modificaciondesuperficiede ti o2parafotocatalisis
TRANSCRIPT

I
S
Ha
b
ARRAA
KSMPAH
C
1h
Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20
Contents lists available at SciVerse ScienceDirect
Journal of Photochemistry and Photobiology C:Photochemistry Reviews
journa l h om epa ge: www.elsev ier .com/ locate / jphotochemrev
nvited review
urface modification of TiO2 photocatalyst for environmental applications
yunwoong Parka, Yiseul Parkb, Wooyul Kimb, Wonyong Choib,∗
School of Energy Engineering, Kyungpook National University, Daegu 702-701, Republic of KoreaSchool of Environmental Science and Engineering, Pohang University of Science and Technology (POSTECH), Pohang 790-784, Republic of Korea
a r t i c l e i n f o
rticle history:eceived 1 September 2012eceived in revised form 26 October 2012ccepted 31 October 2012vailable online 16 November 2012
eywords:emiconductor photocatalysisodified TiO2
hotochemical purification
a b s t r a c t
This paper reviews recent studies on the semiconductor photocatalysis based on surface-modified TiO2 ofwhich application is mainly focused on environmental remediation. TiO2 photocatalysis that is based onthe photoinduced interfacial charge transfer has been extensively studied over the past four decades. Agreat number of modification methods of semiconductor photocatalysts have been developed and inves-tigated to accelerate the photoconversion, to enable the absorption of visible light, or to alter the reactionmechanism to control the products and intermediates. In this regard, various modification methods ofTiO2 are classified according to the kind of surface modifiers (metal-loading, impurity doping, inorganicadsorbates, polymer coating, dye-sensitization, charge transfer complexation) and their effects on photo-catalytic reaction mechanism and kinetics are discussed in detail. Modifying TiO2 in various ways not only
dvanced oxidation process (AOP)ydroxyl radicals
changes the mechanism and kinetics under UV irradiation but also introduces visible light activity that isabsent with pure TiO2. Each modification method influences the photocatalytic activity and mechanismin a way different from others and the observed modification effects are often different depending on thetest substrates and conditions even for the same modification method. Better understanding of the modi-fication effects on TiO2 photocatalysis is necessary to obtain reliable results, to assess the photoconversionefficiency more quantitatively, and to further improve the modification methods.
© 2012 Elsevier B.V. All rights reserved.
ontents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22. TiO2 photocatalysis and some important aspects for environmental applications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2.1. Photo-induced charge transfer and oxidants generation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72.2. Photocatalytic activity of TiO2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 82.3. Surface chemistry . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
3. Surface-modified TiO2 photocatalysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93.1. Metal-loaded TiO2 photocatalysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93.2. Doped TiO2 and visible light photocatalytic activity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113.3. Role of inorganic adsorbates . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 123.4. Surface charge modification. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133.5. Dye-sensitization of TiO2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153.6. Charge transfer complexation on TiO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
24. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
∗ Corresponding author. Tel.: +82 54 279 2283.E-mail address: [email protected] (W. Choi).
389-5567/$20.00 © 2012 Elsevier B.V. All rights reserved.ttp://dx.doi.org/10.1016/j.jphotochemrev.2012.10.001

2 tobiology C: Photochemistry Reviews 15 (2013) 1– 20
ff(LEei2
1
msmatcmp(ehv
to cover the fundamental and general aspects of photocatalysis ingreater detail. The interested readers will find a number of rele-
H. Park et al. / Journal of Photochemistry and Pho
Hyunwoong Park received a B.S. in Environmental Sci-ence at Hallym University (Chuncheon, Korea) in 1999 anda Ph.D. degree (advisor: Wonyong Choi) in Environmen-tal Engineering at POSTECH (Pohang, Korea) in 2004. Hespent one and half years at POSTECH as a postdoctoralresearcher and continued his research at Prof. M.R. Hoff-mann group, California Institute of Technology, Pasadena,U.S.A. In 2008 he moved to School of Energy Engineer-ing at Kyungpook National University (Daegu, Korea) asan assistant professor and then was promoted to asso-ciate professor (2012). His primary research fields arephotoelectrochemical generation of solar fuels and elec-trochemical systems for environmental remediation.
Yiseul Park received a B.S. in Chemical Engineering fromPukyong National University (Busan, Korea) in 2003 anda Ph.D. degree (advisor: Prof. Wonyong Choi) in Envi-ronmental Engineering from POSTECH in 2010. After ayear of postdoctoral reasearch at POSTECH, she joinedProf. Kyoung-Shin Choi’s group at Purdue University(now moved to University of Wisconsin, Madison) asa postdoctoral fellow in 2011. Her research interestsinclude photoelectrochemistry for solar energy conver-sion, electrodeposition of metal oxide and synthesis ofphotocatalysts.
Wooyul Kim received a B.S. from Kwangwoon Univer-sity (2006), and a Ph.D. from POSTECH under the directionof Professor Wonyong Choi (2012). He is currently work-ing as a postdoctoral researcher at POSTECH. His researchinterests include photosynthetic and photocatalytic con-version using dye-sensitized and surface-modified TiO2.
Wonyong Choi received a B.S. in Engineering fromSeoul National University (Seoul, Korea) in 1988, anM.S. in Chemistry from POSTECH in 1990, and a Ph.D.in Chemistry (advisor: Prof. Michael R. Hoffmann) fromCalifornia Institute of Technology in 1996. After postdoc-toral research on atmospheric chemistry at Jet PropulsionLaboratory (Pasadena, California; 1996–1998), he joinedthe faculty of School of Environmental Science and Engi-neering, POSTECH as an assistant professor (1998), andthen was promoted to associate professor (2003), andfull professor (2008). He published more than 170 papersin peer-reviewed journals, which were cited over 15,000times to date. He was awarded Young Scientist Award
rom Korean Academy of Science and Technology (2006), Lectureship Awardor Asian and Oceanian Photochemist from Japanese Photochemistry Association2008), Rising Star faculty fund from POSTECH (2011), and Namgo (Jong-Ryulee) chair professorship from POSTECH (2012). Currently, he is serving as anditor of Journal of Hazardous Materials (Elsevier, since 2008). He is also on theditorial advisory board of Energy and Environmental Science (Royal Society of Chem-stry, since 2008) and Journal of Physical Chemistry (American Chemical Society:009–2011).
. Introduction
The industrialized society discharges a wide variety of environ-ental contaminants from residential, commercial and industrial
ources [1,2]. The commercially available chemicals are approxi-ately 30,000 and not much is known regarding their occurrence
nd fate in the environment [3]. The greater challenge lies withhe treatment of newly emerging micropollutants (e.g., pharma-euticals, antibiotics, and pathogens) which are being detectedore often as analytical methods improve [2]. Advanced oxidation
rocesses (AOPs) such as UV/H2O2 [4–7], ozonation [4–6,8–10],
photo)Fenton [4,11–14], �-radiolysis [15], sonolysis [15–17],lectrochemical oxidation [18–24], and photocatalysis [8,25–27]ave been widely and extensively explored to mitigate a greatariety of pollutants present in various environmental media.Scheme 1. Various modification methods of TiO2 photocatalyst.
They are initiated primarily from the formation of reactive andshort-lived oxygen-containing radicals (e.g.,
•OH, HOO
•, O2
•−).Among them, the hydroxyl radical (
•OH) is the most reac-
tive and powerful oxidant (E0 = 2.7 V) which reacts with mostorganic compounds at diffusion-limited rates [28]. AOPs havebeen considered as alternatives to conventional water treat-ment technologies [7,8,16,29]. However, they have yet toovercome the energy efficiencies and cost competitiveness[30–32].
Among many AOPs, TiO2 photocatalysis has received hugeattention as one of the most viable environmental cleanuptechnologies; yet its cost competitiveness is still lower than con-ventional treatment processes and some AOPs [33]. For example,when compared with granular activated carbon (GAC) adsorptionfor indoor air purification, the installation and annual mainte-nance cost of TiO2 photocatalysis was estimated to be 7–10 timeshigher than that of GAC adsorption to achieve a similar treat-ment efficiency [33]. Therefore, photocatalysts need to have higheractivities to be economically competitive as a practical cleanuptechnology. This can be achieved by either enhancing the quantumefficiency of photocatalysis under UV light or introducing visiblelight activity that is absent with pure TiO2. New photocatalyticmaterials with novel composition and structure are being inten-sively sought to attain higher activities and to excel TiO2-basedphotocatalysts but the success cases for practical applications arelimited. From a practical point of view, alternative photocatalyticmaterials that are as “versatile, economical, stable, abundant, andnon-toxic” as TiO2 are hard to be found. One of the most viable andpractical approaches in developing better photocatalysts is to mod-ify TiO2 in various methods (e.g., impurity doping, sensitization,surface modification/complexation, integration with other nano-structured materials, etc.).
With this in mind, this Review attempts to introduce and discussrecent findings and advances in surface-modified TiO2 as an envi-ronmental cleanup photocatalyst. The modification of TiO2 surfacecan be done in a number of ways (see Scheme 1) and the role of sur-face modifier of TiO2 differs greatly depending on the nature of thespecific method. Some recent articles on the surface-modified TiO2are classified according to the kind of methods in Table 1. Most partsof this Review deal with the surface modification methods of TiO2and their effects on the photocatalytic reactions that are relatedwith the transformation of chemical pollutants. This does not mean
vant, excellent review papers or books focusing on the fundamentalaspects before [27,34–39] and after [26,40–47] the mid-1990 (seealso references therein).
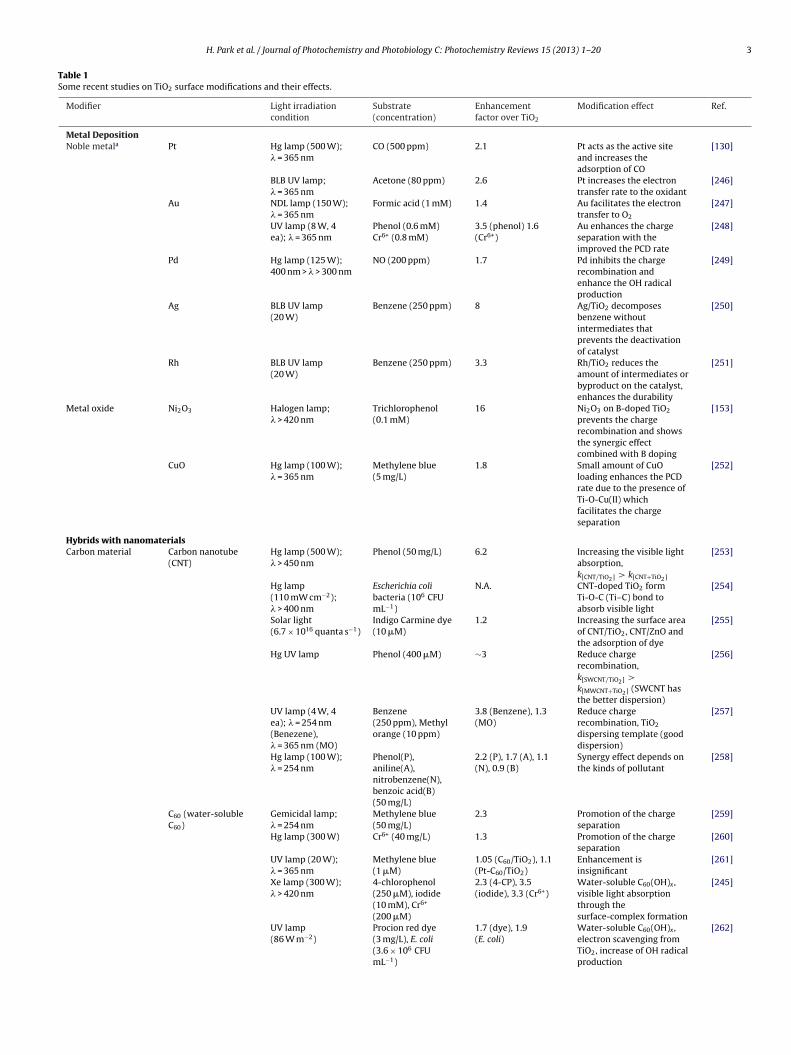
H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20 3
Table 1Some recent studies on TiO2 surface modifications and their effects.
Modifier Light irradiationcondition
Substrate(concentration)
Enhancementfactor over TiO2
Modification effect Ref.
Metal DepositionNoble metala Pt Hg lamp (500 W);
� = 365 nmCO (500 ppm) 2.1 Pt acts as the active site
and increases theadsorption of CO
[130]
BLB UV lamp;� = 365 nm
Acetone (80 ppm) 2.6 Pt increases the electrontransfer rate to the oxidant
[246]
Au NDL lamp (150 W);� = 365 nm
Formic acid (1 mM) 1.4 Au facilitates the electrontransfer to O2
[247]
UV lamp (8 W, 4ea); � = 365 nm
Phenol (0.6 mM)Cr6+ (0.8 mM)
3.5 (phenol) 1.6(Cr6+)
Au enhances the chargeseparation with theimproved the PCD rate
[248]
Pd Hg lamp (125 W);400 nm > � > 300 nm
NO (200 ppm) 1.7 Pd inhibits the chargerecombination andenhance the OH radicalproduction
[249]
Ag BLB UV lamp(20 W)
Benzene (250 ppm) 8 Ag/TiO2 decomposesbenzene withoutintermediates thatprevents the deactivationof catalyst
[250]
Rh BLB UV lamp(20 W)
Benzene (250 ppm) 3.3 Rh/TiO2 reduces theamount of intermediates orbyproduct on the catalyst,enhances the durability
[251]
Metal oxide Ni2O3 Halogen lamp;� > 420 nm
Trichlorophenol(0.1 mM)
16 Ni2O3 on B-doped TiO2
prevents the chargerecombination and showsthe synergic effectcombined with B doping
[153]
CuO Hg lamp (100 W);� = 365 nm
Methylene blue(5 mg/L)
1.8 Small amount of CuOloading enhances the PCDrate due to the presence ofTi-O-Cu(II) whichfacilitates the chargeseparation
[252]
Hybrids with nanomaterialsCarbon material Carbon nanotube
(CNT)Hg lamp (500 W);� > 450 nm
Phenol (50 mg/L) 6.2 Increasing the visible lightabsorption,k[CNT/TiO2] > k[CNT+TiO2]
[253]
Hg lamp(110 mW cm−2);� > 400 nm
Escherichia colibacteria (106 CFUmL−1)
N.A. CNT-doped TiO2 formTi-O-C (Ti–C) bond toabsorb visible light
[254]
Solar light(6.7 × 1016 quanta s−1)
Indigo Carmine dye(10 �M)
1.2 Increasing the surface areaof CNT/TiO2, CNT/ZnO andthe adsorption of dye
[255]
Hg UV lamp Phenol (400 �M) ∼3 Reduce chargerecombination,k[SWCNT/TiO2] >k[MWCNT+TiO2] (SWCNT hasthe better dispersion)
[256]
UV lamp (4 W, 4ea); � = 254 nm(Benezene),� = 365 nm (MO)
Benzene(250 ppm), Methylorange (10 ppm)
3.8 (Benzene), 1.3(MO)
Reduce chargerecombination, TiO2
dispersing template (gooddispersion)
[257]
Hg lamp (100 W);� = 254 nm
Phenol(P),aniline(A),nitrobenzene(N),benzoic acid(B)(50 mg/L)
2.2 (P), 1.7 (A), 1.1(N), 0.9 (B)
Synergy effect depends onthe kinds of pollutant
[258]
C60 (water-solubleC60)
Gemicidal lamp;� = 254 nm
Methylene blue(50 mg/L)
2.3 Promotion of the chargeseparation
[259]
Hg lamp (300 W) Cr6+ (40 mg/L) 1.3 Promotion of the chargeseparation
[260]
UV lamp (20 W);� = 365 nm
Methylene blue(1 �M)
1.05 (C60/TiO2), 1.1(Pt-C60/TiO2)
Enhancement isinsignificant
[261]
Xe lamp (300 W);� > 420 nm
4-chlorophenol(250 �M), iodide(10 mM), Cr6+
(200 �M)
2.3 (4-CP), 3.5(iodide), 3.3 (Cr6+)
Water-soluble C60(OH)x ,visible light absorptionthrough thesurface-complex formation
[245]
UV lamp(86 W m−2)
Procion red dye(3 mg/L), E. coli(3.6 × 106 CFUmL−1)
1.7 (dye), 1.9(E. coli)
Water-soluble C60(OH)x ,electron scavenging fromTiO2, increase of OH radicalproduction
[262]
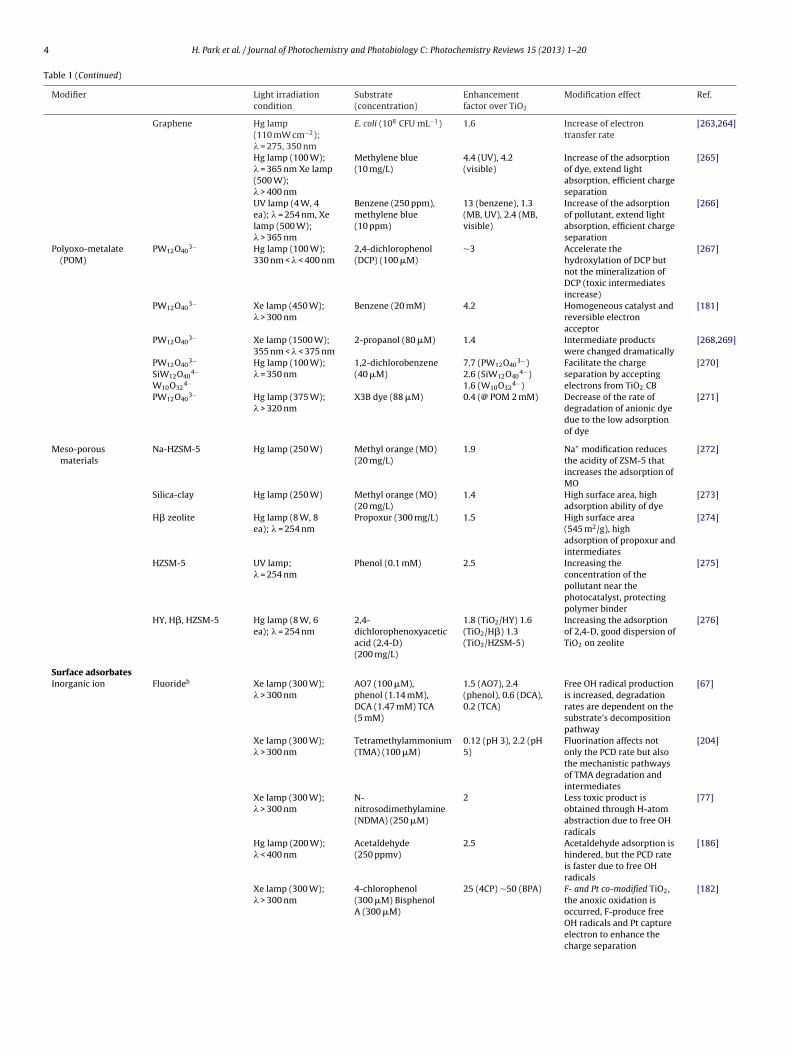
4 H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20
Table 1 (Continued)
Modifier Light irradiationcondition
Substrate(concentration)
Enhancementfactor over TiO2
Modification effect Ref.
Graphene Hg lamp(110 mW cm−2);� = 275, 350 nm
E. coli (108 CFU mL−1) 1.6 Increase of electrontransfer rate
[263,264]
Hg lamp (100 W);� = 365 nm Xe lamp(500 W);� > 400 nm
Methylene blue(10 mg/L)
4.4 (UV), 4.2(visible)
Increase of the adsorptionof dye, extend lightabsorption, efficient chargeseparation
[265]
UV lamp (4 W, 4ea); � = 254 nm, Xelamp (500 W);� > 365 nm
Benzene (250 ppm),methylene blue(10 ppm)
13 (benzene), 1.3(MB, UV), 2.4 (MB,visible)
Increase of the adsorptionof pollutant, extend lightabsorption, efficient chargeseparation
[266]
Polyoxo-metalate(POM)
PW12O403− Hg lamp (100 W);
330 nm < � < 400 nm2,4-dichlorophenol(DCP) (100 �M)
∼3 Accelerate thehydroxylation of DCP butnot the mineralization ofDCP (toxic intermediatesincrease)
[267]
PW12O403− Xe lamp (450 W);
� > 300 nmBenzene (20 mM) 4.2 Homogeneous catalyst and
reversible electronacceptor
[181]
PW12O403− Xe lamp (1500 W);
355 nm < � < 375 nm2-propanol (80 �M) 1.4 Intermediate products
were changed dramatically[268,269]
PW12O403−
SiW12O404−
W10O324−
Hg lamp (100 W);� = 350 nm
1,2-dichlorobenzene(40 �M)
7.7 (PW12O403−)
2.6 (SiW12O404−)
1.6 (W10O324−)
Facilitate the chargeseparation by acceptingelectrons from TiO2 CB
[270]
PW12O403− Hg lamp (375 W);
� > 320 nmX3B dye (88 �M) 0.4 (@ POM 2 mM) Decrease of the rate of
degradation of anionic dyedue to the low adsorptionof dye
[271]
Meso-porousmaterials
Na-HZSM-5 Hg lamp (250 W) Methyl orange (MO)(20 mg/L)
1.9 Na+ modification reducesthe acidity of ZSM-5 thatincreases the adsorption ofMO
[272]
Silica-clay Hg lamp (250 W) Methyl orange (MO)(20 mg/L)
1.4 High surface area, highadsorption ability of dye
[273]
H� zeolite Hg lamp (8 W, 8ea); � = 254 nm
Propoxur (300 mg/L) 1.5 High surface area(545 m2/g), highadsorption of propoxur andintermediates
[274]
HZSM-5 UV lamp;� = 254 nm
Phenol (0.1 mM) 2.5 Increasing theconcentration of thepollutant near thephotocatalyst, protectingpolymer binder
[275]
HY, H�, HZSM-5 Hg lamp (8 W, 6ea); � = 254 nm
2,4-dichlorophenoxyaceticacid (2,4-D)(200 mg/L)
1.8 (TiO2/HY) 1.6(TiO2/H�) 1.3(TiO2/HZSM-5)
Increasing the adsorptionof 2,4-D, good dispersion ofTiO2 on zeolite
[276]
Surface adsorbatesInorganic ion Fluorideb Xe lamp (300 W);
� > 300 nmAO7 (100 �M),phenol (1.14 mM),DCA (1.47 mM) TCA(5 mM)
1.5 (AO7), 2.4(phenol), 0.6 (DCA),0.2 (TCA)
Free OH radical productionis increased, degradationrates are dependent on thesubstrate’s decompositionpathway
[67]
Xe lamp (300 W);� > 300 nm
Tetramethylammonium(TMA) (100 �M)
0.12 (pH 3), 2.2 (pH5)
Fluorination affects notonly the PCD rate but alsothe mechanistic pathwaysof TMA degradation andintermediates
[204]
Xe lamp (300 W);� > 300 nm
N-nitrosodimethylamine(NDMA) (250 �M)
2 Less toxic product isobtained through H-atomabstraction due to free OHradicals
[77]
Hg lamp (200 W);� < 400 nm
Acetaldehyde(250 ppmv)
2.5 Acetaldehyde adsorption ishindered, but the PCD rateis faster due to free OHradicals
[186]
Xe lamp (300 W);� > 300 nm
4-chlorophenol(300 �M) BisphenolA (300 �M)
25 (4CP) ∼50 (BPA) F- and Pt co-modified TiO2,the anoxic oxidation isoccurred, F-produce freeOH radicals and Pt captureelectron to enhance thecharge separation
[182]
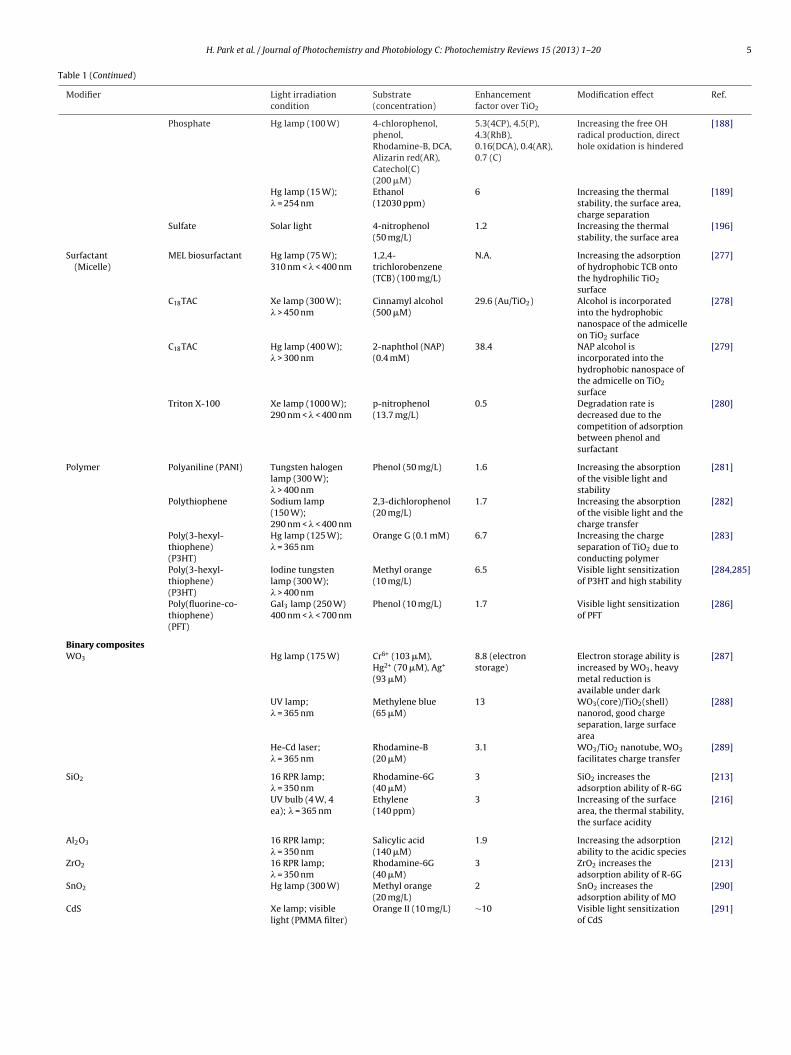
H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20 5
Table 1 (Continued)
Modifier Light irradiationcondition
Substrate(concentration)
Enhancementfactor over TiO2
Modification effect Ref.
Phosphate Hg lamp (100 W) 4-chlorophenol,phenol,Rhodamine-B, DCA,Alizarin red(AR),Catechol(C)(200 �M)
5.3(4CP), 4.5(P),4.3(RhB),0.16(DCA), 0.4(AR),0.7 (C)
Increasing the free OHradical production, directhole oxidation is hindered
[188]
Hg lamp (15 W);� = 254 nm
Ethanol(12030 ppm)
6 Increasing the thermalstability, the surface area,charge separation
[189]
Sulfate Solar light 4-nitrophenol(50 mg/L)
1.2 Increasing the thermalstability, the surface area
[196]
Surfactant(Micelle)
MEL biosurfactant Hg lamp (75 W);310 nm < � < 400 nm
1,2,4-trichlorobenzene(TCB) (100 mg/L)
N.A. Increasing the adsorptionof hydrophobic TCB ontothe hydrophilic TiO2
surface
[277]
C18TAC Xe lamp (300 W);� > 450 nm
Cinnamyl alcohol(500 �M)
29.6 (Au/TiO2) Alcohol is incorporatedinto the hydrophobicnanospace of the admicelleon TiO2 surface
[278]
C18TAC Hg lamp (400 W);� > 300 nm
2-naphthol (NAP)(0.4 mM)
38.4 NAP alcohol isincorporated into thehydrophobic nanospace ofthe admicelle on TiO2
surface
[279]
Triton X-100 Xe lamp (1000 W);290 nm < � < 400 nm
p-nitrophenol(13.7 mg/L)
0.5 Degradation rate isdecreased due to thecompetition of adsorptionbetween phenol andsurfactant
[280]
Polymer Polyaniline (PANI) Tungsten halogenlamp (300 W);� > 400 nm
Phenol (50 mg/L) 1.6 Increasing the absorptionof the visible light andstability
[281]
Polythiophene Sodium lamp(150 W);290 nm < � < 400 nm
2,3-dichlorophenol(20 mg/L)
1.7 Increasing the absorptionof the visible light and thecharge transfer
[282]
Poly(3-hexyl-thiophene)(P3HT)
Hg lamp (125 W);� = 365 nm
Orange G (0.1 mM) 6.7 Increasing the chargeseparation of TiO2 due toconducting polymer
[283]
Poly(3-hexyl-thiophene)(P3HT)
Iodine tungstenlamp (300 W);� > 400 nm
Methyl orange(10 mg/L)
6.5 Visible light sensitizationof P3HT and high stability
[284,285]
Poly(fluorine-co-thiophene)(PFT)
GaI3 lamp (250 W)400 nm < � < 700 nm
Phenol (10 mg/L) 1.7 Visible light sensitizationof PFT
[286]
Binary compositesWO3 Hg lamp (175 W) Cr6+ (103 �M),
Hg2+ (70 �M), Ag+
(93 �M)
8.8 (electronstorage)
Electron storage ability isincreased by WO3, heavymetal reduction isavailable under dark
[287]
UV lamp;� = 365 nm
Methylene blue(65 �M)
13 WO3(core)/TiO2(shell)nanorod, good chargeseparation, large surfacearea
[288]
He-Cd laser;� = 365 nm
Rhodamine-B(20 �M)
3.1 WO3/TiO2 nanotube, WO3
facilitates charge transfer[289]
SiO2 16 RPR lamp;� = 350 nm
Rhodamine-6G(40 �M)
3 SiO2 increases theadsorption ability of R-6G
[213]
UV bulb (4 W, 4ea); � = 365 nm
Ethylene(140 ppm)
3 Increasing of the surfacearea, the thermal stability,the surface acidity
[216]
Al2O3 16 RPR lamp;� = 350 nm
Salicylic acid(140 �M)
1.9 Increasing the adsorptionability to the acidic species
[212]
ZrO2 16 RPR lamp;� = 350 nm
Rhodamine-6G(40 �M)
3 ZrO2 increases theadsorption ability of R-6G
[213]
SnO2 Hg lamp (300 W) Methyl orange(20 mg/L)
2 SnO2 increases theadsorption ability of MO
[290]
CdS Xe lamp; visiblelight (PMMA filter)
Orange II (10 mg/L) ∼10 Visible light sensitizationof CdS
[291]
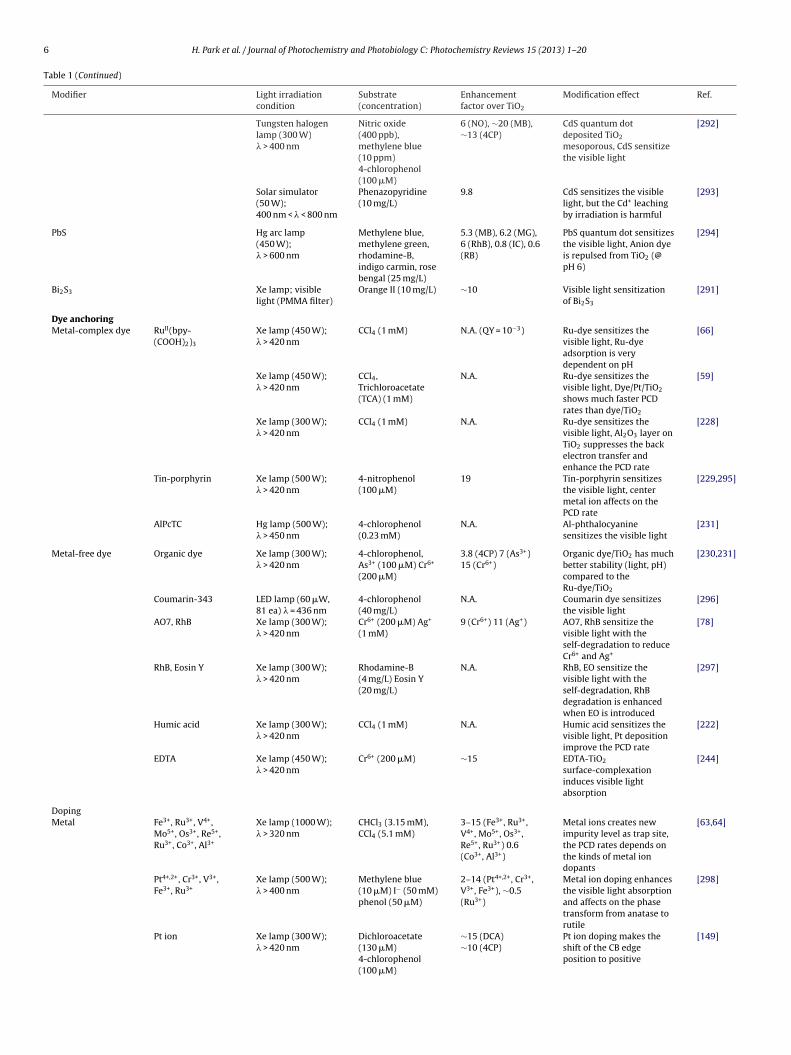
6 H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20
Table 1 (Continued)
Modifier Light irradiationcondition
Substrate(concentration)
Enhancementfactor over TiO2
Modification effect Ref.
Tungsten halogenlamp (300 W)� > 400 nm
Nitric oxide(400 ppb),methylene blue(10 ppm)4-chlorophenol(100 �M)
6 (NO), ∼20 (MB),∼13 (4CP)
CdS quantum dotdeposited TiO2
mesoporous, CdS sensitizethe visible light
[292]
Solar simulator(50 W);400 nm < � < 800 nm
Phenazopyridine(10 mg/L)
9.8 CdS sensitizes the visiblelight, but the Cd+ leachingby irradiation is harmful
[293]
PbS Hg arc lamp(450 W);� > 600 nm
Methylene blue,methylene green,rhodamine-B,indigo carmin, rosebengal (25 mg/L)
5.3 (MB), 6.2 (MG),6 (RhB), 0.8 (IC), 0.6(RB)
PbS quantum dot sensitizesthe visible light, Anion dyeis repulsed from TiO2 (@pH 6)
[294]
Bi2S3 Xe lamp; visiblelight (PMMA filter)
Orange II (10 mg/L) ∼10 Visible light sensitizationof Bi2S3
[291]
Dye anchoringMetal-complex dye RuII(bpy-
(COOH)2)3
Xe lamp (450 W);� > 420 nm
CCl4 (1 mM) N.A. (QY = 10−3) Ru-dye sensitizes thevisible light, Ru-dyeadsorption is verydependent on pH
[66]
Xe lamp (450 W);� > 420 nm
CCl4,Trichloroacetate(TCA) (1 mM)
N.A. Ru-dye sensitizes thevisible light, Dye/Pt/TiO2
shows much faster PCDrates than dye/TiO2
[59]
Xe lamp (300 W);� > 420 nm
CCl4 (1 mM) N.A. Ru-dye sensitizes thevisible light, Al2O3 layer onTiO2 suppresses the backelectron transfer andenhance the PCD rate
[228]
Tin-porphyrin Xe lamp (500 W);� > 420 nm
4-nitrophenol(100 �M)
19 Tin-porphyrin sensitizesthe visible light, centermetal ion affects on thePCD rate
[229,295]
AlPcTC Hg lamp (500 W);� > 450 nm
4-chlorophenol(0.23 mM)
N.A. Al-phthalocyaninesensitizes the visible light
[231]
Metal-free dye Organic dye Xe lamp (300 W);� > 420 nm
4-chlorophenol,As3+ (100 �M) Cr6+
(200 �M)
3.8 (4CP) 7 (As3+)15 (Cr6+)
Organic dye/TiO2 has muchbetter stability (light, pH)compared to theRu-dye/TiO2
[230,231]
Coumarin-343 LED lamp (60 �W,81 ea) � = 436 nm
4-chlorophenol(40 mg/L)
N.A. Coumarin dye sensitizesthe visible light
[296]
AO7, RhB Xe lamp (300 W);� > 420 nm
Cr6+ (200 �M) Ag+
(1 mM)9 (Cr6+) 11 (Ag+) AO7, RhB sensitize the
visible light with theself-degradation to reduceCr6+ and Ag+
[78]
RhB, Eosin Y Xe lamp (300 W);� > 420 nm
Rhodamine-B(4 mg/L) Eosin Y(20 mg/L)
N.A. RhB, EO sensitize thevisible light with theself-degradation, RhBdegradation is enhancedwhen EO is introduced
[297]
Humic acid Xe lamp (300 W);� > 420 nm
CCl4 (1 mM) N.A. Humic acid sensitizes thevisible light, Pt depositionimprove the PCD rate
[222]
EDTA Xe lamp (450 W);� > 420 nm
Cr6+ (200 �M) ∼15 EDTA-TiO2
surface-complexationinduces visible lightabsorption
[244]
DopingMetal Fe3+, Ru3+, V4+,
Mo5+, Os3+, Re5+,Ru3+, Co3+, Al3+
Xe lamp (1000 W);� > 320 nm
CHCl3 (3.15 mM),CCl4 (5.1 mM)
3–15 (Fe3+, Ru3+,V4+, Mo5+, Os3+,Re5+, Ru3+) 0.6(Co3+, Al3+)
Metal ions creates newimpurity level as trap site,the PCD rates depends onthe kinds of metal iondopants
[63,64]
Pt4+,2+, Cr3+, V3+,Fe3+, Ru3+
Xe lamp (500 W);� > 400 nm
Methylene blue(10 �M) I− (50 mM)phenol (50 �M)
2–14 (Pt4+,2+, Cr3+,V3+, Fe3+), ∼0.5(Ru3+)
Metal ion doping enhancesthe visible light absorptionand affects on the phasetransform from anatase torutile
[298]
Pt ion Xe lamp (300 W);� > 420 nm
Dichloroacetate(130 �M)4-chlorophenol(100 �M)
∼15 (DCA)∼10 (4CP)
Pt ion doping makes theshift of the CB edgeposition to positive
[149]
H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20 7
Table 1 (Continued)
Modifier Light irradiationcondition
Substrate(concentration)
Enhancementfactor over TiO2
Modification effect Ref.
Bi ion Halogen lamp(1000 W);� > 420 nm
Phenol (0.6 mM),Cr6+ (0.8 mM)
9.5 (phenol) 11.2(Cr6+)
Bi ion doping enhances thevisible light absorption andreduces the chargerecombination
[299]
Non-metal N Fluorescent light� > 420 nm
Acetaldehyde(485 ppm)
6 N doping narrows the bandgap of TiO2 and enhancesthe PCD rate under visiblelight
[150]
Xe lamp (190 W);� > 385 nm
Tributyltin(10 mg/L)
1.9 Visible light absorbance isincreased
[300]
Hg lamp (200 W);� > 365 nmTungsten halogenlamp (500 W);visible light
Methyl orange(20 mg/L)
1.7 (UV)1.6 (Visible)
N doping retards the phasetransition and increasesthe visible light absorption
[301]
C Xe lamp (150 W);� > 455 nm
4-chlorophenol(0.25 mM)Acetaldehyde(A)(5 vol%)
3.7 (4CP)9.2 (A)
C doping shifts the flatband potential to positivedirection and increases thevisible light absorption
[302]
Xe lamp;530 nm > � > 400 nm
2-propanol(300 ppm)
N.A. Ti–C bond forms aseparated new doping leveland increases the visiblelight absorption
[303]
Xe lamp (300 W);� > 420 nm
4-chlorophenol(0.25 mM)Iodide (10 mM)
2.7 (4CP)12 (Iodide)
C doping is availablewithout extra carbonprecursor using sol–gelmethod
[171]
S Xe lamp (1000 W);� > 440 nm
Methylene blue(50 mM)2-propanol(10 vol%)
11 (MB) 12(2-propanol)
S doping increases thevisible light absorption
[304]
B Halogen lamp;� > 420 nm
Trichlorophenol(0.1 mM)
4 B doping increases thevisible light absorption
[153]
Co-doping B/C Tungsten halogenlamp (1000 W);� > 420 nm
Acid orange 7(AO7) (20 mg/L)
2.8 B,C co-doping producesmore oxygen vacancies andinhibit the chargerecombination
[305]
N/F Tungsten halogenlamp (1000 W);� > 420 nm
Acid orange 7(AO7) (50 mg/L)
7.2 N,F co-doping shows thesynergy effect through theband gap narrowing by Ndoping and the reductionof charge recombination byF doping
[306]
Ce/N Fluorescent lamp(30 W); � > 420 nm
Methylene blue(15 mg/L)
2.8 Ce, N co-doping shows thesynergy effect andeffectively inhibits thecharge recombination
[307]
Pt/N Xe lamp (300 W);� > 420 nm
4-chlorophenol(0.1 mM)
12 Pt,N co-doping shows thesynergy effect through theretardation of phasetransition by N-doping andthe increase of visible light
[177]
in Tabnated
2e
2
brb(s(ob
a Detailed summary of metal deposit effects on TiO2 photocatalysis is presented
b Photophysicochemical properties and photocatalytic activities of surface-fluori
. TiO2 photocatalysis and some important aspects fornvironmental applications
.1. Photo-induced charge transfer and oxidants generation
TiO2 is a wide-bandgap semiconductor with 3.0–3.2 eV ofandgap energy (Eg) that requires the excitation light wavelengthange shorter than ca. 400 nm (Eg = hc/� ∼= 1240/�) (Fig. 1). Theandgap excitation creates electrons (eCB
−) in conduction bandCB) and holes (hVB
+) in valence band (VB) in femtosecond timecale (path 1 in Fig. 1), which are subsequently trapped in 100 ps
shallow trap) to 10 ns (deep trap) and recombines with eachther in the range of 10–100 ns (path 2). Environmental applica-ility of semiconductor photocatalysts is directly related with theabsorption by Pt-doping
le 3. TiO2 is presented in Table 4.
interfacial charge transfers of (trapped) holes and electrons, whichoccur more slowly in 100 ns and ms ranges, respectively (paths3, 4 vs. 8, 9) [27]. The interfacial charge transfers either directlyoxidize/reduce contaminants (paths 3 and 8) or generate reactiveoxidants such as hydroxyl radicals and superoxides (paths 4 and9). The transfers of trapped electrons and trapped holes take placeslowly [48] and the lifetime of trapped electrons in the presenceof O2 has been reported to be about 0.33 ms [49]. This means thatthe slow interfacial charge transfer limits the overall photocatalyticreaction rate. The slow interfacial charge transfers are most sen-sitively influenced by the surface properties of semiconductors.
The modification of the surface affects this charge transfer processvia various mechanisms which include the creation of additionalsurface trap or charge reservoir sites, the change of the intrinsic
8 H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20
Fig. 1. Schematic illustration for energetics and primary reaction mechanism ofTiO2 photocatalysis. 1: Bandgap (Eg) excitation and electron–hole charge pair cre-ation, 2: the charge pair recombination, 3: electron transfer to electron acceptor (A:usually dissolved oxygen), 4: electron transfer to oxygen molecule, 5: formation ofhydroxyperoxyl radical via a reductive pathway, 6: formation of hydrogen perox-ide, 7: formation of hydroxyl radical, 8: hole transfer to electron donor (D: usuallyorganic pollutants), 9: hole transfer to surface hydroxyl group to generate OH rad-ical, 10: hydroxyl radical-mediated oxidation of organic substrate. Note that thee0
ctsco
t(od(totboaltccenprhFtmrsupc[
lahih
Table 2Some selected substrates widely tested in TiO2 photocatalysis.
Substrates References
Aromatics Phenol [18–20,67,147,182,308,116]4-chlorophenol (4-CP) [171,245,309]Toluene [310,311]Polychlorinateddibenzo-p-dioxins(PCCD)
[146]
Benzene [312]Bisphenol A [182,313]
Organic acids Trichloroacetic acid(TCA)
[84,149,224,226]
Dichloroacetic acid(DCA)
[149,226]
Stearic acid [183,314,315]
Amines Tetramethylammonium(TMA)
[84,185,210]
Alkyl amines [77,131]
Cholor-hydrocarbons Chloroform [316,317]Tetrachlorocarbon [66,224,226,316–318]
Dyes Acid orange 7 [67,219,319]Rhodamine-B [151,219,320]Methylene blue [219,321–324]
Alcohols Methanol [325]Inorganics Cr(VI), Chromate [171,230,245]
As(III) [326–329]Ammonia [76,330]Carbon monoxide [129]
Carbonyl Acetone [331]Acetaldehyde [152,186]
nergy levels (right arrow) shift according to the Nernst equation: E(VNHE) = E0(@pH)–0.059pH.
harge transfer rate, the introduction of visible light sensitizer, andhe change of the substrate adsorption/desorption. The modifiedurface and its effects on the interfacial charge transfer and photo-atalysis for environmental purification would be the main themef this review.
The band positions of TiO2 are suitable for achieving the redoxransformation of environmental pollutants. The CB energy levelECB = −0.51 V at pH 7) lies slightly above the reduction potentialf oxygen (E0(O2/O2
•−) = −0.33 V) that is an ubiquitous and pre-ominant oxidant (or electron acceptor) in environmental mediaair and water) (Fig. 1). Most metal oxide semiconductors havehe similar EVB potential edge because VB is primarily composedf O 2p atomic orbitals [50]. Nevertheless, TiO2 is consideredhe most active among the metal oxide semiconductors proba-ly because of the proper position of ECB for the facile transferf eCB
− to O2. For example, WO3 is not working efficiently asn oxidizing photocatalyst mainly because the CB position isower (more positive) than the oxygen reduction potential. In fact,he CB electron transfer to dissolved oxygen molecule has beenonsidered as the rate-determining step in semiconductor photo-atalysis [49,51–56]. Therefore, the presence of alternative strongerlectron acceptor often enhances the photocatalytic reactions sig-ificantly [57]. The photoinduced reduction of O2 lead to theroduction of superoxide radical anion (O2
•−) and hydroperoxyladical (E0(O2/HO2
•) = −0.45 V at pH 7) and its further reduction toydrogen peroxide (E0(HOO•/H2O2 = +1.007 V at pH 7) (paths 4–6 inig. 1) [58]. Hydrogen peroxide may generate the hydroxyl radicalhrough its reaction with the CB electron (path 7). Some substrates
ay undergo the reductive transformation through their directeaction with CB electrons (path 3). A number of electron-acceptingubstrates have been tested for their reductive transformationsing TiO2 photocatalysis, which include chlorinated organic com-ounds (e.g., CCl4 [59–66], trichloroacetic acid [67]), nitrogenousompounds [68–77], and toxic metals ions (e.g., Cr(VI) [65,78], Ag(I)79]).
On the other hand, the TiO2 VB (EVB = +2.69 V at pH 7) places farower (more positive) than the oxidation potentials of most organic
nd inorganic compounds (electron donors). The highly oxidativeoles can be transferred to the surface-adsorbed water or hydrox-de groups (i.e., titanol group) with forming the surface-boundydroxyl radical (
•OHsurf) (or free hydroxyl radical,
•OHfree, after
Others Polyvinyl chloride [332]Soot [85]
desorption from the surface). Some strongly adsorbed molecules(e.g., formate, oxalate, acetate) tend to be oxidized directly by h+
VBwhile weakly adsorbed ones (e.g., chlorinated ethane, chlorophe-nol, iodide) can be more favorably degraded by OH radicals[80–86]. Free
•OH is known to be more reactive than the surface-
bound analogue due to the reorganization energy [87]. A varietyof organic/inorganic pollutants and biological organisms can bedegraded, transformed, and inactivated by the synergic action ofelectrons, holes, hydroxyl radicals, and other oxidant radicals [88].As for the organic contaminants, the photo-generated oxidants likeholes and hydroxyl radicals usually initiate the degradation and fur-ther react with their degradation intermediates, which lead to thefull mineralization of the organics. Some representative studies ofTiO2 photocatalytic transformation are listed in Table 2 accordingto the kind of test substrates.
2.2. Photocatalytic activity of TiO2
TiO2 has two main crystalline phases of rutile and anatase. It isoften said that the latter has higher photocatalytic activities thanthe former probably because of the higher ECB of anatase by ∼0.2 eV(i.e., higher driving force of electron transfer to O2) (Fig. 1). Asdescribed above, the ECB position is very critical for the overallphotocatalysis since it controls the rate-limiting electron transferrate and consequently the charge recombination rate. However, ithas often been reported that the photocatalytic activities of anataseand rutile are greatly affected by the kind of substrates and thatrutile exhibits higher activities than anatase for some reactions[79,89–92]. For example, Ryu and Choi compared eight commercial
TiO2 samples (including anatase, rutile, and the mixed phase) forthe photocatalytic degradation of 19 substrates (phenols, organicacids, amines, chlorohydrocarbons, dyes, inorganic ions, etc.) [79].They observed that the measured photocatalytic activities are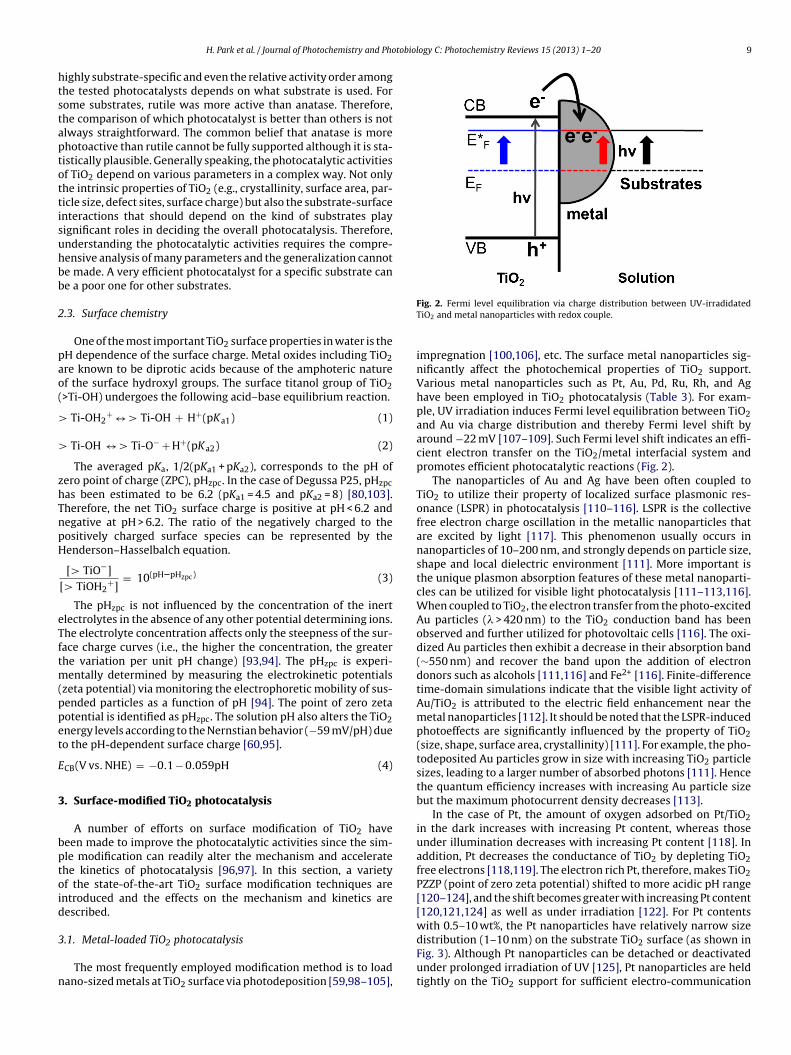
tobiology C: Photochemistry Reviews 15 (2013) 1– 20 9
htstaptottisuhbb
2
pao(
>
>
zhTnpH
eTftm(ppet
E
3
bptoid
3
n
H. Park et al. / Journal of Photochemistry and Pho
ighly substrate-specific and even the relative activity order amonghe tested photocatalysts depends on what substrate is used. Forome substrates, rutile was more active than anatase. Therefore,he comparison of which photocatalyst is better than others is notlways straightforward. The common belief that anatase is morehotoactive than rutile cannot be fully supported although it is sta-istically plausible. Generally speaking, the photocatalytic activitiesf TiO2 depend on various parameters in a complex way. Not onlyhe intrinsic properties of TiO2 (e.g., crystallinity, surface area, par-icle size, defect sites, surface charge) but also the substrate-surfacenteractions that should depend on the kind of substrates playignificant roles in deciding the overall photocatalysis. Therefore,nderstanding the photocatalytic activities requires the compre-ensive analysis of many parameters and the generalization cannote made. A very efficient photocatalyst for a specific substrate cane a poor one for other substrates.
.3. Surface chemistry
One of the most important TiO2 surface properties in water is theH dependence of the surface charge. Metal oxides including TiO2re known to be diprotic acids because of the amphoteric naturef the surface hydroxyl groups. The surface titanol group of TiO2>Ti-OH) undergoes the following acid–base equilibrium reaction.
Ti-OH2+ ↔ > Ti-OH + H+(pKa1) (1)
Ti-OH ↔ > Ti-O− + H+(pKa2) (2)
The averaged pKa, 1/2(pKa1 + pKa2), corresponds to the pH ofero point of charge (ZPC), pHzpc. In the case of Degussa P25, pHzpc
as been estimated to be 6.2 (pKa1 = 4.5 and pKa2 = 8) [80,103].herefore, the net TiO2 surface charge is positive at pH < 6.2 andegative at pH > 6.2. The ratio of the negatively charged to theositively charged surface species can be represented by theenderson–Hasselbalch equation.
[> TiO−][> TiOH2
+]= 10(pH–pHzpc) (3)
The pHzpc is not influenced by the concentration of the inertlectrolytes in the absence of any other potential determining ions.he electrolyte concentration affects only the steepness of the sur-ace charge curves (i.e., the higher the concentration, the greaterhe variation per unit pH change) [93,94]. The pHzpc is experi-
entally determined by measuring the electrokinetic potentialszeta potential) via monitoring the electrophoretic mobility of sus-ended particles as a function of pH [94]. The point of zero zetaotential is identified as pHzpc. The solution pH also alters the TiO2nergy levels according to the Nernstian behavior (−59 mV/pH) dueo the pH-dependent surface charge [60,95].
CB(V vs. NHE) = −0.1 − 0.059pH (4)
. Surface-modified TiO2 photocatalysis
A number of efforts on surface modification of TiO2 haveeen made to improve the photocatalytic activities since the sim-le modification can readily alter the mechanism and acceleratehe kinetics of photocatalysis [96,97]. In this section, a varietyf the state-of-the-art TiO2 surface modification techniques arentroduced and the effects on the mechanism and kinetics areescribed.
.1. Metal-loaded TiO2 photocatalysis
The most frequently employed modification method is to loadano-sized metals at TiO2 surface via photodeposition [59,98–105],
Fig. 2. Fermi level equilibration via charge distribution between UV-irradidatedTiO2 and metal nanoparticles with redox couple.
impregnation [100,106], etc. The surface metal nanoparticles sig-nificantly affect the photochemical properties of TiO2 support.Various metal nanoparticles such as Pt, Au, Pd, Ru, Rh, and Aghave been employed in TiO2 photocatalysis (Table 3). For exam-ple, UV irradiation induces Fermi level equilibration between TiO2and Au via charge distribution and thereby Fermi level shift byaround −22 mV [107–109]. Such Fermi level shift indicates an effi-cient electron transfer on the TiO2/metal interfacial system andpromotes efficient photocatalytic reactions (Fig. 2).
The nanoparticles of Au and Ag have been often coupled toTiO2 to utilize their property of localized surface plasmonic res-onance (LSPR) in photocatalysis [110–116]. LSPR is the collectivefree electron charge oscillation in the metallic nanoparticles thatare excited by light [117]. This phenomenon usually occurs innanoparticles of 10–200 nm, and strongly depends on particle size,shape and local dielectric environment [111]. More important isthe unique plasmon absorption features of these metal nanoparti-cles can be utilized for visible light photocatalysis [111–113,116].When coupled to TiO2, the electron transfer from the photo-excitedAu particles (� > 420 nm) to the TiO2 conduction band has beenobserved and further utilized for photovoltaic cells [116]. The oxi-dized Au particles then exhibit a decrease in their absorption band(∼550 nm) and recover the band upon the addition of electrondonors such as alcohols [111,116] and Fe2+ [116]. Finite-differencetime-domain simulations indicate that the visible light activity ofAu/TiO2 is attributed to the electric field enhancement near themetal nanoparticles [112]. It should be noted that the LSPR-inducedphotoeffects are significantly influenced by the property of TiO2(size, shape, surface area, crystallinity) [111]. For example, the pho-todeposited Au particles grow in size with increasing TiO2 particlesizes, leading to a larger number of absorbed photons [111]. Hencethe quantum efficiency increases with increasing Au particle sizebut the maximum photocurrent density decreases [113].
In the case of Pt, the amount of oxygen adsorbed on Pt/TiO2in the dark increases with increasing Pt content, whereas thoseunder illumination decreases with increasing Pt content [118]. Inaddition, Pt decreases the conductance of TiO2 by depleting TiO2free electrons [118,119]. The electron rich Pt, therefore, makes TiO2PZZP (point of zero zeta potential) shifted to more acidic pH range[120–124], and the shift becomes greater with increasing Pt content[120,121,124] as well as under irradiation [122]. For Pt contentswith 0.5–10 wt%, the Pt nanoparticles have relatively narrow sizedistribution (1–10 nm) on the substrate TiO2 surface (as shown in
Fig. 3). Although Pt nanoparticles can be detached or deactivatedunder prolonged irradiation of UV [125], Pt nanoparticles are heldtightly on the TiO2 support for sufficient electro-communication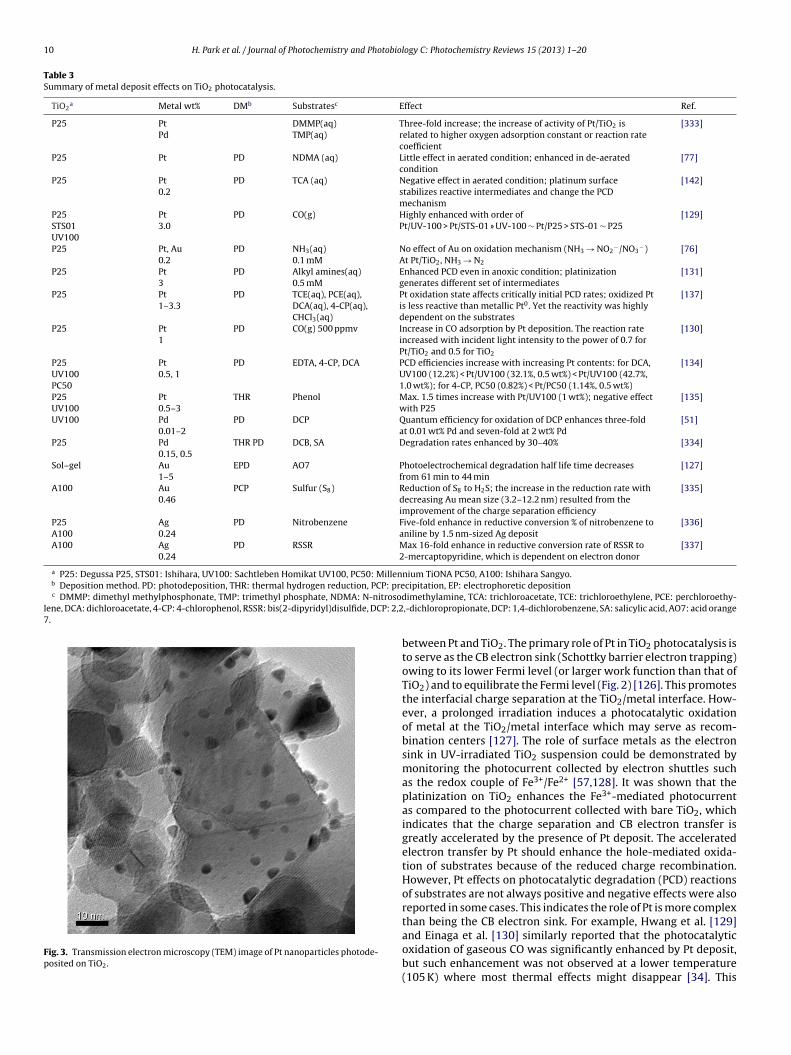
10 H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20
Table 3Summary of metal deposit effects on TiO2 photocatalysis.
TiO2a Metal wt% DMb Substratesc Effect Ref.
P25 PtPd
DMMP(aq)TMP(aq)
Three-fold increase; the increase of activity of Pt/TiO2 isrelated to higher oxygen adsorption constant or reaction ratecoefficient
[333]
P25 Pt PD NDMA (aq) Little effect in aerated condition; enhanced in de-aeratedcondition
[77]
P25 Pt0.2
PD TCA (aq) Negative effect in aerated condition; platinum surfacestabilizes reactive intermediates and change the PCDmechanism
[142]
P25STS01UV100
Pt3.0
PD CO(g) Highly enhanced with order ofPt/UV-100 > Pt/STS-01 » UV-100 ∼ Pt/P25 > STS-01 ∼ P25
[129]
P25 Pt, Au0.2
PD NH3(aq)0.1 mM
No effect of Au on oxidation mechanism (NH3 → NO2−/NO3
−)At Pt/TiO2, NH3 → N2
[76]
P25 Pt3
PD Alkyl amines(aq)0.5 mM
Enhanced PCD even in anoxic condition; platinizationgenerates different set of intermediates
[131]
P25 Pt1–3.3
PD TCE(aq), PCE(aq),DCA(aq), 4-CP(aq),CHCl3(aq)
Pt oxidation state affects critically initial PCD rates; oxidized Ptis less reactive than metallic Pt0. Yet the reactivity was highlydependent on the substrates
[137]
P25 Pt1
PD CO(g) 500 ppmv Increase in CO adsorption by Pt deposition. The reaction rateincreased with incident light intensity to the power of 0.7 forPt/TiO2 and 0.5 for TiO2
[130]
P25UV100PC50
Pt0.5, 1
PD EDTA, 4-CP, DCA PCD efficiencies increase with increasing Pt contents: for DCA,UV100 (12.2%) < Pt/UV100 (32.1%, 0.5 wt%) < Pt/UV100 (42.7%,1.0 wt%); for 4-CP, PC50 (0.82%) < Pt/PC50 (1.14%, 0.5 wt%)
[134]
P25UV100
Pt0.5–3
THR Phenol Max. 1.5 times increase with Pt/UV100 (1 wt%); negative effectwith P25
[135]
UV100 Pd0.01–2
PD DCP Quantum efficiency for oxidation of DCP enhances three-foldat 0.01 wt% Pd and seven-fold at 2 wt% Pd
[51]
P25 Pd0.15, 0.5
THR PD DCB, SA Degradation rates enhanced by 30–40% [334]
Sol–gel Au1–5
EPD AO7 Photoelectrochemical degradation half life time decreasesfrom 61 min to 44 min
[127]
A100 Au0.46
PCP Sulfur (S8) Reduction of S8 to H2S; the increase in the reduction rate withdecreasing Au mean size (3.2–12.2 nm) resulted from theimprovement of the charge separation efficiency
[335]
P25A100
Ag0.24
PD Nitrobenzene Five-fold enhance in reductive conversion % of nitrobenzene toaniline by 1.5 nm-sized Ag deposit
[336]
A100 Ag0.24
PD RSSR Max 16-fold enhance in reductive conversion rate of RSSR to2-mercaptopyridine, which is dependent on electron donor
[337]
a P25: Degussa P25, STS01: Ishihara, UV100: Sachtleben Homikat UV100, PC50: Millennium TiONA PC50, A100: Ishihara Sangyo.b Deposition method. PD: photodeposition, THR: thermal hydrogen reduction, PCP: prec DMMP: dimethyl methylphosphonate, TMP: trimethyl phosphate, NDMA: N-nitrosod
lene, DCA: dichloroacetate, 4-CP: 4-chlorophenol, RSSR: bis(2-dipyridyl)disulfide, DCP: 2,27.
Fig. 3. Transmission electron microscopy (TEM) image of Pt nanoparticles photode-posited on TiO2.
cipitation, EP: electrophoretic depositionimethylamine, TCA: trichloroacetate, TCE: trichloroethylene, PCE: perchloroethy-
,-dichloropropionate, DCP: 1,4-dichlorobenzene, SA: salicylic acid, AO7: acid orange
between Pt and TiO2. The primary role of Pt in TiO2 photocatalysis isto serve as the CB electron sink (Schottky barrier electron trapping)owing to its lower Fermi level (or larger work function than that ofTiO2) and to equilibrate the Fermi level (Fig. 2) [126]. This promotesthe interfacial charge separation at the TiO2/metal interface. How-ever, a prolonged irradiation induces a photocatalytic oxidationof metal at the TiO2/metal interface which may serve as recom-bination centers [127]. The role of surface metals as the electronsink in UV-irradiated TiO2 suspension could be demonstrated bymonitoring the photocurrent collected by electron shuttles suchas the redox couple of Fe3+/Fe2+ [57,128]. It was shown that theplatinization on TiO2 enhances the Fe3+-mediated photocurrentas compared to the photocurrent collected with bare TiO2, whichindicates that the charge separation and CB electron transfer isgreatly accelerated by the presence of Pt deposit. The acceleratedelectron transfer by Pt should enhance the hole-mediated oxida-tion of substrates because of the reduced charge recombination.However, Pt effects on photocatalytic degradation (PCD) reactionsof substrates are not always positive and negative effects were alsoreported in some cases. This indicates the role of Pt is more complexthan being the CB electron sink. For example, Hwang et al. [129]
and Einaga et al. [130] similarly reported that the photocatalyticoxidation of gaseous CO was significantly enhanced by Pt deposit,but such enhancement was not observed at a lower temperature(105 K) where most thermal effects might disappear [34]. This
H. Park et al. / Journal of Photochemistry and Photobiol
Fc
st
srsao[wmacmattPsimmP
aPdrpcswfttoPDtoIoP
aeaTa
ig. 4. Substrate-mediated charge recombination on Pt/TiO2 in the photocatalyticonversion of TCE and phenol.
uggests that the thermal catalytic role of Pt is also important inhe overall photocatalytic behavior of platinized TiO2 [76,84,131].
The reported Pt effects in the PCD of a specific substrate areometimes inconsistent. Zhao et al. [132] reported that the PCDate of Rhodamine-B (RB) dye was three-fold increased by Pt depo-ition, whereas Muradov observed that the platinization did notlter the PCD rate of RB significantly [133]. The Pt-enhanced PCDf phenolic compounds was not observed in several studies as well134–136]. The disagreeing effects of platinization can be relatedith various factors such as the state of Pt (size, oxidation state,orphology) and the specific interaction between the substrate
nd Pt (adsorption, charge transfer). For example, the interfacialharge transfer involved with the intermediates generated on Ptay complicate the overall Pt effect. Any substrate or intermedi-
te that reacts sequentially with an electron and a hole may nullifyhe photocatalytic reaction by serving as an external recombina-ion center. As Fig. 4 shows, quinone compounds generated ont/TiO2 from the reaction of holes with phenolic compounds mayubsequently react with CB electrons to regenerate phenols; thentermediate (e.g., hydroquinone)-mediated charge recombination
ight reduce or nullify the effect of the Pt deposit. Table 3 sum-arizes some selected reports regarding the metal effects on TiO2
CD.The inconsistent and different effects of Pt are likely to be
scribed to different redox behaviors depending on substrates andt oxidation states. It has been shown that the oxidation state of Pteposits is a very important factor in determining the initial PCDates of chlorinated organic compounds. For example, Lee and Choirepared Ptox/TiO2 and Pt0/TiO2 under different photodepositiononditions and compared initial PCD rates for TCE [137]. Despiteimilar Pt content, Ptox/TiO2 has no activity for TCE degradationhereas Pt0/TiO2 is active. In addition, the reactivity of Pt0/TiO2
or TEC is almost the same as the bare TiO2. The insignificant reac-ivity of Ptox/TiO2 for TCE degradation was attributed to role of Ptox
hat acts as a charge recombination center through redox cyclesf adsorbed TCE (Fig. 4). As a result, the literature reports on theCD of TCE on Pt/TiO2 are often inconsistent. Chen et al. [138] andriessen and Grassian [139] reported that the platinization dras-
ically reduced the PCD rate of TCE while Crittenden et al. [140]bserved no significant retardation in PCD of TCE by platinization.n this case, TCE may also induce the external charge recombinationn the platinized TiO2 as illustrated in Fig. 4, which complicates thet effect.
Surface Pt deposits often alter photocatalytic degradation mech-nism of substrates by interfering reaction pathways. As an
− −
xample, NH3, which is oxidized to NO2 and NO3 on bare TiO2nd Au/TiO2, can be converted selectively into N2 on Pt/TiO2 [76].he transition metals like platinum, palladium, and rhodium havestronger affinity for ammonia than gold, silver, and copper [141].
ogy C: Photochemistry Reviews 15 (2013) 1– 20 11
Adsorption energy of atomic nitrogen on noble metals is estimatedthat the Pt surface (−394 kJ/mol) has a moderate affinity for N atomfor the production of dinitrogen, whereas it is too high on Ru andRh or too weak on Au and Ag to produce any active intermediates[141]. This interaction of Pt with lone-pair electron on the N atomalso catalyzes the anoxic degradation of alkylamines, which leadsto the generation of products that are different from those formedon bare TiO2 [131]. In particular, N-alkylated amines (e.g., (CH3)3Nproduced from (CH3)2NH) as well as dealkylated amines are gen-erated in a deaerated Pt/TiO2 suspension. This anoxic N-alkylationpathway is enabled only with neutral alkylamines and not withalkylammonium cations since the ammonium cations do not carrylone-pair electrons on the N atom and do not interact with Pt sur-face. The surface platinization of TiO2 also changes the degradationpathway of trichloroacetate (CCl3CO2
−; TCA). The PCD of TCA isinitiated by the CB electron transfer (reductive pathway) and theresulting dichloroacetate radical (
•CCl2CO2
−) reacts rapidly withdissolved oxygen, leading to a complete degradation with no stableintermediates produced [97,142]. In this regard, dissolved oxygen isrequired essentially for the PCD of TCA and the anoxic degradationpathway is not significant on bare TiO2. However, the anoxic PCD ofTCA becomes significant on Pt/TiO2 since the Pt surface can stabilizethe degradation intermediate, dichlorocarbene (:CCl2), which canbe further degraded without the need of O2. Therefore, the degra-dation of TCA on bare TiO2 requires the presence of O2 while thaton Pt/TiO2 can proceed without O2.
3.2. Doped TiO2 and visible light photocatalytic activity
The primary purposes of doping TiO2 are to retard the fastcharge recombination and enable visible light absorption by creat-ing defect states in the band gap. In the former case, the CB electronsor VB holes are trapped in the defect sites, inhibiting the recombi-nation and enhancing the interfacial charge transfer. In the lattercase, the electronic transitions from VB to defect states (intrabandstates or mid-gap level) or from defect states to CB are allowedunder sub-bandgap irradiation. Dopants are usually classified intometal ions (transition metals and noble metals) and non-metal ions,and the selection of dopant is very crucial in determining overallphotocatalytic activities.
Doping with transition metals such as Fe(III), Ru(III), V(IV),Mo(V), Os(III), Re(V), Rh(III) ions substantially increased photocat-alytic activity for the degradation of CHCl3 under UV irradiation,whereas doping with Co(III) and Al(III) decreased the photoactivity[63,64]. Noble metal ions as dopants have received much less atten-tions as compared to transition metals. As discussed above section,noble metals such as Pt, Au, and Ag are usually deposited on the TiO2surface as a metallic nanoparticle (not as an ionic dopant) to serveas both a CB electron trap and a cocatalyst. There are only a fewreports that studied the effects of noble metal ion dopant on TiO2PCD. For example, Li and Li [143] observed that Au(III)-doped TiO2had visible light activity for methylene blue degradation. Lettmannet al. [144] reported that TiO2 doped with Ru(III), Rh(III), Pt(IV),and Ir(III) exhibited visible light activity for the degradation of4-chlorophenol (4-CP). Burgeth and Kisch also investigated thephotocatalytic degradation of 4-CP under visible irradiation on TiO2modified with chloride complexes of Pt, Ir, Rh, Au, Pd, Co, and Ni[145]; yet their approach is a kind of surface anchoring of the metalcomplexes, not doping into TiO2 lattice. Kim et al. [149] synthe-sized Pt ion-doped TiO2 and Pt0/TiO2 with the same Pt content andcompared them for the analysis of Pt using high-resolution trans-mission electron microscopy and energy-dispersive X-ray analysis;
no Pt signals were seen with Pt ion-doped TiO2 whereas clear Ptsignals were observed with Pt0/TiO2. This is indicative of good dis-persion of Pt dopants in the TiO2 lattice without aggregation in thesurface region. Choi et al. [147,148] also demonstrated the visible
1 tobio
lftc−i
snpicor[wD[fdgNtomorl
ecohceoaatbwotcbomduamcucnd4pTFitCrit
2 H. Park et al. / Journal of Photochemistry and Pho
ight (� > 400 nm) activity of Pt-doped TiO2 (primarily rutile phase)or the degradation of methylene blue, the oxidation of iodide, andhe oxidative degradation of phenol. It was found that the Pt dopingaused the positive shift of TiO2 flat band potential from −0.17 V to0.12 V (vs. NHE) indicating the CB edge position in Pt-doped TiO2
s slightly lowered [149].Doping TiO2 with non-metallic elements has been also exten-
ively studied since Asahi et al. reported the visible light activity ofitrogen-doped TiO2 for the degradation of volatile organic com-ounds [150]. Carbon [151], sulfur [152], and boron [153] doping
n TiO2 have been successively studied for their visible light photo-atalysis. Yet their visible light activities for the degradation ofrganic substances were rather controversial and contradictoryesults were reported in some cases. For example, Gole et al.154] prepared TiO2 − xNx (using alkylammonium as a precursor),hich successfully degraded methylene blue under visible light.espite the same method employed, however, Mrowetz et al.
155] could not observe the visible light activity of TiO2 − xNx
or the degradation of formic acid. The selective reactivity of N-oped TiO2 is ascribed to the limited oxidation power of holesenerated in the mid-gap states (associated with N dopants).akamura et al. [156] raised the similar controversy in a photoelec-
rochemical study. They argued that the photocatalytic oxidationf organic compounds on nitrogen-doped TiO2 under visible illu-ination is mainly induced by surface intermediates of water
xidation or the reductive activation of oxygen, and not by directeactions with holes trapped at the nitrogen-induced mid-gapevel.
Carbon-doped TiO2 (C-TiO2) is also controversial. Since Khant al. reported water splitting with an unprecedent conversion effi-iency of 11% using a chemically modified n-type TiO2 that wasbtained by combusting Ti metal in natural gas flame [157], C-TiO2as been prepared through various synthetic methods includingarbon-mounting [158,159] and carbon-coating [158,160]. Inter-stingly, the state of impurity carbons in the TiO2 lattice has beenften interpreted differently. There are two states of carbon dopant:n anion that substitutes oxygen in the lattice [157,161–163] and
cation that occupies an interstitial lattice site [164–167]. In addi-ion, the oxidation states of carbon dopant are associated with itsonding state: −4 (carbides with Ti–C bond) and +4 (carbonatesith C–O bond) [168]. Flame pyrolysis of Ti metal sheet, annealing
f TiC powders, or ion-assisted electron beam evaporation favourshe formation of Ti–C bond [162,163] while sol–gel processes witharbon precursors and high temperature reactions of TiO2 with car-on precursors form C–O bond (carbonate) [164–167,169]. Thus, itsxidation state seems to be strongly dependent on the preparationethod and condition. Both carbon states may be even co-present
epending on the preparation condition. Although carbon is a ubiq-itous impurity, the addition of external carbon precursors (e.g.,lkylammonium, urea, glucose) was needed to make C-TiO2 inost reported cases. However, recent studies found that C-TiO2
ould be obtained from a conventional sol–gel synthesis withoutsing external carbon precursor [170,171]. In these cases, organicarbons contained in titanium alkoxide precursors were sponta-eously incorporated into the oxide lattice as an impurity dopanturing synthesis. Its photocatalytic activities for the conversion of-CP and iodide were sensitively dependent on calcination tem-erature and were the highest when obtained at 250 ◦C (Fig. 5).he photocurrent collected in the suspension of C-TiO2@250 usinge3+/Fe2+ redox shuttle was also significantly enhanced under vis-ble light, which confirms that the visible light-induced electronransfer occurs on C-TiO2. However, the visible light activity of
-TiO2 was selective. C-TiO2 activated by visible light did noteact with methanol (a common hole scavenger) at all while UV-lluminated C-TiO2 reacted with methanol [171]. This also indicateshat the holes generated in the mid-gap states (associated withlogy C: Photochemistry Reviews 15 (2013) 1– 20
C dopants) are less energetic than the VB holes as in the case ofN-TiO2.
Doped TiO2 with more than two elements has also been stud-ied to further increase the photocatalytic activity under UV andvisible light and to make the doped TiO2 more stable throughcharge compensation. There has been a large number of studiesthat tried various doping combinations of metal/metal (e.g., Sb/Cr[172–174], Pt/Cr [148]), non-metal/non-metal (e.g., N/F [175], N/S[176]), and metal/non-metal pairs (e.g., Pt/N [177]). The combi-natorial method, therefore, has been adopted to rapidly screenthe optimal combinations [178,179]. Choi et al. [148] synthesizedsol–gel derived TiO2 codoped with platinum, chromium, vana-dium, nickel, and found that 0.3 atom% Pt-Cr-TiO2 and 0.3 atom%Cr-V-TiO2 showed the highest visible light photocatalytic activitywith respect to MB degradation and iodide oxidation, respec-tively. However, none of the codoped TiO2 samples were foundto have enhanced photocatalytic activity for phenol degradationwhen compared to their single-doped TiO2 counterparts. The vis-ible light activity of codoped TiO2 is also substrate-specific as theUV-light activity of bare TiO2 is [84]. Therefore, the dopant effectobserved with a specific substrate may not be generalized to othersubstrates. As an example of the metal/non-metal doped TiO2, Kimand Lee [177] prepared Pt-N-TiO2, which exhibited enhanced pho-tocatalytic activities for the degradation of 4-CP under UV andvisible light irradiation. Pt-N-TiO2 also showed higher photocat-alytic activities for the oxidation of gaseous acetaldehyde and thereduction of aqueous Cr(VI) to Cr(III) [180].
3.3. Role of inorganic adsorbates
The inorganic anions adsorbed on photocatalyst surface ofteninfluence the photocatalytic activity significantly. Non-metallicinorganic anions such as fluoride [67,77,181–186], phosphate[187–194], and sulfate [195,196] do not undergo direct photo-catalytic reactions on illuminated semiconductor surface but maychange the photocatalytic activity and mechanism through modi-fying the adsorption of substrates, surface acidity, surface charge,and surface functional groups. For example, carbonate ions affectphotocatalytic water splitting [197,198] and PCD of substrates[199,200] by altering the TiO2 surface charge as well as interactingwith photo-generated oxidants (mostly OH radical). Carbonate ionswere reported to effectively suppress the back reaction of H2 andO2 (i.e., H2O formation) in Pt/TiO2-photocatalyzed water splitting[197]; they also enhanced the adsorption of aniline by ca. 25% andthereby its photodecomposition rate by 1.4 times [200].
Among those inorganic anions as a surface modifier of TiO2,the surface-fluorinated TiO2 (F-TiO2) has been the most intensivelystudied. The surface fluorination is a typical ligand exchange reac-tion between fluoride and hydroxyl groups on TiO2 surface in water(Fig. 6 and reaction (5)).
> Ti-OH + F− ↔ > Ti-F + OH−(pKF = 6.2) (5)
Note that the substitution reaction of TiO2 surface hydroxylgroup (titanol group) by fluoride (or fluoride adsorption) has beenoften adopted for estimating the surface OH site density of metaloxides including TiO2, �-F2O3, �-FeOOH, and Al2O3 [201–203].In the case of TiO2, Torrents and Stone reported that each sub-stituted/adsorbed fluoride occupies 0.33 nm2 on the TiO2 surface(i.e., 3–4 F− ions per 1 nm × 1 nm TiO2 surface) [201]. Vohra et al.calculated TiO2 surface species with varying pH and [NaF] using theMINTEQA2 software and estimated that around 95% of the titanolgroups were displaced with fluoride, predominantly forming >Ti-F
at [NaF] = 10 mM in the pH range of 3–5 [204]. This surface fluori-nation reduced the degree of TiO2 surface charge and shifted thePZZP of TiO2 from pH 6 to less than pH 3 [67]. Since the fluorina-tion is a very mild surface treatment method, neither bulk crystal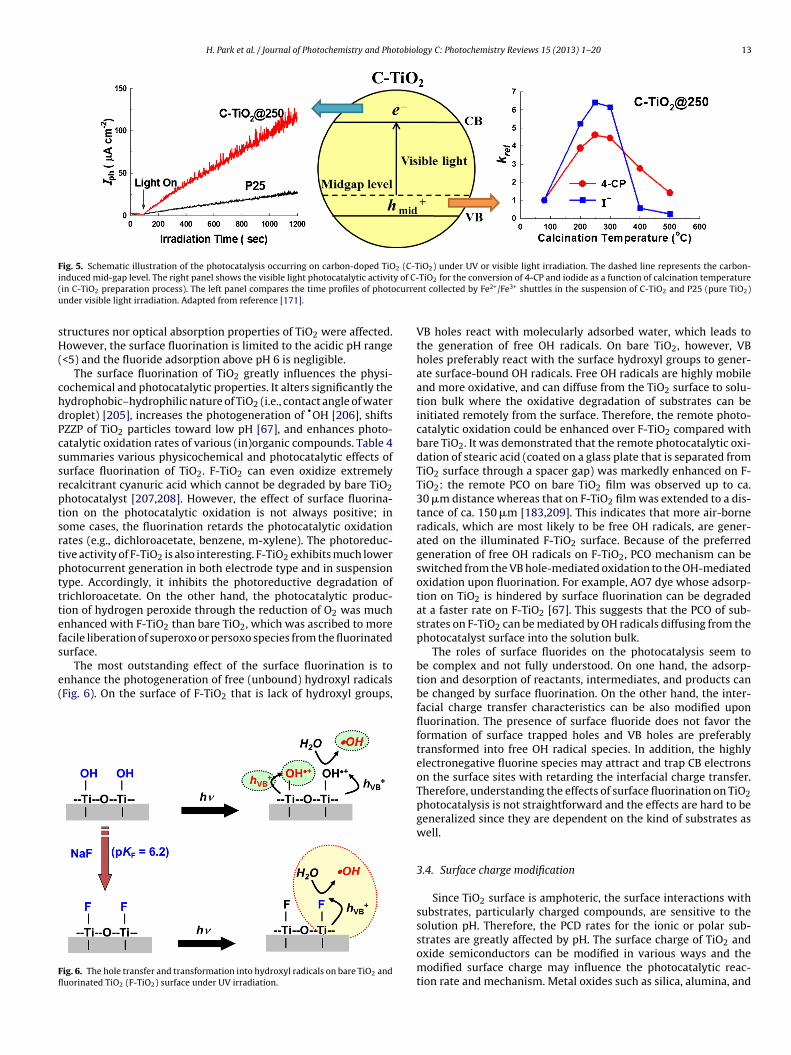
H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20 13
Fig. 5. Schematic illustration of the photocatalysis occurring on carbon-doped TiO2 (C-TiO2) under UV or visible light irradiation. The dashed line represents the carbon-i y of C-( curreu
sH(
chdPcssrptsrtptttefs
e(
Ffl
nduced mid-gap level. The right panel shows the visible light photocatalytic activitin C-TiO2 preparation process). The left panel compares the time profiles of photonder visible light irradiation. Adapted from reference [171].
tructures nor optical absorption properties of TiO2 were affected.owever, the surface fluorination is limited to the acidic pH range
<5) and the fluoride adsorption above pH 6 is negligible.The surface fluorination of TiO2 greatly influences the physi-
ochemical and photocatalytic properties. It alters significantly theydrophobic–hydrophilic nature of TiO2 (i.e., contact angle of waterroplet) [205], increases the photogeneration of
•OH [206], shifts
ZZP of TiO2 particles toward low pH [67], and enhances photo-atalytic oxidation rates of various (in)organic compounds. Table 4ummaries various physicochemical and photocatalytic effects ofurface fluorination of TiO2. F-TiO2 can even oxidize extremelyecalcitrant cyanuric acid which cannot be degraded by bare TiO2hotocatalyst [207,208]. However, the effect of surface fluorina-ion on the photocatalytic oxidation is not always positive; inome cases, the fluorination retards the photocatalytic oxidationates (e.g., dichloroacetate, benzene, m-xylene). The photoreduc-ive activity of F-TiO2 is also interesting. F-TiO2 exhibits much lowerhotocurrent generation in both electrode type and in suspensionype. Accordingly, it inhibits the photoreductive degradation ofrichloroacetate. On the other hand, the photocatalytic produc-ion of hydrogen peroxide through the reduction of O2 was muchnhanced with F-TiO2 than bare TiO2, which was ascribed to moreacile liberation of superoxo or persoxo species from the fluorinatedurface.
The most outstanding effect of the surface fluorination is tonhance the photogeneration of free (unbound) hydroxyl radicalsFig. 6). On the surface of F-TiO2 that is lack of hydroxyl groups,
ig. 6. The hole transfer and transformation into hydroxyl radicals on bare TiO2 anduorinated TiO2 (F-TiO2) surface under UV irradiation.
TiO2 for the conversion of 4-CP and iodide as a function of calcination temperaturent collected by Fe2+/Fe3+ shuttles in the suspension of C-TiO2 and P25 (pure TiO2)
VB holes react with molecularly adsorbed water, which leads tothe generation of free OH radicals. On bare TiO2, however, VBholes preferably react with the surface hydroxyl groups to gener-ate surface-bound OH radicals. Free OH radicals are highly mobileand more oxidative, and can diffuse from the TiO2 surface to solu-tion bulk where the oxidative degradation of substrates can beinitiated remotely from the surface. Therefore, the remote photo-catalytic oxidation could be enhanced over F-TiO2 compared withbare TiO2. It was demonstrated that the remote photocatalytic oxi-dation of stearic acid (coated on a glass plate that is separated fromTiO2 surface through a spacer gap) was markedly enhanced on F-TiO2: the remote PCO on bare TiO2 film was observed up to ca.30 �m distance whereas that on F-TiO2 film was extended to a dis-tance of ca. 150 �m [183,209]. This indicates that more air-borneradicals, which are most likely to be free OH radicals, are gener-ated on the illuminated F-TiO2 surface. Because of the preferredgeneration of free OH radicals on F-TiO2, PCO mechanism can beswitched from the VB hole-mediated oxidation to the OH-mediatedoxidation upon fluorination. For example, AO7 dye whose adsorp-tion on TiO2 is hindered by surface fluorination can be degradedat a faster rate on F-TiO2 [67]. This suggests that the PCO of sub-strates on F-TiO2 can be mediated by OH radicals diffusing from thephotocatalyst surface into the solution bulk.
The roles of surface fluorides on the photocatalysis seem tobe complex and not fully understood. On one hand, the adsorp-tion and desorption of reactants, intermediates, and products canbe changed by surface fluorination. On the other hand, the inter-facial charge transfer characteristics can be also modified uponfluorination. The presence of surface fluoride does not favor theformation of surface trapped holes and VB holes are preferablytransformed into free OH radical species. In addition, the highlyelectronegative fluorine species may attract and trap CB electronson the surface sites with retarding the interfacial charge transfer.Therefore, understanding the effects of surface fluorination on TiO2photocatalysis is not straightforward and the effects are hard to begeneralized since they are dependent on the kind of substrates aswell.
3.4. Surface charge modification
Since TiO2 surface is amphoteric, the surface interactions withsubstrates, particularly charged compounds, are sensitive to thesolution pH. Therefore, the PCD rates for the ionic or polar sub-
strates are greatly affected by pH. The surface charge of TiO2 andoxide semiconductors can be modified in various ways and themodified surface charge may influence the photocatalytic reac-tion rate and mechanism. Metal oxides such as silica, alumina, and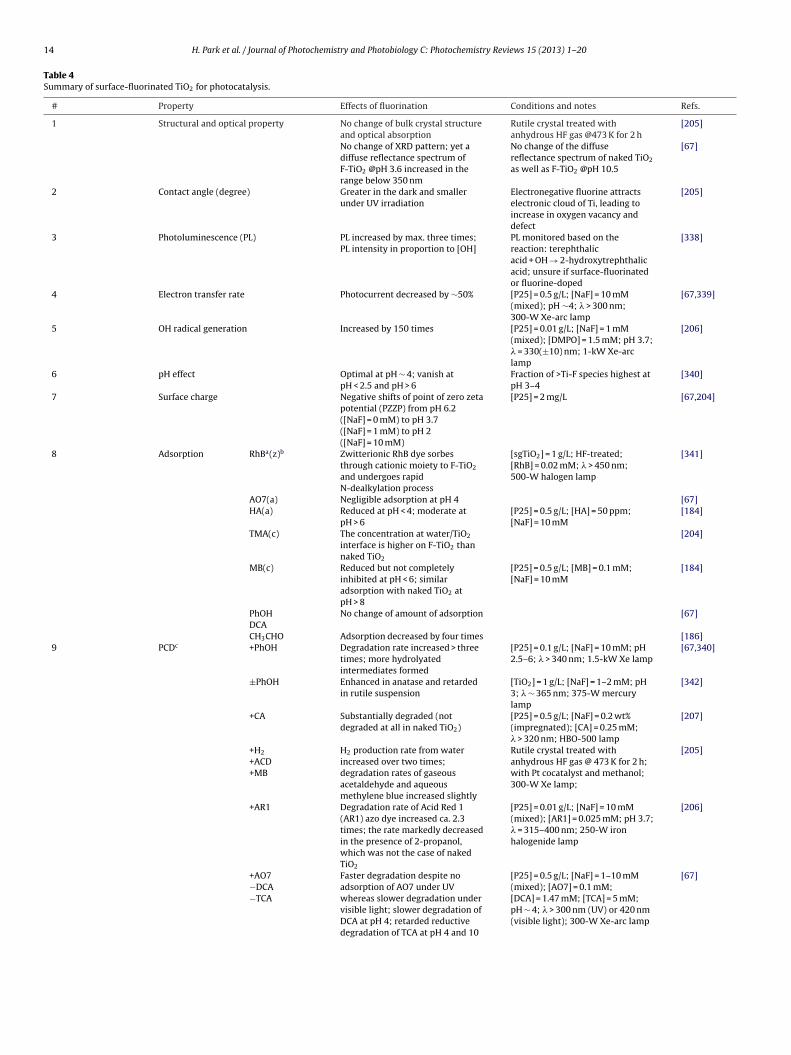
14 H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20
Table 4Summary of surface-fluorinated TiO2 for photocatalysis.
# Property Effects of fluorination Conditions and notes Refs.
1 Structural and optical property No change of bulk crystal structureand optical absorption
Rutile crystal treated withanhydrous HF gas @473 K for 2 h
[205]
No change of XRD pattern; yet adiffuse reflectance spectrum ofF-TiO2 @pH 3.6 increased in therange below 350 nm
No change of the diffusereflectance spectrum of naked TiO2
as well as F-TiO2 @pH 10.5
[67]
2 Contact angle (degree) Greater in the dark and smallerunder UV irradiation
Electronegative fluorine attractselectronic cloud of Ti, leading toincrease in oxygen vacancy anddefect
[205]
3 Photoluminescence (PL) PL increased by max. three times;PL intensity in proportion to [OH]
PL monitored based on thereaction: terephthalicacid + OH → 2-hydroxytrephthalicacid; unsure if surface-fluorinatedor fluorine-doped
[338]
4 Electron transfer rate Photocurrent decreased by ∼50% [P25] = 0.5 g/L; [NaF] = 10 mM(mixed); pH ∼4; � > 300 nm;300-W Xe-arc lamp
[67,339]
5 OH radical generation Increased by 150 times [P25] = 0.01 g/L; [NaF] = 1 mM(mixed); [DMPO] = 1.5 mM; pH 3.7;� = 330(±10) nm; 1-kW Xe-arclamp
[206]
6 pH effect Optimal at pH ∼ 4; vanish atpH < 2.5 and pH > 6
Fraction of >Ti-F species highest atpH 3–4
[340]
7 Surface charge Negative shifts of point of zero zetapotential (PZZP) from pH 6.2([NaF] = 0 mM) to pH 3.7([NaF] = 1 mM) to pH 2([NaF] = 10 mM)
[P25] = 2 mg/L [67,204]
8 Adsorption RhBa(z)b Zwitterionic RhB dye sorbesthrough cationic moiety to F-TiO2
and undergoes rapidN-dealkylation process
[sgTiO2] = 1 g/L; HF-treated;[RhB] = 0.02 mM; � > 450 nm;500-W halogen lamp
[341]
AO7(a) Negligible adsorption at pH 4 [67]HA(a) Reduced at pH < 4; moderate at
pH > 6[P25] = 0.5 g/L; [HA] = 50 ppm;[NaF] = 10 mM
[184]
TMA(c) The concentration at water/TiO2
interface is higher on F-TiO2 thannaked TiO2
[204]
MB(c) Reduced but not completelyinhibited at pH < 6; similaradsorption with naked TiO2 atpH > 8
[P25] = 0.5 g/L; [MB] = 0.1 mM;[NaF] = 10 mM
[184]
PhOHDCA
No change of amount of adsorption [67]
CH3CHO Adsorption decreased by four times [186]9 PCDc +PhOH Degradation rate increased > three
times; more hydrolyatedintermediates formed
[P25] = 0.1 g/L; [NaF] = 10 mM; pH2.5–6; � > 340 nm; 1.5-kW Xe lamp
[67,340]
±PhOH Enhanced in anatase and retardedin rutile suspension
[TiO2] = 1 g/L; [NaF] = 1–2 mM; pH3; � ∼ 365 nm; 375-W mercurylamp
[342]
+CA Substantially degraded (notdegraded at all in naked TiO2)
[P25] = 0.5 g/L; [NaF] = 0.2 wt%(impregnated); [CA] = 0.25 mM;� > 320 nm; HBO-500 lamp
[207]
+H2
+ACD+MB
H2 production rate from waterincreased over two times;degradation rates of gaseousacetaldehyde and aqueousmethylene blue increased slightly
Rutile crystal treated withanhydrous HF gas @ 473 K for 2 h;with Pt cocatalyst and methanol;300-W Xe lamp;
[205]
+AR1 Degradation rate of Acid Red 1(AR1) azo dye increased ca. 2.3times; the rate markedly decreasedin the presence of 2-propanol,which was not the case of nakedTiO2
[P25] = 0.01 g/L; [NaF] = 10 mM(mixed); [AR1] = 0.025 mM; pH 3.7;� = 315–400 nm; 250-W ironhalogenide lamp
[206]
+AO7−DCA−TCA
Faster degradation despite noadsorption of AO7 under UVwhereas slower degradation undervisible light; slower degradation ofDCA at pH 4; retarded reductivedegradation of TCA at pH 4 and 10
[P25] = 0.5 g/L; [NaF] = 1–10 mM(mixed); [AO7] = 0.1 mM;[DCA] = 1.47 mM; [TCA] = 5 mM;pH ∼ 4; � > 300 nm (UV) or 420 nm(visible light); 300-W Xe-arc lamp
[67]

H. Park et al. / Journal of Photochemistry and Photobiology C: Photochemistry Reviews 15 (2013) 1– 20 15
Table 4 (Continued)
# Property Effects of fluorination Conditions and notes Refs.
�Toluene−Benzene−Xylene
No change of toluene degradationrate; degradation rates of benzeneand m-xylene decreased
HF-treated P25; 100-W blacklight [343]
±TMA Higher degradation rates at pH 5–7and lower rates at pH < 5 andpH > 7 for TMA ion
[P25] = 0.5 g/L; [NaF] = 5–20 mM;[TMA] = 0.1 mM; � > 300 nm;300-W Xe-arc lamp
[185]
+CH3CHO Increased by 1.5–2.5 times despitemuch lower adsorption
[186]
10 H2O2 production Highly enhanced; dependent onpH and hole scavengers; noformation without fluoride, holescavengers, and dissolved oxygen;surface superoxo or peroxo speciesreadily liberated from ≡Ti-F
[P25] = 0.5 g/L; [HCOOH] = 10 m;[HF] + [F−] = 10 mM; air-saturated;40-W fluorescent lamp
[344,345]
11 Principle mechanism Reaction switch from directelectron (DET) transfer to OHradical-mediated (OH) process
For phenol, DET 90% and OH 10% innaked TiO2, whereas OH ∼ 100% inF-TiO2
[67,340]
Mobile, free OH radical generatedand diffused
OH radicals traveled as far as∼150 �m
[183]
Singlet oxygen-mediated PCD No measurement of singletoxygen; simply postulated themechanism from a similar reaction
[207]
Fluoride in the Helmholtz layersenhances desorption ofsurface-bound OH radicals througha F–H bond
[342]
a RhB: rhodamine-B, AO7: acid oragne7, HA: humic acid, TMA: tetramethylammonium, MB: methylele blue, PhOH: phenol, DCA: dichloroacetic acid, CA: cyanuric acid,A
zi6paepdtwtadSfeaceddPJs[ste
cctswtwt
CD: acetaldehyde, AR1: acid red 1, TCA: trichloroacetic acid.b (a): anionic, (c): cationic, (z): zwitterionicc + PCD enhanced, − PCD decreased, �: PCD not changed significantly
irconia are the common inorganic surface charge modifiers. Load-ng silica on TiO2 particles shifts PZZP of TiO2 to a lower value (pH
→ 3) and affects the electrophoretic mobilities of suspended TiO2article [185,210,211]. Thus silica-loaded TiO2 particles carry neg-tive surface charge in a wider pH range (pH > 3) and have strongerlectrostatic interaction with cationic substrates. The enhancedhotocatalytic activities of silica-loaded TiO2 have been frequentlyemonstrated for various cationic substrates. Vohra et al. reportedhat photocatalytic degradation of (CH3)4N+ was highly enhancedhen TiO2 was loaded with 18 wt% silica [210]. Chun et al. found
hat the cationic dyes have different adsorption modes on SiO2/TiO2s compared to the similarly structured anionic dyes, and the degra-ation products were changed [211]. Anderson and Bard preparediO2/TiO2 (30/70) via a sol–gel method and achieved a three-old enhanced PCD of cationic rhodamine-6G dye [212,213]. Tadat al. coated TiO2 surface with SiOx monolayer, which exhibited
three-fold activity for the photocatalytic degradation of cationicetylpyridinium bromide surfactant [214]. On the other hand, theffects of metal oxide-loading on TiO2 for the photocatalytic degra-ation of anionic and neutral substrates are often negative and varyepending on the cases. Vohra and Tanaka did not obtain enhancedCD for acetate (anion) and phenol (neutral) [185]. Conversely,ung and Park reported that PCD of trichloroethylene, a neutralubstrate, was accelerated on SiO2/TiO2 [215]. Anderson and Bard212,213] reported that SiO2/TiO2 and Al2O3/TiO2 photocatalystshowed reduced PCD rates for phenol and salicylic acid as comparedo bare TiO2. Fu et al. observed that the photocatalytic oxidation ofthylene was enhanced by loading silica and zirconia on TiO2 [216].
Zeolites can be also coupled with TiO2 to modify the surfaceharge of the composites because the aluminosilicate frameworkarries the negative charge (balanced by counter cations present inhe zeolite pores) resulting from the substitution of Al3+ into theilica structure. Zhang et al. [217] encapsulated the titania clusters
ithin the cavities of FAU-type zeolites and investigated their pho-ocatalytic activities for the degradation of charged substrates inater. Owing to the negative charges of the zeolite framework,
he degradation rates for the cationic substrates on the hybrid
photocatalysts were markedly higher than those for the anionicsubstrates. The combination of the photocatalytic activity of tita-nia clusters encapsulated in zeolites and the negative charges ofthe zeolite framework favors the selective degradation of cationicsubstrates in water. Incidentally, TiO2 loaded on silica mesoporoussupport of MCM-41 also exhibited a similar charge modificationeffect [218]. The acidic silicic framework of MCM-41 provides theenvironment of the negative surface charge on the hybrid cata-lyst. As a result, the adsorption of cationic substrate, (CH3)4N+, onTiO2/MCM-41 was much higher than that on the bare TiO2 andits photocatalytic degradation was highly enhanced on the hybridcatalyst over a wide range of pH.
The surface charge of semiconductor photocatalysts can be alsomodified by coating with ionic polymer. Nafion, an anionic perflu-orinated polymer with sulfonate groups, is an ideal resin for thispurpose because it is chemically and photochemically inert. Thenafion-coated TiO2 (Nf/TiO2) is expected to be stable under UV irra-diation because the perfluorinated carbon backbone is unreactivewith OH radicals [219,220]. The cationic species can be easily ion-exchanged into the nafion resin (see Fig. 7). As a result, the enhancedPCDs of cationic substrates on Nf/TiO2 were observed for tetram-ethylammonium under UV light and cationic dyes (methylene blueand rhodamine-B) under visible light [219,220]. In addition, it wasfound that the sensitized degradation of RhB followed a differ-ent path when the surface of TiO2 was coated with Nafion. TheN-de-ethylation of RhB that leads to the generation of rhodamine-110 was a prevailing path with Nf/TiO2, whereas the cleavage ofthe chromophoric ring structure was dominant with pure TiO2.The presence of Nafion layer may change the reaction mechanismas well. On the other hand, Nf/TiO2 was also utilized as a sup-port of cationic dye sensitizers that anchors ruthenium complexes(Ru(bpy)3
2+) through ion-exchange [200].
3.5. Dye-sensitization of TiO2
Coupling of (in)organic dyes to TiO2 surface has attractedgreat attention as a sensitization method for visible light driven
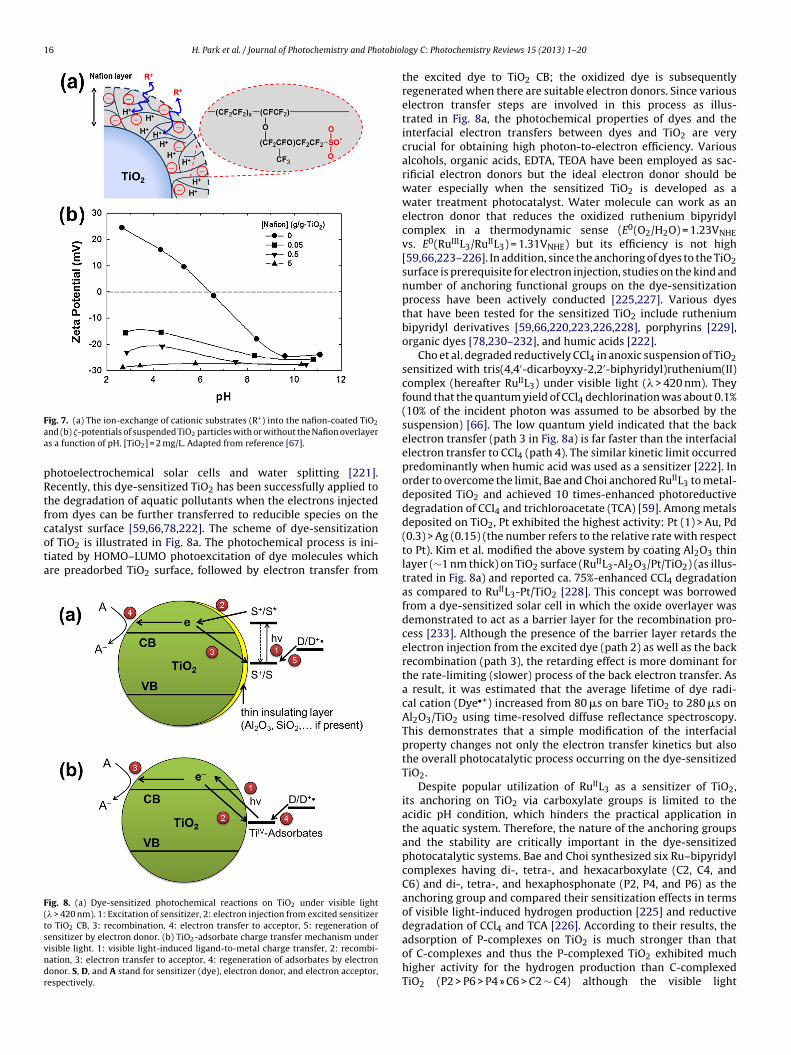
16 H. Park et al. / Journal of Photochemistry and Photobio
Faa
pRtfcota
F(tsvndr
ig. 7. (a) The ion-exchange of cationic substrates (R+) into the nafion-coated TiO2
nd (b) �-potentials of suspended TiO2 particles with or without the Nafion overlayers a function of pH. [TiO2] = 2 mg/L. Adapted from reference [67].
hotoelectrochemical solar cells and water splitting [221].ecently, this dye-sensitized TiO2 has been successfully applied tohe degradation of aquatic pollutants when the electrons injectedrom dyes can be further transferred to reducible species on the
atalyst surface [59,66,78,222]. The scheme of dye-sensitizationf TiO2 is illustrated in Fig. 8a. The photochemical process is ini-iated by HOMO–LUMO photoexcitation of dye molecules whichre preadorbed TiO2 surface, followed by electron transfer fromig. 8. (a) Dye-sensitized photochemical reactions on TiO2 under visible light� > 420 nm). 1: Excitation of sensitizer, 2: electron injection from excited sensitizero TiO2 CB, 3: recombination, 4: electron transfer to acceptor, 5: regeneration ofensitizer by electron donor. (b) TiO2-adsorbate charge transfer mechanism underisible light. 1: visible light-induced ligand-to-metal charge transfer, 2: recombi-ation, 3: electron transfer to acceptor, 4: regeneration of adsorbates by electrononor. S, D, and A stand for sensitizer (dye), electron donor, and electron acceptor,espectively.
logy C: Photochemistry Reviews 15 (2013) 1– 20
the excited dye to TiO2 CB; the oxidized dye is subsequentlyregenerated when there are suitable electron donors. Since variouselectron transfer steps are involved in this process as illus-trated in Fig. 8a, the photochemical properties of dyes and theinterfacial electron transfers between dyes and TiO2 are verycrucial for obtaining high photon-to-electron efficiency. Variousalcohols, organic acids, EDTA, TEOA have been employed as sac-rificial electron donors but the ideal electron donor should bewater especially when the sensitized TiO2 is developed as awater treatment photocatalyst. Water molecule can work as anelectron donor that reduces the oxidized ruthenium bipyridylcomplex in a thermodynamic sense (E0(O2/H2O) = 1.23VNHEvs. E0(RuIIIL3/RuIIL3) = 1.31VNHE) but its efficiency is not high[59,66,223–226]. In addition, since the anchoring of dyes to the TiO2surface is prerequisite for electron injection, studies on the kind andnumber of anchoring functional groups on the dye-sensitizationprocess have been actively conducted [225,227]. Various dyesthat have been tested for the sensitized TiO2 include rutheniumbipyridyl derivatives [59,66,220,223,226,228], porphyrins [229],organic dyes [78,230–232], and humic acids [222].
Cho et al. degraded reductively CCl4 in anoxic suspension of TiO2sensitized with tris(4,4′-dicarboyxy-2,2′-biphyridyl)ruthenium(II)complex (hereafter RuIIL3) under visible light (� > 420 nm). Theyfound that the quantum yield of CCl4 dechlorination was about 0.1%(10% of the incident photon was assumed to be absorbed by thesuspension) [66]. The low quantum yield indicated that the backelectron transfer (path 3 in Fig. 8a) is far faster than the interfacialelectron transfer to CCl4 (path 4). The similar kinetic limit occurredpredominantly when humic acid was used as a sensitizer [222]. Inorder to overcome the limit, Bae and Choi anchored RuIIL3 to metal-deposited TiO2 and achieved 10 times-enhanced photoreductivedegradation of CCl4 and trichloroacetate (TCA) [59]. Among metalsdeposited on TiO2, Pt exhibited the highest activity: Pt (1) > Au, Pd(0.3) > Ag (0.15) (the number refers to the relative rate with respectto Pt). Kim et al. modified the above system by coating Al2O3 thinlayer (∼1 nm thick) on TiO2 surface (RuIIL3-Al2O3/Pt/TiO2) (as illus-trated in Fig. 8a) and reported ca. 75%-enhanced CCl4 degradationas compared to RuIIL3-Pt/TiO2 [228]. This concept was borrowedfrom a dye-sensitized solar cell in which the oxide overlayer wasdemonstrated to act as a barrier layer for the recombination pro-cess [233]. Although the presence of the barrier layer retards theelectron injection from the excited dye (path 2) as well as the backrecombination (path 3), the retarding effect is more dominant forthe rate-limiting (slower) process of the back electron transfer. Asa result, it was estimated that the average lifetime of dye radi-cal cation (Dye•+) increased from 80 �s on bare TiO2 to 280 �s onAl2O3/TiO2 using time-resolved diffuse reflectance spectroscopy.This demonstrates that a simple modification of the interfacialproperty changes not only the electron transfer kinetics but alsothe overall photocatalytic process occurring on the dye-sensitizedTiO2.
Despite popular utilization of RuIIL3 as a sensitizer of TiO2,its anchoring on TiO2 via carboxylate groups is limited to theacidic pH condition, which hinders the practical application inthe aquatic system. Therefore, the nature of the anchoring groupsand the stability are critically important in the dye-sensitizedphotocatalytic systems. Bae and Choi synthesized six Ru–bipyridylcomplexes having di-, tetra-, and hexacarboxylate (C2, C4, andC6) and di-, tetra-, and hexaphosphonate (P2, P4, and P6) as theanchoring group and compared their sensitization effects in termsof visible light-induced hydrogen production [225] and reductivedegradation of CCl4 and TCA [226]. According to their results, the
adsorption of P-complexes on TiO2 is much stronger than thatof C-complexes and thus the P-complexed TiO2 exhibited muchhigher activity for the hydrogen production than C-complexedTiO2 (P2 > P6 > P4 » C6 > C2 ∼ C4) although the visible light
tobiol
aPT
drcdadl
D
m
gttRsmStvsC[d
3
ddtVt
F
ubAofcwcelbccavd[ipseme(
H. Park et al. / Journal of Photochemistry and Pho
bsorbing capabilities are comparable among C- and P-complexes.2 also showed the highest reductive degradation for CCl4 andCA.
The dye-sensitization can be also applied for the self-egradation of dyes and this process can be combined with otheredox processes of inorganic species. For example, the reductiveonversion of heavy metal ions can be coupled with the sensitizedye degradation on TiO2 under visible light irradiation (reactions (6nd 7)). Kyung et al. attempted the simultaneous conversion (oxi-ation of AO7 and Rh-B dyes; reduction of Cr(VI) and Ag(I)) in visible
ight (� > 420 nm)-irradiated TiO2 suspension [78].
ye∗-TiO2 → Dye+•-TiO2 + ecb− (6)
ecb− + Mn+ → M(n − m)+ (7)
The photocatalytic conversion of binary mixtures was syner-istically enhanced in comparison to that of single substrate, andhis synergistic effect was most outstanding in TiO2/Cr(VI)/AO7 sys-em. In addition, the visible light irradiation in TiO2/Ag(I)/AO7 (orh-B) induced the generation of Ag particles via the reduction ofilver ions. As a result, the simultaneous removal of dyes and toxicetal ions can be achieved in the illuminated suspension of TiO2.
ince real wastewaters often contain dye and heavy metal ionsogether, their synergic interaction during the photocatalytic con-ersion process is an interesting issue to be investigated. Chen et al.tudied the effect of transition metal ions (Cu2+, Fe3+, Zn2+, Al3+, andd2+) on the photodegradation of dyes on TiO2 under visible light234]. They found that Cu2+ and Fe3+ influence negatively on theye degradation whereas other metal ions have a minor effect.
.6. Charge transfer complexation on TiO2
Another mechanism of sensitizing TiO2 by visible light is theirect charge transfer between surface adsorbate and semicon-uctor support, where the electron is photo-excited directly fromhe ground-state adsorbate to the semiconductor CB (see Fig. 8b).isible light-irradiated Fe(CN)6
4−/TiO2 system exhibits this chargeransfer mechanism [235].
e(CN)64−/TiO2 + h�(visible) → Fe(CN)6
3−/TiO2 + ecb− (8)
The formation of the charge transfer complex on the TiO2 surfacesually accompanies the appearance of a visible light absorptionand, which is not seen in either the adsorbate or TiO2 alone.romatic compounds with hydroxyl or carboxyl anchoring groupn TiO2 are the common examples. For example, the complexormation between catechol and TiO2 creates a broad new bandentered at ca. 390 nm, which is extended into the visible regionith the onset at ca. 600 nm [236]. The visible light excitation
auses ligand-to-metal charge transfer (LMCT), inducing the directlectron transfer from the organic adsorbates (having no visibleight absorption as an isolated molecule) into TiO2 CB. These adsor-ates include hydroxylated biphenyls [237], aromatic sulfides witharboxyl groups [238,239], substrates with enediol moieties (e.g.,atechol, ascorbic acid), chlorophenols [240,241], and NH3 [242]. As
result of such LMCT on TiO2 surface, pure TiO2 sometimes exhibitsisible light activity for the degradation of organic substrates thato not absorb visible light at all. For example, phenol and 4-CP243], EDTA and formate [244] could be successfully degradedn visible light illuminated (� > 420 nm) aqueous suspension ofure TiO2, which was ascribed to the LMCT between the adsorbedubstrates and TiO2. The decomposition of gaseous NH3 on TiO2
ven at � ∼ 460 nm can be also explained in terms of the LMCTechanism (“in-situ doping” expressed in another way), i.e., directlectron transfer from donor level derived from adsorbed amidoNH2) to the TiO2 CB [242]. Such visible light-induced LMCT can be
ogy C: Photochemistry Reviews 15 (2013) 1– 20 17
applied to the development of visible light active photocatalysts.Recently, Park et al. reported a novel case of C60-based sensitizedphotocatalysis that works through the CT mechanism [245]. In theirwork, a water-soluble fullerol (C60(OH)x) that was anchored to TiO2surface activated TiO2 under visible light irradiation via the surface-complex CT mechanism, which is largely absent in the C60/TiO2system. A theoretical calculation for the electronic energy statesin the (TiO2 cluster + fullerol) model proposed that the photoex-citation can occur from the fullerol HOMO to TiO2 cluster undervisible light irradiation. The absorption spectra of the fullerol rad-ical cations (fullerol
•+) could be obtained by transient absorptionspectroscopic measurement, which supports the LMCT mechanism.
4. Conclusions
Various surface modification methods of TiO2 particles andtheir effects on environmental photocatalytic applications arebriefly reviewed. Although pure TiO2 is a popular photocatalystfor many reasons, it suffers from the low photoefficiency and thelack of visible light activity that hinder its practical applications.The surface-modified TiO2 nanoparticles have been continuouslydeveloped to overcome the demerits for many years. In particular,the hybridization with various emerging nanomaterials (e.g., car-bon nanotubes, fullerenes, graphene, quantum dots, nanoporousmaterials) is being intensively investigated. Each modificationmethod influences the photocatalytic activity and mechanism ina way different from others. However, the effects of the surfacemodification are often inconsistent and even contradictory resultsare reported for the same modification method employed. Thisis largely because the activities of modified TiO2 nanoparticlesdepend on many parameters such as the preparation conditions,the properties of reaction medium, and the kind of substrates.Therefore, the overall photocatalytic behaviors of a specific mod-ified photocatalyst should be understood with a broader view. Inmany cases, the effects of modified photocatalyst are difficult tobe generalized and the optimal conditions of each modificationshould be sought on the basis of case-by-case approach. A bettermodified photocatalyst for one substrate could be worse for oth-ers. Therefore, the best modification method of TiO2 for a specificapplication should be carefully sought based on the comprehensiveunderstanding.
Acknowledgments
This work was supported by KOSEF NRL program (No.R0A-2008-000-20068-0), the KOSEF EPB center (Grant No. R11-2008-052-02002), the Global Frontier R&D Program on Centerfor Multiscale Energy System (2011-0031571), the Basic Sci-ence Research Program (No. 2012R1A2A2A01004517), and KCAP(Sogang Univ.) funded by MEST through NRF (NRF-2011-C1AAA001-2011-0030278).
References
[1] M.A. Shannon, P.W. Bohn, M. Elimelech, J.G. Georgiadis, B.J. Marinas, A.M.Mayes, Nature 452 (2008) 301–310.
[2] R.P. Schwarzenbach, B.I. Escher, K. Fenner, T.B. Hofstetter, C.A. Johnson, U. vonGunten, B. Wehrli, Science 313 (2006) 1072–1077.
[3] D.C.G. Muir, P.H. Howard, Environ. Sci. Technol. 40 (2006) 7157–7166.[4] M. Pera-Titus, V. Garcia-Molina, M.A. Banos, J. Gimenez, S. Esplugas, Appl.
Catal. B 47 (2004) 219–256.[5] K. Ikehata, M.G. El-Din, Ozone-Sci. Eng. 27 (2005) 173–202.[6] K. Ikehata, M.G. El-Din, Ozone-Sci. Eng. 27 (2005) 83–114.[7] F. Wang, D.W. Smith, M.G. El-Din, J. Environ. Eng. Sci. 2 (2003) 413–427.
[8] J.M. Britto, M.D. Rangel, Quim. Nova 31 (2008) 114–122.[9] S. Esplugas, D.M. Bila, L.G.T. Krause, M. Dezotti, J. Hazard. Mater. 149 (2007)631–642.[10] K. Ikehata, N.J. Naghashkar, M.G. Ei-Din, Ozone-Sci. Eng. 28 (2006) 353–414.[11] K. Ikehata, M.G. El-Din, J. Environ. Eng. Sci. 5 (2006) 81–135.

1 tobio
8 H. Park et al. / Journal of Photochemistry and Pho[12] J.J. Pignatello, E. Oliveros, A. MacKay, Crit. Rev. Environ. Sci. Technol. 36 (2006)1–84.
[13] E. Neyens, J. Baeyens, J. Hazard. Mater. 98 (2003) 33–50.[14] R. Aplin, T.D. Waite, Water Sci. Technol. 42 (2000) 345–354.[15] K. Vinodgopal, J. Peller, Res. Chem. Intermed. 29 (2003) 307–316.[16] H. Destaillats, T.M. Lesko, M. Knowlton, H. Wallace, M.R. Hoffmann, Ind. Eng.
Chem. Res. 40 (2001) 3855–3860.[17] H.M. Hung, J.W. Kang, M.R. Hoffmann, Water Environ. Res. 74 (2002)
545–556.[18] H. Park, C.D. Vecitis, W. Choi, O. Weres, M.R. Hoffmann, J. Phys. Chem. C 112
(2008) 885–889.[19] H. Park, C.D. Vecitis, M.R. Hoffmann, J. Phys. Chem. A 112 (2008) 7616–7626.[20] H. Park, C.D. Vecitis, M.R. Hoffmann, J. Phys. Chem. C 113 (2009) 7935–7945.[21] J. Kim, W.J.K. Choi, J. Choi, M.R. Hoffmann, H. Park, Catal. Today (2012),
http://dx.doi.org/10.1016/j.cattod.2012.02.009.[22] H. Park, A. Bak, A.Y. Ahn, J. Choi, M.R. Hoffmann, J. Hazard. Mater. 211–212
(2012) 47–54.[23] S.Y. Yang, Y.S. Choo, S. Kim, S.K. Lim, J. Lee, H. Park, Appl. Catal. B 111–112
(2012) 317–325.[24] S. Kim, S.K. Choi, B.Y. Yoon, S.K. Lim, H. Park, Appl. Catal. B 97 (2010) 135–141.[25] I. Gultekin, N.H. Ince, J. Environ. Manage. 85 (2007) 816–832.[26] K. Demeestere, J. Dewulf, H. Van Langenhove, Crit. Rev. Environ. Sci. Technol.
37 (2007) 489–538.[27] M.R. Hoffmann, S.T. Martin, W. Choi, D.W. Bahnemann, Chem. Rev. 95 (1995)
69–96.[28] G.V. Buxton, C.L. Greenstock, W.P. Helman, A.B. Ross, J. Phys. Chem. Ref. Data
17 (1988) 513–886.[29] Z.H. Liu, Y. Kanjo, S. Mizutani, Sci. Total Environ. 407 (2009) 731–748.[30] R. Bauer, H. Fallmann, Res. Chem. Intermed. 23 (1997) 341–354.[31] O.A.H. Jones, P.G. Green, N. Voulvoulis, J.N. Lester, Environ. Sci. Technol. 41
(2007) 5085–5089.[32] I. Munoz, J. Rieradevall, F. Torrades, J. Peral, X. Domenech, Sol. Energy 79
(2005) 369–375.[33] D.B. Henschel, J. Air Waste Manag. Assoc. 48 (1998) 985–994.[34] A. Linsebigler, C. Rusu, J.T. Yates Jr., J. Am. Chem. Soc. 118 (1996) 5284.[35] M.A. Fox, Acc. Chem. Res. 16 (1983) 314–321.[36] A. Mills, R.H. Davies, D. Worsley, Chem. Soc. Rev. 22 (1993) 417–425.[37] D.F. Ollis, H. Al-Ekabi (Eds.), Photocatalytic Purification and Treatment of
Water and Air, Elsevier, Amsterdam, 1993.[38] M. Schiavello (Ed.), Photocatalysis and Environment: Trends and Applications,
Kluwer Academica Publishers, Dordrecht, 1988.[39] M.A. Fox, M.T. Dulay, Chem. Rev. 93 (1993) 341–357.[40] D.M. Blake, Bibliography of Work on the Photocatalytic Removal of Hazardous
Compounds from Water and Air, National Renewable Energy Laboratory,2001.
[41] T.E. Agustina, H.M. Ang, V.K. Vareek, J. Photochem. Photobiol. C 6 (2005)264–273.
[42] V. Augugliaro, M. Litter, L. Palmisano, J. Soria, J. Photochem. Photobiol. C 7(2006) 127–144.
[43] W. Choi, Catal. Surv. Asia 10 (2006) 16–28.[44] M.J. Esswein, D.G. Nocera, Chem. Rev. 107 (2007) 4022–4047.[45] M. Fagnoni, D. Dondi, D. Ravelli, A. Albini, Chem. Rev. 107 (2007) 2725–2756.[46] A. Fujishima, X.T. Zhang, D.A. Tryk, Surf. Sci. Rep. 63 (2008) 515–582.[47] A. Maldotti, A. Molinari, R. Amadelli, Chem. Rev. 102 (2002) 3811–3836.[48] A.J. Cowan, J.W. Tang, W.H. Leng, J.R. Durrant, D.R. Klug, J. Phys. Chem. C 114
(2010) 4208–4214.[49] H. Gerischer, A. Heller, J. Phys. Chem. 95 (1991) 5261–5267.[50] B. Ohtani, Chem. Lett. 37 (2008) 217–229.[51] C.M. Wang, A. Heller, H. Gerischer, J. Am. Chem. Soc. 114 (1992) 5230–5234.[52] H. Gerischer, A. Heller, J. Electrochem. Soc. 139 (1992) 113–118.[53] J. Kim, W. Choi, Environ. Sci. Technol. 45 (2011) 3183–3184.[54] J. Kim, C.W. Lee, W. Choi, Environ. Sci. Technol. 44 (2010) 6849–6854.[55] H. Park, A. Bak, T.H. Jeon, S. Kim, W. Choi, Appl. Catal. B 115–116 (2012)
74–80.[56] T.H. Jeon, W. Choi, H. Park, J. Phys. Chem. C 115 (2011) 7134–7142.[57] H. Park, W. Choi, J. Phys. Chem. B 107 (2003) 3885–3890.[58] R. Salvador, J. Phys. Chem. C 111 (2007) 17038–17043.[59] E. Bae, W. Choi, Environ. Sci. Technol. 37 (2003) 147–152.[60] W. Choi, M.R. Hoffmann, Environ. Sci. Technol. 29 (1995) 1646–1654.[61] W. Choi, M.R. Hoffmann, J. Phys. Chem. 100 (1996) 2161–2169.[62] W. Choi, M.R. Hoffmann, Environ. Sci. Technol. 31 (1997) 89–95.[63] W. Choi, A. Termin, M.R. Hoffmann, J. Phys. Chem. 98 (1994) 13669–13679.[64] W. Choi, A. Termin, M.R. Hoffmann, Angew. Chem. Int. Ed. Engl. 33 (1994)
1091–1092.[65] Y. Cho, H. Kyung, W. Choi, Appl. Catal. B 52 (2004) 23–32.[66] Y. Cho, W. Choi, C.H. Lee, T. Hyeon, H.I. Lee, Environ. Sci. Technol. 35 (2001)
966–970.[67] H. Park, W. Choi, J. Phys. Chem. B 108 (2004) 4086–4093.[68] C.C. Chen, C.S. Lu, H.J. Fan, W.H. Chung, J.L. Jan, H.D. Lin, W.Y. Lin, Desalination
219 (2008) 89–100.[69] I. Dolamic, T. Burgi, J. Phys. Chem. C 115 (2011) 2228–2234.
[70] Y. Matsushita, N. Ohba, T. Suzuki, T. Ichimura, Catal. Today 132 (2008)153–158.[71] I. Mikami, S. Aoki, Y. Miura, Chem. Lett. 39 (2010) 704–705.[72] H.H. Ou, M.R. Hoffmann, C.H. Liao, J.H. Hong, S.L. Lo, Appl. Catal. B 99 (2010)
74–80.
logy C: Photochemistry Reviews 15 (2013) 1– 20
[73] H.H. Ou, C.H. Liao, Y.H. Liou, J.H. Hong, S.L. Lo, Environ. Sci. Technol. 42 (2008)4507–4512.
[74] M.S. Vohra, S.M. Selimuzzaman, M.S. Al-Suwaiyan, Environ. Technol. 31(2010) 641–654.
[75] X.D. Zhu, S.R. Castleberry, M.A. Nanny, E.C. Butler, Environ. Sci. Technol. 39(2005) 3784–3791.
[76] J. Lee, H. Park, W. Choi, Environ. Sci. Technol. 36 (2002) 5462–5468.[77] J. Lee, W. Choi, J. Yoon, Environ. Sci. Technol. 39 (2005) 6800–6807.[78] H. Kyung, J. Lee, W. Choi, Environ. Sci. Technol. 39 (2005) 2376–2382.[79] J. Ryu, W. Choi, Environ. Sci. Technol. 42 (2008) 294–300.[80] E.R. Carraway, A.J. Hoffman, M.R. Hoffmann, Environ. Sci. Technol. 28 (1994)
786–793.[81] C. Richard, J. Photochem. Photobiol. A 72 (1993) 179–182.[82] Y. Mao, C. Schoneich, K.D. Asmus, J. Phys. Chem. 95 (1991) 10080–10089.[83] R.B. Draper, M.A. Fox, Langmuir 6 (1990) 1396–1402.[84] S. Kim, W. Choi, Environ. Sci. Technol. 36 (2002) 2019–2025.[85] M.C. Lee, W.Y. Choi, J. Phys. Chem. B 106 (2002) 11818–11822.[86] S.K. Choi, S. Kim, S.K. Lim, H. Park, J. Phys. Chem. C 114 (2010) 16475–16480.[87] D. Lawless, N. Serpone, D. Meisel, J. Phys. Chem. 95 (1991) 5166–5170.[88] J. Lee, J. Kim, W. Choi, TiO2 photocatalysis for the redox conversion of aquatic
pollutants, in: P.G. Tratnyek, T.J. Grundl, S.B. Haderlein (Eds.), ACS SymposiumSeries, Vol. 1071, American Chemical Society, Washington, 2011, p. 199.
[89] B. Tryba, M. Toyoda, A.W. Morawski, R. Nonaka, M. Inagaki, Appl. Catal. B 71(2007) 163–168.
[90] Y.K. Du, J. Rabani, J. Phys. Chem. B 107 (2003) 11970–11978.[91] N. Serpone, G. Sauve, R. Koch, H. Tahiri, P. Pichat, P. Piccinini, E. Pelizzetti, H.
Hidaka, J. Photochem. Photobiol. A 94 (1996) 191–203.[92] F. Sabin, T. Turk, A. Vogler, J. Photochem. Photobiol. A 63 (1992) 99–106.[93] D.A. Dzombak, F.M.M. Morel, Surface Complexation Modelling: Hydrous Fer-
ric Oxide, Wiley-Interscience, New York, 1990.[94] W. Stumm, J.J. Morgan, Aquatic Chemistry: Chemical Equilibria and Rates in
Natural Waters, Wiley-Interscience, New York, 1996.[95] M. Gratzel, Heterogeneous Photochemical Electron Transfer Reactions, CRC
press, Boca Raton, FL, 1987.[96] P.V. Kamat, D. Meisel, Curr. Opin. Colloid Interface Sci. 7 (2002) 282–287.[97] W. Choi, J. Lee, S. Kim, S. Hwang, M.C. Lee, T.K. Lee, J. Ind. Eng. Chem. 9 (2003)
96–101.[98] A. Dobosz, A. Sobczynski, Monatsh. Chem. 132 (2001) 1037–1045.[99] C. Hu, Y.H. Tang, Z. Jiang, Z.P. Hao, H.X. Tang, P.K. Wong, Appl. Catal. A 253
(2003) 389–396.[100] J.S. Jang, S.H. Choi, H.G. Kim, J.S. Lee, J. Phys. Chem. C 112 (2008) 17200–17205.[101] A.C. Li, Z.P. Cui, W.J. Li, Rare Metal Mater. Eng. 38 (2009) 303–307.[102] X.C. Ren, Z.F. Shi, L.N. Kong, Chin. J. Catal. 27 (2006) 815–822.[103] C. Yogi, K. Kojima, T. Takai, N. Wada, J. Mater. Sci. 44 (2009) 821–827.[104] F.X. Zhang, S. Miao, Y.L. Yang, X. Zhang, J.X. Chen, N.J. Guan, J. Phys. Chem. C
112 (2008) 7665–7671.[105] H.M. Zhu, B.F. Yang, J. Xu, Z.P. Fu, M.W. Wen, T. Guo, S.Q. Fu, J. Zuo, S.Y. Zhang,
Appl. Catal. B 90 (2009) 463–469.[106] C.S. Mei, S.H. Zhong, J. Inorg. Mater. 20 (2005) 1396–1402.[107] N. Chandrasekharan, P.V. Kamat, J. Phys. Chem. B 104 (2000) 10851–10857.[108] M. Jacob, H. Levanon, P.V. Kamat, Nano Lett. 3 (2003) 353–358.[109] V. Subramanian, E.E. Wolf, P.V. Kamat, J. Am. Chem. Soc. 126 (2004)
4943–4950.[110] N. Zhang, S. Liu, X. Fu, Y.-J. Xu, J. Phys. Chem. C 115 (2011) 9136–9145.[111] E. Kowalska, O.O.P. Mahaney, R. Abe, B. Ohtani, Phys. Chem. Chem. Phys. 12
(2010) 2344–2355.[112] W. Hou, Z. Liu, P. Pavaskar, W.H. Hung, S.B. Cronin, J. Catal. 277 (2011)
149–153.[113] K. Yu, Y. Tian, T. Tatsuma, Phys. Chem. Chem. Phys. 8 (2006) 5417–5420.[114] A.A. Ismail, D.W. Bahnemann, I. Bannat, M. Wark, J. Phys. Chem. C 113 (2009)
7429–7435.[115] M. Maicu, M.C. Hidalgo, G. Colon, J.A. Navio, J. Photochem. Photobiol. A 217
(2011) 275–283.[116] Y. Tian, T. Tatsuma, J. Am. Chem. Soc. 127 (2005) 7632–7637.[117] M. Rycenga, C.M. Cobley, J. Zeng, W. Li, C.H. Moran, Q. Zhang, D. Qin, Y. Xia,
Chem. Rev. 111 (2011) 3669–3712.[118] H. Courbon, J.-M. Herrmann, P. Pichat, J. Phys. Chem. 88 (1984) 5210–5214.[119] J.-M. Herrmann, P. Pichat, J. Catal. 78 (1982) 425.[120] J. Kiwi, M. Gratzel, J. Phys. Chem. 88 (1984) 1302–1307.[121] D.N. Furlong, D. Wells, W.H.F. Sasse, J. Phys. Chem. 89 (1985) 626–632.[122] W.W. Dunn, Y. Aikawa, A.J. Bard, J. Am. Chem. Soc. 103 (1981) 3456–3459.[123] D. Duonghong, E. Borgarello, M. Gratzel, J. Am. Chem. Soc. 103 (1981)
4685–4690.[124] N. Jaffrezic-Renault, P. Pichat, A. Foissy, R. Mercier, J. Phys. Chem. 90 (1986)
2733–2738.[125] D. Lahiri, V. Subramanian, B.A. Bunker, P.V. Kamat, J. Chem. Phys. 124 (2006)
204720.[126] M. Jakob, H. Levanon, P.V. Kamat, Nano Lett. 3 (2003) 353–358.[127] V. Subramanian, E. Wolf, P.V. Kamat, J. Phys. Chem. B 105 (2001)
11439–11446.[128] H. Park, J. Lee, W. Choi, Catal. Today 111 (2006) 259–265.
[129] S. Hwang, M.C. Lee, W. Choi, Appl. Catal. B 46 (2003) 49–63.[130] H. Einaga, M. Harada, S. Futamura, T. Ibusuki, J. Phys. Chem. B 109 (2003) 9290.[131] J. Lee, W. Choi, Environ. Sci. Technol. 38 (2004) 4026–4033.[132] W. Zhao, C. Chen, X. Li, J. Zhao, H. Hidaka, N. Serpone, J. Phys. Chem. B 106(2002) 5022.

tobiol
[[
[[[[[[[
[[[[[
[[[[[[[[
[
[[[
[
[
[
[[[
[[[[[[
[
[
[
[
[[
[[
[[
[[[[[[[
[
[[
[[[[[[[
H. Park et al. / Journal of Photochemistry and Pho
133] N.Z. Muradov, Sol. Energy 52 (1994) 283.134] D. Hufschmidt, D. Bahnemann, J.J. Testa, C.A. Emilio, M.I. Litter, J. Photochem.
Photobiol. A 148 (2002) 223.135] B. Sun, V. Vorontsov, P.G. Smirniotis, Langmuir 19 (2003) 3151.136] M. Trillas, J. Peral, X. Domenech, Appl. Catal. B 5 (1995) 377.137] J. Lee, W. Choi, J. Phys. Chem. B 109 (2005) 7399–7406.138] J. Chen, D.F. Ollis, W.H. Rulkens, H. Bruning, Water Res. 33 (1999) 661.139] M.D. Driessen, V.H. Grassian, J. Phys. Chem. B 102 (1988) 1418.140] J.C. Crittenden, J.C. Liu, D.W. Hand, D.L. Perram, Water Res. 31 (1997) 429.141] A.C.A. de Vooys, M.T.M. Koper, R.A. van Santen, J.A.R. van Veen, J. Electroanal.
Chem. 506 (2001) 127.142] S. Kim, W. Choi, J. Phys. Chem. B 106 (2002) 13311–13317.143] X.Z. Li, F.B. Li, Environ. Sci. Technol. 35 (2001) 2381–2387.144] C. Lettmann, H. Hinrichs, W.F. Maier, Angew. Chem. Int. Ed. 40 (2001) 3160+.145] G. Burgeth, H. Kisch, Coord. Chem. Rev. 230 (2002) 41–47.146] W. Choi, S.J. Hong, Y.-S. Chang, Y. Cho, Environ. Sci. Technol. 34 (2000)
4810–4815.147] K. Naeem, F. Ouyang, Phys. B 405 (2010) 221–226.148] J. Choi, H. Park, M.R. Hoffmann, J. Mater. Res. 25 (2010) 149–158.149] S. Kim, S.J. Hwang, W. Choi, J. Phys. Chem. B 109 (2005) 24260–24267.150] R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki, Y. Taga, Science 293 (2001) 269.151] S.S. Thind, G. Wu, A. Chen, Appl. Catal. B 111–112 (2012) 38–45.152] J. Xu, W. Wang, S. Sun, L. Wang, Appl. Catal. B 111–112 (2012) 126–132.153] W. Zhao, W. Ma, C. Chen, J. Zhao, Z. Shuai, J. Am. Chem. Soc. 126 (2004) 4782.154] J.L. Gole, J.D. Stout, C. Burda, Y.B. Lou, X.B. Chen, J. Phys. Chem. B 108 (2004)
1230–1240.155] M. Mrowetz, W. Balcerski, A.J. Colussi, M.R. Hoffmann, J. Phys. Chem. B 108
(2004) 17269–17273.156] R. Nakamura, T. Tanaka, Y. Nakato, J. Phys. Chem. B 108 (2004) 10617–10620.157] S.U.M. Khan, M. Al-Shahry, W.B. Ingler Jr., Science 297 (2002) 2243.158] B. Tryba, T. Tsumura, M. Janus, A.W. Morawski, M. Inagaki, Appl. Catal. B 50
(2004) 177–183.159] T. Torimoto, Y. Okawa, N. Takeda, H. Yoneyama, J. Photochem. Photobiol. A
103 (1997) 153–157.160] T. Tsumura, N. Kojjitani, M. Toyoda, M. Inagaki, J. Mater. Chem. 12 (2002)
1391–1396.161] S.W. Hsu, T.S. Yang, T.K. Chen, M.S. Wong, Thin Solid Films 515 (2007)
3521–3526.162] Y. Choi, T. Umebayashi, M. Yoshikawa, J. Mater. Sci. 39 (2004) 1837–1839.163] H. Irie, Y. Watanabe, K. Hashimoto, Chem. Lett. 32 (2003) 772–773.164] W.J. Ren, Z.H. Ai, F.L. Jia, L.Z. Zhang, X.X. Fan, Z.G. Zou, Appl. Catal. B 69 (2007)
138–144.165] Y.Z. Li, D.S. Hwang, N.H. Lee, S.J. Kim, Chem. Phys. Lett. 404 (2005) 25–29.166] T. Ohno, T. Tsubota, K. Nishijima, Z. Miyamoto, Chem. Lett. 33 (2004) 750–751.167] S. Sakthivel, H. Kisch, Angew. Chem. Int. Ed. 42 (2003) 4908–4911.168] C.D. Valentine, G. Pacchioni, A. Selloni, Chem. Mater. 17 (2005) 6656–6665.169] Y.K. Kim, H. Park, Appl. Catal. B 125 (2012) 530–537.170] D.S. Wang, L.B. Xiao, Q.Z. Luo, X.Y. Li, J. An, Y.D. Duan, J. Hazard. Mater. 192
(2011) 150–159.171] Y. Park, W. Kim, H. Park, T. Tachikwawa, T. Majima, W. Choi, Appl. Catal. B 91
(2009) 355–361.172] C. Di Valentin, G. Pacchioni, H. Onishi, A. Kudo, Chem. Phys. Lett. 469 (2009)
166–171.173] T. Ikeda, T. Nomoto, K. Eda, Y. Mizutani, H. Kato, A. Kudo, H. Onishi, J. Phys.
Chem. C 112 (2008) 1167–1173.174] J.P. Yan, Z.L. Tang, S.H. Luo, Z.T. Zhang, Rare Metal Mater. Eng. 32 (2003)
507–510.175] D. Li, H. Haneda, S. Hishita, N. Ohashi, Chem. Mater. 17 (2005) 2588–2595.176] J.A. Rengifo-Herrera, K. Pierzchala, A. Sienkiewicz, L. Forro, J. Kiwi, J.E. Moser,
C. Pulgarin, J. Phys. Chem. C 114 (2010) 2717–2723.177] S. Kim, S.K. Lee, J. Photochem. Photobiol. A 203 (2009) 145–150.178] M. Woodhouse, G.S. Herman, B.A. Parkinson, Chem. Mater. 17 (2005)
4318–4324.179] M. Seyler, K. Stoewe, W.F. Maier, Appl. Catal. B 76 (2007) 146–157.180] W. Kim, H. Kim, N. Lakshminarasimhan, T. Tachikawa, T. Majima, H. Park, W.
Choi, in preparation.181] H. Park, W. Choi, Catal. Today 101 (2005) 291–297.182] J. Kim, J. Lee, W. Choi, Chem. Commun. (2008) 756–758.183] J.S. Park, W. Choi, Langmuir 20 (2004) 11523–11527.184] J. Kim, W. Choi, H. Park, Res. Chem. Intermed. 36 (2010) 127–140.185] M.S. Vohra, K. Tanaka, Water Res. 37 (2003) 3992–3996.186] H. Kim, W. Choi, Appl. Catal. B 69 (2006) 127–132.187] K.J.A. Raj, A.V. Ramaswamy, B. Viswanathan, J. Phys. Chem. C 113 (2009)
13750–13757.188] D. Zhao, C.C. Chen, Y.F. Wang, H.W. Ji, W.H. Ma, L. Zang, J.C. Zhao, J. Phys. Chem.
C 112 (2008) 5993–6001.189] L. Korosi, S. Papp, I. Bertoti, I. Dekany, Chem. Mater. 19 (2007) 4811–4819.190] L. Lin, W. Lin, J.L. Xie, Y.X. Zhu, B.Y. Zhao, Y.C. Xie, Appl. Catal. B 75 (2007)
52–58.191] L. Korosi, I. Dekany, Colloid Surf. A 280 (2006) 146–154.192] J.C. Yu, L.Z. Zhang, Z. Zheng, J.C. Zhao, Chem. Mater. 15 (2003) 2280–2286.
193] P.A. Connor, A.J. McQuillan, Langmuir 15 (1999) 2916–2921.194] J. Kim, W. Choi, Appl. Catal. B 106 (2011) 39–45.195] P. Mohapatra, K.M. Parida, J. Mol. Catal. A 258 (2006) 118–123.196] S.K. Samantaray, P. Mohapatra, K. Parida, J. Mol. Catal. A 198 (2003) 277–287.197] K. Sayama, H. Arakawa, J. Chem. Soc. Faraday Trans. 93 (1997) 1647–1654.ogy C: Photochemistry Reviews 15 (2013) 1– 20 19
[198] H. Arakawa, K. Sayama, Catal. Surv. Jpn. 4 (2000) 75–80.[199] X. Quan, J.F. Niu, S. Chen, J.W. Chen, Y.Z. Zhao, F.L. Yang, Chemosphere 52
(2003) 1749–1755.[200] A. Kumar, N. Mathur, J. Colloid Interface Sci. 300 (2006) 244–252.[201] A. Torrents, A.T. Stone, Environ. Sci. Technol. 27 (1993) 1060–1067.[202] L. Sigg, W. Stumm, Colloid Surf. 2 (1981) 101–117.[203] A.J. Hoffman, E.R. Carraway, M.R. Hoffmann, Environ. Sci. Technol. 28 (1994)
776–785.[204] M.S. Vohra, S. Kim, W. Choi, J. Photochem. Photobiol. A 160 (2003) 55–60.[205] J.W. Tang, H.D. Quan, J.H. Ye, Chem. Mater. 19 (2007) 116–122.[206] M. Mrowetz, E. Selli, Phys. Chem. Chem. Phys. 7 (2005) 1100–1102.[207] A. Janczyk, E. Krakowska, G. Stochel, W. Macyk, J. Am. Chem. Soc. 128 (2006)
15574.[208] Y.-C. Oh, W.S. Jenks, J. Photochem. Photobiol. A 162 (2004) 323–328.[209] J.S. Park, W. Choi, Chem. Lett. 34 (2005) 1630–1631.[210] M.S. Vohra, J. Lee, W. Choi, J. Appl. Electrochem. 35 (2005) 757–763.[211] H. Chun, W. Yizhong, T. Hongxiao, Appl. Catal. B 35 (2001) 95–105.[212] C. Anderson, A.J. Bard, J. Phys. Chem. B 101 (1997) 2611–2616.[213] C. Anderson, A.J. Bard, J. Phys. Chem. 99 (1995) 9882–9885.[214] H. Tada, Y. Kubo, M. Akazawa, S. Ito, Langmuir 14 (1998) 2936–2939.[215] K.Y. Jung, S.B. Park, Korean J. Chem. Eng. 18 (2001) 879.[216] X.Z. Fu, L.A. Clark, Q. Yang, M.A. Anderson, Environ. Sci. Technol. 30 (1996)
647–653.[217] G. Zhang, W. Choi, S.H. Kim, S.B. Hong, J. Hazard. Mater. 188 (2011) 198–205.[218] S. Artkla, W. Kim, W. Choi, J. Wittayakun, Appl. Catal. B 91 (2009) 157–164.[219] H. Park, W. Choi, J. Phys. Chem. B 109 (2005) 11667–11674.[220] H. Park, W. Choi, Langmuir 22 (2006) 2906–2911.[221] M. Gratzel, Nature 414 (2001) 338–344.[222] Y. Cho, W. Choi, J. Photochem. Photobiol. A 148 (2002) 129–135.[223] Y. Cho, Y. Park, W. Choi, J. Ind. Eng. Chem. 14 (2008) 315–321.[224] E. Bae, W. Choi, Environ. Sci. Technol. 37 (2002) 147–152.[225] E. Bae, W. Choi, J. Phys. Chem. B 110 (2006) 14792–14799.[226] E. Bae, W. Choi, J.W. Park, H.S. Shin, S.B. Kim, J.S. Lee, J. Phys. Chem. B 108
(2004) 14093–14101.[227] H. Park, E. Bae, J.J. Lee, J. Park, W. Choi, J. Phys. Chem. B 110 (2006) 8740–8749.[228] W. Kim, T. Tachikwawa, T. Majima, W. Choi, J. Phys. Chem. C 113 (2009)
10603–10609.[229] W. Kim, J. Park, H.J. Jo, H.J. Kim, W. Choi, J. Phys. Chem. C 112 (2008) 491–499.[230] Y. Park, S.H. Lee, S.O. Kang, W. Choi, Chem. Commun. 46 (2010) 2477–2479.[231] S.K. Choi, H.S. Yang, J.H. Kim, H. Park, Appl. Catal. B 121–122 (2012)
206–213.[232] S.K. Choi, S. Kim, J. Ryu, S.K. Lim, H. Park, Photochem. Photobiol. Sci. 11 (2012)
1437–1444.[233] E. Palomares, J.N. Clifford, S.A. Haque, T. Lutz, J.R. Durrant, Chem. Commun.
(2002) 1464.[234] C.C. Chen, X.Z. Li, W.H. Ma, J.C. Zhao, H. Hidaka, N. Serpone, J. Phys. Chem. B
106 (2002) 318–324.[235] M. Yang, D.W. Thompson, G.J. Meyer, Inorg. Chem. 41 (2002) 1254–1262.[236] Y.H. Wang, K. Hang, N.A. Anderson, T.Q. Lian, J. Phys. Chem. B 107 (2003)
9434–9440.[237] T. Tachikawa, S. Tojo, M. Fujitsuka, T. Majima, Langmuir 20 (2004) 2753–2759.[238] T. Tachikawa, A. Yoshida, S. Tojo, A. Sugimoto, M. Fujitsuka, T. Majima, Chem.
Eur. J. 10 (2004) 5345–5353.[239] T. Tachikawa, S. Tojo, M. Fujitsuka, T. Majima, J. Phys. Chem. B 108 (2004)
5859–5866.[240] T. Rajh, L.X. Chen, K. Lukas, T. Liu, M.C. Thurnauer, D.M. Tiede, J. Phys. Chem.
B 106 (2002) 10543–10552.[241] A.P. Xagas, M.C. Bernard, A. Hugot-Le Goff, N. Spyrellis, Z. Loizos, P. Falaras, J.
Photochem. Photobiol. A 132 (2000) 115–120.[242] T. Shishido, K. Teramura, T. Tanaka, Catal. Surv. Asia 15 (2011) 240–258.[243] S. Kim, W. Choi, J. Phys. Chem. B 109 (2005) 5143–5149.[244] G. Kim, W. Choi, Appl. Catal. B 100 (2010) 77–83.[245] Y. Park, N.J. Singh, K.S. Kim, T. Tachikawa, T. Majima, W. Choi, Chem. Eur. J. 15
(2009) 10843–10850.[246] Y. Ku, K.Y. Tseng, C.M. Ma, Int. J. Chem. Kinet. 40 (2008) 209–216.[247] M.V. Dozzi, L. Prati, P. Canton, E. Selli, Phys. Chem. Chem. Phys. 11 (2009)
7171–7180.[248] H. Li, Z. Bian, J. Zhu, Y. Huo, Y. Lu, J. Am. Chem. Soc. 129 (2007) 4538–4539.[249] J.B. Zhong, Y. Lu, W.D. Jiang, Q.M. Meng, X.Y. He, J.Z. Li, Y.Q. Chen, J. Hazard.
Mater. 168 (2009) 1632–1635.[250] H. Einaga, React. Kinet. Catal. Lett. 88 (2006) 357–362.[251] H. Einaga, T. Ibusuki, S. Futamura, Environ. Sci. Technol. 38 (2004) 285–289.[252] G. He, J. Hua, S.C. Wei, J.H. Li, X.H. Liang, E. Luo, Appl. Surf. Sci. 255 (2008)
442–445.[253] G. An, W. Ma, Z. Sun, Z. Liu, B. Han, S. Miao, Z. Miao, K. Ding, Carbon 45 (2007)
1795–1801.[254] O. Akhavan, R. Azimirad, S. Safa, M.M. Larijani, J. Mater. Chem. 20 (2010)
7386–7392.[255] K. Byrappa, A.S. Dayananda, C.P. Sajan, B. Basavalingu, M.B. Shayan, K. Soga,
M. Yoshimura, J. Mater. Sci. 43 (2008) 2348–2355.[256] Y. Yao, G. Li, S. Ciston, R.M. Lueptow, K.A. Gray, Environ. Sci. Technol. 42 (2008)
4952–4957.[257] Y.J. Xu, Y. Zhuang, X. Fu, J. Phys. Chem. C 114 (2010) 2669–2676.[258] C.G. Silva, J.L. Faria, Appl. Catal. B 101 (2010) 81–89.[259] J. Lin, R. Zong, M. Zhou, Y. Zhu, Appl. Catal. B 89 (2009) 425–431.[260] S. Mu, Y. Long, S.Z. Kang, J. Mu, Catal. Commun. 11 (2010) 741–744.

2 tobio
0 H. Park et al. / Journal of Photochemistry and Pho[261] W.C. Oh, W.B. Ko, J. Ind. Eng. Chem. 15 (2009) 791–797.[262] V. Krishna, N. Noguchi, B. Koopman, B. Moudgil, J. Colloid Interface Sci. 304
(2006) 166–171.[263] O. Akhavan, E. Ghaderi, J. Phys. Chem. C 113 (2009) 20214–20220.[264] V. Krishna, D. Yanes, W. Imaram, A. Angerhofer, B. Koopman, B. Moudgil, Appl.
Catal. B 79 (2008) 376–381.[265] H. Zhang, X. Lv, Y. Li, Y. Wang, J. Li, ACS Nano 4 (2010) 380–386.[266] Y. Zhang, Z.R. Tang, X. Fu, Y.J. Xu, ACS Nano 4 (2010) 7303–7314.[267] C. Chen, P. Lei, H. Ji, W. Ma, J. Zhao, H. Hidaka, N. Serpone, Environ. Sci. Technol.
38 (2004) 329–337.[268] G. Marci, E. Garcia-Lopez, L. Palmisano, Catal. Today 144 (2009) 42–47.[269] G. Marci, E. Garcia-Lopez, L. Palmisano, D. Carriazo, C. Martin, V. Rives, Appl.
Catal. B 90 (2009) 497–506.[270] R.R. Ozer, J.L. Ferry, Environ. Sci. Technol. 35 (2001) 3242–3246.[271] K. Lv, Y. Xu, J. Phys. Chem. B 110 (2006) 6204–6212.[272] P. Guo, X. Wang, H. Guo, Appl. Catal. B 90 (2009) 677–687.[273] F. Li, Y. Jiang, M. Xia, M. Sun, B. Xue, X. Ren, J. Hazard. Mater. 165 (2009)
1219–1223.[274] M. Mahalakshmi, S. Vishnu Priya, B. Arabindoo, M. Palanichamy, V. Muruge-
san, J. Hazard. Mater. 161 (2009) 336–343.[275] M. Noorjahan, V.D. Kumari, M. Subrahmanyam, P. Boule, Appl. Catal. B 47
(2004) 209–213.[276] M.V. Shankar, S. Anandan, N. Venkatachalam, B. Arabindoo, V. Murugesan,
Chemosphere 63 (2006) 1014–1021.[277] S. Horikoshi, D. Minami, S. Ito, H. Sakai, D. Kitamoto, M. Abe, N. Serpone, J.
Photochem. Photobiol. A 217 (2011) 141–146.[278] S.I. Naya, A. Inoue, H. Tada, J. Am. Chem. Soc. 132 (2010) 6292–6293.[279] H. Tada, H. Matsui, F. Shiota, M. Nomura, S. Ito, M. Yoshihara, K. Esumi, Chem.
Commun. (2002) 1678–1679.[280] E. Pramauro, A.B. Prevot, M. Vincenti, R. Gamberini, Chemosphere 36 (1998)
1523–1542.[281] X. Li, D. Wang, G. Cheng, Q. Luo, J. An, Y. Wang, Appl. Catal. B 81 (2008)
267–273.[282] H.C. Liang, X.Z. Li, Appl. Catal. B 86 (2009) 8–17.[283] B. Muktha, D. Mahanta, S. Patil, G. Madras, J. Solid State Chem. 180 (2007)
2986–2989.[284] D. Wang, J. Zhang, Q. Luo, X. Li, Y. Duan, J. An, J. Hazard. Mater. 169 (2009)
546–550.[285] Y. Zhu, Y. Dan, Sol. Energy Mater. Sol. Cells 94 (2010) 1658–1664.[286] L. Song, R. Qiu, Y. Mo, D. Zhang, H. Wei, Y. Xiong, Catal. Commun. 8 (2007)
429–433.[287] D. Zhao, C.C. Chen, C.L. Yu, W.H. Ma, J.C. Zhao, J. Phys. Chem. C 113 (2009)
13160–13165.[288] W. Smith, Y.P. Zhao, Catal. Commun. 10 (2009) 1117–1121.[289] I. Paramasivam, Y.C. Nah, C. Das, N.K. Shrestha, P. Schmuki, Chem. Eur. J. 16
(2010) 8993–8997.[290] L. Zhang, F. Lv, W. Zhang, R. Li, H. Zhong, Y. Zhao, Y. Zhang, X. Wang, J. Hazard.
Mater. 171 (2009) 294–300.[291] Y. Bessekhouad, D. Robert, J. Weber, J. Photochem. Photobiol. A 163 (2004)
569–580.[292] G.S. Li, D.Q. Zhang, J.C. Yu, Environ. Sci. Technol. 43 (2009) 7079–7085.[293] A.H. Zyoud, N. Zaatar, I. Saadeddin, C. Ali, D. Park, G. Campet, H.S. Hilal, J.
Hazard. Mater. 173 (2010) 318–325.[294] C. Ratanatawanate, Y. Tao, K.J. Balkus Jr., J. Phys. Chem. C 113 (2009)
10755–10760.[295] M.Y. Duan, J. Li, G. Mele, C. Wang, X.F. Lu, G. Vasapollo, F.X. Zhang, J. Phys.
Chem. C 114 (2010) 7857–7862.[296] J.P. Ghosh, C.H. Langford, G. Achari, J. Phys. Chem. A 112 (2008) 10310–10314.[297] M.C. Yin, Z.S. Li, J.H. Kou, Z.G. Zou, Environ. Sci. Technol. 43 (2009) 8361–8366.
[298] J. Choi, H. Park, M.R. Hoffmann, J. Phys. Chem. C 114 (2010) 783–792.[299] S. Sajjad, S.A.K. Leghari, F. Chen, J.L. Zhang, Chem. Eur. J. 16 (2010)13795–13804.[300] S. Bangkedphol, H.E. Keenan, C.M. Davidson, A. Sakultantimetha, W. Sirisak-
soontorn, A. Songsasen, J. Harzard. Mater. 184 (2010) 533–537.
logy C: Photochemistry Reviews 15 (2013) 1– 20
[301] Y.K. Lai, J.Y. Huang, H.F. Zhang, V.P. Subramaniam, Y.X. Tang, D.G. Gong, L.Sundar, L. Sun, Z. Chen, C.J. Lin, J. Harzard. Mater. 184 (2010) 855–863.
[302] S. Sakthivel, H. Kisch, Angew. Chem. Int. Ed. Engl. 42 (2003) 4908–4911.[303] T. Umebayashi, T. Ymaki, S. Tanaka, K. Asai, Chem. Lett. 32 (2003) 330.[304] T. Ohno, M. Akiyoshi, T. Umebayashi, K. Asai, T. Mitsui, M. Matsumura, Appl.
Catal. A 265 (2004) 115–121.[305] Y.M. Wu, M.Y. Xing, J.L. Zhang, F. Chen, Appl. Catal. B 97 (2010) 182–189.[306] Y.M. Wu, M.Y. Xing, B.Z. Tian, J.L. Zhang, F. Chen, Chem. Eng. J. 162 (2010)
710–717.[307] T. Yu, X. Tan, L. Zhao, Y.X. Yin, P. Chen, J. Wei, Chem. Eng. J. 157 (2010)
86–92.[308] S. Murcia-Lopez, M.C. Hidalgo, J.A. Navio, Appl. Catal. B 404 (2011) 59–67.[309] W. Zhao, Y. Sun, F.N. Castellano, J. Am. Chem. Soc. 130 (2008) 12566–12567.[310] F. Dong, S. Guo, H. Wang, X.F. Li, Z.B. Wu, J. Phys. Chem. C 115 (2011)
13285–13292.[311] X. Li, L. Wang, X. Lu, J. Hazard. Mater. 177 (2010) 639–647.[312] M. Kong, Y. Li, X. Chen, T. Tian, P. Fang, F. Zheng, X. Zhao, J. Am. Chem. Soc.
133 (2011) 16414–16417.[313] C.S. Guo, M. Ge, L. Liu, G. Gao, Y. Feng, Y. Wang, Environ. Sci. Technol. 44 (2010)
419–425.[314] Q.C. Xu, Y.H. Ng, Y. Zhang, J.S.C. Loo, R. Amal, T.T.Y. Tan, Chem. Commun. 47
(2011) 8641–8643.[315] Q.C. Xu, D.V. Wellia, Y.H. Ng, R. Amal, T.T.Y. Tan, J. Phys. Chem. C 115 (2011)
7419–7428.[316] S.O. Obare, T. Ito, M.H. Balfour, G.J. Meyer, Nano Lett. 3 (2003) 1151–1153.[317] S.O. Obare, T. Ito, G.J. Meyer, Environ. Sci. Technol. 39 (2005) 6266–6272.[318] J.R. Stromberg, J.D. Wnuk, R.A.F. Pinlac, G.J. Meyer, Nano Lett. 6 (2006)
1284–1286.[319] J. Zhong, F. Chen, J. Zhang, J. Phys. Chem. C 114 (2010) 933–939.[320] N. Zhang, S. Liu, X. Fu, Y.J. Xu, J. Phys. Chem. C 115 (2011) 9136–9145.[321] X. Chen, L. Liu, P.Y. Yu, S.S. Mao, Science 331 (2011) 746–750.[322] M.E. Kurtoglu, T. Longenbach, K. Sohlberg, Y. Gogotsi, J. Phys. Chem. C 115
(2011) 17392–17399.[323] Y. Wang, R. Shi, J. Lin, Y. Zhu, Appl. Catal. B 100 (2010) 179–183.[324] V.D. Binas, K. Sambani, T. Maggos, A. Katsanaki, G. Kiriakidis, Appl. Catal. B
113–114 (2012) 79–86.[325] A.S. Polo, M.C. Santos, R.F.B. De Souza, W.A. Alves, J. Power Sour. 196 (2011)
872–876.[326] J. Ryu, W. Choi, Environ. Sci. Technol. 38 (2004) 2928–2933.[327] J. Ryu, W. Choi, Environ. Sci. Technol. 40 (2006) 7034–7039.[328] J. Ryu, W. Choi, Environ. Sci. Technol. 41 (2007) 6313–6314.[329] H. Lee, W. Choi, Environ. Sci. Technol. 36 (2002) 3872–3878.[330] H. Yuzawa, T. Mori, H. Itoh, H. Yoshida, J. Phys. Chem. C 116 (2012) 4126–4136.[331] W. Choi, J.Y. Ko, H. Park, J.S. Chung, Appl. Catal. B 31 (2001) 209–220.[332] S.M. Cho, W. Choi, J. Photochem. Photobiol. A 143 (2001) 221–228.[333] E.A. Kozlova, A.V. Vorontsov, Appl. Catal. B 63 (2006) 114–123.[334] J. Papp, H.S. Shen, R. Kershaw, K. Dwight, A. Wold, Chem. Mater. 5 (1993)
284–288.[335] T. Kiyonaga, T. Mitsui, M. Torikoshi, M. Takekawa, T. Soejima, H. Tada, J. Phys.
Chem. B 110 (2006) 10771–10778.[336] H. Tada, T. Ishida, A. Takao, S. Ito, Langmuir 20 (2004) 7898–7900.[337] H. Tada, K. Teranishi, S. Ito, Langmuir 15 (1999) 7084–7087.[338] J.G. Yu, W.G. Wang, B. Cheng, B.L. Su, J. Phys. Chem. C 113 (2009) 6743–6750.[339] D. Monllor-Satoca, R. Gomez, J. Phys. Chem. C 112 (2008) 139–147.[340] C. Minero, G. Mariella, V. Maurino, E. Pelizzetti, Langmuir 16 (2000)
2632–2641.[341] Q. Wang, C.C. Chen, D. Zhao, W.H. Ma, J.C. Zhao, Langmuir 24 (2008)
7338–7345.[342] Y. Xu, K. Lv, Z. Xiong, W. Leng, W. Du, D. Liu, X. Xue, J. Phys. Chem. C 111 (2007)
19024–19032.[343] M. Lewandowski, D.F. Ollis, J. Catal. 217 (2003) 38–46.[344] V. Maurino, C. Minero, G. Mariella, E. Pelizzetti, Chem. Commun. (2005)
2627–2629.[345] M. Mrowetz, E. Selli, New J. Chem. 30 (2006) 108–114.