new stable synthetic bacteriochlorins for photodynamic therapy of melanoma
TRANSCRIPT

New stable synthetic bacteriochlorins for photodynamic therapy of melanoma.
Pawel Mroza,b, Ying-Ying Huanga,b, Sahar Janjuaa,c, Timur Zhiyentayeva,d, Christian Ruziée, K.
Eszter Borbase, Dazhong Fane, Michael Krayere, Thiagarajan Balasubramanianf, Eun Kyung Yangg, Hooi Ling Keeg, Dewey Holteng, Jonathan S. Lindseye, and Michael R Hamblina,b,h*
a Wellman Center for Photomedicine, Massachusetts General Hospital, Boston MA; b Department of Dermatology, Harvard Medical School, Boston, MA;
c Aga Khan Medical College, Karachi, Pakistan; d Chemistry Department, M.V. Lomonosov Moscow State University, Moscow, Russia;
e Department of Chemistry, North Carolina State University, Raleigh, NC f NIRvana Pharmaceuticals, Inc., Raleigh, NC;
g Department of Chemistry, Washington University, St. Louis, MO; h Harvard-MIT Division of Health Sciences and Technology, Cambridge, MA
ABSTRACT
Photodynamic therapy (PDT) has been successfully used to treat many malignancies, and has afforded highly encouraging results in skin cancers such as basal cell carcinoma. However, pigmented melanoma remains a notable exception from the range of tumors treated by PDT largely due to the fact that melanin has high absorption of light in wavelength regions where most clinically approved photosensitizers (PS) absorb light (600-690 nm). Moreover, melanoma cells sequester exogenous molecules including photosensitizers inside melanosomes. The aforementioned drawbacks of the clinically used PS have motivated us to search for new classes of PS with improved spectral properties, such as bacteriochlorins (BC) to be used in PDT of melanoma. To overcome the PDT-resistance mechanisms of melanoma, particularly the high optical absorption of melanin, three near-infrared (NIR) absorbing synthetic stable BC were used in PDT treatment of melanoma. Dose and fluence dependent cell killing, intracellular localization (particularly in melanosomes), and correlation between the melanin level and cell death were examined. Intracellular melanosomes are ruptured after illumination as shown by electron microscopy. The best in vitro performing BC were tested upon delivery in micellar nanoparticles against a mouse pigmented melanoma. Two of the BC were effective at significantly lower concentrations (<0.5 μM) than common photosensitizers in present use.
Keywords: melanoma, photodynamic therapy, multidrug resistance, melanosomes, bacteriochlorins *E-mail: [email protected]
1. INTRODUCTION Malignant melanoma is a cancer that arises from melanocytes, the specialized pigmented cells that are found predominantly in the skin. It is the most deadly form of skin cancer. Although melanoma accounts for only 4% of skin cancer cases, it causes 79% of all skin cancer related deaths.1 If diagnosed early, melanoma can be cured by surgical resection with about 80% effectiveness. However, once metastases occur, it is largely refractory to existing therapies and has a very poor prognosis, with a median survival rate of 6 months, and a 5-year survival rate of less than 5%.2 International concern is engendered by the relatively rapid increase in diagnosis of melanoma, an increase that has been termed an epidemic given that the number of melanoma cases worldwide is increasing faster than any other cancer, and in 2002 was six times higher than in 1950.3 In 2007 there were 108,230 new cases of melanoma diagnosed of which 48,290 were noninvasive and 59,940 were invasive, and 8110 deaths were expected.4 Melanoma is the leading cause of cancer death in adults between 20 and 40 years old in the USA.
Melanins are the principal surface pigments in vertebrates and, in humans, play a major role in photoprotection. Although melanin has a mainly protective function in the skin, the process of melanogenesis represents a potential
Photodynamic Therapy: Back to the Future, edited by David H. Kessel, Proc. of SPIE Vol. 7380,73802S · © 2009 SPIE · CCC code: 1605-7422/09/$18 · doi: 10.1117/12.823060
Proc. of SPIE Vol. 7380 73802S-1

cellular hazard and is confined to special membrane-limited organelles (melanosomes) in a set of specialized cells – melanocytes, which synthesize the pigment and transfer it to recipient cells. Once made the melanosomes are moved along arm-like structures called dendrites and transferred to keratinocytes as a cap that protects the nucleus of the cell. In humans keratinocytes and melanocytes are grouped together (in the ratio of 36:1) to form an epidermal melanin unit (EMU).5 Numerous stimuli are able to alter melanogenesis or the production of melanin by cultured melanocytes, although the method by which such stimuli work is not fully understood. Vitamin D metabolites, retinoids, α-MSH, diacylglycerol analogues, thymidine dinucleotides, UV irradiation and chemotherapeutics can trigger melanogenesis and in turn, pigmentation.6 Malignant melanocytes tend to exhibit up-regulated melanogenesis and have defective melanosomes.7 Melanomas can vary from non-pigmented tumors that have no melanin whatsoever, through moderately pigmented to highly pigmented tumors. It is thought that the level of melanin produced is proportional to the degree of differentiation and inversely proportional to the growth rate.8
Cutaneous melanoma remains a management challenge.9 Among cytotoxic agents, dacarbazine remains the benchmark, since it produces responses in 15 to 20 percent of patients, and the median duration of the response is four months.10 Dacarbazine, carmusitine, paclitaxel, temozolomide and cisplatin have shown limited single-agent activity in metastatic disease10 probably due to melanoma cells having low levels of spontaneous apoptosis in vivo, and therefore being particularly resistant to these therapies.11 In response to these challenges, a variety of regimens combining dacarbazine with other cytotoxic agents, tamoxifen, or interferon alfa have been proposed and have shown promising response rates in single-institution phase 2 trials and potential survival advantages in small phase 3 trials.12 Only interferon alfa-2b has been shown to have a reproducible benefit.13 However, high-dose interferon alfa-2b has many side effects;14 therefore, other potential adjuvant therapies have been proposed. These include vaccines15-19 or high-dose bolus interleukin-220 alone or in combination focused with dacarbazine and cisplatin (i.e., biochemotherapy). An interesting approach of Naylor and colleagues21 employed topical 5% imiquimod cream and irradiation of skin metastases with a CW 810-nm laser. It is proposed that laser irradiation of the pigmented lesions produced local thermal damage and tumor destruction with antigen release, and the immune system was activated by the imiquimod to widen the response to distant non-irradiated lesions.
PDT uses a non-toxic dye molecule or photosensitizer (PS) that is activated to an excited singlet state when it absorbs a photon of an appropriate wavelength of visible light.22 This excited PS can then undergo a transition to a long-lived excited triplet state that allows energy transfer to the (triplet) ground state of molecular oxygen to produce the highly cytotoxic singlet oxygen (Type II reaction), or else allows electron transfer involving ground-state oxygen (Type I reaction) with the ultimate formation of products such as the superoxide radical anion or hydroxyl radicals. The reactive oxygen species (ROS) formed by either energy or electron transfer can oxidize important biological molecules such as proteins, lipids and nucleic acids.
There are three main mechanisms that make PDT an effective anti-cancer procedure: (i) direct tumor killing by ROS, (ii) tumor-associated vascular damage, and (iii) activation of anti-tumor immune response.
The relative contributions of all these mechanisms to the overall anti-tumor effects of PDT are difficult to unambiguously establish. The prevailing view is that all three mechanisms are necessary for the optimal tumor damage. Vascular damage tends to be more pronounced when light is delivered at early time points after PS injection, when more of the PS is still localized in the tumor blood vessels.
Bacteriochlorins are tetrapyrrolic macrocycles that contain alternating pyrrole and pyrroline (i.e., reduced pyrrole) rings. The macrocycle structure occurs naturally in photosynthetic pigments (bacteriochlorophylls a and b) found in purple photosynthetic bacteria.23 The presence of the reduced rings in the tetrapyrrole macrocycle has a pronounced effect on the absorption spectra, particularly in comparison with the spectra of the fully unsaturated porphyrin compounds. Bacteriochlorins and bacteriopurpurins (i.e., bacteriochlorins that contain a fifth ring fused to the tetrapyrrolic macrocycle) have intense absorption bands in the region of 720-850 nm. The large absorption peak in the NIR spectrum that is typical of bacteriochlorins is considered to be ideal for maximizing light penetration through tissue, given that both absorption and scattering of light in the 700-800 nm region are minimal. During the past decade, several naturally occurring or naturally derived bacteriochlorins have been evaluated in PDT applications, and some of them have shown significant in vivo efficacy. The most frequently employed BC for PDT has been the Pd-containing bacteriopheophorbide derivative known as TOOKAD, padoporfin or WST09 that is in clinical trials for prostate cancer.24 A related water-soluble Pd-bacteriochlorin derivative called WST11 has been tested for PDT of mouse
Proc. of SPIE Vol. 7380 73802S-2
melanoma xenografts.25 A recent report showed the application of bacteriochlorin p6 as an effective PS for in vitro and in vivo treatment of RIF tumors.26
Bacteriochlorins derived from naturally occurring bacteriochlorophyll have the following limitations: 1. Limited synthetic malleability owing to the presence of substituents about the perimeter of the macrocycle. 2. Susceptibility to unwanted dehydrogenation (yielding the chlorin or porphyrin which lacks the NIR absorption). 3. The requirement for harsh conditions and/or tedious steps for the modification of functional groups that are
already present in the macrocycle. To overcome the aforementioned limitations, a de novo synthetic pathway to stable bacteriochlorins has been developed.27 A key design feature of the synthetic bacteriochlorins is a geminal dimethyl group in each reduced, pyrroline ring. The geminal dimethyl group in each pyrroline ring locks-in the bacteriochlorin chromophore and precludes dehydrogenation or tautomerization processes, thereby affording a stable macrocycle. This structural feature dramatically increases the chemical stability and eliminates susceptibility to air oxidation.
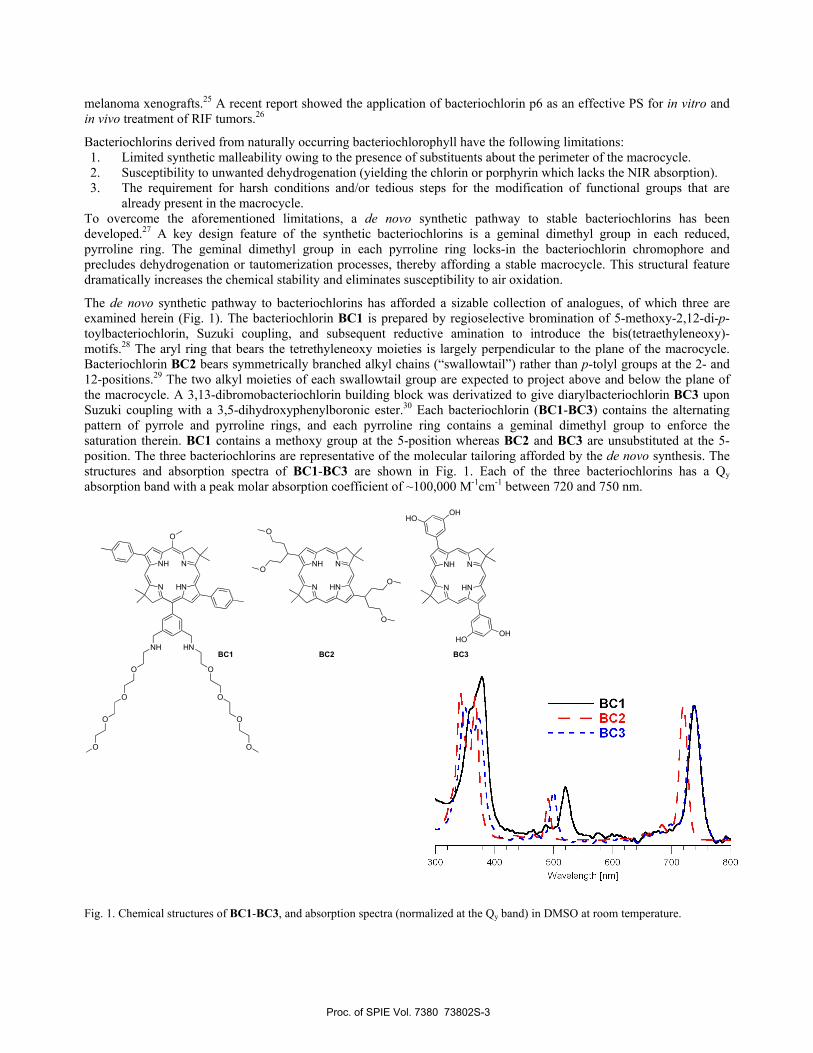
The de novo synthetic pathway to bacteriochlorins has afforded a sizable collection of analogues, of which three are examined herein (Fig. 1). The bacteriochlorin BC1 is prepared by regioselective bromination of 5-methoxy-2,12-di-p-toylbacteriochlorin, Suzuki coupling, and subsequent reductive amination to introduce the bis(tetraethyleneoxy)-motifs.28 The aryl ring that bears the tetrethyleneoxy moieties is largely perpendicular to the plane of the macrocycle. Bacteriochlorin BC2 bears symmetrically branched alkyl chains (“swallowtail”) rather than p-tolyl groups at the 2- and 12-positions.29 The two alkyl moieties of each swallowtail group are expected to project above and below the plane of the macrocycle. A 3,13-dibromobacteriochlorin building block was derivatized to give diarylbacteriochlorin BC3 upon Suzuki coupling with a 3,5-dihydroxyphenylboronic ester.30 Each bacteriochlorin (BC1-BC3) contains the alternating pattern of pyrrole and pyrroline rings, and each pyrroline ring contains a geminal dimethyl group to enforce the saturation therein. BC1 contains a methoxy group at the 5-position whereas BC2 and BC3 are unsubstituted at the 5-position. The three bacteriochlorins are representative of the molecular tailoring afforded by the de novo synthesis. The structures and absorption spectra of BC1-BC3 are shown in Fig. 1. Each of the three bacteriochlorins has a Qy absorption band with a peak molar absorption coefficient of ~100,000 M-1cm-1 between 720 and 750 nm.
Fig. 1. Chemical structures of BC1-BC3, and absorption spectra (normalized at the Qy band) in DMSO at room temperature.
N HN
NNH
O
NH HN
O
O
O
O
O
O
O
O
N HN
NNH
O
OO
O
N HN
NNH
HOOH
HOOH
BC1 BC2 BC3
Proc. of SPIE Vol. 7380 73802S-3

The synthetic bacteriochlorins have several advantages over conventional PS, including naturally occurring bacteriochlorins and their derivatives: (i) exceptional resiliency toward degradation, (ii) a sufficiently concise and robust synthesis to allow exploitation for biomedical applications, (iii) characteristic superior absorption spectra that avoid interference by the black pigment melanin and provide deep tissue penetration of incident light, (iv) ready tunability of photophysical and redox characteristics via manipulation of the peripheral substituents (and central metal ion), which may require changes at only one or two sites to achieve a substantial effect.31,32
In this report we tested the three bacteriochlorins in combination with 730-nm illumination from a diode laser and compared the BC with the clinically approved PS Photofrin (absorbs at 630 nm) and with LuTex (an established NIR absorbing PS). We used a human melanoma cell line Cmel (strongly pigmented) and a set of B16 mouse melanoma variants (B16G4F-weakly pigmented, B16F1-pigmented and B16F10-highly pigmented and B16F10-GFP, the latter of which contains the green fluorescent protein (GFP) useful for microscopy imaging studies).
2. MATERIALS AND METHODS 2.1 Photosensitizers
Bacteriochlorins BC1-BC3 were obtained as described above. Photofrin (PF) was obtained from QLT, Inc., and lutetium texaphyrin (LuTex) was obtained from Pharmacyclics, Inc. For studies involving cell cultures, each PS was dissolved in DMSO at a concentration of 5 mM and stored in the dark at room temperature. In each experiment, the stock solution was diluted into complete medium to obtain the desired final concentration.
2.2 Cell Culture
Mouse B16F1 and B16F10 melanoma cells were obtained from ATCC (Manassas, VA). Mouse B16-G4F cells were a gift from Dr. Eberle from the Department of Biomedicine, University Hospital in Basel, Switzerland. Human melanoma cell line C-mel was a gift of Dr. Tsao from Wellman Center for Photomedicine, MGH, Boston, USA.
All cells were cultured in RPMI1600 medium (Gibco Invitrogen, Carlsbad, CA) with L-glutamine and NaHCO3 supplemented with heat inactivated fetal bovine serum 10% (vol/vol), penicillin (100 U/mL) and streptomycin (100 µg/mL) (all from Sigma, St. Louis, MO) at 37 °C in a 5% CO2 humidified atmosphere in 75 cm2 flasks (BD Falcon, San Jose, CA). To increase melanin production, cells were cultured in Low-Glucose DMEM medium for 7 days before being used for experiments. B16F10-GFP cells are transduced cells that stably express GFP protein and were cultured in constant presence of 500 µg/mL G418 antibiotic (Sigma). When the cells reached 80% confluence, they were washed with PBS (Sigma) and harvested with 2 mL of 0.25% Trypsin-EDTA solution (Sigma). Cells were centrifuged and counted in trypan blue (Sigma) and plated at 5000 cells/well in flat-bottomed 96 well plates (Fisher). Cells were allowed to attach for 24 h.
2.3 Light Source
Illumination of cells and mice utilized a 730-nm laser light (Pharmacyclics, Sunnyvale, CA) for BC and LuTex, and a non-coherent white light source (Lumacare, Newport Beach, CA) fitted with a light guide containing a band pass filter (630 ± 10 nm) for PF adjusted to give a uniform spot of 4 cm in diameter with an irradiance of 100 mW/cm2 as measured with a power meter (model DMM 199 with 201 Standard head, Coherent, Santa Clara, CA).
2.4 In vitro PDT Experiments
PS were added at different concentrations to cells in fresh complete medium for 24 h incubation periods. The DMSO concentration in the medium was less than 0.5%. After incubation the medium was replaced with 200 µL of fresh medium and PDT with 730-nm light was performed. For experiments where the light dose was varied, fluences of 0 (dark toxicity) to 20 J/cm2 were used, and 4 wells (one group) were illuminated at one time. Controls were cells with no treatment and cells with light alone at the highest fluence or with BC alone. For experiments where the PS concentration was varied, a fixed fluence of 10 J/cm2 delivered at the same irradiance was used. Here additional groups of cells were incubated with all the concentrations of PS but no illumination was used to determine the dark toxicity. At the completion of the illumination the plates were returned to the incubator for 24 h before initiating further studies. We used a MTT colorimetric assay [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] that measures
Proc. of SPIE Vol. 7380 73802S-4

mitochondrial reductase activity and correlates well with colony-forming assays as a measure of cell viability, as has been described previously.33-36 The absorbance for MTT assay was read at 560 nm.
2.5 Fluorescence Microscopy – Intracellular Localization
5x105 B16F10 cells were plated on 35 mm dishes and allowed overnight to attach. The next day 1 µM of BC1, BC2 or BC3 in culture medium was added and incubated for 24 h. Cells were washed in PBS and 5 µg/mL of lysotracker or mitotracker (LysoTracker Green DND-26, MitoTracker Green FM, Molecular Probes Invitrogen) was added and incubated for 30 min at 37 °C. For melanosomal co-localization, cells were incubated with the appropriate BC for 24 h, washed with PBS and fixed for 20 min in Cytofix/Cytoperm buffer (BD Biosciences). Next, cells were incubated with anti-TRP1 antibody37 (Abcam, Cambridge, MA) for 1 h at room temperature, washed with PBS and further incubated with FITC-conjugated anti-mouse antibody (Sigma) for 20 min.
Cells were again washed in PBS and 5-10 min later a Leica DMR confocal laser fluorescence microscope (Leica Mikroskopie und Systeme GmbH, Wetzler, Germany) with excitation by a 488 nm argon laser and emission with either a bandpass filter (525 ± 10 nm), or a 580-nm longpass filter and a 63x1.20 NA water immersion lens was used to image the cells at a resolution of 1024x1024 pixels. Images were acquired using TCS NT software (Version 1.6.551, Leica Lasertechnik, Heidelberg, Germany).
2.6 Isolation of Melanosomes from Melanoma Cells.
5x105 B16F10 cells were plated on 35 mm dishes and allowed overnight to attach. Next a 5 μM solution of BC2 was added to cells, and the cells were incubated for 24 h. As controls we used the same cells without BC2 incubation. Then the cells were scraped into PBS solution in the presence of 0.5 mM protease inhibitor. The cells were pelleted by centrifugation (500g, 5 min), and the D-PBS was discarded. The cells were resuspended in the lysis buffer (10 mM Tris/10 mM KCl/150 mM MgCl2, pH 6.7) and incubated on ice for 5 min. The samples were subjected to one freeze/thaw cycle and then dounce homogenized (30 strokes). A sucrose buffer (10 mM Tris/10 mM KCl/150 mM MgCl2/34.2% sucrose, pH 6.7; 1/3 volume) was added to the lysate. First nuclei (1,000g, 5 min), then mitochondria (5,000g, 10 min) were removed by centrifugation. The resultant supernatant was further centrifuged to collect melanosomes (20,000g, 15 min). The melanosome-containing pellet was resuspended in the sucrose buffer (0.25 M sucrose/10 mM Tris, pH 7.0).38
The fluorescence images of vials containing the isolated melanosomes in sucrose buffer were taken with a multi-spectral fluorescence imaging camera system (CRI Maestro)39 with the excitation at 488 nm and collected fluorescence at >700 nm. The images were than analyzed by applying false colored red by the Maestro software.
To measure the uptake of BC2 in melanosomes, the isolated melanosomes were lysed with 1N NaOH/1%SDS and the fluorescence was measured using excitation at 402 nm and detection at 730 nm with a plate reader (Molecular Devices, Sunnyvale CA).
2.7 Preparation of BC3 in Cremophor.
The solution of BC3 in ethanol was prepared by dissolving 1 mg of BC3 in 1 mL of dried ethanol. The solution of Cremophor in ethanol was prepared by dissolving 48 mg of Cremophor in 1 mL of dried ethanol. 200 μL of BC3 solution and 525 μL of Cremophor solution were mixed in a round-bottomed flask. 1 mL of dried ethanol was added to this mixture. The solvent was removed by rotary evaporation at room temperature for 15 min. Then the film was completely dissolved under sterile conditions in 1 mL of sterile 5% dextrose solution and then used for injections in the mouse-model studies.
2.8 Animal tumor model.
C57/BL6 mice (6-8 weeks old) were purchased from Charles River Laboratories (Boston MA) and housed in a pathogen-free environment in the Massachusetts General Hospital (MGH) animal facility. All experiments were carried out according to a protocol approved by the Subcommittee on Research Animal Care (IACUC) at MGH and were in accord with NIH guidelines. Mice were inoculated with 350,000 of B16F10-GFP cells subcutaneously into the depilated right thigh. Two orthogonal dimensions (a and b) of the tumor were measured 2-3 times a week with vernier calipers. Tumor volumes were calculated as follows, volume = 4π/3 x {(a+b)/4}3. When tumors reached a diameter of 5-7 mm (7-9 days after inoculation), PDT was performed.
Proc. of SPIE Vol. 7380 73802S-5
2.9 PDT and tumor response.
Mice bearing B16F10-GFP were anaesthetized with i.p. injection of 87.5 mg/kg of ketamine and 12.5 mg/kg xylazine. The BC3 in Cremophor (5 mg/kg) was administered i.v. via the supraocular plexus. Control mice received Cremophor solution in 5% dextrose only. Fifteen minutes after BC3 injection, 730-nm illumination was performed using a homogeneous spot of 0.9 cm diameter that covered the tumor and a margin of normal tissue. The laser provided power density of 100 mW/cm2, and a fluence of 120 J/cm2 was delivered. The mice were sacrificed when any of the tumor diameters exceeded 1.5 cm.
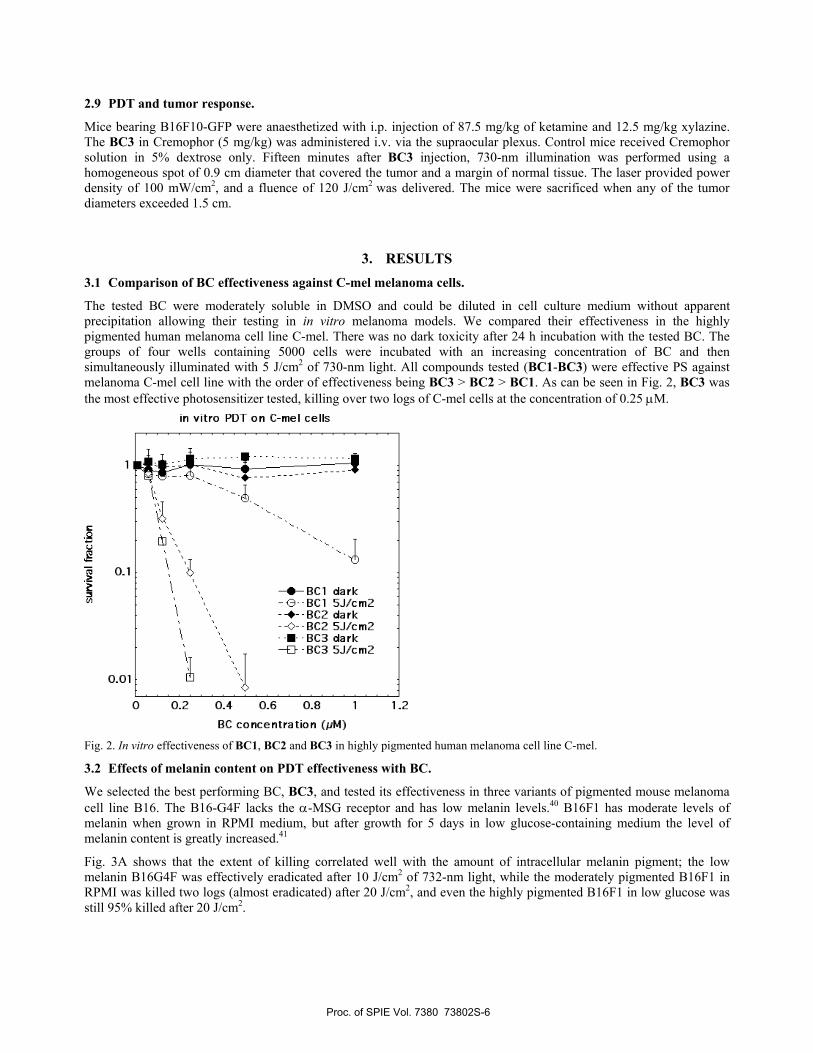
3. RESULTS 3.1 Comparison of BC effectiveness against C-mel melanoma cells.
The tested BC were moderately soluble in DMSO and could be diluted in cell culture medium without apparent precipitation allowing their testing in in vitro melanoma models. We compared their effectiveness in the highly pigmented human melanoma cell line C-mel. There was no dark toxicity after 24 h incubation with the tested BC. The groups of four wells containing 5000 cells were incubated with an increasing concentration of BC and then simultaneously illuminated with 5 J/cm2 of 730-nm light. All compounds tested (BC1-BC3) were effective PS against melanoma C-mel cell line with the order of effectiveness being BC3 > BC2 > BC1. As can be seen in Fig. 2, BC3 was the most effective photosensitizer tested, killing over two logs of C-mel cells at the concentration of 0.25 μM.
Fig. 2. In vitro effectiveness of BC1, BC2 and BC3 in highly pigmented human melanoma cell line C-mel.
3.2 Effects of melanin content on PDT effectiveness with BC.
We selected the best performing BC, BC3, and tested its effectiveness in three variants of pigmented mouse melanoma cell line B16. The B16-G4F lacks the α-MSG receptor and has low melanin levels.40 B16F1 has moderate levels of melanin when grown in RPMI medium, but after growth for 5 days in low glucose-containing medium the level of melanin content is greatly increased.41
Fig. 3A shows that the extent of killing correlated well with the amount of intracellular melanin pigment; the low melanin B16G4F was effectively eradicated after 10 J/cm2 of 732-nm light, while the moderately pigmented B16F1 in RPMI was killed two logs (almost eradicated) after 20 J/cm2, and even the highly pigmented B16F1 in low glucose was still 95% killed after 20 J/cm2.
Proc. of SPIE Vol. 7380 73802S-6
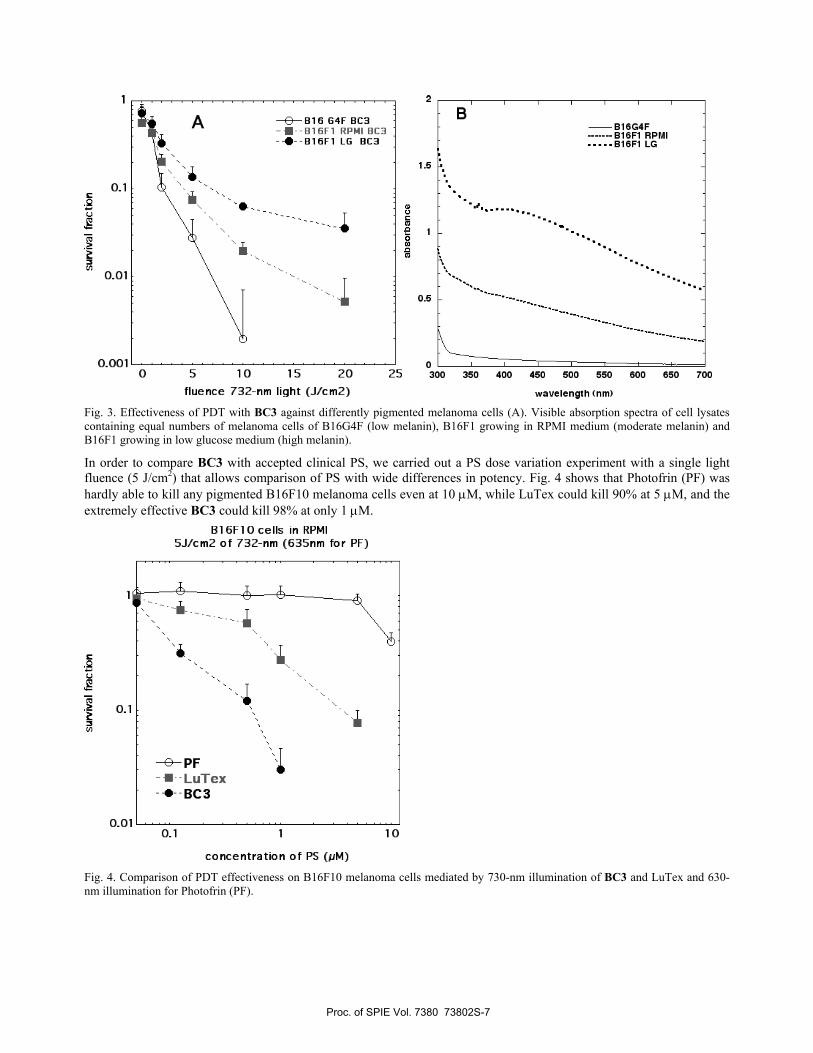
Fig. 3. Effectiveness of PDT with BC3 against differently pigmented melanoma cells (A). Visible absorption spectra of cell lysates containing equal numbers of melanoma cells of B16G4F (low melanin), B16F1 growing in RPMI medium (moderate melanin) and B16F1 growing in low glucose medium (high melanin).
In order to compare BC3 with accepted clinical PS, we carried out a PS dose variation experiment with a single light fluence (5 J/cm2) that allows comparison of PS with wide differences in potency. Fig. 4 shows that Photofrin (PF) was hardly able to kill any pigmented B16F10 melanoma cells even at 10 μM, while LuTex could kill 90% at 5 μM, and the extremely effective BC3 could kill 98% at only 1 μM.
Fig. 4. Comparison of PDT effectiveness on B16F10 melanoma cells mediated by 730-nm illumination of BC3 and LuTex and 630-nm illumination for Photofrin (PF).
Proc. of SPIE Vol. 7380 73802S-7
3.3 Intracellular localization of the BC
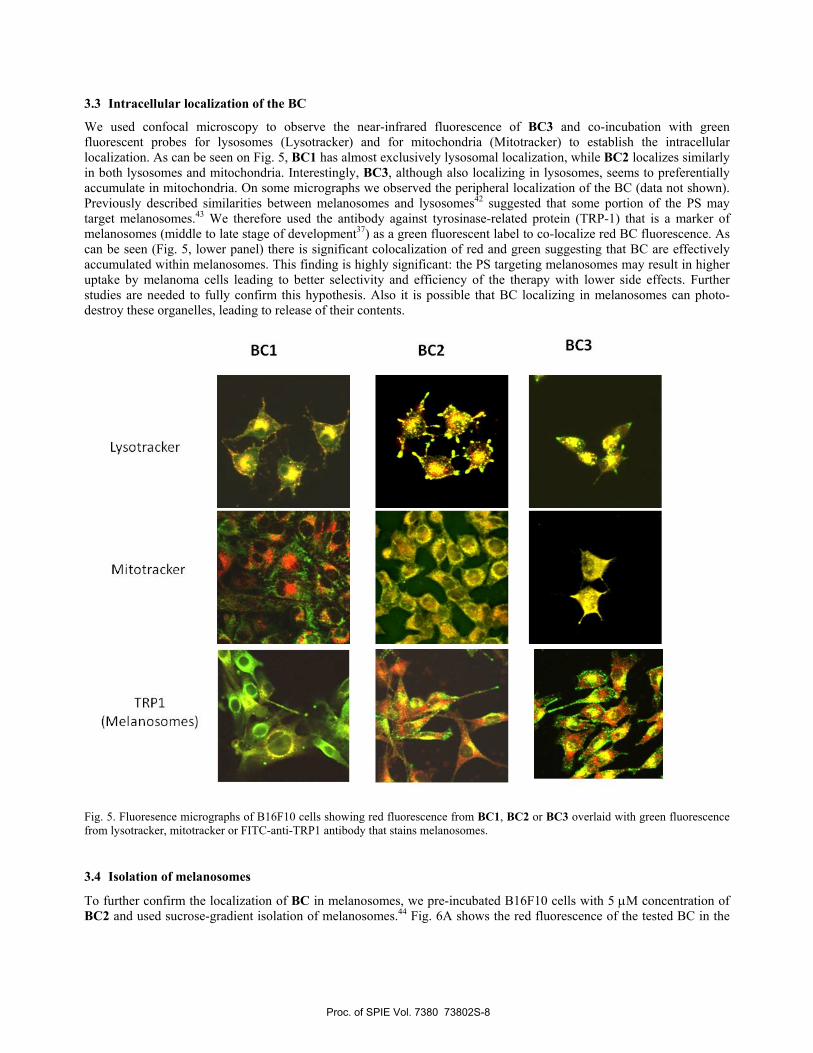
We used confocal microscopy to observe the near-infrared fluorescence of BC3 and co-incubation with green fluorescent probes for lysosomes (Lysotracker) and for mitochondria (Mitotracker) to establish the intracellular localization. As can be seen on Fig. 5, BC1 has almost exclusively lysosomal localization, while BC2 localizes similarly in both lysosomes and mitochondria. Interestingly, BC3, although also localizing in lysosomes, seems to preferentially accumulate in mitochondria. On some micrographs we observed the peripheral localization of the BC (data not shown). Previously described similarities between melanosomes and lysosomes42 suggested that some portion of the PS may target melanosomes.43 We therefore used the antibody against tyrosinase-related protein (TRP-1) that is a marker of melanosomes (middle to late stage of development37) as a green fluorescent label to co-localize red BC fluorescence. As can be seen (Fig. 5, lower panel) there is significant colocalization of red and green suggesting that BC are effectively accumulated within melanosomes. This finding is highly significant: the PS targeting melanosomes may result in higher uptake by melanoma cells leading to better selectivity and efficiency of the therapy with lower side effects. Further studies are needed to fully confirm this hypothesis. Also it is possible that BC localizing in melanosomes can photo-destroy these organelles, leading to release of their contents.
Fig. 5. Fluoresence micrographs of B16F10 cells showing red fluorescence from BC1, BC2 or BC3 overlaid with green fluorescence from lysotracker, mitotracker or FITC-anti-TRP1 antibody that stains melanosomes.
3.4 Isolation of melanosomes
To further confirm the localization of BC in melanosomes, we pre-incubated B16F10 cells with 5 μM concentration of BC2 and used sucrose-gradient isolation of melanosomes.44 Fig. 6A shows the red fluorescence of the tested BC in the
Proc. of SPIE Vol. 7380 73802S-8
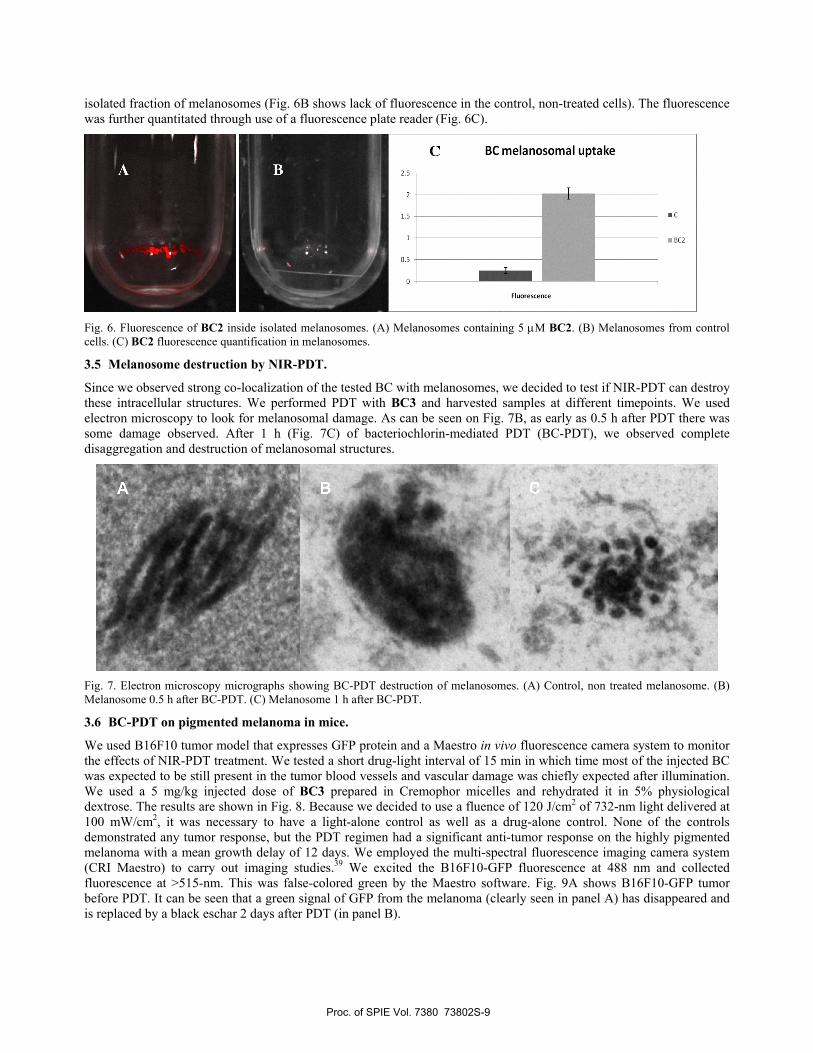
isolated fraction of melanosomes (Fig. 6B shows lack of fluorescence in the control, non-treated cells). The fluorescence was further quantitated through use of a fluorescence plate reader (Fig. 6C).
Fig. 6. Fluorescence of BC2 inside isolated melanosomes. (A) Melanosomes containing 5 μM BC2. (B) Melanosomes from control cells. (C) BC2 fluorescence quantification in melanosomes.
3.5 Melanosome destruction by NIR-PDT.
Since we observed strong co-localization of the tested BC with melanosomes, we decided to test if NIR-PDT can destroy these intracellular structures. We performed PDT with BC3 and harvested samples at different timepoints. We used electron microscopy to look for melanosomal damage. As can be seen on Fig. 7B, as early as 0.5 h after PDT there was some damage observed. After 1 h (Fig. 7C) of bacteriochlorin-mediated PDT (BC-PDT), we observed complete disaggregation and destruction of melanosomal structures.
Fig. 7. Electron microscopy micrographs showing BC-PDT destruction of melanosomes. (A) Control, non treated melanosome. (B) Melanosome 0.5 h after BC-PDT. (C) Melanosome 1 h after BC-PDT.
3.6 BC-PDT on pigmented melanoma in mice.
We used B16F10 tumor model that expresses GFP protein and a Maestro in vivo fluorescence camera system to monitor the effects of NIR-PDT treatment. We tested a short drug-light interval of 15 min in which time most of the injected BC was expected to be still present in the tumor blood vessels and vascular damage was chiefly expected after illumination. We used a 5 mg/kg injected dose of BC3 prepared in Cremophor micelles and rehydrated it in 5% physiological dextrose. The results are shown in Fig. 8. Because we decided to use a fluence of 120 J/cm2 of 732-nm light delivered at 100 mW/cm2, it was necessary to have a light-alone control as well as a drug-alone control. None of the controls demonstrated any tumor response, but the PDT regimen had a significant anti-tumor response on the highly pigmented melanoma with a mean growth delay of 12 days. We employed the multi-spectral fluorescence imaging camera system (CRI Maestro) to carry out imaging studies.39 We excited the B16F10-GFP fluorescence at 488 nm and collected fluorescence at >515-nm. This was false-colored green by the Maestro software. Fig. 9A shows B16F10-GFP tumor before PDT. It can be seen that a green signal of GFP from the melanoma (clearly seen in panel A) has disappeared and is replaced by a black eschar 2 days after PDT (in panel B).
Proc. of SPIE Vol. 7380 73802S-9
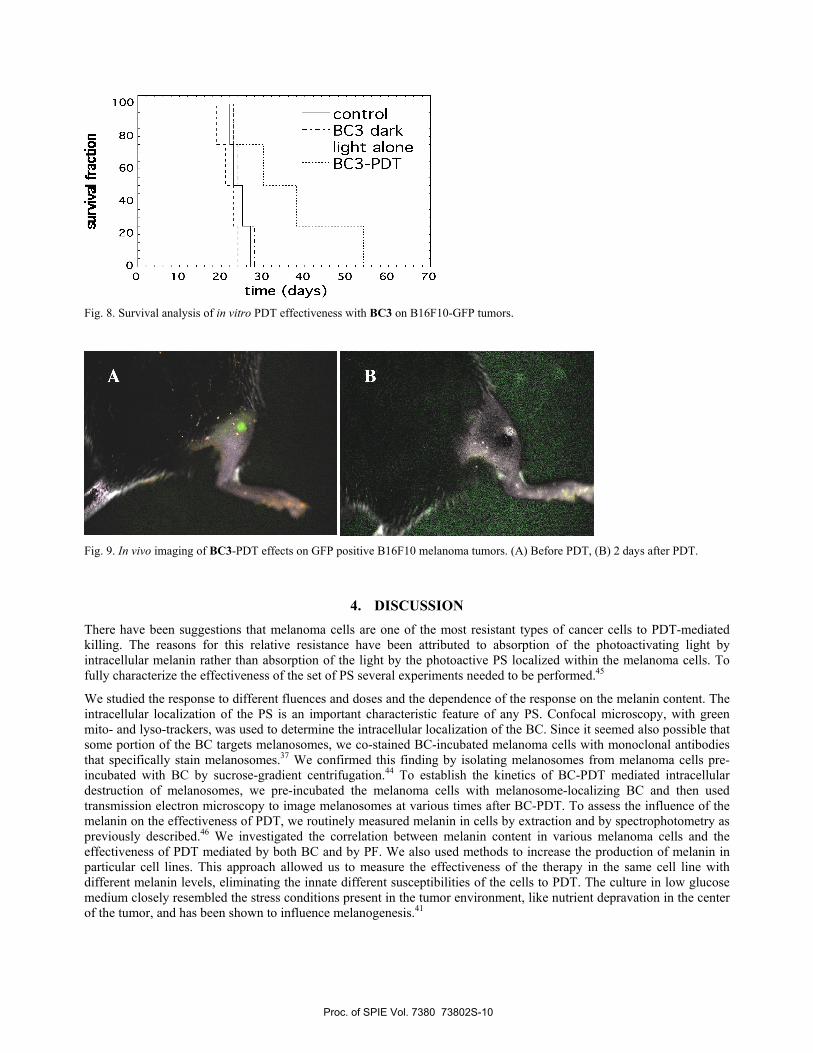
Fig. 8. Survival analysis of in vitro PDT effectiveness with BC3 on B16F10-GFP tumors.
Fig. 9. In vivo imaging of BC3-PDT effects on GFP positive B16F10 melanoma tumors. (A) Before PDT, (B) 2 days after PDT.
4. DISCUSSION There have been suggestions that melanoma cells are one of the most resistant types of cancer cells to PDT-mediated killing. The reasons for this relative resistance have been attributed to absorption of the photoactivating light by intracellular melanin rather than absorption of the light by the photoactive PS localized within the melanoma cells. To fully characterize the effectiveness of the set of PS several experiments needed to be performed.45
We studied the response to different fluences and doses and the dependence of the response on the melanin content. The intracellular localization of the PS is an important characteristic feature of any PS. Confocal microscopy, with green mito- and lyso-trackers, was used to determine the intracellular localization of the BC. Since it seemed also possible that some portion of the BC targets melanosomes, we co-stained BC-incubated melanoma cells with monoclonal antibodies that specifically stain melanosomes.37 We confirmed this finding by isolating melanosomes from melanoma cells pre-incubated with BC by sucrose-gradient centrifugation.44 To establish the kinetics of BC-PDT mediated intracellular destruction of melanosomes, we pre-incubated the melanoma cells with melanosome-localizing BC and then used transmission electron microscopy to image melanosomes at various times after BC-PDT. To assess the influence of the melanin on the effectiveness of PDT, we routinely measured melanin in cells by extraction and by spectrophotometry as previously described.46 We investigated the correlation between melanin content in various melanoma cells and the effectiveness of PDT mediated by both BC and by PF. We also used methods to increase the production of melanin in particular cell lines. This approach allowed us to measure the effectiveness of the therapy in the same cell line with different melanin levels, eliminating the innate different susceptibilities of the cells to PDT. The culture in low glucose medium closely resembled the stress conditions present in the tumor environment, like nutrient depravation in the center of the tumor, and has been shown to influence melanogenesis.41
A B
Proc. of SPIE Vol. 7380 73802S-10

Other mechanisms that may also be involved in melanoma resistance to PDT include the overexpression of certain multi-drug export pumps such as the ATP-binding cassette transporters (ABCA9, ABCB5, ABCC2, and ABCD1) that are overexpressed in melanoma cells.47,48 It has been recently reported that certain PS based on tetrapyrrole structures such as protoporphyrin IX and pyropheophorbide are substrates of ABCG2.49 Another reason for melanoma resistance to PDT may be the known ability of melanin to act as an anti-oxidant and to quench reactive oxygen species. It has been postulated that the ability of melanin to quench excited states of photosensitizers and singlet oxygen, and to scavenge reactive radicals, is an important factor in the protective action of melanin in pigmented tissues against oxidative damage. The mechanism of quenching of excited states of positively charged porphyrin molecules bound to melanin was recently determined by femtosecond absorption and picosecond emission spectroscopy.50 It was concluded that such an ionic binding facilitates an ultrafast energy transfer from the porphyrin molecule (in its lowest singlet excited state) to melanin. The excited-state energy is then rapidly converted into heat.
Despite the aforementioned reasons for PDT ineffectiveness against melanoma, there have been reports of PDT applications for melanoma treatment in both animals and in humans. In animals, both non-pigmented amelanotic cell lines51 and pigmented melanoma cell lines52 have been used to grow tumors in mice,53 hamsters54 and rabbits.55 Biolo and Jori used a liposome-delivered Si(IV)-naphthalocyanine (SiNc) that absorbed at 776 nm to treat pigmented B16 melanomas in mice.56,57 Woodburn et al employed the lutetium texaphyrin PS known as PCI-0123 that absorbs at 732 nm to treat pigmented B16 melanomas in mice.58 Scherz’s group used the Pd-bacteriochlorin known as WST11 to treat M2R melanoma xenografts in nude mice.25 Clinical studies have been carried out in ocular melanomas (choroidal, uveal and iris located tumors).59,60 There have been a few reports of PDT being tested in cutaneous melanomas in human patients, including a study from Russia in which chlorin(e6)61 was injected intravenously at 5 mg/kg and 660-nm light was delivered twice after 1 h and 24 h.
Effective PDT of melanoma clearly requires PS that absorb strongly in the NIR region (>700 nm), enabling use of NIR light, which affords the best penetration of tissues. The presented selection of new stable bacteriochlorins tested here in vitro and in vivo for PDT activity against a spectrum of melanoma cells containing varying amounts of melanin strongly suggest that the proposed synthetic BC may be a promising new class of anti-melanoma PS. Apart from strong absorption in the NIR, the BC are characterized by (i) a good yield of triplet state formation, (ii) efficient production of ROS, (iii) robust synthesis affording ample quantities of products for experimentation, and (iv) the ability to tune these characteristics. These compounds when combined with 730-nm light are significantly better at killing both pigmented and non-pigmented melanoma cells compared to the FDA-approved Photofrin and 635-nm light. Moreover, the BC examined herein, particularly BC2 and BC3, are effective at significantly lower concentrations (BC < 0.5 μM vs PF > 6 μM), thereby reducing side effects. The suggested localization in melanosomes and their subsequent photo-destruction by BC-PDT shows promise for overcoming resistance of melanoma to chemotherapy due to drug sequestration in these organelles.
The lack of development of PDT as a therapy for melanoma is thought to be due to ineffectiveness of PDT using presently available PS and optical quenching of the activating light by the melanin pigment. The results presented here show high promise for future clinical application of synthetic bacteriochlorins in PDT of pigmented melanoma. The ability to tailor at will the groups at the perimeter of the macrocycle augurs well for the use of synthetic bacteriochlorins in a wide variety of biomedical applications.
ACKNOWLEDGMENTS This work was supported by grants from the NIH (R01GM36238 to J.S.L. and R01AI050875 to M.R.H.), a Burroughs-Wellcome fellowship to M.K., and the Jimmy V. NCSU Cancer Therapeutics Training Program. P.M. and T.B. were supported by a grant (R41AI072854) from the National Institute of Allergy and Infectious Diseases to NIRvana Pharmaceuticals, Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the NIH. We are grateful to Pharmacyclics, Inc. for the gift of LuTex, and to QLT, Inc. for the gift of Photofrin.
Proc. of SPIE Vol. 7380 73802S-11

REFERENCES
[1] Balch, C. M., Buzaid, A. C., Soong, S.-J., Atkins, M. B., Cascinelli, N., Coit, D. G., Fleming, I. D., Gershenwald, J. E., Houghton, A., Jr., Kirkwood, J. M., McMasters, K. M., Mihm, M. F., Morton, D. L., Reintgen, D. S., Ross, M. I., Sober, A., Thompson, J. A. and Thompson, J. F., "Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma," J. Clin. Oncol. 19(16), 3635-48 (2001).
[2] Cummins, D. L., Cummins, J. M., Pantle, H., Silverman, M. A., Leonard, A. L. and Chanmugam, A., "Cutaneous malignant melanoma," Mayo Clin. Proc. 81(4), 500-507 (2006).
[3] Hallberg, O., "A theory and model to explain the skin melanoma epidemic," Melanoma Res. 16(2), 115-118 (2006).
[4] American Academy of Dermatology. "Incidence Of Melanoma On The Rise," ScienceDaily 4 August 2007. 15 May 2009 <http://www.sciencedaily.com- /releases/2007/08/070803145951.htm>.
[5] Nordlund, J. J., "The melanocyte and the epidermal melanin unit: an expanded concept," Dermatol. Clin. 25(3), 271-281 (2007).
[6] Gilchrest, B. A., Park, H. Y., Eller, M. S. and Yaar, M., "Mechanisms of ultraviolet light-induced pigmentation," Photochem. Photobiol. 63(1), 1-10 (1996).
[7] Riley, P. A., "Melanogenesis and melanoma," Pigment Cell Res. 16(5), 548-552 (2003). [8] Suzuki, I., Cone, R. D., Im, S., Nordlund, J. and Abdel-Malek, Z. A., "Binding of melanotropic hormones to the
melanocortin receptor MC1R on human melanocytes stimulates proliferation and melanogenesis," Endocrinology 137(5), 1627-33 (1996).
[9] Tsao, H., Atkins, M. B. and Sober, A. J., "Management of cutaneous melanoma," New Engl. J. Med. 351(10), 998-1012 (2004).
[10] Tarhini, A. A. and Agarwala, S. S., "Cutaneous melanoma: available therapy for metastatic disease," Dermatol. Ther. 19(1), 19-25 (2006).
[11] Soengas, M. S. and Lowe, S. W., "Apoptosis and melanoma chemoresistance," Oncogene 22(20), 3138-3151 (2003).
[12] Atkins, M. B., "Cytokine-based therapy and biochemotherapy for advanced melanoma," Clin. Cancer Res. 12(7), 2353s-2358s (2006).
[13] Kirkwood, J. M., Strawderman, M. H., Ernstoff, M. S., Smith, T. J., Borden, E. C. and Blum, R. H., "Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684," J. Clin. Oncol, 14(1),7-17 (1996).
[14] Kirkwood, J. M., Ibrahim, J. G., Sondak, V. K., Richard, J., Flaherty, L. E., Ernstoff, M. S., Smith, T. J., Rao, U., Steele, M. and Blum, R. H., "High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190," J. Clin. Oncol. 18(12), 24444-24458 (2000).
[15] Livingston, P. O., Wong, G. Y., Adluri, S., Tao, Y., Padavan, M., Parente, R., Hanlon, C., Calves, M. J., Helling, F., Ritter, G., Oettgen, H. F. and Lloyd, J. O., "Improved survival in stage III melanoma patients with GM2 antibodies: a randomized trial of adjuvant vaccination with GM2 ganglioside," J. Clin. Oncol. 12(5), 1036-1044 (1994).
[16] Bystryn, J. C., Zeleniuch-Jacquotte, A., Oratz, R., Shapiro, R. L., Harris, M. N. and Roses, D. F., "Double-blind trial of a polyvalent, shed-antigen, melanoma vaccine," Clin. Cancer Res. 7(7), 1882-1887 (2001).
[17] Hsueh, E. C., Gupta, R. K., Qi, K. and Morton, D. L., "Correlation of specific immune responses with survival in melanoma patients with distant metastases receiving polyvalent melanoma cell vaccine," J. Clin. Oncol. 16(9), 2913-2920 (1998).
[18] Berd, D., Maguire, H. C., Jr., Schuchter, L. M., Hamilton, R., Hauck, W. W., Sato, T. and Mastrangelo, M. J., "Autologous hapten-modified melanoma vaccine as postsurgical adjuvant treatment after resection of nodal metastases," J. Clin. Oncol. 15(6), 2359-2370 (1997).
[19] Mitchell, M. S., "Perspective on allogeneic melanoma lysates in active specific immunotherapy," Semin. Oncol. 25(6), 623-635 (1998).
[20] Kalaaji, A. N., "Cytokine therapy in advanced melanoma," J. Drugs Dermatol. 6(4), 374-378 (2007). [21] Naylor, M. F., Chen, W. R., Teague, T. K., Perry, L. A. and Nordquist, R. E., "In situ photoimmunotherapy: a
tumour-directed treatment for melanoma," Br. J. Dermatol. 155(6), 1287-1292 (2006). [22] Castano, A. P., Demidova, T. N. and Hamblin, M. R., "Mechanisms in photodynamic therapy: part one-
photosensitizers, photochemistry and cellular localization," Photodiagn. Photodyn. Ther. 1(4), 279-293 (2004)
Proc. of SPIE Vol. 7380 73802S-12

[23] Lapouge, K., Naveke, A., Gall, A., Ivancich, A., Seguin, J., Scheer, H., Sturgis, J. N., Mattioli, T. A. and Robert, B., "Conformation of bacteriochlorophyll molecules in photosynthetic proteins from purple bacteria," Biochemistry 38(34), 11115-11121 (1999).
[24] Trachtenberg, J., Weersink, R. A., Davidson, S. R., Haider, M. A., Bogaards, A., Gertner, M. R., Evans, A., Scherz, A., Savard, J., Chin, J. L., Wilson, B. C. and Elhilali, M., "Vascular-targeted photodynamic therapy (padoporfin, WST09) for recurrent prostate cancer after failure of external beam radiotherapy: a study of escalating light doses," BJU Int. 102(5), 556-562 (2008).
[25] Mazor, O., Brandis, A., Plaks, V., Neumark, E., Rosenbach-Belkin, V., Salomon, Y. and Scherz, A., "WST11, a novel water-soluble bacteriochlorophyll derivative; cellular uptake, pharmacokinetics, biodistribution and vascular-targeted photodynamic activity using melanoma tumors as a model," Photochem. Photobiol. 81(2), 342-351 (2005).
[26] Chen, Y., Potter, W. R., Missert, J. R., Morgan, J. and Pandey, R. K., "Comparative in vitro and in vivo studies on long-wavelength photosensitizers derived from bacteriopurpurinimide and bacteriochlorin p6: fused imide ring enhances the in vivo PDT efficacy," Bioconjug. Chem. 18(5), 1460-1473 (2007).
[27] Kim, H. J. and Lindsey, J. S., "De novo synthesis of stable tetrahydroporphyrinic macrocycles: bacteriochlorins and a tetradehydrocorrin," J. Org. Chem. 70(14), 5475-5486 (2005).
[28] Fan, D., Taniguchi, M. and Lindsey, J. S., "Regioselective 15-bromination and functionalization of a stable synthetic bacteriochlorin," J. Org. Chem. 72(14), 5350-5357 (2007).
[29] Borbas, K. E., Ruzié, C. and Lindsey, J. S., "Swallowtail bacteriochlorins. Lipophilic absorbers for the near-infrared," Org. Lett. 10(10), 1931-1934 (2008).
[30] Ruzié, C., Krayer, M., Balasubramanian, T. and Lindsey, J. S., "Tailoring a bacteriochlorin building block with cationic, amphipathic, or lipophilic substituents," J. Org. Chem. 73(15), 5806-5820 (2008).
[31] Taniguchi, M., Cramer, D. L., Bhise, A. D., Kee, H. L., Bocian, D. F., Holten, D. and Lindsey, J. S., "Accessing the near-infrared spectral region with stable, synthetic, wavelength-tunable bacteriochlorins," New J. Chem. 32, 947–958 (2008).
[32] Kee, H. L., Kirmaier, C., Tang, Q., Diers, J. R., Muthiah, C., Taniguchi, M., Laha, J. K., Ptaszek, M., Lindsey, J. S., Bocian, D. F. and Holten, D., "Effects of substituents on synthetic analogs of chlorophylls. Part 2: Redox properties, optical spectra and electronic structure," Photochem Photobiol. 83(5), 1125-1143 (2007).
[33] He, P., Ahn, J. C., Shin, J. I., Hwang, H. J., Kang, J. W., Lee, S. J. and Chung, P. S., "Enhanced apoptotic effect of combined modality of 9-hydroxypheophorbide alpha-mediated photodynamic therapy and carboplatin on AMC-HN-3 human head and neck cancer cells," Oncol. Rep. 21(2), 329-334 (2009).
[34] Lobner, D., "Comparison of the LDH and MTT assays for quantifying cell death: validity for neuronal apoptosis?," J. Neurosci. Methods 96(2), 147-152 (2000).
[35] Merlin, J. L., Azzi, S., Lignon, D., Ramacci, C., Zeghari, N. and Guillemin, F., "MTT assays allow quick and reliable measurement of the response of human tumour cells to photodynamic therapy," Eur. J. Cancer 28A(8-9), 1452-1458 (1992).
[36] Hamblin, M. R., Miller, J. L. and Ortel, B., "Scavenger-receptor targeted photodynamic therapy," Photochem. Photobiol. 72(4), 533-540 (2000).
[37] Orlow, S. J., Boissy, R. E., Moran, D. J. and Pifko-Hirst, S., "Subcellular distribution of tyrosinase and tyrosinase-related protein-1: implications for melanosomal biogenesis," J. Invest. Dermatol. 100(1), 55-64 (1993).
[38] Chen, K. G., Valencia, J. C., Lai, B., Zhang, G., Paterson, J. K., Rouzaud, F., Berens, W., Wincovitch, S. M., Garfield, S. H., Leapman, R. D., Hearing, V. J. and Gottesman, M. M., "Melanosomal sequestration of cytotoxic drugs contributes to the intractability of malignant melanomas," Proc. Natl. Acad. Sci. USA 103(26), 9903-9907 (2006).
[39] Su, J., Zhang, J., Liu, L., Huang, Y. and Mason, R. P., "Exploring feasibility of multicolored CdTe quantum dots for in vitro and in vivo fluorescent imaging, "J. Nanosci. Nanotechnol. 8(3), 1174-1177 (2008).
[40] Solca, F. F., Chluba-de Tapia, J., Iwata, K. and Eberle, A. N., "B16-G4F mouse melanoma cells: an MSH receptor-deficient cell clone," FEBS Lett. 322(2), 177-180 (1993).
[41] Nakayasu, M., Saeki, H., Tohda, H. and Oikawa, A., "Effects of sugars on melanogenesis in cultured melanoma cells," J. Cell Physiol. 92(1), 49-55 (1977).
[42] Orlow, S. J., "Melanosomes are specialized members of the lysosomal lineage of organelles," J. Invest. Dermatol. 105(1), 3-7 (1995).
[43] Kuroda, T. S., Itoh, T. and Fukuda, M., "Functional analysis of slac2-a/melanophilin as a linker protein between Rab27A and myosin Va in melanosome transport," Methods Enzymol. 403, 419-431 (2005).
Proc. of SPIE Vol. 7380 73802S-13

[44] Hong, L., Garguilo, J., Anzaldi, L., Edwards, G. S., Nemanich, R. J. and Simon, J. D., "Age-dependent photoionization thresholds of melanosomes and lipofuscin isolated from human retinal pigment epithelium cells," Photochem. Photobiol. 82(6), 1475-1481 (2006).
[45] Mroz, P., Pawlak, A., Satti, M., Lee, H., Wharton, T., Gali, H., Sarna, T. and Hamblin, M. R., "Functionalized fullerenes mediate photodynamic killing of cancer cells: Type I versus Type II photochemical mechanism," Free Radic. Biol. Med. 43(5), 711-719 (2007).
[46] Zadlo, A., Rozanowska, M. B., Burke, J. M. and Sarna, T. J., "Photobleaching of retinal pigment epithelium melanosomes reduces their ability to inhibit iron-induced peroxidation of lipids," Pigment Cell Res. 20(1), 52-60 (2007).
[47] Szakacs, G. and Gottesman, M. M., "Comparing solid tumors with cell lines: implications for identifying drug resistance genes in cancer," Mol. Interv. 4(6), 323-325 (2004).
[48] Chen, K. G., Szakacs, G., Annereau, J. P., Rouzaud, F., Liang, X. J., Valencia, J. C., Nagineni, C. N., Hooks, J. J., Hearing, V. J. and Gottesman, M. M., "Principal expression of two mRNA isoforms (ABCB 5alpha and ABCB 5beta) of the ATP-binding cassette transporter gene ABCB 5 in melanoma cells and melanocytes," Pigment Cell Res. 18(2), 102-112 (2005).
[49] Liu, W., Baer, M. R., Bowman, M. J., Pera, P., Zheng, X., Morgan, J., Pandey, R. A. and Oseroff, A. R., "The tyrosine kinase inhibitor imatinib mesylate enhances the efficacy of photodynamic therapy by inhibiting ABCG2," Clin. Cancer Res. 13(8), 2463-2470 (2007).
[50] Liu, Y., Hong, L., Wakamatsu, K., Ito, S., Adhyaru, B., Cheng, C. Y., Bowers, C. R. and Simon, J. D., "Comparison of structural and chemical properties of black and red human hair melanosomes," Photochem. Photobiol. 81(1), 135-144 (2005).
[51] Dellian, M., Richert, C., Gamarra, F. and Goetz, A. E., "Photodynamic eradication of amelanotic melanoma of the hamster with fast acting photosensitizers," Int. J. Cancer. 65(2), 246-248 (1996).
[52] Busetti, A., Soncin, M., Jori, G. and Rodgers, M. A., "High efficiency of benzoporphyrin derivative in the photodynamic therapy of pigmented malignant melanoma," Br. J. Cancer 79(5-6), 821-824 (1999).
[53] Canti, G., Franco, P., Marelli, O., Cubeddu, R., Taroni, P. and Ramponi, R., "Comparative study of the therapeutic effect of photoactivated hematoporphyrin derivative and aluminum disulfonated phthalocyanines on tumor bearing mice," Cancer Lett. 53(2-3), 123-127 (1990).
[54] Leunig, M., Leunig, A., Lankes, P. and Goetz, A. E., "Evaluation of photodynamic therapy-induced heating of hamster melanoma and its effect on local tumour eradication," Int. J. Hyperthermia 10(2), 297-306 (1994).
[55] Schmidt-Erfurth, U., Bauman, W., Gragoudas, E., Flotte, T. J., Michaud, N. A., Birngruber, R. and Hasan, T., "Photodynamic therapy of experimental choroidal melanoma using lipoprotein-delivered benzoporphyrin," Ophthalmology 101(1), 89-99 (1994).
[56] Biolo, R., Jori, G., Soncin, M., Pratesi, R., Vanni, U., Rihter, B., Kenney, M. E. and Rodgers, M. A., "Photodynamic therapy of B16 pigmented melanoma with liposome-delivered Si(IV)-naphthalocyanine," Photochem. Photobiol. 59(3), 362-365 (1994).
[57] Biolo, R., Jori, G., Soncin, M., Rihter, B., Kenney, M. E. and Rodgers, M. A., "Effect of photosensitizer delivery system and irradiation parameters on the efficiency of photodynamic therapy of B16 pigmented melanoma in mice," Photochem. Photobiol. 63(2), 224-228 (1996).
[58] Woodburn, K. W., Fan, Q., Kessel, D., Luo, Y. and Young, S. W., "Photodynamic therapy of B16F10 murine melanoma with lutetium texaphyrin," J. Invest. Dermatol. 110(5), 746-751 (1998).
[59] Mennel, S., Barbazetto, I., Meyer, C. H., Peter, S. and Stur, M., "Ocular photodynamic therapy--standard applications and new indications (part 1). Review of the literature and personal experience," Ophthalmologica 221(4), 216-226 (2007).
[60] Favilla, I., Favilla, M. L., Gosbell, A. D., Barry, W. R., Ellims, P., Hill, J. S. and Byrne, J. R., "Photodynamic therapy: a 5-year study of its effectiveness in the treatment of posterior uveal melanoma, and evaluation of haematoporphyrin uptake and photocytotoxicity of melanoma cells in tissue culture," Melanoma Res. 5(5), 355-364 (1995).
[61] Sheleg, S. V., Zhavrid, E. A., Khodina, T. V., Kochubeev, G. A., Istomin, Y. P., Chalov, V. N. and Zhuravkin, I. N., "Photodynamic therapy with chlorin e(6) for skin metastases of melanoma," Photodermatol. Photoimmunol. Photomed. 20(1), 21-26 (2004).
Proc. of SPIE Vol. 7380 73802S-14