theoretical investigation of ti-doped zirconia surfaces
TRANSCRIPT

Electronic Structure and Redox Properties of the Ti-Doped Zirconia (111) Surface
Hasani R. Chauke,† Phathutshedzo Murovhi,† Phuti E. Ngoepe,† Nora H. de Leeuw,‡ andRicardo Grau-Crespo*,‡
Materials Modelling Centre, UniVersity of Limpopo, PriVate Bag X1106, SoVenga, 0727, South Africa, andDepartment of Chemistry, UniVersity College London, 20 Gordon Street, London WC1H 0AJ, U.K.
ReceiVed: April 8, 2010; ReVised Manuscript ReceiVed: July 28, 2010
We have applied density functional theory calculations with Hubbard corrections (DFT+U) to investigatethe structural, electronic, and redox properties of Ti-substituted zirconia (111) surfaces. The calculations showthat titanium dopants are likely to accumulate at the oxide surface, where an isolated dopant is 0.25 eV morestable than in the bulk. We have investigated in detail the relative distribution of dopants and oxygen vacanciesat the surface and report the most stable configurations for each composition. It is found that the formationenergy of oxygen vacancies decreases substantially in titanium-substituted surfaces with respect to undopedsurfaces. The analysis of the electronic structure of the doped and reduced surfaces reveals that, when an Ovacancy is created around an isolated Ti dopant, a Ti4+f Ti2+ reduction takes place, with the reduced cationin a high-spin configuration. However, if the vacancy is created in the vicinity of a pair of dopants, each Tiatom adopts a +3 oxidation state with an additional decrease in the vacancy formation energy.
I. Introduction
Zirconia (ZrO2) is one of the most important ceramic materialsin modern technology. Its versatility arises from a combinationof convenient mechanical properties (high flexural strength andgood fracture toughness), high-temperature stability, and other,more specific functional properties.1 For example, its high ionicconductivity, in particular when doped with lower-valencecations, leads to applications in gas sensors2 and solid oxidefuel cells,3 while its high permittivity of around 20 makes it apromising material for gate dielectrics.4,5 Zirconia is also widelyused as a support material in catalysis6-8 and as an inert cruciblein metal casting,9 and the negligible radioactivity of thecomponent ions means that it can also be employed safely asimplant material in biomedical applications.10
The interface of zirconia with metals plays a significantrole in many of these applications.11 In particular, theinteraction of zirconia ceramics with titanium, which is thefocus of the present work, significantly affects the surfaceproperties of titanium alloys obtained by precision casting.It is well-known that Ti melts can react strongly with zirconiasurfaces to form a chemically differentiated interface.12 Someauthors have pointed out that Ti can react with oxygen fromthe zirconia surface to form titanium oxide,13 and also thatzirconium can enter the titanium lattice at ZrO2/Ti inter-faces.14 On the ceramic side, experimental studies have shownthat contact with titanium melts during the casting processproduces a complex interface including an oxygen-deficientzirconia surface ZrO2-x.9
Zirconia/titanium interfaces are also of interest in somebiomedical applications. For example, both titanium alloys andzirconia ceramics can be used as dental implant materials, andtherefore coupling both materials to give a convenient combina-tion of properties could be a promising approach.15 To this end,Correia et al. have used electron microscopy and different
spectroscopic techniques to investigate the composition ofZrO2/Ti joints formed at high temperatures (1600-1770 K) andhave identified several phases at the interface, including reducedzirconia and Ti-doped reduced zirconia with tetragonal structure,as well as other mixed Ti-Zr oxides.16
Besides the unintentional mixing at the Ti/ZrO2 interfacesdescribed above, there are also a number of applications whereTi is purposely doped into the ceramic material to achievecertain target properties. For example, Ti-doped zirconia, withthe cubic fluorite structure stabilized by Y3+ atoms, has beeninvestigated as an anode material for solid oxide fuel cells.17
The solubility of Ti in stabilized cubic zirconia has been testedby Feighery et al.,18 who found that up to 18 atom % Ti4+ canbe incorporated into ZrO2 at 1773 K. It has been suggested thatTi doping can improve the dielectric properties of zirconiumand hafnium oxides for applications in gate capacitors.19,20
TiO2-ZrO2 mixed oxides also find a number of importantapplications in heterogeneous catalysis.21
It is therefore clear that a fundamental understanding of theeffect of titanium doping on the properties of zirconia surfaceswill be useful for the development of these applications. Wereport here the results of a theoretical investigation into theproperties of a Ti-doped zirconia surface. We focus on theelectronic and redox properties of the surface as a function ofTi content and attempt to correlate our results with experimentalobservations. We will show that the presence of Ti at the surfacedrastically changes the chemical behavior of the surface, whichbecomes easily reducible due to the flexible oxidation states ofthe Ti centers.
II. Methodology
We have used quantum-mechanical calculations based on thedensity functional theory (DFT) with periodic boundary condi-tions to investigate the doped and undoped zirconia surfaces.Our structural model is based on the geometry of cubic zirconia,which is the stable polymorph of pure ZrO2 only at hightemperatures (above ∼2600 K).22 However, the cubic phase canbe stabilized at much lower temperatures by doping with lower-
* To whom correspondence should be addressed. E-mail: [email protected].
† University of Limpopo.‡ University College London.
J. Phys. Chem. C 2010, 114, 15403–15409 15403
10.1021/jp103181q 2010 American Chemical SocietyPublished on Web 08/24/2010

valence cations, e.g., Y3+ or Ca2+, and this is the phase presentin many ceramic applications (e.g., refs 23-26). We note thatdoping only with Ti does not seem to stabilize the cubic phaseof zirconia,27 although for the sake of simplicity in this studywe will ignore the presence of other dopants apart from Ti inthe bulk or the surface of the material. Cubic zirconia adoptsthe fluorite (CaF2) structure, which has a face-centered cubicunit cell with space group Fm3m. In this structure, each cationis coordinated to eight equivalent nearest-neighbor anions atthe corners of a cube, while each anion is tetrahedrallycoordinated to four cations.
The (111) surface has been found to be the most stable surfaceof zirconia in a number of theoretical studies, using bothinteratomic potentials and quantum-mechanical methods,28-30
and has been observed in the experimental characterization ofthin films of zirconia.31 Oxygen termination is required for thesurface to be nonpolar,28 which in a fluorite-structured crystalis also more stable than a cation-terminated reconstructed dipolar(111) surface.32 The cubic zirconia (111) surface is representedin our calculations by oxygen-terminated slabs, which repeatperiodically in the direction perpendicular to the surface,separated by a vacuum gap of ∼15 Å. Based on the results ofprevious work, each slab contains nine atomic layers (threeO-Zr-O trilayers), which was shown to be sufficient to obtainconvergence of the surface properties.33 Parallel to the surface,the supercell consists of a 2 × 2 array of hexagonal surfaceunit cells. Each unit cell contains one ZrO2 unit at the surface,and therefore the three-layer simulation supercell contains 36atoms in total, with four oxygen ions at each surface (Figure1). The same slab model has been used previously in theinvestigation of the c-ZrO2(111) and the isostructural CeO2(111)surfaces at the DFT level.33-36 Based on the results ofconvergence tests in previous work,33 the positions of the fouratomic layers at the bottom of the slab were kept fixed at thebulk positions, while the first five layers of the top face, wherewe dope the metallic atoms, were fully relaxed.
All calculations were carried out using the VASP code,37-40
which uses a basis set of plane waves to solve the Kohn-Sham equations with periodic boundary conditions. We haveused the generalized gradient approximation (GGA), with adensity functional built from the Perdew and Zunger41 localfunctional, and the gradient corrections by Perdew et al.42
In order to improve the description of the Ti d electrons, weuse a Hubbard-type correction for these orbitals followingthe so-called DFT+U (or GGA+U) approach, which acts byaltering the one-electron potential locally for the specifiedorbitals of the metal atoms (e.g., Ti d orbitals), reducing thehybridization with the ligands (e.g., O atoms).43-45 Previous
work has shown that the introduction of this correction (oralternatively, a fraction of exact exchange as calculated fromHartree-Fock theory) is essential for the description of theelectronic, magnetic, and surface properties of many transitionmetal and rare earth compounds.46-51 We have employed aneffective Hubbard parameter Ueff ) 3 eV for the Ti d orbitals,which has been found by Nolan et al. to be the optimal valueto reproduce the experimental electronic structure of Ti-dopedTiO2 surfaces, using the same functional and general settingsas in this work.52 Moreover, we will show below that thechoice of Ueff values within certain limits does not changesignificantly the conclusions.
The interaction between the valence electrons and the corewas described with the projected augmented wave (PAW)method53 in the implementation of Kresse and Joubert.54 Thecore electrons, up to 3s in Ti, 4p in Zr, and 1s in O, were keptfrozen in their atomic reference states. The number of planewaves in VASP is controlled by a cutoff energy, which in oursurface calculations was set to Ecut ) 415 eV. The cell parameterwas obtained for the cubic zirconia bulk crystal using anincreased cutoff (500 eV) to minimize Pulay errors, and a 4 ×4 × 4 k-point mesh to sample the reciprocal space. The resultingcell parameter was a ) 5.130 Å, which is less than 1% abovethe experimental value of 5.090 Å (this value is an extrapolationto 0 K using the thermal expansion data in ref 55 as given inref 56). Structural optimizations of the surface model were thenperformed with fixed cell parameters using a conjugate gradientstechnique with an iterative relaxation of the atomic positionsuntil the forces on the atoms were all less than 0.01 eV/Å. Forall geometry optimizations at the surface, a 3 × 3 × 1 k-pointmesh was used, while at the final single-point runs for thecalculation of the electronic density of states, a denser mesh of5 × 5 × 1 k-points was employed.
In order to calculate reaction energies, the energies of the O2
molecule, Ti metal, and bulk ZrO2 were obtained with VASP,using the same oxygen PAW potential, cutoff energy, and otherprecision parameters as in the surface calculations. The O2
molecule was calculated as a spin triplet, and we obtained anequilibrium bond distance d[O-O] ) 1.235 Å, similar toprevious reports.57,58 It should be noted that GGA calculationstend to overbind the O2 molecule and therefore vacancyformation energies predicted within this approximation aretypically too low.59 The cell parameter of bulk cubic zirconiawas optimized to a ) 5.130 Å, which is less than 1% abovethe experimental value of 5.090 Å (this value is an extrapolationto 0 K using the thermal expansion data in ref 55 as given inref 56).
The charge states of the ions at the surface were discussedon the basis of a Bader analysis, which consists of integratingthe electron density in a region defined for each atom in sucha way that the density gradient flux through the dividing surfacesis zero.60 An algorithm and a program developed for this purposeby Henkelman et al. have been employed.61,62 Differences inBader charges and the analysis of the electronic density of states(DOS) allow us to assign approximate oxidation states to thedopant ions, so we can identify them as Ti4+, Ti3+, or Ti2+ evenif their Bader charges are different from the formal values. Wehave also used an electron localization function (ELF), asdefined by Becke and Edgecombe63 and Silvi and Savin,64 toillustrate the localization of excess electrons upon the creationof oxygen vacancies at the surface. The ELF measures electronlocalization within a scale from 0 (low) to 1 (high), and it is aconvenient way to examine the electron redistribution uponanion vacancy formation in solids.35,65
Figure 1. Slab model used to represent the O-terminated ZrO2(111)surface. Red (dark) and blue (light) spheres correspond to oxygen andzirconium atoms, respectively.
15404 J. Phys. Chem. C, Vol. 114, No. 36, 2010 Chauke et al.

III. Results and Discussion
Ti Doping of Fully Oxidized Zirconia and Dopant Seg-regation at the (111) Surface. First, we consider the Zrsubstitution by Ti in the fully oxidized bulk and (111) surfaceof zirconia. In this case, Ti enters the lattice with the sameformal oxidation state as the original Zr ions, and therefore nocharge compensation is required upon substitution. For both thebulk and the surface model the overall composition is thenZr11TiO24, which corresponds to 8.33% Ti doping. In order tomake a precise comparison, the bulk simulation cell has thesame orientation of its axes as the surface model, except thatthe vacuum gap was left at zero.
Our calculations show that in a direct comparison Ti dopingof the (111) surface is 0.25 eV more stable than in the bulk.This energy corresponds to the absolute value of the segregationenthalpy of isolated Ti dopants to that surface, if we neglectany effects from the interaction of the dopant with its imagesin neighboring cells. These effects should be small consideringthat (i) the same lateral interaction is present in both the bulkand the surface model and therefore cancels out when obtainingthe energy difference, (ii) the images are separated by morethan 7 Å, and (iii) the dopant has the same charge state as theoriginal cation and therefore no long-range effects should beexpected.
The value of the segregation enthalpy ∆Hseg ) -0.25 eV(negative sign indicating higher stability of substitution at thesurface) allows us to estimate the relative Ti occupancy ofsurface and bulk sites at thermodynamic equilibrium, assuminga low dopant concentration, using the expression:
where kB ) 8.6173 × 10-5 eV/K is Boltzmann’s constant. Forexample, at T ) 1000 K the equilibrium Ti occupancy of the(111) surface cation sites of zirconia is expected to be ∼18 timeshigher than the Ti occupancy of bulk cation sites. Therefore,we can expect that whenever a Ti-doped zirconia ceramic issubjected to temperatures high enough to allow dopant diffusion,the Ti dopants will accumulate at the surface.
The oxidation state of Ti4+ at the surface is confirmed by theanalysis of the projection of the electronic density of states(DOS) on the Ti 3d orbitals (Figure 2a), where it is clear thatthe d levels of the dopant lie above the Fermi energy. Theprojected DOS for the doped bulk is not shown in the figure,but it looks very similar.
In order to gain insight into the energetics of the dopingprocess of the zirconia surface with Ti (which we will considercomes from titanium metal, because of the practical importanceof Ti/ZrO2 interfaces), we have calculated the energy of theformal reaction:
where the substituted Zr cation is displaced to a bulk positionin the zirconia phase. This reaction is very exothermic (-8.59eV) even before applying a correction for the overbinding ofthe O2 molecule in the GGA approximation, which would makethe reaction energy more negative. If we wanted to estimatethe reaction free energy, the most important correction wouldcome from substituting the energy of the oxygen molecule for
its chemical potential at the pressure and temperature of interest.This would make the reaction free energy less negative but notby much; for example, at 900 K and 1 bar of oxygen partialpressure the correction is of around 2 eV, according to valuestaken from thermodynamic tables.66 This simple analysissuggests that the incorporation of Ti from the metallic phaseinto the zirconia (111) surface is thermodynamically a highlyfavorable process.
Effect of Ti Doping on the Formation of Oxygen Vacanciesat the Surface. We now discuss the effect of Ti dopants on theredox properties of the surface. First, we review the formationof neutral oxygen vacancies at the perfect, undoped zirconiasurface, as discussed previously in ref 35. Two positions of theoxygen vacancies were investigated, one in the outmost oxygenlayer Ou (up) and the other in the subsurface oxygen layer Od
(down) in the top ZrO2 trilayer (Figure 1). The vacancyformation energies (VFE), corresponding to the energy changein the reaction
where 0 denotes an oxygen vacancy, were calculated to be 5.85eV for the Ou vacancy and 5.38 eV for the Od vacancy, inagreement with the values obtained in ref 35 (small differencesof less than 0.7% are due to slightly different calculationsettings). The high values of the VFEs are expected from thewell-known low reducibility of zirconia. Zr3+ cations have beendetected by some authors in zirconia-related materials (e.g., ref67), but this is not a very stable species. Upon creation of thevacancy the electrons prefer to remain localized at the vacancysite, forming an F center, and the vacancy defect is said to be
[ Tisurf
Tibulk] ) e-∆Hseg/kBT (1)
Zr12O24(slab) + Ti(bulk) + O2(gas) f Zr11TiO24(slab) +ZrO2(bulk) (2)
Figure 2. Projection of the electronic partial density of states (PDOS)on the Ti 3d orbital (a) for the fully oxidized surface with one Ti dopantper cell (Zr11TiO24), (b) for the surface with one Ti dopant and oneoxygen vacancy per cell (Zr11TiO230), and (c) for the surface with twoTi dopants and one oxygen vacancy per cell (Zr10Ti2O230).
Zr12O24(slab) f Zr12O230(slab) +1/2O2(gas) (3)
Ti-Doped Zirconia (111) Surface J. Phys. Chem. C, Vol. 114, No. 36, 2010 15405

neutral as it has the same charge state as the original O2- ion atthat site. An illustration of this electron distribution in a vacancyon an undoped zirconia surface is given in Figure 3a, wherethe electron localization function (ELF) is plotted in a planethat contains some oxygen anions and the vacancy site. TheELF is high in the region of the oxygen vacancy, showing thatthe electrons remain trapped in the defect site, and the Zr atomsare thus not changing their oxidation states.
The presence of a Ti atom at the surface drastically changesthe picture given above. We have investigated the formation ofoxygen vacancies at the Ti-doped surface following the reaction
and considering four different relative positions of the vacancywith respect to the dopant: (i) Ou vacancy nearest-neighbor (NN)to Ti (there are three symmetrically equivalent configurationsof this type), (ii) Od vacancy nearest-neighbor (NN) to Ti (thereare also three symmetrically equivalent configurations of thistype), (iii) Ou vacancy next nearest-neighbor (NNN) to Ti, and(iv) Od vacancy next nearest-neighbor (NNN) to Ti.
Our results indicate that, for all configurations, a reductionof the Ti species takes place, which is illustrated for case (iv)in the projected DOS in Figure 2b, where a clear peak of Ti dcharacter appears below the Fermi level. The ELF plot of Figure3b shows the localization of the excess electrons at the dopantsite. In contrast with the undoped surface, no electrons are leftat the vacancy position, making the oxygen vacancy defectpositively charged with respect to the original anion site. Sinceno modification of the electronic states of the Zr ions appearsto occur either, it is safe to conclude that the two excess electronsare located at the dopant, which adopts a formal Ti2+ chargestate. The reduction of the Ti center is confirmed by the Baderanalysis, showing Ti charges between 1.6 and 1.8 au, dependingon the position of the vacancy, well below the values in thefully oxidized surface (2.33 au) and bulk (2.40 au). The DOSand ELF plots of Figures 2b and 3b correspond to the moststable configuration, which is the one with a subsurface (Od)vacancy in NNN position with respect to the dopant. In thisconfiguration, the Ti dopant is connected to 3 Od ions at 2.27Å, and to one other O atom in the layer below, but theneighboring Ou atoms shift away from the dopant to distancesof 2.57 Å (Figure 4). The local geometry around the dopantand the vacancy is different for other configurations, but in allcases the electronic picture is similar.
For all geometric configurations, we identified two types ofspin solutions, one with two unpaired electrons per cell and
one without unpaired electrons, which correspond to the high-spin (HS) and low-spin (LS) states, respectively, of the Ti2+
ion with 3d2 electronic configuration. All the vacancy formationenergies are listed in Table 1. Regardless of the vacancyposition, we find that the magnetic (HS) state of the dopant isalways more stable, by up to ∼0.5 eV, than the nonmagneticLS configuration. The magnetic ground state of Ti2+ is consistentwith the state of this dopant cation in other fluorite-typecrystals.68,69
An interesting aspect of the reduction of the surface in thepresence of Ti dopants is the decrease in the magnitude of theoxygen vacancy formation energy. For the most stable positionof the vacancy with respect to the dopant, this energy is 3.66eV, down from 5.38 eV at the undoped surface. Clearly, theability of Ti centers to accommodate the excess electrons makesit less energetically expensive to remove a neutral oxygen,similarly to what has been observed for gold-doped zirconiasurfaces, although the reducibility of the surface does notincrease as drastically upon Ti doping as in the case of Audoping.35 Other recent computational studies have described theincrease in reducibility of oxide surfaces when doped with ionsthat have a flexible oxidation state.70-72 It is therefore notsurprising that a Ti-doped reduced zirconia region has beenobserved experimentally at the interface between ceramiczirconia and metallic titanium.
It is also interesting that for the most stable configurationthe vacancy is not in an NN position with respect to the dopant,where the dopant-defect electrostatic interaction would be mostfavorable. However, the positioning of the vacancy in an NNNsite is not that surprising considering previous results in zirconiaand similar materials. For example, it has been calculated thatin Ca-stabilized zirconia oxygen vacancies prefer NNN insteadof NN sites.73 It has also been reported recently that at the ceria(111) surface, which is isostructural with the surface investigatedin this work, the most favorable relative position of an oxygenvacancy is in a NNN position with respect to the reduced Cecation.74 In all these cases, lattice relaxation effects dominateover electrostatics to determine the position of the anionvacancy.
Redox Properties of the Surface at Higher Ti Concentra-tions. Considering that high levels of Ti doping of ZrO2 havebeen reported in some experimental studies (e.g., 27 mol % inref 27), and that, according to our results, strong Ti segregationto the surface is expected, we investigate now the incorporationof a second Ti ion and its effect on the redox behavior of the
Figure 3. Electron localization function (ELF) plot for the (111) surfaceof zirconia with one oxygen (Od type) vacancy (a) in the absence ofdopants and (b) when a Ti dopant is present at the surface.
Zr11TiO24(slab) f Zr11TiO230(slab) +1/2O2(gas) (4)
Figure 4. The most stable configuration of the zirconia (111) surfacewith one Ti dopant and one oxygen vacancy (Zr11TiO230). For clarity,only the atoms in the top trilayer are represented. Ti-O distances aregiven in angstroms.
15406 J. Phys. Chem. C, Vol. 114, No. 36, 2010 Chauke et al.

(111) surface. Analogously to eq 2, we can calculate the energyrequired for incorporating this second dopant as
Because of the 2 × 2 periodicity of our slab, in the absence ofother defects there is only one possible configuration for thedouble Ti/Zr substitution at the surface layer, which results inalternating chains of only Ti and only Zr atoms across thesurface. This second substitution is slightly more exothermicthan the first one (-8.77 eV for eq 5 compared to -8.59 for eq2). We have also tried the incorporation of the second Ti dopantin the subsurface cation layer, but this is less stable than thesurface substitution by 0.12 eV. Before creating any oxygenvacancy, the two Ti atoms in the surface are in a formal +4oxidation state, as was the case for one dopant per cell, andwith a very similar Bader charge of ∼2.3 au.
The creation of a vacancy at this heavily doped surface isrepresented by the equation
(if we include all the atoms in the slab model). Again, we needto consider four different positions of the vacancy with respectto the Ti atoms: (i) Ou vacancy NN to both Ti atoms, (ii) Od
vacancy NN to both Ti atoms, (iii) Ou vacancy NN to one Tiatom (which we call TiA) and NNN to the other (TiB), and (iv)Od vacancy NN to TiA and NNN to TiB. The vacancy formationenergies depend significantly on the position of the vacancy,as can be seen in Table 1. The lowest energy configuration isagain the one with a subsurface (Od) vacancy NN to one Ti(case iv, see Figure 5). In this case, the vacancy formationenergy is 3.13 eV, which is more than half an electronvolt lowerthan for an isolated dopant (3.66 eV).
In order to understand this effect from dopant accumulation,we consider the electronic structure of the atoms at the surface.Despite the asymmetry of the vacancy position with respect tothe dopants, both Ti ions seem to adopt the same formaloxidation state Ti3+, instead of forming a (Ti4+, Ti2+) pair, asconfirmed by the projected DOS in Figure 2c. Each Ti atomcontributes a 3d peak below the Fermi level, showing that bothTi atoms are reduced. These peaks are smaller than the oneobserved for the Ti2+ ion in the surface with one isolated dopant(Figure 2b), as expected from a change from 3d2 to 3d1
occupancies. This interpretation is also supported by the Baderanalysis, which yields effective charges of 2.06 and 2.09, whichare intermediate between those calculated for Ti4+ (2.3-2.4)
and for Ti2+ (1.6-1.7). In order to check whether the electronicpicture described here is affected by the size of the simulationcell, we performed a test calculation using a 3 × 3 extensionof the surface (slab composition Zr25Ti2O530), using the moststable relative configuration of the vacancy with respect to thedopants as obtained from the 2 × 2 calculations, but we obtainedthe same electronic redistribution, with very similar Badercharges on the Ti centers. Our results therefore strongly indicatethat when more than one Ti dopant is available near the oxygenvacancy site, the excess electrons distribute themselves overseparate Ti centers, forming Ti3+ species, instead of moving tothe same Ti center to form a Ti2+ cation.
We also found that all the other configurations, with differentrelative positions of the vacancy with respect to the dopants,led to the same qualitative picture (Ti4+ reduced to Ti3+) as thelowest-energy solution. Since Ti3+ has a magnetic momentassociated with its 3d1 electron, we have attempted to obtainthe magnetic coupling between the two dopants by calculatingthe energies associated with both ferromagnetic (FM) andantiferromagnetic (AFM) orientation of the spin moments. Forthe configurations with vacancies in Ou positions, we managedto obtain both FM and AFM solutions, but the differences inenergies between them were too small (1-2 meV), comparedwith the general precision of the DFT calculations, to allowany quantitative description of the coupling (other than themagnetic coupling being very weak). In the case of vacanciesin Od position, we did not manage to obtain AFM solutions atall. This is not indicative of any strong magnetic coupling butresults from the coexistence of other electronic solutions withthe same equipartition of up and down electrons. For example,for the configuration with an Od vacancy NN to one Ti atom,the lowest-energy solution described above was obtained with
TABLE 1: Vacancy Formation Energies (VFE) at the Pure and Ti-Substituted (111) Surface of Cubic Zirconiaa
reduction process description of resulting surface VFE (eV)
Zr12O24(slab) f Zr12O230(slab) +1/2O2(gas) Ou vacancy 5.85
Od vacancy 5.38Zr11TiO24(slab) f Zr11TiO230(slab) +
1/2O2(gas) Ou vacancy NN to HS (LS) Ti 4.41 (4.93)Od vacancy NN to HS (LS) Ti 4.40 (4.31)Ou vacancy NNN to HS (LS) Ti 4.60 (5.05)Od vacancy NNN to HS (LS) Ti 3.66 (4.11)
Zr10Ti2O24(slab) f Zr10Ti2O230(slab) +1/2O2(gas) Ou vacancy NN to both Ti 3.28
Od vacancy NN to both Ti 3.48Ou vacancy NN to TiA and NNN to TiB 3.68Od vacancy NN to TiA and NNN to TiB 3.13
a The most stable configuration for each composition is marked in bold.
Zr11TiO24(slab) + Ti(bulk) + O2(gas) f Zr10Ti2O24(slab) +ZrO2(bulk) (5)
Zr10Ti2O24(slab) f Zr10Ti2O230(slab) +1/2O2(gas) (6)
Figure 5. The most stable configuration of the zirconia (111) surfacewith 2 Ti/Zr substitutions and one oxygen vacancy (Zr10Ti2O230). Forclarity, only the atoms in the top trilayer are represented. Ti-O distancesare given in angstroms.
Ti-Doped Zirconia (111) Surface J. Phys. Chem. C, Vol. 114, No. 36, 2010 15407
FM alignment between the Ti spins (as reflected in the absenceof spin-down d contributions in Figure 2c). Trying to force anAFM by fixing to zero the difference between up and downelectrons instead led to a nonmagnetic solution with one Ti4+
ion and one low-spin Ti2+ ion, which was ∼0.7 eV higher inenergy than the ground state. The possibility of such an excitedstate is clear from the projected DOS diagram of Figure 2c: theoccupied d orbital closer to the Fermi level corresponds to a Tiatom (B) which is different from the Ti atom (A) that contributesto the bottom of the conduction band, and therefore the lowestenergy d-d excitation corresponds to the TiA
3+ + TiB3+f TiA
2+
+ TiB4+ electron transfer.
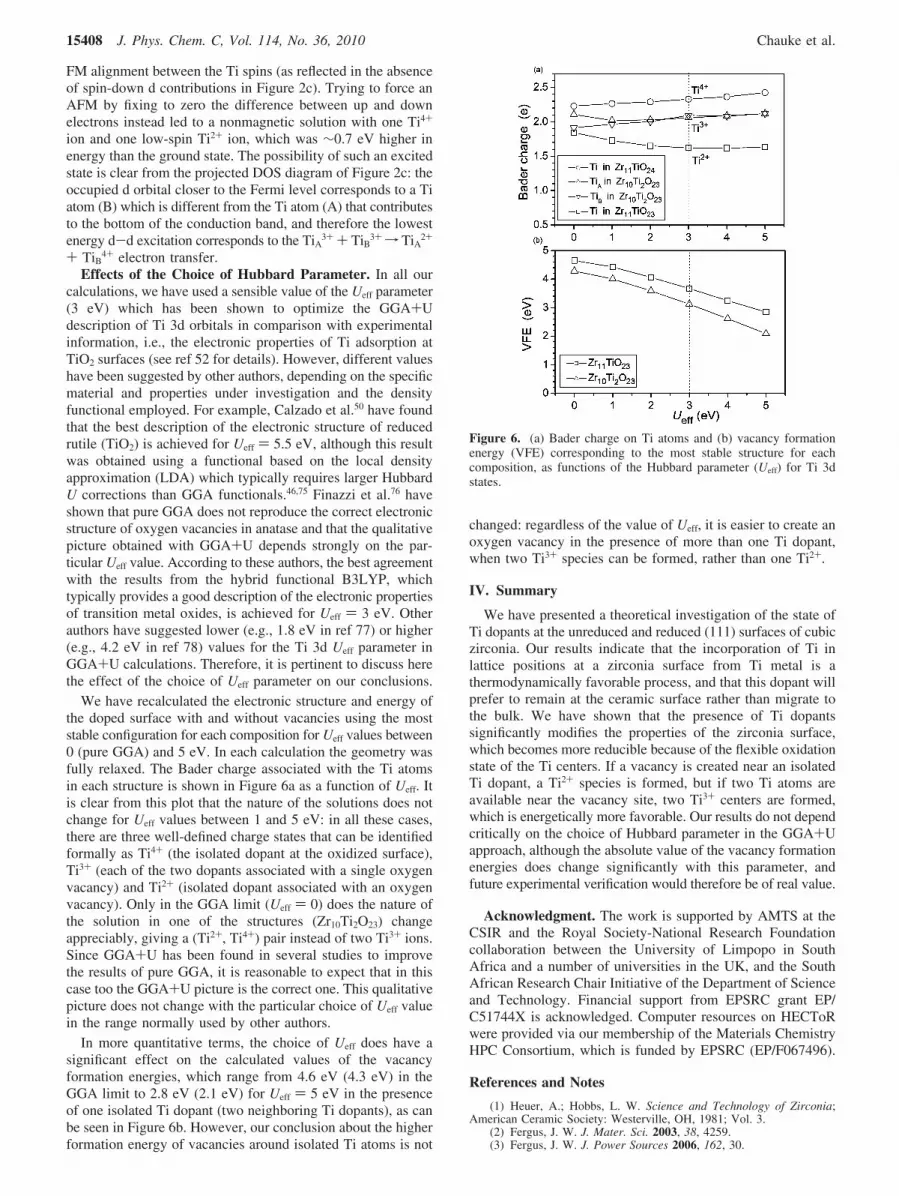
Effects of the Choice of Hubbard Parameter. In all ourcalculations, we have used a sensible value of the Ueff parameter(3 eV) which has been shown to optimize the GGA+Udescription of Ti 3d orbitals in comparison with experimentalinformation, i.e., the electronic properties of Ti adsorption atTiO2 surfaces (see ref 52 for details). However, different valueshave been suggested by other authors, depending on the specificmaterial and properties under investigation and the densityfunctional employed. For example, Calzado et al.50 have foundthat the best description of the electronic structure of reducedrutile (TiO2) is achieved for Ueff ) 5.5 eV, although this resultwas obtained using a functional based on the local densityapproximation (LDA) which typically requires larger HubbardU corrections than GGA functionals.46,75 Finazzi et al.76 haveshown that pure GGA does not reproduce the correct electronicstructure of oxygen vacancies in anatase and that the qualitativepicture obtained with GGA+U depends strongly on the par-ticular Ueff value. According to these authors, the best agreementwith the results from the hybrid functional B3LYP, whichtypically provides a good description of the electronic propertiesof transition metal oxides, is achieved for Ueff ) 3 eV. Otherauthors have suggested lower (e.g., 1.8 eV in ref 77) or higher(e.g., 4.2 eV in ref 78) values for the Ti 3d Ueff parameter inGGA+U calculations. Therefore, it is pertinent to discuss herethe effect of the choice of Ueff parameter on our conclusions.
We have recalculated the electronic structure and energy ofthe doped surface with and without vacancies using the moststable configuration for each composition for Ueff values between0 (pure GGA) and 5 eV. In each calculation the geometry wasfully relaxed. The Bader charge associated with the Ti atomsin each structure is shown in Figure 6a as a function of Ueff. Itis clear from this plot that the nature of the solutions does notchange for Ueff values between 1 and 5 eV: in all these cases,there are three well-defined charge states that can be identifiedformally as Ti4+ (the isolated dopant at the oxidized surface),Ti3+ (each of the two dopants associated with a single oxygenvacancy) and Ti2+ (isolated dopant associated with an oxygenvacancy). Only in the GGA limit (Ueff ) 0) does the nature ofthe solution in one of the structures (Zr10Ti2O23) changeappreciably, giving a (Ti2+, Ti4+) pair instead of two Ti3+ ions.Since GGA+U has been found in several studies to improvethe results of pure GGA, it is reasonable to expect that in thiscase too the GGA+U picture is the correct one. This qualitativepicture does not change with the particular choice of Ueff valuein the range normally used by other authors.
In more quantitative terms, the choice of Ueff does have asignificant effect on the calculated values of the vacancyformation energies, which range from 4.6 eV (4.3 eV) in theGGA limit to 2.8 eV (2.1 eV) for Ueff ) 5 eV in the presenceof one isolated Ti dopant (two neighboring Ti dopants), as canbe seen in Figure 6b. However, our conclusion about the higherformation energy of vacancies around isolated Ti atoms is not
changed: regardless of the value of Ueff, it is easier to create anoxygen vacancy in the presence of more than one Ti dopant,when two Ti3+ species can be formed, rather than one Ti2+.
IV. Summary
We have presented a theoretical investigation of the state ofTi dopants at the unreduced and reduced (111) surfaces of cubiczirconia. Our results indicate that the incorporation of Ti inlattice positions at a zirconia surface from Ti metal is athermodynamically favorable process, and that this dopant willprefer to remain at the ceramic surface rather than migrate tothe bulk. We have shown that the presence of Ti dopantssignificantly modifies the properties of the zirconia surface,which becomes more reducible because of the flexible oxidationstate of the Ti centers. If a vacancy is created near an isolatedTi dopant, a Ti2+ species is formed, but if two Ti atoms areavailable near the vacancy site, two Ti3+ centers are formed,which is energetically more favorable. Our results do not dependcritically on the choice of Hubbard parameter in the GGA+Uapproach, although the absolute value of the vacancy formationenergies does change significantly with this parameter, andfuture experimental verification would therefore be of real value.
Acknowledgment. The work is supported by AMTS at theCSIR and the Royal Society-National Research Foundationcollaboration between the University of Limpopo in SouthAfrica and a number of universities in the UK, and the SouthAfrican Research Chair Initiative of the Department of Scienceand Technology. Financial support from EPSRC grant EP/C51744X is acknowledged. Computer resources on HECToRwere provided via our membership of the Materials ChemistryHPC Consortium, which is funded by EPSRC (EP/F067496).
References and Notes
(1) Heuer, A.; Hobbs, L. W. Science and Technology of Zirconia;American Ceramic Society: Westerville, OH, 1981; Vol. 3.
(2) Fergus, J. W. J. Mater. Sci. 2003, 38, 4259.(3) Fergus, J. W. J. Power Sources 2006, 162, 30.
Figure 6. (a) Bader charge on Ti atoms and (b) vacancy formationenergy (VFE) corresponding to the most stable structure for eachcomposition, as functions of the Hubbard parameter (Ueff) for Ti 3dstates.
15408 J. Phys. Chem. C, Vol. 114, No. 36, 2010 Chauke et al.

(4) Thompson, D. P.; Dickins, A. M.; Thorp, J. S. J. Mater. Sci. 1992,27, 2267.
(5) Zhao, X. Y.; Vanderbilt, D. Phys. ReV. B 2002, 65, 064512.(6) Antolini, E.; Gonzalez, E. R. Solid State Ionics 2009, 180, 746.(7) Reddy, B. M.; Patil, M. K. Curr. Org. Chem. 2008, 12, 118.(8) Gelin, P.; Primet, M. Appl. Catal., B 2002, 39, 1.(9) Lin, K. F.; Lin, C. C. J. Mater. Sci. 1999, 34, 5899.
(10) Manicone, P. F.; Iommetti, P. R.; Raffaelli, L. J. Dent. 2007, 35,819.
(11) Munoz, M. C.; Gallego, S.; Beltran, J. I.; Cerda, J. Surf. Sci. Rep.2006, 61, 303.
(12) Economos, G.; Kingery, W. D. J. Am. Ceram. Soc. 1953, 36, 403.(13) Wang, L. D.; Oki, T. J. Ceram. Soc. Jpn. 1995, 103, 680.(14) Ruh, R. J. Ceram. Soc. Jpn. 1963, 46, 301.(15) Gahlert, M.; Rohling, S.; Wieland, M.; Sprecher, C. M.; Kniha,
H.; Milz, S. Clin. Oral Implant. Res. 2009, 20, 1247.(16) Correia, R. N.; Emiliano, J. V.; Moretto, P. J. Mater. Sci. 1998,
33, 215.(17) Fergus, J. W. Solid State Ionics 2006, 177, 1529.(18) Feighery, A. J.; Irvine, J. T. S.; Fagg, D. P.; Kaiser, A. J. Solid
State Chem. 1999, 143, 273.(19) Dutta, G.; Hembrarn, K.; Rao, G. M.; Waghmare, U. V. J. Appl.
Phys. 2008, 103, 016102.(20) Triyoso, D. H.; Hegde, R. I.; Zollner, S.; Ramon, M. E.; Kalpat,
S.; Gregory, R.; Wang, X. D.; Jiang, J.; Raymond, M.; Rai, R.; Werho, D.;Roan, D.; White, B. E.; Tobin, P. J. J. Appl. Phys. 2005, 98, 054104.
(21) Reddy, B. M.; Khan, A. Catal. ReV.sSci. Eng. 2005, 47, 257.(22) Masahiro, Y. Am. Ceram. Soc. Bull. 1988, 67, 1950.(23) Lemonnier, S.; Grandjean, S.; Robisson, A. C.; Jolivet, J. P. Dalton
Trans. 2010, 39, 2254.(24) Darby, R. J.; Farnan, I.; Kumar, R. V. AdV. Appl. Ceram. 2009,
108, 506.(25) Kilo, M. Cation transport in stabilised zirconias. In Defects and
Diffusion in Ceramics: An Annual RetrospectiVe VII; Trans Tech Publica-tions Inc.: Zurich, Switzerland, 2005; Vol. 242-244; p 185.
(26) Suarez, G.; Garrido, L. B.; Aglietti, E. F. Mater. Chem. Phys. 2008,110, 370.
(27) Troitzsch, U. J. Am. Ceram. Soc. 2006, 89, 3201.(28) Christensen, A.; Carter, E. A. Phys. ReV. B 1998, 58, 8050.(29) Gennard, S.; Cora, F.; Catlow, C. R. A. J. Phys. Chem. B 1999,
103, 10158.(30) Balducci, G.; Kaspar, J.; Fornasiero, P.; Graziani, M.; Islam, M. S.
J. Phys. Chem. B 1998, 102, 557.(31) Paulidou, A.; Nix, R. M. Phys. Chem. Chem. Phys. 2005, 7, 1482.(32) de Leeuw, N. H.; Cooper, T. G. J. Mater. Chem. 2003, 13, 93.(33) Grau-Crespo, R.; Hernandez, N. C.; Sanz, J. F.; De Leeuw, N. H.
J. Phys. Chem. C 2007, 111, 10448.(34) Branda, M. M.; Castellani, N. J.; Grau-Crespo, R.; de Leeuw, N. H.;
Hernandez, N. C.; Sanz, J. F.; Neyman, K. M.; Illas, F. J. Chem. Phys.2009, 131, 094702.
(35) Grau-Crespo, R.; Hernandez, N. C.; Sanz, J. F.; de Leeuw, N. H.J. Mater. Chem. 2009, 19, 710.
(36) Hernandez, N. C.; Grau-Crespo, R.; de Leeuw, N. H.; Sanz, J. F.Phys. Chem. Chem. Phys. 2009, 11, 5246.
(37) Kresse, G.; Furthmuller, J. Comput. Mater. Sci. 1996, 6, 15.(38) Kresse, G.; Furthmuller, J. Phys. ReV. B 1996, 54, 11169.(39) Kresse, G.; Hafner, J. Phys. ReV. B 1993, 48, 13115.(40) Kresse, G.; Hafner, J. J. Phys.sCondens. Matter 1994, 6, 8245.(41) Perdew, J. P.; Zunger, A. Phys. ReV. B 1981, 23, 5048.(42) Perdew, J. P.; Chevary, J. A.; Vosko, S. H.; Jackson, K. A.;
Pederson, M. R.; Singh, D. J.; Fiolhais, C. Phys. ReV. B 1992, 46, 6671.
(43) Anisimov, V. I.; Zaanen, J.; Andersen, O. K. Phys. ReV. B 1991,44, 943.
(44) Liechtenstein, A. I. Phys. ReV. B 1995, 52, R5467.(45) Dudarev, S. L.; Botton, G. A.; Savrasov, S. Y.; Humphreys, C. J.;
Sutton, A. P. Phys. ReV. B 1998, 57, 1505.(46) Rohrbach, A.; Hafner, J.; Kresse, G. J. Phys.sCondens. Mat. 2003,
15, 979.(47) Grau-Crespo, R.; Cora, F.; Sokol, A. A.; de Leeuw, N. H.; Catlow,
C. R. A. Phys. ReV. B 2006, 73, 035116.(48) Nolan, M.; Parker, S. C.; Watson, G. W. Surf. Sci. 2005, 595, 223.(49) Wilson, E. L.; Grau-Crespo, R.; Pang, C. L.; Cabailh, G.; Chen,
Q.; Catlow, C. R. A.; Brown, W. A.; de Leeuw, N. H.; Thornton, G. J.Phys. Chem. C 2008, 112, 10918.
(50) Calzado, C. J.; Hernandez, N. C.; Sanz, J. F. Phys. ReV. B 2008,77, 045118.
(51) Graciani, J.; Plata, J. J.; Sanz, J. F.; Liu, P.; Rodriguez, J. A.J. Chem. Phys. 2010, 132, 104703.
(52) Nolan, M.; Elliott, S. D.; Mulley, J. S.; Bennett, R. A.; Basham,M.; Mulheran, P. Phys. ReV. B 2008, 77, 235424.
(53) Blochl, P. E. Phys. ReV. B 1994, 50, 17953.(54) Kresse, G.; Joubert, D. Phys. ReV. B 1999, 59, 1758.(55) Aldebert, P.; Traverse, J. P. J. Am. Ceram. Soc. 1985, 68, 34.(56) Stefanovich, E. V.; Shluger, A. L.; Catlow, C. R. A. Phys. ReV. B
1994, 49, 11560.(57) Grau-Crespo, R.; Moreira, I. D. R.; Illas, F.; de Leeuw, N. H.;
Catlow, C. R. A. J. Mater. Chem. 2006, 16, 1943.(58) Sun, G. Y.; Kurti, J.; Rajczy, P.; Kertesz, M.; Hafner, J.; Kresse,
G. J. Mol. Struct.sTheochem 2003, 624, 37.(59) Wang, L.; Maxisch, T.; Ceder, G. Phys. ReV. B 2006, 73, 195107.(60) Bader, R. F. W. Atoms in Molecules: A Quantum Theory; Oxford
University Press: London, 1994.(61) Sanville, E.; Kenny, S. D.; Smith, R.; Henkelman, G. J. Comput.
Chem. 2007, 28, 899.(62) Henkelman, G.; Arnaldsson, A.; Jonsson, H. Comput. Mater. Sci.
2006, 36, 354.(63) Becke, A. D.; Edgecombe, K. E. J. Chem. Phys. 1990, 92, 5397.(64) Silvi, B.; Savin, A. Nature 1994, 371, 683.(65) Mori-Sanchez, P.; Recio, J. M.; Silvi, B.; Sousa, C.; Pendas, A. M.;
Luana, V.; Illas, F. Phys. ReV. B 2002, 66, 075103.(66) Chase, M. W. J. NIST-JANAF Thermochemical Tables, 4th ed.;
American Institute of Physics: New York, 1998.(67) Punnoose, A.; Seehra, M. S. Catal. Lett. 2002, 78, 157.(68) Yunusov, N. B.; Zentsov, V. P. Phys. Status Solidi BsBasic Res.
1978, 88, 87.(69) Hoffmann, S. K.; Lijewski, S.; Goslar, J.; Ulanov, V. A. J. Magn.
Reson. 2010, 202, 14.(70) Shapovalov, V.; Metiu, H. J. Catal. 2007, 245, 205.(71) Chretien, S.; Metiu, H. Catal. Lett. 2006, 107, 143.(72) Roldan, A.; Boronat, M.; Corma, A.; Illas, F. J. Phys. Chem. C
2010, 114, 6511.(73) Dwivedi, A.; Cormack, A. N. Philos. Mag. A 1990, 61, 1.(74) Ganduglia-Pirovano, M. V.; Da Silva, J. L. F.; Sauer, J. Phys. ReV.
Lett. 2009, 102, 026101.(75) Loschen, C.; Carrasco, J.; Neyman, K. M.; Illas, F. Phys. ReV. B
2007, 75, 035115.(76) Finazzi, E.; Di Valentin, C.; Pacchioni, G.; Selloni, A. J. Chem.
Phys. 2008, 129, 154113.(77) Palacios, P.; Sanchez, K.; Conesa, J. C.; Wahnon, P. Phys. Status
Solidi AsAppl. Mater. Sci. 2006, 203, 1395.(78) Morgan, B. J.; Watson, G. W. Surf. Sci. 2007, 601, 5034.
JP103181Q
Ti-Doped Zirconia (111) Surface J. Phys. Chem. C, Vol. 114, No. 36, 2010 15409