zwiĄzki - pg.gda.pl · elektronowej i geometrii związków aromatycznych. zgodnie z tą regułą...
TRANSCRIPT

AROMATYCZNE ZWIĄZKI
HETEROCYKLICZNE

Aromatyczne związki heterocykliczne(pięcio- lub sześcioczłonowe) – cykliczne związki
zawierające atom inny niż atom węgla,
np. N, O, S oraz spełniające regułę Hückla.
Reguła Hückla opisuje wymagania dotyczące struktury elektronowej i geometrii związków aromatycznych.
Zgodnie z tą regułą związkami aromatycznymi są płaskie związki pierścieniowe, w których znajduje się 2, 6, 10, 14, 18…. sprzężonych elektronów p (określonej wzorem
4n + 2, gdzie n jest liczbą naturalną = 0,1,2,3,4…).

Przykłady
NH
N
NH
N N
N
N N
N NH
N
pirol imidazol pirydyna chinolina
pirymidyna puryna
N
akrydyna

Pięcioczłonowe aromatyczne związki heterocyklicznepirol, furan i tiofen są bezbarwnymi, trudno rozpuszczalnymi w wodzie cieczami, natomiast rozpuszczają się w większości rozpuszczalników
organicznych. Znajdują się w wielu związkach naturalnych: kwasy nukleinowe, aminokwasy, alkaloidy purynowe, alkaloidy sporyszu czy
aminy biogenne.
O S
furan tiofen
NH
pirol

OCH2
ON
N
N
N
NH2
XO
PO
O
O
CH2 O
X
N
N
NH2
O
O
PO
O
O
CH2 O
X
N
NH
O
OR
O
PO
O
O
DNA: X = H; R = CH3
RNA: X = OH; R = H

Alkaloidy purynowe
N
N NH
N
puryna
N
N NH
N
kofeina
O
O
O
H3C
CH3
CH3
N
NH
CH2CHCOOH
histydyna (His, H)
CH2CHCOOH
tryptofan (Trp, W)
Aminokwasy
NH2
NH2
NH

HN
N
CH2CH2
NH2
histamina
Aminy biogenne
NH
HOCH2CH2NH2
serotonina
Alkaloidy sporyszu
HN
NCH3
COOH
H
kwas lizerginowy
N
OO
N
H
H
H
H
strychnina

W aromatycznych związkach heterocyklicznych wolna para elektronowa na heteroatomie, tak jak elektrony na
atomach węgla sp2 zajmuje orbital p, prostopadły do płaszczyzny pierścienia.
Z Z Z Z Z
Wzory mezomeryczne wynikające ze sprzężenia elektronów π i wolnej pary elektronowej przypominają wzory
mezomeryczne benzenu, nie są one jednak równocenne.

Cząsteczki heterocykli, w przeciwieństwie do cząsteczki benzenu, wykazują moment dipolowy (m), ponieważ w położeniach α i β
zgromadzony jest ładunek ujemny, a na heteroatomie dodatni. Np. furan wykazuje moment dipolowy równy 0.7 D skierowany zgodnie
z oczekiwaniem, tzn. z biegunem dodatnim na atomie tlenu.
O O
O O
furan
furan tetrahydrofuran
m = 0.7 D 1.7 D
-
-
-
+
++
-
-
tetrahydrofuran

■ Otrzymywanie – ogrzewanie związków 1,4-dikarbonylowych z odpowiednimi reagentami.
OH3C CH3
NH3C CH3
SH3C CH3
H
2,4-dimetylofuran
2,4-dimetylopirol
2,4-dimetylotiofen
H3C CH3O O
P2O5
(NH4)2CO3
100oC
P4S10
heksa-2,5-dion

Pirol
O
furan
NH
pirol
NH3 aq
Al2O3, 400 oC
(OH)CH CH(OH)
(OH)HC
NH4OOC
CH(OH)
COONH4
- 2 C
O 2, -
NH 3
, - H
OH
galaktaronian amonukwas śluzowy
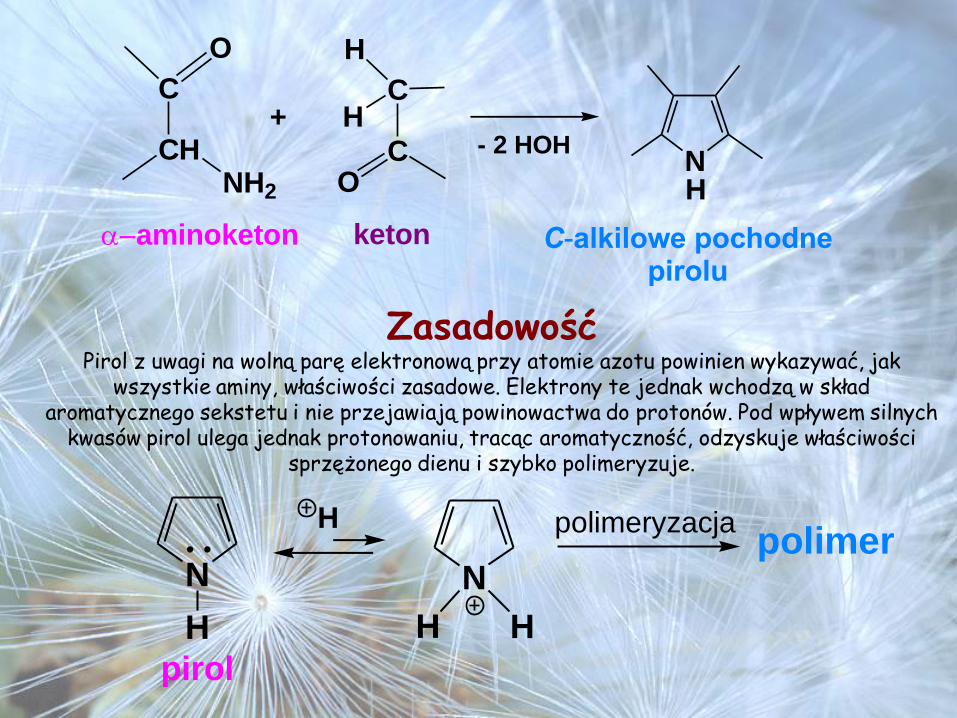
NH
C
O
CHNH2
C
H
C
H
O
- 2 HOH
C-alkilowe pochodnepirolu
aminoketon keton
+
ZasadowośćPirol z uwagi na wolną parę elektronową przy atomie azotu powinien wykazywać, jak
wszystkie aminy, właściwości zasadowe. Elektrony te jednak wchodzą w skład aromatycznego sekstetu i nie przejawiają powinowactwa do protonów. Pod wpływem silnych
kwasów pirol ulega jednak protonowaniu, tracąc aromatyczność, odzyskuje właściwości sprzężonego dienu i szybko polimeryzuje.
N N
H H H
H
pirol
polimerpolimeryzacja

KwasowośćAtom wodoru związany z atomem azotu pirolu jest znacznie bardziej kwaśny (pKa=17.5) niż w amoniaku (pKa=36). Pirol tworzy sole z wodorotlenkami metali alkalicznych, a ze związkami Grignarda reaguje, tak jak kwasy czy związki o
właściwościach kwasowych.
pirol
NH
KOH
CH3MgBr
N
N
K HOH
CH4
MgBr metan
+
+
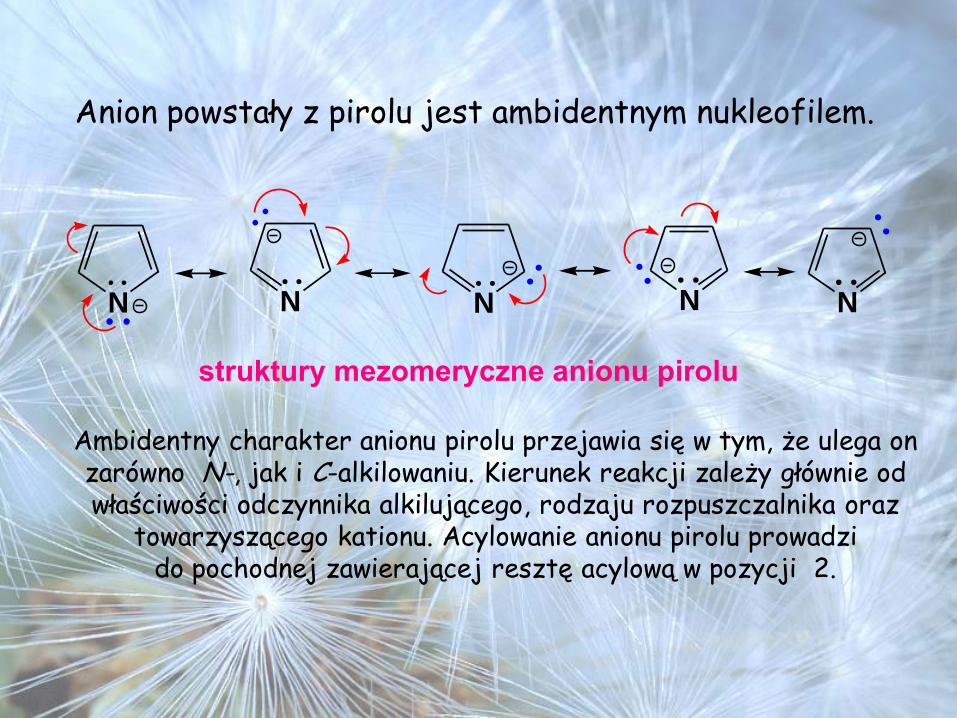
Anion powstały z pirolu jest ambidentnym nukleofilem.
N N N N N
struktury mezomeryczne anionu pirolu
Ambidentny charakter anionu pirolu przejawia się w tym, że ulega on zarówno N-, jak i C-alkilowaniu. Kierunek reakcji zależy głównie od właściwości odczynnika alkilującego, rodzaju rozpuszczalnika oraz
towarzyszącego kationu. Acylowanie anionu pirolu prowadzi do pochodnej zawierającej resztę acylową w pozycji 2.

N
Me
N
H
N
H
N
H
N MgBr
Me
Me
C CH3
O
AcClTosOMe
N
Me
N-metylopirol
2-metylopirol
3-metylopirol
N-metylopirol
2-acetylopirol
Mel

Furan● dekarbonylowanie (eliminacja CO) furfuralu:
HOCH CHOH
HOCH2 CH(OH)
CH
- HOHO CHO O
H,
- CO
, kat.
furanfurfuralpentoza
O
Tiofen
1,3-butadien- H2S S
tiofen
S, 600 oC
etyn
S, 300
oC
+
n-butan
560 oC
S
P4S10
HO OHO O
kwasbursztynowy

Silne zasady, np. BuLi, PhNa są w stanie oderwać protony z pierścienia N-alkilopirolu, furanu i tiofenu. Deprotonacji w pierwszej kolejności ulega proton w pozycji 2, a następnie 4. Deprotonowane heterocykle (aniony) wchodzą w typowe reakcje, np. acylowania czy alkilowania.
+O - C6H6 O O2. H
1. CO2
furan kwas furano-2-karboksylowy
PhNa Na
fenylosód
COOH
S
BuLi
S S2. H
1. CO2
3-metylotiofen kwas 4-metylotiofeno-2-karboksylowy
HOOC
CH3CH3
Li
CH3

Nasycone, pięcioczłonowe związki heterocykliczne
NH
NH
pirol
H2/Ni
pirolidyna
O O
H2/Ni
tetrahydrofuran (THF)furan
S
tetrahydrotiofen1,4-dibromo-butan
BrBr
Na2S
pKa 14 pKa 3

Substytucja elektrofilowa (SE)Pirol, furan i tiofen ulegają SE znacznie łatwiej niż benzen.
Obecność heteroatomu w pierścieniu aktywuje te związki w reakcjach SE. Halogenowanie, nitrowanie, sulfonowanie czy
acylowanie heterocykli pięcioczłonowych prowadzi się w niskich temperaturach żeby zapewnić selektywność reakcji (ograniczyć
podstawienie kolejnych atomów wodoru). Monopodstawienie następuje w pozycji 2, pod warunkiem, że nie jest ona zajęta.
Szereg reaktywności: tiofen > pirol > furan.

O+ Br2
0 oC
dioksan
O Br+ HBr
furan 2-bromofuran
O+ SO3
0 oC
pirydyna
O SO3H
kwas furano-2--sulfonowy
furan
Halogenowanie – nie wymaga stosowania katalizatora.
Sulfonowanie – czynnikiem sulfonującym jest tritlenek siarki. Kwas siarkowy powoduje polimeryzację pirolu i furanu.

NH
NH
NO2HNO3
0 oC
Ac2O+
pirol 2-nitropirol
2-nitropirol (61%)
NH
NH
NO2
AcONO2-10 oC
+
pirol
+NH
NO2
3-nitropirol (13%)
2-nitriotiofen
S+ AcONO2
0 oC S NO2
tiofen
Nitrowanie – w mieszaninie nitrującej zamiast kwasu siarkowego
stosuje się bezwodnik octowy, którego głównym zadaniem jest wiązanie wydzielającej się wody. Można również wykorzystać octan nitrozylu.

O Br
Br2
dioksan
O NO2
HNO3Ac2O
O CCH3
CH3COClSnCl4
O
H2 / Pd
O
O SO3H
SO3
pirydyna
furan
2-nitrofuran tetrahydrofuran 2-acetylofuran
2-bromofurankwas
furano-2-sulfonowy
O

ZZ: O; S lub NH
atak napozycję 2
atak napozycję 3
Z E
H
Z E
H
Z E
H
Z
E
H
Z
E
H
E
Do substytucji dochodzi w pozycji 2.
Dla adduktu w pozycji 3 można
napisać tylko 2 struktury
mezomeryczne, przy czym druga
jest niekorzystna energetycznie,
ponieważ ładunek dodatni jest
zlokalizowany na heteroatomie,
czyli bardziej elektroujemnym
atomie niż atom węgla.

W indolu, analogu pirolu do substytucji SE dochodzi w pozycji 3 pierścienia heterocyklicznego, ponieważ addukt w tej pozycji zapewnia pełny sekstet elektronów π w pierścieniu benzenowym.
NH
atak napozycję 2
atak napozycję 3
E
NH
E
H
NH
E
H
NH
E
H
NH
E
H
NH
HE
NH
HE
indol

Acylowanie – prowadzi się typowymi odczynnikami acylującymi, a więc chlorkami kwasowymi i bezwodnikami, można jednak stosować inne niż chlorek glinu – katalizatory.
furan
O O
Ac2O
BF3 CCH3
O
2-acetylofuran
S S
PhCOCl
SnCl4 C
O
2-benzoilotiofen
tiofen

Sprzęganie – podstawienie grupą azową następuje w najbardziej reaktywnym miejscu,
czyli w pozycji 2.
NH
NH
SnCl4
N N Cl
N N
pirol 2-(fenyloazo)pirol

BARWNIKI PIROLOWE
Barwniki dipirylometenowe – reakcja metanalu z pirolem prowadzi do produktów kondensacji.Wykorzystywane są one jako barwniki, tzw. barwniki dipirylometenowe (zabarwienie żółte lub pomarańczowe).
N CH2OHH
NH
+ CH2OOH
2-hydroksymetylopirolpirol

W środowisku kwaśnym powstały alkohol 1o reaguje z drugą cząsteczką pirolu lub jego homologiem tworząc dipirylometan.
N CH2OHH
N CH3H
N CH2H
H3C
CH3
+
H3C
N CH3H
CH3
H3C
CH3
H
2,5-dimetylopirol2-hydroksymetylo-3,5-dimetylopirol
2,5,2',5'-tetrametylo-pirylometan
[O]
N CHH
N CH3
CH3
H3C
CH3
2,5,2',5'-tetrametylopirylometen

N CHH
N R
R
R
R
N CHH
NH
R
R
R
R
NH
CH N R
H
R
R
R
.. ..
HBr
dipirylometeny - są silnymi zasadami
i z kwasami dają łatwo krystalizujące sole
bromek dipirylometeniowy
Br
....

Synteza indygo – ciemnobłękitny barwnik występujący zarówno naturalnie (liście indygowca barwierskiego – rośliny występującej w Indiach,
także rdestu ptasiego), jak i otrzymywany syntetycznie. W XIX wieku opracowano metodę
syntezy indygo z kwasu antranilowego i chlorooctowego.

N
COOH
NH2
+ ClCH2COOH2. H+
1. Na2CO3COOH
NH
CH2COOH
kwas antranilowy
N-alkilowanie
o-karboksy-N-fenyloglicyna
N
O
OH
H
NN
O OH
H HN
OH
H
COOK- CO2
dekarbo-ksylacja
cyklizacja KOH 260 oC
indoksyl
O2utlenianie
indygo
kwas chlorooctowy
C
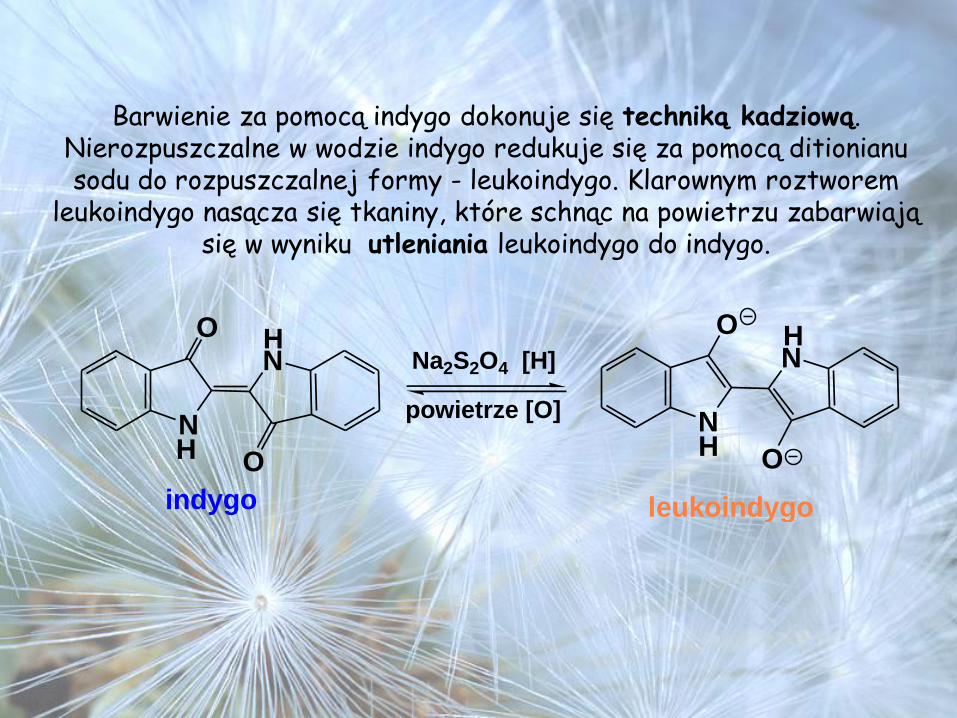
Barwienie za pomocą indygo dokonuje się techniką kadziową. Nierozpuszczalne w wodzie indygo redukuje się za pomocą ditionianu sodu do rozpuszczalnej formy - leukoindygo. Klarownym roztworem
leukoindygo nasącza się tkaniny, które schnąc na powietrzu zabarwiają się w wyniku utleniania leukoindygo do indygo.
N
N
O
OH
H
indygo
N
N
O
OH
HNa2S2O4 [H]
powietrze [O]
leukoindygo

Sześcioczłonowe aromatyczne związki heterocykliczne
N O O
pirydyna piran piran
OO O
R
R
R R
X
dialkilowe pochodne piranów
kation piryliowy
(tlenowe analogi, nietrwałe)

Pirydyna – bezbarwna ciecz o nieprzyjemnym,
charakterystycznym zapachu, miesza się z wodą w każdymstosunku. Jest silnie higroskopijna tworząc z wodą monohydratróżniący się właściwościami fizykochemicznymi od czystejpirydyny. Wykazuje właściwości słabo zasadowe, z mocnymikwasami tworzy krystaliczne sole pirydyniowe. Aromatycznośćpirydyny wynika z planarnej budowy jej cząsteczki i sprzężenia 6elektronów π. Pięć atomów węgla i jeden atom azotu w pierścieniupirydyny mają hybrydyzację sp2. Wolna para elektronów atomuazotu znajduje się na orbitalu sp2, a więc inaczej niż w pirolu i niebierze udziału w sprzężeniu.Pierścień pirydynowy występuje w wielu ważnych związkachorganicznych, np. kokainie, atropinie, witaminie PP czy B6.

N
N
H
H
H H
H wolna para
na orbitalu sp2
pirydyna
.
...
.
Otrzymywanie pirydyny – głównym źródłem pirydyny jest smoła pogazowa (jej zawartość dochodzi do 0.1%). Wyodrębnia się ją za pomocą destylacji. We frakcji zwanej olejem lekkim pirydynie towarzyszą jej homologi: metylopirydyny (zwane pikolinami), dimetylopirydyny oraz trimetylopirydyny (tzw. lutydyny i kolidyny).
Kąty pomiędzy wiązaniami C-C i C-N wynoszą po 120o, wiązania
tworzące pierścień są jednakowej długości - 1.39 Å.
N N
wzory mezomeryczne pirydyny

Reakcje substytucji elektrofilowej (SE)Pirydyna jest znacznie mniej aktywna w reakcjach substytucjielektrofilowej, nie tylko w porównaniu z aromatycznymi heterocyklamipięcioczłonowymi, ale również w porównaniu z benzenem. Reakcje SE
pirydyny wymagają drastycznych warunków (wysokiej temperatury). Ponadtoreakcje SE biegną często w środowisku kwaśnym, co powoduje protonowanieatomu azotu i utworzenie ładunku dodatniego, a wiec jeszcze silniejsządezaktywację.
pirydyna
reaktywność w reakcji SE
benzen
N
jon pirydyniowy
H
N

N
300oC
Br2
H2SO4, 220OC
SO3, HgSO4
NaNO3, 370OC
HNO3
RX lub RCOX
AlCl3
N
Br
pirydyna
+ HBr
N
SO3H
N
NO2
kwas 3-pirydynosulfonowy
3-nitropirydyna
+ HOH
brak reakcji
3-bromopirydyna
(w reakcjach Friedla-Craftsa pirydyna jest nieaktywna)

NH
N
pirydyna
pKb = 2.8pKb = 8.8
piperydyna
pirydyna jest słabszą zasadą niż piperydyna
NN
+
+
++
+
Hpirydyna
protonowana
jon pirydyniowy
m = 2.26 D

SE zachodzi w pozycji 3, ponieważ
pozycja 2 i 4 są dezaktywowane. We
wzorach mezomerycznych po ataku
odczynnika elektrofilowego na pozycję
2 i 4 obserwuje się ładunek dodatni na
atomie azotu, co jest niekorzystne, z
uwagi na to, że atom azotu jest
bardziej elektroujemny od atomu
węgla.N
E
H
N
E
H
N
E
H
N
E
H
N
E
H
N
E
H
N
E H
N
E H
N
E H
E
2 3 4
N

Reakcje substytucji nukleofilowej (SN) –pozycje 2, 4 i 6 są podatne na atak silnych nukleofili.
N
Na+OEt-
Cl
N
OC2H5
EtOH
4-chloropirydyna 4-etoksypirydyna
N
NaNH2
NNH3
2-bromopirydyna 2-aminopirydyna
Br NH2
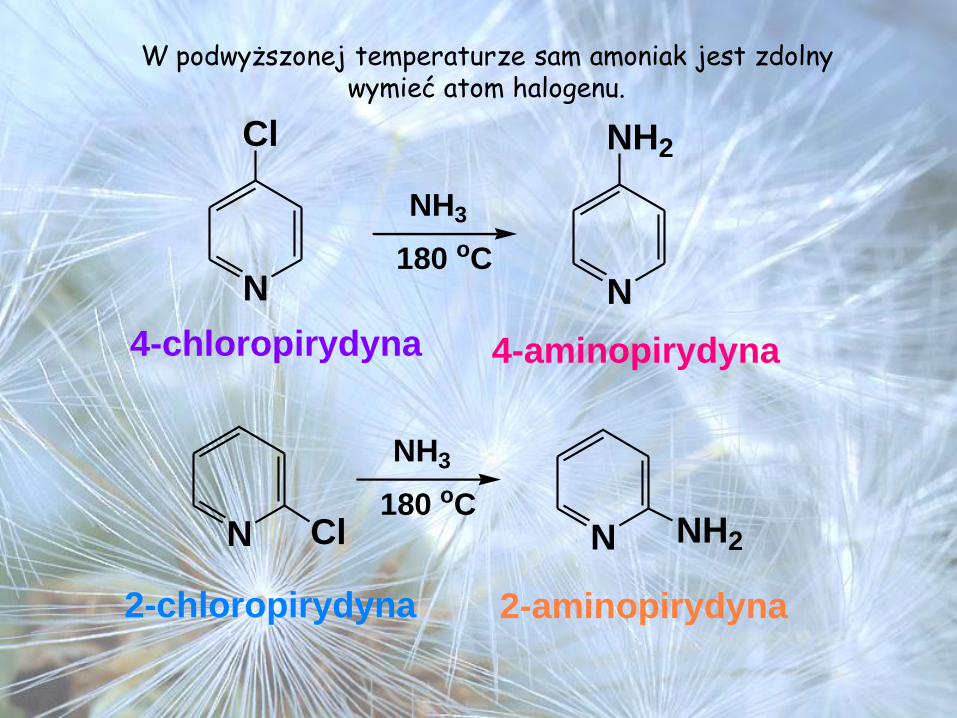
W podwyższonej temperaturze sam amoniak jest zdolny wymieć atom halogenu.
N
NH3
Cl
N
NH2
180 oC
4-chloropirydyna 4-aminopirydyna
N
NH3
N180 oC
2-chloropirydyna 2-aminopirydyna
Cl NH2
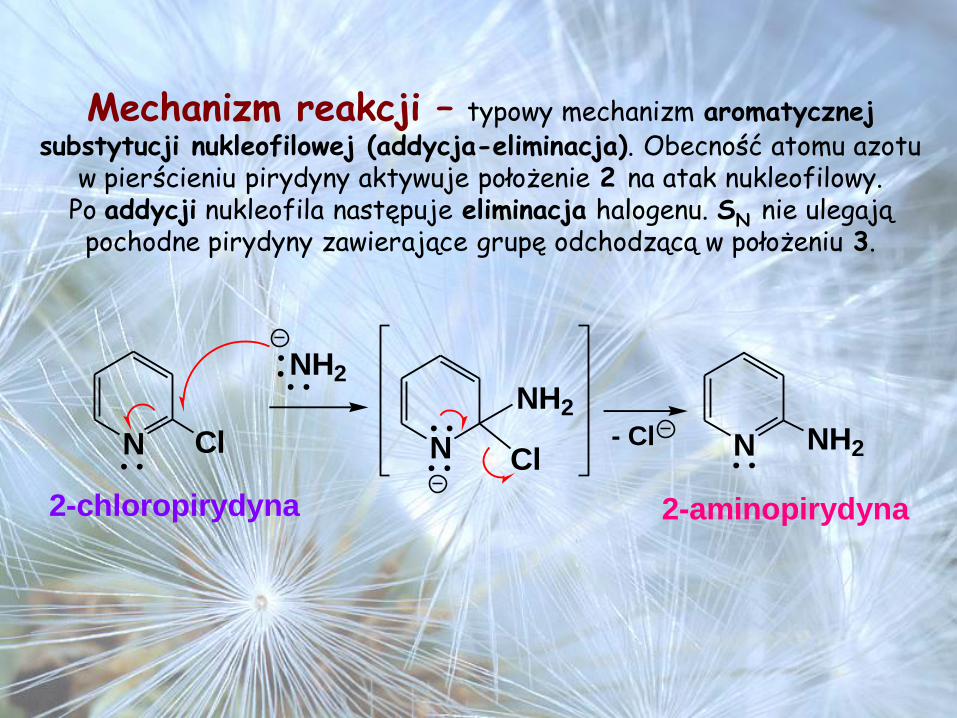
Mechanizm reakcji – typowy mechanizm aromatycznej substytucji nukleofilowej (addycja-eliminacja). Obecność atomu azotu
w pierścieniu pirydyny aktywuje położenie 2 na atak nukleofilowy. Po addycji nukleofila następuje eliminacja halogenu. SN nie ulegają
pochodne pirydyny zawierające grupę odchodzącą w położeniu 3.
N Cl N
NH2
N NH2
NH2
Cl- Cl
2-chloropirydyna 2-aminopirydyna

PytanieJaki produkt otrzymasz w reakcji 3-bromopirydyny z amidkiem sodu ?
Odpowiedź
N
BrH
NH2
N
Br
3-bromopirydyna
- BrN
NH2
N
NH2
+
N
NH2
N
NH2
+
N
NH2
NH3
4-aminopirydyna
3-aminopirydyna

Reakcja Cziczibabina – pod wpływem silnych nukleofili dochodzi do podstawienia atomu wodoru w samej pirydynie.
N
1. NaNH2
2. HOHN NH2
H2+
N
PhLi
100oCN
pirydyna
2-fenylopirydyna
pirydyna 2-aminopirydyna

Mechanizm reakcji
N
NH2
N
NH2
N NH
H+
H
- H+
Hpirydyna
- H2
N NH2 N NHN NH
HOH
2-aminopirydyna
anion aminopirydyniowy

Reakcja 2-pikoliny z aldehydami – atomy α-wodoru w alkilopirydynach mają właściwości
kwasowe i pod wpływem zasad ulegają odszczepieniu. Powstały w ten sposób karboanion reaguje ze
związkami karbonylowymi jako donor elektronów dając hydroksypochodne, które łatwo tracą
cząsteczkę wody tworząc nowe wiązanie C=C, sprzężone z aromatycznymi elektronami π.

N CH3N CH
N CH2
N CH2
N CH
NaOH
HC
O
CH
2-pikolina1-fenylo-2-(2-pirydylo)eten
OH
HC
O
CH
H OH
- HOH
1-fenylo-1-hydroksy-2-(2-pirydylo)etan

Hydroksypirydyny
N OH
2-hydroksypirydyna
N
3-hydroksypirydyna
OH
N
4-hydroksypirydyna
OH
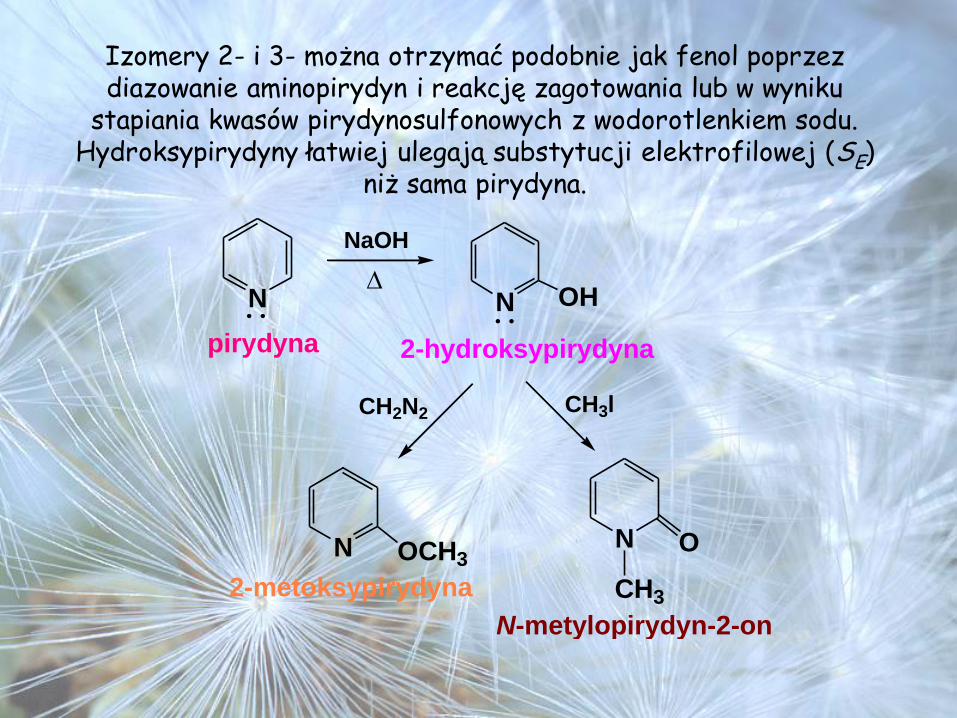
Izomery 2- i 3- można otrzymać podobnie jak fenol poprzez diazowanie aminopirydyn i reakcję zagotowania lub w wyniku
stapiania kwasów pirydynosulfonowych z wodorotlenkiem sodu. Hydroksypirydyny łatwiej ulegają substytucji elektrofilowej (SE)
niż sama pirydyna.
N N OH
NaOH
pirydyna 2-hydroksypirydyna
N OCH3N O
CH2N2 CH3l
2-metoksypirydyna
N-metylopirydyn-2-on
CH3

N N
pirydyn-2-on
2-hydroksypirydyna
(pirydyn-2-ol)
OH O
H
N N
pirydyn-4-on
4-hydroksypirydyna
(pirydyn-4-ol)
H
OH O
równowagatautomeryczna

Utlenianie pirydyny i jej pochodnych – pirydyna jest odporna na działanie wielu utleniaczy, łatwo jednak można utlenić jej łańcuch boczny.
N CH3 N COOH
KMnO4
kwas 2-pikolinowy2-pikolina
N
KMnO4 KMnO4
CH3
N
COOH
N
N
H
CH3
3-pikolinanikotynakwas nikotynowy
(kwas 3-pikolinowy)

Pirydyna w obecności nadkwasów ulega utlenieniu i zostaje przekształcona w 1-tlenek pirydyny (N-tlenek pirydyny).
N
pirydyna
CH3COOOH
N
1-tlenek pirydyny
O
- CH3COOH
Reakcje substytucji elektrofilowej 1-tlenku pirydyny zachodzą w pozycjach 2 i 4 (inaczej niż w pirydynie).
N
O
N
O
1-tlenek pirydyny
N
O
N
O

Z N-tlenku pirydyny
można zsyntetyzować
wiele pochodnych
trudnych do otrzymania
bezpośrednio z pirydyny.
Powrót z N-tlenku
pirydyny (lub tlenku jej
pochodnych) do pirydyny
(lub jej pochodnych)
dokonuje się w reakcji
z PCl3.
N
HNO3
PCl3
[O]
N
O
N
O
NO2
N
O
N
O
CH3
N
NO2
H2SO4100oC PhMgBr
CH3l
l
1-tlenek4-nitropirydyny
1-tlenek2-fenylopirydyny
jodek1-metoksy-
pirydyniowy
4-nitropirydyna
pirydyna1-tlenek pirydyny
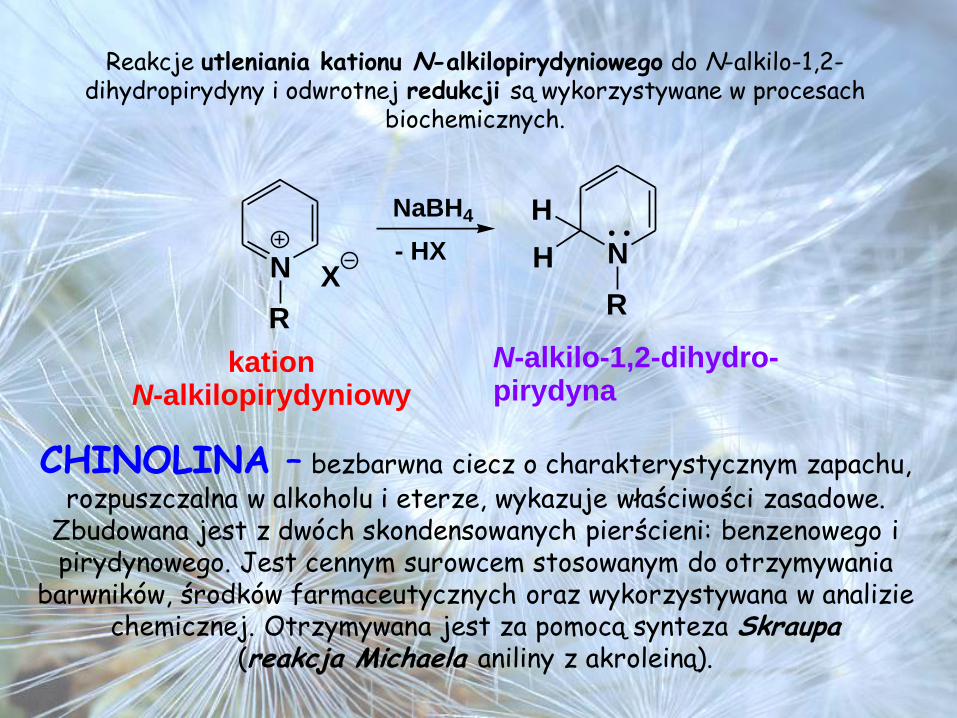
Reakcje utleniania kationu N-alkilopirydyniowego do N-alkilo-1,2-dihydropirydyny i odwrotnej redukcji są wykorzystywane w procesach
biochemicznych.
N
R
X
NaBH4
- HX N
R
H
H
kationN-alkilopirydyniowy
N-alkilo-1,2-dihydro-pirydyna
CHINOLINA – bezbarwna ciecz o charakterystycznym zapachu, rozpuszczalna w alkoholu i eterze, wykazuje właściwości zasadowe.
Zbudowana jest z dwóch skondensowanych pierścieni: benzenowego i pirydynowego. Jest cennym surowcem stosowanym do otrzymywania
barwników, środków farmaceutycznych oraz wykorzystywana w analizie chemicznej. Otrzymywana jest za pomocą synteza Skraupa
(reakcja Michaela aniliny z akroleiną).

+
CH2
OH
CH CH2
OH OH
H2SO4CH2=CH-CHO 2 HOH
glicerolakroleina
+
H
C
O
CHH
CH2NH2
NH
HC
O
N
HO H
H
anilina akroleina4-hydroksy-1,2,3,4-tetrahydrochinolina
NH
- HOH
NO2
NH2
- HOH, -N
chinolina(jest słabszą zasadą niż anilina) 1,2-dihydrochinolina

Substytucja elektrofilowa (SE) chinoliny – pierścień benzenowy jest bardziej podatny na reakcje SE niż pirydynowy, dlatego też do podstawienia atomów wodoru elektrofilami dochodzi w części homocyklicznej.
Chinolina w reakcjach SE jest mniej aktywna niż benzen.
+
N N N
Br2
H2SO4
Br
Brchinolina 5-bromochinolina8-bromochinolina
(powstają w jednakowej ilości)
+
N N
HNO3
H2SO4
NO2
NO25-nitrochinolina8-nitrochinolina
(10 : 1)

Przykładowe pytania1. Podaj nazwy następujących związków:
O CH2CH3S S
Cl
N
Cl
Br H3C
NH
Cl
Br
N
O COOH NH
Br
N
C N
O CH2CH3
CH2CH3

2. Uszereguj następujące związki według malejącej
reaktywności w reakcji SE: furan, pirydyna, benzen,
pirol, tiofen
3. Jakie produkty (wzór + nazwa) otrzymasz w reakcji
pirydyny z następującymi reagentami:
• 1. amidek sodu; 2. H2O
• fenylolit, temp. 100o
• wodorotlenek potasu
• butylolit, temp. 110o
• chlorowodór
• jodek metylu
4. Dlaczego pirol nie tworzy w roztworach wodnych soli z
kwasami, a pirydyna tworzy?
5. Uzasadnij, w jakim położeniu będzie zachodziła reakcja
SE w tiofenie.
6. Narysuj izomery metylopirydyny.
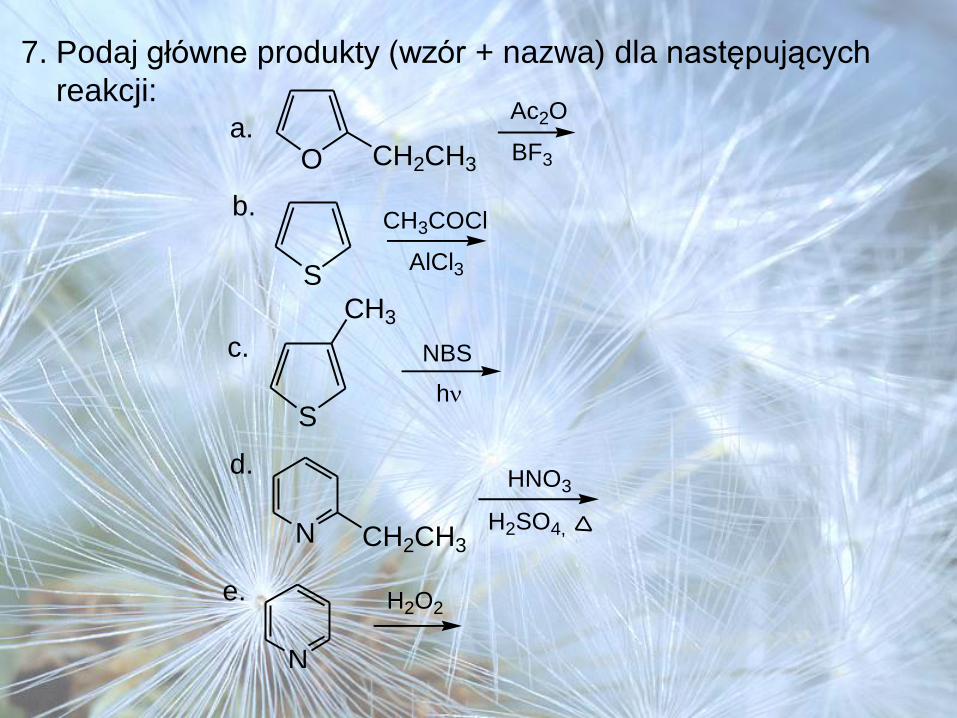
7. Podaj główne produkty (wzór + nazwa) dla następujących
reakcji:a.
O CH2CH3
Ac2O
BF3
S
b.CH3COCl
AlCl3
S
c. NBS
h
CH3
N CH2CH3
d.HNO3
H2SO4,
N
e. H2O2

8. Uzasadnij dlaczego 2-aminopirydyna ulega reakcji
nitrowania i sulfonowania (w pozycji 5) znacznie
łatwiej niż pirydyna.
9. Wyjaśnij dlaczego 4-chloropirydyna jest bardziej
podatna na reakcję SN niż 3-chloropirydyna.
10. N-Tlenek pirydyny w reakcji z bromkiem benzylu
tworzy bromek benzyloksypirydyniowy, który pod
wpływem mocnej zasady daje benzaldehyd i pirydynę.
Zaproponuj mechanizm tej reakcji.

Dziękuję za uwagę