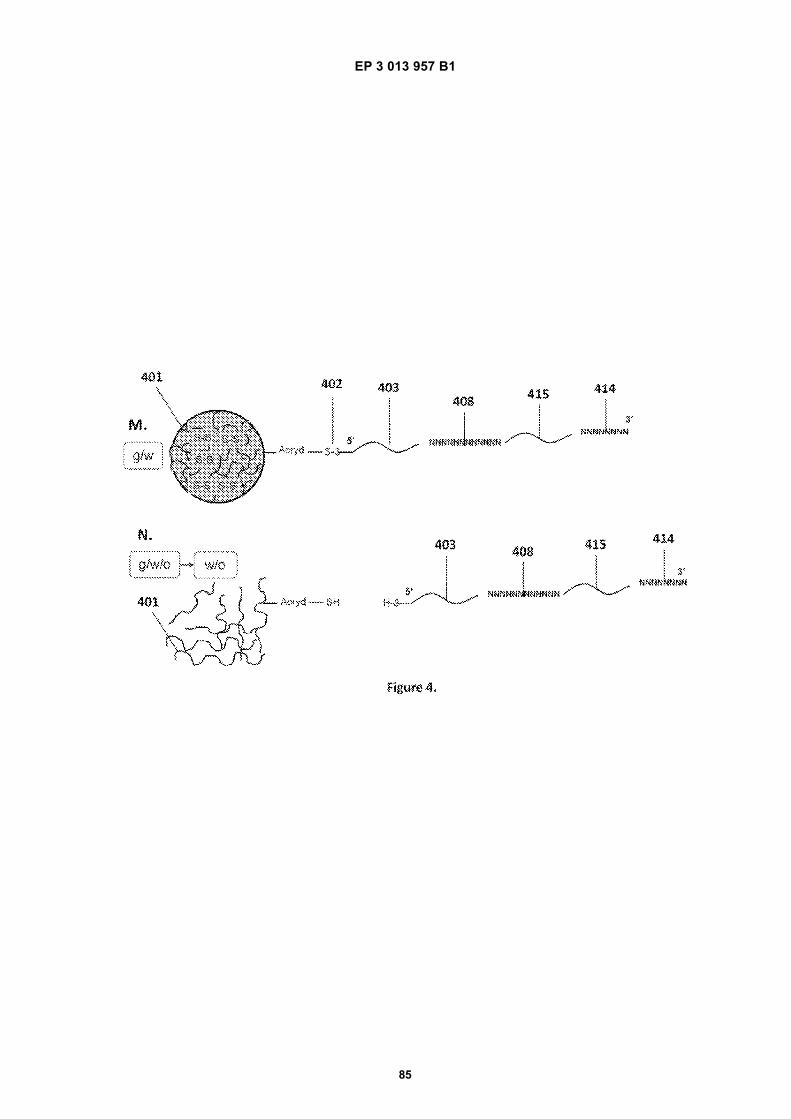
EP 3 013 957 B1
85

EP 3 013 957 B1
86

EP 3 013 957 B1
87

EP 3 013 957 B1
88

EP 3 013 957 B1
89

EP 3 013 957 B1
90
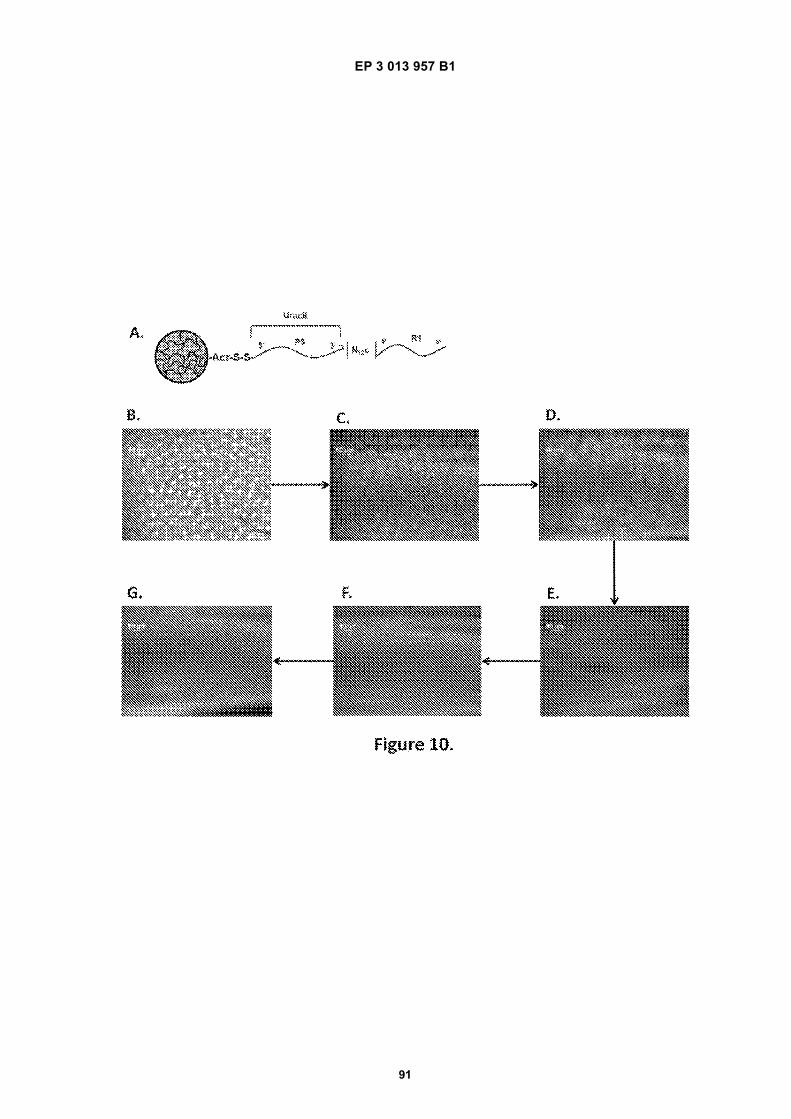
EP 3 013 957 B1
91

EP 3 013 957 B1
92

EP 3 013 957 B1
93

EP 3 013 957 B1
94
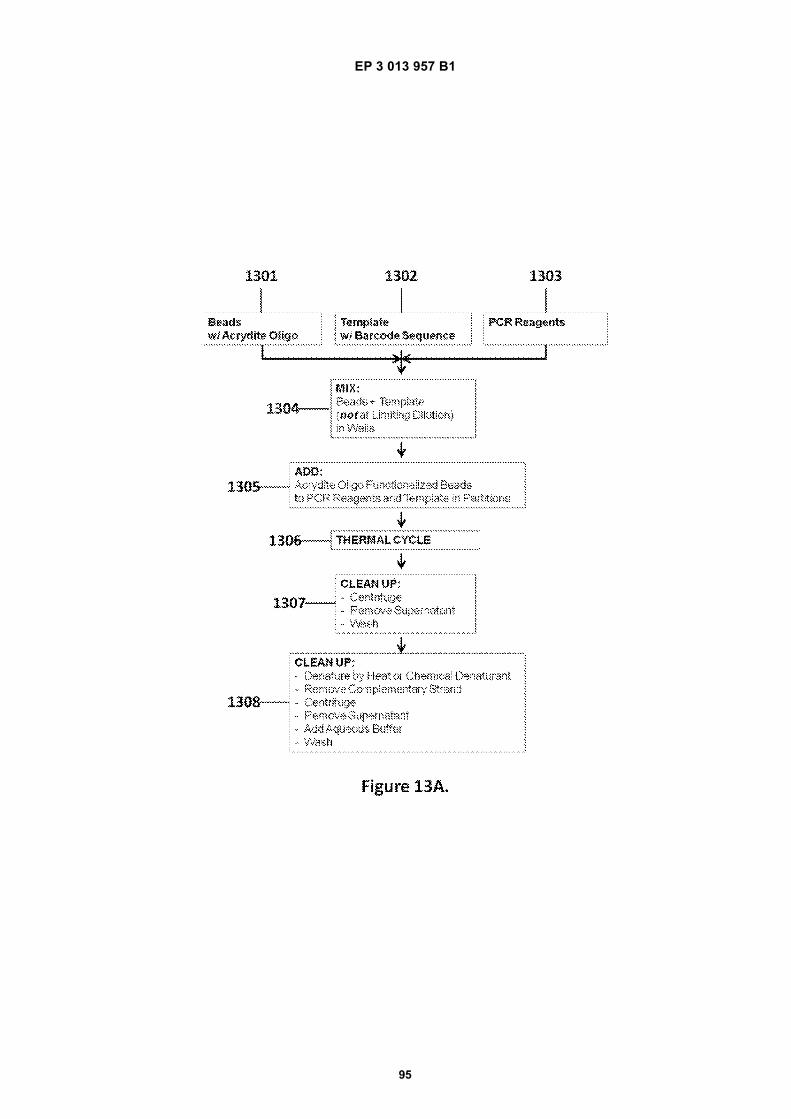
EP 3 013 957 B1
95
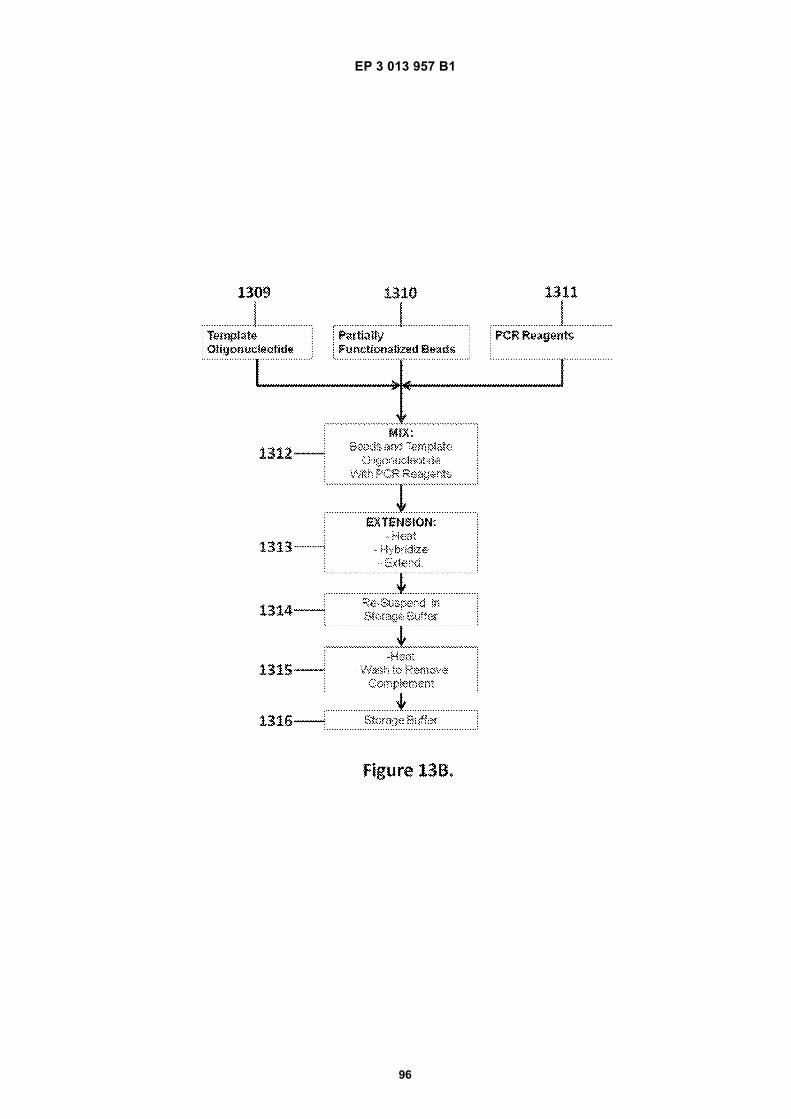
EP 3 013 957 B1
96

EP 3 013 957 B1
97

EP 3 013 957 B1
98
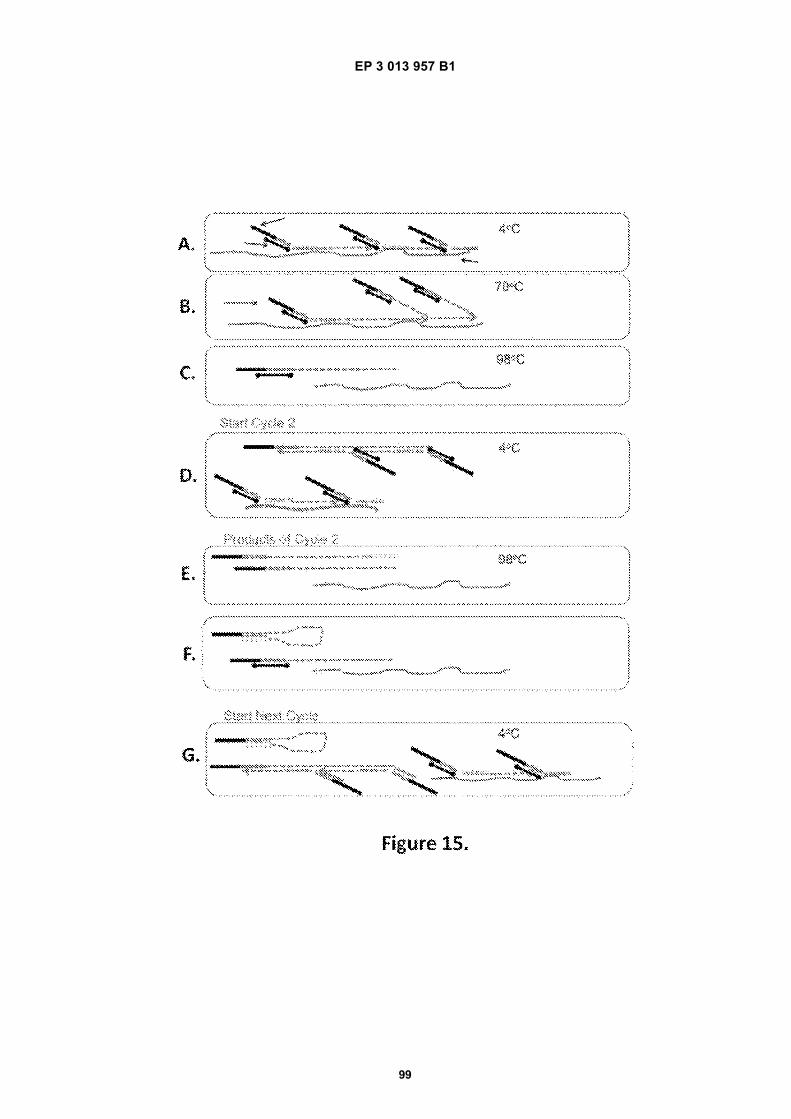
EP 3 013 957 B1
99

EP 3 013 957 B1
100

EP 3 013 957 B1
101

EP 3 013 957 B1
102

EP 3 013 957 B1
103

EP 3 013 957 B1
104

EP 3 013 957 B1
105

EP 3 013 957 B1
106
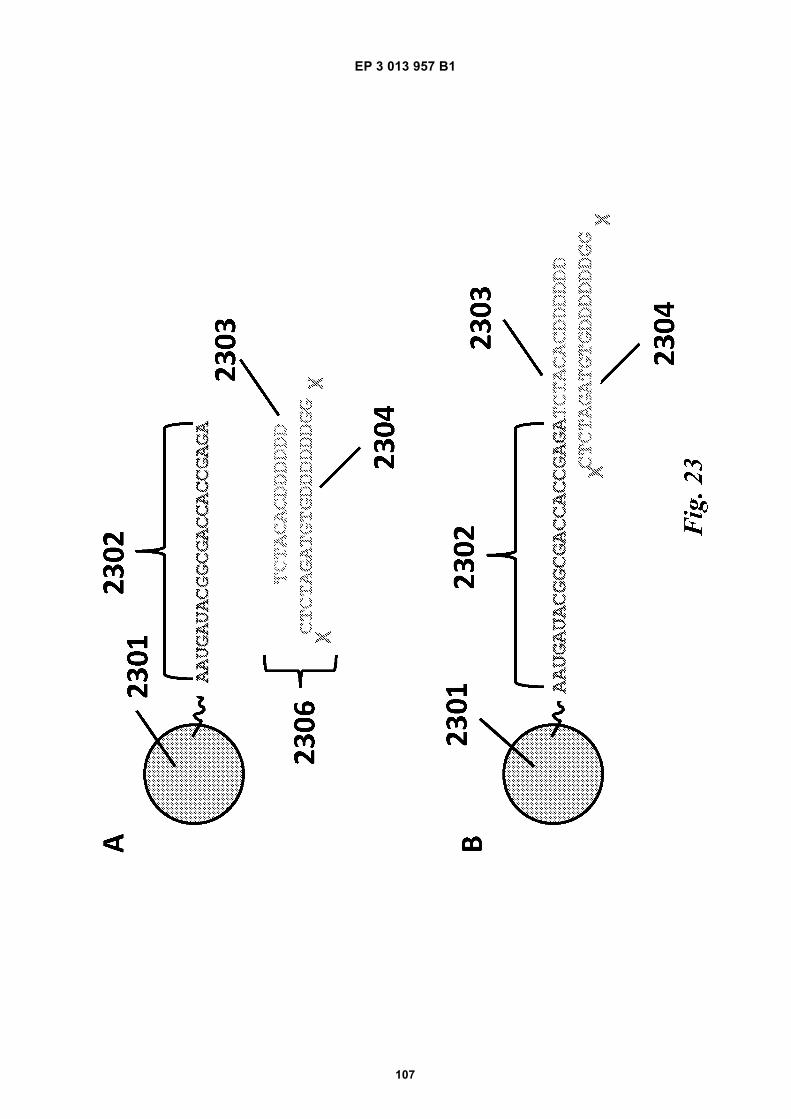
EP 3 013 957 B1
107

EP 3 013 957 B1
108

EP 3 013 957 B1
109
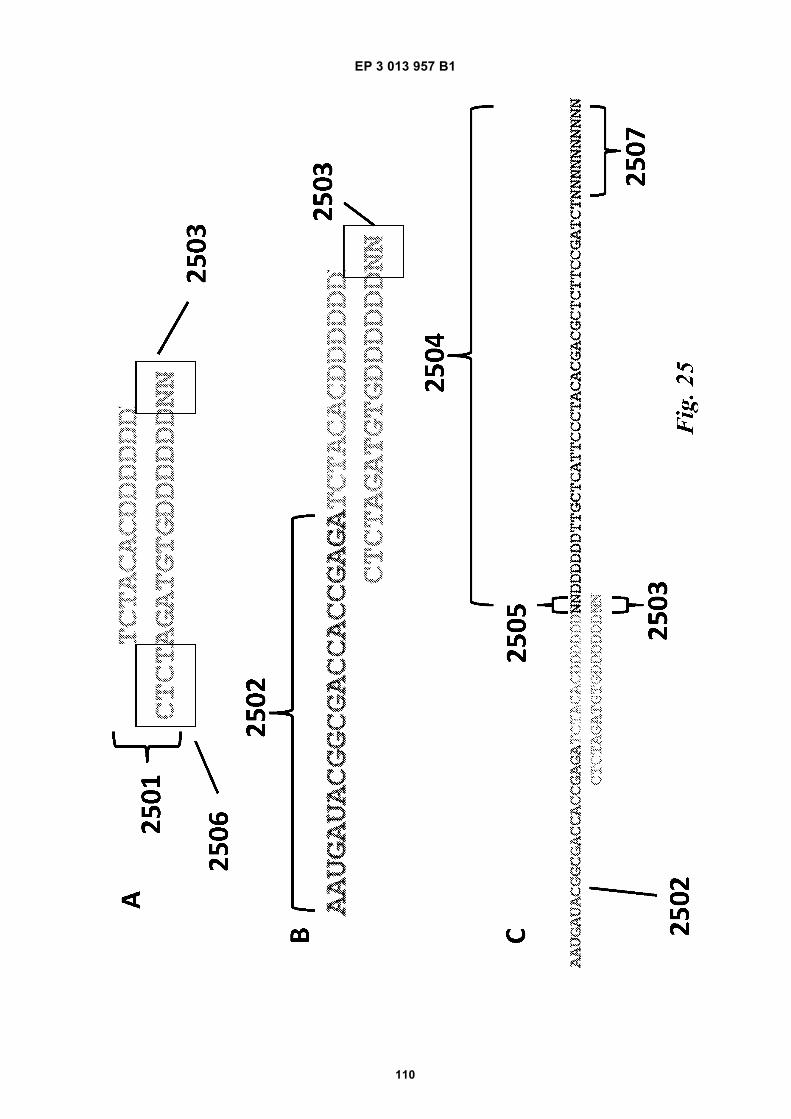
EP 3 013 957 B1
110
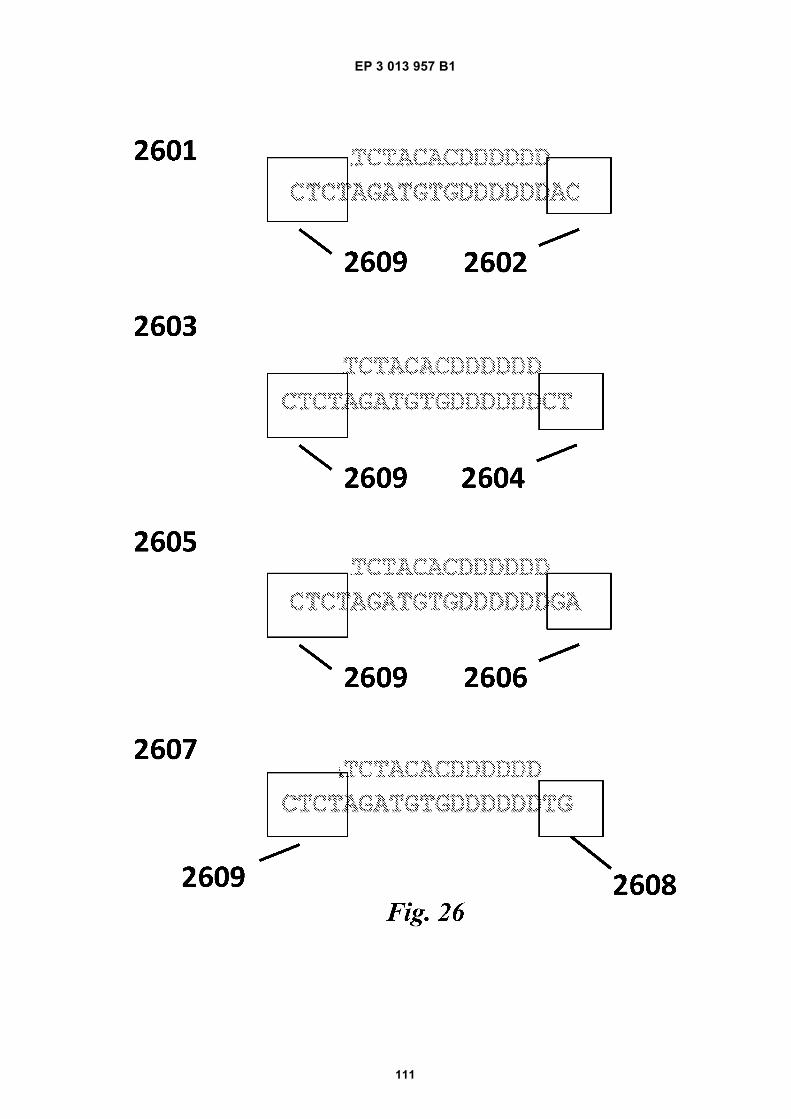
EP 3 013 957 B1
111

EP 3 013 957 B1
112
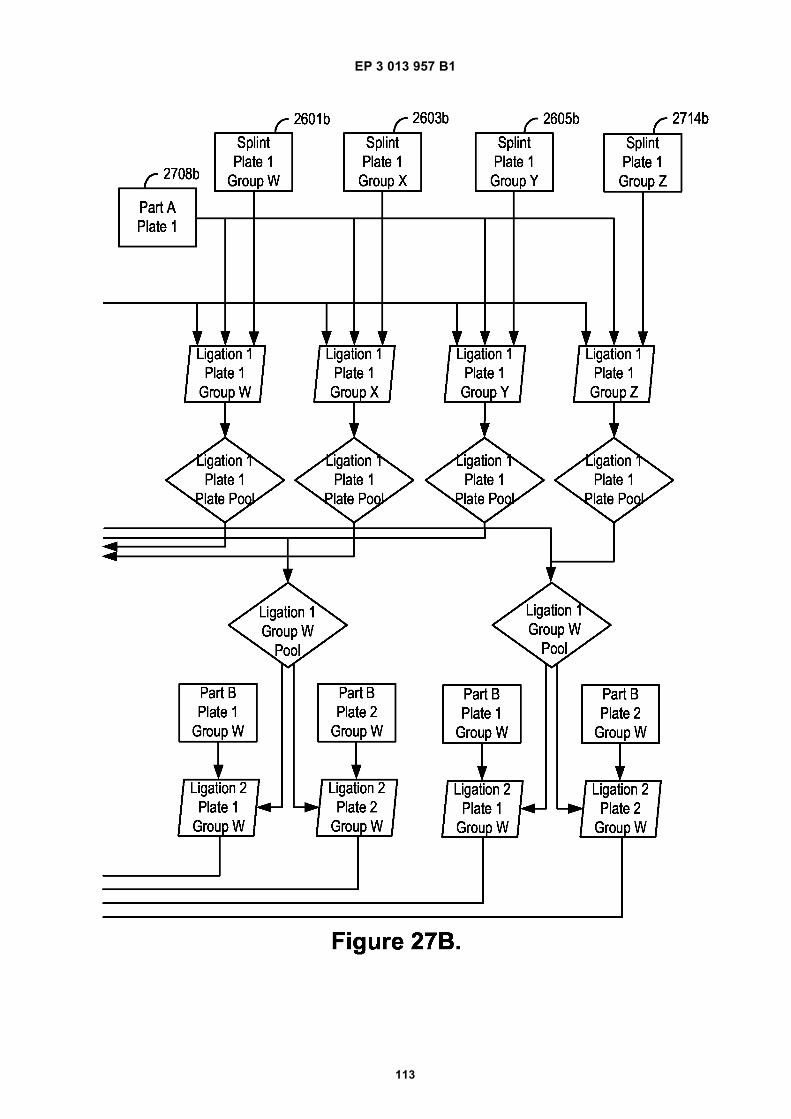
EP 3 013 957 B1
113

EP 3 013 957 B1
114
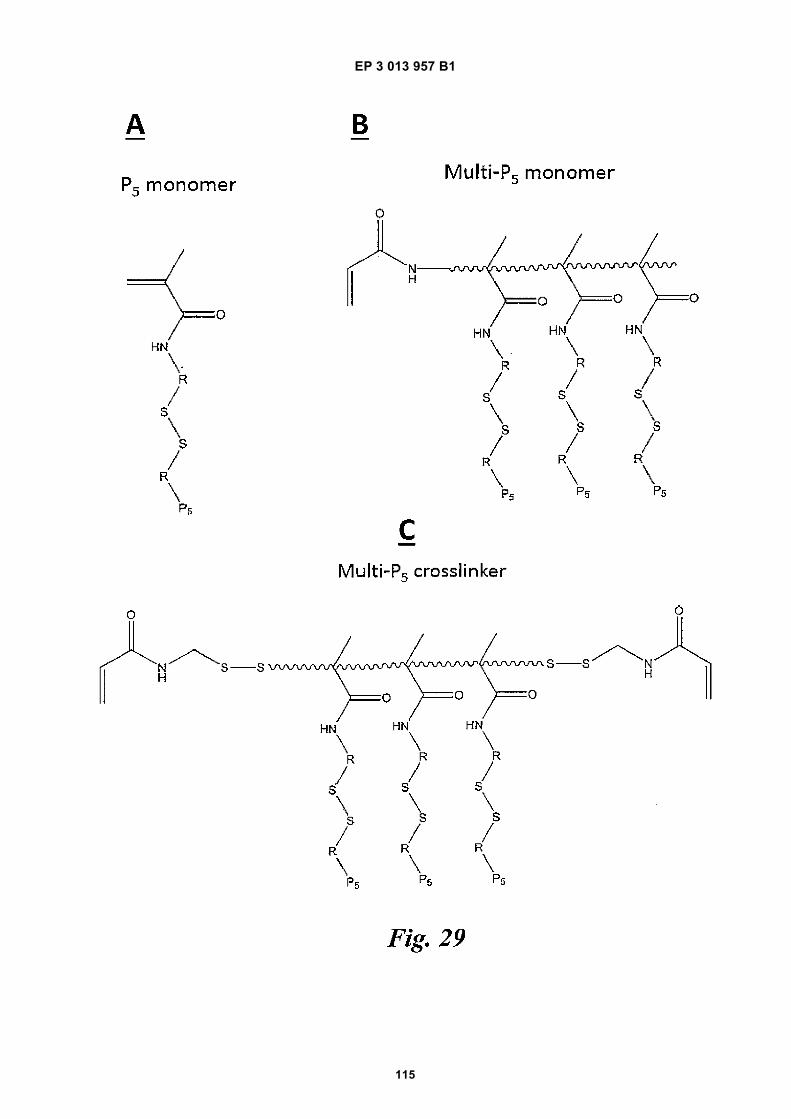
EP 3 013 957 B1
115
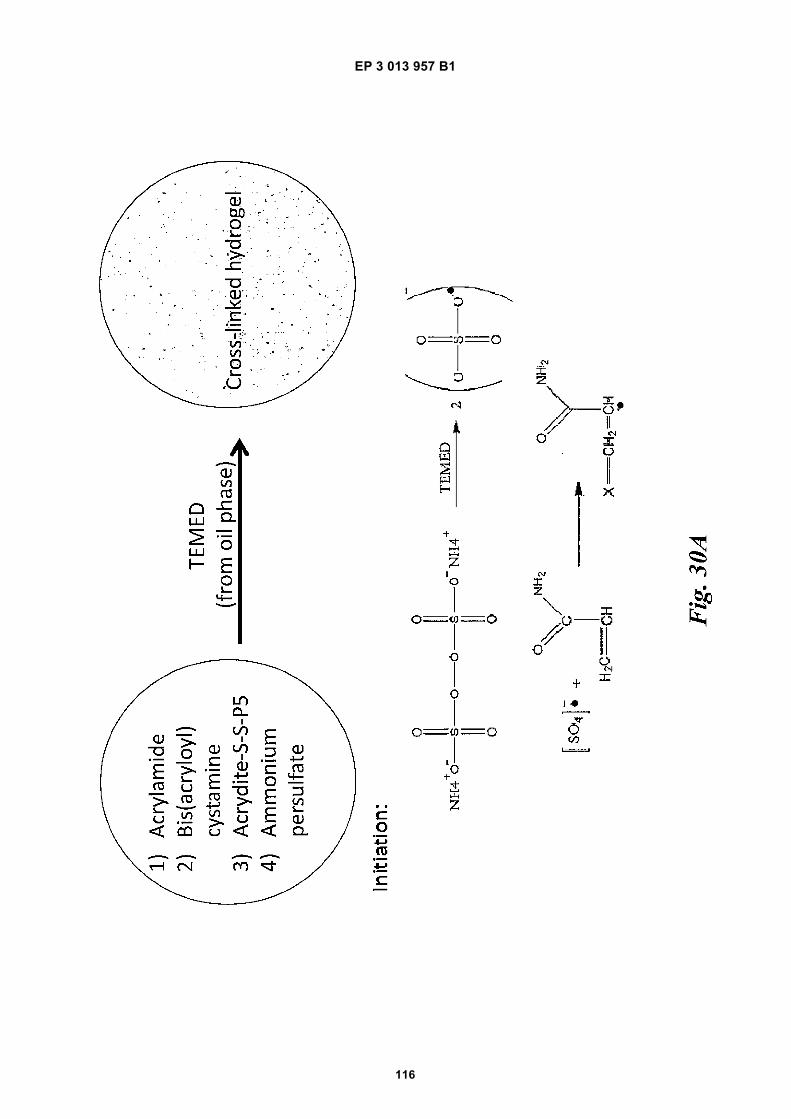
EP 3 013 957 B1
116

EP 3 013 957 B1
117

EP 3 013 957 B1
118

EP 3 013 957 B1
119

EP 3 013 957 B1
120

EP 3 013 957 B1
121

EP 3 013 957 B1
122

EP 3 013 957 B1
123
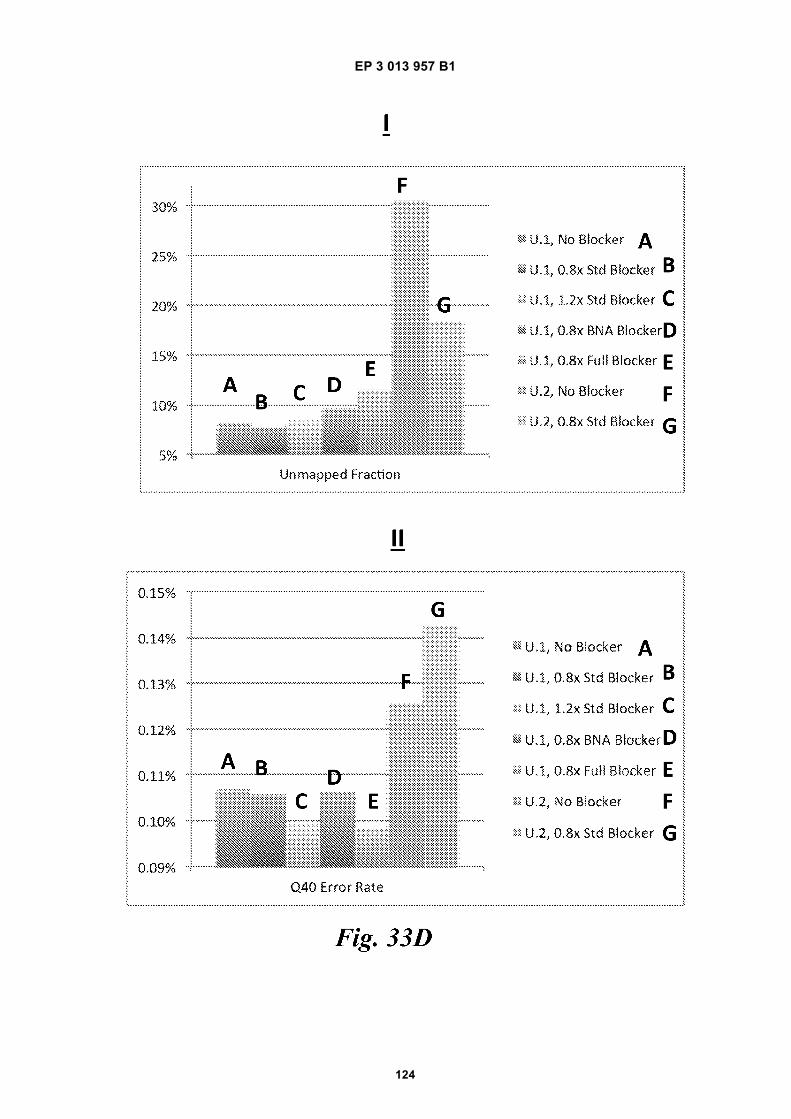
EP 3 013 957 B1
124

EP 3 013 957 B1
125
EP 3 013 957 B1
126
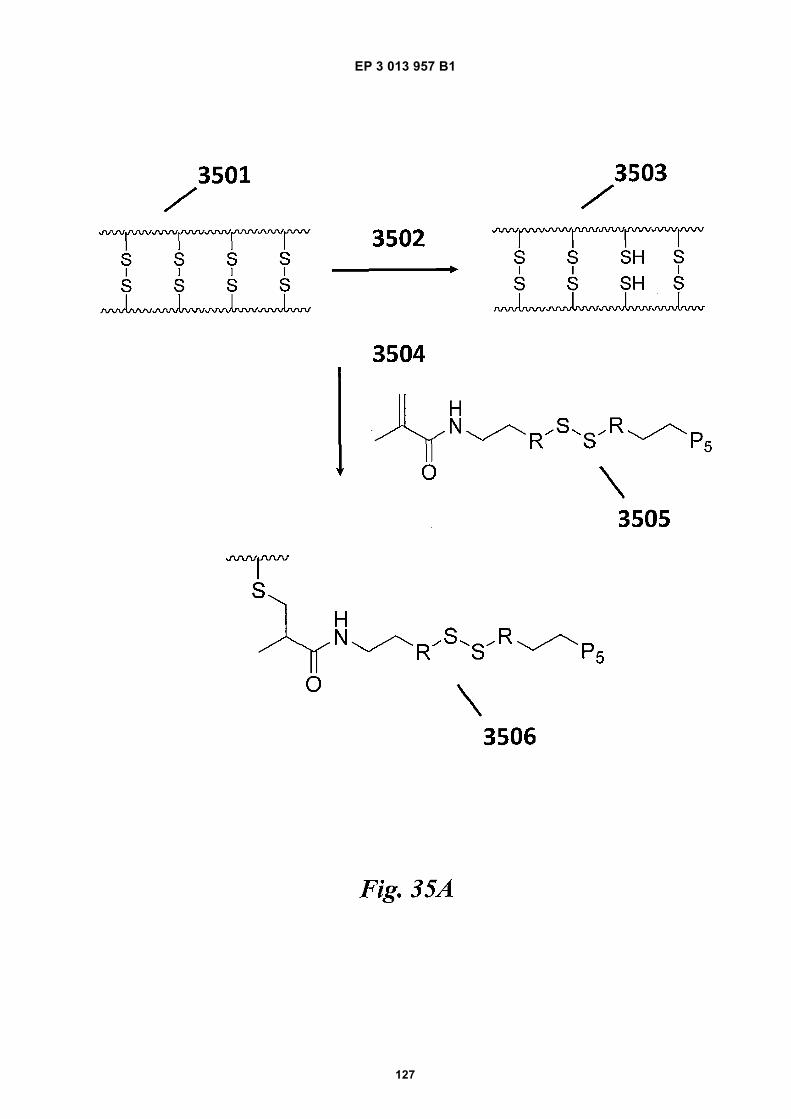
EP 3 013 957 B1
127
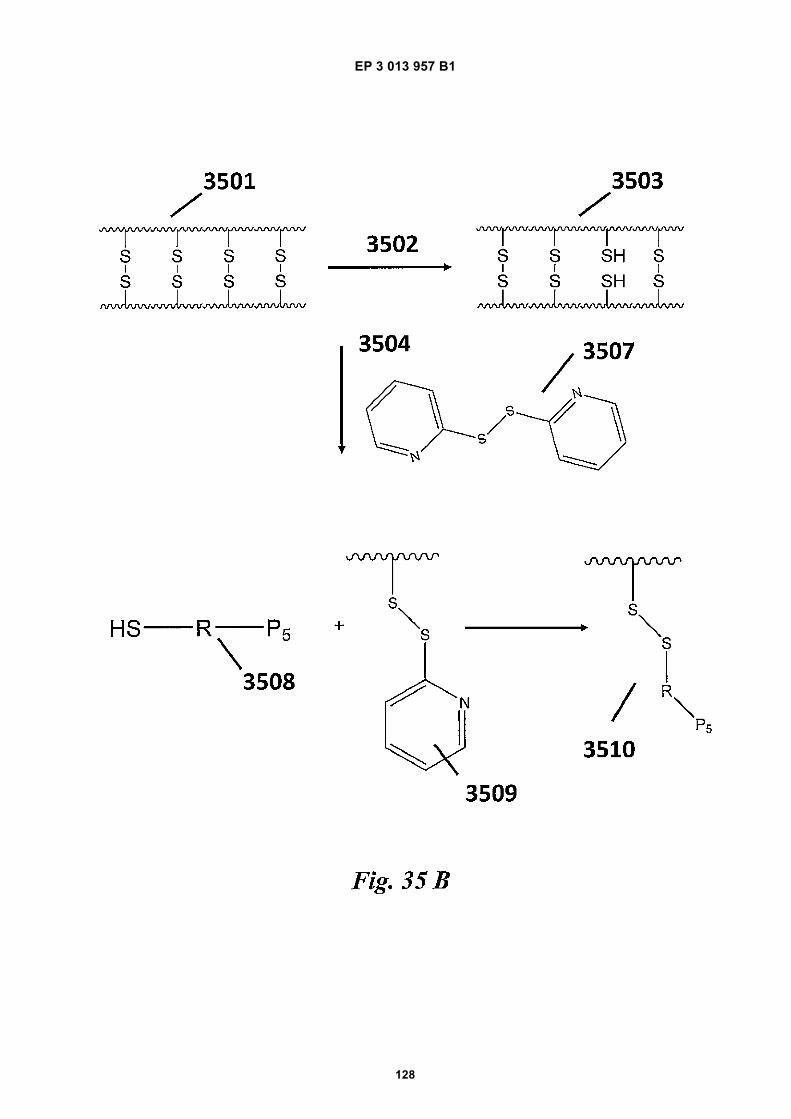
EP 3 013 957 B1
128

EP 3 013 957 B1
129
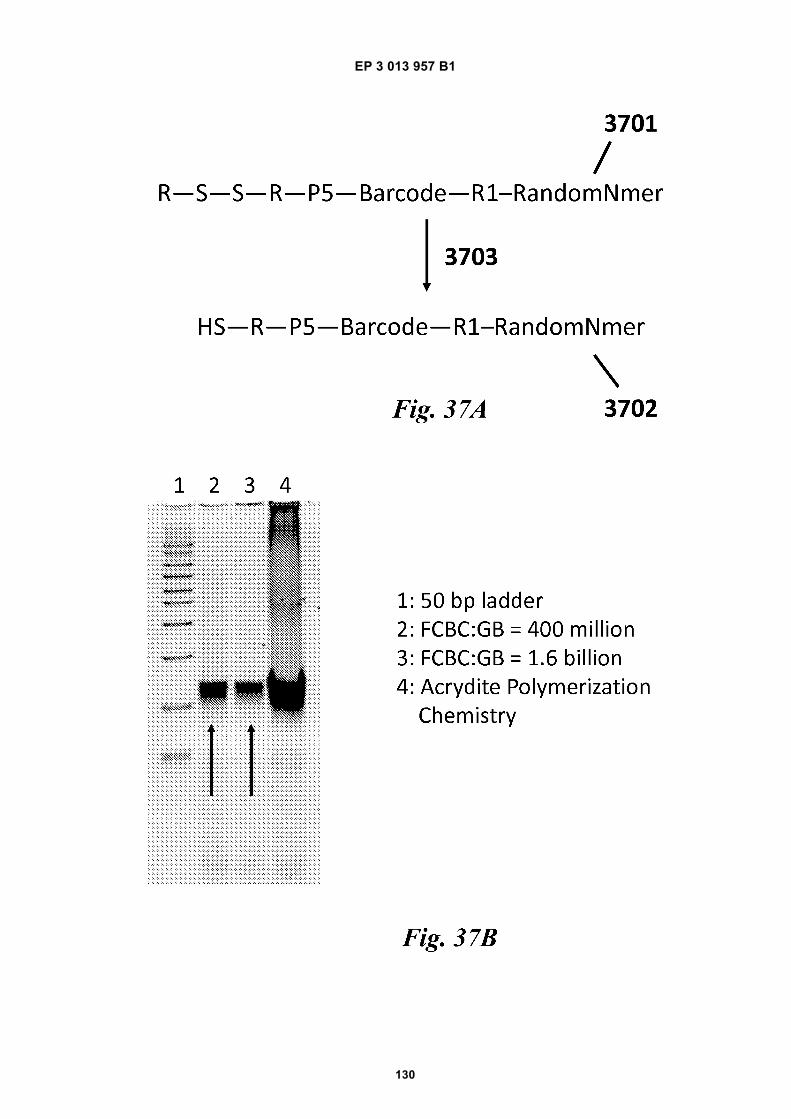
EP 3 013 957 B1
130

EP 3 013 957 B1
131

EP 3 013 957 B1
132

EP 3 013 957 B1
133

EP 3 013 957 B1
134

EP 3 013 957 B1
135
REFERENCES CITED IN THE DESCRIPTION
This list of references cited by the applicant is for the reader’s convenience only. It does not form part of the Europeanpatent document. Even though great care has been taken in compiling the references, errors or omissions cannot beexcluded and the EPO disclaims all liability in this regard.
Patent documents cited in the description
• WO 2010115154 A [0002]• WO 2012048341 A [0002]• US 20120316074 A [0002]
• US 20120211084 A, Weitz [0068]• US 61977804 A [0069] [0193]• US 17597314 A [0079]
Non-patent literature cited in the description
• WEIZMANN et al. Nature Methods, 2006, vol. 3 (7),545-550 [0068]
• GARTECKI et al. Applied Physics Letters, 2004, vol.85 (13), 2649-2651 [0068]
• FISHER et al. Genome Biol., 2011, vol. 12 (1), R1[0326]