prenatal ethanol exposure, generalized learning impairment, and medial prefrontal cortical deficits...
TRANSCRIPT

Prenatal ethanol exposure, generalized learning impairment, and medial
prefrontal cortical deficits in rats
Sheila M. Mihalick*, James E. Crandall, Jason C. Langlois,Jason D. Krienke, William V. Dube1
Psychological Sciences Division, University of Massachusetts Medical School-Shriver Center, 200 Trapelo Road, Waltham, MA 02452-6319, USA
Received 16 October 2000; received in revised form 19 April 2001; accepted 12 July 2001
Abstract
Prenatal ethanol exposure may cause neurological damage and subsequent mental retardation in humans, with learning deficits similar to
those following damage to the prefrontal cortex. This study examined cognitive dysfunction and cortical damage after prenatal exposure to
ethanol using a chronic administration model. Pregnant Sprague–Dawley rats received one of three diets during gestation: a liquid diet
containing 35% ethanol-derived calories (ETOH), an isocaloric liquid diet (ISO), or standard chow (CHOW). Subjects were obtained from
ETOH dams with blood alcohol concentrations (BACs) above 90 mg/dl and corresponding ISO and CHOW controls (one male pup/litter;
n = 6 pups/group). At approximately 90 days of age, subjects began training on a series of unique auditory discrimination problems using a
successive go/no-go procedure. A criterion of 85% accuracy determined when a rat continued to the next problem. Subjects completed a
varying number of problems within a 30-session limit, after which all rats were tested on a tone/click discrimination and reversal. Subjects
were then sacrificed and neuronal number in the medial prefrontal cortex (mPFC) was estimated by the optical fractionator method. Prenatal
ethanol exposure induced significant cell loss in the mPFC, which was associated with significantly impaired reversal learning. Poor
performance by ETOH subjects on the tone/click reversal indicates a transfer of training deficit that may reflect failures of inhibitory control.
D 2001 Elsevier Science Inc. All rights reserved.
Keywords: FAS; Prenatal ethanol; Reversal learning; Rats; Transfer of training; Auditory discrimination; Prefrontal cortex
1. Introduction
Prenatal exposure to alcohol may result in Fetal Alcohol
Syndrome (FAS), with symptoms including craniofacial
dysmorphism and central nervous system damage (reviewed
in Ref. [75]). The behavioral profile of FAS in humans
includes developmental delays [3,4,44], problems with
inhibitory control [48], and poor performance on tests of
intelligence, working memory, and academic achievement
(e.g., Ref. [14,75,77]). FAS is one of the leading known
causes of human mental retardation [2].
The neurological damage primarily associated with FAS
includes microencephaly with disorganization of the cereb-
ral cortex, cerebellum, and basal ganglia as indicated by
neuroglial heterotopias and major white matter tract dys-
genesis (reviewed in Ref. [13]). It is not known exactly how
alcohol disrupts neurodevelopment, but it could impair any
or all of the following processes: cell proliferation and
death, neuronal migration, neurite outgrowth, synaptogene-
sis, and myelination (reviewed in Refs. [33,86]). The few
human FAS brains that have been analyzed show neuro-
pathologic features indicating disruption of cytogenesis and/
or neuronal migration [29,68].
Efforts to model FAS in rodents have generated much data
from many different paradigms, creating an extensive but
diffuse picture of prenatal ethanol effects in the rat. In general,
the data obtained from rodent FASmodels correspond well to
the human profile. Rats exposed prenatally to ethanol typ-
ically show developmental delays (e.g., Ref. [42]), inhibitory
deficits (e.g., Ref. [64]), and impairments on tests of spatial
ability (e.g., Ref. [28]), workingmemory (e.g., Ref. [61]), and
reversal learning (e.g., Ref. [45]). The neuropathologic fea-
tures observed in rodent FAS models include disrupted
neuronal and glial development in the cerebellum, hippocam-
pus, and olfactory bulb (e.g., Refs. [9,23,26,88]).
0892-0362/01/$ – see front matter D 2001 Elsevier Science Inc. All rights reserved.
PII: S0892 -0362 (01 )00168 -4
* Corresponding author. Tel.: +1-607-255-1839.
E-mail addresses: [email protected] (S.M. Mihalick),
[email protected] (W.V. Dube).1 Also corresponding author.
Neurotoxicology and Teratology 23 (2001) 453–462

Several lines of evidence suggest the potential value of
research on ethanol-related damage to the medial prefrontal
cortex (mPFC). First, continuing studies by Miller
[51,52,54], Miller and Kuhn [55], Miller and Potempa
[56], and Miller and Robertson [57] indicate that prenatal
ethanol alters many aspects of typical cortical development
by interfering with cell cycle kinetics, neuronal migration,
glial genesis and transformation, cortical connectivity, and
cell number [24,25,27,32,59]. Second, the cortex is vulner-
able to ethanol toxicity. Ethanol decreases cortical thickness
[42], kills cortical cells in vitro [73], and alters the in vivo
expression of proteins involved in cortical neuron death [43].
Finally, the functional deficits that are observed after
damage to mPFC resemble those seen in FAS and/or mental
retardation. Prefrontal damage is consistent with deficits in
generalized learning, strategic and organizational functions,
and flexibility to changing environmental demands (e.g.,
Ref. [41]). For example, Winocur and Moscovitch [93]
demonstrated that rats with lesions of mPFC were impaired
on transfer of training from one version of a maze to
another, but not on learning that depended on maze-specific
information. As another example, in an animal model of
experimental phenylketonuria in rats, Strupp et al. [78]
found no deficiency in learning during initial training with
different types of problems. There was, however, a defi-
ciency in the transfer of learning that was detected only
when the animals were given new exemplars of each type of
problem. The potential relevance of this finding to mPFC
comes from studies by Diamond et al. [15] linking experi-
mental PKU in rats to neurotransmitter abnormalities spe-
cifically in the prefrontal cortex. Strupp et al. noted that their
finding corresponds with the human literature. Humans with
mental retardation may not be impaired relative to typically
developing individuals on simple tests with salient stimuli,
but they are more likely to exhibit impairments on more
complex tests that require transfer of training [10,11,39,50].
Previous studies with rat models of prenatal ethanol
exposure have not specifically examined the possible asso-
ciation between cortical damage and cognitive deficits.
Although some works on prenatal ethanol do relate behav-
ioral deficits and altered gene expression in PFC [60], there
have been no attempts to link learning impairments and
cortical injury per se. The present experiment was designed
to determine whether prenatal ethanol adversely affects cell
number in rat mPFC and whether exposure leads to cogni-
tive deficits consistent with mPFC damage.
This study used a prenatal chronic exposure model and
examined transfer of training in ethanol-exposed rats, iso-
caloric controls, and ad libitum chow-fed controls. The
initial training was a series of two-choice discrimination
problems with a different pair of auditory stimuli for each
problem. The transfer test was a discrimination reversal
problem in which the positive and negative stimulus func-
tions for a specific pair of stimuli were reversed. Positive
transfer has been reported in several species between these
two tasks (e.g., Refs. [35,37,38,69–72,83]). The major
dependent measures were performance on initial discri-
mination learning, on the transfer test, and neuronal number
in mPFC as estimated by the optical fractionator stereo-
logical method [90].
2. Methods
Mature nulliparous Sprague–Dawley albino females
obtained from Charles River Laboratories (Wilmington,
MA, USA) were timed-mated with mature males. Upon
detection of a vaginal plug (gestation day, or GD, 1),
females were isolated and randomly assigned to the diet
administration groups described below. Dams were housed
in individual cages, with ad libitum access to water, on a
12-h light/dark cycle with lights on at 7 a.m.
2.1. Diet administration
Animals in ethanol and isocaloric groups (described
below) were fed Bio-Serv high-protein liquid diet (Bio-Serv
1265SP; Bio-Serv, Frenchtown, NJ, USA). The diet was
presented in graduated feeding tubes (Bio-Serv 9010) over-
night from 6 p.m. to 9 a.m. Standard lab chow was
unavailable for these groups from GD2 through GD20. On
GD1, all dams assigned to receive the ethanol or isocaloric
diets were fed 30 ml of the liquid control diet in addition to
ad libitum chow. This procedure was done to help minimize
the potential effects of food neophobia (e.g., Ref. [30]).
From GD2 to GD20, dams received the appropriate liquid
diet (ethanol or isocaloric control) only. On GD21, liquid
diet was discontinued and ad libitum chow reinstated.
2.1.1. Ethanol exposure
Nine dams in the ethanol diet group (ETOH) were given
a 100-ml mixture of one-third ethanol diet and two-thirds
isocaloric diet (12% ethanol-derived calories, or EDC, and
23% maltose dextrin-derived calories, or MDC) on GD2–3;
two-thirds ethanol diet and one-third isocaloric diet (23%
EDC and 12% MDC) on GD4–6; and ethanol diet only
(35% EDC) on GD7–20.
2.1.2. Isocaloric control
Each of the nine dams in the isocaloric control group
(ISO) was yoked to an ETOH dam and pair-fed the
isocaloric control diet (35% MDC) on a milliliter per
kilogram weight basis.
2.1.3. Standard diet control
All nine dams in the standard diet control group
(CHOW) had ad libitum access to standard chow.
2.2. Blood alcohol concentration (BAC)
On GD16, 50 ml of blood was collected from the tails of
ETOH dams between 7:30 and 8:00 a.m. (e.g., Ref. [47];
S.M. Mihalick et al. / Neurotoxicology and Teratology 23 (2001) 453–462454

previous research indicates that the highest rate of consump-
tion for liquid diet presented at 7:00 p.m. is between 3:00
and 7:00 a.m. [91]). Tails of ISO and CHOW dams were
also nicked, but no blood was collected. The GD chosen to
measure BAC was consistent with previous work [1,31,47,
53,62,66,92].
BAC was determined by an enzymatic spectrophotomet-
ric assay for alcohol concentration (Sigma Diagnostics kit
no. 332; Sigma, St. Louis, MO). Samples were read at
340 nm UV using a Beckman DU 650 spectrophotometer.
BAC ranged from 16.3 to 159.20 mg/dl.
Three ETOH dams with BAC less than 90 mg/dl were
eliminated from the experiment along with their correspond-
ing pair-fed ISO dams. Individual differences in pattern of
intake may affect peak blood alcohol level [65]. Previous
studies of behavioral or cognitive deficits following prenatal
ethanol administration via 35% EDC liquid diet typically
report BAC levels of 100–120 mg/dl [1,47,92] (also see
Ref. [30]). The 90 mg/dl cutoff point was chosen for
consistency with previous research.
The six remaining ETOH dams provided experimental
subjects; their mean ethanol consumption for GD7–20 was
11.48 g/kg/day, and mean BAC was 116.98 mg/dl.
2.3. Subjects
Experimental subjects consisted of one male pup selected
at random from each of the six ETOH, pair-fed ISO, and
CHOW litters (n= 6 pups/group). One pup per litter was
chosen to avoid artificially constrained variance due to litter
effects. Females were not used as subjects to avoid potential
confounds arising from fluctuations in learning that may
correspond to estrus-related changes in brain plasticity
[84,85].
On postnatal day (PD) 1, the pups were placed with
lactating, chow-fed foster mothers. Thus, litters were not
preserved and pups were distributed among foster dams so
that each new litter consisted of two ETOH, two ISO, two
CHOW, and three foster pups. Subjects were weaned on
PD21 and housed two per cage.
Subjects were isolated at PD85 and reduced to 80–85%
of their free-feeding weights by food restriction. Weights
were monitored daily and maintained at 85 ± 5% of free-
feeding weight by supplemental feedings. Water was always
available in the home cages. Behavioral testing commenced
at approximately PD90. Each animal’s session was run
during the light phase at approximately the same time each
day, 6 days/week.
2.4. Apparatus and stimuli
Each subject was tested in its home cage (46�26�21 cm), which was placed inside a sound-attenuating
enclosure before each session. The testing apparatus was
mounted on a plastic panel that replaced the cage top. Two
stainless steel columns (14� 6� 3 cm) were suspended
from the panel into opposite ends of the cage. The column
at one end was fitted with a response lever (Lever 1), an
audio speaker (Panasonic Micro Speaker EAS-45P36S;
Panasonic, Kent, WA, USA) behind a steel screen, and a
steel cup into which 45-mg food pellets (Bio-Serv F0165)
were dispensed. The other column was fitted only with a
response lever (Lever 2). Macintosh computers presented
and recorded all session events (for details, see Ref. [18]).
Auditory stimuli for discrimination problems were 12
digitized sound effects, sampled music, and synthesized
complex wave forms (details available from the authors).
Continuously repeating segments of 0.4 s duration were
presented via the Macintosh external speaker circuit at 72 dB
SPL measured 2 cm from the response lever.
2.5. Preliminary training
Preliminary training is outlined very briefly in this
section; specific details are available from the authors.
Pretraining consisted of three phases. In Phase 1, animals
habituated to the apparatus and pellet dispenser. In Phase 2,
animals were trained to press the lever adjacent to the pellet
magazine (Lever 1) to produce pellets. In Phase 3, animals
were trained to perform a two-press sequence: Pressing
Lever 2 produced continuously repeating cycles of an
auditory stimulus (different from those used in the sub-
sequent experiment), then pressing Lever 1 terminated the
stimulus and produced a pellet. After animals completed 60
two-lever sequences, they advanced to the successive dis-
crimination condition.
2.6. Successive discrimination condition
Using a successive go/no-go procedure, subjects were
tested on a series of auditory discrimination problems. Each
problem consisted of a novel pair of stimuli, and none of the
stimulus pairings was repeated within groups. Across
groups, the specific stimuli assigned to each ETOH subject
were also assigned to its corresponding ISO subject and one
CHOW subject.
Subjects produced auditory stimuli by pressing Lever 2.
On positive (go) trials, the auditory stimulus (S+) was
presented continuously until the subject pressed Lever 1
and a pellet was dispensed. A correct response was defined
as pressing Lever 1 within 5 s of the S+ stimulus onset.
Presses that occurred after 5 s were reinforced, but not scored
as correct responses. On negative (no-go) trials, the stimulus
(S� ) was presented continuously for 5 s and then termi-
nated without a reinforcer. Lever 1 presses had no pro-
grammed consequences. A correct response was recorded
if Lever 1 was not pressed during the S� presentation (for
further details, see Ref. [17]).
Sessions consisted of 120 trials, 60 S+ and 60 S�, presen-
ted randomly with the restriction that the same stimulus was
presented no more than three times consecutively. The
learning criterion for this and all subsequent phases of testing
S.M. Mihalick et al. / Neurotoxicology and Teratology 23 (2001) 453–462 455

was at least 85% correct responses for two consecutive
sessions. After each subject met this criterion, it was given
a new problem. The successive discrimination condition
continued in this manner for 30 sessions.
2.7. Discrimination reversal condition
After completing the successive discrimination con-
dition, all subjects were presented with a novel discrimina-
tion problem. The stimuli for this problem were a 2-kHz
tone and a 10-Hz click (tone/click), with S+/S� assign-
ments counterbalanced within groups. After subjects met the
learning criterion for this problem, the reinforcement con-
tingencies for both stimuli were reversed in the next session.
Thus, the previous S+ became the S� , and vice versa. The
reversal condition continued for a maximum of 30 sessions.
Subjects were eliminated if they failed to reach at least one
criterion (i.e., if they failed to learn the first tone/click
problem) within 30 sessions.
2.8. Stereological methods
2.8.1. Histology
After completing the behavioral tests, subjects were
deeply anesthetized (10 mg/kg body weight xylazine and
80 mg/kg body weight ketamine) and perfused intracar-
dially with room temperature physiological saline followed
by 200 ml of 4% paraformaldehyde in 0.1 M phosphate
buffer (pH 7.2 at 4 �C). Brains were removed, cryopro-
tected in the same fixative with 30% sucrose at 4 �C, andcut at a thickness of 60 mm in the coronal plane on a
freezing sledge microtome (AO). All sections containing
the prefrontal cortex [21,81] from the frontal pole through
the beginning of the hippocampus were mounted serially
onto gelatin-subbed slides and stained for Nissl substance
with cresyl violet, differentiated, dehydrated, and cover-
slipped with Permount.
2.8.2. Delineation of prefrontal cortex
The mPFC (Fig. 1) was distinguished from surrounding
areas on the basis of cytoarchitectonic criteria of the
prelimbic, dorsal anterior cingulate, and medial precentral
areas [81]. The cardinal feature of the mPFC is an absent
granular layer IV. The rostral boundary is the most rostral
part of the forceps minor of the corpus callosum. The
transition in characteristics of layers I–III and the distinct
features of layers VIa and VIb distinguish the prelimbic area
from the more ventrally located infralimbic area. Dorsal to
the prelimbic area is the dorsal anterior cingulate area
characterized by a more uniform cellular distribution and
the radiations of the shoulder of the corpus callosum.
Dorsolateral to this area along the dorsomedial edge of the
hemisphere is the medial precentral area. It can be distin-
guished from the lateral precentral area based on the
presence of layer IV, a laminated layer V containing more
and larger neurons, a thinner layer VIa with smaller, darker,
and more densely packed cells, and a cell-sparse region
separating layer VIa and VIb, all traits present in the lateral
precentral area. Caudally, the more homogeneous and
denser layer VIa of the ventral anterior cingulate region
distinguishes it dorsally from the dorsal anterior cingulate
area and rostrally from the prelimbic area. The caudal
boundary of mPFC with the retrosplenial cortex is deter-
mined by the appearance of a cell-dense layer II and a
clearly demarcated layer V from a cell-poor layer III in the
most ventral part of the cingulate cortex.
2.8.3. Stereology
Neuronal number was estimated by the optical fractio-
nator method [89,90]. This unbiased stereological approach
involves counting neurons with optical dissectors in a
Fig. 1. Sample low-power photomicrograph of a coronal section through
medial prefrontal cortex stained with cresyl violet. The cortical layers II and
V (where neurons were counted) are delineated with white bars (right).
S.M. Mihalick et al. / Neurotoxicology and Teratology 23 (2001) 453–462456

uniform systematic sample that comprises a known fraction
of the volume of the region being analyzed. It is unaffected
by tissue shrinkage or expansion that occurs during any
tissue preparation. This particular scheme was selected
because it results in mean coefficients of error � .06 and
avoids the need to define explicitly the superficial and deep
boundaries of thin neuronal layers [90].
All slides were coded prior to quantitative procedures
so that experimental treatment group information was not
available until all microscopic analyses were completed.
Video images taken with a Sony camera (Sony, New York,
NY, USA) attached to a Zeiss Axioplan microscope with
100�, 1.4 n.a. oil immersion objective were input to a
Zeiss/Kontron Image Analysis System. Gray level images
were normalized and contour-enhanced before a stand-
ardized counting frame was superimposed.
A neuron was counted if the first recognizable profile of
a neuronal nucleus in focus was within the counting frame.
The 10-mm depth of the counting frame was measured in
0.5-mm steps by the Z-stepping motor of the microscope
automated control unit. The top of the counting frame was
marked from the top of the section by first determining the
first edge of a profile to come into focus and then descend-
ing 10 additional microns before neuronal profiles were
considered to be inside or outside the counting frame.
Total neuronal number in layers II and V of mPFC was
estimated using the following formula:
N ¼X
Q� t
h
1
asf
1
ssf
where N = number of neurons;P
Q � = total number of
neurons counted; t= section thickness (27.50 mm); h = height
of dissector sample (10 mm); asf = area sample fraction = area
of counting frame/area of stepping motor step [(30 mm)2/
(200 mm)2]; and ssf = section sampling fraction = (1/4).
For every fourth section through mPFC (ssf = 1/4),
eight separate measurements of section thickness were
determined by using the 0.5-mm Z-step motor to focus on
the top and subsequently the bottom of each section,
employing the 100� oil immersion objective. Six sections
per animal were used to calculate the mean thickness
(t = 27.50 mm), with a mean coefficient of error = 0.03.
Neuronal counts in each layer were made from a square
counting frame (30� 30 mm) that was sampled every
200 mm within each layer.
3. Results
3.1. Subjects
Table 1 shows characteristics of litters born to dams in
each of the three diet groups. Number of pups did not differ
as a function of sex or treatment (2� 3 ANOVA, P > .05, all
factors). A two-way analysis of variance (ANOVA) indi-
cated that male pups weighed more than female pups on
PD1 [F(1,30) = 8.18, P=.0076], and that mean litter weight
differed according to diet condition [F(2,30) = 4.47, P=.02].
Fisher’s post hoc tests showed that litters exposed prenatally
to ethanol weighed significantly less than litters born to
chow-fed control dams (P < .05), a difference potentially
related to the absolute number of pups per litter. However,
subjects in all groups gained weight steadily (Fig. 2), with
statistically equivalent weight gains in all groups by PD21
(one-way ANOVA, P>.05).
3.2. Pretraining
A one-way ANOVA revealed no significant differences
in the number of sessions to complete pretraining. ETOH,
CHOW, and ISO groups required means of 3.7, 2.5, and 3.0
days, respectively.
3.3. Discrimination learning
3.3.1. Successive discrimination condition
All subjects met the learning criterion for the first
discrimination problem in three to six sessions. No
subject required more than six sessions. A one-way
ANOVA showed that treatment groups did not differ
on the mean number of sessions to acquire the first
discrimination (Fig. 3). Individual subjects learned a
varying number of successive discriminations within the
Table 1
Litter characteristics on PD1
Group Pups Mean n Mean body weight (g)
ETOH Males 8.33 (0.34) 5.93 (0.07)
Females 6.83 (0.24) 5.71 (0.06)
ISO Males 5.83 (0.27) 6.23 (0.08)
Females 7.50 (0.39) 5.80 (0.09)
CHOW Males 5.17 (0.30) 6.56 (0.04)
Females 6.50 (0.46) 6.06 (0.04)
Standard errors of the mean are indicated in parentheses.
Fig. 2. Mean body weight of pups used as subjects in each group, measured
from birth (PD1) to weaning (PD21). S.E.M. values are too small to be
depicted in this figure. All subjects had equivalent weight gains by PD21
(ANOVA, P> .05).
S.M. Mihalick et al. / Neurotoxicology and Teratology 23 (2001) 453–462 457
30-session limit, but a one-way ANOVA determined that
this was unrelated to treatment (P>.05).
3.3.2. Reversal condition: initial discrimination
Treatment groups tended to differ in their proficiency to
acquire a simple discrimination after 30 sessions of experi-
ence with discrimination problems (Fig. 4). One CHOW rat
failed to meet the learning criterion for the tone/click
problem within the 30-session limit, so it was eliminated
from this and all subsequent analyses (i.e., n = 5 for group
CHOW). All other subjects met the learning criterion for the
tone/click problem in two to eight sessions. A one-way
ANOVA revealed a nonsignificant trend for treatment differ-
ences on the number of sessions to learn this problem
[F(2,14) = 3.11, P=.0762].
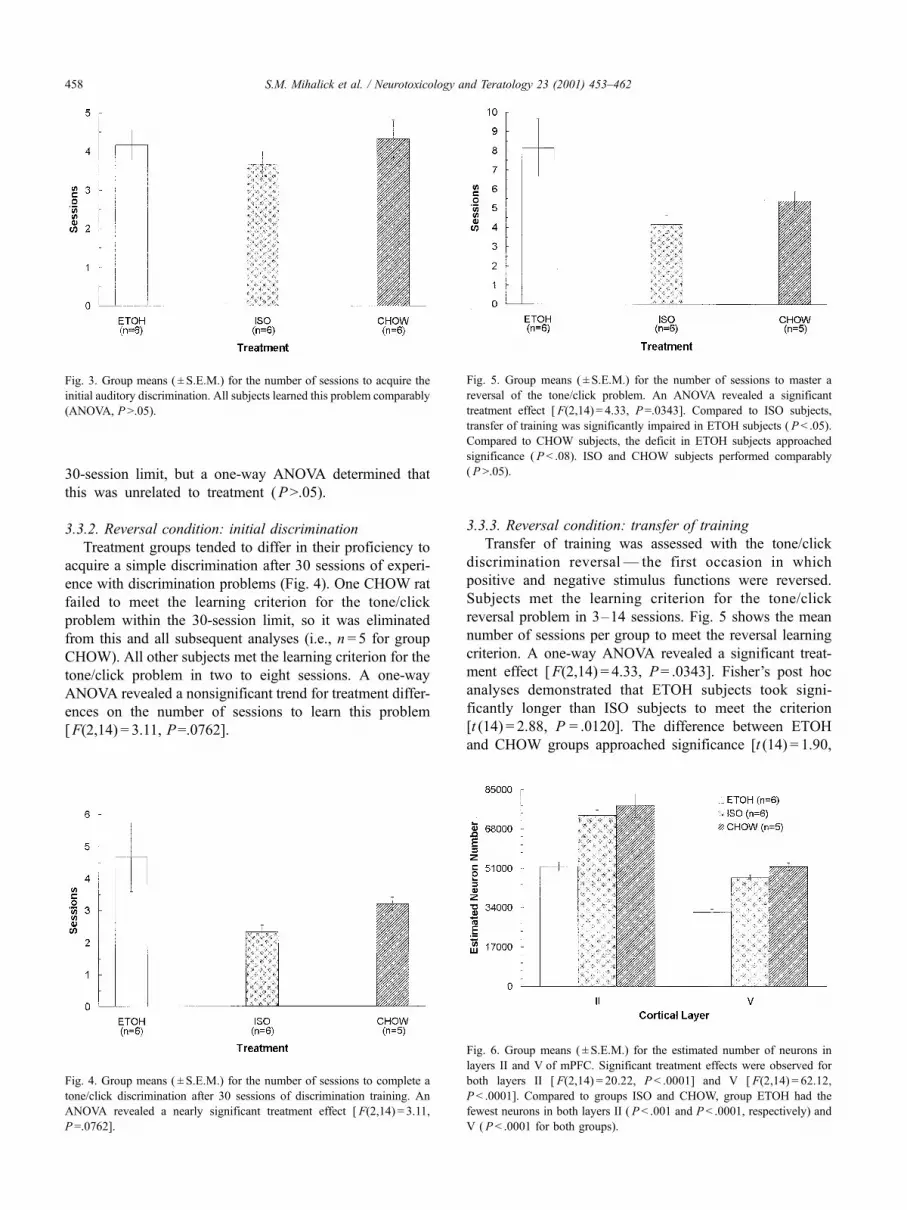
3.3.3. Reversal condition: transfer of training
Transfer of training was assessed with the tone/click
discrimination reversal — the first occasion in which
positive and negative stimulus functions were reversed.
Subjects met the learning criterion for the tone/click
reversal problem in 3–14 sessions. Fig. 5 shows the mean
number of sessions per group to meet the reversal learning
criterion. A one-way ANOVA revealed a significant treat-
ment effect [F(2,14) = 4.33, P= .0343]. Fisher’s post hoc
analyses demonstrated that ETOH subjects took signi-
ficantly longer than ISO subjects to meet the criterion
[t (14) = 2.88, P = .0120]. The difference between ETOH
and CHOW groups approached significance [t (14) = 1.90,
Fig. 3. Group means ( ± S.E.M.) for the number of sessions to acquire the
initial auditory discrimination. All subjects learned this problem comparably
(ANOVA, P >.05).
Fig. 4. Group means ( ± S.E.M.) for the number of sessions to complete a
tone/click discrimination after 30 sessions of discrimination training. An
ANOVA revealed a nearly significant treatment effect [ F(2,14) = 3.11,
P=.0762].
Fig. 5. Group means ( ± S.E.M.) for the number of sessions to master a
reversal of the tone/click problem. An ANOVA revealed a significant
treatment effect [ F(2,14) = 4.33, P=.0343]. Compared to ISO subjects,
transfer of training was significantly impaired in ETOH subjects ( P < .05).
Compared to CHOW subjects, the deficit in ETOH subjects approached
significance ( P < .08). ISO and CHOW subjects performed comparably
( P >.05).
Fig. 6. Group means ( ± S.E.M.) for the estimated number of neurons in
layers II and V of mPFC. Significant treatment effects were observed for
both layers II [ F(2,14) = 20.22, P < .0001] and V [ F(2,14) = 62.12,
P < .0001]. Compared to groups ISO and CHOW, group ETOH had the
fewest neurons in both layers II ( P< .001 and P < .0001, respectively) and
V ( P < .0001 for both groups).
S.M. Mihalick et al. / Neurotoxicology and Teratology 23 (2001) 453–462458

P=.0781], and performance did not differ between ISO
and CHOW groups.
3.4. Prefrontal cortex measures
Prenatal exposure to ethanol significantly affected the
number of neurons in layers II and Vof mPFC (Fig. 6). One-
way ANOVAs revealed significant treatment differences in
both layers II [ F(2,14) = 20.22, P < .0001] and V
[F(2,14) = 62.12, P < .0001]. Fisher’s post hoc tests com-
paring estimates of neuron number in layer II showed that
ETOH subjects had the fewest neurons when compared to
both ISO [t (14) = 5.09, P=.0002] and CHOW [t (14) = 5.78,
P < .0001] groups. Similarly, estimates for layer V were
lowest for ETOH subjects when compared to both ISO
[t (14) = 8.39, P < .0001] and CHOW [t (14) = 10.45,
P < .0001] groups. Table 2 displays the mean neuronal count
and coefficients of error for each group.
3.5. Brain–behavior relationships
Multiple regressions were calculated to determine
whether performance on any learning measure could be
predicted by treatment group membership or estimated
neuron number in layer II or V of mPFC. A systematic
relationship emerged with the number of sessions required
to learn the tone/click reversal problem [model F(3,13) =
4.14, P=.0289]. The predictor variables accounted for a
large proportion of the variation in performance, with
neuron number in layer V explaining the most (partial
r2 = 33%). A reduced number of neurons in layers II
and V was correlated with a greater number of sessions
to complete the transfer test (rp=� .48, P=.0499 and
rp=� .61, P=.0088, respectively).
4. Discussion
The results provide evidence for selective cognitive
deficits consequent to prenatal ethanol exposure. All sub-
jects performed similarly during both pretraining and
acquisition of the initial discrimination problem. When the
tone/click problem was introduced, a treatment-related dif-
ference in performance approached significance. This trend
suggests that groups may have differed in their ability to
generalize learning within task type, but that the effect may
have been too subtle to be detected with the sample sizes in
this study.
More pronounced deficits became evident on the tone/
click discrimination reversal, which ethanol-exposed sub-
jects learned more slowly than controls. There was a
significant difference between ETOH and ISO groups, the
most important comparison for determining the effects of
ethanol, and a nearly significant difference between ETOH
and CHOW groups. The fact that ISO and CHOW groups
did not differ indicates a lack of nutritional effects on
performance. These data support the hypothesis that prena-
tal ethanol exposure impairs the ability to transfer learning
across task types.
The ETOH deficit on this transfer-of-training test shows
an inflexibility at applying previous experience to a new but
related type of problem. This pattern of emerging deficits is
consistent with the human mental retardation literature
showing that individuals with mental retardation may learn
simple discriminations readily with adequate preparation
and appropriate procedures (e.g., Refs. [19,49,74]), but are
increasingly likely to show cognitive impairments as the
stimuli, contingencies, or relevant training histories grow
more complex [10,11,39,50].
The observed impairments cannot be attributed to
ethanol-induced deficits in motivation, perception, or motor
coordination. All subjects performed comparably during
pretraining, showing that they could move between the
levers within the specified time limits and depress the levers
with sufficient force. Equivalent performance between
groups during the successive discrimination condition indi-
cates that the stimuli were equally discriminable to all
subjects, and that the learning deficits shown by ETOH
subjects later in the experiment were not due to hearing
disorders sometimes associated with prenatal ethanol expo-
sure [12,36].
In addition to the across-task generalization issue, the
transfer test results are also consistent with an impairment
in adaptive response inhibition. While this interpretation is
speculative, the present results are consistent with those
from other studies showing that rats exposed prenatally to
ethanol display learning impairments on tasks requiring
behavioral inhibition. These tasks include tests of avoid-
ance [1,22,63,67,80] and operant DRL schedules that
require low rates of temporally spaced responding
[16,82]. Although accurate performance on all discrimina-
tion problems requires withholding responses to the S�stimulus, reversal problems present the greatest challenge
in this regard because accurate performance directly con-
flicts with recent stimulus–response–consequence contin-
gencies. Thus, ethanol-exposed rats in the current study
may have shown poorer reversal learning because of
difficulty overcoming their prepotent response bias. The
human FAS literature also is in accord. Children prenatally
exposed to alcohol may exhibit executive function deficits
Table 2
Mean neuronal counts and coefficients of error (CE) for stereological
measures of mPFC
Group Layer Mean count Mean CE
ETOH II 51,770.40 0.06
V 31,990.20 0.05
ISO II 73,829.80 0.06
V 47,049.20 0.06
CHOW II 78,046.32 0.06
V 51,672.72 0.06
S.M. Mihalick et al. / Neurotoxicology and Teratology 23 (2001) 453–462 459

related to response inhibition (reviewed in Ref. [34]) and
behavioral control [48].
This study also demonstrated that prenatal ethanol
resulted in reduced cell number in layers II and V of the
mPFC. Moreover, there was a significant correlation
between layer V damage and poor performance on the
transfer test. Thus, it seems likely that the cortical injury
caused by early ethanol exposure contributed to the
observed learning deficits. However, the neurological dam-
age probably was not restricted to the mPFC areas examined
in this study. For instance, early ethanol has adverse effects
on the hippocampus [5,8,26] and the cholinergic system
[7,64]. Moreover, structures important for reversal learning,
such as the amygdala [40] or the nucleus accumbens [76],
also may have been damaged.
The relative contributions of mPFC and non-mPFC
structures to reversal learning cannot be determined from
the data in the current study. However, the observed pattern
of behavioral deficits mirrors that obtained after localized
lesions to the prelimbic and/or infralimbic subregions of the
mPFC incorporated by the limbic circuit [6,20,46]. This
functional similarity suggests that impairments in reversal
learning were at least partly related to the damage sustained
within mPFC.
The neurodevelopmental processes that may have been
affected adversely during the relatively lengthy but critical
period of prenatal exposure in this study include: cell
division, cell commitment, neuronal migration, neurite out-
growth, and glial cell transformation [24,25,27,32,59]. The
timing of exposure appears critical to the CNS region that
will be impacted [32,58,87]. Further research using a
parametric binge model with ethanol exposures timed to
coincide with more discrete stages of cortical histogenesis
may provide additional insight into specific neurodevelop-
mental mechanisms affected by prenatal ethanol exposure.
Acknowledgments
Data collection and manuscript preparation were
supported by NIH grants AA 10688 and HD 04147. We
thank William J. McIlvane, Tom Callahan, Kevin Farren,
and Gerson Tomanari for their contributions to the
development of the project; Darlene Butler for assistance
with histology; Stuart Tobet for assistance with image
analysis; Peter McCaffery for help with spectrophotometry;
and Camilla Symonowicz for assistance with animal
breeding and husbandry.
References
[1] E.L. Abel, In utero alcohol exposure and developmental delay of
response inhibition, Alcohol.: Clin. Exp. Res. 6 (1982) 369–376.
[2] E.L. Abel, An update on incidence of FAS: FAS is not an equal
opportunity birth defect, Neurotoxicol. Teratol. 17 (1995) 437–443.
[3] I. Autti-Ramo, M.L. Granstrom, The psychomotor development
during the first year of life of infants exposed to intrauterine alcohol
of various duration. Fetal alcohol exposure and development, Neuro-
pediatrics 22 (1991) 59–64.
[4] I. AuttiRamo, M.L. Granstrom, The effect of intrauterine alcohol ex-
position in various durations on early cognitive development, Neuro-
pediatrics 22 (1991) 203–210.
[5] D.E. Barnes, D.W. Walker, Prenatal ethanol exposure permanently
reduces the number of pyramidal neurons in rat hippocampus, Brain
Res. 227 (1981) 333–340.
[6] J.T. Becker, D.S. Olton, C.A. Anderson, E.R.P. Breitinger, Cognitive
mapping in rats: The role of the hippocampal and frontal systems in
retention and reversal, Behav. Brain Res. 3 (1981) 1–22.
[7] A.C. Black, L.W. Goolsby, G.A. Cohen, H.E. Young, Effects of pre-
natal ethanol exposure on the hippocampal neurochemistry of albino
rats at 90 days of postnatal age, Am. J. Obstet. Gynecol. 173 (1995)
514–519.
[8] D.J. Bonthius, J.R. West, Permanent neuronal deficits in rats
exposed to alcohol during the brain growth spurt, Teratology 44
(1991) 147–163.
[9] D.J. Bonthius, J.R. West, Early postnatal alcohol exposure acutely and
permanently reduces the number of granule cells and mitral cells in
the rat olfactory bulb: A stereological study, J. Comp. Neurol. 324
(1992) 557–566.
[10] P.H. Brooks, C. McCauley, E. Merrill, Cognition and mental retarda-
tion, in: F.J. Menolascino, J.A. Stark (Eds.), Preventive and Curative
Intervention in Mental Retardation, Brooks, Baltimore, 1987,
pp. 295–320.
[11] J.C. Campione, A.L. Brown, R.A. Ferrara, Mental retardation and
intelligence, in: R.J. Sternberg (Ed.), Handbook of Human Intelli-
gence, Cambridge Univ. Press, Cambridge, 1982, pp. 392–492.
[12] M.W. Church, J.A. Kaltenbach, Hearing, speech, language, and ves-
tibular disorders in the Fetal Alcohol Syndrome: A literature review,
Alcohol.: Clin. Exp. Res. 21 (1997) 495–512.
[13] S.K. Clarren, Neuropathology in fetal alcohol syndrome, in: J.R. West
(Ed.), Alcohol and Brain Development, Oxford Univ. Press, New
York, 1986, pp. 158–166.
[14] C.D. Coles, R.T. Brown, I.E. Smith, K.A. Platzman, S. Erickson,
A. Falek, Effects of prenatal alcohol exposure at school age: I.
Physical and cognitive development, Neurotoxicol. Teratol. 13
(1991) 357–367.
[15] A. Diamond, V. Ciaramitaro, E. Donner, S. Djali, M.B. Robinson,
An animal model of early-treated PKU, J. Neurosci. 14 (1994)
3072–3082.
[16] C.D. Driscoll, J.S. Chen, E.P. Riley, Operant DRL performance in rats
following prenatal alcohol exposure, Neurobehav. Toxicol. 2 (1980)
201–211.
[17] W.V. Dube, T.D. Callahan, W.J. McIlvane, Serial reversals of concur-
rent auditory discriminations in rats, Psychol. Rec. 43 (1993) 429–440.
[18] W.V. Dube, T.D. Callahan, W.J. McIlvane, C.K. Deutsch, M.D.
Ullman, O. Koul, R.H. McCluer, Auditory discrimination reversal
learning and assessment of behavioral teratogenesis in rats, Behav.
Processes 37 (1996) 197–207.
[19] W.V. Dube, R.W. Serna, Reevaluation of a programmed method to
teach generalized identity matching to sample, Res. Dev. Disabil. 19
(1998) 347–379.
[20] A.T. Ferry, X.-C.M. Lu, J.L. Price, Effects of excitotoxic lesions in the
ventral striatopallidal – thalamocortical pathway on odor reversal
learning: Inability to extinguish an incorrect response, Exp. Brain
Res. 131 (2000) 320–335.
[21] P.L. Gabbott, B.G. Dickie, R.R. Vaid, A.J. Headlam, S.J. Bacon, Local
circuit neurones in the medial prefrontal cortex (areas 25, 32 and 24b)
in the rat: Morphology and quantitative distribution, J. Comp. Neurol.
377 (1997) 465–499.
[22] P.V. Gallo, J. Weinberg, Neuromotor development and response in-
hibition following prenatal ethanol exposure, Neurobehav. Toxicol.
Teratol. 4 (1982) 505–513.
S.M. Mihalick et al. / Neurotoxicology and Teratology 23 (2001) 453–462460

[23] C.R. Goodlett, D.J. Bonthius, E.A. Wasserman, J.R. West, An
animal model of central nervous dysfunction associated with fetal
alcohol exposure: Behavioral and neuroanatomical correlates, in:
I. Garmezano, E.A. Wasserman (Eds.), Learning and Memory:
Behavioral and Biological Processes, Erlbaum, Inglewood, NJ,
1992, pp. 183–208.
[24] C.R. Goodlett, S.D. Peterson, K.R. Lundahl, A.D. Pearlman, Binge-
like alcohol exposure of neonatal rats via intragastric intubation induces
both Purkinje cell loss and cortical astrogliosis, Alcohol.: Clin. Exp.
Res. 21 (1997) 1010–1017.
[25] A. Granato, M. Santarelli, A. Sbriccoli, D. Minciacchi, Multifaceted
alterations of the thalamo-cortical– thalamic loop in adult rats prena-
tally exposed to ethanol, Anat. Embryol. (Berlin) 191 (1995) 11–23.
[26] P.L. Greene, J.L. Diaz-Granados, A. Amsel, Blood ethanol concen-
tration from early postnatal exposure: Effects on memory-based learn-
ing and hippocampal neuroanatomy in infant and adult rats, Behav.
Neurosci. 106 (1992) 51–61.
[27] C. Guerri, J. Renau-Piqueras, Alcohol, astroglia, and brain develop-
ment, Mol. Neurobiol. 15 (1997) 65–81.
[28] J.L. Hall, M.W. Church, R.F. Berman, Radial arm maze deficits in rats
exposed to alcohol during midgestation, Psychobiology 22 (1994)
181–185.
[29] J.H. Hannigan, What research with animals is telling us about alcohol-
related neurodevelopmental disorder, Pharmacol., Biochem. Behav. 55
(1996) 489–499.
[30] J.H. Hannigan, E.L. Abel, M.L. Kruger, ‘‘Population’’ characteristics
of birthweight in an animal model of alcohol-related developmental
effects, Neurotoxicol. Teratol. 15 (1993) 97–105.
[31] M.B. Heaton, J.J. Mitchell, M. Paiva, D.W. Walker, Ethanol-induced
alterations in the expression of neurotrophic factors in the developing
rat central nervous system, Dev. Brain Res. 121 (2000) 97–107.
[32] C. Ikonomidou, P. Bittigau, M.J. Ishimaru, D.F. Wozniak, C. Koch,
K. Genz, M.T. Price, V. Stefovska, F. Horster, T. Tenkova, K. Dik-
ranian, J.W. Olney, Ethanol-induced apoptotic neurodegeneration and
fetal alcohol syndrome, Science 287 (2000) 1056–1060.
[33] D.G. Jones, Influence of ethanol on neuronal and synaptic maturation
in the central nervous system morphological investigations, Prog.
Neurobiol. 31 (1988) 171–197.
[34] K. Kaemingk, A. Paquette, Effects of prenatal alcohol exposure on
neuropsychological functioning, Dev. Neuropsychol. 15 (1999)
111–140.
[35] A.C. Kamil, T.B. Jones, A. Pietrewicz, J.E. Mauldin, Positive transfer
from successive reversal training to learning set in blue jays (Cyano-
citta cristata), J. Comp. Physiol. Psychol. 91 (1977) 79–86.
[36] W.M. Kaneko, E.P. Riley, C.L. Ehlers, Electrophysiological and be-
havioral findings in rats prenatally exposed to alcohol, Alcohol 10
(1993) 169–178.
[37] M.E. Kaufman, W.I. Gardner, Transfer of training of learning sets in
mental defectives: I. Discrimination reversal, Am. J. Ment. Defic. 73
(1969) 801–803.
[38] M.E. Kaufman, M.W. Peterson, Acquisition of a conditional discrim-
ination learning set by normal and mentally retarded children, Am. J.
Ment. Defic. 69 (1965) 865–870.
[39] M.E. Kaufman, H.J. Prehm, A review of research on learning sets and
transfer of training in mental defectives, in: N.R. Ellis (Ed.), Interna-
tional Review of Research in Mental Retardation, vol. 2, Academic
Press, New York, 1966, pp. 123–149.
[40] R.W. Kentridge, C. Shaw, J.P. Aggleton, Amygdaloid lesions and
stimulus – reward associations in the rat, Behav. Brain Res. 42
(1991) 57–66.
[41] B. Kolb, Animal models for human PFC-related disorders, in:
H.B.M. Uylings, C.G. Van Eden, J.P.C. De Bruin, M.A. Corner,
M.G.P. Feenstra (Eds.), Progress in Brain Research, vol. 85, Elsevier,
Amsterdam, 1990, pp. 501–519.
[42] L.A. Kotkoskie, S. Norton, Cerebral cortical morphology and behav-
ior in rats following acute prenatal ethanol exposure, Alcohol.: Clin.
Exp. Res. 13 (1989) 776–781.
[43] P.E. Kuhn, M.W. Miller, Expression of p53 and ALZ-50 immuno-
reactivity in rat cortex: Effect of prenatal exposure to ethanol, Exp.
Neurol. 154 (1998) 418–429.
[44] B. Larroque, M. Kaminski, P. Dehaene, D. Subtil, M.J. Delfosse,
D. Querleu, Moderate prenatal alcohol exposure and psychomotor
development at preschool age, Am. J. Public Health 85 (1995)
1654–1661.
[45] M.H. Lee, A. Rabe, Infantile handling eliminates reversal learning
deficit in rats prenatally exposed to alcohol, Alcohol 18 (1999)
49–53.
[46] L. Li, J. Shao, Restricted lesions to ventral prefrontal subareas block
reversal learning but not visual discrimination learning in rats, Phy-
siol. Behav. 65 (1998) 371–379.
[47] N.J. Lobaugh, T. Wigal, P.L. Greene, J.L. Diaz-Granados, A. Amsel,
Effects of prenatal ethanol exposure on learned persistence and hippo-
campal neuroanatomy in infant, weanling and adult rats, Behav. Brain
Res. 44 (1991) 81–86.
[48] S.N. Mattson, A.M. Goodman, C. Caine, D.C. Delis, E.P. Riley,
Executive functioning in children with heavy prenatal alcohol
exposure, Alcohol.: Clin. Exp. Res. 23 (1999) 1808–1815.
[49] W.J. McIlvane, Stimulus control analysis and nonverbal instructional
methods for people with intellectual disabilities, in: N.W. Bray (Ed.),
International Review of Research in Mental Retardation, vol. 18,
Academic Press, San Diego, 1992, pp. 55–109.
[50] W.J. McIlvane, M.F. Cataldo, On the clinical relevance of animal
models for the study of human mental retardation, Ment. Retard.
Dev. Disabil. Res. Rev. 2 (1996) 188–196.
[51] M.W. Miller, Effects of prenatal exposure to alcohol on the distribu-
tion and time of origin of corticospinal neurons in the rat, J. Comp.
Neurol. 257 (1987) 372–382.
[52] M.W. Miller, Effects of prenatal exposure to ethanol on neocortical
development: II. Cell proliferation in the ventricular and subventricu-
lar zones of the rat, J. Comp. Neurol. 287 (1989) 326–338.
[53] M.W. Miller, Effect of early exposure to ethanol on the protein and
DNA contents of specific brain regions in the rat, Brain Res. 734
(1996) 286–294.
[54] M.W. Miller, Effects of prenatal exposure to ethanol on callosal pro-
jection neurons in rat somatosensory cortex, Brain Res. 766 (1997)
121–128.
[55] M.W. Miller, P.E. Kuhn, Cell cycle kinetics in fetal rat cerebral cortex:
Effects of prenatal treatment with ethanol assessed by a cumulative
labeling technique with flow cytometry, Alcohol.: Clin. Exp. Res. 19
(1995) 233–237.
[56] M.W. Miller, G. Potempa, Numbers of neurons and glia in mature rat
somatosensory cortex: Effects of prenatal exposure to ethanol,
J. Comp. Neurol. 293 (1990) 92–102.
[57] M.W. Miller, S. Robertson, Prenatal exposure to ethanol alters the
postnatal development and transformation of radial glia to astrocytes
in the cortex, J. Comp. Neurol. 337 (1993) 253–266.
[58] S.M. Mooney, R.M. Napper, J.R. West, Long-term effect of postnatal
alcohol exposure on the number of cells in the neocortex of the rat: A
stereological study, Alcohol.: Clin. Exp. Res. 20 (1996) 615–623.
[59] D.B. Moore, M.A. Quintero, A.C. Ruygrok, D.W. Walker, M.B. Hea-
ton, Prenatal ethanol exposure reduces parvalbumin-immunoreactive
GABAergic neuronal number in the adult rat cingulate cortex, Neuro-
sci. Lett. 249 (1998) 25–28.
[60] A.H. Nagahara, R.J. Handa, Fetal alcohol exposure alters the induc-
tion of immediate early gene mRNA in the rat prefrontal cortex after
an alternation task, Alcohol.: Clin. Exp. Res. 19 (1995) 1389–1397.
[61] A.H. Nagahara, R.J. Handa, Fetal alcohol exposure produces delay-
dependent memory deficits in juvenile and adult rats, Alcohol.: Clin.
Exp. Res. 21 (1997) 710–715.
[62] J.A. Osborn, C. Yu, K. Gabriel, J. Weinberg, Fetal ethanol effects on
benzodiazepine sensitivity measured by behavior on the elevated plus-
maze, Pharmacol., Biochem. Behav. 60 (1998) 625–633.
[63] V.D. Petkov, E.R. Konstantinova, V.V. Petkov, J.V. Vaglenova, Learn-
ing and memory in rats exposed pre- and postnatally to alcohol. An
S.M. Mihalick et al. / Neurotoxicology and Teratology 23 (2001) 453–462 461

attempt at pharmacological control, Methods Find. Exp. Clin. Phar-
macol. 13 (1991) 43–50.
[64] E.P. Riley, S. Barron, J.H. Hannigan, Response inhibition deficits
following prenatal alcohol exposure: A comparison to the effects of
hippocampal lesions in rats, in: J.R. West (Ed.), Alcohol and Brain
Development, Oxford Univ. Press, New York, 1986, pp. 71–102.
[65] E.P. Riley, L.S. Meyer, Considerations for the design, implementation,
and interpretation of animal models of fetal alcohol effects, Neuro-
behav. Toxicol. Teratol. 6 (1984) 97–101.
[66] E.P. Riley, N.R. Shapiro, E.A. Lochry, Nose-poking and head-dipping
behaviors in rats prenatally exposed to alcohol, Pharmacol., Biochem.
Behav. 11 (1979) 513–519.
[67] G.A. Rockwood, E.P. Riley, Effects of scopolamine on spontaneous
alternation and shuttle avoidance in rats exposed to alcohol in utero,
Alcohol 2 (1985) 575–579.
[68] T.M. Roebuck, S.N. Mattson, E.P. Riley, A review of the neuro-
anatomical findings in children with fetal alcohol syndrome or
prenatal exposure to alcohol, Alcohol.: Clin. Exp. Res. 22 (1998)
339–344.
[69] A.M. Schrier, Transfer by macaque monkeys between the learning
set and repeated reversal tasks, Percept. Mot. Skills 23 (1966)
787–792.
[70] A.M. Schrier, Transfer between the repeated reversal and learning
set tasks: A reexamination, J. Comp. Physiol. Psychol. 87 (1974)
153–156.
[71] R.J. Schusterman, Transfer effects of successive discrimination-rever-
sal training in chimpanzees, Science 137 (1962) 422–423.
[72] R.J. Schusterman, Successive discrimination-reversal training and
multiple discrimination training in one-trial learning by chimpanzees,
J. Comp. Physiol. Psychol. 58 (1964) 153–156.
[73] G.K. Seabold, J. Luo, M.W. Miller, Effect of ethanol on neurotrophin-
mediated cell survival and receptor expression in cultures of cortical
neurons, Brain Res. Dev. Brain Res. 108 (1998) 139–145.
[74] M. Sidman, L.T. Stoddard, Programming perception and learning
for retarded children, in: N.R. Ellis (Ed.), International Review of
Research in Mental Retardation, vol. 2, Academic Press, New York,
1966, pp. 151–208.
[75] K.J. Smith, M.J. Eckardt, The effects of prenatal alcohol on the central
nervous system, Recent Dev. Alcohol. 9 (1991) 151–164.
[76] C.E. Stern, R.E. Passingham, The nucleus accumbens in monkeys
(Macaca fascicularis): III. Reversal learning, Exp. Brain Res. 106
(1995) 239–247.
[77] A.P. Streissguth, H.M. Barr, P.D. Sampson, Moderate prenatal alcohol
exposure: Effects on child IQ and learning problems at age 7 1/2
years, Alcohol.: Clin. Exp. Res. 14 (1990) 662–669.
[78] B.J. Strupp, M. Bunsey, D.A. Levitsky, K. Hamberger, Deficient
cumulative learning: An animal model of retarded cognitive develop-
ment, Neurotoxicol. Teratol. 16 (1994) 71–79.
[79] J.D. Thomas, S.P. Weinert, S. Sharif, E.P. Riley, MK-801 administra-
tion during ethanol withdrawal in neonatal rat pups attenuates ethanol-
induced behavioral deficits, Alcohol.: Clin. Exp. Res. 21 (1997)
1218–1225.
[80] J. Vaglenova, V.V. Petkov, Fetal alcohol effects in rats exposed pre-
and postnatally to a low dose of ethanol, Alcohol.: Clin. Exp. Res. 22
(1998) 697–703.
[81] C.G. van Eden, H.B.M. Uylings, Cytoarchitectonic development of
the prefrontal cortex in the rat, J. Comp. Neurol. 241 (1985) 253–267.
[82] N.S. Vigliecca, S. Fulginiti, S.A. Minetti, Acute ethanol exposure
during pregnancy in rats: Effects upon a multiple learning task,
Alcohol 6 (1989) 63–68.
[83] J.M. Warren, Reversal learning and the formation of learning sets by
cats and monkey, J. Comp. Physiol. Psychol. 61 (1966) 421–428.
[84] S.G. Warren, A.G. Humphreys, J.M. Juraska, W.T. Greenough, LTP
varies across the estrous cycle: Enhanced synaptic plasticity in pro-
estrus rats, Brain Res. 703 (1995) 26–30.
[85] S.G. Warren, J.M. Juraska, Spatial and nonspatial learning across the
rat estrous cycle, Behav. Neurosci. 111 (1997) 259–266.
[86] J.R. West, W.J. Chen, N.J. Pantazis, Fetal alcohol syndrome: The
vulnerability of the developing brain and possible mechanism of dam-
age, Metab. Brain Dis. 9 (1994) 291–322.
[87] J.R. West, C.R. Goodlett, Teratogenic effects of alcohol on brain
development, Ann. Med. 22 (1990) 319–325.
[88] J.R. West, C.R. Goodlett, D.J. Bonthius, K.M. Hamre, B.L. Marcus-
sen, Cell population depletion associated with fetal alcohol brain
damage: Mechanisms of BAC-dependent cell loss, Alcohol.: Clin.
Exp. Res. 14 (1990) 813–818.
[89] M. West, New stereological methods for counting neurons, Neurobiol.
Aging 14 (1993) 275–285.
[90] M.J. West, L. Slomianka, H.J.G. Gundersen, Unbiased stereological
estimation of the total number of neurons in the subdivisions of the rat
hippocampus using the optical fractionator, Anat. Rec. 231 (1991)
482–497.
[91] S.G. Wiener, W.J. Shoemaker, L.Y. Koda, F.E. Bloom, Interaction of
ethanol and nutrition during gestation: Influence on maternal and off-
spring development in the rat, J. Pharmacol. Exp. Ther. 216 (1981)
572–579.
[92] T. Wigal, A. Amsel, Behavioral and neuroanatomical effects of pre-
natal, postnatal, or combined exposure to ethanol in weanling rats,
Behav. Neurosci. 104 (1990) 116–126.
[93] G. Winocur, M. Moscovitch, Hippocampal and prefrontal contribu-
tions to learning and memory: Analysis of lesion and aging effects on
maze learning in rats, Behav. Neurosci. 104 (1990) 544–551.
S.M. Mihalick et al. / Neurotoxicology and Teratology 23 (2001) 453–462462