spinal dysraphism: mr imaging rationale
TRANSCRIPT

J. Neuroradiol., 2004, 31, 3-24© Masson, Paris, 2004Review
SPINAL DYSRAPHISM: MR IMAGING RATIONALE
A. ROSSI (1), A. CAMA (2), G. PIATELLI (2), M. RAVEGNANI (2), R. BIANCHERI (1), P. TORTORI-DONATI (1)
(1) Department of Pediatric Neuroradiology,(2) Department of Neurosurgery, G. Gaslini Children’s Research Hospital, Largo G. Gaslini 5, 16147 Genoa, Italy.
SUMMARY
Spinal cord development occurs through the three consecutive periods of gastrulation (weeks 2-3), primary neurulation(weeks 3-4), and secondary neurulation (weeks 5-6). Spinal cord malformations derive from defects in these early embryonicstages, and are collectively called spinal dysraphisms. Spinal dysraphisms may be categorized clinically into open and closed,based on whether the abnormal nervous tissue is exposed to the environment or covered by skin. Open spinal dysraphismsinclude myelomeningocele and other rare abnormalities such as myelocele, hemimyelomeningocele, and hemimyelocele,and are always associated with a Chiari II malformation. Closed spinal dysraphisms are further divided into two subsetsbased on whether a subcutaneous mass is present in the low back. Closed spinal dysraphisms with mass comprise lipomye-locele, lipomyelomeningocele, meningocele, and myelocystocele. Closed spinal dysraphisms without mass comprise simpledysraphic states (tight filum terminale, filar and intradural lipomas, persistent terminal ventricle, and dermal sinuses) andcomplex dysraphic states. The latter category involves abnormal notochordal development, either in the form of failed mid-line integration (ranging from complete dorsal enteric fistula to neurenteric cysts and diastematomyelia) or of segmentalagenesis (caudal agenesis and spinal segmental dysgenesis). Magnetic resonance imaging is the imaging modality of choicefor evaluation of this complex group of disorders.
Key words: spinal cord, dysraphism, MR imaging.
RÉSUMÉ
Dysraphies spinales : aspects en IRMLe développement de la moelle épinière se déroule au cours de 3 phases successives : la gastrulation (2-3e semaine), la
neurulation primitive (3e et 4e semaines) et la neurulation secondaire (5e et 6e semaines). Les malformations de la moelleproviennent de défauts survenus lors de ces phases et sont dénommées dysraphies spinales (vertébromédullaires). Les dysra-phies spinales peuvent être subdivisés en dysraphies fermés ou ouverts selon que le tissu nerveux anormal est recouvert ou nonpar la peau. Les dysraphies comprennent les myéloméningocèles et d’autres anomalies rares telles que les myélocèles, les hémi-myéloméningocèles et les hémimyélocèles. Ils sont souvent associés à une malformation de Chiari de type II. Les dysraphiesfermées sont subdivisées en deux groupes selon que une masse sous-cutanée est présente ou absente à la partie basse du dos.Les dysraphies spinaux fermés avec masse regroupent les lipomyélocèles, les lipomyéloméningocèles, les méningocèles et lesmyélocystocèles. Les dysraphies fermées sans masse regroupent les états dysraphiques simples (tight filum terminale, lipomesintraduraux, persistance de ventricule terminal, sinus dermique) et des états dysraphiques complexes. Cette dernière catégoriecorrespond à un développement anormal de la notocorde, soit des défauts d’intégration de la ligne médiane (allant de la fistuleentérine dorsale complète au kyste neurentérique et diasthématomyélie) ou l’agénésie segmentaire (agénésie caudale et dysgé-nésie segmentaire spinale). L’imagerie par résonance magnétique est la méthode de choix pour le diagnostic de ces affections.
Mots-clés : moelle, dysraphies, IRM.
INTRODUCTION
Congenital malformations of the spinal cord arecollectively termed spinal dysraphisms. Theseconditions are usually diagnosed at birth or in earlyinfancy, but some may be discovered in older chil-dren or adults. Because of its multiplanar imagingand tissue characterization capabilities, magneticresonance imaging (MRI) has greatly improved thediagnosis of these disorders and has enhanced thepossibility of earlier and case-tailored treatment.Classification is based on a rational correlation ofclinical, neuroradiological, and embryological infor-mation. Use of classification schemes may provehelpful in making a diagnosis in daily clinical prac-tice [51]. The present paper attempts to summarizethe basic concepts about normal and deranged spi-nal cord embryogenesis, to describe the principal
malformations, and to offer a practical approach toneuroradiological decision-making.
EMBRYOLOGY
Spinal dysraphisms result from derangementoccurring during a limited period of time duringearly embryogenesis, i.e., between gestationalweeks 2 and 6. The relevant embryogenetic stepsare represented by gastrulation (weeks 2-3), primaryneurulation (weeks 3-4), and secondary neurulation(weeks 5-6) [50].
During gastrulation, the bilaminar embryonicdisk, formed by epiblast (future ectoderm) andhypoblast (future endoderm), is converted into atrilaminar disk with formation of an interveningthird layer, the mesoderm. This process begins byday 14 or 15 when a stripe of thickened epiblastcomposed by totipotential cells, the so-called primi-tive streak, appears caudally in the midline of thedorsal surface of the embryo. The Hensen’s node is
Reprint request: A. ROSSI, address abovee-mail: [email protected]

4 A. ROSSI et al.
the knob-like cranial termination of the primitivestreak. Epiblastic cells start migrating toward theprimitive streak and pass inward at the Hensen’snode to the interface of epiblast and primitive endo-derm. Waves of epiblastic cells migrating laterallyalong the interface form the interposed mesoderm,whereas cells migrating along the midline form thenotochord. The notochord, the foundation of theaxial skeleton, extends throughout the entire lengthof the future vertebral column. From the mesodermsurrounding the neural tube and notochord, the skulland vertebral column, and the membranes of thebrain and spinal cord are developed.
According to traditional views, the notochordinduces the overlying ectoderm to differentiateinto neuroectoderm, although recent studies sup-port the hypothesis that the default state of theectoderm is in fact neural ectoderm [27]. Estab-lishment of the neural plate marks the beginningof primary neurulation. On about day 18, the neu-ral plate starts bending, forming paired neuralfolds that progressively increase in size andapproach each other to eventually fuse in the mid-line to form the neural tube. According to classicalembryological theories, neural tube closure occursfirst at the level of the fourth somite (future cran-iocervical junction) and proceeds both cephaladand caudad in a zipperlike fashion. The cranialend of the neural tube (rostral neuropore) closesat day 25, whereas the caudal end (caudal neu-ropore) closes at day 27 or 28. Closure of the pos-terior neuropore terminates primary neurulation.
The segment of the spine and spinal cord caudadto somite 32 is formed by secondary neurulation,that begins immediately after completion of pri-mary neurulation and proceeds until approximatelygestational day 48. During secondary neurulation,an additional part of the neural tube is laid downcaudad to the caudal neuropore by the so-calledtail bud, a mass of cells deriving from the caudalportion of the primitive streak. The spinal cordformed by secondary neurulation differs from theprimary neural tube in many ways. Most notably,while the primary neural tube results from anupfolding of the lateral borders of the neural platewhich join at the midline, the secondary neuraltube is formed by an infolding of the neural plate,creating an initially solid medullary cord that sub-sequently becomes cavitated [9, 55]. The secondaryneural tube eventually results in the tip of theconus medullaris and the filum terminale, which isa fibroconnectival structure practically devoid ofneural elements. This has traditionally beenexplained through the concept of retrogressive dif-ferentiation, in which a combination of regression,degeneration, and further differentiation wouldoccur. However, such process has not been clearlydemonstrated in humans, and it is possible that alack of proliferation, rather than regression ordegeneration, contributes to the eventual rudimen-tary character of the filum terminale (Dr M. Catala,personal communication). A slight expansion of thecentral canal within the conus medullaris, calledterminal ventricle, represents the eventual remnantof the secondary neural tube lumen. It is usuallyundetectable by MRI in normal individuals.
TERMINOLOGY
Open and closed spinal dysraphisms
The term dysraphism etymologically refers todefective neural tube closure and, therefore, shouldapply to abnormalities of primary neurulation only.However, it is used to refer to congenital spinal corddisorders in general. Due to the common embryo-logical origin, caudal spinal anomalies are alsoincluded in this group.
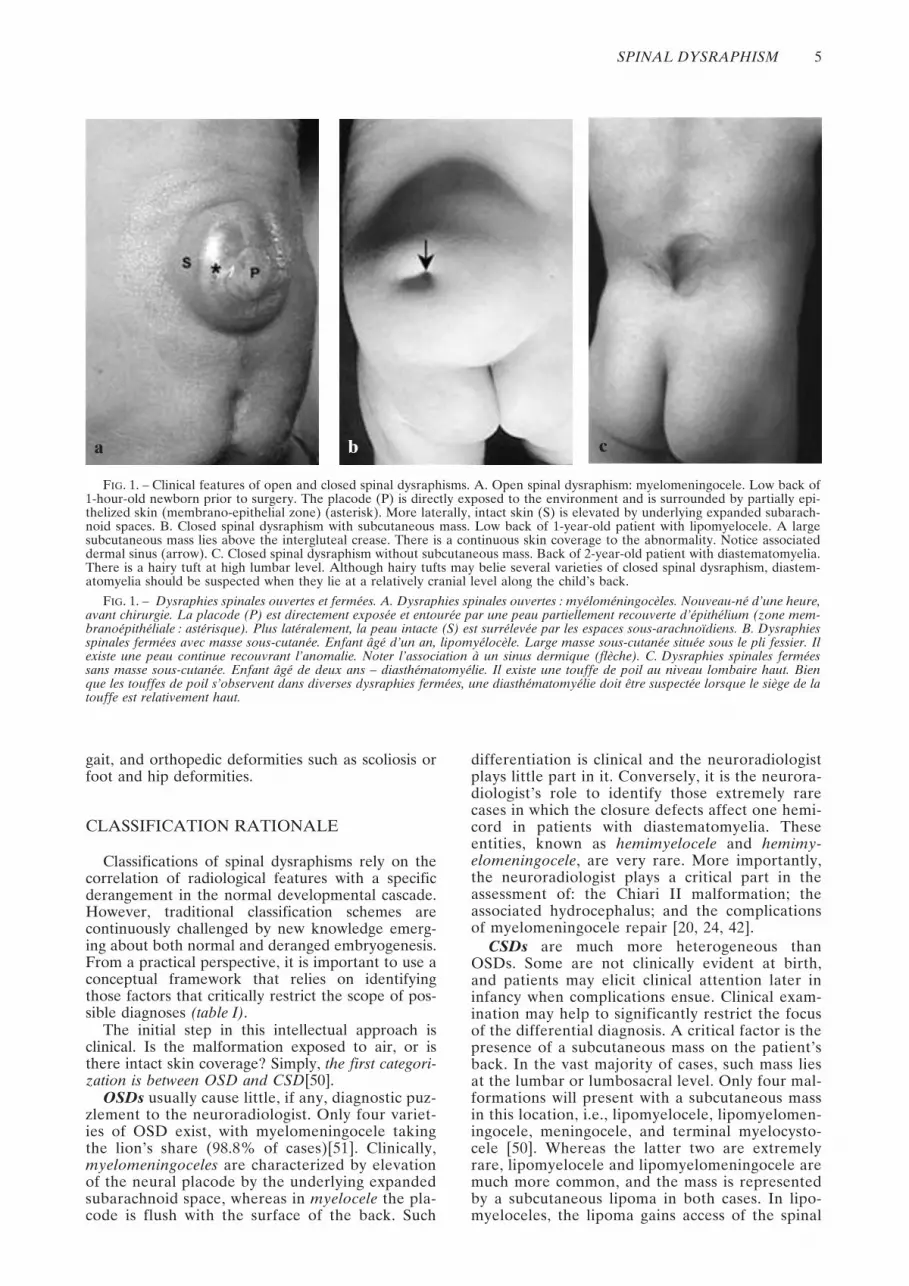
Spinal dysraphisms are categorized into open spi-nal dysraphisms (OSD) and closed spinal dysra-phisms (CSD) [50] (figure 1). In OSDs, the nervoustissue is exposed to the environment through acongenital bony defect. Conversely, CSDs are cov-ered by skin (i.e., there is no exposed neural tissue),although they are belied by cutaneous birthmarks inas many as 50% of cases [14]. We therefore suggestthat the synonymous term “occult spinal dysra-phisms” be abandoned, as it implies true, completeabsence of tell-tale signs of the underlying abnor-mality [51].
Spina bifida
The term “spina bifida” refers to defective fusionof posterior spinal bony elements [18] but was, andstill is, widely used as a synonym of spinal dysra-phisms. The terms “spina bifida aperta” or “cystica”and “spina bifida occulta” were used in the past torefer to OSD and CSD, respectively [40], but theseterms are no longer in widespread use and we do notencourage their utilization.
Placode
The placode is a segment of non-neurulatedembryonic neural tissue, i.e., frozen at the neuralplate stage. A placode is found in all OSDs as wellas in several types of CSDs. It is exposed to the envi-ronment in the former, and covered by the integu-ments in the latter. A placode may be categorizedinto terminal and segmental depending on location.A terminal placode lies at the caudal end of the spi-nal cord and may in turn be either apical or parietal,depending on whether the defect involves the apexproper or a longer segment of the cord [51]. A seg-mental placode lies at an intermediate level alongthe spinal cord; caudad to the abnormality the cordregains normal morphology and structure [50].
Tethered cord syndrome
Many believe that the tethered cord syndrome(TCS) is some sort of malformation. Instead, it is aclinical syndrome [53, 54] that may ensue as acomplication of myelomeningocele repair or as thepresentation of several forms of CSD, including spi-nal lipomas, the tight filum terminale, diastemato-myelia, and caudal agenesis. TCS involves tractionon a low-lying conus medullaris with progressiveneurologic deterioration due to metabolic derange-ment. The clinical picture includes motor and sen-sory dysfunction, muscle atrophy, decreased orhyperactive reflexes, urinary incontinence, spastic
SPINAL DYSRAPHISM 5
gait, and orthopedic deformities such as scoliosis orfoot and hip deformities.
CLASSIFICATION RATIONALE
Classifications of spinal dysraphisms rely on thecorrelation of radiological features with a specificderangement in the normal developmental cascade.However, traditional classification schemes arecontinuously challenged by new knowledge emerg-ing about both normal and deranged embryogenesis.From a practical perspective, it is important to use aconceptual framework that relies on identifyingthose factors that critically restrict the scope of pos-sible diagnoses (table I).
The initial step in this intellectual approach isclinical. Is the malformation exposed to air, or isthere intact skin coverage? Simply, the first categori-zation is between OSD and CSD[50].
OSDs usually cause little, if any, diagnostic puz-zlement to the neuroradiologist. Only four variet-ies of OSD exist, with myelomeningocele takingthe lion’s share (98.8% of cases)[51]. Clinically,myelomeningoceles are characterized by elevationof the neural placode by the underlying expandedsubarachnoid space, whereas in myelocele the pla-code is flush with the surface of the back. Such
differentiation is clinical and the neuroradiologistplays little part in it. Conversely, it is the neurora-diologist’s role to identify those extremely rarecases in which the closure defects affect one hemi-cord in patients with diastematomyelia. Theseentities, known as hemimyelocele and hemimy-elomeningocele, are very rare. More importantly,the neuroradiologist plays a critical part in theassessment of: the Chiari II malformation; theassociated hydrocephalus; and the complicationsof myelomeningocele repair [20, 24, 42].
CSDs are much more heterogeneous thanOSDs. Some are not clinically evident at birth,and patients may elicit clinical attention later ininfancy when complications ensue. Clinical exam-ination may help to significantly restrict the focusof the differential diagnosis. A critical factor is thepresence of a subcutaneous mass on the patient’sback. In the vast majority of cases, such mass liesat the lumbar or lumbosacral level. Only four mal-formations will present with a subcutaneous massin this location, i.e., lipomyelocele, lipomyelomen-ingocele, meningocele, and terminal myelocysto-cele [50]. Whereas the latter two are extremelyrare, lipomyelocele and lipomyelomeningocele aremuch more common, and the mass is representedby a subcutaneous lipoma in both cases. In lipo-myeloceles, the lipoma gains access of the spinal
b c
FIG. 1. – Clinical features of open and closed spinal dysraphisms. A. Open spinal dysraphism: myelomeningocele. Low back of1-hour-old newborn prior to surgery. The placode (P) is directly exposed to the environment and is surrounded by partially epi-thelized skin (membrano-epithelial zone) (asterisk). More laterally, intact skin (S) is elevated by underlying expanded subarach-noid spaces. B. Closed spinal dysraphism with subcutaneous mass. Low back of 1-year-old patient with lipomyelocele. A largesubcutaneous mass lies above the intergluteal crease. There is a continuous skin coverage to the abnormality. Notice associateddermal sinus (arrow). C. Closed spinal dysraphism without subcutaneous mass. Back of 2-year-old patient with diastematomyelia.There is a hairy tuft at high lumbar level. Although hairy tufts may belie several varieties of closed spinal dysraphism, diastem-atomyelia should be suspected when they lie at a relatively cranial level along the child’s back.
FIG. 1. – Dysraphies spinales ouvertes et fermées. A. Dysraphies spinales ouvertes : myéloméningocèles. Nouveau-né d’une heure,avant chirurgie. La placode (P) est directement exposée et entourée par une peau partiellement recouverte d’épithélium (zone mem-branoépithéliale : astérisque). Plus latéralement, la peau intacte (S) est surrélevée par les espaces sous-arachnoïdiens. B. Dysraphiesspinales fermées avec masse sous-cutanée. Enfant âgé d’un an, lipomyélocèle. Large masse sous-cutanée située sous le pli fessier. Ilexiste une peau continue recouvrant l’anomalie. Noter l’association à un sinus dermique (flèche). C. Dysraphies spinales ferméessans masse sous-cutanée. Enfant âgé de deux ans – diasthématomyélie. Il existe une touffe de poil au niveau lombaire haut. Bienque les touffes de poil s’observent dans diverses dysraphies fermées, une diasthématomyélie doit être suspectée lorsque le siège de latouffe est relativement haut.
a

6 A. ROSSI et al.
canal through a wide bony spina bifida andattaches to the neural placode, i.e., the placode-lipoma interface lies within the spinal canal.Conversely, in lipomyelomeningoceles, ballooningof the subarachnoid spaces pushes the neural pla-code out of the spinal canal, i.e., the placode-lipoma interface lies outside the spinal canal.Therefore, the simple assessment of location ofthe placode-lipoma interface permits one to makea confident diagnosis [51].
The diagnosis of CSD without a subcutaneousmass also benefits from a thorough clinical assess-ment. Several birthmarks belie underlying dysraphicstates. Among these, focal hirsutism is significantlyassociated with CSDs. When a hairy tuft lies rela-tively cephalad along the child’s back, diastemato-myelia is likely [51]. Of the cutaneous birthmarks,the least sensitive in predicting underlying malfor-mation is the capillary hemangioma. However, cap-illary hemangiomas of the lumbar region areassociated with CNS pathology in greater than 10%of cases. Dorsal dimples or ostia indicate a dermalsinus. All dermal sinuses above the gluteal creaseshould be presumed to violate the subarachnoidspace until proven otherwise. Conversely, skin pitslocated within the intergluteal cleft need no furtherinvestigation as they are related to simple sacrococ-cygeal cysts or fistulas. Caudal agenesis may bebetrayed by a rudimentary tail, lower limb abnor-malities, or anorectal malformations. Imperforateanus is associated with surgically correctable intra-
dural pathology in at least 10% of patients. Allpatients with imperforate anus should undergo imag-ing studies to rule out the intradural pathology.Although these clinical information are critical infocusing one’s attention to a specific subset of dysra-phism, it is in the category of CSD without a subcu-taneous mass that the neuroradiologist will bechallenged most severely.
OPEN SPINAL DYSRAPHISMS
Myelomeningocele and Myelocele
Myelomeningocele represents 98.8% of all OSDs[51]. In myelomeningocele, the placode protrudestogether with the meninges through a bony defect inthe midline of the back, therefore being exposed tothe environment. Because of expansion of theunderlying subarachnoid space, the surface of theplacode is elevated above the skin surface. This dis-tinguishes myelomeningoceles from the far lesscommon myelocele, or myeloschisis, in which theplacode is flush with the cutaneous surface. As thedegree of motor deficit is affected by direct injury tothe exposed placode during delivery, cesarian sec-tion is typically performed. Because placode ulcera-tion and infection are leading causes of mortality inthe untreated newborn, affected patients are oper-ated on soon after birth. The subsequent clinical pic-ture includes sensorimotor deficits of the lowerextremities, bowel and bladder incontinence, hind-brain dysfunction, intellectual and psychological dis-turbances. Additionally, it is estimated that 18-40%of individuals with OSDs are allergic to latex. Aller-gic responses can vary from mild to anaphylaxis, andoccur when latex products touch the skin or mucousmembranes. Latex-free devices must be used in theMR environment when dealing with these patients.
Embryologically, both myelomeningoceles andmyeloceles result from faulty primary neurulation[3], with persistence of a portion of non-neurulatedplacode. The vast majority of OSDs involve the lum-bosacral spine, and the placode is terminal. How-ever, purely lumbar, thoracolumbar, and thoracicmyelomeningoceles exist, in which case the placodeis segmental. The exposed surface of the placoderepresents the would-be inner surface of the spinalcord and is covered by a rich network of small andfriable vessels. The cutaneous ectoderm remains in alateral position, resulting in a midline skin defect.Because the mesenchyme cannot migrate behind theneural tube, bones, cartilage, muscles and ligamentsare forced to develop anterolaterally to the neuraltissue, and therefore will appear everted. The ven-tral surface of the placode is formed by the would-be external surface of the spinal cord, from whichthe nerve roots originate. These nerve roots courseobliquely through the subarachnoid space to reachtheir corresponding neural foramina.
Owing to the need for prompt surgical operation,these newborns only very rarely undergo MRI studiesprior to surgery. However, we suggest a preopera-tive MRI investigation should be performed when-ever possible, in order to obtain: (1) anatomiccharacterization of the various components of the
TABLE I. – Clinical-neuroradiological classification of spinal dysraphisms
Open spinal dysraphisms• Myelomeningocele• Myelocele• Hemimyelomeningocele• Hemimyelocele
Closed spinal dysraphismsWith subcutaneous massLumbosacral
• Lipomas with dural defect— Lipomyelomeningocele— Lipomyelocele
• Terminal myelocystocele• MeningoceleCervico-thoracic• Myelocystocele• Skin-covered myelomeningocele (limited
dorsal myeloschisis)• Meningocele
Without subcutaneous massSimple dysraphic states• Intradural lipoma• Filar lipoma• Tight filum terminale• Persistent terminal ventricle• Dermal sinus
Complex dysraphic states1. Disorders of midline notochordal
integrationa) Diastematomyeliab) Neurenteric cystsc) Dorsal enteric fistula
2. Disorders of notochordal formationa) Caudal agenesisb) Segmental spinal dysgenesis
SPINAL DYSRAPHISM 7
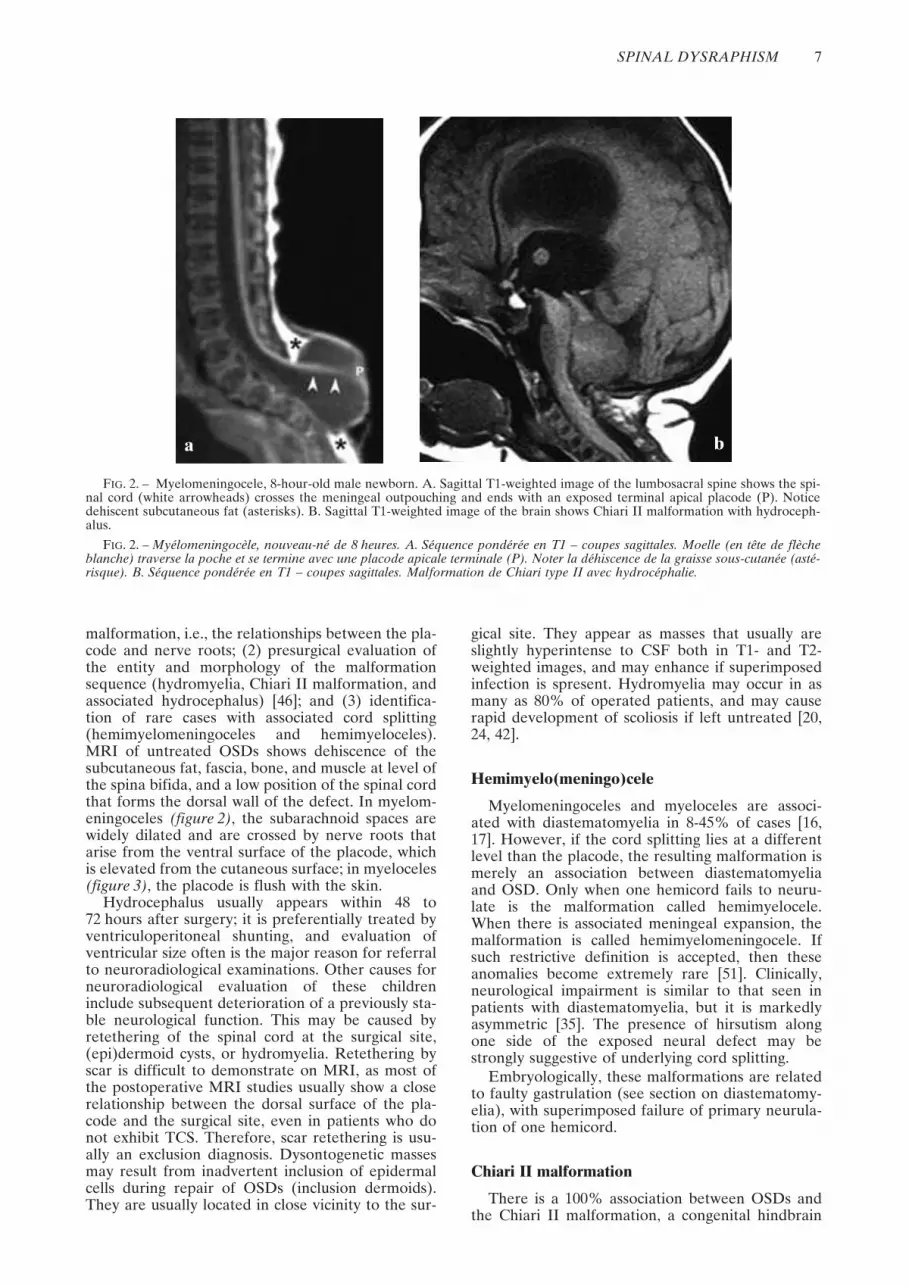
malformation, i.e., the relationships between the pla-code and nerve roots; (2) presurgical evaluation ofthe entity and morphology of the malformationsequence (hydromyelia, Chiari II malformation, andassociated hydrocephalus) [46]; and (3) identifica-tion of rare cases with associated cord splitting(hemimyelomeningoceles and hemimyeloceles).MRI of untreated OSDs shows dehiscence of thesubcutaneous fat, fascia, bone, and muscle at level ofthe spina bifida, and a low position of the spinal cordthat forms the dorsal wall of the defect. In myelom-eningoceles (figure 2), the subarachnoid spaces arewidely dilated and are crossed by nerve roots thatarise from the ventral surface of the placode, whichis elevated from the cutaneous surface; in myeloceles(figure 3), the placode is flush with the skin.
Hydrocephalus usually appears within 48 to72 hours after surgery; it is preferentially treated byventriculoperitoneal shunting, and evaluation ofventricular size often is the major reason for referralto neuroradiological examinations. Other causes forneuroradiological evaluation of these childreninclude subsequent deterioration of a previously sta-ble neurological function. This may be caused byretethering of the spinal cord at the surgical site,(epi)dermoid cysts, or hydromyelia. Retethering byscar is difficult to demonstrate on MRI, as most ofthe postoperative MRI studies usually show a closerelationship between the dorsal surface of the pla-code and the surgical site, even in patients who donot exhibit TCS. Therefore, scar retethering is usu-ally an exclusion diagnosis. Dysontogenetic massesmay result from inadvertent inclusion of epidermalcells during repair of OSDs (inclusion dermoids).They are usually located in close vicinity to the sur-
gical site. They appear as masses that usually areslightly hyperintense to CSF both in T1- and T2-weighted images, and may enhance if superimposedinfection is spresent. Hydromyelia may occur in asmany as 80% of operated patients, and may causerapid development of scoliosis if left untreated [20,24, 42].
Hemimyelo(meningo)cele
Myelomeningoceles and myeloceles are associ-ated with diastematomyelia in 8-45% of cases [16,17]. However, if the cord splitting lies at a differentlevel than the placode, the resulting malformation ismerely an association between diastematomyeliaand OSD. Only when one hemicord fails to neuru-late is the malformation called hemimyelocele.When there is associated meningeal expansion, themalformation is called hemimyelomeningocele. Ifsuch restrictive definition is accepted, then theseanomalies become extremely rare [51]. Clinically,neurological impairment is similar to that seen inpatients with diastematomyelia, but it is markedlyasymmetric [35]. The presence of hirsutism alongone side of the exposed neural defect may bestrongly suggestive of underlying cord splitting.
Embryologically, these malformations are relatedto faulty gastrulation (see section on diastematomy-elia), with superimposed failure of primary neurula-tion of one hemicord.
Chiari II malformation
There is a 100% association between OSDs andthe Chiari II malformation, a congenital hindbrain
b
FIG. 2. – Myelomeningocele, 8-hour-old male newborn. A. Sagittal T1-weighted image of the lumbosacral spine shows the spi-nal cord (white arrowheads) crosses the meningeal outpouching and ends with an exposed terminal apical placode (P). Noticedehiscent subcutaneous fat (asterisks). B. Sagittal T1-weighted image of the brain shows Chiari II malformation with hydroceph-alus.
FIG. 2. – Myélomeningocèle, nouveau-né de 8 heures. A. Séquence pondérée en T1 – coupes sagittales. Moelle (en tête de flècheblanche) traverse la poche et se termine avec une placode apicale terminale (P). Noter la déhiscence de la graisse sous-cutanée (asté-risque). B. Séquence pondérée en T1 – coupes sagittales. Malformation de Chiari type II avec hydrocéphalie.
a

8 A. ROSSI et al.
anomaly characterized by a small posterior fossawith caudal displacement of the vermis, brainstem,and fourth ventricle (figure 2) [6]. McLone andKnepper [25] proposed a theory to explain thisconsistent association. Normally, the medial walls ofthe primitive central canal of the neural tube (“neuro-cele”) appose and occlude the neurocele transientlyduring primary neurulation. Failure to occlude theneurocele allows free downward CSF flow. There-fore, CSF leaks freely through the spinal defect intothe amniotic sac because the neural tube remainsnon-neurulated. This results in chronic CSFhypotension within the developing neural tube.Consequently, the rhombencephalic vesicle fails toexpand, causing lack of induction of the perineuralmesenchyme of the posterior cranial fossa. Both thecerebellum and brain stem eventually are forced todevelop within a smaller than normal posterior fossaand consequently herniate through both the tento-rial groove and the foramen magnum [29].
CLOSED SPINAL DYSRAPHISMS
CSDs with subcutaneous mass
These abnormalities are characterized by a skin-covered mass belying the underlying malformation.Most often, the mass lies at the lumbosacral levelright above the intergluteal crease, and the corre-sponding anomalies are represented by four entities:the quite common lipomas with dural defect (lipo-
myelocele and lipomyelomeningocele), and the dis-tinctly uncommon terminal myelocystocele andmeningocele [50]. Differential diagnosis basicallyincludes sacrococcygeal teratomas, whose location ismore caudal, i.e., at or below the intergluteal crease.Cervico-thoracic CSDs with associated subcutaneousmasses are distinctly uncommon.
Lipomas with dural defect: lipomyelocele and lipomyelomeningocele
Lipomyeloceles and lipomyelomeningoceles arecharacterized by a midline subcutaneous fatty massright above the intergluteal crease, usually extendingasymmetrically into one buttock [30]. Because themass is clinically evident at birth, the diagnosis usuallyis obtained before neurological deterioration ensues.Infants who are not treated before 6 months of ageusually develop hypostenia and hypotrophy of mus-cles of both lower limbs, gait disturbances, urinaryincontinence, and paresthesias. These clinical featuresprogress over time if the child is left untreated.
Histologically, the mass is composed by clusters ofmature adipocytes separated by collagenous bands, usu-ally associated with other tissues such as striated muscle,cartilage, bone, nerve cells, ependyma, and aberrantneuroglial tissue. Sometimes, these aberrant tissues coa-lesce to form hamartomatous masses that have beendesignated “dysraphic hamartomas”. Although congen-ital intraspinal lipomas are anatomically stable [26], theymay grow as part of the normal increase of adipose tis-sue throughout childhood [24], other than in particularconditions such as obesity or pregnancy.
Embryologically, spinal lipomas are abnormalities ofprimary neurulation. They are traditionally believed toresult from focal premature disjunction of the cutane-ous ectoderm from the neuroectoderm, allowing mes-enchyme to access the interior of the neural tube,where it will be induced to form fat by contacting theependymal lining [27, 30]. Other embryological mecha-nism have been invoked, involving abnormalities of thedorsal mesoderm that could either be primitive or sec-ondary to defective induction from the neural tube [8].
The anatomic position of the connection betweenthe spinal cord and the lipoma (i.e., the placode-lipoma interface) varies depending on the size of thelipoma and the degree of expansion of the subarach-noid spaces. In lipomyelocele (synonym: lipomy-eloschisis) (figure 4), the placode-lipoma interface lieswithin or at the edge of the spinal canal. Both thebony defect and the subcutaneous fat that extend intothe spinal canal and attach to the cord are clearlydemonstrated by MRI. The placode-lipoma interfacemay extend over several vertebral levels. It may besmooth and regular, or large and irregular, withstripes of fat permeating the exterior of the spinalcord and even penetrating into the central canal.Hydromyelia usually is present in these cases. Thesize of the spinal canal may be increased dependingon the size of the lipoma, but the size of the subarach-noid space ventral to the cord is consistently normal.
Lipomyelomeningoceles may produce a constella-tion of MRI features. Individual cases typically differfrom one another, depending on the relative size ofthe meningocele and lipoma as well as the orienta-tion of the placode. However, the placode-lipoma
FIG. 3. – Myelocele, 12-hour-old newborn. Sagittal T1-weighted images shows exposed, slightly funnel-shaped pla-code (P) lying flush with the skin surface. There is dehiscenceof subcutaneous fat (asterisks). The lack of expansion of sub-arachnoid spaces is the only difference from the much morecommon myelomeningocele.
FIG. 3. – Myélocèle, nouveau-né de 12 heures. Séquencepondérée en T1 – coupes sagittales. Placode en forme d’enton-noir (P). Il existe une déhiscence de la graisse sous-cutanée(astérisque). L’absence d’expansion des espaces sous-arachnoï-diens est la seule différence avec la plus commune myélomé-ningocèle.

SPINAL DYSRAPHISM 9
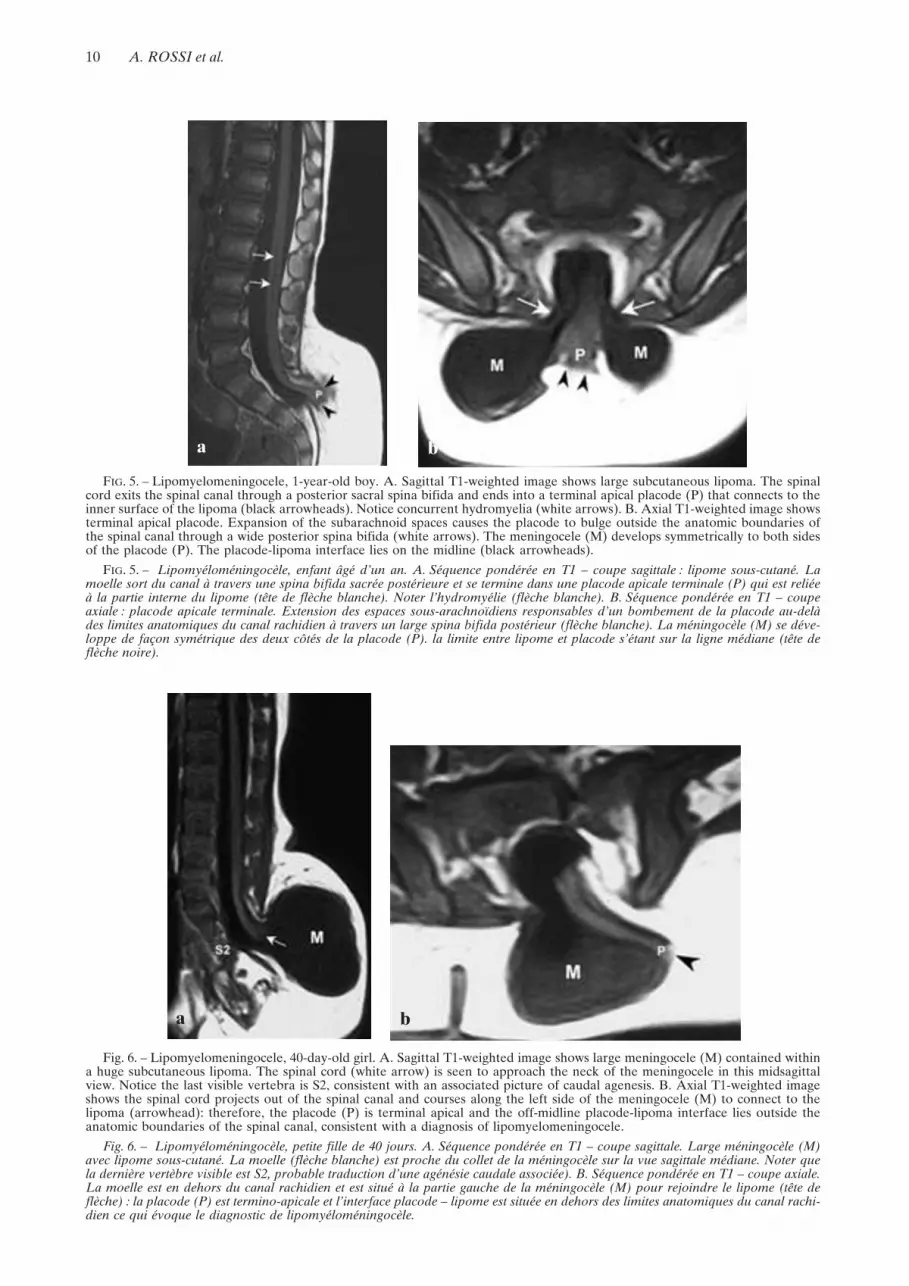
interface lies outside the anatomic boundaries of thespinal canal in all cases. An archetypal condition inwhich the placode-lipoma interface lies exactly alongthe midline within the meningocele is not the rulebut, rather, an exception [50] (figure 5). In mostinstances, the placode is stretched and rotated eccen-trically towards the lipoma on one side, whereas themeninges herniate on the opposite side (figure 6). Insuch an instance, the spinal roots emerging from theside facing the meningocele are generally longer andmay be at risk for surgical trauma, whereas thoselying on the side of the lipoma are shorter and causespinal cord tethering. Different from lipomyeloceles,the spinal canal is dilated due to expansion of thesubarachnoid spaces ventral to the cord.
Meningocele
The classic posterior meningocele results from her-niation of a CSF-filled sac lined by dura and arach-noid through a posterior bony spina bifida (figure 7).It is commonly lumbar or sacral in location, but tho-racic and even cervical meningoceles may be found.In general, spinal meningoceles are less common thanis usually believed (2.4% of all CSDs) [1]. Theirembryogenetic origin is unknown, but they mightresult from ballooning of the meninges through a pos-terior spina bifida due to the relentless effect of CSFpulsations. Although both nerve roots and, morerarely, a hypertrophied filum terminale may coursewithin the meningocele, by definition no part of thespinal cord enters the sac. The spinal cord itself iscompletely normal structurally, although it usually istethered to the neck of sacral meningoceles.
Anterior meningoceles almost always are presacralin location and are consistently found within the set-ting of caudal agenesis [23]. They are usually disco-vered in older children or even in adults when MRIis performed for low back pain, urinary inconti-nence, or constipation.
Terminal myelocystocele
Terminal myelocystoceles involve expansion of thecentral canal of the caudal spinal cord produced byterminal hydromyelia (“syringocele”), surrounded byan expanded dural sheath (“meningocele”). A subcu-taneous lipoma is usually associated, so that theabnormality could perhaps be better defined as a “lipo-myelocystocele”. The inner terminal cyst communi-cates with the central canal of the spinal cord(figure 8), whereas the outer dural sac communicateswith the subarachnoid space. The outer and innerfluid spaces usually do not communicate [5, 26, 36].Patients with terminal myelocystoceles typically haveno bowel or bladder control and poor lower-extremityfunction [6]. The prognosis is mainly related to asso-ciated anomalies, usually belonging to the OEISconstellation (omphalocele, exstrophy of the cloaca,imperforate anus, spinal anomalies) [30, 31].
The embryological origin of the terminal myelocys-tocele has not yet been determined. Current theoriespostulate a perturbation of CSF dynamics [5, 26, 36].Inability of CSF to exit from the early neural tubecould cause the terminal ventricle to balloon into a cystthat disrupts the overlying mesenchyme. Therefore, theterminal myelocystocele could be viewed as a severe,disruptive variety of a persistent terminal ventricle [50].
a b
FIG. 4. – Lipomyelocele, 1-month-old girl. A. Sagittal T1-weighted image shows large subcutaneous lipoma with fatty tissuecreeping through a wide posterior bony spina bifida into the spinal canal to connect with the placode (arrowheads). Although thesize of the subcutaneous lipoma may not seem huge, the amount of fat is way too large for a newborn. Moreover, the mass isflattened as the child lies supine. B. Axial T1-weighted image demonstrates the placode (P) - lipoma (L) interface into the spinalcanal.
FIG. 4. – Lipomyélocèle, petite fille âgée d’1 mois. A. Séquence pondérée en T1 – coupe sagittale. Lipome sous-cutané importantavec tissu graisseux allant à travers un large spina bifida postérieur dans le canal rachidien rejoindre la placode (tête de flèche). Bienque la taille des lipomes sous-cutanés ne semble pas très importante, la quantité de graisse est trop peu importante pour un nouveau-né. De plus, la masse est aplatie en décubitus. B. Séquence pondérée en T1 – Coupe axiale : placode (P) – lipome (L).
10 A. ROSSI et al.
b
FIG. 5. – Lipomyelomeningocele, 1-year-old boy. A. Sagittal T1-weighted image shows large subcutaneous lipoma. The spinalcord exits the spinal canal through a posterior sacral spina bifida and ends into a terminal apical placode (P) that connects to theinner surface of the lipoma (black arrowheads). Notice concurrent hydromyelia (white arrows). B. Axial T1-weighted image showsterminal apical placode. Expansion of the subarachnoid spaces causes the placode to bulge outside the anatomic boundaries ofthe spinal canal through a wide posterior spina bifida (white arrows). The meningocele (M) develops symmetrically to both sidesof the placode (P). The placode-lipoma interface lies on the midline (black arrowheads).
FIG. 5. – Lipomyéloméningocèle, enfant âgé d’un an. A. Séquence pondérée en T1 – coupe sagittale : lipome sous-cutané. Lamoelle sort du canal à travers une spina bifida sacrée postérieure et se termine dans une placode apicale terminale (P) qui est reliéeà la partie interne du lipome (tête de flèche blanche). Noter l’hydromyélie (flèche blanche). B. Séquence pondérée en T1 – coupeaxiale : placode apicale terminale. Extension des espaces sous-arachnoïdiens responsables d’un bombement de la placode au-delàdes limites anatomiques du canal rachidien à travers un large spina bifida postérieur (flèche blanche). La méningocèle (M) se déve-loppe de façon symétrique des deux côtés de la placode (P). la limite entre lipome et placode s’étant sur la ligne médiane (tête deflèche noire).
a
a
Fig. 6. – Lipomyelomeningocele, 40-day-old girl. A. Sagittal T1-weighted image shows large meningocele (M) contained withina huge subcutaneous lipoma. The spinal cord (white arrow) is seen to approach the neck of the meningocele in this midsagittalview. Notice the last visible vertebra is S2, consistent with an associated picture of caudal agenesis. B. Axial T1-weighted imageshows the spinal cord projects out of the spinal canal and courses along the left side of the meningocele (M) to connect to thelipoma (arrowhead): therefore, the placode (P) is terminal apical and the off-midline placode-lipoma interface lies outside theanatomic boundaries of the spinal canal, consistent with a diagnosis of lipomyelomeningocele.
Fig. 6. – Lipomyéloméningocèle, petite fille de 40 jours. A. Séquence pondérée en T1 – coupe sagittale. Large méningocèle (M)avec lipome sous-cutané. La moelle (flèche blanche) est proche du collet de la méningocèle sur la vue sagittale médiane. Noter quela dernière vertèbre visible est S2, probable traduction d’une agénésie caudale associée). B. Séquence pondérée en T1 – coupe axiale.La moelle est en dehors du canal rachidien et est situé à la partie gauche de la méningocèle (M) pour rejoindre le lipome (tête deflèche) : la placode (P) est termino-apicale et l’interface placode – lipome est située en dehors des limites anatomiques du canal rachi-dien ce qui évoque le diagnostic de lipomyéloméningocèle.
b

SPINAL DYSRAPHISM 11
CSDs without subcutaneous massSimple dysraphic states
This subset of abnormalities is heterogeneousfrom an embryological perspective, as it involvesdefects of both primary and secondary neurulation.
However, they may be grouped from a clinicalstandpoint, as they represent the most commonabnormalities found in relatively older children whousually do not have significant low-back cutaneousstigmata but complain with TCS [45, 47].
Intradural and intramedullary lipoma
Intradural and intramedullary lipomas do not dif-fer from lipomas with dural defects both in patholog-ical and embryological terms. However, they arecontained within an intact dural sac. Intradural lipo-mas lie along the midline in the groove formed bythe dorsal surface of the unapposed folds of the pla-code, and may bulge posteriorly in the subarachnoidspaces elevating the pia mater. Large lipomas maydisplace the cord laterally, resulting in an off-midlineplacode-lipoma interface. In rare instances, lipomasare completely intramedullary. Intradural lipomasare commonly located at lumbosacral level (figure 9)and usually present with TSC, whereas cervicotho-racic lipomas generally produce insidious signs ofspinal cord compression (figure 10). At MRI, lipo-mas appear as masses that are isointense to subcuta-neous fat on all sequences, including those acquiredwith fat suppression (figure 10) [50].
Filar lipoma
Filar lipoma is an elementary anomaly of sec-ondary neurulation characterized by fibrolipoma-tous thickening of the filum terminale. Theoccurrence of incidental fat within the filum termi-nale in the normal adult population is estimated tobe 1.5 to 5% of unselected MRI studies [4, 52].Therefore, it may be considered an anatomic vari-ant if there are no signs of TCS. The exact embryo-
a b c
FIG. 7. – Meningocele, 7-month-old girl. A,B. Sagittal T1-weighted images show large sacral cerebrospinal fluid-filled mass(M). The overlying skin is continuous. The conus medullaris is low. C. T1-weighted images show associated lipomatous filum ter-minale (arrows).
FIG. 7. – Méningocèle, petite fille de 7 mois. A, B. Séquence pondérée en T1 – coupe sagittale. Large masse sacrée contenant duliquide cérébrospinal (M). La peau est continue. Le cône médullaire est en position basse. C. Séquence pondérée en T1. Lipome dufilum terminale (flèche).
FIG. 8. – Terminal myelocystocele. Sagittal T1-weightedimage shows large subcutaneous syringocele continuous withhydromyelia (arrow) through a wide spina bifida. Courtesy P.D. Barnes, Boston, MA, USA
FIG. 8. – Myélocystocèle terminal. Séquence pondérée enT1 – coupe sagittale. Syringocèle sous-cutané continu avechydromyélie (fleche) à travers de large spina bifida. Clichésdus à l’obligeance de P. D. Barnes, Boston, MA, USA

12 A. ROSSI et al.
logical mechanisms by which lipomas of the filumarise remain unknown, but impaired canalization ofthe tail bud and persistence of cells capable ofmaturing into adipocytes are likely to be involved[52]. MRI detects fatty tissue within a thickened
filum terminale as a stripe of increased signal inten-sity on sagittal T1-weighted images (figure 11).Because the filum frequently lies slightly off themidline, axial and coronal T1-weighted images areuseful to make the diagnosis.
b
FIG. 9. – Intradural lipoma, 2-month-old girl. A. Sagittal T1-weighted image shows low-lying spinal cord tethered (small arrow-heads) to the anterior surface of a lumbosacral lipoma (L). The lipoma is not continuous with the subcutaneous fat. Noticeconcurrent dermal sinus (large arrowhead). B. Axial T1-weighted image shows the placode (P) -lipoma (L) interface (arrow-heads). The lipoma is intradural and clearly separated from the subcutaneous fat.
FIG. 9. – Lipome intradural, fille de 2 mois. A. Séquence pondérée en T1 – coupe sagittale. Moelle attachée à la face antérieured’un lipome lombosacré (L). Le lipome n’est pas en continuité avec la graisse sous-cutanée. Noter l’existence d’un sinus dermique(flèche large). B. Séquence pondérée en T1 – coupe axiale. Interface lipome (L) – placode (P) (tête de flèche). Le lipome est intra-dural est séparé de la graisse sous-cutanée.
a
a
FIG. 10. – Intradural cervical lipoma. A. Sagittal T1-weighted image show huge intradural lipoma that fills the spinal canalalmost completely. The spinal cord is compressed. The spinal canal is enlarged, with scalloped posterior vertebral walls. B. Fat-suppressed sagittal T1-weighted image confirms the fatty composition of the mass. Courtesy Prof. U. Aydingoz, Hacettepe, Uni-versity, Turkey.
FIG. 10. – Lipome cervical intradural. A. Séquence pondérée en T1 – Coupe sagittale. Large lipome intradural occupant la quasi-totalité du canal rachidien. La moelle est comprimée et le canal élargi avec scalping postérieur des corps vertébraux. B. Séquencepondérée en T1 avec suppression de graisse. Coupe sagittale. Confirmation du caractère graisseux de la masse. Clichés dus à l’obli-geance. Courtesy Prof. U. Aydingoz, Hacettepe, University, Turkey.
b
SPINAL DYSRAPHISM 13
Tight filum terminale
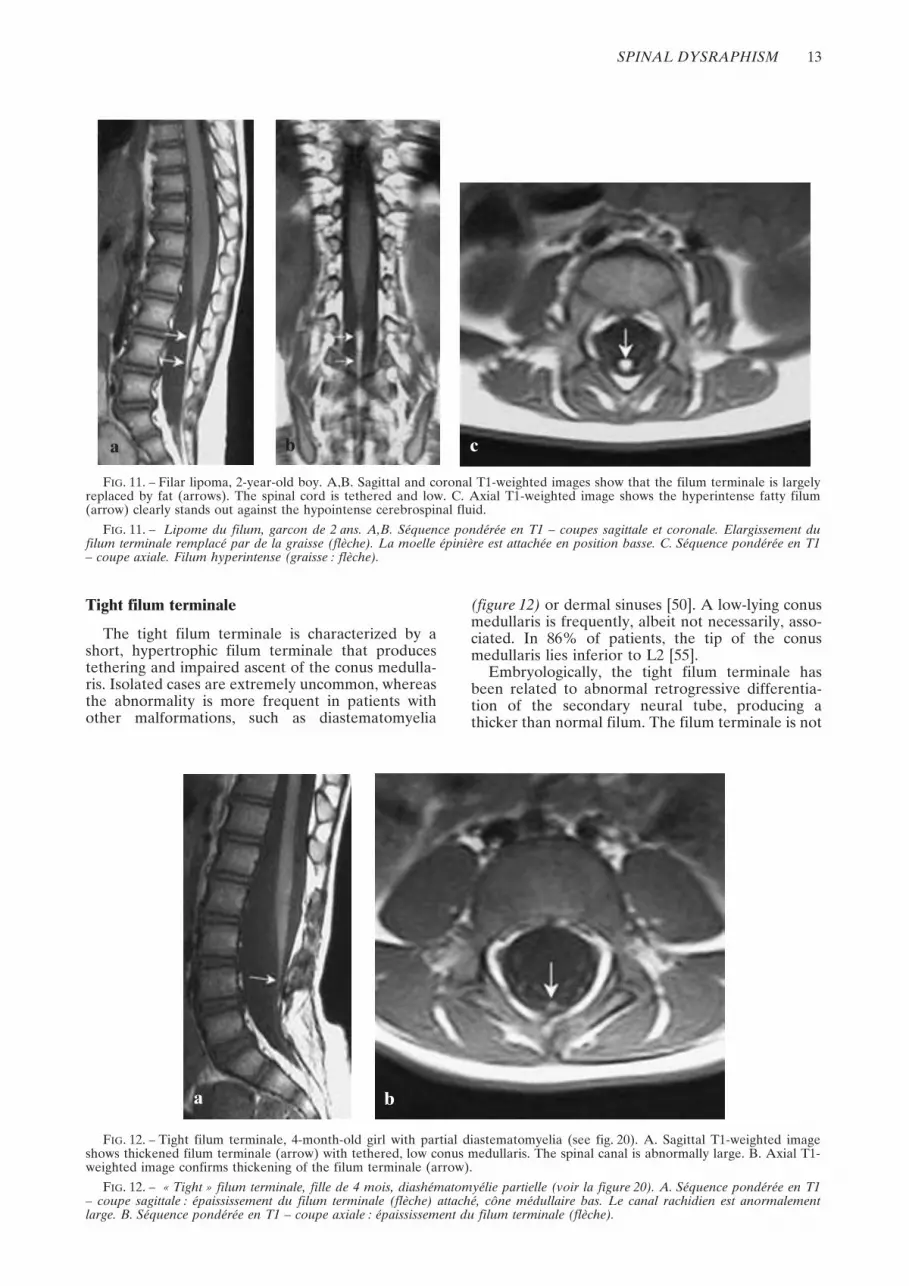
The tight filum terminale is characterized by ashort, hypertrophic filum terminale that producestethering and impaired ascent of the conus medulla-ris. Isolated cases are extremely uncommon, whereasthe abnormality is more frequent in patients withother malformations, such as diastematomyelia
(figure 12) or dermal sinuses [50]. A low-lying conusmedullaris is frequently, albeit not necessarily, asso-ciated. In 86% of patients, the tip of the conusmedullaris lies inferior to L2 [55].
Embryologically, the tight filum terminale hasbeen related to abnormal retrogressive differentia-tion of the secondary neural tube, producing athicker than normal filum. The filum terminale is not
c
FIG. 11. – Filar lipoma, 2-year-old boy. A,B. Sagittal and coronal T1-weighted images show that the filum terminale is largelyreplaced by fat (arrows). The spinal cord is tethered and low. C. Axial T1-weighted image shows the hyperintense fatty filum(arrow) clearly stands out against the hypointense cerebrospinal fluid.
FIG. 11. – Lipome du filum, garcon de 2 ans. A,B. Séquence pondérée en T1 – coupes sagittale et coronale. Elargissement dufilum terminale remplacé par de la graisse (flèche). La moelle épinière est attachée en position basse. C. Séquence pondérée en T1– coupe axiale. Filum hyperintense (graisse : flèche).
a b
a b
FIG. 12. – Tight filum terminale, 4-month-old girl with partial diastematomyelia (see fig. 20). A. Sagittal T1-weighted imageshows thickened filum terminale (arrow) with tethered, low conus medullaris. The spinal canal is abnormally large. B. Axial T1-weighted image confirms thickening of the filum terminale (arrow).
FIG. 12. – « Tight » filum terminale, fille de 4 mois, diashématomyélie partielle (voir la figure 20). A. Séquence pondérée en T1– coupe sagittale : épaississement du filum terminale (flèche) attaché, cône médullaire bas. Le canal rachidien est anormalementlarge. B. Séquence pondérée en T1 – coupe axiale : épaississement du filum terminale (flèche).

14 A. ROSSI et al.
>2 mm in diameter in normal individuals [57], butthe exact thickness of the filum terminale may bedifficult to measure at MRI.
Dermal sinus
The dermal sinus is an epithelium-lined fistula thatextends inward from the skin surface and can connectwith the CNS and its meningeal coating. It is foundmore frequently in the lumbosacral region, althoughcervical, thoracic, and occipital locations are possible[1]. On clinical examination, a midline dimple or pin-point ostium is found (figure 1), often in associationwith hairy nevus, capillary hemangioma, or hyperpig-mented patches. The cutaneous opening of a dermalsinus tract differs from that of a sacrococcygeal fistula.Dermal sinus tracts are found above the interglutealcleft and usually are directed superiorly. By compari-son, sacrococcygeal pits are found within the natal cleftextending either straight down or inferiorly. They areanatomically located below the level of the cul-de-sacof the subarachnoid space and do not require furtherimaging evaluation [56]. Although the cutaneousabnormality usually is evident at birth, some patientsare not referred to medical attention until they developcomplications such as local infection or meningitis andabscesses that may result from bacteria invading theCNS along the dermal sinus tract.
Embryologically, dermal sinus tracts are tradition-ally believed to result from focal incomplete disjunc-tion of the neuroectoderm from the cutaneousectoderm [16]. Dermal sinuses are easily recognized onmidsagittal MR images as a thin hypointense stripewithin the subcutaneous fat (figure 9 and 13), whereasthey are more difficult to detect on axial scans. Theintrathecal portion of the tract usually is not detectable
on MRI, which makes it difficult to assess the trueextent of the tract itself and, particularly, whether itpierces the dura and involves the CNS. In a consider-able percentage of cases, dermal sinuses are associatedwith dermoids, generally located at level of the caudaequina or near the conus medullaris and probablyresulting from encystment of part of the dermal sinus
a b
FIG. 14. – Dermal sinus with dermoid, 8-year-old girl. A. Slightly parasagittal T2-weighted image shows sacral dermal sinuscoursing obliquely downward in subcutaneous fat (arrow). B. Midsagittal T2-weighted image shows huge dermoid in the thecalsac (arrowheads), extending upward to the tip of the conus medullaris. The mass gives slightly lower signal than cerebrospinalfluid and is outlined by a thin low-signal rim.
FIG. 14. – Sinus dermique avec kyste dermoïde, fillette de 8 ans. A. Séquence pondérée en T2 – coupe parasagittale. Sinus dermi-que sacré de trajet oblique vers le bas dans la graisse sous-cutanée (flèche). B. Séquence pondérée en T2. Kyste dermoïde dans lesac tecal (tête de flèche) s’étendant vers le haut jusqu’au cône médullaire. Le signal de la masse est légèrement plus bas que celui duliquide cérébrospinal et est entouré par un fin anneau en hyposignal.
FIG. 13. – A. Thoracic demal sinus, 2-month-old boy. Sag-ittal T1-weighted image shows dermal sinus coursingobliquely through the subcutaneous fat (arrow).
FIG. 13. – Sinus dermique thoracique, garcon âgé de 2 mois.A. Séquence pondérée en T1 – coupe sagittale : sinus dermiquede trajet oblique dans la graisse sous-cutanée (flèche).

SPINAL DYSRAPHISM 15
tract (figure 14). This association was found in 11.3% ofcases in our series [51], but may be higher. Dermoidsshow variable MRI features depending on their con-tent. Some portions may be hyperintense on T1-weighted images, but the mass may be isointense toCSF on both T1- and T2-weighted images and, there-fore, may be difficult to discern. Infected dermoidsexhibit intense contrast enhancement that may be ring-like if an abscess develops.
Persistent terminal ventricle
The “fifth ventricle” of the historic scientific litera-ture [21] is a small ependyma-lined cavity within theconus medullaris that always is identifiable on post-mortem examinations but must achieve a certain sizeto be visible at MRI (figure 15) [10]. Embryologically,it is related to incomplete regression of the terminalventricle during secondary neurulation, with preser-vation of its continuity with the central canal of therostral spinal cord. The latter point is critical becausefailure of regression of the terminal ventricle associ-ated with inefficient connection to the central canalabove may produce a terminal myelocystocele, whichis a much more severe abnormality.
By itself, the persistent terminal ventricle is asymp-tomatic; however, cases have been reported in whicha huge cystic dilation was associated with low-backpain, sciatica, and bladder disorders [44]. It is unclearwhether these “terminal ventricle cysts” are develop-mental variants or result from pathological changesleading to obstruction of the terminal ventricle [10].Differentiation with hydromyelia is based on the loca-tion immediately above the filum terminale, whereasintramedullary tumors are excluded by the lack ofgadolinium enhancement. The size of the “cyst” usu-ally remains unchanged on follow-up scans.
Complex dysraphic states
Because gastrulation is characterized by thedevelopment of the notochord, spinal dysraphismsoriginating during this period will characteristicallyshow a complex picture in which not only the spinalcord, but also other organs deriving from or inducedby the notochord, are severely abnormal. Therefore,disorders of gastrulation are also called complex dys-raphic states [13]. In the vast majority of cases, theseabnormalities are covered by skin, and no tell-talesubcutaneous masses are present. The only excep-tions are hemimyelocele and hemimyelomeningo-cele, two exceedingly rare abnormalities that weredescribed in the “open spinal dysraphism” section.
Failures of notochordal development have beencategorized in two subsets: (1) disorders of midlinenotochordal integration, which result in longitudinalsplitting, and (2) disorders of notochordal formation,which result in the absence of a certain notochordalsegment [50]. A thorough discussion of all complexdysraphic states is beyond the scope of this paper.Only the main entities will be dealt with here.
Disorders of midline notochordal integrationEmbryologically, prospective notochordal cells are
derived from the Hensen’s node, and stream in equalnumbers from both sides of the node past the primitivepit to migrate between the ectoderm and the endodermin the midline. Midline integration [51] is the processby which the two paired notochordal anlagen fuse inthe midline to form a single notochordal process. Thecause of failed midline notochordal integration hasbeen the source of continuing debate among authors,and several possible explanations have been proposed[17]. The eventual malformation depends on the sever-ity of the abnormality and the outcome of the repair
b
FIG. 15. – Persistent terminal ventricle. A. Sagittal T1-weighted image shows faint hypointensity within the conus medullaris(arrow). There is concurrent filar lipoma (arrowheads). B. Axial T1-weighted image confirms intramedullary cavity involving theconus medullaris (arrow), at the anatomic site of the terminal ventricle.
FIG. 15. – Persistance du ventricule terminal. A. Séquence pondérée en T1 – coupe sagittale. Hyposignal discret dans le cônemédullaire (flèche). Présence d’un lipome (tête de flèche). B. Séquence pondérée en T1 – coupe axiale. Cavité de siège intramédul-laire (flèche).
a

16 A. ROSSI et al.
efforts of the embryo. Several malformations belong tothis wide group [51]. Only the most important entitieswill be dealt with here.
Neurenteric cysts
The most severe form of failed midline noto-chordal integration is dorsal enteric fistula, in whichan abnormal canal connecting the skin surface withthe bowel crosses the intervening space between aduplicated spine [51]. Complete dorsal enteric fistulahas only exceptionally been reported in the litera-ture. Neurenteric cysts, usually located anterior tothe spinal cord in close connection with vertebralabnormalities, probably represent localized forms ofdorsal enteric fistula. They are lined with a mucin-secreting, cuboidal or columnar epithelium thatresembles the alimentary tract [51]. Their content isvariable, and the chemical composition may be simi-lar to CSF in some cases. The typical location isintradural in the cervicothoracic spine anterior tothe cord (figure 16); however, neurenteric cysts alsomay be found in the lumbar spine and even in theposterior fossa.
Embryologically, neurenteric cysts are related toendodermal differentiation of primitive streak rem-nants that remain trapped between a split notochord.Owing to the common underlying embryologicalmechanism, neurenteric cysts frequently are associ-ated with diastematomyelia, in which case they maybe located in the cleft between the two hemicords.
On MRI (figure 16), neurenteric cysts usually areisointense to hyperintense relative to CSF on longrelaxation time sequences. On T1-weighted images,
they appear isointense or slightly hyperintense toCSF, consistent with high-protein content. Absenceof contrast enhancement is the rule; however, wehave seen one case of a neurenteric cyst thatenhanced following intravenous gadolinium adminis-tration [51].
Diastematomyelia
The most frequent form of failed midline integra-tion of the notochord is diastematomyelia. Diastem-atomyelia (literally, split cord) refers to a variablyelongated separation of the spinal cord in two usu-ally symmetric halves. Whether this malformationrepresents true cord splitting or cord duplication hasbeen the subject of enduring debate. In fact, there isa continuous spectrum of abnormality rangingbetween partially cleft cord in a single dural tube atone end and completely duplicated cord within dualdural tubes with intervening bony spur at the otherend. The term “split cord malformations” (SCM)has been suggested to describe this malformativecontinuum [33, 35]. We believe this is just an Englishtranslation of the original Greek derived term,diastematomyelia. We therefore encourage to retainthe traditional denomination, which also has theadvantage of being widely recognized and acceptedin the literature.
Abnormal midline integration results in two pairednotochordal processes separated by intervening primi-tive streak cells. Each heminotochord induces a sep-arate neural plate. The resulting malformationdepends on the developmental fate of the interven-ing primitive streak tissue. If it further developstoward bone and cartilage, the two hemicords even-tually will be contained into two individual duralsacs separated by an osteocartilaginous spur. Con-versely, if the primitive streak tissue is reabsorbed orleaves a thin fibrous septum, the two hemicordseventually will lie within a single dural tube. Thisrepresents the foundation of classification ofdiastematomyelia into two groups [33].
Diastematomyelia type I consists of two hemi-cords contained within individual dural tubes, sepa-rated by a bone or osteocartilaginous septum thatextends from the vertebral body to the neural arches[33]. This rigid median septum is entirely extradural.Clinically, patients usually present with scoliosis andTCS. A hairy tuft lying high along the child’s back isa very reliable clinical marker of diastematomyelia[1]. Vertebral anomalies are the rule and includebifid lamina, widened interpediculate distance,hemivertebrae, bifid vertebrae, fused vertebrae, andnarrowing of the intervertebral disk space. Scoliosisalso is common and is seen in 30-60% of these indi-viduals.
The radiological hallmark [28] is the osseous orosteocartilaginous septum with resulting doubledural tubes, each containing a hemicord (figure 17).Although in the archetypal case the spur isosseous and connects the vertebral body to theneural arch along a midsagittal plane, “atypical”spurs are common. The spur may course obliquely,and may be complete or incomplete, in which case itmay originate either from the vertebral body or fromthe neural arch. In some cases, it divides the spinal
FIG. 16. – Neurenteric cyst. Sagittal PD-weighted imageshows an intradural cyst ventral to the spinal cord at the C7-T2 level. Reproduced with permission from Martin AJ, Pen-ney CC. Spinal neurenteric cyst. Arch Neurol 2001;58:126-127. Copyrighted 2001, American Medical Association.
FIG. 16. – Kyste neurentérique. Séquence en densité de pro-tons – coupe sagittale. Kyste intradural situé en avant de lamoelle en regard de C7-T2. Avec la permission de Martin AJ,Penney CC. Spinal neurenteric cyst. Arch Neurol 2001;58:126-127. Copyrighted 2001, American Medical Association.

SPINAL DYSRAPHISM 17
canal unequally, and the two hemicords will beasymmetric. In young children, the spur may bemostly cartilagineous and progressively ossifies asthe child grows. In most cases of diastematomyeliatype I, the cleft is located at the thoracic or lumbarlevel and lies at the caudal end of the cord splitting.The two hemicords usually surround the spur tightlybefore fusing with each other to form a normal spi-nal cord below, whereas rostrally the splitting ismuch more elongated. Therefore, there is a cranio-caudal sequence of partial clefting, completediastematomyelia with single dural tube, anddiastematomyelia with dual dural tubes (figure 18).Hydromyelia is a common associated finding andmay involve the normal cord both above and belowthe splitting, as well as one or both hemicords(figure 18) [41]. In the vast majority of cases, thehemicords fuse again below the spur to form a nor-mal cord. In rare cases the cleft may be terminal, inwhich case two hemicones and hemifilum (generallyhypertrophic and tethered) are present. Failure ofneurulation of one hemicord produces a hemimyelo-cele or hemimyelomeningocele.
Diastematomyelia type II has a common duraltube housing both hemicords without interveningrigid median septum [33, 28. Three variants exist:presence of an intervening fibrous septum, absenceof a septum, and partial cord splitting [50].
A midline, nonrigid, fibrous septum sometimes isdetected at surgery. In these cases, clinical signs ofTCS may appear, and indeed the assumption thatdiastematomyelia type II does not produce TCS isincorrect [35]. These septa may be identified on axialand coronal T2-weighted images as thin hypointensestripes interposed between the two hemicords(figure 19). Absence of a septum is the most
common occurrence in diastematomyelia type II(figure 20). Although the diagnosis is relativelystraightforward both on axial and coronal MRimages, diastematomyelia type II may be difficult toappreciate on sagittal MR images, where the onlytell-tale sign is an apparent thinning of the spinalcord that results from partial averaging with theintervening subarachnoid space between the twohemicords (figure 19 and 20) [50]. In rare cases, thecleft is partial and the splitting incomplete; these arethe mildest forms of diastematomyelia (figure 21)[49].
Hydromyelia may be present with the same featuresas in diastematomyelia type I. The conus medullaris istypically low, and there is a strong association withtight filum terminale and filar lipomas. Associatedvertebral anomalies are usually milder than in typeI, and are represented by butterfly vertebrae in mostcases. Posterior spina bifida is often present, whereasscoliosis is usually absent.
Disorders of notochordal formationProgrammed cell death, or apoptosis, is a process
of cell elimination that occurs during normal develop-ment and represents a crucial phenomenon in varioussteps of embryogenesis. During abnormal gastrula-tion, prospective notochordal cells that are wronglyspecified in terms of their rostrocaudal positionalencoding are eliminated. Eventually, fewer cells oreven no cell will form the notochord at a given abnor-mal segmental level. The consequences of such a seg-mental notochordal paucity are manifold and affectthe development of the spinal column and spinal cordas well as of other organs that rely on the notochord astheir inductor. If the prospective notochord is depleted,a wide array of segmental vertebral malformations
a b
FIG. 17. – Diastematomyelia type I. A. Axial CT scan shows bony spur (S) separating the spinal canal into two halves, eachcontaining a separate dural sac (asterisks). The spur articulates with the vertebral body anteriorly (arrowheads), whereas poste-riorly there is a bony spina bifida. B. Axial T1-weighted image shows the spur (S) and the dual dural tubes, each containing ahemicord (hc).
FIG. 17. – Diasthématomyélie de type I. A. Scanner – coupe axiale. Epine osseuse (S) séparant le canal rachidien en deux partiescontenant chacune un sac dural (astérisque). L’épine s’attache sur le corps vertébral en avant (tête de flèche). Pas de spina bifida.B. Séquence pondérée en T1 – coupe axiale. Epine (S) et sacs duraux contenant une hémimoelle (hc).

18 A. ROSSI et al.
b d
FIG. 18. – Diastematomyelia type I with hydromyelia, 1-year-oldgirl. A. Axial CT scan shows obliquely coursing bony spur (S), sep-arating the abnormally large spinal canal into two asymmetric parts.There is associated left posterior spina bifida. B. Parasagittal, C. mid-sagittal, and D. coronal T1-weighted images shows asymmetric bonyspur (S) containing high-signal bone marrow and projecting into thespinal canal. The spur is located at the bottom end of the cord split-ting, and each hemicord contains hydromyelic cavities (arrowheads).There also is hydromyelia involving the spinal cord both above andbelow the splitting (arrows). Multiple vertebral segmentation defectsinvolving the lumbar spine and sacrum are revealed by rudimentaryintervertebral disks. E-G. Axial T2-weighted images show the mal-formation sequence from cephalad to caudad: E hydromyelia (aster-isks) with initial cord separation, F split spinal cord (hc) within singledural sac associated with right hydromyelia (asterisk), and G splitspinal cord with dual dural sacs and intervening bony spur (S). Axialviews also show the right hemicord has one paramedian set of nerveroots connected to the bony spur (thin arrow, G); anterior and pos-terior set of nerve roots is also seen (arrowheads, G).
FIG. 18. – Diasthématomyélie type I avec hydromyélie, fillette de 1 an. A. Scanner – coupe axiale. Epine osseuse séparantun canal anormalement large en deux parties asymétriques. Spina bifida postérieure gauche associée. B. Séquence pondéréeen T1 – coupes parasagittale (B), sagittale moyenne (C) et frontale (D). Epine osseuse asymétrique (S) contenant un hyper-signal (moelle osseuse). L’épine est située au sommet du dédoublement médullaire. Chaque hémimoelle contient une cavitéhydromyélique (tête de flèche). Il existe une hydromyélie dans la moelle située au-dessus et au-dessous du dédoublement(flèche). De multiples défauts de segmentation vertébraux atteignant le rachis lombaire, le sacrum, sont révélés par l’existencede disques intervertébraux rudimentaires. E-G. Séquence pondérée en T2 – coupe axiale (fig. 18E à 18G). (E) Hydromyélie(astérisque). (F) Dédoublement médullaire (hc) (astérisque) avec sac duraux et épine osseuse (S). Ces vues axiales montrentdes racines nerveuses, en rapport avec l’hémimoelle droite et connectée à l’épine osseuse (flèche fine, G) ; des racines antérieu-res et postérieures sont également observées (tête de flèche, G).
a c
e g
h

SPINAL DYSRAPHISM 19
including segmentation defects, indeterminate orblock vertebrae, or absence of several vertebrae, willresult. Because of lack of neural induction andabsence of a floor plate, fewer prospective neuroecto-dermal cells, or even no cell at all, will be induced toform the neural tube in the pathological segment [48].The resulting malformation essentially depends onthe segmental level and the extent of the abnormalityalong the longitudinal embryonic axis, with subse-quent interference on the processes of primary and/orsecondary neurulation. In the vast majority of cases,the abnormality involves the caudal extremity of theembryo, resulting in the caudal agenesis constellation.Much more rarely, the abnormality involves an inter-mediate notochordal segment, thereby resulting insegmental spinal dysgenesis [48].
Caudal agenesis
Caudal agenesis (CA) is a heterogeneous constella-tion of anomalies comprising total or partial agenesis ofthe spinal column, anal imperforation, genital anoma-lies, bilateral renal dysplasia or aplasia, pulmonary
hypoplasia, and lower limb abnormalities [15]. CA isalso commonly, albeit inappropriately, called caudalregression syndrome; etymologically, the term “caudalagenesis” should be preferred, as “caudal regression”implies a concept of excessive regression of the embry-onic tail that cannot be adequately applied in tail-lessanimals, such as humans (M. Catala, personal commu-nication). Agenesis of the sacrococcygeal spine may bepart of syndromic complexes such as OEIS (omphalo-cele, cloacal exstrophy, imperforate anus, and spinaldeformities) [7], VACTERL (vertebral abnormality,anal imperforation, cardiac anomalies, tracheoeso-phageal fistula, renal abnormalities, limb deformities)[45], and the Currarino triad (partial sacral agenesis,anorectal malformation, and presacral mass: teratomaand/or meningocele) [11, 12, 19]. There is a definiteassociation with maternal diabetes mellitus (1% of off-spring of diabetic mothers). CA in humans can beinherited as an autosomal dominant condition [9].
The congenital spectrum of vertebral abnormalityin CA may range from agenesis of the coccyx toabsence of the sacral, lumbar, and lower thoracicvertebrae, but the vast majority of these anomalies
a b c
d
FIG. 19. – Diastematomyelia type II with fibrous septum,11-year-old girl. A. The only tell-tale sign of the abnormality onthis sagittal T2-weighted image is an apparent thinning of thespinal cord (arrow), which actually results from the interveningsubarachnoid space between the two hemicords. The spinalcanal is enlarged. There are concurrent vertebral segmentationdefects with rudimentary intervertebral disks (arrowheads). B.Coronal and C. axial T2-weighted images show the two hemi-cords (hc) are contained within a single dural sac, which isdivided into two halves by an intervening hypointense band(arrowhead). A fibrous septum was found at surgery.
FIG. 19. – Diasthématomyélie de type II avec septumfibreux, fillette de 11 ans. A. Séquence pondérée en T2 – coupesagittale. Aspect d’amincissement de la moelle épinière (flèche)résultant de l’interposition d’espace sous-arachnoïdien entre lesdeux hémimoelles. Elargissement du canal rachidien. Défautsde segmentations vertébraux avec disques intervertébraux rudi-mentaires. B. Coupe coronale et C. coupe axiale sequence pon-dérée en T2. Les deux hémimoelles (hc) sont contenues dansun simple sac dural divisé en deux parties par une bandehypointense (tête de flèche). Le septum fibreux a été retrouvé àl’intervention chirurgicale.

20 A. ROSSI et al.
involve only the sacrum and coccyx. The sacrummay be totally or subtotally absent, with S1 throughS4 present in individual cases. Sacral aplasia may beasymmetric, with resulting total or subtotal hemis-acrum that may, in turn, be unilateral or bilateral[34]. The heterogeneous spectrum of vertebral mal-
formation requires anteroposterior and lateral plainradiographs for full appreciation. These radiographicstudies constitute an essential part of the neuroradi-ological workup.
Traditionally, CA has been categorized into twotypes depending on the location and shape of the
b c
FIG. 20. – Diastematomyelia type II without septum, 13-year-old girl. A. Sagittal T2-weighted image shows a low spinal cordwith apparent focal thinning (arrow) resulting from partial averaging with the intervening subarachnoid space between the twohemicords. Hydromyelia (H) involves the cord above the splitting. There is associated tight filum terminale (arrowhead). B. Coro-nal T2-weighted image shows split hemicords (hc) and cranial hydromyelia (H). C. Axial T2-weighted image clearly shows thetwo hemicords (hc) contained into a single dural tube, with no intervening septum.
FIG. 20. – Diasthématomyélie de type II sans septum, fillette de 13 ans. A. Séquence pondérée en T2 – coupe sagittale. Moelle épi-nière basse avec aspect d’amincissement (flèche) résultant de la présence d’espace sous-arachnoïdien entre les deux hémimoelles.Hydromyélie (H) située dans la moelle au-dessus du dédoublement. Tight filum terminale (tête de flèche). B. Séquence pondérée enT2 –coupe frontale. Hémimoelle (hc) et hydromyélie crâniale (H). C. Séquence pondérée en T2 – coupe axiale. Les deux hémimoel-les (hc) sont contenues dans un même sac dural sans septum.
a
a b
FIG. 21. – Diastematomyelia type II with partial splitting, 4-month-old girl. Same case as fig. 12. A. Sagittal T2-weighted imageshows faint thinning of the conus medullaris (arrowhead), which has a low position due to associated tight filum terminale.B. Axial T2-weighted image shows incomplete cord splitting (arrow).
FIG. 21. – Diasthématomyélie de type II avec dédoublement partiel, fillette de 4 mois. Même cas que celui de la fig. 12. A. Séquencepondérée en T2 – coupe sagittale. Amincissement modéré des cônes médullaires (tête de flèche) en position basse du fait de l’existenced’un tight filum terminale. B. Séquence pondérée en T2 – coupe axiale. Dédoublement incomplet de la moelle (flèche).

SPINAL DYSRAPHISM 21
conus medullaris: either high and abrupt (type I) orlow and tethered (type II). Although these two typeswere believed to be embryologically related to disor-dered primary or secondary neurulation, respec-tively [32], both are actually consistent with anearlier abnormality of gastrulation [50]. Segmentalmaldevelopment of the caudal notochord and axial-paraxial mesoderm results in an abnormality thatinterferes with either secondary neurulation alone,or both primary and secondary neurulation,depending on the longitudinal extent of the originalnotochordal damage [50]. Therefore, the crucialembryological watershed between the two varietiesis the interface between primary and secondary neu-rulation (i.e., the junction between the true noto-chord and the tail bud), corresponding to the caudalend of the future neural plate. This site has been thesource of continuing debate among authors: recentdata suggest that it corresponds to S3 through S5[31]. Based on this theory, the degree of spinal cordaplasia correlates with the severity of the spinal mal-formation, with a greater degree of vertebral aplasiain type I than in type II [32].
Type I CA (figure 22 and 23). If not only the tailbud, but also part of the true notochord fails todevelop, interference is generated with both theprocesses of primary and secondary neurulation[50]. Depending on the severity of the originaldamage, the eventual degree of vertebral aplasiawill range from absence of the coccyx and lowermidsacrum to aplasia of all coccygeal, sacral,
lumbar, and lower thoracic vertebrae (figure 22).However, the last vertebra is L5 through S2 in themajority of patients. Owing to the same embryolog-ical mechanism, there is aplasia of the caudalmetameres of the spinal cord. This results in anabrupt spinal cord terminus that nearly always isclub or wedge shaped (figure 23) [2]. The spinalcord terminus is high-lying (most often oppositeT12) in most cases, but it may lie opposite to L1 ina minority of cases (figure 22). The cauda equinaalso is frequently abnormal, and the nerve rootshave an abnormal course that has been termed the“double bundle shape” [34]. The thecal sac tapersbelow the cord terminus, and ends at an unusuallyhigh level (figure 23). Associated caudal anomalies,such as anterior meningoceles and teratomas, maybe present, although much less frequently than intype II CA. Unlike in type II, the cord is not teth-ered to these caudal anomalies. This accounts for thenegligible proportion of TCS with progressive neuro-logical deterioration in these patients, contrary topatients with type II CA. Generally, these patientshave a stable neurological defect due to their“fixed” spinal cord dysplasia [34], and their motordeficit tends to parallel the extent of the bonyabnormality. Conversely, sensory findings are muchless well predictable from the radiographic appear-ance.
Type II CA (figure 24 and 25). If the whole or apart of the tail bud fails to develop but the true noto-chord is unaffected, primary neurulation occurs nor-
a b
FIG. 22. – Caudal agenesis, type I: lumbar/thoracolumbaragenesis, 8-year-old girl. A. Radiograph, anteroposterior viewshows agenesis of the lumbar and sacrococcygeal spine withilioiliac approximation and reduced pelvic diameters. Only arudiment of T12 articulating with an abnormal pair of ribs(arrowheads) is visible. B. Sagittal T1-weighted image showsthe spinal cord terminates abruptly opposite T9 (arrow).
FIG. 22. – Agénésie caudale de type I. Agénésie lombaire/thoracolombaire, fillette de 8 ans. A. Radiographie standard.Agénésie des rachis lombaire et sacrococcygien. Réduction dudiamètre pelvien. La vertèbre T12 est rudimentaire et s’articuleavec une des côtes anormales (tête de flèche). B. Séquence pon-dérée en T1 – coupe sagittale. Terminaison de moelle épinière,brutale en regard de T9 (flèche).
FIG. 23. – Caudal agenesis, type I. 8-year-old boy. A. SagittalT1-weighted image and B. sagittal T2-weighted image showsubtotal sacrococcygeal agenesis, with a rudiment of S1 as thelast visible vertebra, articulating with medialized ileum (I). Thecord terminus is blunt and lies opposite the lower half of L1(arrow), a somewhat atypically “low” position for type I CA.There is terminal hydromyelia and “double bundle” arrange-ment of the nerve roots of the cauda equina. The dural sactapers abruptly and ends abnormally high (arrowheads).
FIG. 23. – Agénésie caudale type I, garcon de 8 ans.A. Séquence pondérée en T1 – coupe sagittale et pondérée en T2coupe sagittale. Agénésie subtotale sacrococcygienne. Aspect rudi-mentaire de S2 (dernière vertèbre visible) s’articulant avec l’iliaque(I) en position médiale.
a b

22 A. ROSSI et al.
mally, and there is interference only with the processof secondary neurulation [50]. There is a lesserdegree of vertebral dysgenesis than in type I, with upto S4 present as the last vertebra. As a consequence,only the most caudal portion of the conus medullaris(corresponding to the metameres formed by second-ary neurulation) is absent. In these cases, partialagenesis of the conus is difficult to recognize,because the conus itself is stretched caudally andtethered to a tight filum, lipoma (figure 24), terminalmyelocystocele, or lipomyelomeningocele (figure 6).In some cases, the cord tapers progressively to tetherto the neck of an anterior sacral meningocele. Insuch cases, the anomaly may be discovered in laterchildhood or adolescence, when the patient developsconstipation, urinary incontinence, dysmenorrhea,dyspareunia, or back pain. Teratomas or other cau-dal tumors can be found in a minority of patients.Low-back masses must be differentiated from over-growing fatty tissue that is sometimes present atlevel of the buttocks in these patients. More oftenthan in type I CA, imaging studies performed inthese patients may be difficult to interpret, especiallywhen looking for small teratomatous masses alongthe walls of anterior meningoceles, in the presacralspace, or deep within the pelvic cavity. In thesecases, presaturation slabs must not be placed ante-rior to the spinal column in order not to miss presac-ral malformed components.
Segmental spinal dysgenesis
The clinical-radiological definition of SSDincludes (1) segmental agenesis or dysgenesis of thelumbar or thoracolumbar spine; (2) segmental
abnormality of the underlying spinal cord and nerveroots; (3) congenital paraplegia or paraparesis; and(4) congenital lower limb deformities [48]. Segmen-tal vertebral anomalies may involve the thoracolum-bar, lumbar, or lumbosacral spine. As is the casewith CA, the embryogenesis of SSD may be relatedto genetically induced notochordal derangementduring gastrulation involving an intermediate, ratherthan the caudalmost, segment of the notochord [48].
In the most severe cases, the spinal cord at thelevel of the abnormality is thoroughly absent, andthe bony spine is focally aplastic. As a result, thespine and spinal cord are “cut in two” (figure 25),with resulting acute angle kyphosis. Between the twospinal segments, the spinal canal is extremely nar-rowed or even totally interrupted. The lower spinalcord segment is invariably bulky and low-lying [48].A horseshoe kidney is typically lodged in theconcavity of the kyphosis [50]. Newborns with severeSSD are paraplegic at birth and invariably showhypotrophic and deformed lower limbs withequinocavovarus feet.
In less severe cases, there is focal hypoplasia ofthe spinal cord, which will therefore appear nar-rower than normal on MRI studies (figure 26) [48].There is no disconnection of either the spinal cord orthe spine, although bony stenosis of the spinal canaland minor vertebral abnormalities affect the patho-logical segment.
FIG. 24. – Caudal agenesis, type II. 3-month-old boy. SagittalT1-weighted image shows the spinal cord is low and tethered(arrow) to an intradural lipoma (L). The vertebral anomaly isless severe than in type I, with S3 present in this case.
FIG. 24. – Agénésie caudale type II, garcon de 3 mois.Séquence pondérée en T2 – coupe sagittakle. Moelle en positionbasse (flèche), lipome intradural (L). Les anomalies vertébralessont moins sévères que dans le type I ; S3 est présent dans ce cas.
FIG. 25. – Segmental spinal dysgenesis, 2-month-old girl.Sagittal T2-weighted image shows acute thoracolumbarkyphosis with complete interruption of the spinal column.There are two completely separated spinal cord segments; theupper ends several vertebral levels above the gibbus (whitearrowhead) and shows hydromyelia, whereas the lower isbulky and low (arrows). Notice extreme narrowing of spinalcanal at the gibbus apex (black arrowheads).
FIG. 25. – Dysgénésie spinale segmentaire, fillette de 2 mois.Séquence pondérée en T2 – coupe sagittale. Cyphose thoraco-lombaire avec interruption complète du rachis. Il existe deux seg-ments de moelle totalement séparés : la portion supérieure setermine à quelques niveaux vertébraux sous le gibbus (tête deflèche blanche et la partie inférieure est volumineuse (flèche). Ànoter le rétrécissement du canal rachidien faissant passer le gib-bus par bosse à hauteur du sommet de la cyphose (flèche noire).

SPINAL DYSRAPHISM 23
CONCLUSION
Although the MR picture in patients with spinalcord malformations may appear complicated andpuzzling even to the experienced observer, webelieve that a rational approach focusing on a corre-lation among clinical, embryological, and neuroradio-logical data greatly facilitates the diagnosis.Neuroradiologists should seek the maximum degreeof collaboration with neurosurgeons and other spe-cialists involved in the management of spina bifidapatients in order to improve their diagnostic capabili-ties and to provide more useful information for themanagement of these children.
ACKNOWLEDGEMENTS
The authors are indebted to Dr Martin Catala(Faculté de Médecine Pitié-Salpêtrière, Paris, France)for providing insight into the more recent embryo-logical theories on spinal cord development.
RÉFÉRENCES
[1] BARKOVICH AJ, EDWARDS MSB, COGEN PH. MR evaluationof spinal dermal sinus tracts in children. AJNR Am J Neuro-radiol 1991; 12: 123-129.
[2] BARKOVICH AJ, RAGHAVAN N, CHUANG SH. MR of lumbo-sacral agenesis. AJNR Am J Neuroradiol 1989; 10: 1223-1231.
[3] BRENINGSTALL GN, MARKER SM, TUBMAN DE. Hydrosyrin-gomyelia and diastematomyelia detected by MRI in myelo-meningocele. Pediatr Neurol 1992; 8: 267-271.
[4] BROWN E, MATTHES JC, BAZAN C 3rd, JINKINS JR. Preva-lence of incidental intraspinal lipoma of the lumbosacralspine as determined by MRI. Spine 1994; 19: 833–836.
[5] BYRD SE, HARVEY C, DARLING CF. MR of terminal myelo-cystoceles. Eur J Radiol 1995; 20: 215-220.
[6] CAMA A, TORTORI-DONATI P, PIATELLI GL, FONDELLI MP,ANDREUSSI L. Chiari complex in children. Neuroradiologicaldiagnosis, neurosurgical treatment and proposal of a newclassification (312 cases). Eur J Pediatr Surg 1995; 5 (Suppl1): 35-38.
[7] CAREY JC, GREENBAUM B, HALL BD. The OEIS complex(omphalocele, exstrophy, imperforate anus, spinal defects).Birth Defects Orig Artic Ser 1978; 14: 253-263.
[8] CATALA M. Embryogenesis. Why do we need a new expla-nation for the emergence of spina bifida with lipoma? ChildsNerv Syst 1997; 13: 336-340.
[9] CATALA M. Genetic control of caudal development. Clin Ge-net 2002; 61: 89-96.
[10] COLEMAN LT, ZIMMERMAN RA, RORKE LB. Ventriculus ter-minalis of the conus medullaris: MR findings in children.AJNR Am J Neuroradiol 1995; 16: 1421-1426.
[11] CURRARINO G, COLN D, VOTTELER T. Triad of anorectal, sa-cral, and presacral anomalies. AJR Am J Roentgenol 1981;137: 395-398.
[12] DIAS MS, AZIZKHAN RG. A novel embryogenetic mecha-nism for Currarino’s triad: inadequate dorsoventral separa-tion of the caudal eminence from hindgut endoderm. PediatrNeurosurg 1998; 28: 223-229.
[13] DIAS MS, WALKER ML. The embryogenesis of complex dys-raphic malformations: a disorder of gastrulation? PediatrNeurosurg 1992; 18: 229-253.
[14] DROLET B. Birthmarks to worry about. Cutaneous markersof dysraphism. Dermatol Clin 1998; 16: 447-453.
[15] DUHAMEL B. From the mermaid to anal imperforation: thesyndrome of caudal regression. Arch Dis Child 1961; 36: 152-155.
[16] ELTON S, OAKES WJ. Dermal sinus tracts of the spine. Neu-rosurg Focus [serial online]. January 2001; 10: Article 4.Available at: http: //www.neurosurgery.org/focus/jan01/10-1-4.pdf. Accessed May 22, 2003.
[17] FARIS JC, CROWE JE. The split notochord syndrome. J Pe-diatr Surg 1975; 10: 467-472.
[18] FRENCH BN. The embryology of spinal dysraphism. ClinNeurosurg 1983; 30: 295-340.
[19] GUDINCHET F, MAEDER P, LAURENT T, MEYRAT B, SCHNY-DER P. Magnetic resonance detection of myelodysplasia inchildren with Currarino triad. Pediatr Radiol 1997; 27: 903-907.
[20] HERMAN JM, MCLONE DG, STORRS BB, DAUSER RC. Ana-lysis of 153 patients with myelomeningocele or spinal lipomareoperated upon for a tethered cord. Pediatr Neurosurg 1993;19: 243-249.
[21] KERNOHAN JW. The ventriculus terminalis: its growth anddevelopment. J Comp Neurol 1924; 38: 10-125.
[22] KNITTLE JL, TIMMERS K, GINSBERG-FELLNER F, BROWN RE,KATZ DP. The growth of adipose tissue in children and ado-lescents. Cross-sectional and longitudinal studies of adiposecell number and size. J Clin Invest 1979; 63: 269-246.
[23] LEE KS, GOWER DJ, MCWHORTER JM, ALBERTSON DA.The role of MR imaging in the diagnosis and treatment ofanterior sacral meningocele. Report of 2 cases. J Neurosurg1988; 69: 628-631.
[24] MCLONE DG, DIAS MS. Complications of myelomeningo-cele closure. Pediatr Neurosurg 1991-92; 17: 267-273.
[25] MCLONE DG, KNEPPER PA. The cause of Chiari II malfor-mation: a unified theory. Pediatr Neurosci 1989; 15: 1-12.
[26] MCLONE DG, NAIDICH TP. Terminal myelocystocele. Neu-rosurgery 1985; 16: 36-43.
[27] NAIDICH TP, BLASER SI, DELMAN BN, MCLONE DG, DIAS MS,ZIMMERMAN RA, RAYBAUD CA, BIRSCHANSKY SB, ALT-MAN NR, BRAFFMAN BH. Embryology of the spine and spi-nal cord. ASNR 2002: 3-13.
[28] NAIDICH TP, HARWOOD-NASH DC. Diastematomyelia. PartI. Hemicords and meningeal sheaths. Single and double ara-
FIG. 26. – Segmental spinal dysgenesis, 8-year-old boy.Sagittal T1-weighted image shows indeterminate vertebrae atthe upper lumbar level resulting in congenital kyphosis with-out complete disconnection of the spine. The spinal cord atthe dysgenesis level is thin (arrowheads). The conus med-ullaris is bulky and low (arrows).
FIG. 26. – Dysgénésie spinale segmentaire. Séquence pondé-rée en T1 – coupe sagittale montrant l’existence de vertebrasnon déterminées dans la region lombaire supérieure resultantd’une cyphose congénitale sans disconnexion totale du rachis.La moelle au niveau de la dysgénésie est fine (flèche). Le cônemédullaire est épaissi et bas (flèche).

24 A. ROSSI et al.
chnoid and dural tubes. AJNR Am J Neuroradiol 1983; 4:633-636.
[29] NAIDICH TP, MCLONE DG, FULLING F. The Chiari II malfor-mation. Part IV. The hindbrain deformity. Neuroradiology1983; 25: 179-197.
[30] NAIDICH TP, MCLONE DG, MUTLEUR S. A new understan-ding of dorsal dysraphism with lipoma (lipomyeloschisis): ra-diological evaluation and surgical correction. AJNR Am JNeuroradiol 1983; 4: 103-116.
[31] NIEVELSTEIN RAJ, HARTWIG NG, VERMEJI-KEERS C,VALK J. Embryonic development of the mammalian caudalneural tube. Teratology 1993; 48: 21-31.
[32] NIEVELSTEIN RAJ, VALK J, SMIT LME, VERMEJI-KEERS C.MR of the caudal regression syndrome: embryologic impli-cations. AJNR Am J Neuroradiol 1994; 15: 1021-1029.
[33] PANG D, DIAS MS, AHAB-BARMADA M. Split cord malfor-mation. Part I: a unified theory of embryogenesis for doublespinal cord malformations. Neurosurgery 1992; 31: 451-480.
[34] PANG D. Sacral agenesis and caudal spinal cord malforma-tions. Neurosurgery 1993; 32: 755-779.
[35] PANG D. Split cord malformation. Part II: clinical syndrome.Neurosurgery 1992; 31: 481-500.
[36] PEACOCK WJ, MUROVIC JA. Magnetic resonance imaging inmyelocystoceles. Report of two cases. J Neurosurg 1989; 70:804-807.
[37] PIERRE-KAHN A, ZERAH M, RENIER D, CINALLI G, SAINTE-ROSE C, LELLOUCH-TUBIANA A, BRUNELLE F, LE MER-RER M, GIUDICELLI Y, PICHON J, KLEINKNECHT B, NATAF F.Congenital lumbosacral lipomas. Childs Nerv Syst 1997; 13:298-334.
[38] PROP N, FRENSDORF EL. A postvertebral endodermal cystassociated with axial deformities: a case showing the “endo-dermal-ectodermal adhesion syndrome”. Pediatrics 1967; 39:555-562.
[39] RAGHAVAN N, BARKOVICH AJ, EDWARDS M, NORMAN D.MR imaging in the tethered spinal cord syndrome. AJNRAm J Neuroradiol 1989; 10: 27-36.
[40] SATTAR MT, BANNISTER CM, TURNBULL IW. Occult spinaldysraphism–The common combination of lesions and the cli-nical manifestations in 50 patients. Eur J Pediatr Surg 1996;6 (Suppl I): 10-14.
[41] SCHLESINGER AE, NAIDICH TP, QUENCER RM. Concurrenthydromyelia and diastematomyelia. AJNR Am J Neurora-diol 1986; 7: 473-477.
[42] SCOTT RM, WOLPERT SM, BARTOSHESKY LF, ZIMBLER S,KLAUBER GT. Dermoid tumors occurring at the site of pre-vious myelomeningocele repair. J Neurosurg 1986; 65: 779-783.
[43] SCOTTI G, HARWOOD-NASH DC. Congenital thoracic dermalsinus: diagnosis by computer assisted metrizamide myelogra-phy. J Comput Assist Tomogr 1980; 4: 675-677.
[44] SIGAL R, DENYS A, HALIMI P, SHAPEERO L, DOYON D,BOUDGHÈNE F. Ventriculus terminalis of the conus medulla-ris: MR imaging in four patients with congenital dilatation.AJNR Am J Neuroradiol 1991; 12: 733-737.
[45] SMITH NM, CHAMBERS HM, FURNESS ME, HAAN EA. TheOEIS omplex omphalocele-exstrophy-imperforate anus-spi-nal defects: recurrence in sibs. J Med Genet 1992; 29: 730-732.
[46] TORTORI-DONATI P, CAMA A, FONDELLI MP, ROSSI A. Lemalformazioni di Chiari. In: Tortori-Donati P, Taccone A,Longo M, eds. Malformazioni cranio-encefaliche. Neurora-diologia. Turin: Minerva Medica; 1996: 209-236.
[47] TORTORI-DONATI P, CAMA A, ROSA ML, ANDREUSSI L,TACCONE A. Occult spinal dysraphism: neuroradiologicalstudy. Neuroradiology 1990; 31: 512-522.
[48] TORTORI-DONATI P, FONDELLI MP, ROSSI A, RAYBAUD CA,CAMA A, CAPRA V. Segmental spinal dysgenesis. Neurora-diologic findings with clinical and embryologic correlation.AJNR Am J Neuroradiol 1999; 20: 445-456.
[49] TORTORI-DONATI P, FONDELLI MP, ROSSI A. Anomaliecongenite del midollo spinale. In: Simonetti G, Del MaschioA, Bartolozzi C, Passariello R, eds. Trattato Italiano di Ri-sonanza Magnetica. Naples: Idelson-Gnocchi; 1998: 517-553.
[50] TORTORI-DONATI P, ROSSI A, BIANCHERI R, CAMA A. Ma-gnetic resonance imaging of spinal dysraphism. Top MagnReson Imaging 2001; 12: 375-409.
[51] TORTORI-DONATI P, ROSSI A, CAMA A. Spinal dysraphism:a review of neuroradiological features with embryologicalcorrelations and proposal for a new classification. Neurora-diology 2000; 42: 471-491.
[52] UCHINO A, MORI T, OHNO M. Thickened fatty filum termi-nale: MR imaging. Neuroradiology 1991; 33: 331-333.
[53] WARDER DE, OAKES WJ. Tethered cord syndrome and theconus in a normal position. Neurosurgery 1993; 33: 374–378.
[54] WARDER DE, OAKES WJ. Tethered cord syndrome: the low-lying and normally positioned conus. Neurosurgery 1994; 34:597–600.
[55] WARDER DE. Tethered cord syndrome and occult spinaldysraphism. Neurosurg Focus [serial online]. January 2001;10: Article 1. Available at http: //www.neurosurgery.org/fo-cus/jan01/10-1-1.pdf. Accessed November 20, 2001.
[56] WEPRIN BE, OAKES WJ. Coccygeal pits. Pediatrics 2000; 105:E69.
[57] YUNDT KD, PARK TS, KAUFMAN BA. Normal diameter of fi-lum terminale in children: in vivo measurement. Pediatr Neu-rosurg 1997; 27: 257-259.