possible mechanisms of neurodegeneration in schizophrenia
TRANSCRIPT

Abstract Brain morphological alterations in schizo-
phrenic patients have led to the neurodevelopmental
hypothesis of schizophrenia. On the other hand, a
progressive neurodegenerative process has also been
suggested and some follow-up studies have shown
progressive morphological changes in schizophrenic
patients. Several neurotransmitter systems have been
suggested to be involved in this disorder and some of
them could lead to neuronal death under certain
conditions. This review discusses some of the bio-
chemical pathways that could lead to neurodegener-
ation in schizophrenia showing that neuronal death
may have a role in the etiology or natural course of
this disorder.
Keywords Dopamine � Antipsychotics �Nitric oxide � NMDA antagonist � Excitotoxicity �Apoptosis
Introduction
The etiology of schizophrenia has not been clearly
understood although several hypotheses have been
developed. Morphological alterations in the brains
from schizophrenic patients have been found, mainly in
the cerebellum [1–4], frontal and temporal lobes [5, 6],
nucleus accumbens [7, 8], mediodorsal thalamus [7]
and other brain regions [9, 10] as compared to control
subjects, although inconsistent results have also been
reported [1, 11, 12]. Evidence of neurodegeneration
has been found in some studies ([1, 13]; but see also
[14, 15]). Discrepancies may arise from the heteroge-
neity of the disorder.
In this regard, there is an intense discussion about
possible neurodegeneration in schizophrenia which
raises the question of which biochemical pathway
could be underlying such a degenerative process. In
this review, the reader will explore the possible
mechanisms of neurodegeneration that could be
involved in schizophrenia. The neurobiological evi-
dence about the developmental theory will not be
discussed since it has been extensively reviewed in the
literature [16].
Brain morphological abnormalities in schizophrenia
An altered neurodevelopment may account for the
morphological alterations in the brains from schizo-
phrenic patients but does not explain that at least some
of those alterations show a progressive course [1, 11],
which suggests an active neurodegenerative process.
On the other hand, both hypotheses (neurodevelop-
ment and neurodegeneration) may be related to each
I. Perez-Neri � S. Montes � C. Rıos (&)Department of Neurochemistry, National Institute ofNeurology and Neurosurgery, Insurgentes Sur 3877 Col. LaFama. Tlalpan, 14269 Mexico City, Mexicoe-mail: [email protected]
J. Ramırez-BermudezDepartment of Clinical Research, National Institute ofNeurology and Neurosurgery, Insurgentes Sur 3877 Col. LaFama. Tlalpan, 14269 Mexico City, Mexico
J. Ramırez-BermudezDepartment of Psychiatry, National Institute of Neurologyand Neurosurgery, Insurgentes Sur 3877 Col. La Fama.Tlalpan, 14269 Mexico City, Mexico
Neurochem Res (2006) 31:1279–1294
DOI 10.1007/s11064-006-9162-3
123
OVERVIEW
Possible Mechanisms of Neurodegeneration in Schizophrenia
Ivan Perez-Neri Æ Jesus Ramırez-Bermudez ÆSergio Montes Æ Camilo Rıos
Accepted: 31 August 2006 / Published online: 28 September 2006� Springer Science+Business Media, LLC 2006

other. Reduced neurotrophic factor signaling, for
example, could lead to both an altered development
of the central nervous system (CNS) and neuronal
death because neurotrophic factors are involved in
CNS development and neuronal survival. So, hypoth-
eses for neurodegeneration and an altered CNS devel-
opment in schizophrenia are not mutually exclusive
and they even could be complementary.
The major brain morphological abnormalities
observed in schizophrenic patients are the loss of cortical
gray matter, the reduced volume of the amygdala, the
hippocampus [17], the frontal and temporal lobes [1, 5,
11, 18] and ventricular enlargement [1, 5, 11, 17, 19]. A
complete description of those alterations is beyond the
scope of this review. Some of the pathological findings
may be observed since the earlier psychotic episodes
and thus they are not likely to be due to chronic
antipsychotic medication [1, 11] and they are more
evident in patients who have suffered multiple epi-
sodes of the disease [5]. It has also been reported a
general decrease of brain mass from the onset of the
disorder [17, 20–22]. Some follow-up neuroimaging
studies have reported a progressive ventricular
enlargement in schizophrenic patients [1, 11]. This
finding may also be observed during physiological
ageing [23] but in schizophrenia it occurs earlier than in
normal subjects.
The interpretation of pathological findings in schizo-
phrenia should be taken with caution since several
factors (such as reduced neuronal size or neuropil,
changes in glial cell number or volume) may be
responsible for the reduced volume observed in some
brain regions in schizophrenic patients. Neuronal loss
is one of those factors. Reduced number of neurons has
been reported in the anterior cingulate cortex [10],
nucleus accumbens [1], hippocampus [1, 10] and
thalamus [24–26] from people who died with schizo-
phrenia. Other studies have failed to replicate those
results [1, 10] but it should be considered that total
neuron numbers could mask significant decreases in
the number of specific cell types within a brain nucleus.
In fact, reduced number in specific cell groups such as
striatal cholinergic interneurons [27], cortical parval-
bumin-containing and calbindin-containing c-aminobu-
tyric acid-(GABA)-ergic cells [28], non-pyramidal
neurons in hippocampal sector CA2 [29], cortical
NADPH-diaphorase positive neurons [30, 31] and
hypothalamic nitric oxide synthase-containing neurons
[32], has also been reported in schizophrenia. The
medication status of the patients included in those
studies is a confounding factor although reduced
number of hippocampal neurons is not likely to be
due to neuroleptic exposure at the time of death [29]
and could be associated to the reduced brain regional
volume in first-episode schizophrenics [1, 11] and thus
neuronal loss in schizophrenia may be related to
disease rather than to medication.
If neurodegeneration is involved in the pathophys-
iology of schizophrenia, biochemical or histopatholog-
ical changes indicative of cell death are expected to be
present in schizophrenic brains. Gliosis has been
reported in schizophrenia for at least 20 years [1].
Glial proliferation has been found in the subiculum
and orbitofrontal cortex of the brains from people with
schizophrenia and dementia compared to those from
people with schizophrenia without dementia, indepen-
dently of neuroleptic exposure, although none of those
subgroups were significantly different from control
samples [13], suggesting that a neurodegenerative
process leading to gliosis could be present in a subset
of schizophrenic patients (those with severe cognitive
impairment). It is also possible that astrocytosis is an
epiphenomenon to schizophrenia but it remains to be
determined if the co-morbidity of dementia is respon-
sible for neuronal death (leading to gliosis) in this
disorder or if the pathophysiology of schizophrenia
could in some patients lead to neurodegeneration
manifested as dementia.
However, most studies have failed to find gliosis in
schizophrenic brains [14, 15, 25] at least partially due to
methodological differences between them [1]. Apop-
totic mechanisms have also been suggested to occur in
schizophrenia [8]. In this regard, significantly reduced
content of the antiapoptotic protein Bcl-2 [33] and
increased Bax/Bcl-2 ratio (associated to susceptibility
for apoptotic death) in Brodmann’s Area (BA) 21 [34]
and ultrastructural changes in oligodendroglial cells
suggestive of apoptotic death [35] have been found in
the brains from people who died with schizophrenia.
Those results are not likely to be due to antipsychotic
medication [33–35].
In spite of the discrepancies between studies, brain
histopathological findings in schizophrenia suggest that
neurodegeneration may occur in a subset of patients
with this disorder.
Glutamatergic hypofunction
Excitotoxicity
An excitotoxic hypothesis for schizophrenia has been
recently reviewed [36]. Some post-mortem studies have
found either increased or decreased expression of
ionotropic glutamatergic receptors in some brain
regions from people who died with schizophrenia
1280 Neurochem Res (2006) 31:1279–1294
123

[18, 36–39]. Those results may be influenced by
antipsychotic drugs since they increase the expression
of those receptors [40]. Increased expression could be
a compensatory mechanism related to the glutamater-
gic hypofunction suggested to occur in schizophrenia
[40, 41] since this effect has also been reported
following N-methyl-D-aspartate receptor (NMDAR)
blockade in rats [42] and primary forebrain cultures
[43].
Decreased expression of ionotropic glutamatergic
receptors found in some studies could be related to
neurotoxicity. Brake and co-workers [44] have reported
that excitotoxic lesions of the prefrontal cortex of
neonate rats lead to an increased dopaminergic
response in the nucleus accumbens when they reach
adulthood. This neurochemical response is similar to
that suggested to occur at the onset of schizophrenia.
Glutamatergic neurons in the medial prefrontal
cortex project to the nucleus accumbens [45] and
reduce dopamine release in this limbic region through
action on metabotropic glutamate receptors (mGluR)
[46, 47] although activation of NMDAR leads to the
opposite effect [48]. In fact, both increases and
decreases in dopamine release have been reported
using different agonists for some mGluR subtypes, as
well as different experimental conditions [49]. On the
other hand, reduced binding to glutamate uptake
sites, indicative of glutamatergic innervation, has been
reported to be decreased in the nucleus accumbens
and other brain regions (unchanged in BA 9 [50]) of
schizophrenic brains, independently of chronic halo-
peridol or clozapine treatment [51]. Excitatory amino
acid transporters are also reduced at the mRNA level
in the ventral striatum in schizophrenia brains post-
mortem [52]. Thus, diminished prefrontal cortical
projections to the nucleus accumbens possibly due
to neuronal death may increase dopamine release in
this nucleus by disinhibition from glutamatergic
modulation.
Mitochondrial dysfunction
It seems contradictory to suggest the involvement of
excitotoxic cell death in schizophrenia when cerebro-
spinal fluid (CSF) glutamate concentration has been
reported to be reduced [53, 54] or unchanged [55, 56] in
schizophrenic patients compared to controls. But it
should be considered that deficiencies in energetic
metabolism, which have also been suggested to occur
in schizophrenia [57, 58], may sensitize neurons to the
physiological glutamate concentrations in the extracel-
lular fluid leading to excitotoxic cell death [59]. This is
related to the fact that reduced ATP availability during
mitochondrial dysfunction decreases Na+/K+ ATPase
activity which in turn maintains membrane potential,
leading to a prolonged depolarization and extruding
magnesium from the NMDAR channel, thus increasing
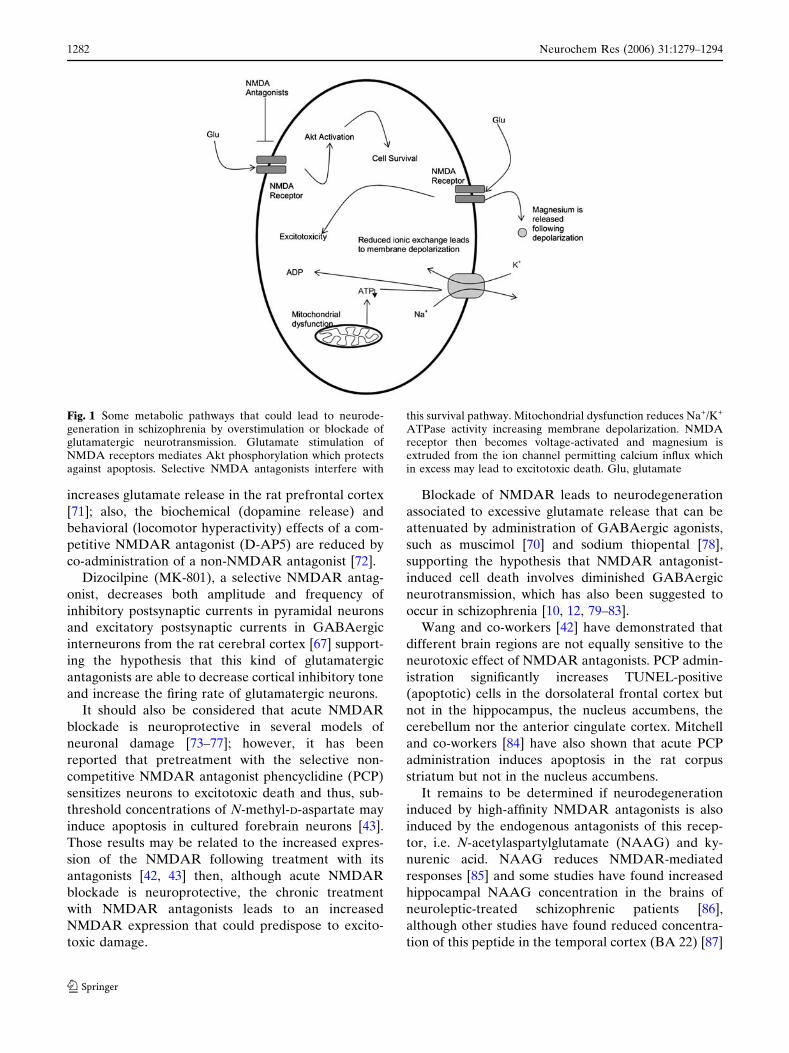
the receptor’s activity (Fig. 1) [57].
Reduced Na+/K+ ATPase activity during mitochon-
drial dysfunction may also alter glutamate uptake since
an energy failure leading to ATP depletion and
intracellular sodium accumulation leads to the reverse
transport of glutamate [60–62]. Regarding schizophre-
nia, decreased glutamate uptake in subcortical brain
regions is likely to occur due to decreased expression
of glutamate transporters [51, 52], possibly associated
to neuroleptic medication [63], although it may be
further reduced if mitochondrial function is compro-
mised.
Other studies further support that mitochondrial
dysfunction is associated to excitotoxicity. Mitochon-
drial toxin-induced neurotoxicity in vitro is attenuated
with co-incubation with a non-competitive NMDAR
antagonist [64] although in vivo studies are not
completely consistent with those results [65]. Reduced
mitochondrial volume and number in oligodendroglial
cells have been reported in both prefrontal cortex (BA
10) and caudate nucleus from schizophrenic brains [35]
although those results should be taken with caution
since they only suggest a reduced mitochondrial
function. In addition, other studies supporting
mitochondrial dysfunction have been reported in
schizophrenia, as discussed in a later section.
GABAergic dysfunction and NMDAR antagonism
neurotoxicity
In the cerebral cortex, glutamatergic neurons stimulate
GABAergic interneurons through NMDAR [66, 67].
Those GABAergic interneurons inhibit pyramidal
neurons in the frontal cortex [66, 68, 69]. Some authors
have suggested that NMDAR blockade reduces corti-
cal inhibitory tone, thus increasing the firing rate of
glutamatergic neurons leading to excitotoxic death at
postsynaptic (i.e. GABAergic) neurons through non-
NMDAR [36, 70]. The association between NMDAR
hypofunction and neurodegeneration by cortical disin-
hibition was proposed at least 10 years ago [66]. If a
similar mechanism occurs in schizophrenia it should be
expected to produce an increased NMDAR and
decreased non-NMDAR expressions. In fact, those
results have been found in some studies [37].
Support for the hypothesis that NMDAR antago-
nism leads to increased glutamatergic function through
non-NMDAR arise from studies showing that
ketamine, a non-competitive NMDAR antagonist,
Neurochem Res (2006) 31:1279–1294 1281
123
increases glutamate release in the rat prefrontal cortex
[71]; also, the biochemical (dopamine release) and
behavioral (locomotor hyperactivity) effects of a com-
petitive NMDAR antagonist (D-AP5) are reduced by
co-administration of a non-NMDAR antagonist [72].
Dizocilpine (MK-801), a selective NMDAR antag-
onist, decreases both amplitude and frequency of
inhibitory postsynaptic currents in pyramidal neurons
and excitatory postsynaptic currents in GABAergic
interneurons from the rat cerebral cortex [67] support-
ing the hypothesis that this kind of glutamatergic
antagonists are able to decrease cortical inhibitory tone
and increase the firing rate of glutamatergic neurons.
It should also be considered that acute NMDAR
blockade is neuroprotective in several models of
neuronal damage [73–77]; however, it has been
reported that pretreatment with the selective non-
competitive NMDAR antagonist phencyclidine (PCP)
sensitizes neurons to excitotoxic death and thus, sub-
threshold concentrations of N-methyl-D-aspartate may
induce apoptosis in cultured forebrain neurons [43].
Those results may be related to the increased expres-
sion of the NMDAR following treatment with its
antagonists [42, 43] then, although acute NMDAR
blockade is neuroprotective, the chronic treatment
with NMDAR antagonists leads to an increased
NMDAR expression that could predispose to excito-
toxic damage.
Blockade of NMDAR leads to neurodegeneration
associated to excessive glutamate release that can be
attenuated by administration of GABAergic agonists,
such as muscimol [70] and sodium thiopental [78],
supporting the hypothesis that NMDAR antagonist-
induced cell death involves diminished GABAergic
neurotransmission, which has also been suggested to
occur in schizophrenia [10, 12, 79–83].
Wang and co-workers [42] have demonstrated that
different brain regions are not equally sensitive to the
neurotoxic effect of NMDAR antagonists. PCP admin-
istration significantly increases TUNEL-positive
(apoptotic) cells in the dorsolateral frontal cortex but
not in the hippocampus, the nucleus accumbens, the
cerebellum nor the anterior cingulate cortex. Mitchell
and co-workers [84] have also shown that acute PCP
administration induces apoptosis in the rat corpus
striatum but not in the nucleus accumbens.
It remains to be determined if neurodegeneration
induced by high-affinity NMDAR antagonists is also
induced by the endogenous antagonists of this recep-
tor, i.e. N-acetylaspartylglutamate (NAAG) and ky-
nurenic acid. NAAG reduces NMDAR-mediated
responses [85] and some studies have found increased
hippocampal NAAG concentration in the brains of
neuroleptic-treated schizophrenic patients [86],
although other studies have found reduced concentra-
tion of this peptide in the temporal cortex (BA 22) [87]
Fig. 1 Some metabolic pathways that could lead to neurode-generation in schizophrenia by overstimulation or blockade ofglutamatergic neurotransmission. Glutamate stimulation ofNMDA receptors mediates Akt phosphorylation which protectsagainst apoptosis. Selective NMDA antagonists interfere with
this survival pathway. Mitochondrial dysfunction reduces Na+/K+
ATPase activity increasing membrane depolarization. NMDAreceptor then becomes voltage-activated and magnesium isextruded from the ion channel permitting calcium influx whichin excess may lead to excitotoxic death. Glu, glutamate
1282 Neurochem Res (2006) 31:1279–1294
123

and the CSF [88] from schizophrenic patients. Differ-
ences in the regions studied may explain such discrep-
ancies. On the other hand, kynurenic acid
concentration has been shown to be increased in BA
9 of schizophrenic brains [89] and the CSF from young
male schizophrenic patients, most of them drug-naıve
[90].
Antiapoptotic signaling
Although the biochemical pathways leading to the
neurotoxic effects of NMDAR antagonists have not
been completely elucidated, evidence of apoptotic
death induced by those antagonists suggests the
participation of this subtype of glutamatergic receptor
in antiapoptotic signaling. It has been reported that
glutamate concentrations below the threshold for
excitotoxic death induce Akt (protein kinase B,
PKB) phosphorylation [91, 92], most likely through
calcium/calmodulin dependent kinase (CaMK) kinase,
inducing the activation of this protein [93]. Akt
activation is part of an antiapoptotic pathway [93–95]
and thus NMDAR blockade may interfere with the
signaling for cell survival (Fig. 1).
The gene encoding Akt has been linked to schizo-
phrenia in European [96, 97] and Japanese populations
[98]. Furthermore, Akt protein levels are decreased in
lymphocytes, frontal cortex and hippocampus from
patients with schizophrenia independently of age,
gender and haloperidol treatment [96] suggesting that
Akt-mediated signaling pathways are altered in this
disorder.
Reduced expression of brain-derived neurotrophic
factor (BDNF) and its receptor [80] could also contrib-
ute to an enhanced susceptibility for apoptotic death in
schizophrenia since this neurotrophin possesses neuro-
protective properties [99]. Reduced BDNF content has
been reported in the prefrontal cortex (BA’s 9 [80] and
46 [100]) from schizophrenic brains and reduced [101] or
unchanged [102] in serum from schizophrenic patients.
All those results are likely to be independent of
antipsychotic medication. Glutamatergic hypofunction
could lead to reduced BDNF expression since activation
of non-NMDAR leads to a 10-fold increase in BDNF
mRNA content in hippocampal neurons [103]. Also,
kynurenic acid (an endogenous antagonist of glutama-
tergic receptors [104, 105]) blocks the increased mRNA
BDNF content induced by the activation of glutama-
tergic receptors in hippocampal neurons [103], and thus
glutamatergic hypofunction could also be related to the
decreased BDNF expression.
In summary, mitochondrial dysfunction has been
suggested to occur in schizophrenia [57, 58] and may
lead to an increased susceptibility for excitotoxic
damage [59] (Fig. 1). On the other hand, NMDAR
antagonism leads to neurodegeneration and GABAer-
gic neurotransmission may have protective effects
against those insults [70, 78]; however, it has been
hypothesized to be decreased in this disorder [12,
79–81, 83] and may contribute to excitotoxic damage
by disinhibiting pyramidal neurons [36, 66–70]. Fur-
thermore, glutamatergic hypofunction could also lead
to decreased antiapoptotic signaling mediated by Akt
(Fig. 1) [93–95] and BDNF [106] since both are
regulated by glutamatergic receptors [91, 92, 103].
Dopaminergic overstimulation
Mitochondrial dysfunction and oxidative stress
Evidence supporting dopaminergic hypofunction in the
cerebral cortex and hyperfunction in subcortical brain
regions in schizophrenia has been elsewhere reviewed
[10, 107].
Dopaminergic overstimulation leads to cell death
under certain conditions (Fig. 2) [58, 108–112]. Intra-
striatal injection of dopamine induces parenchymal
damage [113] that may be associated to mitochondrial
dysfunction since dopamine inhibits mitochondrial
respiration [114–116]. In fact, mitochondrial cyto-
chrome c oxidase activity has been shown to be
reduced in the caudate nucleus from schizophrenic
brains [117]. Since some typical antipsychotics inhibit
mitochondrial respiration [118–120], it remains to be
determined if cytochrome c activity in schizophrenia is
influenced by neuroleptic drugs.
On the other hand, an energetic failure may
decrease the activity of the dopamine transporter
(DAT) maintaining the neurotransmitter in the
extracellular fluid for a longer time. Drugs impairing
ATP synthesis, which may also induce cell death [i.e.
1-methyl-4-phenylpyridinium (MPP+)], significantly
diminish dopamine uptake in vitro by a mechanism
that is not dependent on competition for the DAT or
synaptosomal integrity [121]. Similar results have
been obtained with other mitochondrial toxins [122].
However, the mechanism underlying those effects is
still to be determined since this inhibition of dopa-
mine uptake is neither dependent on ATP depletion
nor reactive oxygen species (ROS) generation [122].
Also, in vivo studies are not completely consistent
with those in vitro results. Administration of the
mitochondrial toxin 3-nitropropionic acid to rats
increases dopamine turnover [65] which is not con-
sistent with inhibition of dopamine uptake since
Neurochem Res (2006) 31:1279–1294 1283
123
blockade of DAT reduces the concentration of
dopamine metabolites [123].
Some iron (III)–catechol complexes are superoxide
anion scavengers [124] which may be underlying the
protective effect of dopamine against iron-induced
lipid peroxidation [114]; however, dopamine-induced
toxicity is associated to the production of ROS [111,
125, 126]. This is due to an interaction between iron (a
lipid peroxidant [77, 127, 128]) and hydrogen peroxide
(the product of monoamine oxidase (MAO) activity
[129, 130]) leading to oxidative damage [131, 132] since
it may be independently reduced by iron chelation
[114], catalase activity [115] or MAO inhibition
[114–116].
Dopaminergic receptor-induced cell death
and excitotoxicity
Selective dopamine D1 receptor agonists potentiate
serum deprivation-induced apoptosis [133] and induce
activation of caspase 3 [134], although neuroprotective
effects of selective D1 (but not D2) receptor agonists
against FeCl2 treatment or glutathione depletion have
also been reported [133]; thus, stimulation of D1
receptors may lead to neuroprotection or toxicity
depending on the neurotoxic insult (oxidative stress
vs serum-deprivation).
Incubation of cell cultures with high dopamine
concentrations (above 100 lM) reduces intracellular
ATP levels [58] and concentrations above 200 lM
reduce cell viability [58, 111]. Cell death may be
prevented by addition of selective D1 receptor antag-
onists [135]. In contrast, D2 receptor agonists have
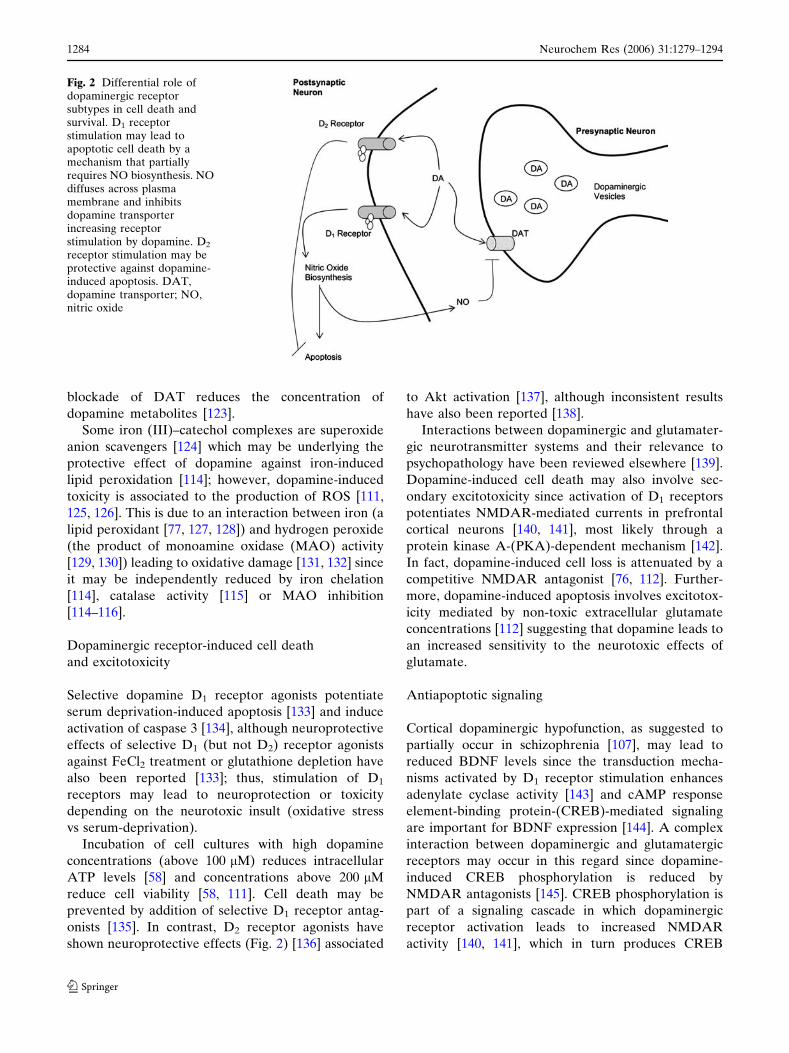
shown neuroprotective effects (Fig. 2) [136] associated
to Akt activation [137], although inconsistent results
have also been reported [138].
Interactions between dopaminergic and glutamater-
gic neurotransmitter systems and their relevance to
psychopathology have been reviewed elsewhere [139].
Dopamine-induced cell death may also involve sec-
ondary excitotoxicity since activation of D1 receptors
potentiates NMDAR-mediated currents in prefrontal
cortical neurons [140, 141], most likely through a
protein kinase A-(PKA)-dependent mechanism [142].
In fact, dopamine-induced cell loss is attenuated by a
competitive NMDAR antagonist [76, 112]. Further-
more, dopamine-induced apoptosis involves excitotox-
icity mediated by non-toxic extracellular glutamate
concentrations [112] suggesting that dopamine leads to
an increased sensitivity to the neurotoxic effects of
glutamate.
Antiapoptotic signaling
Cortical dopaminergic hypofunction, as suggested to
partially occur in schizophrenia [107], may lead to
reduced BDNF levels since the transduction mecha-
nisms activated by D1 receptor stimulation enhances
adenylate cyclase activity [143] and cAMP response
element-binding protein-(CREB)-mediated signaling
are important for BDNF expression [144]. A complex
interaction between dopaminergic and glutamatergic
receptors may occur in this regard since dopamine-
induced CREB phosphorylation is reduced by
NMDAR antagonists [145]. CREB phosphorylation is
part of a signaling cascade in which dopaminergic
receptor activation leads to increased NMDAR
activity [140, 141], which in turn produces CREB
Fig. 2 Differential role ofdopaminergic receptorsubtypes in cell death andsurvival. D1 receptorstimulation may lead toapoptotic cell death by amechanism that partiallyrequires NO biosynthesis. NOdiffuses across plasmamembrane and inhibitsdopamine transporterincreasing receptorstimulation by dopamine. D2
receptor stimulation may beprotective against dopamine-induced apoptosis. DAT,dopamine transporter; NO,nitric oxide
1284 Neurochem Res (2006) 31:1279–1294
123

phosphorylation mediated by CaMK [146]; then,
CREB activation by this mechanism may be altered
during glutamatergic hypofunction. On the other hand,
reduced BDNF levels could lead to an increased
subcortical dopaminergic response since chronic BDNF
depletion in heterozygote knock-out mice leads to an
age-related increase in striatal dopamine content [147,
148] which is likely to reflect reduced dopamine release
to produce sensitization of postsynaptic receptors, since
those mice show an increased sensitivity to the loco-
motor-stimulating effect of methamphetamine [148].
On the other hand, dopamine may also alter
antiapoptotic signaling mediated by Akt as phosphor-
ylation of this protein is reduced by stimulation
[149, 150] while increased by blockade [150] of D2
dopaminergic receptors, which is consistent with a
PKA-dependent Akt activation mechanism [93, 151].
Akt knock-out mice showed increased sensitivity to the
disrupting effects of amphetamine on prepulse inhibi-
tion of the acoustic startle response [96] which is a
behavioral model to study sensorimotor gating in
schizophrenia [152–154].
In summary, dopamine may induce cell death by
mechanisms that may be dependent or independent on
dopaminergic receptors (Fig. 2) [58, 108–113]. The
mitochondrial respiratory chain is altered in the brains
of at least a subset of schizophrenic patients [117] and
may be inhibited by dopamine [114–116]. Mitochon-
drial dysfunction leads in turn to dopaminergic over-
activity by decreasing dopamine uptake [121, 122].
Also, dopamine catabolism by MAO leads to the
generation of hydrogen peroxide [129, 130] which
reacts with free iron (II) leading to oxidative damage
[131, 132]. Activation of D1 receptors initiates a
signaling cascade to produce an increased NMDAR
activity [140, 141] and secondary excitotoxicity [76,
112] mediated by non-toxic extracellular glutamate
concentrations [112]. Since BDNF expression is med-
iated by CREB [144] which is activated by dopami-
nergic receptors, reduced cortical dopaminergic
neurotransmission may alter trophic factor signaling
which is important for neuronal survival [99]. Further-
more, BDNF deficiency increases dopaminergic
response [148]. On the other hand, subcortical dopa-
minergic overstimulation of D2 receptors decreases
Akt activation [149, 150] which has antiapoptotic
properties [93, 94].
Reduced nitric oxide biosynthesis
Nitric oxide (NO) is synthesized by the enzyme NO
synthase (NOS) after NMDAR activation [155, 156].
This is due to the fact that the N-terminus of type I
NOS (nNOS) encodes a PDZ domain that interacts
with PSD 95, which in turn associates to the C-
terminus of the NR1/NR2 subunits of the NMDAR
[157–159].
NO is associated to both neurotoxic and neuropro-
tective effects [160]. NO is responsible for the gluta-
mate-induced activation of guanylate cyclase [155, 161]
which is considered its physiological target, but is
associated to glutamate neurotoxicity [156, 162] and
dopamine-induced cell-death.
D1 receptor stimulation may induce cell death
through a mechanism that requires, at least in part,
NOS activation [135] (Fig. 2). Since dopamine recep-
tor stimulation increases NMDAR activity and
excitotoxicity (Section ‘‘Dopaminergic receptor-
induced cell death and excitotoxicity’’), this mecha-
nism could be involved in dopamine-induced NO
biosynthesis.
The genes encoding NOS [163] and NOS binding
proteins (CAPON) [164] have been linked to
schizophrenia in Chinese but not British [165]
populations. NO is able to reduce dopamine reup-
take [166, 167] increasing the extracellular concen-
tration of this neurotransmitter and thus increasing
the activation of its receptors, which may lead to
cell death (Fig. 2).
Some studies have found evidence for a significant
reduction in NO biosynthesis measured as constitu-
tive NOS activity in post-mortem prefrontal cortex
(BA 9) [168] and nitrite plus nitrate concentration in
the CSF [169, 170] and plasma [171] from patients
with schizophrenia. These results are not likely to be
due to antipsychotic medication since CSF nitrite/
nitrate concentrations were reported in first-episode
schizophrenics [169, 170]. In this regard, other studies
have led to inconsistent results [172–176] but meth-
odological issues regarding the analysis of nitrite and
nitrate in biological samples should be considered
[177–179].
Since excessive NO concentrations are associated
to neuronal damage [135, 156, 162] and inhibition of
the mitochondrial respiratory chain [180, 181],
reduced NOS activity in schizophrenia may be
neuroprotective against certain mechanisms of neu-
ronal damage. However, reduced NO biosynthesis
may lead to some susceptibility for neuronal death
since NO has also been associated to neuroprotective
effects [160, 182, 183] related to an increased
expression of the antiapoptotic proteins Bcl-2 and
Bcl-xL, reduced serum deprivation-induced caspase-3
activity [184], Akt activation [185] and to antioxidant
mechanisms [183].
Neurochem Res (2006) 31:1279–1294 1285
123

Oxidative stress
ROS have been implicated in the pathophysiology of
most neurodegenerative disorders [128, 186–192].
Some studies have suggested that oxidative stress
may also play a role in schizophrenia [193], especially
associated to the development of tardive dyskinesia
[88]. Increased 8-hydroxy 2¢-deoxyguanosine, a marker
of oxidative DNA damage, has been found in the
hippocampus from schizophrenic brains [194]. Markers
for cell cycle activation were found in the same study
[194] as have also been found in apoptotic cells [195–
197], although the influence of antipsychotic treatment
could not be completely ruled out.
Reduced total glutathione content has been
reported in the CSF and prefrontal cortex of schizo-
phrenic patients, most of them drug-naıve [54], which
may reflect reduced antioxidant defenses and oxidative
stress. Plasma peroxidation products have been shown
to be increased in first-episode (drug-naıve) schizo-
phrenic patients and were significantly and positively
correlated with the Brief Psychiatric Rating Scale
negative symptom scores [198]. Other studies have
failed to find a significant difference in the concentra-
tion of oxidation products in the CSF [88] and
fibroblasts [171] from medicated schizophrenic
patients.
Both increased plasma xanthine oxidase activity
[175], which generates superoxide anions, and
decreased superoxide dismutase (SOD) activity
[175, 199] have been found in the plasma from neuro-
leptic-treated schizophrenic patients, suggesting the
presence of oxidative stress which may lead to cell
death [200], although CSF SOD activity in schizophrenic
patients was reported not significantly different com-
pared to control levels [88]. Polymorphisms in the SOD
gene have been linked to schizophrenia [201]. SOD is an
antioxidant enzyme with neuroprotective effects in
neurodegenerative challenges such as glutamate excito-
toxicity [202], 1-methyl-4-phenyl-1,2,3,6-tetrahydro-
pyridine [203] and MPP+ [204] neurotoxicities.
Catalase and glutathione peroxidase activities have
also been shown to be decreased in schizophrenic
patients [199]. On the other hand, Sirota and
co-workers [205] have found that neutrophils isolated
from schizophrenic patients release an increased
amount of superoxide radicals in response to stimula-
tion compared to neutrophils isolated from control
patients; they also found a positive and statistically
significant correlation between neutrophil superoxide
anion generation and negative symptom scores [205].
Other studies have found that glutathione peroxidase
activity correlates with psychotic symptom severity in
patients with schizophrenia [206]. Results from both
studies are independent of pharmacological treatment.
The presence of oxidative stress in schizophrenia
should be taken carefully since it has been demon-
strated haloperidol-induced ROS generation in neuro-
nal cultures [207] that may be associated to
mitochondrial complex I inhibition [118–120]. Similar
results are obtained by chronic administration of
risperidone [119] and fluphenazine [120]. Both inhibi-
tion [119] and a lack of effect [120] on complex I
activity have been reported for clozapine. In this
regard, oxidative stress in schizophrenic patients could
be associated to pharmacological treatment; however,
studies with first-episode, drug-naıve patients suggest
that oxidative stress may play a role in the pathophys-
iology of schizophrenia.
On the other hand, oxidative stress has been
associated to impaired learning [190] and may be
involved in the cognitive deficit of patients with
schizophrenia; this may involve reduced glutamatergic
neurotransmission since the oxidation of the redox-
sensitive site in the NMDAR reduces the activation of
this receptor [105, 208, 209].
However, although ROS generation has been asso-
ciated to the induction of cell death in several studies,
the possible impairment in the oxidant/antioxidant
balance in schizophrenic patients should be taken
carefully since some free radicals have important
physiological roles [210, 211].
Neuroprotective effect of atypical antipsychotics
Some neuroimaging studies have observed that brain
morphological abnormalities are less evident when
schizophrenic patients are taking pharmacological
treatment [11].
In contrast to typical antipsychotics such as halo-
peridol which have been associated to ROS generation
[207] and other neurotoxic effects [212], atypical
antipsychotics such as clozapine and olanzapine have
shown antiapoptotic effects in several studies. In spite
of the possible haloperidol-induced oxidative stress,
this drug has been shown to increase Akt phosphor-
ylation [96] which may signal an antiapoptotic pathway
[93–95]. Furthermore, both haloperidol and sulpiride
increase Nerve Growth Factor mRNA [213] and
protein content [214] in the hippocampus, the striatum
and the nucleus accumbens; thus, those drugs may
enhance trophic factor signaling.
Olanzapine protects against methamphetamine-in-
duced neurotoxicity [215] and reduces apoptotic cell
death induced by trophic factor withdrawal [216].
1286 Neurochem Res (2006) 31:1279–1294
123

Wang and co-workers [42] showed that olanzapine
pretreatment significantly decreases the number of
apoptotic cells and the expression of the proapoptotic
protein Bax induced by PCP administration to neonate
rats. Similar results have been reported for the chronic
administration of quetiapine [217]. Furthermore, both
clozapine and olanzapine increase Bcl-2 protein and
mRNA levels in the frontal cortex and hippocampus
[218, 219]; this protein is protective against neuronal
death induced by several mechanisms [116, 220–222]
and has been shown to be decreased in the temporal
cortex of brains from patients who died with schizo-
phrenia [33]. Quetiapine attenuates PCP-induced
increased Bax and decreased Bcl-XL levels in the
posterior cingulate cortex [217]. Furthermore, olanza-
pine reduces the methamphetamine-induced decrease
in the levels of Bcl-2 in the caudate/putamen [215].
The antiapoptotic activity of olanzapine and zipr-
asidone involves Akt phosphorylation [216] and may
also involve trophic factor signaling since chronic
administration of those drugs increase BDNF expres-
sion in the rat hippocampus [219]. BDNF has been
associated to cell survival [223] and has been shown to
be decreased in schizophrenia (See section ‘‘Glutama-
tergic hypofunction’’ under subsection ‘‘Antiapoptotic
signaling’’) independently of pharmacological treat-
ment since BDNF levels are not different in BA 9 and
46 in haloperidol-treated monkeys respect to their
controls [80].
Other trophic factors are also modulated by atypical
antipsychotics. The administration of clozapine
increases Fibroblast Growth Factor-2 mRNA levels
in the rat prefrontal [224] and parietal [225] cortices,
striatum and nucleus accumbens [225].
The neuroprotective properties of some atypical
antipsychotics (such as clozapine, ziprasidone, olanza-
pine and quetiapine) may be related to their agonistic
properties on 5-HT1A receptors [226–229] since selec-
tive agonists of this subtype of serotoninergic receptor
protect against excitotoxic damage [73, 230–232],
MPP+ neurotoxicity [73], traumatic [233] and ischemic
brain injury [234, 235]; also, those drugs have been
shown to reduce caspase 3 activation [73], stauro-
sporine-induced apoptosis [236, 237], serum depriva-
tion-induced cell death [238] and to increase BDNF
mRNA and protein levels [239].
The involvement of serotoninergic neurotransmis-
sion in the pathophysiology of schizophrenia and the
modulatory role of antipsychotic drugs on serotonin-
ergic receptors have been previously reviewed [240,
241]. Reduced activation of 5-HT1A receptors could
represent some susceptibility for neuronal death and
may lead to an increased expression of this receptor as
has been reported in BA 46 of brains from people who
died with schizophrenia [242].
Since excessive extracellular glutamate concentra-
tions may lead to neuronal damage, some of the
neuroprotective effects of atypical antipsychotics may
be associated to the inhibitory effect of 5-HT1A
receptor activation on evoked glutamate release
[243]. Haloperidol leads to an increased basal gluta-
mate concentration in the striatum and nucleus
accumbens [244]. Also, the chronic administration of
clozapine increases depolarization-induced glutamate
release in the nucleus accumbens [244].
In other studies [245], it has been reported that the
administration of the antipsychotic zotepine reduces
the neuronal vacuolization associated to the neuro-
toxic effect of dizocilpine. On the other hand,
chlorpromazine and trifluoperazine reduce NADPH-
induced lipid peroxidation [246], suggesting that at
least some atypical antipsychotics protect against
oxidative stress.
For those reasons it may be hypothesized that at least
part of the therapeutic effect of atypical antipsychotics
may be due to their neuroprotective effect against
neurodegeneration in patients with schizophrenia.
Conclusions
For decades, schizophrenia has been considered by
most clinicians and researchers as a non-degenerative
disease; however, some follow-up neuroimaging stud-
ies show progressive morphological alterations sugges-
tive of degeneration occurring at least in a subset of
schizophrenic patients. As the development of schizo-
phrenia does not exclude the possibility of comorbid
degenerative diseases, it must be determined if
neurodegeneration in some schizophrenic patients is
associated to schizophrenia itself or if it is just an
independent process, or if those degenerative disorders
actually diagnosed as schizophrenia are indeed, as
suggested by Harrison [1], ‘‘an as yet unrecognized
novel neurodegenerative disorder’’. Discussion about
the possible neurodegeneration in schizophrenia has
both diagnostic and therapeutic implications.
On the other hand, the hypothesis of neurodegen-
eration in schizophrenia should be supported on
biochemical mechanisms that explain cell death in
the context of this disorder. Scientific literature about
this issue shows that the main developed hypotheses
regarding the pathophysiology of schizophrenia
involve possible mechanisms of neurodegeneration,
and thus neuronal death may be associated to the
etiology and/or the course of this disorder.
Neurochem Res (2006) 31:1279–1294 1287
123

Acknowledgements We thank Dr Mario Trevino-Villegas(CINVESTAV-IPN) for his valuable comments about thisreview. I. Perez-Neri receives a grant from the NationalCouncil for Science and Technology (CONACyT, No. 186343).
References
1. Harrison P (1999) The neuropathology of schizophrenia: acritical review of the data and their interpretation. Brain122:593–624
2. Katsetos C, Hyde T, Herman M (1997) Neuropathology ofthe cerebellum in schizophrenia-an update: 1996 and futuredirections. Biol Psychiatry 42:213–224
3. Wassink T, Andreasen N, Nopoulos P et al (1999)Cerebellar morphology as a predictor of symptom andpsychosocial outcome in schizophrenia. Biol Psychiatry45:41–48
4. Bersani G, Venturi P, Taddei I et al (1994) Vermalcerebellar atrophy and hypofrontality in schizophrenia.Schizophr Res 11:133
5. Lieberman J (1999) Is schizophrenia a neurodegenerativedisorder? A clinical and neurobiological perspective. BiolPsychiatry 46:729–739
6. Perlman W, Weickert C, Akil M et al (2004) Postmorteminvestigations of the pathophysiology of schizophrenia:the role of susceptibility genes. J Psychiatry Neurosci29:287–293
7. O’Donnell P, Grace A (1999) Disruption of information withincortical-limbic circuits and the pathophysiology of schizophre-nia. In: Tamminga CA (ed) Schizophrenia in a molecular age.American Psychiatric Press, Washington, pp 117–118
8. Jarskog L, Glantz L, Gilmore J et al (2005) Apoptoticmechanisms in the pathophysiology of schizophrenia. ProgNeuropsychopharmacol Biol Psychiatry 29:846–858
9. Bogerts B, Hantsch J, Herzer M (1983) A morphometricstudy of the dopamine-containing cell groups in the mes-encephalon of normals, Parkinson patients, and schizo-phrenics. Biol Psychiatry 18:951–969
10. Benes F (2000) Emerging principles of altered neuralcircuitry in schizophrenia. Brain Res Rev 31:251–269
11. Lieberman J, Chakos M, Wu H et al (2001) Longitudinalstudy of brain morphology in first episode schizophrenia.Biol Psychiatry 49:487–499
12. Lewis D (2000) GABAergic local circuit neurons andprefrontal cortical dysfunction in schizophrenia. Brain ResRev 31:270–276
13. Arnold S, Franz B, Trojanowski J et al (1996) Glial fibrillaryacidic protein-immunoreactive astrocytosis in elderlypatients with schizophrenia and dementia. Acta Neuropa-thol 91:269–277
14. Damadzic R, Bigelow L, Krimer L et al (2001) A quanti-tative immunohistochemical study of astrocytes in theentorhinal cortex in schizophrenia, bipolar disorder andmajor depression: absence of significant astrocytosis. BrainRes Bull 55:611–618
15. Falke E, Han L, Arnold S (2000) Absence of neurodegen-eration in the thalamus and caudate of elderly patients withschizophrenia. Psychiatry Res 93:103–110
16. Green MF (1998) Schizophrenia from a neurocognitiveperspective. Allyn & Bacon, Boston
17. Barkataki I, Kumari B, Das M et al (2006) Volumetricstructural brain abnormalities in men with schizophrenia orantisocial personality disorder. Behav Brain Res 169:239–247
18. Deakin J, Simpson D (1997) A two-process theory ofschizophrenia: evidence from studies in post mortem brain.J Psychiatr Res 31:277–295
19. Lauer C, Krieg J (1998) Slow-wave sleep and ventricularsize: a comparative study in schizophrenia and majordepression. Biol Psychiatry 44:121–128
20. De Lisi L, Sakuma M, Tew W et al (1997) Schizophrenia asa chronic active brain process: a study of brain structuralchange subsequent to the onset of schizophrenia. PsychiatryRes 74:129–140
21. Gur R, Cowell P, Turetsky B et al (1998) A follow upmagnetic resonance imaging study of schizophrenia:relationship of neuroanatomical changes to clinical andneurobehavioral measures. Arch Gen Psychiatry55:145–152
22. Rapoport J, Giedd J, Blumenthal J et al (1999) Progressivecortical change during adolescence in childhood-onsetschizophrenia: a longitudinal magnetic resonance imagingstudy. Arch Gen Psychiatry 56:649–654
23. Anderton B (2002) Ageing of the brain. Mech Ageing Dev123:811–817
24. Byne W, Buchsbaum M, Mattiace L et al (2002) Post-mortem assessment of thalamic nuclear volumes in subjectswith schizophrenia. Am J Psychiatry 159:59–65
25. Popken G, Bunney W, Potkin S et al (2000) Subnucleus-specific loss of neurons in medial thalamus of schizophren-ics. Proc Natl Acad Sci USA 97:9276–9280
26. Young K, Manaye K, Liang C et al (2000) Reduced numberof mediodorsal and anterior thalamic neurons in schizo-phrenia. Biol Psychiatry 47:944–953
27. Holt D, Herman M, Hyde T et al (1999) Evidence for adeficit in cholinergic interneurons in the striatum in schizo-phrenia. Neuroscience 94:21–31
28. Beasley C, Zhang Z, Patten I et al (2002) Selective deficitsin prefrontal cortical GABAergic neurons in schizophreniadefined by the presence of calcium-binding proteins. BiolPsychiatry 52:708–715
29. Benes F, Kwok E, Vincent S et al (1998) A reduction ofnonpyramidal cells in sector CA2 of schizophrenics andmanic depressives. Biol Psychiatry 44:88–97
30. Akbarian S, Bunney W, Potkin S et al (1993) Altereddistribution of nicotinamide-adenine dinucleotide phos-phate-diaphorase cells in frontal lobe of schizophrenicsimplies disturbances of cortical development. Arch GenPsychiatry 50:169–177
31. Akbarian S, Vinuela A, Kim J et al (1993) Distorteddistribution of nicotinamide-adenine dinucleotide phos-phate-diaphorase neurons in temporal lobe of schizophren-ics implies anomalous cortical development. Arch GenPsychiatry 50:178–187
32. Bernstein H, Stanarius A, Baumann B et al (1998) Nitricoxide synthase-containing neurons in the human hypothal-amus: reduced number of immunoreactive cells in theparaventricular nucleus of depressive patients and schizo-phrenics. Neuroscience 83:867–875
33. Jarskog L, Gilmore J, Selinger E et al (2000) Cortical Bcl-2protein expression and apoptotic regulation in schizophre-nia. Biol Psychiatry 48:641–650
34. Jarskog L, Selinger E, Lieberman J et al (2004) Apoptoticproteins in the temporal cortex in schizophrenia: high Bax/Bcl-2 ratio without caspase 3 activation. Am J Psychiatry161:109–115
35. Uranova N, Orlovskaya D, Vikhreva O et al (2001)Electron microscopy of oligodendroglia in severe mentalillness. Brain Res Bull 55:597–610
1288 Neurochem Res (2006) 31:1279–1294
123

36. Deutsch S, Rosse R, Schwartz B et al (2001) A revisedexcitotoxic hypothesis of schizophrenia: therapeutic impli-cations. Clin Neuropharmacol 24:43–49
37. Ulas J, Cotman C (1993) Excitatory amino acid receptors inschizophrenia. Schizophr Bull 19:105–117
38. Meador-Woodruff J, Hogg A, Smith R (2001) Striatalionotropic glutamate receptor expression in schizophrenia,bipolar disorder, and major depressive disorder. Brain ResBull 55:631–640
39. Gao X, Sakai K, Roberts R et al (2000) Ionotropicglutamate receptors and expression of N-methyl-D-aspar-tate receptor subunits in subregions of human hippocam-pus: effects of schizophrenia. Am J Psychiatry 157:1141–1149
40. Heresco-Levy U (2003) Glutamatergic neurotransmissionmodulation and the mechanisms of antipsychotic atypical-ity. Prog Neuropsychopharmacol Biol Psychiatry 27:1113–1123
41. Walker E, Kestler L, Bollini A et al (2004) Schizophrenia:etiology and course. Annu Rev Psychol 55:401–430
42. Wang C, McInnis J, Ross-Sanchez M et al (2001) Long-termbehavioral and neurodegenerative effects of perinatalphencyclidine administration: implications for schizophre-nia. Neuroscience 107:535–550
43. Wang C, Kaufmann J, Sanchez-Ross M et al (2000)Mechanisms of N-methyl-D-aspartate-induced apoptosis inphencyclidine-treated cultured forebrain neurons. J Phar-macol Exp Ther 294:287–295
44. Brake W, Flores G, Francis D et al (2000) Enhancednucleus accumbens dopamine and plasma corticosteronestress responses in adult rats with neonatal excitotoxiclesions to the medial prefrontal cortex. Neuroscience96:687–695
45. Morgane P, Galler J, Mokler D (2005) A review of systemsand networks of the limbic forebrain/limbic midbrain. ProgNeurobiol 75:143–160
46. Taber M, Fibiger H (1995) Electrical stimulation of theprefrontal cortex increases dopamine release in the nucleusaccumbens of the rat: modulation by metabotropic gluta-mate receptors. J Neurosci 15:3896–3904
47. Feenstra M, Botterblom M, van Uum J (1998) Localactivation of metabotropic glutamate receptors inhibitsthe handling-induced increased release of dopamine inthe nucleus accumbens but not that of dopamine ornoradrenaline in the prefrontal cortex: comparison withinhibition of ionotropic receptors. J Neurochem 70:1104–1113
48. Howland J, Taepavarapruk P, Phillips A (2002) Glutamatereceptor-dependent modulation of dopamine efflux in thenucleus accumbens by basolateral, but not central, nucleusof the amygdala in rats. J Neurosci 22:1137–1145
49. Cartmell J, Schoepp D (2000) Regulation of neurotrans-mitter release by metabotropic glutamate receptors.J Neurochem 75:889–907
50. Scarr E, Beneyto M, Meador-Woodruff JH et al (2005)Cortical glutamatergic markers in schizophrenia. Neuro-psychopharmacology 30:1521–1531
51. Aparicio-Legarza MI, Cutts AJ, Davis B et al (1997)Deficits of [3H]D-aspartate binding to glutamate uptakesites in striatal and accumbens tissue in patients withschizophrenia. Neurosci Lett 232:13–16
52. McCullumsmith RE, Meador-Woodruff JH (2002) Striatalexcitatory amino acid transporter transcript expression inschizophrenia, bipolar disorder, and major depressivedisorder. Neuropsychopharmacology 26:368–375
53. Kim J, Kornhuber H, Schmid-Burgk W et al (1980) Lowcerebrospinal fluid glutamate in schizophrenic patients and anew hypothesis on schizophrenia. Neurosci Lett 20:379–382
54. Do KQ, Trabesinger AH, Kristen-Kruger M et al (2000)Schizophrenia: glutathione deficit in cerebrospinal fluid andprefrontal cortex in vivo. Eur J Neurosci 12:3721–3728
55. Korpi E, Kaufmann C, Marnela K et al (1987) Cerebrospi-nal fluid amino acid concentrations in chronic schizophre-nia. Psychiatry Res 20:337–345
56. Gattaz W, Gasser T, Beckmann H (1985) Multidimensionalanalysis of the concentrations of 17 substances in the CSF ofschizophrenics and controls. Biol Psychiatry 20:360–366
57. Ben-Shachar D (2002) Mitochondrial dysfunction in schizo-phrenia: a possible linkage to dopamine. J Neurochem83:1241–1251
58. Ben-Shachar D, Zuk R, Gazawi H et al (2004) Dopaminetoxicity involves mitochondrial complex I inhibition: impli-cations to dopamine-related neuropsychiatric disorders.Biochem Pharmacol 67:1965–1974
59. Kornhuber J, Seller M (1997) Psychotogenicity and N-methyl-D-aspartate receptor antagonism: implications for neuropro-tective pharmacotherapy. Biol Psychiatry 41:135–144
60. Camacho A, Massieu L (2006) Role of glutamate trans-porters in the clearance and release of glutamate duringischemia and its relation to neuronal death. Arch Med Res37:11–18
61. Danbolt N (2001) Glutamate uptake. Prog Neurobiol65:1–105
62. Longuemare MC, Rose CR, Farrell K et al (1999)K+-induced reversal of astrocyte glutamate uptake islimited by compensatory changes in intracellular Na+.Neuroscience 93:285–292
63. Nanitsos EK, Nguyen KTD, St’astny F et al (2005)Glutamatergic hypothesis of schizophrenia: involvement ofNa+/K+-dependent glutamate transport. J Biomed Sci12:975–984
64. Zeevalk G, Derr-Yellin E, Nicklas W (1995) NMDAreceptor involvement in toxicity to dopamine neurons invitro caused by the succinate dehydrogenase inhibitor3-nitropropionic acid. J Neurochem 64:455–458
65. Beal M, Brouillet E, Jenkins B et al (1993) Neurochemicaland histologic characterization of striatal excitotoxic lesionsproduced by the mitochondrial toxin 3-nitropropionic acid.J Neurosci 13:4181–4192
66. Olney J, Farber N (1995) Glutamate receptor dysfunction inschizophrenia. Arch Gen Psychiatry 52:998–1007
67. Li Q, Clarck S, Lewis D et al (2002) NMDA receptorantagonists disinhibit rat posterior cingulated and retro-splenial cortices: a potential mechanism of neurotoxicity.J Neurosci 22:3070–3080
68. Benes F, Berretta S (2001) GABAergic interneurons:implications for understanding schizophrenia and bipolardisorder. Neuropsychopharmacology 25:1–27
69. Keverne E (1999) GABA-ergic neurons and the neurobi-ology of schizophrenia and other psychoses. Brain Res Bull48:467–473
70. Farber N, Jiang X, Dikranian K et al (2003) Muscimolprevents NMDA antagonist neurotoxicity by activatingGABAA receptors in several brain regions. Brain Res993:90–100
71. Moghaddam B, Adams B, Verma A et al (1997) Activationof glutamatergic neurotransmission by ketamine: a novelstep in the pathway from NMDA receptor blockade todopaminergic and cognitive disruptions associated to theprefrontal cortex. J Neurosci 17:2921–2927
Neurochem Res (2006) 31:1279–1294 1289
123

72. Feenstra M, Botterblom M, van Uum J (2002) Behavioralarousal and increased dopamine efflux after blockade ofNMDA-receptors in the prefrontal cortex are dependent onactivation of glutamatergic neurotransmission. Neurophar-macology 42:752–763
73. Madhavan L, Freed W, Anantharam V et al (2003)5-Hydroxytryptamine 1A receptor activation protectsagainst N-methyl-D-aspartate-induced apoptotic cell deathin striatal and mesencephalic cultures. J Pharmacol ExpTher 304:913–923
74. Santamarıa A, Rıos C (1993) MK-801, an N-methyl-D-aspartate receptor antagonist, blocks quinolinic acid-in-duced lipid peroxidation in rat corpus striatum. NeurosciLett 159:51–54
75. Santamarıa A, Rıos C, Solıs-Hernandez F et al (1996)Systemic DL-kynurenine and probenecid pretreatmentattenuates quinolinic acid-induced neurotoxicity in rats.Neuropharmacology 35:23–28
76. Lieb K, Andrae J, Reisert I et al (1995) Neurotoxicity ofdopamine and protective effects of the NMDA receptorantagonist AP-5 differ between male and female dopami-nergic neurons. Exp Neurol 134:222–229
77. Jara-Prado A, Ortega-Vazquez A, Ruano L et al (2003)Homocysteine-induced brain lipid peroxidation: effects ofNMDA receptor blockade, antioxidant treatment, andnitric oxide synthase inhibition. Neurotox Res 5:237–244
78. Jevtovic-Todorovic V, Wozniak D, Powell S et al (2001)Propofol and sodium thiopental protect against MK-801induced neuronal necrosis in the posterior cingulated/retrosplenial cortex. Brain Res 913:185–189
79. Akbarian S, Kim J, Potkin S et al (1995) Gene expressionfor glutamic acid decarboxylase is reduced without loss ofneurons in prefrontal cortex of schizophrenics. Arch GenPsychiatry 52:258–266
80. Hashimoto T, Bergen S, Nguyen Q et al (2005) Relation-ship of brain-derived neurotrophic factor and its receptorTrkB to altered inhibitory prefrontal circuitry in schizo-phrenia. J Neurosci 25:372–383
81. Woo T, Whitehead R, Melchitzky D et al (1998) A subclassof prefrontal c-aminobutyric acid axon terminals are selec-tively altered in schizophrenia. Proc Natl Acad Sci USA95:5341–5346
82. Bird E, Barnes J, Iversen L et al (1977) Increased braindopamine and reduced glutamic acid decarboxylase andcholine acetyl transferase activity in schizophrenia andrelated psychosis. Lancet 2:1157–1159
83. Dean B, Hussain T, Hayes W et al (1999) Changes inserotonin2A and GABAA receptors in schizophrenia: stud-ies on the human dorsolateral prefrontal cortex. J Neuro-chem 72:1593–1599
84. Mitchell I, Cooper A, Griffiths M et al (1998) Phencyclidineand corticosteroids induce apoptosis of a subpopulation ofstriatal neurons: a neural substrate for psychosis? Neuro-science 84:489–501
85. Grunze H, Rainnie D, Hasselmo M et al (1996) NMDA-dependent modulation of CA1 local circuit inhibition.J Neurosci 16:2034–2043
86. Tsai G, Passani L, Slusher B et al (1995) Abnormalexcitatory neurotransmitter metabolism in schizophrenicbrains. Arch Gen Psychiatry 52:829–836
87. Nudmamud S, Reynolds L, Reynolds G (2003) N-acetyl-aspartate and N-acetylaspartylglutamate deficits in supe-rior temporal cortex in schizophrenia and bipolardisorder: a postmortem study. Biol Psychiatry 53:1138–1141
88. Tsai G, Goff D, Chang R et al (1998) Markers ofglutamatergic neurotransmission and oxidative stress asso-ciated to tardive dyskinesia. Am J Psychiatry 155:1207–1213
89. Schwarcz R, Rassoulpour A, Wu H et al (2001) Increasedcortical kynurenate content in schizophrenia. Biol Psychi-atry 50:521–530
90. Erhardt S, Blennow K, Nordin C et al (2001) Kynurenicacid levels are elevated in the cerebrospinal fluid of patientswith schizophrenia. Neurosci Lett 313:96–98
91. Manabe S, Lipton S (2003) Divergent NMDA signalsleading to proapoptotic and antiapoptotic pathways in therat retina. Invest Ophth Vis Sci 44:385–392
92. Sutton G, Chandler LJ (2002) Activity-dependent NMDAreceptor-mediated activation of protein kinase B/Akt incortical neuronal cultures. J Neurochem 82:1097–1105
93. Datta S, Brunet A, Greenberg M (1999) Cell survival: a playin three Akts. Gene Dev 13:2905–2927
94. Dan H, Sun M, Kaneko S et al (2004) Akt phosphorylationand stabilization of X-linked inhibitor of apoptosis protein(XIAP). J Biol Chem 279:5405–5412
95. Brunet A, Datta S, Greenberg M (2001) Transcription-dependent and -independent control of neuronal survival bythe PI3K-Akt signaling pathway. Curr Op Neurobiol11:297–305
96. Emamian E, Hall D, Birnbaum M et al (2004) Convergentevidence for impaired AKT1-GSK3b signaling in schizo-phrenia. Nat Genet 36:131–137
97. Schwab S, Hoefgen B, Hanses C et al (2005) Furtherevidence for association of variants in the AKT1 gene withschizophrenia in a sample of European sib-pair families.Biol Psychiatry 58:446–450
98. Ikeda M, Iwata N, Suzuki T et al (2004) Association ofAKT1 with schizophrenia confirmed in a Japanese popula-tion. Biol Psychiatry 56:698–700
99. Morse J, Wiegand S, Anderson K et al (1993) Brain-derivedneurotrophic factor (BDNF) prevents the degeneration ofmedial septal cholinergic neurons following fimbria tran-section. J Neurosci 73:4146–4156
100. Weickert C, Hyde T, Lipska B et al (2003) Reduced brain-derived neurotrophic factor in prefrontal cortex of patientswith schizophrenia. Mol Psychiatry 8:592–610
101. Toyooka K, Asama K, Watanabe Y et al (2002) Decreasedlevels of brain-derived neurotrophic factor in serum ofchronic schizophrenic patients. Psychiatry Res 110:249–257
102. Shimizu E, Hashimoto K, Watanabe H et al (2003) Serumbrain-derived neurotrophic factor (BDNF) levels in schizo-phrenia are indistinguishable from controls. Neurosci Lett351:111–114
103. Zafra F, Hengerer B, Leibrock J et al (1990) Activitydependent regulation of BDNF and NGF mRNAs in the rathippocampus is mediated by non-NMDA glutamate recep-tors. EMBO J 9:3545–3550
104. Schwarcz R, Pellicciari R (2002) Manipulation of brainkynurenines: glial targets, neuronal effects, and clinicalopportunities. J Pharmacol Exp Ther 303:1–10
105. Stone T, Addae J (2002) The pharmacological manipulationof glutamate receptors and neuroprotection. Eur J Phar-macol 447:285–296
106. Torrey F, Barci B, Webster M et al (2005) Neurochemicalmarkers for schizophrenia, bipolar disorder, and majordepression in postmortem brains. Biol Psychiatry 57:252–260
107. Davis K, Kahn R, Ko G et al (1991) Dopamine inschizophrenia: a review and reconceptualization. Am JPsychiatry 148:1474–1486
1290 Neurochem Res (2006) 31:1279–1294
123

108. Walkinshaw G, Waters C (1995) Induction of apoptosis incatecholaminergic PC12 cells by L-DOPA: implications forthe treatment of Parkinson’s disease. J Clin Invest 95:2458–2464
109. Luo Y, Hattori A, Munoz J et al (1999) Intrastriataldopamine injection induces apoptosis through oxidation-involved activation of transcription factors AP-1 and NF-jBin rats. Mol Pharmacol 56:254–264
110. Luo Y, Umegaki H, Wang X et al (1998) Dopamine inducesapoptosis through an oxidation-involved SAPK/JNK acti-vation pathway. J Biol Chem 273:3756–3764
111. Offen D, Ziv I, Sternin H et al (1996) Prevention ofdopamine-induced cell death by thiol antioxidants: possibleimplications for treatment of Parkinson’s disease. ExpNeurol 141:32–39
112. Zhang J, Price J, Graham D et al (1998) Secondaryexcitotoxicity contributes to dopamine-induced apoptosisof dopaminergic neuronal cultures. Biochem Biophys ResCommun 248:812–816
113. Filloux F, Townsend J (1993) Pre- and postsynaptic neuro-toxic effects of dopamine demonstrated by intrastriatalinjection. Exp Neurol 119:79–88
114. Ben-Shachar D, Zuk R, Glinka Y (1995) Dopamineneurotoxicity: inhibition of mitochondrial respiration.J Neurochem 64:718–723
115. Berman S, Hastings T (1999) Dopamine oxidation altersmitochondrial respiration and induces permeability transi-tion in brain mitochondria: implications for Parkinson’sdisease. J Neurochem 73:1127–1137
116. Cohen G, Farooqui R, Kesler N (1997) Parkinson disease: anew link between monoamine oxidase and mitochondrialelectron flow. Proc Natl Acad Sci USA 94:4890–4894
117. Cavelier L, Jazin E, Eriksson I et al (1995) Decreasedcytochrome-c oxidase activity and lack of age-relatedaccumulation of mitochondrial DNA deletions in the brainsof schizophrenics. Genomics 29:217–224
118. Balijepalli S, Boyd M, Ravindranath V (1999) Inhibition ofmitochondrial complex I by haloperidol: the role of thioloxidation. Neuropharmacology 38:567–577
119. Balijepalli S, Kenchappa R, Boyd M et al (2001) Proteinthiol oxidation by haloperidol results in inhibition ofmitochondrial complex I in brain regions: comparison withatypical antipsychotics. Neurochem Int 38:425–435
120. Prince J, Yassin M, Oreland L (1997) Neuroleptic-inducedmitochondrial enzyme alterations in the rat brain. J Phar-macol Exp Ther 280:261–267
121. Fonck C, Baudry M (2003) Rapid reduction of ATPsynthesis and lack of free radical formation by MPP+ inrat brain synaptosomes and mitochondria. Brain Res975:214–221
122. Maragos W, Zhu J, Chesnut M et al (2002) Mitochondrialtoxin inhibition of [3H]dopamine uptake into rat striatalsynaptosomes. Biochem Pharmacol 63:1499–1505
123. Chen L, He M, Sibille E et al (1999) Adaptive changes inpostsynaptic dopamine dynamics in mice lacking mono-amine oxidase B. J Neurochem 73:647–655
124. Zhao Z, Khan S, O’Brien P (1998) Catecholic ironcomplexes as cytoprotective superoxide scavengers againsthypoxia:reoxygenation injury in isolated hepatocytes.Biochem Pharmacol 56:825–830
125. Baez S, Segura-Aguilar J, Widersten M et al (1997)Glutathione transferases catalyze the detoxication of oxi-dized metabolites (o-quinones) of catecholamines and mayserve as an antioxidant system preventing degenerativecellular processes. Biochem J 324:25–28
126. Hrometz S, Brown A, Nichols D et al (2004) 3,4-Methy-lenedioxymethamphetamine (MDMA, ecstasy)-mediatedproduction of hydrogen peroxide in an in vitro model: therole of dopamine, the serotonin-reuptake transporter, andmonoamine oxidase-B. Neurosci Lett 367:56–59
127. Santamarıa A, Santamarıa D, Dıaz-Munoz M et al (1997)Effects of Nx-nitro-L-arginine and L-arginine on quinolinicacid-induced lipid peroxidation. Toxicol Lett 93:117–124
128. Rıos C, Tapia R (1987) Changes in lipid peroxidationinduced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridineand 1-methyl-4-phenylpyridinium in mouse brain homogen-ates. Neurosci Lett 77:321–326
129. Shih J, Chen K, Ridd M (1999) Monoamine oxidase: fromgenes to behavior. Annu Rev Neurosci 22:197–217
130. Kopin I (1985) Catecholamine metabolism: basic aspectsand clinical significance. Pharmacol Rev 37:333–364
131. Crichton R, Wilmet S, Legssyer R et al (2002) Molecularand cellular mechanisms of iron homeostasis and toxicity inmammalian cells. J Inorg Biochem 91:9–18
132. Liochev S, Fridovich I (1997) How does superoxidedismutase protect against tumor necrosis factor: a hypoth-esis informed by effect of superoxide on ‘‘free’’ iron. FreeRad Biol Med 23:668–671
133. Noh J, Gwag B (1997) Attenuation of oxidative neuronalnecrosis by a dopamine D1 agonist in mouse cortical cellcultures. Exp Neurol 146:604–608
134. Mitchell E, Snyder-Keller A (2003) Blockade of D1dopaminergic transmission alleviates c-fos induction andcleaved caspase-3 expression in the brains of rat pupsexposed to prenatal cocaine or perinatal asphyxia. ExpNeurol 182:64–74
135. Chen J, Wersinger C, Sighu A (2003) Chronic stimulation ofD1 dopamine receptors in human SK-N-MC neuroblastomacells induce nitric oxide synthase activation and cytotoxic-ity. J Biol Chem 278:28089–28100
136. Double K, Halliday G, Henderson J et al (2003) Thedopamine receptor agonist lisuride attenuates iron-medi-ated dopaminergic neurodegeneration. Exp Neurol184:530–535
137. Nair V, Sealfon S (2003) Agonist-specific transactivation ofphosphoinositide 3-kinase signaling pathway mediated bythe dopamine D2 receptor. J Biol Chem 278:47053–47061
138. Griffiths M, Cooper A, Barber D et al (2000) Pharmaco-logical mechanisms mediating phencyclidine-induced apop-tosis of striatopallidal neurons: the roles of glutamate,dopamine, acetylcholine and corticosteroids. Brain Res855:1–10
139. Pralong E, Magistretti P, Stoop R (2002) Cellular perspec-tives on the glutamate-monoamine interactions in limbiclobe structures and their relevance for some psychiatricdisorders. Prog Neurobiol 67:173–202
140. Chen G, Greengard P, Yan Z (2004) Potentiation ofNMDA receptor currents by dopamine D1 receptors inprefrontal cortex. Proc Natl Acad Sci USA 101:2596–2600
141. Wirkner K, Krause T, Koles L et al (2004) D1 but not D2dopamine receptors or adrenoceptors mediate dopamine-induced potentiation of N-methyl-d-aspartate currents inthe rat prefrontal cortex. Neurosci Lett 372:89–93
142. Skeberdis V, Chevaleyre V, Lau C et al (2006) Proteinkinase A regulates calcium permeability of NMDA recep-tors. Nat Neurosci 9:501–510
143. Zafra F, Lindholm D, Castren E et al (1992) Regulation ofbrain-derived neurotrophic factor and nerve growth factormRNA in primary cultures of hippocampal neurons andastrocytes. J Neurosci 12:4793–4799
Neurochem Res (2006) 31:1279–1294 1291
123

144. Conti A, Cryan J, Dalvi A et al (2002) cAMP responseelement-binding protein is essential for the upregulation ofbrain-derived neurotrophic factor transcription, but not thebehavioral or endocrine responses to antidepressant drugs.J Neurosci 22:3262–3268
145. Konradi C, Leveque J, Hyman S (1996) Amphetamine anddopamine-induced immediate early gene expression instriatal neurons depends on postsynaptic NMDA receptorsand calcium. J Neurosci 16:4231–4239
146. Dudman J, Eaton M, Rajadhyaksha A et al (2003)Dopamine D1 receptors mediate CREB phosphorylationvia phosphorylation of the NMDA receptor at Ser897-NR1.J Neurochem 87:922–934
147. Dluzen D, Story G, Xu K et al (1999) Alterations innigroestriatal dopaminergic function within BDNF mutantmice. Exp Neurol 160:500–507
148. Dluzen D, Gao X, Story G et al (2001) Evaluation ofnigroestriatal dopaminergic function in adult +/+ and +/–BDNF mutant mice. Exp Neurol 170:121–128
149. Beaulieu J, Sotnikova T, Marion S et al (2005) An Akt/b-Arrestin 2/PP2A signaling complex mediates dopaminergicneurotransmission and behavior. Cell 122:261–273
150. Beaulieu J, Sotnikova T, Yao W et al (2004) Lithiumantagonizes dopamine-dependent behaviors mediated by anAKT/glycogen synthase kinase 3 signaling cascade. ProcNatl Acad Sci USA 101:5099–5104
151. Filippa N, Sable C, Filloux C et al (1999) Mechanism ofprotein kinase B activation by cyclic AMP-dependentprotein kinase. Mol Cell Biol 19:4989–5000
152. Imre G, Fokkema D, Den Boer J et al (2006) Dose–response characteristics of ketamine effect on locomotion,cognitive function and central neuronal activity. Brain ResBull 69:338–345
153. Wolf R, Dobrowolny H, Matzke K et al (2006) Prepulseinhibition is different in two inbred mouse strains (CPB-Kand BALB/cJ) with different hippocampal NMDA receptordensities. Behav Brain Res 166:78–84
154. Yee B, Keist R, von Boehmer L et al (2005) A schizophre-nia-related sensorimotor deficit links a3-containingGABAA receptors to a dopamine hyperfunction. Proc NatlAcad Sci USA 102:17154–17159
155. Bredt DS, Snyder SH (1989) Nitric oxide mediates gluta-mate-linked enhancement of cGMP levels in the cerebel-lum. Proc Natl Acad Sci USA 86:9030–9033
156. Nelson EJ, Connolly J, McArthur P (2003) Nitric oxide andS-nitrosylation: excitotoxic and cell signaling mechanism.Biol Cell 95:3–8
157. Brenman JE, Bredt DS (1997) Synaptic signaling by nitricoxide. Curr Op Neurobiol 7:374–378
158. Kone BC, Kuncewicz T, Zhang W et al (2003) Proteininteractions with nitric oxide synthases: controlling the righttime, the right place, and the right amount of nitric oxide.Am J Physiol Renal Physiol 285:F178–F190
159. O’Brien RJ, Lau LF, Huganir RL (1998) Molecularmechanisms of glutamate receptor clustering at excitatorysynapses. Curr Op Neurobiol 8:364–369
160. Fiscus RR (2002) Involvement of cyclic GMP and proteinkinase G in the regulation of apoptosis and survival inneural cells. Neurosignals 11:175–190
161. Knowles RG, Palacios M, Palmer R et al (1989) Formation ofnitric oxide from L-arginine in the central nervous system: atransduction mechanism for stimulation of the solubleguanylate cyclase. Proc Natl Acad Sci USA 86:5159–5162
162. Dawson VL, Dawson TM, London ED et al (1991) Nitricoxide mediates glutamate neurotoxicity in primary corticalcultures. Proc Natl Acad Sci USA 88:6368–6371
163. Shinkai T, Ohmori O, Nakamura J (2002) Allelic associa-tion of the neuronal nitric oxide synthase (NOS1) gene withschizophrenia. Mol Psychiatry 7:560–563
164. Zheng Y, Li H, Qin W et al (2005) Association of thecarboxyl-terminal PDZ ligand of neuronal nitric oxidesynthase gene with schizophrenia in the Chinese Hanpopulation. Biochem Biophys Res Commun 328:809–815
165. Puri V, McQuillin A, Thirumalai S et al (2005) Failure toconfirm allelic association between markers at the CAPONgene locus and schizophrenia in a British sample. BiolPsychiatry 59:195–197
166. Mortensen O, Amara S (2003) Dynamic regulation of thedopamine transporter. Eur J Pharmacol 479:159–170
167. Pogun S, Baumann M, Kuhar M (1994) Nitric oxide inhibits[3H]dopamine uptake. Brain Res 641:83–91
168. Xing G, Chavko M, Zhang LX et al (2002) Decreasedcalcium-dependent constitutive nitric oxide synthase(cNOS) activity in prefrontal cortex in schizophrenia anddepression. Schizophr Res 58:21–30
169. Ramirez J, Garnica R, Boll MC et al (2004) Low concen-tration of nitrite and nitrate in the cerebrospinal fluid fromschizophrenic patients: a pilot study. Schizophr Res 68:357–361
170. Perez-Neri I, Ramırez J, Montes S et al (2005) Reducednitric oxide biosynthesis in schizophrenia alters citrullinemetabolism in the central nervous system. Program No.116.4. Abstract Viewer/Itinerary Planner. Society for Neu-roscience, Washington, DC
171. Das I, Ramchand C, Gliddon A et al (1998) Nitric oxide,free radicals and polyamines may have a role in themembrane pathology of schizophrenia. Neuropsychobiolo-gy 37:65–67
172. Yanik M, Vural H, Kocyigit A et al (2003) Is the arginine-nitric oxide pathway involved in the pathogenesis ofschizophrenia? Neurobiology 47:61–65
173. Zoroglu S, Herken H, Yurekli M et al (2002) Thepossible pathophysiological role of plasma nitric oxideand adrenomedullin in schizophrenia. J Psychiatry Res36:309–315
174. Srivastava N, Barthwal M, Dalal P et al (2001) Nitritecontent and antioxidant enzyme levels in the blood ofschizophrenia patients. Psychopharmacology 158:140–145
175. Akyol O, Herken H, Uz E et al (2002) The indices ofendogenous oxidative and antioxidative processes in plasmafrom schizophrenic patients: the possible role of oxidant/antioxidant imbalance. Prog Neuropsychopharmacol BiolPsychiatry 26:995–1005
176. Das I, Khan N, Puri B et al (1995) Elevated platelet calciummobilization and nitric oxide synthase activity may reflectabnormalities in schizophrenic brain. Biochem Biophys ResCommun 212:375–380
177. Ellis G, Adatia I, Yazdanpanah M et al (1998) Nitrite andnitrate analyses: a clinical biochemistry perspective. ClinBiochem 31:195–220
178. Tsikas D (2005) Analysis of the L-arginine/nitric oxidepathway: the unique role of mass spectrometry. Curr PharmAnal 1:15–30
179. Tsikas D (2005) Methods of quantitative analysis of thenitric oxide metabolites nitrite and nitrate in humanbiological fluids. Free Rad Res 39:797–815
180. Bolanos JP, Peuchen S, Heales S et al (1994) Nitric oxide-mediated inhibition of the mitochondrial respiratory chainin cultured astrocytes. J Neurochem 63:910–916
181. Brookes PS, Levonen AL, Shiva S et al (2002) Mitochon-dria: regulators of signal transduction by reactive oxygenand nitrogen species. Free Rad Biol Med 33:755–764
1292 Neurochem Res (2006) 31:1279–1294
123

182. Chiueh C, Andoh T, Chock P (2005) Roles of thioredoxin innitric oxide-dependent preconditioning-induced toleranceagainst MPTP neurotoxin. Toxicol Appl Pharmacol207:S96–S102
183. Khan M, Jatana M, Elango C et al (2006) Cerebrovascularprotection by various nitric oxide donors in rats afterexperimental stroke. Nitric Oxide 15:114–124
184. Figueroa S, Lopez E, Arce C et al (2005) SNAP, a NOdonor, induces cellular protection only when corticalneurons are submitted to some aggression process. BrainRes 1034:25–33
185. Tejedo JR, Cahuana GM, Ramırez R et al (2004) Nitricoxide triggers the phosphatidylinositol 3-kinase/Akt sur-vival pathway in insulin-producing RINm5F cells by arous-ing Src to activate insulin receptor substrate-1.Endocrinology 145:2319–2327
186. Perez-Severiano F, Escalante B, Rıos C (1998) Nitric oxidesynthase inhibition prevents acute quinolinate-inducedstriatal neurotoxicity. Neurochem Res 23:1297–1302
187. Perez-Severiano F, Rıos C, Segovia J (2000) Striataloxidative damage parallels the expression of a neurologicalphenotype in mice transgenic for the mutation of Hunting-ton’s disease. Brain Res 862:234–237
188. Perez-Severiano F, Santamarıa A, Pedraza-Chaverri J et al(2004) Increased formation of reactive oxygen species, butnot changes in glutathione peroxidase activity, in striata ofmice transgenic for the Huntington’s disease mutation.Neurochem Res 29:729–733
189. Rodrıguez E, Mendez-Armenta M, Villeda-Hernandez Jet al (1999) Dapsone prevents morphological lesions andlipid peroxidation induced by quinolinic acid in rat corpusstriatum. Toxicology 139:111–118
190. Perez-Severiano F, Salvatierra-Sanchez R, Rodrıguez-PerezM et al (2004) S-Allylcysteine prevents amyliod-b peptide-induced oxidative stress in rat hippocampus and ameliorateslearning deficits. Eur J Pharmacol 489:197–202
191. Alcaraz-Zubeldia M, Montes S, Rios C (2001) Participationof manganese-superoxide dismutase in the neuroprotectionexerted by copper sulfate against 1-methyl 4-phenylpyrid-inium neurotoxicity. Brain Res Bull 55:277–279
192. Rojas P, Rios C (1993) Increased striatal lipid peroxidationafter intracerebroventricular MPP+ administration to mice.Pharmacol Toxicol 72:364–368
193. Mahadik S, Evans D, Lal H (2001) Oxidative stress and roleof antioxidant and x-3 essential fatty acid supplementationin schizophrenia. Prog Neuropsychopharmacol Biol Psychi-atry 25:463–493
194. Nishioka N, Arnold S (2004) Evidence for oxidative DNAdamage in the hippocampus of elderly patients with chronicschizophrenia. Am J Geriatr Psychiatry 12:167–175
195. Kruman I, Wersto R, Cardozo-Pelaez F et al (2004) Cellcycle activation linked to neuronal cell death initiated byDNA damage. Neuron 41:549–561
196. Copani A, Uberti D, Sortino M et al (2001) Activation ofcell-cycle-associated proteins in neuronal death: a manda-tory or dispensable path? Trends Neurosci 24:25–31
197. Bossenmeyer-Pourie C, Lievre V, Grojean S et al (2002)Sequential expression patterns of apoptosis- and cell cycle-related proteins in neuronal response to severe or mildtransient hypoxia. Neuroscience 114:869–882
198. Mahadik S, Mukherjee S, Scheffer R et al (1998) Elevatedplasma lipid peroxides at the onset of nonaffective psycho-sis. Biol Psychiatry 43:674–679
199. Ranjekar P, Hinge A, Hedge M et al (2003) Decreasedantioxidant enzymes and membrane essential polyunsatu-
rated fatty acids in schizophrenic and bipolar mood disorderpatients. Psychiatry Res 121:109–122
200. Klein J, Ackerman S (2003) Oxidative stress, cell cycle, andneurodegeneration. J Clin Invest 111:785–793
201. Akyol O, Yanik M, Elyas H et al (2005) Associationbetween Ala-9Val polymorphism of Mn-SOD gene andschizophrenia. Prog Neuropsychopharmacol Biol Psychiatry29:123–131
202. Schwartz P, Reaume A, Scott R et al (1998) Effects of over-and under-expression of Cu,Zn-superoxide dismutase onthe toxicity of glutamate analogs in transgenic mousestriatum. Brain Res 789:32–39
203. Przedborski S, Kostic V, Jackson-Lewis V et al (1992)Transgenic mice with increased Cu/Zn-superoxide dismu-tase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neurosci12:1656–1667
204. Alcaraz-Zubeldia M, Rojas P, Boll C et al (2001) Neuro-protective effect of acute and chronic administration ofcopper (II) sulfate against MPP+ neurotoxicity in mice.Neurochem Res 26:59–64
205. Sirota P, Gavrieli R, Wolach B (2003) Overproduction ofneutrophil radical oxygen species correlates with negativesymptoms in schizophrenic patients: parallel studies onneutrophil chemotaxis, superoxide production and bacteri-cidal activity. Psychiatry Res 121:123–132
206. Yao J, Reddy R, Van Kammen D (1999) Human plasmaglutathione peroxidase and symptom severity in schizo-phrenia. Biol Psychiatry 45:1512–1515
207. Sagara Y (1998) Induction of reactive oxygen species inneurons by haloperidol. J Neurochem 71:1002–1012
208. Smythies J (1999) Redox mechanisms at the glutamatesynapse and their significance: a review. Eur J Pharmacol370:1–7
209. Hynd M, Scott H, Dodd P (2004) Glutamate-mediatedexcitotoxicity and neurodegeneration in Alzheimer’sdisease. Neurochem Int 45:583–595
210. Droge W (2002) Free radicals in the physiological control ofcell function. Physiol Rev 82:47–95
211. Klann E, Thiels E (1999) Modulation of protein kinases andprotein phosphatases by reactive oxygen species: implica-tions for hippocampal synaptic plasticity. Prog Neuropsy-chopharmacol Biol Psychiatry 23:359–376
212. Dean C (2006) Antipsychotic-associated neuronal changesin the brain: toxic, therapeutic, or irrelevant to the long-term outcome of schizophrenia? Prog Neuropsychophar-macol Biol Psychiatry 30:174–189
213. Ozaki T, Mui K, Yamagami M (1999) Comparison of theeffects of dopamine D1 and D2 receptor antagonists onnerve growth factor mRNA expression. Eur J Pharmacol369:133–143
214. Ozaki T (2000) Comparative effects of dopamine D1 and D2
receptor antagonists on nerve growth factor protein induc-tion. Eur J Pharmacol 402:39–44
215. He J, Xu H, Yang Y et al (2004) Neuroprotective effects ofolanzapine on methamphetamine-induced neurotoxicity areassociated with an inhibition of hyperthermia and preven-tion of Bcl-2 decrease in rats. Brain Res 1018:186–192
216. Bradley R, Dwyer D (2004) Olanzapine produces trophiceffects in vitro and stimulates phosphorylation of Akt/PKB,ERK1/2, and the mitogen-activated protein kinase p38.Brain Res 1011:58–68
217. He J, Xu H, Yang Y et al (2006) The effects of chronicadministration of quetiapine on the phencyclidine-inducedreference memory impairment and decrease of Bcl-XL/Bax
Neurochem Res (2006) 31:1279–1294 1293
123

ratio in the posterior cingulate cortex in rats. Behav BrainRes 168:236–242
218. Bai O, Zhang H, Li X (2004) Antipsychotic drugs clozapineand olanzapine upregulate bcl-2 mRNA and protein in ratfrontal cortex and hippocampus. Brain Res 1010:81–86
219. Luo C, Xu H, Li X (2004) Post-stress changes in BDNFimmunoreactivities in hippocampal neurons: effect ofchronic administration of olanzapine. Brain Res1025:194–202
220. Zhong L, Sarafian T, Kane D et al (1993) bcl-2 inhibitsdeath of central neural cells induced by multiple agents.Proc Natl Acad Sci USA 90:4533–4537
221. Susin S, Zamzami N, Castedo M et al (1996) Bcl-2 inhibitsthe mitochondrial release of an apoptogenic protease. J ExpMed 184:1331–1341
222. Danial N, Korsmeyer S (2004) Cell death: critical controlpoints. Cell 116:205–219
223. Eaton M, Whittemore S (1996) Autocrine BDNF secretionenhances the survival and serotonergic differentiation ofraphe neuronal precursor cells grafted into the adult ratCNS. Exp Neurol 140:105–114
224. Maragnoli M, Fumagalli F, Gennarelli M et al (2004)Fluoxetine and olanzapine have synergistic effects in themodulation of fibroblast growth factor 2 expression withinthe rat brain. Biol Psychiatry 55:1095–1102
225. Rivas M, Molteni R, Tascedda F et al (1999) Selectivemodulation of fibroblast growth factor-2 expression in therat brain by the atypical antipsychotic clozapine. Neuro-pharmacology 38:1075–1082
226. Ichikawa J, Ishii H, Bonaccorso S et al (2001) 5-HT2A andD2 receptor blockade increases cortical DA release via5-HT1A receptor activation: a possible mechanism ofatypical antipsychotic-induced cortical dopamine release.J Neurochem 76:1521–1531
227. Newman-Tancredi A, Gavaudan S, Conte C et al (1998)Agonist and antagonist action of antipsychotic agents at5-HT1A receptors: a [35S]GTPcS binding study. EurJ Pharmacol 355:245–256
228. Rollema H, Lu Y, Schmidt A et al (1997) Clozapineincreases dopamine release in prefrontal cortex by 5-HT1A
receptor activation. Eur J Pharmacol 338:R3–R5229. Rollema H, Lu Y, Schmidt A et al (2000) 5-HT1A receptor
activation contributes to ziprasidone-induced dopaminerelease in the rat prefrontal cortex. Biol Psychiatry48:229–237
230. Oosterink B, Korte S, Nyakas C et al (1998) Neuroprotec-tion against N-methyl-D-aspartate-induced excitotoxicity inrat magnocellular nucleus basalis by the 5-HT1A receptoragonist 8-OH-DPAT. Eur J Pharmacol 358:147–152
231. Cosi C, Waget A, Rollet K et al (2005) Clozapine,ziprasidone and aripiprazole against kainic acid-inducedlesion of the striatum in mice, in vivo: role of 5-HT1A
receptor activation. Brain Res 1043:32–41232. Harkany T, Mulder J, Horvath K et al (2001) Oral post-
lesion administration of 5-HT1A receptor agonist repinotanhydrochloride (BAY X 3702) attenuates NMDA-induceddelayed neuronal death in rat magnocellular nucleusbasalis. Neuroscience 108:629–642
233. Kline A, Yu J, Horvath E et al (2001) The selective 5HT1A
receptor agonist repinotan HCl attenuates histopathologyand spatial learning deficits following traumatic brain injuryin rats. Neuroscience 106:547–555
234. Schaper C, Zhu Y, Kouklei M et al (2000) Stimulation of5-HT1A receptors reduces apoptosis after transient fore-brain ischemia in the rat. Brain Res 883:41–50
235. Alessandri B, Tsuchida E, Bullock R (1999) The neuropro-tective effect of a new serotonin receptor agonist, BAYX3702, upon focal ischemic brain damage caused by acutesubdural hematoma in the rat. Brain Res 845:232–235
236. Suchanek B, Struppeck H, Fahrig T (1998) The 5-HT1A
receptor agonist BAY x 3702 prevents staurosporine-induced apoptosis. Eur J Pharmacol 355:95–101
237. Ahlemeyer B, Breier H, Semkova I et al (2000) S-100bprotects cultured neurons against glutamate- and stauro-sporine-induced damage and is involved in the antiapop-totic action of the 5 HT1A-receptor agonist, Bay x 3702.Brain Res 858:121–128
238. Ahlemeyer A, Glaser A, Schapker C et al (1999) The5-HT1A receptor agonist Bay x 3702 inhibits apoptosisinduced by serum deprivation in cultured neurons. EurJ Pharmacol 370:211–216
239. Galter D, Unsicker K (2000) Sequential activation of the5-HT1A serotonin receptor and TrkB induces the seroto-nergic neuronal phenotype. Mol Cell Neurosci 15:446–455
240. Aghajanian GK, Marek GJ (2000) Serotonin model ofschizophrenia: emerging role of glutamate mechanisms.Brain Res Rev 31:302–312
241. Meltzer HY, Li Z, Kaneda Y et al (2003) Serotoninreceptors: their key role in drugs to treat schizophrenia.Prog Neuropsychopharmacol Biol Psychiatry 27:1159-1172
242. Burnet P, Eastwood S, Harrison P (1997) [3H]WAY-100635for 5-HT1A receptor autoradiography in human brain: acomparison with [3H]8-OH-DPAT and demonstration ofincreased binding in the frontal cortex in schizophrenia.Neurochem Int 30:565–574
243. Mauler F, Fahring T, Horvath E et al (2001) Inhibition ofevoked glutamate release by the neuroprotective 5-HT1A
receptor agonist BAY x 3702 in vitro and in vivo. Brain Res888:150–157
244. Yamamoto B, Cooperman M (1994) Differential effects ofchronic antipsychotic drug treatment on extracellular glu-tamate and dopamine concentrations. J Neurosci 14:4159–4166
245. Okamura N, Hashimoto K, Kanahara N et al (2003)Protective effect of the antipsychotic drug zotepine ondizocilpine-induced neuropathological changes in rat retro-splenial cortex. Eur J Pharmacol 461:93–98
246. Khatua A, Bhattacharyya M (2001) NADPH-inducedoxidative damage of rat liver microsomes: protective roleof chlorpromazine and trifluoperazine. Pol J Pharmacol53:629–634
1294 Neurochem Res (2006) 31:1279–1294
123