lrg1 promotes angiogenesis by modulating endothelial tgf-b signalling
TRANSCRIPT

ARTICLEdoi:10.1038/nature12345
LRG1 promotes angiogenesis bymodulating endothelial TGF-b signallingXiaomeng Wang1, Sabu Abraham1, Jenny A. G. McKenzie1, Natasha Jeffs1, Matthew Swire1, Vineeta B. Tripathi1,Ulrich F. O. Luhmann2, Clemens A. K. Lange2,3,4, Zhenhua Zhai5, Helen M. Arthur5, James W. B. Bainbridge2,3, Stephen E. Moss1*& John Greenwood1*
Aberrant neovascularization contributes to diseases such as cancer, blindness and atherosclerosis, and is the consequenceof inappropriate angiogenic signalling. Although many regulators of pathogenic angiogenesis have been identified, ourunderstanding of this process is incomplete. Here we explore the transcriptome of retinal microvessels isolated frommouse models of retinal disease that exhibit vascular pathology, and uncover an upregulated gene, leucine-richalpha-2-glycoprotein 1 (Lrg1), of previously unknown function. We show that in the presence of transforming growthfactor-b1 (TGF-b1), LRG1 is mitogenic to endothelial cells and promotes angiogenesis. Mice lacking Lrg1 develop a mildretinal vascular phenotype but exhibit a significant reduction in pathological ocular angiogenesis. LRG1 binds directly tothe TGF-b accessory receptor endoglin, which, in the presence of TGF-b1, results in promotion of the pro-angiogenicSmad1/5/8 signalling pathway. LRG1 antibody blockade inhibits this switch and attenuates angiogenesis. These studiesreveal a new regulator of angiogenesis that mediates its effect by modulating TGF-b signalling.
The formation of new blood vessels by angiogenesis is a key feature ofseveral diseases including age-related macular degeneration, prolifera-tive diabetic retinopathy (PDR), atherosclerosis, rheumatoid arthritisand cancer. The factors that promote neovascularization have beenthe subject of extensive research, with the vascular endothelial growthfactors (VEGFs) and their receptors emerging as master regulators1–3.Despite the prominent role of VEGF, other factors contribute to neoan-giogenesis through coordinated crosstalk that is often highly context-dependent4–6. Such complexity is exemplified in TGF-b1 signalling,which can switch from being mostly angiostatic to pro-angiogenic7.What regulates this switch is not fully understood, but activation ofthe pro-angiogenic pathway involves TGF-b type II receptor (TbRII)recruitment of the predominantly endothelial TGF-b type I receptoractivin receptor-like kinase-1 (ALK1), which in turn initiates activa-tion of the transcription factors Smad1, 5 and 8, resulting in a pro-angiogenic phenotype7–10. The regulation of this differential signallingis contingent on several factors including the concentration of TGF-b,its bioavailability and the presence or absence of other regulatory fac-tors such as bone morphogenic proteins and accessory receptors suchas endoglin (ENG) and betaglycan (also known as TGF-b type IIIreceptor)11.
Our incomplete understanding of the role of the fine-tuning ofangiogenesis suggests that additional modulators have yet to be iden-tified. Our objective in this study, therefore, was to identify new reg-ulators of pathogenic angiogenesis that may lead to the developmentof more effective treatment strategies.
Retinal vascular expression of LRG1To identify new regulators of neovascularization we exploited three mousemutants that exhibit marked remodelling of the retinal vasculature (Sup-plementary Fig. 1 and Supplementary Videos 1–4). Genome-wide trans-criptome analysis of retinal microvessel fragments isolated from the retinaldegeneration 1 (rd1) mouse, the very low density lipoprotein receptor
(VLDLR) knockout mouse (Vldlr2/2), the Grhl3ct/J curly tail mouse(Jackson Laboratory) and appropriate wild-type control mice yielded 62genes that were differentially regulated but common to all three retinaldisease models (Supplementary Table 1). When ranked according to foldchange, a gene encoding a secreted glycoprotein of unknown function,namely Lrg1, emerged as the most significantly upregulated. LRG1 is ahighly conserved member of the leucine-rich repeat family of proteins,many of which are involved in protein–protein interactions, signalling andcell adhesion (Supplementary Fig. 2a, b).
Validation of the microarray data revealed that in the retina, LRG1 isrestricted almost exclusively to the vasculature, is expressed undernormal conditions and is upregulated during retinal vascular remodel-ling in the three mouse models of retinal disease (Fig. 1a–d andSupplementary Fig. 3). However, LRG1 expression was not restrictedto the retina, as we also observed LRG1 staining in the choriocapillarisof the mouse eye (Supplementary Fig. 4a). Consistent with the dataobtained in the mouse, we observed low levels of constitutive LRG1expression in normal adult human retinal vessels and weakly, but notexclusively, in vessels in other human tissues including breast, skin andintestine (Supplementary Fig. 4b).
We next investigated whether the Lrg1 transcript is also increasedin the retinae of models of choroidal and retinal neovascularization.Choroidal neovascularization (CNV) was induced in wild-type mice,and 1 week after laser injury we observed a significant increase in Lrg1transcript levels in both the retina and retinal pigment epithelium(RPE)/choroid (Fig. 1e, f). We then examined intra-retinal/pre-retinalneovascularization in the mouse model of oxygen-induced retino-pathy (OIR), which displays hypoxia-driven retinal angiogenesis. Atpostnatal day (P) 17, during the ischaemic proliferative phase of OIRwhen neovascularization is most prevalent, Lrg1 transcript levels werealso upregulated (Fig. 1g). However, at the end of the hyperoxic phase(P12), Lrg1 messenger RNA was significantly reduced. Indeed, thepattern of Lrg1 expression at the two time points observed mirrored
*These authors contributed equally to this work.
1Departmentof Cell Biology,UCL Institute of Ophthalmology, London EC1V 9EL, UK. 2Department of Genetics,UCL Institute of Ophthalmology, London EC1V 9EL, UK. 3NIHR BiomedicalResearch Centre forOphthalmology, Moorfields Eye Hospital, London EC1V 2PD, UK. 4University Eye Hospital Freiburg, Freiburg 79106, Germany. 5Institute of Genetic Medicine, Newcastle University, Newcastle NE1 3BZ, UK.
3 0 6 | N A T U R E | V O L 4 9 9 | 1 8 J U L Y 2 0 1 3
Macmillan Publishers Limited. All rights reserved©2013
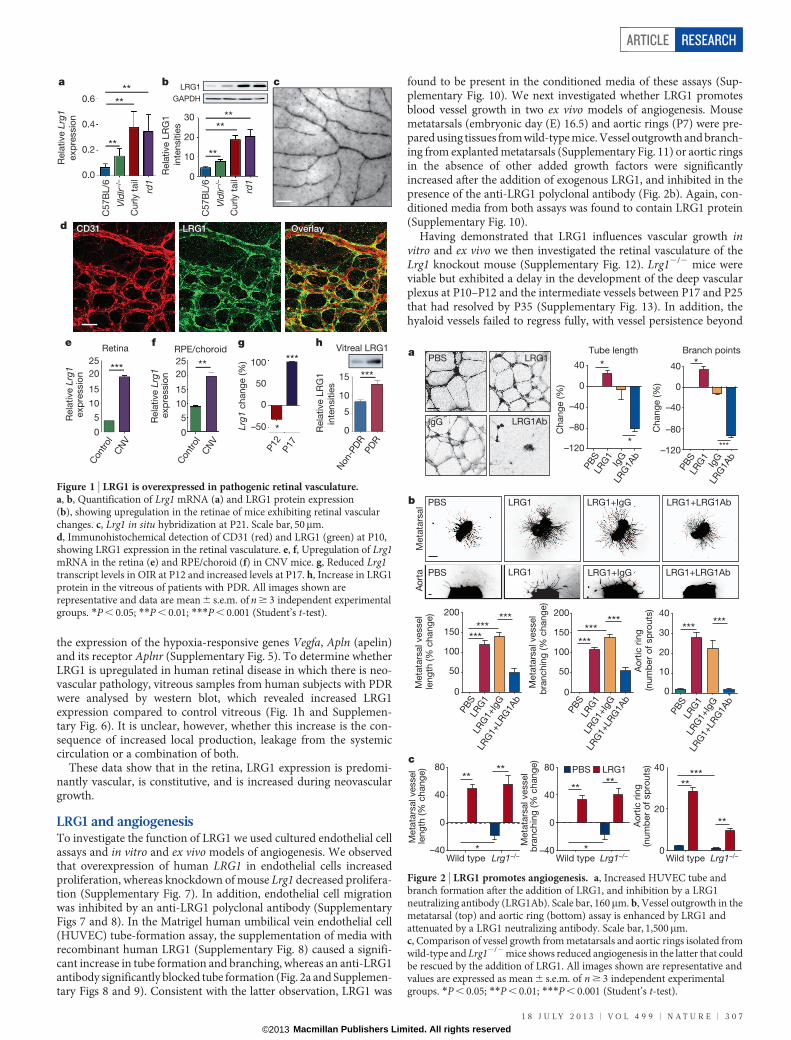
the expression of the hypoxia-responsive genes Vegfa, Apln (apelin)and its receptor Aplnr (Supplementary Fig. 5). To determine whetherLRG1 is upregulated in human retinal disease in which there is neo-vascular pathology, vitreous samples from human subjects with PDRwere analysed by western blot, which revealed increased LRG1expression compared to control vitreous (Fig. 1h and Supplemen-tary Fig. 6). It is unclear, however, whether this increase is the con-sequence of increased local production, leakage from the systemiccirculation or a combination of both.
These data show that in the retina, LRG1 expression is predomi-nantly vascular, is constitutive, and is increased during neovasculargrowth.
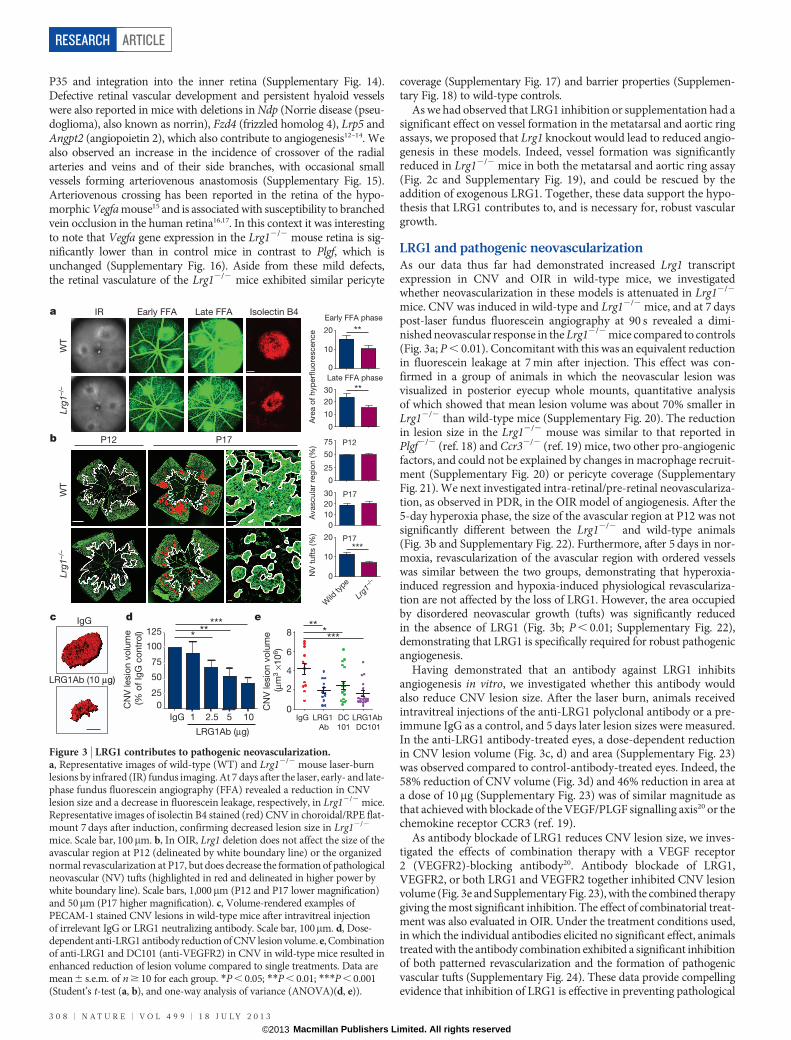
LRG1 and angiogenesisTo investigate the function of LRG1 we used cultured endothelial cellassays and in vitro and ex vivo models of angiogenesis. We observedthat overexpression of human LRG1 in endothelial cells increasedproliferation, whereas knockdown of mouse Lrg1 decreased prolifera-tion (Supplementary Fig. 7). In addition, endothelial cell migrationwas inhibited by an anti-LRG1 polyclonal antibody (SupplementaryFigs 7 and 8). In the Matrigel human umbilical vein endothelial cell(HUVEC) tube-formation assay, the supplementation of media withrecombinant human LRG1 (Supplementary Fig. 8) caused a signifi-cant increase in tube formation and branching, whereas an anti-LRG1antibody significantly blocked tube formation (Fig. 2a and Supplemen-tary Figs 8 and 9). Consistent with the latter observation, LRG1 was
found to be present in the conditioned media of these assays (Sup-plementary Fig. 10). We next investigated whether LRG1 promotesblood vessel growth in two ex vivo models of angiogenesis. Mousemetatarsals (embryonic day (E) 16.5) and aortic rings (P7) were pre-pared using tissues from wild-type mice. Vessel outgrowth and branch-ing from explanted metatarsals (Supplementary Fig. 11) or aortic ringsin the absence of other added growth factors were significantlyincreased after the addition of exogenous LRG1, and inhibited in thepresence of the anti-LRG1 polyclonal antibody (Fig. 2b). Again, con-ditioned media from both assays was found to contain LRG1 protein(Supplementary Fig. 10).
Having demonstrated that LRG1 influences vascular growth invitro and ex vivo we then investigated the retinal vasculature of theLrg1 knockout mouse (Supplementary Fig. 12). Lrg12/2 mice wereviable but exhibited a delay in the development of the deep vascularplexus at P10–P12 and the intermediate vessels between P17 and P25that had resolved by P35 (Supplementary Fig. 13). In addition, thehyaloid vessels failed to regress fully, with vessel persistence beyond
ColIV
c
d
hf
Lrg1 Overlay
ge
a b
****
Rela
tive L
rg1
exp
ressio
n
0.6
0.4
0.2
0.0
C5
7B
L/6
Vld
lr–/–
Cu
rly t
ail
rd1
**
**** LRG1
GAPDH
Rela
tive L
RG
1
in
ten
sitie
s
30
20
10
0
**
****
Con
trol
CN
V
25
20
15
10
5Rela
tive L
rg1
exp
ressio
n
Retina
0
***25
20
15
10
5
0
**
RPE/choroid
Rela
tive L
rg1
exp
ressio
n
Lrg1
chang
e (%
) 100
50
0
–50
P12
P17
***
* Rela
tive L
RG
1
inte
nsitie
s ***15
10
5
0
Non
-PD
RPD
R
Vitreal LRG1
Vld
lr–/–
Cu
rly t
ail
rd1
CD31CD31 LRG1LRG1 OverlayOverlayCD31 LRG1 Overlay
Con
trol
CN
VC
57
BL
/6
Figure 1 | LRG1 is overexpressed in pathogenic retinal vasculature.a, b, Quantification of Lrg1 mRNA (a) and LRG1 protein expression(b), showing upregulation in the retinae of mice exhibiting retinal vascularchanges. c, Lrg1 in situ hybridization at P21. Scale bar, 50mm.d, Immunohistochemical detection of CD31 (red) and LRG1 (green) at P10,showing LRG1 expression in the retinal vasculature. e, f, Upregulation of Lrg1mRNA in the retina (e) and RPE/choroid (f) in CNV mice. g, Reduced Lrg1transcript levels in OIR at P12 and increased levels at P17. h, Increase in LRG1protein in the vitreous of patients with PDR. All images shown arerepresentative and data are mean 6 s.e.m. of n $ 3 independent experimentalgroups. *P , 0.05; **P , 0.01; ***P , 0.001 (Student’s t-test).
Meta
tars
al
a
b
40
0
–40
–80
–120
40
0
–40
–80
–120
Tube length Branch points
Chang
e (%
)
Chang
e (%
)
PBS
LRG
1Ig
GLR
G1A
b
PBS
LRG
1Ig
GLR
G1A
b
***
**
Meta
tars
al vessel
leng
th (%
chang
e) 200
150
100
50
0
PBS
LRG
1LR
G1+
IgG
LRG
1+LR
G1A
b
******
***
Meta
tars
al vessel
bra
nchin
g (%
chang
e)
200
150
100
50
0
PBS
LRG
1LR
G1+
IgG
LRG
1+LR
G1A
b
******
***40
30
20
10
0
******
PBS
LRG
1LR
G1+
IgG
LRG
1+LR
G1A
b
Ao
rtic
rin
g(n
um
ber
of
sp
routs
)
Aort
a
PBS LRG1 LRG1+IgG LRG1+LRG1Ab
PBS LRG1+IgGLRG1 LRG1+LRG1Ab
Wild type
40
20
0
Ao
rtic
rin
g(n
um
ber
of
sp
routs
)
*****
80
40
0
–40
**
*
**
Wild type Lrg1–/–
PBS LRG180
40
0
–40Wild type Lrg1–/–
**
*
**
c
Meta
tars
al vessel
leng
th (%
chang
e)
Meta
tars
al vessel
bra
nchin
g (%
chang
e)
*
**
PBS LRG1
LRG1AbIgG
Lrg1–/–
Figure 2 | LRG1 promotes angiogenesis. a, Increased HUVEC tube andbranch formation after the addition of LRG1, and inhibition by a LRG1neutralizing antibody (LRG1Ab). Scale bar, 160mm. b, Vessel outgrowth in themetatarsal (top) and aortic ring (bottom) assay is enhanced by LRG1 andattenuated by a LRG1 neutralizing antibody. Scale bar, 1,500mm.c, Comparison of vessel growth from metatarsals and aortic rings isolated fromwild-type and Lrg12/2 mice shows reduced angiogenesis in the latter that couldbe rescued by the addition of LRG1. All images shown are representative andvalues are expressed as mean 6 s.e.m. of n $ 3 independent experimentalgroups. *P , 0.05; **P , 0.01; ***P , 0.001 (Student’s t-test).
ARTICLE RESEARCH
1 8 J U L Y 2 0 1 3 | V O L 4 9 9 | N A T U R E | 3 0 7
Macmillan Publishers Limited. All rights reserved©2013
P35 and integration into the inner retina (Supplementary Fig. 14).Defective retinal vascular development and persistent hyaloid vesselswere also reported in mice with deletions in Ndp (Norrie disease (pseu-doglioma), also known as norrin), Fzd4 (frizzled homolog 4), Lrp5 andAngpt2 (angiopoietin 2), which also contribute to angiogenesis12–14. Wealso observed an increase in the incidence of crossover of the radialarteries and veins and of their side branches, with occasional smallvessels forming arteriovenous anastomosis (Supplementary Fig. 15).Arteriovenous crossing has been reported in the retina of the hypo-morphic Vegfa mouse15 and is associated with susceptibility to branchedvein occlusion in the human retina16,17. In this context it was interestingto note that Vegfa gene expression in the Lrg12/2 mouse retina is sig-nificantly lower than in control mice in contrast to Plgf, which isunchanged (Supplementary Fig. 16). Aside from these mild defects,the retinal vasculature of the Lrg12/2 mice exhibited similar pericyte
coverage (Supplementary Fig. 17) and barrier properties (Supplemen-tary Fig. 18) to wild-type controls.
As we had observed that LRG1 inhibition or supplementation had asignificant effect on vessel formation in the metatarsal and aortic ringassays, we proposed that Lrg1 knockout would lead to reduced angio-genesis in these models. Indeed, vessel formation was significantlyreduced in Lrg12/2 mice in both the metatarsal and aortic ring assay(Fig. 2c and Supplementary Fig. 19), and could be rescued by theaddition of exogenous LRG1. Together, these data support the hypo-thesis that LRG1 contributes to, and is necessary for, robust vasculargrowth.
LRG1 and pathogenic neovascularizationAs our data thus far had demonstrated increased Lrg1 transcriptexpression in CNV and OIR in wild-type mice, we investigatedwhether neovascularization in these models is attenuated in Lrg12/2
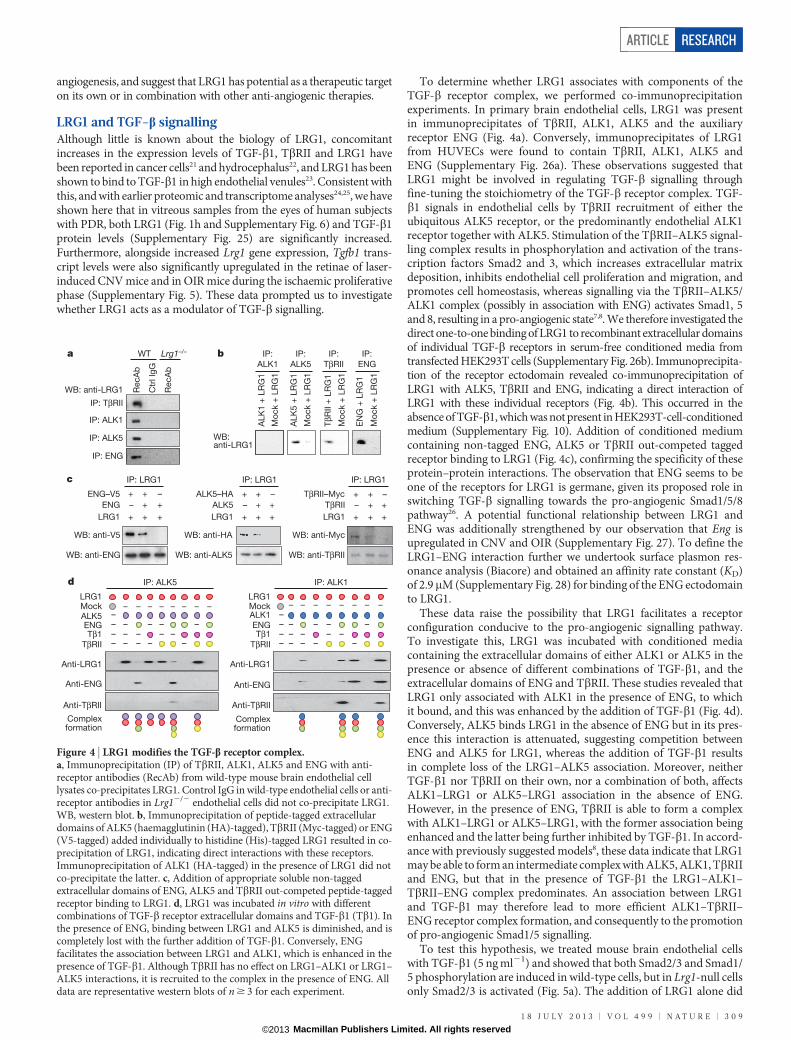
mice. CNV was induced in wild-type and Lrg12/2 mice, and at 7 dayspost-laser fundus fluorescein angiography at 90 s revealed a dimi-nished neovascular response in the Lrg12/2 mice compared to controls(Fig. 3a; P , 0.01). Concomitant with this was an equivalent reductionin fluorescein leakage at 7 min after injection. This effect was con-firmed in a group of animals in which the neovascular lesion wasvisualized in posterior eyecup whole mounts, quantitative analysisof which showed that mean lesion volume was about 70% smaller inLrg12/2 than wild-type mice (Supplementary Fig. 20). The reductionin lesion size in the Lrg12/2 mouse was similar to that reported inPlgf2/2 (ref. 18) and Ccr32/2 (ref. 19) mice, two other pro-angiogenicfactors, and could not be explained by changes in macrophage recruit-ment (Supplementary Fig. 20) or pericyte coverage (SupplementaryFig. 21). We next investigated intra-retinal/pre-retinal neovasculariza-tion, as observed in PDR, in the OIR model of angiogenesis. After the5-day hyperoxia phase, the size of the avascular region at P12 was notsignificantly different between the Lrg12/2 and wild-type animals(Fig. 3b and Supplementary Fig. 22). Furthermore, after 5 days in nor-moxia, revascularization of the avascular region with ordered vesselswas similar between the two groups, demonstrating that hyperoxia-induced regression and hypoxia-induced physiological revasculariza-tion are not affected by the loss of LRG1. However, the area occupiedby disordered neovascular growth (tufts) was significantly reducedin the absence of LRG1 (Fig. 3b; P , 0.01; Supplementary Fig. 22),demonstrating that LRG1 is specifically required for robust pathogenicangiogenesis.
Having demonstrated that an antibody against LRG1 inhibitsangiogenesis in vitro, we investigated whether this antibody wouldalso reduce CNV lesion size. After the laser burn, animals receivedintravitreal injections of the anti-LRG1 polyclonal antibody or a pre-immune IgG as a control, and 5 days later lesion sizes were measured.In the anti-LRG1 antibody-treated eyes, a dose-dependent reductionin CNV lesion volume (Fig. 3c, d) and area (Supplementary Fig. 23)was observed compared to control-antibody-treated eyes. Indeed, the58% reduction of CNV volume (Fig. 3d) and 46% reduction in area ata dose of 10mg (Supplementary Fig. 23) was of similar magnitude asthat achieved with blockade of the VEGF/PLGF signalling axis20 or thechemokine receptor CCR3 (ref. 19).
As antibody blockade of LRG1 reduces CNV lesion size, we inves-tigated the effects of combination therapy with a VEGF receptor2 (VEGFR2)-blocking antibody20. Antibody blockade of LRG1,VEGFR2, or both LRG1 and VEGFR2 together inhibited CNV lesionvolume (Fig. 3e and Supplementary Fig. 23), with the combined therapygiving the most significant inhibition. The effect of combinatorial treat-ment was also evaluated in OIR. Under the treatment conditions used,in which the individual antibodies elicited no significant effect, animalstreated with the antibody combination exhibited a significant inhibitionof both patterned revascularization and the formation of pathogenicvascular tufts (Supplementary Fig. 24). These data provide compellingevidence that inhibition of LRG1 is effective in preventing pathological
LRG1Ab (10 μg)
P12
**
a
b
c
P17
WT
Lrg1
–/–
IR Early FFA Late FFA Isolectin B4Early FFA phase
Late FFA phase
20
10
0
30
20
10
0
Are
a o
f h
yp
erfl
uo
rescen
ce
WT
Lrg1
–/–
75
50
25
0
Avascu
lar
reg
ion
(%
)
30
20
10
0
20
10
0NV
tu
fts (%
)
Wild
type
Lrg1
–/–
IgG e125
100
75
50
25
0
IgG 1 2.5 5 10
LRG1Ab (μg)
**
CN
V lesio
n v
olu
me
(% o
f Ig
G c
on
tro
l) ****
d
2
4
6
8
0LRG1
Ab
DC
101
CN
V lesio
n v
olu
me
(μm
3 ×
10
6)
IgG LRG1Ab
DC101
****
**
**
P12
P17
***
P17
Figure 3 | LRG1 contributes to pathogenic neovascularization.a, Representative images of wild-type (WT) and Lrg12/2 mouse laser-burnlesions by infrared (IR) fundus imaging. At 7 days after the laser, early- and late-phase fundus fluorescein angiography (FFA) revealed a reduction in CNVlesion size and a decrease in fluorescein leakage, respectively, in Lrg12/2 mice.Representative images of isolectin B4 stained (red) CNV in choroidal/RPE flat-mount 7 days after induction, confirming decreased lesion size in Lrg12/2
mice. Scale bar, 100mm. b, In OIR, Lrg1 deletion does not affect the size of theavascular region at P12 (delineated by white boundary line) or the organizednormal revascularization at P17, but does decrease the formation of pathologicalneovascular (NV) tufts (highlighted in red and delineated in higher power bywhite boundary line). Scale bars, 1,000mm (P12 and P17 lower magnification)and 50mm (P17 higher magnification). c, Volume-rendered examples ofPECAM-1 stained CNV lesions in wild-type mice after intravitreal injectionof irrelevant IgG or LRG1 neutralizing antibody. Scale bar, 100mm. d, Dose-dependent anti-LRG1 antibody reduction of CNV lesion volume. e, Combinationof anti-LRG1 and DC101 (anti-VEGFR2) in CNV in wild-type mice resulted inenhanced reduction of lesion volume compared to single treatments. Data aremean 6 s.e.m. of n $ 10 for each group. *P , 0.05; **P , 0.01; ***P , 0.001(Student’s t-test (a, b), and one-way analysis of variance (ANOVA)(d, e)).
RESEARCH ARTICLE
3 0 8 | N A T U R E | V O L 4 9 9 | 1 8 J U L Y 2 0 1 3
Macmillan Publishers Limited. All rights reserved©2013
angiogenesis, and suggest that LRG1 has potential as a therapeutic targeton its own or in combination with other anti-angiogenic therapies.
LRG1 and TGF-b signallingAlthough little is known about the biology of LRG1, concomitantincreases in the expression levels of TGF-b1, TbRII and LRG1 havebeen reported in cancer cells21 and hydrocephalus22, and LRG1 has beenshown to bind to TGF-b1 in high endothelial venules23. Consistent withthis, and with earlier proteomic and transcriptome analyses24,25, we haveshown here that in vitreous samples from the eyes of human subjectswith PDR, both LRG1 (Fig. 1h and Supplementary Fig. 6) and TGF-b1protein levels (Supplementary Fig. 25) are significantly increased.Furthermore, alongside increased Lrg1 gene expression, Tgfb1 trans-cript levels were also significantly upregulated in the retinae of laser-induced CNV mice and in OIR mice during the ischaemic proliferativephase (Supplementary Fig. 5). These data prompted us to investigatewhether LRG1 acts as a modulator of TGF-b signalling.
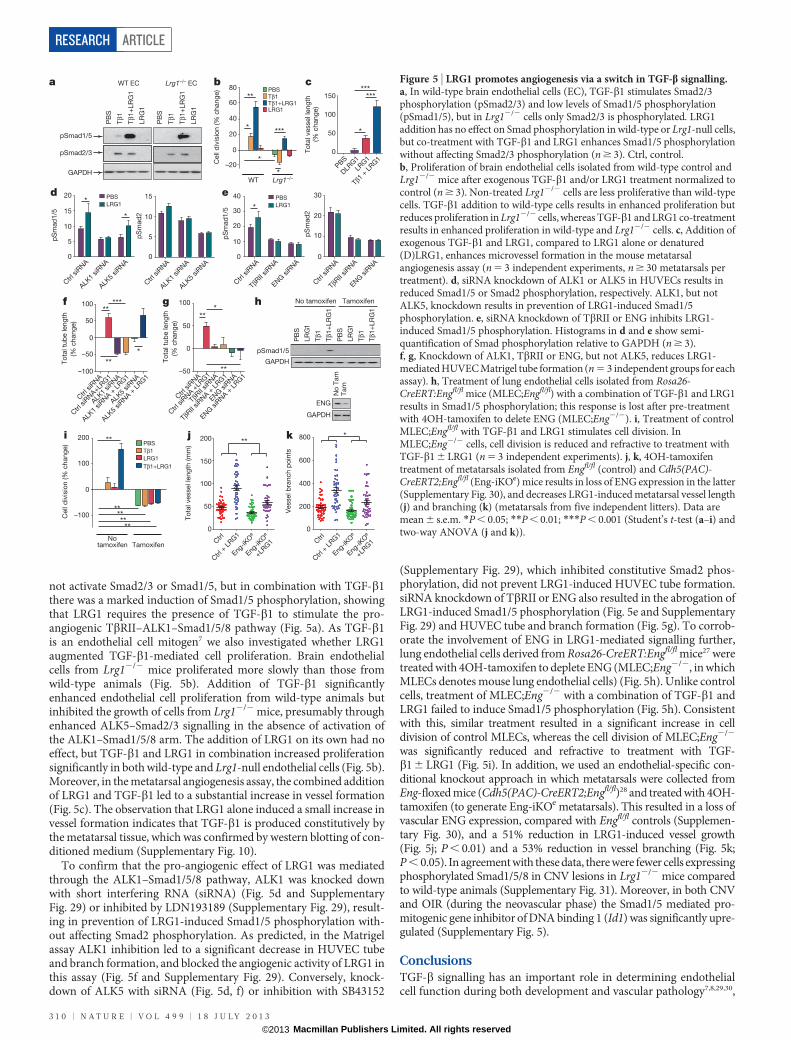
To determine whether LRG1 associates with components of theTGF-b receptor complex, we performed co-immunoprecipitationexperiments. In primary brain endothelial cells, LRG1 was presentin immunoprecipitates of TbRII, ALK1, ALK5 and the auxiliaryreceptor ENG (Fig. 4a). Conversely, immunoprecipitates of LRG1from HUVECs were found to contain TbRII, ALK1, ALK5 andENG (Supplementary Fig. 26a). These observations suggested thatLRG1 might be involved in regulating TGF-b signalling throughfine-tuning the stoichiometry of the TGF-b receptor complex. TGF-b1 signals in endothelial cells by TbRII recruitment of either theubiquitous ALK5 receptor, or the predominantly endothelial ALK1receptor together with ALK5. Stimulation of the TbRII–ALK5 signal-ling complex results in phosphorylation and activation of the trans-cription factors Smad2 and 3, which increases extracellular matrixdeposition, inhibits endothelial cell proliferation and migration, andpromotes cell homeostasis, whereas signalling via the TbRII–ALK5/ALK1 complex (possibly in association with ENG) activates Smad1, 5and 8, resulting in a pro-angiogenic state7,8. We therefore investigated thedirect one-to-one binding of LRG1 to recombinant extracellular domainsof individual TGF-b receptors in serum-free conditioned media fromtransfected HEK293T cells (Supplementary Fig. 26b). Immunoprecipita-tion of the receptor ectodomain revealed co-immunoprecipitation ofLRG1 with ALK5, TbRII and ENG, indicating a direct interaction ofLRG1 with these individual receptors (Fig. 4b). This occurred in theabsence of TGF-b1, which was not present in HEK293T-cell-conditionedmedium (Supplementary Fig. 10). Addition of conditioned mediumcontaining non-tagged ENG, ALK5 or TbRII out-competed taggedreceptor binding to LRG1 (Fig. 4c), confirming the specificity of theseprotein–protein interactions. The observation that ENG seems to beone of the receptors for LRG1 is germane, given its proposed role inswitching TGF-b signalling towards the pro-angiogenic Smad1/5/8pathway26. A potential functional relationship between LRG1 andENG was additionally strengthened by our observation that Eng isupregulated in CNV and OIR (Supplementary Fig. 27). To define theLRG1–ENG interaction further we undertook surface plasmon res-onance analysis (Biacore) and obtained an affinity rate constant (KD)of 2.9mM (Supplementary Fig. 28) for binding of the ENG ectodomainto LRG1.
These data raise the possibility that LRG1 facilitates a receptorconfiguration conducive to the pro-angiogenic signalling pathway.To investigate this, LRG1 was incubated with conditioned mediacontaining the extracellular domains of either ALK1 or ALK5 in thepresence or absence of different combinations of TGF-b1, and theextracellular domains of ENG and TbRII. These studies revealed thatLRG1 only associated with ALK1 in the presence of ENG, to whichit bound, and this was enhanced by the addition of TGF-b1 (Fig. 4d).Conversely, ALK5 binds LRG1 in the absence of ENG but in its pres-ence this interaction is attenuated, suggesting competition betweenENG and ALK5 for LRG1, whereas the addition of TGF-b1 resultsin complete loss of the LRG1–ALK5 association. Moreover, neitherTGF-b1 nor TbRII on their own, nor a combination of both, affectsALK1–LRG1 or ALK5–LRG1 association in the absence of ENG.However, in the presence of ENG, TbRII is able to form a complexwith ALK1–LRG1 or ALK5–LRG1, with the former association beingenhanced and the latter being further inhibited by TGF-b1. In accord-ance with previously suggested models8, these data indicate that LRG1may be able to form an intermediate complex with ALK5, ALK1, TbRIIand ENG, but that in the presence of TGF-b1 the LRG1–ALK1–TbRII–ENG complex predominates. An association between LRG1and TGF-b1 may therefore lead to more efficient ALK1–TbRII–ENG receptor complex formation, and consequently to the promotionof pro-angiogenic Smad1/5 signalling.
To test this hypothesis, we treated mouse brain endothelial cellswith TGF-b1 (5 ng ml21) and showed that both Smad2/3 and Smad1/5 phosphorylation are induced in wild-type cells, but in Lrg1-null cellsonly Smad2/3 is activated (Fig. 5a). The addition of LRG1 alone did
a b
c
WT
RecA
b
Ctr
l Ig
G
RecA
b
WB: anti-LRG1
Lrg1–/–
d
WB:
ALK
1 +
LR
G1
Mo
ck +
LR
G1
Mo
ck +
LR
G1
Mo
ck +
LR
G1
Mo
ck +
LR
G1
ALK
5 +
LR
G1
EN
G +
LR
G1
TβR
II +
LR
G1
anti-LRG1
IP:
ALK1
IP:
ALK5
IP:
ENG
IP:
TβRII
–
+
+
ENG–V5
ENG
LRG1
IP: LRG1
WB: anti-V5
WB: anti-ENG
ALK5–HA
ALK5
LRG1
WB: anti-HA
WB: anti-ALK5
TβRII–Myc
TβRII
LRG1
WB: anti-Myc
WB: anti-TβRII
–+ +
+ +
++ +
–
–+ +
+ +
++ +
–
+ +
+
+ +
–
IP: LRG1 IP: LRG1
IP: TβRII
IP: ALK1
IP: ALK5
IP: ENG
IP: ALK5
Mock
Tβ1ENG
ALK5
LRG1+
++–
– + +–– –
++ + + + + + + +– –– –++ + +
TβRII
+++ +
– – –– – –
+– – ––
+–
++– +
–+
+
––
+
Anti-LRG1
Anti-ENG
Anti-TβRII
Mock
IP: ALK1
Tβ1ENG
ALK1
LRG1 ++ ++
++
+–
–
+ +
++ +
+ +–– –
– – –– – –
+ + + +– –– –++ + +
+– – ––
+–
++– +
–+
+
––
+
Anti-LRG1
Anti-ENG
Anti-TβRII
TβRII
Complexformation
Complexformation
Figure 4 | LRG1 modifies the TGF-b receptor complex.a, Immunoprecipitation (IP) of TbRII, ALK1, ALK5 and ENG with anti-receptor antibodies (RecAb) from wild-type mouse brain endothelial celllysates co-precipitates LRG1. Control IgG in wild-type endothelial cells or anti-receptor antibodies in Lrg12/2 endothelial cells did not co-precipitate LRG1.WB, western blot. b, Immunoprecipitation of peptide-tagged extracellulardomains of ALK5 (haemagglutinin (HA)-tagged), TbRII (Myc-tagged) or ENG(V5-tagged) added individually to histidine (His)-tagged LRG1 resulted in co-precipitation of LRG1, indicating direct interactions with these receptors.Immunoprecipitation of ALK1 (HA-tagged) in the presence of LRG1 did notco-precipitate the latter. c, Addition of appropriate soluble non-taggedextracellular domains of ENG, ALK5 and TbRII out-competed peptide-taggedreceptor binding to LRG1. d, LRG1 was incubated in vitro with differentcombinations of TGF-b receptor extracellular domains and TGF-b1 (Tb1). Inthe presence of ENG, binding between LRG1 and ALK5 is diminished, and iscompletely lost with the further addition of TGF-b1. Conversely, ENGfacilitates the association between LRG1 and ALK1, which is enhanced in thepresence of TGF-b1. Although TbRII has no effect on LRG1–ALK1 or LRG1–ALK5 interactions, it is recruited to the complex in the presence of ENG. Alldata are representative western blots of n $ 3 for each experiment.
ARTICLE RESEARCH
1 8 J U L Y 2 0 1 3 | V O L 4 9 9 | N A T U R E | 3 0 9
Macmillan Publishers Limited. All rights reserved©2013
not activate Smad2/3 or Smad1/5, but in combination with TGF-b1there was a marked induction of Smad1/5 phosphorylation, showingthat LRG1 requires the presence of TGF-b1 to stimulate the pro-angiogenic TbRII–ALK1–Smad1/5/8 pathway (Fig. 5a). As TGF-b1is an endothelial cell mitogen7 we also investigated whether LRG1augmented TGF-b1-mediated cell proliferation. Brain endothelialcells from Lrg12/2 mice proliferated more slowly than those fromwild-type animals (Fig. 5b). Addition of TGF-b1 significantlyenhanced endothelial cell proliferation from wild-type animals butinhibited the growth of cells from Lrg12/2 mice, presumably throughenhanced ALK5–Smad2/3 signalling in the absence of activation ofthe ALK1–Smad1/5/8 arm. The addition of LRG1 on its own had noeffect, but TGF-b1 and LRG1 in combination increased proliferationsignificantly in both wild-type and Lrg1-null endothelial cells (Fig. 5b).Moreover, in the metatarsal angiogenesis assay, the combined additionof LRG1 and TGF-b1 led to a substantial increase in vessel formation(Fig. 5c). The observation that LRG1 alone induced a small increase invessel formation indicates that TGF-b1 is produced constitutively bythe metatarsal tissue, which was confirmed by western blotting of con-ditioned medium (Supplementary Fig. 10).
To confirm that the pro-angiogenic effect of LRG1 was mediatedthrough the ALK1–Smad1/5/8 pathway, ALK1 was knocked downwith short interfering RNA (siRNA) (Fig. 5d and SupplementaryFig. 29) or inhibited by LDN193189 (Supplementary Fig. 29), result-ing in prevention of LRG1-induced Smad1/5 phosphorylation with-out affecting Smad2 phosphorylation. As predicted, in the Matrigelassay ALK1 inhibition led to a significant decrease in HUVEC tubeand branch formation, and blocked the angiogenic activity of LRG1 inthis assay (Fig. 5f and Supplementary Fig. 29). Conversely, knock-down of ALK5 with siRNA (Fig. 5d, f) or inhibition with SB43152
(Supplementary Fig. 29), which inhibited constitutive Smad2 phos-phorylation, did not prevent LRG1-induced HUVEC tube formation.siRNA knockdown of TbRII or ENG also resulted in the abrogation ofLRG1-induced Smad1/5 phosphorylation (Fig. 5e and SupplementaryFig. 29) and HUVEC tube and branch formation (Fig. 5g). To corrob-orate the involvement of ENG in LRG1-mediated signalling further,lung endothelial cells derived from Rosa26-CreERT:Engfl/fl mice27 weretreated with 4OH-tamoxifen to deplete ENG (MLEC;Eng2/2, in whichMLECs denotes mouse lung endothelial cells) (Fig. 5h). Unlike controlcells, treatment of MLEC;Eng2/2 with a combination of TGF-b1 andLRG1 failed to induce Smad1/5 phosphorylation (Fig. 5h). Consistentwith this, similar treatment resulted in a significant increase in celldivision of control MLECs, whereas the cell division of MLEC;Eng2/2
was significantly reduced and refractive to treatment with TGF-b1 6 LRG1 (Fig. 5i). In addition, we used an endothelial-specific con-ditional knockout approach in which metatarsals were collected fromEng-floxed mice (Cdh5(PAC)-CreERT2;Engfl/fl)28 and treated with 4OH-tamoxifen (to generate Eng-iKOe metatarsals). This resulted in a loss ofvascular ENG expression, compared with Engfl/fl controls (Supplemen-tary Fig. 30), and a 51% reduction in LRG1-induced vessel growth(Fig. 5j; P , 0.01) and a 53% reduction in vessel branching (Fig. 5k;P , 0.05). In agreement with these data, there were fewer cells expressingphosphorylated Smad1/5/8 in CNV lesions in Lrg12/2 mice comparedto wild-type animals (Supplementary Fig. 31). Moreover, in both CNVand OIR (during the neovascular phase) the Smad1/5 mediated pro-mitogenic gene inhibitor of DNA binding 1 (Id1) was significantly upre-gulated (Supplementary Fig. 5).
ConclusionsTGF-b signalling has an important role in determining endothelialcell function during both development and vascular pathology7,8,29,30,
a b
100
50
0
–50
–100
**
***T
ota
l tu
be leng
th
(% c
hang
e)
Ctrl
siR
NA
ALK1
siRNA
ALK5
siRNA
Ctrl
siR
NA+LR
G1
ALK1
siRNA +
LRG1
ALK5
siRNA +
LRG1
***
***
80
60
40
20
0
–20Cell
div
isio
n (%
chang
e)
WT Lrg1–/–
**
*
*
*
PBSTβ1Tβ1+LRG1LRG1
PBS
DLR
G1
LRG1
Tβ1
+ LRG1
150
100
50
0To
tal vessel le
ng
th
(% c
hang
e)
***
*
***
pSmad1/5
WT EC
PB
S
Tβ1
Tβ1
+LR
G1
LR
G1
PB
S
Tβ1
Tβ1
+LR
G1
LR
G1
Lrg1–/– EC
pSmad2/3
GAPDH
c
e
f g
Ctrl
siR
NA
ALK1
siRNA
ALK5
siRNA
15
10
5
0
15
10
5
0
20
Ctrl
siR
NA
ALK1
siRNA
ALK5
siRNA
*
*
pS
mad
1/5
pS
mad
2
PBS
LRG1
d
Ctrl
siR
NA
TβRII
siRNA
ENG siR
NA
Ctrl
siR
NA +
LRG1
TβRII
siRNA +
LRG1
ENG siR
NA +
LRG1
100
50
0
–50
To
tal tu
be leng
th
(% c
hang
e)
Ctrl
siR
NA
TβRII
siRNA
ENG siR
NA
30
40
20
10
0
30
20
10
0
pS
mad
1/5
pS
mad
2
PBS
LRG1
Ctrl
siR
NA
TβRII
siRNA
ENG siR
NA
*
h**
*
**GAPDH
No tamoxifen TamoxifenP
BS
PB
S
Tβ1
LR
G1
LR
G1
Tβ1
Tβ1
+LR
G1
Tβ1
+LR
G1
pSmad1/5
ENG
GAPDH
No
Tam
Tam
i 200
100
0
–100
PBS
Tβ1
LRG1
Tβ1+LRG1
tamoxifen Tamoxifen
Cell
div
isio
n (%
chang
e)
**
********
No
j
To
tal vessel le
ng
th (m
m)
50
100
150
200
Ctrl
Ctrl
+ L
RG1
Eng-iK
Oe
Eng-iK
Oe
+LRG1
0
k**
Vessel b
ranch p
oin
ts
200
400
600
800
Ctrl
Ctrl
+ L
RG1
Eng-iK
Oe
Eng-iK
Oe
+LRG1
0
*
Figure 5 | LRG1 promotes angiogenesis via a switch in TGF-b signalling.a, In wild-type brain endothelial cells (EC), TGF-b1 stimulates Smad2/3phosphorylation (pSmad2/3) and low levels of Smad1/5 phosphorylation(pSmad1/5), but in Lrg12/2 cells only Smad2/3 is phosphorylated. LRG1addition has no effect on Smad phosphorylation in wild-type or Lrg1-null cells,but co-treatment with TGF-b1 and LRG1 enhances Smad1/5 phosphorylationwithout affecting Smad2/3 phosphorylation (n $ 3). Ctrl, control.b, Proliferation of brain endothelial cells isolated from wild-type control andLrg12/2 mice after exogenous TGF-b1 and/or LRG1 treatment normalized tocontrol (n $ 3). Non-treated Lrg12/2 cells are less proliferative than wild-typecells. TGF-b1 addition to wild-type cells results in enhanced proliferation butreduces proliferation in Lrg12/2 cells, whereas TGF-b1 and LRG1 co-treatmentresults in enhanced proliferation in wild-type and Lrg12/2 cells. c, Addition ofexogenous TGF-b1 and LRG1, compared to LRG1 alone or denatured(D)LRG1, enhances microvessel formation in the mouse metatarsalangiogenesis assay (n 5 3 independent experiments, n $ 30 metatarsals pertreatment). d, siRNA knockdown of ALK1 or ALK5 in HUVECs results inreduced Smad1/5 or Smad2 phosphorylation, respectively. ALK1, but notALK5, knockdown results in prevention of LRG1-induced Smad1/5phosphorylation. e, siRNA knockdown of TbRII or ENG inhibits LRG1-induced Smad1/5 phosphorylation. Histograms in d and e show semi-quantification of Smad phosphorylation relative to GAPDH (n $ 3).f, g, Knockdown of ALK1, TbRII or ENG, but not ALK5, reduces LRG1-mediated HUVEC Matrigel tube formation (n 5 3 independent groups for eachassay). h, Treatment of lung endothelial cells isolated from Rosa26-CreERT:Engfl/fl mice (MLEC;Engfl/fl) with a combination of TGF-b1 and LRG1results in Smad1/5 phosphorylation; this response is lost after pre-treatmentwith 4OH-tamoxifen to delete ENG (MLEC;Eng2/2). i, Treatment of controlMLEC;Engfl/fl with TGF-b1 and LRG1 stimulates cell division. InMLEC;Eng2/2 cells, cell division is reduced and refractive to treatment withTGF-b1 6 LRG1 (n 5 3 independent experiments). j, k, 4OH-tamoxifentreatment of metatarsals isolated from Engfl/fl (control) and Cdh5(PAC)-CreERT2;Engfl/fl (Eng-iKOe) mice results in loss of ENG expression in the latter(Supplementary Fig. 30), and decreases LRG1-induced metatarsal vessel length(j) and branching (k) (metatarsals from five independent litters). Data aremean 6 s.e.m. *P , 0.05; **P , 0.01; ***P , 0.001 (Student’s t-test (a–i) andtwo-way ANOVA (j and k)).
RESEARCH ARTICLE
3 1 0 | N A T U R E | V O L 4 9 9 | 1 8 J U L Y 2 0 1 3
Macmillan Publishers Limited. All rights reserved©2013

and its activity is regulated at several levels from gene expression tocontrol of extracellular bioavailability. The multiplicity of regulatorymechanisms together with variable combinations of receptors/co-receptors creates complex patterns of TGF-b activity that define itscontext-dependent effects. In particular, the balance between theALK5 and ALK1 signalling pathways is considered to be central indetermining the angiogenic switch, with ENG being proposed as akey regulatory molecule in promoting signalling through the ALK1pathway26,30,31. In searching for mediators of vascular remodelling inthe diseased/damaged retina we have discovered a new regulator ofTGF-b signalling. The data presented here support a hypothesis thatLRG1 activates the TGF-b angiogenic switch by binding to the access-ory receptor ENG and, in the presence of TGF-b1, promotes signallingvia the TbRII–ALK1–Smad1/5/8 pathway (Supplementary Fig. 32).Moreover, our evidence suggests that LRG1 may have a more domi-nant role in disorganized pathological rather than developmental/physiological angiogenesis. Although in the retina this is clearly sup-ported by our in vivo data, the ex vivo and in vitro studies indicatethat LRG1 angiogenic activity is not restricted to the eye. The modu-lating effect of LRG1 on TGF-b1 signalling is the first demonstration,to our knowledge, of a definitive function for LRG1 and raises theintriguing possibility that it may influence other major biological pro-cesses in which TGF-b has a role, such as neoplasia32 and the immuneresponse33. Inhibition of LRG1, which we show here causes a shift awayfrom angiogenic signalling, could prevent pathogenic activation ofthis pathway, while leaving homeostatic TGF-b signalling unperturbed.From these studies we suggest, therefore, that LRG1 is a highly pro-mising therapeutic target for controlling pathogenic angiogenesis inocular disease, and potentially in other diseases such as cancer andatherosclerosis.
METHODS SUMMARYMicrovessel global gene expression analysis was undertaken using Affymetrixmouse 430.2 gene arrays. CNV and OIR were induced in mice as described inthe Methods. All other methods are described in the Methods.
Full Methods and any associated references are available in the online version ofthe paper.
Received 26 August 2011; accepted 3 June 2013.
1. Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel, D. V. & Ferrara, N. Vascularendothelial growth factor is a secreted angiogenic mitogen. Science 246,1306–1309 (1989).
2. Carmeliet, P. et al. Abnormal blood vessel development and lethality in embryoslacking a single VEGF allele. Nature 380, 435–439 (1996).
3. Ferrara, N. et al. Heterozygous embryonic lethality induced by targetedinactivation of the VEGF gene. Nature 380, 439–442 (1996).
4. Holderfield, M. T. & Hughes, C. C. Crosstalk between vascular endothelial growthfactor, notch, and transforming growth factor-b in vascular morphogenesis. Circ.Res. 102, 637–652 (2008).
5. Chung, A. S. & Ferrara, N. Developmental and pathological angiogenesis. Annu.Rev. Cell Dev. Biol. 27, 563–584 (2011).
6. Carmeliet, P. & Jain, R. K. Molecular mechanisms and clinical applications ofangiogenesis. Nature 473, 298–307 (2011).
7. Pardali, E., Goumans,M. J.& ten Dijke,P. Signaling bymembers of the TGF-b familyin vascular morphogenesis and disease. Trends Cell Biol. 20, 556–567 (2010).
8. Goumans, M. J., Liu, Z. & ten Dijke, P. TGF-b signaling in vascular biology anddysfunction. Cell Res. 19, 116–127 (2009).
9. Cunha, S. I. et al. Genetic and pharmacological targeting of activin receptor-likekinase1 impairs tumorgrowthandangiogenesis. J. Exp.Med.207,85–100(2010).
10. Cunha, S. I. & Pietras, K. ALK1 as an emerging target for antiangiogenic therapy ofcancer. Blood 117, 6999–7006 (2011).
11. ten Dijke, P. & Arthur, H. M. Extracellular control of TGFb signalling in vasculardevelopment and disease. Nature Rev. Mol. Cell Biol. 8, 857–869 (2007).
12. Xu, Q. et al.Vasculardevelopment in the retinaand innerear: controlby Norrin andFrizzled-4, a high-affinity ligand-receptor pair. Cell 116, 883–895 (2004).
13. Ye, X. et al. Norrin, Frizzled-4, and Lrp5 signaling in endothelial cells controls agenetic program for retinal vascularization. Cell 139, 285–298 (2009).
14. Hackett, S. F., Wiegand, S., Yancopoulos, G. & Campochiaro, P. A. Angiopoietin-2playsan important role in retinal angiogenesis.J.Cell. Physiol.192,182–187 (2002).
15. Haigh, J. J. et al. Cortical and retinal defects caused by dosage-dependentreductions in VEGF-A paracrine signaling. Dev. Biol. 262, 225–241 (2003).
16. Zhao, J., Sastry, S. M., Sperduto, R. D., Chew, E. Y. & Remaley, N. A. Arteriovenouscrossing patterns in branch retinal vein occlusion. The Eye Disease Case-ControlStudy Group. Ophthalmology 100, 423–428 (1993).
17. Kumar, B. et al. The distribution of angioarchitectural changes within the vicinity ofthe arteriovenous crossing in branch retinal vein occlusion. Ophthalmology 105,424–427 (1998).
18. Rakic, J. M. et al. Placental growth factor, a member of the VEGF family, contributesto the development of choroidalneovascularization. Invest. Ophthalmol. Vis. Sci. 44,3186–3193 (2003).
19. Takeda, A. et al. CCR3 is a target for age-related macular degeneration diagnosisand therapy. Nature 460, 225–230 (2009).
20. VandeVeire, S.et al. Furtherpharmacological and genetic evidence for the efficacyof PIGF inhibition in cancer and eye disease. Cell 141, 178–190 (2010).
21. Sun, D., Kar, S. & Carr, B. I. Differentially expressed genes in TGF-b1 sensitive andresistant human hepatoma cells. Cancer Lett. 89, 73–79 (1995).
22. Li, X., Miyajima, M., Jiang, C. & Arai, H. Expression of TGF-bs and TGF-b type IIreceptor in cerebrospinal fluid of patients with idiopathic normal pressurehydrocephalus. Neurosci. Lett. 413, 141–144 (2007).
23. Saito, K. et al. Gene expression profiling of mucosal addressin cell adhesionmolecule-11 high endothelial venule cells (HEV) and identification of a leucine-rich HEV glycoprotein as a HEV marker. J. Immunol. 168, 1050–1059 (2002).
24. Spirin, K. S. et al. Basement membrane and growth factor gene expression innormal and diabetic human retinas. Curr. Eye Res. 18, 490–499 (1999).
25. Gao, B. B., Chen, X., Timothy, N., Aiello, L. P. & Feener, E. P. Characterization of thevitreous proteome in diabetes without diabetic retinopathy and diabetes withproliferative diabetic retinopathy. J. Proteome Res. 7, 2516–2525 (2008).
26. Lebrin, F. et al. Endoglin promotes endothelial cell proliferation and TGF-b/ALK1signal transduction. EMBO J. 23, 4018–4028 (2004).
27. Anderberg, C. et al. Deficiency for endoglin in tumor vasculature weakens theendothelial barrier tometastatic dissemination. J. Exp.Med. 210, 563–579 (2013).
28. Mahmoud,M.et al. Pathogenesisof arteriovenousmalformations in theabsenceofendoglin. Circ. Res. 106, 1425–1433 (2010).
29. Bobik, A. Transforming growth factor-betas and vascular disorders. Arterioscler.Thromb. Vasc. Biol. 26, 1712–1720 (2006).
30. ten Dijke, P., Goumans, M. J. & Pardali, E. Endoglin in angiogenesis and vasculardiseases. Angiogenesis 11, 79–89 (2008).
31. Ray, B. N., Lee, N. Y., How, T. & Blobe, G. C. ALK5 phosphorylation of the endoglincytoplasmic domain regulates Smad1/5/8 signaling and endothelial cellmigration. Carcinogenesis 31, 435–441 (2010).
32. Lynch, J. et al. MiRNA-335 suppresses neuroblastoma cell invasiveness by directtargeting of multiple genes from the non-canonical TGF-b signalling pathway.Carcinogenesis 33, 976–985 (2012).
33. Gregory, A. D., Capoccia, B. J., Woloszynek, J. R. & Link, D. C. Systemic levels ofG-CSF and interleukin-6 determine the angiogenic potential of bone marrowresident monocytes. J. Leukoc. Biol. 88, 123–131 (2010).
Supplementary Information is available in the online version of the paper.
Acknowledgements This project was supported by grants from the Lowy MedicalResearch Foundation, the Medical Research Council, The Wellcome Trust, UCLBusiness (Proof of Concept Grant) and the Rosetrees Trust. J.W.B.B. is supported by aNIHRResearchProfessorship.H.M.A. is supported byaBritish Heart FoundationSeniorFellowship. We would also like to thank M. Gillies for his role in initiating the originalproject, P. Luthert and C. Thaung for human tissue samples and advice on humanpathology specimens, S. Perkins and R. Nan for assistance with the surface plasmonresonance analysis, and P. ten Dijke for discussions and advice.
Author Contributions The project was conceived by J.G., S.E.M. and X.W. Experimentsweredesigned by J.G., S.E.M., X.W. andS.A. Microarrays wereperformed byJ.A.G.M. andqPCR reactions by X.W. X.W. and S.A. characterized the Lrg1 knockout mice and LRG1antibody. X.W. performed all the metatarsal assays (except in Fig. 5j, k), aortic ringassays and Matrigel assays, carried out all the biochemical and molecular biology workand analysed the data. S.A. and X.W. undertook the immunohistochemistry andgenerated the OIR mouse model. U.F.O.L., C.A.K.L., S.A., X.W. andJ.W.B.B. performed theCNV experiments, and S.A. and X.W. analysed the data. J.W.B.B. provided human vitrealsamples. Z.Z. and H.M.A. generated MLEC;Engfl/fl cells and X.W. performed proliferationassay and biochemical analysis. Z.Z., S.A. and H.M.A. carried out the metatarsal assayson Eng knockout mice. V.B.T. performed the Biacore experiments. N.J. and M.S.provided assistance and technique support. X.W., S.A., J.G. and S.E.M. produced thefigures, and J.G. and S.E.M. wrote the text, with all authors contributing to the finalmanuscript. J.G. and S.E.M. provided leadership throughout the project.
Author Information Reprints and permissions information is available atwww.nature.com/reprints. The authors declare no competing financial interests.Readers are welcome to comment on the online version of the paper. Correspondenceand requests for materials should be addressed to J.G. ([email protected]) orS.E.M. ([email protected]).
ARTICLE RESEARCH
1 8 J U L Y 2 0 1 3 | V O L 4 9 9 | N A T U R E | 3 1 1
Macmillan Publishers Limited. All rights reserved©2013

METHODSAnimals. C75BL/6J mice were purchased from Harlan Laboratories. rd1 (ref. 34),Vldlr2/2 (ref. 35) and Grhl3ct/J curly tail mice were purchased from the JacksonLaboratory. Lrg12/2 mice were generated by the University of California Daviesknockout mouse project (KOMP) repository (http://www.komp.org/ and Supplemen-tary Fig. 12). Rosa26-CreERT;Engfl/fl and Cdh5(PAC)-CreERT2;Engfl/fl mice have beenpreviously described27,28. All procedures were performed in accordance with the UKAnimals (Scientific Procedures) Act and with the Association for Research in Visionand Ophthalmology Statement for the Use of Animals in Ophthalmic and VisionResearch and the Animal Welfare and the Ethical Review Bodies of the UCL Instituteof Ophthalmology and Newcastle University.Vessel isolation and gene expression analysis. Mouse retinal vessels from wild-type C57BL/6J (15 weeks), Vldlr2/2 (16 weeks), rd1 (18 weeks) and Grhl3ct/J curlytail (13 weeks) mice were isolated as described elsewhere36. RNA was extractedfrom the enriched microvascular preparations and processed for whole-genomemicroarray analysis as previously described36. Twelve mice were used per strainper RNA extraction. This was repeated four times, providing RNA for four chipsper animal model.Quantitative PCR (qPCR). RNA was extracted using Trizol (Invitrogen) followedby an RNeasy clean-up (QIAGEN). RNA was reverse transcribed using theQuantiTect Reverse Transcription Kit (QIAGEN) and PCR was conducted withQuantiTect PowerSybr Green (Applied Biosystems) using a 7900HT Fast Real-TimePCR System (Applied Biosystems); samples were normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). Primers used in this study are listed inSupplementary Table 2. Student’s t-test was performed to determine statistical sig-nificance between test groups.SDS–PAGE and western blotting. Proteins were separated by SDS–PAGE. Gelswere either stained using Coomassie-blue or transferred onto a Hybond-P PVDFmembrane (GE Healthcare). Blots were probed with phospho-Smad1/5 antibody(rabbit monoclonal antibody, NEB), phospho-Smad2 antibody (rabbit monoclo-nal antibody, NEB), TGF-b1 antibody (mouse monoclonal, R&D systems), TbRIIantibody (mouse monoclonal, R&D systems), ALK1 antibody (rabbit polyclonal,Santa Cruz Biotechnology), ALK5 antibody (rabbit polyclonal, Abcam), endoglinantibody (mouse monoclonal, R&D Systems), LRG1 antibody (HPA001888,rabbit polyclonal, Sigma) or GAPDH antibody (mouse monoclonal, Novus),followed by horseradish peroxidase (HRP)-conjugated secondary antibodies(GE Healthcare) or HRP-conjugated Protein A (GE Healthcare). Densitometrywas performed using ImageJ software (National Institutes of Health). Student’st-test was performed to determine statistical significance between test groups.RNA in situ hybridization. Eyes were fixed in 2% (w/v) paraformaldehyde (PFA)in PBS for 2 min and dissected in 23 PBS. Retinae were flattened and fixed in100% ice-cold methanol overnight at 220 uC. After recovery from methanol,retinae were re-fixed for 10 min in 4% PFA and washed in PBS before digestionfor 10 min in proteinase K (80mg ml21 in PBS). Retinae were re-fixed for 5 min in4% PFA and 0.2% glutaraldehyde in PBS. After a brief wash in PBS, retinae werepre-incubated in hybridization buffer (50% formamide, 53 SSC, 50 mg ml21
transfer RNA, 1% SDS, 50 mg ml21 heparin) for 1 h at 65 uC. Denatured RNAprobes were incubated with retinae at 65 uC overnight. Primers used to generateRNA probes by PCR are listed in Supplementary Table 2. Probes were labelledwith digoxigenin (DIG)-UTP using a DIG RNA labelling kit (Roche Diagnostics).Signal was developed with alkaline phosphatase-conjugated anti-digoxigenin Fabfragments, according to the manufacturer’s instructions.Immunohistochemistry. Retinal/RPE whole-mounts were fixed and stained aspreviously described37, and incubated overnight with antibodies against humanLRG1 (HPA001888), mouse PECAM-1 (rat monoclonal, BD Biosciences), mousecollagen IV (rabbit polyclonal, AbD Serotec), human collagen IV (goat polyclonal,Millipore), rat NG2 (rabbit polyclonal, Millipore), mouse F4/80 (rat monoclonal,AbD Serotec), mouse endoglin (rat monoclonal, Santa Cruz Biotechnology) orhuman VE-cadherin (mouse monoclonal, Santa Cruz Biotechnology), and iden-tified with Alexa 488, Alexa 594 or Alexa 647 secondary antibodies (Invitrogen) orFITC-GSL (DyLight 594) isolectin B4 (Vector Labs). Retinae were flat-mounted inMowiol and examined by epifluorescence (Leica DM IRB inverted research micro-scope or Olympus SZX16 research stereo zoom microscope) or confocal (CarlZeiss LSM 510 or 710) microscopy. For quantifying the retinal vascular area,raw image data were processed with Photoshop CS4.3. Three-dimensional ren-dering of confocal Z-stacks was carried out using Imaris 7.5 software (BitplaneAG). The retinal vasculature was analysed through automatic surface renderingaided by manual threshold adjustment so that only blood vessels were included foranalysis. Imaris Key Frame Animation was used for movie generation.Human tissue. Vitreous and plasma samples were collected from patients havingsurgery for PDR or epiretinal membrane. Human tissue arrays were obtainedfrom Pantomics and stained with rabbit anti-LRG1 antibody (Sigma). The study
followed the ethical guidelines of the Declaration of Helsinki. Institutional ReviewBoards granted approval for allocation and biochemical analysis of specimens.Cells and cell culture. Pooled HUVECs were purchased from Lonza and culturedaccording to suppliers instructions. HEK293T cells were purchased fromInvitrogen and cultured as recommended. Mouse primary brain endothelial cellswere isolated, purified and cultured as previously described for rat38. The immor-talized Lewis rat brain microvascular endothelial cell line GPNT was grown aspreviously described39. MLECs were isolated from Rosa26-CreERT;Engfl/fl micecarrying the Immortomouse transgene, and were collected and cultured as previ-ously described27. Cells were pre-treated with 1 mM 4OH-tamoxifen for 48 h inculture to generate ENG-depleted cells (MLEC;Eng2/2) and untreated cellsserved as controls (MLEC;Engfl/fl).Generation of LRG1 polyclonal antibody. Rabbits were immunized with puri-fied full-length His-tagged human LRG1 protein (Covalab). Pre-immune serawere collected to produce control IgG. Antisera were collected after 3 months andantibody was purified by HiTrap Protein G FF column (GE Healthcare) andconcentrated and desalted using HiPrep 26/10 Desalting (GE Healthcare).Matrigel HUVEC tube formation assay. HUVECs were grown on growth factor-reduced Matrigel (BD Biosciences) as described elsewhere40. The 96-well plateswere coated with Matrigel-containing diluent (control) or LRG1 (20mg ml21),rabbit polyclonal antibody against LRG1 (C10-54, 100 nM), rabbit IgG (100 nM),ALK1 inhibitor (LDN 193189, Axon Medchem BV, 100 nM) or ALK5 inhibitor(SB43152, Sigma, 10mM), and allowed to polymerize in the incubator at 37 uC for45 min. Tube formation was visualized using an Olympus SZX16 Research stereo-microscope and analysed by counting the number of branch points and total tubelength per well using ImageJ. Three independent experiments were carried out andeach was performed in triplicate. Student’s t-test was performed to determinestatistical significance between test groups.Metatarsal angiogenesis assay. The metatarsal angiogenesis assay was carriedout as described41. Metatarsal bones were isolated from E16.5 wild-type control orLrg12/2 littermate mice and treated with TGF-b1 (5 ng ml21, R&D systems),LRG1 (20mg ml21), anti-LRG1 polyclonal antibody (100 nM) or rabbit IgG(100 nM) as indicated. Medium was replaced every 2 days. At day 10 of culture,the explants were fixed and stained for PECAM-1 (rat monoclonal, BDBiosciences) and visualized under an Olympus SZX16 Research stereo-zoommicroscope. After image processing in Photoshop CS4 to mask the cartilage,the length of PECAM-1-positive tubular structures and the number of branchpoints were determined by Imaris 7.5 software (Bitplane) using automatic fila-ment tracing with manual threshold corrections. Statistical data were importedinto Excel (Microsoft) for calculating total vessel length and the number of branchpoints. A least three independent experiments were carried out, comprising aminimum of 30 metatarsals for each treatment. Student’s t-test was performed todetermine statistical significance between test groups.
To investigate the effect of ENG depletion on the pro-angiogenic effect ofLRG1, metatarsals from Cdh5(PAC)-CreERT2;Engfl/fl mice28 were used to gene-rate endothelial-specific depletion of ENG after addition of 1 mM 4OH-tamoxifen3 days after metatarsal bone isolation, when neovessels began to emerge. Onday 4, LRG1 was added to a final concentration of 20mg ml21 and the media(including LRG1 and 4OH-tamoxifen supplements) was refreshed every otherday until day 12. ENG depletion was confirmed using an anti-ENG antibody(E-Bioscience). Separate experiments using control metatarsals confirmed that1 mM 4OH-tamoxifen per se did not affect neovessel formation in the metatarsalangiogenesis assay. Analysis of angiogenesis was carried out as described above.Metatarsals from five independent litters was used and two-way ANOVA wasperformed to determine statistical significance between test groups.Aortic ring angiogenesis assay. The aortic ring angiogenesis assay was performedusing a modified method described previously42. Diameter rings (1 mm) weresliced from aortae of P7 wild-type control or Lrg12/2 littermate mice. Aortic ringswere then placed in a 96-well plate coated with a rat tail collagen I gel (BD Bio-sciences) containing LRG1 (20mg ml21), anti-LRG1 polyclonal antibody (100 nM)or rabbit IgG (100 nM) as indicated, and cultured in DMEM supplemented with2.5% FBS containing relevant compounds. Medium was replaced every 2 days. Atday 10 of culture, the explants were fixed, stained for GSL isolectin IB4 (VectorLabs) and visualized under an Olympus SZX16 Research stereo-zoom microscope.The number of sprouts was counted manually. Three independent experimentswere carried out with a mean of $15 aortic rings being analysed for each treatment.Student’s t-test was performed to determine statistical significance between testgroups.Mouse model of CNV. CNV was induced as described elsewhere36,43. In the anti-LRG1 antibody blockade, study animals received an intravitreal injection ofimmediately after the laser burn. Antibody at a concentration of 1, 2.5, 5 or10 mg (each in 1 ml) of the anti-LRG1 polyclonal antibody was delivered to oneeye and a pre-immune IgG, serving as control, delivered to the contralateral eye.
RESEARCH ARTICLE
Macmillan Publishers Limited. All rights reserved©2013

Five (in the case of antibody treatment) or seven days after injury, CNV lesionswere imaged as described before36,43,44. Mice were then killed for retina and RPEflat-mount preparation and mRNA extraction. The CNV lesions were visualizedafter FITC-conjugated GSL isolectin B4 (Vector Labs) and mouse PECAM-1 (ratpolyclonal, BD Biosciences) staining using an Olympus SZX16 Research stereo-zoom microscope and a Zeiss LSM 710 confocal microscope. Student’s t-test wasperformed to determine the statistical significance between wild-type and Lrg1knockout mice. One-way ANOVA was used to test statistical significancebetween antibody treatment groups.Mouse model of OIR. Nursing mothers and neonatal mice were placed in a 75%oxygen supply chamber from P7 to P12 and exposed to a standard 12 h light–darkcycle as previously described45. The extent of vaso-obliteration was determined inretinal flat-mounts at P12, and the extent of normal vessel regrowth and neovascu-larization were evaluated at P17 as previously described46. Retinae were also recov-ered for mRNA extraction and analysis at P12 and P17. The effect of antibodyblockade on retinal revascularisation and neovascular tuft formation was carriedout by delivering anti-LRG1 blocking antibody (50 mg kg21 intraperitoneal in100ml at P13 and P15), anti-VEGFR2 blocking antibody (DC101, 12.5 mg kg21
intraperitoneal at P13 and P15) or a combination of the two, followed by assessmentof the vasculature at P17. Student’s t-test was performed to determine the statisticalsignificance between wild-type and Lrg1 knockout mice. One-way ANOVA wasused to test statistical significance between antibody treatment groups.Co-immunoprecipitation. Primary mouse brain endothelial cells from wild-typeor Lrg12/2 littermate mice, GPNT cells or HUVECs were lysed in RIPA buffer(50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholateand 1% Nonidet P-40). Soluble peptide-tagged extracellular domains of TbRII(Myc-tagged), ALK1 (HA-tagged), ALK5 (HA-tagged) and ENG (V5-tagged), aswell as full-length LRG1 (His-tagged), were generated in separate cultures oftransfected HEK293T cells, and serum-free media containing the individual pro-teins was collected after 5 days. Non-tagged secreted extracellular domains of theTGF-b receptors were also generated in an identical manner. Media containingindividual extracellular domains of a TGF-b receptor were incubated with mediacontaining LRG1 in the presence or absence of TGF-b1 at 4 uC with rotationbefore immunoprecipitation. After pre-clearing, cell lysates or recombinant pro-tein mixtures were incubated with TGF-b receptor antibodies or anti-LRG1antibody-conjugated protein G beads at 4 uC overnight and then fractionatedby SDS–PAGE and blotted. The membranes were probed with antisera asdescribed earlier.Proliferation assay. Mouse primary brain endothelial cells from Lrg12/2 andwild-type mice were cultured in EGM2 media supplemented with puromycin(5 mg ml21) until sub-confluent, followed by 48 h serum starvation in EBM2medium. Cells were stimulated with TGF-b1 (5 ng ml21), LRG1 (20mg ml21)or TGF-b1 plus LRG1 in EBM2 medium at 37 uC. After 3 h, cells were fixed andstained with an antibody to Ki67 (mouse monoclonal antibody, Dako) to detectproliferating cells. The proliferation rate was evaluated as the percentage of Ki67-positive cells of the total endothelial cell number per well. Student’s t-test wasperformed to determine the statistical significance between treatment groups.Molecular biological methods. The coding sequence of human LRG1(NM_052972) carrying a 63His tag or HA tag at the 39 end and Kozak consensussequence at the 59 end was cloned into pcDNA3.1 (Invitrogen) at the HindIII/XhoIsites to form pcDNA-LRG1-His or pcDNA-LRG1-HA (Supplementary Fig. 8).The coding sequence of human LRG1 was cloned into a pEGX4T1 GST expressionvector (GE Healthcare) at the BamHI/SalI site to form glutathione S-transferase(GST)–ENG. The extracellular domain of human TbRII (NM_001024847.2) car-rying a Myc tag, ALK1 (NM_000020.2) carrying a HA tag, ALK5 (NM_004612.2)carrying a HA tag, ENG (NM_001114753.1) carrying a V5 tag at the 39 end andKozak consensus sequence at the 59 end were cloned into pcDNA3.1 at HindIII/EcoRI sites. The recombinant human proteins were expressed in HEK293T cells(Invitrogen). siRNA oligonucleotides (SASI_Rn01_00111211 (Sigma)) were usedfor Lrg1 gene knockdown in GPNT cells, and siRNA oligonucleotides (ON-TARGETplus SMARTpools, Thermo Scientific) were used for knockdown inHUVECs of ALK1 (L-005302-00-0005), ALK5 (L-003929-00-0005), ENG (L-011026-00-0005) and TBRII (L-001000-00-50), and control siRNA (D-001810-10-05) was used as a negative control for knockdown in HUVECs.
Lipofectamine 2000 transfection reagent (Invitrogen) was used for transfectionof mammalian cells. Oligofectamine 2000 transfection reagent (Invitrogen) wasused for siRNA knockdown in GPNT cells. GeneFECTOR transfection reagent
(VennNova) was used for siRNA knockdown in HUVECs. PCR and qPCRprimer sequences are shown in Supplementary Table 2.Purification of recombinant proteins. LRG1–His was expressed in HEK293Tcells and purified using HisPrep FF16/10 column (GE Healthcare) and bufferexchanged into PBS using HiPrep 26/10 Desalting (GE Healthcare) according tomanufacturer’s instruction. GST–ENG was expressed in BL21-competent cellsand purified using glutathione Sepharose 4B (GE Healthcare) and eluted inelution buffer (50 mM Tris-HCl, 10 mM reduced glutathione, pH 8.0) accordingto the manufacturer’s instruction. Denatured LRG1–His protein was generatedby boiling at 100 uC for 15 min.Surface plasmon resonance. All surface plasmon resonance experiments werecarried out on a BiacoreT200 instrument (GE Healthcare). LRG1 was covalentlyimmobilized via primary amino groups on a CM5 sensor chip as per manufac-turer’s instructions (specific contact time 20 s at a flow rate of 10 ml min21; LRG1concentration at 25 mg ml21 diluted using 10 mM sodium acetate, pH 5.0). Theamount of immobilized LRG1 corresponded to 2,000 response units in flow cell 2.Flow cell 1 on the same sensor chip, reserved for control runs, was treatedidentically but without LRG1 immobilization. For all SPR measurements, GST-tagged ENG was diluted in running buffer (13 PBS, pH 7.2). The association wasmonitored by injecting different concentrations (1–50 nM) of the analyte (ENG)into channels 1 and 2, starting with the lowest analyte concentration. All experi-ments were conducted in triplicates at 25 uC at a flow rate of 30 ml min21. Theassociation time for ligand–analyte steady state binding was optimised to 180 sand a subsequent 300 s were allowed for dissociation. Between injections thesensor chip surface was regenerated with glycine-HCl, pH 2.0, at a flow rate of30 ml min21 for 30 s. All curves were corrected for nonspecific binding by sub-traction of control curves obtained from injection of the analyte through theblank flow cell 1. The affinity and dissociation constants were calculated fromthe plots of the steady-state binding as a function of protein concentration, usingthe Biacore T200 evaluation software and a homogenous 1:1 Langmuir bindingkinetic model. The analysis provided values for the dissociation affinity constant(KD), the association rate constant (Ka) and the dissociation rate constant (Kd).Statistical analyses. Data are represented as mean 6 s.e.m. Statistical analyseswere performed by Student’s t-test or one-way ANOVA followed by Tukey/Bonferroni post-test analysis or two-way ANOVA as appropriate, using Prism5 (GraphPAD Software Inc.). *P , 0.05; **P , 0.01; ***P , 0.001. Each repre-sents significant statistical comparisons among the listed (x axis) experimentalgroups.
34. Blanks, J. C. & Johnson, L. V. Vascular atrophy in the retinal degenerative rd mouse.J. Comp. Neurol. 254, 543–553 (1986).
35. Heckenlively, J. R. et al. Mouse model of subretinal neovascularization withchoroidal anastomosis. Retina 23, 518–522 (2003).
36. McKenzie, J. A. et al. Apelin is required for non-neovascular remodelling in theretina. Am. J. Pathol. 108, 399–409 (2012).
37. Fruttiger, M. Development of the mouse retinal vasculature: angiogenesis versusvasculogenesis. Invest. Ophthalmol. Vis. Sci. 43, 522–527 (2002).
38. Abbott, N. J., Hughes, C. C., Revest, P. A. & Greenwood, J. Development andcharacterisation of a rat brain capillary endothelial culture: towards an in vitroblood–brain barrier. J. Cell Sci. 103, 23–37 (1992).
39. Romero, I. A. et al. Changes in cytoskeletal and tight junctional proteins correlatewith decreased permeability induced by dexamethasone in cultured rat brainendothelial cells. Neurosci. Lett. 344, 112–116 (2003).
40. Arnaoutova, I. & Kleinman, H. K. In vitro angiogenesis: endothelial cell tubeformation on gelled basement membrane extract. Nature Protocols 5, 628–635(2010).
41. Deckers, M. et al. Effect of angiogenic and antiangiogenic compounds on theoutgrowth of capillary structures from fetal mouse bone explants. Lab. Invest. 81,5–15 (2001).
42. Nicosia, R. F. & Ottinetti, A. Growth of microvessels in serum-free matrix culture ofrat aorta. A quantitative assay of angiogenesis in vitro. Lab. Invest. 63, 115–122(1990).
43. Balaggan, K. S. et al. EIAV vector-mediated delivery of endostatin or angiostatininhibits angiogenesis and vascular hyperpermeability in experimental CNV. GeneTher. 13, 1153–1165 (2006).
44. Toma, H. S., Barnett, J. M., Penn, J. S. & Kim, S. J. Improved assessment of laser-induced choroidal neovascularization. Microvasc. Res. 80, 295–302 (2010).
45. Smith, L. E. et al. Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis.Sci. 35, 101–111 (1994).
46. Connor, K. M. et al. Quantification of oxygen-induced retinopathy in the mouse: amodel of vessel loss, vessel regrowth and pathological angiogenesis. NatureProtocols 4, 1565–1573 (2009).
ARTICLE RESEARCH
Macmillan Publishers Limited. All rights reserved©2013

W W W. N A T U R E . C O M / N A T U R E | 1
SUPPLEMENTARY INFORMATIONdoi:10.1038/nature12345
Vldlr -/-
50 m
C57/BL6
PECAM-1DAPI
RD1Curly tail
Supplementary Figure 1. Retinal vasculature of C57/BL6 wild type mouse and threemouse models of retinal disease. Imaris rendered representative 3D images of PECAM-1 stained retinal vasculature (red) of control C57/BL6 mouse and of the vascular abnormalities(highlighted by arrow heads) in Vldlr-/-, Curly tail (Grhl3ct/J) and RD1 mice. In each image asingle X-Y section stained for nuclei (blue) is incorporated to show the relative positioning ofthe nuclear layers (see also Supplementary movies 1-4).

SUPPLEMENTARY INFORMATION
2 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
a
b
Supplementary Figure 2. Predicted structure and species homology of LRG1. a, Predicted ribbon structure (left) and space-filling model (right) of mouse LRG1 (342 amino acids) obtained through the Robetta full-chain protein structure prediction server(http://robetta.bakerlab.org). The leucine-rich C-terminal (LRC) contains mainly β strands and is connected to the leucine-rich repeats(LRR) by loops. Each LRR contains a β strand and an α helix with the structural units arranged in parallel with a curved β sheet liningthe inner circumference and the α helices flanking the outer circumference. The LRC and LRR domain contain 3 potentialglycosylation sites with a further 4 sites in the linker region between the LRR and the signal peptide. Images generated courtesy ofProfessor C.M. Marson (UCL, London). b, Alignment of the Lrg1 amino-acid sequence between different vertebrate species obtainedthrough the EMBL-EBI European Bioinformatics Institute.
W W W. N A T U R E . C O M / N A T U R E | 3
SUPPLEMENTARY INFORMATION RESEARCH
α-LRG1
α-GAPDH
WT
Lrg1
+/-
Lrg1
-/-
50
37
a
Pre-immune sera Pre-absorbed -LRG1 sera
Don
key
anti-
rabb
itIs
olec
tin B
4
WT Lrg1-/-
LRG1 polyclonal antibody LRG1 polyclonal antibody
bSense probe Antisense probe
c
kDa
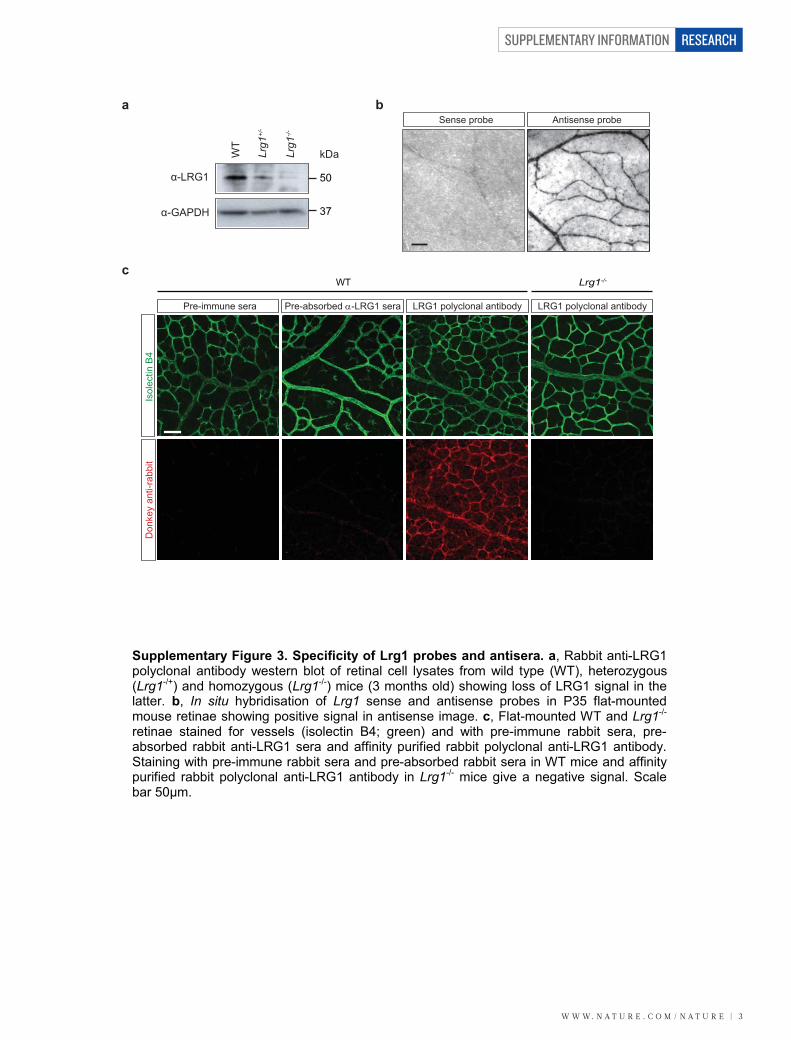
Supplementary Figure 3. Specificity of Lrg1 probes and antisera. a, Rabbit anti-LRG1 polyclonal antibody western blot of retinal cell lysates from wild type (WT), heterozygous (Lrg1-/+) and homozygous (Lrg1-/-) mice (3 months old) showing loss of LRG1 signal in the latter. b, In situ hybridisation of Lrg1 sense and antisense probes in P35 flat-mounted mouse retinae showing positive signal in antisense image. c, Flat-mounted WT and Lrg1-/- retinae stained for vessels (isolectin B4; green) and with pre-immune rabbit sera, pre-absorbed rabbit anti-LRG1 sera and affinity purified rabbit polyclonal anti-LRG1 antibody. Staining with pre-immune rabbit sera and pre-absorbed rabbit sera in WT mice and affinity purified rabbit polyclonal anti-LRG1 antibody in Lrg1-/- mice give a negative signal. Scale bar 50μm.
SUPPLEMENTARY INFORMATION
4 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
VE-Cadherin DAPI
LRG1Isolectin B4
WT
mou
se c
horo
id
Merge
LRG1Collagen IV Merge
Hum
an b
reas
tH
uman
ski
nH
uman
inte
stin
e
a
b
DAPI
aniteR
Hum
an re
tina
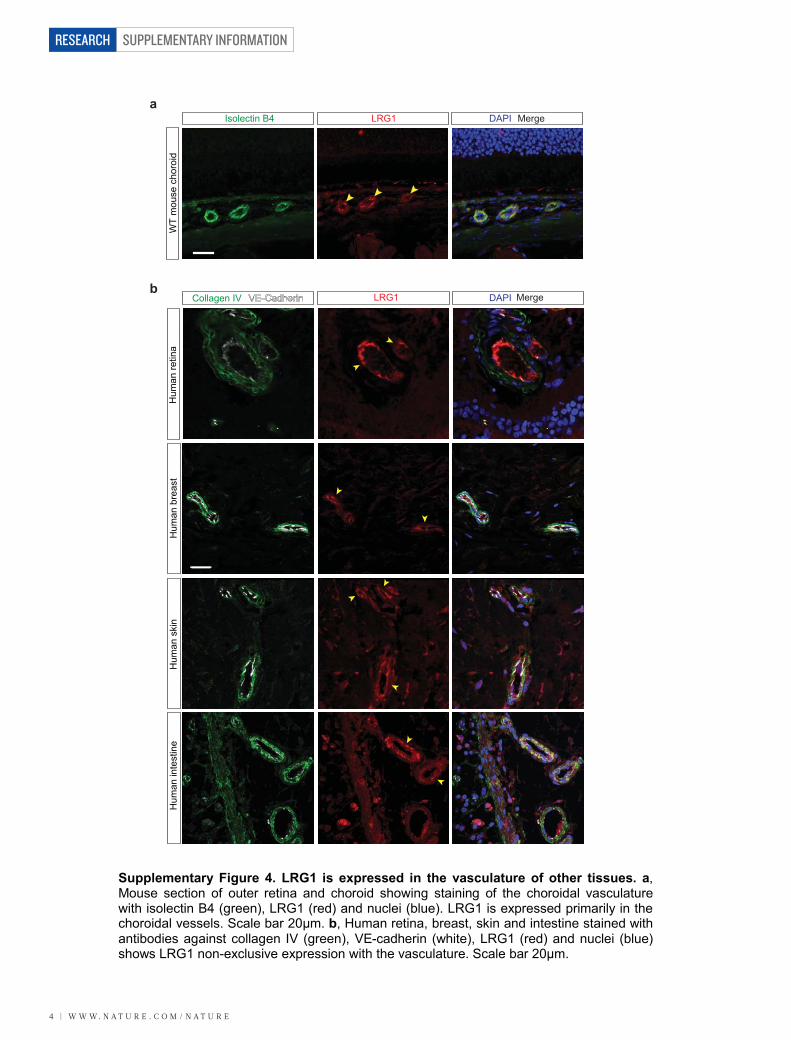
Supplementary Figure 4. LRG1 is expressed in the vasculature of other tissues. a, Mouse section of outer retina and choroid showing staining of the choroidal vasculature with isolectin B4 (green), LRG1 (red) and nuclei (blue). LRG1 is expressed primarily in the choroidal vessels. Scale bar 20μm. b, Human retina, breast, skin and intestine stained with antibodies against collagen IV (green), VE-cadherin (white), LRG1 (red) and nuclei (blue) shows LRG1 non-exclusive expression with the vasculature. Scale bar 20μm.
W W W. N A T U R E . C O M / N A T U R E | 5
SUPPLEMENTARY INFORMATION RESEARCH
Pai1
% o
f cha
nge 100
50
0
-50
P12P17
P12P17
P12P17
P12P17
P12P17
P12P17
% o
f cha
nge
% o
f cha
nge
% o
f cha
nge
% o
f cha
nge
% o
f cha
nge
Id1Tgfb1
Vegf AplnAplnr
100
50
0
-50
100
50
0
-50
200
100
0
-100
200
150
100
50
0
-50
-100
800
600
400
200
0
-200
****** ***
******
*
***
*
*
a
b
Contro
lCNV
Contro
lCNV
Contro
lCNV
Pai1Id1Tgfb1
20
15
10
5
0
Rel
ativ
e in
tens
ities 5
4
32
1
0
Rel
ativ
e in
tens
ities 15
10
5
0
Rel
ativ
e in
tens
ities*** ***
*
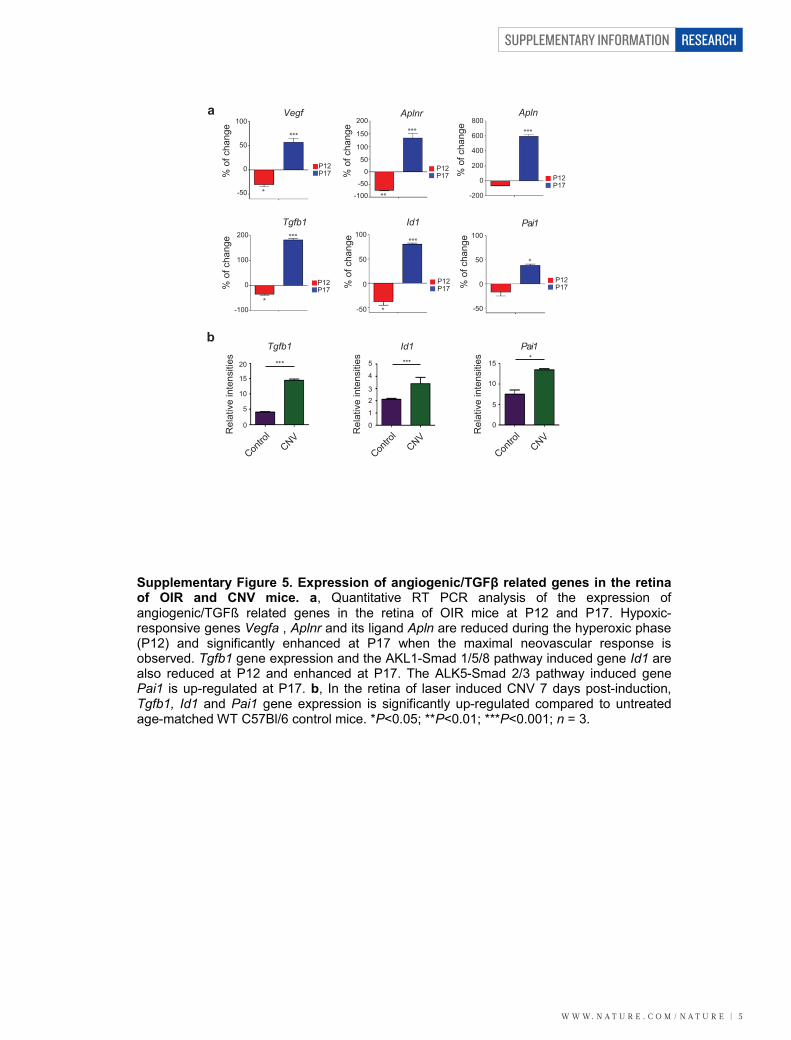
Supplementary Figure 5. Expression of angiogenic/TGFβ related genes in the retina of OIR and CNV mice. a, Quantitative RT PCR analysis of the expression of angiogenic/TGFß related genes in the retina of OIR mice at P12 and P17. Hypoxic-responsive genes Vegfa , Aplnr and its ligand Apln are reduced during the hyperoxic phase (P12) and significantly enhanced at P17 when the maximal neovascular response is observed. Tgfb1 gene expression and the AKL1-Smad 1/5/8 pathway induced gene Id1 are also reduced at P12 and enhanced at P17. The ALK5-Smad 2/3 pathway induced gene Pai1 is up-regulated at P17. b, In the retina of laser induced CNV 7 days post-induction,Tgfb1, Id1 and Pai1 gene expression is significantly up-regulated compared to untreated age-matched WT C57Bl/6 control mice. *P<0.05; **P<0.01; ***P<0.001; n = 3.
SUPPLEMENTARY INFORMATION
6 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
b
a
α-LRG1
PDR Ctl PDR CtlPDR Ctl
α-LRG1
PDR Ctl
PDR Ctl PDR Ctl
PDR Ctl PDR Ctl
PDR Ctl
1
1
1
1
1
1
1
1
2 2
2 2
2 2
2 2
3 3
3 3
3 3
3 3
4 4
4 4
4 4
4 4
5 5
5 5
5 5
5 5
6 6
6 6
6 6
6 6
7 7
7 7
7 7
7 7
8 8
8 8
8 8
8 8
9 9
9 9
9 9
9 9
10 10
10 10
10 10
10 10
***R
elat
ive
inte
nsiti
es15
10
5
0Ctl PDR
Ctl PDR
Rel
ativ
ein
tens
ities
1510
50
2025
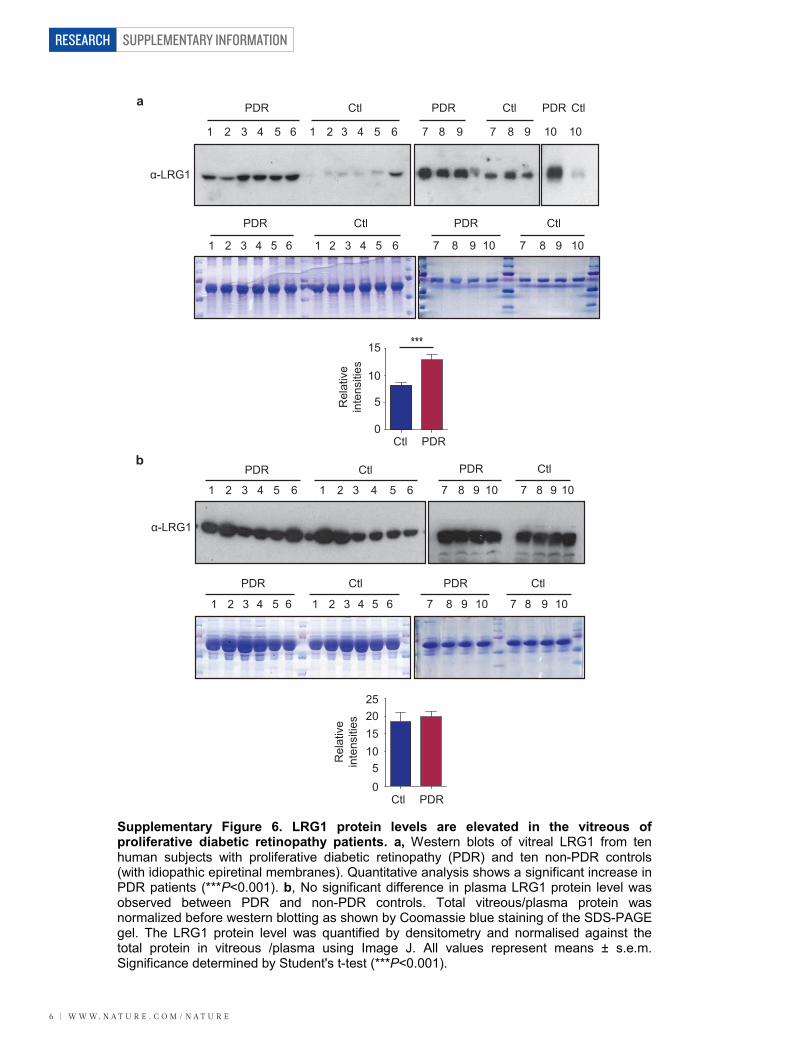
Supplementary Figure 6. LRG1 protein levels are elevated in the vitreous of proliferative diabetic retinopathy patients. a, Western blots of vitreal LRG1 from ten human subjects with proliferative diabetic retinopathy (PDR) and ten non-PDR controls (with idiopathic epiretinal membranes). Quantitative analysis shows a significant increase in PDR patients (***P<0.001). b, No significant difference in plasma LRG1 protein level was observed between PDR and non-PDR controls. Total vitreous/plasma protein was normalized before western blotting as shown by Coomassie blue staining of the SDS-PAGE gel. The LRG1 protein level was quantified by densitometry and normalised against the total protein in vitreous /plasma using Image J. All values represent means ± s.e.m. Significance determined by Student's t-test (***P<0.001).
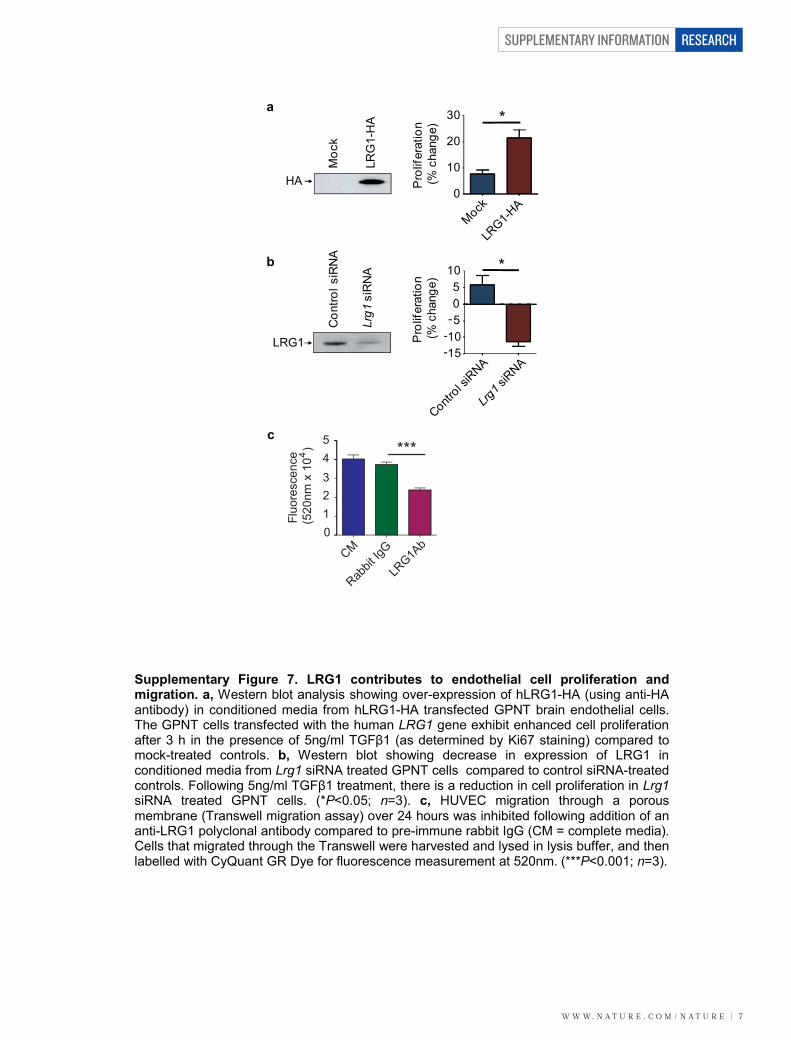
W W W. N A T U R E . C O M / N A T U R E | 7
SUPPLEMENTARY INFORMATION RESEARCH
*
0
10
20
30
Pro
lifer
atio
n (%
cha
nge)
*
-15-10-505
10
Pro
lifer
atio
n (%
cha
nge)
a
b
HA
LRG
1-H
A
Moc
kC
ontro
l siR
NA
Lrg1
siR
NA
LRG1
c
CM
Rabbit
IgG
LRG1A
b
543210
***
Fluo
resc
ence
(5
20nm
x 1
0 )4
LRG1-H
AMock
Control s
iRNA
Lrg1 s
iRNA
Supplementary Figure 7. LRG1 contributes to endothelial cell proliferation and migration. a, Western blot analysis showing over-expression of hLRG1-HA (using anti-HA antibody) in conditioned media from hLRG1-HA transfected GPNT brain endothelial cells. The GPNT cells transfected with the human LRG1 gene exhibit enhanced cell proliferation after 3 h in the presence of 5ng/ml TGFβ1 (as determined by Ki67 staining) compared to mock-treated controls. b, Western blot showing decrease in expression of LRG1 in conditioned media from Lrg1 siRNA treated GPNT cells compared to control siRNA-treated controls. Following 5ng/ml TGFβ1 treatment, there is a reduction in cell proliferation in Lrg1 siRNA treated GPNT cells. (*P<0.05; n=3). c, HUVEC migration through a porous membrane (Transwell migration assay) over 24 hours was inhibited following addition of an anti-LRG1 polyclonal antibody compared to pre-immune rabbit IgG (CM = complete media). Cells that migrated through the Transwell were harvested and lysed in lysis buffer, and then labelled with CyQuant GR Dye for fluorescence measurement at 520nm. (***P<0.001; n=3).
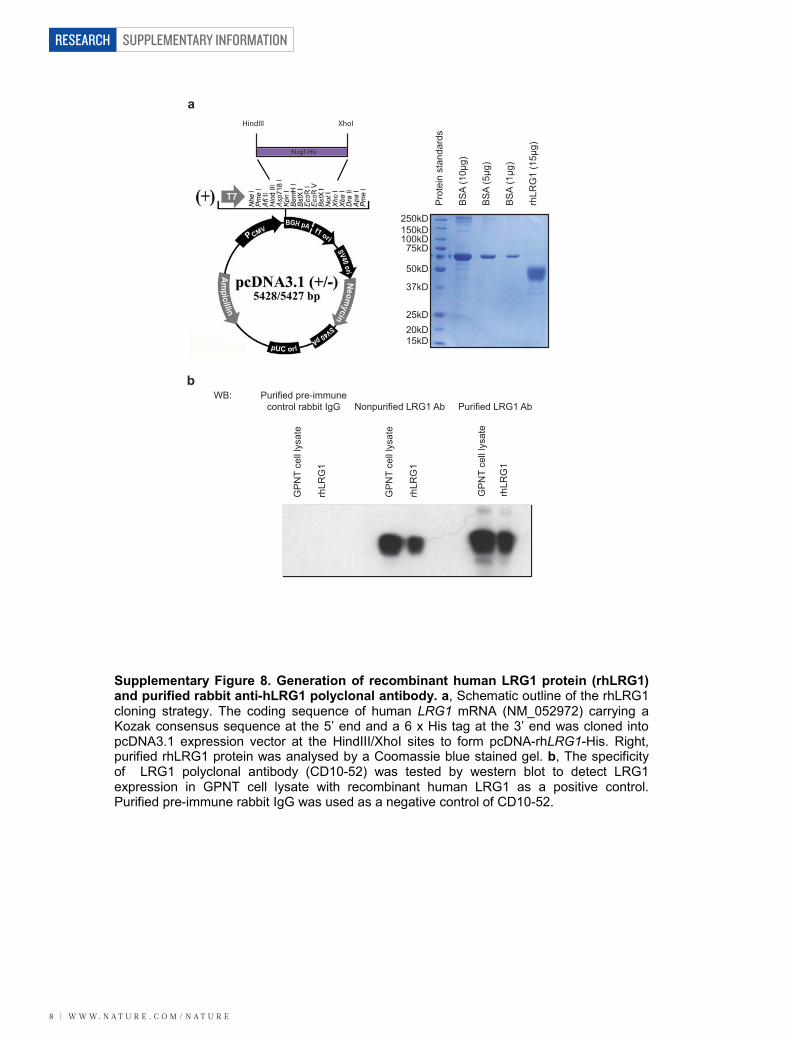
SUPPLEMENTARY INFORMATION
8 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
Purified pre-immune control rabbit IgG Nonpurified LRG1 Ab Purified LRG1 Ab
GP
NT
cell
lysa
te
rhLR
G1
GP
NT
cell
lysa
te
rhLR
G1
GP
NT
cell
lysa
te
rhLR
G1
WB:
HindIII XhoI
hLrg1-His
a
b
Pro
tein
sta
ndar
ds
BS
A (1
0μg)
BS
A (5μg
)
BS
A (1μg
)
rhLR
G1
(15μ
g)
250kD150kD100kD
75kD
50kD
37kD
25kD20kD15kD
Supplementary Figure 8. Generation of recombinant human LRG1 protein (rhLRG1) and purified rabbit anti-hLRG1 polyclonal antibody. a, Schematic outline of the rhLRG1 cloning strategy. The coding sequence of human LRG1 mRNA (NM_052972) carrying a Kozak consensus sequence at the 5’ end and a 6 x His tag at the 3’ end was cloned into pcDNA3.1 expression vector at the HindIII/XhoI sites to form pcDNA-rhLRG1-His. Right, purified rhLRG1 protein was analysed by a Coomassie blue stained gel. b, The specificity of LRG1 polyclonal antibody (CD10-52) was tested by western blot to detect LRG1 expression in GPNT cell lysate with recombinant human LRG1 as a positive control. Purified pre-immune rabbit IgG was used as a negative control of CD10-52.
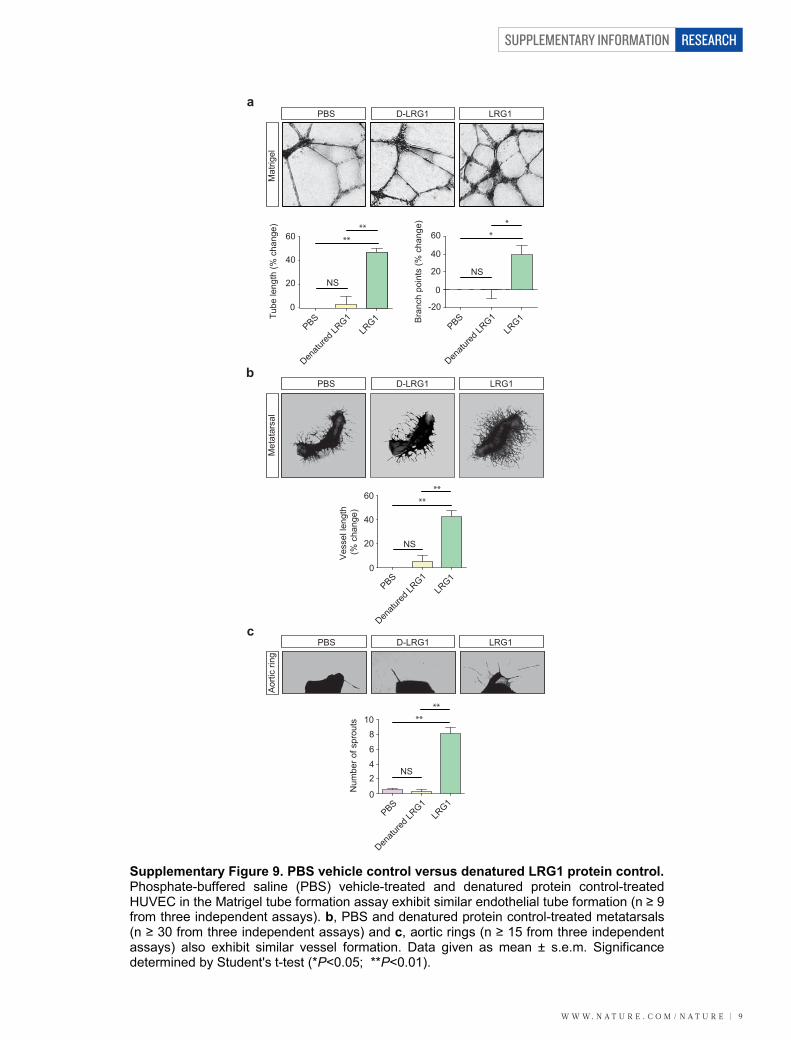
W W W. N A T U R E . C O M / N A T U R E | 9
SUPPLEMENTARY INFORMATION RESEARCH
a
b
c
60
40
20
0
-20
60
40
20
0
PBS
Denatu
red LR
G1LR
G1 Bra
nch
poin
ts (%
cha
nge)
Tube
leng
th (%
cha
nge) **
PBS D-LRG1 LRG1
**
NS
**
NS
PBS
Denatu
red LR
G1LR
G1
60
40
20
0
Ves
sel l
engt
h (%
cha
nge)
PBS
Denatu
red LR
G1LR
G1
Num
ber o
f spr
outs 10
8
6
42
0
PBS
Denatu
red LR
G1LR
G1
PBS D-LRG1 LRG1
PBS D-LRG1 LRG1
****
NS
****
NS
Mat
rigel
Met
atar
sal
Aor
tic ri
ng
Supplementary Figure 9. PBS vehicle control versus denatured LRG1 protein control. Phosphate-buffered saline (PBS) vehicle-treated and denatured protein control-treated HUVEC in the Matrigel tube formation assay exhibit similar endothelial tube formation (n ≥ 9 from three independent assays). b, PBS and denatured protein control-treated metatarsals (n ≥ 30 from three independent assays) and c, aortic rings (n ≥ 15 from three independent assays) also exhibit similar vessel formation. Data given as mean ± s.e.m. Significance determined by Student's t-test (*P<0.05; **P<0.01).

SUPPLEMENTARY INFORMATION
1 0 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
LRG1
TGFβ1
WB: Mou
se a
ortic
ring
Mou
se m
etat
arsa
l
HU
VE
C
Mou
se p
rimar
y br
ain
endo
thel
ial c
ells
Mou
se lu
ng e
ndot
helia
l cel
ls
HE
K 2
93 s
erum
-free
cul
ture
med
ium
HE
K 2
93 5
day
con
ditio
ned
seru
m fr
ee m
ediu
m
TGFβ1
WB:
a
b
Supplementary Figure 10. LRG1 and TGFß-1 expression in conditioned media. a,Western blot analysis showing the presence of LRG1 and TGFß-1 in conditioned mediaharvested from mouse aortic ring, mouse metatarsal, mouse primary brain endothelial cellcultures, immortalised mouse lung endothelial cell cultures (from Rosa26-CreERT:Engfl/fl
mouse) and HUVEC Matrigel assays. b, Western blot analysis showing the absence of TGFß-1 in HEK 293 cell culture medium and following 5 days of cell conditioning.
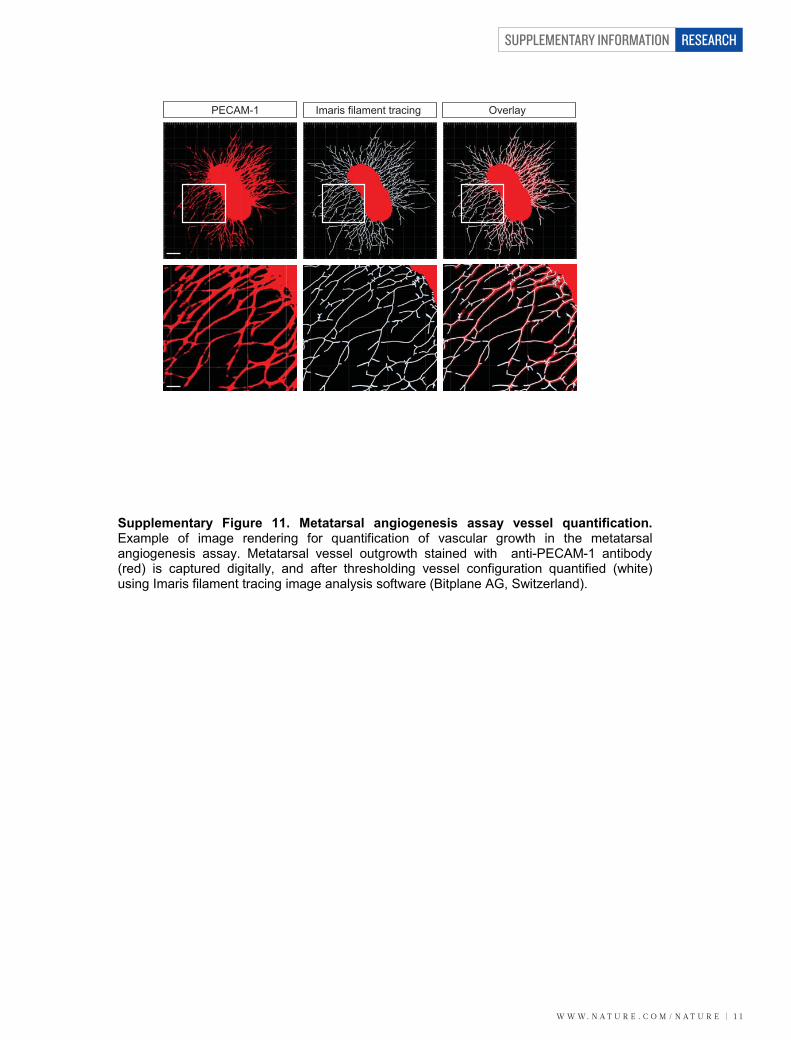
W W W. N A T U R E . C O M / N A T U R E | 1 1
SUPPLEMENTARY INFORMATION RESEARCH
PECAM-1 Imaris filament tracing Overlay
Supplementary Figure 11. Metatarsal angiogenesis assay vessel quantification. Example of image rendering for quantification of vascular growth in the metatarsal angiogenesis assay. Metatarsal vessel outgrowth stained with anti-PECAM-1 antibody (red) is captured digitally, and after thresholding vessel configuration quantified (white) using Imaris filament tracing image analysis software (Bitplane AG, Switzerland).

SUPPLEMENTARY INFORMATION
1 2 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
TUF/TUR
NeoFwd/SD
SU/LacZRev
GAPDH
Wild
type
Lrg1
+/-
Lrg1
-/-
wild ty
pe
Lrg1+
/-
Lrg1-/-
60%
40%
20%
0
Distribution of Lrg1-/- mice
27.45%
48.04%
24.52%
n=102
Forward Primer
Reverse Primer Length WT HET KO
SU LacZRev 367 bp - + +
TUF TUR 76 bp + + -
NeoFwd SD 307 bp - + +
a
b
c
Supplementary Figure 12. Genotyping the Lrg1-/- mouse. a, Strategy for PCR-based genotyping Lrg1 knockout mice (from KOMP). b, The sizes of expected PCR products for wild type (WT), heterozygote (HET) and null mutant (KO). c. Genotyping examples of WT, heterozygous and homozygous mice and their ratio in the progeny of heterozygous breeding pairs.
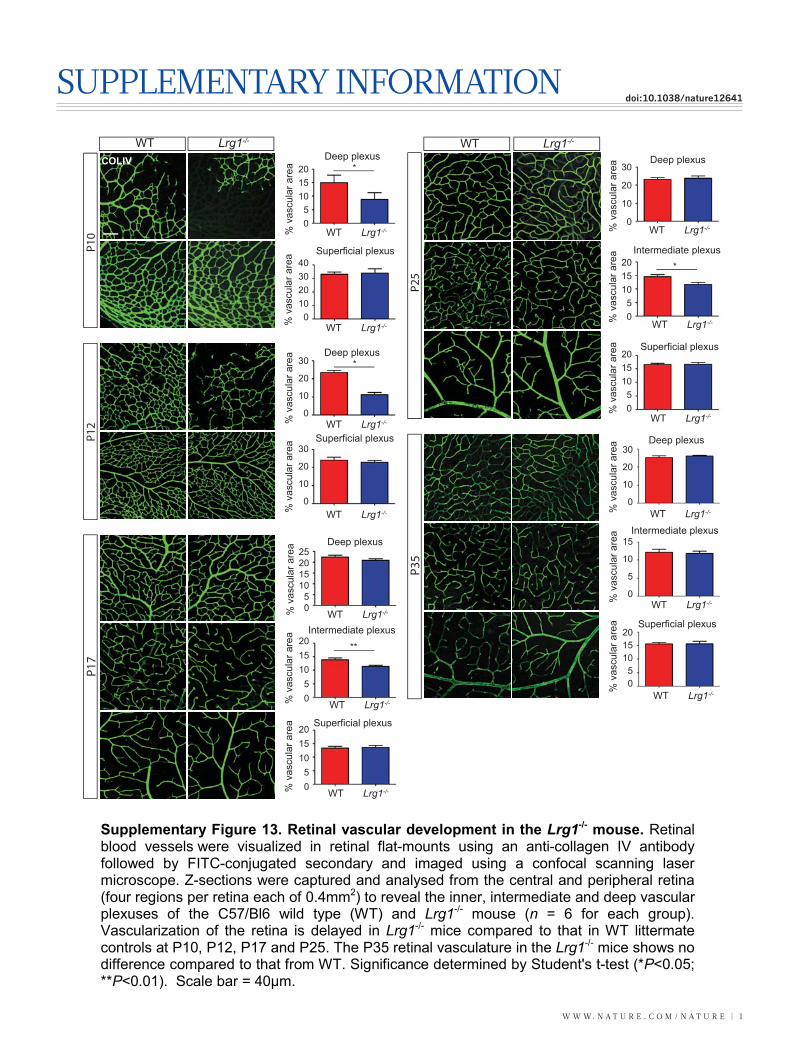
W W W. N A T U R E . C O M / N A T U R E | 1
SUPPLEMENTARY INFORMATIONdoi:10.1038/nature12641
P17
P35
WT Lrg1-/-
P10
P12
COLIV Deep plexusDeep plexus
Deep plexus
Superficial plexus
Superficial plexus
Superficial plexus Intermediate plexus
201510
50
201510
50
201510
50
40302010
0
30
20
10
0
30
20
10
0
30
20
10
0
% v
ascu
lar a
rea *
*
**
Deep plexus
Superficial plexus
Intermediate plexus201510
50
25201510
50
201510
50
**
*
WT Lrg1-/-
% v
ascu
lar a
rea
% v
ascu
lar a
rea
% v
ascu
lar a
rea
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
% v
ascu
lar a
rea
% v
ascu
lar a
rea
% v
ascu
lar a
rea
P25
% v
ascu
lar a
rea
% v
ascu
lar a
rea
% v
ascu
lar a
rea
P25
b
Deep plexus
Superficial plexus
Intermediate plexus
201510
50
30
20
10
0
15
10
5
0
% v
ascu
lar a
rea
% v
ascu
lar a
rea
% v
ascu
lar a
rea
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
Supplementary Figure 13. Retinal vascular development in the Lrg1-/- mouse. Retinal blood vessels were visualized in retinal flat-mounts using an anti-collagen IV antibody followed by FITC-conjugated secondary and imaged using a confocal scanning laser microscope. Z-sections were captured and analysed from the central and peripheral retina (four regions per retina each of 0.4mm2) to reveal the inner, intermediate and deep vascular plexuses of the C57/Bl6 wild type (WT) and Lrg1-/- mouse (n = 6 for each group). Vascularization of the retina is delayed in Lrg1-/- mice compared to that in WT littermate controls at P10, P12, P17 and P25. The P35 retinal vasculature in the Lrg1-/- mice shows no difference compared to that from WT. Significance determined by Student's t-test (*P<0.05; **P<0.01). Scale bar = 40μm.
P17
P35
WT Lrg1-/-
P10
P12
COLIV Deep plexusDeep plexus
Deep plexus
Superficial plexus
Superficial plexus
Superficial plexus Intermediate plexus
201510
50
201510
50
201510
50
40302010
0
30
20
10
0
30
20
10
0
30
20
10
0
% v
ascu
lar a
rea *
*
**
Deep plexus
Superficial plexus
Intermediate plexus201510
50
25201510
50
201510
50
**
*
WT Lrg1-/-
% v
ascu
lar a
rea
% v
ascu
lar a
rea
% v
ascu
lar a
rea
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
% v
ascu
lar a
rea
% v
ascu
lar a
rea
% v
ascu
lar a
rea
P25
% v
ascu
lar a
rea
% v
ascu
lar a
rea
% v
ascu
lar a
rea
P25
b
Deep plexus
Superficial plexus
Intermediate plexus
201510
50
30
20
10
0
15
10
5
0
% v
ascu
lar a
rea
% v
ascu
lar a
rea
% v
ascu
lar a
rea
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
WT Lrg1-/-
Supplementary Figure 13. Retinal vascular development in the Lrg1-/- mouse. Retinal blood vessels were visualized in retinal flat-mounts using an anti-collagen IV antibody followed by FITC-conjugated secondary and imaged using a confocal scanning laser microscope. Z-sections were captured and analysed from the central and peripheral retina (four regions per retina each of 0.4mm2) to reveal the inner, intermediate and deep vascular plexuses of the C57/Bl6 wild type (WT) and Lrg1-/- mouse (n = 6 for each group). Vascularization of the retina is delayed in Lrg1-/- mice compared to that in WT littermate controls at P10, P12, P17 and P25. The P35 retinal vasculature in the Lrg1-/- mice shows no difference compared to that from WT. Significance determined by Student's t-test (*P<0.05; **P<0.01). Scale bar = 40μm.
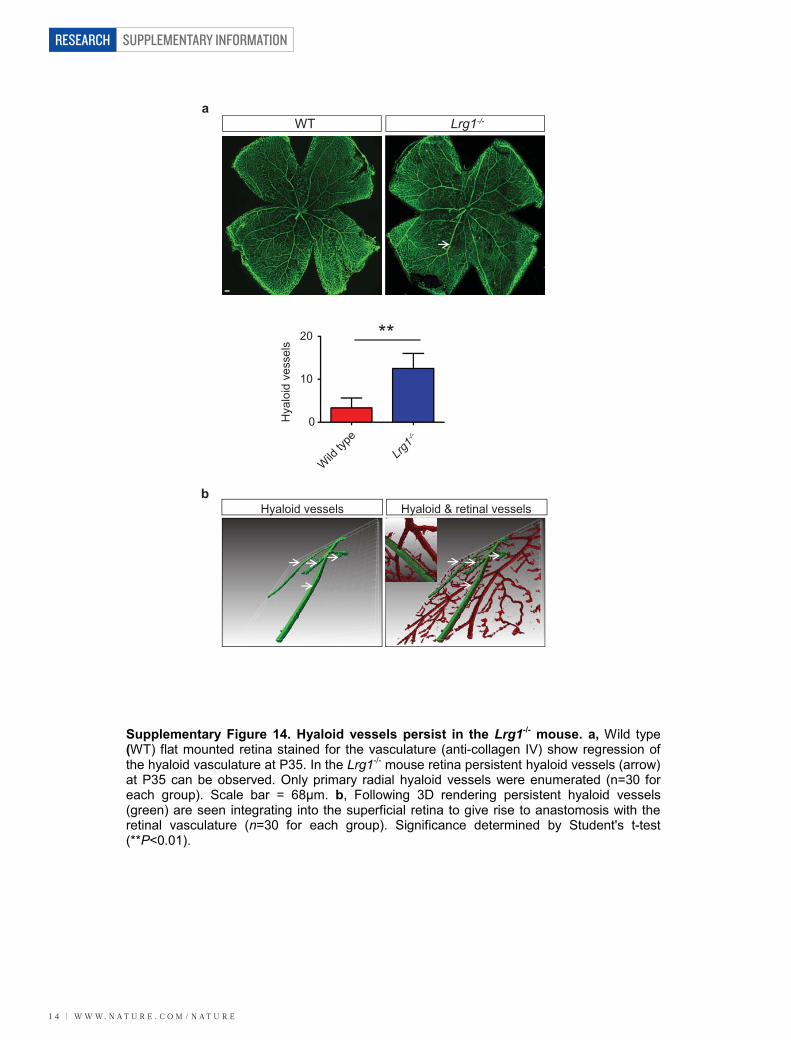
SUPPLEMENTARY INFORMATION
1 4 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
**
Hya
loid
ves
sels
20
10
0
Wild
type
Lrg1-/-
a
b
WT Lrg1-/-
Hyaloid vessels Hyaloid & retinal vessels
Supplementary Figure 14. Hyaloid vessels persist in the Lrg1-/- mouse. a, Wild type (WT) flat mounted retina stained for the vasculature (anti-collagen IV) show regression of the hyaloid vasculature at P35. In the Lrg1-/- mouse retina persistent hyaloid vessels (arrow) at P35 can be observed. Only primary radial hyaloid vessels were enumerated (n=30 for each group). Scale bar = 68μm. b, Following 3D rendering persistent hyaloid vessels (green) are seen integrating into the superficial retina to give rise to anastomosis with the retinal vasculature (n=30 for each group). Significance determined by Student's t-test (**P<0.01).
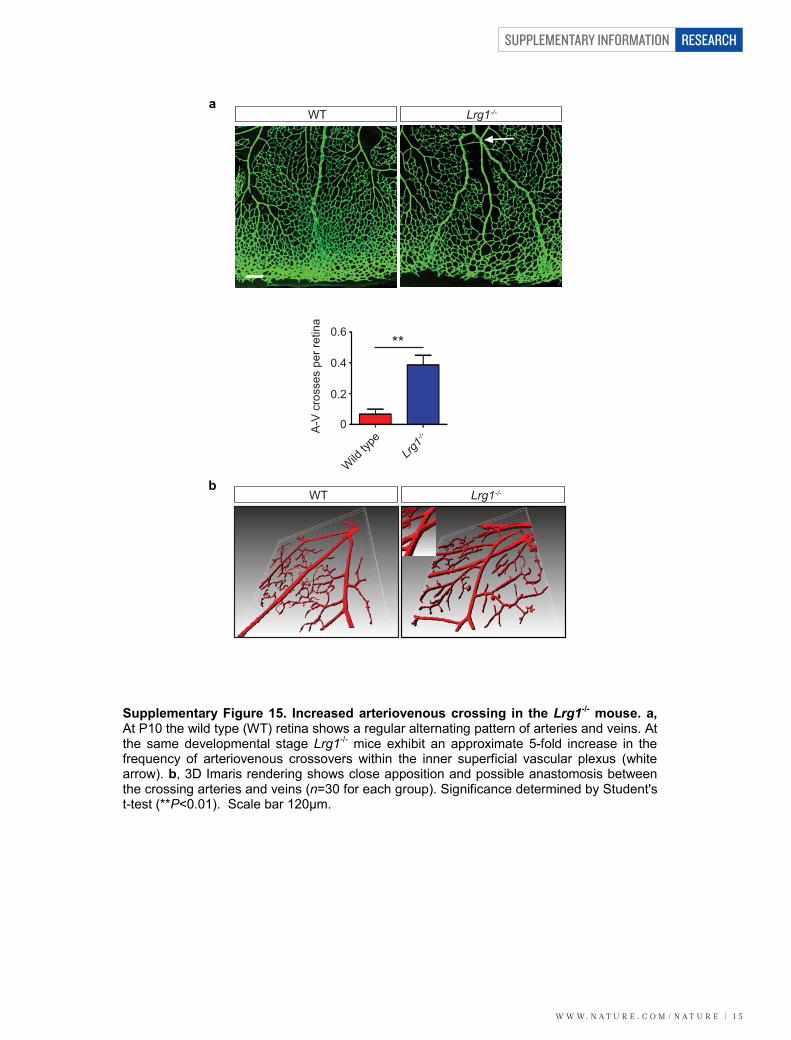
W W W. N A T U R E . C O M / N A T U R E | 1 5
SUPPLEMENTARY INFORMATION RESEARCH
WT Lrg1-/-
**
A-V
cro
sses
per
retin
a0.6
0.4
0.2
0
Wild
type
Lrg1-/-
a
b WT Lrg1-/-
Supplementary Figure 15. Increased arteriovenous crossing in the Lrg1-/- mouse. a, At P10 the wild type (WT) retina shows a regular alternating pattern of arteries and veins. At the same developmental stage Lrg1-/- mice exhibit an approximate 5-fold increase in the frequency of arteriovenous crossovers within the inner superficial vascular plexus (white arrow). b, 3D Imaris rendering shows close apposition and possible anastomosis between the crossing arteries and veins (n=30 for each group). Significance determined by Student's t-test (**P<0.01). Scale bar 120μm.
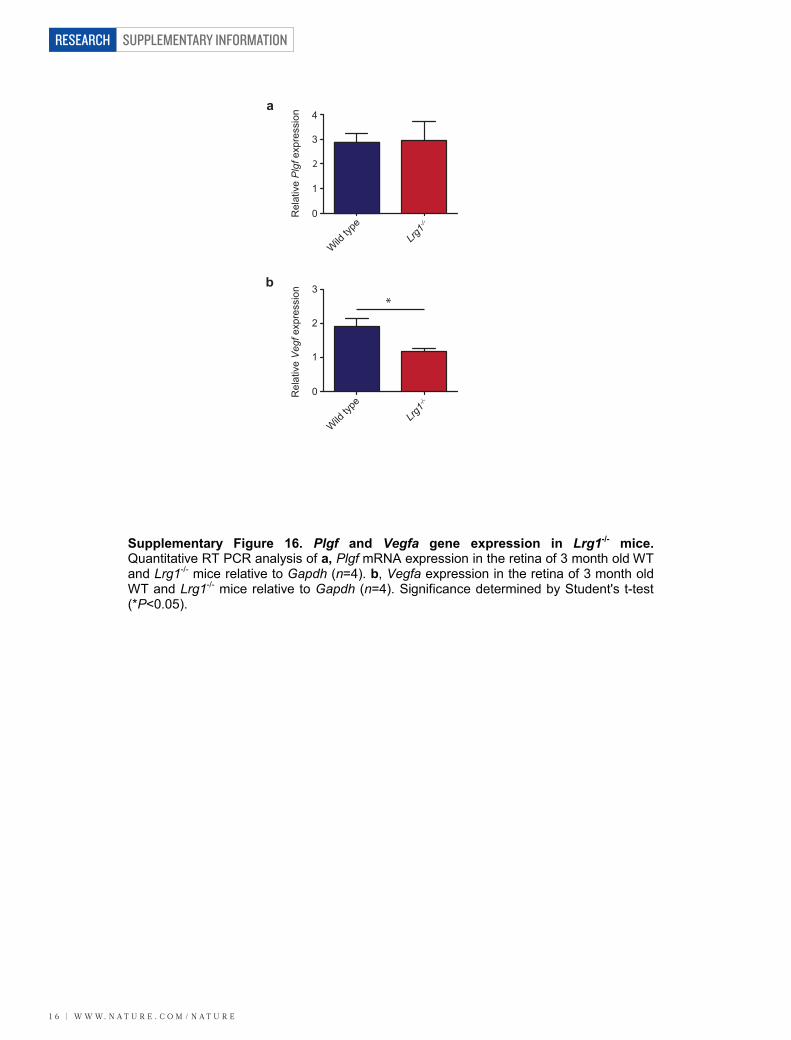
SUPPLEMENTARY INFORMATION
1 6 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
4
3
2
1
0
Wild ty
peLrg
1-/-
Rel
ativ
e P
lgfe
xpre
ssio
n
Wild ty
peLrg
1-/-
Rel
ativ
e V
egfe
xpre
ssio
n 3
2
1
0
*
a
b
Supplementary Figure 16. Plgf and Vegfa gene expression in Lrg1-/- mice. Quantitative RT PCR analysis of a, Plgf mRNA expression in the retina of 3 month old WT and Lrg1-/- mice relative to Gapdh (n=4). b, Vegfa expression in the retina of 3 month old WT and Lrg1-/- mice relative to Gapdh (n=4). Significance determined by Student's t-test (*P<0.05).
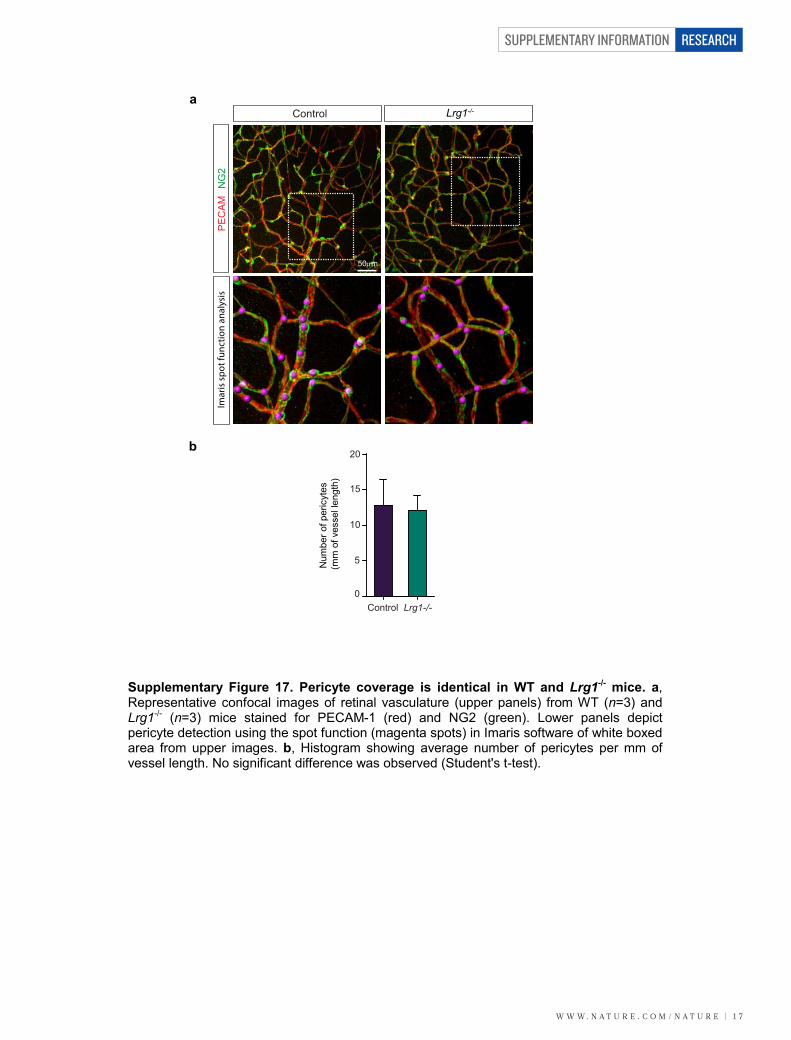
W W W. N A T U R E . C O M / N A T U R E | 1 7
SUPPLEMENTARY INFORMATION RESEARCH
20
15
10
5
0Control Lrg1-/-
Imar
is s
pot f
unct
ion
anal
ysis
NG
2M
AC
EP
Lrg1-/-Controla
b
Num
ber o
f per
icyt
es
(mm
of v
esse
l len
gth)
50m
Supplementary Figure 17. Pericyte coverage is identical in WT and Lrg1-/- mice. a,Representative confocal images of retinal vasculature (upper panels) from WT (n=3) and Lrg1-/- (n=3) mice stained for PECAM-1 (red) and NG2 (green). Lower panels depict pericyte detection using the spot function (magenta spots) in Imaris software of white boxed area from upper images. b, Histogram showing average number of pericytes per mm of vessel length. No significant difference was observed (Student's t-test).
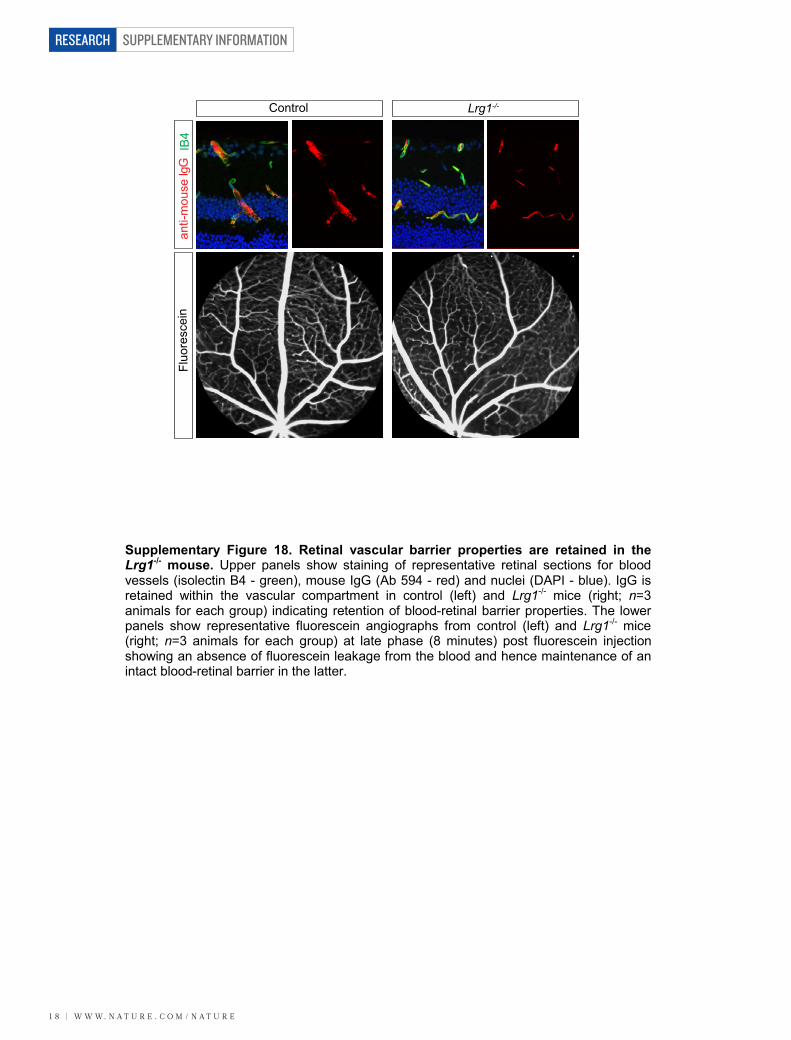
SUPPLEMENTARY INFORMATION
1 8 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
Control Lrg1-/-
anti-
mou
se Ig
GIB
4Fl
uore
scei
n
Supplementary Figure 18. Retinal vascular barrier properties are retained in the Lrg1-/- mouse. Upper panels show staining of representative retinal sections for blood vessels (isolectin B4 - green), mouse IgG (Ab 594 - red) and nuclei (DAPI - blue). IgG is retained within the vascular compartment in control (left) and Lrg1-/- mice (right; n=3 animals for each group) indicating retention of blood-retinal barrier properties. The lower panels show representative fluorescein angiographs from control (left) and Lrg1-/- mice (right; n=3 animals for each group) at late phase (8 minutes) post fluorescein injection showing an absence of fluorescein leakage from the blood and hence maintenance of an intact blood-retinal barrier in the latter.

W W W. N A T U R E . C O M / N A T U R E | 1 9
SUPPLEMENTARY INFORMATION RESEARCH
PBS LRG1
LRG1PBS
PBS
LRG1PBS
LRG1
Wild type
Lrg1-/-
Met
atar
sal
Aor
ta
Met
atar
sal
Aor
ta
b
a
Supplementary Figure 19. LRG1 promotes angiogenesis in WT mice and rescues angiogenesis in Lrg1-/- mice. a, Examples of vessel outgrowth in the metatarsal (top row) and aortic ring (bottom row) assay showing enhanced angiogenesis following LRG1 treatment. b, Examples of vessel outgrowth in metatarsals (top row) and aortic rings (bottom row) from Lrg1-/- mice showing rescue of LRG1 induced angiogenesis. See Figure 2 for quantification. Scale bar = 1,500μm.
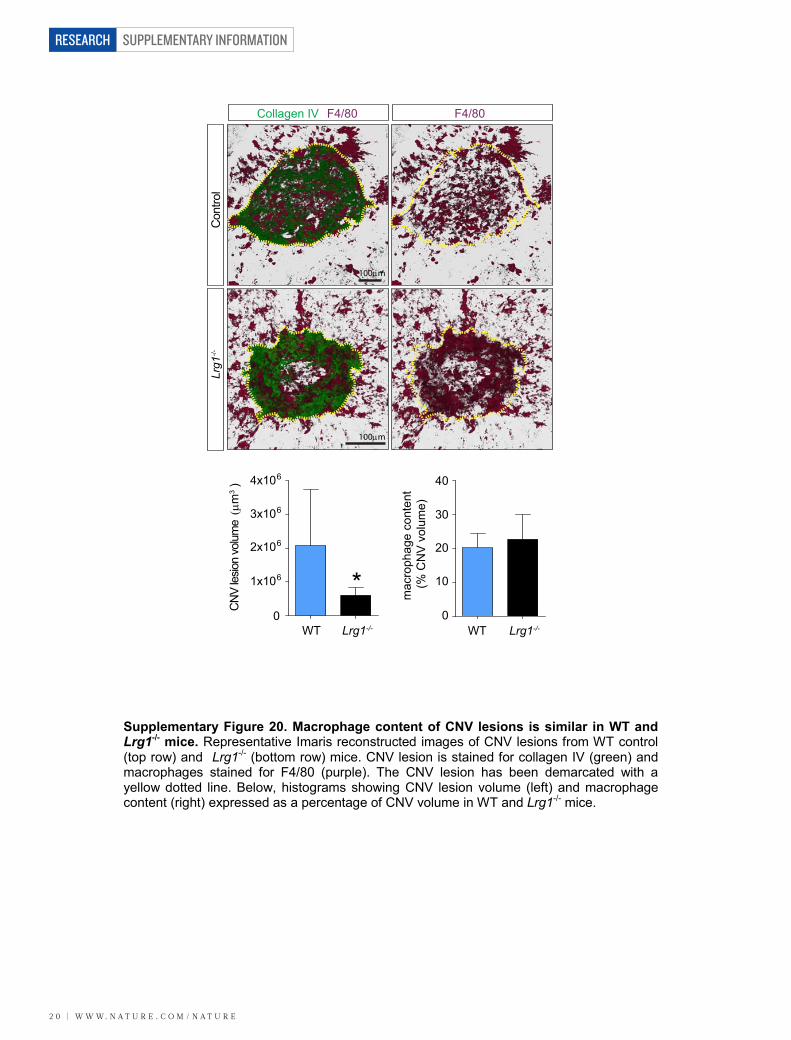
SUPPLEMENTARY INFORMATION
2 0 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
100m
100m
Collagen IV F4/80
lortnoC
1grL-/-
WT Lrg1-/-
CN
V le
sion
volu
me
m )
3
mac
roph
age
cont
ent
(% C
NV
vol
ume)
1x106
2x106
3x106
4x106
0WT Lrg1-/-
*0
10
20
30
40
F4/80
Supplementary Figure 20. Macrophage content of CNV lesions is similar in WT and Lrg1-/- mice. Representative Imaris reconstructed images of CNV lesions from WT control (top row) and Lrg1-/- (bottom row) mice. CNV lesion is stained for collagen IV (green) and macrophages stained for F4/80 (purple). The CNV lesion has been demarcated with a yellow dotted line. Below, histograms showing CNV lesion volume (left) and macrophage content (right) expressed as a percentage of CNV volume in WT and Lrg1-/- mice.
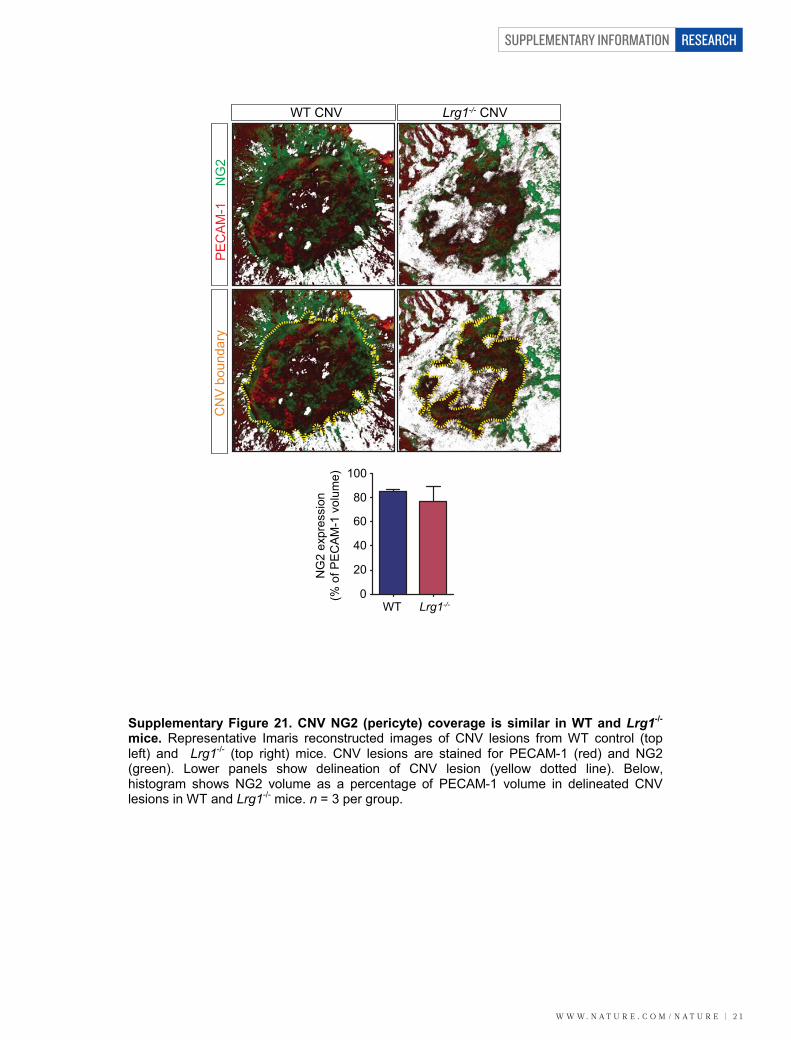
W W W. N A T U R E . C O M / N A T U R E | 2 1
SUPPLEMENTARY INFORMATION RESEARCH
WT CNV Lrg1-/- CNV
PE
CA
M-1
NG
2C
NV
bou
ndar
y
100
80
60
40
20
0WT Lrg1-/-
NG
2 ex
pres
sion
(% o
f PE
CA
M-1
vol
ume)
Supplementary Figure 21. CNV NG2 (pericyte) coverage is similar in WT and Lrg1-/-
mice. Representative Imaris reconstructed images of CNV lesions from WT control (top left) and Lrg1-/- (top right) mice. CNV lesions are stained for PECAM-1 (red) and NG2 (green). Lower panels show delineation of CNV lesion (yellow dotted line). Below, histogram shows NG2 volume as a percentage of PECAM-1 volume in delineated CNV lesions in WT and Lrg1-/- mice. n = 3 per group.
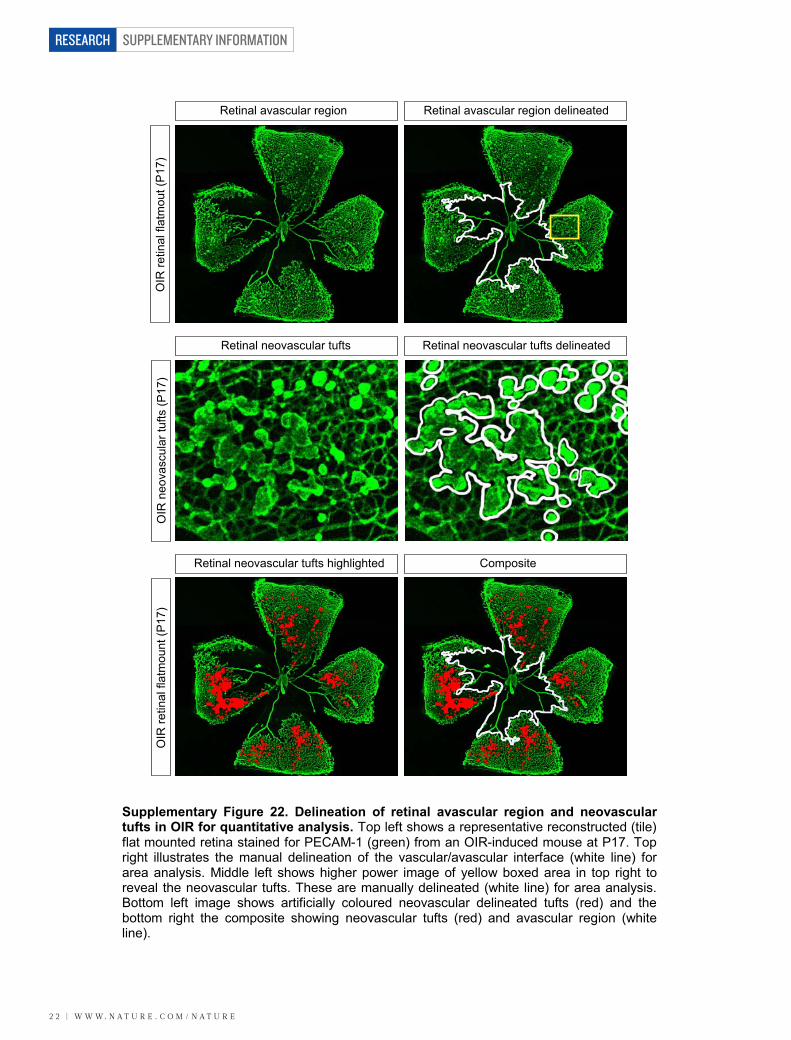
SUPPLEMENTARY INFORMATION
2 2 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
Retinal avascular region Retinal avascular region delineated
Retinal neovascular tufts Retinal neovascular tufts delineated
Retinal neovascular tufts highlighted Composite
OIR
retin
al fl
atm
out (
P17
)O
IR n
eova
scul
ar tu
fts (P
17)
OIR
retin
al fl
atm
ount
(P17
)
Supplementary Figure 22. Delineation of retinal avascular region and neovascular tufts in OIR for quantitative analysis. Top left shows a representative reconstructed (tile) flat mounted retina stained for PECAM-1 (green) from an OIR-induced mouse at P17. Top right illustrates the manual delineation of the vascular/avascular interface (white line) for area analysis. Middle left shows higher power image of yellow boxed area in top right to reveal the neovascular tufts. These are manually delineated (white line) for area analysis. Bottom left image shows artificially coloured neovascular delineated tufts (red) and the bottom right the composite showing neovascular tufts (red) and avascular region (white line).
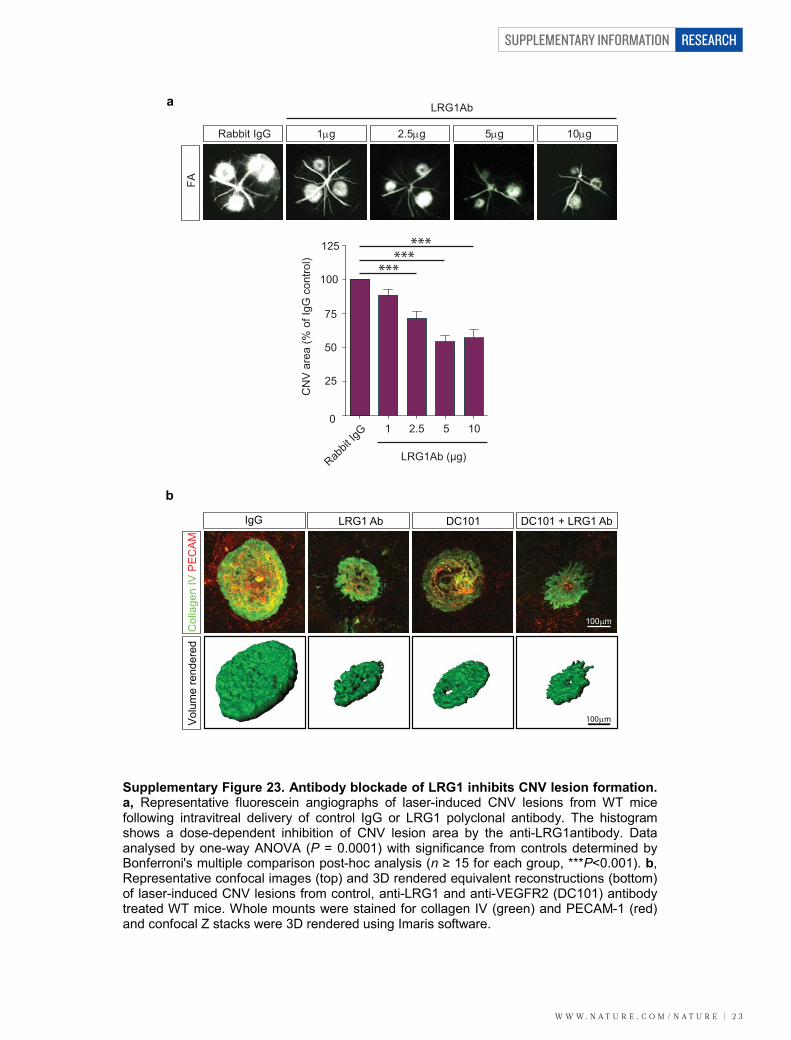
W W W. N A T U R E . C O M / N A T U R E | 2 3
SUPPLEMENTARY INFORMATION RESEARCH
DC101 DC101 + LRG1 AbIgG LRG1 Ab
b
Rabbit IgG
LRG1Ab
1g
a
125
100
75
50
25
0
CN
V a
rea
(% o
f IgG
con
trol)
Rabbit
IgG 2.51 5 10
******
Col
lage
n IV
PE
CA
M
100m
100m
Vol
ume
rend
ered
2.5g 5g 10g
FA
LRG1Ab (μg)
***
Supplementary Figure 23. Antibody blockade of LRG1 inhibits CNV lesion formation. a, Representative fluorescein angiographs of laser-induced CNV lesions from WT mice following intravitreal delivery of control IgG or LRG1 polyclonal antibody. The histogram shows a dose-dependent inhibition of CNV lesion area by the anti-LRG1antibody. Data analysed by one-way ANOVA (P = 0.0001) with significance from controls determined by Bonferroni's multiple comparison post-hoc analysis (n ≥ 15 for each group, ***P<0.001). b, Representative confocal images (top) and 3D rendered equivalent reconstructions (bottom) of laser-induced CNV lesions from control, anti-LRG1 and anti-VEGFR2 (DC101) antibody treated WT mice. Whole mounts were stained for collagen IV (green) and PECAM-1 (red) and confocal Z stacks were 3D rendered using Imaris software.
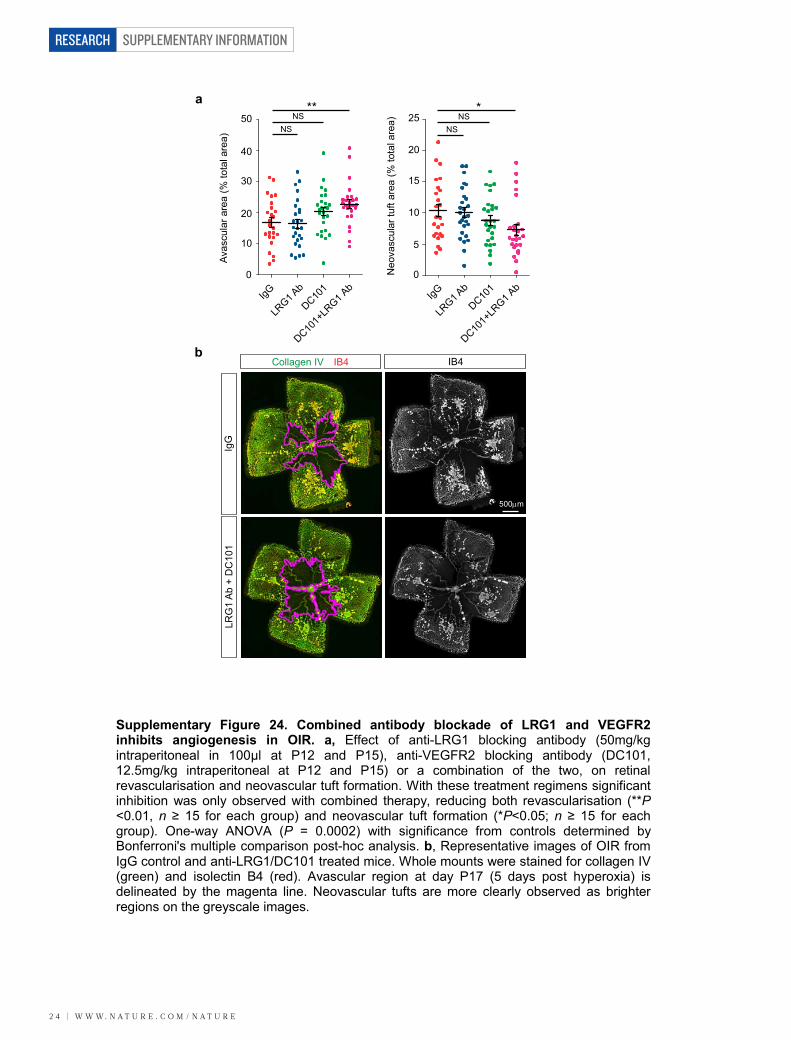
SUPPLEMENTARY INFORMATION
2 4 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
a
b
IgG
LRG1 A
b
DC101
DC101+
LRG1 A
b0
5
10
15
20
25
Neo
vasc
ular
tuft
area
(% to
tal a
rea)
0
10
20
30
40
50
Avas
cula
r are
a (%
tota
l are
a)
IgG
LRG1 A
b
DC101
DC101+
LRG1 A
b
Collagen IV IB4
IgG
LRG
1 A
b +
DC
101
IB4
500m
**NS
NS
*NS
NS
Supplementary Figure 24. Combined antibody blockade of LRG1 and VEGFR2 inhibits angiogenesis in OIR. a, Effect of anti-LRG1 blocking antibody (50mg/kg intraperitoneal in 100μl at P12 and P15), anti-VEGFR2 blocking antibody (DC101, 12.5mg/kg intraperitoneal at P12 and P15) or a combination of the two, on retinal revascularisation and neovascular tuft formation. With these treatment regimens significant inhibition was only observed with combined therapy, reducing both revascularisation (**P ˂0.01, n ≥ 15 for each group) and neovascular tuft formation (*P<0.05; n ≥ 15 for each group). One-way ANOVA (P = 0.0002) with significance from controls determined by Bonferroni's multiple comparison post-hoc analysis. b, Representative images of OIR from IgG control and anti-LRG1/DC101 treated mice. Whole mounts were stained for collagen IV (green) and isolectin B4 (red). Avascular region at day P17 (5 days post hyperoxia) is delineated by the magenta line. Neovascular tufts are more clearly observed as brighter regions on the greyscale images.
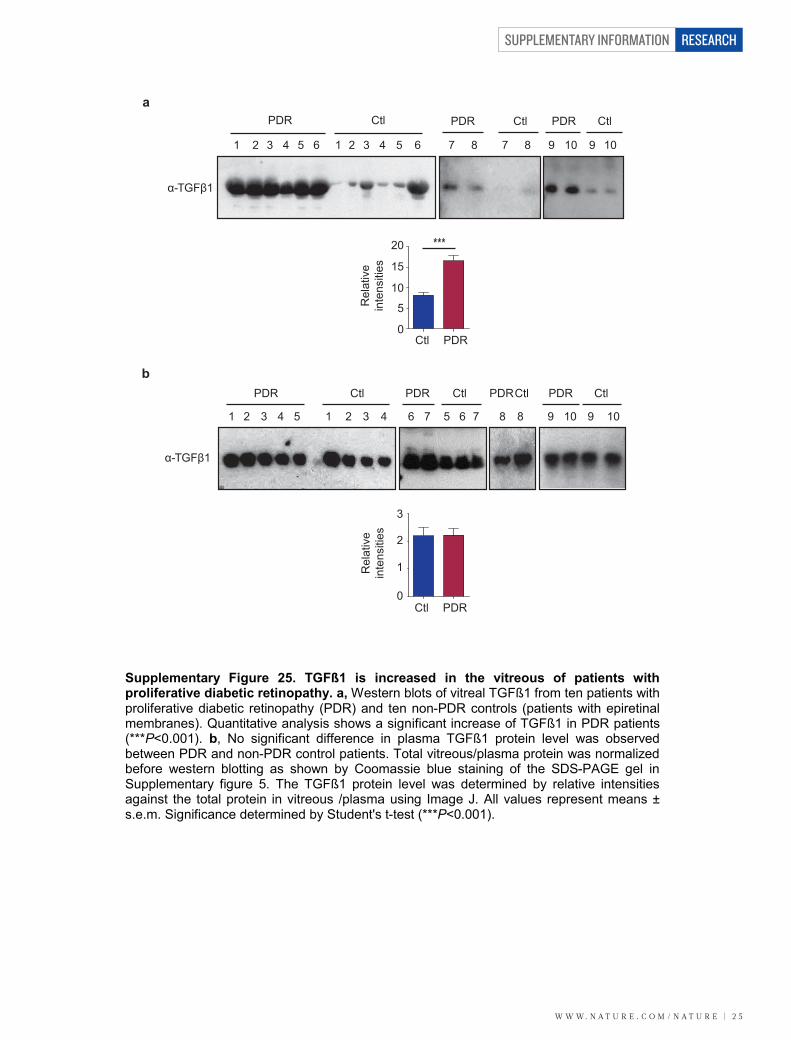
W W W. N A T U R E . C O M / N A T U R E | 2 5
SUPPLEMENTARY INFORMATION RESEARCH
a
α-TGFβ1
PDR Ctl PDR Ctl PDRCtl PDR Ctl
1 12 23 34 45 56 67 7 8 8 9 910 10
Rel
ativ
ein
tens
ities
Ctl PDR
1
0
3
2
α-TGFβ1
***
Rel
ativ
ein
tens
ities
Ctl PDR
15
10
5
0
20
PDR Ctl
1 12 23 34 45 56 6
PDR CtlPDR Ctl
7 78 8 99 10 10
b
Supplementary Figure 25. TGFß1 is increased in the vitreous of patients with proliferative diabetic retinopathy. a, Western blots of vitreal TGFß1 from ten patients with proliferative diabetic retinopathy (PDR) and ten non-PDR controls (patients with epiretinal membranes). Quantitative analysis shows a significant increase of TGFß1 in PDR patients (***P<0.001). b, No significant difference in plasma TGFß1 protein level was observed between PDR and non-PDR control patients. Total vitreous/plasma protein was normalized before western blotting as shown by Coomassie blue staining of the SDS-PAGE gel in Supplementary figure 5. The TGFß1 protein level was determined by relative intensities against the total protein in vitreous /plasma using Image J. All values represent means ± s.e.m. Significance determined by Student's t-test (***P<0.001).

SUPPLEMENTARY INFORMATION
2 6 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
Moc
k
ALK
5
ALK
1
EN
G
Moc
k
Moc
k
TβR
IIM
ock
LRG
1M
ock
WB:α-HA
WB:α-HA
WB:α-Myc
WB:α-V5
WB:α-His
LRG
1Ab
Rbt
IgG
IP: LRG1WB: α-TβRII
IP: LRG1WB: α-ALK1
IP: LRG1WB: α-ALK5
IP: LRG1WB: α-ENG
a
b
Supplementary Figure 26. Binding of LRG1 to TGFβ receptors and generation of peptide-tagged LRG1 and TGFβ receptor extracellular domains. a, Western blots showing that immunoprecipitation of LRG1 with polyclonal rabbit antibody from HUVEC lysates co-immunoprecipitates the TGFβ receptors TβRII, ALK1, ALK5 and endoglin (ENG). (Representative of n ≥ 3 for each assay). b, Western blot of peptide-tagged recombinant extracellular domains of TGFβ receptors TβRII (Myc-tagged), ALK1 (HA-tagged), ALK5 (HA-tagged) and ENG (V5-tagged) and His-tagged LRG1 in transfected HEK 293 cell medium. (Representative western blots; n ≥ 3 for each experiment).
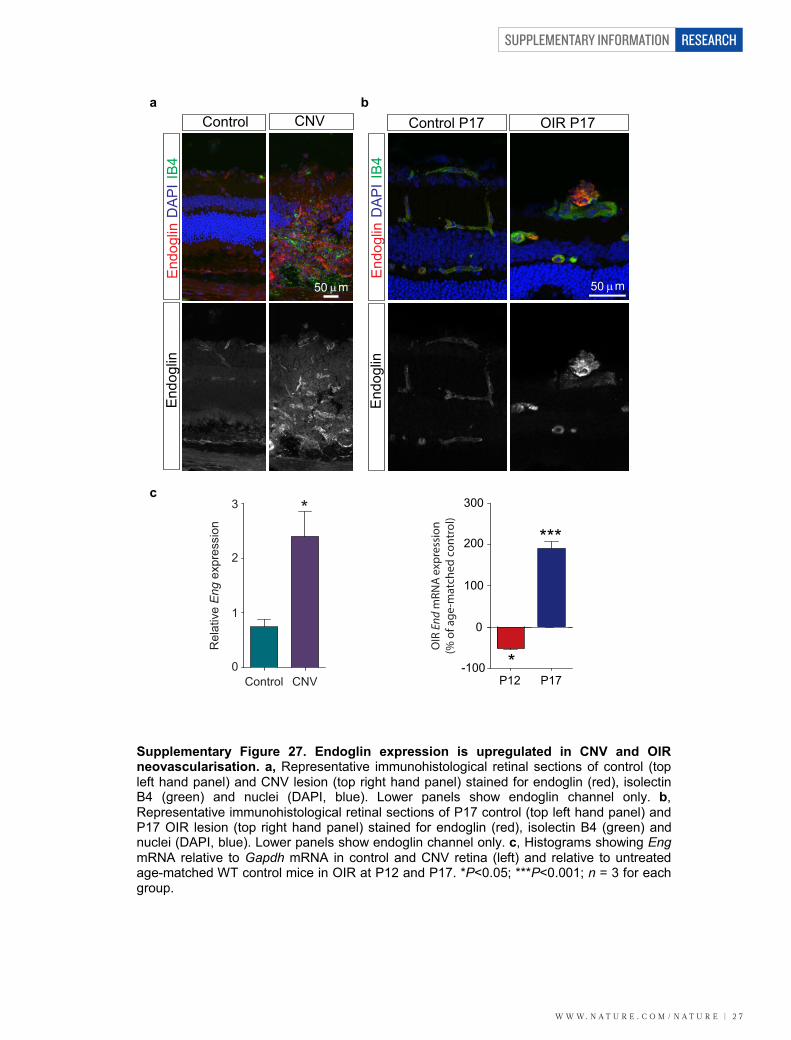
W W W. N A T U R E . C O M / N A T U R E | 2 7
SUPPLEMENTARY INFORMATION RESEARCH
CNVControl
3
2
1
0
Rel
ativ
e E
ng e
xpre
ssio
n
*
Control CNV
End
oglin
DA
PII
B4
50 m
End
oglin
Control P17 OIR P17
End
oglin
DA
PII
B4
End
oglin
OIR
End
mRN
A e
xpre
ssio
n(%
of a
ge-m
atch
ed c
ontr
ol)
P12 P17
300
200
100
0
-100 *
***
50 m
a b
c
Supplementary Figure 27. Endoglin expression is upregulated in CNV and OIR neovascularisation. a, Representative immunohistological retinal sections of control (top left hand panel) and CNV lesion (top right hand panel) stained for endoglin (red), isolectin B4 (green) and nuclei (DAPI, blue). Lower panels show endoglin channel only. b,Representative immunohistological retinal sections of P17 control (top left hand panel) and P17 OIR lesion (top right hand panel) stained for endoglin (red), isolectin B4 (green) and nuclei (DAPI, blue). Lower panels show endoglin channel only. c, Histograms showing EngmRNA relative to Gapdh mRNA in control and CNV retina (left) and relative to untreated age-matched WT control mice in OIR at P12 and P17. *P<0.05; ***P<0.001; n = 3 for each group.
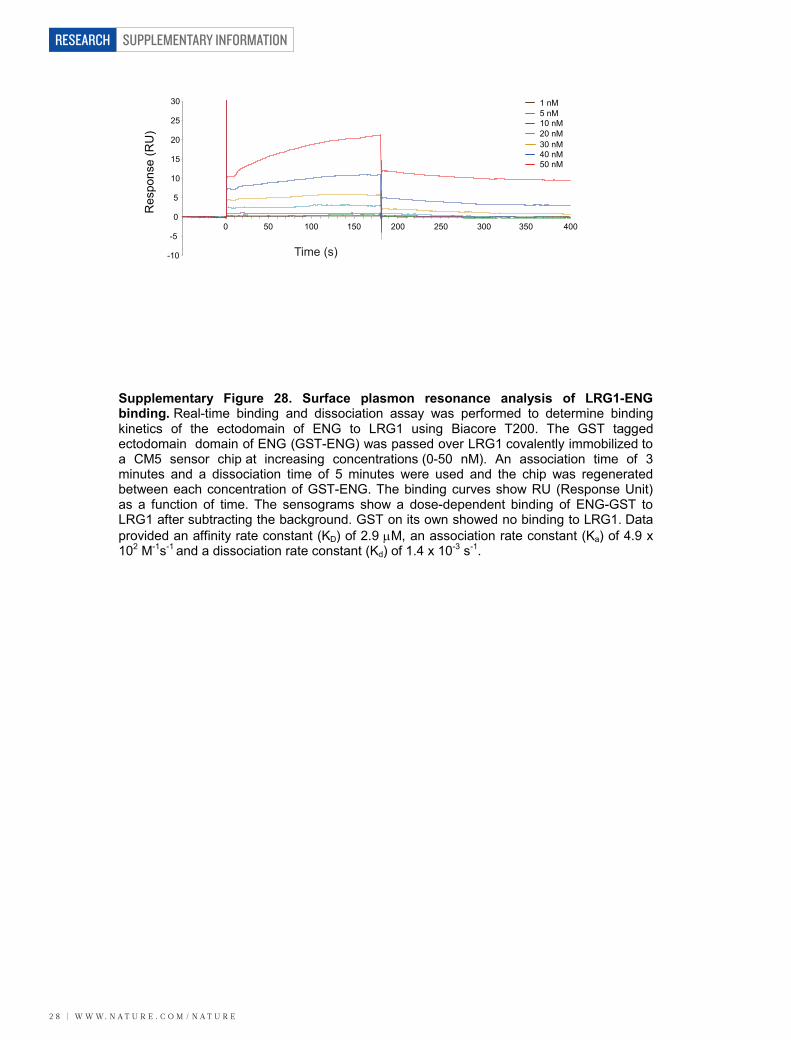
SUPPLEMENTARY INFORMATION
2 8 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
-10
-5
0
5
10
15
20
25
30
0 50 250200150 400350300100
1 nM
50 nM40 nM30 nM20 nM10 nM5 nM
Res
pons
e (R
U)
Time (s)
Supplementary Figure 28. Surface plasmon resonance analysis of LRG1-ENG binding. Real-time binding and dissociation assay was performed to determine binding kinetics of the ectodomain of ENG to LRG1 using Biacore T200. The GST tagged ectodomain domain of ENG (GST-ENG) was passed over LRG1 covalently immobilized to a CM5 sensor chip at increasing concentrations (0-50 nM). An association time of 3 minutes and a dissociation time of 5 minutes were used and the chip was regenerated between each concentration of GST-ENG. The binding curves show RU (Response Unit) as a function of time. The sensograms show a dose-dependent binding of ENG-GST to LRG1 after subtracting the background. GST on its own showed no binding to LRG1. Data provided an affinity rate constant (KD) of 2.9 M, an association rate constant (Ka) of 4.9 x 102 M-1s-1 and a dissociation rate constant (Kd) of 1.4 x 10-3 s-1.
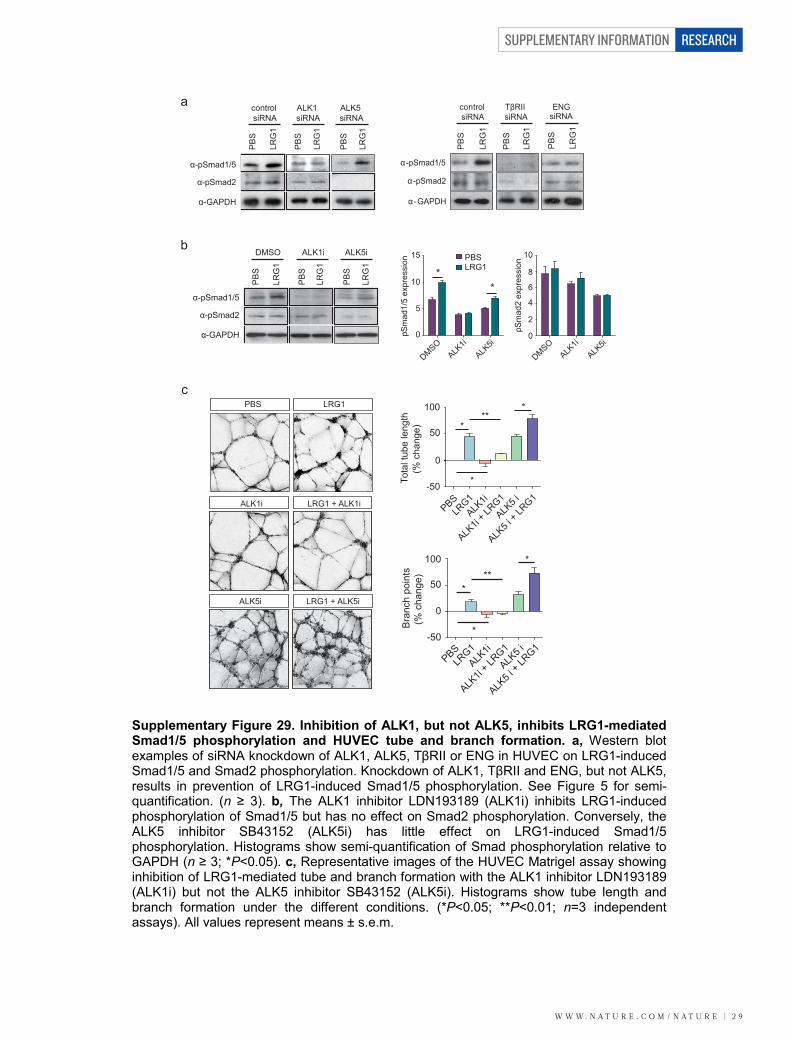
W W W. N A T U R E . C O M / N A T U R E | 2 9
SUPPLEMENTARY INFORMATION RESEARCH
α-pSmad1/5
α-GAPDH
PB
S
LRG
1
PB
S
LRG
1
PB
S
LRG
1
DMSO ALK1i ALK5i
α-pSmad2
bPBSLRG1
15
10
5
0pSm
ad1/
5 ex
pres
sion
DMSOALK
1i
ALK5i
DMSOALK
1i
ALK5i
pSm
ad2
expr
essi
on
8
10
2
0
4
6*
*
100
50
0
-50Tota
l tub
e le
ngth
(% c
hang
e) *
*
*
PBSALK
1i
ALK5 i
LRG1
ALK1i
+ LRG1
ALK5 i
+ LR
G1
**
100
50
0
-50
PBSALK
1i
ALK5 i
LRG1
ALK1i
+ LRG1
ALK5 i
+ LR
G1
*
*
* Bra
nch
poin
ts(%
cha
nge) **
PBS
ALK1i
LRG1
LRG1 + ALK1i
ALK5i LRG1 + ALK5i
c
control siRNA
ALK1 siRNA
ALK5 siRNA
-pSmad1/5
α-GAPDH
PB
S
LRG
1
PB
S
LRG
1
PB
S
LRG
1
α-pSmad2
control siRNA
TβRII siRNA
PB
S
LRG
1
PB
S
LRG
1
-pSmad1/5
GAPDH
-pSmad2
-
α
α
ENGsiRNA
PB
S
LRG
1
α α
a
Supplementary Figure 29. Inhibition of ALK1, but not ALK5, inhibits LRG1-mediated Smad1/5 phosphorylation and HUVEC tube and branch formation. a, Western blot examples of siRNA knockdown of ALK1, ALK5, TβRII or ENG in HUVEC on LRG1-induced Smad1/5 and Smad2 phosphorylation. Knockdown of ALK1, TβRII and ENG, but not ALK5, results in prevention of LRG1-induced Smad1/5 phosphorylation. See Figure 5 for semi-quantification. (n ≥ 3). b, The ALK1 inhibitor LDN193189 (ALK1i) inhibits LRG1-induced phosphorylation of Smad1/5 but has no effect on Smad2 phosphorylation. Conversely, the ALK5 inhibitor SB43152 (ALK5i) has little effect on LRG1-induced Smad1/5 phosphorylation. Histograms show semi-quantification of Smad phosphorylation relative to GAPDH (n ≥ 3; *P<0.05). c, Representative images of the HUVEC Matrigel assay showing inhibition of LRG1-mediated tube and branch formation with the ALK1 inhibitor LDN193189 (ALK1i) but not the ALK5 inhibitor SB43152 (ALK5i). Histograms show tube length and branch formation under the different conditions. (*P<0.05; **P<0.01; n=3 independent assays). All values represent means ± s.e.m.
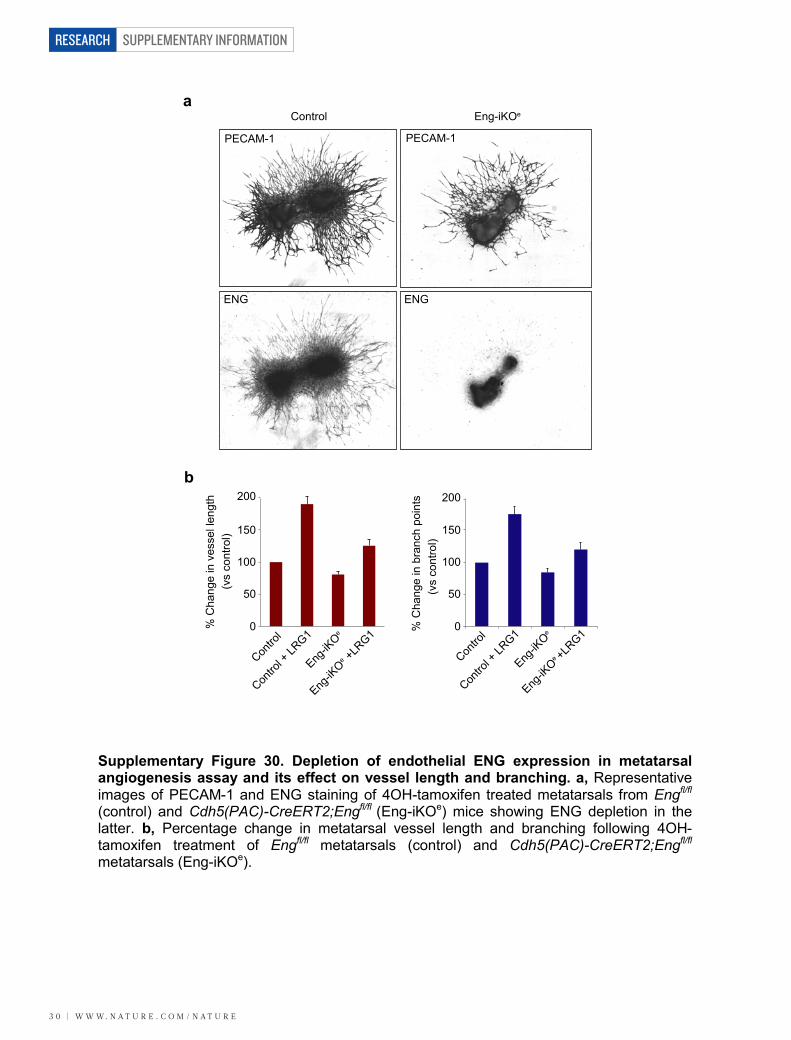
SUPPLEMENTARY INFORMATION
3 0 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
Contro
l
Contro
l + LR
G1
Eng-iK
Oe
Eng-iK
Oe +L
RG1% C
hang
e in
bra
nch
poin
ts
Contro
l
Contro
l + LR
G1
Eng-iK
Oe
Eng-iK
Oe +L
RG1
% C
hang
e in
ves
sel l
engt
h
0
50
100
150
200
(vs
cont
rol)
(vs
cont
rol)
a
b
Eng-iKOeControl
PECAM-1 PECAM-1
ENG ENG
0
50
100
150
200
Supplementary Figure 30. Depletion of endothelial ENG expression in metatarsal angiogenesis assay and its effect on vessel length and branching. a, Representative images of PECAM-1 and ENG staining of 4OH-tamoxifen treated metatarsals from Engfl/fl
(control) and Cdh5(PAC)-CreERT2;Engfl/fl (Eng-iKOe) mice showing ENG depletion in the latter. b, Percentage change in metatarsal vessel length and branching following 4OH-tamoxifen treatment of Engfl/fl metatarsals (control) and Cdh5(PAC)-CreERT2;Engfl/fl
metatarsals (Eng-iKOe).
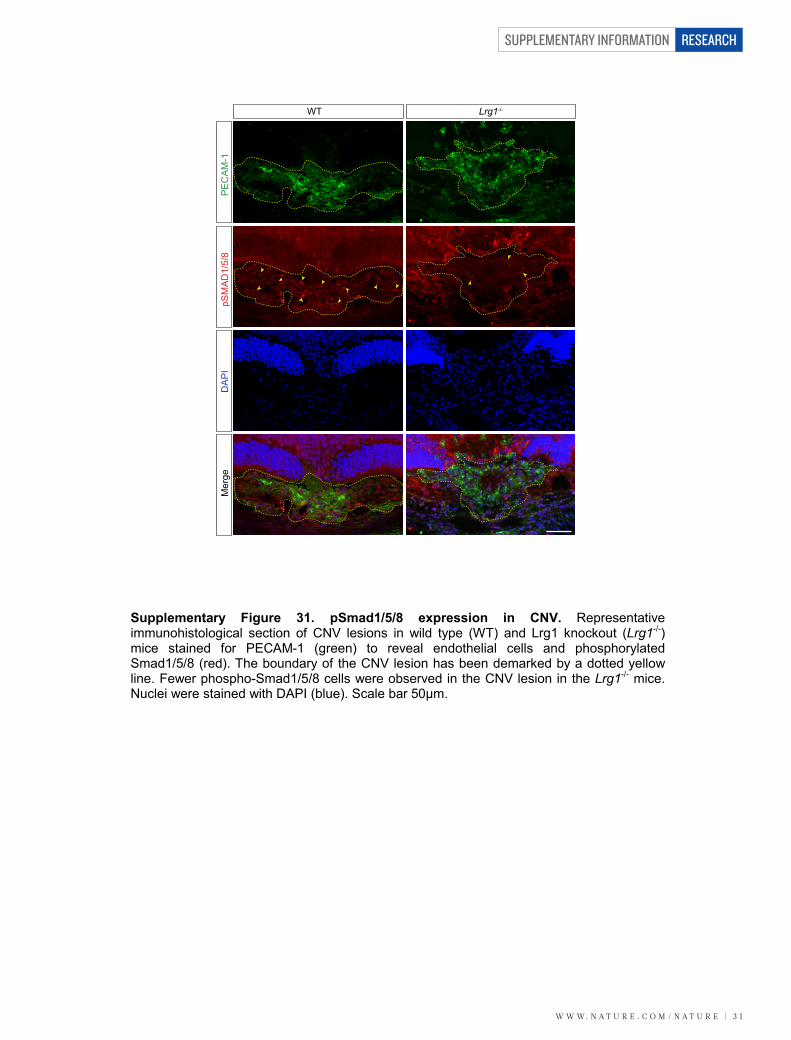
W W W. N A T U R E . C O M / N A T U R E | 3 1
SUPPLEMENTARY INFORMATION RESEARCH
WT Lrg1-/-
DA
PI
pSM
AD
1/5/
8P
EC
AM
-1M
erge
Supplementary Figure 31. pSmad1/5/8 expression in CNV. Representative immunohistological section of CNV lesions in wild type (WT) and Lrg1 knockout (Lrg1-/-) mice stained for PECAM-1 (green) to reveal endothelial cells and phosphorylated Smad1/5/8 (red). The boundary of the CNV lesion has been demarked by a dotted yellow line. Fewer phospho-Smad1/5/8 cells were observed in the CNV lesion in the Lrg1-/- mice. Nuclei were stained with DAPI (blue). Scale bar 50μm.
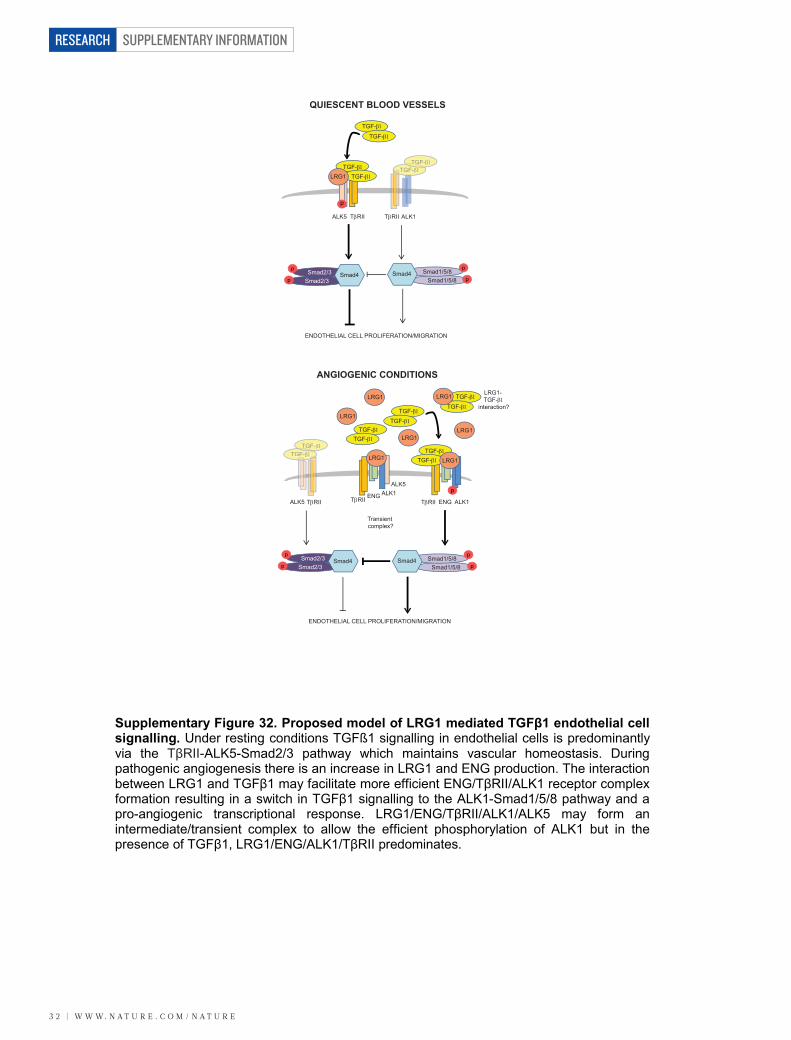
SUPPLEMENTARY INFORMATION
3 2 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
TGF-TGF-
p
p
p
p
p
p
p
p
p
p
QUIESCENT BLOOD VESSELS
ANGIOGENIC CONDITIONS
TGF-
TGF-
TGF-TGF-
TRII
LRG1
ALK5 TRII ALK1
Smad2/3Smad2/3
Smad4 Smad4 Smad1/5/8Smad1/5/8
ENDOTHELIAL CELL PROLIFERATION/MIGRATION
LRG1
LRG1
LRG1
LRG1 LRG1
TRIIALK5 TRIIALK1
TRII
ALK5
ENG ALK1ENG
Transient complex?
TGF-
TGF-TGF-
TGF-TGF-
TGF-
Smad2/3Smad2/3
Smad4 Smad4 Smad1/5/8Smad1/5/8
ENDOTHELIAL CELL PROLIFERATION/MIGRATION
LRG1
TGF-
TGF-TGF-TTGTG
TGF-
LRG1 LRG1- TGF-
interaction?
Supplementary Figure 32. Proposed model of LRG1 mediated TGFβ1 endothelial cell signalling. Under resting conditions TGFß1 signalling in endothelial cells is predominantly via the TβRII-ALK5-Smad2/3 pathway which maintains vascular homeostasis. During pathogenic angiogenesis there is an increase in LRG1 and ENG production. The interaction between LRG1 and TGFβ1 may facilitate more efficient ENG/TβRII/ALK1 receptor complex formation resulting in a switch in TGFβ1 signalling to the ALK1-Smad1/5/8 pathway and a pro-angiogenic transcriptional response. LRG1/ENG/TβRII/ALK1/ALK5 may form an intermediate/transient complex to allow the efficient phosphorylation of ALK1 but in the presence of TGFβ1, LRG1/ENG/ALK1/TβRII predominates.
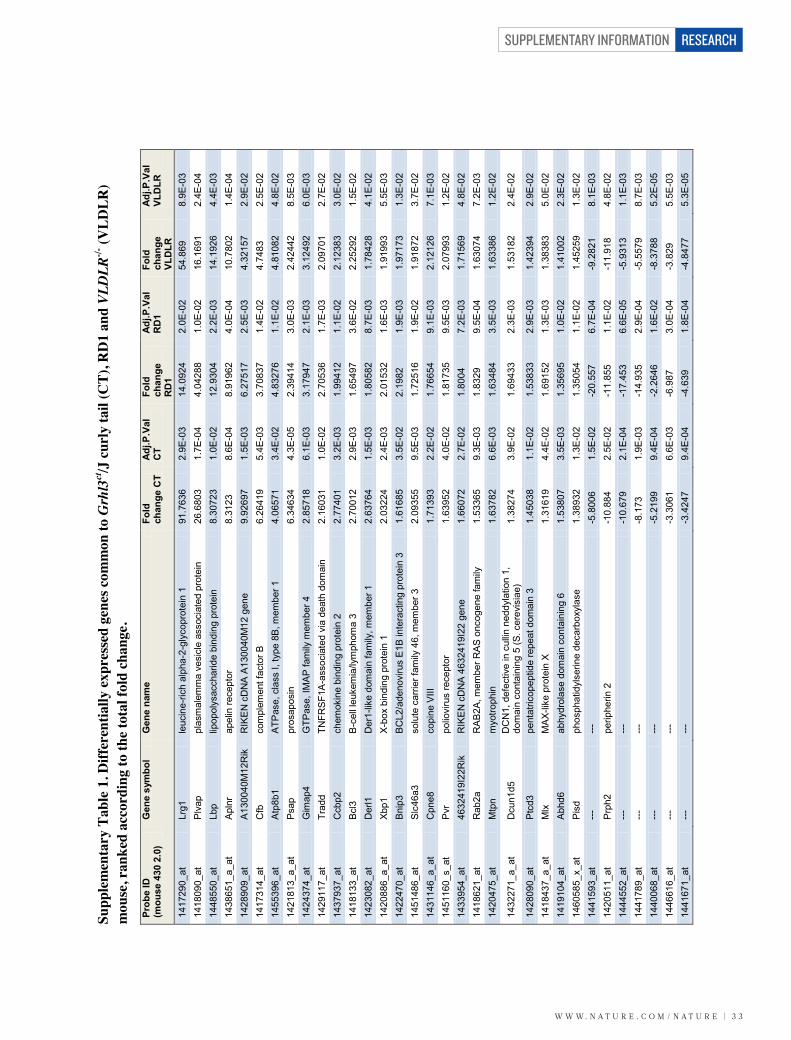
W W W. N A T U R E . C O M / N A T U R E | 3 3
SUPPLEMENTARY INFORMATION RESEARCH
Prob
e ID
(m
ouse
430
2.0
) G
ene
sym
bol
Gen
e na
me
Fold
ch
ange
CT
Adj.P
.Val
C
T Fo
ld
chan
ge
RD
1
Adj.P
.Val
R
D1
Fold
ch
ange
VL
DLR
Adj.P
.Val
VL
DLR
1417
290_
at
Lrg1
le
ucin
e-ric
h al
pha-
2-gl
ycop
rote
in 1
91
.763
6 2.
9E-0
3 14
.092
4 2.
0E-0
2 54
.869
8.
9E-0
3 14
1809
0_at
P
lvap
pl
asm
alem
ma
vesi
cle
asso
ciat
ed p
rote
in
26.6
803
1.7E
-04
4.04
288
1.0E
-02
16.1
691
2.4E
-04
1448
550_
at
Lbp
lipop
olys
acch
arid
e bi
ndin
g pr
otei
n 8.
3072
3 1.
0E-0
2 12
.930
4 2.
2E-0
3 14
.192
6 4.
4E-0
3 14
3865
1_a_
at
Apl
nr
apel
in re
cept
or
8.31
23
8.6E
-04
8.91
962
4.0E
-04
10.7
802
1.4E
-04
1428
909_
at
A13
0040
M12
Rik
R
IKE
N c
DN
A A
1300
40M
12 g
ene
9.92
697
1.5E
-03
6.27
517
2.5E
-03
4.32
157
2.9E
-02
1417
314_
at
Cfb
co
mpl
emen
t fac
tor B
6.
2641
9 5.
4E-0
3 3.
7083
7 1.
4E-0
2 4.
7483
2.
5E-0
2 14
5539
6_at
A
tp8b
1 A
TPas
e, c
lass
I, ty
pe 8
B, m
embe
r 1
4.06
571
3.4E
-02
4.83
276
1.1E
-02
4.81
082
4.8E
-02
1421
813_
a_at
P
sap
pros
apos
in
6.34
634
4.3E
-05
2.39
414
3.0E
-03
2.42
442
8.5E
-03
1424
374_
at
Gim
ap4
GTP
ase,
IMA
P fa
mily
mem
ber 4
2.
8571
8 6.
1E-0
3 3.
1794
7 2.
1E-0
3 3.
1249
2 6.
0E-0
3 14
2911
7_at
Tr
add
TNFR
SF1
A-a
ssoc
iate
d vi
a de
ath
dom
ain
2.16
031
1.0E
-02
2.70
536
1.7E
-03
2.09
701
2.7E
-02
1437
937_
at
Ccb
p2
chem
okin
e bi
ndin
g pr
otei
n 2
2.77
401
3.2E
-03
1.99
412
1.1E
-02
2.12
383
3.0E
-02
1418
133_
at
Bcl
3 B
-cel
l leu
kem
ia/ly
mph
oma
3 2.
7001
2 2.
9E-0
3 1.
6549
7 3.
6E-0
2 2.
2529
2 1.
5E-0
2 14
2308
2_at
D
erl1
D
er1-
like
dom
ain
fam
ily, m
embe
r 1
2.63
764
1.5E
-03
1.80
582
8.7E
-03
1.78
428
4.1E
-02
1420
886_
a_at
Xb
p1
X-bo
x bi
ndin
g pr
otei
n 1
2.03
224
2.4E
-03
2.01
532
1.6E
-03
1.91
993
5.5E
-03
1422
470_
at
Bni
p3
BC
L2/a
deno
viru
s E
1B in
tera
ctin
g pr
otei
n 3
1.61
685
3.5E
-02
2.19
82
1.9E
-03
1.97
173
1.3E
-02
1451
486_
at
Slc
46a3
so
lute
car
rier f
amily
46,
mem
ber 3
2.
0935
5 9.
5E-0
3 1.
7251
6 1.
9E-0
2 1.
9187
2 3.
7E-0
2 14
3114
6_a_
at
Cpn
e8
copi
ne V
III
1.71
393
2.2E
-02
1.76
654
9.1E
-03
2.12
126
7.1E
-03
1451
160_
s_at
P
vr
polio
viru
s re
cept
or
1.63
952
4.0E
-02
1.81
735
9.5E
-03
2.07
993
1.2E
-02
1433
954_
at
4632
419I
22R
ik
RIK
EN
cD
NA
463
2419
I22
gene
1.
6607
2 2.
7E-0
2 1.
8004
7.
2E-0
3 1.
7156
9 4.
8E-0
2 14
1862
1_at
R
ab2a
R
AB
2A, m
embe
r RA
S on
coge
ne fa
mily
1.
5336
5 9.
3E-0
3 1.
8329
9.
5E-0
4 1.
6307
4 7.
2E-0
3 14
2047
5_at
M
tpn
myo
troph
in
1.63
782
6.6E
-03
1.63
484
3.5E
-03
1.63
386
1.2E
-02
1432
271_
a_at
D
cun1
d5
DC
N1,
def
ectiv
e in
cul
lin n
eddy
latio
n 1,
do
mai
n co
ntai
ning
5 (S
. cer
evis
iae)
1.
3827
4 3.
9E-0
2 1.
6943
3 2.
3E-0
3 1.
5318
2 2.
4E-0
2
1428
090_
at
Ptc
d3
pent
atric
opep
tide
repe
at d
omai
n 3
1.45
038
1.1E
-02
1.53
833
2.9E
-03
1.42
394
2.9E
-02
1418
437_
a_at
M
lx
MA
X-lik
e pr
otei
n X
1.31
619
4.4E
-02
1.69
152
1.3E
-03
1.38
383
5.0E
-02
1419
104_
at
Abh
d6
abhy
drol
ase
dom
ain
cont
aini
ng 6
1.
5380
7 3.
5E-0
3 1.
3569
5 1.
0E-0
2 1.
4100
2 2.
3E-0
2 14
6058
5_x_
at
Pis
d ph
osph
atid
ylse
rine
deca
rbox
ylas
e 1.
3893
2 1.
3E-0
2 1.
3505
4 1.
1E-0
2 1.
4525
9 1.
3E-0
2 14
4159
3_at
---
---
-5
.800
6 1.
5E-0
2 -2
0.55
7 6.
7E-0
4 -9
.282
1 8.
1E-0
3 14
2051
1_at
P
rph2
pe
riphe
rin 2
-1
0.88
4 2.
5E-0
2 -1
1.85
5 1.
1E-0
2 -1
1.91
8 4.
8E-0
2 14
4455
2_at
---
---
-1
0.67
9 2.
1E-0
4 -1
7.45
3 6.
6E-0
5 -5
.931
3 1.
1E-0
3 14
4178
9_at
---
---
-8
.173
1.
9E-0
3 -1
4.93
5 2.
9E-0
4 -5
.557
9 8.
7E-0
3 14
4006
8_at
---
---
-5
.219
9 9.
4E-0
4 -2
.264
6 1.
6E-0
2 -8
.378
8 5.
2E-0
5 14
4661
6_at
---
---
-3
.306
1 6.
6E-0
3 -6
.987
3.
0E-0
4 -3
.829
5.
5E-0
3 14
4167
1_at
---
---
-3
.424
7 9.
4E-0
4 -4
.639
1.
8E-0
4 -4
.847
7 5.
3E-0
5
Supp
lem
enta
ry T
able
1. D
iffer
entia
lly e
xpre
ssed
gen
es c
omm
on to
Grh
l3ct/J
cur
ly ta
il (C
T), R
D1
and
VLD
LR-/-
(VLD
LR)
mou
se, r
anke
d ac
cord
ing
to th
e to
tal f
old
chan
ge.
SUPPLEMENTARY INFORMATION
3 4 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
Prob
e ID
(m
ouse
430
2.0
) G
ene
sym
bol
Gen
e na
me
Fold
ch
ange
CT
Adj.P
.Val
C
T Fo
ld
chan
ge
RD
1
Adj.P
.Val
R
D1
Fold
ch
ange
VL
DLR
Adj.P
.Val
VL
DLR
1445
746_
at
Eif4
h E
ukar
yotic
tran
slat
ion
initi
atio
n fa
ctor
4H
, m
RN
A (c
DN
A c
lone
MG
C:1
1689
IM
AG
E:39
6210
4)
-3.8
752
1.9E
-03
-4.5
091
6.7E
-04
-4.4
588
7.4E
-04
1458
068_
at
---
---
-3.4
222
1.1E
-02
-4.9
557
1.7E
-03
-3.0
991
3.7E
-02
1447
024_
at
---
---
-3.5
262
5.0E
-03
-4.1
03
1.6E
-03
-3.7
124
6.5E
-03
1457
477_
at
Mbn
l2
mus
cleb
lind-
like
2 -2
.456
1 3.
9E-0
2 -5
.953
4 9.
0E-0
4 -2
.921
8 4.
1E-0
2 14
5968
7_x_
at
---
---
-2.6
253
3.2E
-02
-5.0
14
1.6E
-03
-3.0
497
3.7E
-02
1442
548_
at
---
---
-3.1
449
1.2E
-02
-3.8
378
3.0E
-03
-3.3
423
1.9E
-02
1436
892_
at
Spr
ed2
spro
uty-
rela
ted,
EV
H1
dom
ain
cont
aini
ng 2
-2
.950
3 2.
4E-0
3 -4
.722
6 1.
8E-0
4 -2
.611
7 6.
8E-0
3 14
6056
7_at
R
fx7
regu
lato
ry fa
ctor
X, 7
-2
.818
3 5.
5E-0
3 -3
.544
4 1.
3E-0
3 -3
.880
6 1.
1E-0
3
1442
278_
at
Kdm
5b
Jum
onji,
AT
rich
inte
ract
ive
dom
ain
1B (R
bp2
like)
(Jar
id1b
), m
RN
A
-3.0
173
1.8E
-02
-4.1
552
3.0E
-03
-3.0
153
4.1E
-02
1429
113_
at
2900
092E
17R
ik
/// P
rrt2
RIK
EN
cD
NA
290
0092
E17
gen
e ///
pro
line-
rich
trans
mem
bran
e pr
otei
n 2
-3.3
018
2.6E
-02
-3.1
771
1.6E
-02
-3.5
922
4.4E
-02
1442
411_
at
---
---
-3.1
871
6.6E
-03
-3.2
468
3.2E
-03
-2.5
439
4.1E
-02
1442
735_
at
---
---
-2.7
302
2.0E
-02
-3.1
033
6.0E
-03
-2.7
454
4.6E
-02
1437
965_
at
Hea
tr1
HE
AT
repe
at c
onta
inin
g 1
-2.1
302
3.1E
-02
-3.8
366
1.2E
-03
-2.5
485
2.5E
-02
1458
798_
at
---
---
-2.8
396
2.1E
-04
-2.7
643
1.8E
-04
-2.2
725
7.4E
-04
1444
599_
at
Her
c4
hect
dom
ain
and
RLD
4
-3.2
621
9.4E
-04
-2.1
305
4.6E
-03
-2.3
084
8.6E
-03
1440
755_
at
---
---
-2.1
872
1.1E
-02
-2.8
937
1.6E
-03
-2.1
518
2.7E
-02
1446
954_
at
---
---
-2.1
791
8.6E
-04
-2.5
859
1.8E
-04
-2.4
363
1.1E
-04
1435
984_
at
Zfp4
0 Zi
nc fi
nger
pro
tein
40
(Zfp
40),
mR
NA
-2
.012
5 1.
4E-0
2 -1
.704
5 2.
6E-0
2 -3
.063
7 1.
1E-0
3 14
2628
7_at
A
txn7
at
axin
7
-1.8
399
1.0E
-02
-2.1
381
2.0E
-03
-1.9
469
1.2E
-02
1419
236_
at
Hel
b he
licas
e (D
NA
) B
-1.6
318
1.2E
-02
-2.4
282
3.8E
-04
-1.7
659
1.1E
-02
1428
996_
at
4833
426J
09R
ik
RIK
EN
cD
NA
483
3426
J09
gene
-1
.823
3.
2E-0
2 -1
.605
9 4.
7E-0
2 -2
.361
1.
0E-0
2 14
3297
8_at
90
3060
7L02
Rik
R
IKE
N c
DN
A 9
0306
07L0
2 ge
ne
-2.0
972
3.5E
-03
-1.8
484
4.8E
-03
-1.8
23
2.2E
-02
1442
071_
at
Abc
e1
ATP
-bin
ding
cas
sette
, sub
-fam
ily E
(OAB
P),
mem
ber 1
-1
.710
6 1.
4E-0
2 -2
.335
7 9.
3E-0
4 -1
.640
2 4.
9E-0
2
1445
438_
at
Ddh
d1
DD
HD
dom
ain
cont
aini
ng 1
-1
.798
5 6.
8E-0
3 -2
.130
5 1.
2E-0
3 -1
.695
9 2.
3E-0
2
1445
436_
at
BC
0598
42
PR
ED
ICTE
D: M
us m
uscu
lus
cDN
A s
eque
nce
BC
0598
42 (B
C05
9842
), m
RN
A
-2.0
196
1.8E
-03
-1.9
107
1.6E
-03
-1.6
305
2.3E
-02
1455
611_
at
Pia
s1
prot
ein
inhi
bito
r of a
ctiv
ated
STA
T 1
-1.8
771
1.8E
-03
-1.4
53
1.2E
-02
-1.6
163
1.3E
-02
1440
331_
at
Kds
r 3-
keto
dihy
dros
phin
gosi
ne re
duct
ase
-1.6
376
3.1E
-03
-1.4
473
6.7E
-03
-1.4
862
1.9E
-02
1442
443_
at
---
---
-1.4
444
2.4E
-02
-1.5
212
6.7E
-03
-1.5
421
2.3E
-02
1437
919_
at
Bdp
1 B
dou
ble
prim
e 1,
sub
unit
of R
NA
pol
ymer
ase
III tr
ansc
riptio
n in
itiat
ion
fact
or II
IB
-1.4
457
1.1E
-02
-1.4
95
3.7E
-03
-1.3
805
4.7E
-02
Supp
lem
enta
ry T
able
1 (c
ontin
ued)
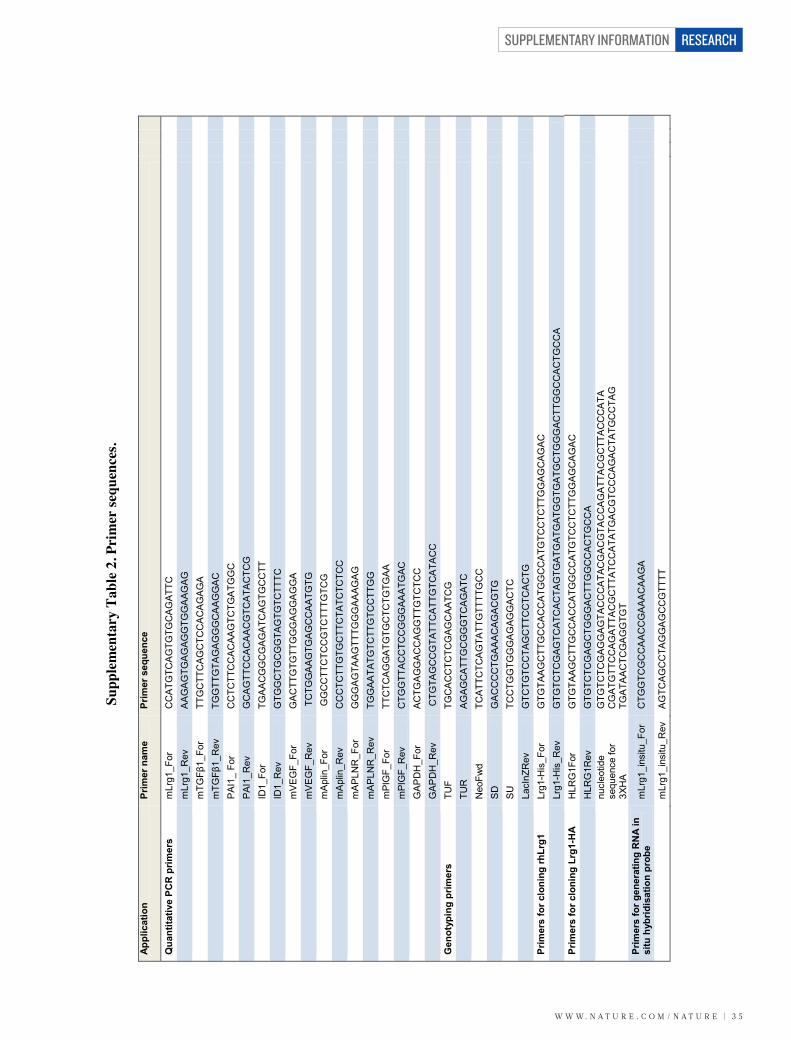
W W W. N A T U R E . C O M / N A T U R E | 3 5
SUPPLEMENTARY INFORMATION RESEARCH
Appl
icat
ion
Prim
er n
ame
Prim
er s
eque
nce
Qua
ntita
tive
PCR
prim
ers
mLr
g1_F
or
CC
ATG
TCA
GTG
TGC
AG
ATTC
m
Lrg1
_Rev
A
AG
AG
TGA
GAG
GTG
GA
AGA
G
mTG
Fβ1_Fo
r TT
GC
TTC
AGC
TCC
AC
AG
AG
A
mTG
Fβ1_Rev
TG
GTT
GTA
GA
GG
GC
AA
GG
AC
P
AI1_
For
C
CTC
TTC
CA
CAA
GTC
TGA
TGG
C
PAI
1_R
ev
GC
AG
TTC
CA
CA
AC
GTC
ATA
CTC
G
ID1_
For
TGA
AC
GG
CG
AG
ATC
AGTG
CC
TT
ID1_
Rev
G
TGG
CTG
CG
GTA
GTG
TCTT
TC
mVE
GF_
For
GA
CTT
GTG
TTG
GG
AG
GA
GG
A
m
VEG
F_R
ev
TC
TGG
AA
GTG
AG
CC
AA
TGTG
m
Apl
in_F
or
GG
CC
TTC
TCC
GTC
TTTG
TCG
m
Apl
in_R
ev
CC
CTC
TTG
TGC
TTC
TATC
TCTC
C
mAP
LNR
_For
G
GG
AG
TAAG
TTTG
GG
AAAG
AG
m
APLN
R_R
ev
TGG
AATA
TGTC
TTG
TCC
TTG
G
mP
lGF_
For
TTC
TCA
GG
ATG
TGC
TCTG
TGAA
m
PlG
F_R
ev
CTG
GTT
AC
CTC
CG
GG
AAA
TGAC
G
AP
DH
_For
A
CTG
AG
GA
CC
AGG
TTG
TCTC
C
GA
PD
H_R
ev
CTG
TAG
CC
GTA
TTC
ATTG
TCA
TAC
C
G
enot
ypin
g pr
imer
s TU
F TG
CA
CC
TCTC
GAG
CA
ATC
G
TUR
A
GA
GC
ATT
GC
GG
GTC
AG
ATC
N
eoFw
d TC
ATT
CTC
AG
TATT
GTT
TTG
CC
S
D
GA
CC
CC
TGAA
AC
AGA
CG
TG
SU
TC
CTG
GTG
GG
AG
AGG
AC
TC
LacI
nZR
ev
GTC
TGTC
CTA
GC
TTC
CTC
AC
TG
Pr
imer
s fo
r clo
ning
rhLr
g1
Lrg1
-His
_For
G
TGTA
AG
CTT
GC
CA
CC
ATG
GC
CA
TGTC
CTC
TTG
GA
GC
AG
AC
Lr
g1-H
is_R
ev
GTG
TCTC
GAG
TCAT
CA
CTA
GTG
ATG
ATG
ATG
GTG
ATG
CTG
GG
ACTT
GG
CC
AC
TGC
CA
Pr
imer
s fo
r clo
ning
Lrg
1-H
A
HLR
G1F
or
GTG
TAA
GC
TTG
CC
AC
CA
TGG
CC
ATG
TCC
TCTT
GG
AG
CA
GA
C
HLR
G1R
ev
GTG
TCTC
GAG
CTG
GG
AC
TTG
GC
CA
CTG
CC
A
nu
cleo
tide
sequ
ence
for
3XH
A
GTG
TCTC
GAG
GA
GTA
CC
CAT
ACG
AC
GTA
CC
AG
ATT
AC
GC
TTA
CC
CA
TA
CG
ATG
TTC
CA
GAT
TAC
GC
TTAT
CC
ATA
TGA
CG
TCC
CA
GA
CTA
TGC
CTA
G
TGA
TAA
CTC
GA
GG
TGT
Prim
ers
for g
ener
atin
g R
NA
in
situ
hyb
ridis
atio
n pr
obe
mLr
g1_i
nsitu
_For
C
TGG
TCG
CC
AA
CC
GA
AAC
AAG
A
m
Lrg1
_ins
itu_R
ev
AG
TCA
GC
CTA
GG
AG
CC
GTT
TT
Supp
lem
enta
ry T
able
2. P
rim
er se
quen
ces.
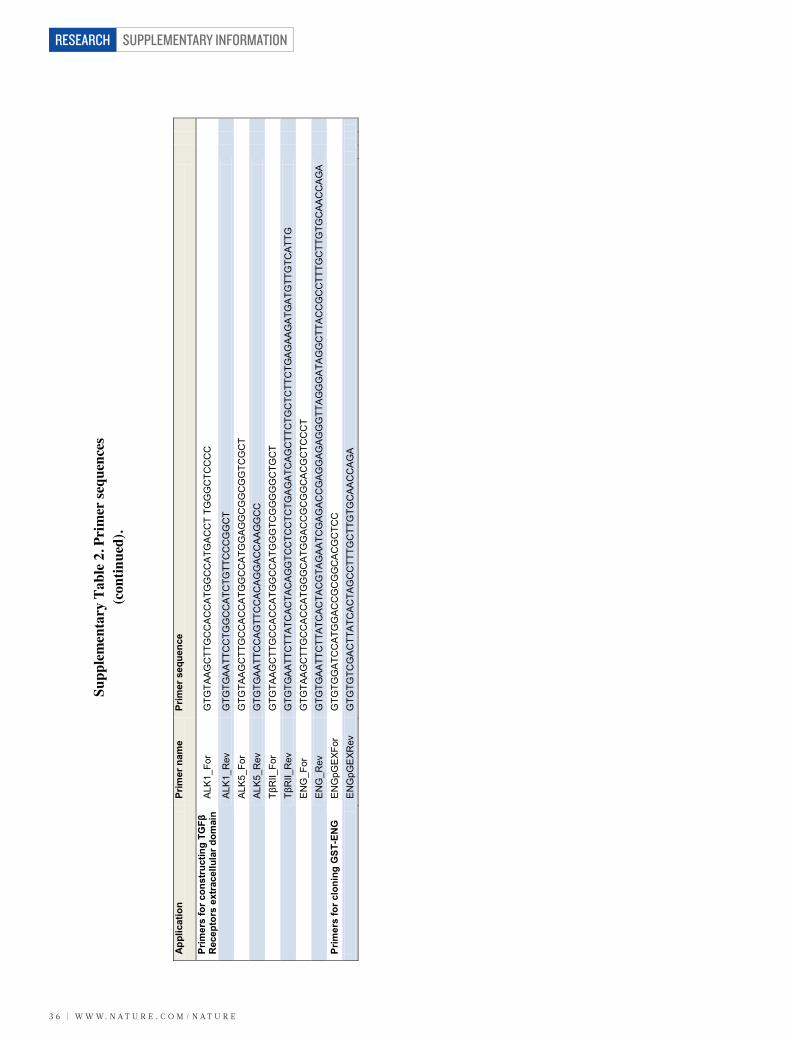
SUPPLEMENTARY INFORMATION
3 6 | W W W. N A T U R E . C O M / N A T U R E
RESEARCH
Appl
icat
ion
Prim
er n
ame
Prim
er s
eque
nce
Prim
ers
for c
onst
ruct
ing
TGFβ
R
ecep
tors
ext
race
llula
r dom
ain
ALK
1_Fo
r G
TGTA
AG
CTT
GC
CA
CC
ATG
GC
CA
TGA
CC
T TG
GG
CTC
CC
C
A
LK1_
Rev
G
TGTG
AA
TTC
CTG
GC
CA
TCTG
TTC
CC
GG
CT
ALK
5_Fo
r G
TGTA
AG
CTT
GC
CA
CC
ATG
GC
CA
TGG
AGG
CG
GC
GG
TCG
CT
ALK
5_R
ev
GTG
TGA
ATT
CC
AG
TTC
CA
CA
GG
AC
CA
AG
GC
C
TβRII_Fo
r G
TGTA
AG
CTT
GC
CA
CC
ATG
GC
CA
TGG
GTC
GG
GG
GC
TGC
T
Tβ
RII_Rev
G
TGTG
AA
TTC
TTA
TCA
CTA
CA
GG
TCC
TCC
TCTG
AG
ATC
AGC
TTC
TGC
TCTT
CTG
AG
AAG
ATG
ATG
TTG
TCA
TTG
E
NG
_For
G
TGTA
AG
CTT
GC
CA
CC
ATG
GG
CA
TGG
AC
CG
CG
GC
AC
GC
TCC
CT
EN
G_R
ev
GTG
TGA
ATT
CTT
ATC
AC
TAC
GTA
GAA
TCG
AG
AC
CG
AG
GAG
AG
GG
TTA
GG
GA
TAG
GC
TTA
CC
GC
CTT
TGC
TTG
TGC
AAC
CA
GA
Prim
ers
for c
loni
ng G
ST-E
NG
E
NG
pGE
XFor
G
TGTG
GA
TCC
ATG
GA
CC
GC
GG
CA
CG
CTC
C
EN
GpG
EXR
ev
GTG
TGTC
GA
CTT
ATC
AC
TAG
CC
TTTG
CTT
GTG
CAA
CC
AG
A
Supp
lem
enta
ry T
able
2. P
rim
er se
quen
ces
(con
tinue
d).