neurovascular coupling in hippocampus is mediated via diffusion by neuronal-derived nitric oxide
TRANSCRIPT

Author's Accepted Manuscript
Neurovascular coupling in hippocampus ismediated via diffusion by neuronal-derivednitric oxide
Cátia F. Lourenço, Ricardo M. Santos, Rui M.Barbosa, Enrique Cadenas, Rafael Radi, JoãoLaranjinha
PII: S0891-5849(14)00237-8DOI: http://dx.doi.org/10.1016/j.freeradbiomed.2014.05.021Reference: FRB12030
To appear in: Free Radical Biology and Medicine
Received date: 12 February 2014Revised date: 19 May 2014Accepted date: 23 May 2014
Cite this article as: Cátia F. Lourenço, Ricardo M. Santos, Rui M. Barbosa,Enrique Cadenas, Rafael Radi, João Laranjinha, Neurovascular coupling inhippocampus is mediated via diffusion by neuronal-derived nitric oxide, FreeRadical Biology and Medicine, http://dx.doi.org/10.1016/j.freeradbiomed.2014.05.021
This is a PDF file of an unedited manuscript that has been accepted forpublication. As a service to our customers we are providing this early version ofthe manuscript. The manuscript will undergo copyediting, typesetting, andreview of the resulting galley proof before it is published in its final citable form.Please note that during the production process errors may be discovered whichcould affect the content, and all legal disclaimers that apply to the journalpertain.
www.elsevier.com/locate/freerad-
biomed

� �
�
1�
�
Neurovascular�coupling�in�hippocampus�is�mediated�via�diffusion�by�neuronal�1�
derived�nitric�oxide�2�
Cátia�F.�Lourenço1,�Ricardo�M.�Santos1,�Rui�M.�Barbosa1,�Enrique�Cadenas2,�Rafael�Radi3,�João�3�
Laranjinha1*�4�
�5�1Faculty� of� Pharmacy� and� Center� for� Neurosciences� and� Cell� Biology,� University� of� Coimbra,� Health�6�Sciences�Campus,�Azinhaga�de�Santa�Comba,�3000�548�Coimbra,�Portugal2�7�2Department�of�Pharmacology�and�Pharmaceutical�Sciences,�School�of�Pharmacy,�University�of�Southern�8�California,�Los�Angeles,�CA�90089,�USA.�9�3Department� of� Biochemistry� and� Center� for� Free� Radical� and� Biomedical� Research,� Facultad� de�10�Medicina,�Universidad�de�la�Republica,�Montevideo,�Uruguay.�11�
�12�
*To�whom�correspondence�should�be�addressed:�João�Laranjinha,�Center�for�Neurosciences�13�and�Cell�Biology�and�Faculty�of�Pharmacy,�University�of�Coimbra,�Health�Sciences�Campus,�14�Azinhaga�de�Santa�Comba,�3000�548�Coimbra,�Portugal.�E�mail:�[email protected]�15�
�16��17�Abbreviated�title:�Nitric�oxide�mediates�the�neurovascular�coupling�18��19�Conflict�of�Interest:��The�authors�declare�no�competing�financial�interests�20��21�Abstract:��22�
�23�
The� coupling� between� neuronal� activity� and� cerebral� blood� flow� (CBF)� is� essential� for� normal� brain�24�
function.� The� mechanisms� behind� such� neurovascular� coupling� process� remain� elusive,� mainly� due� to�25�
difficulties� to�probe�dynamically� the� functional�and�coordinated� interaction�between�neurons�and� the�26�
vasculature� in� vivo.� Direct� and� simultaneous� measurements� of� nitric� oxide� (�NO)� dynamics� and� CBF�27�
changes�in�hippocampus�in�vivo�support�that�during�glutamatergic�activation�nNOS�derived��NO�induces�28�
a�time,�spatially,�and�amplitude�coupled�increase�in�the�local�CBF,�later�followed�by�a�transient�increase�29�
in� local� O2� tension.� These� events� are� dependent� on� the� activation� of� NMDA�glutamate� receptor� and�30�

2�
�
nNOS,� without� a� significant� contribution� of� endothelial�derived� �NO� and� astrocyte�neuron� signaling�1�
pathways.�Upon�diffusion�of��NO�from�active�neurons,�the�vascular�response�encompasses�the�activation�2�
of�soluble�guanylate�cyclase.�Hence,�in�the�hippocampus,�neurovascular�coupling�is�mediated�by�nNOS�3�
derived��NO�via�a�diffusional�connection�between�active�glutamatergic�neurons�and�blood�vessels.�4�
�5�
Introduction�6�
�7�
Neuronal�activity�imposes�a�need�for�blood�flow�carrying�substrates�in�order�for�the�brain�to�maintain�its�8�
functional�and�structural�integrity[1].�Intensive�research�during�the�last�decades�aimed�to�untangle�the�9�
underlying�mechanisms�that�couple�the�neuronal�activity�to�cerebral�blood�flow�(CBF)�–�neurovascular�10�
coupling[2].�Strong�evidences� indicate� that�astrocytes�are�critical�contributors� to� the�process,�bridging�11�
neurons� and� blood� vessels,� although� several� questions� still� persist[3]� and� disparate� temporal� events�12�
demand� reconciliation[4].� Other,� non�astrocytic� physiological� mechanisms� have� been� proposed� to�13�
underlie�the�coupling�between�neuronal�activity�and�the�changes�in�CBF[5�7].�In�this�regard,�the�concept�14�
that��NO,�produced�upon�neuronal�activation,�reaches�blood�vessels�by�diffusion�from�neurons,�inducing�15�
vasodilation,� has� been� a� tempting� suggestion[8�12].� In� neurons,� �NO� is� produced� upon� glutamatergic�16�
activation� by� the� neuronal� isoform� of� nitric� oxide� synthase� (nNOS),� an� enzyme� physically� and�17�
functionally� coupled� to� the� NMDA� glutamate� receptors� [13,� 14].� Because� �NO� is� highly� diffusible� and�18�
overcomes� specific� ligand�receptor� interaction,� its� biological� action� is� critically� determined� by� its�19�
concentration�and�temporal�dynamics,�as�well�as�by�the�distribution�of�the�potential�targets;�by�this�way,�20�
the�profile�of��NO�change�in�time�and�space�is�translated�into�a�biological�action�[15].�These�unorthodox�21�
properties� of� �NO� have� raised� difficulties� to� directly� substantiate� its� role� as� a� diffusible� messenger� in�22�
neurovascular�coupling,�as�well�as�in�other�signaling�pathways.��23�

3�
�
Pharmacological� approaches� have� provided� insights� on� the� involvement� of� �NO� in� neurovascular�1�
coupling�[10,�12,�16,�17],�but�also�opposite�evidences�[18�20].�It�has�also�been�suggested�that,�although�2�
�NO�is�required,�it�does�not�directly�mediate�the�neuron�to�vessels�signaling,�at�least�in�somatosensory�3�
cortex[21].� A� step� forward� was� provided� by� the� in� vivo� measurements� of� �NO� dynamics,� during�4�
activation�of� rat� somatosensory�cortex�and� the�observation� that�a� transient� increase� in� �NO�preceded�5�
CBF�change[22],�however�without�clarifying�the�source�of��NO�and�the�interdependency�of�both�events.�6�
Thus,� in� spite� of� the� intensive� research� on� �NO� over� the� last� decades,� its� role� in� the� neurovascular�7�
coupling�still�remains�elusive.�8�
In�this�work�we�aimed�to�study�the�neuronal�derived��NO�dynamics�in�connection�with�CBF�changes�in�9�
hippocampus,�identifying�the�pathway�for��NO�production�and�uncovering�the�underlying�mechanism�for�10�
vasodilation.� We� used� an� array� incorporating� a� �NO�selective� microelectrode[23],� a� microinjection�11�
pipette� and� a� laser� Doppler� probe� assembled� in� a� pre�defined� geometry,� that� allowed� to� monitor�12�
neurovascular� coupling� upon� local� glutamate� stimulation� within� �NO� diffusional� spread.� On� basis� of� a�13�
real�time� and� in� vivo� sequence� comprising� glutamate� stimulation,� �NO� production� and� CBF� (and� O2)�14�
increase,�we�demonstrate� that,� in�hippocampus,�neurovascular�coupling� is�mediated�by�nNOS�derived�15�
�NO�via�a�diffusional�connection.�16�
�17�
Materials�and�Methods�18�
�19�
Array� for� �NO�and�CBF�measurements.�The� �NO� sensors� were� fabricated� as� previously� described[24].�20�
Briefly,�a�single�carbon�fiber�(30�μm�Ø�Textron�Lowell,�MA)�was�encased�in�a�glass�capillary�and�pulled�in�21�
a�vertical�puller.�The�protruding�carbon�fiber�was�cut�to�a�tip�length�of�200±50�μm.�The�electrical�contact�22�
between� the� carbon� fiber� and� a� copper� wire� was� achieved� by� using� conductive� silver� paint� (RS,�23�

4�
�
Northants,� U.K.).� To� improve� their� analytical� properties� for� in� vivo� measurements� of� �NO,� the� sensors�1�
were� coated� with� Nafion®� (5%� solution,� 2� coatings� with� 4� minutes� drying� at� 170°C)� and� with� o�2�
phenylenediamine� (o�PD� 5� mM� solution� was� electropolymerized� at� a� constant� potential� of� +0.7� V� vs�3�
Ag/AgCl�during�30�minutes).�Each�sensor�was�evaluated�for��NO�sensitivity�and�selectivity�against�major�4�
interferents�(ascorbate,�nitrite,�noradrenaline�and�dopamine)�by�constant�voltage�amperometry�at�+0.9�5�
V�vs�Ag/AgCl�using�a�FAST�16�high�speed�electrochemical�system�(Quanteon,�LLC,�Nicholasville,�KY)�in�a�6�
two�electrode� configuration.� The� sensors� used� in� this� study� presented� an� average� sensitivity� of�7�
258±92pA/μM� and� selectivity� ratios� of� 28673:1,� 5732:1,� 239:1� and� 213:1� against� ascorbate,� nitrite,�8�
noradrenaline� and� dopamine,� respectively.� Cerebral� blood� flow� was� measured� using� a� laser� Doppler�9�
flowmeter� device� (Periflux� system� 5000,� Perimed,� Sweden)� to� which� a� needle� probe� (PF411;� outer�10�
diameter,� 450� �m;� fiber� separation,� 150� �m;� wavelength,� 780� nm)� was� attached.� Calibration� of� the�11�
probe�was�done�routinely�using�PF1001�motility�standard�(Perimed,�Sweden)�to�equalize�the�perfusion�12�
values�among� the�different� recordings.�The� time� constant�was� set� to�0.03� s�and� the� signal�processing�13�
unit�used�a�bandwidth�of�32�Hz.�Laser�Doppler�flowmetry�measures�CBF� in�arbitrary�units� (a.u.)�and� is�14�
therefore�used�for�measuring�relative�changes�in�CBF.�The��NO�sensor�and�the�Laser�Doppler�probe�were�15�
assembled�to�an�ejection�micropipette�using�sticky�wax�in�the�configuration�schematized�in�Figure�1.�The�16�
micropipette� was� filled� with� solutions� using� a� syringe� fitted� with� a� flexible� microfilament� (MicroFil,�17�
World�Precision�Instruments,�UK)�previously�to�brain�insertion.��18�
�19�
Array� for� O2� and� CBF� measurements.� To� record� O2� fluctuations� the� array� configuration� included� a�20�
micropipette�and�a�carbon�fiber�microelectrode�similar�to�that�used�for� �NO�recording.�Such�electrode�21�
was�evaluated�for�O2�sensitivity�by�constant�voltage�amperometry�at��0.8�V�vs�Ag/AgCl�using�a�FAST�16�22�

5�
�
high�speed�electrochemical� system� (Quanteon,� LLC,�Nicholasville,�KY)� in�a� two�electrode�configuration�1�
essentially�as�previously�described.�2�
�3�
In�vivo�experimental�setup.��All�the�procedures�used�in�this�study�were�performed�in�accordance�with�4�
the� European� Union� Council� Directive� for� the� Care� and� Use� of� Laboratory� animals,� 2010/63/EU,�5�
implemented� under� the� supervision� and� approval� of� the� local� Institutional� Animal� Care� and� Use�6�
Committee� of� the� animal� facility� of� Center� for� Neurosciences� and� Cell� Biology� University� of� Coimbra�7�
(ORBEA)�and� licensed�by�the�national�regulatory�office�(Direcção�Geral�de�Alimentação�e�Veterinária—8�
DGAV).� The� animals� were� submitted� to� surgery� under� anesthesia� and� body� temperature� and� blood�9�
pressure�was�controlled�during�the�experiments�as�detailed�below.�At�the�end�of� the�experiments�the�10�
animals�were�sacrificed�by�cervical�displacement�while�still�under�anesthesia.�Forty�five�male�Wistar�rats�11�
(8�10�weeks;�weight�294�±�31�g)�were�anaesthetized�by�an� intraperitoneal� injection�of�urethane� (1.25�12�
g/Kg)�and�placed� in�a�stereotaxic�apparatus�(Stoelting�Co.,�USA).�Body�temperature�was�maintained�at�13�
37ºC�using�a�deltaphase�isothermal�pad�(BrainTree�Scientific,�MA,�USA)�and�controlled�through�a�rectal�14�
thermometer.�An�incision�was�made�with�the�scalp�and�the�skin�was�reflected�to�expose�the�surface�of�15�
the�skull,�allowing�for�drilling�a�hole�(3x4�mm)�in�the�surface�overlying�the�hippocampus.�Another�hole�16�
(~1� mm� diameter)� was� drilled� in� a� site� remote� from� the� recording� area� for� insertion� of� an� Ag/AgCl�17�
reference�electrode�(200�μm�diameter).�After�removing�the�dura�matter,�the�array�was�inserted�into�the�18�
rat�hippocampus�according�to�coordinates�calculated�based�on�the�rat�brain�atlas�of�Paxinos�and�Watson�19�
(2007)� using� the� tip� of� the� microelectrode� as� reference� in� the� bregma� (anteroposterior� �4.1� � mm;�20�
mediolateral��2.8�mm�and�dorsoventral��3.7�mm).�After�the�insertion�of�the�array�into�the�hippocampus�21�
it�was�allowed�to�stabilize�for�30�minutes�before�the�beginning�of�the�experiment.�22�
�23�

6�
�
Experimental�Design/�Drug�application.�Solutions,�L�glutamate�20�mM,�NMDA�(N�methyl�D�aspartate)�1�
0.1� mM,� �NO� solution� 0.1� mM� or� tACPD� (trans�1�amino�cyclopentane�1,3�dicarboxylic� acid)� 0.1�1� mM�2�
(25�nL)�in�saline�buffer�(NaCl�0.9%,�pH�7.4),�were�locally�applied�from�the�tip�of�the�micropipette�using�a�3�
Picospritzer� III� (Parker� Hannifin� Corp.,� General� Valve� Operation,� USA).� A� �NO� solution� 0.1� mM� was�4�
prepared�by�diluting�a�saturated��NO�solution,�prepared�as�previously�described�[25],� in�deoxygenated�5�
saline�buffer.�Stimulations�were�performed�by�pressure�pulses�of�1s�at�7�15�psi�with�a�minimum�interval�6�
of� 15� minutes� of� recover.� Typically� three� initial� responses� were� obtained� before� pharmacology�7�
modulation� to� achieve� a� steady� state� level.� MK�801� (1� mg/Kg� in� saline),� 7�Nitroindazole� (50� mg/Kg� in�8�
DMSO),�L�NIO�(40�mg/Kg�in�DMSO),�acetylsalicylic�acid�(300�mg/Kg�in�saline)�and�caffeine�(50�mg/kg�in�9�
saline)�were�injected�intraperitoneally.�A�cocktail�of�MPEP�and�LY367385�(100�nmol�each)�was�injected�in�10�
the�lateral�ventricle�using�a�micropipette.�ODQ�(25�pmol�in�DMSO�0.5%)�and�sodium�fluoroacetate�(20�11�
μmol)�were� locally�applied�by�an�extra�micropipette�attached� to� the�previous�described�array� in�close�12�
proximity� to� the� laser� Doppler� probe.� The� effects� were� evaluated� after� 10� to� 30� min� for� ODQ� and�13�
MPEP/LY36785� and� sodium� fluoroacetate,� and� after� 20� to� 40� minutes� for� the� remaining� and� the�14�
responses�compared�with� the�corresponding�control�experiments� (with�vehicles).� �Blood�pressure�was�15�
non�invasively�measured�in�the�tail�using�a�LE5001�system�(Letica,�Scientific�Instruments).�Arterial�blood�16�
gases�and�pH�were�evaluated�in�a�Rapidlab®�1260�Blood�Gas�Analyzer�(Siemens�Healthcare�Diagnostics)�17�
from�blood�samples�collected�from�the�femoral�artery�4�to�7�h�after�anesthetic�injection.��18�
�19�
Data�Analysis�and�Statistics.�The��NO�and�CBF�recordings�were�synchronized�using�OriginPro�7.5�based�20�
on� markers� extracted� from� the� respective� software.� The� remaining� analyses� were� performed� with�21�
GraphPad�Prism�5.�Data�are�presented�as�mean�±�SEM.�Statistical�analysis�of� the�data�was�performed�22�
using�Student’s�t�test.�The� �NO�and�CBF�signals�were�characterized�in�terms�of�1)��NO�peak�flux�of�the�23�

7�
�
signal,�based�on�the�conversion�of�the�amperometric�currents�to��NO�fluxes�according�to�Faraday’s�law�1�
(I=n.F.�,� in� which� I� corresponds� to� the� amperometric� current,� n� corresponds� to� the� one� electron� per�2�
molecule�exchanged�for�the�oxidation�of��NO,�F�corresponds�to�the�Faraday�constant�and���is�the�flux)�3�
or�amplitude�change�(the�increase�of�CBF�beyond�the�CBF�basal�levels,�considered�100%�in�the�absence�4�
of�stimuli),� respectively;�2)�Trise,� the�time� in�seconds�necessary�to�reach�the�maximum�amplitude�after�5�
stimulation,�3)�Ttotal,�the�time�in�seconds�from�the�stimulation�point�to�return�to�basal�levels.��6�
�7�
�8�
Results�9�
�10�
Nitric�oxide�and�cerebral�blood�flow�changes:�coupling�in�space,�time,�and�amplitude�11�
We�have�previously�reported�that�a�controlled�and�localized�glutamate�stimulus�in�the�rat�hippocampus�12�
promotes�an�instantaneous�and�transient�elevation�of��NO�concentration�levels�through�the�activation�of�13�
ionotropic�glutamate�receptors[23].�By�upgrading�such�experimental�strategy,�simultaneously�measuring�14�
local�CBF,�we�observed�that�a�transient��NO�increase�induced�by�glutamate�ejection�(0.5�nmol,�25�nL,�1s),�15�
was�followed,�seconds�later,�by�a�transient�change�in�CBF�(Figure�1).��NO�production�was�characterized�16�
by�a�flux�of�3.5±1.8�fmol.s�1�with�a�time�rise�of�22±3�s�and�a�total�duration�of�64±4�s�(54�peaks�analyzed�17�
from�15� individual�experiments).�The�CBF�started�to� increase�7±2�s�after�stimulation�reaching�122±5%�18�
beyond�the�basal�level�after�62±3�s�and�returning�to�basal�levels�after�216±15�s.�19�
Likewise,� the�specific�activation�of�NMDA�glutamate�receptor,�using�the�synthetic�agonist�N�methyl�D�20�
aspartate�(NMDA),�resulted�in��NO�and�CBF�dynamics�similar�to�those�obtained�with�glutamate�and�with�21�
the�same�temporal�correlation,�thus�imparting�specificity�to�the�pathway�leading�to��NO�production.�The�22�
ejection�of�NMDA�(2.5�pmol)� resulted� in�a�transient� �NO�production�characterized�by�a� flux�of�3.5±0.5�23�

8�
�
fmol.s�1�with�a�time�rise�of�19±4�s�and�a�total�duration�of�78±5�s,�that�was�followed�by�an�increase�of�CBF�1�
that� lasted� 296±46s� and� reached� the� maximum� of� 104±10%� beyond� the� basal� level� after� 73±9� s� (12�2�
peaks�analyzed�from�5�individual�experiments).�Local�pressure�ejection�of�the�vehicle�(NaCl�0.9%)�caused�3�
no�change�in�either�the�baseline�oxidation�current�or�the�CBF�levels�(Supplementary�Figure�2).�4�
The� CBF� changes� induced� by� glutamatergic� activation� in� hippocampus� were� independent� of� arterial�5�
blood�pressure,�blood�gases�or�pH,�which�remained�stable�and�within�the�physiological�range�during�the�6�
time�window�in�which�experiments�were�performed,�as�randomly�assessed.�On�average,�the�values�for�7�
pH,�paCO2�and�paO2�were�7.39±0.07;�41±7�mmHg�and�108±31�mmHg,�respectively.��8�
�9�
Exogenous�nitric�oxide�mimics�glutamate�induced�CBF�changes�10�
The�temporal�coupling�between�endogenous� �NO�signal�and�the� increase� in�CBF�was�mimicked�by� the�11�
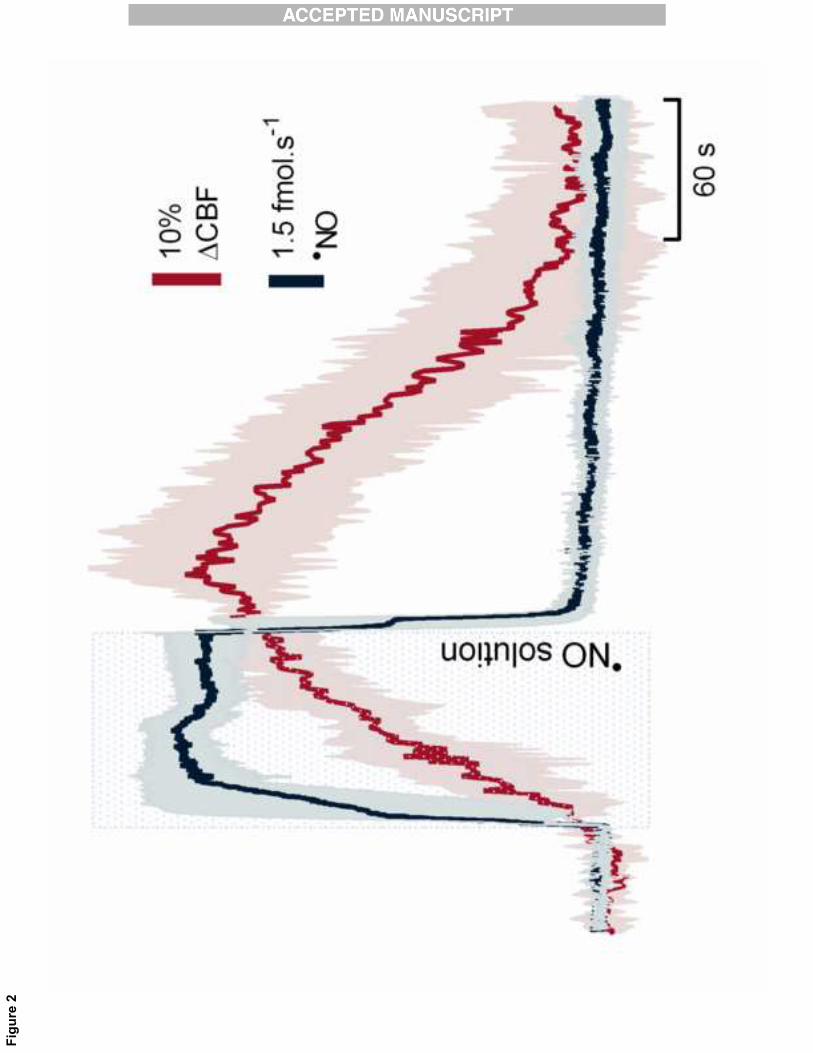
local� ejection� of� a� �NO� solution.� Figure� 2� shows� that� when� �NO� is� exogenously� applied,� a� temporal�12�
correlation�is�observed�between�the��NO�signal�and�CBF�change�that�is�similar�to�that�observed�for�the�13�
endogenous�glutamate�dependent�production�of��NO�(CBF�started�to�increase�8±2�s�after��NO�ejection,�14�
lasting�for�282±60�s�whereas��NO�temporary� increase�was�65�±�12�s,�n=3).�Nevertheless,�and�although�15�
the� �NO� flux� achieved� is� higher� when� exogenously� added� (11.3±2.7� fmol.s�1� versus� 3.8±1.6� fmol.s�1�16�
achieved�via�glutamate�stimulus),�the�CBF�change�from�the�baseline�is�weaker�than�that�observed�upon�17�
endogenous��NO�production�(55±11%�versus�122±5%�beyond�the�basal�level).�In�this�regard,�it�has�to�be�18�
considered�the�peculiar�nature�of��NO�volume�signaling�in�the�brain[26]:�following�activation�of�a�volume�19�
of� tissue,� comprising� numerous� synaptic� �NO� sources,� small� dispersed� �NO� sources� of� low� individual�20�
efficacy�can�cooperate�to�originate�an�extensive�and�strong�volume�signal,�inducing�an�increase�in�CBF.�21�
We� have� recently� shown� that� the� decay� of� �NO� when� produced� endogenously� in� hippocampus� via�22�
activation� of� NMDA� glutamate� receptors� is� much� slower� than� when� ejected� exogenously[23,� 27].�23�

9�
�
Therefore,�one�could�expect�a� lower�diffusional�spread�due�the� fast�decay�achieved�upon� �NO�release�1�
from�a�single�point�in�the�tip�of�the�pipette,�as�compared�with�glutamate�stimulus�and,�consequently,�a�2�
lower�effect�on�the�regional�CBF.�3�
�4�
Glutamate�induced�CBF�changes�correlate�with�oxygen�tension�changes�5�
The� local� transient� change� in� CBF� elicited� by� neuronal��NO� is� expected� to� translate� into� correlated�6�
changes�in�local�O2�tension.�By�measuring�O2�tensions,�we�observed�that�the�local�transient�changes�in�7�
CBF�elicited�by�glutamate�stimulation�are�followed,�seconds�later,�by�a�correlated�transient�elevation�in�8�
O2�tension�(Figure�3,�R2=0.69,�p=0.0016,�n=12�from�3�individual�experiments).�This�observation�supports�9�
the�prediction�that�the��NO�signal�is�sequentially�followed�by�CBF�increase�and�by�a�transitory�elevation�10�
in� O2� tension.� It� should� be� remarked� that� the� geometrical� configuration� of� the� array� and� the� physical�11�
properties�of�carbon�fiber�microelectrodes�and�LDF�probe�impose�that�the�measurement�of��NO/O2�and�12�
CBF�are�preformed�within�a�volume�of�hippocampal�tissue�likely�comprising�a�few�hundreds�of�microns�13�
of� diameter.� Within� this� volume� of� tissue,� the� fine� correlation� observed� between� O2� tension� and� CBF�14�
events,� measured� at� separate� locations� within� �NO� diffusion� field,� further� supports� the� validity� of� the�15�
approach�used�to�study�the�relationship�between��NO�and�CBF.�16�
�17�
Neuronal�derived��NO�is�the�mediator�of�neurovascular�coupling�in�hippocampus�18�
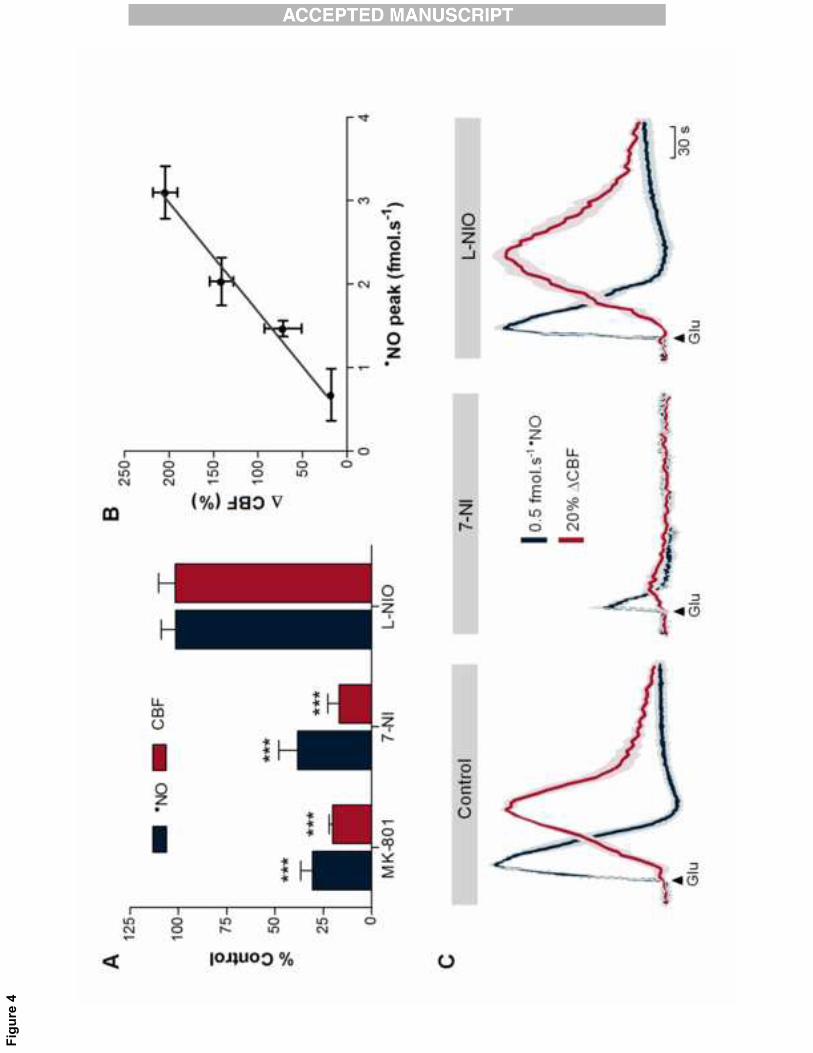
The�neurovascular�coupling�process�is�expected�to�be�prone�to�modulation�by�functional� interferences�19�
along� the� pathway� of� nNOS�dependent� �NO� production,� in� particular� at� the� level� of� NMDA� glutamate�20�
receptor� and� nNOS.� Among� several� potential� pharmacological� tools,� we� have� selected� a� glutamate�21�
NMDA� receptor� blocker� (MK�801),� a� selective� nNOS� inhibitor� (7�NI)� and� a� selective� eNOS� inhibitor� (L�22�
NIO).� The� results� obtained� are� summarized� in� Figure� 4A.� The� responses� obtained� following� drugs�23�

10�
�
injection� were� compared� with� the� corresponding� control� experiments� (with� vehicles).� None� of� the�1�
vehicles�used�promote�any�significant�effect�in��NO�or�CBF�responses�(Supplementary�Figure�2).�Blocking�2�
of� the� NMDA� receptor� elicited� a� significant� inhibition� of� �NO� production� (73±8%,� p=0.0017,� n=3)� that�3�
was�accompanied�by�a�dramatic�decrease�of�glutamate�induced�CBF�(74±3%,�p=0.0008,�n=3).�Likewise,�4�
the� inhibition�of�the�neuronal� isoform�of�NOS�by�7�NI� induced�a�significant� inhibition�of�both� �NO�and�5�
CBF�responses� to�glutamate� (62±10�and�83±6%,�p=0.0002�and�p=0.0025,� respectively,�n=4),�as�well�as�6�
the� decrease� in� CBF� basal� levels� (27±7%).� Conversely,� no� significant� effects� were� found� on� either�7�
induced�� or� basal� levels� upon� inhibition� of� the� endothelial� NOS� isoform� with� L�NIO� (Figure� 4C).�8�
Nevertheless,�it�was�observed�an�increase�in�mean�arterial�blood�pressure�of�the�animals�after�the�L�NIO�9�
injection,�supporting�its�efficacy�in�terms�of�eNOS�inhibition�(n=4,�MABP�90.3±1.1�to�114.9±2.5�mmHg).��10�
The�modulation�of�the�NMDA�receptor:nNOS�pathway�showed�that,�in�hippocampus,�the�increase�in�CBF�11�
was�dependent�on��NO�signal�amplitude�(Figure�4B).�Within�the�range�tested,�the�relationship�between�12�
�NO�and�CBF�showed�to�be�linear�above�a�threshold�of�0.43�fmol.s�1,�which�is�coherent�with�the�concept�13�
of� a� linear� relationship� between� neuronal� activity� and� vascular� responses� provided� by� neuroimaging�14�
studies[28].� However,� it� should� be� pointed� out� that� below� the� threshold� our� data� does� not� identify�15�
whether�the�relationship�is�linear�or�a�gating�process�is�operative.�16�
�17�
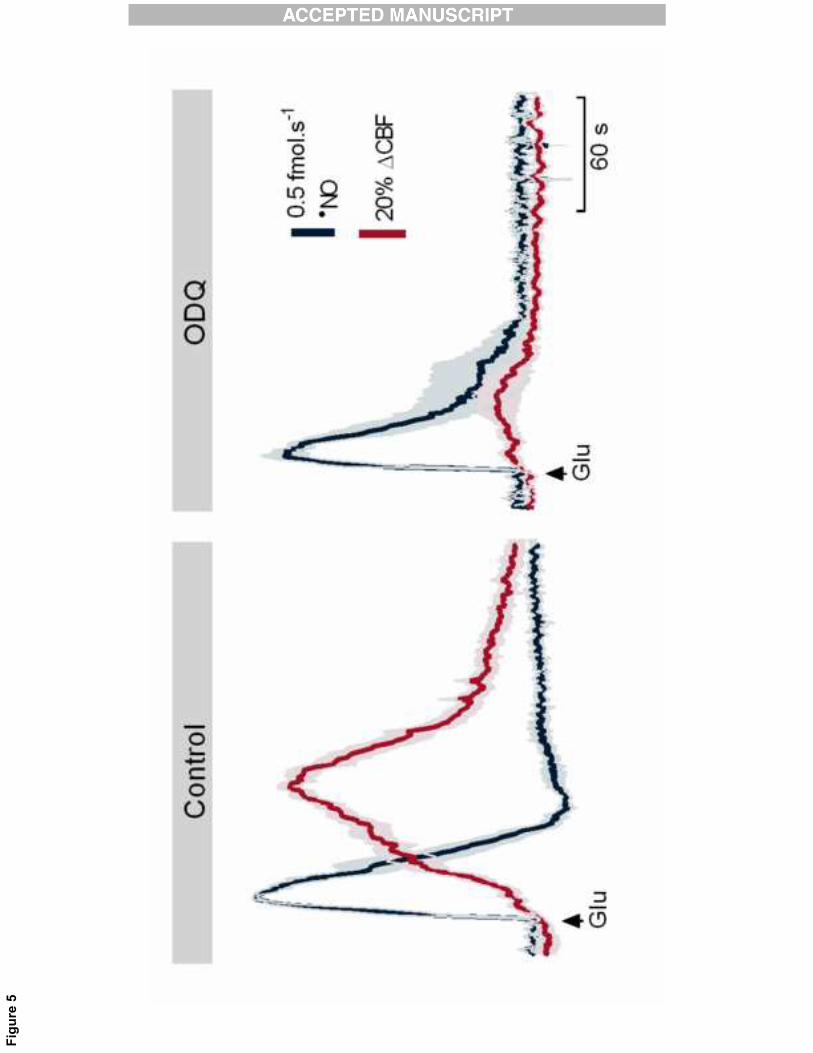
The��NO�mediated�neurovascular�coupling�encompasses�the�activation�of�soluble�guanylate�cyclase�18�
The� classical� pathway� for� vasodilation� mediated� by� �NO� involves� the� activation� of� soluble� guanylate�19�
cyclase� (sGC)� in� smooth� muscle� cells,� which� via� cGMP�dependent� protein� kinases,� promotes� the�20�
dephosphorylation�of�myosin�light�chains�and�thus�vasodilation[15].�In�order�to�unravel�the�mechanism�21�
by�which�neuronal�derived��NO�promoted�the�increase�in�CBF,�the�effect�of�ODQ,�a�selective�heme�site�22�
inhibitor� of� soluble� guanylate� cyclase,� was� evaluated.� Following� the� local� application� of� ODQ� in�23�

11�
�
hippocampus,� glutamate� induced� a� typical� �NO� concentration� dynamics� but� the� CBF� changes� were�1�
significantly� reduced� (67±7%,� p=0.002,� n=4)� (Figure� 5).� The� slight� inhibition� of� �NO� signal� observed� is�2�
likely� related� to� the� unspecific� inhibition� of� nNOS� by� ODQ[29]� (nNOS� being� an� hemeprotein� might� be�3�
affected� by� ODQ),� and� not� a� consequence� of� cGMP� decrease� in� neurons,� as� �NO� concentration� was�4�
resumed� in� the� following� stimulations� while� CBF� remained� inhibited,� progressively� returning� to� the�5�
control�values�obtained� in� the�absence�of�ODQ.�The�uncoupling�observed�between� �NO�concentration�6�
dynamics�and�CBF�changes�in�the�presence�of�ODQ�strongly�supports�that��NO�produced,�and�diffusing�7�
from�neurons,�reaches�vascular�smooth�muscle�cells�and�triggers�vasodilation�via�sGC�activation.�8�
�9�
The�astrocytic�pathway�is�not�explicitly�involved�in�the��NO�mediated�neurovascular�coupling��10�
Given�the�commonly�accepted�paradigm�of�astrocytes�bridging�neurons�and�vascular�cells�encompassing�11�
metabotropic� glutamate� receptors� activation� and� subsequent� production� of� arachidonic� acid�12�
metabolites[2,� 6],� we� addressed� the� potential� contribution� of� the� astrocytic� bridge� to� neurovascular�13�
coupling�in�hippocampus�under�our�experimental�conditions.�For�that�purpose,�a�cocktail�of�antagonists�14�
of�metabotropic�glutamate�receptors�(LY367385�and�MPEP)�was�injected�intracerobroventricularly�in�the�15�
rat� brain� to� inhibit� mGluR1� and� mGluR5,� respectively.� If� glutamate�triggered� [Ca2+]i� elevations� in�16�
astrocytes�are�crucial�for�neurovascular�coupling,�their�specific�inhibition�should�hamper�the�increase�of�17�
CBF�induced�by�glutamate�via�the�astrocytes.�However,�the�inhibition�of�astrocyte�mGluR�receptors�did�18�
not� elicit� any� alteration� in� the� recorded� �NO� and� CBF� signals� (Figure� 6A).� Both,� inhibition� of�19�
cyclooxygenase� activity� by� acetylsalicylic� acid� –downstream� in� the� astrocytic� pathway–� and� the�20�
impairment�of�astrocytic�metabolism�linked�to�the�inhibition�of�mitochondrial�aconitase�via�metabolism�21�
of�sodium�fluoroacetate��which�is�preferentially�uptaked�by�glial�cells[30]��had�no�effect�either.�Also,�no�22�
contribution� could� be� assigned� to� adenosine� receptors,� argued� to� intermediate� a� complex� signaling�23�

12�
�
process� of� neuronal� activity�induced� increase� in� cerebral� vasodilation� [31,� 32],� as� caffeine,� a�1�
nonselective� antagonist� of� adenosine� receptors[33]� also� failed� to� modulate� both� �NO� and� CBF� (Figure�2�
6A).�As�an�additional�control,�the�specific�activation�of�mGLuR�receptors�by�the�mGluR�agonist�tACPD�–3�
known�to�promote�Ca2+�elevations�in�astrocyte�somata�and�related�pathways[5]–�did�not�elicit�changes�4�
in�either��NO�production�or�CBF�(Figure�6B).�5�
�6�
�NO�dependent�CBF�increase�is�region�specific�7�
The� regional� occurrence� of� the� diffusive� connection� established� by� �NO� between� neurons� and� vessels�8�
without� the� intermediacy� of� cellular� systems� between� glutamatergic� synapses� and� vessels� in� brain�9�
regions�other�than�hippocampus�was�assessed,�for�it�has�been�previously�reported�that�the�mechanisms�10�
underlying�neurovascular�coupling�may�be�region�specific.�With�that�intent�the�experimental�approach�11�
used� in�hippocampus�was�applied� to�probe�cerebral�cortex�with� the�tip�of� the�array�positioned� in� the�12�
deeper� layers,�where�nNOS�immunoreactivity� is�predominant[34].�Whereas�data�support�the�coupling,�13�
the�profiles�of��NO�and�CBF�change�do�not�reproduce�the�phenomenon�observed�in�hippocampus�in�all�14�
its�dimensions�(Figure�7).�In�fact,�in�cerebral�cortex,�as�compared�with�hippocampus,�glutamate�induced�15�
a�weaker�production�of��NO�(0.46±0.14�fmol.s�1)�followed�by�a�less�intense�increase�in�CBF�(46±6%)�(12�16�
peaks� analyzed� from� 4� individual� experiments)� but,� quantitatively,� the� response� of� �NO� and� CBF� to�17�
glutamate�in�cerebral�cortex�was�not�proportionally�attenuated;�while��NO�production�in�cerebral�cortex�18�
was� 87�4%� lower� than� in� hippocampus� and� the� reduction� of� CBF� change� was� only� 62�5%� lower.� We�19�
would� predict� that� distinct� neuroanatomic� regions� (encompassing� variations� in� nNOS� expression,�20�
vascularization�density,�mean�distance�between�glutamatergic�neurons�and�arterioles,��NO�inactivation�21�
mechanisms)� exhibited� different� quantitative� relationships� between� �NO� and� CBF� signals� but�22�
maintaining� a� temporal� and� local� correlation,� as� observed.� Therefore,� the� kinetics� of� neuronal��NO�23�

13�
�
dependent� CBF� changes� may� be� intrinsically� distinct� in� hippocampus� as� compared� with� other� brain�1�
regions.�2�
Discussion�3�
The�mechanisms�that�regulate�the�synergy�between�cerebral�microcirculation�and�local�neuronal�activity�4�
have�been�debated�over�a�century�without�clear�conclusions,�in�part�because�of�the�severe�experimental�5�
limitations�to�measure�the�process�in�real�time�in�the�natural�working�environment.�The�identification�of�6�
�NO� as� a� diffusible� vasodilator� produced� at� active� neurons,� via� ionotropic� glutamate� receptors�7�
dependent�pathways,�led�to�the�hypothesis�that�it�could�be�the�mediator�coupling�the�brain�activity�to�8�
CBF[9]�but,�paradoxically,� it� introduced�a� further�difficulty.� In� fact,�because� �NO� is�small,�hydrophobic,�9�
and�overcomes�storage�and�specific�interaction�with�receptors,�it�conveys�information�associated�to�its�10�
concentration�dynamics,�a�parameter�difficult�to�measure�in�vivo.�11�
By�simultaneously�and�direct�measuring��NO�and�CBF�dynamics�in�the�hippocampus,�this�work�supports�12�
that� �NO,� generated� by� nNOS� during� glutamatergic� activation,� induces� a� coupled� increase� in� the� local�13�
CBF�(Figure�8).�Coherently,��NO�and�CBF�events�precede�an�increase�of�O2�tension�from�a�background,�a�14�
critical�prediction�of� the� neurovascular� coupling�concept.�The�observation� that�both� MK�801�and�7�NI�15�
individually� elicited� a� significant� inhibition� of� �NO� production,� and� that� this� effect� was� translated� into�16�
CBF� changes,� clearly� identifies� the� pathway� that� paves� the� neurovascular� coupling� process� in� the�17�
hippocampus�and�recognizes�nNOS�derived��NO�as�the�coupler�molecule.�Data�showing�the�abolishment�18�
of� CBF� changes� in� spite� of� the� strong� �NO� signal� under� conditions� of� sGC� inhibition� with� ODQ,�19�
mechanistically�established�the�pathway�for��NO�dependent�CBF�changes�in�the�rat�hippocampus�by�the�20�
canonic�cGMP�dependent�vasodilation�pathway�and��NO�as�a�critical�messenger.�Furthermore,�data�also�21�
supports�that�nNOS�derived��NO�participates�in�the�maintenance�of�resting�CBF,�as�7�NI�administration�22�
also� promoted� a� decrease� in� hippocampal� CBF� basal� levels.� It� should� be� noted� that� glutamate�23�

14�
�
microinjections� (within� the� range� used)� and� electric� stimulation� induce� equivalent� cerebrovascular�1�
effects,�as�shown�previously�in�cerebellar�cortex�upon�electric�stimulation�of�parallel�fibers[35].�2�
The�pattern�of��NO�production�and�CBF�changes�measured�accomplishes�the�fundamental�criteria�of�the�3�
neurovascular� coupling� concept:� a� temporal,� amplitude� and� spatial� relationship[36].� Besides� the�4�
temporal� and� amplitude� correlation� evident� in� the� occurrence� of� both� dynamics,� data� show� that� the�5�
increase� in� CBF� is� limited� in� space,� affecting� just� the� area� of� increased� neuronal� activity,� the�6�
hippocampus,� for� if� the� laser�Doppler�probe� is�placed� in� the�overlay�cerebral� cortex�no�CBF�change� is�7�
observed�in�response�to�glutamate.��8�
The�strategy�established�in�this�work�also�complies�with�the�criteria�for��NO�signaling�in�the�framework�9�
of� the� coupling� inasmuch� as� stimulation� of� dispersed� glutamatergic� synapses� via� nNOS� generates� a�10�
volume�signaling�that�encompasses�the�diffusion�of��NO�from�neurons�to�nearby�arterioles�to�induce�an�11�
increase� in� CBF,� thus� establishing� that,� upon� eliciting� neuronal� activation,� �NO� from� nNOS� launches� a�12�
diffusible� connection� between� neurons� and� the� vasculature.� The� concept� of� �NO�mediated� diffusible�13�
connection�is�supported�by�two�observations:�first,�in�hippocampal�slices,�in�the�absence�of�a�functional�14�
vascular�system�to�remove��NO,�the�diffusion�field�of��NO�was�measured�as�being�close�to�400�μm[37];�15�
second,�the�mean�distance�between�arterioles�and�nerve�fibers�along�the�longitudinal�axis�of�pyramidal�16�
layer� neurons� in� the� CA1� region� of� hippocampus� is� circa� 150� μm[38],� i.e.,� within� the� range� of� �NO�17�
diffusional� spread.� Furthermore,� mathematical� modeling� of� the� physicochemical� process� of� �NO�18�
production� from� an� active� neuron� in� cerebellar� slices� estimated� that� the� range� of� �NO� concentration�19�
reaching� the� nearby� blood� vessels� rise� drastically,� exceeding� c.a.� 100� fold� its� basal� levels� during�20�
stimulated�neuronal�activity[39].��21�
Among�the�wide�collection�of�proposed�mechanisms,�a�generally�accepted�paradigm�for�neurovascular�22�
coupling�supports�the�view�of�astrocytes�carrying�the�neuronal�signal�by�bridging�neurons�and�vascular�23�

15�
�
cells[6].�Astrocytes�are�known�to�respond�to�glutamate�via�metabotropic�glutamate�receptors�(mGluR)�1�
activation,�involving�a�transient�increase�in�[Ca2+]i�that�ends�up�in�the�synthesis�of�vasoactive�substances�2�
upon� activation� of� phospholipase� A2� and� arachidonic� acid� production.� The� vasoactive� compounds�3�
include�prostaglandin�E2� (via�cycloxygenase)[5]�and/or�epoxyeicosatrienoic�acids� (via�cytochrome�P450�4�
epoxygenase)[7,� 40].� Accordingly,� it� has� been� speculated� that� �NO� levels� may� modulate� the�5�
cerebrovascular� response� to� mGluR� activation� by� regulating� the� enzymatic� conversion� of� arachidonic�6�
acid�via�cytochrome�P450�to�either�vasodilators�or�vasoconstrictors[41].�However,�our�results�show�that,�7�
in�hippocampus,�this�pathway�has�minor�if�any�contribution�in�the�neurovascular�coupling�promoted�by�8�
glutamatergic�activation,�supporting�that�this�process�is�developed�by�different�mechanisms�depending�9�
on� the� brain� region.� Also,� it� should� also� be� considered� that� glutamate�dependent� astrocyte� signaling�10�
may�change� during�development.�Recently,� it�was� demonstrated� that�astrocytic� mGluR5� expression� is�11�
developmentally�regulated,�being�undetectable� in�adulthood,�and�that,� in�adult�mice,�astrocytes�failed�12�
to� respond� to� the� mGluR5� agonist� tACPD� with� significant� Ca2+� increase[3].� Also,� it� is� of� note� that�13�
functional� NMDA� receptors[42]� and� constitutive� NOS� expression[43]have� been� found� in� cultured�14�
astrocytes.�Therefore,�and�although�the�abolishment�of�astrocytic�metabolism�by�sodium�fluoroacetate�15�
did�not�affect�the�recorded�dynamics,�we�cannot�fully�exclude�the�potential�involvement�of�astrocytes�in�16�
the�NMDA�nNOS�pathway,�leading�to�CBF�changes.�Further�supporting�the�notion�of�region�specificity�of�17�
neurovascular� coupling� process[44],� we� observed� that� glutamatergic� activation� in� cerebral� cortex,� as�18�
compared� with� hippocampus,� resulted� in� a� significant� lower� �NO� production� of� translated� into� less�19�
dramatic�CBF�changes.�The� �NO/CBF�ratio�was,�however,� lower� in�the�cortex,� in�agreement�to�the� less�20�
outstanding�role�attributed�to�the��NO�in�this�brain�region[18�20].��Also,�the�latency�of�CBF�response�is�21�
slower� in� the� cerebral� cortex� than� in� hippocampus,� which,� in� turn,� is� longer� than� that� commonly�22�
reported� by� studies� using� electric� stimulation� [22,� 45,� 46].� Apart� the� stimulation� paradigm,� these�23�

16�
�
variations�can�be�related�with�differences� in�the�neurovascular�coupling�process�and�vascular�network�1�
among� regions.� In� fact,� Lauritzen� group� has� observed� slower� CBF� responses� in� cerebellum� [47,� 48]� as�2�
compared� with� the� somatosensory� system� [46].� These� observations� reinforce� the� notion� that� the�3�
kinetics� of� neuronal��NO� dependent� CBF� changes� may� be� intrinsically� distinct� in� hippocampus,� as�4�
compared�with�other�brain�regions.�5�
Following�the�formulation�of�the�hypothesis�of��NO�as�a�candidate�to�mediate�neurovascular�coupling[9]�6�
intensive� research� has� attempted� to� elucidate� its� role� in� the� process.� All� the� studies,� however,� were�7�
largely�based� in� the�pharmacological�modulation�of�NOS�activity�and,�expectedly,�controversial� results�8�
were�collected.�Assuming�as�plausible�that�different�pathways�and�molecules�might�be� involved� in�the�9�
neurovascular� coupling� phenomenon,� and� that� the� relative� contribution� of� these� pathways� can� vary�10�
among� regions,� we� should� however� be� aware� that� the� use� of� such� pharmacological� tools� without�11�
additional�verification�of�their�efficacy�can�give�rise�to�misleading�results.�For�instance,�it�was�shown�that�12�
7�NI,�a�widely�used�selective�nNOS�inhibitor,�did�not�maximally�inhibit�nNOS�activity�and�did�not�affect�13�
all�brain� regions�at� the�same�extent[49].�Additionally,�methodological�variables�such�as� the�anesthetic�14�
used� seem� to� contribute� to� the� discrepancy� of� the� results,� because� their� possible� effects� on�15�
neurotransmission� and� hemodynamics[50].� Therefore,� for� a� messenger� that� conveys� information�16�
associated�with�its�concentration�dynamics,�its�direct,�localized�and�quantitative�measurement�in�vivo�is�17�
mandatory�to�progress�in�the�understanding�of�its�role�in�the�mechanisms�of�neurovascular�coupling,�a�18�
critical� pathway� for� proper� brain� function.� Indeed,� a� study� in� cerebellar� slices,� by� simultaneously�19�
measuring��NO�dynamics�and�capillaries�local�enlargements�associated�to�treatment�with�NOS�inhibitor�20�
or�with�tetrodotoxin,�suggested�that��NO�from�neuronal�origin�is�responsible�for�the�vast�majority�of�the�21�
vasomotor�response�in�cerebellum[51].�22�

17�
�
Finally,� from� a� pure� conceptual� viewpoint,� the� mechanism� of� neuronal�derived� �NO,� mediating�1�
neurovascular� coupling�via�volume�signaling,�may� be�a�non�canonical� fashion� to�underlie�a�process�of�2�
vital�importance�for�the�brain�to�preserve�its�structural�and�functional�integrity.�However,�although�the�3�
mechanistic� link� to� match� blood� supply� with� the� metabolic� demands� imposed� by� increased� neuronal�4�
activity�is�based�in�diffusion�and�characterized�by�lack�of�specific�recognition�by�receptors,�it�still�remains�5�
a�highly�and�intrinsically�controlled�mechanism.�Because�red�blood�cells�constitutes�the�major�pathway�6�
for� �NO� inactivation� in� the� brain[27],� the� increase� in� the� cerebral� blood� flow� triggered� by� neuronal�7�
derived��NO�constitute�itself�a�mechanism�to�shape�the��NO�concentration�dynamics�and�interrupt�the�8�
signaling�pathway.��9�
�10�
References��11�
[1] Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev 12�Neurosci 5:347-360; 2004. 13�
[2] Attwell, D.; Buchan, A. M.; Charpak, S.; Lauritzen, M.; Macvicar, B. A.; Newman, E. A. Glial 14�and neuronal control of brain blood flow. Nature 468:232-243; 2010. 15�
[3] Sun, W.; McConnell, E.; Pare, J. F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; 16�Nedergaard, M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. 17�Science 339:197-200; 2013. 18�
[4] Petzold, G. C.; Murthy, V. N. Role of astrocytes in neurovascular coupling. Neuron 71:782-797; 19�2011. 20�
[5] Zonta, M.; Angulo, M. C.; Gobbo, S.; Rosengarten, B.; Hossmann, K. A.; Pozzan, T.; 21�Carmignoto, G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. 22�Nat Neurosci 6:43-50; 2003. 23�
[6] Carmignoto, G.; Gomez-Gonzalo, M. The contribution of astrocyte signalling to neurovascular 24�coupling. Brain Res Rev; 2009. 25�
[7] Harder, D. R.; Alkayed, N. J.; Lange, A. R.; Gebremedhin, D.; Roman, R. J. Functional 26�hyperemia in the brain: hypothesis for astrocyte-derived vasodilator metabolites. Stroke 29:229-234; 27�1998. 28�

18�
�
[8] Faraci, F. M. nNOS-containing perivascular nerves: stranger by the minute. Circ Res 91:7-8;1�2002. 2�
[9] Iadecola, C. Regulation of the cerebral microcirculation during neural activity: is nitric oxide the 3�missing link? Trends Neurosci 16:206-214; 1993. 4�
[10] Meng, W.; Tobin, J. R.; Busija, D. W. Glutamate-induced cerebral vasodilation is mediated by 5�nitric oxide through N-methyl-D-aspartate receptors. Stroke 26:857-862; discussion 863; 1995. 6�
[11] Northington, F. J.; Tobin, J. R.; Koehler, R. C.; Traystman, R. J. In vivo production of nitric 7�oxide correlates with NMDA-induced cerebral hyperemia in newborn sheep. Am J Physiol 269:H215-8�221; 1995. 9�
[12] Pelligrino, D. A.; Gay, R. L., 3rd; Baughman, V. L.; Wang, Q. NO synthase inhibition modulates 10�NMDA-induced changes in cerebral blood flow and EEG activity. Am J Physiol 271:H990-995; 1996. 11�
[13] Garthwaite, J.; Boulton, C. L. Nitric oxide signaling in the central nervous system. Annu Rev 12�Physiol 57:683-706; 1995. 13�
[14] Sun, H. S.; Doucette, T. A.; Liu, Y.; Fang, Y.; Teves, L.; Aarts, M.; Ryan, C. L.; Bernard, P. B.; 14�Lau, A.; Forder, J. P.; Salter, M. W.; Wang, Y. T.; Tasker, R. A.; Tymianski, M. Effectiveness of PSD95 15�inhibitors in permanent and transient focal ischemia in the rat. Stroke 39:2544-2553; 2008. 16�
[15] Pacher, P.; Beckman, J. S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. 17�Physiol Rev 87:315-424; 2007. 18�
[16] Akgoren, N.; Fabricius, M.; Lauritzen, M. Importance of nitric oxide for local increases of blood 19�flow in rat cerebellar cortex during electrical stimulation. Proc Natl Acad Sci U S A 91:5903-5907; 1994. 20�
[17] Faraci, F. M.; Breese, K. R. Nitric oxide mediates vasodilatation in response to activation of N-21�methyl-D-aspartate receptors in brain. Circ Res 72:476-480; 1993. 22�
[18] Adachi, K.; Takahashi, S.; Melzer, P.; Campos, K. L.; Nelson, T.; Kennedy, C.; Sokoloff, L. 23�Increases in local cerebral blood flow associated with somatosensory activation are not mediated by NO. 24�The American journal of physiology 267:H2155-2162; 1994. 25�
[19] Greenberg, J. H.; Sohn, N. W.; Hand, P. J. Nitric oxide and the cerebral-blood-flow response to 26�somatosensory activation following deafferentation. Experimental brain research. Experimentelle 27�Hirnforschung 129:541-550; 1999. 28�
[20] Wang, Q.; Kjaer, T.; Jorgensen, M. B.; Paulson, O. B.; Lassen, N. A.; Diemer, N. H.; Lou, H. C. 29�Nitric oxide does not act as a mediator coupling cerebral blood flow to neural activity following 30�somatosensory stimuli in rats. Neurological research 15:33-36; 1993. 31�
[21] Lindauer, U.; Megow, D.; Matsuda, H.; Dirnagl, U. Nitric oxide: a modulator, but not a mediator, 32�of neurovascular coupling in rat somatosensory cortex. Am J Physiol 277:H799-811; 1999. 33�

19�
�
[22] Buerk, D. G.; Ances, B. M.; Greenberg, J. H.; Detre, J. A. Temporal dynamics of brain tissue 1�nitric oxide during functional forepaw stimulation in rats. NeuroImage 18:1-9; 2003. 2�
[23] Lourenço, C. F.; Santos, R.; Barbosa, R. M.; Gerhardt, G.; Cadenas, E.; Laranjinha, J. In vivo 3�modulation of nitric oxide concentration dynamics upon glutamatergic neuronal activation in the 4�hippocampus. Hippocampus 21:622-630; 2011. 5�
[24] Santos, R. M.; Lourenco, C. F.; Piedade, A. P.; Andrews, R.; Pomerleau, F.; Huettl, P.; Gerhardt, 6�G. A.; Laranjinha, J.; Barbosa, R. M. A comparative study of carbon fiber-based microelectrodes for the 7�measurement of nitric oxide in brain tissue. Biosens Bioelectron 24:704-709; 2008. 8�
[25] Barbosa, R. M.; Lopes Jesus, A. J.; Santos, R. M.; Pereira, C. L.; Marques, C. F.; Rocha, B. S.; 9�Ferreira, N. R.; Ledo, A.; Laranjinha, J. Preparation, standardization and measurement of nitric oxide 10�solutions. Global Journal of Analytical Chemistry 2:272-284; 2011. 11�
[26] Steinert, J. R.; Kopp-Scheinpflug, C.; Baker, C.; Challiss, R. A.; Mistry, R.; Haustein, M. D.; 12�Griffin, S. J.; Tong, H.; Graham, B. P.; Forsythe, I. D. Nitric oxide is a volume transmitter regulating 13�postsynaptic excitability at a glutamatergic synapse. Neuron 60:642-656; 2008. 14�
[27] Santos, R. M.; Lourenco, C. F.; Pomerleau, F.; Huettl, P.; Gerhardt, G. A.; Laranjinha, J.; 15�Barbosa, R. M. Brain nitric oxide inactivation is governed by the vasculature. Antioxid Redox Signal 1416�1011-1021; 2011. 17�
[28] Li, B.; Freeman, R. D. High-resolution neurometabolic coupling in the lateral geniculate nucleus. 18�J Neurosci 27:10223-10229; 2007. 19�
[29] Feelisch, M.; Kotsonis, P.; Siebe, J.; Clement, B.; Schmidt, H. H. The soluble guanylyl cyclase 20�inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one is a nonselective heme protein inhibitor of nitric 21�oxide synthase and other cytochrome P-450 enzymes involved in nitric oxide donor bioactivation. Mol 22�Pharmacol 56:243-253; 1999. 23�
[30] Fonnum, F.; Johnsen, A.; Hassel, B. Use of fluorocitrate and fluoroacetate in the study of brain 24�metabolism. Glia 21:106-113; 1997. 25�
[31] Iliff, J. J.; D'Ambrosio, R.; Ngai, A. C.; Winn, H. R. Adenosine receptors mediate glutamate-26�evoked arteriolar dilation in the rat cerebral cortex. Am J Physiol Heart Circ Physiol 284:H1631-1637; 27�2003. 28�
[32] Xu, H. L.; Pelligrino, D. A. ATP release and hydrolysis contribute to rat pial arteriolar dilatation 29�elicited by neuronal activation. Exp Physiol 92:647-651; 2007. 30�
[33] Fredholm, B. B. Adenosine receptors as drug targets. Exp Cell Res 316:1284-1288; 2010. 31�
[34] Gotti, S.; Sica, M.; Viglietti-Panzica, C.; Panzica, G. Distribution of nitric oxide synthase 32�immunoreactivity in the mouse brain. Microsc Res Tech 68:13-35; 2005. 33�

20�
�
[35] Yang, G.; Iadecola, C. Glutamate microinjections in cerebellar cortex reproduce cerebrovascular 1�effects of parallel fiber stimulation. Am J Physiol 271:R1568-1575; 1996. 2�
[36] Moore, C. I.; Cao, R. The hemo-neural hypothesis: on the role of blood flow in information 3�processing. J Neurophysiol 99:2035-2047; 2008. 4�
[37] Ledo, A.; Barbosa, R. M.; Gerhardt, G. A.; Cadenas, E.; Laranjinha, J. Concentration dynamics of 5�nitric oxide in rat hippocampal subregions evoked by stimulation of the NMDA glutamate receptor. Proc6�Natl Acad Sci U S A 102:17483-17488; 2005. 7�
[38] Lovick, T. A.; Brown, L. A.; Key, B. J. Neurovascular relationships in hippocampal slices: 8�physiological and anatomical studies of mechanisms underlying flow-metabolism coupling in 9�intraparenchymal microvessels. Neuroscience 92:47-60; 1999. 10�
[39] Oleinick, A. I.; Amatore, C.; Guille, M.; Arbault, S.; Klymenko, O. V.; Svir, I. Modelling release 11�of nitric oxide in a slice of rat's brain: describing stimulated functional hyperemia with diffusion-reaction 12�equations. Math Med Biol 23:27-44; 2006. 13�
[40] Iliff, J. J.; Wang, R.; Zeldin, D. C.; Alkayed, N. J. Epoxyeicosanoids as mediators of neurogenic 14�vasodilation in cerebral vessels. Am J Physiol Heart Circ Physiol 296:H1352-1363; 2009. 15�
[41] Mulligan, S. J.; MacVicar, B. A. Calcium transients in astrocyte endfeet cause cerebrovascular 16�constrictions. Nature 431:195-199; 2004. 17�
[42] Lalo, U.; Pankratov, Y.; Kirchhoff, F.; North, R. A.; Verkhratsky, A. NMDA receptors mediate 18�neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci 26:2673-2683; 2006. 19�
[43] Oka, M.; Wada, M.; Yamamoto, A.; Itoh, Y.; Fujita, T. Functional expression of constitutive 20�nitric oxide synthases regulated by voltage-gated Na+ and Ca2+ channels in cultured human astrocytes. 21�Glia 46:53-62; 2004. 22�
[44] Devonshire, I. M.; Papadakis, N. G.; Port, M.; Berwick, J.; Kennerley, A. J.; Mayhew, J. E.; 23�Overton, P. G. Neurovascular coupling is brain region-dependent. NeuroImage 59:1997-2006; 2012. 24�
[45] Devor, A.; Dunn, A. K.; Andermann, M. L.; Ulbert, I.; Boas, D. A.; Dale, A. M. Coupling of total 25�hemoglobin concentration, oxygenation, and neural activity in rat somatosensory cortex. Neuron 39:353-26�359; 2003. 27�
[46] Norup Nielsen, A.; Lauritzen, M. Coupling and uncoupling of activity-dependent increases of 28�neuronal activity and blood flow in rat somatosensory cortex. The Journal of physiology 533:773-785; 29�2001. 30�
[47] Mathiesen, C.; Caesar, K.; Akgoren, N.; Lauritzen, M. Modification of activity-dependent 31�increases of cerebral blood flow by excitatory synaptic activity and spikes in rat cerebellar cortex. The 32�Journal of physiology 512 ( Pt 2):555-566; 1998. 33�

21�
�
[48] Offenhauser, N.; Thomsen, K.; Caesar, K.; Lauritzen, M. Activity-induced tissue oxygenation 1�changes in rat cerebellar cortex: interplay of postsynaptic activation and blood flow. The Journal of 2�physiology 565:279-294; 2005. 3�
[49] Kalisch, B. E.; Connop, B. P.; Jhamandas, K.; Beninger, R. J.; Boegman, R. J. Differential action 4�of 7-nitro indazole on rat brain nitric oxide synthase. Neuroscience letters 219:75-78; 1996. 5�
[50] Franceschini, M. A.; Radhakrishnan, H.; Thakur, K.; Wu, W.; Ruvinskaya, S.; Carp, S.; Boas, D. 6�A. The effect of different anesthetics on neurovascular coupling. NeuroImage 51:1367-1377; 2010. 7�
[51] Rancillac, A.; Rossier, J.; Guille, M.; Tong, X. K.; Geoffroy, H.; Amatore, C.; Arbault, S.; 8�Hamel, E.; Cauli, B. Glutamatergic Control of Microvascular Tone by Distinct GABA Neurons in the 9�Cerebellum. J Neurosci 26:6997-7006; 2006. 10�
11�
�12�
Figure�Legends:�13�
�14�
Figure� 1:� Coupling� between� �NO� concentration� dynamics� and� CBF� changes� upon� glutamatergic�15�
activation�in�the�hippocampus.�A.�Representative�recording�of�the�simultaneous�measurements�of��NO�16�
concentration� dynamics� (bottom,� blue� line)� and� CBF� changes� (top,� red� line)� in� the� hippocampus� of�17�
urethane�anesthetized�rat�over�a�period�of�five�stimulations.�Glutamate�was�locally�applied�at�the�times�18�
indicated� by� the� arrows� (0.5� nmol,� 1� s).� B.� Detail� of� the� temporal� correlation� of� �NO� concentration�19�
dynamics� and� CBF� changes� evoked� by� local� glutamate� stimulation.� C.� Array� used� to� measure�20�
simultaneously� •NO� concentration� dynamics� and� CBF� changes� in� the� rat� hippocampus.� The� array�21�
comprises� a� •NO�selective� microelectrode,� a� microinjection� pipette� and� a� laser� Doppler� flow� probe�22�
assembled�in�a�pre�defined�geometry.�23�
�24�

22�
�
Figure�2:�Exogenous� �NO�mimics�glutamate�induced�CBF�changes.�Average�recordings�of�CBF�changes�1�
(red� line)� in� response�to�exogenous�and� localized� �NO�application� in� the�hippocampus� (blue� line).� �NO�2�
solution�(100�μM�in�saline)�was�locally�applied�by�pressure�ejection�(5�psi,�90�s,�1�μL).�3�
�4�
Figure� 3:� The� CBF� change� coupled� to� �NO� dynamics� is� followed� by� a� transient� increase� in� local� O2�5�
tension.�Average� recordings� of� simultaneous� measurements� of� O2� (green� line)� and� CBF� changes� (red�6�
line)� in� the� hippocampus� of� urethane�anesthetized� rat� and� temporal� correlation� with� �NO� dynamics�7�
(blue� line).� Insert� shows� the� correlation� between� the� amplitudes� of� O2� and� CBF� changes� (R2=0.69,�8�
p=0.0016,�n=12�from�3�individual�experiments).�9�
�10�
Figure� 4:� Glutamate�induced� �NO� production� and� CBF� changes� are� dependent� on� the� activation� of�11�
NMDA� receptor� and� nNOS.� A.� Effect� of� MK�801� (1� mg/kg,� NMDA� receptor� blocker),� 7�NI� (50� mg/Kg,�12�
selective�nNOS�inhibitor)�and�L�NIO�(40�mg/Kg,�selective�eNOS�inhibitor).�Data�represents�mean�±�SEM.�13�
Statistical�analysis�was�performed�by�Student’s�t�test�in�relation�to�control�experiments�(***p<�0.001).�B.�14�
Plot�of�the�linear�relationship�between�the�amplitude�of��NO�production�and�CBF�changes�obtained�by�15�
glutamatergic�activation.�The�linear�regression�showed�a�R2=0.98�and�a�slope�of�78.9%�CBF/fmol.s�1�NO�16�
(X� intercept=0.438).�C.�Average� recordings� of� the� 7�NI� and� L�NIO� effects� on� the� �NO� production� (blue�17�
line)�and�CBF�changes�(red�line)�elicited�by�glutamate�in�the�hippocampus.�18�
�19�
Figure� 5:� Neuronal� derived��NO�mediates� the� neurovascular� coupling� through� activation� of� soluble�20�
guanylate� cyclase.� Effect� ODQ,� a� selective� heme�site� inhibitor� of� soluble� guanylate� cyclase,� on�21�

23�
�
glutamate�induced��NO�signals�(blue�line)�and�CBF�changes�(red�line)�in�the�rat�hippocampus.�ODQ�was�1�
locally�applied�through�an�additional�pipette�attached�to�the�array�in�the�proximity�of�the�laser�Doppler�2�
probe�(25�pmol,�0.5�μL).�3�
�4�
Figure� 6:� The� astrocytic� pathway� is� not� explicitly� involved� in� the� �NO�mediated� neurovascular�5�
coupling.�A.�Effect�of�LY367385/MPEP�(100��M�each,�antagonists�of�metabotropic�glutamate�receptors,�6�
mGluR1�and�mGluR5,�respectively,�n=6),�acetylsalicylic�acid�(ASA,�300�mg/kg,�COX�inhibitor,�n=5),�sodium�7�
fluoroacetate� (SFA,� 20� μmol� locally,� glial� toxin,� n=5)� and� caffeine� (CAF,� 50� mg/kg,� nonselective�8�
antagonist�of�adenosine�receptors,�n=3)�on��NO�signals�and�CBF�changes.�Data�represents�mean�±�SEM.�9�
Statistical�analysis�was�performed�by�Student’s�t�test�in�relation�to�control�experiments�(performed�with�10�
vehicles).� B.� Representative� recording� of� tACPD� injection,� an� agonist� of� mGluR,� in� �NO� dynamics�11�
(bottom,� blue� line)� and� CBF� changes� (top,� red� line)� in� the� hippocampus� of� urethane�anesthetized� rat.�12�
tACPD� was� locally� pressure� ejected� at� the� time� indicated� by� the� arrows� (25�,� 50�,� and� 100� pmol,�13�
respectively).�14�
�15�
Figure� 7:� Coupling� between� �NO� concentration� dynamics� and� CBF� changes� upon� glutamatergic�16�
activation� in� the� cortex.� Average� recordings� of� the� simultaneous� measurement� of� �NO� concentration�17�
dynamics� (bottom,� blue� line)� and� CBF� changes� (top,� red� line)� in� the� cerebral� cortex� of� urethane�18�
anesthetized�rat�upon�glutamate�stimulation�(0.5�nmol,�1�s).�19�
�20�

24�
�
Figure� 8:�Nitric� oxide� is� a� critical� diffusible�messenger� between� active� neurons� and� the� local� blood�1�
vessels� in� hippocampus.� The� scheme� depicts� the� cellular� relationships� and� the� sequence� of� events�2�
underlying�the�neurovascular�coupling�mediated�by��NO�upon�glutamatergic�stimulation.��NO�transitorily�3�
produced� by� neurons� upon� stimulation� on� glutamate� NMDA� receptors� diffuses� towards� neighboring�4�
blood�vessels�where,�following�the�interaction�with�soluble�GC,�triggers�a�sequence�of�events�comprising�5�
vasodilation�and�a�transient�and�localized�increase�in�O2�tension.�6�
Highlights�7�
� Nitric�oxide�is�as�a�mediator�of�the�neurovascular�coupling�in�hippocampus�8�� Nitric�oxide�induces�a�time,�spatial�and�amplitude�coupled�increase�of�local�CBF�9�� Both�NO�and�CBF�dynamics�are�dependent�on�the�activation�of�NMDA�receptors�and�nNOS�10�� The�CBF�increase�mediated�by�neuronal�derived�NO�depends�on�the�activation�of�sGC��11�
�12�
�13�

Gra
phic
al A
bstr
act (
for r
evie
w)

Figu
re 1
Figu
re 2

Figu
re 3
Figu
re 4
Figu
re 5

Figu
re 6

Figu
re 7

Figu
re 8