regulación molecular de los procesos biologicos
DESCRIPTION
Temario completo de Regulación Molecular de los Procesos Biologicos de la Universidad de Murcia.TRANSCRIPT

1
2014
Facultad de Biología Grado en Biología 1855: Regulación Mole-
cular de los procesos Biológicos.
[REGULACIÓN MOLECULAR DE LOS
PROCESOS BIOLÓGICOS] Adiv al Erbos

2

3
Bloque II: Regulación Molecular De Los Procesos Celulares
Tema 1: Aspectos metabólicos del Citosol ............................................. 13
1 Importancia del citosol en el metabolismo (Funciones) ..................................... 13
2 Genoma y proteoma: Origen de la diversidad estructural y funcional de las
proteínas ............................................................................................................ 15
2.1 Splicing alternativo ........................................................................................................ 16
2.2 Modificaciones post-traduccionales ............................................................................. 17
3 Ribosomas ....................................................................................................... 17
4 Síntesis de proteínas e inhibidores ..................................................................... 1
5 Chaperonas moleculares .................................................................................... 2
5.1 El descubrimiento de las Chaperonas ............................................................................. 3
5.2 El plegamiento de las proteínas ...................................................................................... 4
5.3 Funciones de las chaperonas .......................................................................................... 5
5.4 Clasificación de las HSP ................................................................................................... 6
5.5 Modo de acción de algunas chaperonas ......................................................................... 7
5.5.1 HSP70 y co-chaperonas (HSP20 y HSP40) ............................................................................... 7
5.5.2 HSP90 ..................................................................................................................................... 8
5.5.3 HSP60 (GroEL y GroES) ........................................................................................................... 9
5.6 Resumen ........................................................................................................................ 11
6 Cambios post-traduccionales (CPT) ................................................................... 13
6.1 Cambios post-traduccionales reversibles ...................................................................... 13
6.1.1 Fosforilación/defosforilación ................................................................................................ 13
6.1.2 Modificación de los dominios N-terminales de las histonas ................................................. 18
6.1.3 Otras modificaciones covalentes reversibles ........................................................................ 20
6.2 Cambios post-traduccionales irreversibles ................................................................... 21
6.2.1 Adición de restos hidrofóbicos .............................................................................................. 21
6.2.2 Glicosilación .......................................................................................................................... 25
6.2.3 Cambios por proteólisis limitada .......................................................................................... 25
6.2.4 Otros cambios post-traduccionales irreversibles .................................................................. 25
Tema 2: Tráfico citosol-núcleo y citosol-mitocondria ............................. 27
1. Tráfico citosol-núcleo ...................................................................................... 27
1.1. El poro nuclear ........................................................................................................ 28
1.2. La lámina nuclear ......................................................................................................... 30
1.3. Tráfico de proteínas a través del poro nuclear ............................................................ 31

4
1.3.1. Modelo de importación a través del poro nuclear .............................................................. 32
1.3.2. Modelo de exportación a través del poro nuclear ............................................................... 33
1.3.3. Modelo simplificado de importación/exportación de proteínas y mRNA ............................ 33
1.4. El transporte a través del poro en función del tamaño de la carga ............................. 34
1.5. Diversidad de carioferinas ............................................................................................ 34
1.6. Complejos protéicos asociados a la distrofina ............................................................. 35
2. Tráfico citosol-mitocondria y cloroplastos .................................................... 36
2.1. El genoma mitocondrial .......................................................................................... 36
2.2. Destinos de las proteínas importadas a los orgánulos energéticos ........................ 38
2.3 Transporte a través de la membrana mitocondrial ...................................................... 39
2.3.1. Señal de localización mitocondrial ...................................................................................... 39
2.3.2. Modelo general de transporte a la matriz mitocondrial ..................................................... 41
2.3.4. Modelo general de transporte a la membrana mitocondrial interna ................................. 42
2.3.5. Modelo general de transporte al espacio intermembrana .................................................. 43
2.3.6. Modelo general de transporte a la membrana mitocondrial externa ................................. 44
2.4. Transporte citosol-cloroplasto ..................................................................................... 45
3. Tráfico de proteínas al peroxisoma ............................................................... 46
4. Tráfico de proteínas a los plastidios ................................................................. 46
Anexo I: Dinámica del complejo TIM ................................................................... 47
Anexo II: Señales de localización mitocondrial ..................................................... 48
Anexo III: Genes y Parkinson ............................................................................... 49
Tema 3: Proteolisis intracelular ............................................................. 53
1. Procesos regulados por la proteolisis ............................................................... 53
2. El ciclo de los compuestos nitrogenados y la vida media de las proteínas ......... 54
3. Proteólisis intracelular .................................................................................... 55
3.1 Proteólisis intracelular en procariotas .......................................................................... 55
3.2 Proteólisis intracelular en eucariotas ............................................................................ 56
4. Sistema ubiquitina-proteasoma (UPS) de degradación proteica ....................... 57
4.1 La ubiquitina .................................................................................................................. 57
4.2 Sistema de adición de ubiquitina .................................................................................. 58
4.2.1 Variabilidad en la actuación del complejo E2-E3 .................................................................. 59
4.3 Destino de las unidades o polímeros de ubiquitina ...................................................... 61
4.4 Las proteínas de deconjugación de ubiquitina E4 ......................................................... 61
4.4.1 Regulación de p53, el equilibrio E3/E4 ................................................................................. 62

5
4.5 Sistema ERAD de control de calidad y señal UPR .......................................................... 63
4.6 Relación con el ciclo celular del sistema UPS ................................................................ 64
4.7 Motivos de reconocimiento para los complejos E2-E3 ................................................. 65
4.7.1 Caja de destrucción de ciclinas ............................................................................................. 65
4.7.2 Secuencias PEST .................................................................................................................... 65
5. El proteasoma ................................................................................................. 65
5.1 Relación entre el Alzheimer y el proteosoma ............................................................... 66
5.2 Inhibidores del proteasoma en el tratamiento contra el cáncer .................................. 68
Tema 4: Regulación del ciclo celular ...................................................... 71
1 Introducción al ciclo celular .............................................................................. 71
1.1 Transducción de la señal de división y cáncer ............................................................... 71
2 Regulación del ciclo celular ............................................................................... 72
2.1 El factor de maduración de los oocitos (MPF) .............................................................. 72
2.1.1 Regulación de MPF: ejemplo de cantidad de ciclinas y nivel de fosforilación ...................... 73
2.2 Proteínas inhibidoras de complejos ciclina-CDK (INKs) ................................................. 74
2.3 Visión en conjunto de la actuación de los complejos Ciclina-CDK ................................ 74
2.4 La proteína del retinoblastoma (RB) y E2F .................................................................... 76
2.5 Puntos de control del ciclo celular ................................................................................ 76
2.6 Visión global de la regulación del ciclo celular .............................................................. 78
3 La silimarina un potencial anticancerígeno ....................................................... 80
Tema 5: Retículo endoplasmático I ........................................................ 81
1 Biogénesis de lípidos en el retículo endoplasmático .......................................... 81
1.1 Biosíntesis de lípidos y fosfolípidos ............................................................................... 81
1.2 Composición lipídica de las membranas celulares ........................................................ 82
1.3 Biosíntesis y movimientos de lípidos a través de las membranas celulares ................. 84
1.3.1 Topología de la síntesis de glicoesfingolípidos de mamíferos .............................................. 86
1.3.2 Síntesis de fosfolípidos .......................................................................................................... 86
1.4 Enfermedades ligadas al retículo endoplasmático ....................................................... 88
2 Las proteínas citocromo P450 ........................................................................... 89
2.1 Modo de acción y características del citocromo P450 .................................................. 90
2.1.1 Modo de acción de Citocromo P450 reductasa .................................................................... 92
2.1.2 Modo de acción de la adrenoxina reductasa ........................................................................ 92
2.2 Otras funciones de P450 ............................................................................................... 92
2.3 Enfermedades relacionadas con el citocromo P450 ..................................................... 94

6
Anexo I: Aislamiento de fracciones subcelulares .................................................. 96
Ejemplo: Aislamiento, caracterización y análisis proteico de la fracción microsomal del
músculo de ratón ................................................................................................................ 97
Tema 6: Retículo endoplasmático II ..................................................... 101
1 Transporte al retículo endoplasmático ............................................................ 101
1.1 El péptido líder o péptido señal .................................................................................. 101
1.2 Asociación del ribosoma al retículo endoplasmático .................................................. 103
1.2.1 Partícula de reconocimiento de la señal (SRP) ................................................................... 104
1.2.2 Componentes de los translocones y proteínas asociadas ................................................... 105
1.3 Transporte post-traduccional en Escherichia coli ....................................................... 106
1.4 Respuesta a proteínas mal plegadas (UPR) ................................................................. 106
1.5 Topología de las proteínas de membrana ................................................................... 108
1.5.1 Incorporación de proteínas de tipo I en la bicapa .............................................................. 109
1.5.2 Incorporación de proteínas de tipo II, III y IV en la bicapa .................................................. 109
Tema 7: Glicosilación de proteínas en el retículo endoplasmático y en las
cisternas del Golgi ............................................................................... 111
1 Las glicoproteínas ........................................................................................... 111
1.1 Funciones de la glicosilación de proteínas ............................................................ 111
2 La glicosilación de proteínas ........................................................................... 112
2.1 Introducción a los glúcidos .......................................................................................... 112
2.2 Donadores activados de azucares para la glicosilación de proteínas ......................... 113
2.3 Introducción a la N-glicosilación y la O-glicosilación ................................................... 114
3 El proceso de N-glicosilación y el plegamiento en el retículo ........................... 116
3.1 La formación del oligosacárido precursor ................................................................... 117
3.2 Transferencia del oligosacárido precursor .................................................................. 119
3.3 Modificaciones generales sobre el oligosacárido precursor ....................................... 120
3.4 Control de calidad en el retículo endoplasmático ...................................................... 121
3.4.1 Maquinaria del ERAD ......................................................................................................... 125
3.5 Chaperonas del retículo endoplasmático.................................................................... 125
3.6 Respuesta a proteínas mal plegadas (UPR) ................................................................. 126
3.6.1 Regulación de los receptores por BiP.................................................................................. 127
3.7 Otras proteínas que ayudan al plegamiento en el retículo endoplasmático .............. 128
3.8 Ensamblado de las subunidades proteicas en el retículo endoplasmático ................. 130
3.9 Modificaciones de la N-glicosilación a nivel del aparato de Golgi .............................. 130

7
3.10 Modificaciones en el Golgi con destino al lisosoma .................................................. 133
3.10.1 Receptores de manosa-6-fosfato...................................................................................... 134
3.11 N-glicosilación en procariotas ................................................................................... 135
4 El proceso de O-glicosilación ........................................................................... 136
4.1 O-glicosilación de proteoglicanos con xilosa ............................................................... 137
4.1.3 Sindecanos y glipicanos como moduladores de la acción proteica .................................... 138
4.2 O-glicosilación de N-acetilglucosmaina en el citosol .................................................. 139
4.2.1 Regulación/actuación de OGT mediante O-glicosilación de N-acetilglucosamina ............. 140
4.2.2 Relación de la O-glicosilación con N-acetilglucosamina y enfermedades .......................... 142
5 Glicosil fosfatidilinositol (GPI) ......................................................................... 143
5.1 Síntesis de glucosil fosfatidilinosiltol ........................................................................... 144
5.2 Beneficios de la utilización del enlace por GPI ............................................................ 145
6 Funciones de los glicoconjugados ................................................................... 146
Anexo I: Destino de las proteínas en función del oligosacárido N-unido y el
plegamiento ..................................................................................................... 147
Tema 8: Tráfico vesicular ..................................................................... 149
1 Proceso general de gemación y fusión de membranas .................................... 149
1.1 Proceso de gemación ................................................................................................. 150
1.2 Proceso de fusión ........................................................................................................ 152
2 Circulación de vesículas entre el retículo endoplasmático y el aparato de Golgi
......................................................................................................................... 154
Bloque III: Regulación Molecular De La Acción Hormonal
Tema 1: Hormonas liposolubles y receptores intracelulares ................ 157
1 Acción hormonal ............................................................................................ 158
1.1 Proteínas carrier de hormonas liposolubles y fracción libre ....................................... 158
1.2 Características de la interacción hormona-receptor................................................... 160
2 Agonistas y antagonistas de los receptores de andrógenos ............................. 160
3 Modelo general del receptor de hormonas liposolubles .................................. 161
3.1 Elementos de respuesta, coactivadores y localización de receptores ........................ 162
3.1.1 Co-activadores .................................................................................................................... 163
3.1.2 Elementos de respuesta para los receptores de hormonas esteroideas ............................. 163
3.1.3 Localización de receptores de hormonas liposolubles ........................................................ 164
4 El receptor de glucocorticoides humano ......................................................... 164
4.1 Motivo de dedos de cinc en el receptor de glucocorticoides ..................................... 164

8
4.2 Estructura y variantes del receptor de glucocorticoides ............................................ 165
4.3 Modelo funcional del receptor de hGR ....................................................................... 166
5 Modelos de actuación de los receptores nucleares (NR) .................................. 167
5.1 Desensibilización de receptores .................................................................................. 168
5.2 Modelo de regulación del receptor de ácido retinoico (RAR) ..................................... 169
5.3 Modelo de regulación del complejo receptor de hormonas esteroideas (SRC) ......... 169
5.4 Estructura y dominios estructurales del receptor de vitamina D ............................... 170
5.5 Acciones genómicas y no genómicas de los receptores de estrógenos ...................... 171
Tema 2: Hormonas hidrosolubles y transducción de señales ............... 173
1 Transmisión de señales .................................................................................. 173
2 Características de la interacción hormona-receptor ........................................ 173
2.1 Cinética interacción hormona-receptor ...................................................................... 174
3 Modelos de transducción de la señal .............................................................. 176
4 Sistema adenilato ciclasa y AMPc ................................................................... 177
4.1 Receptores 7tm acoplados a proteínas G ................................................................... 177
4.2 Estructura de las proteínas G triméricas y modelo de actuación ............................... 178
4.3 Sistema Adenilato ciclasa, AMPc y PKA ....................................................................... 180
4.4 Desensibilización de receptores .................................................................................. 182
5 Sistema de fosfolipasa C, IP3 y DAG ................................................................. 182
5.1 Sistema de la fosfolipasa C (variante β) ...................................................................... 183
5.2 Sistema fosfolipasa C (Variante γ) ............................................................................... 184
5.3 Variantes de PKC ......................................................................................................... 185
5.4 Control de la cantidad de calcio libre en el citosol ..................................................... 186
5.4.1 Calmodulina y Calcio-Calmodulina quinasa ....................................................................... 186
5.4.2 Proteínas reguladas por calcio y calmodulina .................................................................... 187
5.4.3 Canales de calcio receptores de IP3 .................................................................................... 188
5.4.4 Evaluación experimental de los aumentos de calcio .......................................................... 188
6 Sistema de guanilato ciclasa ........................................................................... 189
Tema 3: Factores de proliferación celular. Origen del cáncer ............... 191
1 Receptores con actividad Y-quinasa ................................................................ 191
1.1 Modelo de activación de receptores con actividad Y-quinasa .................................... 192
1.2 Estructura de los factores de crecimiento .................................................................. 193
1.2.1 El factor de crecimiento transformante.............................................................................. 193
1.2.2 Eritropoyetina (EPO) ........................................................................................................... 193

9
1.2.3 Factor de crecimiento epidermal ........................................................................................ 194
1.2.4 Factor de crecimiento derivado de fibroblastos ................................................................ 194
1.3 Fosforilación de tirosinas y dominios .......................................................................... 194
1.4 Rutas de señalización en las que intervienen Y-quinasas ........................................... 197
1.4.1 La insulina como factor de crecimiento .............................................................................. 197
1.4.2 La fosforilación de IRS-1 también activa la vía de PKB ....................................................... 198
1.4.3 Múltiples cascadas a partir del receptor de insulina .......................................................... 198
1.4.4 Detalles de la activación de la ruta de las MAPKs .............................................................. 199
1.4.5 Secuencia de transducción para EPO a través de JAK (receptor de citoquinas) ................. 201
1.4.6 AKT1 (=PKB) y PTEN ........................................................................................................... 203
2 Vías de señalización ....................................................................................... 205
2.1 Receptores con actividad enzimática intrínseca o asociada ....................................... 206
2.2 Ruta de transducción para TGFβ-Smad ....................................................................... 207
2.3 Rutas de transducción que involucran proteólisis ...................................................... 208
2.3.1 Ruta de Wnt........................................................................................................................ 208
2.3.2 Ruta de Hh .......................................................................................................................... 209
2.3.3 Ruta de NFκB ...................................................................................................................... 210
2.3.4 Ruta de Notch/Delta ........................................................................................................... 211
3 Anomalías que pueden producir diversas clases de cáncer .............................. 212
Anexo I: Ruta de CREB ....................................................................................... 213
Anexo II: Rutas de transducción de señales en la célula ..................................... 214
Bloque IV: Regulación Molecular De La Neurotransmisión
Tema 1: Canales iónicos que responden al voltaje ............................... 217
1 Extracción y purificación de canales ................................................................ 217
2 Tránsito de iones a través de canales .............................................................. 219
3 Estructura de los canales dependientes de voltaje .......................................... 220
4 Dinámica temporal en la apertura de canales. ................................................ 223
5 Paso de iones a través de los canales .............................................................. 223
6 Experimentos electrofisiológicos con el mutante Shaker ................................. 225
Tema 2: Transmisión colinérgica .......................................................... 227
1 Proteínas implicadas en la transmisión colinérgica .......................................... 227
1.1 Transportadores de colina (ChTs) ......................................................................... 227
1.2 Síntesis de acetilcolina y transportador vesicular (VAChT) ......................................... 227
1.3 Liberación de acetilcolina al espacio sináptico ........................................................... 229

10
1.4 Receptores muscarínicos (m-AChR) ............................................................................ 230
2 Drogas que afectan a la sinapsis colinérgica .................................................... 231
3 Colinesterasas (AChE y BuChE) ........................................................................ 231
Tema 3: Receptores ionotrópicos ........................................................ 235
1 Sistema glutamatérgico .................................................................................. 235
2 Sistema GABAérgico ....................................................................................... 237
3 Receptor de glicina ......................................................................................... 238
Tema 4: Transmisión adrenérgica y receptores metabotrópicos .......... 239
1 Ruta de biosíntesis de catecolaminas .............................................................. 239
2 Rutas dopaminérgicas .................................................................................... 240
3 Inactivación de catecolaminas ........................................................................ 241
4 Sistema dopaminérgico .................................................................................. 241
5 Sistema adrenérgico ....................................................................................... 242
6 Sistema serotoninérgico ................................................................................. 242
Tema 5: Transmisión peptidérgica ....................................................... 245
1 Encefalinas ..................................................................................................... 245
2 Síntesis y acciones de los neuropéptidos......................................................... 245
3 Estructura génica de los neuropeptidos .......................................................... 246
4 Receptores de neuropeptidos ......................................................................... 247

11
Bloque II: Regulación Molecular De Los
Procesos Celulares
TEMA 1 Aspectos Metabólicos del Citosol
La construcción del ribosoma y su regulación. Destino celular de las proteínas producidas en ribosomas libres. Chaperonas moleculares: familias y diversidad funcional. Cambios post-traduccionales en las proteínas sintetizadas en ribosomas libres.
TEMA 2 Transporte de Material del Citosol al Núcleo, del Núcleo al Citosol y del Citosol a la Mitocondria
Señales peptídicas en proteínas destinadas al núcleo. Mecanismo molecular del transporte de proteínas y polinucleótidos por el poro nuclear. Proteínas Ran, importinas y exportinas. Motivos estructurales de las proteínas destinadas a la mitocondria. Translocones y ubicación de las proteínas en los compartimentos mitocondriales. El genoma mitocondrial, mutaciones y patolo-gías derivadas. Efectos de los antibióticos sobre la actividad mitocondrial.
TEMA 3 Proteolisis Intracelular
Recambio de proteínas. Degradación lisosomal y no lisosomal. Ubiquitina y proteínas relacio-nadas. El sistema ubiquitina/proteasoma y su implicación en la actividad celular y el cáncer. Proteasomas: estructura, función y aspectos evolutivos. Proteasas que se activan por calcio. Patologías derivadas del mal funcionamiento de las vías degradativas.
TEMA 4 Regulación del Ciclo Celular
Estapas del ciclo y su regulación. Ciclinas, proteín-quinasas y proteín-fosfatasas. Mecanismo de acción de los factores de crecimiento. Puntos de control de DNA dañado. Fallos en la regu-lación del ciclo y cáncer.
TEMA 5 Funciones del Retículo Endoplásmico-I
Obtención y caracterización de fracciones subcelulares. El retículo endoplásmico. Biogénesis de fosfolípidos, ceramidas y esteroides. El colectivo de proteínas citocromo P450. Su mplica-ción en la síntesis de lípidos y en la eliminación de tóxicos.
TEMA 6 Funciones del Retículo Endoplásmico-II
Síntesis de proteínas en ribosomas del retículo endoplásmico. Propiedades estructurales y funcionales del péptido líder y su receptor. Estructura y función de los translocones del retículo. Inserción de proteínas transmembranales en la bicapa lipídica.
TEMA 7 Glicosilación de Proteínas en el Retículo Endoplásmico y en las Cisternas del Golgi
Glicosilación de proteínas. Justificación. Glicosilación en el citosol. Síntesis del oligoglicano base en el retículo endoplásmico. Síntesis e incorporación a las proteínas de glicosil-fosfatidil inositol. Control del plegamiento de las proteínas y salida hacia el lisosoma o al Golgi. Glicosil-transferasas del Golgi. Clases de oligoglucanos. Lectinas vegetales y animales. Desglicosila-ción química y enzimática. Exo- y endo-glicosidasas. Glicómica y sus aplicaciones en medici-na.
TEMA 8 Tráfico Vesicular
Proteínas implicadas en la gemación. Mecanimo molecular de la fusión de vesículas con las membranas. Biogénesis del lisosoma. Exocitosis. Producción y regeneración de vesículas si-nápticas. Endocitosis inducida por receptores. Endosomas tempranos y tardíos. Transcitosis.

12

13
¿Dónde hay más volumen relativo de citosol, en un hepatocito o en miofibra muscular?
El volumen relativo de citosol es mayor en un hepatocito porque en la fibra muscular, aun-
que el volumen celular es mayor, este está ocupado por el retículo sarcoplásmico.
Tema 1: Aspectos metabo licos del Citosol
El citosol es el medio que rodea a todos los componentes subcelulares (orgánulos, ribosomas,
gránulos de glucógeno, enzimas, proteínas del citoesqueleto y vesículas de triglicéridos). El
volumen que ocupa el citosol depende de:
- El tipo celular pudiendo representar el 50% del volumen celular, pero gran parte de este estará ocupado por vesículas del retículo endoplasmático, del complejo de Golgi… lo que depende del tipo de célula.
- El estado celular, ya que por ejemplo, una célula del sistema inmune no tiene el
mismo desarrollo de retículo endoplásmico cuando está estimulada para producir an-
ticuerpos que cuando no está estimulada, por lo que también depende del estado ce-
lular.
La concentración de proteína presente en el citosol es muy alta, del orden del 20% (P/v, 20mg/1ml o 20g/100ml). Debido a esa gran concentración de proteínas el citosol tiene un as-
pecto de gel estructurado.
1 Importancia del citosol en el metabolismo (Funciones) El citosol es importantísimo en el metabolismo intermediario ya que gran parte de todas las
reacciones fisiológicas de la célula se van a producir en él.
La glicolisis, gluconeogénesis, la biosíntesis de ácidos grasos (importante para la incorporación
de restos hidrofóbicos a las proteínas de membrana), la biosíntesis de aminoácidos, las inter-
conversiones de aminoácidos, la biosíntesis de nucleótidos, la biosíntesis de azucares e inter-
conversiones de azúcares, la biosíntesis y degradación del glucógeno, la biosíntesis de triglicé-
ridos (la degradación de ácidos grasos se realiza en la mitocondria).
Los granos de glucógeno se sitúan en el citosol y asociados a ellos están las enzimas implicadas
en su síntesis y degradación. El propio gránulo de glucógeno tiene una cubierta proteica (esas
enzimas de síntesis (glucógeno sintetasa) y degradación (glucógeno fosforilasa). Las unidades
que salen de la degradación del glucógeno son glucosa-1-fosfato.
El metabolismo del glucógeno está fuertemente regulado por hormonas, unas que favorecen
la degradación y movilización de glucógeno para aportar glucosa en situaciones en las que se
necesite una elevada concentración de glucosa en sangre, y hay otras que, después de la co-
mida, cuando los niveles de glucosa son muy altos en sangre, pues ese exceso de glucosa se
acumula en forma de glucógeno. Y otras proteínas que responden a las señales reguladoras de
acumulación o degradación de glucógeno.

14
Por ejemplo, para que una proteína incorpore un resto hidrofóbico ha de ser sintetizada por
los ribosomas libres del citosol. Esto se debe a que la formación de geranil-pirofosfato y far-
nesi-pirofosfato, a partir del isopentenil-pirofofasto y el dimetilalil-pirofosfato (que proceden
del ácido mevalonico) se produce en el citosol. Por lo tanto la única vía de incorporación de
poli-isoprenos o ácidos grasos a las proteínas va a ser mediante su síntesis citosólica.
También hay gotitas de grasa, que son TAGs. Las enzimas de síntesis (triglicéridos sintetasa) y
degradación (triglicéridos lipasa) están siempre cerca de los TAGs, para que la acción sea in-
mediata y no tengan que ir buscando esas enzimas por el citosol.
Otro de los aspectos importantes del citosol es la presencia del citoesqueleto. En el citosol
tiene lugar la remodelación del citoesqueleto que por una parte determina el movimiento de
algunos tipos celulares, y por otra determina la posición relativa de los orgánulos subcelula-
res, lo que afecta a la eficiencia y rapidez de la respuesta a los estímulos.
La red del citoesqueleto proporciona las vías por las que van a circular los elementos del cito-
sol gracias a los motores moleculares (dineinas y quinesinas). Este sistema de conducción (ac-
tina, tubulina,…) contribuye a la eficiencia en el acoplamiento de la vesícula desde la mem-
brana de salida hacia la membrana de llegada, garantizando, además, la complementariedad
(las vesículas del retículo endoplasmático van a ser conducidas a la cara proximal del retículo
de Golgi y no a la cara distal).
Puesto que la red del citoesqueleto regula la actividad celular normal, los fallos en la composi-
ción, el funcionamiento o en la dinámica de los componentes pueden favorecer los problemas
de supervivencia celular o incluso la transformación en célula neoplásica. El caso más extremo
es la metástasis o posibilidad de que una célula migre de un tejido afectado mediante la sepa-
ración, entrada al sistema circulatorio y deposición posterior en el tejido aceptor.
Otros de los aspectos por los que el citosol tiene una gran importancia en el metabolismo es
por la presencia de ribosomas. En eucariotas solo existe un tipo de ribosomas que en función
de la proteína en síntesis se van a encontrar libres o acoplados al retículo endoplasmático.
El destino de una proteína está determinado por la secuencia de aminoácidos que contiene en
el extremo amino-terminal (N-terminal) donde puede situarse una señal de tránsito que la
dirigirá:
- Si se trata de un ribosoma libre al citosol, la mitocondria, el núcleo, el peroxisoma o el
cloroplasto.
- Si se trata de un ribosoma asociado al RE, al propio retículo, al complejo de Golgi, a la
membrana plasmática, al lisosoma o exportación por exocitosis (constitutiva o regula-
da).
Al ser sintetizadas, las proteínas pueden portar un péptido señal que provoca el acopla-
miento al RE a través de un receptor de la señal, que posibilita que el péptido sea introduci-
do en el RE mediante un translocon.

15
La albumina es una parte fundamental del plasma sanguíneo que interviene en el transporte de
hormonas (Triyodotironina, tiroxina,…) y que es una secreción constitutiva en el hígado. Por el con-
trario, la secreción de insulina desde las células islotes del páncreas es una secreción regulada.
2 Genoma y proteoma: Origen de la diversidad estructural y fun-
cional de las proteínas Entre 1990 y 2001, se realizó un proyecto con el fin de conocer la secuencia de bases de todo
el DNA humano (Proyecto Genoma Humano). El ser humano tiene 3.000millones de pares de
bases (aprox. 25.000 genes) y 46 cromosomas
No hay relación entre el tamaño del genoma (en pares de bases) y el número de genes y
número de cromosomas entre las distintas especies animales y vegetales. No hay tal corres-
pondencia, lo que determina la plasticidad y las capacidades de una célula y la posibilidad de
establecer comunicación con otras células son los genes.
La unidad funcional es la proteína y la unidad informativa es el gen. La ventaja evolutiva y la
capacidad de supervivencia de una célula es cuanto mayor cuanto mayor sea la diversidad de
las proteínas.
Organismo Tamaño (pb) Nº de genes Nº cromosomas
Homo sapiens (humano) 3.274 millones ~25.000 46
Mus musculus (ratón) 3.420 millones ~25.000 40
Canis familiaris (perro) 2.385 millones ~20.000 78
Gallus gallus (gallo) 1.050 millones ~18.000 66
Danio rerio (pez cebra) 1.563 millones ~22.000 48
Drosophila melanogaster (mosca de la fruta) 168 millones ~13,000 8
Oryza sativa (arroz) 487 millones ~44.000 24
Caenorhabditis elegans (gusano) 103 millones ~19.000 12
Saccharomyces cerevisiae (levadura) 12 millones ~6.000 32

16
Una variante del splicing alternativo puede localizarse en el citosol, mientras que la otra se
localice en la membrana plasmática. Aunque ambas se sintetizaran mediante ribosomas li-
bres, la variante de la membrana plasmática podría estar recibiendo un resto de ácido palmíti-
co o poli-isopreno, adquiriendo una naturaleza anfifílica y por lo tanto siendo incorporada en
la cara citosólica de la membrana plasmática. En caso de que el resto hidrofóbico fuera incor-
porado en el retículo endoplasmático esta quedará orientada hacia la matriz extracelular.
El conjunto de genes de un organismo señala la información que puede expresar la célula (la
capacidad que tiene), la variedad de proteínas expresa la capacidad de la célula para hacer
frente a una situación concreta, la variedad de proteínas es altamente variable. Los genes son
los mismos en todas las células, pero la composición de proteínas (el proteoma de la célula)
varía y depende del tipo celular, del organismo, del momento del desarrollo del organismo, del
estado celular en ese momento, etc. El proteoma va cambiando para ir adaptándose a cada
situación, pero los genes son siempre los mismos.
No se sabe cuántas proteínas es capaz de fabricar el ser humano (100.000 proteínas distintas),
pero esto tiene mucho interés, ya que comparando las proteínas de una persona sana con otra
enferma, podríamos ver qué proteínas son causantes de esa enfermedad o qué proteínas, en
su ausencia, causan esa enfermedad.
Se sospecha que la mayoría de los 25.000 genes sufren splicing alternativo (90%). Se calcula
que debe de haber unas 100.000 proteínas distintas, ya que por cada splicing suelen formarse
unas 4 variantes de la proteína.
Hasta la fecha se han identificado 20.000 proteínas distintas, que son las más abundantes.
Como cada año se van mejorando las técnicas de identificación y análisis de proteínas, por
tanto cada año se irán descubierto cada vez más proteínas. Unas 1.000 proteínas distintas se
han identificado en la mitocondria. En el suero sanguíneo se han encontrado 3000 proteínas y
en el líquido cefalorraquídeo unas 1000 proteínas.
La diversidad de proteínas aparece por resultado de Splicing alternativo (ensamblado alterna-
tivo de los exones y eliminación de intrones) y transformaciones (o modificaciones) post-
traduccionales.
2.1 Splicing alternativo El Splicing alternativo es la capacidad de producir variantes de la misma proteína que proce-
den del ensamblado de exones comunes y exones no comunes.
En un mismo tipo celular pueden
coexistir dos variantes distintas de
la misma proteína puesto que pue-
den presentar diferente afinidad
por el ligando. El plegamiento de
estas variantes será diferente y el
sitio de unión al ligado también.
Esto hace que dos variantes res-
pondan de diferente manera a un
mismo ligando o a concentraciones
diferentes del mismo.
Este fenómeno de maduración alternativa también puede provocar una variante con idéntica
funcionalidad pero diferente localización.

17
2.2 Modificaciones post-traduccionales Otro de los aspectos que aumentan extraordinariamente la diversidad de las proteínas pro-
ducidas a partir de un número limitado de genes son las modificaciones post-traduccionales,
que pueden ser reversibles o irreversibles.
Los cambios post-traduccionales reversibles son:
- La fosforilación (Quinasas/Fosfatasas)
- Acetilación (acetilasas/desacetilasas o acetiltransferasas) como en el caso de las histo-
nas y la expresión de genes.
- Metilación (metilasas/desmetilasas o metiltransferasas)
- Oxidación (oxidasas/reductasas)
- Adición de ubiquitina
Este tipo de cambios provocan cambios conformacionales en la estructura de las proteínas
modulando o modificando su actividad. Además según sea la estructura tridimensional, la pro-
teína estará capacitada para alcanzar su destino o no.
Los cambios post-traduccionales irreversibles son:
- Adición de restos hidrofóbicos como el ácido palmítico (16C) o poli-isoprenos. Por
ejemplo la proteína del oncogén RAS se encuentra asociada a la membrana y manda
señales para la proliferación y división celular.
- Glicosilación entre la que podemos distinguir dos clases, la O-glicosilación (adición de
un oligosacárido mediante interacciones con los grupos hidroxilo de serina y/o treoni-
na) y N-glicosilación (adición de los restos al grupo amino de asparragina o glutamina)
- Proteólisis
3 Ribosomas Uno de los compuestos más abundantes en el citosol son los ribosomas que varían dependien-
do de si la célula es eucariota o procariota.
- Ribosomas Procariotas (70S, 2.800 kD)
o Subunidad 50S
rRNAs 23S y 5S
34 proteínas
o Subunidad 30S
rRNA 16S
21 proteínas
- Ribosomas Eucariotas (80S 4.500 kD)
o Subunidad 60S
rRNAs 28S, 5.8S y 5S
45 proteínas
o Subunidad 40S
rRNA 18S
33 proteínas
Una proteína que pierde los restos añadidos durante los cambios post-traduccionales irre-
versibles pierde su actividad biológica al perder su estructura tridimensional. En ciertos
casos esta proteína puede llegar a ser tóxica dando lugar a agregados como es el caso de
los priones.

En los organismos procariotas encontramos unos
20.000 ribosomas, mientras que en una célula eucario-
ta podemos encontrar unos 10.000.000 lo que supone
mucho más de la mitad. Estos ribosomas están com-
puestos por dos subunidades indicadas en la página
anterior, cada una de las cuales compuestas por varios
rRNAs y proteínas asociadas (55 en procariotas y 78 en
ecuariotas).
La síntesis de estas proteínas asociadas a ribosomas tiene que estar muy controlada por dos
motivos. En primer lugar por la energía necesaria para sintetizar una proteína, y en segundo
lugar porque hay que producir las copias necesarias de acuerdo con la cantidad de rARN dis-
ponible. Si esta síntesis no estuviese controlada tendríamos dos problemas fundamentales:
- Las proteínas que se asocian con el ribosoma pueden llegar a tener unos 80 kD, lo que
supone un despilfarro energético.
- La sobreproducción de cualquiera de ellas supone problemas, ya que a grandes con-
centraciones de una proteína, esta puede desplazar a otras del lugar que le corres-
ponde, a pesar de tener menos afinidad por el sitio de unión. Esto provoca ribosomas
no operativos o de baja eficiencia.
En el cuadro se representan las interacciones de las proteínas relacionadas con el ARNr 16S de
la subunidad pequeña del ribosoma procariota (S =
Small). Podemos clasificar estas proteínas en función de
su relación entre ellas y el ARN.
- Las proteínas de la primera fila son aquellas que
interaccionan directamente con el ARNr.
- Las proteínas de la segunda fila son aquellas
que interaccionan con e ARNr y con las proteínas
de primera línea.
- Las proteínas de la tercera fila, no interaccionan
con el ARNr, sino que se asocian con las proteí-
nas anteriores.
Por ello en estas condiciones, el desplazamiento que podría provocar una desregulación de la
expresión de las proteínas del ribosoma afectaría gravemente a la funcionalidad ya que, si por
ejemplo la proteína S20 desplazara a S7, ninguna de las que interaccionan con S7 podrían unir-
se. Por ello en los procariotas, las proteínas están organizadas en operones.
Un operón es un conjunto de genes que se van a expresar de manera coordinada, es decir, que
la expresión del primer gen conlleva la expresión de los genes que le siguen. Todas las proteí-
nas que intervienen en la biosíntesis de las subunidades del ribosoma están agrupadas en ope-
rones de 2 o 3 proteínas.

1
Además la primera de las proteínas expresadas por el operón tiene la facultad de unirse a su
lugar en el ARNr correspondiente y de actuar como un represor de su propio operón unién-
dose al mensajero. Por ello, cuando todos los sitios correspondientes de los ARNr sean ocupa-
dos se inhibirá la expresión de más subunidades.
4 Síntesis de proteínas e inhibidores Uno de los aspectos más importantes para el control de patógenos y la investigación básica es
la inhibición de la síntesis de proteínas en procariotas y eucariotas.
Se conocen una serie de inhibidores que afectan a la síntesis de proteínas en procariotas,
mitocondrias y cloroplastos, ya que estos orgánulos proceden de los procariotas (teoría endo-
simbiotica) y presentan los mismos ribosomas.
- El Cloranfenicol inhibe a la peptidiltransferasa, una ribozima (ARN con acción catalíti-
ca no proteica) que transfiere la cadena polipeptídica desde el sitio peptidil (P) al sitio
aminoacídico, para que se vaya formando la cadena.
- La Puromicina es un análogo estructural del Tyr-tRNA (aminoacil de tRNA), de manera
que se engaña al ribosoma, ya que incorpora a la puromicina creyendo que es la Tyr-
tRNA y por tanto ya no puede continuar la elongación de la cadena polipeptídica.
También puede afectar a eucariotas.
- La Tetraciclina y la Estreptomicina se unen a la Subunidad 30S y provoca la lectura
errónea del mRNA.
- La Eritromicina se liga al rRNA 23S y detiene la translocación del ribosoma a lo largo
del mRNA.
En cuanto a los Eucariotas:
- La Ciclohexidimida actúa como el cloranfenicol en procariotas.
La utilización de Ciclohexidina nos permite determinar si la recuperación de actividad de una
proteína, previamente inhibida, se produce por la síntesis de novo o mediante su recuperación a
partir de un pool o reservorio. Para ello se comienza el ensayo inhibiendo a la enzima en cues-
tión (p.e. AChE con isopropil-pirofosfato) y a continuación se bloquea la síntesis de proteínas
añadiendo Clicloheximida al cultivo. Si tras un tiempo la actividad proteica se recupera querrá
decir que hay una reserva de proteína inactiva que ha sido movilizada por acción de las chape-
ronas, pero si no se recupera, querrá decir que la recuperación se debía a la expresión del gen.
Además la combinación en experimentos secuenciales con Ciclohexmida, Cloranfenicol y ambos,
nos permiten determinar si una proteína procede del genoma nuclear, mitocondrial o de un
reservorio.
El ritmo de síntesis de proteínas es de 15 aa/s, es decir, que el ribosoma se desplaza sobre la
cadena de mRNA a una velocidad de 45 b/s. Por ejemplo, una proteína de 75.000 Da, consi-
derando que la masa promedio de un aminoácido es de 100 Da, contiene unos 750 aminoáci-
dos. Por lo tanto serán necesarios 50 segundos para sintetizar una proteína. Pero puesto que
se forma un polisoma en el que varios ribosomas leen el mismo mensajero al mismo tiempo
con cierto desfase entre ellos, en 100 segundos no tendremos 2 copias, sino muchísimas más.

2
Puesto que las mitocondrias se ven afectadas por los inhibidores procarióticos, los efectos del
consumo de estos compuestos sobre una persona dependerán de la cantidad, del genoma y
el proteoma del individuo, así como de la naturaleza del antibiótico y del tejido.
Cualquier agente antibacteriano que pueda afectar a nuestras mitocondrias afecta al resto de
la célula también. Dependiendo de la naturaleza química de estos compuestos, tendrán mayor
o menos dificultad para atravesar la membrana (cuanto más polar sea, pasará con menos faci-
lidad; cuanto menos polar sea, pasará con mayor facilidad). También depende de la naturaleza
de la membrana mitocondrial externa e interna, que tienen distinta permeabilidad.
Las células que se dividen muy activamente tienen muchas más mitocondrias, por lo son mu-
cho más vulnerables a estos inhibidores. La vida media de una mitocondrial en una célula nor-
mal es de 5 o 6 días. Por eso, para que se afectaran de manera notable las células del organis-
mo, tendría que someterse a un tratamiento con antibióticos durante unos 5, 6 o 7 días, para
afectar gravemente a las células. Si el tratamiento es de un par de días, por ejemplo, es posible
que solo el 20 o 30% de las mitocondrias de una célula se vean afectadas, pero aun así la célula
seguiría funcionando más o menos de forma aceptable (con el 80% de mitocondrias funcio-
nando). Pero si por ejemplo se ven afectadas más del 50-60% de las mitocondrias, pues la célu-
la se verá gravemente afectada por esa disminución de mitocondrias.
Los tratamientos con antibióticos bacterianos tienen que ser lo más breves posible para que
no afecte mucho a las células y además, que no se cree resistencia por parte de las bacterias.
Los tratamientos con antibióticos pueden causar trastornos en tejidos del epitelio intestinal,
apareciendo diarreas, se muere la flora intestinal, y algunos antibióticos pueden ocasionar
estado de anemia, porque se dañan células sanguíneas que sufren un recambio rápido y nece-
sitan multiplicar sus mitocondrias.
También hay que destacar que cuando estamos sometidos a un tratamiento con antibióticos,
nos sentimos débiles y sin fuerzas, porque con el antibiótico se ve reducido la síntesis de ATP
en las mitocondrias.
5 Chaperonas moleculares En la construcción del ribosoma, intervenía
los RNA ribosomales y además un montón de
proteínas, donde unas 55 para el ribosoma
bacteriano y en torno a 78 para el ribosoma
de eucariotas.
Se puede utilizar la Eritromicina, porque no puede cruzar (o casi no puede entrar) la
membrana mitocondrial interna.
Tetraciclinas: No pueden pasar a través de la membrana plasmática de las células de la
médula ósea, porque se crea el complejo tetraciclina-Ca, debido a que en el hueso hay una
gran concentración de calcio (los huesos están formados por fosfatocálcico).

3
Estas proteínas se sintetizan en ribosomas libres, y después entran al núcleo (a través de los
poros nucleares), llegando al nucleolo (que es la fábrica de ribosomas). A continuación se for-
man las partículas pre-ribosomales (uniéndose al RNA ribosomal), que salen por los poros nu-
cleares hacia el citosol. Una vez fuera, terminan la maduración, uniéndose al RNA mensajero
ambas subunidades ribosomales (pequeña y grande) y empieza la síntesis de proteínas.
Para que todo esto ocurra de forma rápida y eficiente, hacen falta unas proteínas de ayuda
que son las chaperonas.
La secuencia de aminoácidos de una proteína determina su estructura tridimensional, lo que
quiere decir, que al concluirse la síntesis de la cadena polipeptídica la proteína debería estar
plegada. Pero lo que sucede en realidad es que los determinantes estructurales se pueden
situar en el extremo central o en el carboxilo, y por lo tanto, la proteína no pueda plegarse
hasta que se haya completado la síntesis.
Cuando una proteína presenta los determinantes estructurales en la secuencia N-terminal,
esta se irá plegando conforme sea sintetizada pero si se sitúan más adelante, la proteína co-
menzará a plegarse de forma errónea formando un ovillo. Para evitar este fenómeno, las cha-
peronas van a mantener la estructura de la proteína en síntesis desplegada hasta que los
determinantes estructurales permitan el plegamiento correcto.
5.1 El descubrimiento de las Chaperonas En 1960, Anfinsen puso de manifiesto que la información necesaria para la estructura de una
proteína se encontraba en la secuencia de aminoácidos en sus experimentos con la ribonu-
cleasa, pero los datos fueron malinterpretados.
El experimento consistía en ver como la actividad de la ribonucleasa se desplomaba al añadir
urea (agente desnaturalizante) al medio. Si ahora se introducía la mezcla en una bolsa de diáli-
sis, la urea abandonaba el medio y al poco tiempo la ribonucleasa recuperaba su actividad
biológica.
Este experimento desembocó en un dogma de la biología molecular que posterior mente tuvo
que ser matizado: La secuencia de aminoácidos de una proteína determina la estructura tridi-
mensional.
Pero las condiciones en el interior celular son más complejas que in vitro. En primer lugar no
hay un solo tipo de proteínas, sino miles, y en segundo lugar un mensajero está siendo tradu-
cido a la vez por muchos ribosomas, por lo que varias copias de la misma proteína se están
sintetizando progresivamente y muy próximas, por lo que si presenta dominios hidrofóbicos,
estas podrán asociarse para formar agregados.
Por lo tanto, en muchas ocasiones, será necesaria la participación de otras proteínas, denomi-
nadas chaperonas, para proteger las regiones hidrofóbicas e impedir estos procesos. Las cha-
peronas, al igual que las demás proteínas, van a actuar reduciendo la energía necesaria, o
facilitando el plegamiento, pero sin intervenir al final de la reacción.

4
Si se introduce un cultivo de linfocitos en una cabina de flujo a 42 °C durante 10 minutos,
aunque la célula sigue viviendo, muchas de sus proteínas comienzan a desnaturalizarse, lo
que provoca las señales de estrés térmico que desembocan en la sobreexpresión de chape-
ronas para favorecer el replegamiento y la recuperación de proteínas.
El nombre de Heat shock protein (HSP) o proteínas de choque térmico, que reciben las chape-
ronas, proviene de la observación de su síntesis cuando se somete un cultivo celular a una
temperatura mayor a la que presenta un funcionamiento óptimo.
En condiciones normales siempre existe una expresión basal de chaperonas, ya que, como
veremos más adelante, estas van a tener muchas más funciones. Para ejercer dichas funciones
precisan de un aporte de ATP, puesto que va a ser su hidrólisis la que produzca la variación de
energía libre necesaria para dar lugar a procesos desfavorables termodinámicamente.
En general, las chaperonas van a ser inducibles por todos los factores que faciliten el unfol-
ding1 como:
- Variaciones de temperatura.
- Disolventes orgánicos (etanol, propiodioxano,…)
- Elementos metálicos (Zn, Pb,…)
Otra de las funciones que desarrollan las chaperonas es el mantenimiento de la cadena de
aminoácidos desplegada para aquellas proteínas cuyo destino son los orgánulos celulares, y
que por lo tanto precisan atravesar translocones con un diámetro de poro pequeño (TON y PIN
en la mitocondria). Esta es una función tan importante que aquellas levaduras que no sinteti-
zan chaperonas son inviables.
Finalmente, los fallos en la síntesis de ciertas chaperonas están asociados con enfermedades
como el Alzheimer, la fibrosis quística o la mucolipidosis (acumulo de material lipídico en los
lisosomas). Quizás no es el agente causal, pero es un factor de riesgo para el desarrollo de la
enfermedad.
5.2 El plegamiento de las proteínas En el entorno celular las proteínas se sintetizan como cadenas de aminoácidos desplegadas
cuyo estado de energía interna es máximo y por lo tanto muy inestable. Por ello existe la
tendencia natural de reducir esta energía mediante el plegamiento que puede realizarse co-
rrecta o incorrectamente, dando lugar a diferentes estados conformacionales.
La parte izquierda de la gráfica de la siguiente página representa como una proteína desplega-
da va a ir adquiriendo diferentes estructuras conformacionales de plegamiento parcial, hasta
alcanzar el estado nativo en el que posee una estructura tridimensional que le permite llevar a
cabo su actividad catalítica. Cada uno de los sucesivos estados presentan una menor energía
interna, y por lo tanto una mayor estabilidad en los que la proteína se estabiliza per se median-
te interacciones intramoleculares.
1 Pérdida de estructura nativa, desnaturalización.

5
Las proteínas admiten cierta flexibilidad conformacional ya que cuando estas interaccio-
nan con el sustrato sufren un cambio conformacional y por lo tanto la estructura nativa no
es un estado fijo e inmóvil. Esta flexibilidad es lo que se conoce como ajuste inducido.
En algunas ocasiones las proteínas se pliegan de manera errónea cayendo en un estado de
energía interna inferior por el que necesitarán de la participación de las chaperonas para al-
canzar el estado de transición y superar la barrera energética que les impide alcanzar la estruc-
tura nativa. Las chaperonas actúan por tanto como catalizadores que reducen la energía nece-
saria para pasar de un estado a otro, facilitando la adquisición de la estructura nativa e impi-
diendo que fruto del plegamiento incorrecto las proteínas comiencen a desarrollar interaccio-
nes intermoleculares para estabilizarse.
Muchas proteínas, en su variación por los estados parcialmente plegados, comienzan a desa-
rrollar interacciones intermoleculares con proteínas de la misma clase que conducen a la for-
mación de agregados, tal y como se repre-
senta a la derecha de la gráfica inferior.
Los estados de agregación son peligrosos
ya que suelen ser tóxicos para muchos
tipos celulares, aunque las chaperonas
van a ser capaces de actuar sobre aquellos
que no sean insolubles, recuperándose
las proteínas.
Los agregados pueden ser amorfos, pero
también pueden ser oligómeros muy es-
tructurados y estos pueden polimerizarse
para formar fibrillas amiloides muy inso-
lubles. Todos estos estados serán reversi-
bles, siempre y cuando las chaperonas
sean capaces de acceder a ellos.
5.3 Funciones de las chaperonas Como ya se puede intuir, las chaperonas presentan muchas funciones en el entorno celular.
1) Facilitan el plegamiento del polipéptido para alcanzar la estructura nativa.
2) Previenen el plegamiento en aquellas proteínas cuyo destino no es el citosol y precisa
atravesar translocones.
3) Importantes en las interacciones proteína-proteína, proteína-RNA y proteína-DNA
(histonas).
4) Renaturalización de proteínas parcialmente desnaturalizadas.
5) Conversión de agregados en unidades proteicas.
6) Colaboración a la proteólisis participando de manera activa en el ciclo de la ubiquiti-
na-proteosoma y en la autofagia.
7) Formación de agregados con diversos receptores, como el de cortisol, que se sitúa en
el citosol secuestrado por una chaperona que tapa su sitio de reconocimiento nuclear

6
hasta que la llegada del cortisol produce un cambio conformacional que libera el sitio y
permite su importación al núcleo y la búsqueda de su elemento de respuesta (secuen-
cia de nucleótidos).
8) Transporte de proteínas por translocones como en el caso de las chaperonas SecA en
procariotas y BiP en eucariotas. La proteína de unión o Binding protein (BiP) facilita la
entrada de proteínas al lumen del retículo endoplasmático en el transporte post-
traduccional (no confundir con la introducción co-traduccional mencionada anterior-
mente). BiP es por tanto una chaperona que va a empujar proteínas sintetizadas, me-
diante gasto de ATP, al interior del retículo.
9) Control de calidad (plegamiento correcto) a través de los distintos pasos hasta el des-
tino final de una proteína. Las chaperonas son capaces de forzar a las proteínas a con-
seguir su estructura nativa aunque sea necesaria la desnaturalización completa para
volver a realizar el plegamiento.
Además, las chaperonas participan en la progresión del ciclo celular, mantenimiento de los
telómeros, apoptosis, transducción de señales mitóticas, transporte vesicular, inmunidad inna-
ta y degradación proteica.
Conforme aumenta la edad, disminuye la capacidad de síntesis de chaperonas, lo que genera
problemas con la proteostasis (homesotasis del proteoma) dando lugar a neurodegeneración,
demencias, diabetes tipo-2, patologías por depósitos de material en lisosomas, fibrosis quísti-
ca, patologías cardiovasculares y cáncer.
5.4 Clasificación de las HSP El número de chaperonas que se conoce es enorme y está en constante aumento conforme
aumenta las investigaciones al respecto. La tabla con algunas de estas chaperonas puede ser
consultada en el documento anexo donde se indica la localización tisular, celular, los sustratos
y algunas enfermedades relacionadas con cada chaperona.
En cualquier caso se han agrupado en varias familias en función del tamaño molecular en kDa.
- HPS20
- HSP40
- HSP60
- HSP70
- HSP90
- HSP100
Para establecer la identidad (parecido) entre las chaperonas de una misma familia se elige una
como prototipo o estándar y se calcula el parecido de secuencia con respecto a las demás.
Puesto que se encuentra en todos los orgánulos debieron aparecer en un momento temprano
de la evolución, y puesto que están representadas por muchas familias, su función es muy
importante ya que nos encontramos con un sistema de redundancia.
Esta diversidad también puede explicarse si se considera que la diana de acción son miles de
proteínas con multitud de dominios estructurales diferentes. Cuando una chaperona detecta

7
una proteína parcialmente desplegada que ha dejado expuesto un dominio hidrofóbico, desa-
rrolla su actividad catalítica golpeando a la proteína hasta que recupera su forma nativa. De
esta manera se evita la formación de agregados insolubles. Pero esto puede pasar con cual-
quier proteína, por lo que es necesario que el centro activo sea flexible para abarcar una gran
cantidad de motivos. Además, la combinación de varias chaperonas aportará una cierta especi-
ficidad, pero relativa, para establecer una unión de baja afinidad que permitirá la salida poste-
rior de la proteína.
5.5 Modo de acción de algunas chaperonas Veamos ahora el modo de acción de algunas chaperonas representativas de la mayoría casos
en los que van a participar.
Una proteína desplegada pasa por varios estados de plegamiento parcial (estados de Molten o
glóbulos de modulo fundido) en los que
presenta varias alternativas favorables
termodinámicamente. Por un lado la
proteína puede alcanzar la estructura
nativa, que es el camino favorecido
termodinámicamente (en menor medi-
da la reversión), por otro lado, puede
formar agregados cuya reversión es
muy desfavorable.
En cualquier caso, el plegamiento es un proceso espontáneo y termodinámicamente favora-
ble (ΔG < 0), pero el hecho de que el proceso ocurra no conlleva un tiempo definido y por ello
en muchas ocasiones las chaperonas van a intervenir para que el plegamiento se produzca
rápidamente.
Además, es muy probable que las proteínas que presenten los determinantes estructurales
en los extremos C-terminales precisen de la colaboración de las chaperonas.
5.5.1 HSP70 y co-chaperonas (HSP20 y HSP40)
La HSP70 es una chaperona que presenta tres dominios funcionales:
- Dominio de unión a nucleótidos (ATP/ADP) o nucleotide binding domain con actividad
ATPasa.
- Dos dominios de unión a proteínas con estados de baja y alta afinidad.
El dominio de unión a nucleótidos, en
verde, presenta una actividad ATPasa
basal pero la unión a la proteína diana
por medio de los dos dominios de unión,
en gris y amarillo, provoca un cambio
conformacional que provoca la actividad
ATPasa. La hidrólisis de ATP a ADP provo-
ca un tirón molecular que aumenta la
afinidad de los dominios de unión por la

8
proteína diana que es “golpeada” una y otra vez hasta alcanzar su estado nativo. Por lo tanto
la HSP70-ADP presenta una alta afinidad por la proteína diana.
Para que se pueda llevar a cabo cada ciclo la proteína NEF (Nucleotide Exchange Factor) inter-
cambia el ADP por ATP, haciendo que los dominios de unión a proteínas de HSP70 cambien de
alta a baja afinidad. HSP70-ATP es el estado de baja afinidad por la proteína diana.
Si la proteína sigue presentando regiones hidrofóbicas al medio, y por ende, está mal plegada,
entonces aún presenta la afinidad suficiente por los dominios de unión a proteínas y permane-
ce en lugar activo para recibir un nuevo “golpe”. Pero si ha adquirido su estructura nativa en-
tonces el cambio de HSP70 a un estado de baja afinidad le permite liberarse.
La HSP40 y HSP20 son co-chaperonas que median en el reconocimiento de proteínas mal ple-
gadas, caracterizadas por la exposición de regiones de al menos 8-10 o 12 aminoácidos hidro-
fóbicos2, transportándolas hasta las proximidades de la HSP70 ejecutora y además promueven
su actividad ATPasa.
Si observamos un ejemplo en el que interviene la proteína J (co-chaperona), cuando esta in-
teracciona con una proteína
desplegada o mal plegada (1), se
une a ella y la traslada hasta la
HSP70, formándose un complejo
ternario (2) entre la proteína J,
la proteína diana y HSP70.
A continuación se produce la
hidrolisis de ATP en HSP70 (3) lo
que provoca su aumento de
afinidad por la proteína diana y
un descenso de afinidad por la
proteína J.
Ahora NEF interviene y se une a HSP70 (4) provocando la salida del ADP (5) y la entrada de ATP
(6) con el consiguiente cambio conformacional, pasando a un estado de baja afinidad y favore-
ciendo además la salida de NEF (7).
Este ciclo puede tener éxito y conseguirse la estructura nativa de la proteína o puede fracasar.
Cuando tras varios intentos no se consigue plegar correctamente a la proteína esta es deriva-
da hacia la degradación a través del sistema de ubiquitina-proteosoma o a través de la auto-
fagia.
5.5.2 HSP90
La HSP90 (Apo-HSP90) es un dímero que presenta tres dominios funcionales:
- Dominio de unión C-terminal (CD) por donde los monómeros se unen a través de sus
extremos C-terminales y en ocasiones es denominado pinza molecular.
2 Arginina y lisina

9
- Dos dominios (uno por monómero) intermedios de unión a proteínas (MD).
- Dos dominios (uno por monómero) N-terminales de unión a nucleótidos (ND).
Esta chaperona presenta tres estados conformacionales diferentes en función de los ligandos a
los que se encuentre unida.
La primera conformación o con-
formación de alta afinidad es la
que se produce cuando la HSP90
no interacciona con sus ligandos
(ATP/ADP y una proteína desple-
gada). Se encuentra en un estado
relajado con los monómeros
unidos a través de CD separados
y exponiendo al medio algunos
residuos hidrofóbicos con alta
afinidad por las proteínas mal
plegadas.
La segunda conformación o de baja afinidad se produce cuando HSP90 incorpora ATP en los
dominios ND y una proteína desplegada en los dominios MD. La estructura se cierra y el sus-
trato queda secuestrado por interacciones no covalentes de baja afinidad.
La tercera conformación o conformación compacta se produce cuando otras proteínas como
AHA1 interaccionan con HSP90 de baja afinidad, provocando la aproximación de los dominios
ND con ATP unido, que dimerizan y “aplastan” a la proteína que se encuentra unida en MD.
Finalmente se produce la hidrólisis del ATP a ADP +Pi que provoca la vuelta al estado de alta
afinidad inicial, la separación de los dominios ND y la liberación de la proteína plegada. Poste-
riormente se libera el ADP + Pi y la chaperona vuelve al estado más desplegado de alta afini-
dad.
Tal y como se puede observar en la imagen, el paso de un estado a otro está muy regulado
por proteínas que comprueban que la proteína unida a HSP90 necesita o no plegarse impi-
diendo o permitiendo el avance del proceso.
5.5.3 HSP60 (GroEL y GroES)
La familia de chaperonas HSP60, a diferencia de las anteriores, forman estructuras poliméricas
de oligómeros (Chaperoninas3) que en procariotas dan lugar a una estructura don forma de
barril compuesto por:
- GroEL que es un cilindro con 14 subunidades proteicas (HSP60) cuyo interior da lugar a
dos cámaras, una superior y otra inferior separadas por una barrera.
- GroES es una especie de tapadera con 7-8 subunidades de HSP60 que interacciona con
los huecos de las cámaras de GroEL.
3 Existe una tendencia a denominar Chaperonas a las monoméricas y Chaperoninas a las poliméricas.

10
Cuando una proteína resulta resistente al plegamiento por HSP70, entonces esta se la cede a
GroEL, mediante un intercambio de ADP por ATP (cambio de alta afinidad a baja afinidad), que
la introduce en la cámara abierta (cis).
Puesto que cada complejo presenta dos cámaras están actuaran de manera cíclica y alternativa
por lo que mientras que una cámara puede presentar ATP unido, la otra podrá presentar ADP
o el sitio vacío.
Al mismo tiempo que la proteína va entrando en la cámara cis de GroEL, se promueve la salida
de 7 ADP en la cámara trans y la separación de su unidad GroES, al mismo tiempo que 7 ATP se
unen a la cámara cis y promueve la unión de GroES.
Ahora la actividad ATPasa de las subunidades HSP60 se activa e hidroliza el ATP provocando un
cambio conformacional que presionan la proteína dentro de la cámara cis hasta conseguir su
estructura nativa. En este estado con ADP, la cámara cis tiene una alta afinidad por la proteína,
pero la unión del ATP a la cámara trans provoca la salida del ADP y de GroES de la cámara cis.
Puesto que la cámara presenta el interior tapizado por aminoácidos hidrofóbicos, en el caso de
que la proteína esté plegada, la salida de GroES provoca la expulsión inmediata de la proteína.
Cabe destacar que la etapa más larga del ciclo es el plegamiento de la proteína.

11
5.6 Resumen Hemos visto en los apartados anteriores como las chaperonas participan en el plegamiento de
las proteínas, pero no hay que olvidar, que esta es solo una de sus acciones.
En la imagen se ha realizado una síntesis de
la importancia de las chaperonas en el ple-
gamiento de proteínas en procariotas.
Cuando la cadena polipeptídica comienza a
emerger del ribosoma esta se encuentra
con unas chaperonas unidas que mantienen
la estructura desplegada en el caso de que
se trate de una región hidrofóbica.
A partir de aquí se abren dos posibilidades.
Por un lado la proteína puede sintetizarse
completamente y plegarse per se, como
ocurre en el 70% de los casos. Por otro lado
la proteína necesitará de la participación de
chaperonas que la mantengan desplegada
hasta encontrarse con HSP70 (20%) o que
además necesiten de la participación de los
complejos GroEL-GroES para finalizar el
plegamiento (10%).
Puesto que es más rentable no utilizar chaperonas, la mayoría de las proteínas se pliegan a la
estructura nativa espontáneamente, seleccionándose evolutivamente la presencia de los
determinantes estructurales en las regiones N-terminales de la secuencia de aminoácidos.
Volviendo a las distintas posibilidades que tiene una proteína para plegarse vamos a integrar lo
visto junto con las vías de degradación. Una proteína recién sintetizada pasa a un estado de
plegamiento parcial del que puede derivar en una proteína con estructura nativa, paso facilita-
do por las chaperonas con una importancia relativa de 180, o puede formar estructuras mal
plegadas o “misfolded” y
agregados protéicos.
Tal y como se muestra en
la imagen, la importancia
relativa de la degrada-
ción de proteínas vía
ubiquitina-proteosoma
es de 600, unas 20 veces
mayor que la degrada-
ción de agregados vía
autofágia que tiene una
importancia relativa de
30.
12
Si una célula tiene problemas para conseguir ATP, ¿se acorta la vida celular?
Puesto que la célula tiene dañada la función mitocondrial y depende de un aporte conti-
nuo de ATP la célula va a sufrir, y uno de los problemas se localizará en la falta de chape-
ronas, lo que provocará un acortamiento de la vida.
La rotenona y el amital son dos toxinas capaces de inhibir el transporte electrónico en la
cadena respiratoria mitocondrial provocando un descenso en la producción de ATP que
afecta gravemente al sistema nervioso. Esto se debe a que la neurona necesita el ATP para
repolarizar la membrana a expensas de la bomba Na/K
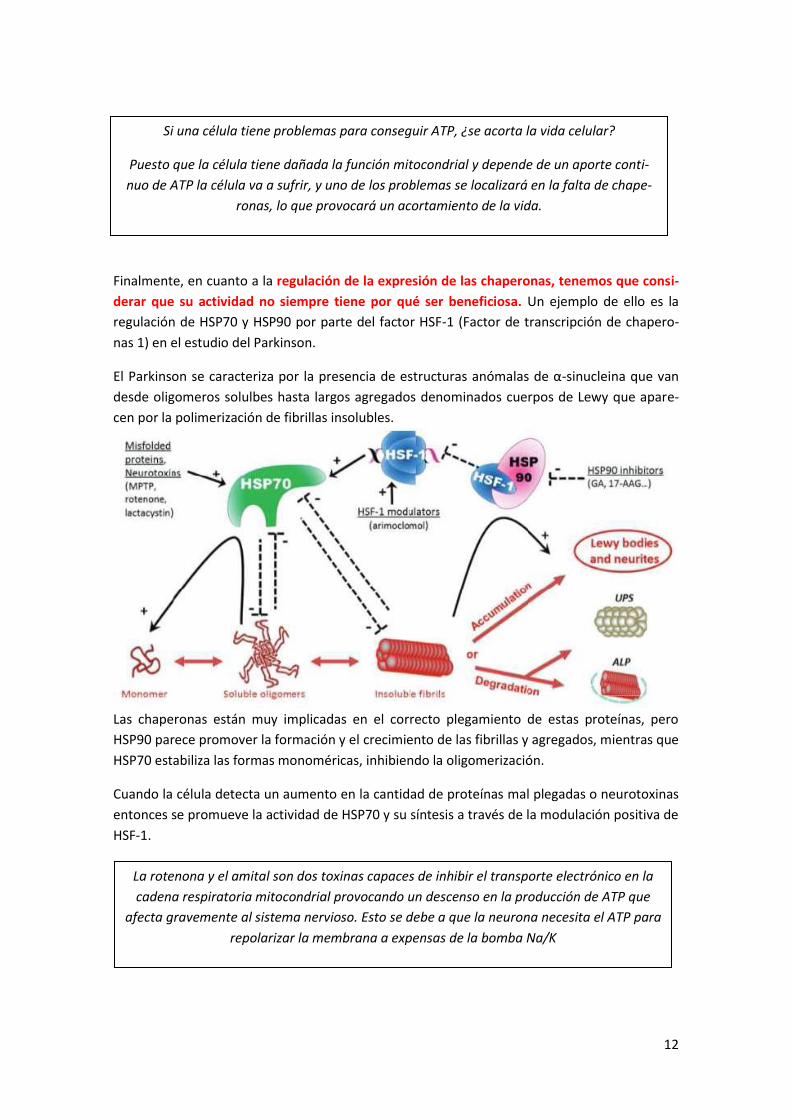
Finalmente, en cuanto a la regulación de la expresión de las chaperonas, tenemos que consi-
derar que su actividad no siempre tiene por qué ser beneficiosa. Un ejemplo de ello es la
regulación de HSP70 y HSP90 por parte del factor HSF-1 (Factor de transcripción de chapero-
nas 1) en el estudio del Parkinson.
El Parkinson se caracteriza por la presencia de estructuras anómalas de α-sinucleina que van
desde oligomeros solulbes hasta largos agregados denominados cuerpos de Lewy que apare-
cen por la polimerización de fibrillas insolubles.
Las chaperonas están muy implicadas en el correcto plegamiento de estas proteínas, pero
HSP90 parece promover la formación y el crecimiento de las fibrillas y agregados, mientras que
HSP70 estabiliza las formas monoméricas, inhibiendo la oligomerización.
Cuando la célula detecta un aumento en la cantidad de proteínas mal plegadas o neurotoxinas
entonces se promueve la actividad de HSP70 y su síntesis a través de la modulación positiva de
HSF-1.

13
Pero HSP90 actúa como un secuestrador de HSF-1, evitando la producción de HSP70, por lo
que parece que promueve la formación de los cuerpos de Lewis.
Por casos como este, la relación entre las distintas chaperonas está siendo objeto de profunda
investigación ya que, por ejemplo, el mantenimiento de la vida útil de p53, el guardián del
genoa, mediante una chaperona específica alargaría la vida.
6 Cambios post-traduccionales (CPT) Los cambios post-traduccionales afectan a la mayoría de las proteínas, sino a todas. Actual-
mente se conocen más de 200 clases de CPTs clasificados reversibles e irreversibles.
El 5% del genoma humano (1200 genes) está dedicado a codificar proteínas implicadas en CPTs
y hay que considerar que esto no hace referencia 1200 genes, ya que si consideramos la posi-
bilidad de la maduración alternativa podríamos estar hablando de unas 5000 proteínas dife-
rentes, lo que nos indica que estos cambios tienen una importancia trascendental para que la
proteína cumpla su función. Actualmente se han identificado 250 genes (1000 proteínas) de-
dicadas a la glicosilación.
Una misma proteína puede sufrir varios CPTs diferentes y una proteína encargada de realizar
los CPTs puede actuar sobre muchos sustratos.
6.1 Cambios post-traduccionales reversibles Los cambios post-traduccionales reversibles son aquellos que modifican la estructura tridimen-
sional de las proteínas modulando su actividad. Entre ellos destacan la fosforila-
dión/defosforilación, acetilación/desacetilación, metilación/desmetilación, oxida-
ción/reducción y adición de ubiquitina. Se trata de modificaciones por enlaces covalentes.
En este ámbito profundizaremos en la fosforilación/defosforilación y en las modificaciones de
las colas N-terminales de las histonas.
6.1.1 Fosforilación/defosforilación
El grado de fosforilación de las proteínas está mediado por quinasas y fosfatasas que regulan el
fosfoproteoma, que hace referencia al espectro de proteínas con fosfato que hay en un tipo
celular en cada momento en función de la circunstancia fisiológica.
En el genoma humano se han anotado genes para más de 500 protein quinasas (PKs) y más
de 150 protein fosfatasas (PPs). El motivo por el que hay menos PPs es porque estas actúan a
través de proteínas adaptadoras que amplían el rango de sustratos sobre los que pueden ac-
tuar. Se cree que estas proteínas actúan sobre más de un tercio del proteoma humano, es
decir, sobre unas 30.000-40.000 proteínas distintas, ya que, por ejemplo la proteín quinasa A
(PKA) es capaz de fosforilar a más de 100 proteínas diferentes.
Existen dos tipos fundamentales de quinasas y sus fosfatasas con proteínas adaptadoras al-
ternativas.
- Las Serín/Treonin (S/T) quinasas/fosfatasas que actúan sobre residuos de serina y
treonina como PKA, PKC, Calcio calmodulina quinasa (CCK)

14
El Mg es un catión divalente que interacciona con las cargas negativas de la molécula de
ATP dando lugar a una geometría que le permite acceder al sitio activo de la quinasa.
La quinasa Abl es una proteína oncogénica con actividad tirosina quinasa (Y-quinasa) que
presenta en su estructura 1 serina, 1 treonina y 9 tirosinas, capaces de ser fosforiladas dando
lugar a unas 2000 variantes. La hiperfosforilación de la quinasa Abl provoca leucemia mieloi-
de crónica cuyo tratamiento actual consiste en la utilización de inhibidores como el Imatinib.
La proteína tau (τ) está asociada a la regulación de la polimerización de la tubilina y presenta
unos 40 sitios de fosforilación en su cadena polipeptídica. La hiperfosforilación de esta proteína
está relacionada con las enfermedades mencionadas porque se impide su estabilización de los
microtúbulos e interrumpe el transporte vesicular. Además comienza a formar placas amiloides.
- Las Tirosin (Y) quinasas/fosfatasas que actúan sobre residuos de tirosina como el re-
ceptor de insulina y Abl. Pueden ser solubles o estar asociadas a la membrana. Todos
los receptores de factores de crecimiento desarrollan esta actividad.
En cuanto a los sustratos de las PKs tenemos que considerar el ATP-Mg y las proteínas sobre
las que actúan.
Quinasas y fosfatasas regulan casi todos (si no todos) los procesos celulares a través de pun-
tos clave en el metabolismo, la transcripción, la traducción, el ciclo celular, la estabilidad de
proteínas, el movimiento celular, la apoptosis, exocitosis regulada, la dinámica del citoesquele-
to, la contracción muscular, la acción hormonal, la transducción de señales, etc.
Patologías asociadas con la pérdida o ganancia de función de quinasas/fosfatasas
Existen muchas patologías asociadas a los fallos relacionados con estas enzimas clave para las
células.
- La diabetes de tipo 2 está producida por un fallo en la cascada de quinasas mediada
por insulina.
- Las mutaciones en los genes para las proteínas SrC (oncogen), Abl (oncogen), fos-
foinositol-3 quinasa (PI3K, cataliza el paso de fosfatidil inositol-4,5-bifosfato a fosfatidil
inositol-3,4,5-trifosfato) y factor activado por Ras (Raf), producen cáncer.
- Procesos inflamatorios, como artritis y asma, se producen por la activación defectuosa
de algunas PKs, como JAK, JNK e IKK.
- La hiperfosforilación de tau en Alzheimer y Parkinson.
- Anomalías en alguna de un total de 30 quinasas pueden llevar a la hipertensión.
Concepto de secuencia consenso o secuon
Si analizamos la secuencia fosforilada por la PKA en las proteínas a las que regula observamos
que siempre son fosforiladas en un residuo de serina o de treonina. Pero en una proteína de
unos 70 kDa con una media de 700 aminoácidos puede haber muchos residuos de serina o
treonina.

15
La unión de una hormona a su receptor es probabilística, es decir, cada vez que la molécula
toca el receptor se activa, pero cuanto mayor sea la concentración de hormona, más toques
de producirán. Este concepto es muy importante ya que las uniones nunca serán de carácter
covalentes.
Por ello la actividad S/T quinasa de PKA y de cualquier quinasa/fosfatasa no se va a realizar
por la mera presencia del resto diana, sino porque este se encuentra situado en un entorno
tal, que la proteína ejecutora pueda reconocer un punto de actuación. Estos entornos no son
más que secuencias de aminoácidos concretas que están expuestas u orientadas de tal manera
que el resto a fosforilar pueda ser reconocido.
Estas secuencias de aminoácidos se conocen como secuencias consenso o secuon, y en el
caso de que en el caso de PKA es
xR[RK]x[ST]B
Donde el aminoácido fosforilado es la serina (S), menos frecuente treonina (T), seguidas de un
resto hidrofóbico y precedidas por cualquier aminoácido (x), una lisina (K) o arginina (R), una
arginina y cualquier aminoácido (x).
Ejemplos de regulación por fosforilación: Cascada de adrenalina en hepatocito
Un ejemplo típico de regulación por fosforilación es la cascada de amplificación de la señal que
produce la adrenalina al interaccionar con su receptor β1-adrenérgico en el hepatocito y que
desencadena la movilización de glucosa al activar la glucogenolisis.
Esta cascada comienza con un aumento de la concentración de adrenalina en las inmediacio-
nes del hepatocito que provoca la interacción (choques) entre la hormona y el receptor. Estos
choques provocan un cambio conformacional que permite la interacción con la proteína G que
desencadena la liberación de la subunidad Gsα.
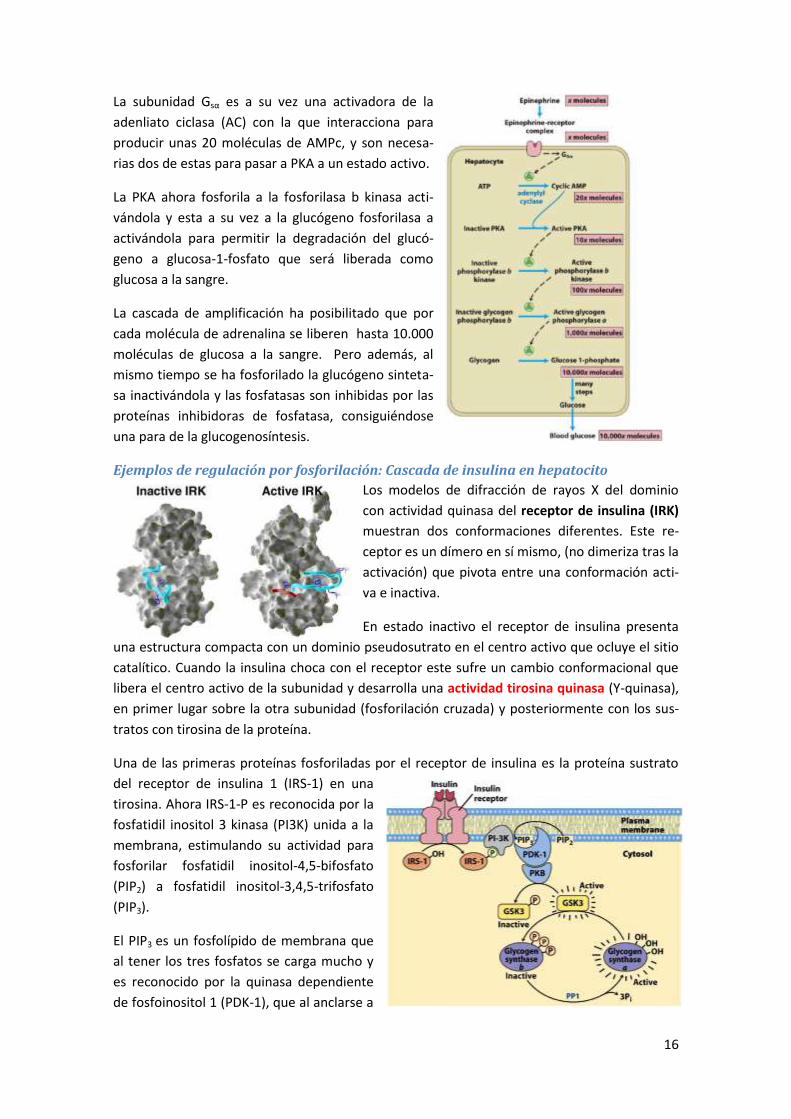
16
La subunidad Gsα es a su vez una activadora de la
adenliato ciclasa (AC) con la que interacciona para
producir unas 20 moléculas de AMPc, y son necesa-
rias dos de estas para pasar a PKA a un estado activo.
La PKA ahora fosforila a la fosforilasa b kinasa acti-
vándola y esta a su vez a la glucógeno fosforilasa a
activándola para permitir la degradación del glucó-
geno a glucosa-1-fosfato que será liberada como
glucosa a la sangre.
La cascada de amplificación ha posibilitado que por
cada molécula de adrenalina se liberen hasta 10.000
moléculas de glucosa a la sangre. Pero además, al
mismo tiempo se ha fosforilado la glucógeno sinteta-
sa inactivándola y las fosfatasas son inhibidas por las
proteínas inhibidoras de fosfatasa, consiguiéndose
una para de la glucogenosíntesis.
Ejemplos de regulación por fosforilación: Cascada de insulina en hepatocito
Los modelos de difracción de rayos X del dominio
con actividad quinasa del receptor de insulina (IRK)
muestran dos conformaciones diferentes. Este re-
ceptor es un dímero en sí mismo, (no dimeriza tras la
activación) que pivota entre una conformación acti-
va e inactiva.
En estado inactivo el receptor de insulina presenta
una estructura compacta con un dominio pseudosutrato en el centro activo que ocluye el sitio
catalítico. Cuando la insulina choca con el receptor este sufre un cambio conformacional que
libera el centro activo de la subunidad y desarrolla una actividad tirosina quinasa (Y-quinasa),
en primer lugar sobre la otra subunidad (fosforilación cruzada) y posteriormente con los sus-
tratos con tirosina de la proteína.
Una de las primeras proteínas fosforiladas por el receptor de insulina es la proteína sustrato
del receptor de insulina 1 (IRS-1) en una
tirosina. Ahora IRS-1-P es reconocida por la
fosfatidil inositol 3 kinasa (PI3K) unida a la
membrana, estimulando su actividad para
fosforilar fosfatidil inositol-4,5-bifosfato
(PIP2) a fosfatidil inositol-3,4,5-trifosfato
(PIP3).
El PIP3 es un fosfolípido de membrana que
al tener los tres fosfatos se carga mucho y
es reconocido por la quinasa dependiente
de fosfoinositol 1 (PDK-1), que al anclarse a

17
la membrana es reconocida por la protein quinasa B (PKB).
La PKB es una también es una S/T quinasa que es capaz de fosforilar a la glucógeno sintetasa
quinasa 3 (GSK3), pasándola a un estado inactivo. Esto permite la desfosforilación de la gluco-
geno sintasa a través de la protein fosfatasa 1 (PP1, presente en todos los tejidos).
Integración de la señal de adrenalina e insulina en hepatocitos
El hecho de que en un momento determinado se active o inhiba la glucogenosíntesis y la glu-
cogenolisis, va a depender de la cantidad de interacciones positivas o negativas que reciba el
conjunto de receptores.
Los hepatocitos presentan receptores de adrenalina e insulina en grandes cantidades, pero va
a ser el número de interacciones de es-
tos con su sustrato (que depende de su
concentración exterior de hormonas) lo
que determine que vía predomina.
Si observamos al conjunto de proteínas
descritas sobre el gránulo de glucógeno,
vemos que todas están asociadas y mo-
duladas por quinasas sensibles a insulina
o adrenalina según los mecanismos des-
critos.
Papel de la insulina como factor de crecimiento y proteínas G
Hemos visto la actuación de la insulina como hormona reguladora de los niveles de glucosa en
sangre, pero esta es además un factor de crecimiento como el factor de crecimiento derivado
de plaquetas, la somatostatina, el factor de crecimiento epidermal, etc.
En cualquier caso, el término factor de crecimiento puede hacer referencia tanto al crecimien-
to de la propia célula, como al de la masa de células en sí, por lo que algunos denominan a
estos últimos factores mitóticos o factores de proliferación.
Al igual que en los apartados anteriores,
la acción de la insulina como factor de
crecimiento comienza con la activación
de la actividad Y-quinasa del receptor que
sufre una fosforilación cruzada en los
restos de tirosina de sus extremos carbo-
xilo terminales.
Al igual que en el caso anterior, el IRS-1
reconoce estos fosfatos y es fosforilado
en varios de sus residuos de tirosina.
Ahora a diferencia de en el caso anterior
o al mismo tiempo, los residuos de fosfa-
to van a poder ser reconocidos por la
proteína de unión a Grb2 (Growth factor

18
receptor-bound protein 2), que cambia de conformación y se une a la proteína Sos (Son of Se-
venless).
La unión de la proteína Sos a Grb2 la capacita para actuar como un intercambiador de GDP
por GTP en Ras unida a la membrana. La proteína Ras-GTP es reconocida por el factor activado
por Ras 1 (Raf-1) y este fosforila a MEK (Mitogen-activated protein kinase kinase) que pasa a
fosforilar a la quinasa regulada por señales externas (ERK).
La ERK fosforilada tiene la capacidad de trasladarse al núcleo porque su fosforilación libera la
señal de localización nuclear que activa el sistema carioferinas (transportadores del poro nu-
clear), y allí comienza la fosforilación de los factores de transcripción diana que se unirán a su
secuencia de reconocimiento activando o inhibiendo la expresión de un gen.
La importancia de las proteínas G radica en su actividad como intercambiadoras de nucleóti-
dos de guanina. Estas son diferenciadas en dos clases:
- Las proteínas G monoméricas como:
o Ras que es una proteína de la cara citoplasmática de la membrana plasmática
y actúa como “pivote” de la transducción de señales al núcleo.
o Rho que está implicada en la dinámica del citoesqueleto.
o Ran que participa en el ciclo de transporte de proteínas citosol-nucleo.
- Las proteínas G triméricas como:
o Gs(α,β,γ) estimuladoras del sistema adenliato ciclasa
o Gi(α,β,γ) Inhibidoras de la adenilato ciclasa.
Las mutaciones en proteínas como Ras que provocan la pérdida de su actividad GTPasa dan
lugar a una activación constante que desregula completamente la expresión de proteínas
desembocando en un cáncer.
6.1.2 Modificación de los dominios N-terminales de las histonas
Las histonas son unas proteínas sintetizadas en los ribosomas libres del citosol que entran al
núcleo para estabilizar a la fibra de DNA polimerizándose en octámeros. Estas proteínas pre-
sentan a pH fisiológico una gran abundancia de cargas positivas en sus extremos N-terminales,
situándose su punto isoeléc-
trico a un pH superior a 10.
Aminoácidos tales como la
lisina (K) o la arginina (R)
proporcionan estas cargas
que son idóneas para estabi-
lizar a la hebra de DNA, car-
gada negativamente por sus
enlaces fosfodiéster a pH
fisiológico.
Uno de los puntos de regulación de la compactación de la cromatina van a ser las modificacio-
nes covalentes reversibles sobre las colas N-terminales de las histonas, tales como la acetila-
ción, metilación y fosforilación.

19
La acetilación de histonas conlleva una neutralización de carga positiva mediante la adi-
ción de un grupo acetilo (acetato), donado por el Acetil-CoA procedente de la mitocondria
a través de la lanzadera de Citrato.
Cuando hay abundancia de Acetil-CoA la histona acetil transferasa (HAT) incorpora un res-
to de acetato al grupo ε-amino de la lisina y permitiendo la expresión de ciertos genes aso-
ciados a un estado de energía disponible.
Cuando los niveles de energía disminuyen, la histona desacetilasa (HDAC) puede realizar la
reacción contraria liberando el resto acetilo y condensándose de nuevo la cromatina.
La cola de una histona es susceptible de sufrir muchas modificaciones diferentes al mismo
tiempo y además un mismo aminoácido puede sufrir varias diferentes. En la imagen se repre-
sentan las diferentes modificaciones que pue-
den sufrir diferentes aminoácidos de una histo-
na en lisinas y argininas (ac = acetilación, me =
metilación, PS = fosforilación y u = ubiquitiniza-
ción).
Esto establece un código de modificaciones de
histonas que van a variar para expresar o silen-
ciar una determinada región génica o van a
facilitar la reparación del DNA. Pero el signifi-
cado de este código todavía no se ha estableci-
do.
Estas modificaciones responden al hecho de que en función de los restos presentes en un
determinado aminoácido y de su entorno, una proteína podrá o no reconocer la secuencia,
favoreciendo o impidiendo la expresión. Por ejemplo, la el factor de transcripción BPTF es ca-
paz de reconocer lisinas acetiladas o metiladas en la cola N-terminal de la histona H3, pero en
esta puede haber muchas lisinas con esta modificación, por lo que lo que reconoce la proteína
es una secuencia que presenta la lisina modificada, no solo el resto.

20
El paso de la estructura compacta (heterocromatina) o abierta (eucromatina) depende de la
acción conjunta de proteínas que “escriben” sobre las histonas, las que “leen” la información
y las que “borran” los cambios. La acción cooperativa de estas proteínas decide cuándo de-
ben expresarse o silenciarse genes y por cuánto tiempo. También regulan los mecanismos de
reparación del DNA.
6.1.3 Otras modificaciones covalentes reversibles
Otras modificaciones covalentes reversibles son:
- Incorporación de ácido adenílico (AMP) en glutamina sintetasa de E. coli por adenilil-
transferasa, una actividad regulada a su vez por uridil-transferasa.
- Adición de metilo (SAM) en restos de ácido glutámico de receptores que regulan la
quimiotaxis bacteriana.
- Adición de ubiquitina a histonas y otras proteínas de manera reversible (histonas) me-
diante unos pocos restos, o de manera irreversibles (degradación proteica).
- Incorporación de ADP-ribosa en histonas, factor de elongación EF2, subunidades αs yαi
de las proteínas Gs y Gi del sistema adenilatociclasa (reversible o irreversible).

21
La molécula de inositol es un alcohol que no debe de ser confundido con un glúcido, a
pesar de que su estructura cíclica recuerde a estos. Nótese que a diferencia de los glúci-
dos, el inositol no presenta una molécula de oxígeno en el ciclo.
6.2 Cambios post-traduccionales irreversibles Entre los cambios post-traduccionales más importantes son:
- Adición de restos hidrofóbicos.
- Glicosilación
- Proteólisis
6.2.1 Adición de restos hidrofóbicos
La observación de la membrana plasmática revela la existencia de dos localizaciones proteicas
de diferente origen.
Las proteínas situadas en la cara
citosólica de la membrana plas-
mática presentan restos de áci-
dos grasos (palmítico, mirístico,
farnesilo o geranil-geranilo) hi-
drofóbicos, mientras que las
proteínas situadas hacia el espa-
cio extracelular (ectoproteínas)
presentan glicosil fosfatidil inosi-
tol (GPI) en su extremo C-
terminal.
Las ectoproteínas van a ser sintetizadas en ribosomas asociados al retículo endoplasmático
donde incorporarán a la fosfoetanolamina C-
terminal varios residuos de un oligosacáridos de
manosa y N-acetil-glucosamina. A continuación
el conjunto proteína-oligosacárido es unido al
fosfatidilinositol (fosfolípido) que porta un par de
diacilglicerol insertos en la membrana, formando
el puente de GPI. La proteína anclada ahora viaja
por las cisternas intracelulares hasta ser incorpo-
rada a la membrana por exocitosis.
En cuando a las proteínas que quedan mirando hacia el interior celular, estas se sintetizan en
ribosomas libres en el citosol donde son modificados por la presencia de secuencias de reco-
nocimiento para transferasas específicas y según el resto hidrofógico que incorporen las vamos
a diferenciar en tres clases:
- Las proteínas que incorporan el ácido palmítico (16C) en una cisteína (C) o serina (S),
siempre interna en la cadena de aminoácidos.
- Las proteínas que incorporan el ácido mirístico (14C) en una glicina (G) N-terminal.

22
El dolicol es una molécula anfipática con un resto alcohol que puede mirar hacia el interior
o el exterior del retículo endoplasmático gracias a los movimientos de flip-flop. Cuando el
dolicol es cargado en la cara citosólica con la N-acetil-glucosamina y la manosa, se interna-
liza para glicosolar proteínas.
- Las proteínas que incorporan restos de poliisopreno como Farnesil (15C) o geranil-
geranilo (20C) en cisteínas (C) C-terminales que posteriormente pueden incorporar un
resto metilo.
La adición de los restos hidrofóbicos aporta un carácter anficílico a la proteína que la desplaza
hasta la membrana más cercana desde donde se desplazará hasta su localización definitiva.
La adición de restos hidrofógicos como cambio post-traduccional irreversible es un proceso
que en humanos afecta a más de 700 proteínas entre las que 500 incorporan poliisoprenos,
100 incorporan ácido palmítico y otras 100 ácido mirístico. En el género Tripanosoma se han
identificado más de 100 proteínas unas a la membrana a través de puentes GPI.
Δ3-Isopentenil pirofosfato y la síntesis de poliisoprenos
El Δ3-Isopentenil pirofosfato (5C) es la molécula precursora de toda la familia de los isoprenos
presentes en gran cantidad de proteínas celulares. Además es el precursor de muchas molécu-
las como el dolicol (relacionado con la N-glicosilación) que acepta los azúcares que serán trans-
feridos a las proteínas.
En el citosol encontramos un equilibrio entre los isómeros de dimetilalil pirofosfato y Δ3-
Isopentenil pirofosfato, condensados por la preniltransferasa (cabeza-cola) para dar lugar al
geranil pirofosfato (10C).
La condensación del ge-
ranil pirofosfato con una
nueva molécula de Δ3-
Isopentenil pirofosfato
da lugar al farnesil piro-
fosfato (15C) o la con-
densación de dos molé-
culas de geranil pirofos-
fato (cabeza-cabeza) da
lugar al geranil-geranilo.
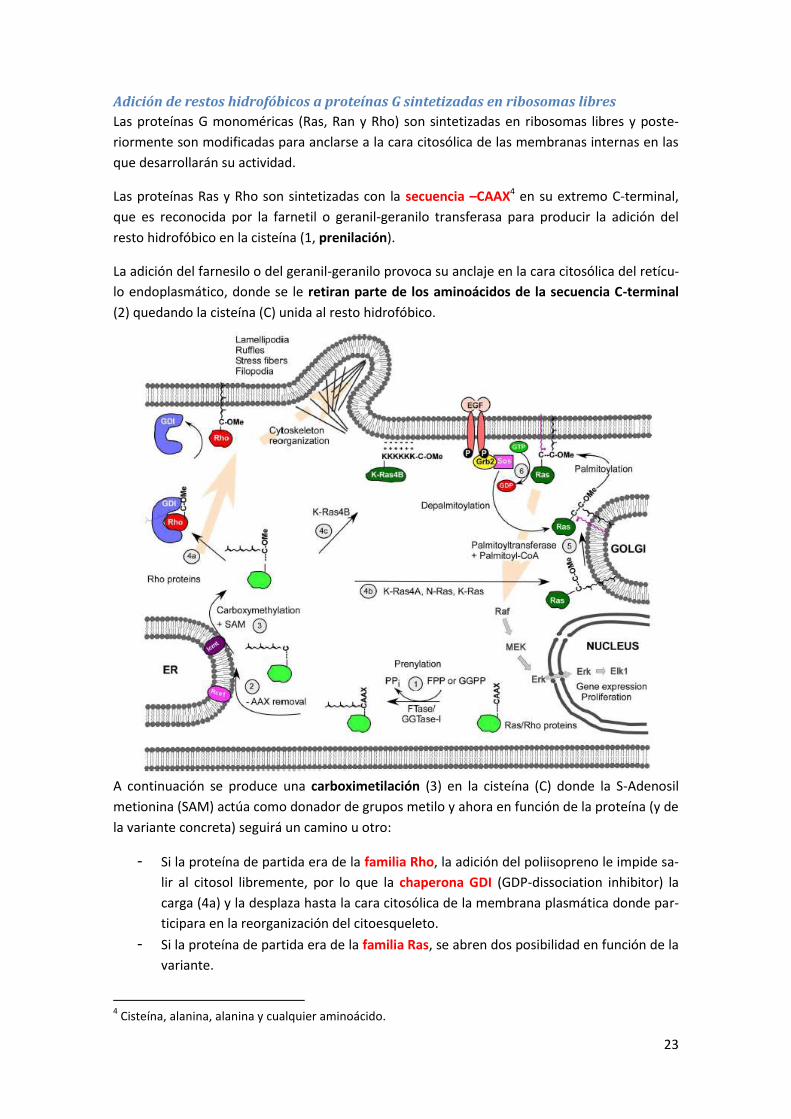
23
Adición de restos hidrofóbicos a proteínas G sintetizadas en ribosomas libres
Las proteínas G monoméricas (Ras, Ran y Rho) son sintetizadas en ribosomas libres y poste-
riormente son modificadas para anclarse a la cara citosólica de las membranas internas en las
que desarrollarán su actividad.
Las proteínas Ras y Rho son sintetizadas con la secuencia –CAAX4 en su extremo C-terminal,
que es reconocida por la farnetil o geranil-geranilo transferasa para producir la adición del
resto hidrofóbico en la cisteína (1, prenilación).
La adición del farnesilo o del geranil-geranilo provoca su anclaje en la cara citosólica del retícu-
lo endoplasmático, donde se le retiran parte de los aminoácidos de la secuencia C-terminal
(2) quedando la cisteína (C) unida al resto hidrofóbico.
A continuación se produce una carboximetilación (3) en la cisteína (C) donde la S-Adenosil
metionina (SAM) actúa como donador de grupos metilo y ahora en función de la proteína (y de
la variante concreta) seguirá un camino u otro:
- Si la proteína de partida era de la familia Rho, la adición del poliisopreno le impide sa-
lir al citosol libremente, por lo que la chaperona GDI (GDP-dissociation inhibitor) la
carga (4a) y la desplaza hasta la cara citosólica de la membrana plasmática donde par-
ticipara en la reorganización del citoesqueleto.
- Si la proteína de partida era de la familia Ras, se abren dos posibilidad en función de la
variante.
4 Cisteína, alanina, alanina y cualquier aminoácido.

24
El mensajero m de la acetilcolinesterasa (AChE) produce una proteína que incorpora GPI
y se sitúa en la cara externa de los rafts lipídicos, junto a los receptores muscarínicos.
La existencia de los tres mensajeros (m, h y t) se justifica por la necesidad de llevar a la
enzima a diferentes espacios.
o Las proteínas K-Ras4A, N-Ras y K-Ras5 viajan en vesículas al Golgi (4b) donde
la palmitoiltransferasa añade un residuo de palmítico (5, palmitolación) y va en
vesículas a la cara citosólica de la membrana plasmática. Pero aquí se estable-
ce una dinámica donde la pérdida del residuo de palmítico (6, despalmitola-
ción) la devuelve al Golgi para ser palmitolada nuevamente.
o Las proteínas K-Ras4B se desplazan directamente del retículo endoplasmático
hasta la cara citosólica de la membrana plasmática gracias a interacciones ió-
nicas de sus cargas positivas con las cabezas polares de los fosfolípidos, carga-
dos negativamente.
Raft lipídico
A diferencia de lo que se pensaba años atrás, la membrana plasmática no presenta una com-
posición homogénea de lípidos y proteínas. En ella encontramos los raft lipídicos o balsas lipí-
dicas que son dominios de membrana muy estructurados.
Los raft lipídicos son dominios muy ricos en colesterol y esfigolípidos, es decir, moléculas an-
fipáticas que se organizan
de manera muy compacta
gracias a que no presen-
tan insaturaciones. Estos
dominios son pobres en
fosfolípidos. En función de
la presencia o no de ca-
veolina, se van a diferen-
ciar dos tipos de raft:
- Los raft caveolares son aquellos cuya proteína estructural es la caveolina.
- Los raft no caveolares son aquellos cuya proteína estructural no es la caveolina, como
por ejemplo la flotilina.
- Además existen otros raft que no presentan proteína estructural pero contienen mu-
cho colesterol entre las cadenas de esfigolípidos saturados.
Las proteínas de membrana solo alcanzan su funcionalidad en el entorno lipídico adecuado, de
la misma manera que el lípido al que se unen solo alcanza sus propiedades termodinámicas y
termotrópicas en su entorno adecuado. Por ello la relación lípido-proteína es indisoluble.
Por alguna razón ciertos grupos de proteínas solo se dirigen a los raft lipídicos como las unidas
a través de GPI.
5 Las variantes K-Ras presentan cuatro residuos de lisina (K) en el extremo C-terminal cargados positiva-
mente a pH fisiológico.

25
Los raft lipídicos son dominios dinámicos que se unen y separan constantemente por lo que
solo cuando interaccionen un dominio con, por ejemplo, el receptor de adrenalina y otro con
la proteína G, se producirá la señal.
6.2.2 Glicosilación
La glicosilación de proteínas como cambios post-traduccionales irreversibles son la N-
glicosilación y la O-glicosilación.
La N-glicosilación se produce en el retículo endoplasmático donde los restos son incorporados
a asparaginas (N) o glutaminas (Q) internas en un único oligosacárido.
La O-glicosilación puede tener lugar tanto en el retículo como en el citosol y consiste en la
introducción de monosacáridos en los restos de serina (S) y/o treonina (T), uno por uno a tra-
vés de interacciones con los grupos hidroxilos.
6.2.3 Cambios por proteólisis limitada
Los cambios post-traduccionales asociados a la proteólisis hacen referencia a la degradación
selectiva de parte de la cadena de aminoácidos con el fin de permitir que esta adquiera su
estructura tridimensional final o pueda desarrollar su actividad catalítica.
El gran ejemplo es la activación de zimógenos, como el pepsinogeno a pepsina, los factores de
coagulación de la sangre, del complemento, apoptosis (Caspasas), proinsulina y otras hormo-
nas polipeptídicas, como las del hipotálamo e hipófisis, encefalinas, endorfinas, neuropéptidos,
etc.
También se incluyen aquí los cortes de péptidos de destino a orgánulos como aquellas que
entran al retículo y cuya secuencia hidrofóbica inicial es cortada, o las señales de tránsito a los
orgánulos energéticos y el peroxisoma.
6.2.4 Otros cambios post-traduccionales irreversibles
Otros cambios irreversibles que afectan a las pueden ser:
- La conversión de formilmetionina en metionina es un proceso típico en procariotas
donde el inicio de la traducción siempre comienza con este aminoácido. De la misma
manera, en eucariotas todas las proteínas comienzan por metioinina, pero luego no la
portan en su secuencia por lo que este resto es retirado en muchos péptidos.
- La adición de grupos prostéticos (hemo, biotina,…) y metales (Cu, Zn, Mn,…) en las pro-
teínas permite que estas puedan realizar su actividad catalítica.
- La hidroxilación de residuos, como la prolina y la lisina en el colágeno aporta resisten-
cia a la fibra.
- La adición de carboxilato (-COO) en la posición γ de restos de glutamato de la pro-
trombina da lugar a un nuevo aminoácido (no proteogénico). Esto aporta dos cargas
negativas importantes en la formación de complejos con calcio que inmovilizan a la
proteína en los sitios con heridas para que se convierta en trombina y el tripsinogeno
en tripsina, provocando la coagulación de la sangre.

26
En la reacción de coagulación interviene la vitamina K1 y K2 que presenta colas isopréticas. La
enzima clave es la carboxilasa que se ve fuertemente inhibida por la warfarina (también un
rodenticida).
En medicina este inhibidor se utiliza en personas que han sufrido una angina de pecho o infar-
to de miocardio para evitar la formación de trombos. Análogamente se ha utilizado el sintrom,
un medicamento que en bajas dosis no realiza su efecto, pero que en dosis altas es muy perju-
dicial. Es muy complicado ajustar la dosis.

27
Tema 2: Tra fico citosol-nú cleo y citosol-mitocondria
El tráfico entre el citosol y los distintos orgánulos subcelulares hace referencia al tránsito de
vesículas o proteínas generadas por los ribosomas libres del citosol y que luego van a ser tras-
ladados hasta sus sitios funcionales ya sea el núcleo, los orgánulos energéticos o el peroxiso-
ma.
1. Tráfico citosol-núcleo El núcleo es el destino de numerosas proteínas sintetizadas en los ribosomas libres del citosol,
como es el caso de las proteínas histónicas, no histónicas, los componentes de la cromatina,
enzimas como las RNA-polimerasas y DNA-polimerasas, factores de transcripción… Todas ellas
presentan en común un dominio de aminoácidos conocido como secuencia de localización
nuclear (NLS, Nuclear localization sequence). La NLS es una secuencia de pequeño tamaño
(≈20 aminoácidos) que puede presentar dos tipos de epítopo reconocido por las proteínas de
transporte:
- Un epítopo secuencial es aquel reconocido como secuencia de aminoácidos lineal. Es-
te tipo de secuencias son reconocidas por los anticuerpos cuando la proteína está des-
naturalizada o plegada de manera que se muestre completamente.
- Un epítopo conformacional es aquel que puede ser reconocido al plegarse la secuen-
cia y por lo tanto no tiene porqué ser lineal, los diferentes aminoácidos pueden estar
repartidos por el péptido y reunirse con el plegamiento para permitir el reconocimien-
to. Este tipo de epítopos no puede ser reconocido por los anticuerpos cuando la pro-
teína está desnaturalizada, ya que esté es reconocido como un motivo tridimensional.
Estas secuencias son muy ricas en ciertos aminoácidos como la lisina (K) y arginina (R), que
según el tipo de epítopo se presentaran continuas o discontinuas, y en muchas ocasiones esta-
rán ocultas hasta la llegada de una señal extracelular que modifique el factor de transcripción
para que muestre la NLS.
Podemos ver varios ejemplos de secuencias NLS clásicas, no clásicas e incluso algunas especia-
les de epítopo conformacional. Tal y como se observa en la secuencia M9, no siempre es tan
fácil distinguir la señal de localización nuclear. Las señales se pueden localizar en cualquier
punto de la proteína excepto en los extremos, ya que, la importina precisa de un cierto apoyo
para transportar a la proteína, por ello generalmente las encontraremos internas en la secuen-
cia de aminoácidos.

28
Las partículas preribosomales presentan del orden de megadaltones, por lo que las
proteínas generadas en el núcleo van a precisar del acoplamiento con exportinas para
salir al citosol. Existen muchas enfermedades derivadas del mal funcionamiento de los
transportadores de determinadas proteínas nucleares.
La existencia de estos dos tipos de NLS nos permite inferir que no es una condición estricta
que la proteína se encuentre desplegada para ingresar al núcleo, como ocurre en la mitocon-
dria. Además, como veremos posteriormente, esta secuencia se puede situar en cualquier
punto de la cadena de aminoácidos, ya que no será cortada al llegar a su lugar de acción por la
dinámica nuclear durante la mitosis celular.
Hoy sabemos que el tránsito de proteínas entre el citosol y el núcleo es bidireccional, es de-
cir, ciertas proteínas ingresan y salen del núcleo en respuesta a factores hormonales gracias a
la presencia de otra señal denominada señal de exportación nuclear (NES, Nuclear export sig-
nal) que las proteínas pueden mostrar alternativamente según las condiciones celulares.
Para poder realizar este transporte la célula cuenta con una compleja batería de proteínas
denominadas genéricamente como carioferinas (importinas, exportinas, Ran…) capaces de
realizar el transporte a través del poro nuclear.
Las complejas interacciones que se producen entre las carioferinas, así como la gran cantidad
de interrogantes planteados sobre el proceso ha llevado al desarrollo de hasta 6 hipótesis so-
bre el transporte de proteínas entre el citosol y el núcleo.
Un hecho muy importante es que las proteínas de localización nuclear siempre van a mante-
ner su secuencia de importación/exportación, a diferencia de lo que ocurre para las proteínas
de localización mitocondrial o del retículo endoplasmático. Esto se debe a que durante la mito-
sis se produce la desorganización de la membrana nuclear, liberándose todo el contenido al
citosol. La conservación de la secuencia, permite el posterior regreso de las proteínas al núcleo
tras la reorganización en las células hijas.
1.1. El poro nuclear El poro nuclear es el complejo proteico que permite el tránsito a través de la membrana nu-
clear. Su luz debe de ser lo suficientemente gran-
de como para permitir el paso de polinucleótidos
acompañados de proteínas y las heteronucleo-
proteínas, así como las partículas preribosomales.
El poro nuclear permite el paso libre de sustancias como el agua, los iones y proteínas meno-
res de 20 kDa a favor de gradiente de concentración. Para las proteínas que tienen un tamaño
de 40-50 kDa también hay un cierto tránsito, pero se imponen resistencias a la difusión, y para
proteínas de 60-70 kDa el poro es totalmente selectivo permitiendo el paso solo a través de la
acción de las carioferinas que reconocen las NLS.
Al núcleo pasan proteínas como las ribonucleoproteinas nucleares heterogéneas (hnRNPs), al
citosol salen las partículas pre-ribosomales y los pre-mRNAs, en conjunto a una velocidad de
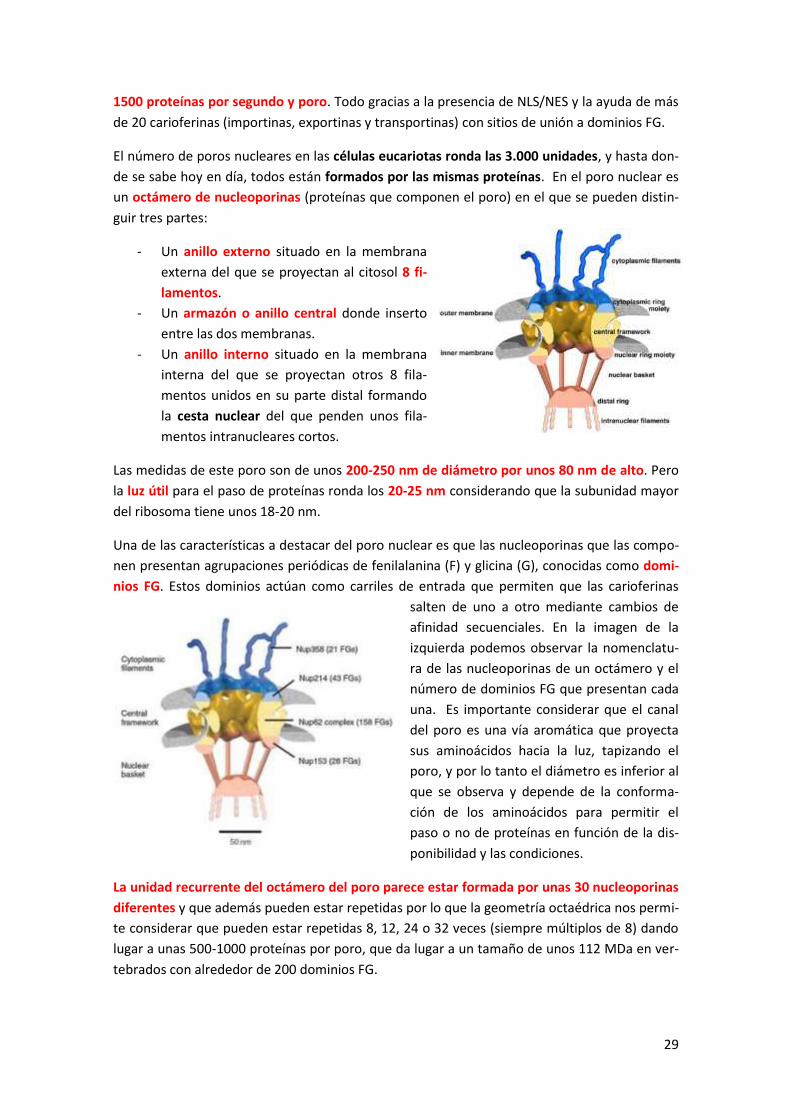
29
1500 proteínas por segundo y poro. Todo gracias a la presencia de NLS/NES y la ayuda de más
de 20 carioferinas (importinas, exportinas y transportinas) con sitios de unión a dominios FG.
El número de poros nucleares en las células eucariotas ronda las 3.000 unidades, y hasta don-
de se sabe hoy en día, todos están formados por las mismas proteínas. En el poro nuclear es
un octámero de nucleoporinas (proteínas que componen el poro) en el que se pueden distin-
guir tres partes:
- Un anillo externo situado en la membrana
externa del que se proyectan al citosol 8 fi-
lamentos.
- Un armazón o anillo central donde inserto
entre las dos membranas.
- Un anillo interno situado en la membrana
interna del que se proyectan otros 8 fila-
mentos unidos en su parte distal formando
la cesta nuclear del que penden unos fila-
mentos intranucleares cortos.
Las medidas de este poro son de unos 200-250 nm de diámetro por unos 80 nm de alto. Pero
la luz útil para el paso de proteínas ronda los 20-25 nm considerando que la subunidad mayor
del ribosoma tiene unos 18-20 nm.
Una de las características a destacar del poro nuclear es que las nucleoporinas que las compo-
nen presentan agrupaciones periódicas de fenilalanina (F) y glicina (G), conocidas como domi-
nios FG. Estos dominios actúan como carriles de entrada que permiten que las carioferinas
salten de uno a otro mediante cambios de
afinidad secuenciales. En la imagen de la
izquierda podemos observar la nomenclatu-
ra de las nucleoporinas de un octámero y el
número de dominios FG que presentan cada
una. Es importante considerar que el canal
del poro es una vía aromática que proyecta
sus aminoácidos hacia la luz, tapizando el
poro, y por lo tanto el diámetro es inferior al
que se observa y depende de la conforma-
ción de los aminoácidos para permitir el
paso o no de proteínas en función de la dis-
ponibilidad y las condiciones.
La unidad recurrente del octámero del poro parece estar formada por unas 30 nucleoporinas
diferentes y que además pueden estar repetidas por lo que la geometría octaédrica nos permi-
te considerar que pueden estar repetidas 8, 12, 24 o 32 veces (siempre múltiplos de 8) dando
lugar a unas 500-1000 proteínas por poro, que da lugar a un tamaño de unos 112 MDa en ver-
tebrados con alrededor de 200 dominios FG.
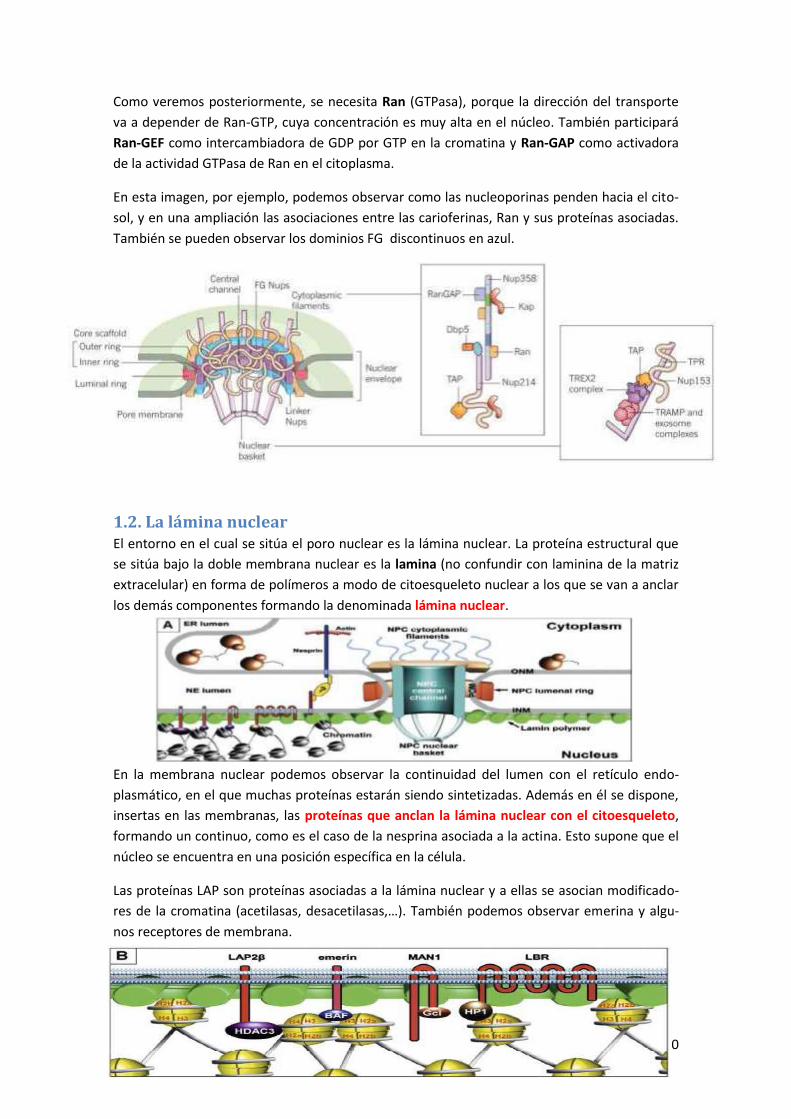
30
Como veremos posteriormente, se necesita Ran (GTPasa), porque la dirección del transporte
va a depender de Ran-GTP, cuya concentración es muy alta en el núcleo. También participará
Ran-GEF como intercambiadora de GDP por GTP en la cromatina y Ran-GAP como activadora
de la actividad GTPasa de Ran en el citoplasma.
En esta imagen, por ejemplo, podemos observar como las nucleoporinas penden hacia el cito-
sol, y en una ampliación las asociaciones entre las carioferinas, Ran y sus proteínas asociadas.
También se pueden observar los dominios FG discontinuos en azul.
1.2. La lámina nuclear El entorno en el cual se sitúa el poro nuclear es la lámina nuclear. La proteína estructural que
se sitúa bajo la doble membrana nuclear es la lamina (no confundir con laminina de la matriz
extracelular) en forma de polímeros a modo de citoesqueleto nuclear a los que se van a anclar
los demás componentes formando la denominada lámina nuclear.
En la membrana nuclear podemos observar la continuidad del lumen con el retículo endo-
plasmático, en el que muchas proteínas estarán siendo sintetizadas. Además en él se dispone,
insertas en las membranas, las proteínas que anclan la lámina nuclear con el citoesqueleto,
formando un continuo, como es el caso de la nesprina asociada a la actina. Esto supone que el
núcleo se encuentra en una posición específica en la célula.
Las proteínas LAP son proteínas asociadas a la lámina nuclear y a ellas se asocian modificado-
res de la cromatina (acetilasas, desacetilasas,…). También podemos observar emerina y algu-
nos receptores de membrana.

31
Si analizamos el gen de la lamina, vemos que por splicing alternativo se da lugar a dos mensa-
jeros (prelamina A y lamina C) que presentan una secuencias de localización nuclear y que por
lo tanto es traducido por ribosomas libres en el citosol, y posteriormente entra en el núcleo.
En cambio la emerina es una proteína integral de la membrana nuclear interna, y es sintetizada
en ribosomas asociados al retículo, donde se interna en la membrana y difunde hasta llegar a
su localización final.
Muchas de las proteínas que yacen en la membrana nuclear van a ser sintetizadas en riboso-
mas asociados al retículo y luego se desplazarán por movimientos laterales hasta alcanzar su
sitio funcional.
1.3. Tráfico de proteínas a través del poro nuclear En el transporte de proteínas a través del poro nuclear van a intervenir las proteínas Ran (pro-
teína G monomérica con actividad GTPasa) y las carioferinas (importinas, exportinas y trans-
portinas) que van a tener sitios de unión a los dominios FG de las nucleoporinas y van a ir sal-
tando de uno al siguiente gracias al aporte de energía de la hidrólisis del GTP (asociado a Ran)
e indirectamente del ATP (no se conoce su participación pero es necesario para que se pro-
duzca el movimiento).
La dirección del transporte depende directamente de Ran-GTP (elevada concentración en el
núcleo), cuya cantidad va a depender de la actividad GTPasa intrínseca, regulada por la proteí-
na activadora Ran-GAP situada en el citosol. Además la proteína Ran-GEF intercambiará el GDP
por GTP al producirse la reentrada de Ran al núcleo. A su vez, estas proteínas van a estar regu-
ladas por diferentes cascadas que omitiremos.
La estructura tridimensional de Ran-GDP muestra una geometría muy compacta con tres bu-
cles. Pero la conformación de Ran-GTP muestra un gran cambio conformacional donde la adi-
ción de las cargas negativas provoca que el bucle C-terminal se despliegue y aleje de la proteí-
na al presentar una gran cantidad de aspartato (D), que a pH fisiologíco presenta carga negati-
va, lo que permite alojar la proteína a la que se une.
Todo esto da lugar a la existencia de múltiples hipótesis para el transporte de proteínas a tra-
vés del poro nuclear.

32
1.3.1. Modelo de importación a través del poro nuclear
La proteína (carga) que tiene que ingresar en el núcleo porta una secuencia NLS que va a ser
reconocida por una importina, en este caso dimérica (α-β), que reconoce la secuencia de
importación (1) y formará un complejo con la carga para pasar el poro nuclear (2). En este
caso la subunidad α presenta una alta afinidad por la NLS, mientras que la subunidad β es la
que tiene la capacidad de reconocer los dominios FG de las nucleoporinas.
Una vez en el interior del núcleo, la alta afinidad de la subunidad β por Ran-GTP provoca la
disociación de la importina (3) que da lugar a un complejo Ran-GTP-β y a la subunidad α-carga
que termina disociándose y liberando la carga dentro del núcleo.
Puesto que la subunidad β es la que interacciona con los dominios FG del poro nuclear, una vez
unida a Ran-GTP, vuelve a atravesar el poro hacia el citosol, donde la proteína Ran-GAP activa
a Ran provocando la hidrólisis del GTP en GDP y la disociación de la subunidad β (5).
Para que la subunidad α abandone el núcleo es necesario que se una a la proteína CAS y a Ran-
GTP (5). Una vez fuera sufrirá la hidrolisis del GTP a GDP y todo el complejo se disociará, que-
dando ambas subunidades listas para comenzar el ciclo.
Cerrado el ciclo de las importinas, es ahora Ran-GDP la que tiene que volver al núcleo. Para
ello el factor de transporte nuclear 2 (NTF2) se une a Ran-GDP y la introduce a través del poro
nuclear (6). Finalmente el intercambio de GDP por GTP realizado por Ran-GEF provoca la diso-
ciación de NTF2 y Ran-GTP vuelve a entrar en el ciclo.
Según este modelo la proteína G Ran no es importante para la entrada de proteínas, pero es
esencial para la salida de las importinas. Además el gradiente de Ran-GTP contribuye a la sali-
da, mientras que el gradiente de Ran-GDP contribuye a la entrada.

33
1.3.2. Modelo de exportación a través del poro nuclear
Al igual que en el caso anterior, para la exportación de una proteína desde el núcleo hasta el
citosol será necesaria la participación de Ran-GTP.
El proceso comienza con la formación de un complejo ternario como consecuencia del recono-
cimiento de la exportina, la NES de la pro-
teína de carga y Ran-GTP. Este complejo
ternario atraviesa el poro nuclear a favor
de gradiente de concentración de Ran-
GTP y su estabilidad se debe a la presen-
cia del GTP.
Cuando el complejo llega al citosol la acti-
vidad GTPasa de Ran es activada por Ran-
GAP, lo que provoca la desestabilización
del complejo ternario y la disociación de
las unidades.
Ahora la exportina (mostrando la señal
NLS) y Ran-GDP entran de nuevo al núcleo
de la misma manera que antes.
1.3.3. Modelo simplificado de importación/exportación de proteínas y mRNA
Una simplificación de la importación de proteínas propuesta en los anteriores apartados hace
referencia a la participación de un complejo binario y no ternario.
Según este una proteína con una NLS va a ser reconocida por una importina que la introducirá
en el núcleo a través del poro nuclear. Pero no va a ser Ran-GTP quien provoque la desestabili-
zación, sino que la propia importina va a ser capaz de unir GTP y liberar la proteína de carga.
Una vez formado el complejo importina-GTP, esta cambia de conformación y sale al citosol,
donde Ran-GAP, provoca la actividad GTPasa de la importina y se libera el GDP.
Durante la síntesis del mRNA, estos van siendo reconocidos por multitud de proteínas que van
formando un complejo alrededor del mensajero, sustituyéndose unos por otros y permitiendo
la posterior salida al citosol donde la proteína Dbp5 permitirá el de ensamblaje de todo el con-
junto y la traducción del mRNA por los ribosomas.

34
1.4. El transporte a través del poro en función del tamaño de la carga El transporte de proteínas a través del poro
nuclear presenta diferentes situaciones en fun-
ción del tamaño de la proteína a transportar.
Cuando la carga son proteínas pequeñas, en un
mismo poro nuclear se puede estar realizando
un transporte bidireccional donde las cargas y
las carioferinas van saltando de un dominio FG
a otro.
Cuando la carga es de un complejo grande rodeado
de carioferinas, esta precisa establecer numerosos
contactos con los filamentos de la nucleoporinas
para poder avanzar y además el transporte solo
puede realizarse en una dirección.
Finalmente, quedan las ribonucleoproteinas muy largas (mRNA + proteínas) envueltas en ca-
rioferinas a modo de cuentas de collar que atraviesan el poro gracias a factores accesorios.
1.5. Diversidad de carioferinas La existencia de una increíble variedad de proteínas que entran y salen del núcleo, sin contar
con los factores de transcripción podría precisar de una ingente cantidad de proteínas para
realizar el transporte. Pero la célula ha desarrollado una estrategia intermedia en la que una
serie de importinas y exportinas, junto con proteínas adaptadoras y colaboradoras pueden
llevar a cabo el proceso aplicando el principio de redundancia.
Según el principio de redundancia una proteína va a poder ser transportada por distintas im-
portinas, de manera que además de no saturarse el sistema, el fallo de una carioferina no va a
provocar una deficiencia del sistema.
Además las carioferinas humanas presentan ortólogos en levaduras, por lo que son un sistema
bastante conservado.

35
1.6. Complejos protéicos asociados a la distrofina Las distrofias musculares son enfermedades generalmente graves cuya base molecular radica
en la extraordinaria fragilidad del sarcolema, la membrana celular de las células musculares
que está sometida a un fuerte estrés mecánico por los ciclos de tensión relajación.
El sarcolema está fuertemente reforzado en sus dos caras mediante una lámina basal exter-
na del músculo, y mediante la red del citoesqueleto en su parte interna, quedando apuntala-
da por una gran diversidad de proteínas estructurales.
La matriz extracelular queda
anclada al sarcolema a través
de la laminina-2 que se une a
los péptidoglicanos (distrogli-
canos α/β) y estos a los sarco-
glicanos (α/β/γ/δ). En la cara
interna del sarcolema los pép-
tidoglicanos mencionados se
unen a la distrofina, desmina y
sincoilinas, que anclan el sar-
colema al citoesqueleto de la
fibra muscular.
La deficiencia en cualquiera de los elementos estructurales, ya sea por mutación o por falta de
estos, provoca un estado patológico ya que todo el conjunto queda debilitado. Por ello se ha-
bla de:
- Enfermedad de Duchenne para la deficiencia en la laminina-2.
- Sarcoglicanopatía si es una deficiencia de sarcoglicanos.
- Distroglicanopatía si es una deficiencia en los distranoglicanos.
Pero además tenemos que considerar las deficiencias en todo el espectro, ya que una mala
glicosilación de las proteínas implicadas también dará lugar a un estado patológico.
El fallo de las proteínas provoca un aumento de la permeabilidad del sarcolema que permite la
salida de los componentes al plasma sanguíneo, así como la entrada. Uno de los indicadores de
lesión muscular más utilizados es la identificación de una alta concentración de creatinina en
plasma.
Pero lo más preocupante desde el punto de vista de la célula muscular es la entrada de calcio,
ya que los cambios en la concentración de calcio son un mensaje que va a activar a las calpai-
nas, las proteasas de la actina y la miosina, destruyendo la masa muscular.
Hoy día se sabe que las mutaciones en la lamina A de la lámina nuclear también dan lugar a
una distrofia muscular debido a que el citoesqueleto como conjunto falla, provocando la rup-
tura del sarcolema. Por lo que, la deficiencia de cualquier proteína estructural del citoesque-
leto puede afectar a la integridad del sarcolema.

36
El intercambio de señales decrece con la edad produciendo un deterioro de la función
mitocondrial que va a favorecer trastornos neurodegenerativos ya que el sistema ner-
vioso central consume mucho ATP.
2. Tráfico citosol-mitocondria y cloroplastos El proteoma mitocondrial está formando por unas 1000 proteínas de las cuales solo 13 están
codificadas por el genoma mitocondrial. Las proteínas restantes son sintetizadas por el núcleo
e importadas a la mitocondria.
El motivo por el que estos genes no han sido transferidos al núcleo se desconocen, pero exis-
ten dos posibilidades:
- No ha pasado suficiente tiempo para una transferencia completa.
- Las proteínas codificadas son demasiado hidrofóbicas como para poder ser sintetiza-
das en el citosol.
En cualquier caso el mitDNA representa el 0,5% del DNA total de una célula y el número de
copias varía mucho en función del número de mitocondrias. Si cada mitocondria tiene 2 o 3
copias estaríamos hablando de unas 80-100 copias del genoma mitocondrial.
La teoría endosimbiótica apoya que el genoma mitocondrial sea DNA circular de doble cadena
desprovisto de histonas, que yace en la matriz mitocondrial y que está controlado por una
herencia maternal.
La importancia de la mitocondria radica en su papel para la supervivencia de la célula partici-
pando en el transporte de electrónico, la fosforilación oxidativa y la respiración celular. Tam-
bién está muy relacionado con el metabolismo como en el caso del ciclo de los ácidos tricarbo-
xilicos, el ciclo de la urea y en la liberación de citocromo C para determinar los procesos apop-
tóticos durante la morfogénesis.
El hecho de que dos genomas codifiquen distintas subunidades de un complejo proteico obliga
a la existencia de una comunicación fluida entre la mitocondria y el núcleo, estableciéndose
lo que se conocen como señales retrógradas, de la mitocondria al núcleo, y las señales anteró-
gradas, del núcleo a la mitocondria.
En el momento en el que la mitocondria lanza una señal S.O.S al núcleo, como el aumento de
los niveles de ROS, este responde mediante un aumento en la producción de SOD, peroxida-
sas, glutatión reducido y afines.
2.1. El genoma mitocondrial El genoma mitocondrial es circular y presenta un tamaño de 16 Kb que codifica:
- 7 subunidades del complejo respiratorio I
- 1 subunidad del complejo respiratorio III
- 3 subunidades del complejo respiratorio IV
- 2 subunidades de la ATP sintasa
- 22 tRNA
- 2 RNA ribosomales

37
En el citosol son necesarios al menos 30 tRNA para sintetizar una proteína ya que hay
una preferencia por ciertos tripletes.
Uno de los factores que más destacan es el hecho de que solo se sintetizan 22 tRNA para 21
aminoácidos existentes (solo él triplete de la serina está degenerado y puede ser codificado
por dos tRNA). Esto se debe a que la síntesis de proteínas en la mitocondria se va a producir
mediante apareamientos no canónicos en los que el reconocimiento del codón se realizará
sobre dos bases, balanceándose la tercera.
Esta traducción tan laxa aporta un menor grado de fidelidad que facilita las mutaciones duran-
te la síntesis de proteínas y además obliga a cambiar el significado de algunos tripletes de
bases, existiendo una divergencia desde el endosimbionte.
En cuanto a los rRNA se codifica uno de 16 S y otro de 12 S que se modifican y ensamblan en
la propia mitocondria, pero todas las
proteínas accesorias son codificadas
por el genoma nuclear, lo que obliga a
la existencia de dos series de proteínas
ribosomales, una de las cuales porta la
secuencia de localización mitocondrial.
La RNA-pol y la DNA-pol también son
importadas.
Existen muchas enfermedades asocia-
das a mutaciones en los genes mito-
condriales que codifican para las
subunidades de los complejos respira-
torios ya que por ejemplo, cuando el
complejo 1 no es funcional solo se pro-
ducen 2 moléculas de ATP a través del
complejo II, en vez de 3.
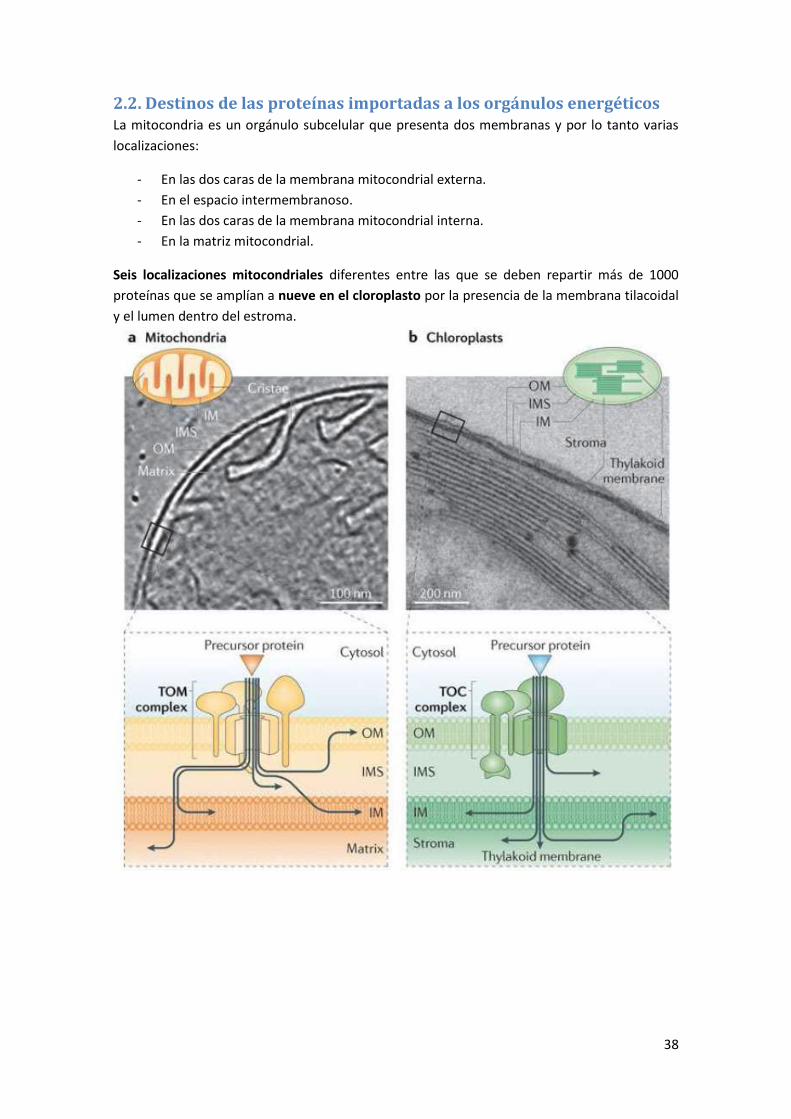
38
2.2. Destinos de las proteínas importadas a los orgánulos energéticos La mitocondria es un orgánulo subcelular que presenta dos membranas y por lo tanto varias
localizaciones:
- En las dos caras de la membrana mitocondrial externa.
- En el espacio intermembranoso.
- En las dos caras de la membrana mitocondrial interna.
- En la matriz mitocondrial.
Seis localizaciones mitocondriales diferentes entre las que se deben repartir más de 1000
proteínas que se amplían a nueve en el cloroplasto por la presencia de la membrana tilacoidal
y el lumen dentro del estroma.
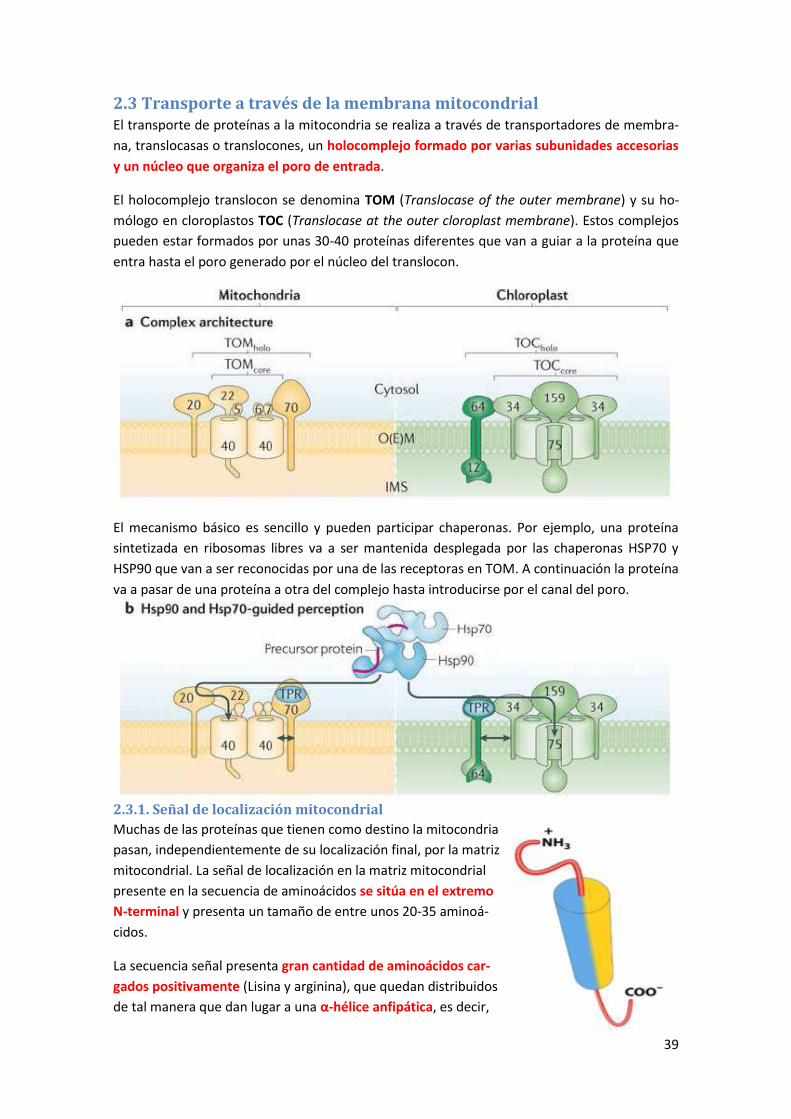
39
2.3 Transporte a través de la membrana mitocondrial El transporte de proteínas a la mitocondria se realiza a través de transportadores de membra-
na, translocasas o translocones, un holocomplejo formado por varias subunidades accesorias
y un núcleo que organiza el poro de entrada.
El holocomplejo translocon se denomina TOM (Translocase of the outer membrane) y su ho-
mólogo en cloroplastos TOC (Translocase at the outer cloroplast membrane). Estos complejos
pueden estar formados por unas 30-40 proteínas diferentes que van a guiar a la proteína que
entra hasta el poro generado por el núcleo del translocon.
El mecanismo básico es sencillo y pueden participar chaperonas. Por ejemplo, una proteína
sintetizada en ribosomas libres va a ser mantenida desplegada por las chaperonas HSP70 y
HSP90 que van a ser reconocidas por una de las receptoras en TOM. A continuación la proteína
va a pasar de una proteína a otra del complejo hasta introducirse por el canal del poro.
2.3.1. Señal de localización mitocondrial
Muchas de las proteínas que tienen como destino la mitocondria
pasan, independientemente de su localización final, por la matriz
mitocondrial. La señal de localización en la matriz mitocondrial
presente en la secuencia de aminoácidos se sitúa en el extremo
N-terminal y presenta un tamaño de entre unos 20-35 aminoá-
cidos.
La secuencia señal presenta gran cantidad de aminoácidos car-
gados positivamente (Lisina y arginina), que quedan distribuidos
de tal manera que dan lugar a una α-hélice anfipática, es decir,

40
una hélice en la que todas las cargas positivas quedan a un lado generando una parte polar y
otra no polar. Pero no todas las proteínas van a portar esta hélice, solo las que van o tienen
que pasar por la matriz mitocondrial.
Para poner de manifiesto esta secuencia se generó una proteína DHFR (típica del citosol) qui-
mérica que contenía una secuencia de localización mitocondrial de 61 aminoácidos procedente
del citocromo B, cuya localización es el espacio intermembrana de la mitocondria.
El experimento reveló que la proteína DHFR quiméri-
ca era transportada hasta el espacio intermembrana,
pero si se eliminaba una parte de la secuencia, dejan-
do solo 35 aminoácidos la proteína era transportada a
la matriz mitocondrial.
Esto supone la existencia de dos señales en la se-
cuencia de localización del citocromo b, una de ellas
(1-35 aa) es la señal de importación a la matriz mito-
condrial, y la otra (36-61 aa) es la señal de localización
en el espacio intermembrana, a donde se dirige tras
pasar por la matriz.
Una vez que la proteína entra en la matriz la secuencia de localización es cortada por protea-
sas y se produce el plegamiento adecuado de la proteína, que ha tenido que permanecer des-
plegada para poder atravesar el translocon TOM y el translocon TIM (Translocase of the inner
membrane).
En la actualidad hay dos teorías sobre el motivo por el que las proteínas pasan por la matriz
mitocondrial para alcanzar su destino final en otra localización:
- La teoría conservativa defiende que para que se produzcan los plegamientos definiti-
vos y las modificaciones adecuadas, todas las proteínas tienen que pasar por la matriz
mitocondrial ya que ahí es donde se sitúa la maquinaria específica de modificación.
- La teoría no conservativa defiende que este paso no es necesario.
La realidad es que unas 100 proteínas siguen el modelo conserva-
tivo y las otras no. Un ejemplo son las proteínas que, una vez
dentro de TIM, son incorporadas a la membrana mitocondrial
interna sin tener que pasar por la matriz mitocondrial.
Otra de las conclusiones que se obtuvieron del experimento an-
terior era la necesidad de que la proteína estuviera desplegada
(parcial o completamente) para atravesar los translocones. Si se
adiciona metotrexato, un potente inhibidor de DHFR por su gran
afinidad, esta se pliega y no entra en la mitocondria a pesar de
portar la secuencia de localización mitocondrial.

41
Este mecanismo de tirón molecular es análogo al que se produce en el transporte
post-traduccional (no confundir con co-traduccional) al lumen del retículo endoplas-
mático.
2.3.2. Modelo general de transporte a la matriz mitocondrial
Según lo explicado para la entrada de proteínas en la matriz mitocondrial van a intervenir cha-
peronas (necesidad de ATP), la secuencia de localización, el receptor de importación en la
membrana externa mitocondrial y los complejos TOM y TIM.
Cuando una proteína es sintetizada
por los ribosomas libres y lleva una
secuencia de localización mitocon-
drial, las chaperonas citosólicas
(Hsc70) mantienen el péptido des-
plegado mediante la hidrólisis de
ATP y lo guían hasta un receptor de
importación en la membrana mito-
condrial externa.
Detectada la secuencia de importa-
ción el receptor se une al complejo
TOM, situando la proteína para
entrar a través del poro (Tom40).
Puesto que la proteína se dirige a la
matriz mitocondrial el complejo Tim
reconoce la secuencia de importa-
ción y en una zona de contacto
entre ambas membranas esta es
transferida a través del poro de
TIM.
El tránsito de la proteína a través de los poros se produce gracias a la existencia de un poten-
cial de membrana generado por la gran concentración de protones en el espacio intermem-
brana. Puesto que la secuencia señal está carga positivamente entra a favor de gradiente por
el poro de TIM, pero para que la proteína completa pase es necesaria la actuación de otro gru-
po de chaperonas de la matriz mitocondrial y motores moleculares (PAM) que tiran de ella.
Una vez dentro de la matriz, la secuencia de localización es cortada por proteasas y se produce
el plegamiento para obtener la proteína nativa.
Al igual que en el citosol, si el plegamiento no se produce de manera adecuada intervendrán
las chaperonas mitocondriales o la proteína será derivada a la degradación por el proteoso-
ma (también presente en la mitocondria).
Este modelo general se vuelve más complejo si consideramos la cantidad de destinos que po-
demos encontrar para una proteína en la mitocondria. Veamos en los siguientes apartados
modelos generales para que la proteína alcance diferentes posiciones.
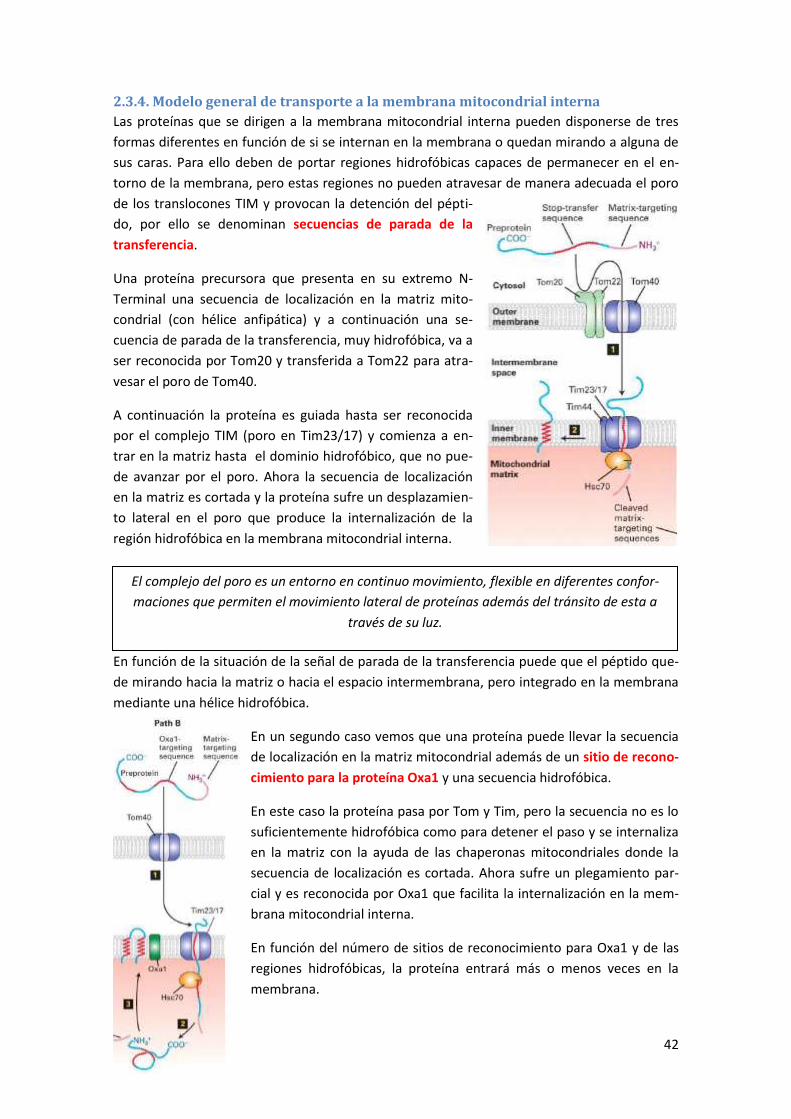
42
El complejo del poro es un entorno en continuo movimiento, flexible en diferentes confor-
maciones que permiten el movimiento lateral de proteínas además del tránsito de esta a
través de su luz.
2.3.4. Modelo general de transporte a la membrana mitocondrial interna
Las proteínas que se dirigen a la membrana mitocondrial interna pueden disponerse de tres
formas diferentes en función de si se internan en la membrana o quedan mirando a alguna de
sus caras. Para ello deben de portar regiones hidrofóbicas capaces de permanecer en el en-
torno de la membrana, pero estas regiones no pueden atravesar de manera adecuada el poro
de los translocones TIM y provocan la detención del pépti-
do, por ello se denominan secuencias de parada de la
transferencia.
Una proteína precursora que presenta en su extremo N-
Terminal una secuencia de localización en la matriz mito-
condrial (con hélice anfipática) y a continuación una se-
cuencia de parada de la transferencia, muy hidrofóbica, va a
ser reconocida por Tom20 y transferida a Tom22 para atra-
vesar el poro de Tom40.
A continuación la proteína es guiada hasta ser reconocida
por el complejo TIM (poro en Tim23/17) y comienza a en-
trar en la matriz hasta el dominio hidrofóbico, que no pue-
de avanzar por el poro. Ahora la secuencia de localización
en la matriz es cortada y la proteína sufre un desplazamien-
to lateral en el poro que produce la internalización de la
región hidrofóbica en la membrana mitocondrial interna.
En función de la situación de la señal de parada de la transferencia puede que el péptido que-
de mirando hacia la matriz o hacia el espacio intermembrana, pero integrado en la membrana
mediante una hélice hidrofóbica.
En un segundo caso vemos que una proteína puede llevar la secuencia
de localización en la matriz mitocondrial además de un sitio de recono-
cimiento para la proteína Oxa1 y una secuencia hidrofóbica.
En este caso la proteína pasa por Tom y Tim, pero la secuencia no es lo
suficientemente hidrofóbica como para detener el paso y se internaliza
en la matriz con la ayuda de las chaperonas mitocondriales donde la
secuencia de localización es cortada. Ahora sufre un plegamiento par-
cial y es reconocida por Oxa1 que facilita la internalización en la mem-
brana mitocondrial interna.
En función del número de sitios de reconocimiento para Oxa1 y de las
regiones hidrofóbicas, la proteína entrará más o menos veces en la
membrana.
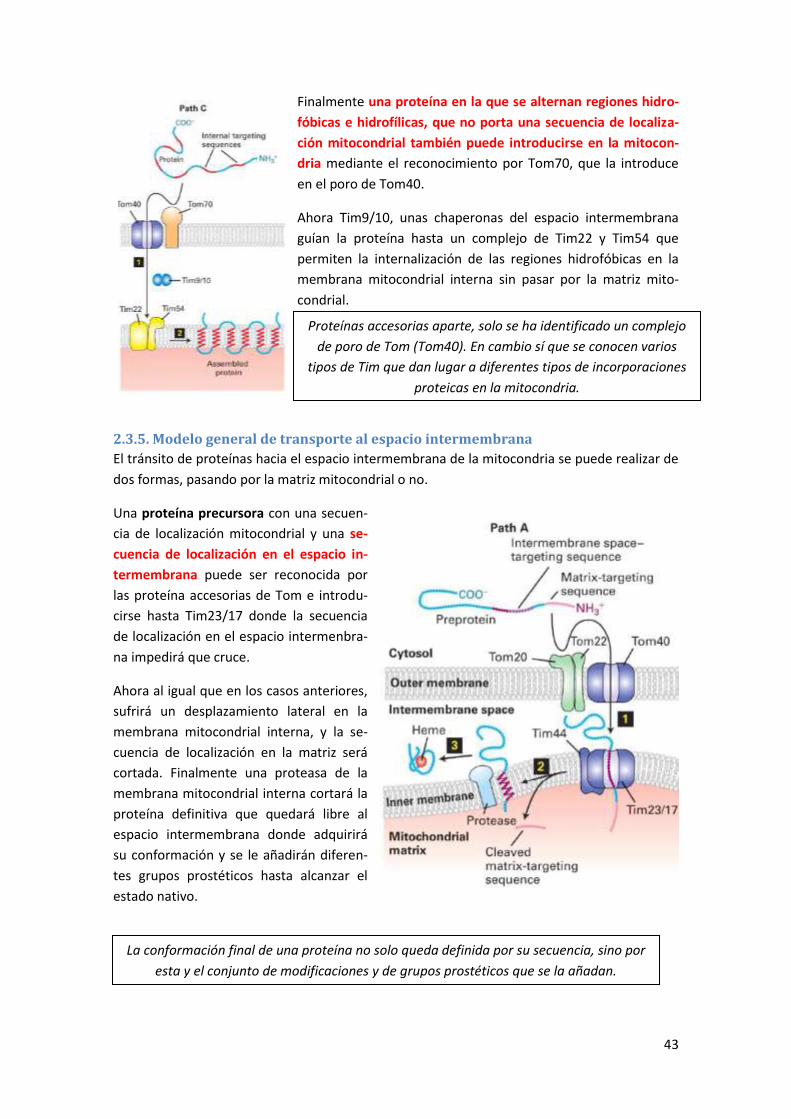
43
Proteínas accesorias aparte, solo se ha identificado un complejo
de poro de Tom (Tom40). En cambio sí que se conocen varios
tipos de Tim que dan lugar a diferentes tipos de incorporaciones
proteicas en la mitocondria.
La conformación final de una proteína no solo queda definida por su secuencia, sino por
esta y el conjunto de modificaciones y de grupos prostéticos que se la añadan.
Finalmente una proteína en la que se alternan regiones hidro-
fóbicas e hidrofílicas, que no porta una secuencia de localiza-
ción mitocondrial también puede introducirse en la mitocon-
dria mediante el reconocimiento por Tom70, que la introduce
en el poro de Tom40.
Ahora Tim9/10, unas chaperonas del espacio intermembrana
guían la proteína hasta un complejo de Tim22 y Tim54 que
permiten la internalización de las regiones hidrofóbicas en la
membrana mitocondrial interna sin pasar por la matriz mito-
condrial.
2.3.5. Modelo general de transporte al espacio intermembrana
El tránsito de proteínas hacia el espacio intermembrana de la mitocondria se puede realizar de
dos formas, pasando por la matriz mitocondrial o no.
Una proteína precursora con una secuen-
cia de localización mitocondrial y una se-
cuencia de localización en el espacio in-
termembrana puede ser reconocida por
las proteína accesorias de Tom e introdu-
cirse hasta Tim23/17 donde la secuencia
de localización en el espacio intermenbra-
na impedirá que cruce.
Ahora al igual que en los casos anteriores,
sufrirá un desplazamiento lateral en la
membrana mitocondrial interna, y la se-
cuencia de localización en la matriz será
cortada. Finalmente una proteasa de la
membrana mitocondrial interna cortará la
proteína definitiva que quedará libre al
espacio intermembrana donde adquirirá
su conformación y se le añadirán diferen-
tes grupos prostéticos hasta alcanzar el
estado nativo.
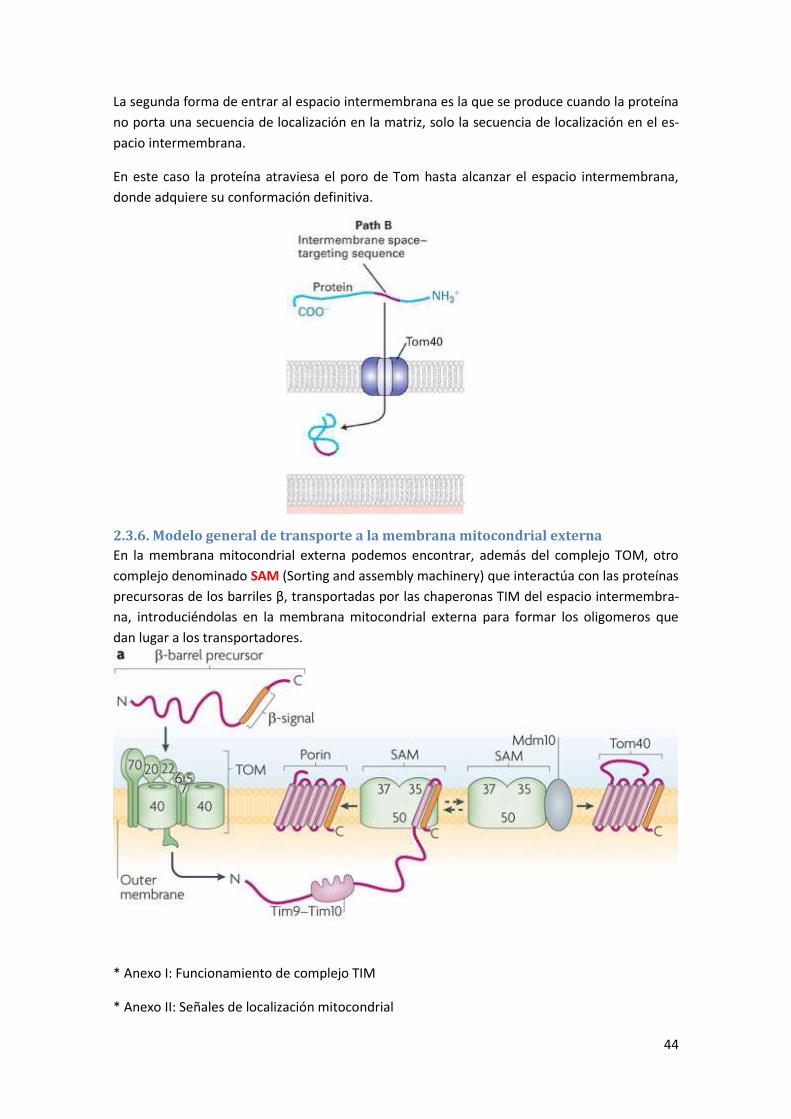
44
La segunda forma de entrar al espacio intermembrana es la que se produce cuando la proteína
no porta una secuencia de localización en la matriz, solo la secuencia de localización en el es-
pacio intermembrana.
En este caso la proteína atraviesa el poro de Tom hasta alcanzar el espacio intermembrana,
donde adquiere su conformación definitiva.
2.3.6. Modelo general de transporte a la membrana mitocondrial externa
En la membrana mitocondrial externa podemos encontrar, además del complejo TOM, otro
complejo denominado SAM (Sorting and assembly machinery) que interactúa con las proteínas
precursoras de los barriles β, transportadas por las chaperonas TIM del espacio intermembra-
na, introduciéndolas en la membrana mitocondrial externa para formar los oligomeros que
dan lugar a los transportadores.
* Anexo I: Funcionamiento de complejo TIM
* Anexo II: Señales de localización mitocondrial

45
2.4. Transporte citosol-cloroplasto El cloroplasto vegetal presenta una cámara extra formada por la membrana tilacoidal que da
lugar a la existencia de otras tantas localizaciones subcelulares.
Se conoce poco sobre las secuencias que determinan la localización de las proteínas que son
transportadas al cloroplasto, pero se ha detectado la existencia de una secuencia de localiza-
ción en el estroma que puede ir seguida de otra secuencia diferente de localización en los tila-
coides.
La plastocianina, una proteína de localización en el lumen tilacoidal porta, en su precursor, las
dos secuencias de localización. Atraviesa los complejos TOC y TIC (homólogos a TOM y TIM)
para alcanzar el estroma y una vez dentro, el precursor de la plastocianina sufre un corte pro-
teolítico que retira la secuencia de localización en el estroma, para que ahora la secuencia de
localización en el tilacoide sea reconocida por la proteína SRP cloroplastica (Signal recognition
particle).
SRP interacciona con un receptor en la membrana del tilacoide e introduce el precursor a tra-
vés de otra proteína poro hasta el lumen tilacoidal, donde se le corta la secuencia de localiza-
ción y adquiere la conformación final.
En el caso de una proteína que liga metales, esta porta la secuencia de localización en el es-
troma y una secuencia de localiza-
ción en el tilacoide que comienza
con dos argininas (R). Cuando atra-
viesa TOC y TIC se le retira la se-
cuencia de importación al estroma
dejando a la vista las argininas y
comienza a unir los metales co-
rrespondientes plegándose y atra-
vesando un complejo en la mem-
brana tilacoidal.
Finalmente el péptido con las argi-
ninas es cortado y la proteína ad-
quiere su estructura final en lumen
tilacoidal.
Es importante considerar que las
proteínas no entran a través de
los complejos poro completamen-
te desplegadas ya que eso cuesta
mucha energía y cómo podemos
observar hay casos en los que la
proteína pasa casi completamente
plegada.
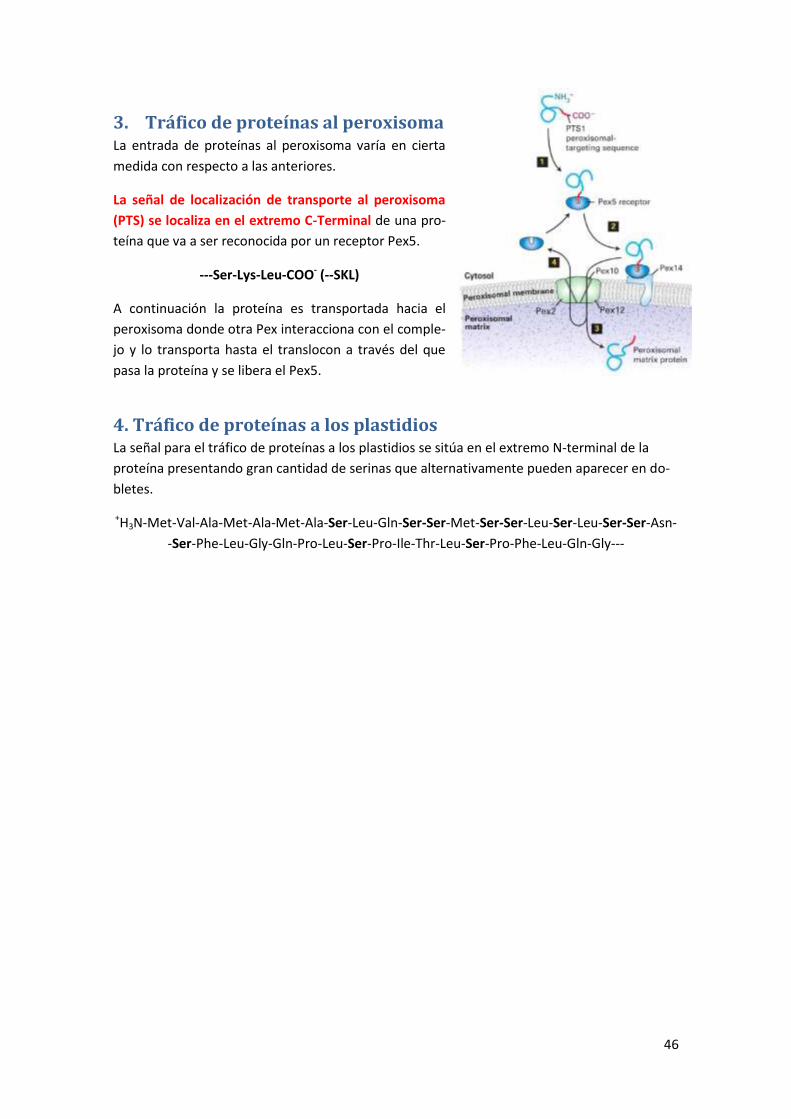
46
3. Tráfico de proteínas al peroxisoma La entrada de proteínas al peroxisoma varía en cierta
medida con respecto a las anteriores.
La señal de localización de transporte al peroxisoma
(PTS) se localiza en el extremo C-Terminal de una pro-
teína que va a ser reconocida por un receptor Pex5.
---Ser-Lys-Leu-COO- (--SKL)
A continuación la proteína es transportada hacia el
peroxisoma donde otra Pex interacciona con el comple-
jo y lo transporta hasta el translocon a través del que
pasa la proteína y se libera el Pex5.
4. Tráfico de proteínas a los plastidios La señal para el tráfico de proteínas a los plastidios se sitúa en el extremo N-terminal de la
proteína presentando gran cantidad de serinas que alternativamente pueden aparecer en do-
bletes.
+H3N-Met-Val-Ala-Met-Ala-Met-Ala-Ser-Leu-Gln-Ser-Ser-Met-Ser-Ser-Leu-Ser-Leu-Ser-Ser-Asn-
-Ser-Phe-Leu-Gly-Gln-Pro-Leu-Ser-Pro-Ile-Thr-Leu-Ser-Pro-Phe-Leu-Gln-Gly---
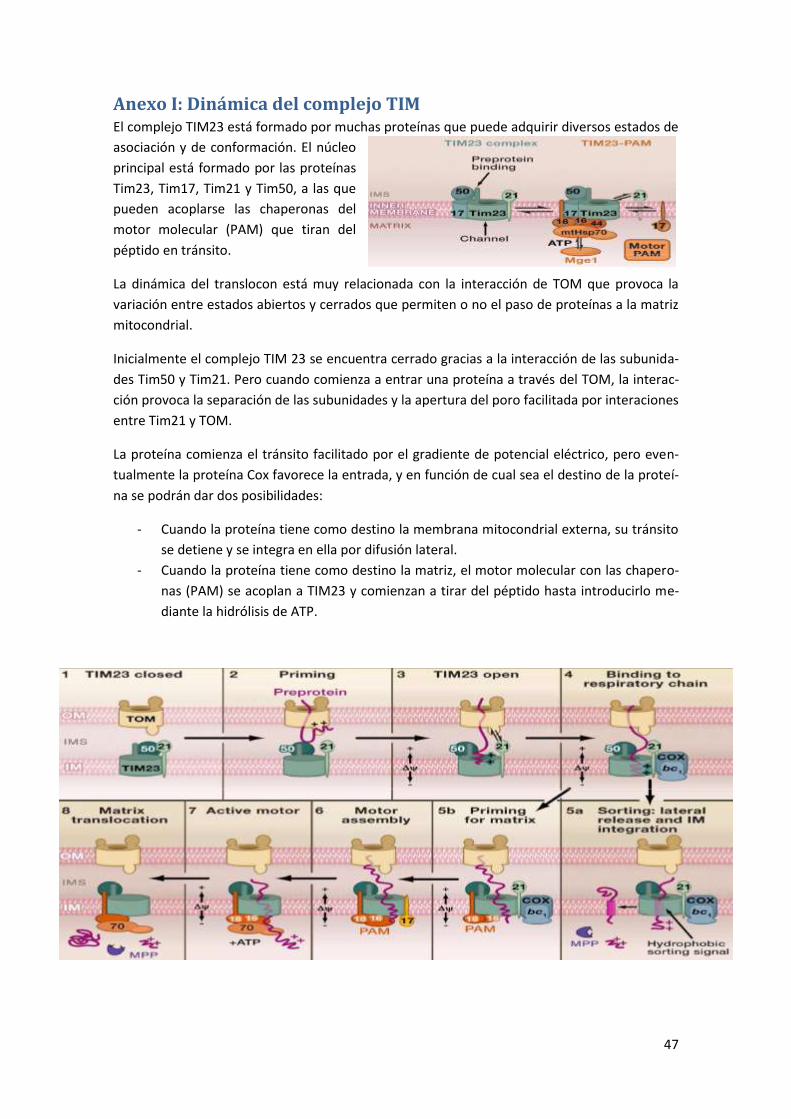
47
Anexo I: Dinámica del complejo TIM El complejo TIM23 está formado por muchas proteínas que puede adquirir diversos estados de
asociación y de conformación. El núcleo
principal está formado por las proteínas
Tim23, Tim17, Tim21 y Tim50, a las que
pueden acoplarse las chaperonas del
motor molecular (PAM) que tiran del
péptido en tránsito.
La dinámica del translocon está muy relacionada con la interacción de TOM que provoca la
variación entre estados abiertos y cerrados que permiten o no el paso de proteínas a la matriz
mitocondrial.
Inicialmente el complejo TIM 23 se encuentra cerrado gracias a la interacción de las subunida-
des Tim50 y Tim21. Pero cuando comienza a entrar una proteína a través del TOM, la interac-
ción provoca la separación de las subunidades y la apertura del poro facilitada por interaciones
entre Tim21 y TOM.
La proteína comienza el tránsito facilitado por el gradiente de potencial eléctrico, pero even-
tualmente la proteína Cox favorece la entrada, y en función de cual sea el destino de la proteí-
na se podrán dar dos posibilidades:
- Cuando la proteína tiene como destino la membrana mitocondrial externa, su tránsito
se detiene y se integra en ella por difusión lateral.
- Cuando la proteína tiene como destino la matriz, el motor molecular con las chapero-
nas (PAM) se acoplan a TIM23 y comienzan a tirar del péptido hasta introducirlo me-
diante la hidrólisis de ATP.

48
Anexo II: Señales de localización mitocondrial

49
Anexo III: Genes y Parkinson El Parkinson (PD) es una enfermedad que afecta al 1% de las personas mayores de 60 años,
caracterizada por la pérdida de neuronas que fabrican dopamina (DA) en los núcleos cerebra-
les y la aparición de cuerpos de Lewy (CL, no en todas las variantes de PD). El tratamiento más
utilizado, pero que pierde eficacia a largo plazo, es suministrar L-DOPA, un análogo de la do-
pamina capaz de atravesar la membrana hematoencefálica.
Las regiones cerebrales afectadas son las que controlan el movimiento y la coordinación por lo
que los enfermos de PD muestran hipocinesia, tremor, bradicinesia, acinesia, rigidez en rueda
dentada e hipertonía muscular.
Conforme avanzan los estudios sobre PD se han establecido ciertos factores ambientales como
la exposición a plaguicidas, disolventes, metales y campos magnéticos, pero hoy día se sabe
que existen ciertos componentes genéticos:
- La α-sinucleína (ASN) es una proteína de 140 aminoácidos, muy importante en la sín-
tesis de dopamina que actúa como inhibidor de la tyr-hidroxilasas clave en la síntesis
de catecolaminas convirtiendo la tirosina en DOPA y ésta a dopamina que puede con-
vertirse en noradrenalina para posteriormente pasar a adrenalina. También interviene
en la transducción de señales, en la dinámica del citoesqueleto, en la plasticidad neu-
ronal, en el aprendizaje, en la memoria,… Las mutaciones en el gen ASN provocan la
formación de fibras y agregados fribrilares (efecto potenciado por ROS) que dan lugar
a la formación de placas que en personas jóvenes y sanas son elminadas por el sistema
ubiquitina-proteosoma (UPS). Pero con el paso del tiempo el sistema comienza a fallar
dando lugar a la aparición de PD, demencia,… o el exceso de ROS y neurotoxinas natu-
rales provocan la pérdida de las neuronas DA.
- La parkina es una E3-ligasa del sistema UPS encargada de la degradación de ASN, por
lo que mutaciones en este gen (hasta 57 en casos de PD temprano en menores de 45
años) derivan en la aparición de PD.
- El oncogen DJ1 es una chaperona de la matriz mitocondrial inducido por estrés oxida-
tivo y que previene la producción de ROS. Mutaciones en este gen provocan el aumen-
to de ROS que desencadena la agregación de ASN derivando en PD.
- La quinasa PINK1 o quinasa 1 inducida por PTEN, es otra proteína de acción protectora
frente al estrés oxidativo cuya mutación ha sido relacionada con la aparición de PD.
Considerando el aumento de los niveles de ROS como un factor determinante en la aparición
de ROS, muchos de los tratamientos actuales han dado gran importancia al uso de terpenoi-
des, aceites esenciales y demás, como agentes inhibidores de la cadena de transporte de elec-
trones.
El correcto funcionamiento de la mitocondria depende de un equilibrio entre los inductores de
ROS (ROS inducers) y los sumideros de ROS (ROS quenchers). Durante la juventud, las células
funcionan normalmente y controlan los niveles de ROS que son necesarios en cierta cantidad
como señales de transducción o segundos mensajeros.
Recordemos que las ROS son capaces de formar peróxidos con los dobles enlaces de los ácidos
grasos insaturados (esenciales como el araquidónico) y pueden dañar el DNA rompiendo las

50
dos hebras. Otro de los efectos negativos es la oxidación de proteínas en restos de cisteína,
arginina y lisina, que provoca un cambio en la estructura tridimensional.
A medida que envejecemos, la capacidad de la célula para manejar los niveles de ROS disminu-
ye, bien por la sobreproducción o porque la comunicación núcleo-mitocondria ya no es eficien-
te, y con esa premisa se está estudiando la relación de ROS con PD, transtornos cardíacos,
cáncer,…
La degeneración mitocondrial y la sobreproducción de ROS conducen en última instancia a la
neurodegenración causante del PD, ya que el sistema nervioso (SN) precisa de grandes aportes
de ATP.
Por ello tanto la mutación de los complejos respiratorios de la cadena de transporte de elec-
trones como su inhibición mediante químicos (MPTP, Rotenona o Paraquat) conducen a una
bajada en la producción de los niveles de ATP, que puede ser atajada mediante inhibidores o
secuestradores de estas sustancias como los triterpenos, creatinina, coenzima Q10 y nicotina-
mida. En caso contrario se genera una disfunción mitocondrial.
Por otro lado, la producción de ROS va a suponer una sobreproducción de ciertos factores que
afectan a la proliferación mitocondrial, impidiendo la transmisión del daño e induciendo la
mitofagia. Si no fuese suficiente el último paso es la apoptosis celular.
Además una sobreproducción de ROS induce a factores de transcripción nucleares que van a
promover la expresión de catalasa, superóxido dismutasa, glutatión reducido,… que disminu-
yen la cantidad y suprimen la neurodegeneración si se alcanza un estado de neuroprotección.
Pero cuando la cantidad de ROS aumenta demasiado se produce la activación de BAX junto a la
liberación de citocromo C que conduce a la apoptosis y por ende a la pérdida de neuronas.
Esta sobreproducción también produce la formación de agregados de ASN, la parkina, el onco-
gen DJ1 y la quinasa PINK1.
En condiciones normales, la parkina se encuentra se encuentra inhibida, pero su unión con
PINK1 la activa y le permite comenzar la degradación de ASN en el citosol. El acoplamiento
con DJ1 permite al conjunto ligarse a la membrana mitocondrial y actuar como un activo pro-
tector frente a la acumulación de ASN. Pero las mutaciones de PINK1 o Parkina provocan neu-
rodegenración al no poder interactuar entre ellas. Además el exceso de ROS también perjudica
la acción del proteosoma, por lo que puede interferir con la degradación de ASN.
Otras proteínas como miro/milton participan en el transporte de mitocondrias a través de los
microtúbulos anterógrado y retrógrado, en los que intervienen como adaptadores de dineina y
quinesina situados en la membrana mitocondrial externa. La mutación de estos genes dan
lugar a mitocondrias defectuosas en relación a la fusión o fisión que conducen a la neurodege-
neración.
También hay que destacar el papel del activador de sirtuina 1 (SIRT1 activator) y el revaratrol,
una agente antioxidante (reductor) de los frutos rojos a base de fenoles que dan lugar a qui-
nonas relativamente inocuas. Las sirtuinas son desacetliasas de lisina dependientes de NAD+
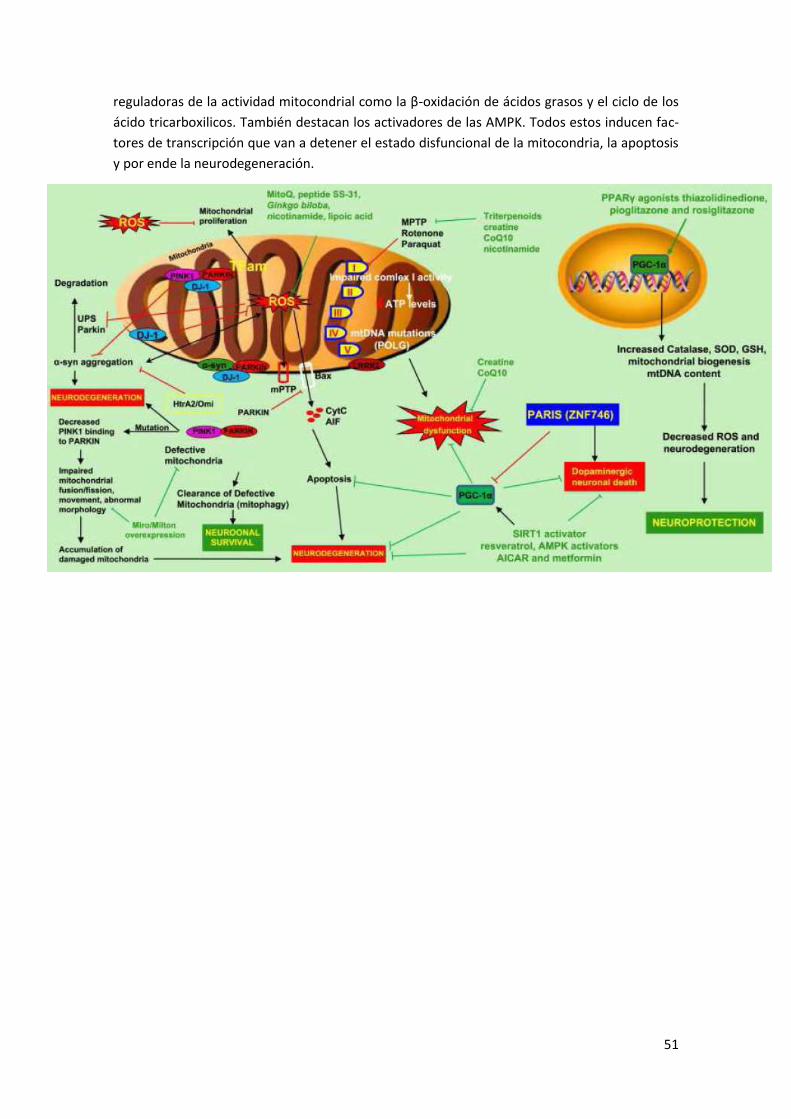
51
reguladoras de la actividad mitocondrial como la β-oxidación de ácidos grasos y el ciclo de los
ácido tricarboxilicos. También destacan los activadores de las AMPK. Todos estos inducen fac-
tores de transcripción que van a detener el estado disfuncional de la mitocondria, la apoptosis
y por ende la neurodegeneración.

52

53
Las proteínas envejecidas tienen que ser degradadas rápidamente ya que representan un
peligro potencial que puede formar agregados, y aquellas que sufren grandes oscilaciones
en el tiempo también para poder regular adecuadamente el metabolismo y el ciclo celular.
Tema 3: Proteolisis intracelúlar
La proteólisis es el proceso de destrucción de proteínas que regula los niveles del proteoma,
tanto en variedad, como en cantidad de proteínas a lo largo del tiempo.
La ventaja evolutiva, con respecto al uso de segundos mensajeros, que tiene la regulación del
proteoma mediante una ruta proteolítica es la inmediatez, pero esto plantea el gran inconve-
niente de que la proteína a degradar debe de estar perfectamente identificada, ya que no
existen estados intermedios, o hay proteína o no la hay.
Por ello van a existir una serie de proteínas encargadas del reconocimiento de la diana a la que
se enlazará ubiquitina y además otra serie de proteínas relacionadas con el proteosoma que
comprobarán que la proteína con ubiquitina es la adecuada, garantizando la degradación de
los objetivos.
1. Procesos regulados por la proteolisis Existen muchísimos proceso regulados por la proteólisis y su importancia radica en la posibili-
dad de la manipulación de la vida media de las proteínas. Las terapias actuales están intentan-
do alargar la vida media de anti-oncogenes a la par que se favorece la degradación de oncoge-
nes.
La proteolisis interviene en factores como:
- La transcripción génica, a través de la degradación de los factores de transcripción. La
modificación de la vida media de los factores de transcripción puede producir un au-
mento o disminución de la señal que pueden desembocar en una ganancia de función
o una pérdida de función.
- Progresión del ciclo celular, a través de las ciclinas. La cantidad de ciclinas está regula-
da por el sistema UPS. En la medida en la que se puedan dirigir las dianas terapéuticas
contra la célula tumoral selectivamente se podrá favorecer o evitar la proliferación y la
apoptosis.
- La organogénesis a través de la degradación proteica que se produce.
- Respuesta inflamatoria a través de la degradación de las pro-citoquinas para evitar
una sobreestimulación.
- El metabolismo del colesterol.
- El procesamiento de antígenos para presentarlos en la membrana plasmática.

54
Para establecer la vida media de una proteína en el torrente circulatorio esta es marcada
y se inyecta a un paciente. A continuación se realizan medidas sucesivas de la cantidad de
proteína para observar como su cantidad va decreciendo.
2. El ciclo de los compuestos nitrogenados y la vida media de las
proteínas Desde la propuesta del modelo del DNA, todos los investigadores se volcaron en establecer el
fundamento del dogma de la biología molecular, las etapas de la replicación, la transferencia
de información del DNA al RNA y la síntesis de proteínas. Pero una vez que se estableció el
proceso, se empezaron a preguntar por qué unas proteínas tenían una vida media corta o lar-
ga.
Aunque las proteínas presenten una vida media larga o corta, todas se degradan para ser re-
novadas. Una persona de unos 70 Kg puede ingerir unos 100 g de proteína diarios y degradar
unos 400 g de proteína endógena, resultado de la proteolisis de proteínas funcionales y no
funcionales.
Esto establece un pool de 500 g de aminoácidos, de los cuales 100 g van a ser excretados y los
restantes serán destinados hacia la síntesis de proteínas que reemplacen a las degradadas.
Por tanto la importancia de la proteolisis tiene que ser transcendental ya que se han identifi-
cado 500-600 proteasas en humanos. La proteolisis limitada (no de degradación completa)
también es necesaria para quitar señales de tránsito o permitir la activación de receptores.
La vida media de una proteína se define como el tiempo que debe transcurrir para que la
cantidad de una proteína se reduzca a la mitad.
Los grupos de investigación, a partir de proteínas marcadas radiactivamente analizaron la vida
de muchas proteínas intentado definir el porqué de la vida media.
Lo que encontraron fue dos grupos de proteínas, unas con una vida media corta (horas como
mucho) y otras de vida media larga (días e incluso meses). El análisis detallado de estas pro-
teínas concluyó que aquellas que presentaban una vida media corta eran principalmente en-
zimas reguladoras de cuya cantidad depende la velocidad de un ciclo metabólico o un proceso
determinado. Es el caso de:
- Ornitina descarboxilasa que media la formación de poliaminas (empaquetan el DNA).
- RNA polimerasa I
- Tirosina aminotransferasa
- Serina deshidratasa
- PEP carboxilasa
- β-hidroxi-β-metilglutaril-CoA deshidrogenasa, productora del ácido mevalónico que
dará colesterol.
Cuyo tiempo de vida media oscila entre las 0,2 h y las 5 h.
Las proteínas de vida media larga como:

55
- Aldolasa
- GAPDH
- Citocromo b
- LDH
- Citocromo c
- Histonas
- Receptores nicotínicos y muscarínicos (otra cosa es que suban o bajen).
Presentan tiempos de vida media que oscilan entre las 118 h e incluso cuatro meses como es
el caso de la hemoglobina.
En definitiva el proteoma va a estar representado por el balance entre la ´síntesis y degrada-
ción de proteínas. Los fallos en cualquiera de los dos extremos van a desencadenar estados
patológicos ya que no se va alcanzar la homeostasis del proteoma.
La vida media de una proteína mutada será más corta porque en el momento en el que el ple-
gamiento cambie, será identificada como aberrante por los sistemas de chaperonas que las
pondrán en marcha hacia la degradación.
El análisis bioinformático de la secuencia de aminoácidos de las proteínas reveló que aquellas
que presentan una vida media larga presentan en sus extremos aminoterminales restos de
metionina, glicina, alanina, serina, treonina o valina, lo que sugiere que estos estabilizan a la
proteína, evitando su degradación a corto plazo
evitando el reconocimiento sencillo por parte del
sistema UPS (ubiquitina-proteasoma). En cambio las
proteínas de vida media corta presentaban isoleu-
cina, glutamina, tirosina, glutamato, prolina, leuci-
na, fenilalanina, asparragina, lisina o arginina.
3. Proteólisis intracelular
3.1 Proteólisis intracelular en procariotas La proteólisis intracelular en procariotas es muy activa debido a la limitación en la cantidad de
aminoácidos a la que están sometidos. Una vez que una proteína cumple su función es rápi-
damente degradada para fabricar otra, ya que no disponen de reservas.
Se han identificado en este sentido, muchas proteasas que actúan por defecto degradando
proteínas de manera continua a menos que se active algún mecanismo para detenerlas. Por lo
tanto en procariotas la vía normal es la síntesis y degradación continua de proteínas, a dife-
rencia de los eucariotas.
Para esta proteólisis se han identificado varias proteasas como Lon y ClP que detectan las pro-
teínas que no son necesarias o las desplegadas y las degradan mediante cortes proteolíticps.
Estas proteínas tienen en muchos casos una doble función, por un lado son chaperonas y por
otro proteasas, en dos dominios diferentes o mediante dos subunidades.

56
Estas proteasas detectan, como chaperonas, una estructura tridimensional aberrante y la en-
caminan hacia la degradación realizando cortes proteolíticos cada 60-70 aminoácidos. Ambas
actividades son ATP dependientes, es decir, se precisa una molécula de ATP para el reconoci-
miento y otra molécula para la proteólisis.
A continuación los fragmentos pequeños son degradados por otros mecanismos que no cues-
tan tanta energía.
3.2 Proteólisis intracelular en eucariotas La célula eucariota presenta dos sistemas de degradación, la denominada vía lisosomal y la no
lisosomal, cuya gran diferencia radica en que la primera funciona a pH ácido y la segunda a pH
neutro.
Para determinar la vía de degradación de una proteína se puede recurrir a dos estrategias en
las que se comienza marcando una proteína:
- Si utilizamos agentes lisosomotrópicos para neutralizar el pH del lisosoma o disipar el
gradiente con respecto al citosol, las catepsinas encargadas de la degradación no pue-
den actuar y por lo tanto si la proteína no es degradada, podemos concluir que su de-
gradación sigue la vía lisosomal. Los agentes lisosomotrópicos son la quinina, la clorpi-
na (antimaláricos) y el cloruro amónico.
- La utilización de células sin lisosomas o con lisosomas poco desarrollados como los
reticulocitos, permite establecer también si una proteína se degrada o no por vía liso-
somal.
En la vía no lisosomal de degradación de proteínas eucarióticas podemos distinguir dos meca-
nismos degradativos:
- El sistema de las calpaínas que funciona a pH neutro y alta concentración de Calcio.
Las calpaínas son proteasas dependientes de calcio que están inactivas a concentra-
ciones normales citosólicas de calcio (0,1 μM), hasta que la apertura de los canales de
calcio dependientes de voltaje del RE produce una salida de calcio, aumentando la
concentración hasta 1-5 μM (10-50 veces más) y activándolas. A continuación el calcio
es secuestrado por sus sumideros (calmodulina). Los fallos que provocan aumentos en
la concentración de calcio están relacionados con distrofias musculares debidas a la
proteólisis descontrolada que se provoca por la activación permanente de las calpaí-
nas.
- El sistema UPS (ubiquitina-proteasoma) que funciona a pH neutro. El sistema comien-
za con el reconocimiento de una proteína a degradar que presenta expuesta una lisina
a la que se va a incorporar una molécula de ubiquitina. A continuación se va formando
un polímero de ubiquitina sobre la primera, y esto sucede en varios puntos al mismo
tiempo que la proteína va siendo envuelta por los polímeros que la dirigen hacia el
proteasoma. En el proteasoma los polímeros de ubiquitina son retirados y la proteína
va siendo desplegada por ATPasas para favorecer su entrada y su degradación.
La vía lisosomal de degradación de proteínas funciona a pH ácido a través de las catepsinas.
En esta vía se degradan proteínas de membrana procedentes de la endocitosis (receptores y

57
ligandos) así como en las estructuras procedentes de la mitofagia y la degradación de orgánu-
los en general.
4. Sistema ubiquitina-proteasoma (UPS) de degradación proteica
4.1 La ubiquitina La ubiquitina es una pequeña proteína de 76 aminoácidos, que recibe el nombre de su ubiqui-
dad ya que está presenten en todos los organismos ecuariotas, desde las levaduras hasta los
seres humanos con una variación de tan solo 3 aminoácidos. El resto de aminoácidos ocupan
las mismas posiciones en la secuencia polipeptídica, siendo una proteína muy conservada evo-
lutivamente. La generación de organismos Knockout para esta es inviable, por lo que el siste-
ma UPS es consustancial con la vida.
En la secuencia de la ubiquitina podemos encontrar 7 lisinas muy importantes denominadas
según su posición como K6, K11, K27, K29, K33, K48 y K63. De estas las lisinas K29 y K48 van a
ser las implicadas en la degradación de la proteína diana.
Estructuralmente esta proteína, que se está convirtiendo en la gran estrella del cáncer, presen-
ta una gran parte de hoja β y una hélice α. Es muy importante recalcar la importancia de la
situación del grupo amino en las lisinas (K) que recordemos, son ε-amino.
Cuando la ubiquitina se empieza polimerizar sobre la proteína diana va a formar un polímero
en el que la ubiquitina entrante se va a enlazar siempre con la misma lisina de la molécula de
ubiquitina precedente. Por lo tanto, el extremo C-terminal de la primera ubiquitina se enlaza
con una de las lisinas expuestas de la proteína a degradar, y
a continuación el extremo C-terminal de otra ubiquitina se
enlaza con la K48 de la primera, y así sucesivamente hasta
formar un polímero de ubiquitina enlazado por K48.
Si observamos el punto de enlace entre las lisinas y la ubi-
quitina vemos que esta última porta una glicina en su ex-
tremo C-terminal que da lugar a un enlace isopeptídico (ε-
amino + carboxilo), que no debe confundirse con un enlace
peptídico (α-amino + carboxilo). El enlace isopeptídico es
una enlace covalente y fuerte que requerirá de enzimas
hidrolizantes para su ruptura.

58
Una proteína cuya conformación esté alterada o se haya solucionado tendrá lisinas expuestas
a las que se unirán las unidades de ubiqutina conduciéndola hacia la degradación. Pero no
todo está determinado, en función de las circunstancias la proteína podrá ser degradada por el
proteasoma, o seguir otros caminos.
4.2 Sistema de adición de ubiquitina De las 100.000 proteínas que se estiman en el proteoma humano podemos suponer que unas
50.000 van a ser degradas por el sistema UPS. Pero no son necesarias otras tantas otras tanas
proteínas para su reconocimiento.
La ubiquitina es una proteína metabólicamente poco activa y que tiene poca tendencia a
reaccionar espontáneamente para formar el puente isopeptídico a través de la glicina del ex-
tremo C-terminal. Por ello precisa ser activada a través de la adición de ácido adenílico (AMP).
La enzima activadora de ubiquitina (E1) cataliza la activación de ubiquitina mediante la utiliza-
ción de ATP y la liberación de pirofosfato inorgánico para formar un intermediario con tenden-
cia a reaccionar con la proteína E1. El mecanismo de reacción comienza con:
1) La activación de la ubiquitina catalizada por E1 que forma un intermedio reactivo
de ubiquitina-AMP.
2) Reacción del intermediario Ubiquitina-AMP con la enzima E1 que da lugar a la for-
mación de un enlace tioester con una cisteína (C) de E1.
A continuación interviene la proteí-
na de conjugación de ubiquitina
(E2) que también es la donadora de
ubiquitina a la proteína diana. Para
que se produzca la reacción E2 y la
lisina (K) de la proteína diana tienen
que aproximarse y por ello la ligasa
de ubiquitina (E3) se encarga de
aproximar a E2-Ubiquitina y la pro-
teína diana, a modo de adaptador.
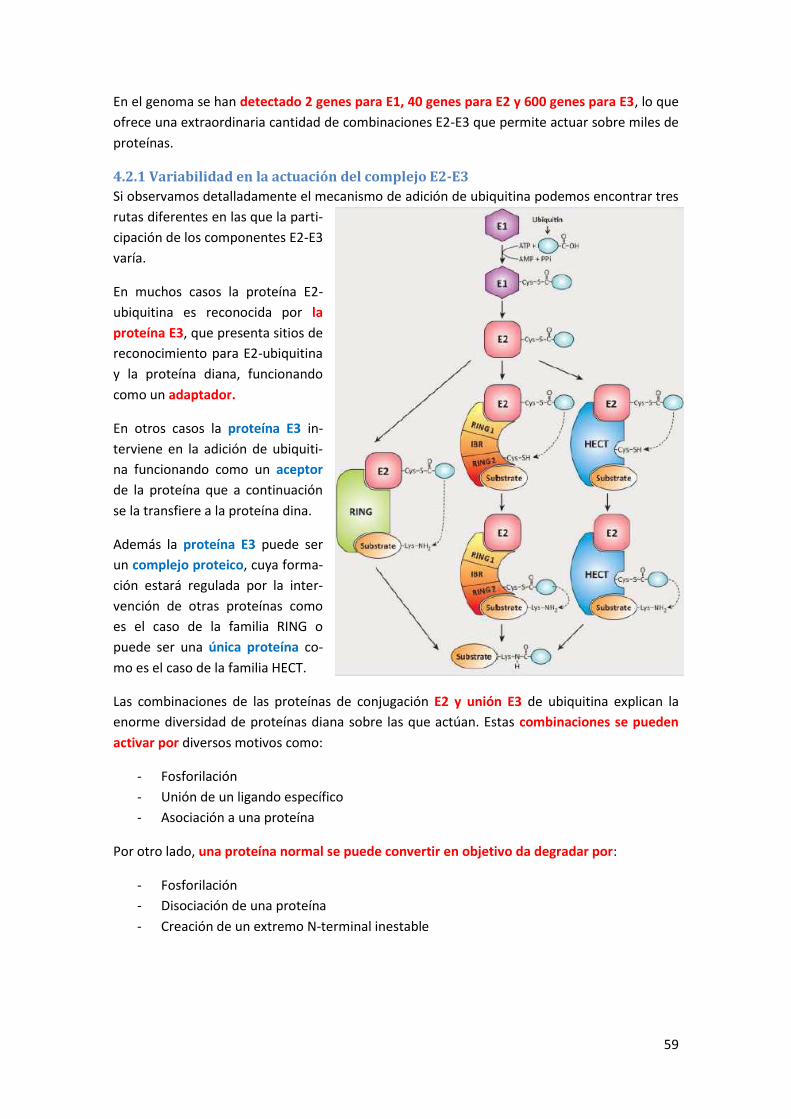
59
En el genoma se han detectado 2 genes para E1, 40 genes para E2 y 600 genes para E3, lo que
ofrece una extraordinaria cantidad de combinaciones E2-E3 que permite actuar sobre miles de
proteínas.
4.2.1 Variabilidad en la actuación del complejo E2-E3
Si observamos detalladamente el mecanismo de adición de ubiquitina podemos encontrar tres
rutas diferentes en las que la parti-
cipación de los componentes E2-E3
varía.
En muchos casos la proteína E2-
ubiquitina es reconocida por la
proteína E3, que presenta sitios de
reconocimiento para E2-ubiquitina
y la proteína diana, funcionando
como un adaptador.
En otros casos la proteína E3 in-
terviene en la adición de ubiquiti-
na funcionando como un aceptor
de la proteína que a continuación
se la transfiere a la proteína dina.
Además la proteína E3 puede ser
un complejo proteico, cuya forma-
ción estará regulada por la inter-
vención de otras proteínas como
es el caso de la familia RING o
puede ser una única proteína co-
mo es el caso de la familia HECT.
Las combinaciones de las proteínas de conjugación E2 y unión E3 de ubiquitina explican la
enorme diversidad de proteínas diana sobre las que actúan. Estas combinaciones se pueden
activar por diversos motivos como:
- Fosforilación
- Unión de un ligando específico
- Asociación a una proteína
Por otro lado, una proteína normal se puede convertir en objetivo da degradar por:
- Fosforilación
- Disociación de una proteína
- Creación de un extremo N-terminal inestable

60
FOS y JUN son dos pro-oncogenes importantes para el correcto funcionamiento de la divi-
sión celular, pero que al sufrir mutaciones se convierten en oncogenes que desencadenan
la aparición de cáncer.
Le denomina C-FOS y C-JUN a las versiones celulares de los pro-oncogenes que por diver-
sos motivos pueden ser incorporados a un virus, denominándose entonces V-FOS y V-JUN.
Puesto que la tasa de mutación en la replicación viral es tan elevada, la probabilidad de
que estos genes muten se hace alta, y al introducir los virus las copias mutadas en la célula
pueden provocar el desarrollo del cáncer.
Las proteínas Fos y Jun forman un factor de transcripción dimérico, denominado Ap1, que
se une a los elementos de respuesta del DNA e induce la expresión de los genes de la divi-
sión celular sacando a la célula de la fase G0.
Estos factores se puede degradar por la vía UPS cuando son fosforilados, por lo que esta-
blecen un buen ejemplo de que la fosforilación puede marcar el destino hacia la degrada-
ción del factor.
Fosforlación de IκB como ejemplo de degradación por el sistema UPS
Veamos el ejemplo de regulación del factor nuclear κB (NFκB, formado por las subunidades
p50 y p65) que abunda en todas las células y en linfocitos B induce la expresión de las cadenas
κ de las inmunoglobulinas.
En el citosol de las células no estimuladas el NFκB se encuentra formando un complejo con la
proteína inhibidora IκB. El complejo NFκB-IκB presenta la secuencia de entrada al núcleo
ocluida, pero cuando se produce la señal pertienente, la quinasa IKK fosforila a la subunidad
IκB inhibidora, provocando su disociación del complejo que ahora muestra la señal de tránsito.
Disociación de IκB como ejemplo de degradación por el sistema UPS
La fosforilación IκB no solo provoca la disociación sino que además convierta al inhibidor en
una diana para la incorporación de ubiquitina que la dirige al proteasoma. Esto se debe a que
el factor inhibidor también porta una señal de degradación tapada por la formación del com-
plejo.
Creacción de un extremo N-terminal inestable
El aminoácido presente en el extremo N-terminal puede afectar a la estabilidad de una proteí-
na, por lo que cortes proteóliticos en esta sección pueden alterar la vida media de la proteína y
convertirla en una diana para el sistema UPS.

61
4.3 Destino de las unidades o polímeros de ubiquitina La incorporación de ubiquitina a una proteína no implica necesariamente su degradación. Una
proteína puede recibir la ubiquitina en número y conformación diferentes que implican dife-
rentes opciones desde la regulación a la degradación.
En primer lugar una proteína puede recibir una unidad de ubiquitina en una lisina (K) o varias,
lo que está relacionado con la regulación de las histonas, las endocitosis y la gemación de los
virus.
Por el contrario, la adición de polímeros en escalera unidos a través de K48 o zigzagueante a
través de K29, sí que se ha relacionado directamente con la degradación proteasomal.
Pero la adición de un polímero lineal unido a través de K63 se ha relacionado como una señal
de daño en el DNA, el tráfico vesicular y la activación de quinasas.
También se ha encontrado la unión del polímero a través de K6, pero presenta una función
desconocida.
Por lo tanto, en función de la conformación que adquiere el polímero de ubiquitina, va a poder
ser reconocido por una u otra proteína que encaminará a la proteína marcada hacia una ruta u
otra.
La ubiquitina no es la única proteína pequeña que sirve para este tipo de regulación. Otras
del mismo tipo como SUMO y NEDD, a través de mecanismos similares a los de E1-E2-E3 con
su propio juego de adaptadores, realizan las mismas funciones añadiendo un nivel más de
complejidad al proceso.
4.4 Las proteínas de deconjugación de ubiquitina E4 Además del sistema de activación, conjugación y ligasa de ubiquitina existe otras 100 proteí-
nas cuya función es la de retirar ubiquitina (E4). Estas proteínas actúan retirando las unidades
de ubiquitina desde los extremos (como lo haría una exopeptidasa) o cortando por el interior
del polímero (como lo haría una endopeptidasa).

62
Como se representa en la ima-
gen, la ligasa de ubiquitina (E3)
puede ir añadiendo unidades
de ubiquitina a la proteína
sustrato y conducirla a la de-
gradación, o puede actuar una
de estas E4, retirando las uni-
dades en el conocido proceso
de deconjugación de ubiquiti-
na.
El motivo de la sofisticación de
este sistema radica en que la
célula debe de estar comple-
tamente segura de que la pro-
teína a degradar es la correcta.
Por ejemplo, el virus del papiloma humano (HPV) es el responsable del cáncer de útero porque
provoca la adición de ubiquitina a p53, el guardián del genoma. La introducción de una proteí-
na propia del virus que funciona como una E3 ligasa de ubiquitina, provoca la adición de ubi-
quitina a p53, uno de los anti-oncogenes más importante por la multitud de funciones que
desempeña. Al bajar los niveles de la proteína aumenta la frecuencia de daños en el DNA,
pero no se produce la apoptosis, por lo que el virus provoca una proliferación incontrolada.
4.4.1 Regulación de p53, el equilibrio E3/E4
La proteína p53 se encuentra en un continuo balance entre la adición de ubiquitina y su retira-
da. La adición de ubiquitina está mediada por la proteína E3 ligasa Mdm2/Mdmx, mientras que
la retirada de unidades está medida por la proteasa específica de ubiquitina 7 (USP7).
Entre las funciones de p53 podemos encontrar que es un factor de transcripción que induce la
propia expresión de su E3 ligasa, por lo que se produce una retroalimentación. Mdm2/Mdmx
también es susceptible de ser degradada por el proceso de adición de ubiqtuina y al igual que
p53, puede ser rescatada por la acción de USP7 o USP2a.
Con vistas al tratamiento del cáncer, se
supone que la perpetuación del efecto de
p53 es un factor clave y beneficioso como
anti-oncogen que es. Por ello el tratamien-
to busca alargar la vida del factor de
transcripción mediante estrategias como:
- Bloqueo de la acción de
Mdm2/MdmX
- Favorecer la acción de USP7
- Evitar el reconocimiento de p53
por el sistema UPS.

63
Por ello se han desarrollado más de 30-40 inhibidores de la acción del proteasoma.
En células no estresadas p53 está inactivo en el citosol. En respuesta a una situación de estrés
como una respuesta oncogénica, un daño en el DNA o la hipoxia, p53 se activa y entra al nú-
cleo donde actua como factor de transcripción para los genes de respuesta al estrés que inhi-
ben la división celular a través de la detención del ciclo, por apoptosis o por necrosis.
Pero la proteína p53 es muy inestable y presenta una vida media corta, por lo que su cantidad
depende de su síntesis y degradación. Como ya hemos mencionado, uno de los genes expresa-
dos es el de la proteína MDM2, una E3-ligasa (p53 está controlada por al menos 5 de estas
ligasas) que desvía a p53 a su degradación y por lo tanto es un oncogen. Pero también entra en
juego ARF, un inhibidor de MDM2, y por tanto es un anti-oncogen.
MDM2 puede formar complejos con p300 e incorporar polímero de ubiquitina a p53, pero
también puede introducir una sola unidad de ubiquitina, desplazando a p53 al citosol, sin de-
gradarla.
4.5 Sistema ERAD de control de calidad y señal UPR Hasta ahora hemos visto el sistema UPS como un ejemplo de proteólisis intracelular, pero
también podemos encontrar otros como el control de calidad del ERAD que va derivar a las
proteínas mal plegadas hacia la proteolisis.
Una proteína cuyo destino es la membrana plasmática o la excreción debe atravesar el retículo
endoplasmático y el aparato de Golgi. Pero antes de dejar el retículo se la somete a un control
de calidad por el que tiene varias posibilidades:
- Si la proteína está bien plegada y responde adecuadamente sigue su camino.
- Si la proteína no está bien plegada se la conduce a la degradación en lo que se conoce
como ruta de ERAD (Endoplasmic-reticulum-associated protein degradation).
En el retículo endoplasmático no hay proteosoma, pero la ruta de ERAD asocia una serie de
chaperonas que van a acerar la proteína mal plegada hasta un translocon que la sacará al cito-
sol donde será degradada por el proteosoma.
Otro caso es en el que se produce como resultado de mutaciones o de condiciones en el que
una gran cantidad de proteínas en el retículo están mal plegadas. En ese momento el retículo

64
La fibrosis quística es una enfermedad incurable de origen genético debido a la mutación
del gen CFTR (27 Exones) que codifica la proteína reguladora de la conductancia tranmem-
brana de la fibrosis quística (1480 aminoácidos). La proteína es una ATPasa de tipo ABC (ATP
binding cassette) que actúa como un canal de cloruro situado en la membrana plasmática.
En situaciones normales la proteína se sintetiza en ribosomas asociados al retículo desde
donde se desplaza al Golgi para glicosilarse y posteriormente es incorporada a la membrana
plasmática. Pero sus mutaciones, entre ellas el frecuente cambio de la fenilalanina 508,
modifica el plegamiento de la proteína y esta es incapaz de superar el control de calidad de
la vía ERAD.
Aunque la proteína no es completamente funcional con esta mutación, se está investigando
la inducción de chaperonas o la utilización de químicos que fuercen el plegamiento en el
retículo de manera que aunque no se pliegue correctamente supere el control de calidad
nota la falta de chaperonas y se activa la ruta UPR (Unfolding protease response) que manda
señales al núcleo para aumentar la producción de chaperonas.
En cualquier caso, las proteínas mal plegadas del retículo que son translocadas hacia el exte-
rior sufren la adición de ubiquitina y son conducidas al proteasoma.
4.6 Relación con el ciclo celular del sistema UPS El proteasoma es importante durante la división celular. El ciclo descansa en unos complejos
llamados ciclinas y quinasas dependientes de ciclinas (CKDs), por lo que todo el ciclo va a estar
controlado por quinasas, fosfatasas y el proteasoma, que controla los niveles de cada una.
Durante la interfase comienza la síntesis de las ciclinas mitóticas que al unirse a su CDK corres-
pondiente forma el factor promotor de la mitosis (MPF) cuya actividad quinasa permite la con-
tinuación del ciclo celular. A continuación se le añade ubiquitina a las ciclina durante la anafa-
se, produciéndose la degradación por
vía UPS y quedando las CDKs inacti-
vas.
La proteína APC es el complejo pro-
motor de la anafase, una E3-ligasa
que se encuentra activa cuando su
unidad Cdh1 está desfosforilada. La
retirada del fosfato está mediada por
la fosfatasa Cdc14 que por tanto es
un regulador negativo de las ciclinas
(su actividad permite la adición de
ubiquitina y la consiguiente bajada de
actividad de MPF). Pero el complejo
ciclina/CDK de G1 es el encargado de
fosforilar la subunidad Cdh1 de APC y
promover la mitosis inactivado a la
E3-ligasa.

65
4.7 Motivos de reconocimiento para los complejos E2-E3
4.7.1 Caja de destrucción de ciclinas
Como hemos visto, las ciclinas están reguladas por síntesis y degradación a través del sistema
UPS. Se ha observado que para que estas sean reconocidas por el sistema de E3-ligasa deben
de llevar una secuencia de en principio 9 aminoácidos que van a ser los determinantes de su
degradación y que se han denominado caja de destrucción de ciclinas, cuya secuencia consen-
so es:
RXXLXXXXD/N
Esta secuencia se encuentra en ciclinas y otras proteínas implicadas como securina (inhibidora
de la separasa de las cormátidas), el factor pVHL (supresor de tumores), y su mutación ocasio-
na la aparición de cáncer.
Esta caja de destrucción de ciclinas es por tanto el motivo estructural reconocido por los
complejos E2-E3, para la adición de ubiquitina a estas proteínas.
4.7.2 Secuencias PEST
Las secuencias PEST (Prolina, glutamato, serina y treonina) parecen estar relacionadas con
proteínas de vida media corta que son degradadas por el sistema UPS.
PEST
Agrupaciones de estas secuencias (hasta 80) parecen ser un motivo estructural que es recono-
cido por los complejos E2-E3 encargados de la adición de ubiquitina.
5. El proteasoma El proteasoma es una estructura supramolecular cilíndrica con un coeficiente de sedimenta-
ción de 26S, formada por un núcleo degradativo de 20S y complejos en los extremos de 19S.
La localización del proteasoma es en el citosol (no parece que haya en el retículo, el lisosoma o
el Golgi) y en la mitocondria, donde se produce la síntesis de proteínas. También hay pro-
teasomas en el núcleo ya que muchos de los factores de transcripción van a ser degradados
ahí.
En cuanto a sus variantes tenemos tres, el inmunoproteasoma, relacionado con el corte de
fragmentos antígenicos para las células Th, el timoproteasoma, relacionado con la diferencia-
ción de los linfocitos T en el timo, y el proteasoma clásico,
La progresión del ciclo celular está determinada por el balance entre reguladores positivos (CDKs)
y reguladores negativos (inhibidores de ciclinas dependientes de quinasas, CKIs). Las ciclinas del
tipo D (D1, D2 y D3) son las primeras expresadas durante el ciclo celular. De estas 3, la ciclina D1
es la que más frecuentemente se sobreexpresa en los casos de cáncer humano. El mecanismo de
sobreexpresión de la ciclina D1 puede deberse a la duplicación génica, la activación transcripcional
y la alteración del sistema de degradación. De estos posibles mecanismos, la inhibición de la de-
gradación vía UPS parece ser la causa primaria de la sobreexpresión en tumores humanos.

66
Estructuralmente el proteasoma está formado en 4 capas de 7 subunidades (28 en total) pre-
sentado dos capas de subunidades β
internas que son las que presentan
la actividad proteolítica, y dos capas
de subunidades α externas que pre-
sentan una treonina (T) nucleofílica
en el extremo N-terminal. Este nú-
cleo central es el que presenta un
coeficiente de sedimentación de 20S
y está presente, con cierta variación
en eucariotas, procariotas y arqueas.
En las tapas de 19S, hay proteínas que reconocen las colas de poliubiquitina, por lo que una
proteína que esté marcada por poliubiquitina se tendrá que pegar ahí. Luego habrá proteínas
que quitan ubiquitina (USP, proteasas específicas de ubiquitina), otras que despliegan la pro-
teína si es que está plegada denominadas ATPasas de clase AAA (ATPasa con varias actividades
biológicas), y luego otras que la van introduciendo tirando de ella. En total hablamos de unas
25 proteínas distintas por lo que en conjunto tendremos 50-55 proteínas.
Una vez que están en el núcleo
proteolítico (zona central de las
subunidades beta), hay subuni-
dades que desarrollan actividad
como la tripsina, otras como la
quimotripsina, etc. Entonces, un
polipéptido más o menos gran-
de lo cortan en varios trozos, y
éstos se irán degradando. El
conjunto tendrá unos 40-50nm.
5.1 Relación entre el Alzheimer y el proteosoma El mal funcionamiento del proteosoma puede estar detrás de enfermedades neurodegenerati-
vas como el Parkinson y el Alzheimer. A propósito del Parkinson hablábamos de la alfa-
sinucleína y la parkina.
Aquí estamos viendo uno de los rasgos histopatológicos que padecen los enfermos de Alz-
heimer, la aparición de estos nudos neurofibrilares, depósitos de localización interna en las
neuronas que son como ovillos de proteína TAU hiperfosforilada con muchos restos de fosfato.
TAU hiperfosforilada adopta unas conformaciones distintas a TAU normal (TAU es una proteína
que está asociada a los microtúbulos y que son reguladores de la dinámica de polimerización
de alfa y beta tubulina).
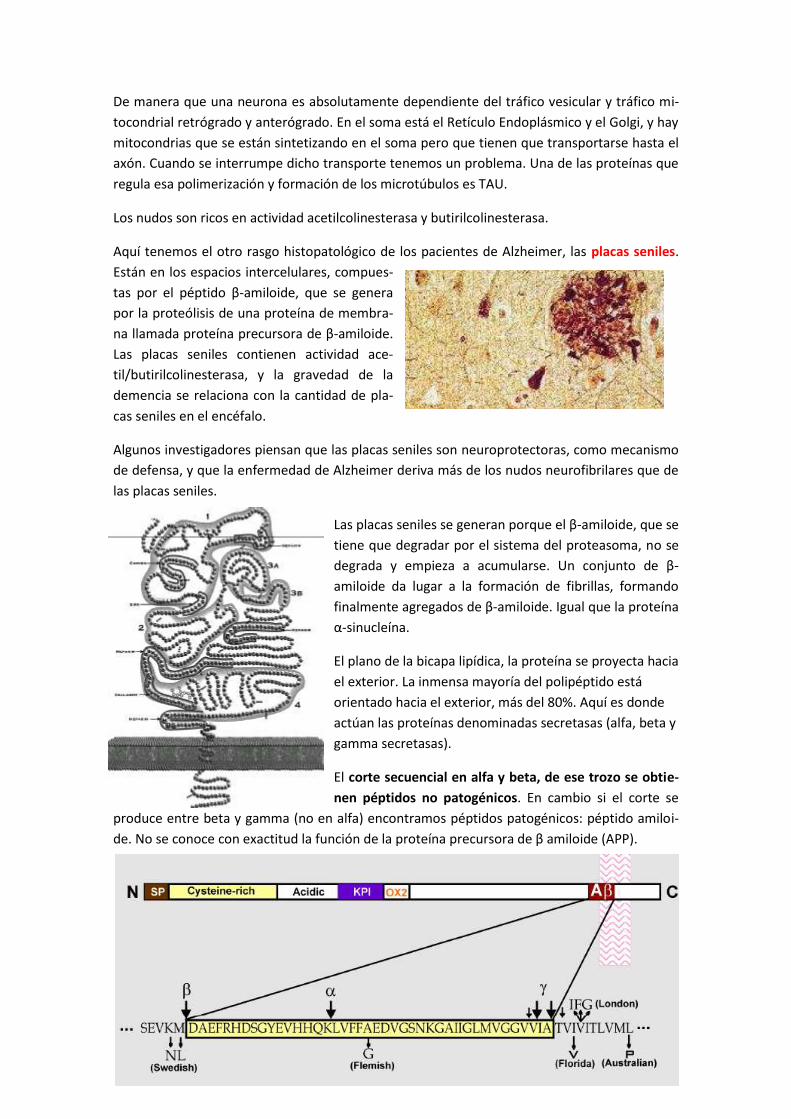
67
De manera que una neurona es absolutamente dependiente del tráfico vesicular y tráfico mi-
tocondrial retrógrado y anterógrado. En el soma está el Retículo Endoplásmico y el Golgi, y hay
mitocondrias que se están sintetizando en el soma pero que tienen que transportarse hasta el
axón. Cuando se interrumpe dicho transporte tenemos un problema. Una de las proteínas que
regula esa polimerización y formación de los microtúbulos es TAU.
Los nudos son ricos en actividad acetilcolinesterasa y butirilcolinesterasa.
Aquí tenemos el otro rasgo histopatológico de los pacientes de Alzheimer, las placas seniles.
Están en los espacios intercelulares, compues-
tas por el péptido β-amiloide, que se genera
por la proteólisis de una proteína de membra-
na llamada proteína precursora de β-amiloide.
Las placas seniles contienen actividad ace-
til/butirilcolinesterasa, y la gravedad de la
demencia se relaciona con la cantidad de pla-
cas seniles en el encéfalo.
Algunos investigadores piensan que las placas seniles son neuroprotectoras, como mecanismo
de defensa, y que la enfermedad de Alzheimer deriva más de los nudos neurofibrilares que de
las placas seniles.
Las placas seniles se generan porque el β-amiloide, que se
tiene que degradar por el sistema del proteasoma, no se
degrada y empieza a acumularse. Un conjunto de β-
amiloide da lugar a la formación de fibrillas, formando
finalmente agregados de β-amiloide. Igual que la proteína
α-sinucleína.
El plano de la bicapa lipídica, la proteína se proyecta hacia
el exterior. La inmensa mayoría del polipéptido está
orientado hacia el exterior, más del 80%. Aquí es donde
actúan las proteínas denominadas secretasas (alfa, beta y
gamma secretasas).
El corte secuencial en alfa y beta, de ese trozo se obtie-
nen péptidos no patogénicos. En cambio si el corte se
produce entre beta y gamma (no en alfa) encontramos péptidos patogénicos: péptido amiloi-
de. No se conoce con exactitud la función de la proteína precursora de β amiloide (APP).

68
5.2 Inhibidores del proteasoma en el tratamiento contra el cáncer Jugando con el proteosoma podemos aumen-
tar la vida media de anti-oncogenes y reducir
la vida media de oncogenes. En el año 2003,
en la FDA de los EEUU, autorizó por primera
vez el uso en clínica, en enfermos de cáncer,
de bortezomib. Es un derivado del ácido bo-
ronico, y actúa como inhibidor del proteoso-
ma. Se utiliza con éxito en el tratamiento de
linfomas y leucemia mieloide crónica, es decir,
para distintas clases de cáncer derivados de la
sangre.
Se están dando pasos agigantados hacia la resolución y mejora de determinados tipos de cán-
cer. Antiguamente la leucemia era sinónimo de condena a muerte, hoy en día la esperanza de
vida de niños y adultos que tienen leucemia, aunque depende del estadio y del tipo de cáncer,
ha aumentado significativamente.
Bortezomib lo que hace es inhibir la actividad proteolítica del proteosoma. Vemos aquí una
proteína que contiene sucesivos restos de
ubiquitina (6 restos de ubiquitina), entonces
lo que pasa es que en la tapa están las en-
zimas que quitan ubiquitina (familia USP,
familia UCHL, familia POH, que son 3 clases
distintas de proteínas que quitan ubiquiti-
na).
Aquí lo que vemos es la usp14 y la uchl5, y
como está inhibido el proteosoma, lo que
van a hacer es bloquea la degradación de la
proteína. Esto nos da una acumulación de
proteínas que llevan pequeñas cadenas de
poliubiquitina, aunque se le han quitado
algunas. Las enzimas que quitan ubiquitinas
pueden quitarlas de una en una, pero sue-
len dejar 2, 3 o 4 ubiquitinas, y con eso es
suficiente para que la proteína recupere
parte de su funcionalidad.
Esto ocurre en un sistema muy proliferativo, pero es que en una leucemia se dividen las células
tumorales muy activamente. Al bloquear la proliferación celular, al final todas las células se
resienten, tanto las tumorales como las sanas. El bortezomid se incluye dentro del cóctel de
químicos que se suministra en la quimioterapia.

69
Otro agente que también se está utilizando
es el AP15, que es otro inhibidor. Este lo que
hace es evitar la acción de USP14 y UCHL5,
en lugar de bloquear. El proteosoma está
activo, pero se suprime la eliminación de
ubiquitina, luego tendríamos proteínas que
conservan la ubiquitina pero que desarrolla,
probablemente, su actividad biológica.
Para el tratamiento de tipos de cáncer que
no sean leucemias o de la sangre se em-
plean otras técnicas y agentes.
En el caso de la proteína ErbB2 (oncogén),
se sobreexpresa en determinadas clases de
cáncer de mama. La POH-1 (enzima que
quita ubiquitinas), alarga la vida de ErbB2
(es una proteína que desarrolla actividad
tirosin-quinasa y que aumenta la prolifera-
ción celular). Se degrada vía proteosoma, su vida media depende del proteosoma, siendo mar-
cada por ubiquitina. Pero en este tipo de tumor también se expresa POH-1, que quita ubiquiti-
na, alargando la vida de este oncogén ErbB2.
En estos tumores, además de la sobreexpresión génica de la proteína ErbB2, se produce un
aumento de la proteína POH1, teniendo al final una cantidad superior de un oncogén (ErbB2)
que hace que la célula normal se convierta en neoplásica.
Si inhibimos la proteína POH-1, estamos favoreciendo la degradación de la proteína oncogéni-
ca, de aquí la importancia de la inhibición de las proteínas que quitan ubiquitinas para el tra-
tamiento de estos cánceres.
En el caso de la proteína UCHL5, se sobreexpresa en ciertos tumores y aumenta la superviven-
cia de las células de carcinoma de pulmón, porque reduce la apoptosis. Cuando se silencia
UCHL5, cae Bcl2 (supervivencia, anti-apoptosis) y sube Bax (pro-apoptosis). El uso de borte-
zomib en leucemias aumenta la cantidad de p53, debido a que aumenta su estabilidad, pero
no por una sobreexpresión.
Luego aumenta CDK-21 y CDK7 (fosfatasas del ciclo celular) y IκB (proteína inhibidora del fac-
tor de transcripción NFκB, que tiene que ver en la proliferación de células inmunes).
La inhibición del proteosoma en ciertos tumores de la sangre, favorece p53 y detiene el ciclo
celular frenando la división, además se mantiene o se estabiliza proteínas que son inhibidores
de factores de transcripción que tienen mucho que ver con la división celular.
¿Hasta dónde podremos llegar con el empleo de inhibidores del proteosoma? Se está investi-
gando y probando en cáncer de colon, pulmón, riñón, etc. Se está jugando con 3 procesos celu-
lares: división celular, apoptosis y autofagia. Cuando hablamos de necrosis y autofagia tene-
mos que diferenciar entre auto/macroautofagia y microautofagia. Se ha visto que hay proteí-

70
nas que sirven para alimentar a las células en momento de ayuno, siendo proteínas que llevan
la secuencia KFERQ.
Si tenemos un cultivo de células de fibroblastos y lo ponemos en ayuno, sin suministro de sue-
ro, las células sobreviven cierto tiempo degradando proteínas que son prescindibles. Las pro-
teínas que se degradan son KFERQ, son reconocidas por chaperonas de la familia HSP70, que
ve esa secuencia o agrupaciones KFERQ y la lleva a las proximidades del lisosoma. Allí se en-
cuentra la proteína de reconocimiento LAMP-2 (proteína de membrana asociada al lisosoma).
La proteína llega con la chaperona, llega al receptor que está en la membrana del lisosoma,
siendo capturada e introducida en el interior del lisosoma.
En ratas en ayunas, las proteínas que se degradan son también las mismas, con esta secuencia.
Por ello, parece que estas secuencias KFERQ sirven para marcar proteínas prescindibles. Esta
degradación selectiva de proteínas ocurre en todos los tejidos, excepto en cerebro y testículo.
Una célula tumoral tiene unas características muy particulares: es absolutamente dependiente
del suministro de sangre, puesto que necesita un aporte continuo de nutrientes debido a que
tiene un metabolismo muy activo, hasta el punto que hacen uso de estas proteínas prescindi-
bles. De esta manera, podemos atacar a la célula tumoral poniéndole cloroquina?, agentes
lisomotrópicos para que no pueda degradar proteínas con el lisosoma, y éstas terminan mu-
riendo, entrando en autofagia.
A través de los antígenos de histocompatibilidad y las diferencias de los receptores que tengan
las células normales y las tumorales, seremos capaces de distinguirlas, evitando así atacar a las
células sanas, centrándonos solamente en las tumorales.

71
Tema 4: Regúlacio n del ciclo celúlar
1 Introducción al ciclo celular El estudio de la regulación del ciclo celular implica el conocimiento de dicho ciclo. La salida de
fase G0 a G1 está promocionada por el aumento de la ciclina D que va formar complejos con
CDK4 y CDK6. A continua-
ción, para el paso de G1 a
fase S se produce un aumen-
to de ciclina E que va a for-
mar complejos con la CDK2 y
la progresión en fase S está
mediada por el aumento de
ciclina A, que forma comple-
jos con la CDK2 también.
Finalmente para entrar en
fase G2 y que se produzca la
mitosis se produce un au-
mento de ciclinas A y B uni-
das a CDK1. Por tanto el
orden de actuación de las
CDKs es 6-4-2-1 y para las
ciclinas es D-E-A-B.
Hoy día se sabe que las CDKs activas son heterodímeros que presentan una subunidad catalí-
tica (quinasa) y una subunidad reguladora (ciclina). Son sus combinaciones las que intervie-
nen a lo largo del ciclo celular, pero ante un defecto en una de ellas, otra puede sustituirla, ya
que es un caso de redundancia.
Cuanta más cantidad de ciclina, más actividad quinasa se produce y más rápida será la veloci-
dad de funcionamiento del ciclo celular.
Pero la complejidad de las relaciones entre cada uno de los protagonistas del ciclo celular hace
que a pesar de conocer las proteínas implicadas, no se conozca el proceso con todo detalle.
1.1 Transducción de la señal de división y cáncer En una célula hay diversos receptores para factores de crecimiento que transmiten señales
positivas o negativas a la célula. Por ejemplo, el factor de crecimiento transformante (TGF) es
un factor de no crecimiento que se opone a la división celular.
En circunstancias normales, una célula va a recibir una señal positiva que va a desencadenar la
actividad Y-kinasa dando lugar a la fosforilación cruzada y a la activación secuencial de las pro-
teínas GrB2, SOS, RAS, RAF, MEK y ERK, muchas de las cuales son quinasas que van a mandar
otras tantas señales fosforilando proteínas.

72
Una de estas proteínas que se van a fosforilar son FOS y JUN que dimerizan y entran al núcleo
para unirse a los elementos de respuesta que van a dar lugar a la expresión de los genes de las
ciclinas de la fase G1 (Ciclina D, CDK6 y CDK4), además del factor E2F.
Para que se produzca toda la cascada el impulso debe de ser lo suficientemente duradero y
por ello cada proteína implicada presentará un tiempo de actuación, permitiendo que los ni-
veles de expresión superen el umbral que permite la progresión del ciclo celular.
Pero hay que tener en cuenta que la sobreproducción también es peligrosa. Las cantidades de
cada proteína tienen que producirse en un momento dado, ser suficientes y a continuación
decaer para progresar de fase. Esto establece varios niveles que pueden llevar a la producción
de cáncer:
- La sobreproducción del factor de crecimiento conduce a una sobreestimulación de to-
da la cascada que puede inducir al cáncer.
- Mutaciones en un receptor de factores de crecimiento son oncogénicas porque hay
variantes truncadas que expresan actividad de manera constitutiva y por lo tanto se
considera al receptor como un pro-oncogén.
- Mutaciones en RAS que aumentan el tiempo de actividad son oncogénicas.
- Mutaciones en cualquiera de las protein kinasas que intervienen (RAF, MEK o ERK)
producen cáncer.
- La sobre expresión de los oncogenes FOS y JUN deriva en cáncer, así como su trans-
formación a V-FOS y V-JUN (viral mutada) que impide su degradación.
- Las mutaciones que provocan una sobreproducción de las quinasas o una atenuación
de las fosfatasa que inhiben la ruta, también es un factor desencadenante de cáncer.
2 Regulación del ciclo celular El ciclo celular está controlado por tres niveles:
- Cantidad de ciclinas y CDKs
- Quinasas y fosfatasas
- Proteínas inhibidoras (INKs)
2.1 El factor de maduración de los oocitos (MPF) El factor promotor de la maduración (MPF) o factor de maduración de los oocitos de sapo fue
descubierto trabajando con oocitos de sapo. Este descubrimiento se realizó al transferir mate-
rial citoplasmático desde un oocito detenido en metafase hasta otro oocito no fertilizado. Este
tratamiento provocaba la mitosis del oocito no fertilizado y el compuesto aislado del citosol
que provocaba este efecto era un complejo de ciclina-CDK que se denominó MPF.

73
Como ya vimos en el tema anterior, el MPF presenta una actividad quinasa y su degradación
está regulada por APC activo, una E3 ligasa, que determina la adición de ubiquitina a la ciclina
del complejo y su consiguiente degradación por la vía UPS.
El complejo APC está activo siempre y cuando su subunidad Cdh1 no presente fosfato, ya que
cuando es fosforilado por el complejo Ciclina-CDK de G1 queda inactivo. La actuación de Cdc14,
una fosfatasa, retira el fosfato de Cdh1, activado a APC y degradando la ciclina.
2.1.1 Regulación de MPF: ejemplo de cantidad de ciclinas y nivel de fosforilación
Si observamos el modelo de regulación del factor promotor de la mitosis constituido por la
ciclina y el CDK, vemos que la quinasa dependiente de ciclina presenta varios restos importan-
tes en su estructura que pueden estar fosforilados:
- La tirosina 15 (Y15) cuya fosforilación es inhibidora.
- La treonina 161 (T161) cuya fosforilación es activadora.
Cuando los niveles de ciclina mitótica aumentan, estas forman los MPF inactivos que van a ser
fosforilados por la quinasa Wee1 en la posición Y15 (inhibidora). A continuación el complejo
MPF va a ser fosforilado en la posición T161 (activadora) por la quinasa CAK, pero permanece-
rá inactivo por la inhibición de Y15.
El estado activo del complejo MPF, que permite fosforilar a sus sustratos, solo se alcanza por la
acción de la fosfatasa Cdc25, que retira el fosfato de la posición Y15 dejando libre el sitio cata-
lítico.
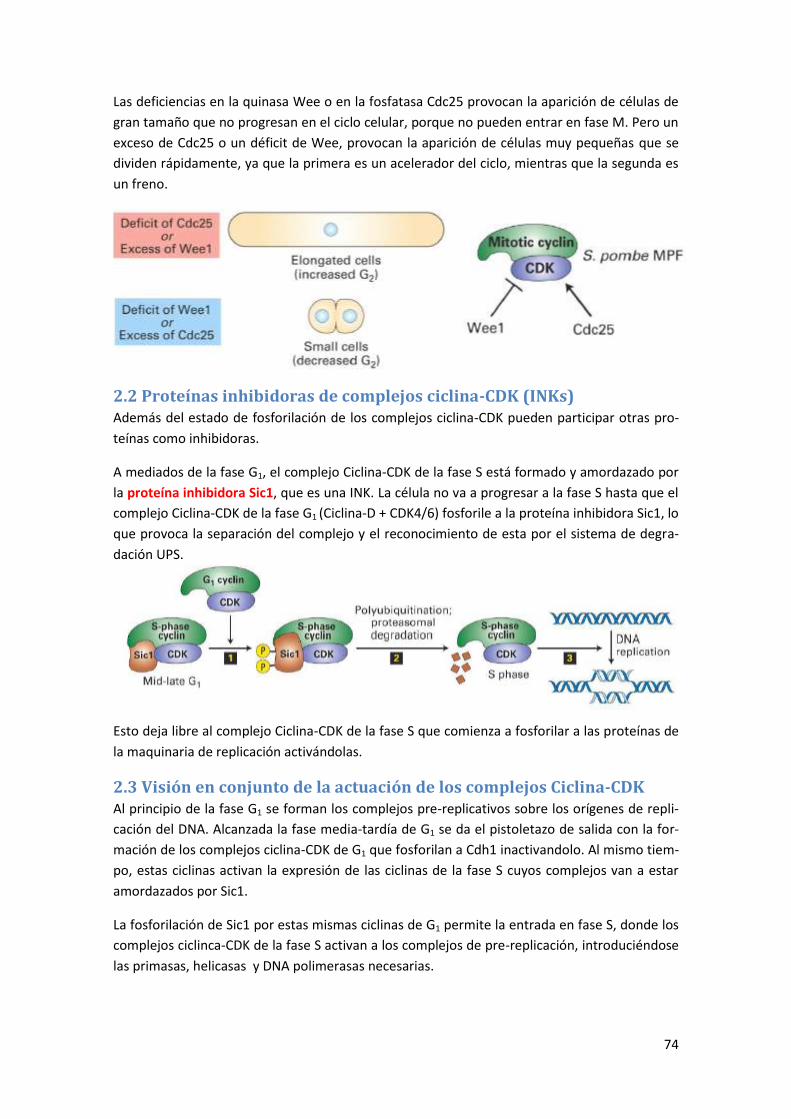
74
Las deficiencias en la quinasa Wee o en la fosfatasa Cdc25 provocan la aparición de células de
gran tamaño que no progresan en el ciclo celular, porque no pueden entrar en fase M. Pero un
exceso de Cdc25 o un déficit de Wee, provocan la aparición de células muy pequeñas que se
dividen rápidamente, ya que la primera es un acelerador del ciclo, mientras que la segunda es
un freno.
2.2 Proteínas inhibidoras de complejos ciclina-CDK (INKs) Además del estado de fosforilación de los complejos ciclina-CDK pueden participar otras pro-
teínas como inhibidoras.
A mediados de la fase G1, el complejo Ciclina-CDK de la fase S está formado y amordazado por
la proteína inhibidora Sic1, que es una INK. La célula no va a progresar a la fase S hasta que el
complejo Ciclina-CDK de la fase G1 (Ciclina-D + CDK4/6) fosforile a la proteína inhibidora Sic1, lo
que provoca la separación del complejo y el reconocimiento de esta por el sistema de degra-
dación UPS.
Esto deja libre al complejo Ciclina-CDK de la fase S que comienza a fosforilar a las proteínas de
la maquinaria de replicación activándolas.
2.3 Visión en conjunto de la actuación de los complejos Ciclina-CDK Al principio de la fase G1 se forman los complejos pre-replicativos sobre los orígenes de repli-
cación del DNA. Alcanzada la fase media-tardía de G1 se da el pistoletazo de salida con la for-
mación de los complejos ciclina-CDK de G1 que fosforilan a Cdh1 inactivandolo. Al mismo tiem-
po, estas ciclinas activan la expresión de las ciclinas de la fase S cuyos complejos van a estar
amordazados por Sic1.
La fosforilación de Sic1 por estas mismas ciclinas de G1 permite la entrada en fase S, donde los
complejos ciclinca-CDK de la fase S activan a los complejos de pre-replicación, introduciéndose
las primasas, helicasas y DNA polimerasas necesarias.
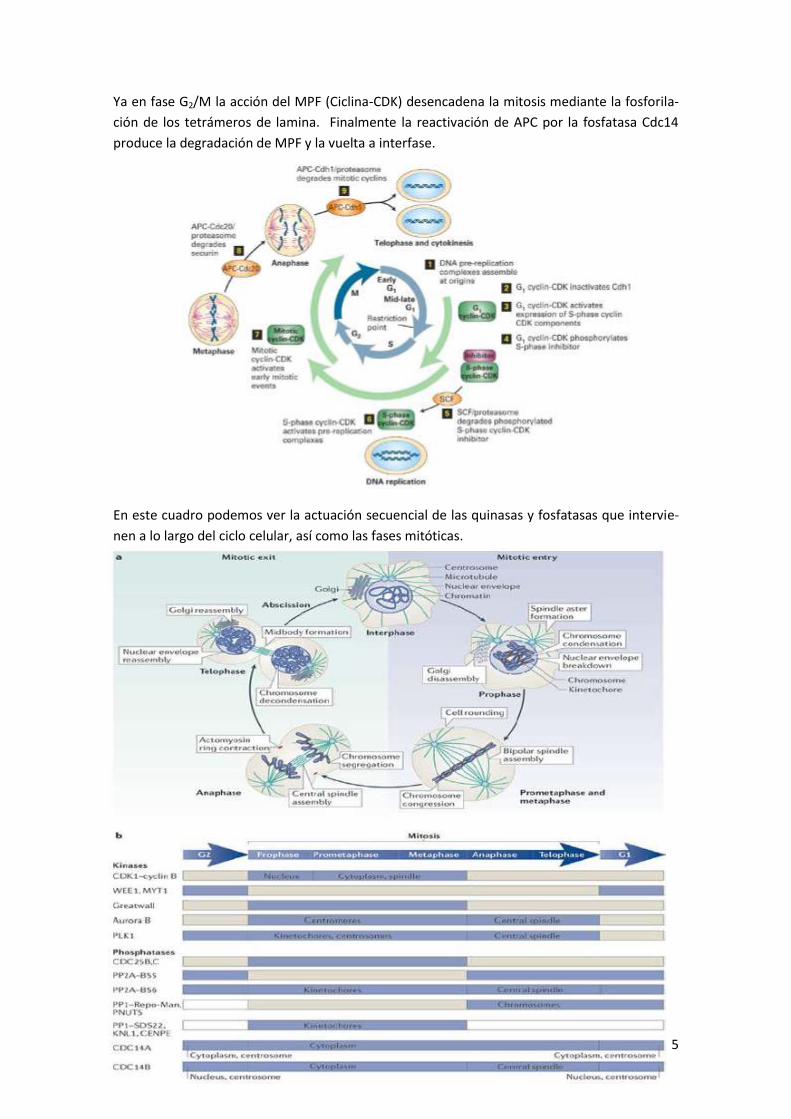
75
Ya en fase G2/M la acción del MPF (Ciclina-CDK) desencadena la mitosis mediante la fosforila-
ción de los tetrámeros de lamina. Finalmente la reactivación de APC por la fosfatasa Cdc14
produce la degradación de MPF y la vuelta a interfase.
En este cuadro podemos ver la actuación secuencial de las quinasas y fosfatasas que intervie-
nen a lo largo del ciclo celular, así como las fases mitóticas.

76
2.4 La proteína del retinoblastoma (RB) y E2F La proteína retinoblastoma (RB) fue el primer anti-oncogen descubierto, ya que su falta o
deficiencia provoca la aparición del retinoblastoma, un cáncer ocular.
La RB es una proteína vigilante que a mediados de la fase G1 se encuentra formando un com-
plejo con el factor de transcripción E2F, actuando como un factor inhibidor de la expresión al
que se asocian proteínas histonas-desacetilasas, promoviendo la compactación del DNA.
Cuando los niveles de ciclina-D + CDK4/6 aumentan, se fosforila a la proteína RB provocando la
disociación de E2F. El factor E2F actúa ahora como un factor de transcripción que se une a sus
elementos de respuesta promoviendo la expresión de varios genes entre los que se encuentra
el mismo (retroalimentación positiva), la ciclina-E y la CDK-2, cuyos complejos mantienen el
nivel de fosforilación de RB (retroalimentación positiva).
La falta o deficiencia de RB provoca una estimulación continua de las ciclinas-E y CDK2 que
conlleva un exceso de división celular.
2.5 Puntos de control del ciclo celular Para una mayor comprensión del funcionamiento de los puntos de control del DNA vamos a
comenzar con la definición de las proteínas implicadas:
- ATM y ATR son dos quinasas que se activan ante un daño en el DNA, la primera cuan-
do se rompen ambas hebras y la segunda cuando se rompe una o hay DNA no replica-
do.
- p53 es un factor de transcripción que induce la expresión de muchísimas proteínas
como p21CIP.
- p21CIP es la proteína inhibidora del ciclo celular que realiza su acción sobre los comple-
jos Ciclina-CDK.
- Chk1 y Chk2 son dos quinasas que frenan la acción de la fosfatasa Cdc25.
- Cdc25 es la fosfatasa que activa a MPF.
- APC es una E3-ligasa de ubiquitina.
En el ciclo celular podemos encontrad cuatro puntos de control para el daño del DNA o que
no esté replicado, que van a estar mediados por la actuación, en primer lugar de los sensores
ATM/ATR.
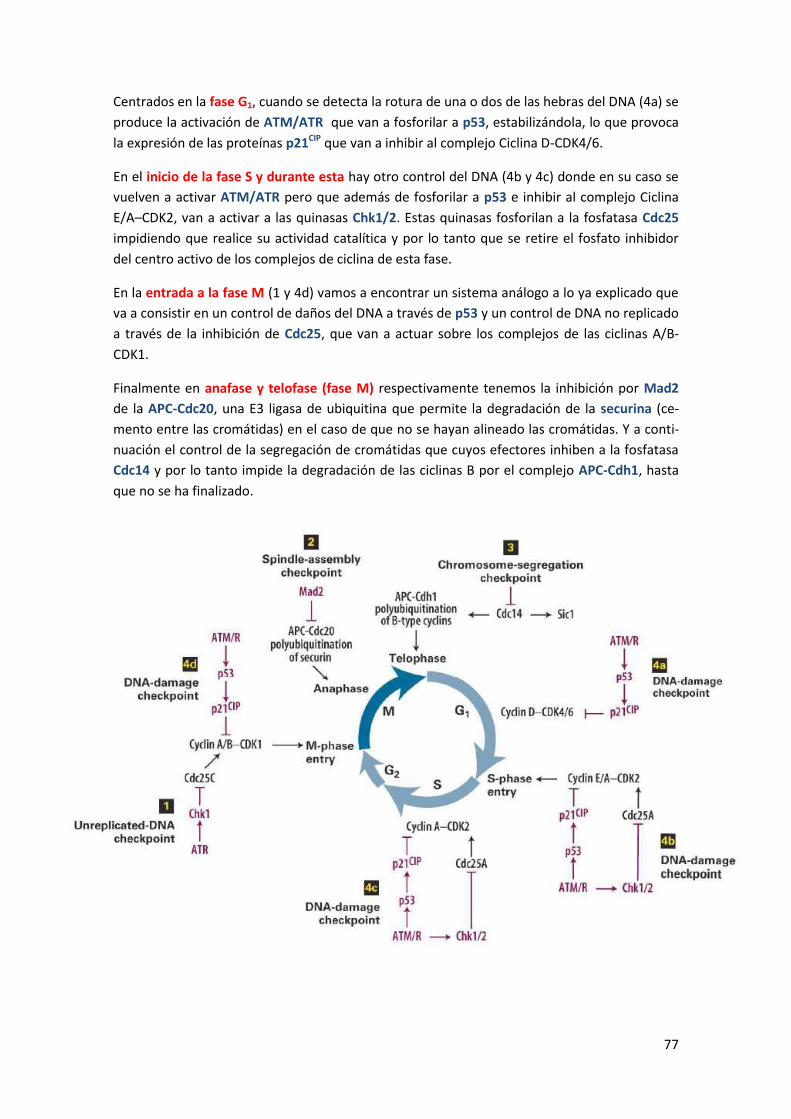
77
Centrados en la fase G1, cuando se detecta la rotura de una o dos de las hebras del DNA (4a) se
produce la activación de ATM/ATR que van a fosforilar a p53, estabilizándola, lo que provoca
la expresión de las proteínas p21CIP que van a inhibir al complejo Ciclina D-CDK4/6.
En el inicio de la fase S y durante esta hay otro control del DNA (4b y 4c) donde en su caso se
vuelven a activar ATM/ATR pero que además de fosforilar a p53 e inhibir al complejo Ciclina
E/A–CDK2, van a activar a las quinasas Chk1/2. Estas quinasas fosforilan a la fosfatasa Cdc25
impidiendo que realice su actividad catalítica y por lo tanto que se retire el fosfato inhibidor
del centro activo de los complejos de ciclina de esta fase.
En la entrada a la fase M (1 y 4d) vamos a encontrar un sistema análogo a lo ya explicado que
va a consistir en un control de daños del DNA a través de p53 y un control de DNA no replicado
a través de la inhibición de Cdc25, que van a actuar sobre los complejos de las ciclinas A/B-
CDK1.
Finalmente en anafase y telofase (fase M) respectivamente tenemos la inhibición por Mad2
de la APC-Cdc20, una E3 ligasa de ubiquitina que permite la degradación de la securina (ce-
mento entre las cromátidas) en el caso de que no se hayan alineado las cromátidas. Y a conti-
nuación el control de la segregación de cromátidas que cuyos efectores inhiben a la fosfatasa
Cdc14 y por lo tanto impide la degradación de las ciclinas B por el complejo APC-Cdh1, hasta
que no se ha finalizado.
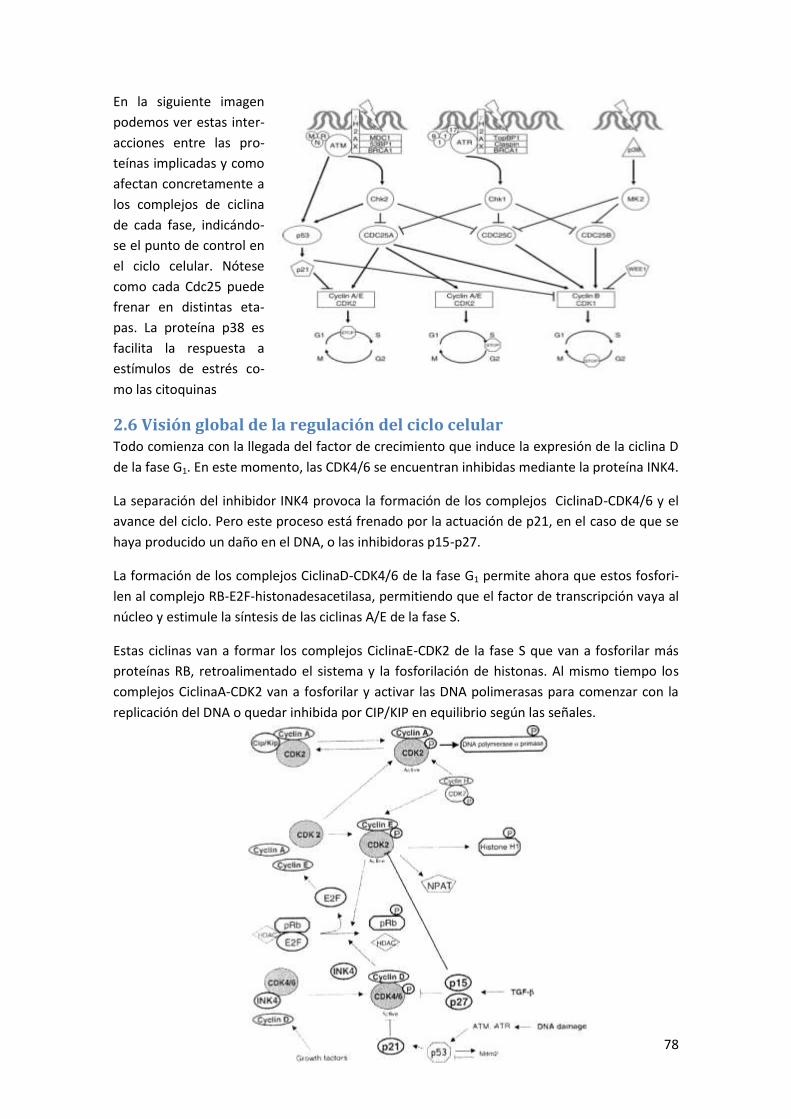
78
En la siguiente imagen
podemos ver estas inter-
acciones entre las pro-
teínas implicadas y como
afectan concretamente a
los complejos de ciclina
de cada fase, indicándo-
se el punto de control en
el ciclo celular. Nótese
como cada Cdc25 puede
frenar en distintas eta-
pas. La proteína p38 es
facilita la respuesta a
estímulos de estrés co-
mo las citoquinas
2.6 Visión global de la regulación del ciclo celular Todo comienza con la llegada del factor de crecimiento que induce la expresión de la ciclina D
de la fase G1. En este momento, las CDK4/6 se encuentran inhibidas mediante la proteína INK4.
La separación del inhibidor INK4 provoca la formación de los complejos CiclinaD-CDK4/6 y el
avance del ciclo. Pero este proceso está frenado por la actuación de p21, en el caso de que se
haya producido un daño en el DNA, o las inhibidoras p15-p27.
La formación de los complejos CiclinaD-CDK4/6 de la fase G1 permite ahora que estos fosfori-
len al complejo RB-E2F-histonadesacetilasa, permitiendo que el factor de transcripción vaya al
núcleo y estimule la síntesis de las ciclinas A/E de la fase S.
Estas ciclinas van a formar los complejos CiclinaE-CDK2 de la fase S que van a fosforilar más
proteínas RB, retroalimentado el sistema y la fosforilación de histonas. Al mismo tiempo los
complejos CiclinaA-CDK2 van a fosforilar y activar las DNA polimerasas para comenzar con la
replicación del DNA o quedar inhibida por CIP/KIP en equilibrio según las señales.

79
En el caso de que se produzca un daño en
el DNA en la fase M, podemos observar el
funcionamiento detallado del sistema del
punto de control. El daño es captado por
ATM/ATR que van a estabilizar a p53,
activando a p21 y esta inhibirá la fosfori-
lación de los complejos CiclinaA/B-CDK1.
Al mismo tiempo se activan Chk1/2 que
fosforilan a la fosfatasa Cdc25 impidiendo
la activación de los complejos de ciclina
mencionados.
Pero cuando nos enfrentamos al tema de
la regulación del ciclo celular hay dema-
siadas incognitas. Un tipo celular no reci-
be una sola clase de un factor de prolife-
ración, sino que presenta una población
de receptores para cada factor que indu-
cen señales contrapuestas o sinérgicas.
La célula procesa esta información, pero cada una de las cascadas que lleva asociadas son muy
complejas y por ello muy difíciles de comprender y de investigar.
Dado que buena parte de la regulación descansa sobre la actuación del proteasoma, en mu-
chos casos se utilizan inhibidores de este o factores citostáticos que prevengan la división celu-
lar.
Un buen ejemplo de esta complejidad se encuentra en
el modelo de la regulación de la activación de p53 por
Chk2 en respuesta a la radiación ionizante. Esta radia-
ción activa a ATM cuando se rompen dos hebras del
DNA. La quinasa ahora fosforila Chk2, pero también
puede actuar sobre p53 aumentando su estabilidad y
produciendo una detención del ciclo celular. Pero la
fosforilación de Chk2 sobre p53 conduce a la apoptosis
celular.
La proteína p53 puede ser modificada por muchas quinasas y acetilasas diferentes que modu-
lan su función. La fosforilación o desfosforilación de varios residuos de serina (S) pueden tener
un gran impacto sobre la estabilidad de p53. Estudios con anticuerpos específicos para resi-
duos fosforilados han establecido que las serinas (S) 6, 9, 15, 20, 33, 37 y 46 de p53 son sitios
de fosforilación en las células con daño en el DNA y la fosforilación de diferentes residuos pre-
senta efectos distintos. Se ha propuesto que la fosforilación de las serinas (s) N-terminales 15,
33 y 37 permiten la modificación de una lisina (K) C-terminal a través de la mejora del reclu-
tamiento de las proteínas co-activadoras p300/CBP/PCAF. En contraposición, se requiere la
fosforilación de S20 para estabilizar a p53 en respuesta a un daño en el DNA. La S20 impide
que Mdm2 se una a p53 y la convierta en una diana del sistema UPS.

80
3 La silimarina un potencial anticancerígeno La silimarina es una molécula con propiedades antitumorales que se extrae del cardo (Silybum
marianum) y cuyos efectos, que quedan descritos en la imagen, favorecen la detención del
ciclo celular, la desaparición de la inflamación, baja la actividad de muchas proteasas (impi-
diendo que una célula anide en un tejido al romper la matriz) y atenúa los factores oncogéni-
cos.

81
Tema 5: Retí cúlo endoplasma tico I
Síntesis de fosfolípidos, ceramidas y esteroides. Citocromo P450
El retículo endoplasmático es el conjunto de elementos de membrana en el interior celular que
ejerce un gran papel en la biosíntesis y glicosilación de proteínas, así como en la detoxificación.
Estos tres aspectos son la clave de su importancia ya que:
- La biosíntesis de proteínas en el retículo marca su destino en la célula.
- Modificaciones de las proteínas como la glicosilación proporciona a la proteína unas
características diferentes (menor vulnerabilidad a proteasas).
- La detoxificación es el proceso gracias al cual se ha mantenido la vida sobre el planeta.
La mayoría de los compuestos son hidrofóbicos por lo que si no se produjera este efec-
to, quedarían adosados a la membrana plasmática impidiendo el funcionamiento de
las proteínas y del sistema de transporte de iones.
1 Biogénesis de lípidos en el retículo endoplasmático El retículo endoplasmático es el lugar donde se sintetizan los lípidos como el colesterol, la car-
diolipina, los esfingolípidos y los fosfolípidos (fosfatidilcolina y fosfatidiletanolamina). También
se sintetizan otros esteroles como los glucocorticoides, los mineralocorticoides y los gestáge-
nos.
1.1 Biosíntesis de lípidos y fosfolípidos La fabrica celular de lípidos y fosfolípidos es el retículo endoplasmático, desde donde van a
pasar a los diferentes orgánulos subcelulares en forma de vesículas de transporte que contie-
nen, además, el material proteico.
Los componentes para la fabricación de los ácidos grasos (Acil-CoA y glicerol fosfato) se locali-
zan en el citosol, pero las enzimas necesarias para su síntesis están adosadas a la cara citosó-
lica de la membrana del retículo endoplasmático.
Recordatorio: La adición de dos restos de ácido graso al glicerol fosfato genera el diacilgli-
cerol fosfato o ácido fosfatídico (PA). Si se le quita el fosfato tendremos el diacilglicerol
(DAG) y si se añade una colina tendremos la fosfatidilcolina, también denominada lecitina.
Partiendo desde la fosfatidilcolina (PC) podemos obtener la lisofosfatidilcolina (LPC) al reti-
rarle el ácido araquidónico (AA). La LPC puede formar ácido lisofosfatídico (LCA) y el AA
dará lugar a los eicosanoides.

82
Obsérvese como los puntos de insaturación se localizan en la posición 2 del glicerol fosfato.
Todos estos elementos son fuertemente hidrofóbicos y presentan una distribución diferente
entre las distintas membranas celulares.
1.2 Composición lipídica de las membranas celulares La composición lipídica de las membranas celulares varía muchísimo, por ello se habla de perfil
lipídico o lipidoma haciendo referencia a esta composición.
La comparación del contenido relativo de los lípidos en las diferentes membranas que se
muestra en la tabla nos indica la existencia de una discontinuidad entre el retículo endoplas-
mático liso y rugoso. Esto se debe a que la cantidad de proteínas varía a lo largo del retículo, el
entorno es diferente, y por ello solo aquellas proteínas que estén solubles podrán difundir a
cualquier cisterna, mientras que las ancladas al citoesqueleto verán su movilidad reducida. Las
diferentes porciones del retículo endoplasmático rugoso presentan un empaquetamiento y
orden especial que determina una composición proteica y lipídica concreta. Por su parte, el
retículo endoplasmático liso también presenta estas características que limitan la difusión de
algunos lípidos y proteínas.
El análisis de los componentes mayoritarios en cada membrana nos revela que:
- La membrana plasmática es siempre la que tiene el mayor contenido relativo de co-
lesterol.
- La membrana mitocondrial interna es la más rica en cardiolipina, el lípido del múscu-
lo cardíaco porque estas células presentan muchas mitocondrias.
Recordatorio: La ceramida puede dar lugar a esfingosina (Sph) y esta a esfingosina-1-
fosfato (S1P). También es el precursor para la síntesis de esfingomielina (SM), ceramida-1-
fosfato (C1P) y Glucosil ceramida (GlcCer).
Recordatorio: el fosfatidilinositol (PI) puede presentarse en varios estados de fosforilación
hasta el fosfatidilinositol-3,4,5-trifosfato (PIP3).
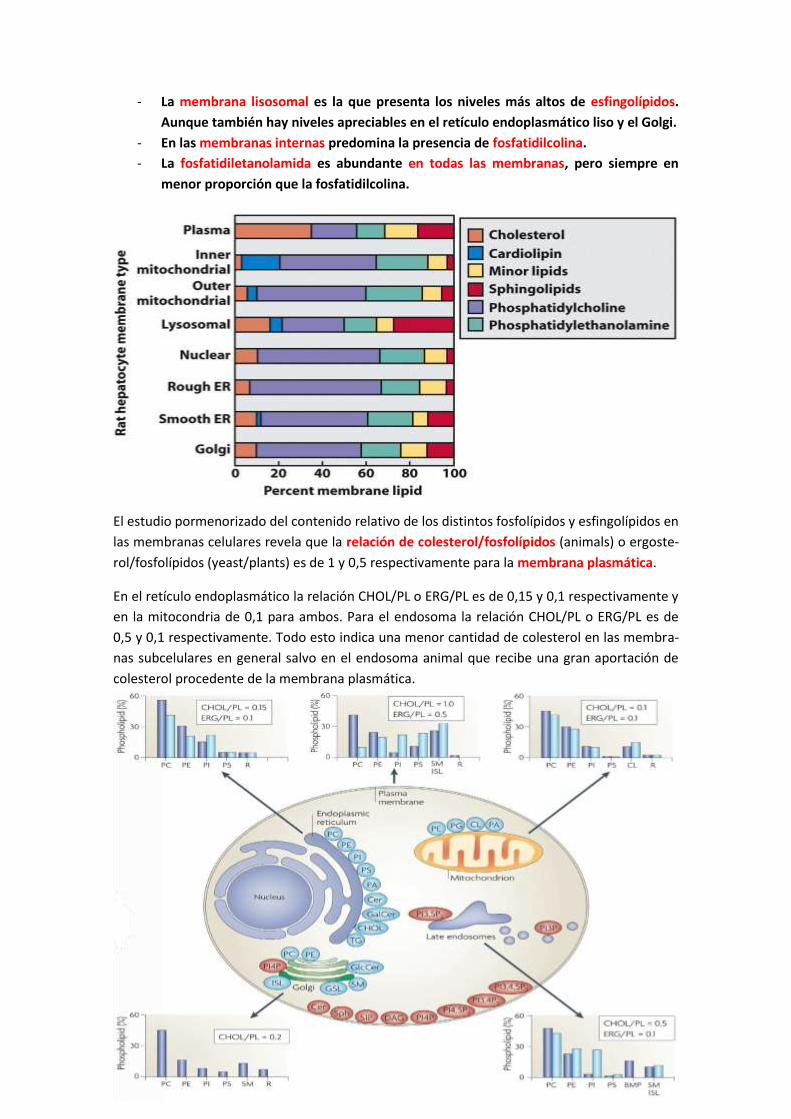
83
- La membrana lisosomal es la que presenta los niveles más altos de esfingolípidos.
Aunque también hay niveles apreciables en el retículo endoplasmático liso y el Golgi.
- En las membranas internas predomina la presencia de fosfatidilcolina.
- La fosfatidiletanolamida es abundante en todas las membranas, pero siempre en
menor proporción que la fosfatidilcolina.
El estudio pormenorizado del contenido relativo de los distintos fosfolípidos y esfingolípidos en
las membranas celulares revela que la relación de colesterol/fosfolípidos (animals) o ergoste-
rol/fosfolípidos (yeast/plants) es de 1 y 0,5 respectivamente para la membrana plasmática.
En el retículo endoplasmático la relación CHOL/PL o ERG/PL es de 0,15 y 0,1 respectivamente y
en la mitocondria de 0,1 para ambos. Para el endosoma la relación CHOL/PL o ERG/PL es de
0,5 y 0,1 respectivamente. Todo esto indica una menor cantidad de colesterol en las membra-
nas subcelulares en general salvo en el endosoma animal que recibe una gran aportación de
colesterol procedente de la membrana plasmática.
BIOGÉNESIS DE LÍPIDOS EN EL RETÍCULO ENDOPLÁSMICO

84
En el retículo endoplasmático vamos a encontrar casi la variedad total de lípidos y fosfolípidos,
ya que es la fábrica de estos. Entre ellos tenemos en orden de abundancia fosfatidilcolina (PC),
fosfatidiletanolamida (PE), fosfatidilinositol (PI), fosfatidilserina (PS) y ácido fosfatídico (PA),
además de otros como galactosilceramida (GalCer), colesterol (CHOL) y triglicéridos varios
(TG).
En las vesículas del aparato de Golgi volvemos a encontrar a la fosfatidilcolina (PC) como el
más abundante, pero también encontramos fosfatidiletanolamida (PE) y en menores cantida-
des fosfatidilinositol (PI), fosfatidilserina (PS), esfingomielina (SM), glucosilceramida (GluCer),
fosfatidilinositol-4-fosfato (PI4P), esfingolípidos (ISL) y glucoesfingolípidos (GLS).
La mitocondria es más restrictiva y la variedad de fosfolípidos que presenta es más limitada
como la fosfatidilcolina (PC), fosfatidiletanolamida (PE), fosfatidilglicerol (PG), cardiolipina (CL)
y ácido fosfatídico (PA).
En el endosoma tardío hay fosfatidilinositol-3,5-bifosfato (PI3,5P2), fosfatidilcolina (PC) fosfati-
diletanolamida (PE), esfingomielina (SM) y bis-monoglicerol-fosfato (BMP), mientras que en
las vesículas que llegan encontramos fosfoinositol-3-fosfato (PI3P).
Finalmente en la membrana plasmática encontramos gran cantidad de fosfatidilinositol en
todas sus variedades en lo que se conocen como fosfoinosítidos. Además también hay cera-
mida (Cer), esfingosina (Sph), esfingosina-1-fosfato (S1P) y diacilglicerol (DAG).
1.3 Biosíntesis y movimientos de lípidos a través de las membranas
celulares A modo de síntesis introductoria, en la cara externa
del retículo endoplasmático tenemos el glicerol-
fosfato al que una acil transferasa le introduce un
resto de ácido graso en la posición 1, y a continuación
otra le mete un segundo resto de ácido graso que
suele ser insaturado. A continuación actúa la fosfatasa
que retira el fosfato de la posición 3 y una CDP-colina
transferasa cataliza la introducción de colina-fosfato y
libera citidina monofosfato (CMP), y da lugar a la fos-
fatidilcolina (PC). Este mismo proceso sucede con la
fosfatidiletanolamida (PE) y la fosfatidilserina (PS).
La mayor parte de los glicerofosfolípidos ensamblados en el retículo endoplasmático son fosfa-
tidilcolina (PC), fosfatidiletanolamida (PE), fosfatidilinositol (PI), fosfatidilserina (PS) y ácido
fosfatídico (PA). El retículo endoplasmático también sintetiza ceramida (Cer), galactoceramida
(GalCer), colesterol (CHOL) y ergosterol (ERG). El retículo endoplasmático y las reservas lipídi-
cas participan en la síntesis de triglicéridos (TG) y de esteres de ácidos grasos.
Puesto que las enzimas implicadas se localizan en la cara externa del retículo, la fosfatidilcolina
(PC) se va acumulando en la monocapa externa, pero también van a haber enzimas (Translo-
casas) que favorecen el movimiento de flip-flop, evitando que la monocapa externa se haga
más extensa que la interna. Pero este movimiento de flip-flop espontaneo está restringido
Major glycerophospholipids assembled in the ER are phosphatidylcholine (PtdCho; PC), phosphatidylethanolamine (PtdEtn; PE), phosphatidylinositol (PtdIns; PI), phosphatidylserine (PtdSer; PS) andphosphatidic acid (PA). The ER synthesizes also Cer, galactosylceramide (GalCer), cholesterol and ergosterol. Both the ER and lipid droplets participate in steryl ester and triacylglycerol (TG) synthesis. The Golgi lumen is the site of synthesis of sphingomyelin (SM), complex glycosphingolipids (GSLs) and yeast inositol sphingolipid (ISL) synthesis. inositol sphingolipid (ISL) synthesis. PtdCho is also synthesized in the Golgi, and may be coupled to protein secretion at the level of its diacylglycerol (DAG) precursor. Approximately 45% of the phospholipid in mitochondria (mostly PtdEtn, PA and cardiolipin (CL)) is autonomously synthesized by the organelle. BMP (bis (monoacylglycero) phosphate) is a major phospholipid in the inner membranes of late endosomes.

85
salvo que una proteína con huecos hidrofógicos y mediante el gasto de ATP, pase los lípidos de
un lado a otro. Este movimiento está facilitado por las flipasas y flopasas.
Además, las scramblasas facilitan el intercambio de fosfolípidos a favor de gradiente, sin gasto
de ATP.
Pero en las cisternas del Golgi no se puede producir estos
movimientos por que no se encuentran estas proteínas. La
fosfatidilcolina (PC) y la esfingomielina (SM) que queda en
la cara interna no puede salir a la externa, porque no hay
transportadores, pero el diacilglicerol (DAG) y la ceramida
(Cer) si. El lumen del Golgi es el lugar de síntesis de la es-
fingomielina (SM), los glicesfingolípidos complejos (GSLs)) y
el inositol esfingolípido (ISL) de levaduras. Las proteínas P4
son las translocasas dependientes de ATP que van a permi-
tir la salida a la monocapa extertna de la fosfoetanolamida
(PE) y la fosfoserina (PS).
Aproximadamente el 45% de los fosfolípidos de la mitocondria son fosfatidiletanolamida, áci-
do fosfatídico y cardiolipina, que son sintetizados autónomamente por el orgánulo.
En la membrana plasmática la fosfatidilcolina (PC), la esfingomielina (SM) y los glicoesfingolí-
pidos complejos (GSLs) no puede pasar a la monocapa interna, mientras que si encontramos
translocasas P4 para fosfatidiletanolamida (PE) y fosfatidilserina (PS). Esto supone la existencia
de una asimetría entre ambas monocapas donde la
fosfaticilcolina es más abundante en la monocapa
externa, mientras que la fosfatidilserina (PS) y la fos-
fatidiletanolamida (PE) son más abundantes en la
monocapa interna. La distribución asimétrica es vital
para la morfología de la membrana, ya que la gran
cabeza de aminocuaternario presente en la fosfatidil-
colina (PC) se tiene que situar en la parte exterior de
la curvatura, mientras que la fosfatidilserina (PS) y la
fosfatidiletanolamida (PE) no presentan impedimen-
tos estéricos en la parte interna de la curvatura.
poner lo de abajoponer lo de abajo
Major glycerophospholipids assembled in the ER are phosphatidylcholine (PtdCho; PC), phosphatidylethanolamine (PtdEtn; PE), phosphatidylinositol (PtdIns; PI), phosphatidylserine (PtdSer; PS) andphosphatidic acid (PA). The ER synthesizes also Cer, galactosylceramide (GalCer), cholesterol and ergosterol. Both the ER and lipid droplets participate in steryl ester and triacylglycerol (TG) synthesis. The Golgi lumen is the site of synthesis of sphingomyelin (SM), complex glycosphingolipids (GSLs) and yeast inositol sphingolipid (ISL) synthesis. inositol sphingolipid (ISL) synthesis. PtdCho is also synthesized in the Golgi, and may be coupled to protein secretion at the level of its diacylglycerol (DAG) precursor. Approximately 45% of the phospholipid in mitochondria (mostly PtdEtn, PA and cardiolipin (CL)) is autonomously synthesized by the organelle. BMP (bis (monoacylglycero) phosphate) is a major phospholipid in the inner membranes of late endosomes.
Major glycerophospholipids assembled in the ER are phosphatidylcholine (PtdCho; PC), phosphatidylethanolamine (PtdEtn; PE), phosphatidylinositol (PtdIns; PI), phosphatidylserine (PtdSer; PS) andphosphatidic acid (PA). The ER synthesizes also Cer, galactosylceramide (GalCer), cholesterol and ergosterol. Both the ER and lipid droplets participate in steryl ester and triacylglycerol (TG) synthesis. The Golgi lumen is the site of synthesis of sphingomyelin (SM), complex glycosphingolipids (GSLs) and yeast inositol sphingolipid (ISL) synthesis. inositol sphingolipid (ISL) synthesis. PtdCho is also synthesized in the Golgi, and may be coupled to protein secretion at the level of its diacylglycerol (DAG) precursor. Approximately 45% of the phospholipid in mitochondria (mostly PtdEtn, PA and cardiolipin (CL)) is autonomously synthesized by the organelle. BMP (bis (monoacylglycero) phosphate) is a major phospholipid in the inner membranes of late endosomes.
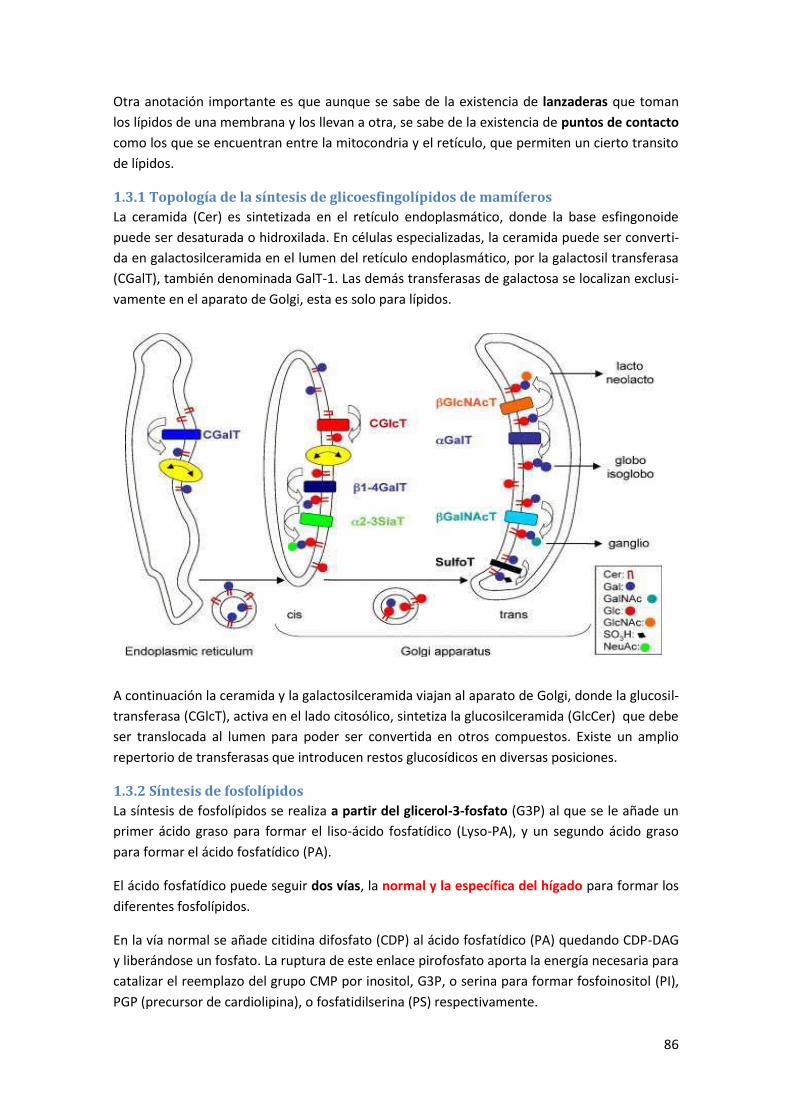
86
Otra anotación importante es que aunque se sabe de la existencia de lanzaderas que toman
los lípidos de una membrana y los llevan a otra, se sabe de la existencia de puntos de contacto
como los que se encuentran entre la mitocondria y el retículo, que permiten un cierto transito
de lípidos.
1.3.1 Topología de la síntesis de glicoesfingolípidos de mamíferos
La ceramida (Cer) es sintetizada en el retículo endoplasmático, donde la base esfingonoide
puede ser desaturada o hidroxilada. En células especializadas, la ceramida puede ser converti-
da en galactosilceramida en el lumen del retículo endoplasmático, por la galactosil transferasa
(CGalT), también denominada GalT-1. Las demás transferasas de galactosa se localizan exclusi-
vamente en el aparato de Golgi, esta es solo para lípidos.
A continuación la ceramida y la galactosilceramida viajan al aparato de Golgi, donde la glucosil-
transferasa (CGlcT), activa en el lado citosólico, sintetiza la glucosilceramida (GlcCer) que debe
ser translocada al lumen para poder ser convertida en otros compuestos. Existe un amplio
repertorio de transferasas que introducen restos glucosídicos en diversas posiciones.
1.3.2 Síntesis de fosfolípidos
La síntesis de fosfolípidos se realiza a partir del glicerol-3-fosfato (G3P) al que se le añade un
primer ácido graso para formar el liso-ácido fosfatídico (Lyso-PA), y un segundo ácido graso
para formar el ácido fosfatídico (PA).
El ácido fosfatídico puede seguir dos vías, la normal y la específica del hígado para formar los
diferentes fosfolípidos.
En la vía normal se añade citidina difosfato (CDP) al ácido fosfatídico (PA) quedando CDP-DAG
y liberándose un fosfato. La ruptura de este enlace pirofosfato aporta la energía necesaria para
catalizar el reemplazo del grupo CMP por inositol, G3P, o serina para formar fosfoinositol (PI),
PGP (precursor de cardiolipina), o fosfatidilserina (PS) respectivamente.
Topology of mammalian
glycosphingolipid (GSL) synthesis.
Ceramide is produced in the ER,
where the sphingoid base can be
desaturated or hydroxylated. In
specialized cells, ceramide can be
converted to GalCer in the ER lumen
by the Gal transferase CGalT, also by the Gal transferase CGalT, also
called GalT-1. The glucosyltransferase
CGlcT, active on the cytosolic side of
the Golgi apparatus, synthesizes
GlcCer which must be translocated to
the Golgi lumen to be converted to
LacCer by the β1,4-galactosyl-
transferase GalT-2 .
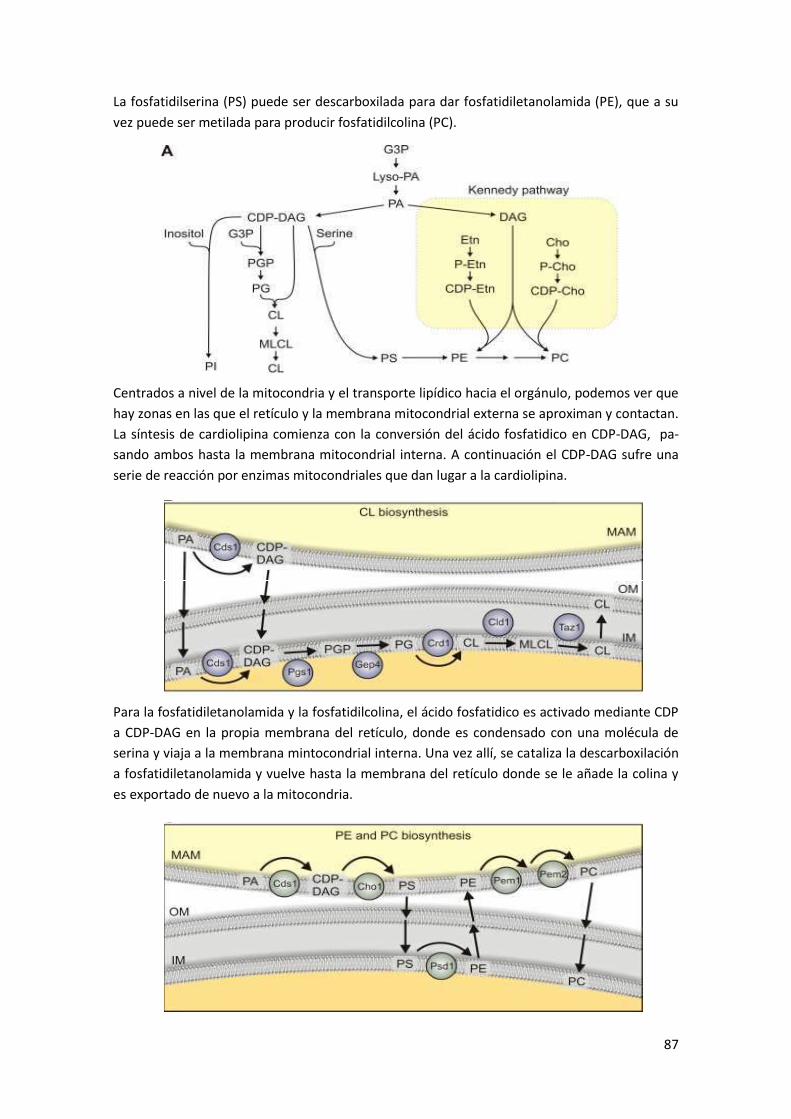
87
La fosfatidilserina (PS) puede ser descarboxilada para dar fosfatidiletanolamida (PE), que a su
vez puede ser metilada para producir fosfatidilcolina (PC).
Centrados a nivel de la mitocondria y el transporte lipídico hacia el orgánulo, podemos ver que
hay zonas en las que el retículo y la membrana mitocondrial externa se aproximan y contactan.
La síntesis de cardiolipina comienza con la conversión del ácido fosfatidico en CDP-DAG, pa-
sando ambos hasta la membrana mitocondrial interna. A continuación el CDP-DAG sufre una
serie de reacción por enzimas mitocondriales que dan lugar a la cardiolipina.
Para la fosfatidiletanolamida y la fosfatidilcolina, el ácido fosfatidico es activado mediante CDP
a CDP-DAG en la propia membrana del retículo, donde es condensado con una molécula de
serina y viaja a la membrana mintocondrial interna. Una vez allí, se cataliza la descarboxilación
a fosfatidiletanolamida y vuelve hasta la membrana del retículo donde se le añade la colina y
es exportado de nuevo a la mitocondria.
Synthesis of phospholipids. G3P, glycerol-3P; Cho,
choline; Etn, ethanolamine; CL, cardiolipin; MLCL, monolyso-CL; P-Cho, P-choline; P-Etn, P-ethanolamine. PA, phosphatidic acid. Cleavage of the pyrophosphate bond in CDP-diacylclycerol (DAG) provides the energetic driving force to catalytically replace CMP with inositol, G3P, or Ser to form PI, PGP or PS, respectively, using specific enzymes. PGP is dephosphorylated to produce PG. CL is synthesized from PG and CDP-DAG with the catalytic cleavage of the pyrophosphate bond in the latter substrate providing the chemical energy to transfer the PA moiety to the vacant primary hydroxyl of PG. PS can be decarboxylated to PE, which in turn can be methylated to yield PC. Alternatively, PE and PC can be to yield PC. Alternatively, PE and PC can be synthesized via an enzymatic cascade known as the Kennedy pathway. (B) Membrane topology and lipid transport events in the synthesis of CL and aminoglycerophospholipids (PE and PC); C) PA synthesized in the ER or mitochondria drive biosynthetic reactions. CDP-DAG may derive from the ER/MAM or be generated at the mitochondrial IM by the action of Cds1. IM, mitochondrial inner membrane; OM, mitochondrial outer membrane.Cds1, CDP-DAG synthase; Cld1, phospholipase A; Crd1, CL synthase; Cho1, Gep4, phosphatidylglycerolphosphate phosphatase; Pem1 and Pem2, P-Etn methyltransferases; Pgs1, phosphatidylglycerolphosphate synthase; Psd1, P-Ser decarboxylase; Taz, transacylase.
Synthesis of phospholipids. G3P, glycerol-3P; Cho, choline; Etn, ethanolamine; CL, cardiolipin; MLCL, monolyso-CL; P-Cho, P-choline; P-Etn, P-ethanolamine. PA, phosphatidic acid. Cleavage of the pyrophosphate bond in CDP-diacylclycerol (DAG) provides the energetic driving force to catalytically replace CMP with inositol, G3P, or Ser to form PI, PGP or PS, respectively, using specific enzymes. PGP is dephosphorylated to produce PG. CL is synthesized from PG and CDP-DAG with the catalytic cleavage of the pyrophosphate bond in the latter substrate providing the chemical energy to transfer the PA moiety to the vacant primary hydroxyl of PG. PS can be decarboxylated to PE, which in turn can be methylated to yield PC. Alternatively, PE and PC can be to yield PC. Alternatively, PE and PC can be synthesized via an enzymatic cascade known as the Kennedy pathway. (B) Membrane topology and lipid transport events in the synthesis of CL and aminoglycerophospholipids (PE and PC); C) PA synthesized in the ER or mitochondria drive biosynthetic reactions. CDP-DAG may derive from the ER/MAM or be generated at the mitochondrial IM by the action of Cds1. IM, mitochondrial inner membrane; OM, mitochondrial outer membrane.Cds1, CDP-DAG synthase; Cld1, phospholipase A; Crd1, CL synthase; Cho1, Gep4, phosphatidylglycerolphosphate phosphatase; Pem1 and Pem2, P-Etn methyltransferases; Pgs1, phosphatidylglycerolphosphate synthase; Psd1, P-Ser decarboxylase; Taz, transacylase.
Synthesis of phospholipids. G3P, glycerol-3P; Cho,
choline; Etn, ethanolamine; CL, cardiolipin; MLCL, monolyso-CL; P-Cho, P-choline; P-Etn, P-ethanolamine. PA, phosphatidic acid. Cleavage of the pyrophosphate bond in CDP-diacylclycerol (DAG) provides the energetic driving force to catalytically replace CMP with inositol, G3P, or Ser to form PI, PGP or PS, respectively, using specific enzymes. PGP is dephosphorylated to produce PG. CL is synthesized from PG and CDP-DAG with the catalytic cleavage of the pyrophosphate bond in the latter substrate providing the chemical energy to transfer the PA moiety to the vacant primary hydroxyl of PG. PS can be decarboxylated to PE, which in turn can be methylated to yield PC. Alternatively, PE and PC can be to yield PC. Alternatively, PE and PC can be synthesized via an enzymatic cascade known as the Kennedy pathway. (B) Membrane topology and lipid transport events in the synthesis of CL and aminoglycerophospholipids (PE and PC); C) PA synthesized in the ER or mitochondria drive biosynthetic reactions. CDP-DAG may derive from the ER/MAM or be generated at the mitochondrial IM by the action of Cds1. IM, mitochondrial inner membrane; OM, mitochondrial outer membrane.Cds1, CDP-DAG synthase; Cld1, phospholipase A; Crd1, CL synthase; Cho1, Gep4, phosphatidylglycerolphosphate phosphatase; Pem1 and Pem2, P-Etn methyltransferases; Pgs1, phosphatidylglycerolphosphate synthase; Psd1, P-Ser decarboxylase; Taz, transacylase.

88
1.4 Enfermedades ligadas al retículo endoplasmático Se conocen unas 10.000 enfermedades monogénicas (que dependen exclusivamente de un
gen) como la fibrosis quística, distrofia muscular de Duchenne, etc. Destacan en el ámbito de
este tema las enfermedades causadas por acumulación de material lipídico en el lisosoma
(mucolipidosis).
Por endocitosis se forman primero los endosomas primarios y luego los endosomas tardíos, y
esos endosomas tardíos terminan en el lisosoma, para la degradación. Entre los materiales a
degradar están los lípidos y las proteínas embebidas, donde son muy abundantes los gangliósi-
dos, sobre todo en la neurona, a nivel del sistema nervioso central.
Hay una serie de enfermedades por la acumulación de material lipídico (gangliósido GM1,
GM2, GM3,etc.), conociéndose por gangliosidosis (para GM1), sialidosis, enfermedad de Gau-
cher,… , dependiendo de la deficiencia de una proteína aparecen las distintas enfermedades,
siendo deficiencias en un solo gen.
Esta acumulación del material lipídico (o también llamadas estas enfermedades de “inclusión
de material lipídico en el lisosoma”), que pueden ser gangliósidos, colesterol, etc. o cualquier
otro material que no se pueda digerir en el lisosoma, provoca su acumulación lo que desem-
boca en el aumento de tamaño y la progresiva pérdida de funcionalidad celular.
Esto les sucede por ejemplo a las neuronas. Los niños que nacen con este tipo de enfermedad
presentan un severo retraso mental, muriendo a los pocos años de edad, dependiendo de la
variante de la enfermedad. Son enfermedades gravísimas que se detectan por amniocentesis.
Si se detecta en los primeros estadios hay tratamiento.
La terapia que se ha ideado para el tratamiento de estas enfermedades es la administración
de chapeoronas (químicas) como inhibidores competitivos que fuerzan el plegamiento de la
proteína, mejorando su actividad catalítica o permitiéndole llegar hasta su destino a pesar
de no ser totalmente funcional.
Una proteína normal se va a plegar correctamente al sintetizarse en el retículo a pH neutro,
con destino al lisosoma. Pero cuando las proteínas mutadas predominan en formas mal plega-
das que no van a ser funcionales.

89
La administración de chaperonas químicas puede forzar un buen plegamiento, suficiente como
para pasar los controles de calidad del retículo y viajar al Golgi, desde donde es transportada al
lisosoma, cuyo pH provoca la disociación de la chaperona. Aunque esto puede suponer una
pérdida de conformación, la proteína va a presentar cierta actividad catalítica, que en muchos
casos será suficiente para realizar su función, ya que sin la chaperona, no habría superado el
ERAD del retículo, o incluso podría haber comenzado a agregarse.
Pero este tipo de tratamientos solo es aplicable a enfermedades monogenéticas cuya causa
se conoce. La aplicación en enfermedades multifactoriales como el Alzheimer o el Parkinson es
inviable.
En cualquier caso es necesario que la chaperona química pueda atravesar la membrana plas-
mática, siendo homólogos no hidrolizables, no transformables, de un sustrato. Si lo que falta
es la sialidasa, porque la proteína está pero no tiene el plegamiento adecuado para tener acti-
vidad biológica, se pone un homólogo del ácido siálico de manera que a través del ajuste indu-
cido se pega al centro activo y obliga al plegamiento de la proteína. Quizás la proteína, una vez
plegada, no adquiera el 100% de su actividad catalítica, pero con que tenga el 20-30% ya es
suficiente, para que el desarrollo del sistema nervioso central del niño afectado sea mediana-
mente normal, dependiendo del grado de recuperación.
2 Las proteínas citocromo P450 Se calcula que hay unos 100.000 xenobióticos, siendo productos de la industria química que se
emplean como aditivos alimentarios, cosméticos, etc. Gracias a estos citocromos p450 se ha
podido mantener la vida en el planeta, ya que aparecen como una adaptación al medio.
Las primeras proteínas del citocromo P450 aparecen para desempeñar una función fisiológica
esencial como la transformación de colesterol en sales biliares, andrógenos y gestágenos, co-
mo enzimas con actividad hidroxilasa. Pero los fenómenos de duplicación génica permitieron
la variación suficiente como para que estos citocromos se fueran especializando además en la
detoxificación, es decir, la eliminación de productos que generalmente se va a conseguir au-
mentando la solubilidad de los tóxicos.
El citocromo P450 recibe su nombre por presentar un grupo prostético hemo que cuando
se reduce con ditionito (agente reductor) y luego se burbujea la disolución resultante con
monóxido de carbono (CO) se forma un complejo que presenta un espectro de absorción
con un máximo entre 446 y 452 nm.
Por ejemplo, el benceno y sus derivados que al ser metabolizados se convierten en fenoles y
difenoles, mucho más hidrosolubles que pueden ser conjugados con ácido cólico en el riñón,
para formar colatos o con el ácido hipúrico, y ser eliminados con la orina.

90
2.1 Modo de acción y características del citocromo P450 En humanos se han detectado unos 57-58 citocromos P450 que en estado basal presentan un
Fe3+ unido. Cuando el sustrato reducido entra en el sitio activo, entonces P450 capta un elec-
trón procedente del NADPH reduciéndose a Fe2+.
Ahora entra una molécula de
oxigeno que provoca la dismuta-
ción de P450 (oxidación) formán-
dose especies inestables del oxí-
geno que van a pivotar en equili-
brio entre varias estructuras de
resonancia y el anión superóxido.
Finalmente se recibe el segundo
electrón procedente del NADPH y
el oxígeno, que ahora tiene dos
cargas negativas, forma el anión
peróxido que va a oxidar el sustra-
to reducido y el NADPH captando
los dos protones para liberar el
sustrato oxidado y H2O.
A nivel celular, hay dos sistemas o grupos de proteínas del citocromo P450, unas localizadas
en el retículo endoplásmico y otras en la mitocondria:
- En el retículo, los electrones pasan del NADPH hasta la proteína NCR (NADPH Cito-
cromo p450 Reductasa, que es una flavoproteína porque tiene como grupos prostéti-
cos FADH2 y FMNH2, que son estables cuando reciben los dos electrones, pero pueden
soltarlos o los dos a la vez o de uno en uno). Esos electrones son cedidos al citocromo
p450, que cuando toma los electrones y pasa al estado reducido. En estado reducido
puede pasar a estado oxidado a través de la reducción del sustrato. La NCR es una de
las pocas proteínas que tiene dos grupos prostéticos, ya que la mayoría de las proteí-
nas tienen uno de estos dos grupos, pero no los dos al mismo tiempo).
- En el sistema mitocondrial los electrones pasan desde el NADPH a la proteína NAdR
(NADPH Adrenodoxina Reductasa, es una flavoproteína que tiene el grupo prostético
FADH2). De ahí los electrones pasan a la adrenodoxina, la cual transfiere los electro-
¿Quién es el aceptor de oxígeno? ¿Dónde entra el primer electrón? ¿Dónde entra el se-
gundo electrón?

91
Los barbitúricos son una familia de fármacos derivados del ácido barbitúrico que actúan como sedan-
tes del sistema nervioso central y producen un amplio esquema de efectos, desde sedación suave
hasta anestesia total. Tienen un alto potencial de adicción y la sobredosis puede causar la muerte
El alcohol se metaboliza a través del sistema P450. El etanol por el citocromo p450 se oxida a acetal-
dehído y luego pasa a acético. Por lo que no debemos consumir medicamentos y alcohol al mismo
tiempo, pues si el citocromo está ocupado trabajando con el alcohol no puede estar trabajando con
el sedante, por lo que si se consumen las dos cosas, estaríamos alargando la acción del sedante.
nes al propio citocromo p450, que luego lo pierde ya al sustrato para pasar del estado
reducido al oxidado (igual que en retículo). La adrenodoxina es un transportador que
tiene cluster de grupos Fe-S asemejándose en cuanto a estructura a la ferredoxina.
Se habla de transporte electrónico microsomal, para aumentar la solubilidad y para fabricar
ciertos metabolitos.
Los miembros de la gran familia de citocromo P450 presentan ho-
mología de la secuencia, debido a que los aminoácidos que “suje-
tan” el grupo hemo son muy similares. (Familias de CYP1 = CYP1A,
CYP1B, etc.).
Para que pertenezcan a la misma familia (CYP1A y CYP1B), la homo-
logía en la secuencia tiene que ser al menos de un 40%. Dentro de
la misma familia (CYP1A) aparecen varias subfamilias, que para que formen parte de esta fami-
lia tienen que presentar una homología de al menos el 50-55%.
Prácticamente todas las familias de P450 las tenemos distribuidas por todos los tejidos, aun-
que hay poco en cerebro y músculo esquelético. Sin embargo, el hígado es muy rico en p450,
debido a que es el órgano que se encarga de la detoxificación.
El perfil de p450 es distinto según el tejido, así como entre distintas especies animales. No
podemos comparar el espectro de absorción de citocromo p450 de animales de distintas espe-
cies, debido a que la exposición a genes diversos es distinta, siendo incluso el perfil de p450
diferente entre personas (individuos de la misma especie). Esto quiere decir, que el efecto de
diversos fármacos (vida media del fármaco en la sangre, por ejemplo), es diferente según el
individuo. Dependiendo del rango de p450 que tenga una persona, la dosis de un fármaco
puede variar, por lo que se habla de medicina personalizada
El citocromo P450 es una proteína inducible (del repertorio de p450 va a ser inducible aquella
variante necesaria para metabolizar un determinado agente). Si a un animal le administramos
una dosis de barbitúrico (sedante, como por ejemplo: veronal), la cantidad de la variante de
p450 necesario para metabolizar el barbitúrico aumenta a nivel transcripcional (es decir, a
nivel de cantidad de mensajero) y postranscripcional (es decir, la vida media del mensajero),
cuanto más resistente sea al tratamiento o a la acción de las ribonucleasas se estarán produ-
ciendo más cantidad de proteínas.

92
2.1.1 Modo de acción de Citocromo P450 reductasa
La citocromo P450 reductasa presen-
ta:
- un dominio de unión a FMN
(azul)
- un dominio de unión a FAD
(amarillo)
- un dominio de conexión (ro-
jo)
- Un dominio de unión a FAD y
NADPH (verde)
Los electrones van a pasar desde el NADPH hasta el FADH2 y luego al FMN, que se los cede al
citocromo P450, reduciéndolo. Estos electrones pueden llegar de dos en dos o de uno en uno,
ya que el citocromo va a poder recibir un electrón del NADPH, pero además otro del citocromo
B5 (adosado a la membrana). El donador de electrones para el citocromo B5 (desde la cito-
cromo B5 reductasa) es el NADH, por lo que en función del balance de poder reductor, los
electrones procederán en principio del NADPH o del NADH.
2.1.2 Modo de acción de la adrenoxina reductasa
Existen tres modelos teóricos para explicar el trasiego de electrones desde la adrenosina re-
ductasa por la adrenoxina, hasta el citocromo P450:
- El modelo binario establece que el trasiego de electrones a través de las proteínas se
produce mediante interacciones binarias entre
dos de las proteínas en cada momento.
- El modelo ternario establece la formación de un
complejo ternario que implica la interacción de
las tres proteínas en el momento del flujo elec-
trónico.
- El modelo cuaternario establece la existencia de
dímeros entre adrenosina reductasa-adrenosina
y adrenosina-P450 que van a interaccionar a
través de la adrenosina para realizar la transfe-
rencia de electrones, formando un complejo
cuaternario.
2.2 Otras funciones de P450 Como ya se comentó al inicio de este apartado, la función primaria de las proteínas P450 era
participar en las reacciones metabólicas como hidroxilasas del retículo endoplasmático y de la
mitocondria.
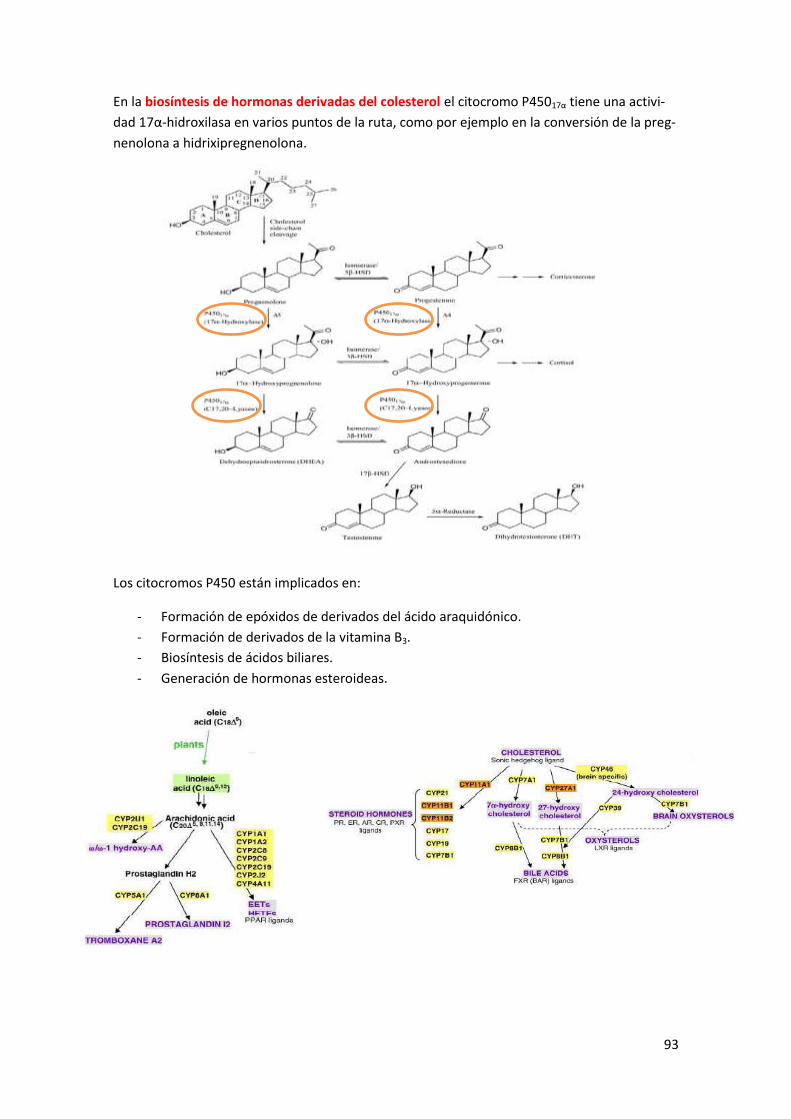
93
En la biosíntesis de hormonas derivadas del colesterol el citocromo P45017α tiene una activi-
dad 17α-hidroxilasa en varios puntos de la ruta, como por ejemplo en la conversión de la preg-
nenolona a hidrixipregnenolona.
Los citocromos P450 están implicados en:
- Formación de epóxidos de derivados del ácido araquidónico.
- Formación de derivados de la vitamina B3.
- Biosíntesis de ácidos biliares.
- Generación de hormonas esteroideas.

94
2.3 Enfermedades relacionadas con el citocromo P450 Existen tres tipos de variantes alélicas por las que una persona puede sufrir una enfermedad
relacionada con los genes de P450:
- Deleción del gen que conduce a la ausencia de un tipo particular de P450.
- Mutación en el gen que pueden dar lugar a:
o Bajo nivel de enzima
o Enzima inestable.
o Enzima con especificidad de sustrato diferente.
- Duplicación del gen que conduce a un aumento de la cantidad de P450 que puede ser
beneficioso o perjudicial.
El hecho de que P450 actué sobre un metabolito no significa que el producto sea más inofen-
sivo que el sustrato, en algunas ocasiones, como el caso de los epoxiderivados del antraceno o
el naftaleno, estos son más perjudiciales y cancerígenos que los compuestos de los que proce-
den.
La siguiente tabla muestra variantes de citocromos que metabolizan medicamentos que se
utilizan para la depresión, psicosis, úlcera, cáncer, problemas de corazón, etc. Hay fármacos
contra distintas enfermedades, que se metabolizan a través de cit. p450.

95
UMs = metabolizadores ultrarápidos (un metabolizador ultrarápido es aquel que tiene copias
múltiples o superenzimas, con el centro activo idóneo para metabolizar determinados fárma-
cos. A lo mejor a ese individuo le tienen que administrar 200 veces la dosis normal.
PMs = metabolizadores pobres (un metabolizador pobre a lo mejor con el 40% de la dosis
normal tiene ya suficiente).
Si hay 57 tipos de cit. p450 y uno de ellos está suprimido, apenas se notará el efecto, ya que,
otro puede desempeñar su función en cierta medida.

96
Anexo I: Aislamiento de fracciones subcelulares El estudio del retículo endoplasmático mediante estudios funcionales requiere del aislamiento
de este. Para ello, la primera consideración consiste en elegir un tejido que presente gran
cantidad de retículo endoplasmático. Un buen tejido sería el hígado de rata, nunca se elegiría
el músculo esquelético, que presenta mucho retículo sarcoplasmático pero apenas retículo
endoplasmático.
El primer paso del procedimiento de aislamiento consiste en el homogeneizado del tejido
(hígado a una concentración del 20% p/v) en potter manual con medio tamponado isotónico
con el interior del retículo (Tris 20 mM pH 7.5 y sacarosa 0.32 M) y antiproteasas para evitar la
degradación de proteínas. Puesto que algunas proteasas calcio-dependientes actúan a muy
baja concentración de calcio (incluso con la del agua destilada), se suelen añadir EDTA (secues-
trador de calcio) para que no se activen.
A continuación, se centrifuga el homogenizado a 1,000 g durante 10 minutos quedando un
precipitado (P1) con el debris celular (membrana con proteínas y restos) y la fracción nuclear, y
un sobrenadante (S1).
Ahora se centrifuga S1 a 10,000 g durante 10-20 minutos, obteniéndose un precipitado (P2)
con la fracción mitocondrial, y un nuevo sobrenadante (S2).
Se centrifuga S2 a 100,000 g durante 40-60 minutos, obteniéndose un precipitado (P3) con la
fracción microsomal (membrana del retículo endoplasmático, el aparato de Golgi y restos de
membrana plasmática) y un sobrenadante (S3). Para separar los componentes de la fracción
microsomal de P3 en fracciones enriquecidas (nunca al 100%) se recurre a la centrifugación en
gradientes de densidad continuos o discontinuos de sacarosa.
Finalmente, si se centrifuga S3 de nuevo a 100,000 g durante 2-3 h se obtiene un precipitado
(P4) que contiene la fracción ribosomal, y un sobrenadante (S4) que contiene los componentes
solubles del citosol.
Una vez separadas las diferentes fracciones subcelulares, tendremos que asegurarnos de que
son aquello que deberían, y para ello se emplean miden concentraciones de enzimas o proteí-
nas que marcan exclusivamente una fracción u orgánulo determinado.

97
Orgánulo Marcador
Retículo endoplasmático Aril sulfatasa, BiP
Aparato de Golgi Galactosil transferasa, COP-I
Matriz mitocondrial Succinato deshidrogenasa
Lisosoma β-hexosaminidasa, β-glucuronidasa
Citosol Lactato deshidrogenasa
Membrana plasmática Na/K-ATPasa (sensible a ouabaína)
Retículo sarcoplasmático Ca-ATPasa
Peroxisoma Catalasa
Balsa lipídica 5’-nucleotidasa, Fosfatasa alcalina
Para determinar la riqueza del orgánulo cuyo marcador se está analizando existen tablas que
establecen las unidades de actividad por miligramo de proteína (U/mg) del marcador para una
fracción pura. Por lo que si estoy analizando la succinato deshidrogenasa de la matriz mito-
condrial (100% = 5,000 U/mg) y obtengo un valor de 4,000 U/mg, tendré una preparación de
matriz mitocondrial al 80%.
Ejemplo: Aislamiento, caracterización y análisis proteico de la fracción
microsomal del músculo de ratón En un caso experimental para distrofia muscular de carácter recesivo se presentan dos rato-
nes, uno silvestre (Lama2+/?) y uno enfermo (Lama2dy/Lama2dy). Fenotípicamente los rato-
nes Lama2dy no pueden producir merosina (la cadena 2 de la laminina) por lo que presentan
una deficiencia de laminina-2 y muestran un distrofia.
Por ello se lleva a cabo un aislamiento de la fracción microsomal del músculo de ratón sano y
enfermo, con el fin de compáralas.
El protocolo comienza con la homogeneización del musculo (cada ratón por separado) en me-
dio isotónico (KCl 100 mM; 40ml/10g de músculo) y se centrifuga 20 minutos a 8.500 g. Se
obtiene un sobrenadante (S1) que contiene principalmente membranas y algo de la fracción
del derbis celular y núcleos. También se obtiene un precipitado (P1) que contiene la fracción
mitocondrial.
A continuación, para extraer la máxima cantidad posible de fracción microsomal, se vuelve a
repetir el homogenizado y la centrifugación sobre P1, obteniéndose un nuevo sobrenadante
(S2).
S1 y S2 se filtran a través de gasas y se someten a una centrifugación de 30 minutos a 100.000
g, obteniéndose un precipitado (P3) que contiene la fracción microsomal. Esta fracción se ho-
mogeniza y lava (KCl 600 mM; 10ml/10g músculo) parar retirar la actina y miosina del aparato
contráctil del músculo y se vuelve a centrifugar 30 minutos a 100.000 g.
El nuevo precipitado (P4) se vuelve a homogenizar y lavar con KCl, se incuba 30 minutos a 4ºC,
y se vuelve a centrifugar en las mismas condiciones.
Finalmente se obtiene un precipitado (P5) que contiene la fracción microsomal del músculo y
se homogeniza en tampón isotónico (Sacarosa 250 mM; 1ml/10g).
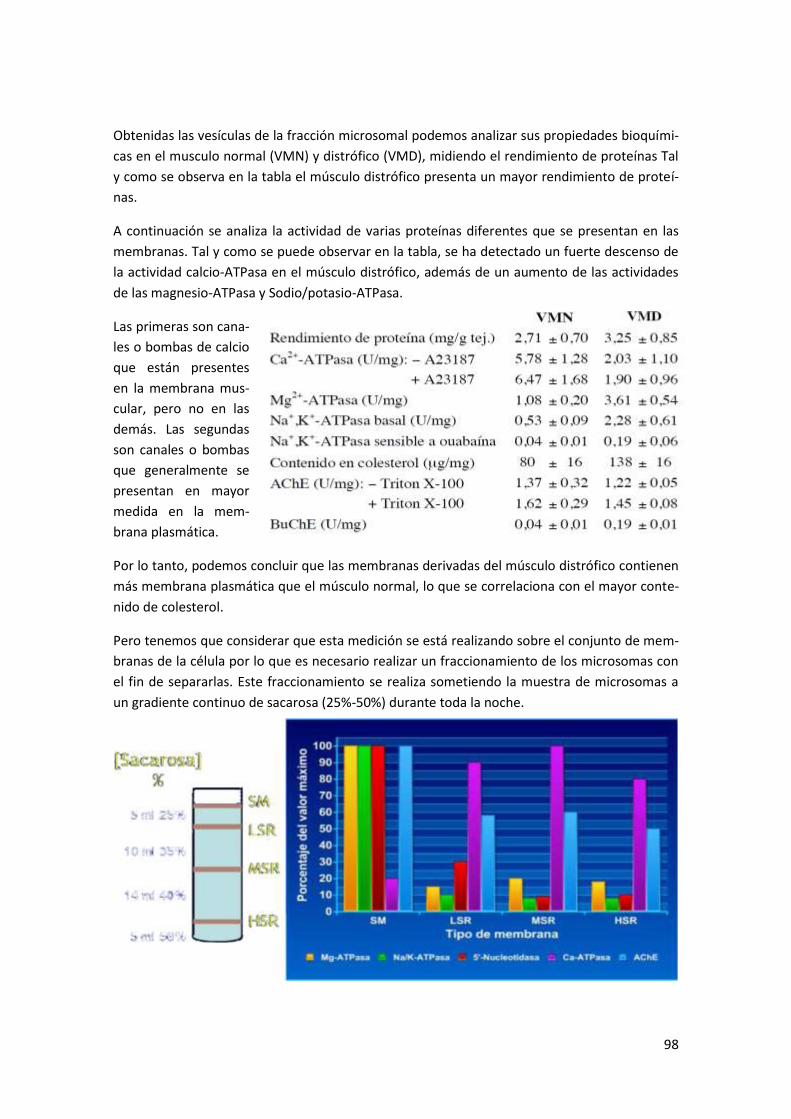
98
Obtenidas las vesículas de la fracción microsomal podemos analizar sus propiedades bioquími-
cas en el musculo normal (VMN) y distrófico (VMD), midiendo el rendimiento de proteínas Tal
y como se observa en la tabla el músculo distrófico presenta un mayor rendimiento de proteí-
nas.
A continuación se analiza la actividad de varias proteínas diferentes que se presentan en las
membranas. Tal y como se puede observar en la tabla, se ha detectado un fuerte descenso de
la actividad calcio-ATPasa en el músculo distrófico, además de un aumento de las actividades
de las magnesio-ATPasa y Sodio/potasio-ATPasa.
Las primeras son cana-
les o bombas de calcio
que están presentes
en la membrana mus-
cular, pero no en las
demás. Las segundas
son canales o bombas
que generalmente se
presentan en mayor
medida en la mem-
brana plasmática.
Por lo tanto, podemos concluir que las membranas derivadas del músculo distrófico contienen
más membrana plasmática que el músculo normal, lo que se correlaciona con el mayor conte-
nido de colesterol.
Pero tenemos que considerar que esta medición se está realizando sobre el conjunto de mem-
branas de la célula por lo que es necesario realizar un fraccionamiento de los microsomas con
el fin de separarlas. Este fraccionamiento se realiza sometiendo la muestra de microsomas a
un gradiente continuo de sacarosa (25%-50%) durante toda la noche.
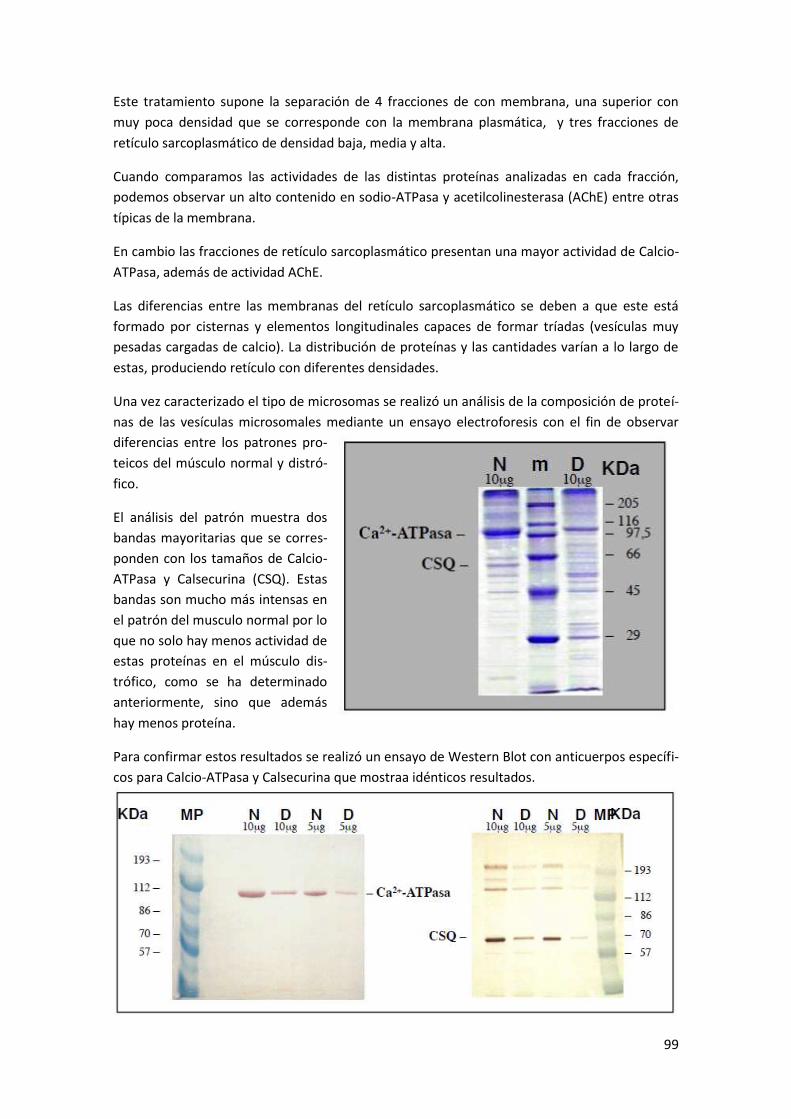
99
Este tratamiento supone la separación de 4 fracciones de con membrana, una superior con
muy poca densidad que se corresponde con la membrana plasmática, y tres fracciones de
retículo sarcoplasmático de densidad baja, media y alta.
Cuando comparamos las actividades de las distintas proteínas analizadas en cada fracción,
podemos observar un alto contenido en sodio-ATPasa y acetilcolinesterasa (AChE) entre otras
típicas de la membrana.
En cambio las fracciones de retículo sarcoplasmático presentan una mayor actividad de Calcio-
ATPasa, además de actividad AChE.
Las diferencias entre las membranas del retículo sarcoplasmático se deben a que este está
formado por cisternas y elementos longitudinales capaces de formar tríadas (vesículas muy
pesadas cargadas de calcio). La distribución de proteínas y las cantidades varían a lo largo de
estas, produciendo retículo con diferentes densidades.
Una vez caracterizado el tipo de microsomas se realizó un análisis de la composición de proteí-
nas de las vesículas microsomales mediante un ensayo electroforesis con el fin de observar
diferencias entre los patrones pro-
teicos del músculo normal y distró-
fico.
El análisis del patrón muestra dos
bandas mayoritarias que se corres-
ponden con los tamaños de Calcio-
ATPasa y Calsecurina (CSQ). Estas
bandas son mucho más intensas en
el patrón del musculo normal por lo
que no solo hay menos actividad de
estas proteínas en el músculo dis-
trófico, como se ha determinado
anteriormente, sino que además
hay menos proteína.
Para confirmar estos resultados se realizó un ensayo de Western Blot con anticuerpos específi-
cos para Calcio-ATPasa y Calsecurina que mostraa idénticos resultados.

100
Las balsas lipídicas son elementos de membrana ricos en colesterol y esfingolípidos, que son
insolubles en detergentes. Por ello, si homogeneizamos la membrana muscular en un medio
con tritón-X100 y se centrifuga en un gradiente de sacarosa, las balsas lipídicas quedan sobre
el gradiente, viajando a través de todo este.
La membrana del músculo normal muestra un solo tipo de balsas lipídicas, mientras que en
músculo distrófico encontramos dos. Si ahora analizamos las diferentes fracciones del gradien-
te, concretamente la presencia de caveolina (proteína estructural de rafts), podemos observar
como en el músculo distrófico hay muchas más poblaciones de rafts.
Por lo tanto, la patología distrófica no solo afecta a los perfiles proteicos, sino también a la
composición lipídica de las membranas en relación a la mayor necesidad de reparaciones que
precisa.
ISOLATION OF LIPID RAFTS, AS TRITON X-100-RESISTANT MEMBRANES, FROM MUSCLES OF NORMAL AND DYSTROPHIC Lama2dyMICE
Caveolin 3
C.J. Vidal: Targeting of AChE to lipid rafts
As for dystrophin, the deficiency of laminin-2 in Lama2dy mice produces an increase in caveolin-3 and, therefore, of caveolae in the sarcolemma
Normal Muscle Dystrophic Muscle
Tube Top

101
Tema 6: Retí cúlo endoplasma tico II
Síntesis de proteínas e inserción en la membrana El retículo endoplasmático no es una entidad estanca, sino que en función del estado celular
va a presentar un mayor o menor desarrollo, relacionado con la producción de proteínas.
El acoplamiento de las subunidades catalíticas y reguladoras de una proteína también se pro-
duce a nivel del retículo endoplasmático.
La importancia del retículo endoplasmático para la síntesis de proteínas es vital, ya que se es-
tima que alrededor del 30% de todas las proteínas (30.000) son sintetizadas en ribosomas
asociados al retículo.
1 Transporte al retículo endoplasmático
1.1 El péptido líder o péptido señal Para que una proteína se sintetizada en el retículo endoplasmático precisa portar una etiqueta
conocida como péptido líder o péptido señal cuya función es asociar el ribosoma que comien-
za la traducción al retículo endoplasmático rugoso.
El descubrimiento de esta secuencia de aminoácidos se produjo en los años 70, cuando los
experimento de ingeniería genética permitieron la síntesis de globinas quiméricas con secuen-
cias de la -galactosidasa de Escherichia coli. Este experimento reveló que la introducción de
esas secuencias dirigían a la globina hacia el retículo, por lo que permitía la asociación de los
ribosomas a las membranas y la transferencia desde un espacio celular a otro.
El experimento clave para comprender la importancia del péptido señal consistió en introducir
el mensajero de una proteína que tiene que ingresar en el retículo, los ribosomas y los mate-
riales necesarios para la síntesis de proteínas en un tubo de ensayo. Tras la síntesis se añadió la
fracción microsomal que contenía el retículo y se determinó el tamaño de la proteína.
Cuando una rata se empieza a intoxicar con barbital se activa el sistema del citocromo p450,
por lo que el retículo se engrosa por la inducción en la síntesis de proteínas. En el caso de los
linfocitos B, su estimulación para la producción de anticuerpos también hace aumentar de
tamaño el retículo, ya que la inmunoglobulina va a adquirir su estructura terciaria en el re-
tículo y va a pasar al Golgi, donde se va a obtener la glicosilación definitiva.

102
Al mismo tiempo, en otro tubo de ensayo se situaron los mismos componentes, pero añadien-
do la fracción microsomal desde el principio, y se midió el tamaño de la proteína.
Los resultados indicaron que la proteína del primer ensayo era más grande que la del segundo,
esto se debe a que al no ingresar la proteína en el retículo, no se corta el péptido señal. En el
segundo caso se produjo la entrada de la proteína al retículo donde inmediatamente se le cor-
to el péptido.
La caracterización del péptido líder que dirige a las proteínas al lumen del retículo endoplas-
mático determinó que se trataba de una secuencia N-terminal con un tamaño de 25-35 ami-
noácidos, cuya naturaleza se correspondía con la organización de un núcleo hidrofóbico que
ocupa entre 10-15 aminoácidos. Esta secuencia es retirada de la proteína al entrar al lumen.
En la actualidad conocemos dos tipos de señales que mandan una proteína al retículo; Por un
lado está la secuencia de importación el retículo, que es el péptido líder mencionado cuya se-
cuencia puede ser:
+H3N-Met-Met-Ser-Phe-Val-Ser-Leu-Leu-Leu-Val-Gly-Ile-Leu-Phe-Trp-Ala-Thr-Glu-Ala-Glu-Gln-Leu-Thr-Lys-Cys-Gly-Val-Phe-Gln-----
Pero también podemos encontrar una secuencia especial de retorno al retículo para aquellas
proteínas que son transportadas en las cisternas hacia el Golgi y tienen que regresar, ya que el
transporte es bidireccional. La secuencia se denomina KDEL y se sitúa en el extremo C-terminal
de todas las proteínas solubles del retículo, para que regresen a este si van al Golgi o si han
escapado al citosol. Cuando las proteínas son de membrana la secuencia es KKXX.
----Lys-Asp-Glu-Leu-COO- (--KDEL)

103
Aunque las dimensiones del péptido líder son muy variables a lo largo de la escala evolutiva,
presenta siempre una división en tres secciones y una serie de características en común:
- Comienza siempre con una metionina (M).
- En la primera sección N-terminal presenta un aminoácido con carga positiva como la
arginina (R) o la lisina (K).
- La segunda sección consta de aminoácidos muy hidrofóbicos como leucina (L) y trip-
tófano (W).
- La tercera sección, muy próxima al punto de corte de la secuencia señal, presenta
siempre un aminoácido muy pequeño como glicina (G), alanina (A) o serina (S) en una
posición inmediatamente anterior al lugar de corte.
1.2 Asociación del ribosoma al retículo endoplasmático La traducción del mensajero que contiene una secuencia señal para el retículo comienza como
la de cualquier otro. Los 30 primeros aminoácidos elongados ya portan el péptido señal, pero
el entorno del túnel del ribosoma los mantienen extendidos hasta que cuando se han traduci-
do los 70-80 primeros aminoácidos, la cadena polipeptídica comienza a salir del túnel al en-
torno citosólico y se pliega espontáneamente para formar el núcleo hidrofóbico.
La aparición del núcleo hidrofóbico (motivo estructural), en el extremo N-terminal de la cade-
na polipeptídica en crecimiento, es la señal necesaria para que el conjunto sea reconocido por
la partícula de reconocimiento de la señal (SRP), que se une al polipéptido e interacciona con
el ribosoma, deteniendo la traducción.
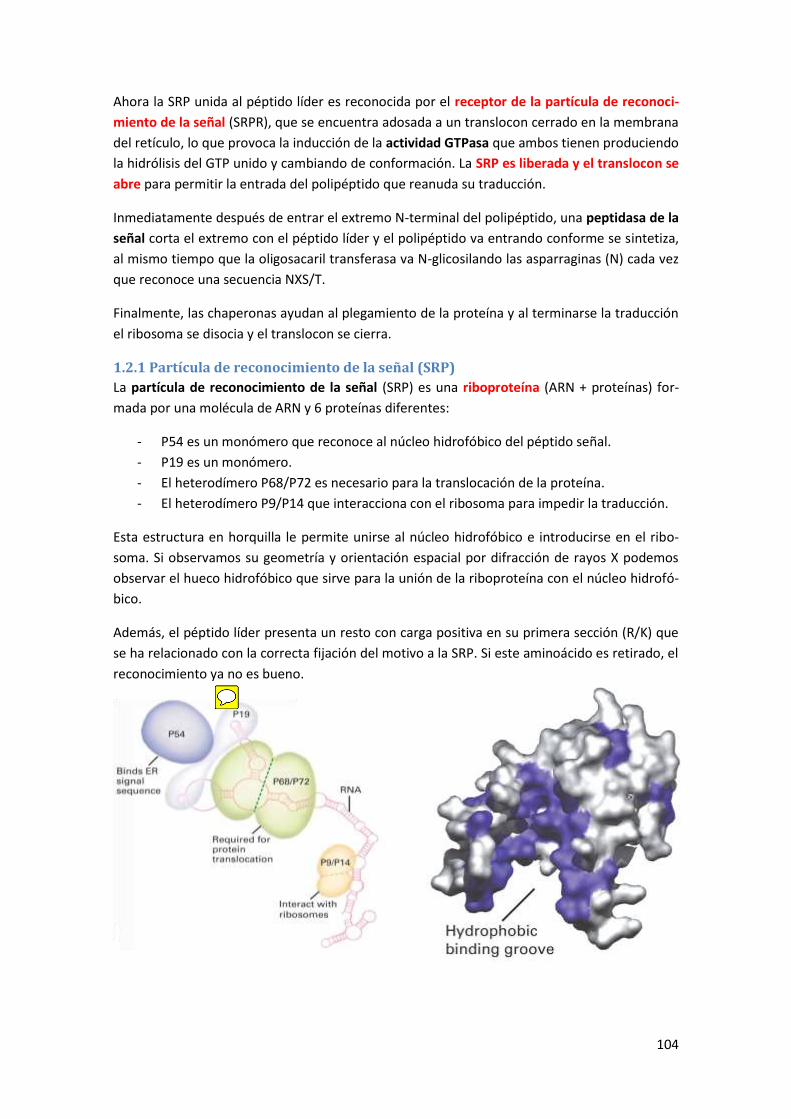
104
Ahora la SRP unida al péptido líder es reconocida por el receptor de la partícula de reconoci-
miento de la señal (SRPR), que se encuentra adosada a un translocon cerrado en la membrana
del retículo, lo que provoca la inducción de la actividad GTPasa que ambos tienen produciendo
la hidrólisis del GTP unido y cambiando de conformación. La SRP es liberada y el translocon se
abre para permitir la entrada del polipéptido que reanuda su traducción.
Inmediatamente después de entrar el extremo N-terminal del polipéptido, una peptidasa de la
señal corta el extremo con el péptido líder y el polipéptido va entrando conforme se sintetiza,
al mismo tiempo que la oligosacaril transferasa va N-glicosilando las asparraginas (N) cada vez
que reconoce una secuencia NXS/T.
Finalmente, las chaperonas ayudan al plegamiento de la proteína y al terminarse la traducción
el ribosoma se disocia y el translocon se cierra.
1.2.1 Partícula de reconocimiento de la señal (SRP)
La partícula de reconocimiento de la señal (SRP) es una riboproteína (ARN + proteínas) for-
mada por una molécula de ARN y 6 proteínas diferentes:
- P54 es un monómero que reconoce al núcleo hidrofóbico del péptido señal.
- P19 es un monómero.
- El heterodímero P68/P72 es necesario para la translocación de la proteína.
- El heterodímero P9/P14 que interacciona con el ribosoma para impedir la traducción.
Esta estructura en horquilla le permite unirse al núcleo hidrofóbico e introducirse en el ribo-
soma. Si observamos su geometría y orientación espacial por difracción de rayos X podemos
observar el hueco hidrofóbico que sirve para la unión de la riboproteína con el núcleo hidrofó-
bico.
Además, el péptido líder presenta un resto con carga positiva en su primera sección (R/K) que
se ha relacionado con la correcta fijación del motivo a la SRP. Si este aminoácido es retirado, el
reconocimiento ya no es bueno.

105
La SRP es una riboproteína con actividad GTPasa que tiene un punto isoeléctrico muy alto
(carga positiva a pH fisiológico), esto se debe a que precisa interaccionar con polinucleótidos
(que tienen carga negativa a pH fisiológico), al igual que las histonas.
1.2.2 Componentes de los translocones y proteínas asociadas
Aunque todo parece indicar que existe más de una clase de translocón en el retículo, se ha
confirmado que el translocón Sec 61 participa en el transporte co-traduccional. Esta relación
se estableció a partir de la utilización de agentes que causaban crosslinking químico entre el
mensajero y el translocon, dando lugar a enlaces covalentes que paralizaban el proceso, lo que
permitía su observación.
Si observamos su tamaño relativo con respecto al ribosoma podemos hacernos una idea de lo
grande que es el ribosoma. Estas proteínas del translocon Sec 61 presentan una gran homolo-
gía a lo largo de la escala evolutiva.
El translocón Sec61, al menos a nivel del retículo endoplasmático, está formado por 4 unidades
formadas por tres péptidos denominados Sec61, Sec61 y Sec61, por tanto doce péptidos
en total que se organizan para formar un barril que permite la existencia del canal.
Otro conjunto de proteínas que pueden formar el translocón, esta vez implicadas en el trans-
porte post-traduccional son la Sec 62, 63, 71, 72 y Ssh1. Estas proteínas mantienen una gran
homología entre ellas.
Cuando una cadena polipeptídica presenta en su extremo N-terminal un péptido líder, este es
reconocido por el complejo del translocón (doble sentido) y es introducido al lumen del retícu-
lo donde se le retira la señal, a menos de que esta se encuentre interna en la secuencia.
Hasta ahora se pensaba que aquello que
entraba en el retículo ya no podía aban-
donarlo, pero esto se ha desmentido ya
que depende del translocón que partici-
pa, por ejemplo en el caso de ERAD. En
cualquier caso, para que una proteína
entre en el retículo mediante transporte
post-traducional es necesario que algo
tire de ella. Las chaperonas BiP son pro-
teínas que, mediante el gasto de ATP, van

106
a pivotar entre conformación de alta y baja afinidad para tirar de la proteína e introducirla en
el lumen del retículo.
Asociado a cualquiera de los dos transloco-
nes se encuentran también las siguientes
proteínas:
- Proteína de membrana asociada a la
cadena en translocación (TRAM,
Translocating chain-associated mem-
brane protein).
- Proteína asociada a la translocación
(TRAP, Translocation-associated pro-
tein) que está formada por cuatro
subunidades () que puede actuar
como una tapadera (LD).
- Complejo oligosacariltransferasa.
- Peptidasa de la señal.
1.3 Transporte post-traduccional en Escherichia coli El transporte post-traduccional en mamíferos es muy similar al que se produce en bacterias. El
translocon de E. coli está formado por 4 subunidades SecA/Y/E/G, donde SecA tiene una fun-
ción análoga a la chaperona BiP, es decir, presenta conformación de alta afinidad unida a ATP y
baja afinidad cuando no lo tiene.
El proceso comienza con el reconocimiento de la señal en la proteína por SecA que estimula la
entrada de ATP y el cambio a la conformación de alta afinidad que agarra y empuja la proteína
hacia el translocon.
Cuando se desarrolla la actividad ATPasa de SecA se produce el cambio de afinidad y se libera a
la proteína para iniciar un nuevo empuje.
1.4 Respuesta a proteínas mal plegadas (UPR) La existencia del control de calidad de las proteínas en el retículo endoplasmático obliga a que
estas estén perfectamente plegadas en su estructura tridimensional óptima antes de partir al
aparato de Golgi.
Ribosome–translocon complexes. (a) Scheme with a
ribosome–translocon complex, illustrating the switching
station function of the translocon. Soluble (secreted)
proteins pass through the translocon assembly, whereas
TM segments of membrane proteins are shunted by
the translocon into the membrane lipid bilayer. (b) Cryo-
EM images of the canine ribosome–translocon (Sec61)
complex with and without TRAP. The small and large
ribosomal subunits are indicated by S and L,
respectively. The luminal domain of TRAP is indicated
by LD. Images 1 and 3 show reconstructions without
TRAP (i.e. with the Sec61 assembly).
Note the apparent hole in the Sec61 assembly in panel 3,
which has been assumed, until recently, to be an aqueous
channel through which the elongating polypeptide
passes. Ménétret et al. suggest that the hole is actually
only a depression due to the packing of a dimer of Sec61
dimers . Images 2 and 4 show reconstructions that
include the TRAP complex as well as the Sec61
assembly.

107
Este control de calidad va a depender en gran medida de los niveles de calcio, ya que el retícu-
lo es uno de los de los depósitos más importantes para la homeostasis del calcio.
La respuesta a proteínas mal plegadas (UPR, Unfolded protein response) es el sistema que
tiene la célula a modo de alarma ante una aumento de proteínas mal plegadas en el retículo
endoplasmático.
El estudio profundo de este tipo de alarmas ha revelado la existencia de tres vías de señaliza-
ción ante el aumento de proteínas mal plegadas en el lumen del retículo endoplasmático:
- En la membrana del retículo hay unos receptores Ire con un extremo citosólico y un
extremo mirando al lumen que en condiciones normales (bajo número de proteínas
mal plegadas) se encuentran unidos a la chaperona BiP.
Cuando la cantidad de proteínas mal plegadas aumenta, las cantidad de chaperonas
BiP unidas al receptor disminuye, porque estas son necesarias para colaborar con el
plegamiento, y esto provoca la dime-
rización de los receptores que sufren
una fosforilación cruzada por su acti-
vidad quinasas en el lumen, y comien-
zan a desarrollar una actividad nu-
cleasa en el dominio citosólico.
En el citosol hay un mRNA inmaduro
(no competente para la traducción)
que gracias a la acción nucleasa de los
receptores es cortado y empalmado
(Splicing) comenzando la traducción
de factores de transcripción de la ex-
presión de chaperonas que van al nú-
cleo.
El retículo es un lugar donde hay una gran concentración de chaperonas, ya que es el lu-
gar donde se produce el plegamiento de la proteína, y en muchos casos las chaperonas
que deciden si una proteína puede o no partir al Golgi van a ser ligantes de calcio. Dos
chaperonas que van a ser muy importantes para el correcto plegado de las proteínas son:
- Disulfuro isómeras (DSI) cuya función consiste en establecer los enlaces disulfuro
correctos entre las cisteínas (C) para que una proteína adquiera su conformación
final. Este proceso sucede mediante prueba y error hasta que la proteína alcanza
el punto de energía interna mínima o máxima estabilidad.
- Cis-Trans prolil isomerasa es una enzima cuya función es la de romper la prolina
(P) y voltearla. La prolina (P) es un aminoácido con un gran anillo pentagonal rígido
que puede plantear problemas estéricos a la hora de que una proteína adquiera
conformaciones helicoidales, por ello en muchas ocasiones se hace necesaria la
isomerización de Cis a Tans mediante la ruptura del enlace.

108
- La activación de otros tipos de receptores llevan a un aumento de factores de tras-
cripción asociados a la expresión de las proteínas del ERAD que dirige a los péptidos
mal plegados al citosol donde son degradados por el proteasoma.
- Otros receptores sensibles a la cantidad de proteínas mal plegadas producen la fosfori-
lación de factores de traducción que detienen o ralentizan la síntesis de proteínas. En
función del nivel de fosforilación se ralentizará más la traducción.
Si después de activarse estas alarmas la situación no varía la célula activa la vía apoptótica
como se produce en el caso de la -sinucleina o cuando la cantidad de proteínas -amiloide va
aumentando, formando fibras y agregados que disparan la apoptosos con la consecuente pér-
dida de neuronas colinérgicas y el desarrollo de Alzheimer.
1.5 Topología de las proteínas de membrana Uno de los grandes problemas de la biología molecular fue la topología de las proteínas de
membrana. Se precisaba un modelo para explicar como estas proteínas se disponían en la
membrana quedando con los extremos hacia un lado u otro si el primer extremo en entrar al
retículo era siempre el N-terminal.
Atendiendo a su disposición en la membrana podemos clasificar a las proteínas de membrana
en cuatro clases fundamentales:
- Las proteínas de tipo I (Glicoforina, LDLR, proteína HA de la gripe, receptor de insulina,
receptor de hormona del crecimiento) son aquellas que presentan el extremo N-
terminal en el lumen del retículo, del Golgi, o en el espacio extracelular (considere el
transito de proteínas a través de los compartimentos celulares), y el extremo C-
terminal en el citosol. Estas proteínas han perdido su péptido líder.
- Las proteínas de tipo II (receptor de asialoglicoproteina, receptor de transferrina, pre-
cursor de sacarosa-isomaltosa, galactosiltransferasa del Golgi, sialiltransferasa del Gol-
gi y proteína HN de la gripe) son aquellas que presentan el dominio N-terminal en el
citosol, mientras que el dominio C-terminal se ha introducido en el compartimento
celular o en el exterior celular.
- Las proteínas de tipo III (Citocromo P450) son aquellas cuyo extremo N-terminal ape-
nas entra en lumen del compartimento celular en el que se encuentre, presentando la
parte mayoritaria de la proteína en la sección C-terminal externa.
- Las proteínas de tipo IV (Receptores acoplados de proteínas G, transportadores de
glucosa, canales de calcio dependientes de voltaje, bombas de moléculas pequeñas
ABC, canal CFTR de cloruro, translocon Sec61, conexina) son aquellas que atraviesan
la bicapa en varias ocasiones quedando el extremo C-terminal fuera cuando el núme-
ro de veces es impar o dentro cuando es par, en cuyo caso quedan ambos extremos en
la misma cara.
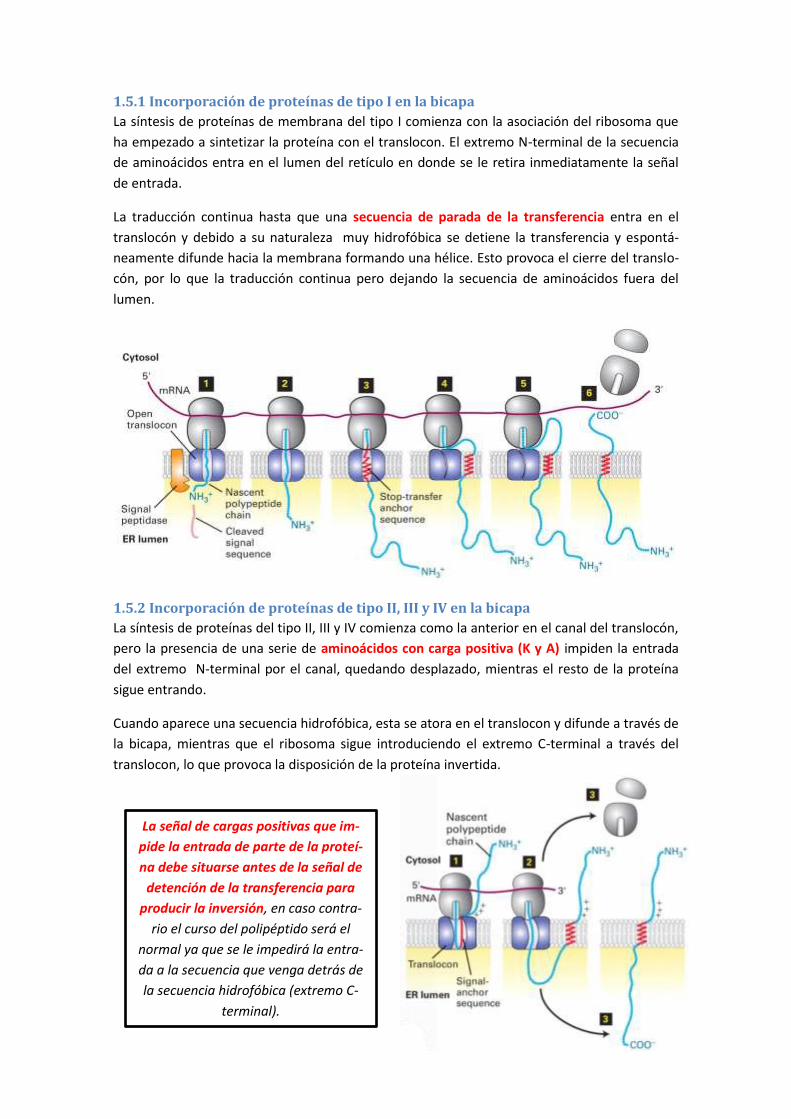
109
1.5.1 Incorporación de proteínas de tipo I en la bicapa
La síntesis de proteínas de membrana del tipo I comienza con la asociación del ribosoma que
ha empezado a sintetizar la proteína con el translocon. El extremo N-terminal de la secuencia
de aminoácidos entra en el lumen del retículo en donde se le retira inmediatamente la señal
de entrada.
La traducción continua hasta que una secuencia de parada de la transferencia entra en el
translocón y debido a su naturaleza muy hidrofóbica se detiene la transferencia y espontá-
neamente difunde hacia la membrana formando una hélice. Esto provoca el cierre del translo-
cón, por lo que la traducción continua pero dejando la secuencia de aminoácidos fuera del
lumen.
1.5.2 Incorporación de proteínas de tipo II, III y IV en la bicapa
La síntesis de proteínas del tipo II, III y IV comienza como la anterior en el canal del translocón,
pero la presencia de una serie de aminoácidos con carga positiva (K y A) impiden la entrada
del extremo N-terminal por el canal, quedando desplazado, mientras el resto de la proteína
sigue entrando.
Cuando aparece una secuencia hidrofóbica, esta se atora en el translocon y difunde a través de
la bicapa, mientras que el ribosoma sigue introduciendo el extremo C-terminal a través del
translocon, lo que provoca la disposición de la proteína invertida.
Incorporation of transmembrane proteins in the bilayer
La señal de cargas positivas que im-
pide la entrada de parte de la proteí-
na debe situarse antes de la señal de
detención de la transferencia para
producir la inversión, en caso contra-
rio el curso del polipéptido será el
normal ya que se le impedirá la entra-
da a la secuencia que venga detrás de
la secuencia hidrofóbica (extremo C-
terminal).

110
Alternancia de secuencias para la producción de los diferentes tipos de proteínas
A partir de las combinaciones de secuencias hidrofóbicas, aminoácidos con carga positivas y
secuencias de parada de la transferencia podemos establecer como queda cada fracción de
una proteína de membrana según su tipo.
El estudio de estos motivos en las proteínas se realizó a través de el establecimiento de los
perfiles de hidrofobicidad de una proteína. Estos se establecen a partir de una secuencia de
aminoácidos sobre la que se va calculando (relativamente) cual es el grado de hidrofobicidad
de los aminoácidos y del entorno que van generando en la secuencia.
La representación de esos valores en un grafica otorga altos valores a aquellos lugares en los
que es muy probable que contenga una de estas regiones transmembrana que quedan atora-
das en los translocones.

111
Tema 7: Glicosilacio n de proteí nas en el retí cúlo en-doplasma tico y en las cisternas del Golgi
La Glicosilación es el proceso de adición de restos de azúcar a las proteínas, pasando a deno-
minarse glicoproteínas. Se trata de un proceso que se produce en el retículo endoplasmático,
en el Golgi y en el citosol.
1 Las glicoproteínas Las glicoproteínas son proteínas que tienen unidos, de modo covalente, uno o varios hidratos
de carbono (oligosacáridos generalmente). A diferencia de los proteoglicanos, la proporción de
azúcares es mucho menor.
Existen muchos tipos de glicoproteínas
que pueden ser de membrana o de
secreción:
- Las glicoproteínas de membra-
na desempeñan funciones rela-
cionadas con la adhesión y el
reconocimiento intercelular. En
la mayor parte de los casos la
Glicosilación de las proteínas de
membrana se producirá en el
dominio extracelular de la
membrana.
- Las glicoproteínas de secreción
son solubles y representan la
mayor parte de las proteínas
del plasma sanguíneo.
1.1 Funciones de la glicosilación de proteínas Los azúcares de las glicoproteínas desempeñan varias funciones destacadas, en muchos casos
por si como hidratos de carbono, y en otros por ser reconocidos mediante una proteína (lecti-
na) de modo específico. Las lectinas son proteínas que reconocen específicamente azúcares, al
mismo tiempo que tienen una función proteica.
Los hidratos de carbono son moléculas hidrofílilicas que van a quedar proyectadas hacia la
superficie de la proteína de una manera móvil (enlaces no fijos). Esto les permite ejercer mu-
chas funciones como:
- Proteger de la proteólisis, ya que los hidratos de carbono no son sustrato de la pro-
teasa. Por ejemplo, la fibronectina glicosilada de la lámina basal es más resistente a la
acción de proteasas.
- Ayudar al plegamiento correcto como función estructural ya que los hidratos de car-
bono N-unidos son añadidos durante la síntesis de la cadena polipeptídica determi-

112
nando en gran medida el modo de plegamiento de la proteína. Los oligosacáridos O-
unidos pueden estabilizar la conformación plegada de una proteína disminuyendo su
libertad conformacional o reduciendo su flexibilidad. Los oligosacáridos también son
marcadores del mecanismo de plegamiento en el retículo, asegurando que las
proteínas no abandonen el retículo
hasta que estén plegadas correc-
tamente (Ciclo calsecuestri-
na/calreticulina).
- Direccionamiento a su destino,
por ejemplo, los restos de mano-
sa-6-fosfato dirigen a la proteína
a los lisosomas.
- Determinar la estabilidad en la
célula (p53)
- Determinar la permanencia en la
sangre (Ceruloplasmina)
- Reconocimiento en superficie ce-
lular (Leucocitos, espermatozoi-
des, bacterias, virus, toxinas).
2 La glicosilación de proteínas
2.1 Introducción a los glúcidos Antes de comenzar con el proceso de glicosilación realicemos un repaso observando la tabla
en la que se muestran sus nombres y la representación utilizada.
Se ha creado una simbología estándar para la representación de azúcares:
- Las hexosas se representan con
círculos.
- Las N-acetilhexosaminas con un
cuadrado.
- Las hexosaminas con un cua-
drado divido diagonalmente en
dos colores.
- Los ácidos urónicos también se
representan con un cuadrado
divido.
El color determina de que azúcar han derivado, por ejemplo, la glucosa se representa con color
azul, por lo que sus derivados tendrán color azul. Además también encontramos los ácidos
siálicos como el ácido N-acetilneuramínico.
Recordemos también que las aldohexosas son azúcares de seis carbonos epímeros de la gluco-
sa, como la manosa y la galactosa. Las aldopentosas como la xilosa son azúcares de cinco car-
bonos epímeros de la ribosa.
Glicosilación de proteínas
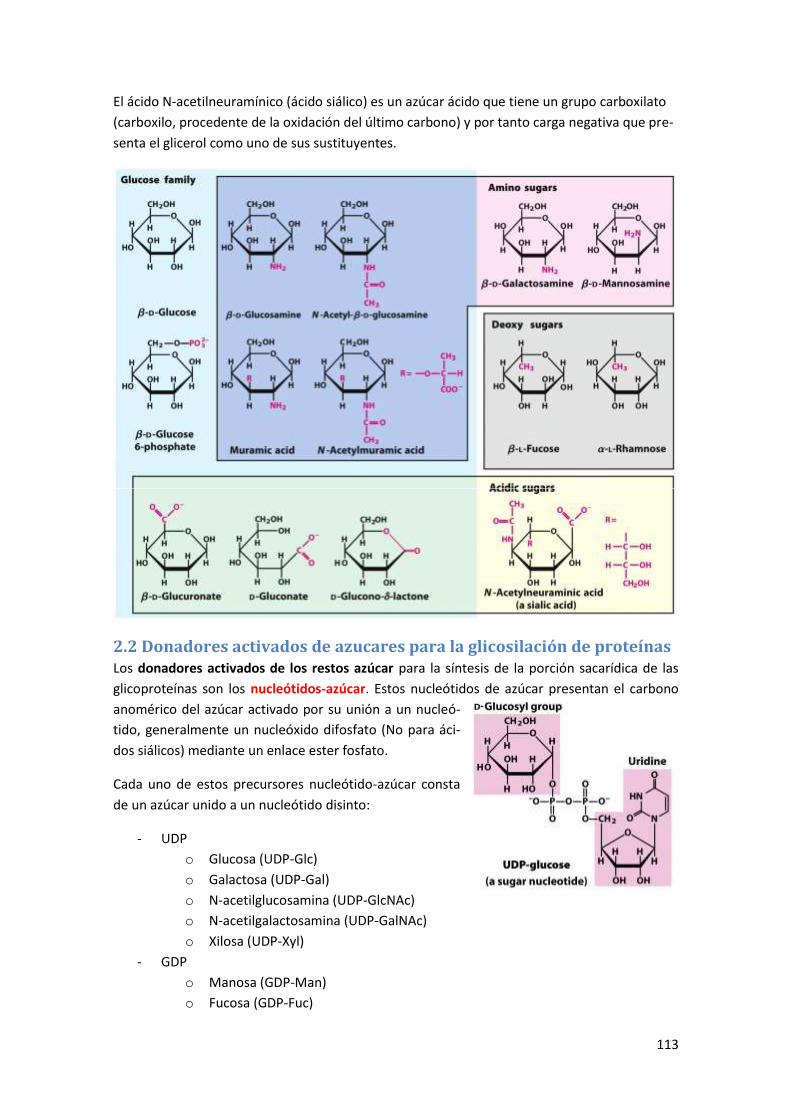
113
El ácido N-acetilneuramínico (ácido siálico) es un azúcar ácido que tiene un grupo carboxilato
(carboxilo, procedente de la oxidación del último carbono) y por tanto carga negativa que pre-
senta el glicerol como uno de sus sustituyentes.
2.2 Donadores activados de azucares para la glicosilación de proteínas Los donadores activados de los restos azúcar para la síntesis de la porción sacarídica de las
glicoproteínas son los nucleótidos-azúcar. Estos nucleótidos de azúcar presentan el carbono
anomérico del azúcar activado por su unión a un nucleó-
tido, generalmente un nucleóxido difosfato (No para áci-
dos siálicos) mediante un enlace ester fosfato.
Cada uno de estos precursores nucleótido-azúcar consta
de un azúcar unido a un nucleótido disinto:
- UDP
o Glucosa (UDP-Glc)
o Galactosa (UDP-Gal)
o N-acetilglucosamina (UDP-GlcNAc)
o N-acetilgalactosamina (UDP-GalNAc)
o Xilosa (UDP-Xyl)
- GDP
o Manosa (GDP-Man)
o Fucosa (GDP-Fuc)
Glicosilación de proteínas
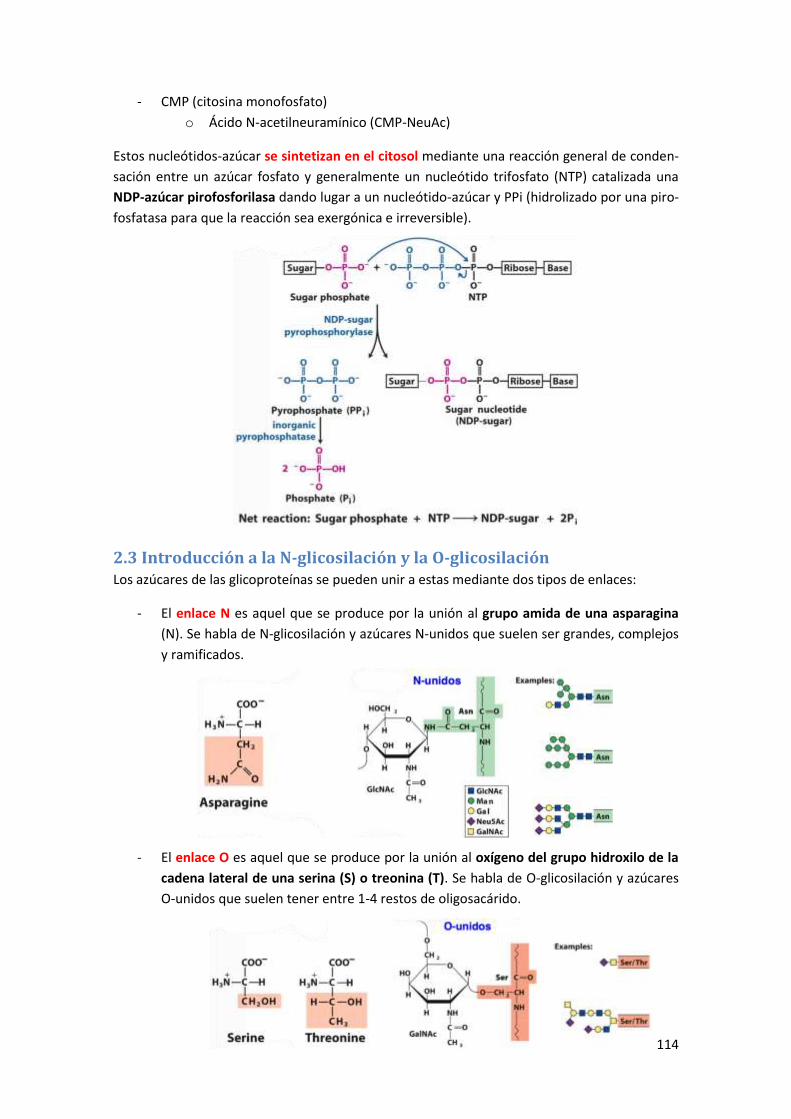
114
- CMP (citosina monofosfato)
o Ácido N-acetilneuramínico (CMP-NeuAc)
Estos nucleótidos-azúcar se sintetizan en el citosol mediante una reacción general de conden-
sación entre un azúcar fosfato y generalmente un nucleótido trifosfato (NTP) catalizada una
NDP-azúcar pirofosforilasa dando lugar a un nucleótido-azúcar y PPi (hidrolizado por una piro-
fosfatasa para que la reacción sea exergónica e irreversible).
2.3 Introducción a la N-glicosilación y la O-glicosilación Los azúcares de las glicoproteínas se pueden unir a estas mediante dos tipos de enlaces:
- El enlace N es aquel que se produce por la unión al grupo amida de una asparagina
(N). Se habla de N-glicosilación y azúcares N-unidos que suelen ser grandes, complejos
y ramificados.
- El enlace O es aquel que se produce por la unión al oxígeno del grupo hidroxilo de la
cadena lateral de una serina (S) o treonina (T). Se habla de O-glicosilación y azúcares
O-unidos que suelen tener entre 1-4 restos de oligosacárido.

115
Esto diferencia a las glicoproteínas de los proteoglicanos que presentan largas cadenas de poli-
sacárido O-unidas.
Vemos el ejemplo de la glicoforina glicosilada (N y O-glicosilada) que presenta todos los restos
de glicosilación en el dominio externo de membrana.
Otro ejemplo de la importancia de la glicosilación es la eritropoyetina (EPO), una hormona
secretada por los riñones cuya función es la estimulación de la producción de eritrocitos. La
EPO es una glicoproteína de 165 aminoácido que presenta N-glicosilación en tres restos de
asparagina (N) y O-glicosilación en una serina (S). La porción de hidratos de carbono represen-
ta un 40% de su peso y su función es la de evitar que la hormona sea retirada prematuramente
de la sangre.
La EPO se utiliza en clínica
para tratar anemias provoca-
das por la quimioterapia con-
tra el cancer. Se obtiene como
un recombinante para uso
terapéutico en un tipo celular
capaz de glicosilar.
También es usada en el mun-
do del deporte ya que al au-
mentar el hematocrito, au-
menta la capacidad de trans-
porte de oxígeno en la sangre
y el rendimiento deportivo,
pero se puede distinguir la
EPO natural de la recombinan-
te analizando el patrón de
glicosilación.
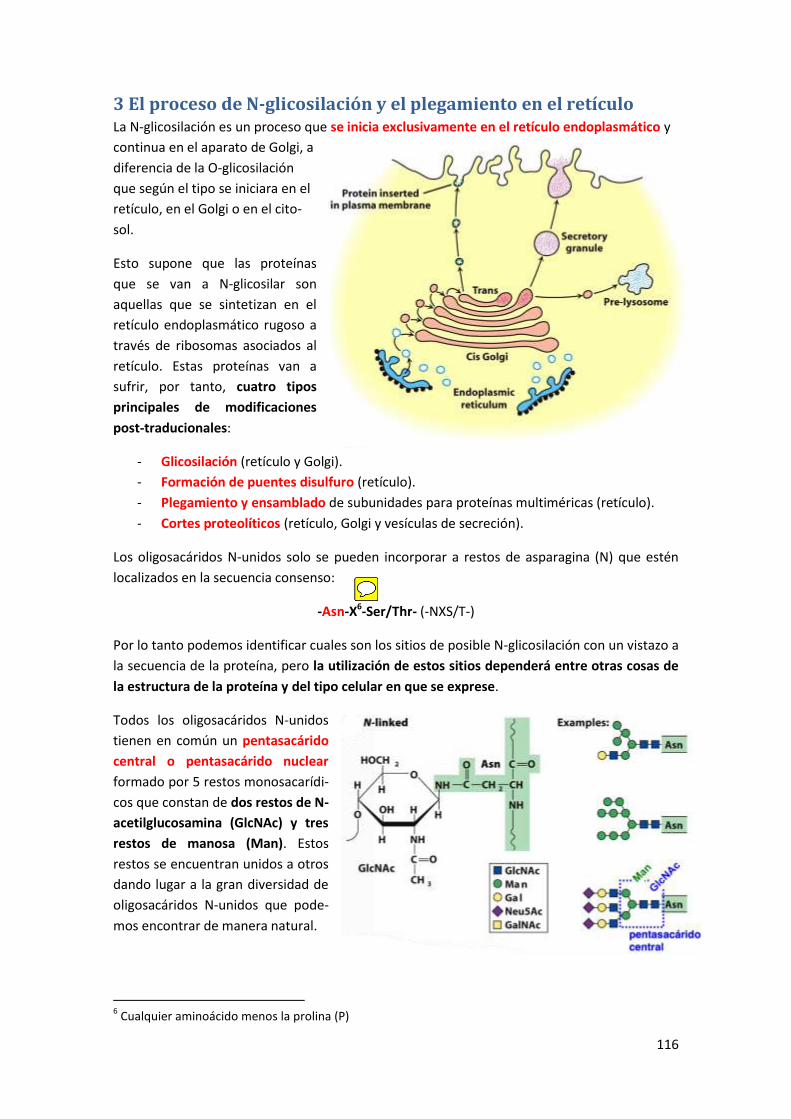
116
3 El proceso de N-glicosilación y el plegamiento en el retículo La N-glicosilación es un proceso que se inicia exclusivamente en el retículo endoplasmático y
continua en el aparato de Golgi, a
diferencia de la O-glicosilación
que según el tipo se iniciara en el
retículo, en el Golgi o en el cito-
sol.
Esto supone que las proteínas
que se van a N-glicosilar son
aquellas que se sintetizan en el
retículo endoplasmático rugoso a
través de ribosomas asociados al
retículo. Estas proteínas van a
sufrir, por tanto, cuatro tipos
principales de modificaciones
post-traducionales:
- Glicosilación (retículo y Golgi).
- Formación de puentes disulfuro (retículo).
- Plegamiento y ensamblado de subunidades para proteínas multiméricas (retículo).
- Cortes proteolíticos (retículo, Golgi y vesículas de secreción).
Los oligosacáridos N-unidos solo se pueden incorporar a restos de asparagina (N) que estén
localizados en la secuencia consenso:
-Asn-X6-Ser/Thr- (-NXS/T-)
Por lo tanto podemos identificar cuales son los sitios de posible N-glicosilación con un vistazo a
la secuencia de la proteína, pero la utilización de estos sitios dependerá entre otras cosas de
la estructura de la proteína y del tipo celular en que se exprese.
Todos los oligosacáridos N-unidos
tienen en común un pentasacárido
central o pentasacárido nuclear
formado por 5 restos monosacarídi-
cos que constan de dos restos de N-
acetilglucosamina (GlcNAc) y tres
restos de manosa (Man). Estos
restos se encuentran unidos a otros
dando lugar a la gran diversidad de
oligosacáridos N-unidos que pode-
mos encontrar de manera natural.
6 Cualquier aminoácido menos la prolina (P)
Glicosilación de proteínas

117
3.1 La formación del oligosacárido precursor El proceso de la N-glicosilación comienza con la síntesis de un oligosacárido de 14 restos, de-
nominado oligosacárido precursor u oligosacárido nuclear (core), sobre el dolicol fosfato, que
una vez completo es transferido en bloque a la proteína. El oligosacárido precursor está con-
servado en eucariotas en general (plantas, animales y hongos).
Este oligosacárido de 14 restos está formado por dos restos de N-acetilglucosamina unidas por
enlaces 1,4, nueve restos de manosa unidas por enlaces 1,2 1,3 y 1,6, y tres restos de
glucosa unidos por enlaces 1,2 y 1,3. Generalmente se abrevia la composición como G3M9.
Si analizamos estructuralmente el oligosacárido lo más cercano a la proteína son los dos restos
de N-acetilglucosamina (GlcNAc a y b) unidos por enlaces 1,4. A continuación la primera ma-
nosa (c) es un punto de bifurcación del que parten dos ramas:
- La rama A con la siguiente
manosa (d) unida por enlace
1,3 y que termina con los
tres restos de glucosa (l, m y
n).
- Las ramas B y C que parten de
la siguiente manosa (e) unida
por enlace 1,6. Esta manosa
(e) es un nuevo punto de bi-
furcación que da lugar a la
rama B por enlace 1,3 a la
siguiente manosa (h), y la ra-
ma C por enlace 1,6 a la si-
guiente manosa (j).
Los enlaces entre las manosas no ramificadas son 1,2, las primeras dos glucosas de la rama A
están unidas por enlace 1,3 (sensibles a glucosidasa II) y la última a través de enlace 1,2
(sensibles a glucosidasa I).
El dolicol fosfato sobre el que se sintetiza el oligosacárido precursor es un lípido especializado
de la membrana del retículo endo-
plasmático de carácter isoprenoide
formado por un número variable de
unidades de isopreno (11-24 unidades
en función de la especie). Este lípido
actúa como transportador y el azúcar
va a quedar unido al grupo fosfato.
Aunque la ruta de síntesis del oligosacárido precursor comienza con el dolicol fosfato, este
quedará unido a través de un puente pirofosfato (grupo pirofosforilo) por lo que en muchos
casos se hablará del dolicol pirofosfato.
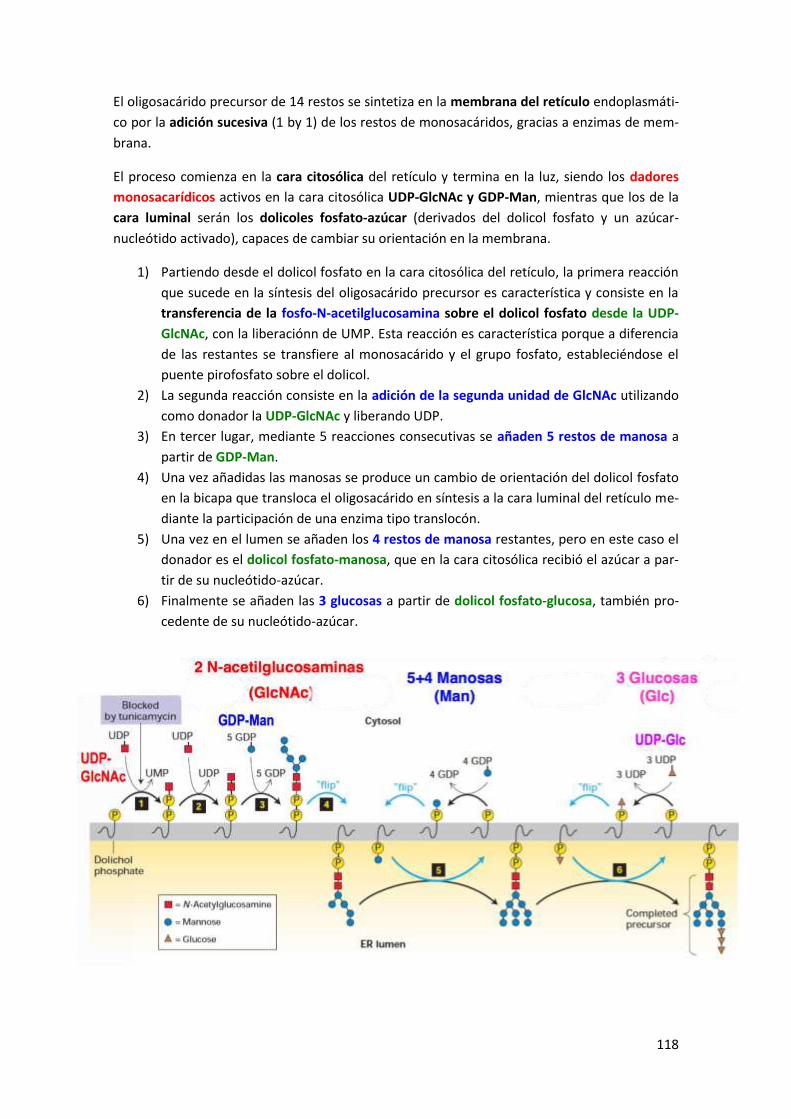
118
El oligosacárido precursor de 14 restos se sintetiza en la membrana del retículo endoplasmáti-
co por la adición sucesiva (1 by 1) de los restos de monosacáridos, gracias a enzimas de mem-
brana.
El proceso comienza en la cara citosólica del retículo y termina en la luz, siendo los dadores
monosacarídicos activos en la cara citosólica UDP-GlcNAc y GDP-Man, mientras que los de la
cara luminal serán los dolicoles fosfato-azúcar (derivados del dolicol fosfato y un azúcar-
nucleótido activado), capaces de cambiar su orientación en la membrana.
1) Partiendo desde el dolicol fosfato en la cara citosólica del retículo, la primera reacción
que sucede en la síntesis del oligosacárido precursor es característica y consiste en la
transferencia de la fosfo-N-acetilglucosamina sobre el dolicol fosfato desde la UDP-
GlcNAc, con la liberaciónn de UMP. Esta reacción es característica porque a diferencia
de las restantes se transfiere al monosacárido y el grupo fosfato, estableciéndose el
puente pirofosfato sobre el dolicol.
2) La segunda reacción consiste en la adición de la segunda unidad de GlcNAc utilizando
como donador la UDP-GlcNAc y liberando UDP.
3) En tercer lugar, mediante 5 reacciones consecutivas se añaden 5 restos de manosa a
partir de GDP-Man.
4) Una vez añadidas las manosas se produce un cambio de orientación del dolicol fosfato
en la bicapa que transloca el oligosacárido en síntesis a la cara luminal del retículo me-
diante la participación de una enzima tipo translocón.
5) Una vez en el lumen se añaden los 4 restos de manosa restantes, pero en este caso el
donador es el dolicol fosfato-manosa, que en la cara citosólica recibió el azúcar a par-
tir de su nucleótido-azúcar.
6) Finalmente se añaden las 3 glucosas a partir de dolicol fosfato-glucosa, también pro-
cedente de su nucleótido-azúcar.
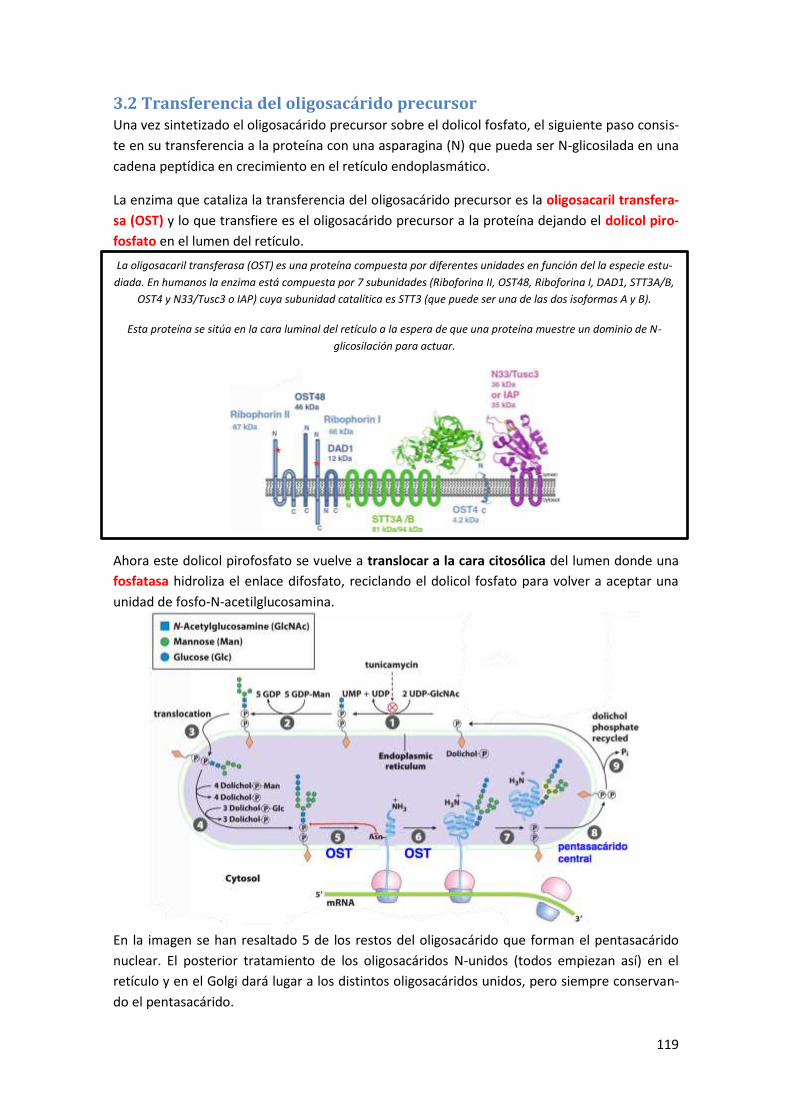
119
3.2 Transferencia del oligosacárido precursor Una vez sintetizado el oligosacárido precursor sobre el dolicol fosfato, el siguiente paso consis-
te en su transferencia a la proteína con una asparagina (N) que pueda ser N-glicosilada en una
cadena peptídica en crecimiento en el retículo endoplasmático.
La enzima que cataliza la transferencia del oligosacárido precursor es la oligosacaril transfera-
sa (OST) y lo que transfiere es el oligosacárido precursor a la proteína dejando el dolicol piro-
fosfato en el lumen del retículo.
Ahora este dolicol pirofosfato se vuelve a translocar a la cara citosólica del lumen donde una
fosfatasa hidroliza el enlace difosfato, reciclando el dolicol fosfato para volver a aceptar una
unidad de fosfo-N-acetilglucosamina.
En la imagen se han resaltado 5 de los restos del oligosacárido que forman el pentasacárido
nuclear. El posterior tratamiento de los oligosacáridos N-unidos (todos empiezan así) en el
retículo y en el Golgi dará lugar a los distintos oligosacáridos unidos, pero siempre conservan-
do el pentasacárido.
La oligosacaril transferasa (OST) es una proteína compuesta por diferentes unidades en función del la especie estu-
diada. En humanos la enzima está compuesta por 7 subunidades (Riboforina II, OST48, Riboforina I, DAD1, STT3A/B,
OST4 y N33/Tusc3 o IAP) cuya subunidad catalítica es STT3 (que puede ser una de las dos isoformas A y B).
Esta proteína se sitúa en la cara luminal del retículo a la espera de que una proteína muestre un dominio de N-
glicosilación para actuar.
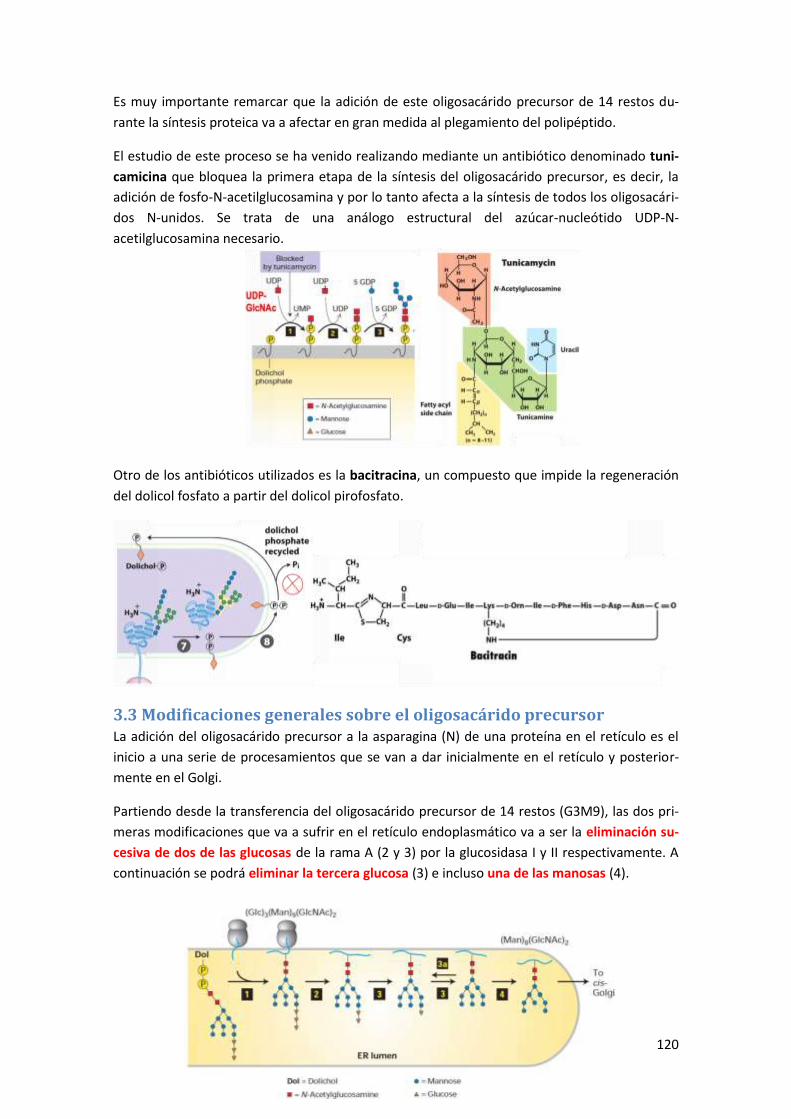
120
Es muy importante remarcar que la adición de este oligosacárido precursor de 14 restos du-
rante la síntesis proteica va a afectar en gran medida al plegamiento del polipéptido.
El estudio de este proceso se ha venido realizando mediante un antibiótico denominado tuni-
camicina que bloquea la primera etapa de la síntesis del oligosacárido precursor, es decir, la
adición de fosfo-N-acetilglucosamina y por lo tanto afecta a la síntesis de todos los oligosacári-
dos N-unidos. Se trata de una análogo estructural del azúcar-nucleótido UDP-N-
acetilglucosamina necesario.
Otro de los antibióticos utilizados es la bacitracina, un compuesto que impide la regeneración
del dolicol fosfato a partir del dolicol pirofosfato.
3.3 Modificaciones generales sobre el oligosacárido precursor La adición del oligosacárido precursor a la asparagina (N) de una proteína en el retículo es el
inicio a una serie de procesamientos que se van a dar inicialmente en el retículo y posterior-
mente en el Golgi.
Partiendo desde la transferencia del oligosacárido precursor de 14 restos (G3M9), las dos pri-
meras modificaciones que va a sufrir en el retículo endoplasmático va a ser la eliminación su-
cesiva de dos de las glucosas de la rama A (2 y 3) por la glucosidasa I y II respectivamente. A
continuación se podrá eliminar la tercera glucosa (3) e incluso una de las manosas (4). N-glicosilación

121
La adición de las glucosas al oligosacárido precursor supone la señal de que este está listo
para ser transferido, por lo que le primer paso tras la transferencia será su retirada. Además
la glucosa proximal a la proteína va a poder ser retirada y añadida cíclicamente, ya que como
veremos va a estar implicada en el plegamiento de la proteína.
Si analizamos el procesamiento del oligosacárido asociado al plegamiento de la proteína en el
retículo endoplasmático, debemos recordar que las proteínas sintetizadas en este espacio
celular van a sufrir procesos de plegamiento, ensamblado de subunidades y formación de
puentes disulfuro.
Este procesamiento debe ofrecer la estructura correcta de la proteína y por ello, asociado a las
modificaciones del oligosacárido precursor se encuentra el sistema de control de calidad del
plegamiento que nos va a asegurar que las proteínas estén correctamente plegadas y ensam-
bladas para partir del retículo al Golgi.
Para que estos procesos de plegamiento se produzcan de manera adecuada destacan tres gru-
pos de proteínas chaperonas como la calnexina, calreticulina y BiP, que se van a unir a las pro-
teínas según se sintetizan, evitando la agregación.
El sistema mejor caracterizado de control de calidad en el retículo va a implicar a calnexina
(CNX), calreticulina (CRT) y la glicosilación de las proteínas.
3.4 Control de calidad en el retículo endoplasmático La eliminación de las dos primeras glucosas del oligosacárido precursor (G1M9) da lugar al
reconocimiento de la proteína N-glicosilada por las chaperonas Calnexina (membrana luminal)
o calreticulina (soluble en el lumen), que también son lectinas.
El reconocimiento por el dominio lectina de estas chaperonas supone una ayuda para el ple-
gamiento correcto de la proteína, al mimos tiempo que se va a retener en el retículo a aquellas
proteínas mal plegadas.
Además Calnexina y calreticulina ayudan con la formación de los puentes disulfuro de las
proteínas recién sintetizadas ya que van a interaccionar con la proteína ERp57 (tioldiosulfuro
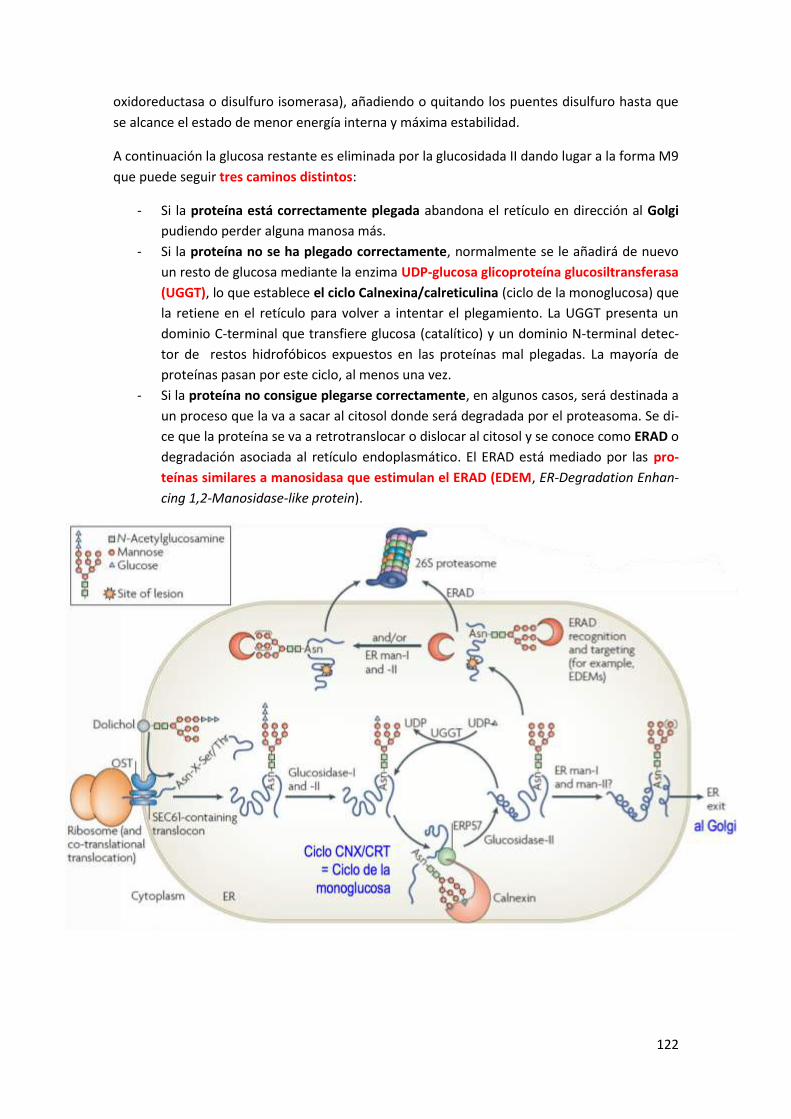
122
oxidoreductasa o disulfuro isomerasa), añadiendo o quitando los puentes disulfuro hasta que
se alcance el estado de menor energía interna y máxima estabilidad.
A continuación la glucosa restante es eliminada por la glucosidada II dando lugar a la forma M9
que puede seguir tres caminos distintos:
- Si la proteína está correctamente plegada abandona el retículo en dirección al Golgi
pudiendo perder alguna manosa más.
- Si la proteína no se ha plegado correctamente, normalmente se le añadirá de nuevo
un resto de glucosa mediante la enzima UDP-glucosa glicoproteína glucosiltransferasa
(UGGT), lo que establece el ciclo Calnexina/calreticulina (ciclo de la monoglucosa) que
la retiene en el retículo para volver a intentar el plegamiento. La UGGT presenta un
dominio C-terminal que transfiere glucosa (catalítico) y un dominio N-terminal detec-
tor de restos hidrofóbicos expuestos en las proteínas mal plegadas. La mayoría de
proteínas pasan por este ciclo, al menos una vez.
- Si la proteína no consigue plegarse correctamente, en algunos casos, será destinada a
un proceso que la va a sacar al citosol donde será degradada por el proteasoma. Se di-
ce que la proteína se va a retrotranslocar o dislocar al citosol y se conoce como ERAD o
degradación asociada al retículo endoplasmático. El ERAD está mediado por las pro-
teínas similares a manosidasa que estimulan el ERAD (EDEM, ER-Degradation Enhan-
cing 1,2-Manosidase-like protein).

123
Un hecho a tener en cuenta sobre este sistema de control de calidad es que no solo se va a
poder reglucosilar la forma 9M del péptido precursor, sino también formas con menos mano-
sas, siempre y cuando mantengan la manosa aceptora de glucosa en la rama A.
Continuando con el procesamiento del oligosacárido precursor tras el ciclo calnexi-
na/calreticulina, pasado un cierto tiempo, la manosidasa I del retículo endoplasmático (ER
ManI) va a eliminar la manosa terminal de la rama B dando lugar al oligosacárido M8B.
Esta nomenclatura indica el número de manosas y aquella rama que es diferente a las restan-
tes por lo que la siguiente forma en la que se pierde la manosa terminal de la rama C sería la
M7A, ya que la rama A es más larga que las dos restantes.
La porción luminal de la calnexina (CNX), análoga a la calreticulina, presenta un dominio de unión a carbohidratos
(domino lectina) que estructuralmente es una sándwich (dos hojas en paralelo), y un brazo largo denominado
brazo P, rico en prolina capaz de interaccionar con la disulfuro isomerasa (ERp57).
Esta disulfuro isomerasa presenta dos centros activos con dos cisteínas (-CXXC-)capaces de catalizar el intercam-
bio de puentes disulfuro.
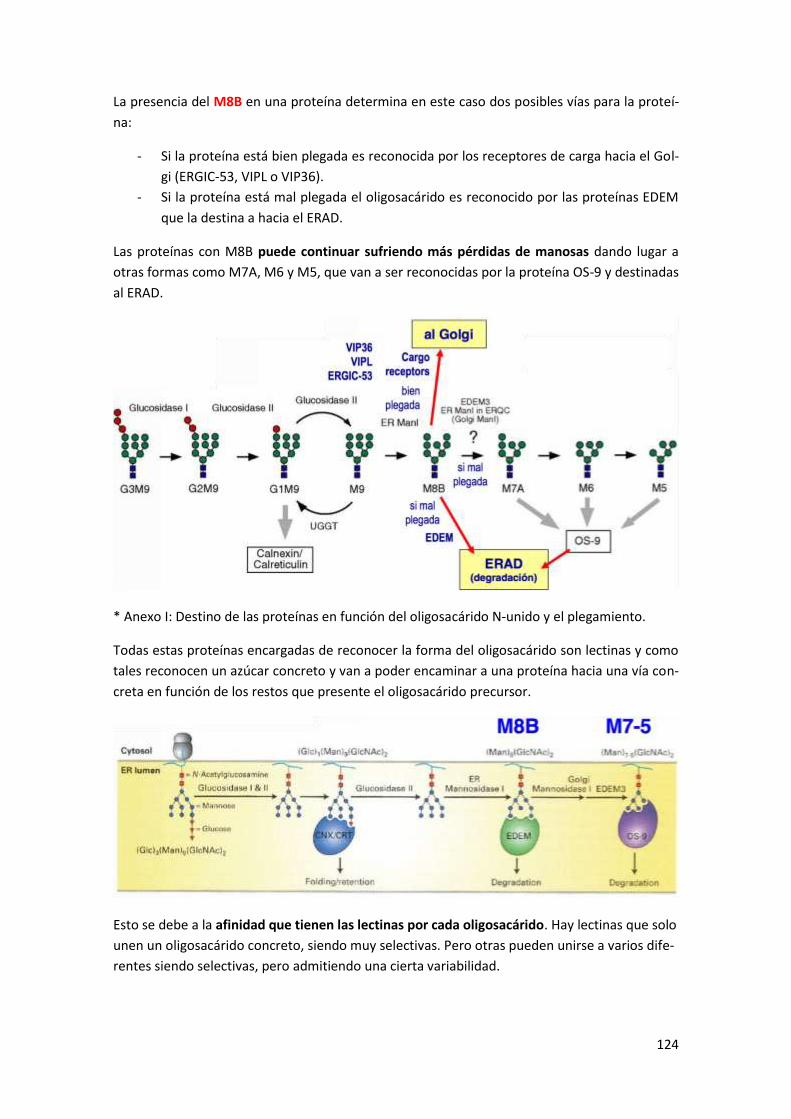
124
La presencia del M8B en una proteína determina en este caso dos posibles vías para la proteí-
na:
- Si la proteína está bien plegada es reconocida por los receptores de carga hacia el Gol-
gi (ERGIC-53, VIPL o VIP36).
- Si la proteína está mal plegada el oligosacárido es reconocido por las proteínas EDEM
que la destina a hacia el ERAD.
Las proteínas con M8B puede continuar sufriendo más pérdidas de manosas dando lugar a
otras formas como M7A, M6 y M5, que van a ser reconocidas por la proteína OS-9 y destinadas
al ERAD.
* Anexo I: Destino de las proteínas en función del oligosacárido N-unido y el plegamiento.
Todas estas proteínas encargadas de reconocer la forma del oligosacárido son lectinas y como
tales reconocen un azúcar concreto y van a poder encaminar a una proteína hacia una vía con-
creta en función de los restos que presente el oligosacárido precursor.
Esto se debe a la afinidad que tienen las lectinas por cada oligosacárido. Hay lectinas que solo
unen un oligosacárido concreto, siendo muy selectivas. Pero otras pueden unirse a varios dife-
rentes siendo selectivas, pero admitiendo una cierta variabilidad.
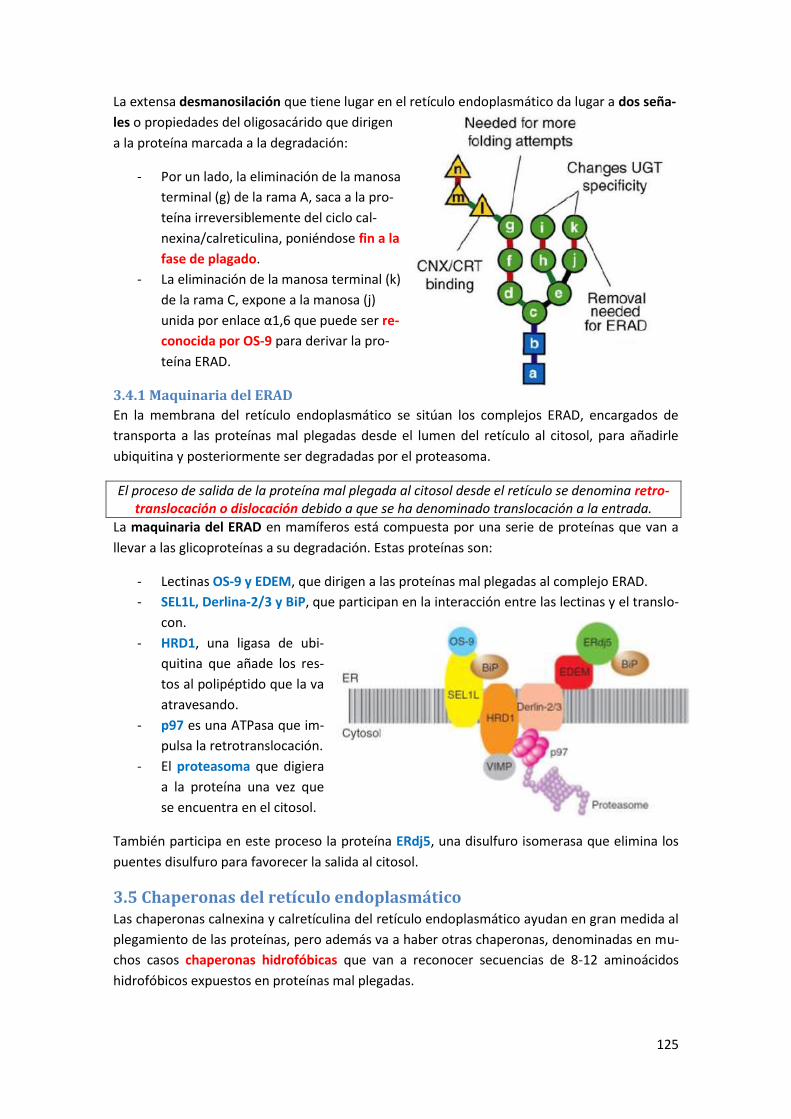
125
La extensa desmanosilación que tiene lugar en el retículo endoplasmático da lugar a dos seña-
les o propiedades del oligosacárido que dirigen
a la proteína marcada a la degradación:
- Por un lado, la eliminación de la manosa
terminal (g) de la rama A, saca a la pro-
teína irreversiblemente del ciclo cal-
nexina/calreticulina, poniéndose fin a la
fase de plagado.
- La eliminación de la manosa terminal (k)
de la rama C, expone a la manosa (j)
unida por enlace α1,6 que puede ser re-
conocida por OS-9 para derivar la pro-
teína ERAD.
3.4.1 Maquinaria del ERAD
En la membrana del retículo endoplasmático se sitúan los complejos ERAD, encargados de
transporta a las proteínas mal plegadas desde el lumen del retículo al citosol, para añadirle
ubiquitina y posteriormente ser degradadas por el proteasoma.
El proceso de salida de la proteína mal plegada al citosol desde el retículo se denomina retro-translocación o dislocación debido a que se ha denominado translocación a la entrada.
La maquinaria del ERAD en mamíferos está compuesta por una serie de proteínas que van a
llevar a las glicoproteínas a su degradación. Estas proteínas son:
- Lectinas OS-9 y EDEM, que dirigen a las proteínas mal plegadas al complejo ERAD.
- SEL1L, Derlina-2/3 y BiP, que participan en la interacción entre las lectinas y el translo-
con.
- HRD1, una ligasa de ubi-
quitina que añade los res-
tos al polipéptido que la va
atravesando.
- p97 es una ATPasa que im-
pulsa la retrotranslocación.
- El proteasoma que digiera
a la proteína una vez que
se encuentra en el citosol.
También participa en este proceso la proteína ERdj5, una disulfuro isomerasa que elimina los
puentes disulfuro para favorecer la salida al citosol.
3.5 Chaperonas del retículo endoplasmático Las chaperonas calnexina y calretículina del retículo endoplasmático ayudan en gran medida al
plegamiento de las proteínas, pero además va a haber otras chaperonas, denominadas en mu-
chos casos chaperonas hidrofóbicas que van a reconocer secuencias de 8-12 aminoácidos
hidrofóbicos expuestos en proteínas mal plegadas.

126
La chaperona BiP de la familia de las
HSP70 del retículo favorece el plega-
miento de las proteínas con ciclos de
plegado y liberación que mantiene a las
proteínas mal plegadas en el retículo
endoplasmático a partir del gasto de
ATP.
Entre las funciones de BiP en el retículo podemos destacar:
- Ayuda al plegamiento en el retículo de las proteínas mal plegadas y mantiene a las
proteínas mal plegadas en el retículo.
- Dirige a las proteínas mal plegadas al ERAD
- Es un regulador de proteínas transductoras transmembrana del estrés del retículo
endoplasmático (PERK, IRE1 y ATF6).
- Interviene en la translocación post-traducional en levaduras.
- Relacionada con los procesos de apoptosis, ya que interacciona con la caspasa 7 del
retículo e impide la activación de proteínas pro-apoptóticas de la familia Bcl2, como
Bax.
Hasta ahora solo hemos hablado del reconocimiento
de las proteínas glicosiladas por las lectinas (EDEM y
OS-9) que las dirigen al ERAD, pero también existe
complejos ERAD para proteínas no glicosiladas en
los que van a intervenir las chaperonas hidrofóbicas
como BiP.
La chaperona BiP es capaz de dirigir a los polipépti-
dos mal plegados hasta los complejos de retrotranlo-
cación donde interaccionan con HERP (análoga a
SEL1L). Además tenemos los elementos comunes
como HRD1 (ligasa de ubiquitina), p97 (ATPasa),
derlina-1 y el proteasoma.
3.6 Respuesta a proteínas mal plegadas (UPR) Un proceso importante en relación al plegamiento en el retículo endoplasmático es la denomi-
nada respuesta a proteínas mal plegadas (UPR, Unfolded protein response) que fue introducido
en el tema anterior.
En ciertas situaciones se puede producir una disfunción del retículo en la que se da un des-
equilibrio entre la capacidad de plegar proteínas del orgánulo y el número de proteínas que
debe plegar. Estas son situaciones como la falta de glucosa e hipoxia, que desencadenan una
falta de energía, o infecciones virales y problemas con la regulación del calcio. El envejeci-
miento también es un factor que afecta a la capacidad de plegar las proteínas que presenta el
retículo.

127
El retículo chequea el nivel de plegamiento y cuando se acumulan proteínas mal plegadas se
pone en marcha la UPR, un complejo sistema de señalización mediado por iniciado por tres
receptores denominados genéricamente como transductores transmembrana del estrés del
retículo endoplasmático:
- Kinasa dependiente de inositol 1(IRE1, Inositol requiring kinase 1)
- Kinasa semejante a PKR del retículo (PERK, PKR7-like ER kinase)
- Factor de transcripción activante 6 (ATF6, Activating transcripction factor 6)
La UPR consta de tres procesos o tipos de respuesta que son la adaptación, alarma y apopto-
sis:
- Durante la fase de adaptación la célula intenta restablecer el equilibrio en el plega-
miento mediante:
o La inhibición de la traducción de un modo general para reducir el número de
proteínas a plegar.
o El aumento de la expresión de chaperonas para aumentar la capacidad de
plegamiento del retículo
o El aumento de la degradación de proteínas mal plegadas por el ERAD.
- La fase de alarma se produce cuando los esfuerzos de la célula por paliar el desequili-
brio fallan. Esta fase se caracteriza por la reducción del bloqueo de la traducción y una
reducción de expresión de las proteínas pro-supervivencia (anti-apoptóticas) como
Bcl2.
- Finalmente la célula entra en apoptosis o autofagia.
3.6.1 Regulación de los receptores por BiP
Los transductores transmembrana del estrés del retículo endoplasmático se encuentran en
condiciones normales unidos a la proteína BiP, impidiendo su activación. Cuando el nivel de
proteínas mal plegada aumenta, BiP es desplazada de los receptores y estos comienzan a
dimerizar y activarse provocando la UPR.
El modo de actuación de IRE1 se explicó deta-
lladamente en el tema anterior, pero a modo
de resumen, recordemos que se encuentra
unido a BiP en la membrana del retículo.
Cuando se producen condiciones de estrés
que desplazan a BiP, dimeriza y sufre una
fosforilación cruzada, comenzando a desarro-
llar actividad RNA-endonucleasa en el domi-
nio citosólico. Esta actividad permite el spli-
cing citosólico del pre-mRNA de XBP1
(XBP1u), una proteína de unión a la caja X
(XBP1s) y por tanto un factor de transcripción
que va al núcleo para aumentar la expresión
7 Protein kinasa activada por ARN de doble cadena (PKR, double-stranded RNA-activated protein kinase)

128
de chaperonas, proteínas implicadas en la degradación de ERAD y proteínas implicadas en la
apoptosis como CHOP.
La activación de PERK también está medida por la sepa-
ración de la proteína BiP en respuesta a un aumento de
proteínas mal plegadas en el retículo, lo que permite su
dimerización y el desarrollo de su actividad fosforilasa
sobre el factor eucariótico de iniciación de la traducción
eIF2α, inactivándolo y produciendo un bloqueo general
de la traducción.
A este bloqueo escapa el factor de traducción activante 4
(ATF4), que es traducido por un proceso independiente
de CAD, por lo que puede ir al núcleo e inducir la expre-
sión de genes que participan en la UPR.
El ATF6 es el único de los tres receptores que no dimeriza. Cuando la
proteína BiP es desplazada se desplaza hacia el Golgi, donde sufre
dos cortes proteólíticos por la S1P y S2P, que liberan un fragmento
citosólico (ATF6f), que es en sí un factor de transcripción que induce
la expresión de los genes propios de la vía.
El mecanismo de actuación de ATF6 es análogo al de otras proteí-nas, como las implicadas en el metabolismo del colesterol donde SREBP, que se situaba en el retículo, se desplazaba hacia el Golgi
donde sufría los corte proteolíticos que liberaban el factor de transcripción que viajaba al núcleo para expresar las proteínas
necesarias en la síntesis de colesterol.
La respuesta a proteínas mal plegadas está implicada en determinadas enfermedades que
incluyen la diabetes, la progresión tumoral y las enfermedades neurodegenerativas como el
Alzheimer, el Parkinson y la esclerosis lateral amniotrófica (ALS).
3.7 Otras proteínas que ayudan al plegamiento en el retículo endo-
plasmático Además de las chaperonas mencionadas hay otras proteínas que también colaboran con el
plegamiento en el retículo endoplasmático, las tiodisulfuro oxidoreductasas (disulfuro isome-
rasas) y las peptidil-prolil isomerasas.
Las tiodisulfuro oxidoreductasas o proteí-
nas disulfuro isomerasas (PDIs) colaboran
con la formación de los puentes disulfuro
correctos entre los restos de cisteína (C) de
las proteínas. Estos puentes se forman por

129
la unión oxidativa de dos grupos tiol y por ello para generarlos será necesario otro puente di-
sulfuro. Los puentes disulfuro son imprescindibles para el establecimiento de la estructura
terciaria y cuaternaria (anticuerpos) de las proteínas. Pero como normal general, estos solo se
puede realizar en el lumen del retículo endoplasmático por lo que solo se van a localizar en
proteínas de secreción y en la porciones exoplásmicas (no citosólica) de las proteínas de mem-
brana.
En función del estado de la proteína disulfuro isomerasa hablaremos de la creación o disocia-
ción de un enlace disulfuro:
- Cuando nos encontramos ante un puente disulfuro incorrecto, la encargada de actuar
es la PDI en su forma reducida (SH HS). El grupo tiol de la PDI ataca al puente disulfuro
incorrecto, lo rompe y se forma un puente disulfuro PDI-Cisteina. El grupo tiol de la
proteína que ahora queda libre ataca a otro de los puentes disulfuro de la proteína, lo
disocia y queda formando un nuevo puente disulfuro. Finalmente el proceso se repite
para liberar la PDI formándose un segundo puente disulfuro intramolecular.
- Para formar un puente disulfuro intramolecular de novo, la encargada de actuar es la
PDI en su forma oxidada (S-S). En este caso se produce una ataque desde el grupo tiol
de la proteína sobre el puente disulfuro de PDI y se genera uno nuevo. A continuación
otro grupo tiol de la proteína ataca a este puente disulfuro quedando uno nuevo so-
bre la proteína y liberándose PDI reducida (SH-HS).
Las PDIs pueden ser oxidadas desde su forma ditiol gracias a la acción de otras proteínas como
ERO1. Estas proteínas no saben si un puente disulfuro es correcto o no, simplemente los van
intercambiando hasta que la estabilidad conformacional de la proteína es tal, que ya no pue-
den actuar.

130
Las PDIs son una familia de proteínas muy diversas que presentan esta función de intercambio
de puentes disulfuro y que pueden presentar uno o varios centros activos, con normalmente,
dos cisteínas (C) separados por dos aminoácidos cuales quiera.
-CxxC-
Otra de las proteínas necesarias para el plegamiento de las proteínas son las peptidil-prolil
isomerasas encargadas de acelerar la isomerización Cis-Trans o Trans-Cis de los enlaces peptí-
dicos donde el segundo aminoácido es una prolina (P) en regiones no plegadas de las proteí-
nas. Esta reacción de isomerización suele ser la etapa limitante en el plegamiento de las pro-
teínas.
Los enlaces peptídicos, en general siem-
pre están en configuración Trans, pero
algunos donde el segundo aminoácido es
una prolina (P) cuyo nitrógeno forma
parte de un anillo será necesaria la iso-
merización para el correcto plegamiento
de la proteína.
3.8 Ensamblado de las subunidades proteicas en el retículo endoplas-
mático Para finalizar con el proceso de plegamiento en el retículo endoplasmático falta comentar que
en este espacio celular tiene lugar el ensamblado de las proteínas oligoméricas, como es el
caso de la hemaglutinina del virus de la gripe.
La hemaglutitina es una proteína trimérica formada por tres cadenas polipeptídicas que se
ensamblan, en primer lugar interaccionando por sus hélices α y finalmente interaccionando
por sus cabezas globulares.
Esta proteína representa un ejemplo de la necesidad de la glicosilación para el adecuado ple-
gamiento ya que si se añade tunicamicina o se muta una de las asparaginas (N) glicosiladas por
una glutamina, el plegamiento no se produce de manera adecuada.
3.9 Modificaciones de la N-glicosilación a nivel del aparato de Golgi El aparato de Golgi es el siguiente orgánulo
que deben atravesar todas las proteínas N-
glicosiladas. El Golgi es un conjunto de sacos
membranosos aplanados que establecen el
principal centro de distribución celular. Pre-
senta un lado Cis al que llegan los materiales
procedentes del retículo y un lado Trans del
que parte las vesículas que llevan los materia-
les a la membrana plasmática, la secreción o
los lisosomas.

131
Durante su paso por el Golgi los oligosacáridos N-unidos a las proteínas van a ser procesados
en una serie de reacciones ordenadas catalizadas por glicosilasas y transferasas que se van a
distribuir conforme al orden de estas
reacciones por las cisternas del aparato:
- Eliminación de manosas en las
cisternas Cis y medias del Golgi
con manosidasas diferentes a
las del retículo.
- Adición de N-acetilglucosamina
(GlcNAc) y fucosa en las cister-
nias medias del Golgi.
- Adición de galactosa (Gal) y
ácido siálico (p.e. Neu5Ac) en
las cisternas Tras del Golgi.
Paralelo a estos procesos se va a produ-
cir la adición de grupos fosfato en la
CGN y TGN para las proteínas lisosoma-
les.
Dado que estas reacciones, en caso de suceder en su totalidad (no suceden siempre) lo hacen
siempre en el mismo orden, vamos a estudiar el proceso completo. Partiendo de la proteína
con G3M9, que llega en el retículo endoplasmático, recordemos que se le retiraban los azúca-
res y una manosa, llegando al aparato de Golgi como M8.
- La primera etapa (2) que se produce en la cara cis del Golgi consiste en la retirada de
tres manosas por la acción de la manosidasa I del Golgi que da lugar a la forma M5.
- La segunda etapa (4) que se puede llevar a cabo al mismo tiempo que la siguiente,
consiste en la retirada de dos manosas más por la manosidasa II del Golgi dando lugar
a un oligosacárido con tres manosas que constituye el pentasacarido nuclear común a
todos las proteínas N-glicosiladas.
- Las tercera etapa (3), que se puede llevar a cabo al mismo tiempo que la anterior con-
siste en la adición de N-acetilglucosamina por la N-acetilglucosamina transferasa I del
golgi en la rama A del péptido precursor.

132
La retirada de la primera de estas manosas por la manosidasa II del retículo convierte a la proteína en
resistente a la acción de la endoglicosidasa H, por lo que podemos detectar las proteínas que han
pasado por el Golgi medio, ya que sus oligosacáridos no serán degradados.
A continuación se pueden añadir galactosa y ácido siálico para dar lugar a los oligosacáridos
complejos. Los donadores monosacarídicos del proceso son los nucleótidos azúcar estudiados
anteriormente que entran al lumen del aparato de Golgi mediante antiporter nucleótido-
azúcar/nucleótido.
Conocer el estado de glicosilación de una proteína supone conocer cómo avanza ésta en la vía
secretora:
- El procedimiento consiste en realizar una electroforesis de la proteína con SDS des-
pués del tratamiento con la endoglicosilasa adecuada. La proteína desglicosilada ten-
drá un tamaño ligeramente menor y por lo tanto avanzará más rápidamente.
- Otro procedimiento consiste en la utilización de lectinas para un determinado azúcar
que se incorpore en una región concreta del Golgi. Por ejemplo, la aglutinina del ger-
men de trigo reconoce a la N-acetilglucosamina añadida en el Golgi medio. La fitohe-
maglutina (lectina de cacahuete) reconoce a los azúcares con galactosa y por lo tanto a
los que pasan a través del Golgi Trans.
El procesamiento de los oligosacáridos N-unidos en el aparato
de Golgi puede dar lugar a tres tipos fundamentales de oligo-
sacáridos maduros:
- N-oligosacáridos ricos en manosa en los que todas sus
ramas acaban en manosas y solo presentan azúcares
que estaban en el precursor de 14 restos.
- N-oligosacáridos complejos en los que no hay manosas
terminales por la adición en todas sus ramas de azúca-
res en el Golgi.
- N-oligosacáridos híbridos que son una mezcla de am-
bos tipos y se forman cuando tras la adición de la pri-
mera N-acetilglucosamina ya no se quitan más mano-
sas.
Pero esto son solo clases fundamentales ya que los oligosacáridos N-unidos pueden ser muy
variados apareciendo los bicatenarios, trecatenarios, tetracaternario y muchas modificaciones
como la adición de fucosa en el núcleo pentasacáridico o incluso N-acetilglucosamina entre las
ramas (bisectriz).
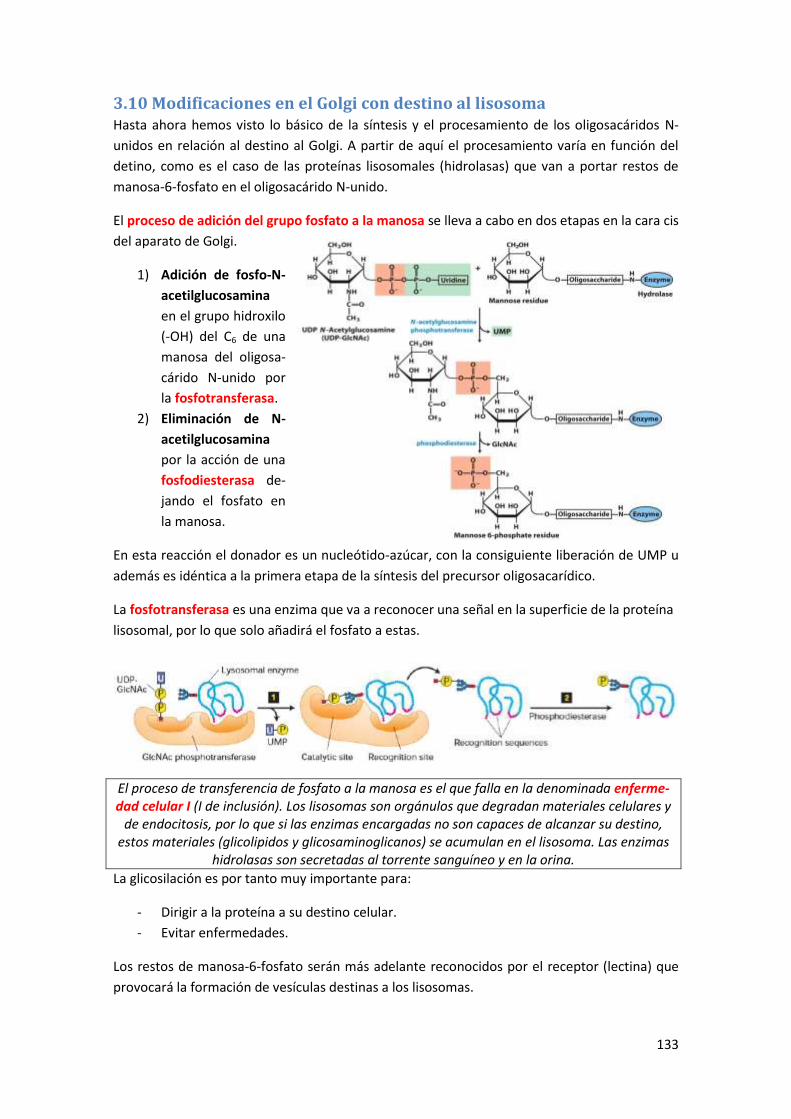
133
3.10 Modificaciones en el Golgi con destino al lisosoma Hasta ahora hemos visto lo básico de la síntesis y el procesamiento de los oligosacáridos N-
unidos en relación al destino al Golgi. A partir de aquí el procesamiento varía en función del
detino, como es el caso de las proteínas lisosomales (hidrolasas) que van a portar restos de
manosa-6-fosfato en el oligosacárido N-unido.
El proceso de adición del grupo fosfato a la manosa se lleva a cabo en dos etapas en la cara cis
del aparato de Golgi.
1) Adición de fosfo-N-
acetilglucosamina
en el grupo hidroxilo
(-OH) del C6 de una
manosa del oligosa-
cárido N-unido por
la fosfotransferasa.
2) Eliminación de N-
acetilglucosamina
por la acción de una
fosfodiesterasa de-
jando el fosfato en
la manosa.
En esta reacción el donador es un nucleótido-azúcar, con la consiguiente liberación de UMP u
además es idéntica a la primera etapa de la síntesis del precursor oligosacarídico.
La fosfotransferasa es una enzima que va a reconocer una señal en la superficie de la proteína
lisosomal, por lo que solo añadirá el fosfato a estas.
El proceso de transferencia de fosfato a la manosa es el que falla en la denominada enferme-dad celular I (I de inclusión). Los lisosomas son orgánulos que degradan materiales celulares y
de endocitosis, por lo que si las enzimas encargadas no son capaces de alcanzar su destino, estos materiales (glicolipidos y glicosaminoglicanos) se acumulan en el lisosoma. Las enzimas
hidrolasas son secretadas al torrente sanguíneo y en la orina.
La glicosilación es por tanto muy importante para:
- Dirigir a la proteína a su destino celular.
- Evitar enfermedades.
Los restos de manosa-6-fosfato serán más adelante reconocidos por el receptor (lectina) que
provocará la formación de vesículas destinas a los lisosomas.
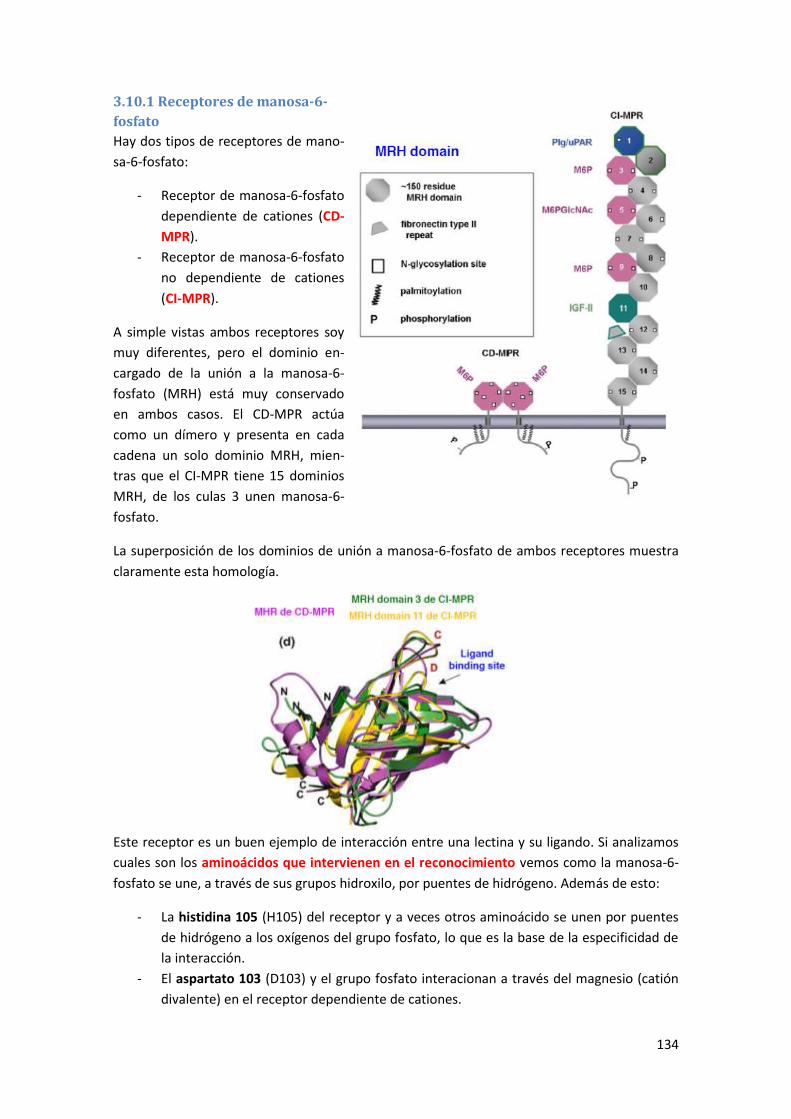
134
3.10.1 Receptores de manosa-6-
fosfato
Hay dos tipos de receptores de mano-
sa-6-fosfato:
- Receptor de manosa-6-fosfato
dependiente de cationes (CD-
MPR).
- Receptor de manosa-6-fosfato
no dependiente de cationes
(CI-MPR).
A simple vistas ambos receptores soy
muy diferentes, pero el dominio en-
cargado de la unión a la manosa-6-
fosfato (MRH) está muy conservado
en ambos casos. El CD-MPR actúa
como un dímero y presenta en cada
cadena un solo dominio MRH, mien-
tras que el CI-MPR tiene 15 dominios
MRH, de los culas 3 unen manosa-6-
fosfato.
La superposición de los dominios de unión a manosa-6-fosfato de ambos receptores muestra
claramente esta homología.
Este receptor es un buen ejemplo de interacción entre una lectina y su ligando. Si analizamos
cuales son los aminoácidos que intervienen en el reconocimiento vemos como la manosa-6-
fosfato se une, a través de sus grupos hidroxilo, por puentes de hidrógeno. Además de esto:
- La histidina 105 (H105) del receptor y a veces otros aminoácido se unen por puentes
de hidrógeno a los oxígenos del grupo fosfato, lo que es la base de la especificidad de
la interacción.
- El aspartato 103 (D103) y el grupo fosfato interacionan a través del magnesio (catión
divalente) en el receptor dependiente de cationes.

135
- El glutamato 133 (E133) es imprescindible para la unión de la manosa, pero al descen-
der el pH al llevar al lisosoma (endosoma) se protona, provocando la pérdida de afini-
dad del receptor por la carga y la consiguiente liberación.
3.11 N-glicosilación en procariotas La N-glicosilación es un proceso que se da, con ciertas variaciones, en todos los reinos eucarió-
ticos y procarioticos.
Una de las diferencias fundamentales es que en bacterias, el aceptor del olicosacárido precur-
sor no es el dolicol fosfato, sino el undecaprenol fosfato.
La N-glicosilación es frecuente en eucariotas y arqueas, no tanto, pero también existe en bac-
terias. La O-glicosilación es más frecuente en eucariotas y bacterias que en arqueas.
La glicosilación en bacterias está reducida, pero puede ser muy importante para la vida como en el caso del patógeno gastrointestintal Campylobacter jejuni donde la glicosilación es lo que le permite ser infectivo.

136
4 El proceso de O-glicosilación El enlace O es aquel que se produce por la unión al oxígeno del grupo hidroxilo de la cadena
lateral de una serina (S) o treonina (T). Se habla de O-glicosilación y azúcares O-unidos que
suelen tener entre 1-4 restos de oligosacárido.
La idea que vamos a tratar es la de la adición de un único resto de azúcar, directamente sobre
la proteína al que posteriormente podrán unirse otros.
El proceso de O-glicosilación se puede dar en tres lugares diferentes en mamíferos:
- En el citosol la O-glicosilación se produce mediante la adición de N-acetiglucosamina
(GlcNAc) en residuos de serina o treonina.
- En el retículo endoplasmático la O-glicosilación se produce por la adición de glucosa,
manosa o fucosa en residuos de serina o treonina, y la adición de galactosa en resi-
duos de hidroxilisina (Hyl) como es el caso del colágeno.
- En el aparato de Golgi la O-glicosilación se produce por la adición de N-
acetilgalactosamina, xilosa y N-acetilglucosamina en residuos de serina o treonina.
Cabe aclarar que a pesar de que la adición de N-acetilglucosamina O-unida se produzca tanto
en el citosol como en el Golgi, se trata de procesos totalmente diferentes y permiten la apari-
ción de estos azúcares en proteínas de secreción o de superficie.
Otro cosa importante es que la adición de N-acetilgalactosamina se va a diferenciar de los
demás procesos de O-glicosilación en que en este caso van a existir muchas enziamas (20)
que intervendrán en el proceso denominadas N-acetilgalactosamina transferasas, mientras
que en los demás procesos de O-glicosilación participaran como mucho un par de enzimas.
Los dos tipos más frecuentes de O-glicosilación con mucha diferencia son:
- Adición de N-acetilgalactosamina (GalNAc) que da lugar a la glicosilación tipo mucina.
Esta glicosilación suele tener entre 1-10 restos de monosacáridos pudiendo presentar
más de 50 estructuras en función de los enlaces y los monosacáridos añadidos.
- Adición de la xilosa presente en los proteoglicanos que presentan largas cadenas de
glicosaminoglicanos (GAGs). Esta glicosilación pude tener más de 100 restos de mono-
sacáridos.

137
Los fallos asociados a la O-glicosilación son causantes de muchas enfermedades, pero muchas de ellas provocan la muerte del animal antes de nacer por su letalidad. Un ejemplo es la defi-ciencia de las enzimas responsables de la manosilación que provocan la aparición de distrofia
muscular. Esta enfermedad se produce porque el -distroglicano tiene azúcares O-unidos que comienzan por manosa, y estos son necesarios para la unión con la lámina basal.
4.1 O-glicosilación de proteoglicanos con xilosa Los proteoglicanos o proteoglucanos son macromoléculas que presentan una o varias cadenas
de polisacáridos denominados glicosaminoglicanos (GAGs) unidos covalentemente a una pro-
teína núcleo.
Los proteoglicanos son componentes
muy importantes de las matrices extra-
celulares y otras proteínas unidas a la
superficie de la membrana plasmática.
Estos glucosaminoglicanos (GAGs) son
polisacáridos en los que alternan gene-
ralmente un resto de ácido urónico
(glucuronico, idurónico) con una N-
acetilexosamina o N-sulfoexosamina
que en muchos casos tienen grupos
sulfato.
Estos GAGs como el condroitin sulfato,
el heparan sulfato, el queratan sulfato
o el dermatan sulfato, tienen muchas
cargas negativas procedentes de los
carboxilatos del ácido urónico y los gru-
pos sulfato. Además la carga positiva
que podría tener la hexosamina (por el
grupo amino) ha sido retirada mediante la adición de un grupo acetato o sulfato.
O-Glicosilación: Proteoglicanos
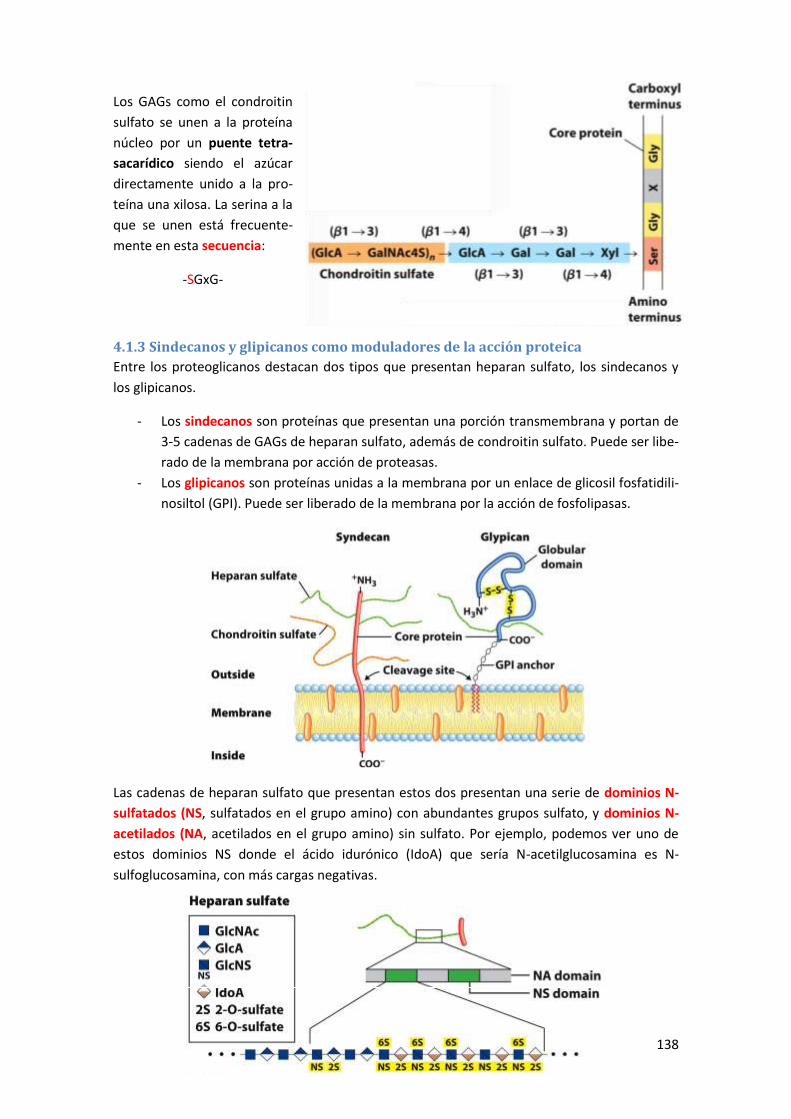
138
Los GAGs como el condroitin
sulfato se unen a la proteína
núcleo por un puente tetra-
sacarídico siendo el azúcar
directamente unido a la pro-
teína una xilosa. La serina a la
que se unen está frecuente-
mente en esta secuencia:
-SGxG-
4.1.3 Sindecanos y glipicanos como moduladores de la acción proteica
Entre los proteoglicanos destacan dos tipos que presentan heparan sulfato, los sindecanos y
los glipicanos.
- Los sindecanos son proteínas que presentan una porción transmembrana y portan de
3-5 cadenas de GAGs de heparan sulfato, además de condroitin sulfato. Puede ser libe-
rado de la membrana por acción de proteasas.
- Los glipicanos son proteínas unidas a la membrana por un enlace de glicosil fosfatidili-
nosiltol (GPI). Puede ser liberado de la membrana por la acción de fosfolipasas.
Las cadenas de heparan sulfato que presentan estos dos presentan una serie de dominios N-
sulfatados (NS, sulfatados en el grupo amino) con abundantes grupos sulfato, y dominios N-
acetilados (NA, acetilados en el grupo amino) sin sulfato. Por ejemplo, podemos ver uno de
estos dominios NS donde el ácido idurónico (IdoA) que sería N-acetilglucosamina es N-
sulfoglucosamina, con más cargas negativas.
O-Glicosilación: Proteoglicanos
O-Glicosilación: Proteoglicanos

139
Los GAGs interaccionan con diversos ligandos modulando su actividad mediante su unión a los
dominios NS.
Una proteína al unirse a un dominio NS
puede cambiar su conformación activa,
como ocurre a la antitrombina cuando
al unirse a un dominio NS específico
(secuencia), puede interaccionar con el
factor Xa, inhibiendo la coagulación
sanguínea.
La unión de dos proteínas a dominios
NS adyacentes les brinda la aproxima-
ción necesaria para interaccionar,
como también le ocurre a la antitrom-
bina y a la trombina, produciendo una
inhibición de la coagulación sanguínea.
Otro tipo de proceso es el que ocurre cuando una proteína se une a los dominios NS de los
proteoglicanos de la superficie de la membrana plasmática, aumentando su concentración
local, como es el caso de la lipoprotein
lipasa. En el metabolismo de los triacilglice-
roles, las células captan los ácidos grasos
cuando el quilomicrón pasa cerca de la
membrana y es degradado por las lipopro-
teín lipasas. Si esta degradación se produce
cerca de la membrana plasmática la reab-
sorción es más eficiente.
Finalmente tenemos el caso de los receptores y
su ligando, como ocurre para el factor de creci-
miento de fibroblastos (FGF). La unión del FGF y
del receptor a dominios NS de los proteoglicanos
aumenta la concentración local de ambos en un
punto, facilitando su interacción y la formación
de complejos.
4.2 O-glicosilación de N-acetilglucosmaina en el citosol La adición de un único resto N-acetilglucosamina O-unida a restos de serina (S) o Treonina (T),
es una modificación post-traduccional muy importante que ocurre en el citosol (también en el
núcleo y en aparato de Golgi, pero con otras enzimas).
La característica más destacada de este tipo de O-glicosilación es que, a diferencia de todas las
anteriores, es reversible, de manera que hay enzimas que la van a realizar y otras que la reti-
ran. La glicosiltransferasa encargada de añadir N-acetilglucosamina es la O-unida-N-
acetilglucosamina transferasa (OGT), y la hexosaminidasa encargada de retirarla es la N-
acetilglucosaminidasa (OGA). Siendo el donador la UDP-N-acetilglucosamina.
O-Glicosilación
O-Glicosilación
O-Glicosilación
O-Glicosilación

140
En el genoma humano se presenta un
único gen para OGT situado en el
cromosoma X, pero que sufre splicing
alternativo para dar tres isoformas.
Se trata de una proteína homodiméri-
ca que presenta:
- Una porción N-terminal reguladora.
- Una porción C-terminal catalítica con la actividad transferasas en cuyo extremo hay un
dominio de unión a PIP3 (PPO, Phosfatidylinositide 3,4,5-trisphosphate targetin do-
main).
La OGA presenta un dominio N-terminal catalítico con actividad hexosaminidasa y un dominio
C-terminal histona acetiltransferasa (homólogo, actividad no comprobada).
Este proceso de O-glicosilación se presenta en gran cantidad de proteínas clave en rutas de
señalización como la propia OGT, SP-1 (factor de transcripción), p53, APP (Alzheimer), RNA
polimerasa II, Tau (Alzheimer) y Clatrina AP-3. Por lo tanto, la adición de N-acetilglucosamina
O-unida debe de ser un componente de regulación importante tal y como la regulación por
fosforilación lo es.
4.2.1 Regulación/actuación de OGT mediante O-glicosilación de N-acetilglucosamina
La actividad de la OGT está controlada a tres niveles:
- Por la concentración de su sustrato UDP-N-acetilgluco-samina, un nucleótido azúcar
cuyos bajos niveles representan un indicador o sensor de una baja concentración de
glucosa por su conexión a través de la ruta de biosíntesis de hexosaminas (HBP).
- Por el dominio PPO que detecta los aumentos de PIP3 en la membrana plasmática tras
la activación de PI3K, lo que supone la translocación a la membrana para actuar sobre
las proteínas de esa zona.
- Por CaMKIV (Proteína kinasa IV dependiente de calcio-calmodulina) que activa a OGT
fosforilandola.

141
La activación de OGT supone la O-glicosilación de proteínas que puede afectar a varios proce-
sos:
- La glicosilación de factores de trans-
cripción como SP-1 supone su trans-
locación al núcleo y la expresión de
proteínas.
- La glicosilación de proteínas afecta a
la estabilidad (vida media), como en
el caso de p53 que supone un aumen-
to de su estabilidad.
- La glicosilación de proteínas también
puede afectar a su fosforilación ya
que si este proceso se realiza sobre
un resto de Serina (S) o treonina (T)
susceptible de ser fosforilado, ambas
modificaciones serán excluyentes.
Un ejemplo de este tipo es el factor CRTC2 (coactivador de CREB en hepatocitos) que permanece en el citosol cuando S70 y S171 se encuentran fosforiladas, pero la adición de N-acetilglucosamina en estos
restos permiten su migración al núcleo donde se expresan los genes de la gluconeogénesis.
N-acetilglucosaminidación y fosforilación como modificaciones excluyentes
El modo en el que la adición de N-acetiglucosamina y la fosforilación son excluyentes depende
de muchos factores y puede suponer varios quebraderos de cabeza para su estudio.
El caso más simple es aquel en el que el res-
to de serina o treonina es el mismo que se
fosforila o se le añade N-aceltilglucosamina
como sucede en C-myc y ER-.
Pero también existen casos en los que la presencia de dos residuos susceptibles alternativa-
mente de estos cambios están muy pró-
ximos en la secuencia, por lo que si uno
de ellos es glucosilado o fosforilado, el
otro no podrá ser modificado, lo que es-
tablece una regulación reciproca como
sucede en el caso de p53 y de la RNA po-
limerasa II.
Esta proximidad entre dos residuos susceptibles tam-
bién se puede dar como resultado de la conformación
proteica y por lo tanto no tienen porque estar próximos
en la secuencia. Es el caso de la CaMKIV.
Finalmente tenemos la posibilidad de que la actividad
de la proteína sea regulada por el estado de modifica-
O-Glicosilación: O-GlcNAc
Hu FEBS L 2010
O-Glicosilación: O-GlcNAc
Hu FEBS L 2010
O-Glicosilación: O-GlcNAc
Hu FEBS L 2010
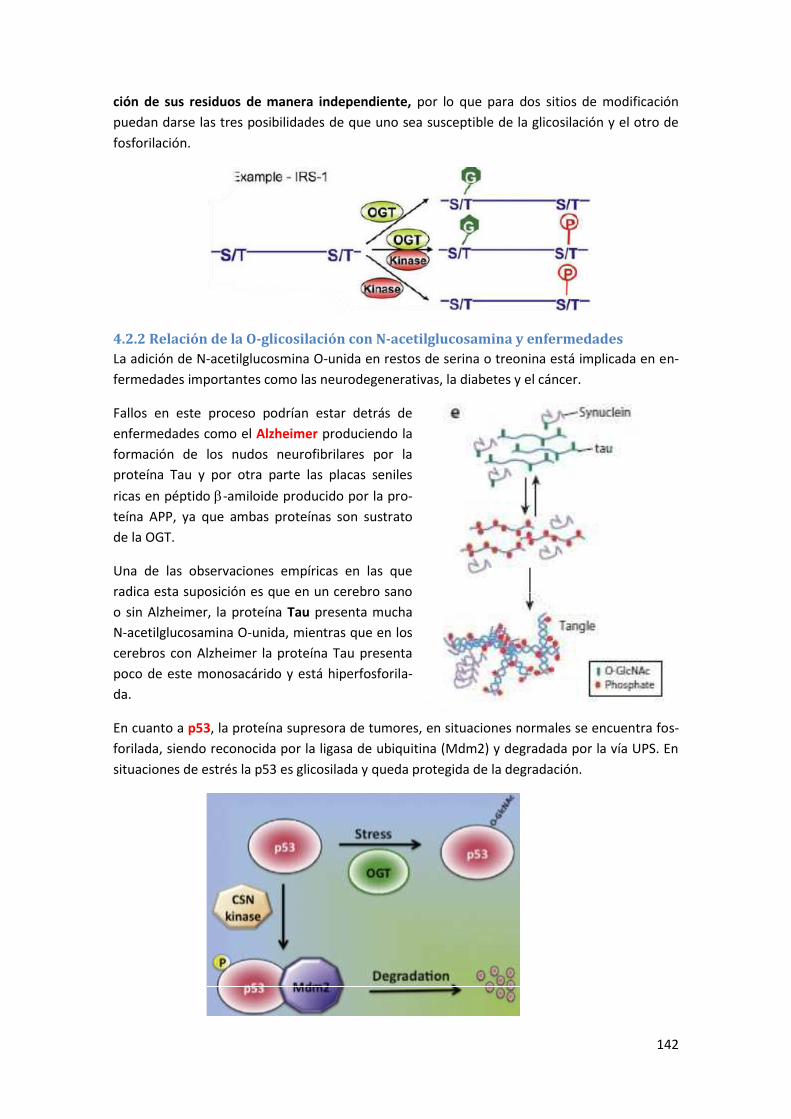
142
ción de sus residuos de manera independiente, por lo que para dos sitios de modificación
puedan darse las tres posibilidades de que uno sea susceptible de la glicosilación y el otro de
fosforilación.
4.2.2 Relación de la O-glicosilación con N-acetilglucosamina y enfermedades
La adición de N-acetilglucosmina O-unida en restos de serina o treonina está implicada en en-
fermedades importantes como las neurodegenerativas, la diabetes y el cáncer.
Fallos en este proceso podrían estar detrás de
enfermedades como el Alzheimer produciendo la
formación de los nudos neurofibrilares por la
proteína Tau y por otra parte las placas seniles
ricas en péptido -amiloide producido por la pro-
teína APP, ya que ambas proteínas son sustrato
de la OGT.
Una de las observaciones empíricas en las que
radica esta suposición es que en un cerebro sano
o sin Alzheimer, la proteína Tau presenta mucha
N-acetilglucosamina O-unida, mientras que en los
cerebros con Alzheimer la proteína Tau presenta
poco de este monosacárido y está hiperfosforila-
da.
En cuanto a p53, la proteína supresora de tumores, en situaciones normales se encuentra fos-
forilada, siendo reconocida por la ligasa de ubiquitina (Mdm2) y degradada por la vía UPS. En
situaciones de estrés la p53 es glicosilada y queda protegida de la degradación.
O-Glicosilación: O-GlcNAc
Ozcan BBA 2010
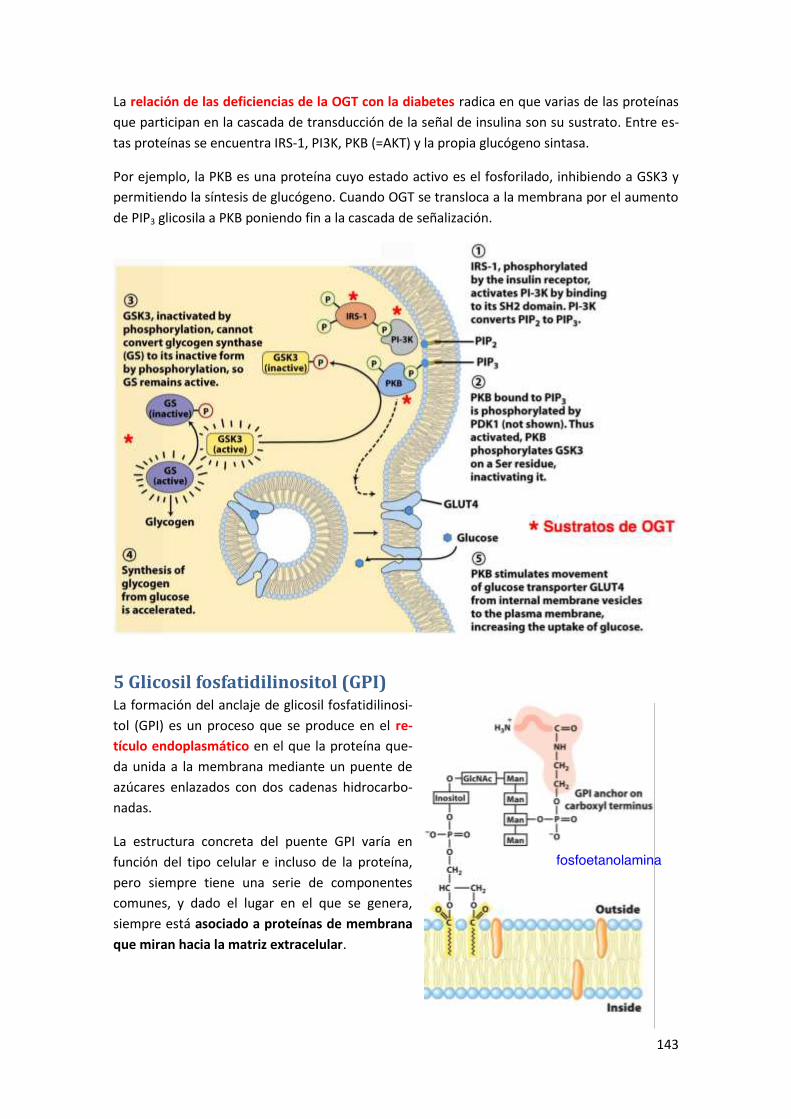
143
La relación de las deficiencias de la OGT con la diabetes radica en que varias de las proteínas
que participan en la cascada de transducción de la señal de insulina son su sustrato. Entre es-
tas proteínas se encuentra IRS-1, PI3K, PKB (=AKT) y la propia glucógeno sintasa.
Por ejemplo, la PKB es una proteína cuyo estado activo es el fosforilado, inhibiendo a GSK3 y
permitiendo la síntesis de glucógeno. Cuando OGT se transloca a la membrana por el aumento
de PIP3 glicosila a PKB poniendo fin a la cascada de señalización.
5 Glicosil fosfatidilinositol (GPI) La formación del anclaje de glicosil fosfatidilinosi-
tol (GPI) es un proceso que se produce en el re-
tículo endoplasmático en el que la proteína que-
da unida a la membrana mediante un puente de
azúcares enlazados con dos cadenas hidrocarbo-
nadas.
La estructura concreta del puente GPI varía en
función del tipo celular e incluso de la proteína,
pero siempre tiene una serie de componentes
comunes, y dado el lugar en el que se genera,
siempre está asociado a proteínas de membrana
que miran hacia la matriz extracelular.
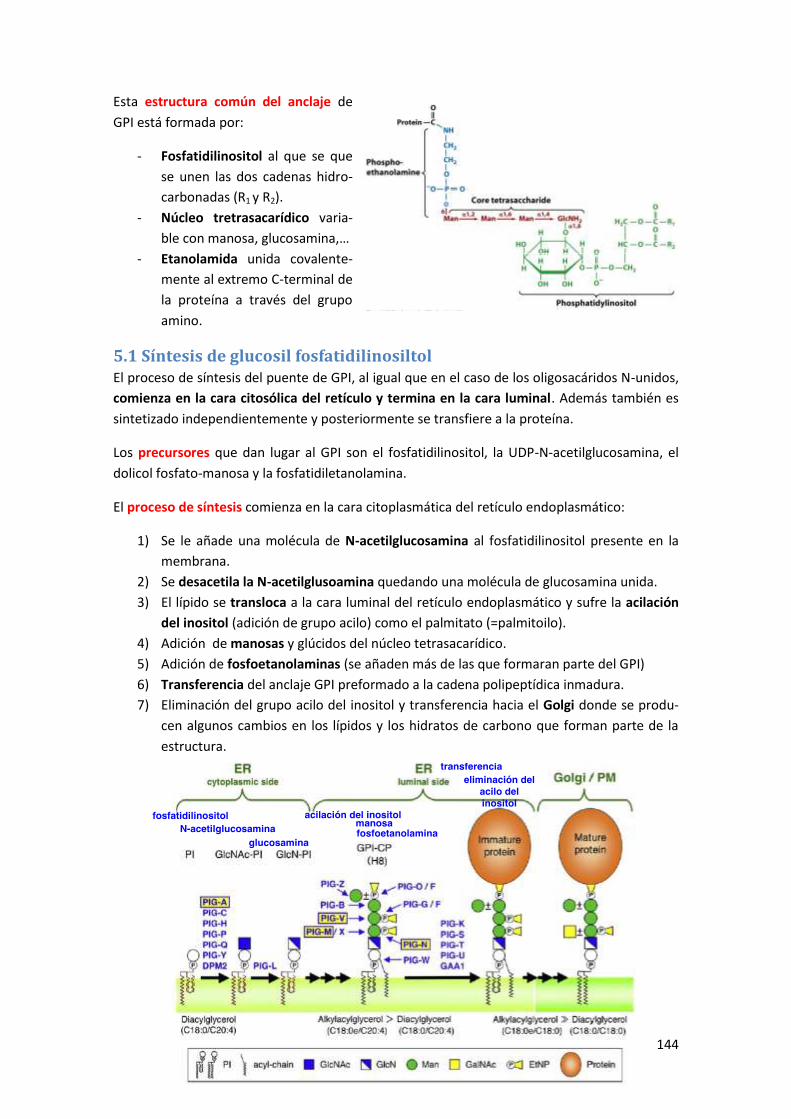
144
Esta estructura común del anclaje de
GPI está formada por:
- Fosfatidilinositol al que se que
se unen las dos cadenas hidro-
carbonadas (R1 y R2).
- Núcleo tretrasacarídico varia-
ble con manosa, glucosamina,…
- Etanolamida unida covalente-
mente al extremo C-terminal de
la proteína a través del grupo
amino.
5.1 Síntesis de glucosil fosfatidilinosiltol El proceso de síntesis del puente de GPI, al igual que en el caso de los oligosacáridos N-unidos,
comienza en la cara citosólica del retículo y termina en la cara luminal. Además también es
sintetizado independientemente y posteriormente se transfiere a la proteína.
Los precursores que dan lugar al GPI son el fosfatidilinositol, la UDP-N-acetilglucosamina, el
dolicol fosfato-manosa y la fosfatidiletanolamina.
El proceso de síntesis comienza en la cara citoplasmática del retículo endoplasmático:
1) Se le añade una molécula de N-acetilglucosamina al fosfatidilinositol presente en la
membrana.
2) Se desacetila la N-acetilglusoamina quedando una molécula de glucosamina unida.
3) El lípido se transloca a la cara luminal del retículo endoplasmático y sufre la acilación
del inositol (adición de grupo acilo) como el palmitato (=palmitoilo).
4) Adición de manosas y glúcidos del núcleo tetrasacarídico.
5) Adición de fosfoetanolaminas (se añaden más de las que formaran parte del GPI)
6) Transferencia del anclaje GPI preformado a la cadena polipeptídica inmadura.
7) Eliminación del grupo acilo del inositol y transferencia hacia el Golgi donde se produ-
cen algunos cambios en los lípidos y los hidratos de carbono que forman parte de la
estructura.
GPI
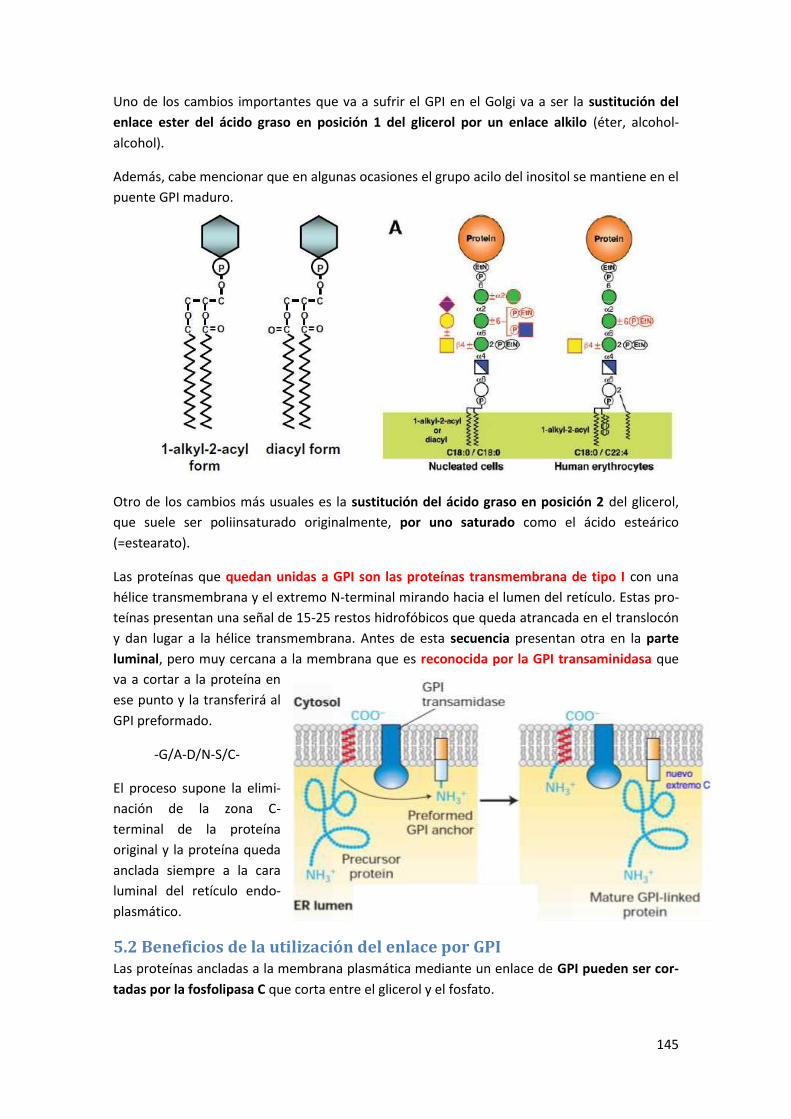
145
Uno de los cambios importantes que va a sufrir el GPI en el Golgi va a ser la sustitución del
enlace ester del ácido graso en posición 1 del glicerol por un enlace alkilo (éter, alcohol-
alcohol).
Además, cabe mencionar que en algunas ocasiones el grupo acilo del inositol se mantiene en el
puente GPI maduro.
Otro de los cambios más usuales es la sustitución del ácido graso en posición 2 del glicerol,
que suele ser poliinsaturado originalmente, por uno saturado como el ácido esteárico
(=estearato).
Las proteínas que quedan unidas a GPI son las proteínas transmembrana de tipo I con una
hélice transmembrana y el extremo N-terminal mirando hacia el lumen del retículo. Estas pro-
teínas presentan una señal de 15-25 restos hidrofóbicos que queda atrancada en el translocón
y dan lugar a la hélice transmembrana. Antes de esta secuencia presentan otra en la parte
luminal, pero muy cercana a la membrana que es reconocida por la GPI transaminidasa que
va a cortar a la proteína en
ese punto y la transferirá al
GPI preformado.
-G/A-D/N-S/C-
El proceso supone la elimi-
nación de la zona C-
terminal de la proteína
original y la proteína queda
anclada siempre a la cara
luminal del retículo endo-
plasmático.
5.2 Beneficios de la utilización del enlace por GPI Las proteínas ancladas a la membrana plasmática mediante un enlace de GPI pueden ser cor-
tadas por la fosfolipasa C que corta entre el glicerol y el fosfato.
Anclaje GPI: Estructura y síntesis
GPI: Estr. y síntesis

146
Además el enlace por puente de GPI permite una difusión más libre en la bicapa, ya que una
proteína anclada por GPI está relacionada con la monocapa externa, mientras que las proteí-
nas transmembrana presentan interacciones con el citoesqueleto, dificultando su difusión.
El anclaje por GPI generalmente dirige a las proteínas a los dominios apicales de la membrana
de las células polarizadas (epiteliales) y en muchos casos se concentran en los rafts lipídicos.
6 Funciones de los glicoconjugados Los glicoconjugados son moléculas que presentan muchos tipos de funciones:
- Los glúcidos son moléculas muy ricas en información ya que las lectinas son proteínas
que reconocen específicamente azúcares en todos lo reinos, incluidos los virus.
o El ejemplo clave es del las selectinas (lecinas que intervienen en el proceso de
reconocimiento y adhesión celular) que van a participar en la extravasación de
leucocitos (selectina-P, Selectina-E y selectina-L) en la implantación del em-
brión en el endometrio (selectina-L) y en la fertilización de óvulos.
o También es muy importante en el reconocimiento intercelular por parte de las
bacterias (Helicobacteri pylori y las úlceras de estómago).
o También hay lectinas virales como la hemaglutinina del virus de la gripe, nece-
saria para la infección.
Dada la gran cantidad de lectinas y su importancia para las interacciones entre células y pató-genos, estas son la diana farmacológica de numerosos tratamiento que van a competir por los receptores de los oligosacáridos con el fin de impedir que el virus reconozca los oligosacáridos,
como en el caso del tamiflu y la relenza, dos medicamentos análogos de los azúcares que reconocen los virus e impiden la infección.
- Los glúcidos también regulan la permanencia de una glicoproteína en el plasma san-
guíneo. La mayoría de las proteínas plasmáticas están glicosiladas con ácido siálico
(ácido N-acetilneuramínico) y conforme envejecen, van perdiendo estos restos glicosí-
dicos por lo que son captadas por el receptor de axialoglicoproteínas del hepatocito
que reconoce las galactosas expuestas en los oligosacáridos produciendo la endocito-
sis y degradación de la proteína.
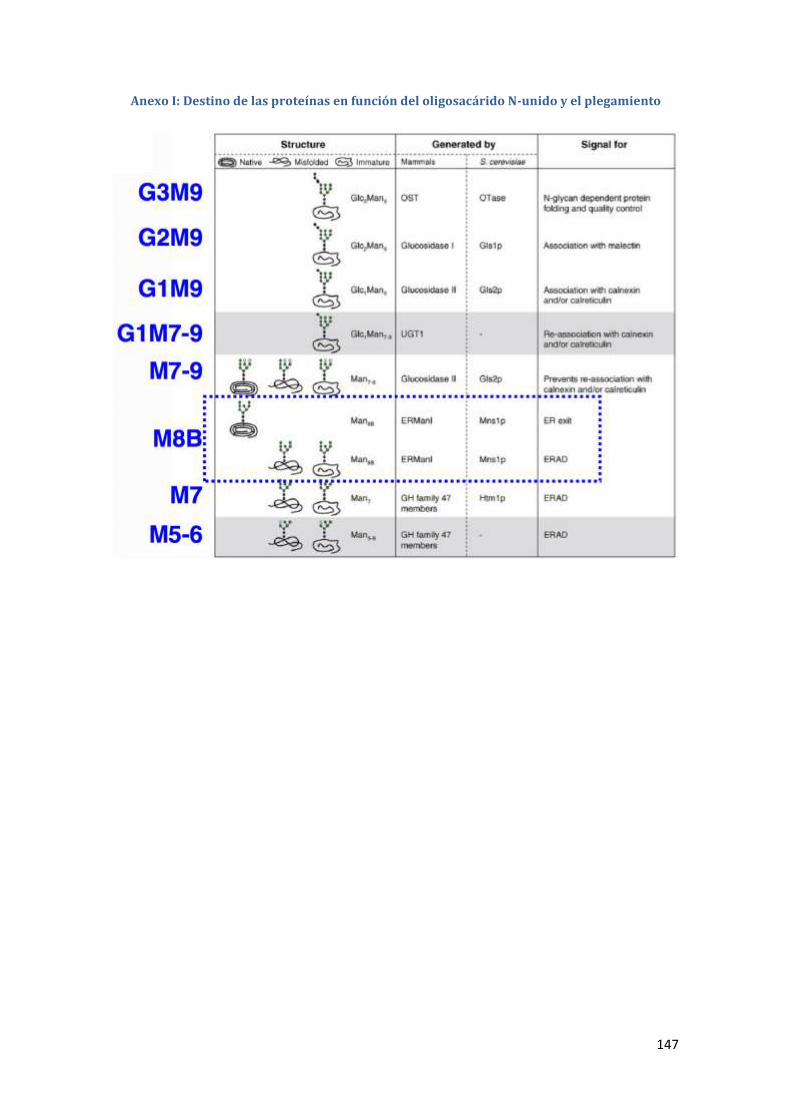
147
Anexo I: Destino de las proteínas en función del oligosacárido N-unido y el plegamiento

148
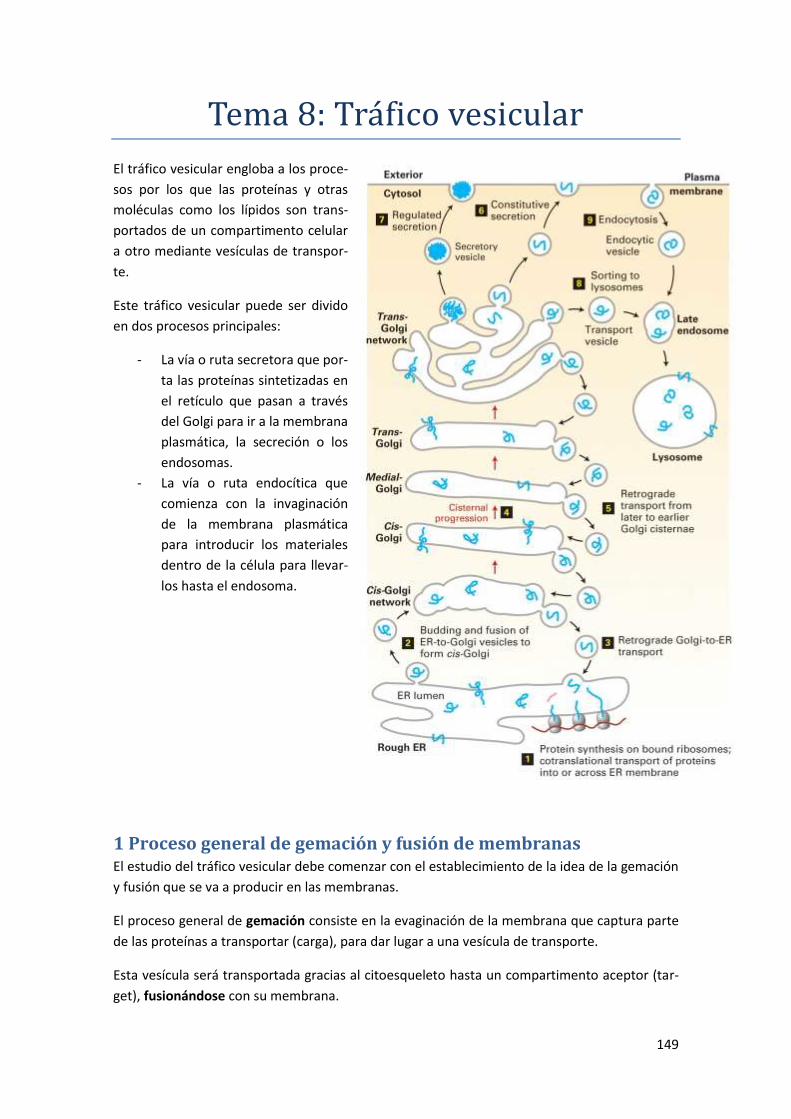
149
Tema 8: Tra fico vesicúlar
El tráfico vesicular engloba a los proce-
sos por los que las proteínas y otras
moléculas como los lípidos son trans-
portados de un compartimento celular
a otro mediante vesículas de transpor-
te.
Este tráfico vesicular puede ser divido
en dos procesos principales:
- La vía o ruta secretora que por-
ta las proteínas sintetizadas en
el retículo que pasan a través
del Golgi para ir a la membrana
plasmática, la secreción o los
endosomas.
- La vía o ruta endocítica que
comienza con la invaginación
de la membrana plasmática
para introducir los materiales
dentro de la célula para llevar-
los hasta el endosoma.
1 Proceso general de gemación y fusión de membranas El estudio del tráfico vesicular debe comenzar con el establecimiento de la idea de la gemación
y fusión que se va a producir en las membranas.
El proceso general de gemación consiste en la evaginación de la membrana que captura parte
de las proteínas a transportar (carga), para dar lugar a una vesícula de transporte.
Esta vesícula será transportada gracias al citoesqueleto hasta un compartimento aceptor (tar-
get), fusionándose con su membrana.
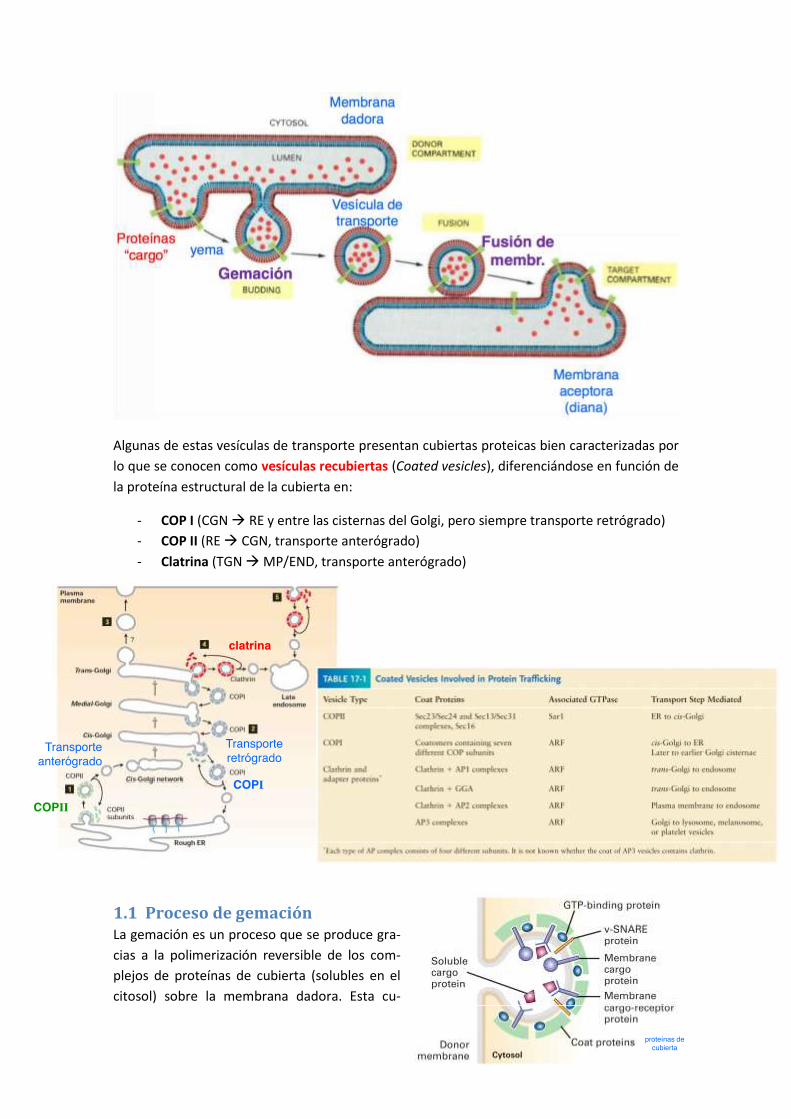
150
Algunas de estas vesículas de transporte presentan cubiertas proteicas bien caracterizadas por
lo que se conocen como vesículas recubiertas (Coated vesicles), diferenciándose en función de
la proteína estructural de la cubierta en:
- COP I (CGN RE y entre las cisternas del Golgi, pero siempre transporte retrógrado)
- COP II (RE CGN, transporte anterógrado)
- Clatrina (TGN MP/END, transporte anterógrado)
1.1 Proceso de gemación La gemación es un proceso que se produce gra-
cias a la polimerización reversible de los com-
plejos de proteínas de cubierta (solubles en el
citosol) sobre la membrana dadora. Esta cu-
Proceso general

151
bierta tiene dos funciones principales:
- Genera la curvatura necesaria para
que se produzca la gemación gra-
cias a su forma y fuerte unión con
los lípidos.
- Selecciona la carga que portará la
vesícula.
El mecanismo de selección de la carga depende de que la proteína a transportar por la vesícula
sea soluble o esté integrada en la membrana:
- Las proteínas integradas en la membrana presentan en su porción citosólica señales
de clasificación (Sorting signals) que van a ser reconocidas por las proteínas de la cu-
bierta.
- Las proteínas solubles en el lumen del orgánulo van a ser reconocidas por receptores
de carga integrados en la membrana, que portan en su porción citosólica señales de
clasificación.
Estas señales de clasificación casi siempre son secuencias de aminoácidos muy cortas. En el
caso de proteínas solubles es por ejemplo la manosa-6-fosfato cuyo receptor de carga va a ser
el receptor de manosa-6-fosfato.
El ensamblado y desensamblado de la cubierta está regulado por la presencia en los tres tipos
de vesículas recubiertas por la presencia de proteínas pequeñas de unión a GTP con actividad
GTPasa que son proteínas monoméricas similares a Ras y forman parte de la superfamilia de
Proceso general
Proceso general

152
las GTPasas de proteínas interruptoras (Switch) que se presentan activas unidas a GTP e inacti-
vas unidas a GDP. Hay dos tipos de estas:
- Sar1 es la que actúa en vesículas recubiertas de COPII.
- ARF es la que actúa en vesículas de COPI y clatrina.
Sar1-GDP es una proteína inactiva y soluble en el citosol que cuando interacciona con un factor
intercambiador de nucleótidos de guanina Sar1-GEF (= Sec12 de la membrana del retículo en-
doplasmático) intercambia GDP por GTP.
Sar1-GTP presenta una conformación abierta que expone su extremo N-terminal hidrofóbico,
el cual se inserta en la bicapa
lipídica citosólica del retículo. Se
forma así el “cimiento” que pro-
mueve la polimerización de la
cubierta de COPII sobre la mem-
brana del retículo que fuerza la
gemación.
Pasado cierto tiempo la actividad
GTPasa de Sar1-GTP hidroliza el
GTP a GDP provocando la disocia-
ción de la cubierta sobre la vesí-
cula ya formada.
1.2 Proceso de fusión Una vez tenemos la vesícula sin cubierta proteica, el siguiente paso consiste en la fusión con la
membrana diana adecuada. Este proceso se conoce como docking de la vesícula o atraque.
En este proceso van a tener mucha importancia las proteínas Rab, otras proteínas pequeñas
de unión a GTP de la familia de las GTPasas de
proteínas interruptoras (Switch).
Rab-GDP se presenta soluble en el citosol,
pero cuando interviene un GEF, sufriendo el
intercmabio de GDP por GTP, Rab-GTP se
ancla a la membrana de la vesícula a través de
un resto isoprenoide.
Rab-GTP es reconocida por el efector de Rab
(receptor) en la membrana de destino y co-
mienza el proceso de fusión mediado por las
proteínas SNARE.
Más adelante, una vez que se produzca la fusión Rab hidrolizará su GTP y se liberara al citosol.

153
Una vez que la vesícula ya está en las proximidades de la membrana, gracias a la acción de Rab
y su efector, para que se produzca la fusión van a intervenir dos tipos de proteínas SNARE:
- v-SNARE están presentes en la vesícula
- t-SNARE están en la membrana diana (target)
Según el proceso de fusión las SNARE que intervienen van a variar, por ejemplo, en la exocito-
sis va a intervenir la v-SNARE del tipo VAMP (Vesicle-
Associated Membrane Protein) concretamente la
VAMP-2 (Sinaptobrevina 2), y las t-SNARE sintaxina y
SNAP-258 (SiNaptosome-Associeated Protein).
Estas proteínas van a interaccionar formando un
complejo SNARE en el que las porciones citosólicas se
enrollan para formar un haz de 4 hélices muy estable
(VAMP-2+Sintaxina+2xSNAP-25) que aproximará las
membranas hasta que se fusionan.
La estabilidad del complejo SNARE de 4 hélices se
debe a la existencia de un patrón repetitivo entre las
diferentes hélices con aminoácidos de características
similares cada 7 restos. Este patrón permite que la
interacción se produzca entre aminoácido hidrofóbi-
cos que quedan mirando hacia el interior de la hélice
y las fuerzas hidrofóbicas los mantengan unidos.
Además se van a formar puentes de hidrógeno muy fuertes entre una arginina (R) de las v-
SNARE y una glutamina (Q) de cada t-SNARE, contribuyendo a la estabilidad del haz. Por ello en
muchas ocasiones se denominará a las t-SNARE como Q-SNARE y a las v-SNARE como R-
SNARE.
8 No confundir con SNAP (Soluble NSF Attachment Protein)
Proceso general: Fusión
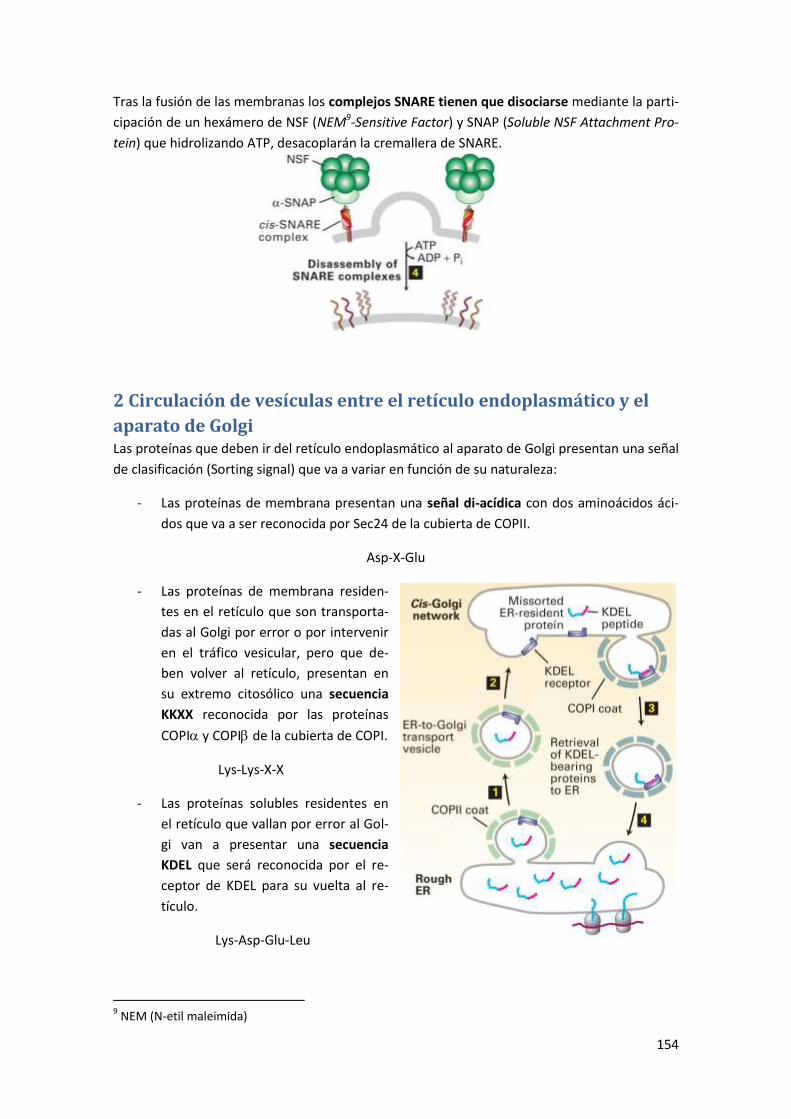
154
Tras la fusión de las membranas los complejos SNARE tienen que disociarse mediante la parti-
cipación de un hexámero de NSF (NEM9-Sensitive Factor) y SNAP (Soluble NSF Attachment Pro-
tein) que hidrolizando ATP, desacoplarán la cremallera de SNARE.
2 Circulación de vesículas entre el retículo endoplasmático y el
aparato de Golgi Las proteínas que deben ir del retículo endoplasmático al aparato de Golgi presentan una señal
de clasificación (Sorting signal) que va a variar en función de su naturaleza:
- Las proteínas de membrana presentan una señal di-acídica con dos aminoácidos áci-
dos que va a ser reconocida por Sec24 de la cubierta de COPII.
Asp-X-Glu
- Las proteínas de membrana residen-
tes en el retículo que son transporta-
das al Golgi por error o por intervenir
en el tráfico vesicular, pero que de-
ben volver al retículo, presentan en
su extremo citosólico una secuencia
KKXX reconocida por las proteínas
COPI y COPI de la cubierta de COPI.
Lys-Lys-X-X
- Las proteínas solubles residentes en
el retículo que vallan por error al Gol-
gi van a presentar una secuencia
KDEL que será reconocida por el re-
ceptor de KDEL para su vuelta al re-
tículo.
Lys-Asp-Glu-Leu
9 NEM (N-etil maleimida)

155
Bloque III: Regulación Molecular De La
Acción Hormonal
TEMA 1 Propiedades Funcionales de las Hormonas Liposolubles
Clasificación y propiedades de las hormonas. Propiedades de la interacción hormona-receptor. Hormonas liposolubles: transporte por el plasma. Alternativas para la activación de receptores intracelulares. Dominios estructurales de los receptores. Elementos de res-puesta a hormonas. Agonistas y antagonistas sintéticos y sus efectos. Co-activadores y co-represores. Acciones genómicas y no genómicas. Diversidad de genes regulados por las hormonas esteroideas.
TEMA 2 Mecanismos de Acción de las Hormonas Hidrosolubles
Transducción de señales hormonales. Receptores transmembranales y síntesis de segun-dos mensajeros. Proteínas G triméricas. AMP cíclico, generación y efectos. Proteín-quinasas y proteín-fosfatasas. Fosfolipasa C: activación y síntesis de inositol-tris-fosfato y diacilglicerol. Variantes de proteín-quinasa C y sus efectos celulares. Calmodulina y sus funciones. GMP cíclico. Óxido nítrico.
TEMA 3 Funciones de los Factores de Proliferación Celular
Factores de proliferación celular. Receptores con actividad tirosín-quinasa intrínseca o diferida. Dominios SH1, SH2 y SH3. MAP quinasas. Receptores con actividad tirosín-fosfatasa. Otros receptores. Origen del cáncer: protooncogenes, oncogenes y anti-oncogenes.

156

157
Tema 1: Hormonas liposolúbles y receptores intracelúlares
La comunicación intercelular, así como la que se produce entre las células y los tejidos, puede
llevarse a cabo mediante cuatro formas fundamentales:
- La comunicación dependiente de contacto es aquella que se produce mediante el con-
tacto directo de dos células a través de proteínas de la membrana que actúan como
señales. Cuando una de estas proteínas de membrana colisiona con el receptor ade-
cuado se produce la transducción de la señal que puede ser transmitida a ambas célu-
las.
- La comunicación paracrina es aquella que se produce a través de un mediador quími-
co liberado por una célula y detectado a nivel local de donde se produce la liberación.
Un ejemplo concreto sería la señalización autocrina en la que la propia célula capta el
estímulo emitido por ella como es el caso de los receptores α-adrenérgicos que son
autoreguladores. Entran en este grupo las prostaglandinas, prostacilcinas, linfoquinas y
citoquinas.
- La comunicación sináptica se caracteriza porque la distancia que recorre el neuro-
transmisor es muy pequeña y los mediadores son liberados al espacio sináptico.
- La comunicación endocrina es aquella en la que el mediado químico viaja largas dis-
tancias a través del torrente sanguíneo.

158
1 Acción hormonal Las hormonas se han clasificado generalmente según su naturaleza química en función del
compuesto del que derivan. Podemos establecer por tanto cuatro clases fundamentales:
- Hormonas derivadas de aminoácidos como
las que derivan de la tirosina (adrenalina) y
la tiroxina (tri- y tetraiodotironina que son
liposolubles).
- Hormonas polipeptídicas que representan la mayor variedad de lo que se entiende
como hormonas. Presentan un tamaño variable desde hormonas de 2-3 aminoácidos
hasta 100-150 aminoácidos. Son la insulina, el glucagón, todas las hormonas del eje del
hipotálamo e hipófisis, la hormona del crecimiento (GH), todas las del tracto gastroin-
testinal… Estas hormonas presentan una naturaleza hidrosoluble y pueden reaccionan
de dos formas:
o Con receptores de la membrana como los receptores de somatotropina, re-
ceptores de LH dando lugar a una transducción de la señal a través de segun-
dos mensajeros que desencadenan la señal a nivel de proteínas.
o Con receptores de la membrana que también pueden actuar a través de se-
gundos mensajeros pero cuyo cambio es a nivel génico y por lo tanto más len-
to.
- Las hormonas de naturaleza esteroidea incluyen a los gluco- y mineralocorticoides,
adrenógenos, estrógenos, gestágenos, el ácido retinóico (vitamina A), el 1-25-dihidroxi
vitamina D2, D3… Se trata de hormonas no hidrosolubles (liposolubles) que van a te-
ner paso libre a través de las membranas que se van a unir a receptores intracelulares.
Los derivados del ácido retinoico que presentan algunos productos de belleza, sobre todo si se usan junto con radiación solar pueden unirse a receptores nucleares que llevan a la transcripción de genes que pueden conllevar efectos perjudiciales.
- Las hormonas eicosanoides derivadas del ácido araquidónico como las protaglandinas,
prostaciclinas y tromoboxanos (derivado del ácido araquidónico) son hormona liposo-
lubles que van a unirse a receptores de membrana desencadenando la liberación de
segundos mensajeros.
La cantidad de hormonas que circulan por el torrente sanguíneo es extraordinariamente baja,
en el mejor de los casos del orden nanomolar (10-9), por lo que para determinar si una persona
presenta un problema hormonal es necesario recurrir a pruebas de radio-inmunoensayos (RIA)
en los que se utilizan anticuerpos y ligandos radiactivos para evaluar las concentraciones hor-
monales.
1.1 Proteínas carrier de hormonas liposolubles y fracción libre Las hormonas liposolubles como las que se vierten desde la tiroides y la corteza adrenal son
muy hidrofóbicas, por lo que van a precisar de proteínas de transporte general o proteínas

159
carrier, como la albúmina u otras más específicas. La regulación de los niveles de este tipo de
proteínas va a afectar a la cantidad de hormona disponible (libre).
La albúmina es una proteína carrier del torrente circulatorio que tiene la capacidad de unir
compuestos tan diversos como la bilirrubina o los derivados del ácido glucurónico gracias a
una estructura con huecos de regiones hidrofóbicas. La unión de hormonas liposolubles a la
albúmina siempre será con una unión o afinidad muy baja, con respecto a proteínas carrier
más específicas.
La globulina ligante de corticoesteroides (CBG, Corticosteroid
binding globulin) es una proteína carrier específica que une al
80% de los 17-hidroxiesteroides. Por ejemplo, el cortisol en
el torrente circulatorio se presenta en un 70% unido a CBG,
un 22% unido a albúmina y un 8% libre que puede formar
agregados y es la que entra en la célula.
La globulina ligante de hormonas sexuales (SHBG, Sexual
hormone-binding globulin) liga específicamente andrógenos y
estradiol, pero con menos afinidad que la albúmina. La canti-
dad de SHBG se reduce durante la pubertad, por lo que el
nivel de testosterona libre (89-87% con albúmina, 10% con
SHBG y 1-3% libre) aumenta provocando la aparición de los
caracteres sexuales secundarios.
La globulina ligante de andrógenos (ABG, Androgen-binding
globulin) suministra la testosterona para la síntesis de proteínas
en espermatozoides, y la globulina ligante de tiroides (TBG,
Thyroid-binding globlulin) transporta las hormonas tiroideas.
La fracción de hormona liposoluble libre en el torrente circula-
torio es la que es capaz de entrar en la célula y unirse a sus
receptores, de manera que, por una parte nos encontramos las hormonas formando un com-
plejo con las proteínas carrier (CH) y la hormona libre (H) que oscila entre el 3-10% del total.
Estas concentraciones se encuentran en realidad en un equilibrio en el que también van a par-
ticipar los receptores (R), ya que serán los que formen complejos con las hormonas (HR), reti-
rando hormona libre del equilibrio y desplazándolo hacia la disociación de la proteína carrier,
de acuerdo con la ley de acción de masas.
Posteriormente la liberación de la hormona por parte del receptor puede invertir el desplaza-
miento para recuperar el equilibrio.
En términos de constante de afinidad y de disociación, la afinidad del receptor por la hormona
es altísima, es decir, su constante de afinidad oscila entre 109-1012 M-1, por lo que el equilibrio
está muy desplazado hacia la formación de complejos hormona-receptor. Por ende, la cons-
tante de disociación es muy baja, del mismo orden pero en negativo, lo que significa que la

160
hormona y el receptor se encuentran muy poco disociados a una determinada concentración
de ambos.
1.2 Características de la interacción hormona-receptor Al igual que la interacción antígeno-anticuerpo, la interacción hormona-receptor es una inter-
acción de alta afinidad (constante de afinidad muy alta, constante de disociación muy baja)
pero de carácter no covalente y se trata de una interacción saturable, al igual que la de una
enzima con su sustrato.
Según los casos, se trata de una interacción muy específica y estereoespecífica, ya que no
presenta la misma afinidad por todos los estereoisómeros.
Es una interacción sensible a los agentes que modifican o cambian la estructura del receptor
o del ligando (hormona). Por ejemplo, un cambio en la secuencia de la somatostatina (poli-
peptídica) cambia su afinidad por el receptor, y las mutaciones en los receptores están relacio-
nadas con muchas patologías, tanto si provocan un aumento como un descenso de la afinidad.
Los receptores presentan ligandos que actúan como agonistas (su unión al receptor mimetiza
el efecto del ligando natural) y otros como antagonistas (ocupan el receptor pero no lo acti-
van).
2 Agonistas y antagonistas de los receptores de andrógenos Los receptores de andrógenos y estrógenos del interior celular, tienen huecos hidrofóbicos
porque el estrógeno es hidrofóbico. Por lo que un determinado plaguicida o un compuesto
aromático se puede unir a estos huecos aunque con baja afinidad. En cualquier caso la ocupa-
ción o no dependerá tanto de la afinidad como de la concentración del compuesto ya que a
altas concentraciones de ciertos plaguicidas los renacuajos tardan 2 días en hacer la metamor-
fosis (Activación de receptores de hormonas sexuales), mientras que lo normal es que tarden
24 días.
En el caso de los receptores de andrógenos uno de sus agonis-
tas naturales la androstenediona (andrógeno). El dianabol (me-
tandrostenolona) es un andrógeno sintético prohibido que se ha
utilizado para el engorde del ganado y el aumento de la masa
muscular. Su abuso se ha relacionado con adenomas hepatoce-
lulares que son el paso previo a un hepatoma o cáncer de híga-
do.
Los esteroides anabolizantes inducen la secreción de testos-terona en hombres, que da lugar a la atrofia del testículo y al aumento de las mamas. En las mujeres, el efecto conlleva una disminución de la ovulación y de la secreción de estrógenos, además de una regresión de la mama.

161
El tamoxifeno y el raloxifeno son reguladores de los receptores de estrógenos que actúan
como antagonistas en unos tejidos y agonistas en otros.
El tamoxifeno es un compuesto utilizado en la terapia contra el cáncer de mama tras la mas-
tectomía total o parcial y la aplicación de la quimioterapia o la radioterapia, para que evitar
que el cáncer reaparezca. Pero a largo plazo, la administración de tamoxifeno se ha relaciona-
do con la inducción del cáncer de útero.
Este efecto, inhibidor por una parte (antagonismo) e induc-
tor por otra (agonismo), se debe a que el receptor de estró-
genos va a desencadenar respuestas diferentes en función
del tejido. En el tejido mamario, el tamoxifeno provoca la
inhibición de una ruta de transducción que culmina con el
reclutamiento de moduladores que promueven la expresión
de oncogenes. Por el contrario, en el útero esta ruta culmina
con el reclutamiento de moduladores impiden la expresión
de oncogenes, por lo que su inhibición provoca la expresión.
Al fin y al cabo el receptor de estrógenos estrogenos es un factor de transcripción que se une
al DNA una vez activado, pero en unos casos será para reclutar Co-activadores y en otros Co-
represores, que según el tipo celular serán comunes o no, desencadenando respuestas dife-
rentes.
3 Modelo general del receptor de hormonas liposolubles Se han caracterizado más de 50 genes humanos para los distintos receptores intracelulares
de hormonas liposolubles y en muchos no se conoce el ligando, es decir, muchos de ellos son
genes putativos (homólogos de secuencia), cuyo funcionamiento no se ha comprobado. Por
ejemplo, se conoce la existencia de receptores de compuestos aromáticos con esta estructura
y que son inductores de cáncer, por no se ha caracterizado el ligando.
Todos los receptores de hormonas liposolubles presentan una estructura general común for-
mada por entre 500 y 1000 aminoácidos que consta de:
- Región o dominio de unión al ligando (225-285 aá) en la porción C-terminal.
- Región o dominio de unión al DNA (65-70 aá) en la porción central.
- Región o dominio de trans-activación (100-500 aá), que es variable, en la porción N-
terminal.

162
Atendiendo a la homología de secuencia (identidad de aminoácidos) de los dominios de los
diferentes receptores, podemos observar que el dominio de unión al DNA está muy conserva-
do (homología 42-94%), el dominio de unión al ligando está algo menos conservado (homolo-
gía 15-57 %) lo que es lógico porque aporta la especificidad en la unión con las diferentes hor-
monas, y finalmente la región de trans-activación no presenta homología.
Es lógico pensar que el dominio de unión a hormonas presente homología ya que por ejemplo, el corti-sol no dista mucho del estradiol o la testosterona, estructuralmente hablando, por lo que la zona que constituye los huecos hidrofóbicos tendrá que presentar semejanzas. En cambio no es lo mismo la estructura de la tiroxina la del ácido retinoico por lo que en ese caso habrá menos homología de se-cuencia entre ambos receptores.
3.1 Elementos de respuesta, coactivadores y localización de receptores El dominio de unión al DNA de los re-
ceptores intracelulares de hormonas
liposolubles se va a unir a los elemen-
tos de respuesta a hormonas en el
DNA (secuencias de pares de bases).
Mientras que el dominio unión al ligan-
do, en el caso del receptor de estróge-
nos, presenta entre sus hélices la de-
nominada hélice 12 que sobresale de la
proteína cuando el ligando no está uni-
do en el hueco.
La unión de la hormona al receptor provoca un cambio de conformación que retrae la hélice
12 y permite que el factor de transcripción (receptor) sea reconocido por los moduladores
(Co-activadores/represores).
Los antagonistas del receptor de estrógenos, como el tamoxifeno, provocan un cambio de conforma-ción parcial que impide que el receptor sea reconocido por los moduladores.
También el receptor de estrógenos al que la unión del estradiol provoca la conformación com-
pacta con capacidad de unión al DNA y los co-activadores.

163
Pero como ya hemos mencionado, el tamoxifeno se une con más afinidad al receptor provo-
cando una conformación plegada, pero diferente, que no es competente para el reclutamiento
de co-activadores.
3.1.1 Co-activadores
Una de estas familias de co-activadores son los de la familia p160 que presentan varios domi-
nios importantes en su secuencia:
- El dominio básico de hélice-bucle-hélice que presenta una estructura de pinza que se
une al DNA.
- El dominio PAS que participa en las interacciones proteína-proteína.
- El dominio de interacción con el receptor de hormonas nucleares que presenta en su
secuencia secuencias de LXXLL que son los que se unirán al receptor hormonal con la
hélice 12 plegada.
- El dominio de interacción con otros co-activadores como P300 y CBP10.
3.1.2 Elementos de respuesta para los receptores de hormonas esteroideas
En la tabla podemos observar los elementos de respuesta a hormonas (HREs, Hormone res-
ponse Elements) a los que se unen
los receptores de hormonas este-
roideas como el receptor de andró-
genos, el receptor de glucocorticoi-
des, el de ácido retinoico (vitamina
A), el de vitamina D y el de las hor-
monas tiroideas. Además de otros
receptores (RX) que forman hete-
rodímeros con los receptores del
ácido retinoico o el de vitamina D.
Si observamos la secuencia del elemento de respuesta (secuencia de pares de bases) los recep-
tores de ácido retinoico, los de vitamina D y los de hormonas tiroideas se unen al mismo
elemento de respuesta, solo diferenciado por el tamaño de la región bisagra. Esto se debe a
que estos receptores dimerizan (Como FOS, JUN, NFκB,…) dando lugar a homodímeros. Pero
10
CBP = CREB binding proteín, CREB = CRE-binding protein y CRE = cAMP response element.

164
cuidado, como hemos mencionado antes, algunos de ellos puede dimerizar con otros recepto-
res y dar lugar a heterodímeros.
Esto establece dos puntos de colisión (unión) con el DNA para los receptores dimerizados, que
en el caso de interactuar con otros co-activadores o co-represores presentarán 4 puntos coli-
sión con el DNA [2x(1 del receptor + 1 por cada co-activador)]. Si no hay dimerización adecua-
da no se produce una fijación adecuada al DNA.
Los experimentos que implican el estudio de receptores y el modo de acción de estos son
complicados porque la cantidad de estos que presenta una célula es muy pequeña. Para identi-
ficarlos hay que llevar a cabo un seguimiento que permita su purificación, requiriéndose en
muchos casos ligandos radiactivos.
Además, es necesario utilizar tejidos con un alto contenido de receptores de esta clase, como
son el tejido mamario, el ovárico o los testículos.
3.1.3 Localización de receptores de hormonas liposolubles
Los receptores de hormonas liposolubles ejercen su acción en el núcleo, pero se van a distri-
buir entre el citosol y el núcleo, según el tipo.
- Los receptores de glucocorticoides se localizan en el citosol y no entran al núcleo has-
ta que unen el ligando.
- Los receptores de andrógenos y estrógenos se sitúan en el núcleo, pero en un estado
no competente, como unidades independientes que precisan del ligando para dimeri-
zar y formar los homodímeros.
- Los receptores heterodiméricos como los de las hormonas tiroideas, los de ácidos re-
tinoicos, el de la vitamina D2 o D3, se sitúan siempre unidos a su elementos de res-
puesta pero asociados a co-represores en ausencia del ligando. Esto se debe a que el
sitio de interacción con el modulador es excluyente, es decir, o tiene unido un co-
represor o tiene unido un co-activador.
4 El receptor de glucocorticoides humano
4.1 Motivo de dedos de cinc en el receptor de glucocorticoides El dominio de unión al DNA del receptor de glucocorticoides presenta un motivo estructural
conocido como dedos de cinc caracterizado por la presencia de moléculas de cinc coordinadas
a través de cuatro restos de cisteína (C).

165
Además, esta estructura alberga el loop que permite la dimerización de los receptores (negri-
ta) cuya detección se realizó porque la mutación de la alanina (A458) a treonina (T) impide la
dimerización o la hace defectiva.
Si no se produce esta dimerización no hay un posicionamiento correcto y fijo sobre el DNA lo
que impide la expresión adecuada de los genes.
4.2 Estructura y variantes del receptor de glucocorticoides El gen del receptor de glucocorticoides humano codifica para una estructura en la que vamos a
encontrar 4 dominios diferentes:
- Dominio NTD (N-terminal domine) en el extremo N-terminal que es regulador.
- Dominio DBD (DNA binding domain) en el centro por el que se produce la unión al
DNA.
- Dominio HR (Hinge región) que es la región bisagra.
- Dominio LBD (Ligand-binding domain) en el extremo C-terminal que es el de unión al
ligando.
En función del proceso de maduración del mensajero podremos encontrar dos variantes (iso-
formas) resultado del empalme alternativo del exón 9α (hGRα) o 9β (hGRβ), que presentan
diferente número de aminoácidos.
Dentro de estos dominios estructurales se han caracterizado varios dominios funcionales que
determinan el modo de actuación de los receptores de glucocorticoides. Estos receptores se
encuentran solubles en el citosol interaccionando con chaperonas en ausencia del ligando.
Cuando la hormona liposolu-
ble difunde al citosol e im-
pacta con el receptor este
muestra sus secuencias de
localizan nuclear (dominio
funcional) y es importado al
núcleo.

166
Además de las dos secuencias de localización, los receptores de glucocorticoides presentan
dominios funcionales de trans-activación (en NTD), de dimerización11 (de DBD-LBD) y una re-
gión de unión a las proteínas de choque térmico (HSPs).
Podemos observar el mecanismo de funcionamiento en el que el receptor de glucocorticoides
se encuentra amordazado en el citosol por las HSPs, hasta que la aparición del ligando lo libera
y entra al núcleo donde dimeriza y se une a sus elementos de respuesta.
El receptor de glucocorticoides, como muchos otros, puede formar homodímeros o hetero-
dímeros con otros factores
de transcripción, pero en
este último caso, es el factor
de transcripción el que de-
termina el elemento de res-
puesta, que será el suyo pro-
pio.
Eventualmente el ligando se
disocia del receptor y las ex-
portinas los vuelven a trans-
portar al citosol.
4.3 Modelo funcional del receptor de hGR El modelo general de actuación de los receptores de hormonas hidrosolubles que se ha men-
cionado con anterioridad es aplicable a todos ellos y se basa en el cambio conformacional so-
bre la hélice 12 que produce la unión del ligando.
Si observamos uno de estos receptores sin el ligando unido, podemos ver como las proteínas
inhibidoras interaccionan con el lugar de unión al ligando y por lo tanto van a competir por
ese lugar, pero tienen menos afinidad.
Cuando el ligando interviene la proteína inhibidora es desplazada y el receptor sufre un cambio
conformacional que lo pliega y le permite unirse al DNA dando lugar a una estructura sobre la
que se ensamblan los co-activadores que van a permitir la transcripción génica.
11
Las mutaciones en las regiones de dimerización afectan negativamente a la funcionalidad del recep-tor.

167
Concretamente podemos observar la estructura del hGRα cuando presenta unido un agonistas
(hélice plegada) y cuando presenta un antagonista (hélice extendida). Esta hélice 12 es crítica
para construcción de la maquinaria de transcripción.
Cuando el receptor de glucocorticoides (GR) une a su ligando forma dímeros que se unen a sus
elementos de respuesta (GREs)
y son reconocidos por co-
activadores como p160, CAF y
p300/CBP, formando conjuntos
de hasta 30-40 proteínas que
representan la maquinaria de
transcripción en la que se intro-
ducen las RNA polimerasas.
Como podemos ver, la regula-
ción de los niveles de GR se
produce a través de la vía UPS.
5 Modelos de actuación de los receptores nucleares (NR) Para entender como los receptores de hormonas liposolubles tenemos que tener en cuenta
que estos receptores nucleares (NR), para permitir la expresión de genes, van a tener que mo-
dificar la estructura de la cromatina a fin de que las polimerasas, helicasas y toda la maquinaria
de transcripción pueda acceder.
Este cambio en la estructura de la
cromatina está mediada por las
proteínas remodeladoras de la
cromatina que van a ser recluta-
das por los receptores unidos a su
elemento de respuesta en el DNA.
Entre estos remodeladores tene-
mos las histonas desmetilasas
(HDMs), las histonas desacetilasas
(HDACs), ATPasas, Histona metil-
transferasas (HMTs), histona ace-

168
tiltransferasas (HATs)… en muchos casos dependientes del gaso de ATP y que en función de la
situación van a actuar como co-activadores o co-represores activando o silenciando la expre-
sión génica.
Por ejemplo la hipoacetilación y la metilación de la histona H3 en la lisina9 (H3K9) se relaciona
con una represión por condensación de la cromatina, mientras que la hiperacetilación o la
metilación en la histona H3 en la lisina 4 (H3K4) se relaciona con expresión por cromatina des-
condensada.
Algunos factores de crecimiento también van a colaborar con esta remodilación mediante la
fosforilación de proteínas que va a llevar a la activación de los receptores.
Por lo tanto, los complejos correguladores pueden estar bajo el control de rutas de señaliza-
ción extra- e intracelulares que perciben los cambios en el entorno y el estado nutricional de
la célula y desencadenan cambios epigenéticos apropiados.
En algunos casos, como en los receptores de hormonas liposolubles heterodiméricos que se
presentan unidos a sus elementos de respuesta pero de forma no competente (las hormonas
tiroideas, los de ácidos retinoicos, el de la vitamina D2 o D3), cuando no presentan ligando, la
hélice 12 actúa como lugar de unión reconocido por los complejos de co-represores que man-
tienen el promotor silenciado.
Cuando llega el ligando, la hélice 12 se pliega, como si de un interruptor se tratase, y los com-
plejos de co-activadores entran uniéndose a las secuencias LXXLL y provocando la remodela-
ción de la cromatina que permite la transcripción.
5.1 Desensibilización de receptores Un concepto importante es la desensibilización de receptores que se produce como respuesta
a una exposición prolongada a un ligando. Llega un momento en el que, a pesar de que se
mantengan los niveles de ligando, no se produce más respuesta.
Los dos motivos que explican este hecho es porque se produce una bajada en la afinidad del
receptor por el ligando o porque la cantidad de receptores disminuye. En muchos casos esta
bajada en el nivel de receptores se debe a que ellos mismos actúan como reguladores negati-
vos de los genes que los codifican, por lo que mientras que se produce la respuesta se reprime
la síntesis de receptores hasta que llega un momento en el que son degradados.
En otras ocasiones el receptor actúa como una señal de expresión de ribonucleasas que de-
gradan al mensajero que codifica el receptor. En cualquier caso, la forma más rápida de des-
ensibilización es la degradación del receptor.
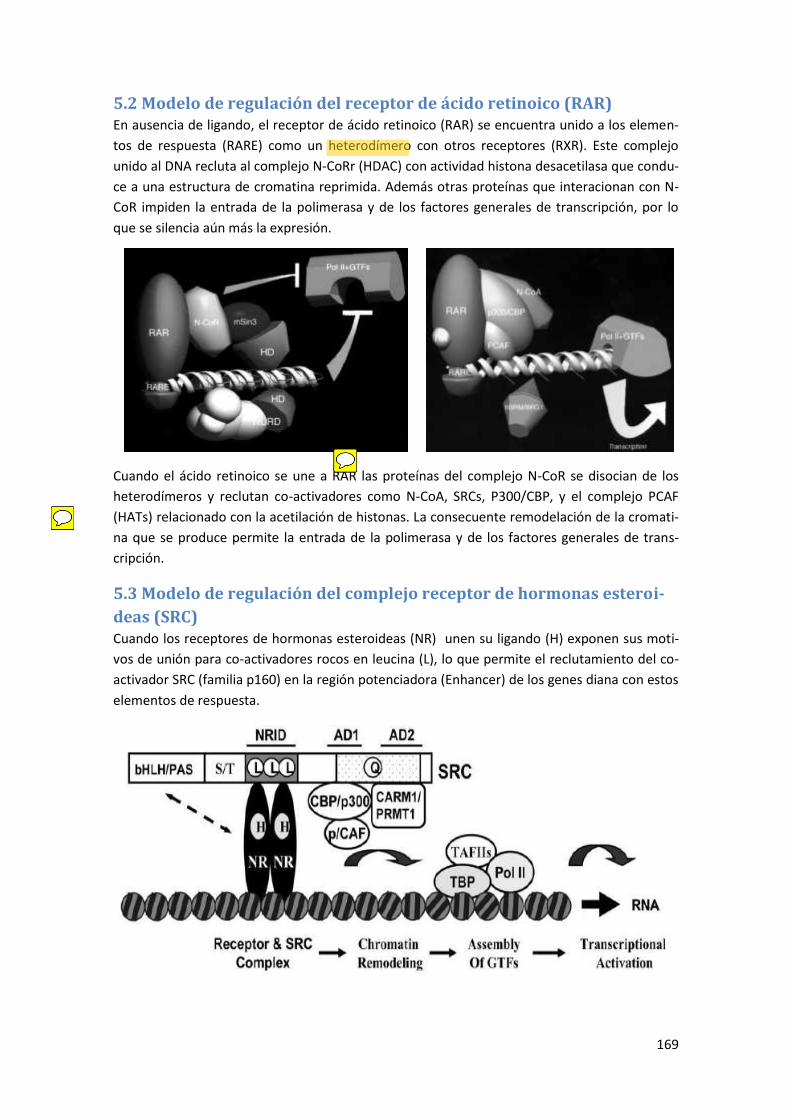
169
5.2 Modelo de regulación del receptor de ácido retinoico (RAR) En ausencia de ligando, el receptor de ácido retinoico (RAR) se encuentra unido a los elemen-
tos de respuesta (RARE) como un heterodímero con otros receptores (RXR). Este complejo
unido al DNA recluta al complejo N-CoRr (HDAC) con actividad histona desacetilasa que condu-
ce a una estructura de cromatina reprimida. Además otras proteínas que interacionan con N-
CoR impiden la entrada de la polimerasa y de los factores generales de transcripción, por lo
que se silencia aún más la expresión.
Cuando el ácido retinoico se une a RAR las proteínas del complejo N-CoR se disocian de los
heterodímeros y reclutan co-activadores como N-CoA, SRCs, P300/CBP, y el complejo PCAF
(HATs) relacionado con la acetilación de histonas. La consecuente remodelación de la cromati-
na que se produce permite la entrada de la polimerasa y de los factores generales de trans-
cripción.
5.3 Modelo de regulación del complejo receptor de hormonas esteroi-
deas (SRC) Cuando los receptores de hormonas esteroideas (NR) unen su ligando (H) exponen sus moti-
vos de unión para co-activadores rocos en leucina (L), lo que permite el reclutamiento del co-
activador SRC (familia p160) en la región potenciadora (Enhancer) de los genes diana con estos
elementos de respuesta.

170
Además SRC interacciona a través de un dominio rico en glutamina (Q) con CBP, p300, CAF,
CARM1 y PRMT1 que son co-activadores comunes dando lugar a la remodelación de la croma-
tina mediante las actividades acetiltransferasa y metiltransferasa que permite el ensamblado
de los factores generales de transcripción y de la RNA polimerasa II.
5.4 Estructura y dominios estructurales del receptor de vitamina D La estructura de los receptores de vitamina D (VDR) es muy parecida a la de los anteriores, ya
que también presenta un dominio de localización nuclear, un dominio de unión con el DNA, un
motivo en dedos de cinc y una zona de unión al ligando.
Las regiones de activación son las que también están comprometidas con la dimerización que
se va a llevar a cabo para formar heterodímeros con los receptores de otras proteinas (RX).
Estos heterodímeros se unen a los elementos de respuesta y reclutan a co-activadores como
CBP, p300, remodeladores de la cromatina, proteinas activadoras de la familia p160… hasta
formar un holoenzima de 30-40 proteínas que da lugar al complejo de transcripción al que se
acopla la RNA polimerasa.
La importancia de la vitamina D radica en que su falta provoca raquitismo ya que es muy im-
portante para la formación del hueso. Su efecto permite retener el fosfato y el calcio necesa-
rios para dar lugar al hidroxiapatito que lo forma.
Además la vitamina D o la 1,25-dihidroxi-vitamina D como hormona está relacionada con la
regulación de la proliferación celular y la apoptosis, ya que es capaz de inducir la expresión de
50-60 genes en distintos tipos celulares.
Esta hormona detiene la progresión del ciclo celular en el paso de G1 a S, bien directamente o
mediante la inducción de factores de crecimiento como el factor de crecimiento transformante
β (TGFβ, factor de no crecimiento).
La detención es producida porque se va a promover la expresión de los inhibidores de los
complejos Ciclina/CDK, como p21 y p27, que contribuyen a la caída de la actividad quinasa y
conducen a la recuperación de la proteína retinoblastoma (RB) formada por p107 y p130, que
se une a E2F. Impidiendo la expresión de ciclinas.

171
También afecta a la proliferación inhibiendo la señalización por RAS bloqueando el receptor
del factor de crecimiento epidérmico (EGFR) que activaba a RAS, Raf, MEK y ERK para la sínte-
sis de los genes anti-apoptóticos y de proliferación.
Finalmente la vitamina D induce la apoptosis directamente vía inhibición de la anti-apoptótica
Bcl2 y mediante la activación de la pro-apoptótica Bax.
5.5 Acciones genómicas y no genómicas de los receptores de estróge-
nos Los receptores de estrógenos pueden dar lugar a acciones directamente sobre el genoma,
como factores de transcripción y no genómicas reclutando complejos proteicos que disparan
cascadas de transducción.
En cuanto a las acciones genómicas el elemento de respuesta del receptor de estrógenos se
denomina ERE, y a él se unen directamente los receptores dimerizados a los cuales se les pue-
den unir posteriormente co-activadores.
Pero también se puede unir a otros factores de transcripción como Fos y Jun (oncogenes que
forman el complejo AP-1), que lo necesitan para poder iniciar la expresión.
En muchas ocasiones estos complejos de factores de transcripción formados van a tener que
ser fosforilados para ser activos y el propio receptor de hormonas esteroideas va a poder for-
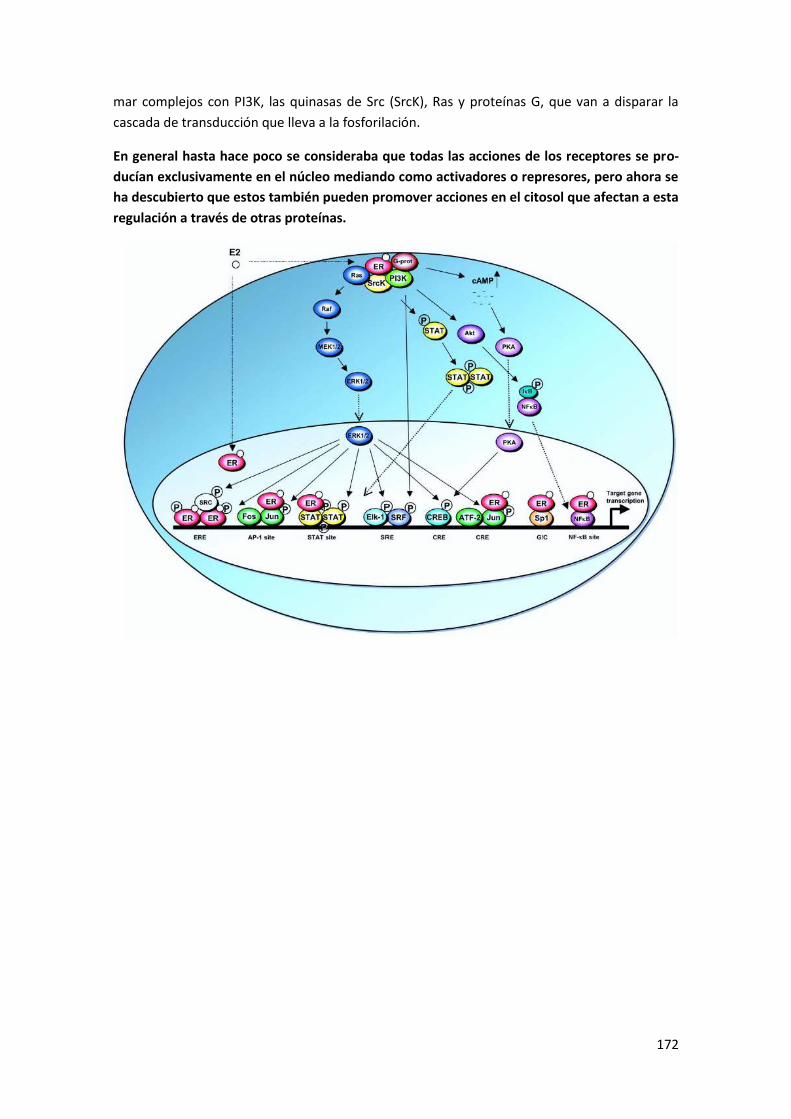
172
mar complejos con PI3K, las quinasas de Src (SrcK), Ras y proteínas G, que van a disparar la
cascada de transducción que lleva a la fosforilación.
En general hasta hace poco se consideraba que todas las acciones de los receptores se pro-
ducían exclusivamente en el núcleo mediando como activadores o represores, pero ahora se
ha descubierto que estos también pueden promover acciones en el citosol que afectan a esta
regulación a través de otras proteínas.

173
Tema 2: Hormonas hidrosolúbles y transdúccio n de sen ales
La inmensa mayoría de las hormonas son de naturaleza polipeptídica y por lo tanto hidrosolu-
bles lo que les impide atravesar las membranas celulares. Esto hace que precisen de recepto-
res de membrana con los que van a colisionar provocando un cambio conformacional en el
receptor que lo va a capacitar para interaccionar con otras proteínas y eventualmente para
exaltar actividades enzimáticas propias del receptor o la producción de segundos mensajeros.
Afortunadamente, el número de segundos mensajeros no es muy elevado por lo que podemos
nombrar AMPc, GMPc, óxido nítrico (NO), diacilglicerol (DAG), cambios en la concentración de
calcio e inositoles trifosfato (IP3).
1 Transmisión de señales La transducción de la señal producida por la colisión entre un ligando y el receptor es la
transmisión de un estímulo que informa a todos los componentes implicados en la respuesta.
Las células responden a gran variedad de estímulos como antígenos, glicoproteínas, señales de
desarrollo, componentes de la matriz extracelular, factores de crecimiento, hormonas, luz,
toque mecánico, neurotransmisores, nutrientes, olores, feromonas y sabores.
Pero en muchos casos, el fin de todas estas señales será la producción de segundos mensaje-
ros que provocarán un cambio en el potencial de membrana que va a llegar hasta regiones
particulares del cerebro para integrar e interpretar las señales.
2 Características de la interacción hormona-receptor Podemos definir cuatro características fundamentales para la interacción entre una hormona y
un receptor:
- Se trata de una interacción específica ya que para cada receptor hay un ligando prefe-
rente, es decir, un ligando que presenta alta afinidad por un receptor concreto, aun-
que este puede ser estimulado por una gran cantidad de moléculas similares como los
receptores α y β adrenérgicos que responden ante adrenalina.
- Es un proceso de amplificación ya que la interacción que provoca la señal presenta
una estequiometria 1:1 pero la activación del receptor produce cambios en más de
una proteína o da lugar a más de un segundo mensajero por lo que la señal se amplifi-
ca. Por ejemplo, la activación de adenilato ciclasa (AC) a través de proteínas G da lugar
a la producción de gran cantidad de AMPc que a su vez activa a muchas moléculas de
protein quinasa A (PKA) y esta fosforila a muchas proteínas.
- Se trata de un proceso sujeto a la desensibilización de receptores, es decir, que se
produce una atenuación de la respuesta cuando la señal permanece en el tiempo o es
excesivamente intensa. En muchos casos la propia activación del receptor desencade-

174
na un mecanismo de feedback o regulación negativa que desencadena la desconexión
del propio receptor o su eliminación de la superficie celular.
- Se trata de un proceso en el que va a producirse una integración de la señal ya que en
la membrana plasmática hay toda una población de receptores que pueden producir
señales positivas o negativas que dan lugar a un aumento o descenso de la actividad
de AC. Por ejemplo, el café disminuye la actividad de la fosfodiesterasa encargada de
la degradación de AMPc.
Los eventos que se pueden producir a lo largo de la transducción de la señal quedan reflejados
a continuación. En muchas ocasiones al principio de la señal suele generarse un conjunto de
proteínas asociadas que se denominan generalmente como relé (Scaffold) que van a actuar
una a continuación de la otra de forma concatenada. Estas proteínas pueden estar reunidas
gracias a la participación de una proteína estructural que las aproxime para aumentar la efi-
ciencia del mecanismo.
A continuación se produce la amplificación de la señal mediada por la producción del segundo
mensajero que va a activar a
numerosas proteínas que a su
vez actuaran sobre otras que
van a estar moduladas por
otros procesos y por lo tanto,
en este punto se produce la
integración de señales positi-
vas y negativas.
Finalmente llegamos a la mo-
dulación de la señal donde se
va a producir un aumento de
proteínas activadoras y un
descenso de las inhibidoras
para que los factores de
transcripción queden libres o
activados y eventualmente
ingresen al núcleo para unirse
a sus elementos de respuesta
en el DNA y se produzca la
transcripción génica.
2.1 Cinética interacción hormona-receptor Los experimentos sobre la interacción ligando-receptor se realizan mediante la utilización de
ligandos radiactivos, como por ejemplo, muscarotoxina radiactiva para el receptor nicotínico.
Generalmente la cinética se representa situando una cantidad creciente de ligando en abscisas
y no es todo radiactivo, sino que se realiza el experimento con una mezcla de ligando radiacti-
vo y no radiactivo cuya concentración puede variar desde 1-100 nM.

175
En 10 tubos de ensayo se introducen las mismas cantidades de proteína (que sabemos que
presentan receptores), como por ejemplo 1 mg, se deja un tubo sin ligando (blanco) y a los
demás se les añaden concentraciones crecientes de ligando.
Se incuban los tubos durante un determinado tiempo y a continuación se pasa el contenido
por un filtro que solo va a dejar pasar las moléculas de bajo peso molecular, es decir, el ligando
no unido.
A continuación pasas los filtros por un con-
tador de centelleo (mide radiactividad)
empezando por el blanco, que nos va a
ofrecer el background inespecífico que
restaremos a las mediciones de receptor
con concentraciones de hormona crecien-
tes, obteniéndose la cantidad de ligando
por miligramo de proteína. Esta curva es la
que se representa como unión total.
Esta unión es saturable, por lo que si se va aumentando la concentración de ligando, llegará un
momento en el que no se ligue más, pero las células o las membranas utilizadas presentan
muchas más proteínas de las que nos interesan y a las que también se puede pegar el ligando.
Por ello se repite el experimento con un antagonista del receptor concreto obteniéndose solo
la señal del ligando que se ha unido inespecíficamente porque los huecos en los receptores van
a estar ocupados por el antagonista.
Se resta este valor a la curva de unión y obtenemos los datos de unión específica.
A partir de estos datos podemos hacer una representación de la hormona ligada al receptor y
la relación entre ligando unido y ligando libre. Esto simplifica el proceso porque cuando tu
realizas un experimento lo que puedes medir es la cantidad de ligando que se ha unido a una
proteína y el resto que ha quedado libre. Por lo que si situamos en ordenadas la cantidad de
ligando (unido y libre), y en abscisas la cantidad de ligando unido, obtendremos una recta de
pendiente – 1/Kd, es decir, la constante de disociación, que cuanto menor sea, más afinidad
tendrá el receptor por la hormona.

176
Podemos observar los valores de la constante
de disociación para diferentes agonsitas natu-
rales de los receptores de adrenalina. La propia
adrenalina presenta Kd=5, el isoproterenol
Kd=0,4 y el antagonista Propranolol Kd=0,0046
por lo que este último es un antagonista increí-
blemente afín al receptor.
3 Modelos de transducción de la señal Existen varios modelos de transducción de la señal que implican a diferentes tipos de recepto-
res y que desencadenan la producción de diferentes actividades.
El caso más estudiado es el de los receptores 7tm que en presencia de ligando se activan y
producen la activación de enzimas que desencadenan la producción de segundos mensajeros a
través de proteínas G triméricas.
Los receptores de factores del crecimiento, diméricos per se o monoméricos que dimerizan,
van a expresar actividad Y-quinasa en presencia del ligando fosforilando a toda una serie de
proteínas que llegarán hasta los factores de transcripción.
Otros clase de receptores son los monómeros que presentan un dominio extracelular de
unión al ligando y un dominio citosólico catalítico. El prototipo es el receptor de la guanilato
ciclasa (GC) de membrana, que ante la interacción con el ligando va a provocar la generación
de GMPc en el corazón, produciendo vasoconstricción y vasodilatación.
Luego también tenemos los canales iónicos que responden a ligandos. Se trata de proteínas
oligoméricas en las que la interacción con el ligando da lugar a una conformación abierta por la
que pueden pasar iones a favor de gradiente de concentración.

177
Quedan los receptores de hormonas liposolubles ya estudiados que atraviesan la membrana y
finalmente los receptores de adhesión como las integrinas que unen moléculas de la matriz y
generan cambios conformacionales que pueden modificar la dinámica del citoesqueleto, como
en el caso de la morfogénesis y la embriogénesis.
4 Sistema adenilato ciclasa y AMPc
4.1 Receptores 7tm acoplados a proteínas G Los receptores 7tm están formados por siete hélices transmembrana que se asocian en su cara
citosólica a proteínas G triméricas que van a desencadenar el proceso que activará a una pro-
teína de membrana inactiva.
Cuando el ligando se une al receptor 7tm se produce un cambio de conformación que afecta a
la proteína G trimérica unida por dos puntos a la membrana. La subunidad Gα sea estimulado-
ra o inhibidora está unida a la membrana mediante un resto de ácido palmítico y la subunidad
Gγ mediante una cola de polisopreno.
La activación de 7tm provoca la separación de Gα que en caso de ser activadora interacciona
con una tercera enzima, como la fosfolipasa C (PLC).
En mamíferos se han identificado 24 genes para la subuindad Gα, 5 para la subunidad Gβ y 11
para la subunidad Gγ, que pueden alternarse para dar lugar a miles de combinaciones.
Dado que se han identificado más de 1000 tipos de receptores 7tm, cada uno de ellos tendrá
asociada una combinación diferente de proteínas G sin necesitar el mismo número de genes.
Este fue uno de los grandes problemas en los años 60, ya que este tipo de receptores parecían
unir miles de ligandos diferentes desencadenando diferentes respuestas y no era posible que
los 7tm tuvieran miles de huecos de unión con ligandos, por lo que hasta el descubrimiento de
las proteínas G como intermediarais entre el receptor y el sistema de transducción, no se llegó
a comprender como se producía el proceso.

178
Los receptores 7tm presentan el extremo N-terminal en el exterior y el C-terminal en el inte-
rior relacionado con la proteína G.
Cuando la proteína G está en reposo presenta una molécula de GDP, pero cuando el receptor
se une al ligando provoca el intercambio de GDP por GTP que provoca la disociación de la
subunidad Gsα (activadora) hasta que la propia actividad GTPasa de la subunidad lo hidroliza y
desconecta el sistema.
Pero no tenemos que olvidar que las subunidades Gβ/γ también tienen actividad biológica, co-
mo en el caso de la motilidad cardiaca donde la adrenalina estimula y la acetilcolina inhibe la
velocidad del ritmo cardíaco. Esto se debe a que los receptores muscarínicos de acetilcolina
están acoplados proteínas Gi que disminuyen la actividad de adenilato cilcasa, pero además las
subunidades β y γ abren canales de potasio que favorecen la hiperpolarización de la membra-
na, atenuando la motilidad cardíaca desde dos puntos.
Para saber si un proceso está mediado por una proteína G se utilizan homólogos no hidroliza-bles de GTP que se unen a las subunidades Gα que van a impedir que el proceso que estamos estudiando pare, por lo que si medimos el aumento del proceso en sí, como la liberación de un compuesto, y vemos que tras añadir el homólogo este se vuelve constitutivo, podremos determinar que hay proteínas G implicadas. Si la estructura del GTP sería GDP-O-P, homólogos no hidrolizables pueden ser GDP-S-P o GDP-NH-P, que presentan un tamaño parecido pero no pueden ser hidrolizados por la célula por presentar nitrógeno o azufre, en vez de oxígeno.
4.2 Estructura de las proteínas G triméricas y modelo de actuación La proteína G monomérica Ras puede ser utilizada co-
mo modelo de la subunidad Gα de las proteínas G tri-
méricas.
En la estructura podemos diferenciar:
- Centro de activación donde se introduce el
GTP-Mg.
- Interruptor “switch” I con un resto de glicina
(Gly60).
- Interruptor “switch” II con un resto de treonina
(Thr35).
- Resto de alanina (Ala146).
Estos aminoácidos son muy importantes porque son
los que se encargan de excluir al ATP del centro de
activación, permitiendo solo la entrada del ATP-Mg.
La unión del GTP a la proteína se produce a través
de la interacción entre los oxígenos del último
fosfato con la treonina y la glicina generando una
estructura muy compacta que puede interaccionar
con adenilato ciclasa, pero en el momento en el que
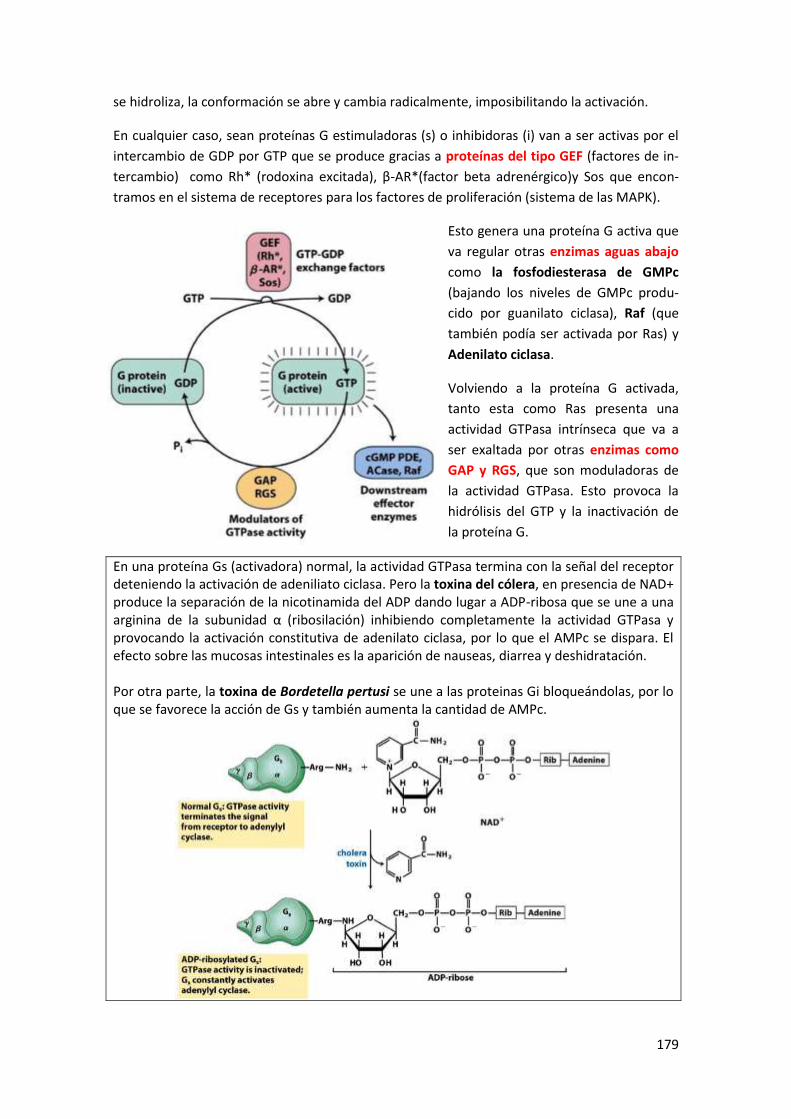
179
se hidroliza, la conformación se abre y cambia radicalmente, imposibilitando la activación.
En cualquier caso, sean proteínas G estimuladoras (s) o inhibidoras (i) van a ser activas por el
intercambio de GDP por GTP que se produce gracias a proteínas del tipo GEF (factores de in-
tercambio) como Rh* (rodoxina excitada), β-AR*(factor beta adrenérgico)y Sos que encon-
tramos en el sistema de receptores para los factores de proliferación (sistema de las MAPK).
Esto genera una proteína G activa que
va regular otras enzimas aguas abajo
como la fosfodiesterasa de GMPc
(bajando los niveles de GMPc produ-
cido por guanilato ciclasa), Raf (que
también podía ser activada por Ras) y
Adenilato ciclasa.
Volviendo a la proteína G activada,
tanto esta como Ras presenta una
actividad GTPasa intrínseca que va a
ser exaltada por otras enzimas como
GAP y RGS, que son moduladoras de
la actividad GTPasa. Esto provoca la
hidrólisis del GTP y la inactivación de
la proteína G.
En una proteína Gs (activadora) normal, la actividad GTPasa termina con la señal del receptor deteniendo la activación de adeniliato ciclasa. Pero la toxina del cólera, en presencia de NAD+ produce la separación de la nicotinamida del ADP dando lugar a ADP-ribosa que se une a una arginina de la subunidad α (ribosilación) inhibiendo completamente la actividad GTPasa y provocando la activación constitutiva de adenilato ciclasa, por lo que el AMPc se dispara. El efecto sobre las mucosas intestinales es la aparición de nauseas, diarrea y deshidratación. Por otra parte, la toxina de Bordetella pertusi se une a las proteinas Gi bloqueándolas, por lo que se favorece la acción de Gs y también aumenta la cantidad de AMPc.

180
En el caso de la motilidad cardíaca vamos a encontrar implicadas a las proteínas G. Por un lado
la acetilcolina frena el ritmo cardíaco, por otro la adrenalina lo acelera.
Existen dos tipos de receptores de acetilcolina:
- Los receptores muscarínicos (M1-5) actúan asociados a proteínas G donde podemos di-
ferenciar entre:
o M1, M3 y M5 que están acoplados a la producción de IP3 mediante aumentos
de calcio.
o M2 y M4 acoplados a proteínas Gi que bajan la actividad de adenilato ciclasa.
El receptor colinérgico muscarínico N2 es el que predomina en la musculatura cardíaca y por
ello su activación provoca una bajada del ritmo cardíaco mediada por Gαi, y además las
subunidades β y γ, se acoplan a canales de potasio que desencadenan una hiperpolarización de
la membrana, relajando el músculo.
- Los receptores nicotínicos son canales iónicos.
Existen multitud de estos sistemas enzimáticos y cada uno de ellos especializado en diferentes
actividades, tal y como se muestra en la tabla donde se indica el tipo de proteína G trimérica y
las subunidades que median en la función.
4.3 Sistema Adenilato ciclasa, AMPc y PKA La enzima adenilato ciclasa es una proteína
monomérica adosada a la membrana plasmáti-
ca que a expensas de ATP-Mg produce AMPc. El
AMPc es una molécula que va a estar regulado
por dos sistemas, la propia adenilato ciclasa,
que lo produce, y la fosfodiesterasa (PDE) de
AMPc que lo degrada a AMP.
Para facilitar la transducción de la señal hasta la
proteinkinasa A (PKA), en muchas ocasiones el
sistema de adenilato ciclasa se encuentra aso-

181
ciado a PKA mediante proteínas de anclaje como AKAP79 (proteína estructural de anclaje de
PKA a adenilato ciclasa) o la gravina (ADAP250) que se asocia a la membrana.
PKA consta de dos subunidades catalíticas (azul) y dos subunidades reguladoras (rojo) que
presentan menor afinidad por las primeras que el AMPc, por ello cuando la concentración au-
menta desde 1 μM a 5 μM, la subunidad catalítica es desplazada por el AMPc, liberándose.
Cada una de las subunidades reguladoras presenta dos sitios de unión para AMPc por lo que
serán necesarias 4 moléculas de
AMPc para provocar el cambio con-
formacional que disminuye la afini-
dad por la subunidad catalítica hasta
en 10.000 veces.
Las subunidades catalíticas de PKA
son activas en el momento en el que
son liberadas, debido a que las
subunidades catalíticas funcionan
como un dominio pseudosustrato
que impedían la actividad enzimática
de PKA.
La PKA regula a un gran número de proteínas a través de su actividad S/T quinasa cuyo sitio de fosforilación se caracteriza por presentar arginina (R) o lisina (K) en posición -1/-2. Para hacer-nos una idea de las señales reguladas a través de AMPc como segundo mensajero tenemos la tabla de la derecha.
El modelo de actuación del sistema comienza con la unión del ligando al receptor que activa a una proteína G que va a interaccionar con adenilato ciclasa. El aumento en la concentración de AMPc provoca la liberación de PKA que va a fosforilar a proteínas como CREB, que se únirá a su elemento de respuesta (CRE) al dimerizar y reclutar co-activadores como CBP y P300. Como podemos ver a propósito del receptor de glucocorticoides, este es un punto en el que ambas vías van a desencadenar el mismo efecto.

182
4.4 Desensibilización de receptores Uno de los aspectos más importantes que ya se han comentado con posterioridad es la desen-
sibilización de los receptores que se produce por una exposición prolongada al ligando y que
desencadena una disminución de la afinidad por el ligando o una disminución del número de
receptores.
La unión del receptor al ligando lo convierte en sustrato de las propias quinasas a las que acti-
va, como PKA, dando lugar a un sistema de retroalimentación negativa. Esta fosforilación se
clasifica como homoérica al ser producida por PKA, o heteromérica cuando se da a través de
una quinasa específica como βARK (quinasa del receptor β-adrenérgico). En cualquier caso, el
reclutamiento de las quinasas dependen de proteínas como las subunidades β y γ de proteí-
nas G disociadas.
Una vez que el receptor es fosfori-
lado su afinidad por la β-arrestina
aumenta, y esta recluta a adaptinas
que van a interaccionar con la cla-
trina para formar vesículas endocí-
ticas que internalizarán los recep-
tores hacia el endosoma temprano.
Es en este momento en el que el
ligando es liberado del receptor, ya
que si se introdujera en la vesícula
endocítica seguiría estimulando al
receptor.
Los receptores permanecerán en
los endosomas hasta ser desfosfori-
lados por protein-fosfatasas mien-
tras la oleada de hormonas dismi-
nuye.
5 Sistema de fosfolipasa C, IP3 y DAG Otro de los segundos mensajeros que van a
actuar en las rutas de transducción celulares es
el inositol 1,4,5-trifosfato (IP3). Se trata de un
ciclohexano hexol (no azúcar, no hay oxígeno
en el ciclo) que presenta seis grupos hidroxilo,
tres de los cuales están enlazados a fosfato.
El IP3 es una molécula muy hidrofílica que va a
actuar como segundo mensajero y no debe ser
confundido con los inositoles-fosfato que pre-
sentan el fosfato en posición 3, que suele estar
relacionada con el anclaje de proteínas.

183
5.1 Sistema de la fosfolipasa C (variante β) La fosfolipasa C se activa mediante la unión de un ligando a los receptores 7tm asociados a
proteínas Gq que provocan el intercambio de GDP por GTP, dando lugar a la separación de la
proteína G.
La proteína Gq activa a la fosfolipasa C que va a
dar lugar a dos segundos mensajeros a partir
del fosfatidilinositol 4,5-bifosfato:
- El IP3 que se va a desplazar hasta los re-
ceptores de IP3 en el retículo endoplas-
mático, que son canales iónicos de cal-
cio que responden a ligando. Cuando se
produce el impacto, los canales se
abren durante un corto periodo de
tiempo provocando una salida masiva
de calcio que va a ser receptado inme-
diatamente por los sistemas pertinen-
tes.
- El diacilglicerol (DAG) que va a difundir
por la cara citosólica de la membrana
plasmática para activar a la proteinqui-
nasa C (PKC) junto con el calcio libera-
do. La vida media del DAG es corta ya
que va a ser rápidamente eliminado por
las fosfolipasas A1 y A2.
Se ha comprobado que la activación de PKC está medida por el calcio gracias a que la adición
de EDTA (acomplejante de cationes divalentes) impide su activación.
En cuanto a las concentraciones de cada uno de estos se-
gundos mensajeros en el caso del IP3 puede variar desde 10-
9 hasta 10-8 mientras que el calcio oscila entre 10-7 (0,1 μM)
hasta 10-6 (1 μM), pudiendo alcanzar en algunas ocasiones 5
μM.
La PKC está implicada en la proliferación celular por lo que la utilización de esteres de forbol,
que mimetizan las acciones del DAG van a provocar una activación muy poderosa del sistema
que inducirá la formación de tumores.

184
Pero estos ésteres presentan una vida media muy larga y por ello sobreactivan a la PKC, como
es el caso de acetato de miristoilforbol. Aunque la naturaleza de sus colas hidrofóbicas y del
propio núcleo es distinta, tiene una gran afinidad por PKC y provoca una proliferación celular
excesiva, lo que demuestra la relación de la quinasa con la iniciación del ciclo celular.
A groso modo, PKC activa a numerosos factores de transcripción como AP1 (Fos y Jun), muy
importantes en el paso de G0 a G1. En la tabla podemos ver algunas de las señales que actúan
a través de la fosfolipasa C, el IP3 y las variaciones en la concentración de calcio.
Además, algunos factores de transcripción como Tubby precisa de la acción de la fosfolipasa
C (no de PKC) para activarse. El factor de transcripción Tubby se encuentra asociado a la mem-
brana plasmática a través del fosfatidilinositol-4,5-bifosfato, por lo que hasta que la fosfolipasa
C no separe el DAG y el IP3, no queda libre, mostrando sus señales de importanción al núcleo.
5.2 Sistema fosfolipasa C (Variante γ) Además de lo ya comentado, existe una variante de la fosfolipasa C, la PLCγ que se va a activar
a través de receptores con actividad Y-quinasa, como el del factor de crecimiento derivado de
plaquetas.
Cuando PDGF impacta con el receptor se produce la fosforilación cruzada que da lugar a la
aparición de restos de Y-fosfato en el dominio citosólico, que pueden ser reconocidos por pro-
teínas como PI3K, GAP y la PLCγ, que se activa, en este caso sin la intermediación de proteínas
Gq.
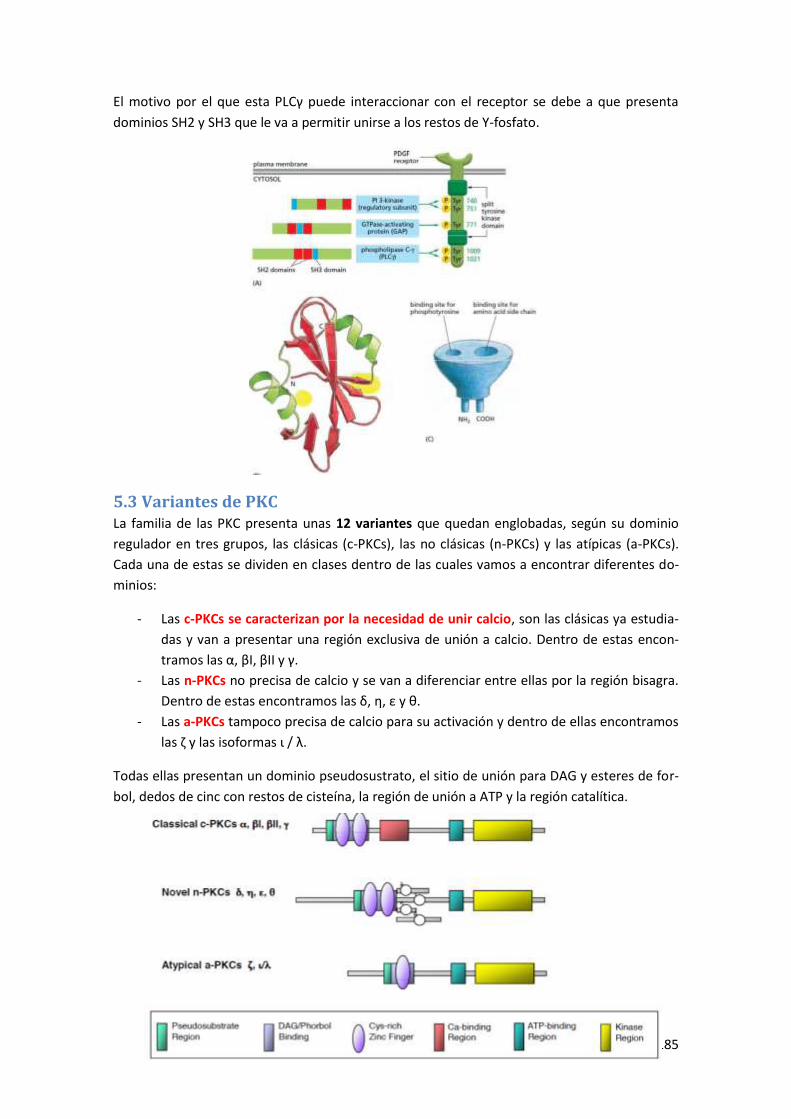
185
El motivo por el que esta PLCγ puede interaccionar con el receptor se debe a que presenta
dominios SH2 y SH3 que le va a permitir unirse a los restos de Y-fosfato.
5.3 Variantes de PKC La familia de las PKC presenta unas 12 variantes que quedan englobadas, según su dominio
regulador en tres grupos, las clásicas (c-PKCs), las no clásicas (n-PKCs) y las atípicas (a-PKCs).
Cada una de estas se dividen en clases dentro de las cuales vamos a encontrar diferentes do-
minios:
- Las c-PKCs se caracterizan por la necesidad de unir calcio, son las clásicas ya estudia-
das y van a presentar una región exclusiva de unión a calcio. Dentro de estas encon-
tramos las α, βI, βII y γ.
- Las n-PKCs no precisa de calcio y se van a diferenciar entre ellas por la región bisagra.
Dentro de estas encontramos las δ, η, ε y θ.
- Las a-PKCs tampoco precisa de calcio para su activación y dentro de ellas encontramos
las ζ y las isoformas ι / λ.
Todas ellas presentan un dominio pseudosustrato, el sitio de unión para DAG y esteres de for-
bol, dedos de cinc con restos de cisteína, la región de unión a ATP y la región catalítica.
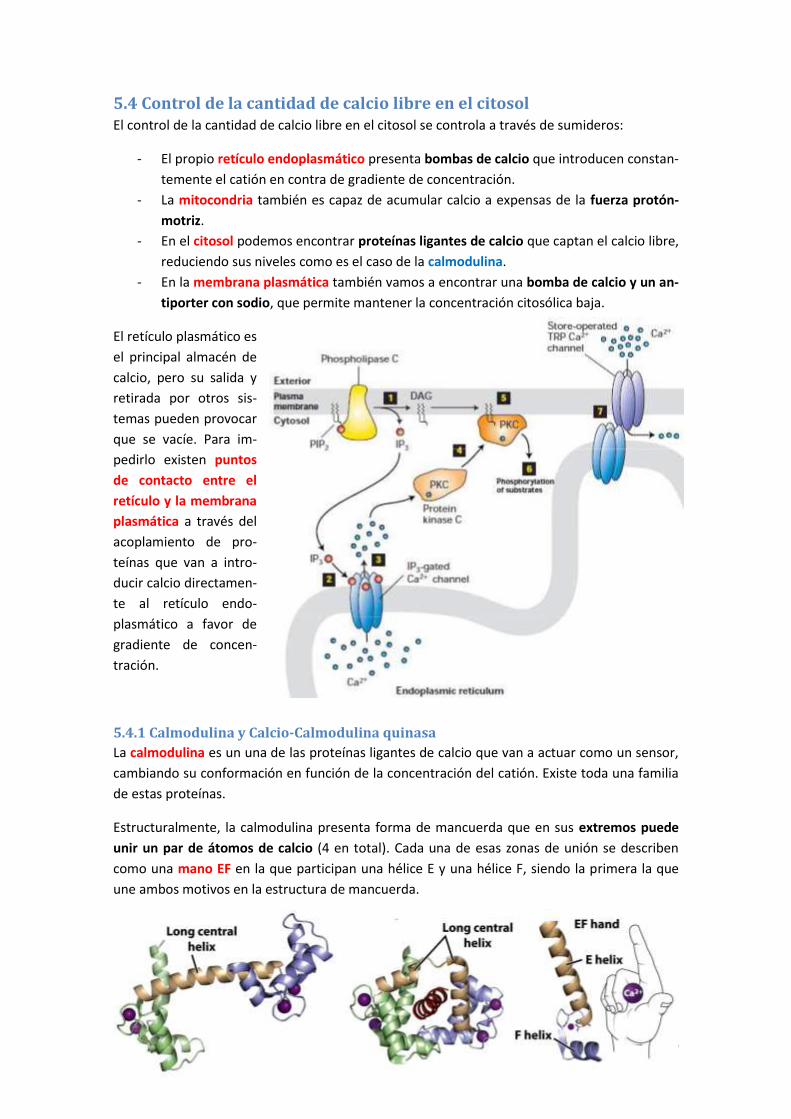
186
5.4 Control de la cantidad de calcio libre en el citosol El control de la cantidad de calcio libre en el citosol se controla a través de sumideros:
- El propio retículo endoplasmático presenta bombas de calcio que introducen constan-
temente el catión en contra de gradiente de concentración.
- La mitocondria también es capaz de acumular calcio a expensas de la fuerza protón-
motriz.
- En el citosol podemos encontrar proteínas ligantes de calcio que captan el calcio libre,
reduciendo sus niveles como es el caso de la calmodulina.
- En la membrana plasmática también vamos a encontrar una bomba de calcio y un an-
tiporter con sodio, que permite mantener la concentración citosólica baja.
El retículo plasmático es
el principal almacén de
calcio, pero su salida y
retirada por otros sis-
temas pueden provocar
que se vacíe. Para im-
pedirlo existen puntos
de contacto entre el
retículo y la membrana
plasmática a través del
acoplamiento de pro-
teínas que van a intro-
ducir calcio directamen-
te al retículo endo-
plasmático a favor de
gradiente de concen-
tración.
5.4.1 Calmodulina y Calcio-Calmodulina quinasa
La calmodulina es un una de las proteínas ligantes de calcio que van a actuar como un sensor,
cambiando su conformación en función de la concentración del catión. Existe toda una familia
de estas proteínas.
Estructuralmente, la calmodulina presenta forma de mancuerda que en sus extremos puede
unir un par de átomos de calcio (4 en total). Cada una de esas zonas de unión se describen
como una mano EF en la que participan una hélice E y una hélice F, siendo la primera la que
une ambos motivos en la estructura de mancuerda.

187
Cuando la concentración de calcio pasa del estado basal (0,1 μM) al entorno de 0,5 μM, la
calmodulina liga cálcio y sufre un cambio conformacional que expone regiones hidrofóbicas
que le van a permitir interaccionar con gran variedad de proteínas como la calcio-calmodulina
quinasa que es una S/T quinasa.
Un ejemplo de acontecimientos relacionados con la calmodulina es la exocitosis de las vesícu-
las sinápticas que depende de la concentración de calcio en la terminal. La concentración de
canales de calcio dependientes de voltaje es muy alta en la terminal sináptica y cuando llega
un potencial de acción estos se abren, aumentando la concentración de calcio. El aumento de
la concentración de calcio es detectado por la calmodulina, que activa a la calcio-calmodulina
quinasa 2 que comienza a fosforilar a las proteínas del citoesqueleto, liberando las vesículas
que se fusionan con la membrana para verter su contenido al espacio sináptico.
Procesos como la secreción de saliva, insulina y otras hormonas como la histamina son tam-
bién dependientes de calcio.
La calcio calmodulina quinasa es una proteína oligomérica con un dominio catalítico y un
dominio inhibitorio que va a presentar memoria.
Cuando la calmodulina con calcio se une a la quinasa, esta sufre un cambio conformacional
que mediante el uso de ATP realiza una primera autofosforilación alcanzando el estado de
activación completa.
Al bajar la concentración de calcio, la calmodulina se disocia, pero la quinasa sigue parcial-
mente activa (50%) debido a la autofosforilación hasta que las fosfatasas correspondientes
retiren el resto de fosfato.
Por lo tanto, la actividad de calcio-calmodulina quinasa va a depender de la concentración de
calcio, la disponibilidad de calmodulina y fosfatasas.
5.4.2 Proteínas reguladas por calcio y calmodulina
Entre las proteínas reguladas por los niveles de calcio a través de la calmodulina podemos ci-
tar:
- Adenilato ciclasa en el cerebro.
- Calcio-calmodulina quinasa I-IV (CaMK I-IV)
- Liberación de calcio desde el retículo sarcoplasmático a través de canales dependien-
tes de calcio.
- Calcineurina (fosfoprotein fosfatasa 2B)
- Fosfodiesterasa de AMPc
- Canales olfativos abiertos por AMPc
- Glutamato descarboxilasa (GlutamatoGABA). El glutamato es, junto a la acetilcolina,
uno de los neurotransmisores excitatorios a nivel de sistema nervioso, pero el GABA es
inhibitorio, por lo que altos niveles de glutamato pueden desencadenar convulsiones,
mientras que altos niveles de GABA conducen a un estado depresivo. La descarboxilasa
utiliza fosfato de piridoxal como cofactor.
- Quinasas y sintasas varias como la óxido nítrico sintasa, que cataliza la formación del
segundo mensajero.

188
- PI3K (oncogén) que fosforila a los fosfolípidos de inositol en posición 3 (anclaje a qui-
nasas dependientes de fosfoinosítidos relacionadas con la supervivencia celular). Por
lo que la fosfatasa correspondiente sería un anti-oncogen.
- Bomba de calcio de la membrana plasmática que va a funcionar o no dependiendo de
la concentración de calcio y por ende de calmodulina.
5.4.3 Canales de calcio receptores de IP3
Los receptores de IP3 situados en la membrana del retículo endoplasmático son canales que se
abren por la unión del ligando. Estos receptores se encuentran agrupados gracias al citoesque-
leto formando islas de canales que están mayoritariamente cerrados a bajas concentraciones
de IP3 por lo que la concentración
de calcio es muy pequeña.
Cuando la concentración de IP3 es
suficientemente alta, toda una de
estas islas de receptores se abre y
se produce un disparo en los nive-
les de calcio. Por los efectos de la
difusión del IP3 estas islas se van
abriendo y cerrando progresiva-
mente dando lugar a una onda de
calcio, que va recorriendo la su-
perficie del retículo.
Finalmente el IP3 va siendo degra-
dado rápidamente por las fosfata-
sas que retiran el fosfato de la
posición 4 o 5, inactivándolo co-
mo segundo mensajero.
5.4.4 Evaluación experimental de los aumentos de calcio
Dado que el mensaje producido al final es el cambio en la concentración de calcio para eva-
luar este cambio en el laboratorio tenemos varias estrategias.
Mediante el uso de electrodos específicos para medir los niveles de calcio podemos evaluar la
capacidad de la fracción microsomal para acumular calcio. Para ello comenzamos calibrando el
electrodo mediante una preparación a la que añadimos calcio y EDTA alternativamente, com-
probando que se produce una bajada al añadir el quelante.
A continuación en una preparación ponemos la fracción microsomal y calcio, y medimos el
nivel de calcio libre con el electrodo. Si el retículo está en condiciones adecuadas, la adición de
ATP-Mg pondrá en marcha las bombas de calcio y se producirá una bajada en la señal.
Estableciendo en que unidades cae la cantidad de calcio libre, podemos determinar el acúmulo
de calcio por cantidad de proteína. Es decir, que si hemos utilizado 0,1 mg de retículo y se ha
producido una caída del orden de 500 nmoles de calcio, la capacidad de acúmulo será de 5
μmol de calcio por cada mg de proteína.
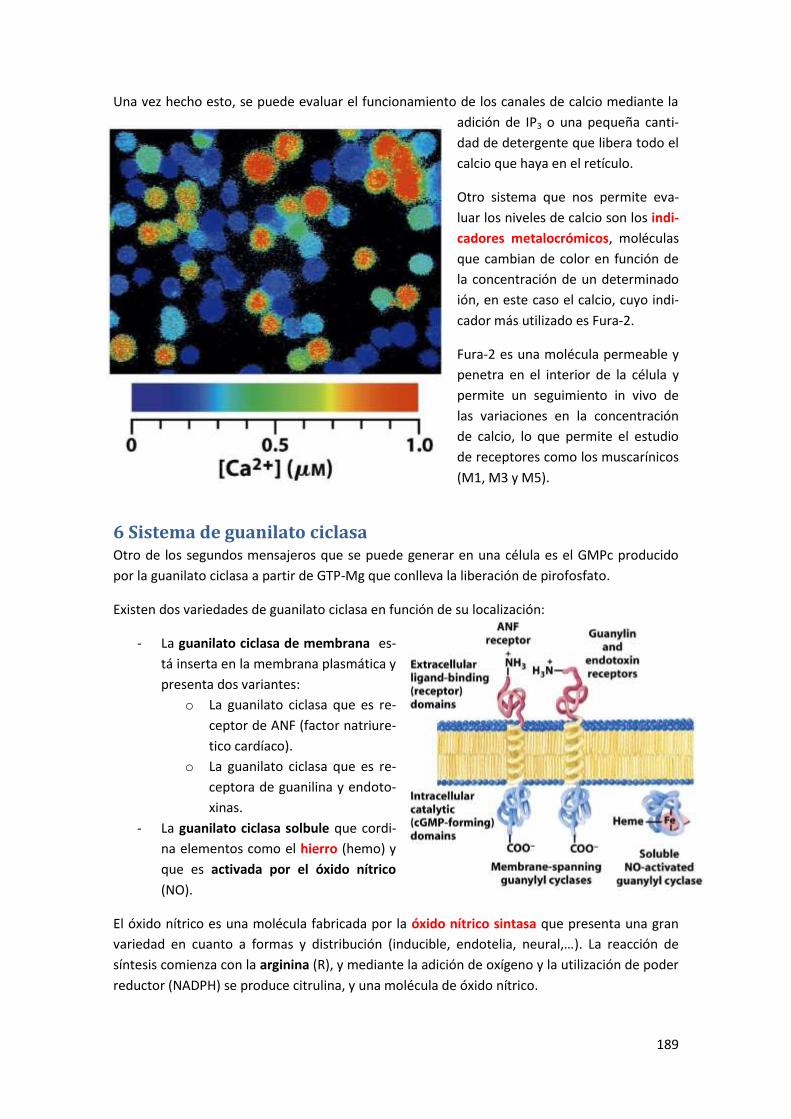
189
Una vez hecho esto, se puede evaluar el funcionamiento de los canales de calcio mediante la
adición de IP3 o una pequeña canti-
dad de detergente que libera todo el
calcio que haya en el retículo.
Otro sistema que nos permite eva-
luar los niveles de calcio son los indi-
cadores metalocrómicos, moléculas
que cambian de color en función de
la concentración de un determinado
ión, en este caso el calcio, cuyo indi-
cador más utilizado es Fura-2.
Fura-2 es una molécula permeable y
penetra en el interior de la célula y
permite un seguimiento in vivo de
las variaciones en la concentración
de calcio, lo que permite el estudio
de receptores como los muscarínicos
(M1, M3 y M5).
6 Sistema de guanilato ciclasa Otro de los segundos mensajeros que se puede generar en una célula es el GMPc producido
por la guanilato ciclasa a partir de GTP-Mg que conlleva la liberación de pirofosfato.
Existen dos variedades de guanilato ciclasa en función de su localización:
- La guanilato ciclasa de membrana es-
tá inserta en la membrana plasmática y
presenta dos variantes:
o La guanilato ciclasa que es re-
ceptor de ANF (factor natriure-
tico cardíaco).
o La guanilato ciclasa que es re-
ceptora de guanilina y endoto-
xinas.
- La guanilato ciclasa solbule que cordi-
na elementos como el hierro (hemo) y
que es activada por el óxido nítrico
(NO).
El óxido nítrico es una molécula fabricada por la óxido nítrico sintasa que presenta una gran
variedad en cuanto a formas y distribución (inducible, endotelia, neural,…). La reacción de
síntesis comienza con la arginina (R), y mediante la adición de oxígeno y la utilización de poder
reductor (NADPH) se produce citrulina, y una molécula de óxido nítrico.
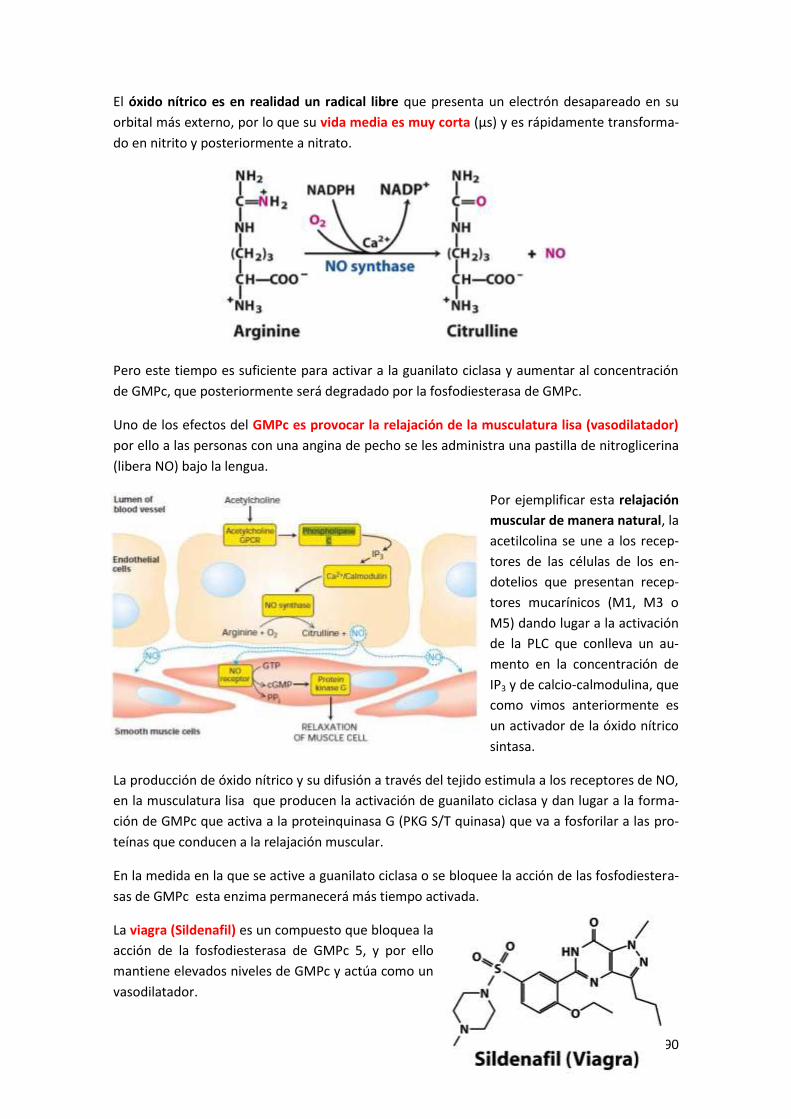
190
El óxido nítrico es en realidad un radical libre que presenta un electrón desapareado en su
orbital más externo, por lo que su vida media es muy corta (μs) y es rápidamente transforma-
do en nitrito y posteriormente a nitrato.
Pero este tiempo es suficiente para activar a la guanilato ciclasa y aumentar al concentración
de GMPc, que posteriormente será degradado por la fosfodiesterasa de GMPc.
Uno de los efectos del GMPc es provocar la relajación de la musculatura lisa (vasodilatador)
por ello a las personas con una angina de pecho se les administra una pastilla de nitroglicerina
(libera NO) bajo la lengua.
Por ejemplificar esta relajación
muscular de manera natural, la
acetilcolina se une a los recep-
tores de las células de los en-
dotelios que presentan recep-
tores mucarínicos (M1, M3 o
M5) dando lugar a la activación
de la PLC que conlleva un au-
mento en la concentración de
IP3 y de calcio-calmodulina, que
como vimos anteriormente es
un activador de la óxido nítrico
sintasa.
La producción de óxido nítrico y su difusión a través del tejido estimula a los receptores de NO,
en la musculatura lisa que producen la activación de guanilato ciclasa y dan lugar a la forma-
ción de GMPc que activa a la proteinquinasa G (PKG S/T quinasa) que va a fosforilar a las pro-
teínas que conducen a la relajación muscular.
En la medida en la que se active a guanilato ciclasa o se bloquee la acción de las fosfodiestera-
sas de GMPc esta enzima permanecerá más tiempo activada.
La viagra (Sildenafil) es un compuesto que bloquea la
acción de la fosfodiesterasa de GMPc 5, y por ello
mantiene elevados niveles de GMPc y actúa como un
vasodilatador.

191
Tema 3: Factores de proliferacio n celúlar. Origen del ca ncer
En 1982 se produjo un hallazgo trascendental que sentaría las bases del estudio del cáncer y
fue el descubrimiento de la proteína Src con actividad Y-quinasa. La proteína Src o proteína
del sarcoma de Rous presenta una variante celular C-Src y una variante de origen viral V-Src.
Cuando un organismo era infectado con el virus portador de V-Src desarrollaba tumores, por lo
que se estableció una relación directa entre la actividad Y-quinasa y la proliferación celular, y
por ende, un punto de estudio clave contra el cáncer.
1 Receptores con actividad Y-quinasa El estudio de los receptores con actividad Y-quinasa revelo que todos estaban formados por un
dominio externo, un dominio transmembrana y un domino citosólico que era el que desarro-
llaba la actividad catalítica Y-quinasa.
El dominio citosólico presenta una actividad intrínseca muy baja que es exaltada cuando el
dominio externo recibe el impacto del ligando, pero este último presenta una gran variabili-
dad pudiendo encontrarse dímeros, como el receptor de insulina (INS-R), otros que dimerizan
como el receptor del factor de crecimiento derivado de plaquetas (PDGF-R) que presenta do-
minios tipo IgG, el receptor del factor de crecimiento de los endotelios vasculares (VEGF-R), el
receptor del factor de crecimiento epidermal (EGF-R). También aparecen dominios externos
ricos en leucina como es el caso del receptor del factor de crecimiento neural (NGF-R). Pero
todos ellos con el mismo dominio citosólico de actividad Y-quinasa.
En estos receptores podemos establecer tres niveles de actividad, por un lado la actividad
basal propia, por otro la actividad que presentan cuando se unen al ligando y finalmente el
nivel de actividad máximo tras producirse la autofosforilación cruzada.
La mayoría de las células se encuentran quiescentes hasta que reciben en sus receptores el
impacto de un factor de crecimiento que desencadena un aumento en la cantidad de ciclinas
mitóticas que permiten el paso de G0 a G1.
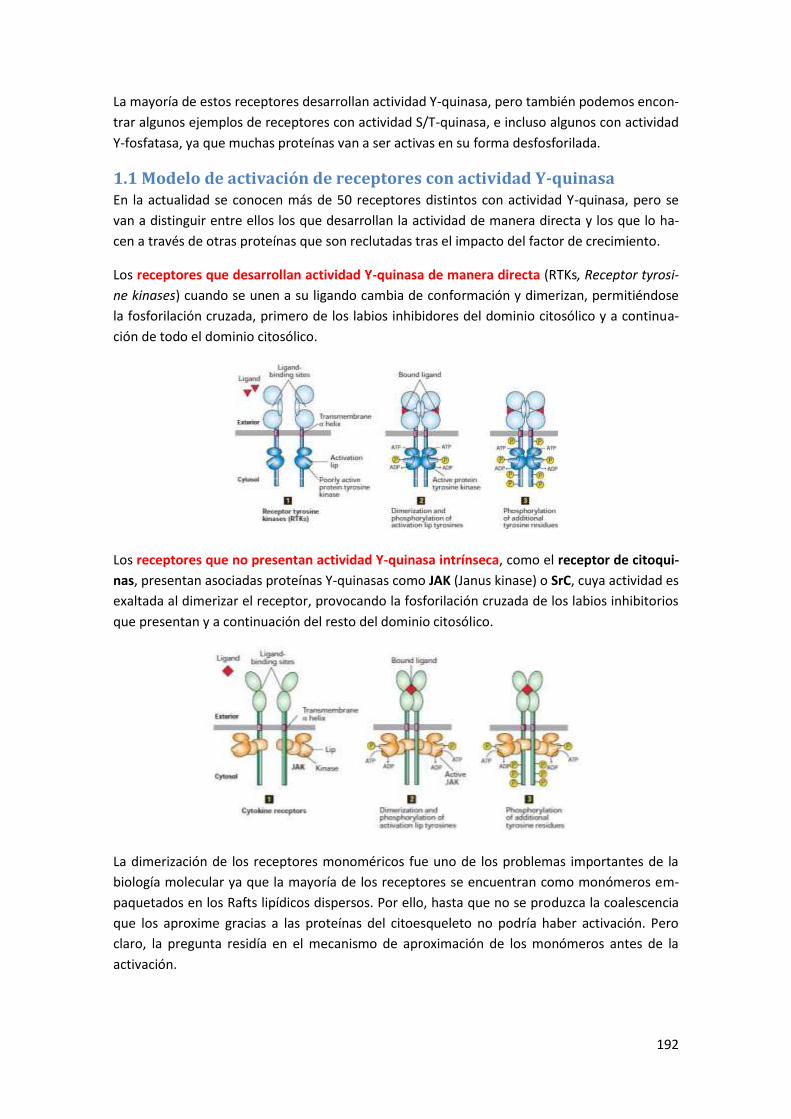
192
La mayoría de estos receptores desarrollan actividad Y-quinasa, pero también podemos encon-
trar algunos ejemplos de receptores con actividad S/T-quinasa, e incluso algunos con actividad
Y-fosfatasa, ya que muchas proteínas van a ser activas en su forma desfosforilada.
1.1 Modelo de activación de receptores con actividad Y-quinasa En la actualidad se conocen más de 50 receptores distintos con actividad Y-quinasa, pero se
van a distinguir entre ellos los que desarrollan la actividad de manera directa y los que lo ha-
cen a través de otras proteínas que son reclutadas tras el impacto del factor de crecimiento.
Los receptores que desarrollan actividad Y-quinasa de manera directa (RTKs, Receptor tyrosi-
ne kinases) cuando se unen a su ligando cambia de conformación y dimerizan, permitiéndose
la fosforilación cruzada, primero de los labios inhibidores del dominio citosólico y a continua-
ción de todo el dominio citosólico.
Los receptores que no presentan actividad Y-quinasa intrínseca, como el receptor de citoqui-
nas, presentan asociadas proteínas Y-quinasas como JAK (Janus kinase) o SrC, cuya actividad es
exaltada al dimerizar el receptor, provocando la fosforilación cruzada de los labios inhibitorios
que presentan y a continuación del resto del dominio citosólico.
La dimerización de los receptores monoméricos fue uno de los problemas importantes de la
biología molecular ya que la mayoría de los receptores se encuentran como monómeros em-
paquetados en los Rafts lipídicos dispersos. Por ello, hasta que no se produzca la coalescencia
que los aproxime gracias a las proteínas del citoesqueleto no podría haber activación. Pero
claro, la pregunta residía en el mecanismo de aproximación de los monómeros antes de la
activación.

193
En esta tabla podemos ver algunas de las proteínas señalizadoras que actúan a través de RTKs.
Proteína señalizadora Receptor Algunas respuestas representativas
Factor de crecimiento epi-dermal (EGF)
EGFR Estimulación de la supervivencia celular, crecimiento y prolifera-ción, o diferenciación de algunos tipos celulares. Actúa como una
señal inductiva en el desarrollo.
Insulina Receptor de
insulina Estimula la utilización de carbohidratos y la síntesis de proteínas.
Factores de crecimiento tipo insulina (IGF1 e IGF2)
IGF receptor -1
Estimula el crecimiento celular y la supervivencia de muchas célu-las.
Factor de crecimiento neu-ral (NGF)
Trk A Estimula la supervivencia y el crecimiento de algunas neuronas.
Factor de crecimiento deri-vado de plaquetas (PDGF)
PDGFR Estimula la supervivencia, crecimiento, proliferación y migración
de varios tipos celulares. Factor estimulador colonial
del macrófagos (MCSF) MCSFR
Estimula la proliferación y diferenciación de monoci-tos/macrófagos.
Factor de crecimiento de fibroblastos
FGFR Estimula la proliferación de varios tipos de células e inhibe la
diferenciación de células precursoras. Actúa como una señal in-ductiva en el desarrollo.
Factor de crecimiento de los endotelios vasculares
(VEGF) VEGFR Estimula la angiogénesis.
1.2 Estructura de los factores de crecimiento
1.2.1 El factor de crecimiento transformante
El factor de crecimiento transformante (TGFβ) es un ligando dimérico cuya estructura facilita la
dimerización de receptores, ya que una de las dos unidades que lo forman se asociará a un
monómero. Presenta un tamaño de entre 50-70 aminoácidos y es resistente a la acción pro-
teolítica. De los nueve restos de cisteína (C) que presenta cada monómero 8 están comprome-
tidos en puentes disulfuro intracate-
narios y uno de ellos une ambos mo-
nómeros mediante un puente interca-
tenario (enlace covalente).
Este tipo de factores se vierten desde
las células secretoras por vía endro-
crina al torrente circulatorio.
1.2.2 Eritropoyetina (EPO)
La eritroproyetina (EPO) presenta una estructura
monomérica con un par de hélices laterales que dan
lugar a una estructura casi simétrica que va a permi-
tir la interacción con dos receptores monoméricos al
mismo tiempo, facilitando el proceso de dimeriza-
ción, a pesar de no ser un dímero en sí.
En cualquier caso, los receptores deben de estar
próximos entre sí.
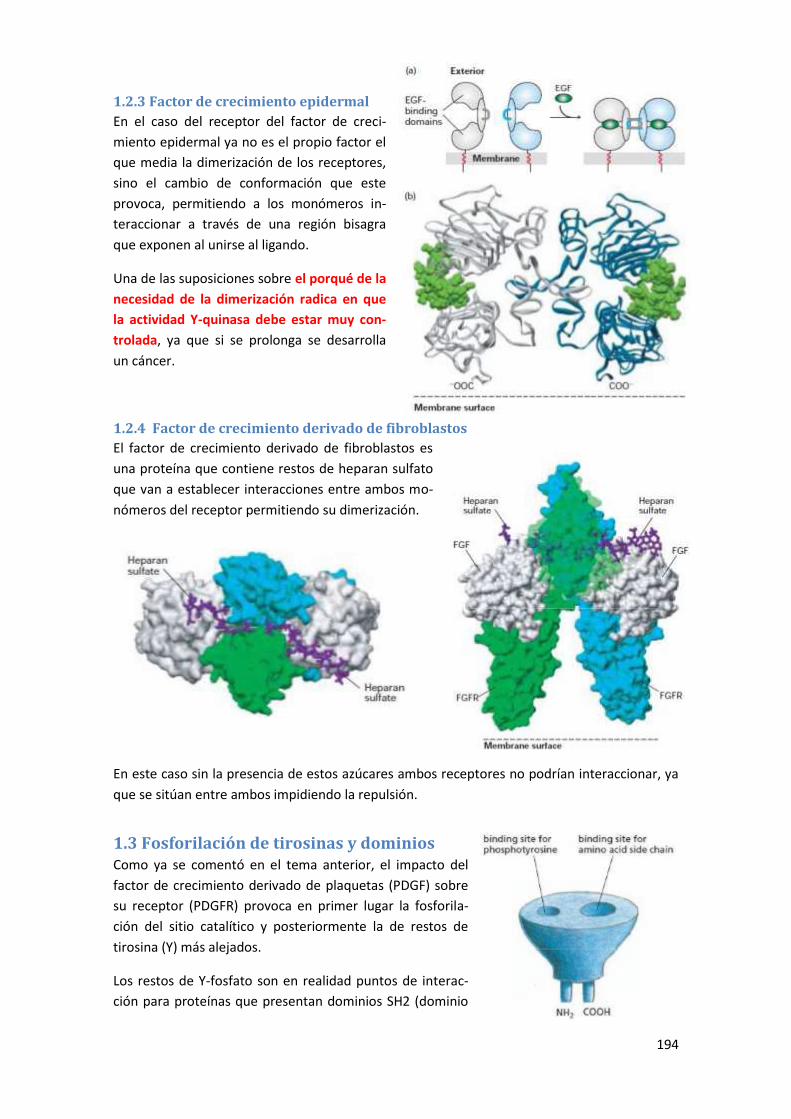
194
1.2.3 Factor de crecimiento epidermal
En el caso del receptor del factor de creci-
miento epidermal ya no es el propio factor el
que media la dimerización de los receptores,
sino el cambio de conformación que este
provoca, permitiendo a los monómeros in-
teraccionar a través de una región bisagra
que exponen al unirse al ligando.
Una de las suposiciones sobre el porqué de la
necesidad de la dimerización radica en que
la actividad Y-quinasa debe estar muy con-
trolada, ya que si se prolonga se desarrolla
un cáncer.
1.2.4 Factor de crecimiento derivado de fibroblastos
El factor de crecimiento derivado de fibroblastos es
una proteína que contiene restos de heparan sulfato
que van a establecer interacciones entre ambos mo-
nómeros del receptor permitiendo su dimerización.
En este caso sin la presencia de estos azúcares ambos receptores no podrían interaccionar, ya
que se sitúan entre ambos impidiendo la repulsión.
1.3 Fosforilación de tirosinas y dominios Como ya se comentó en el tema anterior, el impacto del
factor de crecimiento derivado de plaquetas (PDGF) sobre
su receptor (PDGFR) provoca en primer lugar la fosforila-
ción del sitio catalítico y posteriormente la de restos de
tirosina (Y) más alejados.
Los restos de Y-fosfato son en realidad puntos de interac-
ción para proteínas que presentan dominios SH2 (dominio
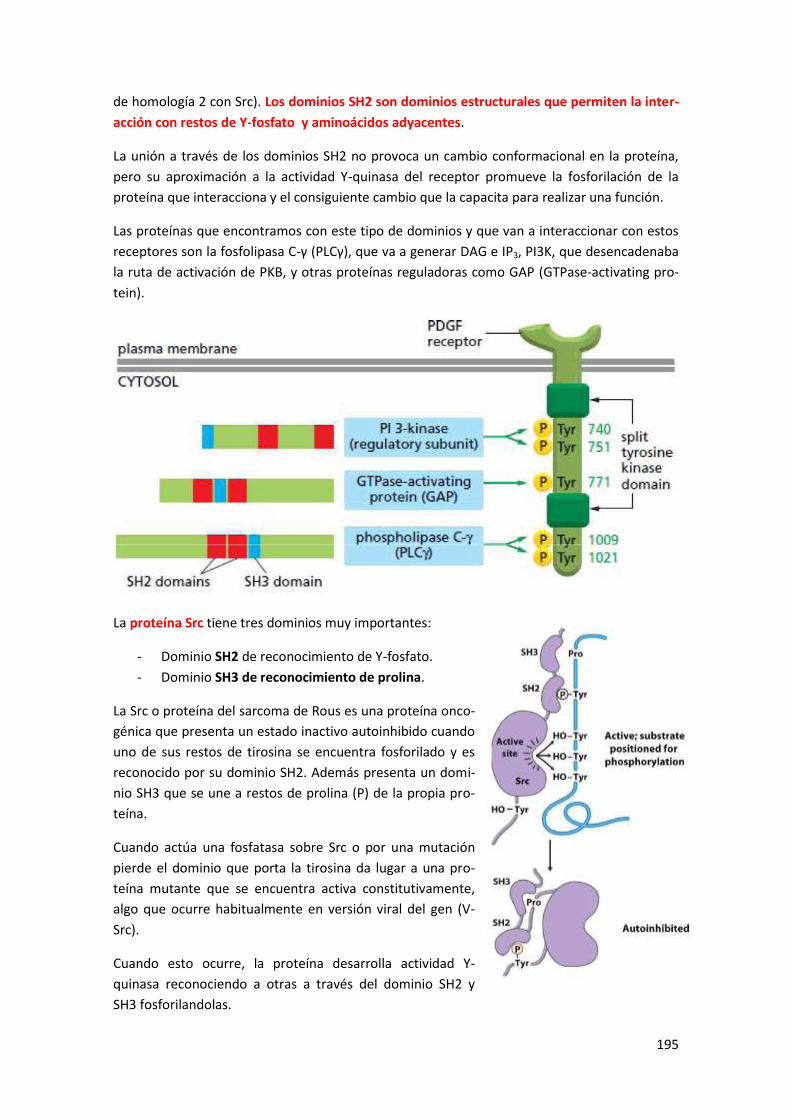
195
de homología 2 con Src). Los dominios SH2 son dominios estructurales que permiten la inter-
acción con restos de Y-fosfato y aminoácidos adyacentes.
La unión a través de los dominios SH2 no provoca un cambio conformacional en la proteína,
pero su aproximación a la actividad Y-quinasa del receptor promueve la fosforilación de la
proteína que interacciona y el consiguiente cambio que la capacita para realizar una función.
Las proteínas que encontramos con este tipo de dominios y que van a interaccionar con estos
receptores son la fosfolipasa C-γ (PLCγ), que va a generar DAG e IP3, PI3K, que desencadenaba
la ruta de activación de PKB, y otras proteínas reguladoras como GAP (GTPase-activating pro-
tein).
La proteína Src tiene tres dominios muy importantes:
- Dominio SH2 de reconocimiento de Y-fosfato.
- Dominio SH3 de reconocimiento de prolina.
La Src o proteína del sarcoma de Rous es una proteína onco-
génica que presenta un estado inactivo autoinhibido cuando
uno de sus restos de tirosina se encuentra fosforilado y es
reconocido por su dominio SH2. Además presenta un domi-
nio SH3 que se une a restos de prolina (P) de la propia pro-
teína.
Cuando actúa una fosfatasa sobre Src o por una mutación
pierde el dominio que porta la tirosina da lugar a una pro-
teína mutante que se encuentra activa constitutivamente,
algo que ocurre habitualmente en versión viral del gen (V-
Src).
Cuando esto ocurre, la proteína desarrolla actividad Y-
quinasa reconociendo a otras a través del dominio SH2 y
SH3 fosforilandolas.

196
En el caso de la glucoqueno sintetasa quinasa 3 (GSK3) va a ser una
serina fosforilada la que se introduzca en el sitio catalítico inacti-
vandola. GSK3 reconoce proteínas a fosforilar en restos de S/T por
la presencia en primer lugar de una serina fosforilada y una serina
susceptible de serlo.
En humanos sea han clasificado más de 90 proteínas con dominios
SH2 y más de 24 con dominios PTB (similares a SH2 pero no homó-
logos). Estos dominios presentan un tamaño de unos 100 aminoáci-
dos que generan un canal para acomodar al resto de Y-fosfato y a
los aminoácidos circundantes (secuon).
Generalmente los dominios SH2 interaccionan con un resto de Y-
fosfato al que se denomina posición 0, y con los siguientes tres
aminoácidos denominados como +1, +2 y +3.
Por ello dentro de los dominios SH2 se encuentran
diversos tipos entre los que encontramos unos que
reconocen aminoácidos cargados negativamente
(Src, Fyn, Hck y Nck), otros que interaccionan con
aminoácido alifáticos en posiciones desde +1-+5
(PLC-C y SHP-2)…
Por ello podemos establecer una multifuncionali-
dad proteica en función de los dominios funcionales
que presenten en su secuencia.
La proteína Grb2 es un adaptador que acopla la
acción del receptor de insulina (IRK) con Sos para activar a la glucógeno sintasa. Esta interac-
ción se produce a través de los dominios SH2 y SH3 que presenta la proteína.
La proteína estructural Shc presenta dominios SH2 y PTB, y como hemos visto Src presenta
dominios SH2, SH3 y un dominio Y-quinasa.
La protéina Shp2 es una Y-fosfatasa que presenta dominios SH2, además del propio de la acti-
vidad catalítica. Por lo que cuando este interacciona con el receptor fosforilado produce una
interrupción de la activación.

197
La proteína Ras-GAP se encarga de exaltar la actividad GTPasa de Ras y presenta además de
los dominios SH2 y SH3, un dominio PH que liga fosfolípidos de inositol con fosfato en posición
3 y el dominio C2 de asociación a los fosfolípidos de la membrana mediante calcio.
La proteína STAT es un factor de transcripción que presenta un dominio SH2, junto con un
dominio de unión al DNA y uno de activación de la transcripción (TA).
La proteína SOCS es una reguladora de la señal que presenta SH2 y un dominio que facilita la
actividad de una E3-ligasa de ubiquitina que se relaciona con la degradación de receptores a
través del sistema UPS.
Finalmente la proteína PLC-γ que produce el IP3 y el DAG como segundos mensajeros está
formada por una gran multitud de dominios como PH, SH2, SH3, C2,…
Debemos entender que la interacción de estos dominios con sus secuencias sustrato o sus
ligandos produce en mayor o menor medida la activación de la enzima y por lo tanto según las
“teclas” que toquemos tendremos unos efectos u otros.
La activación permanente de la fosfolipasa C por parte de los ésteres de forbol desencadenan
cáncer. Pero si en cualquiera de los estímulos que es capaz de percibir PLC-γ se produce un
exceso de activación, también ha a desencadenar el cáncer.
1.4 Rutas de señalización en las que intervienen Y-quinasas
1.4.1 La insulina como factor de crecimiento
Cuando la insulina impacta contra el receptor de insulina IRK induce la autofosforilación cruza-
da en los restos de tirosina del dominio citosólico. Estos restos de Y-fosfato son reconocidos
por diversas proteínas con dominios SH2, como IRS-1 (Sustrato del receptor de insulina 1) que
presenta un dominio tipo PTB, que al interaccionar con el receptor fosforilado acerca a la pro-
teína para ser fosforilada también.
A continuación, Grb2 se une a los restos de Y-fosfato de IRS-1 a través de sus dominios SH2,
siendo competente para interaccionar con Sos a través de los dominios SH3.
La interacción Grb2-Sos permite a este último catalizar el intercambio de GDP por GTP en Ras,
que entonces une y activa a Raf-1.
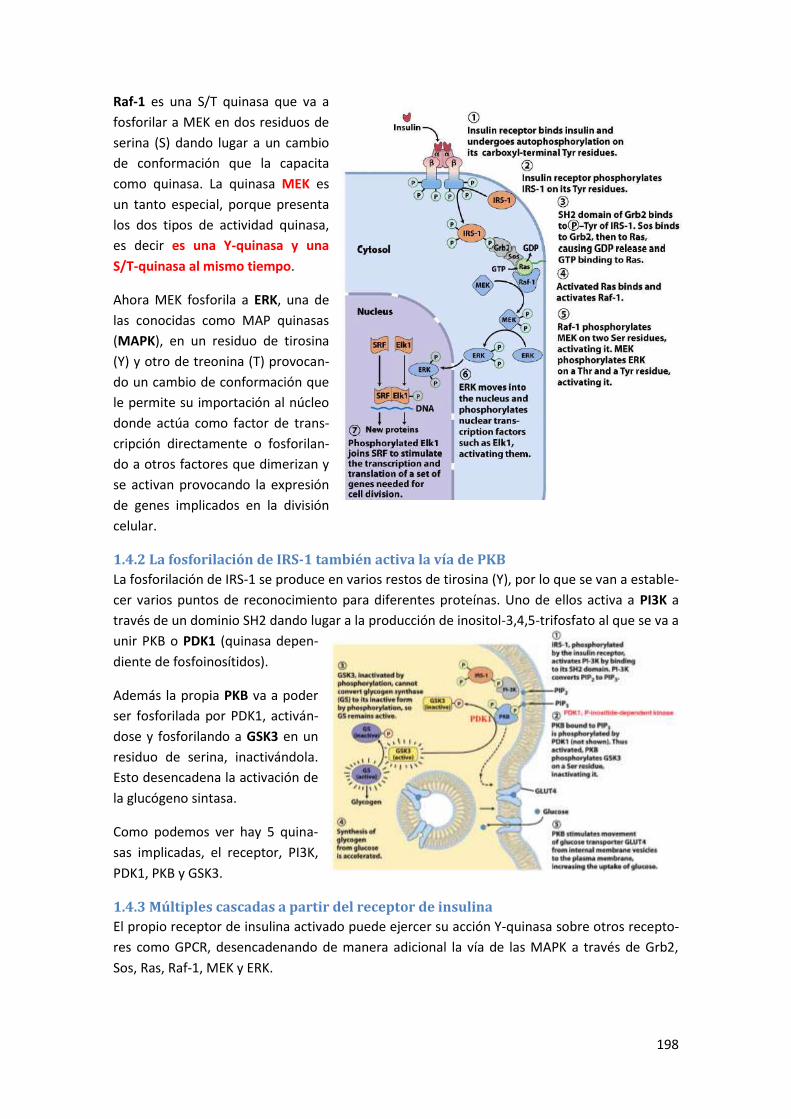
198
Raf-1 es una S/T quinasa que va a
fosforilar a MEK en dos residuos de
serina (S) dando lugar a un cambio
de conformación que la capacita
como quinasa. La quinasa MEK es
un tanto especial, porque presenta
los dos tipos de actividad quinasa,
es decir es una Y-quinasa y una
S/T-quinasa al mismo tiempo.
Ahora MEK fosforila a ERK, una de
las conocidas como MAP quinasas
(MAPK), en un residuo de tirosina
(Y) y otro de treonina (T) provocan-
do un cambio de conformación que
le permite su importación al núcleo
donde actúa como factor de trans-
cripción directamente o fosforilan-
do a otros factores que dimerizan y
se activan provocando la expresión
de genes implicados en la división
celular.
1.4.2 La fosforilación de IRS-1 también activa la vía de PKB
La fosforilación de IRS-1 se produce en varios restos de tirosina (Y), por lo que se van a estable-
cer varios puntos de reconocimiento para diferentes proteínas. Uno de ellos activa a PI3K a
través de un dominio SH2 dando lugar a la producción de inositol-3,4,5-trifosfato al que se va a
unir PKB o PDK1 (quinasa depen-
diente de fosfoinosítidos).
Además la propia PKB va a poder
ser fosforilada por PDK1, activán-
dose y fosforilando a GSK3 en un
residuo de serina, inactivándola.
Esto desencadena la activación de
la glucógeno sintasa.
Como podemos ver hay 5 quina-
sas implicadas, el receptor, PI3K,
PDK1, PKB y GSK3.
1.4.3 Múltiples cascadas a partir del receptor de insulina
El propio receptor de insulina activado puede ejercer su acción Y-quinasa sobre otros recepto-
res como GPCR, desencadenando de manera adicional la vía de las MAPK a través de Grb2,
Sos, Ras, Raf-1, MEK y ERK.
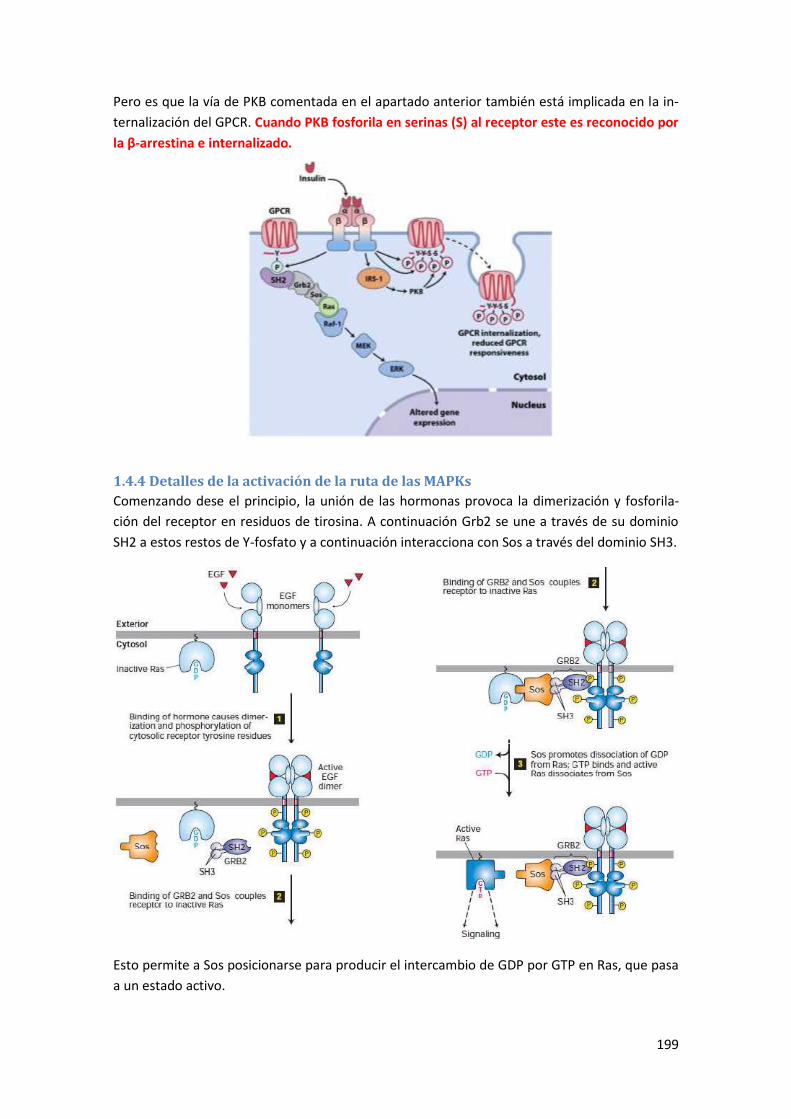
199
Pero es que la vía de PKB comentada en el apartado anterior también está implicada en la in-
ternalización del GPCR. Cuando PKB fosforila en serinas (S) al receptor este es reconocido por
la β-arrestina e internalizado.
1.4.4 Detalles de la activación de la ruta de las MAPKs
Comenzando dese el principio, la unión de las hormonas provoca la dimerización y fosforila-
ción del receptor en residuos de tirosina. A continuación Grb2 se une a través de su dominio
SH2 a estos restos de Y-fosfato y a continuación interacciona con Sos a través del dominio SH3.
Esto permite a Sos posicionarse para producir el intercambio de GDP por GTP en Ras, que pasa
a un estado activo.
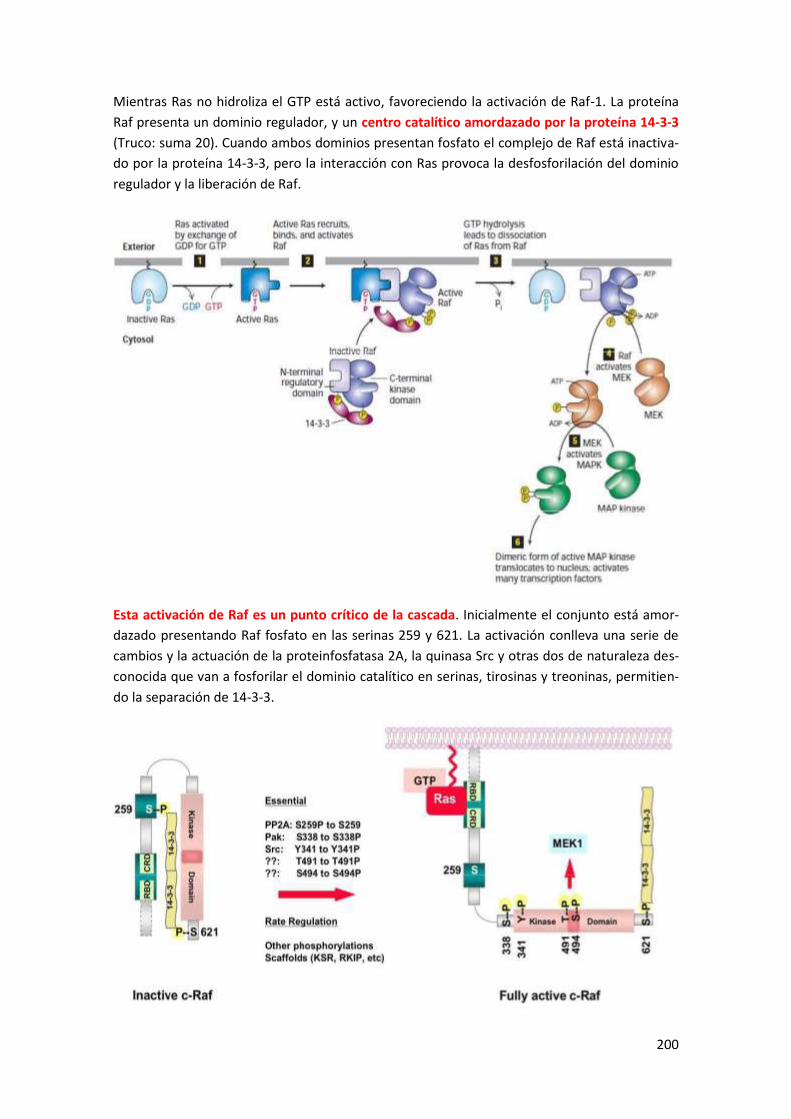
200
Mientras Ras no hidroliza el GTP está activo, favoreciendo la activación de Raf-1. La proteína
Raf presenta un dominio regulador, y un centro catalítico amordazado por la proteína 14-3-3
(Truco: suma 20). Cuando ambos dominios presentan fosfato el complejo de Raf está inactiva-
do por la proteína 14-3-3, pero la interacción con Ras provoca la desfosforilación del dominio
regulador y la liberación de Raf.
Esta activación de Raf es un punto crítico de la cascada. Inicialmente el conjunto está amor-
dazado presentando Raf fosfato en las serinas 259 y 621. La activación conlleva una serie de
cambios y la actuación de la proteinfosfatasa 2A, la quinasa Src y otras dos de naturaleza des-
conocida que van a fosforilar el dominio catalítico en serinas, tirosinas y treoninas, permitien-
do la separación de 14-3-3.

201
La cascada continúa con la
fosforilación de Mek y de las
MAPKs.
La activación de ERK o MAPK se
produce mediante una fosfori-
lación de MEK en Y185 y T183,
situados en el labio de activa-
ción y que promueven el cam-
bio de conformación que per-
mite la entrada al núcleo y el
desarrollo de la actividad cata-
lítica.
Pero ERK puede actuar como
quinasa de otros factores de
transcripción directamente, o
como intermediario.
1.4.5 Secuencia de transducción para EPO a través de JAK (receptor de citoquinas)
El receptor de eritropoyetina (EPO) no presenta actividad Y-quinasa intrínseca, sino que va a
presentar asociado una proteína denominada JAK que será la quinasa. Cuando EPO interaccio-
na con el receptor se produce un cambio conformacional que activa a la proteína JAK, dando
lugar a la fosforilación cruzada en restos de tirosina del propio receptor.
Estos restos de Y-fosfato pueden ser reconoci-
dos por el dominio SH2 de Grb2, desencade-
nando la ruta de las MAPK. Pero también van
a ser reconocidos por la proteína STAT (trans-
ductores de la señal y activadores de la trans-
cripción).
La proteína STAT presenta dominios SH2 que
la aproximan a la actividad Y-quinasa de JAK.
Al fosforilarse estos dominios SH2 le sirven
para dimerizar dando lugar a homodímeros
que exponen secuencias de importación al
núcleo (NLS) donde van a actuar como facto-
res de transcripción.
Además de estos dos sistemas que emanan
del receptor de EPO, también encontramos
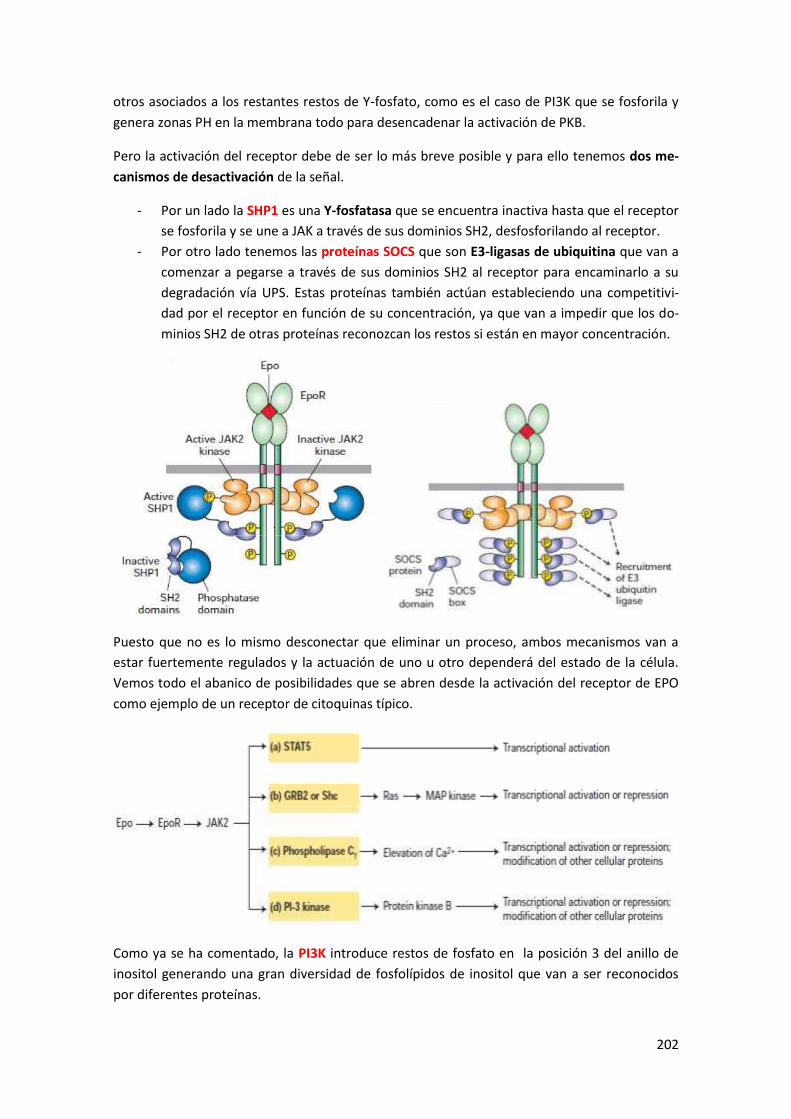
202
otros asociados a los restantes restos de Y-fosfato, como es el caso de PI3K que se fosforila y
genera zonas PH en la membrana todo para desencadenar la activación de PKB.
Pero la activación del receptor debe de ser lo más breve posible y para ello tenemos dos me-
canismos de desactivación de la señal.
- Por un lado la SHP1 es una Y-fosfatasa que se encuentra inactiva hasta que el receptor
se fosforila y se une a JAK a través de sus dominios SH2, desfosforilando al receptor.
- Por otro lado tenemos las proteínas SOCS que son E3-ligasas de ubiquitina que van a
comenzar a pegarse a través de sus dominios SH2 al receptor para encaminarlo a su
degradación vía UPS. Estas proteínas también actúan estableciendo una competitivi-
dad por el receptor en función de su concentración, ya que van a impedir que los do-
minios SH2 de otras proteínas reconozcan los restos si están en mayor concentración.
Puesto que no es lo mismo desconectar que eliminar un proceso, ambos mecanismos van a
estar fuertemente regulados y la actuación de uno u otro dependerá del estado de la célula.
Vemos todo el abanico de posibilidades que se abren desde la activación del receptor de EPO
como ejemplo de un receptor de citoquinas típico.
Como ya se ha comentado, la PI3K introduce restos de fosfato en la posición 3 del anillo de
inositol generando una gran diversidad de fosfolípidos de inositol que van a ser reconocidos
por diferentes proteínas.
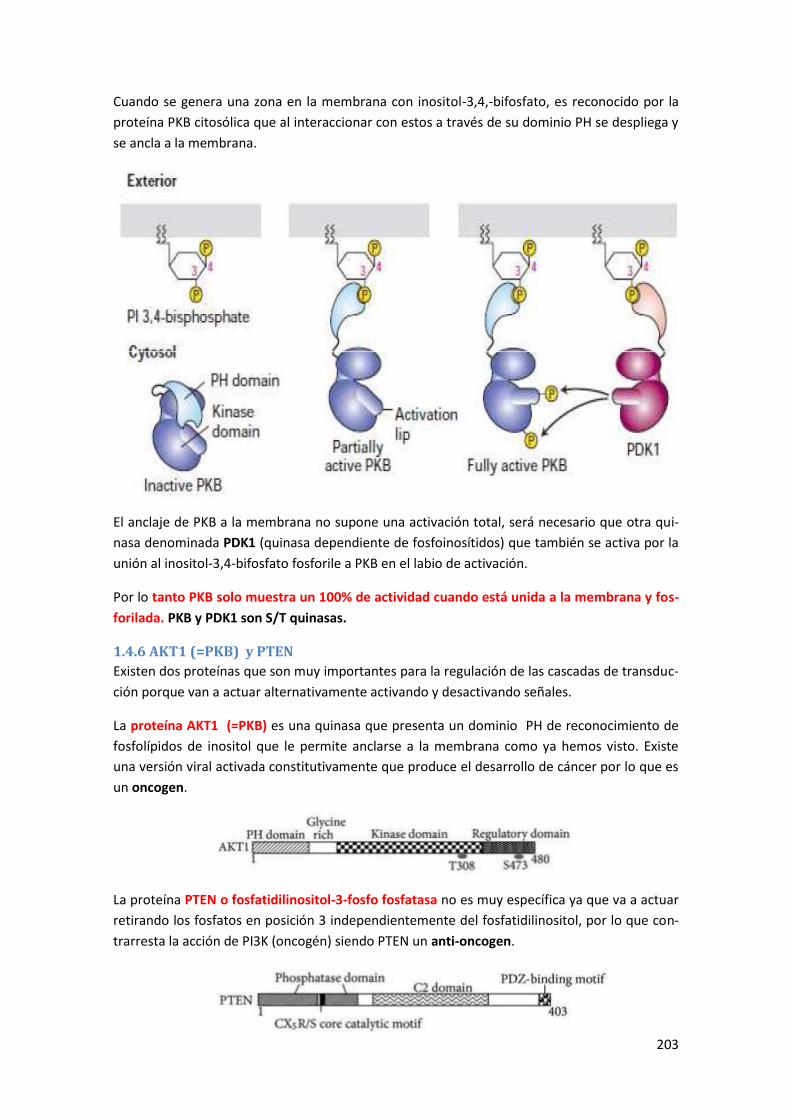
203
Cuando se genera una zona en la membrana con inositol-3,4,-bifosfato, es reconocido por la
proteína PKB citosólica que al interaccionar con estos a través de su dominio PH se despliega y
se ancla a la membrana.
El anclaje de PKB a la membrana no supone una activación total, será necesario que otra qui-
nasa denominada PDK1 (quinasa dependiente de fosfoinosítidos) que también se activa por la
unión al inositol-3,4-bifosfato fosforile a PKB en el labio de activación.
Por lo tanto PKB solo muestra un 100% de actividad cuando está unida a la membrana y fos-
forilada. PKB y PDK1 son S/T quinasas.
1.4.6 AKT1 (=PKB) y PTEN
Existen dos proteínas que son muy importantes para la regulación de las cascadas de transduc-
ción porque van a actuar alternativamente activando y desactivando señales.
La proteína AKT1 (=PKB) es una quinasa que presenta un dominio PH de reconocimiento de
fosfolípidos de inositol que le permite anclarse a la membrana como ya hemos visto. Existe
una versión viral activada constitutivamente que produce el desarrollo de cáncer por lo que es
un oncogen.
La proteína PTEN o fosfatidilinositol-3-fosfo fosfatasa no es muy específica ya que va a actuar
retirando los fosfatos en posición 3 independientemente del fosfatidilinositol, por lo que con-
trarresta la acción de PI3K (oncogén) siendo PTEN un anti-oncogen.
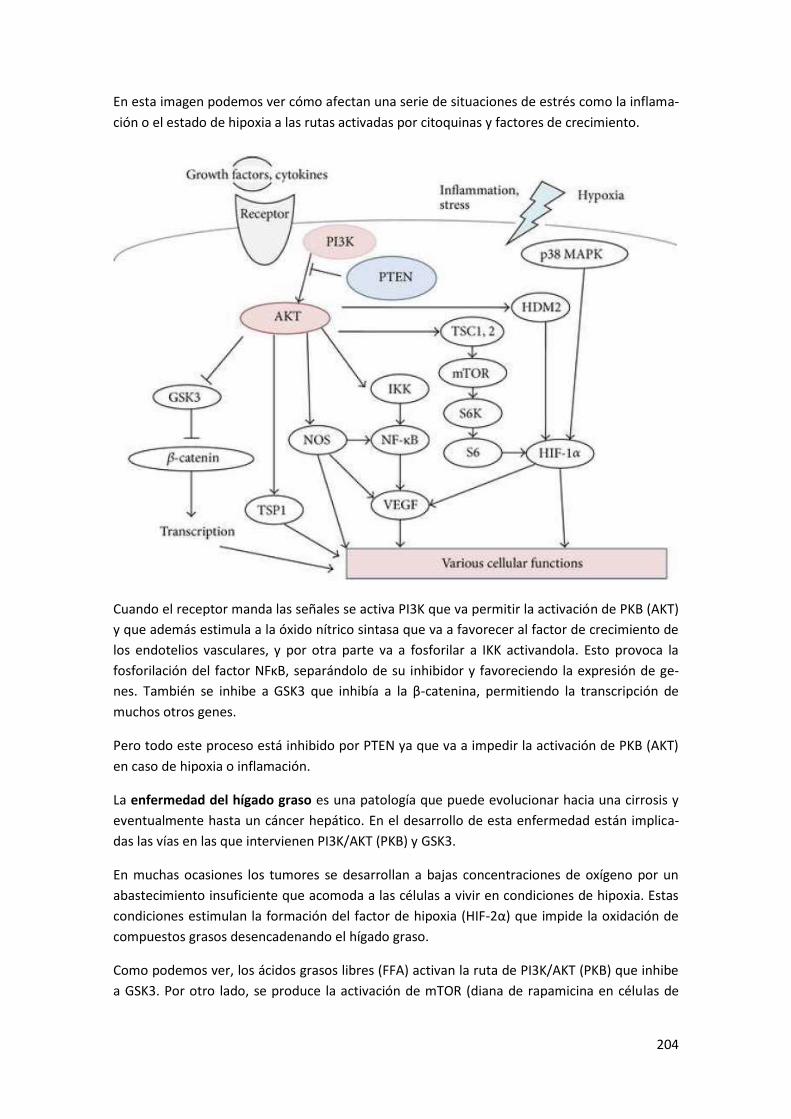
204
En esta imagen podemos ver cómo afectan una serie de situaciones de estrés como la inflama-
ción o el estado de hipoxia a las rutas activadas por citoquinas y factores de crecimiento.
Cuando el receptor manda las señales se activa PI3K que va permitir la activación de PKB (AKT)
y que además estimula a la óxido nítrico sintasa que va a favorecer al factor de crecimiento de
los endotelios vasculares, y por otra parte va a fosforilar a IKK activandola. Esto provoca la
fosforilación del factor NFκB, separándolo de su inhibidor y favoreciendo la expresión de ge-
nes. También se inhibe a GSK3 que inhibía a la β-catenina, permitiendo la transcripción de
muchos otros genes.
Pero todo este proceso está inhibido por PTEN ya que va a impedir la activación de PKB (AKT)
en caso de hipoxia o inflamación.
La enfermedad del hígado graso es una patología que puede evolucionar hacia una cirrosis y
eventualmente hasta un cáncer hepático. En el desarrollo de esta enfermedad están implica-
das las vías en las que intervienen PI3K/AKT (PKB) y GSK3.
En muchas ocasiones los tumores se desarrollan a bajas concentraciones de oxígeno por un
abastecimiento insuficiente que acomoda a las células a vivir en condiciones de hipoxia. Estas
condiciones estimulan la formación del factor de hipoxia (HIF-2α) que impide la oxidación de
compuestos grasos desencadenando el hígado graso.
Como podemos ver, los ácidos grasos libres (FFA) activan la ruta de PI3K/AKT (PKB) que inhibe
a GSK3. Por otro lado, se produce la activación de mTOR (diana de rapamicina en células de
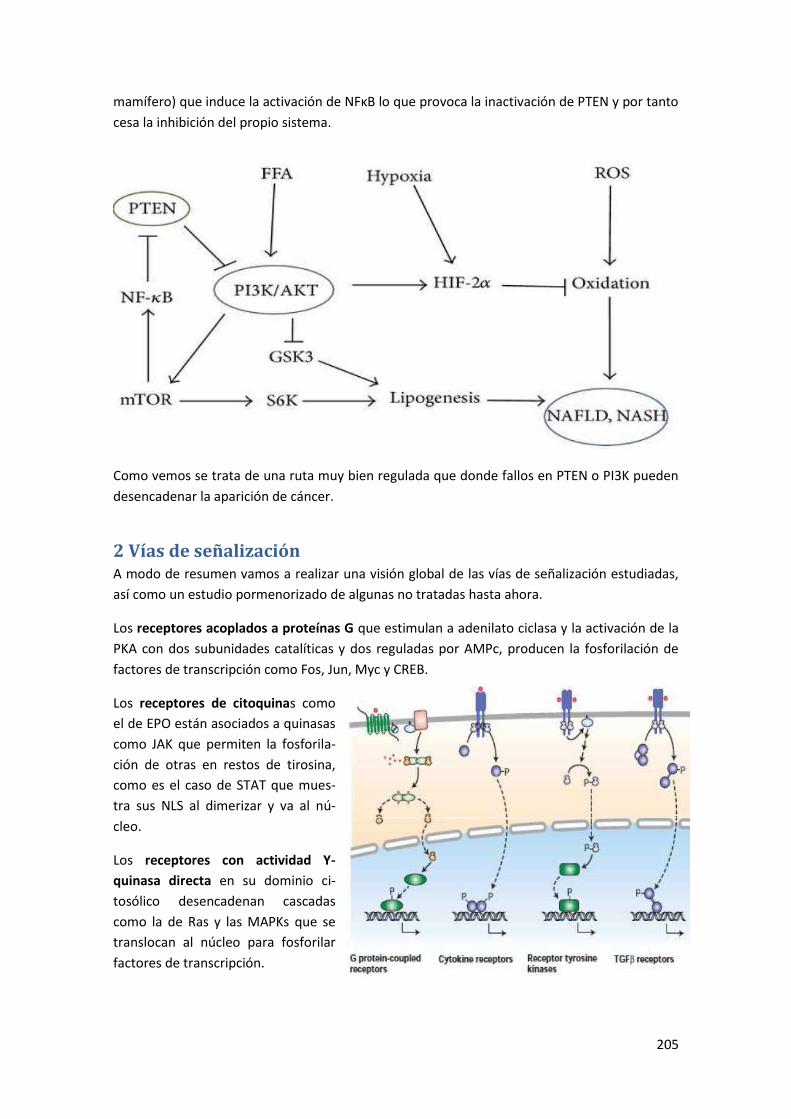
205
mamífero) que induce la activación de NFκB lo que provoca la inactivación de PTEN y por tanto
cesa la inhibición del propio sistema.
Como vemos se trata de una ruta muy bien regulada que donde fallos en PTEN o PI3K pueden
desencadenar la aparición de cáncer.
2 Vías de señalización A modo de resumen vamos a realizar una visión global de las vías de señalización estudiadas,
así como un estudio pormenorizado de algunas no tratadas hasta ahora.
Los receptores acoplados a proteínas G que estimulan a adenilato ciclasa y la activación de la
PKA con dos subunidades catalíticas y dos reguladas por AMPc, producen la fosforilación de
factores de transcripción como Fos, Jun, Myc y CREB.
Los receptores de citoquinas como
el de EPO están asociados a quinasas
como JAK que permiten la fosforila-
ción de otras en restos de tirosina,
como es el caso de STAT que mues-
tra sus NLS al dimerizar y va al nú-
cleo.
Los receptores con actividad Y-
quinasa directa en su dominio ci-
tosólico desencadenan cascadas
como la de Ras y las MAPKs que se
translocan al núcleo para fosforilar
factores de transcripción.

206
Los receptores como los del factor de crecimiento transformante β (TGFβ) son muy especiales
porque van a desarrollar, a diferencia de los anteriores, actividad S/T-quinasa activando a
factores de transcripción como Smad que en función del caso van a reclutar co-activadores o
co-represores.
Estos cuatro tipos de receptores están relacionados con la fosforilación de proteínas que lleva
a una modulación de la expresión génica, pero además podemos encontrar otros tres tipos de
receptores que van a realizar esta modulación a través de la degradación de complejos o pro-
teólisis de proteínas.
Los receptores del tipo Hedgehog (Hh, erizo) son un complejo de dos proteínas que se dispo-
nen en la membrana plasmática, donde una actúa como receptor del ligando y la otra como
colaboradora amordazada por la primera. Cuando el ligando impacta con la proteína recepto-
ra, esta se disocia y la colaboradora comienza a
mandar señales que provocan la disociación de
complejos proteicos cuyas unidades entran al
núcleo como factores de transcripción.
Los receptores Wnt son receptores del tipo
7tm que al unirse al ligando actúan sobre una
proteína que impedía la liberación de factores
de transcripción acoplados a complejos multi-
proteicos. Precisan de una proteína similar a
LDLR para unir el ligando.
Los receptores Notch (muesca) son receptores
que interaccionan con proteínas delta de otras
células favoreciéndose la proteólisis del domi-
nio citosólico mediante presenilinas y conver-
tasas, que marcha al núcleo donde actúa como
un factor de transcripción.
2.1 Receptores con actividad enzimática intrínseca o asociada Veamos algunas características concretas de estos receptores y las actividades en las que están
implicados.
Los receptores para el factor de crecimiento transformante (TGFβ):
- Tienen como ligandos la superfamilia de TGFβ y BMPs, activinas e inhibinas (mamífe-
ros) y Dpp (Drosophila).
- Presentan actividad S/T-quinasa intrínseca en el dominio citosólico.
- Están implicados en la activación de los factores de transcripción Smad.
Los receptores de citoquinas:
- Tienen como ligando los interferones, eritropoyetina, hormona de crecimiento, algu-
nas interleucinas (IL-2 e IL-4) y otras citoquinas.

207
- Estructuralmente están formados por una hélice α transmembrana con un dominio
multi-β extracelular y asociada a las quinasas JAK en la región intracelular.
- Están implicados en la activación directa de los factores de transcripción STAT, la ruta
de PI3K, la ruta de IP3/DAG y la ruta de Ras-MAPK.
Los receptores con actividad tirosina quinasa intrínseca:
- Tienen como ligando insulna, el factor de crecimiento epidermal (EGF), el factor de
crecimiento fibroblástico (FGF), neurotropinas y otros factores de crecimiento.
- El receptor está formado por una hélice α transmembrana con un dominio de activi-
dad Y-quinasa citosólico.
- Participan en la activación de la vía Ras-MAPK, IP3/DAG y PI3K.
Los receptores con actividad guanilato ciclasa:
- Tienen como ligando el factor natriurético atrial y las hormonas peptídicas relaciona-
das.
- Estructuralmente es una hélice α transmembrana con actividad guanilato ciclasa in-
trínseca en el dominio citosólico.
- Están implicados en la generación de GMPc.
Los receptores que desarrollan actividad fosfotirosina fosfatasa (Y-fosfatasa):
- Tienen como ligandos las pleiotropinas y otras hormonas protéicas que van a actuar
como señales no negativas (positivas para la división) inhibiendo al receptor.
- Presentan actividad fosfotirosina fosfatasa en el dominio citosólico inhibida por la
unión de ligandos. Por lo tanto tienen actividad fosfatasa constitutiva garantizando
que no se producirá división celular en ausencia de estímulos.
- Participan en la hidrolisis de los residuos de Y-fosfato que activan varias rutas iniciadas
por Y-quinasas.
Los receptores de las células T:
- Tienen como ligandos pequeños péptidos asociados a los complejos mayores de histo-
compatibilidad (MHC) en la membrana plasmática de macrófagos y otras células pre-
sentadoras de antígenos.
- Se trata de hélices α transmembrana con un gran número de qunasas asociadas al
dominio citosólico y que solo se han localizado en linfocitos T.
- Están implicados en la activación de Y-quinasas citosólicas y las rutas de PI3K, IP3/DAG
y Ras-MAPK.
2.2 Ruta de transducción para TGFβ-Smad En algunas células el factor de crecimiento transformante TGFβ (no crecimiento) es presentado
por un receptor monomérico (RIII) al receptor propiamente dicho (RII), mientras que en otras
la interacción se produce directamente a través de RII, que es un dímero en sí mismo.
La unión del ligando provoca en cualquier caso el reclutamiento de RIII, RII y RI, formando un
heteroligómero que se autofosforila en residuos de S/T.
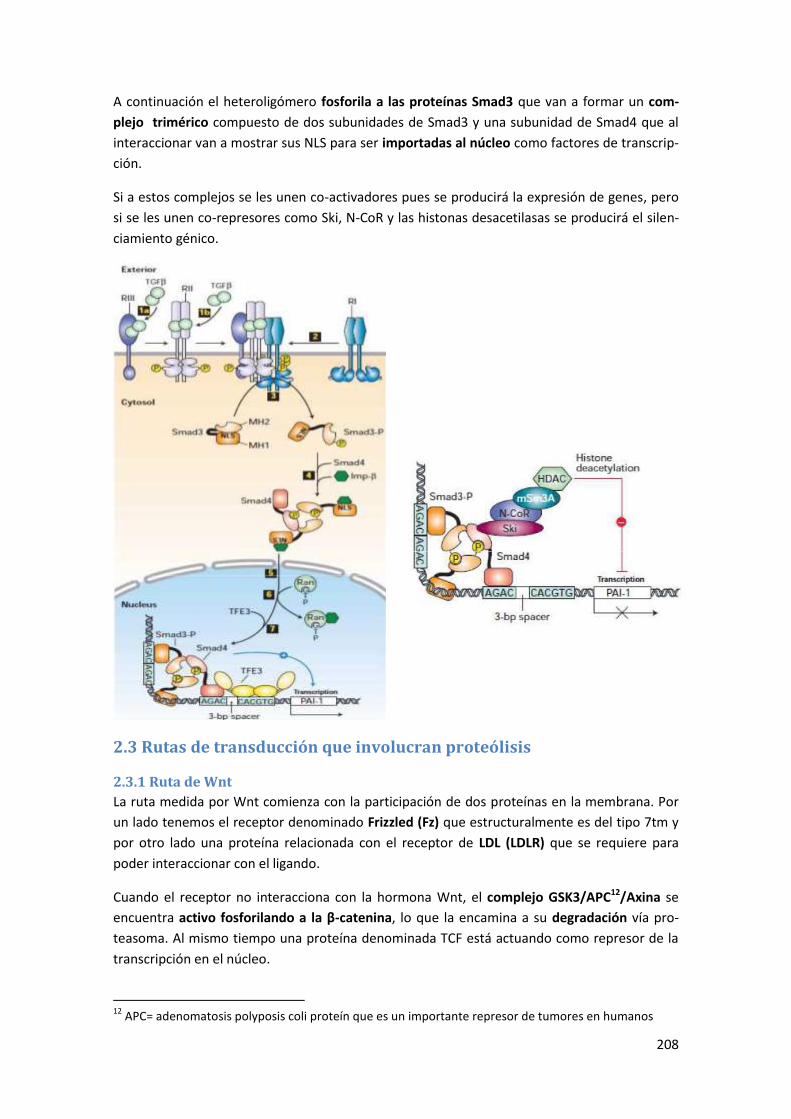
208
A continuación el heteroligómero fosforila a las proteínas Smad3 que van a formar un com-
plejo trimérico compuesto de dos subunidades de Smad3 y una subunidad de Smad4 que al
interaccionar van a mostrar sus NLS para ser importadas al núcleo como factores de transcrip-
ción.
Si a estos complejos se les unen co-activadores pues se producirá la expresión de genes, pero
si se les unen co-represores como Ski, N-CoR y las histonas desacetilasas se producirá el silen-
ciamiento génico.
2.3 Rutas de transducción que involucran proteólisis
2.3.1 Ruta de Wnt
La ruta medida por Wnt comienza con la participación de dos proteínas en la membrana. Por
un lado tenemos el receptor denominado Frizzled (Fz) que estructuralmente es del tipo 7tm y
por otro lado una proteína relacionada con el receptor de LDL (LDLR) que se requiere para
poder interaccionar con el ligando.
Cuando el receptor no interacciona con la hormona Wnt, el complejo GSK3/APC12/Axina se
encuentra activo fosforilando a la β-catenina, lo que la encamina a su degradación vía pro-
teasoma. Al mismo tiempo una proteína denominada TCF está actuando como represor de la
transcripción en el núcleo.
12
APC= adenomatosis polyposis coli proteín que es un importante represor de tumores en humanos
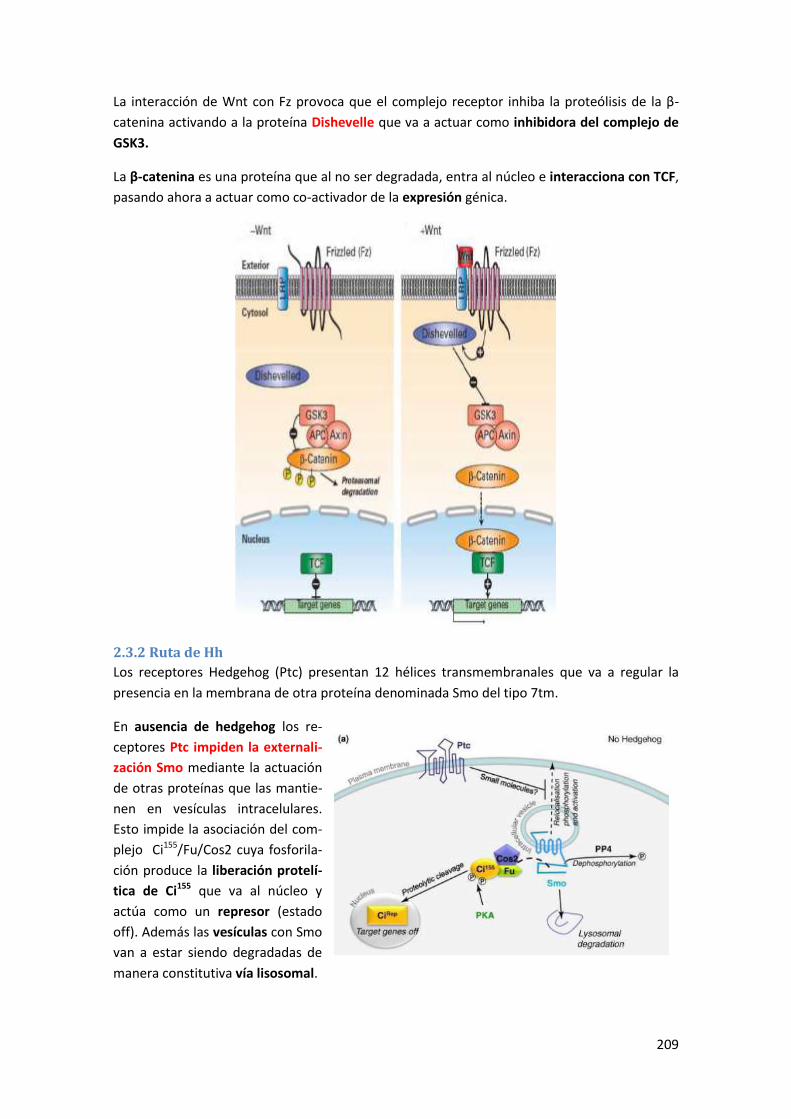
209
La interacción de Wnt con Fz provoca que el complejo receptor inhiba la proteólisis de la β-
catenina activando a la proteína Dishevelle que va a actuar como inhibidora del complejo de
GSK3.
La β-catenina es una proteína que al no ser degradada, entra al núcleo e interacciona con TCF,
pasando ahora a actuar como co-activador de la expresión génica.
2.3.2 Ruta de Hh
Los receptores Hedgehog (Ptc) presentan 12 hélices transmembranales que va a regular la
presencia en la membrana de otra proteína denominada Smo del tipo 7tm.
En ausencia de hedgehog los re-
ceptores Ptc impiden la externali-
zación Smo mediante la actuación
de otras proteínas que las mantie-
nen en vesículas intracelulares.
Esto impide la asociación del com-
plejo Ci155/Fu/Cos2 cuya fosforila-
ción produce la liberación protelí-
tica de Ci155 que va al núcleo y
actúa como un represor (estado
off). Además las vesículas con Smo
van a estar siendo degradadas de
manera constitutiva vía lisosomal.

210
La unión de Hh a Ptc produce la inactivación del receptor y permite la salida de los receptores
a la membrana abriendo tres vías de transducción:
- Por una parte, la interacción de Smo en la membrana con el complejo Ci155/Fu/Cos2,
permite la liberación de Ci que actúa como un factor de transcripción que estimula la
expresión de genes (estado on).
- Por otro lado se abre un sistema similar al sistema de adeniliato ciclasa asociándose
una proteína G trimérica a Cos2 que libera la subunidad α y regula los niveles de
AMPc.
- Finalmente la fosforilación del Smo provoca el reconocimiento por β-arrestina y la in-
ternalización.
El sistema se vuelve mucho más complejo si consideramos que también va a intervenir PKA, y
otros factores.
2.3.3 Ruta de NFκB
La ruta de NFκB se activa mediante ligandos
como el factor de necrosis tumoral α (TNF-α),
interleucina 1 (mamíferos) o la proteína Spätzle
(Drosophila) y desencadena la degradación
dependiente de fosforilación de la proteína
inhibidora IκB que libera al factor de transcrip-
ción propiamente dicho.
Cuando los receptores reciben al ligando se
produce la activación de la quinasa TAK1, que
fosforila a la quinasa IκK activandola. La activa-
ción promueve la fosforilación de IκB que ahora
es reconocida por una E3-Ligasa y degradada
vía UPS, liberando al factor de transcripción
NFκB (heterodímero de p65/p50) que va al
núcleo e induce la transcripción de genes.
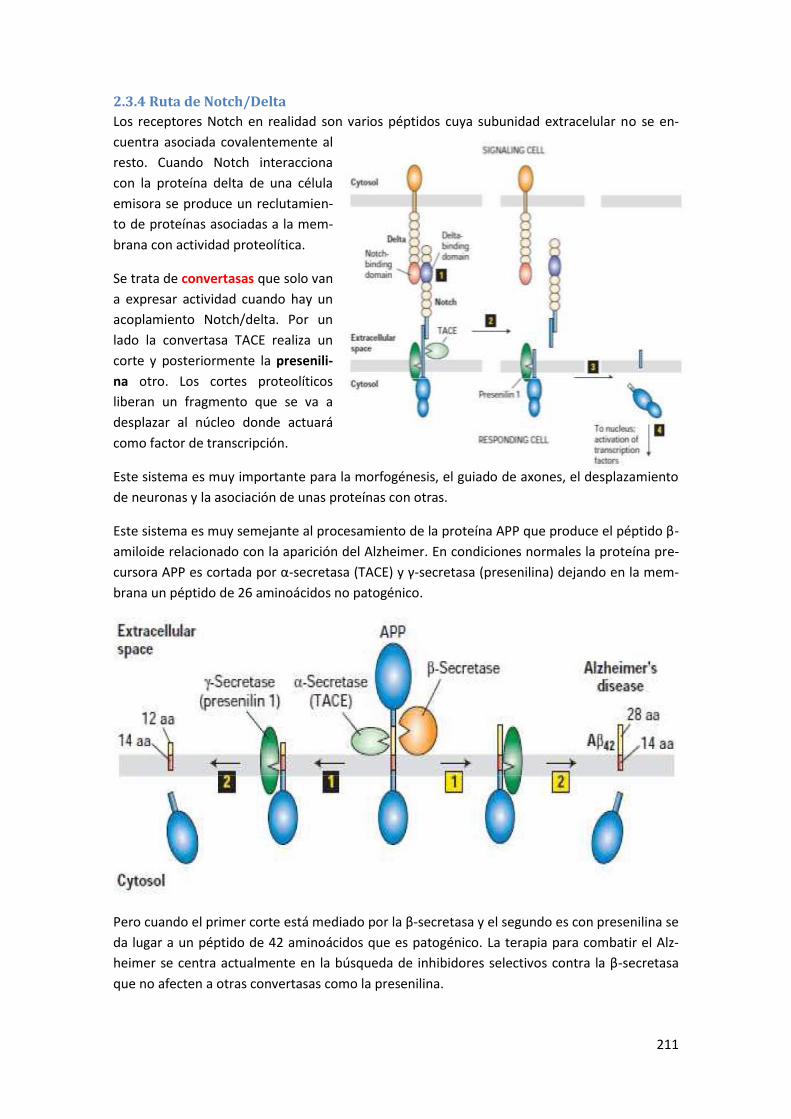
211
2.3.4 Ruta de Notch/Delta
Los receptores Notch en realidad son varios péptidos cuya subunidad extracelular no se en-
cuentra asociada covalentemente al
resto. Cuando Notch interacciona
con la proteína delta de una célula
emisora se produce un reclutamien-
to de proteínas asociadas a la mem-
brana con actividad proteolítica.
Se trata de convertasas que solo van
a expresar actividad cuando hay un
acoplamiento Notch/delta. Por un
lado la convertasa TACE realiza un
corte y posteriormente la presenili-
na otro. Los cortes proteolíticos
liberan un fragmento que se va a
desplazar al núcleo donde actuará
como factor de transcripción.
Este sistema es muy importante para la morfogénesis, el guiado de axones, el desplazamiento
de neuronas y la asociación de unas proteínas con otras.
Este sistema es muy semejante al procesamiento de la proteína APP que produce el péptido β-
amiloide relacionado con la aparición del Alzheimer. En condiciones normales la proteína pre-
cursora APP es cortada por α-secretasa (TACE) y γ-secretasa (presenilina) dejando en la mem-
brana un péptido de 26 aminoácidos no patogénico.
Pero cuando el primer corte está mediado por la β-secretasa y el segundo es con presenilina se
da lugar a un péptido de 42 aminoácidos que es patogénico. La terapia para combatir el Alz-
heimer se centra actualmente en la búsqueda de inhibidores selectivos contra la β-secretasa
que no afecten a otras convertasas como la presenilina.

212
3 Anomalías que pueden producir diversas clases de cáncer La sobreproducción de los factores de proliferación que impulsan la síntesis de ciclinas
mitóticas.
Las cantidades de factores de proliferación están sometidas a una fuerte regulación, pero
la amplificación genética o cualquier vía que produzca un aumento de la cantidad de proteínas,
aumento de su actividad (variantes alélicas) o que alteren las afinidades de los receptores
pueden desencadenar una sobreproducción de ciclinas. Por ejemplo, la sobreproducción de
receptores o las mutaciones que los activan constitutivamente (oncogen Her-B es una variante
truncada de EGFR activada constitutivamente). También la inactivación de las vías de degrada-
ción de las proteínas implicadas.
Sobreproducción de algunos factores de transcripción como C-Fos o C-Jun que median en
la producción de cilcinas que permiten el paso de G0 a G1 (Ciclina D + CDK6/4).
Infección por virus portadores de oncogenes como el virus del papiloma humano que in-
corpora una E3 ligasa de p53.
La reordenación cromosómica durante una división celular puede dejar la expresión de
ciertos genes regulada por potenciadores (enhancers) muy eficientes aumentando la cantidad
del producto.
Conversión de un proto-oncogén en oncogén por mutaciones puntuales como en el caso
de Ras que puede perder su capacidad GTPasa.
Perdida de proteínas con actividad fosfatasa, que actúan sobre proteín-quinasas y fofoinosito-
les.
Anomalías que aumentan la supervivencia celular como las que detienen la apoptosis,
como PKB. Generalmente la proteína Bad está relacionada con la apoptosis porque se une a un
inhibidor de este proceso. Si PKB fosforila a la proteína Bad, esta libera al inhibidor de la apop-
tosis y por lo tanto se inhibe el proceso de muerte celular, lo que puede derivar en tumores.
Pérdida de antioncogenes como p53, Rb, APC, Smad4 (p15), TGFβ-2…
Expresión de telomerasa
Sobreproducción de ciclinas mitóticas.
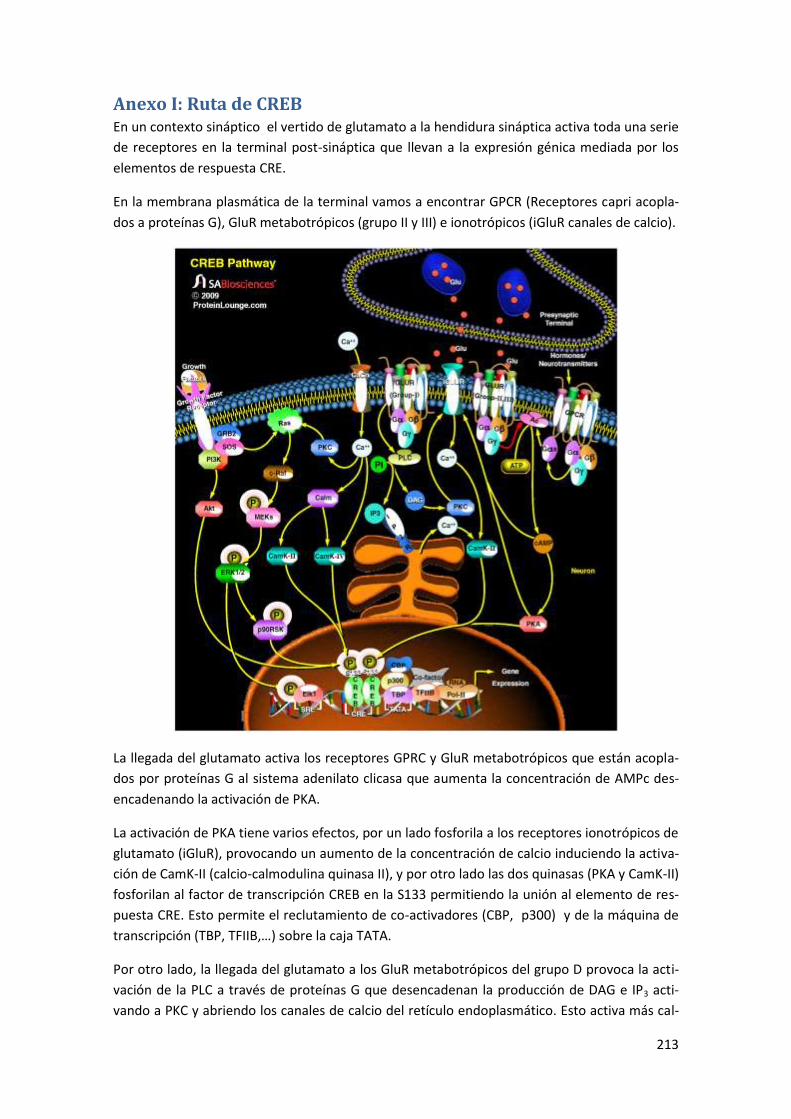
213
Anexo I: Ruta de CREB En un contexto sináptico el vertido de glutamato a la hendidura sináptica activa toda una serie
de receptores en la terminal post-sináptica que llevan a la expresión génica mediada por los
elementos de respuesta CRE.
En la membrana plasmática de la terminal vamos a encontrar GPCR (Receptores capri acopla-
dos a proteínas G), GluR metabotrópicos (grupo II y III) e ionotrópicos (iGluR canales de calcio).
La llegada del glutamato activa los receptores GPRC y GluR metabotrópicos que están acopla-
dos por proteínas G al sistema adenilato clicasa que aumenta la concentración de AMPc des-
encadenando la activación de PKA.
La activación de PKA tiene varios efectos, por un lado fosforila a los receptores ionotrópicos de
glutamato (iGluR), provocando un aumento de la concentración de calcio induciendo la activa-
ción de CamK-II (calcio-calmodulina quinasa II), y por otro lado las dos quinasas (PKA y CamK-II)
fosforilan al factor de transcripción CREB en la S133 permitiendo la unión al elemento de res-
puesta CRE. Esto permite el reclutamiento de co-activadores (CBP, p300) y de la máquina de
transcripción (TBP, TFIIB,…) sobre la caja TATA.
Por otro lado, la llegada del glutamato a los GluR metabotrópicos del grupo D provoca la acti-
vación de la PLC a través de proteínas G que desencadenan la producción de DAG e IP3 acti-
vando a PKC y abriendo los canales de calcio del retículo endoplasmático. Esto activa más cal-
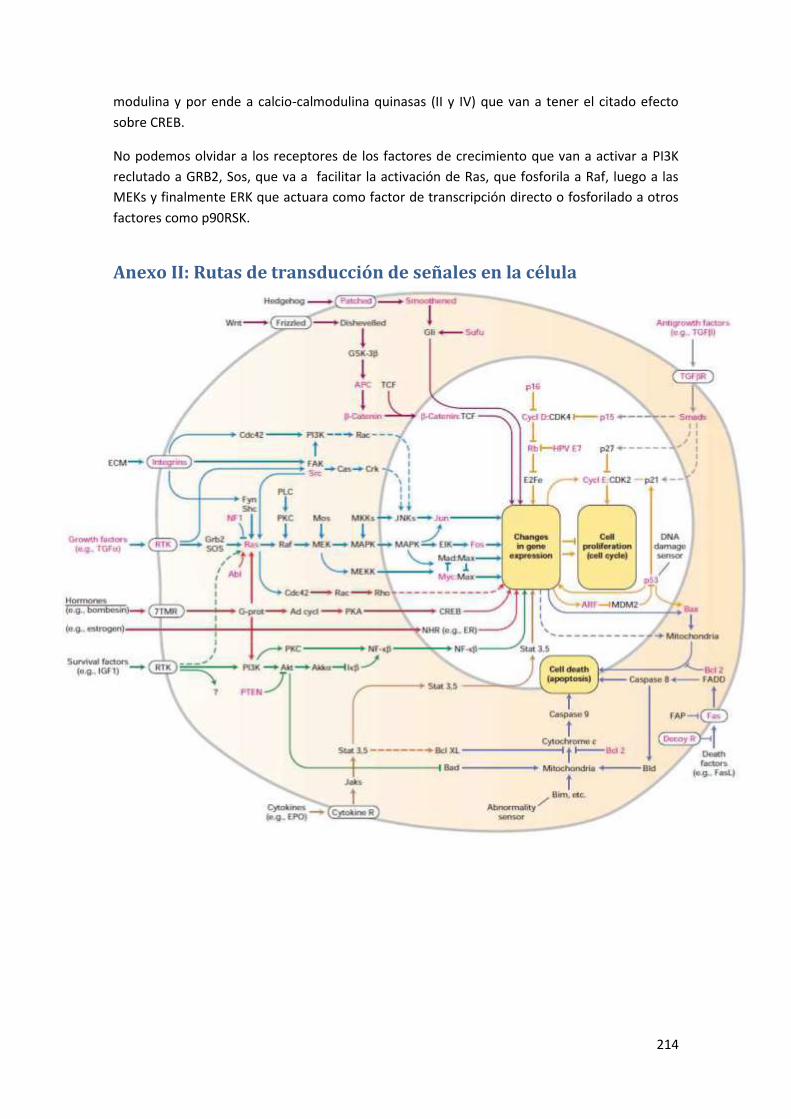
214
modulina y por ende a calcio-calmodulina quinasas (II y IV) que van a tener el citado efecto
sobre CREB.
No podemos olvidar a los receptores de los factores de crecimiento que van a activar a PI3K
reclutado a GRB2, Sos, que va a facilitar la activación de Ras, que fosforila a Raf, luego a las
MEKs y finalmente ERK que actuara como factor de transcripción directo o fosforilado a otros
factores como p90RSK.
Anexo II: Rutas de transducción de señales en la célula

215
Bloque IV: Regulación Molecular De La
Neurotransmisión
TEMA 1 Canales Iónicos Activados por Voltaje
Pruebas electrofisiológicas. Procedimientos de purificación de los canales ióni-cos. Reconstitución en liposomas. Estructura terciaria y caternaria, selectividad iónica y funcio-namiento de los canales de sodio, calcio y potasio. Patologías que originan los fallos genéticos.
TEMA 2 Neurotransmisión Colinérgica
Transporte de colina. Síntesis y transporte vesicular de acetilcolina. Liberación y degradación de acetilcolina: colinesterasas.
TEMA 3 Receptores Ionotrópicos
Aspectos estructurales y funcionales de los receptores ionotrópicos. Receptores excitatorios: colinérgicos nicotínicos y de glutamato. Agonistas y antagonistas. Receptores inhibitorios: de GABA y glicina. Integración funcional de señales que despolarizan e hiperpolarizan la membra-na.
TEMA 4 Neurotransmisión Adrenérgica y Receptores Metabotrópicos
Biosíntesis de catecolaminas: etapas individuales. Liberación, captura e inactivación. Mono-amino oxidasas. Receptores metabotrópicos. Ansiedad, depresión y trastorno bipolar. Estimu-lantes, ansiolíticos, ansiogénicos psicotrópicos.
TEMA 5 Neurotransmisión Peptidérgica
Neuropéptidos: biosíntesis, liberación y degradación. Acciones celulares y fisiológicas. Morfina, heroína, cocaína: adicción, tolerancia y analgesia. Encefalinas y endorfinas. Receptores de opiáceos: clases y efectos. Cannabinoides, receptores y acciones fisiológicas.

216
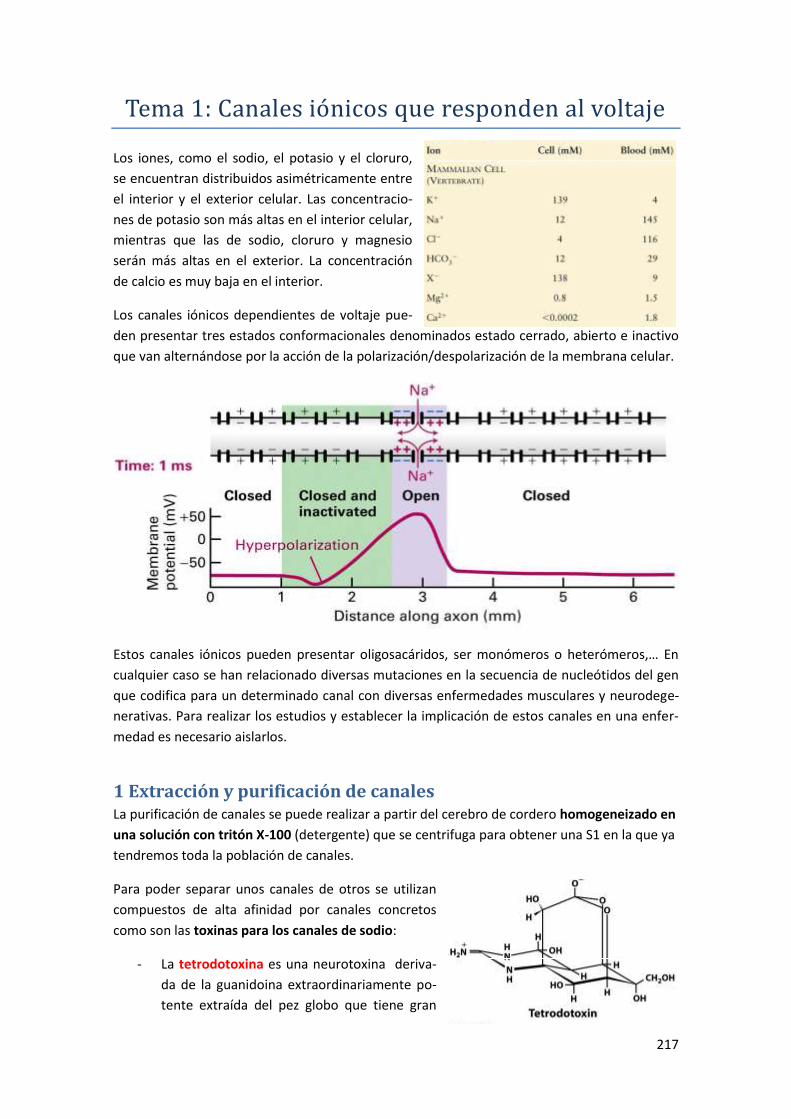
217
Tema 1: Canales io nicos qúe responden al voltaje
Los iones, como el sodio, el potasio y el cloruro,
se encuentran distribuidos asimétricamente entre
el interior y el exterior celular. Las concentracio-
nes de potasio son más altas en el interior celular,
mientras que las de sodio, cloruro y magnesio
serán más altas en el exterior. La concentración
de calcio es muy baja en el interior.
Los canales iónicos dependientes de voltaje pue-
den presentar tres estados conformacionales denominados estado cerrado, abierto e inactivo
que van alternándose por la acción de la polarización/despolarización de la membrana celular.
Estos canales iónicos pueden presentar oligosacáridos, ser monómeros o heterómeros,… En
cualquier caso se han relacionado diversas mutaciones en la secuencia de nucleótidos del gen
que codifica para un determinado canal con diversas enfermedades musculares y neurodege-
nerativas. Para realizar los estudios y establecer la implicación de estos canales en una enfer-
medad es necesario aislarlos.
1 Extracción y purificación de canales La purificación de canales se puede realizar a partir del cerebro de cordero homogeneizado en
una solución con tritón X-100 (detergente) que se centrifuga para obtener una S1 en la que ya
tendremos toda la población de canales.
Para poder separar unos canales de otros se utilizan
compuestos de alta afinidad por canales concretos
como son las toxinas para los canales de sodio:
- La tetrodotoxina es una neurotoxina deriva-
da de la guanidoina extraordinariamente po-
tente extraída del pez globo que tiene gran
Ligandos canal Na: tetrodotoxina, batracotoxina, veratridina, toxinas de escorpión and anémonas100 canales/µ2 axones no mielinizados; 20,000 canales/µ2 axones mielinizadosSelectividad: Na 1; NH3OH 0.9; Li 0.9; NH2NH3 0.6; NH4 0.1; K 0.1; Rb<0.01; CH3NH3< 0.01
K 1; Rb 0.9, NH4 0.1, Li<0.01, Na<0.01
Na –-RVVRVFRIGRILRLIKAAKGIR--Hélice de respuesta al voltaje
Ca –-KILRVLRVLRPLRAINRAKGLK--Hélice de respuesta al voltaje
Ligandoscanal Ca,:dantroleno, nifedipina, nitrendipina
Ligandoscanal K: tetraetilamonio(TEA), dendrotoxina de serpiente mamba
Ligandoscanal Ca,:dantroleno, nifedipina, nitrendipina

218
afinidad por los canales de sodio. Se trata de una molécula hidrosoluble que bloquea
los canales de sodio evitando la despolarización de la membrana.
- La batracotoxina (sapo) y veratridina (planta) son toxinas liposolubles que se unen al
canal de sodio pero por sus dominios intermembrana, alterando el tiempo de transi-
ción entre los estados del canal. No es lo mismo que un canal cambie en 1 s que tar-
de 10-20 min.
- Toxinas de escorpión y anémona.
Para los canales de potasio se utilizan ligandos específicos o toxinas como:
- Tetraetilamonio (TEA)
- Dendrotoxina de serpiente mamba
Para los canales de calcio se utilizan derivados de piridinas sustituidas:
- Dantroleno
- Nifedipina
- Nitredipina
Como ya hemos mencionado en el extracto S1 tendremos toda la población de canales de so-
dio, calcio y potasio. En primer lugar, antes de realizar una purificación específica de los cana-
les de sodio vamos a separar los canales en general del resto de proteínas que producirán
unión inespecífica.
Puesto que los canales de sodio son glicoproteínas, porque están en la membrana plasmática y
podemos presumir que tienen estructura oligomérica, tal vez sean reconocidos por alguna
lectina como LCA (aglutinina de la lenteja). Por lo tanto mediante una matriz de sefarosa-LCA
que reconoce manosas separamos todas las glicoproteínas con manosa terminal y lavamos el
resto. Para liberar las glicoproteínas de la matriz realizamos un lavado con una disolución de
manosa (1M) y se dializa lo obtenido para retirar el exceso de manosa.
A continuación, para realizar un seguimiento a la capacidad de unión de la toxina se añade
tetrodotoxina radiactiva a la disolución, se lava la no unida y se mide con un contador de cen-
telleo la radiactividad total.
En una segunda fase se utiliza una placa de sefarosa-tetrodotoxina para que los canales de
sodio queden específicamente unidos. Finalmente lavamos la sefarosa con una disolución de
tetrodotoxina, obteniéndose una purificación rica en proteínas que ligan esta toxina.
Para comprobar la pureza podemos realizar una electroforesis o para posteriores experimen-
tos inyectarla a un conejo con el fin de generar un antisuero que nos permitirá realizar experi-
mentos como el Western blot.
La purificación de los canales permite la realización de estudios funcionales a través de lipo-
somas a los que se adicionan los canales purificados. Posteriormente mediante la adicción de
sodio radiactivo en este caso, y de toxinas que provocan la despolarización de la membrana
(veratridina) porque pasan los canales del estado cerrado al abierto, pues tendremos que de-
tectar señal en el interior de los liposomas.

219
Por el contrario, la adición de tetrodotoxina retiene los canales de sodio en estado cerrado, lo
que nos permite establecer la cantidad de sodio presente en el liposoma que se considerará
como background inespecífico.
2 Tránsito de iones a través de canales Los estudios mediante liposomas han permitido el establecimiento de los iones capaces de
atravesar cada canal específicamente y la facilidad con la que lo hacen. Los canales iónicos
dependientes de voltaje presen una estructura con alta homología entre ellos ya que se trata
de una familia proteica diseñada para actuar como canales iónicos dependientes de voltaje.
La comparación con otros canales dependientes de ligando (muscarínicos, GABA, Glutama-
to,…) también revela cierta homología, lo que es lógico considerando la similitud topológica y
funcional.
El funcionamiento de estos canales depende de la polarización de la membrana. Cuando se
produce la despolarización los canales pasan del estado cerrado al abierto, permitiendo el
tránsito de iones y a continuación pasan al estado inactivo de manera espontanea. Para que se
produzca la transición del estado inactivo al estado cerrado es necesario que haya repolariza-
ción de la membrana.
Por ello es necesario que la proteína presente dominios que respondan a la despolarización y
al tránsito de iones (ionóforo) que es el canal en sí, muy selectivo y restrictivo. Los canales
dependientes de ligandos presentan un cierto rango siendo unos muy selectivos y otros no,
pero estos canales dependientes de voltaje van a ser todos muy restrictivos.
El tránsito de iones a través de los canales específicos se realiza considerando un 100% de li-
bertad de tránsito para el ión específico.
Na Li NH3OH NH4 NH2NH3 CH3NH3 K Rb
Canal de Na 1 0,9 0,9 0,1 0,6 0,01 0,1 0,01
Canal de K 0,01 0,01 0,1 1 0,9
0
0,2
0,4
0,6
0,8
1
1,2
%
Tránsito de iones

220
A través de los canales de sodio entra por tanto el sodio con un 100% de facilidad, pero tam-
bién:
- El sodio (100%) presenta un diámetro de 5 Å, pero que aumenta ya que pasa a través
del canal solvatado.
- El litio (90%) porque presenta un radio iónico similar en solvatación.
- El hidróxilamonio (NH3OH, 90%) es capaz de atravesar el canal gracias a su capacidad
para formar puentes de hidrógeno con los aminoácidos del canal, mientras que el
amonio NH4 (10%) no presenta electrones desapareados para formar estas interaccio-
nes y no pasa a pesar de presentar menor tamaño.
- La hidracina (NH2NH3, 60%) y el metilamonio (CH3NH3, <1%) a pesar del tener el
mismo tamaño presentan esa gran diferencia de facilidad de paso debido a que la pri-
mera puede establecer puentes de hidrógeno.
- El potasio (10%) y el rubidio (<1%) entran muy mal a través del canal ya que presentan
mayor radio iónico.
A través de los canales de potasio pasa el potasio con un 100% de facilidad en solvatación:
- Rubidio (90%) por presentar un tamaño similar.
- El amonio (10%) en la misma medida que el anterior porque no puede establecer los
enlaces por puentes de hidrógeno.
- El sodio (<1%) y el litio (<1%) no pueden pasar a través del canal a pesar de presentar
un tamaño menor y esto como veremos se debe a las interaciones que se van a esta-
blecer en el canal.
En los axones no mielinizados podemos encontrar una densidad de 100 canales de sodio/2,
mientras que en los axones mielinizados podemos encontrar hasta 20.000 canales/2.
3 Estructura de los canales dependientes de voltaje En general los canales dependientes de voltaje, tanto si son tetrámeros como monómeros va a
estar constituidos por una serie de secuencias cuyo índice de hidrofobicidad es muy alto por lo
que dan lugar a muchas hélices hidrofóbicas que se insertan en la membrana.
Estos canales están formados siempre por 6 hélices hidrofóbicas en las que vamos a diferen-
ciar 4 dominios altamente conservados. El hecho de encontrar esta homología estructural y
secuencial hace pensar que los canales dependientes de voltaje se formaron a partir de un gel
ancestral cuya duplicación ha dado lugar a diferentes variantes. La unión de genes duplicados
habría dado lugar a la formación de los canales monoméricos (tetrámeros de 24 hélices) en los
que la unidad básica se repite.
De las 6 hélices que conforman la unidad de los canales tenemos:
- La hélice 4 con cargas positivas que es el sensor de voltaje.
- Las hélices 5 y 6 que forman el ionóforo, es decir el canal que atraviesan los iones.
- Entre las hélices 5 y 6 hay un segmento o asa que no es igual en todos los casos pero
que son las encargadas de permitir o no el paso de iones según su conformación.
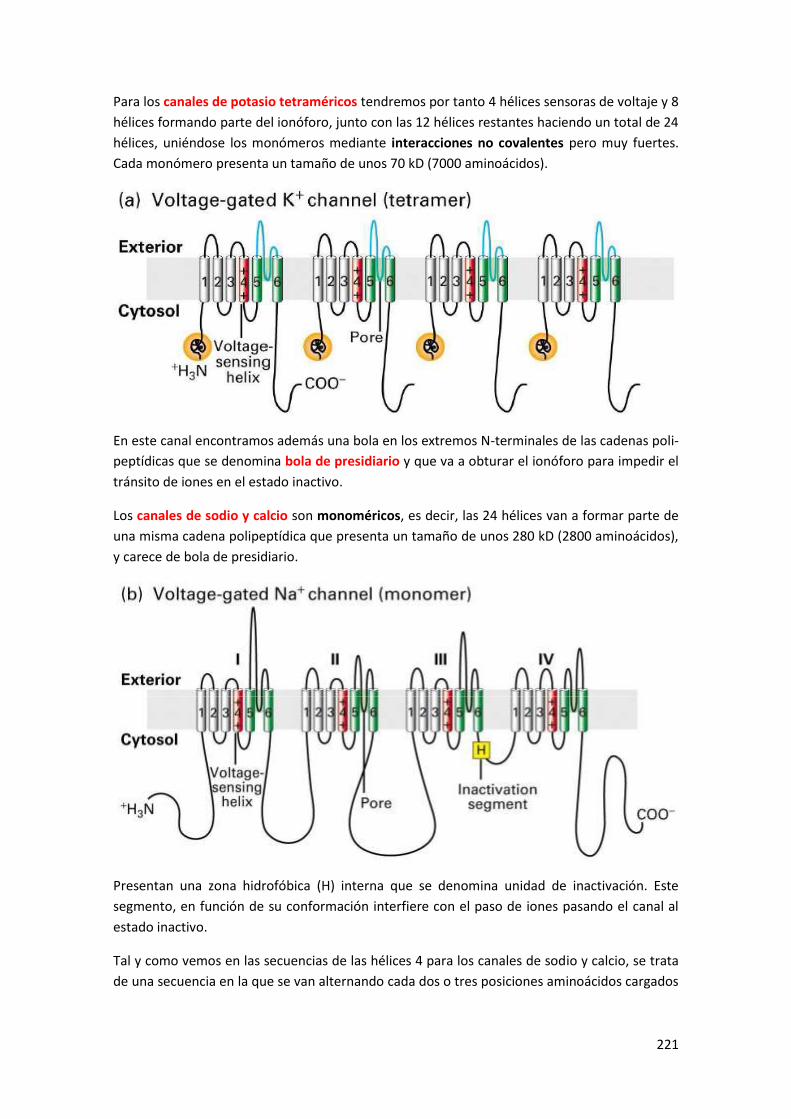
221
Para los canales de potasio tetraméricos tendremos por tanto 4 hélices sensoras de voltaje y 8
hélices formando parte del ionóforo, junto con las 12 hélices restantes haciendo un total de 24
hélices, uniéndose los monómeros mediante interacciones no covalentes pero muy fuertes.
Cada monómero presenta un tamaño de unos 70 kD (7000 aminoácidos).
En este canal encontramos además una bola en los extremos N-terminales de las cadenas poli-
peptídicas que se denomina bola de presidiario y que va a obturar el ionóforo para impedir el
tránsito de iones en el estado inactivo.
Los canales de sodio y calcio son monoméricos, es decir, las 24 hélices van a formar parte de
una misma cadena polipeptídica que presenta un tamaño de unos 280 kD (2800 aminoácidos),
y carece de bola de presidiario.
Presentan una zona hidrofóbica (H) interna que se denomina unidad de inactivación. Este
segmento, en función de su conformación interfiere con el paso de iones pasando el canal al
estado inactivo.
Tal y como vemos en las secuencias de las hélices 4 para los canales de sodio y calcio, se trata
de una secuencia en la que se van alternando cada dos o tres posiciones aminoácidos cargados

222
positivamente a pH fisiológico. Esta hélices son anfipáticas, ya que presentan todas las cargas
en un lado actuando como sensor de los cambios de potencial en la membrana.
Dentro del contexto de los canales identificados cada uno de estos presenta unos requerimien-
tos especiales en función del tejido que van a estar mediados por la presencia o no de subuni-
dades accesorias, pero la unidad básica de 280 kD se mantiene.
Por ejemplo, muchos de los canales son dianas de protein-quinasas regulándose el tiempo de
permanencia en estado abierto en función de la fosforilación.
A nivel genético podemos observar como los canales de sodio y calcio se disponen como uni-
dades repetidas del mismo gen. Además se ha caracterizado la variante Shaker del canal de
potasio (mosca mutante).
En el caso de procariotas, los canales e potasio solo están formados por las dos últimas hélices
y el asa, por lo que es lo mínimo que se necesita para establecerlo y además resulta muy útil
para los estudios funcionales.
Podemos ver el funcionamiento de las hélices sensoras de voltaje representadas en el modelo
de bola de presidiario (aunque se índice canal de sodio, no es correcto). La despolarización de
la membrana provoca el cambio de posición relativa de las hélices en la proteína lo que provo-
ca la transición desde unos estados a otros.
Ligandos canal Na: tetrodotoxina, batracotoxina, veratridina, toxinas de escorpión and anémonas100 canales/µ2 axones no mielinizados; 20,000 canales/µ2 axones mielinizadosSelectividad: Na 1; NH3OH 0.9; Li 0.9; NH2NH3 0.6; NH4 0.1; K 0.1; Rb<0.01; CH3NH3< 0.01
K 1; Rb 0.9, NH4 0.1, Li<0.01, Na<0.01
Na –-RVVRVFRIGRILRLIKAAKGIR--Hélice de respuesta al voltaje
Ca –-KILRVLRVLRPLRAINRAKGLK--Hélice de respuesta al voltaje
Ligandoscanal Ca,:dantroleno, nifedipina, nitrendipina
Ligandoscanal K: tetraetilamonio(TEA), dendrotoxina de serpiente mamba
Ligandoscanal Ca,:dantroleno, nifedipina, nitrendipina
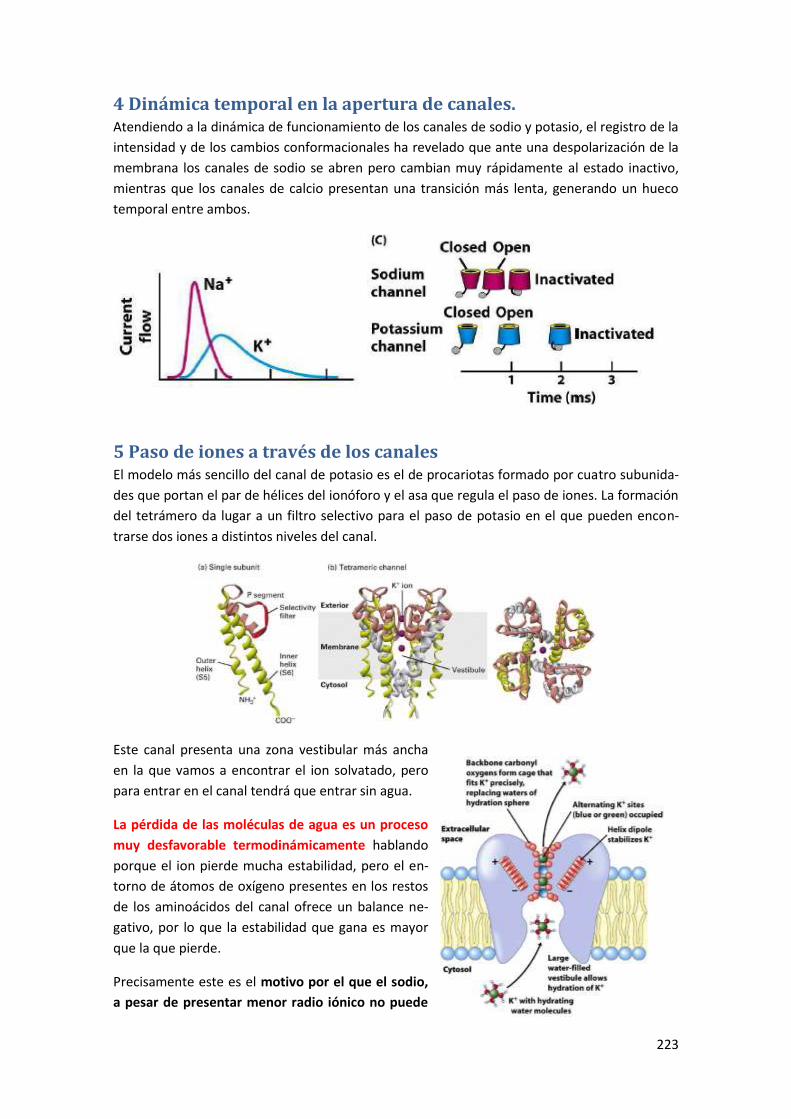
223
4 Dinámica temporal en la apertura de canales. Atendiendo a la dinámica de funcionamiento de los canales de sodio y potasio, el registro de la
intensidad y de los cambios conformacionales ha revelado que ante una despolarización de la
membrana los canales de sodio se abren pero cambian muy rápidamente al estado inactivo,
mientras que los canales de calcio presentan una transición más lenta, generando un hueco
temporal entre ambos.
5 Paso de iones a través de los canales El modelo más sencillo del canal de potasio es el de procariotas formado por cuatro subunida-
des que portan el par de hélices del ionóforo y el asa que regula el paso de iones. La formación
del tetrámero da lugar a un filtro selectivo para el paso de potasio en el que pueden encon-
trarse dos iones a distintos niveles del canal.
Este canal presenta una zona vestibular más ancha
en la que vamos a encontrar el ion solvatado, pero
para entrar en el canal tendrá que entrar sin agua.
La pérdida de las moléculas de agua es un proceso
muy desfavorable termodinámicamente hablando
porque el ion pierde mucha estabilidad, pero el en-
torno de átomos de oxígeno presentes en los restos
de los aminoácidos del canal ofrece un balance ne-
gativo, por lo que la estabilidad que gana es mayor
que la que pierde.
Precisamente este es el motivo por el que el sodio,
a pesar de presentar menor radio iónico no puede
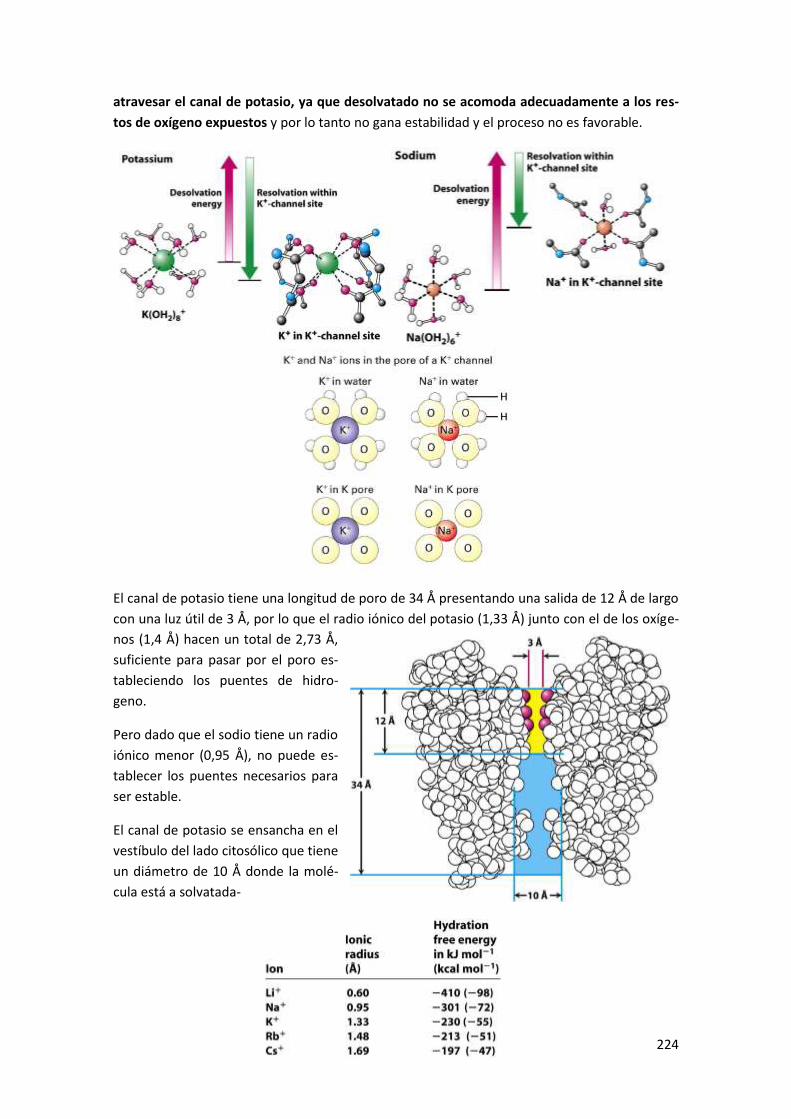
224
atravesar el canal de potasio, ya que desolvatado no se acomoda adecuadamente a los res-
tos de oxígeno expuestos y por lo tanto no gana estabilidad y el proceso no es favorable.
El canal de potasio tiene una longitud de poro de 34 Å presentando una salida de 12 Å de largo
con una luz útil de 3 Å, por lo que el radio iónico del potasio (1,33 Å) junto con el de los oxíge-
nos (1,4 Å) hacen un total de 2,73 Å,
suficiente para pasar por el poro es-
tableciendo los puentes de hidro-
geno.
Pero dado que el sodio tiene un radio
iónico menor (0,95 Å), no puede es-
tablecer los puentes necesarios para
ser estable.
El canal de potasio se ensancha en el
vestíbulo del lado citosólico que tiene
un diámetro de 10 Å donde la molé-
cula está a solvatada-
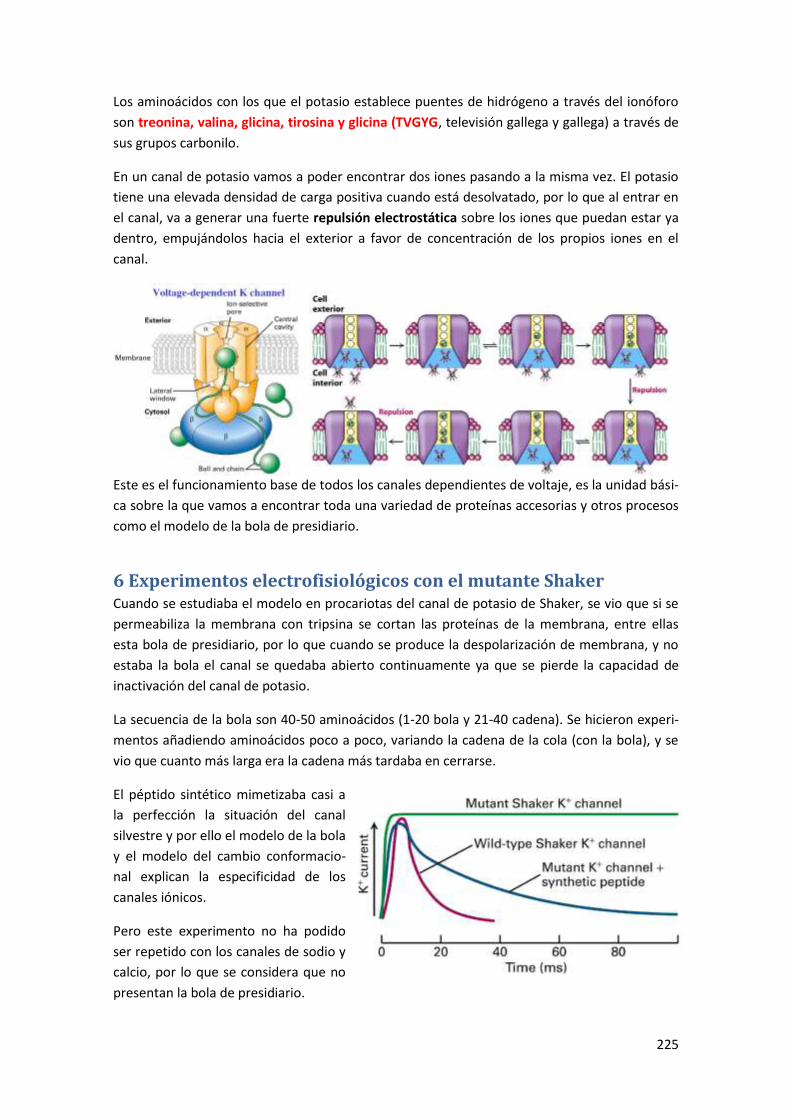
225
Los aminoácidos con los que el potasio establece puentes de hidrógeno a través del ionóforo
son treonina, valina, glicina, tirosina y glicina (TVGYG, televisión gallega y gallega) a través de
sus grupos carbonilo.
En un canal de potasio vamos a poder encontrar dos iones pasando a la misma vez. El potasio
tiene una elevada densidad de carga positiva cuando está desolvatado, por lo que al entrar en
el canal, va a generar una fuerte repulsión electrostática sobre los iones que puedan estar ya
dentro, empujándolos hacia el exterior a favor de concentración de los propios iones en el
canal.
Este es el funcionamiento base de todos los canales dependientes de voltaje, es la unidad bási-
ca sobre la que vamos a encontrar toda una variedad de proteínas accesorias y otros procesos
como el modelo de la bola de presidiario.
6 Experimentos electrofisiológicos con el mutante Shaker Cuando se estudiaba el modelo en procariotas del canal de potasio de Shaker, se vio que si se
permeabiliza la membrana con tripsina se cortan las proteínas de la membrana, entre ellas
esta bola de presidiario, por lo que cuando se produce la despolarización de membrana, y no
estaba la bola el canal se quedaba abierto continuamente ya que se pierde la capacidad de
inactivación del canal de potasio.
La secuencia de la bola son 40-50 aminoácidos (1-20 bola y 21-40 cadena). Se hicieron experi-
mentos añadiendo aminoácidos poco a poco, variando la cadena de la cola (con la bola), y se
vio que cuanto más larga era la cadena más tardaba en cerrarse.
El péptido sintético mimetizaba casi a
la perfección la situación del canal
silvestre y por ello el modelo de la bola
y el modelo del cambio conformacio-
nal explican la especificidad de los
canales iónicos.
Pero este experimento no ha podido
ser repetido con los canales de sodio y
calcio, por lo que se considera que no
presentan la bola de presidiario.

226

227
Tema 2: Transmisio n coline rgica
1 Proteínas implicadas en la transmisión colinérgica Al hablar de la transmisión colinérgica se hace necesario realizar un recordatorio de las proteí-
nas implicadas en el operador colinérico lo que incluye a los transportadores de colina (ChTs),
la colín-acetil transferasa (ChAT) encargada de sintetizar acetilcolina.
Esta molécula, una vez sintetizada, va a ser transportada a las vesículas de la terminal sináptica
por los transportadores vesiculares de ACh (VAChT) para ser vertida al espacio sináptico al
producirse la despolarización de la membrana.
En el espacio sináptico tenemos dos tipos de receptores para ACh, los receptores Nicotínicos
(N-AChR) y los muscarínicos (m-AChR). Finalmente para limpiar el espacio tenemos las coli-
nesterasas (AChE y BuChE).
1.1 Transportadores de colina (ChTs) Los transportadores de colina están presentes en todas las células ya que las colinas son nece-
sarias para la formación de las fosfatidilcolinas que componen las membranas celulares.
Estos transportadores y se van a clasificar en dos tipos en función de su afinidad:
- Los transportadores de colina de baja afinidad se distribuyen en todos los tejidos peri-
féricos.
- Los transportadores de colina de alta afini-
dad se distribuyen a nivel del SNC junco con
los anteriores y se inhiben por hemicholinio-3
y van a depender de sodio para su funciona-
miento ya que se trata de un co-transporte
tipo simporte, es decir, sodio y colina entran
al mismo tiempo. Estos transportadores per-
tenecen a la familia de transportadores de ca-
tecolaminas, siendo todos muy parecidos.
La colina que se encuentra en el torrente sanguíneo, a una concentración 10 mM, va a proce-
der de la dieta (1/2) y de la recaptación que se produce tras la hidrolisis de acetilcolina (1/2).
La presencia de los transportadores de alta afinidad en el SNC asegura el suministro de colina
en momentos de ayuno.
Dado que la recaptación de colina en el espacio sináptico depende de sodio, la inhibición de las
bombas de sodio/potasio de la membrana plasmática mediante ouabaína impedirá el proceso.
1.2 Síntesis de acetilcolina y transportador vesicular (VAChT) Una vez que la colina está en el interior celular esta puede ser unida con acetil-CoA dando
lugar a la acetilcolina (ACh) mediante una reacción bisustrato catalizada por la colina acetil-
transferasa.
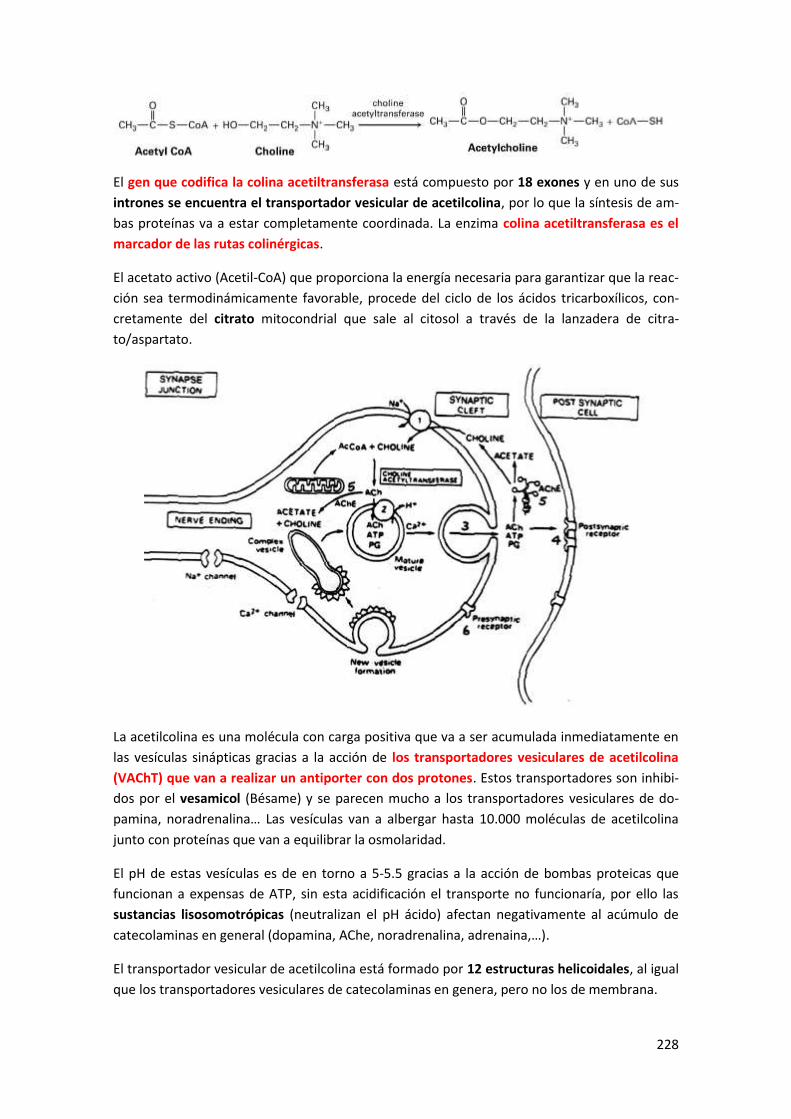
228
El gen que codifica la colina acetiltransferasa está compuesto por 18 exones y en uno de sus
intrones se encuentra el transportador vesicular de acetilcolina, por lo que la síntesis de am-
bas proteínas va a estar completamente coordinada. La enzima colina acetiltransferasa es el
marcador de las rutas colinérgicas.
El acetato activo (Acetil-CoA) que proporciona la energía necesaria para garantizar que la reac-
ción sea termodinámicamente favorable, procede del ciclo de los ácidos tricarboxílicos, con-
cretamente del citrato mitocondrial que sale al citosol a través de la lanzadera de citra-
to/aspartato.
La acetilcolina es una molécula con carga positiva que va a ser acumulada inmediatamente en
las vesículas sinápticas gracias a la acción de los transportadores vesiculares de acetilcolina
(VAChT) que van a realizar un antiporter con dos protones. Estos transportadores son inhibi-
dos por el vesamicol (Bésame) y se parecen mucho a los transportadores vesiculares de do-
pamina, noradrenalina… Las vesículas van a albergar hasta 10.000 moléculas de acetilcolina
junto con proteínas que van a equilibrar la osmolaridad.
El pH de estas vesículas es de en torno a 5-5.5 gracias a la acción de bombas proteicas que
funcionan a expensas de ATP, sin esta acidificación el transporte no funcionaría, por ello las
sustancias lisosomotrópicas (neutralizan el pH ácido) afectan negativamente al acúmulo de
catecolaminas en general (dopamina, AChe, noradrenalina, adrenaina,…).
El transportador vesicular de acetilcolina está formado por 12 estructuras helicoidales, al igual
que los transportadores vesiculares de catecolaminas en genera, pero no los de membrana.
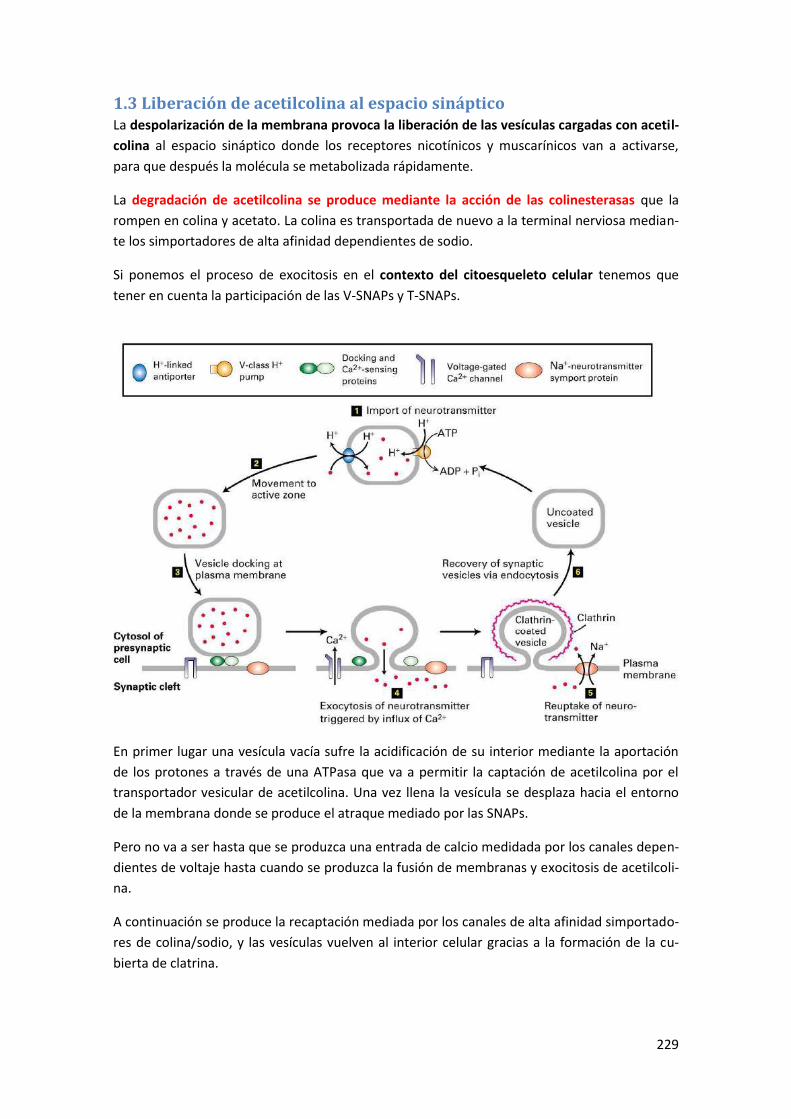
229
1.3 Liberación de acetilcolina al espacio sináptico La despolarización de la membrana provoca la liberación de las vesículas cargadas con acetil-
colina al espacio sináptico donde los receptores nicotínicos y muscarínicos van a activarse,
para que después la molécula se metabolizada rápidamente.
La degradación de acetilcolina se produce mediante la acción de las colinesterasas que la
rompen en colina y acetato. La colina es transportada de nuevo a la terminal nerviosa median-
te los simportadores de alta afinidad dependientes de sodio.
Si ponemos el proceso de exocitosis en el contexto del citoesqueleto celular tenemos que
tener en cuenta la participación de las V-SNAPs y T-SNAPs.
En primer lugar una vesícula vacía sufre la acidificación de su interior mediante la aportación
de los protones a través de una ATPasa que va a permitir la captación de acetilcolina por el
transportador vesicular de acetilcolina. Una vez llena la vesícula se desplaza hacia el entorno
de la membrana donde se produce el atraque mediado por las SNAPs.
Pero no va a ser hasta que se produzca una entrada de calcio medidada por los canales depen-
dientes de voltaje hasta cuando se produzca la fusión de membranas y exocitosis de acetilcoli-
na.
A continuación se produce la recaptación mediada por los canales de alta afinidad simportado-
res de colina/sodio, y las vesículas vuelven al interior celular gracias a la formación de la cu-
bierta de clatrina.

230
Cuando llega un potencial de acción a la terminal sináptica se abren los canales de calcio de-
pendientes de voltaje que provocan la liberación de acetilcolina al espacio sináptico donde los
receptores nicotínicos (ionotrópicos, canales) de acetilcolina que son pentámeros αβ2γδ (SNC
embrionario) o αβ2εδ (musculo embrionario y zonas no sinápticas).
Existen del orden de 11 genes para la subunidad α, 3-4 genes para la subunidad β y un gen
para las restantes. En función de la zona los receptores nicotínicos serán pentámeros homo-
méricos o heteroméricos presentando diferentes afinidades por agonistas y antagonistas.
La subunidad α de estos canales es la
que liga agonistas como la acetilcolina
provocando un cambio conformacional
que pasa el canal al estado abierto pre-
sentando un diámetro de poro de 10 Å
(es menos selectivo y también puede
pasar el calcio).
La entrada de sodio a través de los re-
ceptores ionotrópicos desencadena la
despolarización local de la membrana
que genera un potencial de acción ca-
paz de abrir los canales de sodio de-
pendientes de voltaje y finalmente los tubulotransversos, donde los canales de sodio se aco-
plan a los receptores de rianodina de las cisternas terminales del retículo sarcoplasmático y
provocan la liberación de calcio y la contracción del músculo.
1.4 Receptores muscarínicos (m-AChR) Los receptores muscarínicos de acetilcolina presentan como agonistas la acetilcolina y la mus-
carina. Sus antagonistas son la atropina (belladona) y otros.
La localización de estos canales es en el sistema nervioso central, los ganglios del sistema ner-
vioso periférico, órganos efectores (corazón, pulmón e intestino) y glándulas endocrinas.
Se trata de receptores metabotrópicos, es decir, que todos presentan estructura de receptor
7tm asociado a proteínas G entre los que va-
mos a distinguir dos grupos:
- Los receptores M1, M3 y M5 (impares)
actúan activando a fosfolipasa C gene-
rando IP3 y estimulando el sistema de
PKC (activadores).
- Los receptores M2 y M4 (pares) actúan
reduciendo el sistema de adenilato ci-
clasa y abren los canales de potasio
(inhibidores) favoreciendo la hiperpo-
larización de membranas.

231
2 Drogas que afectan a la sinapsis colinérgica Existen muchas drogas que actúan a nivel de la sinapsis colinérgica ya sea sobre los propios
receptores o aumentan la cantidad de acetilcolina.
La nicotina y la muscarina son agonistas de los receptores nicotínicos y muscarínicos respecti-
vamente. La atropina y la escoplamina son antagonistas de estos receptores y por tanto inhi-
ben la sinapsis.
Los organofosforados se consideran agonistas del sistema porque inhiben la actuación de las
acetilcolinesterasas impidiendo la degradación de acetilcolina y por tanto perpetuando el es-
tímulo.
La toxina botulínica y la toxina del tétanos son antagonistas porque van a impedir la libera-
ción de las vesículas sinápticas, pero el veneno de la viuda negra va a provocar la liberación
masiva de estas vesículas por lo que se considera un agonista.
3 Colinesterasas (AChE y BuChE) Distribución en tejidos, células y fluidos.
- La acetilcolinesterasa (AChE) está distribuida en el sistema nervioso y en los glóbulos
rojos.
- La butirilcolinesterasa (BuChE) se encuentra distribuida por todos los tejidos humanos
salvo en los glóbulos rojos.
Preferencia por substrato e inhibidores.
- La AChE hidroliza específicamente acetilcolina liberando colina y acetato. Es fuerte-
mente inhibida por BW284c51 y poco inhibida por Iso-OMPA.
- La BuChE presenta un centro activo de mayor tamaño que le permite hidrolizar otras
moléculas y puede suplir la acción de AChE, pero su substrato óptimo es la butirilcoli-
na. Es fuertemente inhibida por Iso-OMPA pero poco inhibida por BW284c51.
La e-serina es un inhibidor específico de colinesterasas que permite diferenciar entre coli-
nesterasas y esterasas cualesquiera.
Propiedades cinéticas.
- La AChE es inhibida por un exceso de sustrato.
- La BuChE no se inhibe por exceso de sustrato y es más rápida que AChE.
El centro catalítico de las colinesterasas presenta un resto de glutamato, histidina y serina
muy importantes. La reacción transcurre en dos etapas, primero el glutamato cede elec-
trones a la histidina y esta se los cede a la serina que ataca a la acetilcolina formando un
intermediario carbamil-serina. En segundo lugar una molécula de agua hidroliza el inter-
medio y libera el acetato y la colina.

232
Acciones colinérgicas y no colinérgicas.
- Las acciones colinérgicas de las colinesterasas son aquellas implicadas con la neuro-
transmisión y la sinapsis colinérgica.
- Las acciones no colinérgicas son la hidrólisis de neuropeptidos como la sustancia P, en-
cefalinas y endorfinas. Control de la actividad motora por secreción desde varias re-
giones cerebrales. Están implicadas en la formación de nudos neurofibrilares y placas
amilodes en los cerebros con Alzheimer. Diferenciación y proliferación en tejidos neu-
ronales y no neuronales. Detoxifificación (BuChE).
Toxicidad de los organofosforados.
La toxicidad de los compuestos organofosforados y de los carbamatos (insecticidas) se debe a
que reaccionan con la serina del centro catalítico y provocan la inutilización de las enzimas
disponibles.
Suxametonio y episodios de apnea.
La hipersensibilidad al suxametonio que actúa como inhibidor competitivo de la BuChE en la
placa motora es la primera situación en la que se ha podido demostrar una función biológica
para esta enzima en la detoxificación.
Este compuesto cuando llega a la placa motora de los músculos se une a los receptores de
acetilcolina durante mucho tiempo y provoca la relajación muscular por una despolarización
de membrana continuada. Dado que es un mal sustrato de AChE, bloquea su acción y ralentiza
en gran medida a BuChE.
Por ello se ha utilizado clínicamente como un sedante, o relajante muscular general. Pero en
muchos casos que posteriormente se comprobó que eran pacientes con deficiencias en BuChE,
por mutación o intoxicación de organofosforados, se producía una parálisis mayor de la espe-
rada y apnea, estableciéndose que era BuChE la encargada de la detoxificación.
Variedad de mRNAs para AChE
Todas las formas moléculas de AChE están
codificadas por un solo gen constituido
por 6 exones, donde en función del em-
palme alternativo del 5º o 6º exón se dará
lugar a las formas globulares o asimétricas
respectivamente.
Los mRNAs producidos y las formas a las
que dan lugar se muestran a la derecha.
Formas moléculas de AChE y BuChE
Las formas globulares de AChE proceden del ensamblaje del exón 5 en el mensajero y dan
lugar a proteínas homoméricas cuya naturaleza puede ser hidrofílica o anfifílica en función de
si son unidas a fosfolípidos de la membrana por el producto del gen prima.

233
Existen tres formas hidrofílicas que son el monómero (3-4 S), el dímero (5-6 S) y el tetrámero
(10-11 S) globular, y dos formas anfifílicas, el monómero (3-4 S) y el dímero (5-6 S).
Si el gen prima que codifica la proteína que produce la unión entre las formas globulares y la
membrana no es operativo, por mucho que se exprese el mensajero T para las formas globula-
res anfifílicas todas serán hidrofílicas.
Por otro lado, el mensajero que empalma el exón 6 da lugar a las formas asimétricas de AChe
que forman proteínas heteroméricas en las que varias unidades se van a aunir a través de de
una triple hélice de colágeno a la membrana. Esta hélice está codificada por el gen ColQ.

234

235
Tema 3: Receptores ionotro picos
Los canales ionotrópicos (canales de iones) pueden dividirse en dos clases generales en fun-
ción del estímulo que los abre:
- Los canales iónicos dependientes de voltaje son los canales ya estudiados de sodio,
potasio y calcio que pasan al estado abierto mediante una despolarización de la mem-
brana.
- Los canales iónicos dependientes de transmisores son los clásicos excitatorios de ace-
tilcolina, glutamato y serotonina, o los inhibidores de GABA y glicina que aumentan la
carga negativa en el interior de la célula (hiperpolarización).
Dado que las neuronas presentan toda una población de estos canales, en función de los estí-
mulos que reciba se producirá una hiperpolarización de la membrana o una despolarización,
aumento de calcio…
1 Sistema glutamatérgico El glutamato es un aminoácido que puede actuar como neurotransmisor, pero no puede atra-
vesar la barrera hematoencefálica por lo que debe ser sintetizado en las neuronas a partir de
precursores locales:
- La fuente de glutamato en las neuronas es la glutamina procedente de las células glia-
les, convertida en glutamato por la glutaminasa.
- También puede ser sintetizado por transaminación del 2-oxoglutarato, un intermedia-
rio del ciclo de los ácidos tricarboxílicos que se transforma en α-cetoglutarato. A conti-
nuación el α-cetoglutarato es descarboxilado por la glutamato descarboxilasa dando
lugar al glutamato. En este caso el glutamato se produce a partir de la glucosa metabo-
lizada en las neuronas y representa uno de los motivos del consumo de este glúcido
por el cerebro.
A continuación el glutamato es acumulado en las vesículas sinápticas gracias a la acción del
transportador vesicular de glutamato (VGLUT) que está codificado por 3 genes diferentes.
A continuación el glutamato es vertido al espacio sináptico donde va a activar a tres clases de
receptores y es recuperado del espacio sináptico a través de los transportadores de aminoáci-
dos excitadores (EAATs) de los que hay cinco clases diferentes de alta afinidad que se distribu-
yen en las células de la glía y las terminales presinápticas.
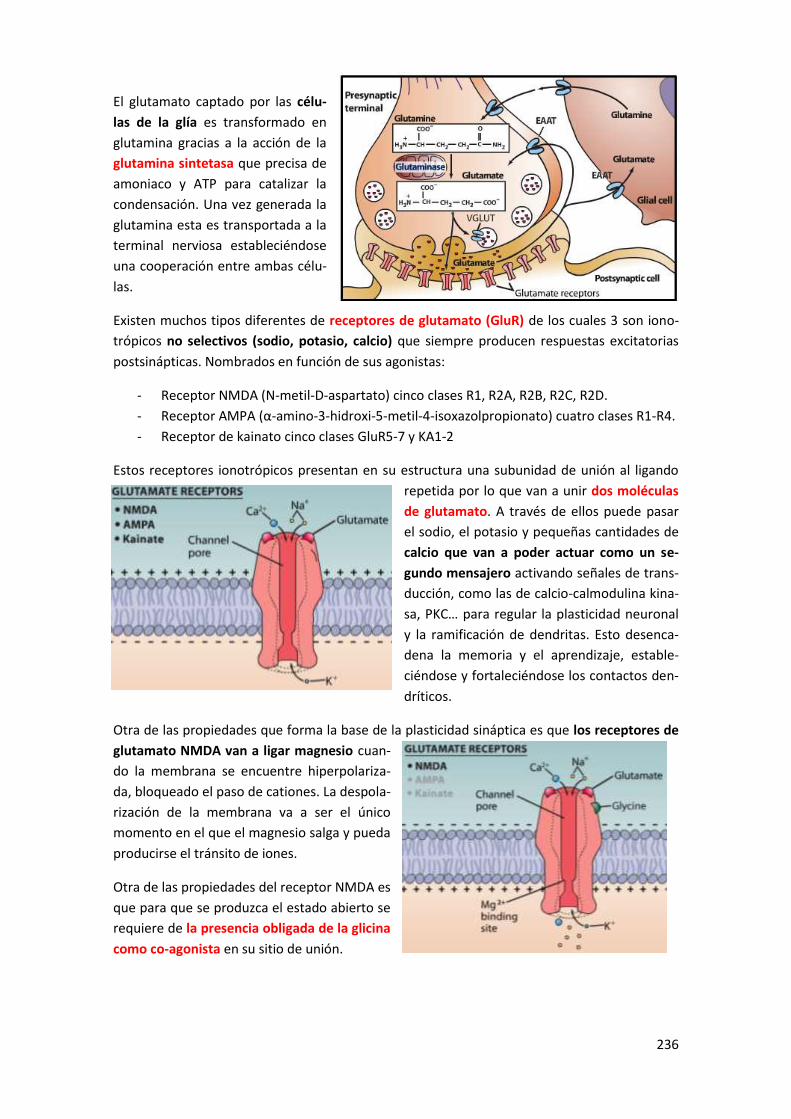
236
El glutamato captado por las célu-
las de la glía es transformado en
glutamina gracias a la acción de la
glutamina sintetasa que precisa de
amoniaco y ATP para catalizar la
condensación. Una vez generada la
glutamina esta es transportada a la
terminal nerviosa estableciéndose
una cooperación entre ambas célu-
las.
Existen muchos tipos diferentes de receptores de glutamato (GluR) de los cuales 3 son iono-
trópicos no selectivos (sodio, potasio, calcio) que siempre producen respuestas excitatorias
postsinápticas. Nombrados en función de sus agonistas:
- Receptor NMDA (N-metil-D-aspartato) cinco clases R1, R2A, R2B, R2C, R2D.
- Receptor AMPA (α-amino-3-hidroxi-5-metil-4-isoxazolpropionato) cuatro clases R1-R4.
- Receptor de kainato cinco clases GluR5-7 y KA1-2
Estos receptores ionotrópicos presentan en su estructura una subunidad de unión al ligando
repetida por lo que van a unir dos moléculas
de glutamato. A través de ellos puede pasar
el sodio, el potasio y pequeñas cantidades de
calcio que van a poder actuar como un se-
gundo mensajero activando señales de trans-
ducción, como las de calcio-calmodulina kina-
sa, PKC… para regular la plasticidad neuronal
y la ramificación de dendritas. Esto desenca-
dena la memoria y el aprendizaje, estable-
ciéndose y fortaleciéndose los contactos den-
dríticos.
Otra de las propiedades que forma la base de la plasticidad sináptica es que los receptores de
glutamato NMDA van a ligar magnesio cuan-
do la membrana se encuentre hiperpolariza-
da, bloqueado el paso de cationes. La despola-
rización de la membrana va a ser el único
momento en el que el magnesio salga y pueda
producirse el tránsito de iones.
Otra de las propiedades del receptor NMDA es
que para que se produzca el estado abierto se
requiere de la presencia obligada de la glicina
como co-agonista en su sitio de unión.

237
Además de estos tres receptores ionotrópicos también
vamos a encontrar receptores metabotrópicos de gluta-
mato (mGluR1-R8) que están asociados a proteínas G que
al disociarse interaccionan directamente con canales ióni-
cos o con proteínas efectoras que producen segundos men-
sajeros que abren o cierran los canales.
Los receptores metabotrópicos producen respuestas más
lentas que los receptores ionotrópicos y sus acciones fisio-
lógicas son muy variadas.
2 Sistema GABAérgico El GABA es un metabolito que se puede obtener a partir de diferentes rutas tal y como se
muestra en la imagen, donde destaca la descarboxilación del glutamato.
Los receptores de GABA y glicina, a diferencia de los de glutamato y AChE, son inhibitodios. El
receptor GABA-A está compuestos por 19 subunidades (α1-6, β1-3, γ1-3, δ, ε, θ, π y ρ1-3)
cuya diversidad sigue aumentando por las variantes de Splicing alternativo que van aparecien-
do y que se localizan a nivel del SNC.
Esto da lugar a una variedad de receptores altísima, al menos 30, cada uno de ellos con pro-
piedades farmacológicas y fisiológicas distintas, además de un modelo característico de expre-
sión. Pero dado que todos favorecen la hiperpolarización de membranas se considera que son
inhibitorios y se han venido empleando agonistas para el tratamiento de crisis convulsivas.
Uno de los agonistas más conocido es
el diazepan, una benzodiacepina. Tam-
bién liga etanol y tiopental, un compo-
nente relacionado con el suero de la
verdad para provocar hipnosis. Dado
que este receptor también puede ligar
hormonas esteroideas, se considera
que está relacionado con los cambios
de humor femeninos.
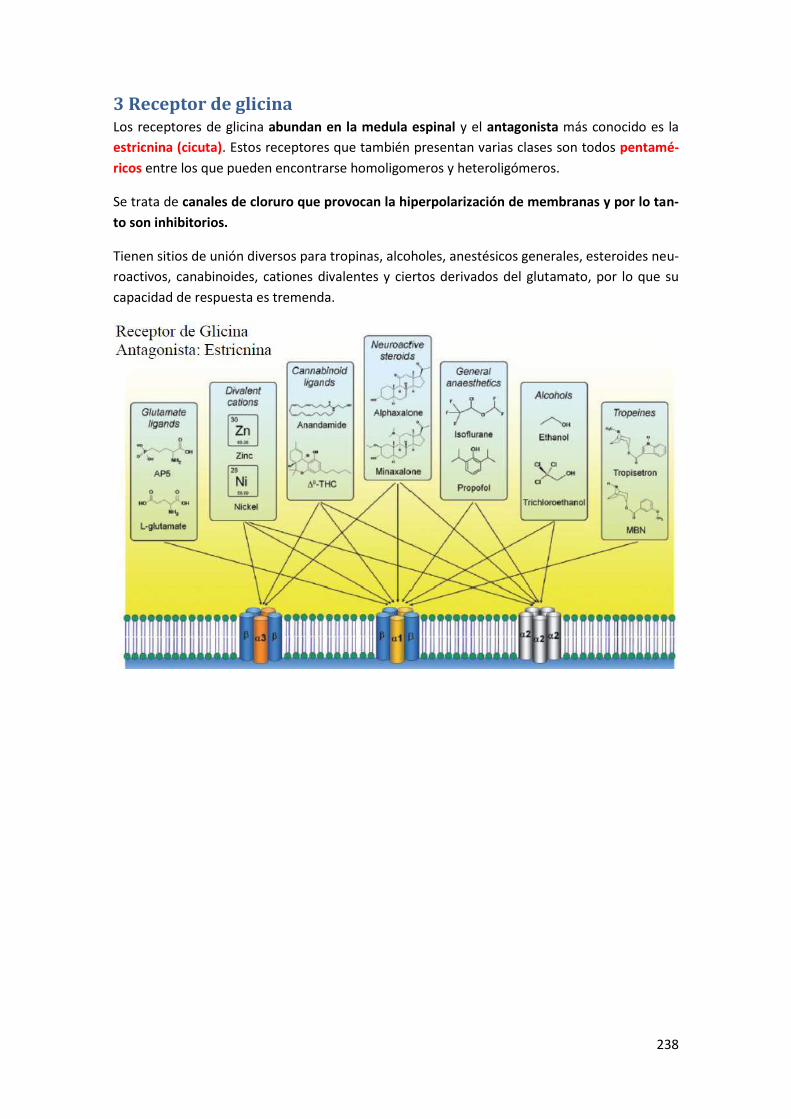
238
3 Receptor de glicina Los receptores de glicina abundan en la medula espinal y el antagonista más conocido es la
estricnina (cicuta). Estos receptores que también presentan varias clases son todos pentamé-
ricos entre los que pueden encontrarse homoligomeros y heteroligómeros.
Se trata de canales de cloruro que provocan la hiperpolarización de membranas y por lo tan-
to son inhibitorios.
Tienen sitios de unión diversos para tropinas, alcoholes, anestésicos generales, esteroides neu-
roactivos, canabinoides, cationes divalentes y ciertos derivados del glutamato, por lo que su
capacidad de respuesta es tremenda.

239
Tema 4: Transmisio n adrene rgica y receptores metabotro picos
1 Ruta de biosíntesis de catecolaminas La ruta de síntesis de catecolaminas comienza con la oxidación de la fenilalanina a tirosina
catalizada por la fenilalanina hidroxilasa
que introduce un átomo de oxígeno (-OH)
en el anillo aromático. La fenilalainina hi-
droxilasa es una metalproteína que re-
quiere hierro para catalizar la reacción y su
deficiencia desencadena la fenilcetonuria.
A continuación se produce la oxidación de la tirosina a DOPA (3,4-dihidroxifenilalanina) catali-
zada por la tirosina hidroxilasa que introduce otro átomo de oxígeno (-OH) en el anillo aromá-
tico. Esta enzima es el punto clave de la regulación de la biosíntesis de catecolaminas porque
la tirosina puede ser necesaria
para muchos otros procesos y
por lo tanto va a estar sometida
a una fuerte regulación. La tiro-
sina hidroxilasa también es una
metalproteína con hierro cuya actividad depende de
- los niveles de tirosina (sustrato).
- oxígeno.
- un cofactor denominado tetrahidrodiocterina (reducida) o dihidrodiocterina (oxida-
da).
- Es fuertemente inhibida por DOPA y altas concentraciones de catecolaminas en el to-
rrente sanguíneo.
- En la región N-terminal presenta un dominio de 150 aminoácidos que activa fuerte-
mente a la proteína por fosforilación en 4 restos de serina (S). Cuando se produce la
despolarización de la membrana y se vierten las catecolaminas, aumentan la cantidad
de calcio-calmoculina y la calcio-calmodulina kinasa que activa la producción de estas
moléculas.
- Se trata de una enzima regulada transinapticamente por lo que cuando una célula re-
cibe inervación colinérgica continuada, aumenta la expresión del gen de la tirosinhi-
droxilasa para compensar la gran estimulación y reponer el contenido vesicular.
A continuación la DOPA es descarboxilada a dopamina mediante la DOPA descarboxilasa o
aminoácido aromático descarboxilasa, una enzima muy poco específica que requiere de fos-
fato de piridoxal (vitamina B6) como cofactor. La deficiencia de esta enzima o cofactor supone
niveles bajos de dopamina asociados con estados depresivos.

240
En muchas neuronas la ruta de síntesis termina aquí, introduciéndose la dopamina en las vesí-
culas sinápticas a través del transportador típico de 12 hélices. Pero en otras neuronas no do-
paminérgicas como las adrenérgicas la dopamina va a continuar la ruta de síntesis hasta dar
lugar a noradrenalina y adrenalina.
A continuación la dopamina es oxidada a noradrenalina (nor = sin grupo metilo) por la do-
pamina-β-hidroxilasa que introduce
oxígeno (-OH) en la posición β del
resto de etanol. La dopamina-β-
hidroxilasa es una cuproproteína
que presenta cobre y precisa de
ácido ascórbico para realizar su
función. La acción de acomplejantes
de cobre como los carbamatos tales
como el disulfiran suprmmimen su actividad aumentando los niveles de dopamina que desen-
cadenan en nauseas, mareos y vómitos por ello el alcoholismo crónico se trata con estos agen-
tes. Esta enzima se utiliza como marcador para el diagnóstico de algunos tipos de cáncer, co-
mo los feocromocitomas, es decir, tumores de la médula adrenal afines a la tinción, y neuro-
blastomas del sistema nervioso.
Al igual que antes, para algunas neuronas la ruta de síntesis finaliza con la formación de nora-
drenalina, pero finalmente otras van a utilizarla para generar adrenalina.
A continuación la noradrenalina es meti-
lada a adrenalina por la feniletanolamina
N-metiltransferasa (FNMT) que introduce
metilo en la posición N. El donador de
grupos metilos es la S-adenosilmetionina
(SAM) y la adrenalina va a ser acumulada
en las vesículas sinápticas.
2 Rutas dopaminérgicas Las rutas dopaminérgicas más importantes
a nivel del sistema nervioso central son la
ruta mesolimbocortical y la mesoestriatal.
Se sabe que esta última ejerce un papel
funcional en la actividad motora y por ello,
los daños de estas neuronas producen tre-
mores y parálisis en la enfermedad de Par-
kinson.
Además de en la coordinación se sabe que
la dopamina está implicada en la motiva-
ción, recompensa y reforzamiento.

241
3 Inactivación de catecolaminas No existen sistemas enzimáticos de degradación de catecolaminas en el espacio sináptico,
como ocurría para ACh, por ello la dopamina, noradrenalina y adrenalina van a ser eliminadas
por difusión y captura desde el espacio sináptico.
Para la degradación de catecolaminas van a intervenir dos sistemas enzimáticos:
- Monoamino oxidasa (MAO) es una flavoproteina con FAD que cataliza una desamina-
ción oxidativa. Se trata de una enzima marcadora de la membrana mitocondrial exter-
na y la reacción que cataliza
consiste en una desamina-
ción (pérdida de N en forma
de amonio) seguida de la in-
troducción de oxígeno (oxi-
dación) dando siempre como
producto final un aldehído.
Puesto que los aldehídos son
muy reactivos porque reac-
cionan con los grupos amino
libres, las oxidasas y reducta-
sas de los tejidos los trans-
forman en ácidos o alcoho-
les como productos finales.
- Catecol-O-metiltransferasa
(COMT) que introduce meti-
lo en los oxígenos de la posi-
ción 3 o 4, es una proteína soluble.
Estas enzimas pueden actuar alternativamente sobre las catecolaminas dando lugar a diferen-
tes productos finales que serán alcoholes o ácidos.
4 Sistema dopaminérgico Agonistas del sistema dopaminérgico como la cocacina y las anfetaminas inhiben a los trans-
portadores de recuperación del neurotransmisor provocando la activación continuada de los
receptores.
Los antagonistas dopaminérgicos como la reserpina inhiben al transportador vesicular de
monoaminas y compuestos como el AMPT (α-metil-ρ-tirosina) inhiben a la tirosin hidroxilasa
impidiendo la síntesis de DOPA.
Los inhibidores de MAO se utilizan para el tra-
tamiento de la depresión, ya que se aumenta el
tiempo de permanencia del neurotransmisor en
el espacio o términal sináptica, ya que el trans-
portador funciona en ambas direcciones en
función de la concentración. Pero MAO presen-

242
ta dos variantes:
- MAO-A tiene como sustrato preferente la noradrenalina y la serotonina. Está distribui-
da a nivel del SNC, el intestino, el hígado y la placenta.
- MAO-B presenta amplio espectro de actuación y se distribuye en la sangre y las pla-
quetas, además del en el SNC.
Por lo tanto compuestos como anfetaminas, cocaína, morfina, nicotina, alcohol, crack y otros
prolongan las acciones de la dopamina aumentando el sentimiento de placer y recompensa,
por lo que se desarrolla adicción.
Recordemos que los transportadores de alta afinidad de las terminales sinápticas son depen-
dientes de sodio y por lo tanto la recuperación de catecolaminas va a estar afectada por oua-
baina y veratridina. Ante una despolarización permanente de la membrana generada por ve-
ratridina, las concentraciones de sodio se equilibran a ambos lados de la membrana y por lo
tanto deja de funcionar el transporte.
El Parkinson supone un déficit de dopamina que se trata con L-DOPA. La dopamina se siblera
en el núcleo accumens, amígdala y corteza anterior, reguladoras de las emociones (circuito de
recompensa y placer).
También abunda en el lóbulo estriado (aprendizaje), hipocampo (memoria) y corteza prefron-
tal (juicio y planificación de actividades).
Todos los receptores de dopamina son metabotrópicos. Existen 5 genes (D1-D5) y parecen
estar acoplados a proteínas G donde unas activan a adenilato ciclasa y otros a PLC.
5 Sistema adrenérgico Los receptores del sistema adrenérgico son sensibles a noradrenalina y adrenalina, y vamos a
encontrar cuatro tipos que se localizan en
posiciones pre- y post-sinápticas. Se nom-
bran como receptores adrenérgicos α1, α2,
β1 y β2 y son todos metabotrópicos.
Muchas de las drogas que afectan a las si-
napsis dopaminérgicas también afectan al
sistema adrenérgico como es el caso de la
reserpina. Además el transportador que
recapta estos neurotransmisores del espacio
sináptico también se fe afectado por cocaí-
na y anfetaminas.
6 Sistema serotoninérgico La indolamina serotonina es sintetizada a partir del aminoácido triptófano mediante dos reac-
ciones sucesivas. En primer lugar la triptófano hidroxilasa cataliza la oxidación del triptófano

243
introduciendo oxígeno (-OH) para formar el 5-hidroxitriptófano. Esta enzima requiere oxígeno
y la tetrahidrobiopterina que se transforma en dihidrobiopterina.
En segundo lugar la descarboxilasa de aminoácidos aromáticos cataliza la descarboxilación de
la molécula generando la 5-hidroxitriptamina, más conocida como serotonina.
La serotonina es sintetizada en muchas regiones cerebrales inervadas por las fibras seroto-
ninérgicas que están implicadas en el control del sueño y la vigilia, el comportamiento y la an-
siedad.
Se ha relacionado con la serotonina el hambre, sueño, actividad sexual, emociones, actividad
autonómica, motora, cognitiva y ritmos circadianos. Las drogas que afectan a la síntesis, libe-
ración o captura de la serotoni-
na se utilizan en el tratamiento
de depresiones, ansiedad y
esquizofrenia.
Hoy en día se utiliza Fenflura-
mina (anorético) para combatir
la obesidad. El sumaptriptan es
muy eficaz contra migrañas y
cefaleas. El prozac o píldora de
la felicidad, es un agonista del
sistema que facilita la liberación
de serotonina y por ello se pro-
voca un estado de bienestar.
La farmacología de los receptores de serotonina es muy amplia y existen 5 clases diferentes,
entre los que encontramos ionotrópicos y metabotrópicos. A diferencia de los estudiados en
este tema que son todos metabotrópicos.

244

245
Tema 5: Transmisio n peptide rgica
Desde los años 50 y 60, los neurofisiólogos se interesaron por responder al motivo de los efec-
tos de la morfina y de los derivados opioides, ya que sus acciones analgésicas tenían que estar
mediadas por algún tipo de receptor.
La utilización de morfina marcada reveló la existencia de un receptor de alta afinidad que de-
bía de responder ante un compuesto interno, que más tarde se clasificarían como encefalinas
y endorfinas.
1 Encefalinas La leucinaencefalina y la metioninencefalina son
dos pentapéptidos de 5 aminoácidos cuyo primer
resto es el de tirosina y su último aminoácido es
leucina y metionina respectivamente. Estos dos
péptidos adoptan la misma conformación espacial
y van a ser reconocidos por el mismo receptor.
2 Síntesis y acciones de los neuropéptidos A raíz del descubrimiento de las encefalinas y endorfinas se vio que había muchos neuropepti-
dos como los del hipotálamo e hipófisis, que además de las acciones hormonales, ejercían su
efecto sobre el sistema nervioso directamente.
Precisamente la inyección de pequeñas cantidades de estos péptidos, como la vasopresina o la
oxitocina, en determinadas regiones del cerebro provocaban cambios que podían involucrar al
comportamiento sexual e incluso a veces inducía polimixia (necesidad de beber agua).
Los neuropéptidos se sintetizan en el soma de las neuronas como proteínas grandes que ma-
duran durante el transporte de las vesículas a la terminal sináptica. Se liberan al espacio sináp-
tico y se unen a los receptores para posteriormente ser degradadas por proteasas. No hay vías
de captura pero en muchas ocasiones los productos de hidrólisis son neuroactivos, llegando a
ser más activos que los precursores. Pero en general no van a presentar actividad.
Entre los neuropeptidos podes encontrar:
- La sustancia P implicada en la percepción del dolor.
- Encefalinas y endorfinas que son analgésicos porque inhiben la liberación de sustancia
P entre otro péptidos.
- Oxitocina
- Vasopresina
- Agiotensina I y II
- Neurotensina
- Gastrina
- NPY, CCK, TRH, LHRH, VIP,…

246
En cuanto a los opiáceos (morfina, heroína y cocaína) y opioides (metilencefalina, leucinence-
falina y endorfina), todos se unen a receptores acoplados a proteínas Gi que reducen la acti-
vidad de adenilato ciclasa.
Un angonista de estos receptores es la metadona (opioide sintético) y una antagonista la nalo-
xona.
Los efectos que provocan son la analgesia, adicción, tolerancia, placer, indiferencia a estímu-
los, inmunosupresión, depresión respiratoria, bradicardia,…
Cuando a una persona se le administra un opiáceo, esta se une al receptor y baja la actividad
de adenilato ciclasa, pero las células reaccionan aumentado la expresión de adenilato ciclasa
debido a la importancia de mantener el sistema de AMPc. Por ello cada vez se desarrolla más
tolerancia a la droga.
Pero si se produce una supresión de la administración, el estímulo que pone en marcha al sis-
tema adenliato ciclasa dispara los niveles de AMPc (al igual que la toxina del cólera) dando
lugar náuseas y diarreas, así como al desarrollo de tremores.
Para saber si una determinada percepción de dolor está relacionada con los receptores de
neuropeptidos se administra el antagonista naloxona. La acupuntura en el tratamiento de do-
lores se basa en la liberación de encefalinas y endorfinas porque las agujas se sitúan en los
puntos de liberación de estos neuropeptidos. Pero si primero administramos naloxona, que
ocupa a los receptores, se impide la atenuación del sistema de adenilato ciclasa que producen
los neuropeptidos y por lo tanto la sensación de dolor continúa.
3 Estructura génica de los neuropeptidos El motivo por el que a los genes de los neuropeptidos se los denomina con el prefijo pre- se
debe a que el producto de los genes se va a obtener a través de ribosomas asociados al retícu-
lo endoplasmático, por lo que portan un péptido líder (pre).
Una vez cortado, el producto de los genes llega a las
vesículas sinápticas donde madura desde proencefa-
lina hasta encefalina.
El gen de la preproencefalina está codifica para cua-
tro moléculas de metioninencefalina, una para leuci-
nencefalina y otros dos péptidos. La proteína sinteti-
zada es cortada por las convertasas o hidrolasas en
puntos flanqueados por arginina (R) o lisina (K).
El gen de la preproopiomelanocortina contiene in-
formación para la hormona adrenocorticorona, pero
se puede cortar para dar lugar a la una gran diversi-
dad de neuropéptidos, en función de las convertasas
presentes en cada tipo neuronal.

247
4 Receptores de neuropeptidos Todos los receptores de neuropepti-
dos son metabotrópicos y están aco-
plados a proteínas G inhibidoras. Su
topolotía es del tipo 7tm con un alto
grado de polimorfismo.
En general se establece que hay tres
clases de receptores de opiáceos
principales denominados μ, δ y κ.
La exposición prolongada de los receptores de opioides a sus agonsitas estimula la vía inhibi-
dora de adenilato ciclasa activando a las proteínas Gi. Precisamente la toxina pertusis se une a
estas subunidades Gα inhibidoras provocando su adenilación e impidiendo que frenen a adeni-
lato ciclasa.
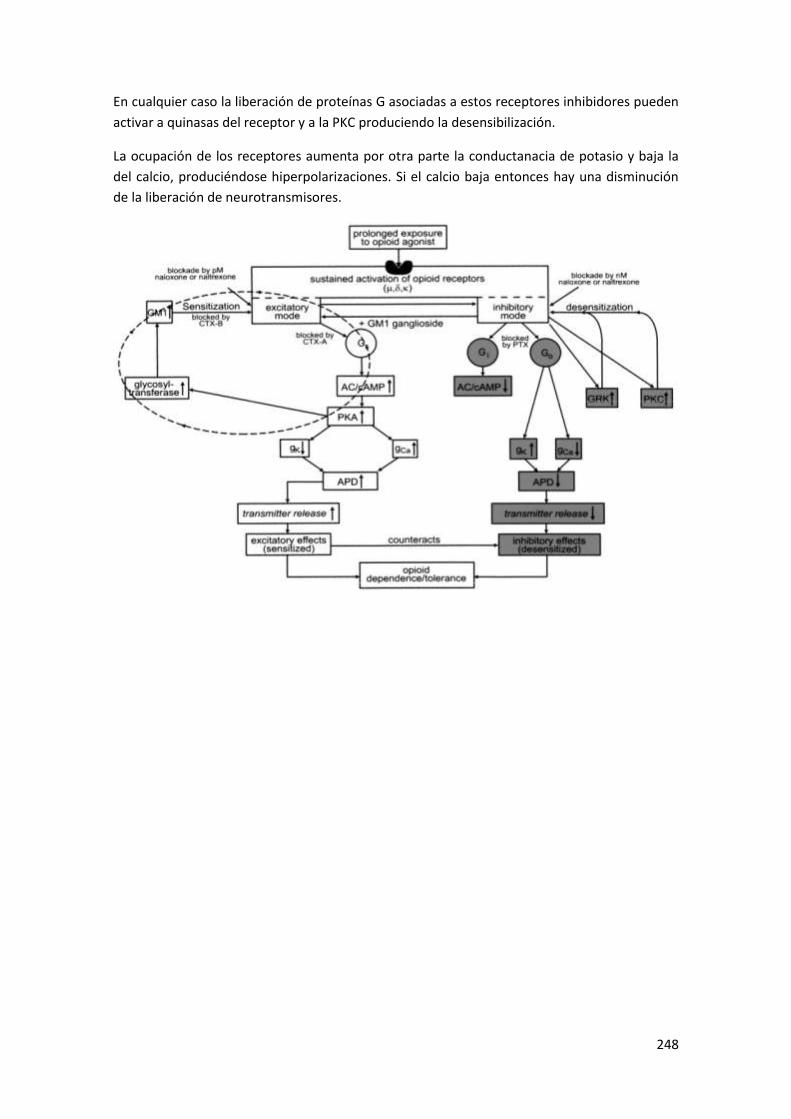
248
En cualquier caso la liberación de proteínas G asociadas a estos receptores inhibidores pueden
activar a quinasas del receptor y a la PKC produciendo la desensibilización.
La ocupación de los receptores aumenta por otra parte la conductanacia de potasio y baja la
del calcio, produciéndose hiperpolarizaciones. Si el calcio baja entonces hay una disminución
de la liberación de neurotransmisores.