biological rationale and current clinical experience with anti-insulin-like growth factor 1 receptor...
TRANSCRIPT

PRINCIPLES & PRACTICE OF ONCOLOGY: RECENT ADVANCES
Biological Rationale and Current Clinical ExperienceWith Anti–Insulin-Like Growth Factor 1 Receptor Monoclonal
Antibodies in Treating SarcomaTwenty Years From the Bench to the Bedside
David Olmos, MD,*† Daniel S. W. Tan, MBBS,†‡ Robin L. Jones, MBBS,*†§ and Ian R. Judson, MBBS*†
Abstract: Two decades have elapsed since insulin-like growth factor-1receptor (IGF-1R) signaling was initially implicated in sarcoma biology tothe first clinical experience of IGF-1R blockade in sarcoma. During these 21years, the IGF pathway and its key mediator IGF-1R have been implicated inthe genesis, growth, proliferation, metastasis, and resistance to conventionaltreatment in several sarcoma subtypes. In addition, IGF-1R has been vali-dated, both in vitro and in vivo, as a target for the treatment of sarcoma.Several radiologic and clinical responses to IGF-1R monoclonal antibodieshave been reported in Ewing sarcoma patients enrolled in early clinicalstudies. Furthermore, these therapies were well tolerated, and thus far severetoxicity has been rare. The early clinical evidence of antitumor activity hassupported the initiation of various phase II clinical trials in Ewing and othersarcoma subtypes, the results of which are eagerly awaited, as well as studiesassessing IGF-1R monoclonal antibodies in combination with traditionalcytotoxics or other targeted therapies. Despite these encouraging results, notall patients benefit from IGF-1R inhibition and consequently there is anurgent need for the identification of predictive markers of response.
Key Words: sarcoma, Ewing sarcoma, insulin-like growth factor-1 receptor,figitumumab, R1507, robatumumab, AMG479, SCH-717454, cixutumumab,IMC-A12
(Cancer J 2010;16: 183–194)
Sarcomas represent a diverse group of tumors that arise fromconnective tissue that account for approximately 1% of all adult
tumors, but represent up to 12% of pediatric malignancies.1–3 Ad-vances have been made in the classification, staging, and multimodaltreatment of these heterogeneous conditions including surgical ad-vances in functional preservation, the use of radiotherapy as adjunct
to other modalities and the identification of active systemic therapiesfor certain sarcoma subtypes.4–6 Despite these advances, prognosisfor these patients remains poor, and many of them will still succumbdue to the development of recurrent or refractory disease.7
The key reasons for the bleak outlook include challenges indetermining histologic classification, as well as the biologic heter-ogeneity of sarcomas. Ongoing efforts to unravel the genetic aber-rations involved in the genesis and progression of sarcomas have ledto improved delineation of subgroups with distinct molecular fea-tures. It is precisely these molecular alterations that cause or drivethese malignancies that have emerged as novel therapeutic targets.Furthermore, these genetic alterations tend to be durable, eg, KITmutations, chromosomal translocations, and coupled with the factthat tumors typically arise from a single molecular lineage, sarcomashave provided an ideal disease model for molecularly targetedtherapies. Indeed, one of the early successes in targeted therapieswas the use of imatinib, a KIT, and platelet-derived growth factorreceptor tyrosine kinase inhibitor, in the treatment of gastrointestinalstromal tumor (GIST).8,9 GIST is now known to be the commonestsarcoma in the gastrointestinal tract, but when imatinib trials beganthe association with activating KIT mutations had only recently beenreported and the disease prevalence was unclear. These early trialshave not only dramatically transformed the management and prog-nosis of GIST but also led to key advances in our understanding ofthe mechanisms of response and resistance to tyrosine kinase inhib-itors (TKIs).
In the last decade, an expanding number of exciting putativetargets have emerged, dramatically changing the therapeutic land-scape in solid tumor management. In sarcoma, a promising class ofnovel targeted agents is the monoclonal antibodies (mAbs) to theinsulin-like growth factor-1 receptor (IGF-1R). IGF-1R was firstlinked with sarcoma biology approximately 20 years ago.10,11 Sincethen, a considerable body of preclinical work has led to the intro-duction of drugs targeting this receptor in early clinical trials. Thepurpose of this manuscript is to review the biologic rationale fortargeting the insulin-like growth factor (IGF) system and the dataand clinical experience from early clinical trials with anti–IGF-1RmAbs in sarcoma.
IGF SIGNALLING SYSTEMIn vertebrates, the IGF system plays a key role in the
growth and development of normal tissues and regulates theoverall growth of organisms.12–14 This system is part of a morecomplex insulin-related signaling network, and in the evolution-ary process, the insulin-related and IGF systems have developedfrom a single, common ancestral receptor15,16 to a more complexsystem that involves 3 ligands (IGF-I, IGF-II, and insulin) and at
From the *Sarcoma Unit, The Royal Marsden NHS Foundation Trust, London;†Drug Development Unit, The Royal Marsden NHS Foundation Trust andThe institute of Cancer Research, Sutton, United Kingdom; and ‡Departmentof Medical Oncology, National Cancer Centre Singapore, Singapore. §R.L.J.is currently at the Fred Hutchinson Cancer Research Center, Seattle, WA.
This work was supported by a translational research fellowship from the SpanishSociety of Medical Oncology (SEOM) (to D.O.) and a Young InvestigatorAward from the American Society of Clinical Oncology (ASCO) Foundation(to D.S.W.T.). The Drug Development Unit of the Royal Marsden NHSFoundation Trust and the Institute of Cancer Research is supported in part bya program grant from Cancer Research U.K. Support was also provided by theExperimental Cancer Medicine Centre (to the Institute of Cancer Research)and the National Institute for Health Research Biomedical Research Centre(jointly to the Royal Marsden NHS Foundation Trust and the Institute ofCancer Research).
Reprints: David Olmos, The Royal Marsden Hospital, Downs Road, Sutton, SM25PT, United Kingdom. E-mail: [email protected].
Copyright © 2010 by Lippincott Williams & WilkinsISSN: 1528-9117/10/1603-0183
The Cancer Journal • Volume 16, Number 3, May/June 2010 www.journalppo.com | 183

least 4 receptors (IGF-1R, IGF-IIR, insulin receptor [IR], andhybrid receptors).17
IGF-1R is activated by IGF-I and IGF-II,18 whereas IGF-2Rbinds only IGF-II; although the latter receptor has no tyrosine-kinaseactivity and does not transduce any signals.19 The IR has 2 isoforms,IR-A and IR-B, the synthesis of which is controlled by alternativesplicing. IR-B, the classic IR, is activated by insulin that in turnregulates glucose uptake and has a low affinity for IGF-I. IR-A is afetal isoform, lacking exon 11, and is activated by IGF-II, leading tosubversion of apoptosis.18,20 Increasing complexity arises from thefact that both functional IGF-1R and IR are dimers and theirmonomers can also form hybrid receptors, ie, half-IGF-1R and ahalf-IR. Functionally, these hybrid receptors can be activated byIGF-I and IGF-II, and their affinity for insulin is an area of greatinterest.18 The levels and activity of IGF-1 are regulated systemi-cally by growth hormone (GH), although the primary regulatoryfactor for IGF-II remains unknown.12 In addition, the IGF systemalso includes 6 binding proteins (insulin-like growth factor bidingproteins [IGFBPs]) and IGFBP proteases.21 Both binding proteinsand proteases play a key role in regulating ligand bioavailability.Hence, the bioavailability of IGF-1 is influenced by IGFBPs, andthat of IGF-II is influenced by both IGFBPs and IGF-IIR,22 whileinsulin has direct access to its receptors. Figures 1A, B illustrate theendocrine, paracrine, and autocrine regulation of the IGF pathway.
The activation of IGF-1R by IGF-I results in proliferation andinhibition of apoptosis.12 More precisely, the binding of the ligand tothe IGF-1R causes phosphorylation of the tyrosine-kinase domainand subsequently the activation of the binding sites of a series ofdocking proteins, including the IR substrates (IRS)-1 to -4 and theSrc homology and collagen domain protein (Shc). Activation ofthese signals result in the downstream cascades involving phospha-tidylinositol-3-kinase-AKT and rat sarcoma viral oncogene ho-molog/murine sarcoma viral oncogene homolog/mitogen-activatedprotein kinase pathways.23–25 Figure 1B illustrates intracellular cas-cade of the IGF pathway.
RATIONALE FOR TARGETING IGF-1R IN SARCOMASRecent comprehensive reviews have addressed the role of the
IGF system in the genesis, maintenance, and progression of differentmalignancies, including sarcoma.26–29 Experimental30–34 and epi-demiogic35–38 studies have linked the dysregulation of the IGF-Isystem with a higher risk of developing cancer. In sarcoma, there isalso extensive experimental and clinical evidence supporting a keyrole of the IGF-1 system in tumorigenesis. Initially, the involvementof the IGF system in sarcoma was postulated on the basis ofepidemiologic data, primarily the higher incidence of some of thesediseases at younger ages. During postnatal linear growth and devel-opment, GH functions are mediated by IGF-1, leading to the pos-tulation that alterations in genes involved in GH and IGF signalingmay result in increased cell growth and risk of tumorigenesis.39
Further support for this association is provided by the observationthat the peak incidence of Ewing sarcoma coincides with the burst ofGH during puberty.40
It has been established that different mechanisms are respon-sible for the increased activity or dependency on the IGF axis invarious sarcoma subtypes, depending on the molecular pathogenesisof the individual subtype (Table 1). For example, Ewing sarcoma,which are characterized in 85% of cases by the translocation EWS/FLI-1 (Ewing sarcoma gene/Friend leukemia virus integration 1),3have been associated with enhanced IGF-1R activity, via an auto-crine/paracrine mechanism, through the inhibitory binding of theEWS/FLI-1 fusion protein to the IGFBP-3 promoter, consequentlyreducing IGBP-3 levels, and thus increasing the level of free IGF-1Rligands.41 In alveolar soft part sarcomas and desmoplastic small
round cell tumors, the respective PAX3-FKHR fusion protein andthe EWS-WT1 fusion gene have been linked to increased IGF-1Rexpression, resulting in enhanced axis signaling.42–46 An increase inIGF-II levels has also been suggested in leiomyosarcomas, synovialsarcoma, and rhabdomyosarcomas. Loss of heterozygosity (LOH)and loss of imprinting (LOI) are the common underlying geneticevents implicated in increased IGF-II transcription in the 2 majorrhabdomyosarcoma subtypes, namely alveolar and embryonal rhab-domyosarcomas47–51; whereas in synovial sarcomas, IGF-II up-regulation seems to be mediated by the characteristic SS18 fusiongenes, which have been shown to induce epigenetic changes in theIGF-II promoter region, rather than LOI that occurs infrequent-ly.52,53 LOI has also been associated with high IGF-II expression insome uterine leiomyosarcomas.54,55 In osteosarcoma, there is noclear evidence that IGF-1 or IGF-II have major involvement in thegrowth and development of this subtype,56 despite the fact that theIGF system plays an important role in the formation and homeosta-sis of bone.40 More recently, a single nucleotide polymorphism inIGF-2R has been found to be associated with an increased risk ofosteosarcoma, possibly implicating IGF-2R in the pathogenesis.57
Finally, in some wild-type and pediatric GIST, gene amplification orincreased expression of IGF-1R has been described, resulting inmuch higher levels of expression of IGF-1R in wild-type comparedwith mutant GIST.58–60 In addition, IGF-I or IGF-II have beenfound to be increased in almost all mutant GIST, with high expres-sion levels associated with a more aggressive phenotype.61
However, despite the strong etiologic link between IGF-1Rand sarcoma, the appeal of IGF-1R–targeting strategies onlyemerged after proof-of-concept preclinical studies demonstratingantitumor efficacy through disruption of IGF-1R–mediated signal-ing. A variety of approaches have been studied, both in vivo and invitro, in different sarcoma subtypes, but especially in Ewing sar-coma and rhabdomyosarcoma models. These include the use ofIGF-1R antisense oligonucleotides,62,63 dominant negative recep-tors,64 mAbs,65–67 and small-molecule TKIs.68–70 Overall, thesestrategies consistently revealed inhibition of sarcoma proliferation,reduced tumorigenesis, tumor invasion and metastasis, and ulti-mately prolonged survival.63–71
THERAPIES AGAINST IGF-1R: IGF-1R MABSEssentially, the strategies for blocking or disrupting IGF-1R
activity in patients may include (a) the reduction of ligand levels orbioactivity or (b) the inhibition of the receptor function usingreceptor-specific antibodies or small-molecule TKIs.27 At the timeof this review, mAbs against IGF-1R represent the most concreteoption in sarcoma with initial promising results in early clinicalstudies and several ongoing phase II studies.
Several anti-IGF-1R mAbs intended for clinical use havebeen developed through the humanization of mouse mAbs, im-munization of genetically engineered mice that produce fullyhuman antibodies, or the selection of specific antibodies fromphage display libraries. These antagonistic IGF-1R mAbs workthrough 2 major mechanisms: first by immediate inhibition ofligand binding, and second by a delayed effect on the down-regulation of IGF-1R. At present, 8 different mAbs (Table 2)have been tested in clinical trials: Figitumumab (also known asCP-751,871, Pfizer), AMG479 (Amgen), R1507 (also known asrobatumumab, Roche), cixutumumab (also known as IMC-A12,ImClone Systems), SCH-717454 (or 19D12, Schering-Plough),MK0646 (or h7C10, Merck), AVE-1642 (Sanofi-Aventis), andBIIB-022 (Biogen Idec).72
These mAbs are IgG1 idiotype66,73–77 with the exception offigitumumab and BIIB022, which are IgG278 and IgG479 class,respectively. There are significant pharmacokinetic and immuno-
Olmos et al The Cancer Journal • Volume 16, Number 3, May/June 2010
© 2010 Lippincott Williams & Wilkins184 | www.journalppo.com

logic differences between IgG1, IgG2, and IgG4 isotypes. First,IgG2 mAbs seem to have longer half-lives than IgG1 and IgG4mAbs.80 This is exemplified by the results of early clinical trials inwhich figitutumab, an IgG2 mAb, has been administered over longerintervals (3- or 4-weekly basis)81–83 compared with others such ascixutumumab (weekly)84 or AMG479 (2-weekly),85 both IgG1
mAbs. These isotypes also differ due to differences in their Fcfragment or their capability to activate the classic complementpathway, the complement-dependent cell-mediated cytotoxicity, andthe antibody-dependent cellular cytotoxicity (ADCC).80 IgG1 mAbsare usually potent activators of classic complement pathway, com-plement-dependent cell-mediated cytotoxicity, and ADCC, whereas
FIGURE 1. Endocrine, paracrine, and autocrine IGF pathway regulation. The endocrine, paracrine, and autocrine regulationof the IGF-1R pathway and therapeutic strategies for its disruption. A, Systemic regulation at the endocrine level. The GH-IGF-IGFBP is directed by the hypothalamus-hypophysis axis in which GH is produced and mediated by the hypothalamus GH re-leasing factors (which include GHRH and somatostatins). In the liver, GH stimulates the production and secretion of both li-gands, IGF-I and IGF-II (the latter is also regulated by genomic imprinting mechanisms), as well as the IGFBPs. The free-ligandlevels are regulated by the presence of 6 different IGFBPs. The disruption of the effector signals in the IGF pathway and endo-crine regulation has been studied at different levels. First, the disruption of the hypothalamus and hypophysis axis, and thusthe GH release inhibition, has been attempted with somatostatin analogues, ie, octreotide in a phase III123; however, this trialfailed to meet the endocrinologic and clinical endpoints. Recently, pegvisomant (Pfizer), a human recombinant GH receptorantagonist, has been tested successfully for the treatment of acromegaly.124 This pegylated recombinant human analogue ofGH can decrease the production and release of IGF-I. Cancer studies have been designed based on this effect. Other strategiesin preclinical development resulting in the reduction of the proportion of free ligand include antiligand mAbs125 or recombi-nant IGFBPs. B, Free-ligand levels at tissue level are also regulated by the presence of the different IGFBPs. Furthermore, differ-ent tissue can also produce IGFBP related proteins (IGFB-rP) which also bind, but with low affinity, to the free ligands. Both,IGFBPs and IGFB-rP levels are regulated by specific proteases. Insulin, IGF-I, and IGF-II bind the different receptors with differ-ent (IGF-1R, IGF-2R, IR-A, IR-B, and hybrid IGF-1R/IR) affinity. Each receptor triggers different intracellular signaling cascades.IGF-1R after binding IGF-I or IGF-II triggers its autophosphorylation which subsequently results in the stimulation of the adap-tator proteins IRS-1 to -4 and Shc. The former activation prompts different signaling cascades through the phosphatidylinosi-tol-3-kinase (PI3K)-AKT and the rat sarcoma viral oncogene homolog/murine sarcoma viral oncogene homolog/MEK/ERKpathways. These activated signaling cascades result in stimulation of the cell cycle and translation, leading to increased prolif-eration and growth and inhibition of apoptosis. IGF-2R ligand binding does not result in intracellular signal activation, butregulates the IGF-II and IGF-I free levels. IGF-1R also has some role in the regulation of glucose metabolism, and IR-A activa-tion has been associated with proliferation, growth, and differentiation. In addition to the above therapeutic strategies, theIGF-1R pathway could be disrupted by using anti-IGF-1R mAbs and TKIs, as described in this review. Another potential strat-egy is represented by the inhibition of downstream intracellular tyrosine kinase proteins, ie, multiple small molecule inhibitorsagainst PI3K, AKT, murine sarcoma viral oncogene homolog, and MEK are under development, with mammalian target ofrapamycin inhibitors being the most representative category in sarcoma.90
The Cancer Journal • Volume 16, Number 3, May/June 2010 IGF-1R Monoclonal Antibodies in Sarcoma
© 2010 Lippincott Williams & Wilkins www.journalppo.com | 185

IgG2 are weak activators or are unable to trigger these immunologicresponses. For example, whereas SCH 71745466 and mk064676
(both IgG1) are associated with ADCC, antibodies such as figitu-mumab (IgG2)78 or BIIB022 (IgG4) do not seem to induce anyimmunologic-mediated responses.
CLINICAL EXPERIENCE WITH IGF-1R MABS INSARCOMA
Recently, final results of 2 early studies involving theexploration of IGF-1R mAbs in sarcoma patients have beenpublished. The larger study, by Olmos et al,81 enrolled 29patients treated with figitumumab at the recommended dose of 20mg/kg every 3 weeks in an unselected cohort of sarcoma patientswith a variety of histologic subtypes (n ! 14). In addition, a 4weekly schedule at 20 mg/kg was explored in a second cohortspecifically for Ewing sarcoma (n ! 15). All but one of thesepatients had received at least 1 line of systemic chemotherapy
(median of 3 lines), and notably 6 teenage or pediatric patients(older than 12 years) were included in the sarcoma expansioncohort. The second study, by Tolcher et al,85 was a phase I studyof AMG479, which included 15 sarcoma patients, including 12Ewing sarcoma treated at the expanded dose levels of 12 and 20mg/kg every 2 weeks. In this study, patients received AMG479on days 1, 15, and 29; and this was followed by a 28-daystreatment-free period before resuming the treatment.
The initial experience with R1507 in sarcoma was firstreported at the AACR-NCI-EORTC Molecular Targets and Can-cer Therapeutics meeting in 2007. Two phase I trials werereported, one of which included a cohort of 4 Ewing sarcomapatients treated at 9 mg/kg weekly, all of whom had a partialresponse (PR) or prolonged stable disease (SD) (!12 weeks).86
The encouraging results of this phase I trial led to the SarcomaAlliance for Research through Collaboration (SARC) 011 study,a phase II study with a planned recruitment figure of approxi-
TABLE 1. Sarcoma Genetics and IGF-1R Pathway Activation
Sarcoma SubtypeCharacteristic
Cytogenetic Alteration Genetic Alteration Biological Function Relevance
Ewing sarcoma t(11;22)(q24;q12) Otherless frequentalterations
Fusion gene EWS/FLI-1Other less common
The resulting fusion oncoproteinacts as an aberranttranscriptional activator withstrong transforming capabilities
Ewing sarcoma cells are verydependent on the IGF-1Rsignaling both in vitro and invivo. EWS-FLI1 decreasedIGFBP-3 levels, which resultsin an IGF-1R activationthrough increased ligandbinding
Synovial sarcoma t(x;18)(p11;q11) Fusion gene SS18-SSX1 orSS18-SSX2
These fusion proteins may functionas transcriptional repressors
IGF-1R and IGF-II are involvedin tumor progression andrelated with the SS18-SSXinduced transformation
RhabdomyosarcomaAlveolar t(2;13)(q35;q14)
t(1;13)(p36;q14)Fusion gene PAX3-FKHR
or PAX7-FKHRThese chimeric transcription
factors increases transcriptionalactivity and target generecognition.
IGF-1R and IGF-II are involvedin the pathogenesis amongother oncogenic events, ie, asecond hit in alveolar subtypefollowing the PAX-FKHR-induced transformation. IGF-IIis increased in bothrhabdomyosarcoma subtypesand functions as an autocrinegrowth factor
Embryonal Often LOH of 11p15.5Usually complexkaryotypes
LOH of IGFII LOH in IGFII alleles resulting inincreased IGFII expression
Desmoplatic smallround cell tumor
t(11;22)(p13;q12) Fusion gene EWS-WT1 This fusion protein acts as atranscriptional activator that failsto suppress tumor growth
IGF-1R transcription is increasedby EWS-WT1 fusedtranscription factor
Gastrointestinalstromal tumors(GIST)
Complex karyotype KIT or PDGFRA activatingmutations
IGF-1R amplification rare(juvenile/childhoodGIST)
Growth factors receptor involvedin the cell signaling
IGF-1R could be expressed inwild-type GIST
IGF-I and -II expressionassociated with worseprognosis
Congenitalfibrosarcoma
t(12;15)(p13;q25) ortriosomy ofchromsome 11
Fusion gene ETV-NTRK3 This chimeric transcripts seems tohave oncogenic activity in bothmesenchymal and hematologicalcells
IGF-1R is required for theETV6-NTRK3 inducedtransformation
Leiomyosarcoma Complex karyotype Occasional LOI of IGFII Epigenetic change resulting in anincreased expression of IGF-IIdue to the loss of the normalgenomic imprinting regulationfor this gene
The PI3K pathway is highlyactivated possibly throughIGF-1R.
IGF-II could also be increase
Osteosarcoma Complex karyotype Activation of IGF-1R by IGF-Istimulates in vitro and in vivogrowth of osteosarcoma cells
Olmos et al The Cancer Journal • Volume 16, Number 3, May/June 2010
© 2010 Lippincott Williams & Wilkins186 | www.journalppo.com
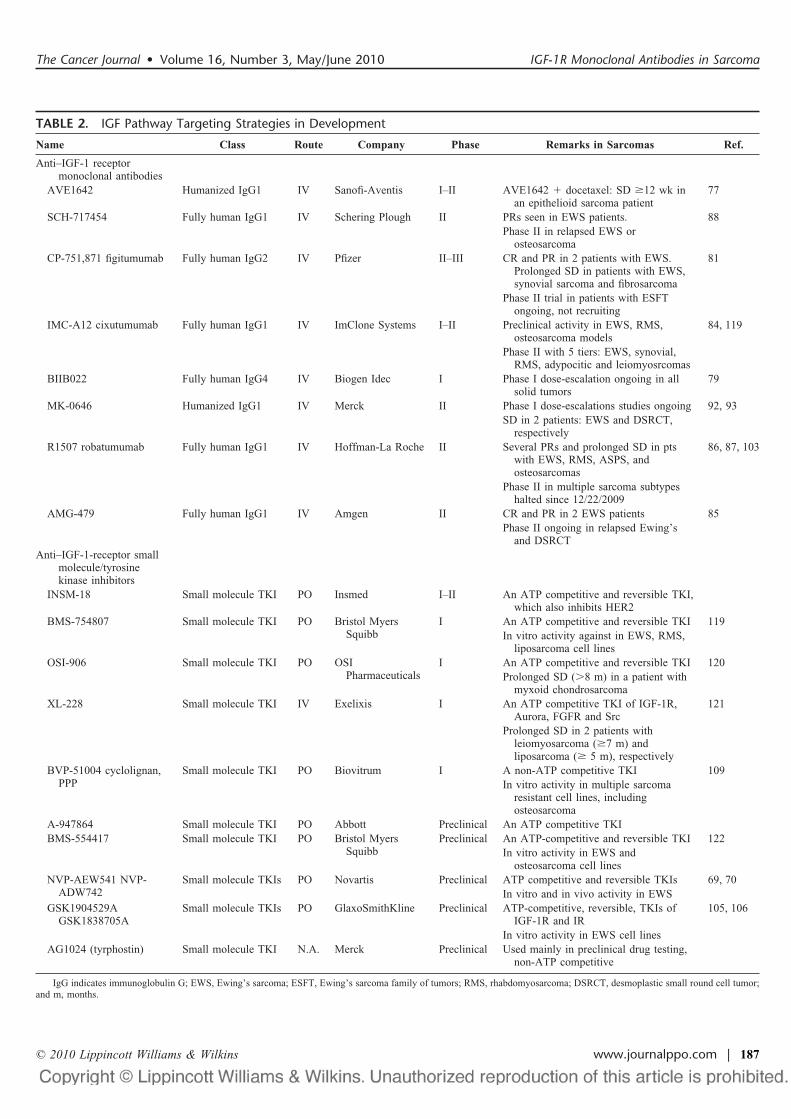
TABLE 2. IGF Pathway Targeting Strategies in Development
Name Class Route Company Phase Remarks in Sarcomas Ref.
Anti–IGF-1 receptormonoclonal antibodies
AVE1642 Humanized IgG1 IV Sanofi-Aventis I–II AVE1642 " docetaxel: SD !12 wk inan epithelioid sarcoma patient
77
SCH-717454 Fully human IgG1 IV Schering Plough II PRs seen in EWS patients. 88Phase II in relapsed EWS or
osteosarcomaCP-751,871 figitumumab Fully human IgG2 IV Pfizer II–III CR and PR in 2 patients with EWS.
Prolonged SD in patients with EWS,synovial sarcoma and fibrosarcoma
81
Phase II trial in patients with ESFTongoing, not recruiting
IMC-A12 cixutumumab Fully human IgG1 IV ImClone Systems I–II Preclinical activity in EWS, RMS,osteosarcoma models
84, 119
Phase II with 5 tiers: EWS, synovial,RMS, adypocitic and leiomyosrcomas
BIIB022 Fully human IgG4 IV Biogen Idec I Phase I dose-escalation ongoing in allsolid tumors
79
MK-0646 Humanized IgG1 IV Merck II Phase I dose-escalations studies ongoing 92, 93SD in 2 patients: EWS and DSRCT,
respectivelyR1507 robatumumab Fully human IgG1 IV Hoffman-La Roche II Several PRs and prolonged SD in pts
with EWS, RMS, ASPS, andosteosarcomas
86, 87, 103
Phase II in multiple sarcoma subtypeshalted since 12/22/2009
AMG-479 Fully human IgG1 IV Amgen II CR and PR in 2 EWS patients 85Phase II ongoing in relapsed Ewing’s
and DSRCTAnti–IGF-1-receptor small
molecule/tyrosinekinase inhibitors
INSM-18 Small molecule TKI PO Insmed I–II An ATP competitive and reversible TKI,which also inhibits HER2
BMS-754807 Small molecule TKI PO Bristol MyersSquibb
I An ATP competitive and reversible TKI 119In vitro activity against in EWS, RMS,
liposarcoma cell linesOSI-906 Small molecule TKI PO OSI
PharmaceuticalsI An ATP competitive and reversible TKI 120
Prolonged SD (#8 m) in a patient withmyxoid chondrosarcoma
XL-228 Small molecule TKI IV Exelixis I An ATP competitive TKI of IGF-1R,Aurora, FGFR and Src
121
Prolonged SD in 2 patients withleiomyosarcoma (!7 m) andliposarcoma (! 5 m), respectively
BVP-51004 cyclolignan,PPP
Small molecule TKI PO Biovitrum I A non-ATP competitive TKI 109In vitro activity in multiple sarcoma
resistant cell lines, includingosteosarcoma
A-947864 Small molecule TKI PO Abbott Preclinical An ATP competitive TKIBMS-554417 Small molecule TKI PO Bristol Myers
SquibbPreclinical An ATP-competitive and reversible TKI 122
In vitro activity in EWS andosteosarcoma cell lines
NVP-AEW541 NVP-ADW742
Small molecule TKIs PO Novartis Preclinical ATP competitive and reversible TKIs 69, 70In vitro and in vivo activity in EWS
GSK1904529AGSK1838705A
Small molecule TKIs PO GlaxoSmithKline Preclinical ATP-competitive, reversible, TKIs ofIGF-1R and IR
105, 106
In vitro activity in EWS cell linesAG1024 (tyrphostin) Small molecule TKI N.A. Merck Preclinical Used mainly in preclinical drug testing,
non-ATP competitive
IgG indicates immunoglobulin G; EWS, Ewing’s sarcoma; ESFT, Ewing’s sarcoma family of tumors; RMS, rhabdomyosarcoma; DSRCT, desmoplastic small round cell tumor;and m, months.
The Cancer Journal • Volume 16, Number 3, May/June 2010 IGF-1R Monoclonal Antibodies in Sarcoma
© 2010 Lippincott Williams & Wilkins www.journalppo.com | 187

mately 300 patients in 5 arms (Ewing sarcoma, osteosarcoma,rhabdomyosarcoma, synovial sarcoma, and others).87 Finally,another preliminary report, in this case with the mAb SCH-717454, was presented by Anderson et al88 in the 2008 AnnualCTOS Meeting. This ongoing study included patients with re-fractory/resistant Ewing sarcoma, and cohorts of other sarcomasincluding osteosarcoma, synovial sarcoma, and rhabdomyosar-coma, treated at a dose of 9 mg/kg every week.
ActivityThe first patient with sarcoma to respond to an IGF-1R mAb
was reported in the 2007 American Society of Clinical Oncologyannual meeting.89 The final results from this Phase I study of
AMG479 included a radiologic complete response (CR) maintainedfor 30 months and an additional unconfirmed PR in Ewing sarcomapatients. No other objectives responses or cases with prolonged SDwere reported.85
A larger series of sarcoma patients were treated in the figi-tumumab study.81 In the 28 patients evaluable for response, 2ongoing radiological objective responses in Ewing sarcoma patientswere observed, which included a pathological CR (21" months) ina 12-year-old boy (as illustrated in Fig. 2) and a confirmed PR (11"months) in a young adult male. In addition, 6 and 4 Ewing sarcomapatients were free of disease progression at 3 and 6 months,respectively (Fig. 3). Furthermore, 5 of these Ewing sarcoma pa-
FIGURE 2. A 12-year-old patient with metastatic Ewing sarcoma (from pelvic bone) treated with figitumumab 20 mg/kg ev-ery 4 weeks. Patient progressed on first-line vincristine, ifosfamide, doxorubicin, and etoposide, and subsequently receivedhigh-dose (HD) melphalan plus busulfan with peripheral blood stem cells rescue achieving an incomplete response (non CR).Further pelvic irradiation with 54 Gy in 30 fractions resulted in complete local control. Patient progressed with new lung me-tastases detected just 8 months after HD and peripheral blood stem cells. A, Baseline computed tomography (CT)-scan at pre-sentation (prefigitumumab) with a target right hilar lesion (3.2 cm in longest Ø) and multiple subcentimetre lung metastaticnodules (data not shown). B, CT scan after 6 cycles of figitumumab, size of hilar mass reduced by about 60%, with remainingcentral calcification. Other lung nodules were completely eradicated (images not shown). C, CT scan after 24 cycles of figitu-mumab showed maintained complete response (white arrow pointed prior target lesion localization). Pathologic complete re-sponse was confirmed after 12 cycles. D, High magnification of a hematoxilin-eosin (H-E) staining showing a uniform popula-tion of small round-to-oval cells with a high nuclear to cytoplasmic ratio typical for Ewing sarcoma. E, High magnification ofstrong membranous immunohistochemical staining for CD99 (also known as MIC2 or single-chain type-1 glycoprotein; a pro-tein nearly universally expressed by Ewing’s sarcomas). F, A representative dual-color fluorescent in situ hybridization EWSbreak apart assay result: the encircled cell shows a pair of probe signals (orange: 5$ EWSR1; green: 3$ EWSR1) split apart dueto an EWS (22q 12) region rearrangement, whereas the other pair are intact with normal fused signals. G, RT-PCR run on 2%agarose gel electrophoresis stained with ethidium bromide. Bands from left to right showed an EWS/FLI-1 type 1 (Exons 7–6fusion) positive control; an EWS/FLI-1 type 2 (Exon 7–5 fusion) positive control; patient sample (tumor mRNA) showing a EWS-FLI-1 type 1 fusion; negative control that did not contain RNA; last band is the DNA marker. "-Actin transcripts were also co-amplified as an internal positive control for each sample. Adapted from Lancet Oncol. 2010;11:129–135.
Olmos et al The Cancer Journal • Volume 16, Number 3, May/June 2010
© 2010 Lippincott Williams & Wilkins188 | www.journalppo.com

tients with prolonged SD had shrinkage of the target tumor lesions.Small decreases in tumor size were also seen in patients withchondrosarcoma, fibrosarcoma, and synovial sarcoma. Overall, thenonprogression rate at 3 months was 34% (95% CI 10–58) and at 6months was 28% (95% CI 5–51) for all sarcoma patients included inthe study. However, being a phase I expansion cohort, the study wasnot powered to formally detect antitumor activity as an end point.81
However, the reported antitumor activity of figitumumab, at least inEwing sarcoma, compares favorably with those of other targetedagents in sarcoma. Preliminary data for the mammalian target ofrapamycin (mTOR) inhibitor, ridaforolimus (previously known asdeforolimus), have shown a nonprogression rate of 30% at 16 weeksin bone sarcomas.90 The mTOR inhibitor was deemed active and aphase III trial comparing ridaforolimus with placebo, as mainte-nance therapy, has recently completed recruitment. Other targetedagents have also been explored, and in a recent trial of varioussarcoma subtypes treated with imatinib, no activity was seen inpatients with Ewing sarcoma, with no objective responses and nopatient remaining on the drug longer than 4 months.91
Supporting the preliminary activity reported with R1507 of 2PRs in the phase I study,86 multiple radiologic responses in Ewingsarcoma and other sarcoma subtypes (including rhabdomyosarcoma,osteosarcoma, and alveolar soft part sarcoma) have been reported inthe initial SARC011 study presentation at the 2009 AmericanSociety of Clinical Oncology Annual Meeting.87 The final results ofthis study are eagerly anticipated. Finally, 3 additional PRs in Ewingsarcoma and a prolonged SD in an osteosarcoma patient wereobserved in the ongoing SCH-717454 study.88
ToxicityIn general, IGF-1R mAbs are well tolerated drugs, the most
common toxicities being mild and occasionally moderate. Severe(grade 3) or life-threatening (grade 4) adverse events are rare. Thecurrent reported adverse event profile, in all patients treated withIGF-1R mAbs in early clinical studies, with the exception offigitumumab in which specific toxicity data for sarcoma have beenreported,81 are summarized in Table 3.
Grade 2 lymphopenia was the most common hematologicadverse event observed with figitumumab81 in sarcoma patients.Other potential grade 3 and 4 hematologic adverse events reported inthe initial Phase I trials included thrombocytopenia.70,85,87,92 Grade3 and 4 nonhematologic adverse events with figitumumab in sar-coma patients included deep venous thrombosis (n ! 1), vomiting(n ! 1), and back pain (n ! 1). Grade 3 fatigue was also describedwith figitumumab in nonsarcoma patients.82,83 Other relevant grade3–4 nonhematologic adverse events described with other IGF-1RmAbs include fatigue,84,85,93 arthralgia,85 chills,93 pneumonitis,92
nausea or vomiting,93 rash and/or pruritus,93 pain,88,93 gastrointesti-nal bleeding,92 and dyspnea.87,88
In addition, hyperglycemia grade 3 (13.9–27.8 mmol/L) hasbeen reported with MK2206,93 IMC-A12,84 and SCH-717454,88
while mild (6.5–8.9 mmol/L) to moderate (9.0–13.9 mmol/L) hy-perglycemia has been described with the other IGF-1R antibod-ies.77,85,87 Mild hyperglycemia was also reported in 4 sarcomapatients treated with figitumumab and an additional patient devel-oped grade 2 hyperglycaemia requiring metformin and glibencam-ide.81 The mechanism for this hyperglycaemia is unclear, althoughIGF-1R may be involved in glucose metabolism via cross-talk andheterodimerization with the IR.94–97 This observation and the in-creased plasma insulin levels reported after treatment with IGF-1RmAbs83,98 suggest compensatory insulin secretion and associatedinsulin resistance, the latter possibly secondary to increased IGF-1and growth-hormone levels.27,99 Other severe laboratory abnormal-ities observed in sarcoma patients included uric acid elevation andtransaminitis.81
Interestingly, despite the expression of IGF-1R in vascularsmooth muscle and endothelial cells100 and the potential cardiotox-icity associated with mAbs, no cardiac toxicity has been reported todate. In the case of sarcoma patients treated with figitumumab, it isnoteworthy that three quarters of the patients were pretreated withanthracyclines and none developed cardiotoxicity.81
Theoretically, IGF-1R mAbs would be expected to have aninhibitory effect on IGF and GH-mediated growth. Thus, IGF-1Rblockade could cause linear and somatic growth delay in a childhoodand teenage population, as supported by the identification of patientswith genetic defects in the IGF-1 axis such as IGF-1 deficiency.101
This potential long-term adverse event is extremely important in themanagement of sarcoma patients, which tend to affect a youngpopulation.1 Current clinical experience is too limited to definitivelyaddress this question.81 Detailed assessments of growth and hor-mone levels have been included in ongoing phase II trials recruitingpediatric and prepubertal teenage patients, and it is hoped that thesestudies will provide insights to the effect of IGF-1R targeted therapyon growth during childhood and puberty.
Ongoing StudiesIn addition to the previously described SARC011 study, other
2 single-agent phase II studies with IGF-1R mAbs have completedrecruitment and final results are anticipated: (1) a study of figitu-mumab in Ewing sarcoma family of tumors (www.clinicaltrials.gov,NCT00560235) with approximately 130 patients, including pediat-ric patients 10 years or older and (2) a study of AMG479 in patientsaged 16 years or older with Ewing sarcoma or desmoplastic small
FIGURE 3. Waterfall plot of best response by RECIST forfigitumumab. Waterfall plot representing responses, in per-cent decrease of tumor size by RECIST criteria, of the targetlesions in the 22 evaluable patients. The white bars representpatients with increase in target lesion size; the gray bars rep-resent patients with a decrease in size of the target lesions.The numbers above/below the bars represent the number oftreatment cycles administered to patients before their with-drawal due to disease progression. ‡PR in soft tissue diseasebut PD with a new bone lesion. *Patients still on study. **Pa-tient with a partial response (PR) after 6 cycles by RECISTdue to residual calcification (Fig. 2), then confirmed patho-logic response. ***Patient with a PR after 2 cycles of treat-ment and radiotherapy to the mediastinum. Adapted fromLancet Oncol. 2010;11:129–135.
The Cancer Journal • Volume 16, Number 3, May/June 2010 IGF-1R Monoclonal Antibodies in Sarcoma
© 2010 Lippincott Williams & Wilkins www.journalppo.com | 189

TABLE 3. Reported Adverse Effects and Maximum Grade With Anti-IGF-1R Monoclonal Antibodies
Toxicity/Adverse Event (NCICTC AE Version 3.0)
Figitumumab CP-751,87181–83,98
RobatumumabR150786, 87 AMG47985
CixututumumabIMC-A1284 SCH-71745488 MK-064692,93
AVE164277 (Only1st Cycle)
Hematologic/bone marrowLymphopenia G2 G3Anemia G2 G2 G1/2Thrombocytopaenia G4 G3 G4
Allergic/hypersensivityHypersensivity reactions G1/2 G1/2
CardiacHypertension G2
Constitutional symptomsAnorexia G2 G2 G2 G1/2 G3Weight loss G2 G2 G1/2Fatigue G3 G2 G3 G1/2 G2 G1/2Pyrexia G1 G3Chills/rigors G2 G3
Dermatology/skinRash/urticaria/eczema G2 G2 G2 G1/2 G2 G3 G1/2Bruising G1Injection site reaction G2Purpura G2
EndocrineHypothyroidism G2
GastrointestinalNausea G2 G2 G2 G2 G3Vomiting G3 G2Diarrhea G2 G2 G3Dysgeusia G1Dyspepsia G2Mucositis/stomatitis G1Constipation G3
Hemorrhage/bleedingAdrenal hemorrhage G3Gastrintestinal bleeding G3
InfectionSkin infection G2Oral/vaginal Candidiasis G2
Lab abnormalitiesHyperglycemia G3 G3 G3 G3 G3 G3 G1/2Elevated ALT/AST G4 G3 G3Elevated GGT G4Hyperbilirubinemia G3Hyperuricemia G4Hypophosphatemia G2Elevated creatinine G2 G1/2Proteinuria G4
LymphaticsPeripheral edema G1/2
MusculoskeletalLimb cramps G2Muscular weakness G2
NeurologyDizziness G1 G1 G1/2Paresthesia G1/2Tremor G1
(Continued)
Olmos et al The Cancer Journal • Volume 16, Number 3, May/June 2010
© 2010 Lippincott Williams & Wilkins190 | www.journalppo.com

round cell tumors (www.clinicaltrials.gov, NCT00563680) with anestimated enrolment of 38 patients. In addition to these studies, 2other phase II trials are still recruiting patients: (1) a study ofSCH-717454 in osteosarcoma and Ewing sarcoma with patientsaged 4 years or older (www.clinicaltrials.gov, NCT00617890) witha planned recruitment of 190 patients; and (2) a study of cixutu-mumab (www.clinicaltrials.gov, NCT00668148) with 185 patients(!12 years) and 5 arms: Ewing sarcoma, rhabdomyosarcoma,leiomyosarcoma, adipocytic sarcomas, and synovial sarcoma.
Despite these promising results and ongoing trials in sarcoma,the disappointing initial results of the randomized phase III trialswith figitumumab and chemotherapy in lung cancer have led topremature study closure, representing a setback to the developmentof IGF-1R mAbs in cancer.102 Indeed, following figitumumab re-sults in lung cancer, Roche103 has recently decided to halt furtherdevelopment of the anti–IGF-1R mAb R1507 in all tumor types. Itremains uncertain if this decision will impact on the final outcomesreport of the SARC011 trial, as it is likely to be pivotal to the futuredevelopment of this family of targeted therapies in sarcoma, espe-cially in Ewing sarcoma.
FUTURE DIRECTIONS IN IGF-1R TARGETING INSARCOMA
Tyrosine Kinase Inhibitors of IGF1-RThere are a number of small molecule TKIs of IGF-1R
currently undergoing clinical evaluation (Table 2). Some of thesesmall molecules also inhibit IR-A, a component of IGF-R hybridreceptors.72 Although this can potentially result in greater antitumoractivity, it may also be associated with a higher incidence ofmetabolic toxicity. From the results of clinical trials of mAbs andTKIs in other tumor types, it is apparent that predicting differencesin efficacy between these 2 classes can be difficult.104 Notably, smallmolecule TKIs do not directly activate the immune response againsttumor cells but may be more effective when activated receptors arelocalized in cytoplasmic caveosomes or endosomes.
Some of these novel IGF-1R TKIs (ie, picropodophylin,GSK183870A, GSK1904529A, BMS-536924, and NVP-AEW541)have already shown promising preclinical activity as single agents or
in combination with different sarcoma models.68–70,105–109 However,clinical differences in sarcoma patients between these 2 mechanismsof IGF-1R blockade currently remain undefined.
IGF-1R mAb CombinationsIGF-1R activation has been associated with chemoresistance
in multiple cancers,110 including some sarcomas such as Ewingsarcoma.11 Indeed, modulation of IGF signaling has been shown toenhance the antitumor activity of cytotoxic drugs in laboratorysarcoma models.111 Thus, a strategy based on the combination offirst- or second-line sarcoma chemotherapy with IGF-1R mAbsseems to be a rational approach in the utilization of these agents.Currently, there are a number of ongoing or planned studies evalu-ating such combinations, including a phase I/II trial of cixutumumabin combination with doxorubicin for advanced and unresectable softtissue sarcomas (www.clinicaltrials.gov, NCT00720174), sponsoredby the National Cancer Institute; and a phase I of SCH-717454 incombination with different commonly used chemotherapies in sar-coma such as vincristine, doxorubicin, and cyclophosphamide orifosfamide and etoposide (www.clinicaltrials.gov, NCT00960063).
Furthermore, clinical studies of rational combinations ofIGF-1R mAbs with other targeted therapies are in progress. Exam-ples of such regimens are the use of mTOR inhibitors in combina-tion with IGF-1R antibodies.112,113 Studies evaluating this approachinclude a trial of temsirolimus with cixutumumab in sarcomas,sponsored by the Memorial Sloan Kettering Cancer Centre and theNational Cancer Institute (www.clinicaltrials.gov, NCT01016015)and a trial of RAD001 (everolimus) in combination with figitumumabsponsored by the Dana-Faber Cancer Institute (www.clinicaltrials.gov,NCT 00927966). Other rational combinations could include regimenswith Heat shock protein 90114 or EGFR/HER2 inhibitors,107 becausethese have been implicated as potential mechanisms of IGF-1Rinhibition resistance in sarcoma cell lines.
Patient SelectionDespite robust preclinical evidence supporting the role of
IGF-1R targeted agents in Ewing sarcoma, clinical results show thatonly a proportion of patients derive significant benefit, with manyprogressing or developing resistance to IGF-1R mAbs quickly.
TABLE 3. (Continued)
Toxicity/Adverse Event (NCICTC AE Version 3.0)
Figitumumab CP-751,87181–83,98
RobatumumabR150786, 87 AMG47985
CixututumumabIMC-A1284 SCH-71745488 MK-064692,93
AVE164277 (Only1st Cycle)
Ocular/earDry eye G2Deafness/discomfort G2
PainArthralgia G3Back pain G3 G2 G3 G2Headache G2 G2 G2Myalgia G2 G1
Renal/urinaryPoliakiuria G2Difficult urination G1
RespiratoryDyspnea G3Pneumonitis G3
VascularDVT/pulmonary embolism G3 G4Cerebrovascular accident G3
The Cancer Journal • Volume 16, Number 3, May/June 2010 IGF-1R Monoclonal Antibodies in Sarcoma
© 2010 Lippincott Williams & Wilkins www.journalppo.com | 191

Although initial reports suggested an association between the EWS/FLI-1 type 1 translocation and response in Ewing sarcoma,85 thepurported predictive value of translocation type has not been ob-served consistently.81 Further evaluation of predictive biomarkersfor IGF-1R targeting drugs needs to be pursued. Defining putativepredictive biomarkers—and eventual companion diagnostics—forIGF-1R therapy should be a priority for the further development ofthese agents. Although IGF-1R71 protein expression was not as-sessed in initial studies, preliminary reports suggested that expres-sion alone might not be sufficiently predictive for clinical benefitfrom IGF-1R targeting drugs; and it is likely that a suite of biomar-kers, both in the host and in the tumor,115 will be required for patientselection. Additional biomarker candidates for study may includeIGF-II,27 IRS-1116,117 or IRS-2117 expression, IGF-1R or IGF-2Rpolymorphisms,39,57 and constitutional activation of other relevantplayers in the IGF-1R downstream pathway.71,114 Larger, correlativestudies, coupled with clinical response data, such as the SARC011parallel biomarker study, which involved collecting tumor tissue andblood samples, will hopefully address some of these questions.
CONCLUSIONSThere is a large amount of preclinical data supporting the use
of agents targeting IGF-1R in sarcomas. This rationale has beenconfirmed by the early report of clinical activity with severalIGF-1R antibodies in Ewing sarcoma, as well as other sarcomasubtypes. However, the benefit of this therapeutic approach does notextend to all patients and there is an urgent need to identifypredictive biomarkers to improve patient selection, as well as toelucidate the mechanisms of resistance to these drugs, therebyfacilitating the development of rational combination regimens. De-spite the former, this novel group of drugs constitutes a promisingtherapy for sarcoma, especially in Ewing sarcoma, and has thepotential to drastically change the outcome of this devastatingdisease.
REFERENCES1. Horner MJ, Ries LA, Krapcho M, et al., eds. SEER Cancer Statistics Review,
1975–2006. Bethesda, MD: National Cancer Institute; 2009. Available at:http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER datasubmission, posted to the SEER web site in 2009.
2. Miller RW, Young JL Jr, Novakovic B. Childhood cancer. Cancer. 1995;75:395–405.
3. Fletcher CDM, Unni KK, Mertens F. World Health Organisation Classifi-cation of Tumours: Pathology and Genetics of Tumours of Soft Tissue andBone. Lyon: IARC Press; 2002.
4. Patel SJ, Lynch JW Jr, Johnson T, et al. Dose-intense ifosfamide/doxorubi-cin/cisplatin based chemotherapy for osteosarcoma in adults. Am J ClinOncol. 2002;25:489–495.
5. Hensley ML, Maki R, Venkatraman E, et al. Gemcitabine and docetaxel inpatients with unresectable leiomyosarcoma: results of a phase II trial. J ClinOncol. 2002;20:2824–2831.
6. Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinas-cidin-743) in advanced pretreated myxoid liposarcomas: a retrospectivestudy. Lancet Oncol. 2007;8:595–602.
7. Hartmann JT. Systemic treatment options for patients with refractory adult-type sarcoma beyond anthracyclines. Anticancer Drugs. 2007;18:245–254.
8. van Oosterom AT, Judson I, Verweij J, et al. Safety and efficacy of imatinib(STI571) in metastatic gastrointestinal stromal tumours: a phase I study.Lancet. 2001;358:1421–1423.
9. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastro-intestinal stromal tumours with high-dose imatinib: randomised trial. Lancet.2004;364:1127–1134.
10. El-Badry OM, Minniti C, Kohn EC, et al. Insulin-like growth factor II actsas an autocrine growth and motility factor in human rhabdomyosarcomatumors. Cell Growth Differ. 1990;1:325–331.
11. Yee D, Favoni RE, Lebovic GS, et al. Insulin-like growth factor I expressionby tumors of neuroectodermal origin with the t(11;22) chromosomal trans-location. A potential autocrine growth factor. J Clin Invest. 1990;86:1806–1814.
12. Jones JI, Clemmons DR. Insulin-like growth factors and their bindingproteins: biological actions. Endocr Rev. 1995;16:3–34.
13. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin andIGF-I receptors. Endocr Rev. 2001;22:818–835.
14. De Meyts P, Whittaker J. Structural biology of insulin and IGF1 receptors:implications for drug design. Nat Rev Drug Discov. 2002;1:769–783.
15. Brogiolo W, Stocker H, Ikeya T, et al. An evolutionarily conserved functionof the Drosophila insulin receptor and insulin-like peptides in growthcontrol. Curr Biol. 2001;11:213–221.
16. Dong MQ, Venable JD, Au N, et al. Quantitative mass spectrometryidentifies insulin signaling targets in C. elegans. Science. 2007;317:660–663.
17. Ryan PD, Goss PE. The emerging role of the insulin-like growth factorpathway as a therapeutic target in cancer. Oncologist. 2008;13:16–24.
18. Belfiore A. The role of insulin receptor isoforms and hybrid insulin/IGF-Ireceptors in human cancer. Curr Pharm Des. 2007;13:671–686.
19. Oates AJ, Schumaker LM, Jenkins SB, et al. The mannose 6-phosphate/insulin-like growth factor 2 receptor (M6P/IGF2R), a putative breast tumorsuppressor gene. Breast Cancer Res Treat. 1998;47:269–281.
20. Cohen P. The twentieth century struggle to decipher insulin signalling. NatRev Mol Cell Biol. 2006;7:867–873.
21. Yu H, Rohan T. Role of the insulin-like growth factor family in cancerdevelopment and progression. J Natl Cancer Inst. 2000;92:1472–1489.
22. Firth SM, Baxter RC. Cellular actions of the insulin-like growth factorbinding proteins. Endocr Rev. 2002;23:824–854.
23. Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int JCancer. 2003;107:873–877.
24. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream.Cell. 2007;129:1261–1274.
25. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways medi-ated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912.
26. Hartog H, Wesseling J, Boezen HM, et al. The insulin-like growth factor 1receptor in cancer: old focus, new future. Eur J Cancer. 2007;43:1895–1904.
27. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. NatRev Cancer. 2008;8:915–928.
28. Kim SY, Toretsky JA, Scher D, et al. The role of IGF-1R in pediatricmalignancies. Oncologist. 2009;14:83–91.
29. Rikhof B, de Jong S, Suurmeijer AJ, et al. The insulin-like growth factorsystem and sarcomas. J Pathol. 2009;217:469–482.
30. Macaulay VM. Insulin-like growth factors and cancer. Br J Cancer. 1992;65:311–320.
31. Angelloz-Nicoud P, Binoux M. Autocrine regulation of cell proliferation bythe insulin-like growth factor (IGF) and IGF binding protein-3 proteasesystem in a human prostate carcinoma cell line (PC-3). Endocrinology.1995;136:5485–5492.
32. Oku K, Tanaka A, Yamanishi H, et al. Effects of various growth factors ongrowth of a cloned human esophageal squamous cancer cell line in aprotein-free medium. Anticancer Res. 1991;11:1591–1595.
33. Frostad S, Bruserud O. In vitro effects of insulin-like growth factor-1(IGF-1) on proliferation and constitutive cytokine secretion by acute my-elogenous leukemia blasts. Eur J Haematol. 1999;62:191–198.
34. Singh P, Dai B, Yallampalli U, et al. Proliferation and differentiation of ahuman colon cancer cell line (CaCO2) is associated with significant changesin the expression and secretion of insulin-like growth factor (IGF) IGF-IIand IGF binding protein-4: role of IGF-II. Endocrinology. 1996;137:1764–1774.
35. Chan JM, Stampfer MJ, Giovannucci E, et al. Plasma insulin-like growthfactor-I and prostate cancer risk: a prospective study. Science. 1998;279:563–566.
36. Hankinson SE, Willett WC, Colditz GA, et al. Circulating concentrations ofinsulin-like growth factor-I and risk of breast cancer. Lancet. 1998;351:1393–1396.
37. Ma J, Pollak MN, Giovannucci E, et al. Prospective study of colorectalcancer risk in men and plasma levels of insulin-like growth factor (IGF)-Iand IGF-binding protein-3. J Natl Cancer Inst. 1999;91:620–625.
38. Yu H, Spitz MR, Mistry J, et al. Plasma levels of insulin-like growth factor-Iand lung cancer risk: a case-control analysis. J Natl Cancer Inst. 1999;91:151–156.
39. Scotlandi K, Picci P. Targeting insulin-like growth factor 1 receptor insarcomas. Curr Opin Oncol. 2008;20:419–427.
40. Gorlick R, Anderson P, Andrulis I, et al. Biology of childhood osteogenicsarcoma and potential targets for therapeutic development: meeting sum-mary. Clin Cancer Res. 2003;9:5442–5453.
41. Prieur A, Tirode F, Cohen P, Delattre O. EWS/FLI-1 silencing and geneprofiling of Ewing cells reveal downstream oncogenic pathways and acrucial role for repression of insulin-like growth factor binding protein 3.Mol Cell Biol. 2004;24:7275–7283.
42. Khan J, Bittner ML, Saal LH, et al. cDNA microarrays detect activation ofa myogenic transcription program by the PAX3-FKHR fusion oncogene.Proc Natl Acad Sci USA. 1999;96:13264–13269.
43. Ayalon D, Glaser T, Werner H. Transcriptional regulation of IGF-I receptor
Olmos et al The Cancer Journal • Volume 16, Number 3, May/June 2010
© 2010 Lippincott Williams & Wilkins192 | www.journalppo.com

gene expression by the PAX3-FKHR oncoprotein. Growth Horm IGF Res.2001;11:289–297.
44. Karnieli E, Werner H, Rauscher FJ III, et al. The IGF-I receptor genepromoter is a molecular target for the Ewing’s sarcoma-Wilms’ tumor 1fusion protein. J Biol Chem. 1996;271:19304–19309.
45. Finkeltov I, Kuhn S, Glaser T, et al. Transcriptional regulation of IGF-Ireceptor gene expression by novel isoforms of the EWS-WT1 fusion protein.Oncogene. 2002;21:1890–1898.
46. Werner H, Idelman G, Rubinstein M, et al. A novel EWS-WT1 gene fusionproduct in desmoplastic small round cell tumor is a potent transactivator ofthe insulin-like growth factor-I receptor (IGF-IR) gene. Cancer Lett. 2007;247:84–90.
47. Pedone PV, Tirabosco R, Cavazzana AO, et al. Mono- and bi-allelicexpression of insulin-like growth factor II gene in human muscle tumors.Hum Mol Genet. 1994;3:1117–1121.
48. Scrable H, Cavenee W, Ghavimi F, et al. A model for embryonal rhabdo-myosarcoma tumorigenesis that involves genome imprinting. Proc NatlAcad Sci USA. 1989;86:7480–7484.
49. Visser M, Sijmons C, Bras J, et al. Allelotype of pediatric rhabdomyosar-coma. Oncogene. 1997;15:1309–1314.
50. Zhan S, Shapiro DN, Helman LJ. Activation of an imprinted allele of theinsulin-like growth factor II gene implicated in rhabdomyosarcoma. J ClinInvest. 1994;94:445–448.
51. Anderson J, Gordon A, Pritchard-Jones K, et al. Genes, chromosomes, andrhabdomyosarcoma. Genes Chromosomes Cancer. 1999;26:275–285.
52. Sun Y, Gao D, Liu Y, et al. IGF2 is critical for tumorigenesis by synovialsarcoma oncoprotein SYT-SSX1. Oncogene. 2006;25:1042–1052.
53. de Bruijn DR, Allander SV, van Dijk AH, et al. The synovial-sarcoma-associated SS18-SSX2 fusion protein induces epigenetic gene (de)regula-tion. Cancer Res. 2006;66:9474–9482.
54. Gloudemans T, Pospiech I, Van Der Ven LT, et al. Expression and CpGmethylation of the insulin-like growth factor II gene in human smoothmuscle tumors. Cancer Res. 1992;52:6516–6521.
55. Vu TH, Yballe C, Boonyanit S, et al. Insulin-like growth factor II in uterinesmooth-muscle tumors: maintenance of genomic imprinting in leiomyomataand loss of imprinting in leiomyosarcomata. J Clin Endocrinol Metab.1995;80:1670–1676.
56. Benini S, Baldini N, Manara MC, et al. Redundancy of autocrine loops inhuman osteosarcoma cells. Int J Cancer. 1999;80:581–588.
57. Savage SA, Woodson K, Walk E, et al. Analysis of genes critical for growthregulation identifies Insulin-like Growth Factor 2 Receptor variations withpossible functional significance as risk factors for osteosarcoma. CancerEpidemiol Biomarkers Prev. 2007;16:1667–1674.
58. Tarn C, Rink L, Merkel E, et al. Insulin-like growth factor 1 receptor is apotential therapeutic target for gastrointestinal stromal tumors. Proc NatlAcad Sci USA. 2008;105:8387–8392.
59. Prakash S, Sarran L, Socci N, et al. Gastrointestinal stromal tumors inchildren and young adults: a clinicopathologic, molecular, and genomicstudy of 15 cases and review of the literature. J Pediatr Hematol Oncol.2005;27:179–187.
60. Agaram NP, Laquaglia MP, Ustun B, et al. Molecular characterization ofpediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14:3204–3215.
61. Braconi C, Bracci R, Bearzi I, et al. Insulin-like growth factor (IGF) 1 and2 help to predict disease outcome in GIST patients. Ann Oncol. 2008;19:1293–1298.
62. Scotlandi K, Maini C, Manara MC, et al. Effectiveness of insulin-likegrowth factor I receptor antisense strategy against Ewing’s sarcoma cells.Cancer Gene Ther. 2002;9:296–307.
63. Shapiro DN, Jones BG, Shapiro LH, et al. Antisense-mediated reduction ininsulin-like growth factor-I receptor expression suppresses the malignantphenotype of a human alveolar rhabdomyosarcoma. J Clin Invest. 1994;94:1235–1242.
64. Scotlandi K, Avnet S, Benini S, et al. Expression of an IGF-I receptordominant negative mutant induces apoptosis, inhibits tumorigenesis andenhances chemosensitivity in Ewing’s sarcoma cells. Int J Cancer. 2002;101:11–16.
65. Scotlandi K, Benini S, Nanni P, et al. Blockage of insulin-like growthfactor-I receptor inhibits the growth of Ewing’s sarcoma in athymic mice.Cancer Res. 1998;58:4127–4131.
66. Kolb EA, Gorlick R, Houghton PJ, et al. Initial testing (stage 1) of amonoclonal antibody (SCH 717454) against the IGF-1 receptor by thepediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:1190–1197.
67. Kalebic T, Tsokos M, Helman LJ. In vivo treatment with antibody againstIGF-1 receptor suppresses growth of human rhabdomyosarcoma and down-regulates p34cdc2. Cancer Res. 1994;54:5531–5534.
68. Scotlandi K, Manara MC, Nicoletti G, et al. Antitumor activity of theinsulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 inmusculoskeletal tumors. Cancer Res. 2005;65:3868–3876.
69. Martins AS, Mackintosh C, Martin DH, et al. Insulin-like growth factor Ireceptor pathway inhibition by ADW742, alone or in combination withimatinib, doxorubicin, or vincristine, is a novel therapeutic approach inEwing tumor. Clin Cancer Res. 2006;12:3532–3540.
70. Manara MC, Landuzzi L, Nanni P, et al. Preclinical in vivo study of newinsulin-like growth factor-I receptor–specific inhibitor in Ewing’s sarcoma.Clin Cancer Res. 2007;13:1322–1330.
71. Cao L, Yu Y, Darko I, et al. Addiction to elevated insulin-like growth factorI receptor and initial modulation of the AKT pathway define the respon-siveness of rhabdomyosarcoma to the targeting antibody. Cancer Res.2008;68:8039–8048.
72. Gualberto A, Pollak M. Emerging role of insulin-like growth factor receptorinhibitors in oncology: early clinical trial results and future directions.Oncogene. 2009;28:3009–3021.
73. Beltran PJ, Mitchell P, Chung YA, et al. AMG 479, a fully humananti-insulin-like growth factor receptor type I monoclonal antibody, inhibitsthe growth and survival of pancreatic carcinoma cells. Mol Cancer Ther.2009;8:1095–1105.
74. Rodon J, Patnaik A, Stein M, et al. A phase I study of q3W R1507, a humanmonoclonal antibody IGF-1R antagonist in patients with advanced cancer(Meeting Abstracts). J Clin Oncol. 2007;25:3590.
75. Burtrum D, Zhu Z, Lu D, et al. A fully human monoclonal antibody to theinsulin-like growth factor I receptor blocks ligand-dependent signaling andinhibits human tumor growth in vivo. Cancer Res. 2003;63:8912–8921.
76. Goetsch L, Gonzalez A, Leger O, et al. A recombinant humanized anti-insulin-like growth factor receptor type I antibody (h7C10) enhances theantitumor activity of vinorelbine and anti-epidermal growth factor receptortherapy against human cancer xenografts. Int J Cancer. 2005;113:316–328.
77. Tolcher AW, Patnaik A, Till E, et al. A phase I study of AVE1642, ahumanized monoclonal antibody IGF-1R (insulin like growth factor1 recep-tor) antagonist, in patients (pts) with advanced solid tumor (ST) (MeetingAbstracts). J Clin Oncol. 2008;26:3582.
78. Cohen BD, Baker DA, Soderstrom C, et al. Combination therapy enhancesthe inhibition of tumor growth with the fully human anti-type 1 insulin-likegrowth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res.2005;11:2063–2073.
79. von Mehren M, Britten C, Piesolor P, et al. A phase 1, dose-escalation studyof BIIB022 (anti-IGF-1R monoclonal antibody) in patients with relapsed orrefractory solid tumours. EJC Suppl. 2009;7:128.
80. Presta LG. Molecular engineering and design of therapeutic antibodies. CurrOpin Immunol. 2008;20:460–470.
81. Olmos D, Postel-Vinay S, Molife LR, et al. Safety, pharmacokinetics, andpreliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871)in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohortstudy. Lancet Oncol. 2010;11:129–135.
82. Haluska P, Worden F, Olmos D, et al. Safety, tolerability, and pharmaco-kinetics of the anti-IGF-1R monoclonal antibody figitumumab in patientswith refractory adrenocortical carcinoma. Cancer Chemother Pharmacol.2010;65:765–773.
83. Haluska P, Shaw HM, Batzel GN, et al. Phase I dose escalation study of theanti insulin-like growth factor-I receptor monoclonal antibody CP-751,871in patients with refractory solid tumors. Clin Cancer Res. 2007;13:5834–5840.
84. Higano CS, Yu EY, Whiting SH, et al. A phase I, first in man study ofweekly IMC-A12, a fully human insulin like growth factor-I receptor IgG1monoclonal antibody, in patients with advanced solid tumors (MeetingAbstracts). J Clin Oncol. 2007;25:3505.
85. Tolcher AW, Sarantopoulos J, Patnaik A, et al. Phase I, pharmacokinetic,and pharmacodynamic study of AMG 479, a fully human monoclonalantibody to insulin-like growth factor receptor 1. J Clin Oncol. 2009;27:5800–5807.
86. Benjamin R, Gore L, Dias C, et al. Activity of R1507, a fully humanizedmonoclonal antibody IGF-1R (insulin-like growth factor receptor) antago-nist, in patients with Ewing’s sarcoma noted in a phase I study. In: 13thAnnual Meeting of the Connective Tissue Oncology Society (CTOS ’07)November 2007; Seattle, Washington, USA; Abstract #932:49.
87. Patel S, Pappo A, Crowley J, et al. A SARC global collaborative phase IItrial of R1507, a recombinant human monoclonal antibody to the insulin-likegrowth factor-1 receptor (IGF1R) in patients with recurrent or refractorysarcomas (Meeting Abstracts). J Clin Oncol 2009;27:10503.
88. Anderson P, Skubitz K, Miller R, et al. Activity of SCH 717454 in subjectswith relapsed osteosarcoma or Ewing’s sarcoma (study P04720). In: 14thAnnual Meeting of the Connective Tissue Oncology Society (CTOS ’08)November 2007; London, United Kingdom; Abstract #35094.
89. Tolcher AW, Rothenberg ML, Rodon J, et al. A phase I pharmacokinetic andpharmacodynamic study of AMG 479, a fully human monoclonal antibodyagainst insulin-like growth factor type 1 receptor (IGF-1R), in advancedsolid tumors (Meeting Abstracts). J Clin Oncol 2007;25:3002.
90. Chawla SP, Tolcher AW, Staddon AP, et al. Survival results with AP23573,a novel mTOR inhibitor, in patients (pts) with advanced soft tissue or bone
The Cancer Journal • Volume 16, Number 3, May/June 2010 IGF-1R Monoclonal Antibodies in Sarcoma
© 2010 Lippincott Williams & Wilkins www.journalppo.com | 193

sarcomas: Update of phase II trial (Meeting Abstracts). J Clin Oncol2007;25:10076.
91. Chugh R, Wathen JK, Maki RG, et al. Phase II multicenter trial of imatinibin 10 histologic subtypes of sarcoma using a Bayesian hierarchical statisticalmodel. J Clin Oncol. 2009;27:3148–3153.
92. Hidalgo M, Tirado Gomez M, et al. A phase I study of MK-0646, ahumanized monoclonal antibody against the insulin-like growth factorreceptor type 1 (IGF1R) in advanced solid tumor patients in a q2 wkschedule (Meeting Abstracts). J Clin Oncol. 2008;26:3520.
93. Atzori F, Tabernero J, Cervantes A, et al. A phase I, pharmacokinetic (PK)and pharmacodynamic (PD) study of weekly (qW) MK-0646, an insulin-likegrowth factor-1 receptor (IGF1R) monoclonal antibody (MAb) in patients(pts) with advanced solid tumors (Meeting Abstracts). J Clin Oncol. 2008;26:3519.
94. Guler HP, Zapf J, Froesch ER. Short-term metabolic effects of recombinanthuman insulin-like growth factor I in healthy adults. N Engl J Med.1987;317:137–140.
95. Clemmons DR. Involvement of insulin-like growth factor-I in the control ofglucose homeostasis. Curr Opin Pharmacol. 2006;6:620–625.
96. Moses AC, Young SC, Morrow LA, et al. Recombinant human insulin-likegrowth factor I increases insulin sensitivity and improves glycemic controlin type II diabetes. Diabetes. 1996;45:91–100.
97. Pennisi P, Gavrilova O, Setser-Portas J, et al. Recombinant human insulin-like growth factor-I treatment inhibits gluconeogenesis in a transgenicmouse model of type 2 diabetes mellitus. Endocrinology. 2006;147:2619–2630.
98. Lacy MQ, Alsina M, Fonseca R, et al. Phase I, pharmacokinetic andpharmacodynamic study of the anti-insulinlike growth factor type 1 Recep-tor monoclonal antibody CP-751,871 in patients with multiple myeloma.J Clin Oncol. 2008;26:3196–3203.
99. del Rincon JP, Iida K, Gaylinn BD, et al. Growth hormone regulation ofp85alpha expression and phosphoinositide 3-kinase activity in adiposetissue: mechanism for growth hormone-mediated insulin resistance. Diabe-tes. 2007;56:1638–1646.
100. Delafontaine P, Song YH, Li Y. Expression, regulation, and function ofIGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. ArteriosclerThromb Vasc Biol. 2004;24:435–444.
101. Laron Z, Anin S, Klipper-Aurbach Y, et al. Effects of insulin-like growthfactor on linear growth, head circumference, and body fat in patients withLaron-type dwarfism. Lancet. 1992;339:1258–1261.
102. Pfizer Ends Late-Stage Lung-Cancer Study. Thomson Reuters; 2009. Avail-able at: http://www.reuters.com/article/idUSTRE5BS3XY20091229. Ac-cessed January 17, 2010.
103. Roche. Genetech Decide to Halt Development of Their IGF-1R Antibody.Available at: http://www.sarctrials.org/public/press91.aspx. Accessed De-cember 22, 2009.
104. Imai K, Takaoka A. Comparing antibody and small-molecule therapies forcancer. Nat Rev Cancer. 2006;6:714–727.
105. Sabbatini P, Rowand JL, Groy A, et al. Antitumor activity ofGSK1904529A, a small-molecule inhibitor of the insulin-like growth fac-tor-I receptor tyrosine kinase. Clin Cancer Res. 2009;15:3058–3067.
106. Sabbatini P, Korenchuk S, Rowand JL, et al. GSK1838705A inhibits theinsulin-like growth factor-1 receptor and anaplastic lymphoma kinase andshows antitumor activity in experimental models of human cancers. MolCancer Ther. 2009;8:2811–2820.
107. Huang F, Greer A, Hurlburt W, et al. The mechanisms of differentialsensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitors. CancerRes. 2009;69:161–170.
108. Friedrichs N, Kuchler J, Endl E, et al. Insulin-like growth factor-1 receptoracts as a growth regulator in synovial sarcoma. J Pathol. 2008;216:428–439.
109. Duan Z, Choy E, Harmon D, et al. Insulin-like growth factor-I receptortyrosine kinase inhibitor cyclolignan picropodophyllin inhibits proliferationand induces apoptosis in multidrug resistant osteosarcoma cell lines. MolCancer Ther. 2009;8:2122–2130.
110. Weroha SJ, Haluska P. IGF-1 receptor inhibitors in clinical trials–earlylessons. J Mammary Gland Biol Neoplasia. 2008;13:471–483.
111. Benini S, Manara MC, Baldini N, et al. Inhibition of insulin-like growthfactor I receptor increases the antitumor activity of doxorubicin and vincris-tine against Ewing’s sarcoma cells. Clin Cancer Res. 2001;7:1790–1797.
112. Kurmasheva RT, Dudkin L, Billups C, et al. The insulin-like growth factor-1receptor-targeting antibody, CP-751,871, suppresses tumor-derived VEGFand synergizes with rapamycin in models of childhood sarcoma. CancerRes. 2009;69:7662–7671.
113. Wan X, Helman LJ. The biology behind mTOR inhibition in sarcoma.Oncologist. 2007;12:1007–1018.
114. Martins AS, Ordonez JL, Garcia-Sanchez A, et al. A pivotal role for heatshock protein 90 in Ewing sarcoma resistance to anti-insulin-like growthfactor 1 receptor treatment: in vitro and in vivo study. Cancer Res. 2008;68:6260–6270.
115. Carden CP, Molife LR, de Bono JS. Predictive biomarkers for targetinginsulin-like growth factor-I (IGF-I) receptor. Mol Cancer Ther. 2009;8:2077–2078.
116. Mukohara T, Shimada H, Ogasawara N, et al. Sensitivity of breast cancercell lines to the novel insulin-like growth factor-1 receptor (IGF-1R)inhibitor NVP-AEW541 is dependent on the level of IRS-1 expression.Cancer Lett. 2009;282:14–24.
117. Byron SA, Horwitz KB, Richer JK, Lange CA, Zhang X, Yee D. Insulinreceptor substrates mediate distinct biological responses to insulin-likegrowth factor receptor activation in breast cancer cells. Br J Cancer.2006;95:1220–1228.
118. Kolb EA, Morton C, Houghton PJ, et al. Pediatric Preclinical TestingProgram (PPTP) evaluation of the fully human anti-IGF-1R antibody IMC-A12. EJC Suppl. 2008;6:176.
119. Carboni JM, Wittman M, Yang Z, et al. BMS-754807, a small moleculeinhibitor of insulin-like growth factor-1R/IR. Mol Cancer Ther.2009;8:3341–3349.
120. Carden CP, Frentzas S, Langham M, et al. Preliminary activity in adreno-cortical tumor (ACC) in phase I dose escalation study of intermittent oraldosing of OSI-906, a small-molecule insulin-like growth factor-1 receptor(IGF-1R) tyrosine kinase inhibitor in patients with advanced solid tumors.J Clin Oncol (Meeting Abstracts). 2009;27:3544.
121. Smith DC, Britten C, Clary DO, Nguyen LT, Woodard P, Hurwitz HI. Aphase I study of XL228, a potent IGF1R/AURORA/SRC inhibitor, inpatients with solid tumors or hematologic malignancies. J Clin Oncol(Meeting Abstracts). 2009;27:3512.
122. Haluska P, Carboni JM, Loegering DA, et al. In vitro and in vivo antitumoreffects of the dual insulin-like growth factor-I/insulin receptor inhibitor,BMS-554417. Cancer Res. 2006;66:362–371.
123. Pollak MN, Chapman JW, Pritchard KI, et al. NCIC-CTG MA14 Trial:Tamoxifen (tam) vs. tam " octreotide (oct) for adjuvant treatment of stageI or II postmenopausal breast cancer. J Clin Oncol (Meeting Abstracts).2008;26:532.
124. Schreiber I, Buchfelder M, Droste M, et al. Treatment of acromegaly withthe GH receptor antagonist pegvisomant in clinical practice: safety andefficacy evaluation from the German Pegvisomant Observational Study. EurJ Endocrinol. 2007;156:75–82.
125. Goya M, Miyamoto S, Nagai K, et al. Growth inhibition of human prostatecancer cells in human adult bone implanted into nonobese diabetic/severecombined immunodeficient mice by a ligand-specific antibody to humaninsulin-like growth factors. Cancer Res. 2004;64:6252–6258.
Olmos et al The Cancer Journal • Volume 16, Number 3, May/June 2010
© 2010 Lippincott Williams & Wilkins194 | www.journalppo.com