myocardial knockdown of m rna-stabilizing
TRANSCRIPT

The FASEB Journal • Research Communication
Myocardial knockdown of mRNA-stabilizing proteinHuR attenuates post-MI inflammatory response andleft ventricular dysfunction in IL-10-null mice
Prasanna Krishnamurthy,1 Erin Lambers, Suresh Verma, Tina Thorne, Gangjian Qin,Douglas W. Losordo, and Raj Kishore1
Feinberg Cardiovascular Research Institute, Division of Cardiology, Northwestern University FeinbergSchool of Medicine, Chicago, Illinois, USA
ABSTRACT Prolonged inflammatory response is as-sociated with left ventricular (LV) dysfunction andadverse remodeling following myocardial infarction(MI). IL-10 inhibits inflammation by suppressingHuR-mediated mRNA stabilization of proinflamma-tory cytokines. Here we report that following MI,IL-10�/� mice showed exaggerated LV dysfunction,fibrosis, and cardiomyocyte apoptosis. Short-hairpinRNA (shRNA)-mediated knockdown of HuR in themyocardium significantly reversed MI-induced LVdysfunctions and LV remodeling. HuR knockdownsignificantly reduced MI-induced cardiomyocyte apop-tosis concomitant with reduced p53 expression.Moreover, HuR knockdown significantly reduced in-farct size and fibrosis area, which in turn was associ-ated with decreased TGF-� expression. In vitro,stable knockdown of HuR in mouse macrophage cellline RAW 264.7 corroborated in vivo data and re-vealed reduced mRNA expression of TNF-�, TGF-�,and p53 following LPS challenge, which was associ-ated with a marked reduction in the mRNA stabilityof these genes. Taken together, our studies suggestthat HuR is a direct target of IL-10, and HuRknockdown mimics anti-inflammatory effects of IL-10.—Krishnamurthy, P., Lambers, E., Verma, S.,Thorne, T., Qin, G., Losordo, D. W., Kishore, R.Myocardial knockdown of mRNA-stabilizing proteinHuR attenuates post-MI inflammatory response andleft ventricular dysfunction in IL-10-null mice. FASEBJ. 24, 2484 –2494 (2010). www.fasebj.org
Key Words: myocardial infarction � cytokines � inflammation� fibrosis � apoptosis
Prolonged inflammation characterized by en-hanced production of proinflammatory cytokinesduring myocardial infarction (MI) leads to left ven-tricular (LV) dysfunction and adverse remodelingchanges such as LV dilation and fibrosis (1–5).Inflammatory cells that infiltrate the injury site se-crete a number of proinflammatory (IL-1�, IL-6,TNF-�, IP-10, MCP-1, etc.) and anti-inflammatory(IL-4, IL-10) cytokines that mediate homeostasiswithin the heart in response to injury (5–7). IL-10, apotent anti-inflammatory cytokine, is a strong deac-
tivator of monocytes and suppressor of various proin-flammatory mediators (8, 9). IL-10 inhibits the syn-thesis of a number of proinflammatory cytokinesimplicated in LV dysfunction, including TNF-�, IL-1�, and IL-6 following MI in mice (10). Previousreports suggest that the IL-10 genotype influences the riskfor cardiovascular events in hemodialysis patients. Thequantitative production of IL-10 is subject to geneticvariation based on polymorphisms in the promoter of itsgene. IL-10 “low-producer” patients have a significantlyenhanced risk of death due to cardiovascular diseasecompared to IL-10 “high-producers” (11). IL-10 inhibitsinflammatory response by suppressing HuR-mediatedmRNA stabilization of proinflammatory cytokines andattenuates post-MI LV dysfunction and remodeling(10). An important mechanism of posttranscriptionalgene regulation of proinflammatory cytokines is therapid degradation of messenger RNAs (mRNAs) sig-naled by AU-rich elements (AREs) in their 3� untrans-lated regions. HuR, an ubiquitously expressed memberof the Hu family of RNA-binding proteins related toDrosophila ELAV, selectively binds AREs and confersstability to ARE-containing mRNAs (12–14). IL-10-knockout (KO) mice display an exaggerated inflamma-tory syndrome including elevated levels of circulatingproinflammatory cytokines including TNF-� (15) anddevelop symptoms of Crohn’s-like disease (16). IL-10inhibits superoxide anion (O2
�) production by down-regulation of the gp9l-phox and p47-phox genes inhuman monocytes (17). We have earlier reported thatsystemic administration of IL-10 inhibits a panel ofproinflammatory cytokine/chemokine mRNA expres-sion in the LV after MI via inhibition of cytokinemRNA-stabilizing protein HuR and suppression of p38MAP kinase (10). Following MI, IL-10-KO mice showedincreased infiltration of inflammatory cells in the bor-der zone of the infarct with LV dysfunction, fibrosis,and cardiomyocyte apoptosis. We tested whether HuRknockdown attenuates these undesirable effects in IL-
1 Correspondence: Feinberg Cardiovascular Research In-stitute, Feinberg School of Medicine, Northwestern Univer-sity, 303 E. Chicago Ave., Chicago. IL 60611, USA. E-mail:R.K., [email protected]; P.K., [email protected]
doi: 10.1096/fj.09-149815
2484 0892-6638/10/0024-2484 © FASEB

10-KO mice. The therapeutic effects of targeting IL-10-sensitive protein HuR on post-MI inflammation, LVdysfunction, and remodeling and the molecular signal-ing that governs these effects remain to be studied.
Although HuR expression increased in LV post-MI,much remains to be understood concerning the HuRprotein-mediated mechanisms controlling mRNA sta-bility of various genes and their effect on the patho-genesis of MI. Given the strong association betweenproinflammatory cytokines and LV dysfunction andremodeling, the potential role of targeted anticyto-kine treatment strategies in MI needs to be fullyevaluated.
Ischemia-induced cardiomyocyte loss plays a prom-inent role in the pathophysiology of cardiac remod-eling after MI (18). Ischemia stimulates p53, leadingto apoptosis of cardiac cells, including myocytes bothin vivo (MI) (18) and in vitro (19). Apoptosis leads tothe disruption of normal myocardial structures, result-ing in replacement of dead cells with excessive deposi-tion of extracellular matrix (ECM; fibrosis) (20). p53 isa well-known proapoptotic factor, and TGF-� promotesfibrogenesis through connective tissue growth factor(CTGF) and SMAD3 signaling during heart failure(21). However, the underlying signaling mechanismsthat may mediate crosstalk between IL-10 and HuR, inthe context of HuR-mediated modulations in eitherTGF-� or p53 signaling, need to be explored. Wehypothesize that shRNA-mediated knockdown of HuRattenuates post-MI LV dysfunction and adverse LVremodeling in IL-10-deficient mice, and therefore mim-ics IL-10 effects. This study was undertaken to elucidatethe effects of HuR knockdown in modulating post-MIinflammation, LV dysfunction and remodeling, and thesignaling mechanisms that regulate HuR mediatedeffects such as apoptosis and fibrosis during LV remod-eling.
METHODS AND MATERIALS
Vertebrate animals
All experiments conform to the protocols approved by theInstitutional Animal Care and Use Committee. Six-week-oldwild-type (WT) and IL-10-KO (IL10tm1Cgn) mice of C57BL/6Jbackground were procured from Jackson Research Labora-tory (Bar Harbor, ME, USA). Although the development ofchronic enterocolitis and other abnormalities have beenreported in the IL-10-KO mice, we did not observe anyphenotypic or behavioral abnormalities in these mice beforeexperimentation. The mice were allowed to acclimatize for10 d under sterile animal management conditions.
Cell culture and reagents
RAW 264.7 cells (mouse mononuclear/macrophage cell line)were cultured in DMEM (Clonitech, Palo Alto, CA, USA) with10% FCS. Cells were stimulated with LPS and/or IL-10 at adose of 10 ng/ml concentration unless otherwise indicated.Recombinant murine and human IL-10 was obtained fromR&D Systems (Minneapolis, MN, USA). LPS was obtainedfrom Sigma-Aldrich (St. Louis, MO, USA). Mouse HuR shRNA
(HshRNA) and a nontarget control shRNA (CshRNA) lentiviralparticles were purchased from Sigma-Aldrich.
MI and study design
Mice were subjected to MI by ligation of the left anteriordescending coronary artery (LAD) as described previously(22). HuR-specific shRNA (4�106 viral particles/mice,HshRNA group) or control, nonspecific shRNA (4�106 viralparticles/mice, CshRNA group), was injected intramyocardi-ally into the LV wall (border zone) at 5 different locations ond 0 immediately after LAD ligation. Subsequently shRNA wasintravenously injected on d 1, 2, 3, 4, 5, and 7 post-MI. Themice in the sham group underwent the same procedureexcept for the LAD ligation. Inflammatory response andcardiomyocyte apoptosis was assessed at 3 d, LV functionalchanges at 14 and 28 d, and structural remodeling at 28 dpost-MI.
Echocardiography
Transthoracic 2-dimensional M-mode echocardiogram wasobtained using the Vevo 770 (VisualSonics, Toronto, ON,Canada) equipped with a 30-MHz transducer. Echocardio-graphic studies were performed before MI (baseline) and at14 and 28 d post-MI on mice anesthetized with a mixture of1.5% isoflurane and oxygen (1 L/min). M-mode tracingswere used to measure LV wall thickness, end-systolic diameter(LVESD), and end-diastolic diameter (LVEDD). The meanvalue of 9 measurements was determined for each sample.Percentage fractional shortening (%FS) and ejection fraction(%EF) were calculated as described previously (23).
Morphometric studies
The hearts were perfused with 30% KCl followed by fixationwith 4% paraformaldehye. Hearts were cut into 3 slices (apex,mid-LV, and base), and frozen sections were made. Themorphometric analysis including infarct size and percent-age fibrosis area (%LV area) was performed on Masson’strichrome-stained tissue sections using ImageJ 1.30 soft-ware (U.S. National Institutes of Health; http://rsb.info.nih.gov/ij/).
Immunohistochemistry
Immunohistochemical detection of HuR was carried outusing the avidin-biotin-DAB complex method on frozen sec-tions as described previously (24). In brief, after an overnightincubation at 4°C with primary monoclonal antibodiesagainst HuR (1:50, Santa Cruz Biotechnology, Santa Cruz,CA, USA), a biotin-conjugated goat anti-rabbit second anti-body (1:250; Vector Laboratories, Burlingame, CA, USA) andsubsequently streptavidin conjugated with horseraddish per-oxidase (HRP, 1:250; Vector Laboratories) were applied. DABperoxidase substrate (Vector Laboratories) was utilized forvisualization, and specimens were counterstained with hemo-toxylin (Vector Laboratories).
Immunofluorescent staining for CD68 on tissue sectionswas performed as described previously (25). Tissue sectionswere permeabilized and stained with anti-CD68 (Serotec,Raleigh, NC, USA) for inflammatory cell infiltration, followedby incubation with respective secondary antibodies. Stainingwithout the primary antibodies was used as control fornonspecific fluorescence. Nuclei were counterstained with 4�,6-diamidino-2-phenylindole (DAPI, 1:5000; Sigma-Aldrich),and sections were examined with a fluorescent microscope
2485HuR KNOCKDOWN ATTENUATES LV DYSFUNCTION AND REMODELING

(Eclipse TE200; Nikon, Tokyo, Japan). Inflammatory cellinfiltration (CD68�) was assessed at 10 randomly selectedhigh-power visual fields (HVFs) in the border zone of in-farcted myocardium and expressed as number per HVF.
Terminal deoxynucleotidyl transferase-mediated dUTP nickend-labeling (TUNEL) staining
TUNEL staining was carried out on 4-�m-thick frozen sec-tions as per the manufacturer’s instructions (Cell DeathDetection Assay; Roche, Indianapolis, IN, USA). Cardiacmyocytes were identified using �-sarcomeric actinin antibod-ies (Sigma Chemicals, St. Louis, MO, USA). DAPI staining wasused to count the total number of nuclei. The index ofapoptosis was calculated as the percentage of apoptoticmyocyte nuclei/total number of nuclei.
Quantitative real-time PCR (Q-PCR)
Gene expression levels of TNF-�, IL-1�, TGF-�, and p53 werequantified in the border zone of infarct as described previ-ously (26). RNA was collected from heart tissue or RAW 264.7cells with RNA STAT-60 (TEL-TEST, Friendswood, TX, USA).Total RNA was reverse transcribed with the iScript cDNAsynthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA), andamplification was performed using the Taqman 7300 (Ap-plied Biosystems, Foster City, CA, USA). Relative mRNAexpression of target genes was normalized to the endogenous18S control gene (Applied Biosystems) and represented asfold change vs. control untreated cells.
Generation of stable HuR-knockdown RAW 264.7 clones bypuromycin selection
RAW 264.7 cells were cultured in DMEM with 10% FBS. Cellswere treated with short-hairpin lentiviral particles againstHuR [5 MOI (multiplicity of infection)] in the presence ofhexadimethrine bromide (8 �g/ml; increases transductionefficiency). After 24 h transduction, the cells were selectedusing puromycin (6 �g/ml; dose determined by titration).Puromycin-resistant HuR-knockdown cell clones were grown,analyzed, and frozen for future use.
RNA protein-binding assay
Immunoprecipitation (IP) of endogenous HuR-mRNA com-plexes, used to assess the association of endogenous HuR withendogenous p53 and TGF-� mRNA, was performed as de-scribed previously (27). Cells were subjected to 254-nm UVlight exposure for 20 min in a stratalinker to cross-linkcellular mRNA-protein complexes. Total cell lysate fromUV-cross-linked cells was used for IP at 4°C, overnight in thepresence of excess (30 �g) IP antibody (IgG1 or anti-HuR;Santa Cruz Biotechnology). RNA bound to immunoprecipi-tated HuR in IP material was isolated and used in Q-PCRreactions to detect the presence of TGF-� or p53 mRNA. Dataare represented as fold change vs. control.
Assessment of mRNA stability
The mRNA stability of TGF-� and p53 mRNA was determinedby actinomycin D chase experiments, following a standardprotocol described elsewhere (28). Briefly, WT and HuR-knockdown RAW 264.7 cell clones were stimulated withLPS (10 ng/ml) in DMEM culture medium. Actinomycin Dwas added to a final concentration of 5 �g/ml to blockfurther transcription. At 0, 30, 60, and 120 min after
actinomycin D treatment, the cells were harvested, andmRNA was quantified by Q-PCR as described above. ThemRNA decay was recorded as the percentage of mRNAremaining over time compared with the amount before theaddition of actinomycin D.
Statistical analyses
Data are presented as means �se. Between 2 groups of mice,an unpaired Student’s t test was performed to determinestatistical significance. When 2 groups were involved,ANOVA with the Tukey’s post hoc test was used to analyze thedata. Values of P 0.05 were considered significant.
RESULTS
HuR knockdown inhibits post-MI LV inflammation
All animal experiments were carried out in accordancewith the Northwestern University Institutional AnimalCare and Use Committee (IACUC)-approved proto-cols. MI was induced in WT and IL-10-deficient miceand followed immediately by intramyocardial injectionsof either lentiviral-control scrambled shRNA (CshRNA)or HuR shRNA (HshRNA). HuR expression was assessedby immunoperoxidase staining for HuR on LV tissuesections at 3 d after MI and shRNA injection (Fig. 1A).The inflammatory cells infiltrated at the border zone ofthe infarct were strongly positive for HuR. However, thenumbers of HuR� cells were higher in the KO mice ascompared to WT mice in the CshRNA-treated group(Fig. 1A, top panels). HuR shRNA significantly knockeddown the expression of HuR in mice of both genotypes(Fig. 1A, bottom panels). HuR staining was observedmostly in inflammatory cells (Fig. 1A, arrows) and in afew degenerating cardiomyocytes (data not shown).mRNA expression of HuR in the myocardium (borderzone) at 3 d post-MI was assessed by Q-PCR. HuRmRNA expression significantly increased in IL-10-KOmice as compared to WT mice, post-MI (P0.01; Fig.1B). HshRNA significantly knocked down HuR mRNAas compared to CshRNA (P0.01; Fig. 1B).
Immunofluorescence staining of CD68� cells oncardiac tissue sections was carried out to study theinflammatory cell infiltration at 3 d post-MI (Fig. 1C).IL-10-KO mice treated with control shRNA (CshRNA)showed increased infiltration of CD68� cells (macro-phage and monocyte) in the border zone of LV infarctas compared to WT mice (P0.01; Fig. 1C, D). How-ever, ShRNA-mediated HuR knockdown significantlyinhibited infiltration of inflammatory cells in the bor-der zone of infarct (P0.01 vs. CshRNA group; Fig. 1C,D). WT mice receiving HshRNA showed reduced infil-tration of inflammatory cells as compared with KOmice (P0.01 vs. KO; Fig. 1C, D).
Resolution of inflammatory cell infiltration by HuRknockdown was corroborated by a significant repres-sion in the expression of a panel of inflammatorycytokines including TNF-�, IL-1�, and monocyte che-moattractant protein-1 (MCP-1, data not shown). Q-PCR for TNF-� and IL-1� showed that the increasedinflammatory cell infiltration was associated with in-
2486 Vol. 24 July 2010 KRISHNAMURTHY ET AL.The FASEB Journal � www.fasebj.org
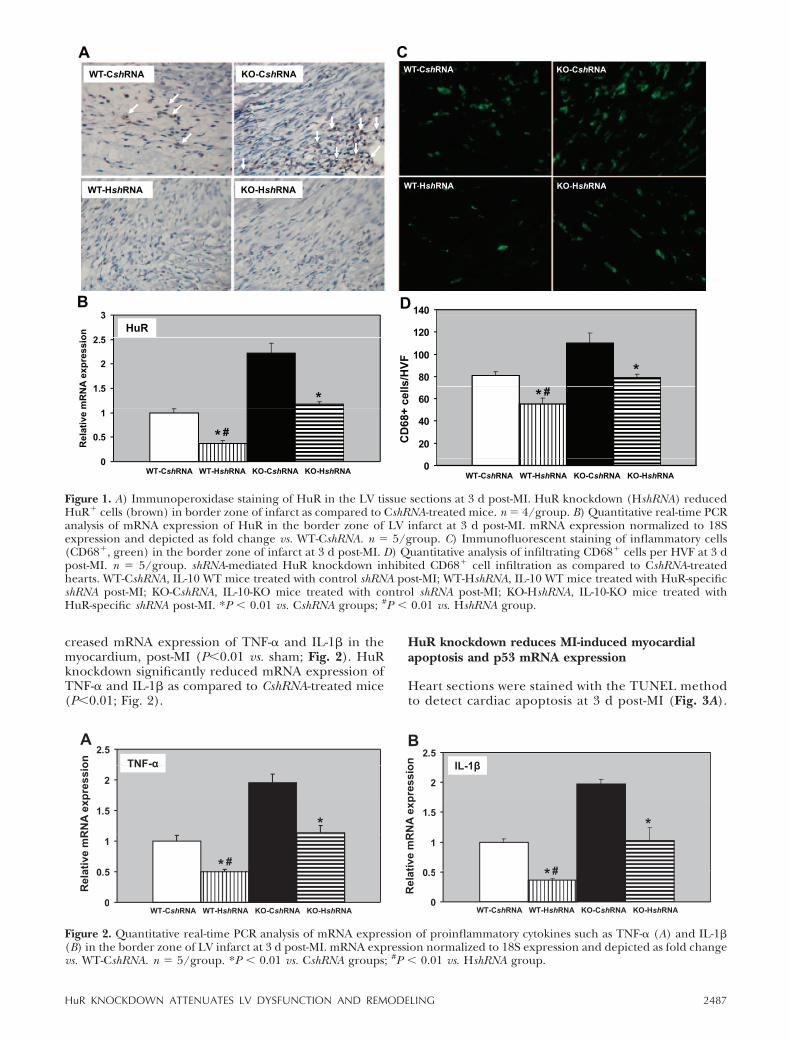
creased mRNA expression of TNF-� and IL-1� in themyocardium, post-MI (P0.01 vs. sham; Fig. 2). HuRknockdown significantly reduced mRNA expression ofTNF-� and IL-1� as compared to CshRNA-treated mice(P0.01; Fig. 2).
HuR knockdown reduces MI-induced myocardialapoptosis and p53 mRNA expression
Heart sections were stained with the TUNEL methodto detect cardiac apoptosis at 3 d post-MI (Fig. 3A).
Figure 1. A) Immunoperoxidase staining of HuR in the LV tissue sections at 3 d post-MI. HuR knockdown (HshRNA) reducedHuR� cells (brown) in border zone of infarct as compared to CshRNA-treated mice. n � 4/group. B) Quantitative real-time PCRanalysis of mRNA expression of HuR in the border zone of LV infarct at 3 d post-MI. mRNA expression normalized to 18Sexpression and depicted as fold change vs. WT-CshRNA. n � 5/group. C) Immunofluorescent staining of inflammatory cells(CD68�, green) in the border zone of infarct at 3 d post-MI. D) Quantitative analysis of infiltrating CD68� cells per HVF at 3 dpost-MI. n � 5/group. shRNA-mediated HuR knockdown inhibited CD68� cell infiltration as compared to CshRNA-treatedhearts. WT-CshRNA, IL-10 WT mice treated with control shRNA post-MI; WT-HshRNA, IL-10 WT mice treated with HuR-specificshRNA post-MI; KO-CshRNA, IL-10-KO mice treated with control shRNA post-MI; KO-HshRNA, IL-10-KO mice treated withHuR-specific shRNA post-MI. *P 0.01 vs. CshRNA groups; #P 0.01 vs. HshRNA group.
Figure 2. Quantitative real-time PCR analysis of mRNA expression of proinflammatory cytokines such as TNF-� (A) and IL-1�(B) in the border zone of LV infarct at 3 d post-MI. mRNA expression normalized to 18S expression and depicted as fold changevs. WT-CshRNA. n � 5/group. *P 0.01 vs. CshRNA groups; #P 0.01 vs. HshRNA group.
2487HuR KNOCKDOWN ATTENUATES LV DYSFUNCTION AND REMODELING

MI increased the number of apoptotic cardiomyocytesat the border zone of infarct as compared to thesham group (WT sham, 0.30�0.02; KO sham,0.39�0.05; P0.01 MI�CshRNA vs. sham; Fig. 3A, B).However, HuR knockdown significantly reduced thenumber of apoptotic cells in the border zone of LVinfarct (P0.01 vs. CshRNA; Fig. 3A, B). It wasinteresting to note that cardiomyocyte apoptosis washigher in IL-10-KO mice as compared to WT mice(P0.01; Fig. 3A, B); however, HuR knockdownsignificantly reduced exaggerated post-MI apoptosisin IL-10-KO mice.
p53 plays an important role in cardiomyocyte apop-tosis following MI (18). We examined p53 mRNA (Fig.3C) and protein expression (Fig. 3D) in the borderzone of infarct at 3 d post-MI. Cardiomyocyte apoptosiswas associated with increased p53 mRNA expressionand protein levels after MI. HuR knockdown reducedp53 mRNA and protein expression levels in the LV(P0.01, CshRNA vs. HuR shRNA; Fig. 3C, D). Thesedata suggest that the HuR-knockdown-mediated de-crease in p53 expression was directly associated with thesuppression of post-MI apoptosis. LPS-induced apopto-sis was studied in control (Csh) and HuR knockdown(Hsh) RAW 264.7 cell clones. We observed reducedapoptosis in HuR knockdown clones as compared tocontrol cells treated with LPS (Supplemental Fig. 1).
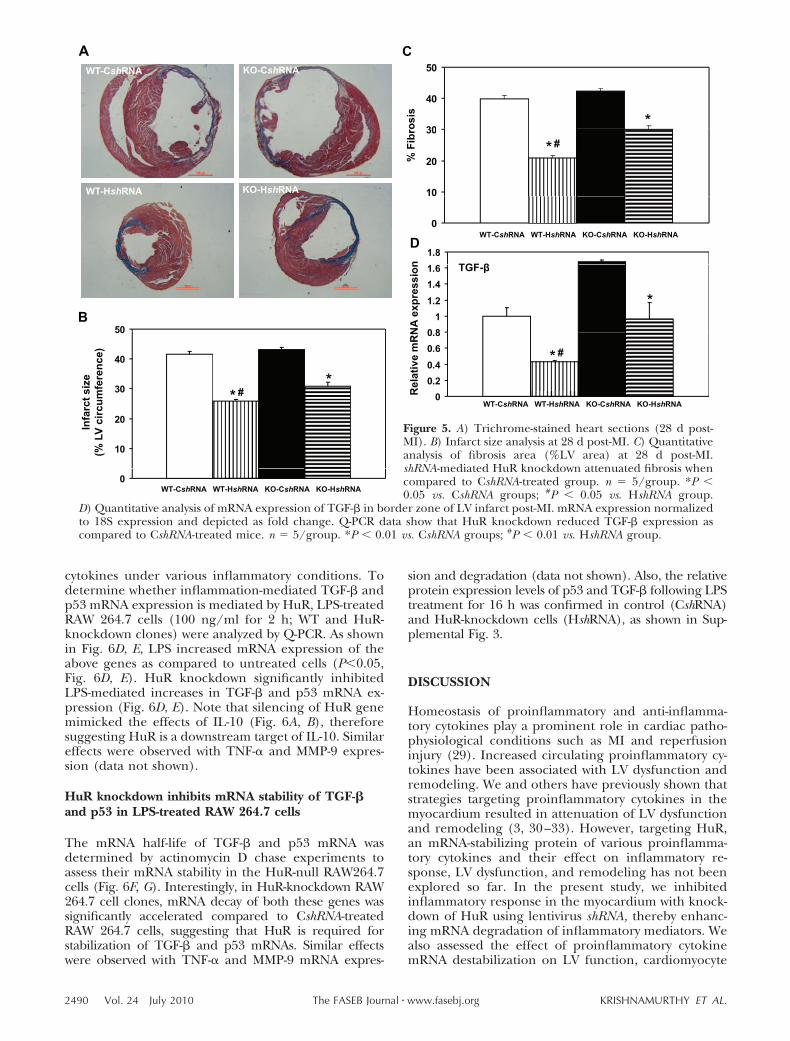
HuR knockdown attenuates post-MI LV dysfunctionand reduces the infarct size
We have earlier reported that IL-10 treatment rescuespost-MI LV functions in mice (10). To assess whetherHuR knockdown in the myocardium may mimic IL-10effects, LV functions were determined by echocardiog-raphy in both WT and IL-10-KO mice injected witheither control or HuR-shRNA, 14 and 28 d after theinduction of MI. M-mode tracings analyzed at 14 (datanot shown) and 28 d post-MI showed similar changes inLV functions (Fig. 4). MI increased LVESD and LVEDD(P0.01 vs. baseline; Fig. 4A, B) and reduced %FS and%EF in CshRNA-treated mice at 28 d post-MI (P0.05vs. baseline; Fig. 4C, D), suggesting LV contractiledysfunctions. HuR knockdown attenuated LV dysfunc-tion, with significantly lowered LVESD and LVEDD(P0.05; Fig. 4A, B), and increased %FS and %EF ascompared to the CshRNA-treated group (P0.05; Fig.4C, D). Heart rates were not significantly differentamong the groups. Infarct size was measured aspercentage of the LV circumference from trichrome-stained sections at 28 d post-MI. There was nodifference in the infarct size/fibrosis in WT and KOmice treated with CshRNA (Fig. 5A, B). HuR knock-down resulted in a significant reduction in the infarctsize as compared to CshRNA-treated mice (P0.01;
Figure 3. A) TUNEL staining for cardiomyocyte apoptosis(red nuclei) in border zone of LV infarct at 3 d post-MI. HuRknockdown attenuated cardiomyocyte apoptosis after MI.B) Quantitative analysis of TUNEL� cardiomyocytes at 3 dpost-MI. shRNA-mediated HuR knockdown reduced cardio-myocyte apoptosis as compared to CshRNA-treated hearts.n � 5/group. C) Quantitative analysis of mRNA expressionof p53 in border zone of LV infarct at 3 d post-MI. mRNAexpression normalized to 18S expression and depicted asfold change. Q-PCR data shows that HuR knockdown re-duced p53 expression as compared to CshRNA-treated mice.
n � 5/group. D) Western blot for p53 protein expression in LV at 3 d post-MI. Equal loading of proteins in each lane isshown by �-actin. n � 3/. *P 0.01 vs. CshRNA groups; #P 0.01 vs. HshRNA group.
2488 Vol. 24 July 2010 KRISHNAMURTHY ET AL.The FASEB Journal � www.fasebj.org

WT-CshRNA, 41.57�1.02; WT-HshRNA, 25.89�0.62;KO-CshRNA, 43.13�0.65; KO-HshRNA, 30.86�1.24;Fig. 5A, B).
HuR knockdown inhibits TGF-� mRNA expressionand attenuates LV fibrosis post-MI
TGF-� plays an important role in post-MI ECM remod-eling. TGF-� expression increases rapidly in the heartpost-MI (7). Quantitative analysis of trichrome-stainedsections indicated increased fibrosis in the LV after 28 dpost-MI in IL-10-KO mice compared to WT mice bothtreated with CshRNA (P0.01 vs. sham; Fig. 5C). It wasinteresting to note that fibrosis was significantly re-duced in the LV following HuR knockdown (P0.05 vs.CshRNA; Fig. 5C). We assessed mRNA expression ofTGF-� in the myocardium in the border zone post-MI(Fig. 5D). Q-PCR analysis indicated that the mRNA ex-pression of TGF-� was increased following MI (P0.01vs. sham, not shown). However, HshRNA-treated miceshowed significantly reduced TGF-� mRNA expressionlevels in the LV (P0.01 vs. CshRNA; Fig. 5D). These dataindicate that HuR knockdown leads to decreased expres-sion of TGF-� mRNA, which in turn correlates withdecreased post-MI fibrosis.
HuR protein physically associates with TGF-� andp53 mRNAs in vivo
Both p53 and TGF-� mRNA harbors ARE elements intheir respective 3�UTRs, sequences to which HuR bindsand stabilizes posttranscription mRNA stability. Real-time PCR analysis for HuR mRNA expression showed
that IL-10 significantly reduced LPS-mediated increasesin HuR mRNA expression (P0.01 vs. LPS-treatedcells; Supplemental Fig. 2). To assess the endogenousassociation of HuR with TGF-� and p53 mRNA, RNA-protein immunoprecipitation experiments were per-formed on whole-cell extracts obtained from cellstreated with LPS and/or IL-10 for 2 h and were UVcross-linked. RNA cross-linked to HuR was immunopre-cipitated by HuR antibodies, isolated from HuR-RNAcomplexes, and assessed for TGF-� and p53 mRNA byQ-PCR. Figure 6A, B shows the association of TGF-� andp53 mRNAs with endogenous HuR. LPS increased theassociation of TGF-� and p53 mRNA to HuR, whileIL-10 inhibited amount of p53 and TGF-� mRNAbound to HuR (Fig. 6A, B). Importantly, the p53 orTGF-� mRNAs were undetectable in nonspecific IgG1IPs (not shown). A similar trend was observed for otherIL-10-sensitive targets TNF-� and MMP-9 (data notshown).
HuR knockdown inhibits mRNA expression of TGF-�and p53 in RAW 264.7 cells
To determine whether HuR mimics IL-10 inhibitoryeffect on p53 and TGF-� mRNA expression, we gener-ated stable HuR-knockdown clones from mouse mono-cyte-macrophage cell line RAW264.7. Cells infectedwith lentiviral HuRshRNA were selected with puromy-cin, and the stable knockdown of HuR was confirmedin 2 of the generated clones by Western blotting, whichshowed complete abrogation of HuR expression ascompared to control shRNA-treated cells (Fig. 6C). HuRbinds and stabilizes a number of proinflammatory
6
7
6
7LVEDD (mm) LVESD (mm)BA
**
4
55
6 ***
2
3
Baseline 7d 14d 28d3
4
Baseline 7d 14d 28dBaseline 7d 14d 28d
50
60
70
25
30
35% FS % EFDC
#* #*
10
20
30
40
WT-CshRNAKO-CshRNAWT-HshRNA5
10
15
20 * *
0
10
Baseline 7d 14d 28d
WT HshRNAKO-HshRNA
0
5
Baseline 7d 14d 28d
Figure 4. Echocardiography: analysis of M-mode tracing of LVEDD (A) and LVESD (B), and %FS (C) and %EF (D) calculations.HuR knockdown improved LV function with significantly lowered LVESD and LVEDD and increased %FS and %EF. n �10/group. *P 0.05 vs. CshRNA groups; #P 0.05 vs. HshRNA group.
2489HuR KNOCKDOWN ATTENUATES LV DYSFUNCTION AND REMODELING
cytokines under various inflammatory conditions. Todetermine whether inflammation-mediated TGF-� andp53 mRNA expression is mediated by HuR, LPS-treatedRAW 264.7 cells (100 ng/ml for 2 h; WT and HuR-knockdown clones) were analyzed by Q-PCR. As shownin Fig. 6D, E, LPS increased mRNA expression of theabove genes as compared to untreated cells (P0.05,Fig. 6D, E). HuR knockdown significantly inhibitedLPS-mediated increases in TGF-� and p53 mRNA ex-pression (Fig. 6D, E). Note that silencing of HuR genemimicked the effects of IL-10 (Fig. 6A, B), thereforesuggesting HuR is a downstream target of IL-10. Similareffects were observed with TNF-� and MMP-9 expres-sion (data not shown).
HuR knockdown inhibits mRNA stability of TGF-�and p53 in LPS-treated RAW 264.7 cells
The mRNA half-life of TGF-� and p53 mRNA wasdetermined by actinomycin D chase experiments toassess their mRNA stability in the HuR-null RAW264.7cells (Fig. 6F, G). Interestingly, in HuR-knockdown RAW264.7 cell clones, mRNA decay of both these genes wassignificantly accelerated compared to CshRNA-treatedRAW 264.7 cells, suggesting that HuR is required forstabilization of TGF-� and p53 mRNAs. Similar effectswere observed with TNF-� and MMP-9 mRNA expres-
sion and degradation (data not shown). Also, the relativeprotein expression levels of p53 and TGF-� following LPStreatment for 16 h was confirmed in control (CshRNA)and HuR-knockdown cells (HshRNA), as shown in Sup-plemental Fig. 3.
DISCUSSION
Homeostasis of proinflammatory and anti-inflamma-tory cytokines play a prominent role in cardiac patho-physiological conditions such as MI and reperfusioninjury (29). Increased circulating proinflammatory cy-tokines have been associated with LV dysfunction andremodeling. We and others have previously shown thatstrategies targeting proinflammatory cytokines in themyocardium resulted in attenuation of LV dysfunctionand remodeling (3, 30–33). However, targeting HuR,an mRNA-stabilizing protein of various proinflamma-tory cytokines and their effect on inflammatory re-sponse, LV dysfunction, and remodeling has not beenexplored so far. In the present study, we inhibitedinflammatory response in the myocardium with knock-down of HuR using lentivirus shRNA, thereby enhanc-ing mRNA degradation of inflammatory mediators. Wealso assessed the effect of proinflammatory cytokinemRNA destabilization on LV function, cardiomyocyte
Figure 5. A) Trichrome-stained heart sections (28 d post-MI). B) Infarct size analysis at 28 d post-MI. C) Quantitativeanalysis of fibrosis area (%LV area) at 28 d post-MI.shRNA-mediated HuR knockdown attenuated fibrosis whencompared to CshRNA-treated group. n � 5/group. *P 0.05 vs. CshRNA groups; #P 0.05 vs. HshRNA group.
D) Quantitative analysis of mRNA expression of TGF-� in border zone of LV infarct post-MI. mRNA expression normalizedto 18S expression and depicted as fold change. Q-PCR data show that HuR knockdown reduced TGF-� expression ascompared to CshRNA-treated mice. n � 5/group. *P 0.01 vs. CshRNA groups; #P 0.01 vs. HshRNA group.
2490 Vol. 24 July 2010 KRISHNAMURTHY ET AL.The FASEB Journal � www.fasebj.org

apoptosis, and remodeling post-MI. The importantfindings of this study are that HuR knockdown mim-icked IL-10 treatment effects and attenuated myocar-dial inflammation, cardiomyocyte apoptosis, and ex-pression of TGF-� and p53 at 3 d post-MI, followed byattenuation of LV dysfunction and remodeling witheffects on fibrosis at 28 d post-MI. The above effectswere suggested to be partly due to HuR-knockdown-
mediated mRNA destabilization of the proinflamma-tory cytokines TGF-� and p53.
Inflammatory cytokines have been implicated inpost-MI LV remodeling and cardiomyocyte hypertro-phy (1) with alterations in fetal gene expression andcontractile abnormalities (2, 3, 31). At baseline, IL-10-KO mice did not show any phenotypical differencesin LV function and remodeling changes as compared
Figure 6. HuR physically binds to TGF-� and p53 mRNAs in vivo. A, B) Cells treated with LPS and/or IL-10 for 2 h were UVcross-linked and lyzed, and RNA-protein complexes were immunoprecipitated in the presence of anti-HuR antibody ornonspecific IgG1. IL-10 inhibited LPS-mediated increases in TGF-� (A) and p53 (B) mRNA association to HuR. n � 3/group.*P 0.01 vs. LPS. C) Western blotting on LPS-treated puromycin-selected HuR-knockdown RAW 264.7 cell clones usingdifferent target constructs (lanes 1 and 2). HuR knockdown (HshRNA) abrogated LPS-stimulated HuR protein expression.�-actin signals served to assess the equal protein loading in all lanes. D, E) Quantitative analysis of mRNA expression of TGF-�(D) and p53 (E) following HuR knockdown. Q-PCR data show that TGF-� and p53 expression increased on LPS treatment ascompared to untreated cells (Unt). HshRNA reduced TGF-� and p53 expression as compared to CshRNA-treated cells. n �3/group. *P 0.05 vs. CshRNA. F, G) mRNA stability of TGF-� (F) and p53 mRNA (G), assessed in HuR-null RAW264.7 cellclones by using actinomycin D (5 �g/ml) chase assays. Total cellular RNA was isolated at times shown; percentage remaininglevels of TGF-� and p53 mRNAs were measured by Q-PCR analysis. Values are means � se from triplicate experiments. *P 0.05 at 120 min.
2491HuR KNOCKDOWN ATTENUATES LV DYSFUNCTION AND REMODELING

to WT mice. However, following MI, infiltration ofCD68� monocyte/macrophages in the border zone ofthe myocardium was higher in IL-10-KO mice as com-pared to WT mice, at 3 d post-MI. Consistent with thisfinding, MI-induced increases in HuR expression werehigher in the KO mice vs. WT mice. Inflammatory cellinfiltration was associated with an increase in mRNAexpression of various proinflammatory cytokines andchemokines (IL-1�, IL-6, TNF-�, IP-10, MCP-1) after MI(10). These “stress-activated” cytokines are produced byvarious cell types in the myocardium, including cardio-myocytes, or by the resident/infiltrating inflammatorycells. The mRNAs encoding most proinflammatorycytokines are short lived, with instability conferredby an ARE in the 3� noncoding region. HuR selectivelybinds AREs and stabilizes mRNAs. IL-10, a potentanti-inflammatory cytokine, has been shown to limit theinfiltration of inflammatory cells in MI (10) and vascu-lar injury models (13). In agreement with the abovestudies, HuR knockdown inhibited inflammatory cellinfiltration and proinflammatory cytokines and chemo-kines (P0.05 vs. MI) in the myocardium. This suggeststhat HuR could be a downstream molecule of IL-10signaling. An earlier report suggests that IL-10 knock-out in an ischemia reperfusion model after LAD liga-tion increases mortality in mice (34). Our laboratoryhas shown that IL-10 injection reduced inflammation-mediated adverse cardiac remodeling and dysfunction(10). The present data targeting HuR (a downstreammolecule) mimic IL-10 effects. We presume that IL-10-KO-induced deleterious effects on the left ventricle,taken together, can be significantly reduced, if notcompletely. In addition, ongoing experiments in ourlaboratory involving IL-10 WT bone marrow transplan-tation in IL-10-KO mice will provide critical insightsthat will further confirm the above effects.
Myocardial expression of proinflammatory cytokinescontributes to depression of contractile performance andadverse LV remodeling (2, 29). In the present study,echocardiography showed increase in LVEDD andLVESD and decrease in %FS and %EF after MI and HuRknockdown attenuated these effects at 28 d post-MI.Proinflammatory cytokine-induced depression of contrac-tile performance might be a direct result of interferencewith myocardial calcium handling (35, 36), myoD degra-dation (37), cardiac fibrosis (38), or cardiomyocyte apop-tosis (18). Although HuR-mediated effects on myoD,MMP-9, and p53 have been reported (27, 37, 39), itseffects on calcium signaling are not clear. The significantup-regulation of proinflammatory cytokines (at 3 d) couldtrigger a second phase of elevated cytokines levels in thenoninfarcted myocardium that promotes interstitial fibro-sis and collagen deposition leading to ventricular dysfunc-tion (29). Most important, a recent study (40) has shownthat transplantation of bone marrow mononuclear cells(BM-MNCs) in infarcted mouse hearts led to a significantimprovement in cardiac function. These BM-MNCs se-creted significant amounts of IL-10, and the cardiacprotection was associated with decreased T-lymphocyteaccumulation, reactive hypertrophy, and myocardial col-lagen deposition. Also, various studies have reported thatactivation of MMPs and p38, and reduced angiogenesis
and STAT-3, have affected remodeling and cardiac dys-function (32, 41–43).
Ischemic-oxidative stress stimulates p53 (proapoptoticfactor), leading to apoptosis of cardiac cells includingmyocytes both in vivo (MI) (18) and in vitro (19). Ische-mia-induced cardiomyocyte cell death (apoptosis) plays aprominent role in the pathophysiology of cardiac remod-eling after MI (18). Several studies have shown involve-ment of inflammatory mediators in progressive myocytesloss due to necrosis and/or apoptosis, suggesting thatthese cytokines are involved in the progression of cardiacremodeling (33, 44). In the present study, MI increasedthe number of apoptotic cells in the border zone ofinfarction, which was significantly higher in IL-10-KOmice as compared to WT mice. Consistent with the abovefinding, p53 mRNA and protein expression was increasedin KO mice vs. WT mice. HuR knockdown attenuatedcardiomyocyte apoptosis associated with decreased p53mRNA expression. In addition to the various proinflam-matory cytokines and MMP-9 mRNA, HuR protein alsobinds and stabilizes p53 mRNA (27). Immunoprecipita-tion of HuR proteins in LPS-treated RAW 264.7 cellsshowed increased association of p53 mRNA with theendogenous HuR protein. However, HuR knockdownsignificantly enhanced decay of p53 mRNA. Taken to-gether, our data suggest that HuR knockdown attenuatescardiomyocyte apoptosis by significantly reducing p53mRNA stability. Also, in addition to HuR, other p53regulators, such as minute double minute 2 and herpesvirus-associated ubiquitin-specific protease (45) mightalso play an important role in the remodeling process.
Apoptosis leads to disruption of normal myocardialstructures, resulting in replacement of dead cells withexcessive deposition of ECM (fibrosis) (20). An underly-ing morphological correlate of LV dysfunction is cardio-myocyte apoptosis and cardiac fibrosis, which leads toincreased stiffness of the heart. Homeostasis of ECM(degradation and accumulation) mediates pathogenesisof LV remodeling. Increased TGF-� production associ-ated with sustained inflammatory response may lead toexcessive extracellular matrix deposition, leading to car-diac fibrosis, resulting in adverse remodeling changes (2,7, 43).
Our study shows that MI increased fibrosis associatedwith increased mRNA expression of TGF-� in the LV.These findings corroborate well with the previous findingthat TGF-� promotes fibrogenesis through CTGF andSMAD3 signaling during heart failure (21). An interestingfinding of the present study is that HuR knockdownsignificantly inhibited fibrosis. Also, IL-10 suppressedHuR expression and had similar effects on fibrosis (10).Therefore, silencing of HuR mimics the effects of IL-10on LV function and remodeling.
Recent studies have suggested that fibrosis might resultfrom proliferation of resident fibroblasts, bone marrow-derived fibroblasts, and epithelial-mesenchymal transition(EMT). TGF-�1, a promoter of cardiac fibrosis, inducedEMT in adult coronary endothelial cells and was mediatedthrough transcription factor Smad3 (20). A synergy ofTNF-� and TGF-� signaling promotes a rapid morpholog-ical conversion of the epithelial cells to the mesenchymalphenotype, and this process is dependent on enhancedp38 MAPK activity (46). Also, earlier reports have sug-
2492 Vol. 24 July 2010 KRISHNAMURTHY ET AL.The FASEB Journal � www.fasebj.org

gested that TGF-� expression in mesangial cells is medi-ated by mRNA-stabilizing factor, HuR (39). In the presentstudy, mRNA decay of TGF-� was accelerated in HuR-knockdown RAW cell clones as compared to CshRNA-treated RAW 264.7 cells, suggesting that HuR stabilizesmRNA of TGF-�. These findings, along with our previousreports of IL-10-mediated inhibition of HuR and p38MAPK, suggest that inflammation might be playing animportant role in endothelial to mesenchymal transition,and the mechanism is yet to be determined from ourongoing experiments.
IL-10 regulation of HuR might be occurring throughvarious signaling pathways. A previous publication fromour laboratory (13) has reported that anti-inflammatorycytokine IL-10 inhibits a panel of proinflammatory cyto-kines through suppression of p38 MAPK. Our data in thiswork suggest that IL-10 exhibits its anti-inflammatoryeffects by inhibiting the mRNA-stabilizing protein HuR.Our previous reports have also shown that IL-10 inhibitsinflammation and attenuates LV remodeling after MI viaactivation of STAT3 and suppression of HuR. The inflam-matory effects were also associated with p38 MAPK expres-sion in the left ventricle (10). There is increasing evidencethat the p38 MAPK cascade is crucial for the control of itsmRNA-destabilizing activity, particularly for the zinc fin-ger protein TTP (another mRNA-stabilizing protein) (47,48). However, it is not clear whether IL-10-mediatedreduction in HuR expression is regulated through p38MAPK and/or STAT3. Recently, Abdelmohsen et al. (49)has reported that microRNA (miR-519) regulates HuRtranslation in several human carcinoma cell lines. How-ever, the role of miR-519 in IL-10-mediated reduction inHuR levels is not yet explored.
In summary, the data presented here suggest that HuRknockdown reduces severity of proinflammatory re-sponses and contributes to improved LV function andremodeling by inhibition of TGF-�-associated fibrosis andp53-associated cardiomyocyte apoptosis after MI. Theeffects are due to HuR-knockdown-mediated destabiliza-tion of TGF-� and p53 mRNA. These results define HuRas a critical player in proinflammatory cytokine-inducedLV dysfunction and remodeling and therefore establish-ing mRNA stability of cytokines as a potential therapeutictarget that could attenuate inflammation-mediated fibro-sis and apoptosis. Also, understanding the effects ofinflammation on transplanted progenitor cells functionand survival in the heart could enhance cell-based thera-peutic approaches in cardiac interventions.
The reported work was supported in part by American HeartAssociation–Davee Foundation scientist development grant(SDG) 0930219N (P.K.) and National Institute of Health grantsAA014575 and HL091983 (R.K.). The authors declare no com-peting interests. All authors contributed substantially to thiswork. R.K. and P.K. conceptualized the experiments. P.K. per-formed all animal surgical procedures and histological analysis.E.L., S.V., and T.T. generated stable clones, performed Westernblots, and provided technical assistance with in vitro experi-ments. P.K. wrote the manuscript, and R.K. edited it. G.Q. andD.W.L. read the manuscript and provided critical appraisal andconceptual insights. All authors discussed the results and impli-cations and commented on the manuscript at all stages.
REFERENCES
1. Sun, M., Chen, M., Dawood, F., Zurawska, U., Li, J. Y., Parker, T.,Kassiri, Z., Kirshenbaum, L. A., Arnold, M., Khokha, R., and Liu,P. P. (2007) Tumor necrosis factor-alpha mediates cardiacremodeling and ventricular dysfunction after pressure overloadstate. Circulation 115, 1398–1407
2. Sun, M., Dawood, F., Wen, W. H., Chen, M., Dixon, I., Kirshen-baum, L. A., and Liu, P. P. (2004) Excessive tumor necrosisfactor activation after infarction contributes to susceptibility ofmyocardial rupture and left ventricular dysfunction. Circulation110, 3221–3228
3. Suzuki, K., Murtuza, B., Smolenski, R. T., Sammut, I. A., Suzuki,N., Kaneda, Y., and Yacoub, M. H. (2001) Overexpression ofinterleukin-1 receptor antagonist provides cardioprotectionagainst ischemia-reperfusion injury associated with reduction inapoptosis. Circulation 104, I308–I303
4. Bujak, M., Ren, G., Kweon, H. J., Dobaczewski, M., Reddy, A.,Taffet, G., Wang, X. F., and Frangogiannis, N. G. (2007)Essential role of Smad3 in infarct healing and in the pathogen-esis of cardiac remodeling. Circulation 116, 2127–2138
5. Frangogiannis, N. G. (2006) Targeting the inflammatory re-sponse in healing myocardial infarcts. Curr. Med. Chem. 13,1877–1893
6. Timmers, L., Sluijter, J. P., van Keulen, J. K., Hoefer, I. E.,Nederhoff, M. G., Goumans, M. J., Doevendans, P. A., vanEchteld, C. J., Joles, J. A., Quax, P. H., Piek, J. J., Pasterkamp, G.,and de Kleijn, D. P. (2008) Toll-like receptor 4 mediatesmaladaptive left ventricular remodeling and impairs cardiacfunction after myocardial infarction. Circ. Res. 102, 257–264
7. Tao, Z. Y., Cavasin, M. A., Yang, F., Liu, Y. H., and Yang, X. P.(2004) Temporal changes in matrix metalloproteinase expres-sion and inflammatory response associated with cardiac ruptureafter myocardial infarction in mice. Life Sci. 74, 1561–1572
8. Frangogiannis, N. G., Mendoza, L. H., Lindsey, M. L., Ballan-tyne, C. M., Michael, L. H., Smith, C. W., and Entman, M. L.(2000) IL-10 is induced in the reperfused myocardium and maymodulate the reaction to injury. J. Immunol. 165, 2798–2808
9. Yao, L., Huang, K., Huang, D., Wang, J., Guo, H., and Liao, Y.(2008) Acute myocardial infarction induced increases in plasmatumor necrosis factor-alpha and interleukin-10 are associatedwith the activation of poly(ADP-ribose) polymerase of circulat-ing mononuclear cell. Int. J. Cardiol. 123, 366–368
10. Krishnamurthy, P., Rajasingh, J., Lambers, E., Qin, G., Losordo,D. W., and Kishore, R. (2009) IL-10 inhibits inflammation andattenuates left ventricular remodeling after myocardial infarc-tion via activation of STAT3 and suppression of HuR. Circ. Res.104, e9–e18
11. Girndt, M., Kaul, H., Sester, U., Ulrich, C., Sester, M., Georg, T.,and Kohler, H. (2002) Anti-inflammatory interleukin-10 geno-type protects dialysis patients from cardiovascular events. KidneyInt. 62, 949–955
12. Chen, C. Y., Xu, N., and Shyu, A. B. (2002) Highly selectiveactions of HuR in antagonizing AU-rich element-mediatedmRNA destabilization. Mol. Cell. Biol. 22, 7268–7278
13. Rajasingh, J., Bord, E., Luedemann, C., Asai, J., Hamada, H.,Thorne, T., Qin, G., Goukassian, D., Zhu, Y., Losordo, D. W.,and Kishore, R. (2006) IL-10-induced TNF-alpha mRNA desta-bilization is mediated via IL-10 suppression of p38 MAP kinaseactivation and inhibition of HuR expression. FASEB J. 20,2112–2114
14. Yarovinsky, T. O., Butler, N. S., Monick, M. M., and Hunning-hake, G. W. (2006) Early exposure to IL-4 stabilizes IL-4 mRNAin CD4� T cells via RNA-binding protein HuR. J. Immunol. 177,4426–4435
15. Cohen, S. L., Moore, A. M., and Ward, W. E. (2004) Interleu-kin-10 knockout mouse: a model for studying bone metabolismduring intestinal inflammation. Inflamm. Bowel Dis. 10, 557–563
16. Scheinin, T., Butler, D. M., Salway, F., Scallon, B., and Feld-mann, M. (2003) Validation of the interleukin-10 knockoutmouse model of colitis: antitumour necrosis factor-antibodiessuppress the progression of colitis. Clin. Exp. Immunol. 133,38–43
17. Kuga, S., Otsuka, T., Niiro, H., Nunoi, H., Nemoto, Y., Nakano,T., Ogo, T., Umei, T., and Niho, Y. (1996) Suppression ofsuperoxide anion production by interleukin-10 is accompanied
2493HuR KNOCKDOWN ATTENUATES LV DYSFUNCTION AND REMODELING

by a downregulation of the genes for subunit proteins ofNADPH oxidase. Exp. Hematol. 24, 151–157
18. Matsusaka, H., Ide, T., Matsushima, S., Ikeuchi, M., Kubota, T.,Sunagawa, K., Kinugawa, S., and Tsutsui, H. (2006) Targeteddeletion of p53 prevents cardiac rupture after myocardialinfarction in mice. Cardiovasc. Res. 70, 457–465
19. Long, X., Boluyt, M. O., Hipolito, M. L., Lundberg, M. S.,Zheng, J. S., O’Neill, L., Cirielli, C., Lakatta, E. G., and Crow,M. T. (1997) p53 and the hypoxia-induced apoptosis of culturedneonatal rat cardiac myocytes. J. Clin. Investig. 99, 2635–2643
20. Zeisberg, E. M., Tarnavski, O., Zeisberg, M., Dorfman, A. L.,McMullen, J. R., Gustafsson, E., Chandraker, A., Yuan, X., Pu,W. T., Roberts, A. B., Neilson, E. G., Sayegh, M. H., Izumo, S.,and Kalluri, R. (2007) Endothelial-to-mesenchymal transitioncontributes to cardiac fibrosis. Nat. Med. 13, 952–961
21. Chuva de Sousa Lopes, S. M., Feijen, A., Korving, J., Korchyn-skyi, O., Larsson, J., Karlsson, S., ten Dijke, P., Lyons, K. M.,Goldschmeding, R., Doevendans, P., and Mummery, C. L.(2004) Connective tissue growth factor expression and Smadsignaling during mouse heart development and myocardialinfarction. Dev. Dyn. 231, 542–550
22. Krishnamurthy, P., Subramanian, V., Singh, M., and Singh, K.(2006) Deficiency of beta1 integrins results in increased myo-cardial dysfunction after myocardial infarction. Heart (Br. Card.Soc.) 92, 1309–1315
23. Finsen, A. V., Christensen, G., and Sjaastad, I. (2005) Echocar-diographic parameters discriminating myocardial infarctionwith pulmonary congestion from myocardial infarction withoutcongestion in the mouse. J. Appl. Physiol. 98, 680–689
24. Hasegawa, H., Kakuguchi, W., Kuroshima, T., Kitamura, T.,Tanaka, S., Kitagawa, Y., Totsuka, Y., Shindoh, M., and Hi-gashino, F. (2009) HuR is exported to the cytoplasm in oralcancer cells in a different manner from that of normal cells. Br.J. Cancer 100, 1943–1948
25. Ii, M., Nishimura, H., Iwakura, A., Wecker, A., Eaton, E.,Asahara, T., and Losordo, D. W. (2005) Endothelial progenitorcells are rapidly recruited to myocardium and mediate protec-tive effect of ischemic preconditioning via “imported” nitricoxide synthase activity. Circulation 111, 1114–1120
26. Qin, G., Kishore, R., Dolan, C. M., Silver, M., Wecker, A.,Luedemann, C. N., Thorne, T., Hanley, A., Curry, C., Heyd, L.,Dinesh, D., Kearney, M., Martelli, F., Murayama, T., Goukassian,D. A., Zhu, Y., and Losordo, D. W. (2006) Cell cycle regulatorE2F1 modulates angiogenesis via p53-dependent transcriptionalcontrol of VEGF. Proc. Natl. Acad. Sci. U. S. A. 103, 11015–11020
27. Zhao, J., Chen, J., Lu, B., Dong, L., Wang, H., Bi, C., Wu, G.,Guo, H., Wu, M., and Guo, Y. (2008) TIP30 induces apoptosisunder oxidative stress through stabilization of p53 messengerRNA in human hepatocellular carcinoma. Cancer Res. 68, 4133–4141
28. Kishore, R., Tebo, J. M., Kolosov, M., and Hamilton, T. A.(1999) Cutting edge: clustered AU-rich elements are the targetof IL-10-mediated mRNA destabilization in mouse macro-phages. J. Immunol. 162, 2457–2461
29. Ono, K., Matsumori, A., Shioi, T., Furukawa, Y., and Sasayama,S. (1998) Cytokine gene expression after myocardial infarctionin rat hearts: possible implication in left ventricular remodeling.Circulation 98, 149–156
30. Li, Y., Takemura, G., Okada, H., Miyata, S., Maruyama, R., Li, L.,Higuchi, M., Minatoguchi, S., Fujiwara, T., and Fujiwara, H.(2006) Reduction of inflammatory cytokine expression andoxidative damage by erythropoietin in chronic heart failure.Cardiovasc. Res. 71, 684–694
31. Mayer, B., Holmer, S. R., Hengstenberg, C., Lieb, W., Pfeifer,M., and Schunkert, H. (2005) Functional improvement in heartfailure patients treated with beta-blockers is associated with adecline of cytokine levels. Int. J. Cardiol. 103, 182–186
32. Wang, M., Tsai, B. M., Turrentine, M. W., Mahomed, Y., Brown,J. W., and Meldrum, D. R. (2005) p38 mitogen activated proteinkinase mediates both death signaling and functional depressionin the heart. Ann. Thorac. Surg. 80, 2235–2241
33. Yeh, C. H., Chen, T. P., Wu, Y. C., Lin, Y. M., and Jing Lin, P.(2005) Inhibition of NFkappaB activation with curcumin atten-uates plasma inflammatory cytokines surge and cardiomyocyticapoptosis following cardiac ischemia/reperfusion. J. Surg. Res.125, 109–116
34. Yang, Z., Zingarelli, B., and Szabo, C. (2000) Crucial role ofendogenous interleukin-10 production in myocardial ischemia/reperfusion injury. Circulation 101, 1019–1026
35. Duncan, D. J., Hopkins, P. M., and Harrison, S. M. (2007)Negative inotropic effects of tumour necrosis factor-alpha andinterleukin-1beta are ameliorated by alfentanil in rat ventricularmyocytes. Br. J. Pharmacol. 150, 720–726
36. Sugishita, K., Kinugawa, K., Shimizu, T., Harada, K., Matsui, H.,Takahashi, T., Serizawa, T., and Kohmoto, O. (1999) Cellularbasis for the acute inhibitory effects of IL-6 and TNF- alpha onexcitation-contraction coupling. J. Mol. Cell. Cardiol. 31, 1457–1467
37. Di Marco, S., Mazroui, R., Dallaire, P., Chittur, S., Tenenbaum,S. A., Radzioch, D., Marette, A., and Gallouzi, I. E. (2005)NF-kappa B-mediated MyoD decay during muscle wasting re-quires nitric oxide synthase mRNA stabilization, HuR protein,and nitric oxide release. Mol. Cell. Biol. 25, 6533–6545
38. Hamid, T., Gu, Y., Ortines, R. V., Bhattacharya, C., Wang, G.,Xuan, Y. T., and Prabhu, S. D. (2009) Divergent tumor necrosisfactor receptor-related remodeling responses in heart failure:role of nuclear factor-kappaB and inflammatory activation.Circulation 119, 1386–1397
39. Huwiler, A., Akool, E.-S., Aschrafi, A., Hamada, F. M.,Pfeilschifter, J., and Eberhardt, W. (2003) ATP potentiatesinterleukin-1 beta-induced MMP-9 expression in mesangial cellsvia recruitment of the ELAV protein HuR. J. Biol. Chem. 278,51758–51769
40. Burchfield, J. S., Iwasaki, M., Koyanagi, M., Urbich, C.,Rosenthal, N., Zeiher, A. M., and Dimmeler, S. (2008) Interleu-kin-10 from transplanted bone marrow mononuclear cells con-tributes to cardiac protection after myocardial infarction. Circ.Res. 103, 203–211
41. Hilfiker-Kleiner, D., Hilfiker, A., Fuchs, M., Kaminski, K.,Schaefer, A., Schieffer, B., Hillmer, A., Schmiedl, A., Ding, Z.,Podewski, E., Podewski, E., Poli, V., Schneider, M. D., Schulz, R.,Park, J. K., Wollert, K. C., and Drexler, H. (2004) Signaltransducer and activator of transcription 3 is required formyocardial capillary growth, control of interstitial matrix depo-sition, and heart protection from ischemic injury. Circ. Res. 95,187–195
42. Li, Z., Ma, J. Y., Kerr, I., Chakravarty, S., Dugar, S., Schreiner, G.,and Protter, A. A. (2006) Selective inhibition of p38alpha MAPKimproves cardiac function and reduces myocardial apoptosis inrat model of myocardial injury. Am. J. Physiol. 291, H1972–H1977
43. Matsumura, S., Iwanaga, S., Mochizuki, S., Okamoto, H., Ogawa,S., and Okada, Y. (2005) Targeted deletion or pharmacologicalinhibition of MMP-2 prevents cardiac rupture after myocardialinfarction in mice. J. Clin. Investig. 115, 599–609
44. Ing, D. J., Zang, J., Dzau, V. J., Webster, K. A., and Bishopric,N. H. (1999) Modulation of cytokine-induced cardiac myocyteapoptosis by nitric oxide, Bak, and Bcl-x. Circ. Res. 84, 21–33
45. Birks, E. J., Latif, N., Enesa, K., Folkvang, T., Luong le, A.,Sarathchandra, P., Khan, M., Ovaa, H., Terracciano, C. M.,Barton, P. J., Yacoub, M. H., and Evans, P. C. (2008) Elevatedp53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc. Res.79, 472–480
46. Bates, R. C., and Mercurio, A. M. (2003) Tumor necrosisfactor-alpha stimulates the epithelial-to-mesenchymal transitionof human colonic organoids. Mol. Biol. Cell 14, 1790–1800
47. Carballo, E., Cao, H., Lai, W. S., Kennington, E. A., Campbell,D., and Blackshear, P. J. (2001) Decreased sensitivity of tristet-raprolin-deficient cells to p38 inhibitors suggests the involve-ment of tristetraprolin in the p38 signaling pathway. J. Biol.Chem. 276, 42580–42587
48. Zhu, W., Brauchle, M. A., Di Padova, F., Gram, H., New, L., Ono,K., Downey, J. S., and Han, J. (2001) Gene suppression bytristetraprolin and release by the p38 pathway. Am. J. Physiol.281, L499–L508
49. Abdelmohsen, K., Srikantan, S., Kuwano, Y., and Gorospe, M.(2008) miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc. Natl. Acad. Sci. U. S. A. 105,20297–20302
Received for publication November 6, 2009.Accepted for publication February 12, 2010.
2494 Vol. 24 July 2010 KRISHNAMURTHY ET AL.The FASEB Journal � www.fasebj.org