development of genomic resources for citrus clementina: characterization of three deep-coverage bac...
TRANSCRIPT

BioMed CentralBMC Genomics
ss
Open AcceResearch articleDevelopment of genomic resources for Citrus clementina: Characterization of three deep-coverage BAC libraries and analysis of 46,000 BAC end sequencesJavier Terol1, M Angel Naranjo1, Patrick Ollitrault2 and Manuel Talon*1Address: 1Centro de Genómica, Instituto Valenciano de Investigaciones Agrarias, Carretera Moncada, Náquera, Km. 4.5 Moncada, Valencia, E46113, Spain and 2CIRAD, UPR 75, Avenue Agropolis, TA A-75/02, 34398 Montpellier, Cedex 5, France
Email: Javier Terol - [email protected]; M Angel Naranjo - [email protected]; Patrick Ollitrault - [email protected]; Manuel Talon* - [email protected]
* Corresponding author
AbstractBackground: Citrus species constitute one of the major tree fruit crops of the subtropical regionswith great economic importance. However, their peculiar reproductive characteristics, low geneticdiversity and the long-term nature of tree breeding mostly impair citrus variety improvement. Inwoody plants, genomic science holds promise of improvements and in the Citrus genera thedevelopment of genomic tools may be crucial for further crop improvements. In this work wereport the characterization of three BAC libraries from Clementine (Citrus clementina), one of themost relevant citrus fresh fruit market cultivars, and the analyses of 46.000 BAC end sequences.Clementine is a diploid plant with an estimated haploid genome size of 367 Mb and 2n = 18chromosomes, which makes feasible the use of genomics tools to boost genetic improvement.
Results: Three genomic BAC libraries of Citrus clementina were constructed through EcoRI, MboIand HindIII digestions and 56,000 clones, representing an estimated genomic coverage of 19.5haploid genome-equivalents, were picked. BAC end sequencing (BES) of 28,000 clones produced28.1 Mb of genomic sequence that allowed the identification of the repetitive fraction (12.5% of thegenome) and estimation of gene content (31,000 genes) of this species. BES analyses identified3,800 SSRs and 6,617 putative SNPs. Comparative genomic studies showed that citrus genehomology and microsyntheny with Populus trichocarpa was rather higher than with Arabidopsisthaliana, a species phylogenetically closer to citrus.
Conclusion: In this work, we report the characterization of three BAC libraries from C.clementina, and a new set of genomic resources that may be useful for isolation of genes underlyingeconomically important traits, physical mapping and eventually crop improvement in Citrus species.In addition, BAC end sequencing has provided a first insight on the basic structure and organizationof the citrus genome and has yielded valuable molecular markers for genetic mapping and cloningof genes of agricultural interest. Paired end sequences also may be very helpful for whole-genomesequencing programs.
Published: 18 September 2008
BMC Genomics 2008, 9:423 doi:10.1186/1471-2164-9-423
Received: 29 January 2008Accepted: 18 September 2008
This article is available from: http://www.biomedcentral.com/1471-2164/9/423
© 2008 Terol et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Page 1 of 12(page number not for citation purposes)

BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
BackgroundCitrus, one of the major fruit tree crops is widely culti-vated throughout the globe and therefore has a tremen-dous economical, social and cultural impact in oursociety. Citrus improvement through traditional tech-niques, however, is highly impaired due to the unusualcombination of biological characteristics of Citrus species,their low genetic diversity and the long-term nature of treebreeding. Citrus are diploid plants with an estimated hap-loid genome size of about 367 Mb and 2n = 18 chromo-somes, which may facilitate the use of genomics tools forcrop improvement. Expressed sequence tag (EST) analysesand molecular marker studies strongly suggest that themain commercial citrus cultivars (oranges, lemons andgrapefruits) are mostly interspecific hybrids and thereforeare heterozygous "species" [1]. In addition, most of thecultivars in these groups, including Clementine varieties,may represent accumulated somatic mutations identifiedover centuries [2].
The development of citrus genomic resources is in itsinfancy although in recent years major efforts and goalsmostly on functional genomics have certainly been under-taken [3]. Critical functional and expression analysesthrough microarrays with several platforms have alsobeen published and analyses of ESTs in public databaseshave been initiated [4,5]. For instance, 401,692 citrusESTs have been deposited at GenBank and are currentlyavailable. This collection constitutes a valuable source forthe direct access to the genes of interest and for the devel-opment of molecular markers for map-based cloning pur-poses or marker-assisted selection programs [6-9].Moreover, genetic linkage maps have been produced withincreasing value and resolution, following the evolutionof new marker systems [10,11]. Genetic transformation incitrus is also available [12] and strategies based ongenome-wide mutagenesis are being explored. Otherinnovative resources such as viral-induced gene silencing(VIGS) are being developed and work in citrus proteomicsis in progress [13,14]. Thus, current advances in citrusresearch include the rapid development of functionalgenomics and molecular biology resources [15] although,on the other hand, basic information on the organizationand structure of citrus genome is lacking. The main chal-lenge for a comprehensive and meaningful description ofgenomes is the integration of the DNA marker-basedgenetic maps with physical maps, and eventually withDNA sequence of the whole genome, the ultimate physi-cal map. For the generation of high-resolution physicalmaps, the construction of BAC libraries containing cloneswith large DNA fragments appears to be indispensable.BAC end sequencing is indeed an important componentof physical map development and can be considered aform of low coverage sequencing [16]. Paired endsequences of BACs form an important part of scaffolding
whole-genome shotgun programs [17] as well as in BACbased genome projects [18,19]. In addition, BAC clonecollections and BAC-based contig maps are powerful toolshaving multiple applications in genomics including posi-tional cloning. The BAC end sequence provides a randomsurvey of the information contents (genes, transposons,repeats) of unsequenced genomes [20-22], and yieldsmolecular markers useful for genetic mapping [23-25],and cloning of genes of agricultural interest [26-28]. Fur-thermore, in many agriculturally important species BACclones and physical maps are being rapidly developedsince they are essential components in linking phenotypictraits to the responsible genetic variation, to integrate thegenetic data, for the comparative analysis of genomes, andto speed up marker-assisted selection (MAS) for breeding.Thus, BAC libraries have become central for physical map-ping, genome analysis, clone based sequencing andsequencing of complex genomes, for both model [18,19]and main crop plants [29-32].
In citrus, two BAC libraries from Poncirus trifoliata [11]and a hybrid of Citrus × Poncirus [10] have been describedin detail. These libraries were constructed as part of a map-based cloning strategy of genes conferring resistance to cit-rus tristeza virus that causes significant economic damageand losses to citrus worldwide. Poncirus is a non domesti-cated genus related to citrus species that produces inediblefruit. However, other efforts to generate BAC citrusresources, for instance in Satsuma or sweet orange havealso been accomplished [3].
In this work, we report the characterization of threegenomic BAC libraries from Clementine (Citrus clemen-tina) mandarin, a cultivar of great [3]economic impor-tance that has been a main target of recent studies [15].The development of these new genomic tools also com-plements the functional genomics platform generated forthis species including an extensive EST collection and a 20k cDNA chip [4,5], expanding further possibilities for iso-lation of genes of agronomical interest [33,34]. This studyprovides the most comprehensive, large insert cloneresource of any Citrus species and reports the analysis of46,339 BAC end sequences offering a first detailed insightinto the sequence composition of the Clementinegenome. The analyses focused on protein coding regions,repeat element composition, microsatellite and singlenucleotide polymorphism contents. Additionally, data ongene homology based comparative genomics with poplarand Arabidopsis are also presented. The annotated BAC-end sequences may well serve as useful resources for phys-ical mapping, positional cloning, genetic marker develop-ment and genome sequencing of C. clementina.
Page 2 of 12(page number not for citation purposes)

BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
Results and discussionBAC library characterizationThree genomic BAC libraries (CCL1, CCER1 and CCH3)were constructed as described in Material and Methods,with DNA from Citrus clementina (var. Clemenules),pEBAC1 as the cloning vector and three different restric-tion enzymes (Table 1). The CCL1 library composed of19,200 clones was generated with MboI partial digestions.EcoRI and HindIII were used for the construction ofCCER1 and CCH3, respectively, and 18,432 clones werepicked from each one. The three libraries contained56,064 BAC clones that were arrayed in 146 384-wellmicrotiter plates. It has been reported that the use of threedifferent restriction enzymes resulted in a more accuratecoverage of the genomes, since the different GC contentsof their recognition sites increases the representation of ahigher number of genomic regions [35]. A single libraryconstructed with one restriction enzyme usually cannotprovide a full coverage of the genome, as the restrictionsites are not uniformly distributed along the genome, andtherefore genomic regions having too many or too fewrestriction sites are not equally represented [36]. Gener-ally, two or more complementary large insert libraries,constructed with different restriction enzymes, have beensuccessfully used for physical mapping of several plantgenomes including those of Arabidopsis[37], japonicarice [38], or soybean [30].
In order to evaluate the average BAC insert size, 362 BACclones (about 120 clones from each library) were ran-domly chosen and the corresponding DNA was extracted,digested with the rare cutter NotI enzyme and analyzed byPFGE. All fragments generated by NotI digestion con-tained the 8.7 kb vector band and various insert fragments(Figure 1). The estimated insert sizes ranged from 10 to330 kb, with an average of 124 kb for CCL1, 132 kb forCCH3 and 127 kb for CCER1. Since the haploid genomesize of C. clementina is in the order of 367 Mb, the librariescoverage is predicted to be 19.5 haploid genome equiva-lents while the probability of finding any specificsequence is greater than 99.999%. It has been estimatedthat the number of clones representing 10× haploid
genomes is adequate for most genome research purposes,including physical mapping [39].
BAC end sequencingA total of 28,032 BAC clones from the three genomiclibraries were selected for end sequencing as described inmaterial and methods and out of this number, 24,221clones (86% success rate) rendered 46,339 BAC endsequences (BESs). The three libraries contributed approx-imately with similar number of reads. The average readlength was 652.8 bp and the genomic raw sequence pro-duced was 28.6 Mb, which corresponds to almost 8% ofthe Clementine genome. Table 2 shows a summary of theBAC end sequencing features. The 46,339 BESs weredeposited at GenBank with accession numbers fromET068227 to ET114565.
Sequence AnnotationChloroplast and mitochondrion DNA analysisIn order to identify extranuclear sequences, BES were firstcompared against the Citrus sinensis chloroplastic [40] andthe Arabidopsis thaliana [41] mitochondrial genomesequences with an stringent threshold of 1e-15. The com-parison indicated that 736 (1.68%) and 46 (0.1%)sequences produced significant matches with the chloro-plastic and mitochondrial genomes, respectively. Chloro-plastic BESs were assembled and the consensus sequencesobtained spanned 101,470 bp, approximately 70% of thechloroplast genome. Chloroplastic and mitochondrialDNA summed up to 480 kb, and therefore, the totalgenomic DNA obtained was 28,1 Mb. Considering theaverage size of the BAC clones, the coverage provided by
Table 1: Genomic Citrus clementina BAC Libraries
Library CCL1 CCER1 CCH3
Vector pECBAC1 pECBAC1 pECBAC1Partial digest enzyme MboI EcoRI HindIIICloning site BamHI EcoRI HindIIIAverage insert size 124 kb 127 kb 132 kbN° of clones 19,200 18,432 18,432Missed wellsa 0.57% 0.37% 0.09%Blue coloniesb 0.78% 1.46% 0.19%
a No bacterial growth detectedb Presence of vector but no insert
Size estimate of BAC clones from the CCER1, CCH3 and CCL1 C. clementina librariesFigure 1Size estimate of BAC clones from the CCER1, CCH3 and CCL1 C. clementina libraries. Bars represent the number of BAC clones in each class. 362 BAC clones were randomly selected from the BAC libraries of C. clementina: CCER1 (light gray), CCH3 (black), and CCL1 (dark gray).
Page 3 of 12(page number not for citation purposes)
BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
the BAC end sequencing was higher than 8.4 genomicequivalents.
Repetitive DNA AnalysisThe repetitive DNA fraction present in Clementine BESsrevealed with RepeatMasker, included 9,618 interspersedrepeats which extended over 2.55 Mb, 8.95% of the totalraw sequence (Table 3). BLASTX search performed toidentify coding regions (see next section), showed that2,173 additional sequences not detected by RepeatMaskeralso presented high significant similarity to transposableelements (TEs) and, therefore, are also part of the repeti-tive DNA fraction of the genome. Thus, the number ofBESs with interspersed repeats rose to 11,791, approxi-
mately a 25% of the total reads, a percentage betweenthose found for Carica papaya (16%) [20] or Musa acumi-nata (36%) [21]. No significant differences between the 3BAC libraries were found when the number of BESs carry-ing repetitive elements was compared (see Additional File1). The sequence length occupied by transposon elements(TEs) including the additional reads identified throughhomology search was in this way increased to 3.58 Mb, afraction corresponding to 12.6% of the total BAC endsequences. This fraction was rather similar to the ratioreported for Brassica rapa (13.8%) in an estimation alsobased on partial sequencing [22]. Comparisons with thepercentages found in fully sequenced genomes showedthat the relative occurrence of TEs in Clementine was also
Table 2: Citrus clementina BAC end sequencing summary
Library CCL1 CCER1 CCH3 Total
Processed BACs 9,216 9,216 9,600 28,032Positive BACs 8581 7262 8378 24,221Success rate 93.1% 78.8% 87.3% 86.4%BES mate pairsa 8036 6401 7681 22,118Genomic equivalentsb 3.1 3.1 3.9 10.1
Reads 16,617 13,663 16,059 46,339Success rate 90% 74% 84% 83%Total raw sequence (bp) 10,102,329 8,783,997 9,676,620 28,562,946Average read length (bp) 638.0 679.6 644.3 652.8Chloro/mito readsc 432 146 204 782Chloro/mito total sequence (bp) 273,452 94,570 111,324 479,346Final genomic sequence (bp)d 9,828,877 8,689,427 9,565,296 28,083,600
a BAC clones with 5' and 3' good quality readsb Genomic equivalents calculated considering the number of clones sequenced and their average lengthc Reads identified as chloroplastic or mitochondrial DNAd Total genomic sequence obtained, without mitochondrial or chloroplastic DNA
Table 3: Repetitive DNA in Citrus clementina BESs identified by Repeat Masker
Number of elements Length (bp) Sequence (%) Number of elements (%)
RetroelementsLINEs
L1/CIN4 360 67,719 0.237% 3.74%LTR elements
Ty1/Copia 3,770 1,142,096 3.999% 39.20%Gypsy/DIRS1 3,917 1,172,005 4.103% 40.73%
Other 265 6,939 0.024% 2.76%Total Retroelements 8,312 2,388,759 8.363% 86.42%
DNA transposonshobo-Activator 346 64,065 0.224% 3.60%Tc1-IS630-Pogo 97 10,162 0.036% 1.01%En-Spm 381 55,814 0.195% 3.96%MuDR-IS905 386 29,983 0.105% 4.01%Tourist/Harbinger 13 1,400 0.005% 0.14%Other 83 5,653 0.020% 0.86%Total DNA transposons 1,306 167,077 0.585% 13.58%
Total interspersed repeats 9,618 2,555,836 8.948%
Page 4 of 12(page number not for citation purposes)
BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
similar to the fractions found in Arabidopsis (10%) [18]and black cottonwood (12.6%) [17] and lower than inrice (35%) [19] and grapevine (38.8%) [42]. Although theproportion of the different TEs largely varied, in all thesespecies, as well as in C. clementina, class I elements(including LINE, Gypsy-like and Copia-like elements)were predominant over class II (including CACTA, MULEand hAT elements). In comparison with the abovesequenced plants, the occurrence of Mutator-like, LINEs,Cacta, and Gypsy elements, represented as the percentageof occupied sequence, was in general lower in the genomeof C. clementina. In contrast, copia-like elements were rel-atively more abundant in Clementine than in Arabidopsis,rice and poplar (Figure 2). The abundance of these ele-ments in citrus has previously been estimated to be rela-tively high (13%) [43], while the data obtained throughpartial sequencing in this study suggested a lower prepon-derance (3.9%; Table III). Copia and gypsy like elements,however, are transcriptionally active in Clementine[43,44] and therefore, could be an important source ofgenetic variability in this species.
In order to identify low complexity repeats, all BESs weresearched against themselves with BLASTN and then classi-fied based on the number of significant hits produced(Figure 3). After filtration, 17,585 BES producing at leastone hit different from themselves were clustered in thisway. The results showed that while a high proportion ofBESs (82%) displayed a low number of hits, 3,221 readsproduced more that 10 hits, suggesting that these BESsmay carry non-coding repetitive sequences, i.e. they maybe interspersed repeats of lower complexity. The amountof sequence occupied by these repeats was estimated to be
1.12 Mb, 3.94% of the analyzed sequence. This propor-tion appears to be moderate in comparison with the23.5% figure reported for poplar [17].
Analysis of coding regionsAfter filtration of mitochondrial, chloroplastic and repeti-tive sequences, the remaining 30,787 BES were analyzedfor coding region identification via homology search. Par-allel searches with BLASTX and BLASTN were performedagainst the non-redundant database (e value cut off of 1e-4) and a database of Citrus ESTs from GenBank (e valuecut off of 1e-15), respectively. The BLASTX search identi-fied 14,030 BES (36% of the total BESs) with significantprotein hits, while the BLASTN search revealed a similarnumber of reads, i.e. 14,023 reads that rendered signifi-cant homology with 40,536 citrus ESTs. Overall, 20,185BESs produced BLASTX and/or BLASN hits, and the 3 BAClibraries rendered a similar number of clones carryingpotential coding regions (see Additional File 1). The totalnumber of BAC clones that produced protein and/or ESThits was 15.658, while 4,527 of them provided hits inboth 5' and 3' ends, an observation that suggested highgene contents in these BAC clones and indeed their possi-ble location at the euchromatin. It was also found that7,868 BES produced hits for both proteins and ESTs,strongly indicating that they contained active transcrip-tion units.
Assuming that each significant BLAST hit corresponds to adifferent transcription unit, the total number of hypothet-ical genes described in the BAC ends was 20,185. Thus,considering the amount of analyzed sequence (28.1 Mb),
Comparative analysis of the most abundant transposable ele-ments from C. clementinaFigure 2Comparative analysis of the most abundant transpos-able elements from C. clementina. Estimates of the amount of specific classes of transposable elements are rep-resented as percentage of occupied sequence in C. clemen-tina. For comparative analysis, data from O. sativa [19], P. trichocarpa [17], and A. thaliana [18] are included.
Putative low complexity repetitive sequences identified through BLASTN search of BESs against themselvesFigure 3Putative low complexity repetitive sequences identi-fied through BLASTN search of BESs against them-selves. The figure represents the frequency distribution of BESs as related to the hit number obtained after BLASTN search of BESs against themselves. BESs producing more than 50 copies were considered to be putative interspersed repeats.
Page 5 of 12(page number not for citation purposes)
BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
the estimated genome size (367 Mb), and the number ofgenomic equivalents analyzed (8.4), the gene contents ofthe genome of Citrus clementina was assessed as 31,000.This gene number is comparable to the estimate reportedfor three species of similar genome size that have beensequenced to completion to date: rice (Oryza sativa), witha genome size of 430 Mb and 41,042 genes identified[45], Black cottonwood with 55,000 genes in 485 Mb[17], and grapevine (Vitis vinifera), with 30,434 genes in487 Mb [42]. Other estimates based on BES analysisobtained for plant species were also analogous. Forinstance, in papaya, the estimated gene contents was35,526 with a 372 Mb genome size [20], and in Chinesecabbage, with a genome of 529 Mb, 43,000 genes werecalculated [22].
Furthermore, total GC content in BESs estimated withEMBOSS was 39% while in coding and non-codingsequences, was 41% and 37%, respectively (Figure 4). Nosignificant differences were found when the GC contentwas compared between the 3 BAC libraries constructed(see Additional File 1). The GC contents reported in otherwoody plants such as Scots Pine (Pinus sylvestris; 39.5%)[46] or yellow-poplar (Liriodendron tulipifera; 41%) [28] aswell as in coding sequences from Solanaceae species i.e.Nicotiana tabacum (40.4%), Solanum tuberosum (39.0%),and Solanum esculentum (39.8%), or the Fabaceae Pisumsativum (39.2%) were also on the same range. The percent-age of GC in Glycine max (46.5%) and A. thaliana (45.4%)was significantly higher [47].
Lastly, Blast2GO [48] was used to analyze the differentfunctions associated with the putative coding regions, andGO terms were assigned to 10,598 sequences. Additionalfile 2 shows the results obtained for the gene ontology cat-
egories Molecular Function and Process. This classifica-tion may be useful to identify and locate genes ofagronomic interest, such as those related to sugar and cel-lulose synthesis [GenBank:ET090832, GenBank:ET070671, GenBank:ET074358], ion transport [GenBank:ET110643, GenBank:ET110643, GenBank:ET074792], or calciummetabolism [GenBank:ET068865, GenBank:ET091143,GenBank:ET084632], for instance.
SSR AnalysisBAC end sequences have proved to be excellent sources toidentify simple sequence repeats (SSRs or microsatellites)in many plant species such as cotton or soybean[23,25,49]. In this work, Sputnik [50] was used to identifya total of 3,814 SSRs longer than 15 bp in the BESs thatdid not carry repetitive sequences. The occurrence of SSRsin the Clementine genome had a frequency of 0.20 SSRper kb, a value almost identical to that reported in a studybased on citrus ESTs (0.19 SSR per kb) [51] and in B. rapa(0.18 SSR per kb) [22]. This frequency, however, waslower than the ratios found in grapevine (0.48 SSR per kb)[42], papaya (0.43 SSRs longer than 12 bp per kb) [20],and A. thaliana (0.33 SSR per kb).
In the Clementine SSR set, there were 758 class I (morethan 10 repeats) and 3,056 class II (less than 10 repeats)SSRs, with di and trinucloetides accounting for almost70% of the SSRs, while tetra and pentanucleotides wereless represented. In general, those motifs containing A/Tnucleotides were far more abundant than G/C richrepeats, specially ATT/TAA and AT/TA tri- and dinucle-otides (Figure 5). A similar distribution was found byJiang et al[8] and Chen et al. [51] in the analysis of 8,218
GC content in BES of Citrus clementina BAC librariesFigure 4GC content in BES of Citrus clementina BAC libraries. Distribution of GC content in coding and non-coding regions of BES of Citrus clementina BAC libraries.
Number of repeats of the most abundant SSRs in BES of Cit-rus clementina BAC librariesFigure 5Number of repeats of the most abundant SSRs in BES of Citrus clementina BAC libraries. Black bars rep-resent the number of repeats found for each SSR motif.
Page 6 of 12(page number not for citation purposes)
BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
and 3,278 citrus SSRs derived from ESTs, respectively. Inthe Clementine genome, microsatellites were morenumerous in non coding sequences (56% of the SSRs)than in putative coding regions (44%), as previouslyreported in papaya [20], Chinese cabbage [22], and Arabi-dopsis[18].
Microsatellites are co-dominant, highly polymorphic, andsimple to use markers that have been successfully used instudies of genetic diversity [7] and genetic mapping in cit-rus [8,52] and in many other plants [25,53-55]. The addi-tional markers reported here will certainly contribute toimprove the coverage of the Citrus genome for many pur-poses, including the development of accurate linkage andgenetic maps.
Contig Assembly and SNP analysisAssembly of BESs that did not contain repetitivesequences was performed with CAP3 [56], and a total of6,461 contigs including 19,057 reads and covering 6.14Mb of sequence were produced. It has been suggested thatC. clementina is an offspring of a C. sinensis × C. reticulatacross and therefore has a heterozygous genome [1]. Inorder to identify possible polymorphisms affecting theassembly of the readings and the construction of the phys-ical map of this species, a BLASTN search of all the contigsand singlets was performed against themselves. One hun-dred thirty sequence pairs that presented a single recipro-cal BLASTN hit and the same protein hit from the previousBLASTX search (identical accession number) were selectedfor further analysis. A total of 81 pairs displayed sequenceidentities higher than 90% and were considered as origi-nated from the same genomic region. These sequenceswere not assembled in the same contig due to the presenceof polymorphisms, similarly to what was reported in thesequencing of the highly heterozygous grapevine variety,Pinot Noir [57].
The contigs generated in the assembly were analyzed inorder to identify single nucleotide polymorphisms (SNPs)in the diploid genome of C. Clementina. SNPs are the mostabundant and powerful polymorphic markers, since theyprovide gene-based markers that can be used in the crea-tion of dense genetic linkage maps [58] and, more impor-tant, in the identification of genes associated with specifictrait loci. BAC end sequences of heterozygous genotypeshave also been successfully utilized in SNP discovery andconstruction of linkage maps [59-61]. PolyBayes [62], asoftware designed to use genomic sequence as a templateand base quality values to discern true allelic variationsfrom sequencing errors, was used to reveal SNP polymor-phisms. The number of putative SNPs identified in theClementine sequences that showed P ≥ 0.9 and SNP depthlower than 10 was 6,617, corresponding to 1.08 SNPs perkb. PolyBayes has been successfully utilized in automated
high throughput identification of SNPs from EST collec-tions, and it has been shown that when using P ≥ 0.95,85% of the predicted SNPs are generally validated experi-mentally [63].
In order to test the accuracy of the SNP prediction carriedout, a total of 30 polymorphisms were randomly chosenfor experimental validation. Primers were designed on theconsensus sequence of the contigs, in order to amplify theregion containing the SNP. Out of 30 genomic regions, 3produced two or more bands in the PCR amplification oryielded sequence reads with a mixture of templates. Thisobservation constitutes a first indication of the level ofheterozygosity of the Clementine genome. Furthermore,the sequence analysis of the remaining SNP candidatesshowed that 24 out of the 27 hypothetical polymor-phisms, an 88.9% success rate, were certainly validated(see Additional File 3). Considering the reliability of theprediction method, more than 5000 putative polymor-phisms with P ≥ 0.95 were found in this work. Transitionswere the most abundant changes (3,546; 53.6%), fol-lowed by transversions (2,162; 32.7%) and indels (909;13.7%) (Table 4). The transition fraction found in poplar(70%) was substantially higher [17]. The predominanttransversion was A/T at a frequency that doubled that ofC/G changes (Table 4), an unexpected observation thatremains to be explained.
It should be noted that the SNP listing reported in thiswork constitutes the first set of putative SNPs identified in
Table 4: SNPs in Citrus clementina BESs identified by PolyBayes
Summary
N° SNPsa 6617P_SNPb 0.984Total seq (kb)c 6139SNP/kb 1.08
Detailed relation of SNPs
Transitions 3546 (53.59%)ag 1781ct 1765
Transversions 2162 (32.67%)ac 541at 713cg 330gt 578
Indels 909 (13.74%)c- 112t- 354g- 108a- 335
a Number of SNPs with P_SNP > 0,9b Average PolyBayes scorec Total length of analyzed sequence
Page 7 of 12(page number not for citation purposes)

BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
any Citrus species, and hence provides a completely newresource for genome analysis in this genera. It is alsoworth mentioning that 4,500 SNPs were located in orclose to putative coding regions, and therefore these 'func-tional SNPs' may provide an inestimable resource for theidentification of genes associated with specific trait loci inaddition to their utility as molecular markers for geneticand comparative mapping, nucleotide diversity analysisand association studies.
Two heterozygous genomes have been sequenced to com-pletion, the Nisqually-1 poplar and the Pinot Noir grape-vine strains. In both cases the frequency ofpolymorphisms found within these heterozygousgenomes, was 2.6 [17] and 4 [57] SNPs per kb, respec-tively. These rates are in contrast with the frequency of1.08 SNPs per kb found in Clementine. The reproductivebiology of the different species could explain these differ-ences. Gametophytic self- and cross-incompatibility, andapomixy would produce low variability within Citrus spe-cies [7], while outcrossing by means of insect and windpollination, which is the norm for poplar and vitis, wouldresult in highly heterozygous cultivars [17,57].
Comparative GenomicsThe non-repetitive fraction of the BESs was also used in aBLASTN search against the complete nucleotide sequenceof the genomes of A. thaliana, P. trichocarpa and O. sativa,with 1e-14 as cut off value. The genomic sequences weredisplayed with chromosomes as single searchable fastasequences. In order to map the BESs unambiguously onthe heterologous complete genomes, only thosesequences producing single significant hits were takeninto account. Table 5 shows that the Populus genome notonly yielded the largest number of significant hits (3-foldmore than Arabidopsis and almost 5-fold than rice), butalso spanned more than twice the length of the sequencedisplaying similarity.
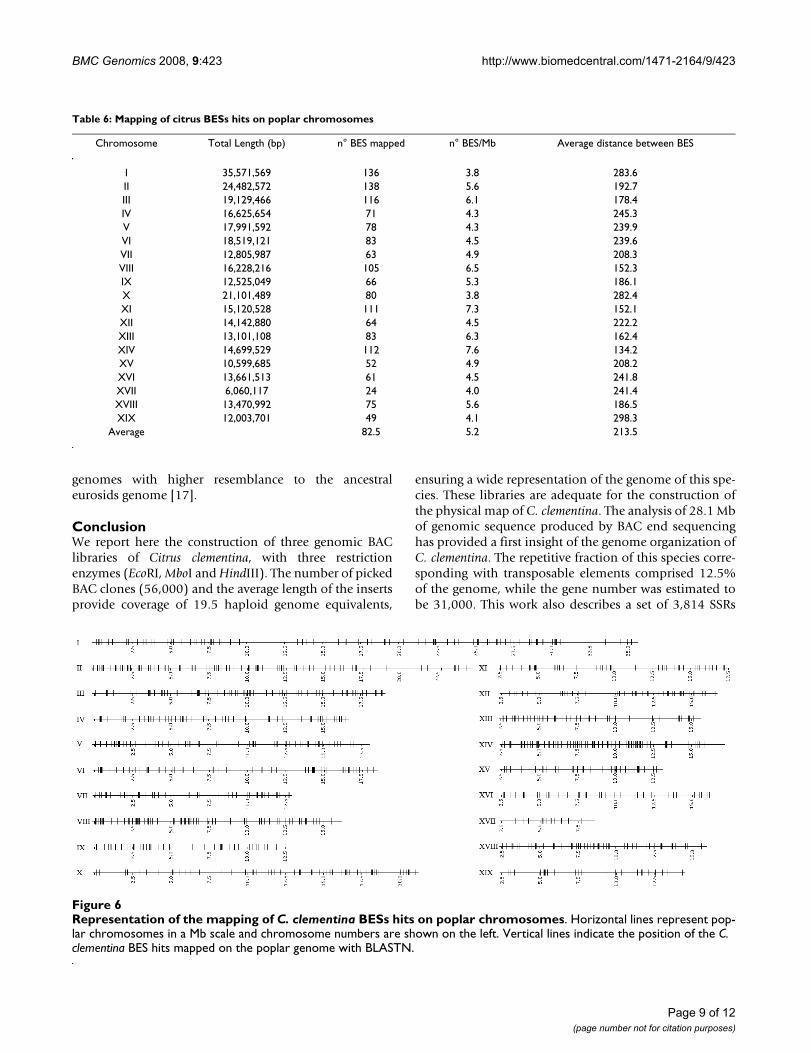
The 1567 BESs that produced significant hits with poplarwere mapped on the chromosomes of this species. Therepresentation drawn in Figure 6 showed that citrussequences were rather uniformly widespread on the 19poplar chromosomes, an observation that can also bededuced from data in Table 6 that in addition shows that
the average number of tags per chromosome was 83 whilethe distance between tags was 213 kb. Considering thatboth species have similar genome size, the uniform distri-bution of the Clementine tags on the poplar chromo-somes may suggest that the citrus genome is convenientlyrepresented in the BAC clone set.
Following the approach of Lai et al. [20], we used forwardand reverse BES read pairs separated by the approximatelength of BAC clone inserts (~120 kb), to analyze themicrosynteny between C. clementina and Arabidopsis, riceand poplar. To be considered as potentially collinear withthe target genome, the citrus mate pairs had to map in theheterologous genome into a region comprised between 10and 300 kb and be also oriented properly. The analyses ofthe sequences identified 108 Clementine BAC end pairsthat met these criteria in poplar, while no one was foundin Arabidopsis or rice. Furthermore, the majority of theseBES pairs mapped on the Populus genome at a distancesimilar to the insert size of the Clementine libraries, sug-gesting the microsynteny between citrus and poplar ishigher than between citrus and Arabidopsis. These resultsare striking since C. clementina and A. thaliana belong toSapindales and Brassicales orders (eurosids II clade) thatprobably split approximately 87 MYA, while P. trichocarpabelongs to Malpighiales order, (eurosids I clade) thatdiverged from eurosids II around 109 MYA [64]. Moreo-ver, similar results were obtained by Lai et al [20] withpapaya, that also exhibited higher level of colinearity withthe poplar than with the Arabidopsis genome despite thatC. papaya is a basal member of the Brassicales. Although adefinitive explanation has not been provided yet, it is cur-rently believed that the genome of A. thaliana has under-gone a recent whole genome duplication, followed bysubsequent gene losses and extensive local gene duplica-tions [18], which might be responsible of the lack ofcolinearity with other eurosid II species. Comparativegenomics with the recently sequenced genome of thegrapevine, provides additional evidence that the genomeof Arabidopsis has been thoroughly rearranged as relatedto an ancient angiosperm genome [42]. The fact thatpapaya, Clementine, grapevine and poplar are long lived,clonally propagated, woody plants, might apparentlycause a deceleration of their molecular clocks, resulting in
Table 5: Genomic BLASTN results with non-repetitive Citrus clementina BESs
Rice Arabidopsis Poplar
Hits 324 443 1,567Total length (kb)a 177.9 235.7 847.5Average e value 1.8E-09 1.4E-06 2.1E-07Average distance between hits (kb) 1,545.4 264.3 213.5Average n° of hits per chromosome 27 88.6 82.5
a Total length of the aligned regions
Page 8 of 12(page number not for citation purposes)
BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
genomes with higher resemblance to the ancestraleurosids genome [17].
ConclusionWe report here the construction of three genomic BAClibraries of Citrus clementina, with three restrictionenzymes (EcoRI, MboI and HindIII). The number of pickedBAC clones (56,000) and the average length of the insertsprovide coverage of 19.5 haploid genome equivalents,
ensuring a wide representation of the genome of this spe-cies. These libraries are adequate for the construction ofthe physical map of C. clementina. The analysis of 28.1 Mbof genomic sequence produced by BAC end sequencinghas provided a first insight of the genome organization ofC. clementina. The repetitive fraction of this species corre-sponding with transposable elements comprised 12.5%of the genome, while the gene number was estimated tobe 31,000. This work also describes a set of 3,814 SSRs
Table 6: Mapping of citrus BESs hits on poplar chromosomes
Chromosome Total Length (bp) n° BES mapped n° BES/Mb Average distance between BES
I 35,571,569 136 3.8 283.6II 24,482,572 138 5.6 192.7III 19,129,466 116 6.1 178.4IV 16,625,654 71 4.3 245.3V 17,991,592 78 4.3 239.9VI 18,519,121 83 4.5 239.6VII 12,805,987 63 4.9 208.3VIII 16,228,216 105 6.5 152.3IX 12,525,049 66 5.3 186.1X 21,101,489 80 3.8 282.4XI 15,120,528 111 7.3 152.1XII 14,142,880 64 4.5 222.2XIII 13,101,108 83 6.3 162.4XIV 14,699,529 112 7.6 134.2XV 10,599,685 52 4.9 208.2XVI 13,661,513 61 4.5 241.8XVII 6,060,117 24 4.0 241.4XVIII 13,470,992 75 5.6 186.5XIX 12,003,701 49 4.1 298.3
Average 82.5 5.2 213.5
Representation of the mapping of C. clementina BESs hits on poplar chromosomesFigure 6Representation of the mapping of C. clementina BESs hits on poplar chromosomes. Horizontal lines represent pop-lar chromosomes in a Mb scale and chromosome numbers are shown on the left. Vertical lines indicate the position of the C. clementina BES hits mapped on the poplar genome with BLASTN.
Page 9 of 12(page number not for citation purposes)

BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
and a collection of the first 6,617 putative SNPs describedin citrus that may be very useful for positional cloning,genetic and comparative mapping, nucleotide diversityanalysis, and association studies. Finally, comparativegenomics through gene homology searches has shownthat, in spite of their taxonomic classification, microsyn-teny between Citrus and Populus is higher than with Arabi-dopsis that is a phylogenetically closer species.
MethodsClementine genotypeCitrus clementina (Clementine mandarin, var. clemenules)developing leaves were used for BAC library construction.
BAC library constructionBAC libraries were constructed from high molecularweight (HMW) genomic DNA processed at AmpliconExpress, (Pullman, Washington) using the methoddescribed in [65]. DNA digestion was performed with var-ying amounts of MboI, EcoRI, and HindIII to identifyappropriate partial digestion conditions. pECBAC1 andcontained two FRT and one oriV elements, thus resultingin the pBAC(FRT-oriV) vector [66]. Ligations were trans-formed into DH10B Escherichia coli cells (Invitrogen) andplated on LB agar with chloramphenicol (30 μg/ml), X-gal(20 mg/ml) and IPTG (0.1 M). Clones were roboticallypicked with a Genomic Solution G3 into 384 well platescontaining LB freezing media. Plates were incubated for18 h, replicated and then frozen at -80°C. The replicatedcopy was used for BAC end sequencing.
Insert size estimationTo estimate insert sizes, 10 μl aliquots of BAC miniprepDNA were digested with 5 U of NotI enzyme for three h at37°C. The digestion products were separated by pulsed-field Weld gel electrophoresis (CHEF-DRIII system, Bio-Rad) in a 1% agarose gel in TBE buffer 0,5×. Insert sizeswere compared to those of the Lambda Ladder PFGMarker (New England Biolabs). Electrophoresis was car-ried out for 18 h at 14°C with an initial switch time of 5s, a Wnal switch time of 15 s, in a voltage gradient of 6 V/cm.
BAC End SequencingBAC clones were inoculated into 96-deep well macro-plates and grown for 20 hs at 37°C. Cells were harvestedby centrifugation and BACs were purified in 96-well platesby a standard alkaline lysis protocol developed by Geno-scope (Paris, France). BAC DNA was precipitated with iso-propanol and washed with 70% ethanol. Sequencing wascarried out on ABI3730 equipment with "Dye Termina-tor" process using ABI kit version 3.1. in the Genoscopefacility.
BioinformaticsThe software phred [67]was used for base calling, andCrossmatch for vector masking. Repetitive DNA was iden-tified with the RepeatMasker software [68], using theviridiplantae section of the RepBase Update [69] as data-base. Assembly was performed with CAP3 [56], using readquality and default parameters. Similarity searches wereperformed with the standalone version of BLAST [51,70],against the NCBI non redundant protein, nucleotide andEST databases available on November 2007 [71]. Parsingof the BLAST results was performed with the Bio::Sear-chIO module from the Bioperl package [72]. Codingsequences were annotated with GO terms using Blast2GO[48]. SPUTNIK [50] was used to identify simple sequencerepeats (SSRs), and POLYBAYES [62] to search for SNPs.
SNP validationDNA extraction was done from leaf tissues of C. clementinacv Nules using the DNeasy® Plant Mini Kit (Qiagen).
PCR amplifications of the samples were performed usinga Mastercycler epgradiend S thermocycler (Eppendorf) in100 μL final volume containing 0.025 U/μL of Pfu DNApolymerase (Fermentas), 0.2 ng/μL of genomic DNA, 0.2mM of each dNTP, 2 mM MgSO4, 75 mM Tris-HCl (pH8.8), 20 mM (NH4)2SO4, 0.2 μM of each primers. Thefollowing PCR program was applied: denaturation at 94°C for 5 min and 35 repeats of the following cycle: 30 s at94°C, 1 min at 55°C or 60°C (according to primers Tm),45 s at 72°C; and final elongation step of 4 min at 72°C.
PCR product purification was done directly or after cut-ting single bands on agarose gel, using respectivelyQIAquick® PCR Purification Kit and QIAquick® Gel Extrac-tion Kit (Qiagen)
Sequencing was carried out on ABI3730 equipment with"Dye Terminator" process using ABI kit version 3.1. SNPswere identified as double peaks by manual inspection ofchromatograms, and subsequently validated
Authors' contributionsJT was involved in BAC library construction and BAC endsequencing, performed the bioinformatic analyses anddrafted the manuscript. MAN performed BAC clone han-dling and was involved in BAC end sequencing. PO car-ried out SNP validation. MT coordinated the project anddrafted the manuscript.
Page 10 of 12(page number not for citation purposes)

BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
Additional material
AcknowledgementsWork at Centro de Genómica was supported by INIA grant RTA04-013, INCO contract 015453 and Ministerio de Educación y Ciencia grant AGL2007-65437-C04-01/AGR.
References1. Nicolosi E, Deng ZN, Gentile A, La Malfa S, Continella G, Tribulato E:
Citrus phylogeny and genetic origin of important species asinvestigated by molecular markers. Theor Appl Genet 2000,100:1155-1166.
2. Gmitter FG Jr: Origin, evolution and breeding of the grape-fruit. Plant Breeding Reviews 1995, 13:345-363.
3. Talon M, Gmitter FG Jr: Citrus Genomics. Int J Plant Genomics2008, 2008:528361.
4. Terol J, Conesa A, Colmenero JM, Cercos M, Tadeo F, Agusti J, et al.:Analysis of 13000 unique Citrus clusters associated with fruitquality, production and salinity tolerance. BMC Genomics 2007,8:31.
5. Forment J, Gadea J, Huerta L, Abizanda L, Agusti J, Alamar S, et al.:Development of a citrus genome-wide EST collection andcDNA microarray as resources for genomic studies. Plant MolBiol 2005, 57:375-391.
6. Chen C, Zhou P, Choi YA, Huang S, Gmitter FG: Mining and char-acterizing microsatellites from citrus ESTs. Theor Appl Genet2006, 112:1248-1257.
7. Barkley NA, Roose ML, Krueger RR, Federici CT: Assessinggenetic diversity and population structure in a citrus germ-plasm collection utilizing simple sequence repeat markers(SSRs). Theor Appl Genet 2006, 112:1519-1531.
8. Jiang D, Zhong GY, Hong QB: Analysis of microsatellites in cit-rus unigenes. Yi Chuan Xue Bao 2006, 33:345-353.
9. Ruiz C, Asins MJ: Comparison between Poncirus and Citrusgenetic linkage maps. Theor Appl Genet 2003, 106:826-836.
10. Deng Z, Tao Q, Chang L, Huang S, Ling P, Yu C, et al.: Constructionof a bacterial artificial chromosome (BAC) library for citrusand identification of BAC contigs containing resistance genecandidates. Theor Appl Genet 2001, 102:1177-1184.
11. Yang ZN, Ye XR, Choi S, Molina J, Moonan F, Wing RA, et al.: Con-struction of a 1.2-Mb contig including the citrus tristeza virusresistance gene locus using a bacterial artificial chromosomelibrary of Poncirus trifoliata (L.) Raf. Genome 2001, 44:382-393.
12. Fagoaga C, Tadeo FR, Iglesias DJ, Huerta L, Lliso I, Vidal AM, et al.:Engineering of gibberellin levels in citrus by sense and anti-sense overexpression of a GA 20-oxidase gene modifies plantarchitecture. J Exp Bot 2007, 58:1407-1420.
13. Katz E, Fon M, Lee YJ, Phinney BS, Sadka A, Blumwald E: The citrusfruit proteome: insights into citrus fruit metabolism. Planta2007, 226:989-1005.
14. Lliso I, Tadeo FR, Phinney BS, Wilkerson CG, Talon M: Proteinchanges in the albedo of citrus fruits on postharvesting stor-age. J Agric Food Chem 2007, 55:9047-9053.
15. Tadeo F, Cercos M, Colmenero-Flores JM, Iglesias DJ, Naranjo MA,Rios G, et al.: Molecular physiology of development and qualityof citrus. Advances in Botanical Research 2008 in press.
16. Town CD: Large-scale DNA sequencing. In The handbook of PlantGenome Mapping Edited by: Meksem K, Kahl G. Wiley-VCH;2005:337-351.
17. Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U,et al.: The Genome of Black Cottonwood, Populus tri-chocarpa (Torr. & Gray). Science 2006, 313:1596-1604.
18. The Arabidopsis Genome Initiative A: Analysis of the genomesequence of the flowering plant Arabidopsis thaliana. Nature2000, 408:796-815.
19. International Rice Genome Sequencing Project: The map-basedsequence of the rice genome. Nature 2005, 436:793-800.
20. Lai C, Yu Q, Hou S, Skelton R, Jones M, Lewis K, et al.: Analysis ofpapaya BAC end sequences reveals first insights into theorganization of a fruit tree genome. Mol Genet Genomics 2006,276:1-12.
21. Cheung F, Town CD: A BAC end view of the Musa Accuminatagenome. BMC Plant Biol 2007, 7:29.
22. Hong CP, Plaha P, Koo DH, Yang TJ, Choi SR, Lee YK, et al.: A Sur-vey of the Brassica rapa Genome by BAC-End SequenceAnalysis and Comparison with Arabidopsis thaliana. Mol Cells2006, 22:300-307.
23. Frelichowski JE Jr, Palmer MB, Main D, Tomkins JP, Cantrell RG, StellyDM, et al.: Cotton genome mapping with new microsatellitesfrom Acala 'Maxxa' BAC-ends. Mol Genet Genomics 2006,275:479-491.
24. Marek LF, Mudge J, Darnielle L, Grant D, Hanson N, Paz M, et al.: Soy-bean genomic survey: BAC-end sequences near RFLP andSSR markers. Genome 2001, 44:572-581.
25. Shultz JL, Samreen K, Rabia B, Jawaad AA, Lightfoot DA: The devel-opment of BAC-end sequence-based microsatellite markersand placement in the physical and genetic maps of soybean.Theor Appl Genet 2007, 114:1081-1090.
26. Coyne CJ, McClendon MT, Walling JG, Timmerman-Vaughan GM,Murray S, Meksem K, et al.: Construction and characterizationof two bacterial artificial chromosome libraries of pea(Pisum sativum L.) for the isolation of economically impor-tant genes. Genome 2007, 50:871-875.
27. Nam W, Penmetsa RV, Endre G, Uribe P, Kim D, Cook DR: Con-struction of a bacterial artificial chromosome library of Med-icago truncatula and identification of clones containingethylene-response genes. Theor Appl Genet 1999, 98:638-646.
28. Liang H, Fang E, Tomkins J, Luo M, Kudrna D, Kim H, et al.: Devel-opment of a BAC library for yellow-poplar (Liriodendron tul-ipifera) and the identification of genes associated with flowerdevelopment and lignin biosynthesis. Tree Genetics & Genomes2007, 3:215-225.
29. Han Y, Gasic K, Marron B, Beever JE, Korban SS: A BAC-basedphysical map of the apple genome. Genomics 2007, 89:630-637.
30. Wu C, Sun S, Nimmakayala P, Santos FA, Meksem K, Springman R, etal.: A BAC- and BIBAC-Based Physical Map of the SoybeanGenome. Genome Res. 2004 Feb;14(2):319-26 2004, 14(2):319-326.
31. Yim YS, Davis GL, Duru NA, Musket TA, Linton EW, Messing JW, etal.: Characterization of Three Maize Bacterial Artificial
Additional File 1Comparative analysis of the 3 BAC libraries. GC content and number of BESs carrying repetitive elements or coding regions are shown for each one of the libraries constructed.Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2164-9-423-S1.doc]
Additional file 2GO Annotations of the coding regions found in BESs. The table shows the GO terms associated with the coding regions identified on the BESs, anno-tated with B2GO. The description of the GO term, as well as the number sequences associated with each term are shown.Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2164-9-423-S2.xls]
Additional File 3SNP Validation summary. The table shows the details concerning the 24 experimentally validated SNPs, indicating the SNP name (SNP_ID), the contig name (CONTIG) and length (CONTIG LENGTH), the consensus contig sequence (CONT SEQUENCE), the position of the SNP in the consensus sequence (SNP POSITION), the probability of the predicted SNP (P_SNP), the alleles found (ALLELE 1, ALLELE 2), and the prim-ers used for genomic DNA amplification (/FORWARD PRIMER, REVERSE PRIMER).Click here for file[http://www.biomedcentral.com/content/supplementary/1471-2164-9-423-S3.xls]
Page 11 of 12(page number not for citation purposes)

BMC Genomics 2008, 9:423 http://www.biomedcentral.com/1471-2164/9/423
Chromosome Libraries toward Anchoring of the PhysicalMap to the Genetic Map Using High-Density Bacterial Arti-ficial Chromosome Filter Hybridization. Plant Physiol 2002,130:1686-1696.
32. Cenci A, Chantret N, Kong X, Gu Y, Anderson OD, Fahima T, et al.:Construction and characterization of a half million cloneBAC library of durum wheat (Triticum turgidum ssp.durum). Theor Appl Genet 2003, 107:931-939.
33. Cercos M, Soler G, Iglesias D, Gadea J, Forment J, Talon M: GlobalAnalysis of Gene Expression During Development and Rip-ening of Citrus Fruit Flesh. A Proposed Mechanism for CitricAcid Utilization. Plant Mol Biol 2006, 62:513-527.
34. Alos E, Cercos M, Rodrigo MJ, Zacarias L, Talon M: Regulation ofcolor break in citrus fruits. Changes in pigment profiling andgene expression induced by gibberellins and nitrate, two rip-ening retardants. J Agric Food Chem 2006, 54:4888-4895.
35. Sun S, Xu Z, Wu C, Ding K, Zhang HB: Genome properties andtheir influences on library construction and physical map-ping. Proceedings of Plant and Animal Genome XI Conference 2003:P77.
36. Ren C, Xu Z, Sun S, Lee MK, Wu C, Scheuring C, et al.: GenomicDNA Libraries and Physical Mapping. In The handbook of plantgenome mapping Edited by: Meksem K, Kahl G. Wiley-VCH;2005:173-213.
37. Chang YL, Tao Q, Scheuring C, Ding K, Meksem K, Zhang HB: Anintegrated map of Arabidopsis thaliana for functional analy-sis of its genome sequence. Genetics 2001, 159:1231-1242.
38. Chen M, Presting G, Barbazuk WB, Goicoechea JL, Blackmon B, FangG, et al.: An integrated physical and genetic map of the ricegenome. Plant Cell 2002, 14:537-545.
39. Xu Z, Sun S, Covaleda L, Ding K, Zhang A, Wu C, et al.: Genomephysical mapping with large-insert bacterial clones by finger-print analysis: methodologies, source clone genome cover-age, and contig map quality. Genomics 2004, 84:941-951.
40. Bausher M, Singh N, Lee SB, Jansen R, Daniell H: The completechloroplast genome sequence of Citrus sinensis (L.) Osbeckvar 'Ridge Pineapple': organization and phylogenetic rela-tionships to other angiosperms. BMC Plant Biology 2006, 6:21.
41. Unseld M, Marienfeld JR, Brandt P, Brennicke A: The mitochondrialgenome of Arabidopsis thaliana contains 57 genes in 366,924nucleotides. Nat Genet 1997, 15:57-61.
42. Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, et al.:The grapevine genome sequence suggests ancestral hexa-ploidization in major angiosperm phyla. Nature 2007,449:463-467.
43. Rico-Cabanas L, Martinez-Izquierdo JA: CIRE1, a novel transcrip-tionally active Ty1-copia retrotransposon from Citrus sinen-sis. Mol Genet Genomics 2007, 277:365-377.
44. Bernet GP, Asins MJ: Identification and genomic distribution ofgypsy like retrotransposons in Citrus and Poncirus. Theor ApplGenet 2003, 108:121-130.
45. Yuan Q, Ouyang S, Liu J, Suh B, Cheung F, Sultana R, et al.: The TIGRrice genome annotation resource: annotating the ricegenome and creating resources for plant biologists. Nucl AcidsRes 2003, 31:229-233.
46. Bogunic F, Muratovic E, Brown SC, Siljak-Yakovlev S: Genome sizeand base composition of five Pinus species from the Balkanregion. Plant Cell Reports 2003, 22:59-63.
47. Carels N, Hatey P, Jabbari K, Bernardi G: Compositional Proper-ties of Homologous Coding Sequences from Plants. J Mol Evol1998, 46:45-53.
48. Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M:Blast2GO: a universal tool for annotation, visualization andanalysis in functional genomics research. Bioinformatics 2005,21:3674-3676.
49. Clement D, Lanaud C, Sabau X, Fouet O, Cunff LL, Ruiz E, et al.: Cre-ation of BAC genomic resources for cocoa (Theobromacacao L.) for physical mapping of RGA containing BACclones. Theor Appl Genet 2004, 108:1627-1634.
50. Abajian C: SPUTNIK. Computer Program 1994.51. Chen C, Zhou P, Choi YA, Huang S, Gmitter FG Jr: Mining and
characterizing microsatellites from citrus ESTs. Theor ApplGenet 2006, 112(7):1248-1257.
52. Kijas JMH, Thomas MR, Fowler JCS, Roose ML: Integration of tri-nucleotide microsatellites into a linkage map of Citrus. TheorAppl Genet 1997, 94:701-706.
53. Varshney RK, Grosse I, H+ U, Siefken R, Prasad M, Stein N, et al.:Genetic mapping and BAC assignment of EST-derived SSRmarkers shows non-uniform distribution of genes in the bar-ley genome. Theor Appl Genet 2006, 113:239-250.
54. Mun JH, Kim DJ, Choi HK, Gish J, Debelle F, Mudge J, et al.: Distri-bution of microsatellites in the genome of Medicago trunca-tula: a resource of genetic markers that integrate geneticand physical maps. Genetics 2006, 172:2541-2555.
55. Feingold S, Lloyd J, Norero N, Bonierbale M, Lorenzen J: Mappingand characterization of new EST-derived microsatellites forpotato (Solanum tuberosum L.). Theor Appl Genet 2005,111:456-466.
56. Huang X, Madan A: CAP3: A DNA sequence assembly pro-gram. Genome Res 1999, 9:868-877.
57. Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, PrussD, et al.: A High Quality Draft Consensus Sequence of theGenome of a Heterozygous Grapevine Variety. PLoS ONE2007, 2:e1326.
58. Rafalski A: Applications of single nucleotide polymorphisms incrop genetics. Curr Opin Plant Biol 2002, 5:94-100.
59. Weil MM, Pershad R, Wang R, Zhao S: Use of BAC end sequencesfor SNP discovery. Methods Mol Biol 2004, 256:1-6.
60. Abe K, Noguchi H, Tagawa K, Yuzuriha M, Toyoda A, Kojima T, et al.:Contribution of Asian mouse subspecies Mus musculusmolossinus to genomic constitution of strain C57BL/6J, asdefined by BAC-end sequence-SNP analysis. Genome Res 2004,14:2439-2447.
61. Yamamoto K, Narukawa J, Kadono-Okuda K, Nohata J, Sasanuma M,Suetsugu Y, et al.: Construction of a Single Nucleotide Poly-morphism Linkage Map for the Silkworm, Bombyx mori,Based on Bacterial Artificial Chromosome End Sequences.Genetics 2006, 173:151-161.
62. Marth GT, Korf I, Yandell MD, Yeh RT, Gu Z, Zakeri H, et al.: A gen-eral approach to single-nucleotide polymorphism discovery.Nat Genet 1999, 23:452-456.
63. Pavy N, Parsons LS, Paule C, MacKay J, Bousquet J: AutomatedSNP detection from a large collection of white spruceexpressed sequences: contributing factors and approachesfor the categorization of SNPs. BMC Genomics 2006,7(174):174.
64. Wikström N, Savolainen V, Chase MW: Evolution of theangiosperms: calibrating the family tree. Proceedings of theRoyal Society B: Biological Sciences 2001, 268:2211-2220.
65. Tao Q, Wang A, Zhang HB: One large-insert plant-transforma-tion-competent BIBAC library and three BAC libraries ofJaponica rice for genome research in rice and other grasses.Theor Appl Genet 2002, 105:1058-1066.
66. Frijters CJ, Zhang Z, Damme Mv, Wang L, Ronald PC, MichelmoreRW: Construction of a bacterial artificial chromosomelibrary containing large Eco RI and Hin dIII genomic frag-ments of lettuce. Theor Appl Genet 1997, 94:390-399.
67. Ewing B, Green P: Base-Calling of Automated SequencerTraces Using Phred. II Error Probabilities. Genome Res 1998,8:186-194.
68. Smit AFA, Hubley R, Green P: RepeatMasker Open-3.0. ComputerProgram 1996.
69. Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichie-wicz J: Repbase Update, a database of eukaryotic repetitiveelements. Cytogenet Genome Res 2005, 110:462-467.
70. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic localalignment search tool. J Mol Biol 1990, 215:403-410.
71. National Center for Biotechnology Information. ElectronicCitation .
72. Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C,et al.: The Bioperl toolkit: Perl modules for the life sciences.Genome Res 2002, 12:1611-1618.
Page 12 of 12(page number not for citation purposes)