localization of the voltage-dependent anion channel-1 ca 2+-binding sites
TRANSCRIPT

A
tcactVMtlt©
K
1
ttMpt[
iarbn
0d
Cell Calcium 41 (2007) 235–244
Localization of the voltage-dependent anionchannel-1 Ca2+-binding sites
Adrian Israelson, Salah Abu-Hamad, Hilal Zaid,Edna Nahon, Varda Shoshan-Barmatz ∗
Department of Life Sciences and The Zlotowski Center for Neuroscience, Ben-Gurion University of the Negev, Beer-Sheva, Israel
Received 6 May 2006; received in revised form 9 June 2006; accepted 12 June 2006Available online 22 August 2006
bstract
Photoreactive azido ruthenium (AzRu) has been recently shown to specifically interact with Ca2+-binding proteins and to strongly inhibitheir Ca2+-dependent activities. Upon UV irradiation, AzRu can bind covalently to such proteins. In this study, AzRu was used to localize andharacterize Ca2+-binding sites in the voltage-dependent anion channel (VDAC). AzRu decreased the conductance of VDAC reconstituted intobilayer while Ca2+, in the presence of 1 M NaCl, but not Mg2+, prevented this effect. AzRu had no effect on mutated E72Q- or E202Q-VDAC1onductance, and [103Ru]AzRu labeled native but not E72Q-VDAC1, suggesting that these residues are required for AzRu interaction withhe VDAC Ca2+-binding site(s). AzRu protected against apoptosis induced by over-expression of native but not E72Q- or E202Q- murineDAC1 in T-REx-293 cells depleted of endogenous hVDAC1. Chymotrypsin and trypsin digestion of AzRu-labeled VDAC followed byALDI-TOF analysis revealed two AzRu-bound peptides corresponding to E72- and E202-containing sequences. These results suggest that
2+
he VDAC Ca -binding site includes E72 and E202, located, according to a proposed VDAC1 topology model, on two distinct cytosolicoops. Furthermore, AzRu protection against apoptosis involves interaction with these residues. Photoreactive AzRu represents an importantool for identifying novel Ca2+-binding proteins and localizing their Ca2+-binding sites.2006 Elsevier Ltd. All rights reserved.
sites; A
ppn(dp
eywords: Voltage-dependent anion channel (VDAC); Porin; Ca2+-binding
. Introduction
Intramitochondrial Ca2+ modulates critical enzymes ofhe TCA cycle, fatty acid oxidation, amino acid catabolism,he F1-ATPase and the adenine nucleotide translocase [1].
itochondria also modulate cytosolic Ca2+ transients or
ulses [2], and play a role in apoptosis through the induc-ion of the mitochondrial permeability transition (PT) pore3]. Mitochondria contain several different systems for trans-Abbreviations: AzRu, azido ruthenium; OMM and IMM, outer andnner mitochondrial membranes; PLB, plannar lipid bilayer; PTP, perme-biliti transition pore; Ru360, ruthenium amine binuclear complex; RuR,uthenium red; SR, sarcoplasmic reticulum; Tricine, N-[2-hydroxy-1,1-is(hydroxy-methyl)-ethyl]-glycine; VDAC, voltage-dependent anion chan-el∗ Corresponding author. Tel.: +972 8 6461336; fax: +972 8 6472992.
E-mail address: [email protected] (V. Shoshan-Barmatz).
tepr[
ttaig
143-4160/$ – see front matter © 2006 Elsevier Ltd. All rights reserved.oi:10.1016/j.ceca.2006.06.005
zido ruthenium (AzRu); Ruthenium red (RuR); Apoptosis
orting Ca2+ including the ruthenium red-sensitive uni-orter and Na+-dependent- and independent-efflux mecha-isms [4]. In addition, recently, a type 1 ryanodine receptormRyR)/Ca2+ release channel, has been identified in car-iac mitochondria [5]. All of these systems mediate trans-ort of Ca2+ across the inner mitochondrial membrane, yethese transport systems has not been identified [4,6]. Sev-ral studies have assigned outer mitochondrial membranerotein, the voltage-dependent anion channel (VDAC), aole in the transport of Ca2+ across the outer membrane7–9].
Mitochondrial Ca2+ overload appears to induce both apop-otic and necrotic cell death [10,11]. Mitochondria respond
o an apoptotic signal by opening the mitochondrial perme-bility transition pore (PTP), a pathway spanning both thenner and outer mitochondrial membranes. PTP was sug-ested to be composed of VDAC, located at the OMM,
2 l Calciu
tilninoasVToat[maeE[fpa[y
tstChsph[ssbasrab
sthutaticisa
tVrlt
2
2
ps(NpnBaorhcfap
2
smGnmogsGGdCGm
2
2
(C 2
36 A. Israelson et al. / Cel
he adenine nucleotide translocase (ANT), located at thenner mitochondrial membrane (IMM), and cyclophilin D,ocated at the matrix [12,13]. The Ca2+-binding compo-ent regulating apoptosis and PTP opening has not yet beendentified, although Ca2+-mediated modulation of the ade-ine nucleotide translocase has been suggested [14]. On thether hand, accumulating evidence suggests that VDAC haskey role in mitochondria-mediated apoptosis (for review
ee Refs. [9,15]) and several lines of evidence indicate thatDAC possesses divalent cation-binding sites: (1) La3+ andb3+ induce VDAC channel closure when assayed in singler multichannel studies [7,9], (2) Ruthenium red (RuR) [7]nd ruthenium amine binuclear complex (Ru360) [8], knowno specifically interact with several Ca2+-binding proteins7,16], induce VDAC channel closure in a time-dependentanner and stabilize the channel in a completely closed state,
nd (3) The inhibitory effect of RuR is prevented by the pres-nce of CaCl2, but is re-established by chelating Ca2+ withGTA, suggesting RuR interaction with a Ca2+-binding site
7]. Recently, the regulation of VDAC gating by Ca2+ and theunction of [Ca2+] as a regulator of VDAC activity, thereby,roviding a novel mechanism for the control of OMM perme-bility to ions and small molecules have been demonstrated17]. The nature of the Ca2+-binding site(s) in VDAC is aset unknown.
Although Ca2+ interacts with a large number of pro-eins, the variety of configurations assumed by Ca2+-bindingites is rather limited. Three structural modules designedo specifically bind Ca2+ are known: the annexin fold, the-2 domain and the well-characterized EF-hand motif. EF-and-containing Ca2+-binding proteins are the best under-tood Ca2+-binding proteins in terms of their organization,acking and response to Ca2+-binding [18]. A given EF-and protein may contain from one to six EF-hand motifs19]. In contrast to the relatively complex EF-hand structure,ome proteins, such as calsequestrin, possess Ca2+-bindingites thought to comprise acidic residues [20]. Other Ca2+-inding motifs remain much less-well described. The primarymino acid sequence of VDAC1, as deduced from its cDNAequence, contains no features of the EF-hand structure, cor-esponding to a high-affinity Ca2+-binding site. However,spartate/glutamate residues are potential low-affinity Ca2+-inding sites.
Despite advances in defining Ca2+-binding proteins, con-iderable experimental difficulties still remain in the localiza-ion of their Ca2+-binding sites. Recently, a novel approachas been developed for such localization, involving these of a photoreactive reagent synthesized in our labora-ory, azido ruthenium (AzRu) [21]. AzRu specifically inter-cts with Ca2+-binding proteins and can bind irreversiblyo Ca2+-binding sites upon UV irradiation. AzRu stronglynhibited the activities of a variety of Ca2+-dependent pro-
esses while having no effect on Ca2+-independent activ-ties [21]. Radioactive [103Ru]AzRu bound covalently andpecifically to Ca2+-binding proteins [21]. Thus, since AzRund Ca2+ share protein-binding sites, AzRu, as a photoreac-(sat
m 41 (2007) 235–244
ive reagent, was employed to identify Ca2+-binding sites inDAC1. The results allowed us to identify two glutamate
esidues E72 and E202, located in two different cytosolicoops, that may form the VDAC Ca2+-binding site(s), or parthereof.
. Experimental
.1. Materials
ATP, CM-cellulose, n-decane, Hepes, leupeptin, mannitol,henylmethyl sulfonyl fluoride (PMSF), soybean asolectin,ucrose, Tris and Triton X-100 were purchased from SigmaSt. Louis, MO). [45Ca] and [103Ru] were purchased fromEN Life Science (Boston, MA). Sephadex LH-20 wasurchased from Amersham Biosciences (Uppsala, Sweden).-Octyl-�-d-glucopyranoside (�-OG) was obtained fromachem AG (Bubendorf, Switzerland). Lauryl-(dimethyl)-mineoxide (LDAO) and ruthenium red (98% pure) werebtained from Fluka (Buchs, Switzerland). Chloroform anduthenium chloride were purchased from Aldrich (Stein-eim, Germany). Hydroxyapatite (Bio-Gel HTP) was pur-hased from Bio-Rad Laboratories (Hercules, CA) and Celiterom Merck (Darmstadt, Germany). Ru360 was synthesizedccording to Ying et al. [22]. AzRu and Az103Ru were pre-ared as previously described [21].
.2. Plasmids and site-directed mutagenesis
mVDAC1 cDNA (obtained from W.J. Craigen, Univer-ity of Houston, Texas, USA) was subcloned into plas-id pEGFP-N1 (Clontech) for construction of a VDAC1-FP- or VDAC1-expressing mammalian vector with aeomycin resistance gene serving as marker. Site-directedutagenesis of mVDAC1 was carried out in vitro by
verlapping PCR amplification. The mutated mVDAC1enes were constructed using the T7 and T3 univer-al primers, together with the following primers: 5′-ACGTTTACACAGAAGTGGAAC-3′ (forward) and 5′-TTCCACTTCTGTGTAAACGTC-3′ (reverse), to intro-uce the E72Q mutation and 5′-GAAGTTGCAGACTG-TGTCAATCTC-3′ (forward) and 5′-GCGAGATTGACA-CAGTCTGCAAC-3′ (reverse), to introduce the E202Qutation. All constructs were confirmed by sequencing.
.3. Cell culture
.3.1. T-REx-293 cellsA transformed primary human embryonal kidney cell line
Invitrogen) grown under an atmosphere of 95% air and 5%O in DMEM supplemented with 10% fetal calf serum
FCS), 2 mM l-glutamine, 1000 U/ml penicillin, 1 mg/mltreptomycin and 5 �g/ml blasticidin. Other cell lines usedre stably transfected derivatives of T-REx-293 that expresshe tetracycline (Tet) repressor.

l Calciu
2
p0
2
ppmcp
2
wp[mviyo(Mcp
2s
wi1asor
2
feft
2
w2iP
wMtMtw
2
fohcO
2
(ww(pa(sewu(wctuww2
2
l[7[tC
2i
A. Israelson et al. / Cel
.3.2. hVDAC1-shRNA T-REx-293 cellsT-REx-293 cells stably transfected with the pSUPERretro
lasmid encoding shRNA targeting hVDAC1 and grown with.5 �g/ml puromycin and 5 �g/ml blasticidin.
.3.3. pc-mVDAC1-hVDAC1-shRNA T-REx-293 cellshVDAC1-shRNA-T-REx-293 cells were transfected with
lasmid mVDAC1, E72Q-mVDAC1 or E202Q-mVDAC1-cDNA4/TO respectively, expressing mVDAC1, E72Q-VDAC1 or E202Q-mVDAC1 under the control of tetra-
ycline. Cells were grown with 200 �g/ml zeocin, 0.5 �g/mluromycin and 5 �g/ml blasticidin.
.4. Expression of mVDAC1 in yeast
The expression of native and mutated murine VDAC genesas conducted in the porinless Saccharomyces cerevisiaeor1- mutant strain M22-2 (MATa, lys2 his4 trp1 ade2 ura3)23] under the control of the yeast porin1 (YVDAC1) pro-oter in a low-copy number plasmid (pSEYC58). S. cere-
isiae M22-2 and its evolved mutant were cultured at 30 ◦Cn selective minimal medium containing ammonium sulfate,east nitrogen base, the required amino acids and 2% glucoser in rich medium containing 1% yeast extract, 2% peptoneYP) with 2% lactate (YPL) or glucose (YPD) at pH 5.5. Yeast
22-2 expressing either native or mutated murine VDAC1ultured in medium containing glucose were used for VDACurification from isolated mitochondria.
.5. Acridine orange (AcOr)/ethidium bromide (EtBr)taining of cells
To determine cell viability, cells were subjected to stainingith 100 �g/ml acridine orange (AcOr) and 100 �g/ml ethid-
um bromide (EtBr) in PBS [24]. Cells were centrifuged at500 × g for 5 min at room temperature and resuspended in25 �l of complete medium, to which 2 �l of AcOr/EtBr
olution were added. The cells were then visualized by flu-rescence microscopy (Olympus IX51) and images wereecorded using an Olympus DP70 camera and a SWB filter.
.6. Membrane preparation
Sarcoplasmic reticulum (SR) membranes were preparedrom rabbit fast twitch skeletal muscle as described by Saitot al. [25]. Mitochondria were isolated from rat liver [7] androm yeast [26] as described previously. Protein concentra-ions were determined by the Lowry procedure [27].
.7. Ca2+ accumulation
Ca2+ uptake by freshly prepared mitochondria (0.5 mg/ml)
as assayed for 1 min at 30 ◦C in the presence of25 mM mannitol, 75 mM sucrose, 120 �M CaCl2 (contain-ng 3 × 104 cpm/nmol [45Ca2+]), 5 mM succinate, 0.1 mMi and 5 mM Hepes/KOH, pH 7.0 [7]. Ca2+ uptake by SRtar
m 41 (2007) 235–244 237
as assayed for 2 min in the presence of 0.1 M KCl, 1.5 mMgCl2, 1.5 mM ATP, 0.5 mM EGTA, 0.5 mM CaCl2 (con-
aining 3 × 104 cpm/nmol [45Ca2+], 50 mM Pi and 20 mMops, pH 6.8 as described previously [28]. Ca2+ uptake was
erminated by rapid Millipore filtration followed by a washith 5 ml of 150 mM KCl.
.8. Purification of VDAC
Native and mutated mVDAC1 were extracted with LDAOrom mitochondria isolated from yeast expressing the nativer mutated protein, and purified by chromatography onydroxyapatite:cilite resin followed by carboxymethyl (CM)-ellulose chromatography, when LDAO was replaced by �-G [9].
.9. VDAC channel recording and analysis
Reconstitution of purified VDAC into a planar lipid bilayerPLB), multi-channel current recording and data analysesere carried out as previously described [7]. Briefly, PLBere prepared from soybean asolectin dissolved in n-decane
50 mg/ml) in a chamber containing 10 mM Hepes/KOH,H 7.4 and 0.5 or 1 M NaCl (cis/trans). Only PLBs withresistance greater than 100 G� were used. Purified VDAC
about 1 ng) was added to the chamber defined as the ciside. After insertion of one or more channels into the PLB,xcess protein was removed by perfusion of the cis chamberith 20 volumes of a same solution. Currents were recordednder voltage-clamp mode using a Bilayer Clamp amplifierWarner Instruments, Hamden, CT). Currents were measuredith respect to the trans side of the membrane (ground). The
urrents were low-pass filtered at 1 kHz, using a Bessal fil-er (Frequency Devices, Haverhill, MA) and digitized on-linesing a Digidata 1200 interface board and pCLAMP 6 soft-are (Axon Instruments). Sigma Plot 2000 scientific softwareas used for data analyses. Experiments were performed at3–25 ◦C.
.10. Photoaffinity labeling with [103Ru]AzRu
Purified proteins were irradiated with a 15-W ultravio-et lamp for 3–4 min in the presence of 100 nM to 1 �M103Ru]AzRu (200 cpm/pmol) in 40 �l of 20 mM Tricine, pH.4. The irradiated samples were separated by SDS-PAGE29], followed by Coomassie staining. The dried gel washen exposed directly to Kodak X-Omat film (Eastman Kodako.).
.11. Matrix-assisted laser desorptiononisation-time-of-flight (MALDI-TOF) analysis
Purified VDAC (25 �l, at 1 mg/ml) was UV-irradiated inhe presence or the absence of 2 �M AzRu (as describedbove), dialyzed against 5 mM Tris/HCl, pH 7.2, for 6 h atoom temperature and dried using a Speed-Vac centrifuge.

2 l Calcium 41 (2007) 235–244
Totita
l−apsaXpap
IvATmet3aG
3
pvi
3p
cpcNgdaaacwwsda
Fig. 1. AzRu induces channel closure of bilayer-reconstituted purifiedVDAC. VDAC was reconstituted into a PLB and currents through VDAC,in response to a voltage step from 0 to −20 mV, were recorded in symmet-rical solutions of 1.0 M NaCl, before and 5 or 10 min after the addition ofAzo
Vv
ViiteAiEsstm15as
3m
[obps[
38 A. Israelson et al. / Cel
he dried samples were re-dissolved in an aqueous solutionf 0.1% trifluoroacetic acid and diluted 1:1 with matrix solu-ion, a saturated solution of �-cyano-4-hydroxycinnamic acidn isopropanol:formic acid:water (2:1:3, v/v). 1 �l of the mix-ure was spotted on the MALDI target plate, air dried, andnalyzed as described below.
To localize the AzRu-binding site in VDAC1, AzRu-abeled VDAC1 (∼25 �g) was precipitated with acetone (at
70 ◦C) to extract detergent that would disrupt the MALDInalysis. The pellet was re-solubilized in 50 mM NH4HCO3,H 8, containing 1 M urea, and incubated for 8 h at 37 ◦C. Theample was then diluted 1:3 with the same buffer without ureand subjected to digestion with trypsin (TPCK-treated, typeIII from bovine pancreas, Sigma), at a ratio of 1:20 enzymerotein (w/w). Digestion was carried out for 24 h at 37 ◦Cfter which aliquots (∼0.6 �g protein) were dried and pre-ared for the MALDI analysis as described previously [30].
MALDI-TOF analysis was performed at the Nationalnstitute for Biotechnology in the Negev, Ben Gurion Uni-ersity, Beer-Sheva, Israel. Mass spectra of VDAC andzRu-VDAC fragments were obtained using a MALDI-OF mass spectrometer Reflex IV (Bruker, Bremen, Ger-any) equipped with a 337 nm nitrogen laser, with delayed
xtraction. The acceleration voltage was 20.0 kV. All spec-ra were obtained in positive-ion mode. For each sample,00–500 laser shots were accumulated in a linear modend processed by XMASS 5.1.5 software (Bruker, Bremen,ermany).
. Results
To localize the Ca2+-binding site(s) of VDAC1, we usedhotoreactive AzRu, specifically inhibited the activities ofarious Ca2+-dependent proteins, but had no effect on Ca2+-ndependent proteins [21].
.1. VDAC1 channel closure induced by AzRu isrevented by Ca2+
Similar to RuR [7] and Ru360 [8], AzRu decreased thehannel conductance of purified VDAC reconstituted into alanar lipid bilayer (Fig. 1). The bilayer-reconstituted VDAChannel activity was measured in symmetric solutions of 1 MaCl on both side of the bilayer in response to a voltageradient of −20 mV, as a function of time. Under these con-itions, VDAC is stable in a long-lived, high-conducting statend remains fully open for up to 2 h of recording. However,ddition of AzRu, RuR or La3+ induced VDAC closure intime-dependent manner, stabilizing the channel in a sub-
onductance state (Fig. 1). AzRu effect on the channel activityas observed only when added to the cis side, in which VDAC
as reconstituted. This suggests that AzRu interacts withite(s) located at the cytosolic face of the protein. AzRu alsoecreased VDAC conductance in multi-channel experimentst all voltages tested between −60 and +60 mV and stabilized
(nst
zRu (20 �M), RuR (20 �M) or La3+ (50 �M). The dashed lines indicateero-current and maximal current levels. The experiments are representativef four to five similar experiments.
DAC conductance at a constant low level, regardless of theoltage gradient applied [21].
To verify the relationship between AzRu inhibition ofDAC conductance and its possible interaction with Ca2+-
nteracting site(s) in VDAC, the effect of Ca2+ on AzRunhibition of VDAC conductance was tested (Fig. 2). Ca2+, inhe presence of as high as 1 M NaCl, prevented the inhibitoryffect of AzRu on VDAC activity (Fig. 2A). The degree ofzRu inhibition of VDAC conductance was dependent on the
ncubation time. Moreover, chelation of the added Ca2+ withGTA re-established AzRu-mediated inhibition, suggestingpecific interaction of AzRu with the VDAC Ca2+-bindingite(s). Ca2+, in a concentration-dependent manner, preventedhe inhibition of VDAC conductance by AzRu with a half-
aximal effect (in the presence of 1 M NaCl) observed at00 �M of Ca2+ (Fig. 2C). In contrast to Ca2+, Mg2+ (up tomM) did not prevent the inhibitory effect of AzRu (Fig. 2And C). The results suggest that AzRu and Ca2+ share theame binding site(s) in VDAC.
.2. [103Ru]AzRu labeled native, but not E72Q-mutated,VDAC1
To localize the VDAC Ca2+-binding site(s), radiolabeled103Ru]AzRu was used as a photoaffinity label. Irradiationf purified VDAC with [103Ru]AzRu resulted in covalentinding of the radiolabeled reagent (or its derivative) to therotein, as revealed by SDS-PAGE followed by Coomassietaining and autoradiography (Fig. 3). VDAC labeling with103Ru]AzRu was inhibited in the presence of Ca2+ or Ru360
Fig. 3A and B). Moreover, [103Ru]AzRu labeled native, butot E72Q-mutated, mVDAC1 (Fig. 3A and B). These resultsuggest that AzRu binds to a Ca2+-binding site and that glu-amate 72 is required for AzRu binding to VDAC.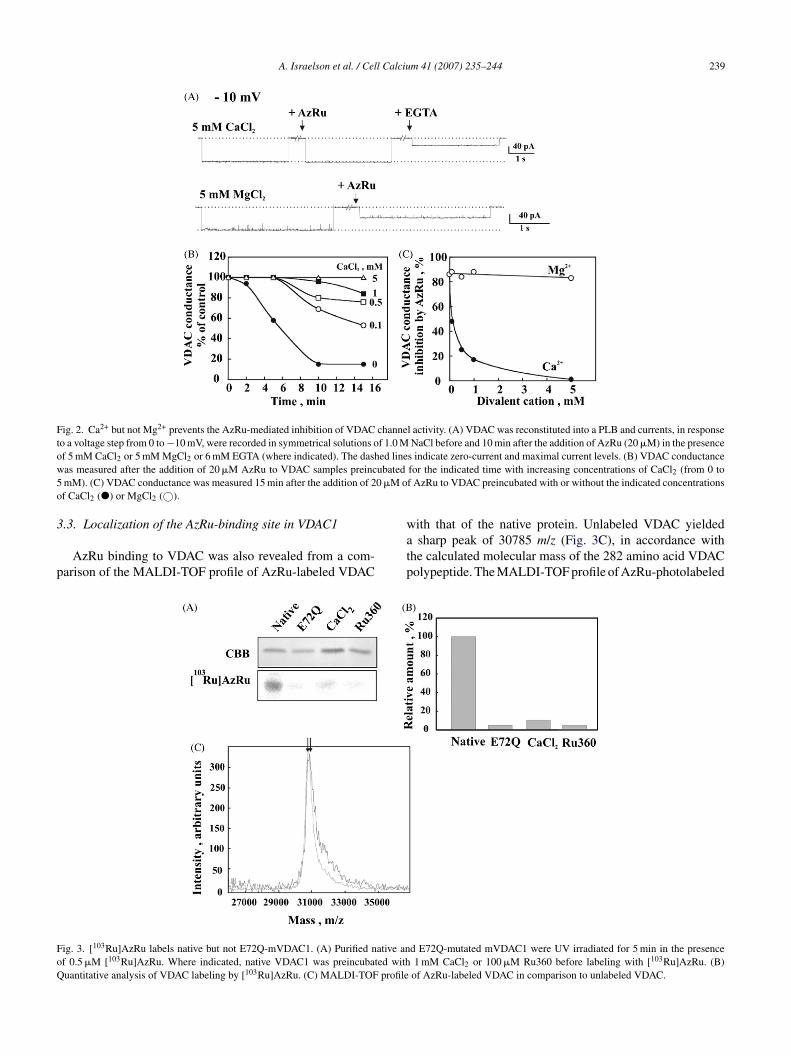
A. Israelson et al. / Cell Calcium 41 (2007) 235–244 239
Fig. 2. Ca2+ but not Mg2+ prevents the AzRu-mediated inhibition of VDAC channel activity. (A) VDAC was reconstituted into a PLB and currents, in responseto a voltage step from 0 to −10 mV, were recorded in symmetrical solutions of 1.0 M NaCl before and 10 min after the addition of AzRu (20 �M) in the presenceo ed linew ubated5 0 �M oo
3
p
FoQ
f 5 mM CaCl2 or 5 mM MgCl2 or 6 mM EGTA (where indicated). The dashas measured after the addition of 20 �M AzRu to VDAC samples preincmM). (C) VDAC conductance was measured 15 min after the addition of 2f CaCl2 (�) or MgCl2 (©).
.3. Localization of the AzRu-binding site in VDAC1
AzRu binding to VDAC was also revealed from a com-arison of the MALDI-TOF profile of AzRu-labeled VDAC
watp
ig. 3. [103Ru]AzRu labels native but not E72Q-mVDAC1. (A) Purified native anf 0.5 �M [103Ru]AzRu. Where indicated, native VDAC1 was preincubated withuantitative analysis of VDAC labeling by [103Ru]AzRu. (C) MALDI-TOF profile
s indicate zero-current and maximal current levels. (B) VDAC conductancefor the indicated time with increasing concentrations of CaCl2 (from 0 tof AzRu to VDAC preincubated with or without the indicated concentrations
ith that of the native protein. Unlabeled VDAC yieldedsharp peak of 30785 m/z (Fig. 3C), in accordance with
he calculated molecular mass of the 282 amino acid VDAColypeptide. The MALDI-TOF profile of AzRu-photolabeled
d E72Q-mutated mVDAC1 were UV irradiated for 5 min in the presence1 mM CaCl2 or 100 �M Ru360 before labeling with [103Ru]AzRu. (B)
of AzRu-labeled VDAC in comparison to unlabeled VDAC.

2 l Calciu
V(AutTwt
taetdattdfamta2tdlt
3E
sAgr(dtmbcbtemt
3oi
ia
esbartec[ciaaeccano
4
Ccbsoabia(CC(minVn[(ibstnai
40 A. Israelson et al. / Cel
DAC showed a broad peak with the average of 30900 m/zFig. 3C). This mass shift of 115 Da between native andzRu-labeled VDAC peaks is compatible with the molec-lar mass of an irradiated AzRu molecule (∼115 Da), andhus could be ascribed to an AzRu derivative bound to VDAC.he broad peak of the AzRu-labeled protein may results fromhether additional Cl− and H2O molecules remain bound to
he modified-Ru-N-VDAC.Next, unlabeled and AzRu-labeled VDAC were subjected
o trypsin or chymotrypsin digestion and MALDI-TOF/MSnalysis. To identify the protein fragments that bind AzRu thelution pattern of the AzRu treated sample was compared tohat of the control sample and to the pattern of fragments pre-icted by in Silico digestion of the untreated VDAC1 usingppropriate software. In the AzRu-labeled VDAC sample,wo peptide masses were detected which could correspondo peptides containing the additional mass of bound AzRuerivatives (see Table 1). The increase in the AzRu bindingragment mass will be of 115–292 Da, dependent whetherdditional Cl− and H2O molecules remain bound to theodified-Ru-N peptide. These AzRu-labeled VDAC pep-
ides were identified as encompassing the sequences of aminocids 61–73 (peptide 1), and amino acids 187–203 (peptide). Peptide 1 contains the amino acid glutamate 72, showno be essential for RuR or AzRu inhibition of VDAC con-uctance ([21,31] and Figs. 1 and 2) and for [103Ru]AzRuabeling of VDAC (Figs. 3A and 3B), while peptide 2 con-ains a glutamate residue at position 202.
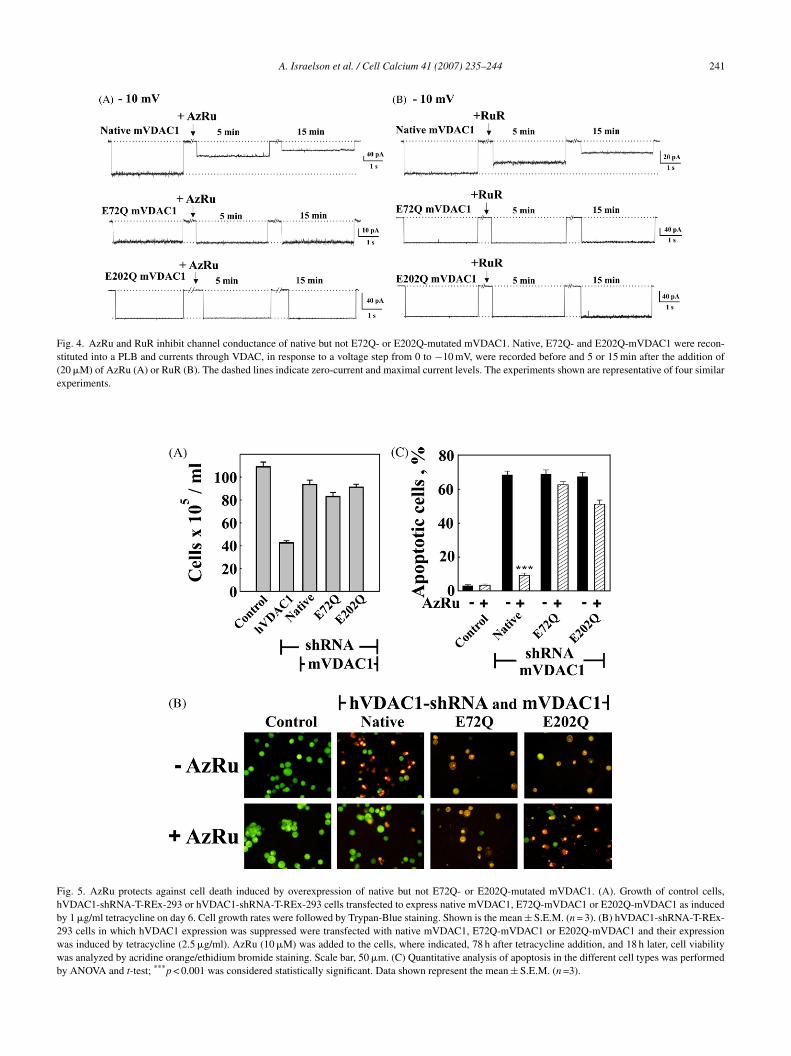
.4. AzRu-induced channel closure of native but not72Q- or E202Q-mutated mVDAC1
The involvement of Glu72 and Glu202 in the Ca2+-bindingite(s) of VDAC was considered by testing the effects ofzRu on the channel activity of purified VDAC1 in whichlutamate 72 or 202 had been replaced by glutamine. Bilayer-econstituted E72Q-mVDAC1 [31] or E202Q-mVDAC1data not shown) showed channel conductance and voltage-ependence as the native protein. RuR was recently showno induce channel closure of native but not E72Q-mutated
VDAC1 [31]. AzRu also induced channel closure of nativeut not E72Q-mutated mVDAC1 (Fig. 4A). Moreover, thehannel activity of E202Q-mutated mVDAC1 was found toe AzRu- or RuR-insensitive (Fig. 4), suggesting that thewo glutamate residues, predicted to be located in two differ-nt VDAC1 cytosolic loops, interact with AzRu or RuR anday, therefore, form the VDAC1 Ca2+ binding site(s) or part
hereof.
.5. AzRu did not protect against cell death induced byver-expression of E72Q-mVDAC1 or E202Q-mVDAC1n VDAC1-shRNA-T-REx-293 cells
In a previous study, we demonstrated that cells express-ng native but not E72Q-mVDAC1 were protected againstpoptotic cell death by RuR [31] or AzRu [21].
tds
m 41 (2007) 235–244
In the current study, a T-REx-293 cell line in whichndogenous human VDAC1 expression was suppressed usingpecific hVDAC1-shRNA was employed [32]. These sta-ly VDAC1-shRNA-expressing T-REx-293 cells prolifer-te extremely slowly, although normal cell growth can beestored by expressing murine VDAC1 under the control ofetracycline (1 �g/ml). By contrast, induction of mVDAC1xpression by high tetracycline levels (2.5 �g/ml) inducedell growth for 3 days followed by apoptotic cell death32]. As observed with the native mVDAC1-expressingells, hVDAC1-shRNA-expressing T-REx-293 cells express-ng either E72Q-mVDAC1 or E202Q-mVDAC1 were alsoble to resume normal cell growth rate (Fig. 5A). Moreover,s was the case with native mVDAC1 over-expression, over-xpression of E72Q- or E202Q-mVDAC1 induced apoptoticell death (∼75%) by the fourth day following high tetracy-line (2.5 �g/ml) treatment (Fig. 5B and C). AzRu protectedgainst cell death induced in this manner in cells expressingative mVDAC1, but not in those expressing either the E72Qr E202Q mutants (Fig. 5B and C).
. Discussion
Several lines of evidence suggest that VDAC possessesa2+-binding site(s) [7,9]. The finding that VDAC channellosure by RuR, a reagent shown to interact with Ca2+-inding proteins, can be prevented by Ca2+ [7,21] stronglyuggests that RuR and Ca2+ share a common binding siter sites. AzRu, a newly-synthesized photoactivable reagent,llows for affinity-labeling and localization of the Ca2+-inding site(s) in VDAC1. AzRu interacts with VDAC andnduces its closure when added to the cis side of the bilayer,n effect that was prevented by Ca2+ but not by Mg2+
Fig. 2), suggesting interaction of AzRu with the VDACa2+-binding site(s). The involvement of glutamate 72 in aa2+-binding site(s) of VDAC is suggested by the following:
1) Glu72 was identified as the interaction site of the carboxyl-odifying reagent DCCD that inhibits VDAC channel activ-
ty [33–35]; (2) replacement of Glu72 by glutamine elimi-ated RuR-, Ru360- [31] and AzRu- (Fig. 4) inhibition ofDAC channel activity; (3) [103Ru]AzRu bound native butot E72Q-mutated VDAC1 (Fig. 3); (4) VDAC labeling with103Ru]AzRu was prevented in the presence of Ca2+ or Ru360Fig. 3); (5) AzRu bound an E72-containing sequence, asdentified by MALDI-TOF analysis (Table 1). AzRu alsoound to a peptide corresponding to an E202-containingequence (Table 1) and replacement of Glu202 with glu-amine eliminated the ability of AzRu to inhibit VDAC chan-el activity (Fig. 4). Furthermore, AzRu-mediated protectiongainst apoptotic cell death was diminished in cells express-ng the E72Q or E202Q VDAC mutant (Fig. 5).
Based on the above findings and previous data, we suggesthat the two glutamate residues E72 and E202, located in twoifferent cytosolic loops, may form the VDAC1 Ca2+-bindingite(s), or part thereof. Alternatively, these residues may sta-
A. Israelson et al. / Cell Calcium 41 (2007) 235–244 241
Fig. 4. AzRu and RuR inhibit channel conductance of native but not E72Q- or E202Q-mutated mVDAC1. Native, E72Q- and E202Q-mVDAC1 were recon-stituted into a PLB and currents through VDAC, in response to a voltage step from 0 to −10 mV, were recorded before and 5 or 15 min after the addition of(20 �M) of AzRu (A) or RuR (B). The dashed lines indicate zero-current and maximal current levels. The experiments shown are representative of four similarexperiments.
Fig. 5. AzRu protects against cell death induced by overexpression of native but not E72Q- or E202Q-mutated mVDAC1. (A). Growth of control cells,hVDAC1-shRNA-T-REx-293 or hVDAC1-shRNA-T-REx-293 cells transfected to express native mVDAC1, E72Q-mVDAC1 or E202Q-mVDAC1 as inducedby 1 �g/ml tetracycline on day 6. Cell growth rates were followed by Trypan-Blue staining. Shown is the mean ± S.E.M. (n = 3). (B) hVDAC1-shRNA-T-REx-293 cells in which hVDAC1 expression was suppressed were transfected with native mVDAC1, E72Q-mVDAC1 or E202Q-mVDAC1 and their expressionwas induced by tetracycline (2.5 �g/ml). AzRu (10 �M) was added to the cells, where indicated, 78 h after tetracycline addition, and 18 h later, cell viabilitywas analyzed by acridine orange/ethidium bromide staining. Scale bar, 50 �m. (C) Quantitative analysis of apoptosis in the different cell types was performedby ANOVA and t-test; ***p < 0.001 was considered statistically significant. Data shown represent the mean ± S.E.M. (n =3).

242 A. Israelson et al. / Cell Calcium 41 (2007) 235–244
F Ru/RuRo 72 ands 1, valin
bRAtbsaan2T
udcPd
hblbtso
srdto
TM
D
T
C
Ncpm
ig. 6. Human VDAC1 membrane topology model showing the proposed Azrganization comprising a single �-helix and 13 �-strands [36]. Glutamateite(s) in VDAC1 (or part thereof), are indicated by circles. In sheep VDAC
ilize VDAC1 in a conformation that is recognized by AzRu,uR and Ru360. The intramolecular interaction of RuR orzRu with E72 and E202 is depicted in Fig. 6 which shows
he topological model of VDAC1 as proposed by Colom-ini [36]. However, when considering a possible oligomerictructure of VDAC as being involved in VDAC1-mediatedpoptosis [37], the interaction of AzRu with glutamate 72nd/or 202 residues located on two VDAC molecules canot be ruled out. In this respect, the interaction of RuR withglutamate residues found in two different subunits of the
ASK-3 potassium channel has been proposed [38].Changes in intracellular Ca2+ concentration not only stim-
late a number of intracellular events, but can also trigger cell
eath pathways, including apoptosis [10]. In isolated mito-hondria, Ca2+ induces membrane permeability transition,TP opening, and release of cytochrome c [9–11,39,40]. Toate, the mitochondrial target of Ca2+-mediated apoptosiswdte
able 1ALDI-TOF/MS of trypsin- or chymotrypsin-digested native and AzRu-labeled V
igested with (A) Masses (m/z) of VDAC peptides (B) Masses (
Observed Theoretical Observed
rypsin 1693.75 1693.82 1970.77
1945.91 1946.00 2184.86
hymotrypsin 1221.56 1221.57 1444.63
ative and AzRu-labeled VDAC were trypsin- or chymotrypsin-digested and suomparison, peptides present only in the AzRu-labeled VDAC sample are proporedicted masses of VDAC digest products. (B) The measured masses of the propass are presented as calculated. (C) The sequences and the locations of the propo
-interacting site. The proposed VDAC1 topology includes a transmembraneGlutamate 202, which interact with AzRu and may form the Ca2+ bindinge is present instead of methionine (indicated by square).
as not yet been identified. The inhibition of PTP openingy RuR, Ru360 and AzRu (when added after Ca2+ accumu-ation in the mitochondria reached a maximal level and justefore PTP opening [7,8,21]) suggests that these reagentsarget a protein other than the Ca2+ uniporter. The presenttudy and other reported results [7–9] indicate the presencef Ca2+-binding site(s) in VDAC.
The physiological function of the VDAC Ca2+-bindingite(s) could not be revealed from experiments using bilayer-econstituted VDAC, although it was recently suggested thatepending on the Ca2+ concentration, VDAC may displayhe full range of potential activities in the cell [17]. More-ver, while mutation of VDAC1 eliminated its interaction
ith RuR or AzRu with VDAC, amino acid replacementid not modify VDAC channel activity in reconstituted sys-em (Fig. 4) or in restore cell growth of hVDAC1-shRNA-xpressing cells (Fig. 5A). On the other hand, the findingDAC
m/z) of AzRu-labeled VDAC peptides (C) Peptide location andSequence
Calculated
1695.77 (+275), RuNCl4(H2O) 63-YRWTEYGLTFTEK-73
1945.36 (+239.5), RuNCl3(H2O) 200-KLETAVNLAWTAGNSNTR-217
1222.13 (+222.5), RuNCl·4(H2O) 71-TEKWNTDNTL-80
bjected to MALDI-TOF analysis (see Section 2). Based on mass (m/z)sed to correspond to AzRu-binding fragments. (A) The observed and theosed AzRu-binding peptides and their masses after subtracting the AzRu
sed AzRu-binding peptides. E72 and E202 are in bold and underlined.

l Calciu
tutaiFdw2
ptfimccirViadts
A
IwM
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
A. Israelson et al. / Cel
hat the anti-apoptotic effect of RuR, Ru360 and AzRu is lostpon expression of E72Q- or E202Q-VDAC1 may suggesthat the proposed VDAC1 Ca2+-binding site is involved inpoptosis regulation. Further support comes from the find-ngs that RuR [31,41–43], Ru360 [31] and AzRu ([21] andig. 5) prevented cell death induced by various stimuli, inifferent cell lines, and that such protection against apoptosisas prevented when VDAC1 was mutated at glutamate 72 or02 (Fig. 5).
According to the favored membrane topology model pro-osed for mammalian VDAC1 [36], the proposed location ofhe Ca2+-binding site(s) in VDAC1 lies in sequence regionsacing the cytosol (Fig. 6). On the other hand, for Ca2+ tonduce PTP opening, the ion must accumulate within the
atrix. Thus, Ca2+ may serve several functions in the mito-hondria, one of which involves the regulation of cytochromerelease, as mediated by the VDAC Ca2+-binding site, shown
n this study and proposed by others [7,9]. The use of aecently-developed system [32] in which endogenous humanDAC1 expression is suppressed by hVDAC1-shRNA and
n which native or mutated murine VDAC1 can be expressedllows for monitoring of Ca2+ transport into the mitochon-ria, exploring the function of VDAC1 in regulating Ca2+-ransport and the function of the proposed Ca2+-bindingite(s) in VDAC1 regulation of cell life and death.
cknowledgments
This research was supported by research grants from thesrael Science Foundation and B.G. Negev Technologies. Weould like to thank Dr. Mark Karpasas for carrying out theALDI-TOF/MS experiments.
eferences
[1] B.J. Nichols, R.M. Denton, Towards the molecular basis for the reg-ulation of mitochondrial dehydrogenases by calcium ions, Mol. Cell.Biochem. 149–150 (1995) 203–212.
[2] M. Bootman, E. Niggli, M. Berridge, P. Lipp, Imaging the hierarchi-cal Ca2+ signalling system in HeLa cells, J. Physiol. 499 (1997) 307–314.
[3] M. Zoratti, I. Szabo, U. De Marchi, Mitochondrial permeability tran-sitions: how many doors to the house? Biochim. Biophys. Acta 1706(2005) 40–52.
[4] T.E. Gunter, L. Buntinas, G.C. Sparagna, K.K. Gunter, The Ca2+ trans-port mechanisms of mitochondria and Ca2+ uptake from physiological-type Ca2+ transients, Biochim. Biophys. Acta 1366 (1998) 5–15.
[5] G. Beutner, V.K. Sharma, L. Lin, S.Y. Ryu, R.T. Dirksen, S.S. Sheu,Type 1 ryanodine receptor in cardiac mitochondria: transducer ofexcitation-metabolism coupling, Biochim. Biophys. Acta 1717 (2005)1–10.
[6] K.K. Gunter, T.E. Gunter, Transport of calcium by mitochondria, J.
Bioenerg. Biomembr. 26 (1994) 471–485.[7] D. Gincel, H. Zaid, V. Shoshan-Barmatz, Calcium binding and translo-cation by the voltage-dependent anion channel: a possible regula-tory mechanism in mitochondrial function, Biochem. J. 358 (2001)147–155.
[
m 41 (2007) 235–244 243
[8] D. Gincel, N. Vardi, V. Shoshan-Barmatz, Retinal voltage-dependentanion channel: characterization and cellular localization, Invest. Oph-thalmol. Vis. Sci. 43 (2002) 2097–2104.
[9] V. Shoshan-Barmatz, D. Gincel, The voltage-dependent anion channel:characterization, modulation, and role in mitochondrial function in celllife and death, Cell Biochem. Biophys. 39 (2003) 279–292.
10] M.J. Berridge, M.D. Bootman, P. Lipp, Calcium–a life and death signal,Nature 395 (1998) 645–648.
11] A.P. Halestrap, E. Doran, J.P. Gillespie, A. O’Toole, Mitochondria andcell death, Biochem. Soc. Trans. 28 (2000) 170–177.
12] M. Zoratti, I. Szabo, The mitochondrial permeability transition,Biochim. Biophys. Acta. 1241 (1995) 139–176.
13] M. Crompton, S. Virji, J.M. Ward, Cyclophilin-D binds strongly tocomplexes of the voltage-dependent anion channel and the adeninenucleotide translocase to form the permeability transition pore, Eur. J.Biochem. 258 (1998) 729–735.
14] N. Brustovetsky, M. Klingenberg, Mitochondrial ADP/ATP carrier canbe reversibly converted into a large channel by Ca2+, Biochemistry 35(1996) 8483–8488.
15] V. Shoshan-Barmatz, A. Israelson, D. Brdiczka, S.S. Sheu, The voltage-dependent anion channel (VDAC): function in intracellular signalling,cell life and cell death, Curr. Pharm. Design 12 (2006) 2249–2270.
16] J.H. Charuk, C.A. Pirraglia, R.A. Reithmeier, Interaction of rutheniumred with Ca2(+)-binding proteins, Anal. Biochem. 188 (1990) 123–131.
17] G. Bathori, G. Csordas, C. Garcia-Perez, E. Davies, G. Hajnoczky,Ca2+-dependent control of the permeability properties of the mito-chondrial outer membrane and VDAC, J. Biol. Chem. 281 (2006)17347–17358.
18] M.R. Nelson, W.J. Chazin, Structures of EF-hand Ca(2+)-binding pro-teins: diversity in the organization, packing and response to Ca2+
binding, Biometals 11 (1998) 297–318.19] A. Lewit-Bentley, S. Rety, EF-hand calcium-binding proteins, Curr.
Opin. Struct. Biol. 10 (2000) 637–643.20] K.H. Krause, M. Milos, Y. Luan-Rilliet, D.P. Lew, J.A. Cox, Ther-
modynamics of cation binding to rabbit skeletal muscle calsequestrin.Evidence for distinct Ca(2+)- and Mg(2+)-binding sites, J. Biol. Chem.266 (1991) 9453–9459.
21] A. Israelson, L. Arzoine, S. Abu-Hamad, V. Khodorkovsky, V. Shoshan-Barmatz, A photoactivable probe for calcium binding proteins, Chem.Biol. 12 (2005) 1169–1178.
22] W.L. Ying, J. Emerson, M.J. Clarke, D.R. Sanadi, Inhibitionof mitochondrial calcium ion transport by an oxo-bridged dinu-clear ruthenium ammine complex, Biochemistry 30 (1991) 4949–4952.
23] E. Blachly-Dyson, J. Song, W.J. Wolfgang, M. Colombini, M. Forte,Multicopy suppressors of phenotypes resulting from the absence ofyeast VDAC encode a VDAC-like protein, Mol. Cell. Biol. 17 (1997)5727–5738.
24] A.J. McGahon, S.J. Martin, R.P. Bissonnette, A. Mahboubi, Y. Shi, R.J.Mogil, W.K. Nishioka, D.R. Green, The end of the (cell) line: methodsfor the study of apoptosis in vitro, Meth. Cell Biol. 46 (1995) 153–185.
25] A. Saito, S. Seiler, A. Chu, S. Fleischer, Preparation and morphology ofsarcoplasmic reticulum terminal cisternae from rabbit skeletal muscle,J. Cell Biol. 99 (1984) 875–885.
26] A.C. Lee, X. Xu, E. Blachly-Dyson, M. Forte, M. Colombini, The roleof yeast VDAC genes on the permeability of the mitochondrial outermembrane, J. Membr. Biol. 161 (1998) 173–181.
27] O.H. Lowry, N.J. Rosebrough, A.L. Farr, R.J. Randall, Protein mea-surement with the Folin phenol reagent, J. Biol. Chem. 193 (1951)265–275.
28] I. Orr, V. Shoshan-Barmatz, Modulation of the skeletal muscle ryan-
odine receptor by endogenous phosphorylation of 160/150-kDa pro-teins of the sarcoplasmic reticulum, Biochim. Biophys. Acta. 1283(1996) 80–88.29] U.K. Laemmli, Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4, Nature 227 (1970) 680–685.

2 l Calciu
[
[
[
[
[
[
[
[
[
[
[
[
[
44 A. Israelson et al. / Cel
30] F. Landry, C.R. Lombardo, J.W. Smith, A method for application ofsamples to matrix-assisted laser desorption ionization time-of-flighttargets that enhances peptide detection, Anal. Biochem. 279 (2000)1–8.
31] H. Zaid, S. Abu-Hamad, A. Israelson, I. Nathan, V. Shoshan Barmatz,The voltage-dependent anion channel-1 modulates apoptotic cell death,Cell Death Differ. 12 (2005) 751–760.
32] S. Abu-Hamad, S. Sivan, V. Shoshan-Barmatz, The expression level ofthe voltage-dependent anion channel controls life and death of the cell,Proc. Natl. Acad. Sci. USA 103 (2006) 5787–5792.
33] I. Shafir, W. Feng, V. Shoshan-Barmatz, Dicyclohexylcarbodiimideinteraction with the voltage-dependent anion channel from sarcoplas-mic reticulum, Eur. J. Biochem. 253 (1998) 627–636.
34] V. Shoshan-Barmatz, N. Hadad, W. Feng, I. Shafir, I. Orr, M. Varsanyi,L.M. Heilmeyer, VDAC/porin is present in sarcoplasmic reticulumfrom skeletal muscle, FEBS Lett. 386 (1996) 205–210.
35] V. Shoshan-Barmatz, A. Israelson, The voltage-dependent anion chan-nel in endoplasmic/sarcoplasmic reticulum: characterization, mod-
ulation and possible function, J. Membr. Biol. 204 (2005) 57–66.36] M. Colombini, VDAC: the channel at the interface between mito-chondria and the cytosol, Mol. Cell. Biochem. 256–257 (2004)107–115.
[
m 41 (2007) 235–244
37] R. Zalk, A. Israelson, E.S. Garty, H. Azoulay-Zohar, V. Shoshan-Barmatz, Oligomeric states of the voltage-dependent anion channeland cytochrome c release from mitochondria, Biochem. J. 386 (2005)73–83.
38] G. Czirjak, P. Enyedi, Ruthenium red inhibits TASK-3 potassium chan-nel by interconnecting glutamate 70 of the two subunits, Mol. Pharma-col. 63 (2003) 646–652.
39] M. Crompton, S. Virji, V. Doyle, N. Johnson, J.M. Ward, The mito-chondrial permeability transition pore, Biochem. Soc. Symp. 66 (1999)167–179.
40] G. Kroemer, N. Zamzami, S.A. Susin, Mitochondrial control of apop-tosis, Immunol. Today 18 (1997) 44–51.
41] J.H. Bae, J.W. Park, T.K. Kwon, Ruthenium red, inhibitor of mito-chondrial Ca2+ uniporter, inhibits curcumin-induced apoptosis via theprevention of intracellular Ca2+ depletion and cytochrome c release,Biochem. Biophys. Res. Commun. 303 (2003) 1073–1079.
42] J.H. Baek, Y.S. Lee, C.M. Kang, J.A. Kim, K.S. Kwon, H.C. Son, K.W.Kim, Intracellular Ca2+ release mediates ursolic acid-induced apoptosis
in human leukemic HL-60 cells, Int. J. Cancer 73 (1997) 725–728.43] W.X. Ding, H.M. Shen, C.N. Ong, Pivotal role of mitochondrialCa(2+) in microcystin-induced mitochondrial permeability transitionin rat hepatocytes, Biochem. Biophys. Res. Commun. 285 (2001)1155–1161.