progressive neurodegeneration in patients with primary immunodeficiency disease on ivig treatment
TRANSCRIPT

Clinical ImmunologyVol. 102, No. 1, January, pp. 19–24, 2002doi:10.1006/clim.2001.5140, available online at http://www.idealibrary.com on
Progressive Neurodegeneration in Patients with PrimaryImmunodeficiency Disease on IVIG Treatment
Ulrike H. M. Ziegner,*,1 Roger H. Kobayashi,* Charlotte Cunningham-Rundles,† Teresa Espanol,‡Anders Fasth,§ Anna Huttenlocher,¶ Paul Krogstad,* Lars Marthinsen,\ Luigi D. Notarangelo,**Srdjan Pasic,** Christian H. L. Rieger,†† Peter Rudge,‡‡ Raman Sankar,* Ann O. Shigeoka,§§
E. Richard Stiehm,* Kathleen E. Sullivan,¶¶ A. David Webster,\\ and Hans D. Ochs***
*Department of Pediatrics, UCLA School of Medicine, Los Angeles, California; ***Department of Pediatrics, University of Washington,Seattle, Washington; †Mount Sinai School of Medicine, New York, New York; ‡Ciutat Sanitaria i Universitaria, Vall d’Hebron, Barcelona,
Spain; §Department of Pediatrics, Goteborg University, Goteborg, Sweden; ¶University of Wisconsin, Madison, Wisconsin; \Departmentof Pediatrics, Halmstad Hospital, Halmstad, Sweden; **Department of Pediatrics, University of Brescia, Spedali Civili, Brescia,
Italy; ††Department of Pediatrics, Ruhr University Bochum, Bochum, Germany; ‡‡Institute of Neurology, National Hospitalfor Neurology and Neurosurgery, London, United Kingdom; §§University of Utah, Salt Lake City, Utah; ¶¶Children’s Hospital
of Philadelphia, Philadelphia, Pennsylvania; and \\Royal Free Hospital School of Medicine, London, United Kingdom
We have identified 14 patients with diverse pri-mary immunodeficiencies who have developed pro-gressive neurodegeneration of unknown etiology.All patients had received immunoglobulin replace-ment therapy for a mean duration of 6.5 years (rangeof 0.5–13.5 years) at the time of first neurologicalsymptoms. Diagnostic tests of blood and cerebrospi-nal fluid analyses included chemistry, cultures, PCRfor viral genomes, and cytology. In addition, neuro-imaging and electrophysiologic studies were per-formed. Brain tissue histology (n 5 5) revealed non-specific encephalitis with microglial infiltration andneuronal loss. Twelve patients died 6 months to 15years (median 4.3 years) after onset of neurologicfindings. No evidence of any infectious disease thatcould have explained our patients’ progressive en-cephalopathy was found either during their life-times or postmortem. These patients may have hadan unusual manifestation of primary immunodefi-ciency diseases, an autoimmune reaction againstneuronal tissue, a yet undefined infectious agent, ora complication of IVIG therapy. To help determinethe etiology of this rare complication, an interna-tional surveillance system for primary immuno-deficiency patients who develop progressive neuro-degeneration of unknown cause is recommended.© 2001 Elsevier Science
Key Words: primary immunodeficiency disease(PID); progressive neurodegeneration; intravenousimmunoglobulin (IVIG); idiopathic encephalopathy.
1 To whom correspondence should be addressed. E-mail:
[email protected].19
INTRODUCTION
In 1995, the World Health Organization (WHO) Sci-entific Group reported 7000 known cases of primaryimmunodeficiency diseases (PID) worldwide (1), anumber that vastly underestimates the true incidence.The majority of these patients have defective B cellfunction (2), resulting in significant antibody deficiencyand the need for lifelong immunoglobulin replacementtherapy. In the United States alone, 17–18 tons ofintravenous immunoglobulin (IVIG) are used per year(3), 30% of which is given to patients with PID.
To produce a single lot of IVIG, plasma units from upto 60,000 donors are pooled (4), and a single lot canserve up to tens of thousands of doses (5). Despite thelarge donor pool, the only infectious agent ever knownto have been transmitted by IVIG infusion is the hep-atitis C virus (HCV), a complication that occurred priorto the availability of serologic methods and polymerasechain reaction (PCR) to identify and eliminate poten-tially HCV-contaminated units (6). Although the trans-mission of Creutzfeldt–Jacob disease (CJD) by bloodproducts has never been confirmed, this possibility re-mains a concern, and the subsequent diagnosis of CJDin a plasma donor was considered a potential safetyhazard, causing the recall of several batches of IVIG(7).
Aseptic meningitis, headaches, and thromboembolicevents, leading to stroke, have been reported as neu-rologic side effects associated with IVIG therapy (8).However, none of these conditions have been reportedas being progressive in nature, and IVIG has emergedas an effective therapy for various neurologic diseases,including demyelinating neuropathies, neuromusculardiseases, myopathies, and certain childhood epilepsies
(9–11).1521-6616/01 $35.00© 2001 Elsevier Science
All rights reserved.

20 ZIEGNER ET AL.
Certain immunodeficient patients are highly suscep-tible to vaccine-associated paralytic poliomyelitis. Fur-thermore, agammaglobulinemic patients are prone tohave chronic enteroviral infections due to the lack ofneutralizing antibodies (12). Chronic enteroviral cen-tral nervous system (CNS) disease, manifesting withheadaches, seizures, hearing loss, lethargy, ataxia,paresthesia, diminished intellectual acuity, loss ofmilestones, and behavioral alterations, seems to pre-cede subsequent dissemination, presenting as der-matomyositis-like syndrome, including myositis-fasci-itis, edema and erythematous rashes, hepatitis,myocarditis, and arthritis. With the introduction ofIVIG in 1982, which allowed a higher IgG trough levelthan that observed during IM immunoglobulin ther-apy, a marked reduction of enteroviral encephalopa-thies was noticed.
In this multicenter study we collected and retrospec-tively analyzed reports of 14 PID patients who haddeveloped progressive neurodegenerative diseasewhile on IVIG therapy without positive detection ofany causative infectious agent. We provide demo-graphic information, clinical presentation, and diag-nostic studies. The intent of this analysis is to alertimmunologists and neurologists of the existence of thisphenomenon and to initiate an early surveillance sys-tem for PID patients on IVIG.
MATERIALS AND METHODS
Patients
Fourteen PID patients, all Caucasians, ages 4 to 62years (median 16 years), qualified for inclusion in thestudy (Table 1). Four patients had X-linked agamma-globulinemia (XLA), five had X-linked hyper-IgM syn-drome (XHIM), one had severe combined immunodefi-ciency (SCID), one combined immunodeficiency (CID),and three patients had common variable immunodefi-ciency (CVID). Seven cases each were identified inEurope and in the U.S.A. Two of fourteen patients werefemale (patients 10 and 11); an expected predominanceof males was due to a high frequency of X-linked in-heritance in the study population.
Two XLA patients had a positive family history, andmutations within the Bruton tyrosine kinase (Btk)gene were identified in three patients. All four XLApatients had low numbers of B lymphocytes and lowserum immunoglobulins. Patients with XHIM had lowserum IgG and IgA and elevated IgM levels (threecases), a positive family history (three cases), and mu-tations of the CD40 ligand (identified in four of the fivepatients). The SCID patient had hypogammaglobuline-mia, lymphopenia (T2, B2, NK1), severely reduced invitro lymphocyte proliferation to mitogens, a positive
family history (brother with SCID), and a mother withrandom X-chromosome inactivation in lymphocytes,suggestive of autosomal recessive inheritance. TheCID patient had residual T cell function. A moleculardiagnosis was available in neither the SCID nor theCID case. The diagnosis of CVID (two of the threeCVID patients were female) was based on low serumimmunoglobulin levels, abnormal antibody responses,depressed isohemagglutinins, and in some cases im-paired cell-mediated immunity, but normal numbers ofcirculating B lymphocytes.
IVIG Exposure
IVIG infusions were started between 2 months and56 years of age (median 1.8 years). The following IVIGbrands were used: Sandoglobulin, Venoglobin-S, Poly-gam S/D, Gammagard S/D, Gamimune N, Endobulin,Octagam, Intraglobin, Polyglobin, Gammonativ, andImmunglobulin Berna. Most patients were exposed totwo or more products, depending on the availability ofthe blood product, and most received the recommendeddose of 400–500 mg/kg every 3 to 4 weeks.
Testing for Infectious Diseases
To explain the neurologic abnormalities, all patientsunderwent extensive testing for infectious agentsknown to cause meningoencephalitis and neurodegen-erative disease. Specimens available from most pa-tients included serum, blood, and cerebrospinal fluid(CSF). Brain tissue was obtained by biopsy (one case)or autopsy (four cases). Because multiple centers andlaboratories were involved and the patients were fre-quently studied over many years, the specimens wereoften analyzed by different techniques. Most patientshad laboratory evaluations for bacterial, viral, and fun-gal infections using cultures, histologic techniques,and more recently PCR. Testing was performed forenteroviruses (EV), including ECHO, coxsackie andpolio strains, Epstein–Barr virus (EBV), cytomegalovi-rus (CMV), parvovirus, herpes simplex virus (HSV) 1and 2, varicella zoster virus (VZV), polyoma viruses,human herpes virus (HHV) 6, 7, and 8, measles, hu-man T cell leukemia/lymphoma virus (HTLV), humanimmunodeficiency virus (HIV), rotavirus, respiratorysyncytial virus (RSV), hepatitis virus A, B, C, and G,mycoplasma, chlamydia, borrelia, toxoplasma, toxo-cara, leishmania, and Giardia lamblia. Not every pa-tient underwent all testing (Table 2). Two patients hadblood tested for CJD by immunoblotting, one each inlaboratories at the NIH and in Madrid, Spain, and allfive brain tissue specimens were histologically evalu-
ated for spongiform encephalitis.
21PID PATIENTS WITH PROGRESSIVE NEURODEGENERATION
Testing for Neurologic Deficits
The neurologic status of each patient was closelymonitored and followed throughout the period of CNSdisease. All patients underwent neuroimaging usingcomputerized tomogram (CT) scanning and/or mag-netic resonance imaging (MRI) and CSF analysis. Inaddition, 13 patients had at least one electroencepha-logram (EEG). In single cases, positron emission to-mography (PET) scans, single proton emission com-puterized tomograms (SPECT), electromyelograms(EMG), and cerebral angiography were performed.Five patients developed vision problems and under-went an extensive ophthalmologic evaluation.
RESULTS
Clinical Course
All 14 PID patients, while on immunoglobulin re-placement therapy, developed progressive neurodegen-erative disease (Table 1). The youngest patient was 16months old, and the oldest was 56 years at onset ofneurologic symptoms (median age 11 years). At thetime the diagnosis of neuropathologic disease was es-tablished, immunoglobulin replacement therapy hadbeen instituted for from 6 months to 13.5 years (me-dian 6.5 years).
Neurologic symptoms were observed in all patients.Eleven developed dementia, including loss of languageskills and memory, and cognitive impairment. Lan-guage deficit affected predominantly expression ratherthan reception and comprehension. Seven patients suf-
TABPat
Patientcase no. Diagnosis Location
Age (years)current or attime of death
Age (at onneu
1a XLA Spain 62a XLA Spain 133 XLA UT, U.S.A. 14 14 XLA NE, U.S.A. 24 15 XHIM PA, U.S.A. 46 XHIM IL, U.S.A. 12 17 XHIM Sweden 16 18 XHIM UT, U.S.A. 25 19a XHIM NY, U.S.A. 29 1
10 CVID Germany 16 111 CVID NY, U.S.A. 59 512a CVID United Kingdom 62 513 SCID Spain 714 CID Italy 11 1
Note. neuro sx, neurological symptoms; tx, treatment.a Case also reported in (14).
fered from ataxia and incoordination. Five patients
TABLE 2ID Testing
Patientcase no. Infectious diseases test results
1 Neg. viral cultures (but started out with 5 days of fever)2 Neg. for CMV, HSV, VZV, adenovirus, EV by culture3 Neg. for EV, EBV, parvovirus, HSV 1 and 2, HHV 6 and
7 (blood, CSF) by PCR; neg. immunoblot for CJD4 Neg. fungal and bacterial cultures; neg. EBV, HSV I, II,
VZV, EV by PCR5 Neg. for EV by RNA-PCR; HIV, CMV, EBV, measles by
viral culture6 Neg. viral and fungal cultures; neg. EV by RNA-PCR7 Neg. cultures for bacteria; neg. CMV,a RSV,
mycobacteria, hepatitis virus A, B, C, G, HIV, HTLV,rotavirus, HSV 112, EBV, toxoplasma, toxocara,leishmania, Giardia lamblia, EV by PCR and culture;neg. for polyomavirus by PCR
8 Neg. viral, bacterial, fungal infections by culture (blood);neg. for EBV by PCR (CSF)
9 Neg. for EV by RNA-PCR; neg. viral cultures10 Neg. for chlamydia, polyomavirus, borrelia, measles,
VZV, toxoplasma, M. tuberculosis, CMV, EBV, EV,mycoplasma, HSV, adenovirus, parvovirus, all testedby PCR in blood and CSF
11 Pos. HCV (but neurologic symptoms arose prior tohepatitis); neg. for EV by RNA-PCR
12 Neg. for EV by RNA-PCR; neg. for polyomaviruses byDNA PCR
13 Neg. for polyomavirus, CMV, HSV 1 and 2, HHV 6, 7,and 8, EV, by PCR and CJD by immunoblot
14 Neg. HSV, CMV, EBV, parvovirus, measles, EV, HIV byPCR in blood and CSF
Note. Neg., negative; Pos., positive.a Postmortem, brain tissue was positive for CMV by PCR; however,
cultures and PCR of blood, urine, bronchioalveolar lavage, and CSF
LE 1ients
years)set ofro sx
Age (years)when Ig txwas started
Time interval (years)between start of Ig txand onset of neuro sx
Time intervalbetween first neuro
sx and death
5 2 3 14 1.5 2.5 91 2 9 34 1 13 Alive1.3 0.2 1.2 31 1 10 15 0.8 14 1.89 10 9 Alive4 0.5 13.5 155 10 6 0.50 47 3 96 56 0.5 65 0.5 4.5 20 9.5 0.6 1
were all negative.

22 ZIEGNER ET AL.
developed clinical seizures, but EEG studies docu-mented abnormalities (epileptiform activity or slowwaves) in 11 of 13 cases studied. Motor deficits ob-served in 7 patients involved spasticity and weakness,mostly affecting the lower extremities and eye muscles.Four patients developed tremor, described as atheto-sis/chorea-like movements in 2 patients.
Five patients experienced an ophthalmopathy withoptic nerve atrophy and a retinitis pigmentosa-likepicture, leading to transient blurred vision and turninginto amblyopia in two patients. Autopsy of one of thepatients with ophthalmopathy revealed partial demy-elinization and gliosis of the optic nerve.
Other symptoms included progressive loss of cogni-tive activities (n 5 3), attention deficit (n 5 3), incon-tinence (n 5 1), tetraparesis (n 5 1), transversemyelitis (n 5 1), mood swings (n 5 1), and aggressionand paranoia (n 5 1).
Patient 7 was treated with intrathecal IVIG, whichdid not change his rapidly progressive neurologic dete-rioration.
Patient 3 was empirically treated with a 3-weekcourse of Pleconaril (3-[3,5-dimethyl-4-[3-(3-methyl-5-isoxazolyl)propoxyl]phenyl]-5-[trifluoromethyl]-1,2,4-oxadiazole; ViroPharma), an investigational anti-picornaviral agent, preventing replication byinhibiting viral uncoating as well as attachment to thehost cell (13). This treatment, approved for compas-sionate use only, had no effect on our patient’s progres-sive deterioration.
To date, progressive neurodegenerative disease hasresulted in the death of 12 patients, between the agesof 4 and 62 years (median age at death 13.5 years).Death occurred 0.5 to 15 years (median 4.3 years) afterthe first neurological symptoms began. The two livingpatients, ages 24 and 25 years, suffer from progressivedementia.
Search for Infectious Agents
Because of the retrospective nature of the study, theextent of the search for an infectious agent varied frompatient to patient (Table 2).
Hepatitis C infection, diagnosed with patient 11, isknown to cause hepatic encephalopathy. However,since the neurologic symptoms had preceded this pa-tient’s hepatitis C infection, this is not a likely expla-nation.
Patient 6 had disseminated histoplasmosis prior tothe CNS disease, but there was no evidence of fungaldisease in CSF cultures and at autopsy.
Postmortem, patient 7 was found to have a positivePCR for CMV in the brain tissue. However, the histol-ogy did show neither inclusion bodies nor evidence ofan inflammatory process that would have been ex-
pected in a patient with XHIM. Furthermore, blood,urine, bronchoalveolar lavage, and CSF, tested byPCR, and cultures for CMV had been repeatedly neg-ative.
Particular attention was given to enteroviral infec-tions, which prior to the IVIG era often caused progres-sive encephalitis leading to dementia, especially in pa-tients with XLA (12). All patients tested negative forenterovirus, including ECHO, coxsackie, and poliovi-rus, by culture or PCR. Histological examination ofbrain tissue from five patients did not show the micro-scopic pattern of spongiform encephalopathy, eliminat-ing CJD.
Neurodiagnostic Tests
CT and/or MRI revealed brain atrophy with promi-nent sulci and enlarged ventricles (Fig. 1) in all 14cases. EEGs showed abnormalities in 11 of 13 patients,consisting of slow waves (n 5 4), epileptiform activity(n 5 4), or both (n 5 3). CSF was abnormal in 6 of 14patients, as demonstrated by elevated protein, ob-served in 3 cases, and pleocytosis, all mononuclearcells, seen in 5 cases; 2 patients had both findings. PETshowed global hypometabolism and SPECT showedmultifocal hypoperfusion in one case. An EMG wasnormal in a patient with ataxia and spasticity of hislower extremities. A brain stem auditory evoked poten-tial and visual evoked potential were abnormal in apatient with ophthalmopathy and motor symptoms.Histologic findings of brain tissue, obtained from 1patient by biopsy and from 4 patients by autopsy, in-cluded reactive astrocytic gliosis, microglial infiltra-tion, patchy demyelinization and neuronal loss, in-volving basal ganglia, brain stem, cortex, and, in 1patient, distinctive loss of Purkinje cells of the cerebel-lar cortex.
DISCUSSION
This retrospective study reports 14 patients withPID who developed progressive neurodegenerative dis-ease of unknown etiology. Because of the antibody de-ficiency, all patients were on immunoglobulin replace-ment therapy for variable time periods prior to theonset of the neurologic symptoms. These cases werefollowed and investigated independently at diversemedical centers in Europe and the United States andwere collected retrospectively by personal communica-tion.
The first description of a similar clinical phenotypeinvolving PID patients was published in 1996 (14),reporting the association of hypogammaglobulinemiaand progressive encephalomyelitis. In four of thesecases, also included in this report, no causative infec-
tious agent could be identified despite thorough testing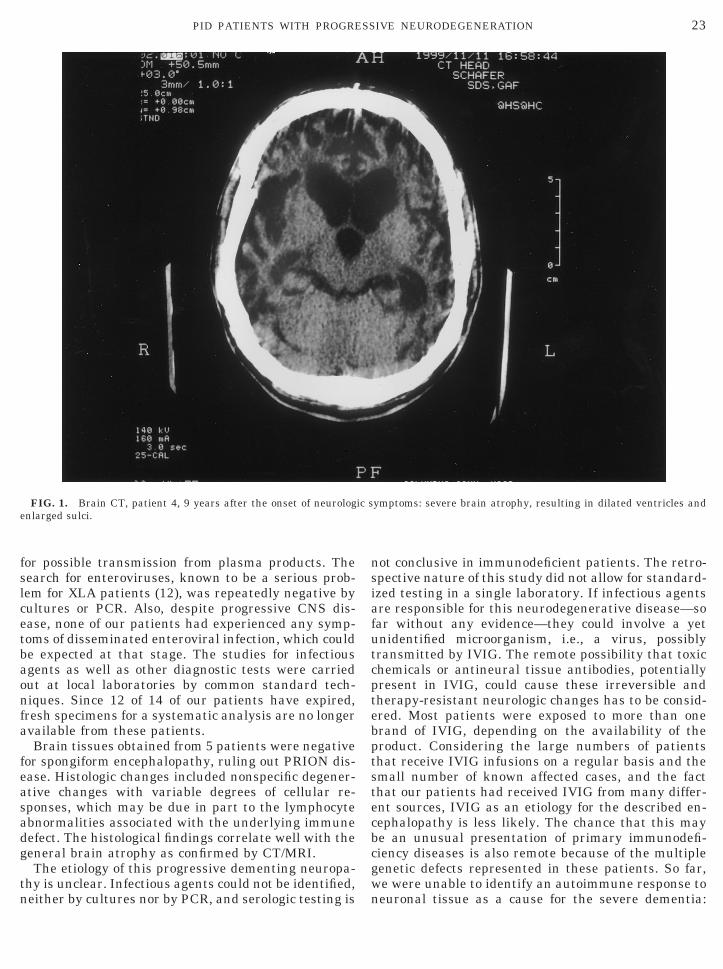
23PID PATIENTS WITH PROGRESSIVE NEURODEGENERATION
for possible transmission from plasma products. Thesearch for enteroviruses, known to be a serious prob-lem for XLA patients (12), was repeatedly negative bycultures or PCR. Also, despite progressive CNS dis-ease, none of our patients had experienced any symp-toms of disseminated enteroviral infection, which couldbe expected at that stage. The studies for infectiousagents as well as other diagnostic tests were carriedout at local laboratories by common standard tech-niques. Since 12 of 14 of our patients have expired,fresh specimens for a systematic analysis are no longeravailable from these patients.
Brain tissues obtained from 5 patients were negativefor spongiform encephalopathy, ruling out PRION dis-ease. Histologic changes included nonspecific degener-ative changes with variable degrees of cellular re-sponses, which may be due in part to the lymphocyteabnormalities associated with the underlying immunedefect. The histological findings correlate well with thegeneral brain atrophy as confirmed by CT/MRI.
The etiology of this progressive dementing neuropa-thy is unclear. Infectious agents could not be identified,
FIG. 1. Brain CT, patient 4, 9 years after the onset of neurologenlarged sulci.
neither by cultures nor by PCR, and serologic testing is
not conclusive in immunodeficient patients. The retro-spective nature of this study did not allow for standard-ized testing in a single laboratory. If infectious agentsare responsible for this neurodegenerative disease—sofar without any evidence—they could involve a yetunidentified microorganism, i.e., a virus, possiblytransmitted by IVIG. The remote possibility that toxicchemicals or antineural tissue antibodies, potentiallypresent in IVIG, could cause these irreversible andtherapy-resistant neurologic changes has to be consid-ered. Most patients were exposed to more than onebrand of IVIG, depending on the availability of theproduct. Considering the large numbers of patientsthat receive IVIG infusions on a regular basis and thesmall number of known affected cases, and the factthat our patients had received IVIG from many differ-ent sources, IVIG as an etiology for the described en-cephalopathy is less likely. The chance that this maybe an unusual presentation of primary immunodefi-ciency diseases is also remote because of the multiplegenetic defects represented in these patients. So far,we were unable to identify an autoimmune response to
ymptoms: severe brain atrophy, resulting in dilated ventricles and
ic sneuronal tissue as a cause for the severe dementia:

24 ZIEGNER ET AL.
Only five patients underwent serologic testing to ruleout autoimmune disease, and none of the obtainedbrain tissue biopsies has been evaluated for immunecomplex deposits.
Although the number of affected patients is smallconsidering the entire population of PID patients, theseriousness of the clinical presentation, the unrespon-siveness to medications, the irreversibility, and theprogression to dementia and death are of concern. Forthese reasons, immunologists and neurologists caringfor such patients will have to develop new strategies tosolve this serious complication of PID. We propose asimple protocol designed to follow such patients, in-cluding sequential testing of at least three specimensfor enteroviruses by PCR and cultures as well as anempiric treatment with Pleconaril. To facilitate activemonitoring of PID patients on IVIG therapy with neu-rological findings, we intend to establish an interna-tional database and registry. A questionnaire for clini-cians has been developed and is available at the URLhttp://www.pid-neurodegeneration.org.
ACKNOWLEDGMENTS
We appreciate the generous support of the Immune DeficiencyFoundation (IDF), which has also provided funds for the moleculartesting of some of the patients. We are indebted to Jackie Fox andKathryn Hennessey for their editorial assistance, Jon Hokanson forhis technical support, and Mike Real for establishing the World WideWeb site. Dr. James F. Bale’s assistance in managing patient AMGis gratefully appreciated. Our special thanks go to the two livingpatients and their families who contributed detailed information bypersonal communication.
REFERENCES
1. Rosen, F. S., Wedgwood, R. J. P., Eibl, M., Griscelli, C., Seligman,M., Aiuti, F., Kishimoto, T., Matsumoto, S., Reznik, I. B., Han-son, L. A., Thompson, R. A., Cooper, M. D., Geha, R. S., Good,R. A., and Waldmann, T. A., Primary immunodeficiency dis-eases: Report of a WHO Scientific Group. Clin. Exp. Immunol.
1(Suppl. 99), 1–24, 1995.2. Conley, M. E., and Stiehm, E. R., Immunodeficiency disorders:General Considerations. In “Immunologic Disorders in Infantsand Children” (E. R. Stiehm, Ed.), pp. 201–252, Saunders, Phil-adelphia, 1996.
3. Lamb, C., “What has caused the current shortages of plasmaderivates and what can be done to correct this situation?” http://www.os.dhhs.gov/partner/bloodsafety/apr28_98.htm, U.S. Foodand Drug Administration, Department of Health and HumanServices, Washington, DC, 1998.
4. Tankersly, D., “What has caused the current shortages of plasmaderivates and what can be done to correct this situation?” http://www.os.dhhs.gov/partner/bloodsafety/apr28_98.htm, U.S. Foodand Drug Administration, Department of Health and HumanServices, Washington, DC, 1998.
5. Milgrom, H., Shortage of intravenous immunoglobulin. Ann. Al-lergy Asthma Immunol. 81, 97–100, 1998.
6. Outbreak of hepatitis C associated with intravenous immuno-logic administration—United States, October 1993–June 1994.MMWR 43(28), 505–521, 1994.
7. Zoon, K., “Disposition of products derived from donors diagnosedwith, or at known risk for, Creutzfeldt–Jakob disease.” U.S. Foodand Drug Administration, Department of Health and HumanServices, Rockville, MD, 1995.
8. Brannagan, T. H., 3rd, Nagle, K. J., Lange, D. J., and Rowland,L. P., Complications of intravenous immune globulin treatmentin neurologic disease. Neurology 47, 674–677, 1996.
9. Dalakas, M. C., Intravenous immune globulin therapy for neu-rologic diseases. Ann. Intern. Med. 126, 721–730, 1997.
10. Van der Meche, F. G. A., Van Doorn, P. A., and Jacobs, B. C.,Inflammatory neuropathies—Pathogenesis and the role of intra-venous immune globulin. J. Clin. Immunol. 15, 63S–69S, 1995.
11. Bushra, J. S., Miller Fisher syndrome: An uncommon acuteneuropathy. J. Emerg. Med. 18, 427–430, 2000.
12. McKinny, R. E., Jr., Katz, S. L., and Wilfert, C. M., Chronicenteroviral encephalitis in agammaglobulinemic patients. Rev.Infect. Dis. 9, 334–356, 1987.
13. Rotbart, H. A., and Webster, A. D., for the Pleconaril TreatmentRegistry Group. Treatment of potentially life-threatening en-terovirus infections with Pleconaril. Clinical Infectious Diseases32, 228–235, 2001.
14. Rudge, P., Webster, A. D. B., Revesz, T., Warner, T., Espanol, T.,Cunningham-Rundles, C., and Hyman, N., Encephalomyelitis in
primary hypogammaglobulinemia. Brain 119, 1–15, 1996.Received August 17, 2001; accepted with revision October 23, 2001