Über die bestimmung von silicium, aluminium, fluor und orthophosphosäure nebeneinander ii
TRANSCRIPT

Th. Millner und F: Künos: Über die Bestimmung usw. 253
werden darf, ebenso wie die Methode der Bestimmung des Queeksilbers als [HgJ 4] [Cu pn211)2).
'Die Verteile unseres Verfahrens gegenüber den klassischen Methoden sind in, allgemeinen Zügen folgende:
1. Die' Einfachheit der Arbeitsweise. Diese geht schon aus der Leichtigkeit, mit der das Komplexsalz [HgJ4] [Cuen~] ausf~llt, hervor, ferner daraus, dass dessen makrokrystalline Form ein schnelles ~iltrieren erlaubt.
2. Die Schnelligkeit, mit der die Bestimmungen ausgeführt werden können, l - - I iß Stunden genügen für eine Queeksilberbestimmung unter allen behandelten Bedingungen.
3. Ihre Genauigkeit, welche aus der befriedigenden Überein- stimmung der Ergebnisse und aus der verhältnismäßig kleinen Grössen- ordnung der ArbeitsIehler hervorgeht, welche dem hohen Molekular- gewicht des Komplexes zu verdanken ist, in welchem das Quecksilber gebunden ist; und endlich,
4. die Möglichkeit, das Quecksilber auch in Gegenwart vieler anderer Elemente aus den verschiedenen analytischen Gruppen zu be- stimmen.
Über die Bestimmung von Silicium, Aluminium, Fluor und 0rthophosphorsäure nebeneinander II.
Von
Th. )Iillner~ bearbeitet mit F. Künos.
[Eingegangen am 17. Februar t933.]
B e s t i m m u n g von F luo r und Or thophosphors /~ure in Gegen- war t von Si l ic ium und A l u m i n i u m in wässr igen Lösungen oder wasser lös l ichen Gemengen, welche A lka l i f l uo r ide , Alka l i s i l i coß luor ide , K r y o l i t h , O r t h o p h o s p h o r s ä u r e oder
o r t h o p h o s p h o r s a u r e Alka l i sa lze e n t h a l t e n .
In einer früheren Arbeit 3) haben wir zur Bestimmung von Silicinm und Aluminium in Gegenwart von Fluor und Orthophosphorsäure in w~ssrigen Lösungen, oder wasserlöslichen Gemengen, welche Alkali- fluoride, Alkalisilicofluõride, Kryolith, Orthophosphors~ure oder ortho- phosphorsaure Alkalisalze enthalten, einen Analysengang angegeben, mit welchem man, sowohl bei grösseren, wie auch bei geringen Mengen, z. ]3. wenigen ~Iilligrammen der Ausgangssubstanz, zu richtigen Werten ge- langen kann.
1) G. Spgcu und P. Sp reu , diese ZtsehrfV. 89, t88 (1932). ~) pn ~-~ propylendiamin. 3) Diese Ztsehrft. 90, 16t (1932).

254 Th. Millner und F. Künos:
Nachstehend geben wir in Fortsetzung dieser unserer früheren Arbeit Methoden zur Fluor- und Oßhophosphorsäurebestimmung in Gegenwart von Silicium und Aluminium an, welche bei wässrigen Lösungen oder wasserlöslichen Gemengen, die Alkalifluoride, Alkalisilicofluoride, Kryo- lith, 0rthophosphorsäure oder orthophosphorsaure Alkalisalze enthalten, auch bei geringen Mengen, z. B. wenigen Milligrammen der Ausgangs- substanz zu richtigen Werten führen.
1. Bemerkungen über die l~luorbestimmung in Gegenwart von Silieium und Aluminium, aber in Abwesenheit von 0rthophosphorsäure.
J. H. de Boer und J. B a s a r t 1) haben eine maßanalytische Bestimm- ung des Fluors (auch in Komplexen und unlöslichen X~luoriden) angegeben. Die Methode beruht darauf, dass Zirkonium mit Oxyanthrachinonen Farblacke bildet, weiter darauf, dass Zirkonium an Fluor-Ionen vlel stärker komplex gebunden wird, als an die Farbstoffe, sodass die Farb. lacke durch die Fluor-Ionen zersetzt werden.
Die Titration geschieht in folgender Weise : von zwei gleichen Mengen einer Zirkoniumoxychloridlösung (mit gleichviel Salzsäure versetzt) wird die eine mit der abgewogenen Menge Fluorid von unbekanntem Fluor- gehalt versetzt, wobei man dafür Sorge trägt, dass stets Zirkoninm im Überschuss anwesend ist. Auch Komplexe und in Wasser unlösliche Fluoride gehen in dieser Weise in Lösung. l~ach Behandlung mit alizarin- sulfosaurem Natrium werden beide, alsdann violettroten Lösungen mit einer genau bekannten Kalinmfluoridlösung bis zur Erreichung genau der. selben 0rangefarbe titriert (die Zwischenfarben sind dabei besser zu ver. gleichen als die gelben Endfarben). Der Unterschied in den zugegebenen Mengen der Finoridlösung entspricht dem X~luorgehalt der eingewogenen Menge des Fluorides.
J. H. de Boer und J. B a s a r t haben die Methode an Fluoridmengen geprüft, welche wenigstens etwa i00 .mg Fluor enthielten. Für solche Fluoridmengen ist die Methode in der beschriebenen Form auch in Gegen. wart von Silicinm und Aluminium ohne weiteres anwendbar. Wir haben nun versucht, die Methode auch zur Bestimmung k le iner Fluormengen (in Gegenwart von Silicinm und Aluminium) anzuwenden, doch gelingt dies nur nach gewissen Abänderungen der von J. H. de Boer und J. B a s a r t angegebenen Ausführungsweise.
J. H. de Boer und J. B a s a r t führten ihre Bestimmungen in stark salzsänrehaltigen Lösungen aus, da die Farblackspaltung bei grossen Säurekonzentrationen gegenüber kleinen Schwankungen der Säure- konzentration ziemlich unempfindlich ist.
1) Ztschrft. f. anorg. Chem. 152, 213 (1926); vergl, diese Z~seb~rft. 69, 355 (t926).

Über die Bestimmung von Silicium, Aluminium usw. 250
Kleine Fluormengen (z. B. I~~0 mg Fluor) kann man unter den von J. H. de B o e r und J. B a s a r t angegebenen Bedingungen nur sehr ungenau oder überhaupt nicht mehr bestimmen, weil man bei den an- gegebenen Konzentrationen der Maßflüssigkeiten im Falle so kleiner Fluormengen nur einen schlecht messbaren, kleinen ùVerbrauch" an K_F-Lösung bekommt. Wir versuchten die Bestimmung kleiner Flnor- mengen unter Beibehaltung der von J. H. de B o e r und J. B a s a r t an- gegebenen starken Salzsi~urekonzentration, aber mit etwa ~[0--15fach verdünnten Mal]flüssigkeiten auszuführen, bekamen so aber keine brauch- baren Farbumschl/*ge. Nach Übergang auf kleine Sa]zs£urekonzentrationen konnten wir mit den verdünnten Messflüssigkeiten brauchbare Farb- umschl/$ge und gut reproduzierbare Mal]werte erreichen, nur mussten wir dafür Sorge tragen, dass nach Beendigung zusammengehöriger Titrationen die in unseren Fgllen nur etwa 25--100 c c m betragenden Gesamtvolumina der t i trierten Flüssigkeiten auf etwa =L 2~o übereinstimmten, da bei kleinen Salzsiiurekonzentrationen die Endpunkteinste]lung der Reaktion sehr empfindlich ist. In dieser Weise gelang es uns, die Anwendbarkeit der Methode bis auf Fluormengen von I mg auszudehnen.
Zu bemerken ist noch, dass die Methode weder in der ursprünglichen noch in der abgeänderten Form für Fluorbestimmungen in K~SiF~ brauchbar ist, da sich K2SiF 6 in Zirkoniumoxychloridlösung nicht auflöst.
2. Die Zusammensetzung der bei Bestimmung kleiner Fluormengen benutzten Lösungen.
Wir benutzten folgende Lösungen:
K F - L ö s u n g A. 2,500 g K F . 2 H20 wurden in Wasser zu ~ 1 gelöst.. ~1 c c m dieser Lösung enthielt 0,506 m g Fluor.
K F - L ö s u n g B. 5,000 g K F . 2 H~O wurden in Wasser zu I l gelöst. i c c m dieser Lösung enthalt i ,012 mg Fluor.
Z i r k o n i u m o x y c h l o r i d l ö s u n g A. Aus t0 g basischem Zirkonium- nitrat wurde das Zirkonium mit Ammõniak als t tydroxyd gefällt ; sodann wurde das Zirkoniumhydroxyd bis zum Verschwinden der Salpeters/~ure- reaktion mit heissem Wasser ausgewaschen, in i i 0 c c m konz. Salzs/~ure (D 1,19) gelöst und die Lösung mit Wasser auß I 1 aufgefüllt.
Z i r k o n i u m o x y c h l o r i d l ö s u n g ]3. Diese wird aus 20 g basischem Zirkoniumnitrat in derselben Weise hergestellt, wie Lösung A, nur wird das Zirkoniumhydrõxyd in ~t20 c c m konz. Salzs/~ure (D ~,i9) gelöst und mit Wasser zu i 1 aufgefüllt.
L ö s u n g v o n a l i z a r i n s u l f o s a u r e m l~Iatr ium ( A l i z a r i n - lösung) . 0,t5 g Alizarinsulfos~ure werden mit Hilfe von I c c m einer 2 n-NaOH-Lösung in Wasser gelöst und auf 500 ccn~ aufgefüllt.

256 Th. 3/iillner und F. Künos:
3. Fluorbestimmung in kleinen l)Iengen von Na~SiF« nach der Zirkon-Alizarinmethode.
Zur Priffung der ]3rauchbarkeit der abgeßnderten Zirkon-Alizarin- methode bei kleinen Fluormengen haben wir an mehreren kleinen Na~SiF 6- Proben Fluorbestimmungen durchgeführt.
Den Gang der ]3estimmung und die Berechnung des Resultates zeigen wir zunächst an ~Iand eines Beispieles.
Es ist notwendig, vor der endgültigen ]3estimmung den ungeß~hren Fluorgehalt der Analysenprobe in einer Vorprobe ganz roh zu bestimmen.
Es soll der Fluorgehalt von i0,0 m g z. ]3. eines unbekannten Fluorids bes t immt werden, dessen Vorprobe einen ungefi~hren Fluorgehalt von 50- -60% ergab. (Das unbekannte Fluorid kann z. ]3. Na2SiF 6 sein; '10,0 m g NaeSiF 6 enthalten theoretisch 6,06 m g Fluor.)
Zu diesem Zweck wird nach Zugabe von 0,5 c c m Alizarinlösung und darauffolgendem 2 Minuten langem Warten der Verbrauch von ~0,0 c c m
Zirkoniumoxychlorid]ösung A an KF-Lösung A bestimmt. Es sei hier- bei ein Verbrauch von 30,0 c c m gefunden worden. Das Gesamtvolumen der t i trierten Flüssigkeit stellt sich dann auf 40,0 c c m .
Nun muss die zweite Titration in Anwesenheit der 10,0 m g des unbekannten Fluorides so durchgeführt werden, dass nach beendigtet Titration das Gesamtvolumen der t i trierten Flüssigkeit ebenfalls auf ungefähr 40 c c m kommt.
Hierzu wird die eingewogene Menge (10,0~ng) des unbekannten Fluorids in genau i0,0 c c m Zirkoniumoxyehloridiösung A aufgelöst. Nunmehr ist einfach auszurechnen, dass die in der eingewogenen Fluorid- menge enthaltene Fluormenge von etwa 5- -6 m g einem Verbrauch an KF-Lösung-A von etwa t0 - -12 c c m entspricht» dass also beim Titrieren dieser zweiten i0,0 c c m Zirkoniumoxychloridlösung A nur mehr etwa 30,0 - - t0 ~- 20 c c m K_F-Lösung A verbraucht werden. Das wiirde aber nur zu einem Gesamtvolumen der t i trierten Flüssigkeit von etwa 30 ccm Bühren. Um nun zu einem Gesamtvolumen von etwa 40 c c m nach be- endigter Titration zu kommen, müssen zu den t0,0 c c m der Zirkonoxy- chloridlösung A im voraus weitere t0 - -12 c c m Wasser zugesetzt werden.
Nach Auflösen der Einwage in der Zirkoninmoxychloridiösung müssen also in diesem Falle der Lösung etwa t t c c m Wasser zugesetzt werden. Nach Zugabe von 0~5 c c m Alizarinlösung und nach "2 Minuten langem Warten wird die Lösung mit K_F-Lösung A auf dieselbe Farbe wie die Pendantbest immung titriert. Findet man auf diese Weise einen Verbrauch an KF-Lösung A von t8,0 c c m (mit einer Genauigkeit von etwa 0,3 c c m ) , so berechnet sich der Fluorgehalt der eingewogenen Probe Ms ~qui:¢alent mit 3 0 , 0 - t8,0 ~ i2,0 ± 0 , 5 c c m KF-Lösung A, also zu

Über die Bestimmung von SilicKtm, Aluminium usw. 257
12,0 . 0,506 = 6,06 mg Fluor mit einer Genanigkeit von etwa :~ 0,2 bis =~0,3 mg Fluor.
Die Fluorbestimmungen an Na2SiF«-Proben haben wir mit Einwagen von 2,0 bis 50,0 mg Na2SiF 6 durchgeführt. Das Einwägen geschah innerhalb der Fehlergrenzen von ~ 0 , 2 m g . Jede Bestimmung haben wir zweimal wiederholt. Oberhalb der Menge von 20 mg 2ga2SiF « haben wir zu den ]~estimmungen Zirkoniumoxyehloridlösung B und K_F-Lösung :B benutzt. Die erhaltenen Resultate haben wir in untenstehender Tabelle zusammen- gestellt:
Tabe l l e ]:. F l u o r b e s t i m m u n g in ~ a t r i u m s i l i c o ß l u o r i d n a c h der Z i r k o n i u m -
Al i za r inme~hode .
Fluorgehaltin drei ]Vlittelwert der ]Fehler für den Na2SiF6 Fluorgehal~ unabhängigen Fluor- I ?¢Iit~elwert der Einwage berechneß Bestimmungen Fluor-
gefunden bes~immungen bestimmungen mg mg mg mg mg
2,0
3,0
4,0
t,21
t,81
i i
2,42
! 5,0 i 3,03
10,0
20,0
30,0
50,0
6,06
12,13
18,20
30,32
1,1 t,Π1,0
1,7 ~1,9 1,7
2,2 2A 2,2
3,3 3,0 3g
6,4 6,t 5,8
11,9 t2,0 t2,1
t8,4: 18,4 18,2
30,6 30,4: 30,4
- - 0 , 1
1,8 ± 0,0
2,2
3 , t
6,1
12,0
18,3
30,5
- - 0 , 2
@ 0,t
4- 0,0
- -0 , t
-I- 0,1
-}- 0,2
Ztschrft, f. a.nal. Chem. 92, 7. m 8. Heft. 17

258 Th. Millner und F. Künos:
4. Flnorbestimmung in kleinen Mengen von NaaAIF 6 (Kryolith) nach der Zirkonium-Alizarinmethode.
Auch an Meinen Na3AIF«(Kryolith)-Proben haben wir die Brauchbar- keit der abge/~nderten Zirkonium-Alizarinmethode geprüft. Die Fluor- best immungen haben wir in gleicher Weise, wie bei Na2SiF« durchgeführt. Das Einwägen der Proben von 2,0 bis 50,0 mg Kryoli th geschah innerhalb der Fehlergrenzen von :j: 0,2 mg. Jede Bestimmung haben wir zweimal wiederholt. Oberhalb der Menge von 20 mg Kryol i th haben wir zu den Best immungen Zirkoniumoxychloridlösung B und KF-Lösung B benutzt. Die erhaltenen Resultate haben wir in Tabelle I I zusammengestellt.
T a b e l l e I I . F l u o r b e s M m m u n g in K r y o l i ~ h n a c h der Z i r k o n i u m -
A l i z a r i n m e t h o d e .
Naß lF« (Kryoli~h) Einwage
mg
2,0
3,0
4,0
5,0
10,0
20,0
30,0
50,0
Fluorgehalt indrei Fluorgehalt unabhängigen berechnet Bes~immungen
gefunden mg mg
1,09 t,l 1,1 1,2
t,60 1,6 t,6 1,5
2,18 2,0 2,1 2,3
2,70 2,8 2,5 2,8
5,42 5,4 5,3 5,0
10,90 t0,7 11,2 10,7
«6,28 16,0 16,0 t6,2
27,14 26,7 26,7 26,9
Mittclwer$ der Fehler für den Fluor Mittelwer$ der
- Fluor- bes~lmmungen bestimmungen
mg mg
1,1
1,6
2,1
2,7
5 j 2 •
10,9
t6,1
26,8
B 0,0
B 0,0
- - 0 , 1
B 0,0
- - 0 , 2
B 0,0
--0,2
- - 0 , 3

Über die Bestimmung von Silieiu~m, Aluminium usw. 259
Die Zahlen der Tabellen I und I I zeigen, dass man mit der abge- änderten Zirkonium-Alizarinmethode sowohl in Gegenwart von Silicinm, wie auch in Gegenwart von Aluminium brauchbare Fluorbestimmungen ausführen kann.
ö. Bemerkungen über die l~luorbestimmung nach der Zirkonium-Alizarin- methode in Gegenwart von Silieium und Aluminium und bei gleichzeitiger
Anwesenheit von 0rthophosphorsäure. Die Fluorbestimmung nach der Zirkonium-Alizarinmethode wird
durch anwesende PhosphorsEure gestört, da der Zirkonium-Alizarin- Farblack von Orthophosphorsäure unter Bildung von Zirkoninmphosphat zersetzt wird. Wir haben demzufolge nach einer Methode zur Trennung der Orthophosphorsäure von Fluor gesucht, welche gestattet, nach er- folgter Trennung das Fluor nach der Zirkonium-Alizarinmethode zu bestimmen.
Man kann nach Angaben von F. P. T r e a d w e l l und A. A. K o c h 1} aus Lösungen, welche ~'luor und Orthophosphorsäure gleichzeitig ent- halten, den Orthophosphorsäuregehalt ohne Fluorverluste abscheiden, indem man zu der sorgfältig neutrahsiert~n Lösung eine, auf Phosphor- säure bezogen, überschüssige Menge von Silbernitratlösung zusetzt und den entstandenen Silberphosphatniederschlag entfernt. In dieser von übersehfissigem Silbernitrat mit Natriumchlorid befreiten Lösung ist dann die zu bestimmende Fluormenge restlos vorhanden und kann nach irgend einer der bekannten Fluorbestimmungsmethoden ermittelt werden.
Wir konnten bei Proben, die neben Fluor nur Silicium und Ortho- phosphorsäure, aber kein Aluminium enthielten, die Phosphorsäure- abtrennungsmethode von F. P. T r e a d w e l l und A. A. K o c h mit gutem Erfolg anwenden und bekamen bei den nach der Phosphorsäureabtrennung vorgenommenen Fluorbestimmungen mittels der Zirkoninm-Alizarin- methode richtige Fluòrwerte.
Ist aber neben Fluor und Orthophosphorsäure auch Aluminium vor- handen, so entstehen bei der im Abschnitt 6 beschriebenen -4mwendung der Methode von F. P. T r e a d w e l l und A. A. K o c h grosse Fluorverluste, ganz unabhängig davon, ob gleichzeitig auch Silicium vorhanden ist oder nicht. Die Ursache dieser Fluorverhiste haben wir darin gefunden, dass bei der Abseheidung der Orthophosphorsäure aus neutraler Lösung der abgeschiedene Silberphosph tniederschlag beträehthche Mengen von Alu- minium, aber auch gleichzeitig von ]~~luor mit sich reisst. Man kann in solchen Silberphosphatniedersehlägen den Gehalt an mitgerissenem Fluör leicht mit der Probe von F. t~eigl und P. K r u m h o l z 2) und den Gehalt an mitgerissenem Aluminium z. B. mittels der Eriochromeyaninreaktion
1) Diese Ztschrft. 48, 485 (1904). 2) Ber. Deutsch. Chem. Ges. 62, t138 (1929); vergl, diese Ztschrft. 79,
213 (t930); 3/iikrochemie, Pregl-Festschrift S. 83 (7929). 17"

260 Th. i~~illner und F. ]~ünos:
von E. E e g r i w e 1) nachweisen2). Dieses Mitreissen von F l u o r durch Sf lberphospha t i s t an die Anwesenhei t von A lumin ium gebunden . Aus Proben , welche F luor , Ortbophosphors/~ure u n d Silicium, abe r kein 2Auminium en tha l t en , scheidet sich un te r dense lben Bedingungen das Sf lberphospha t ohne ncrmenswer tes Mitreissen von F l u o r ab.
Die Schwierigkei t , d ie durch das Mitausf£Ilen v o n F l u o r m i t dem Sf lberphospha tn iedersch lag en t s t and , haben wir durch doppe l tes Ausfi~llen des S i lbe rphospha tes zu umgehen versucht . Lös t m a n das S i lbe rphospha t nach dem ers ten Abscheiden noch e inmal in v e r d ü n n t e r Sa lpe te r säure u n d scheidet es aus dieser Lösung aufs neue ab, so k a n n m a n in den vere in ig ten F i l t r a t e n be ider Fä l l ungen (al lerdings nur mi t m/~ßiger Genauigkei t ) das gesamte F l u o r mi t t e l s der Z i rkon ium-Al iza r inmethode f inden.
6. F luorbes t immung in geringen Mengen von Na2SiF s nach der Z i rkon ium- Al izär inmethode bei Anwesenhei t von 0r thophosphorsäure .
W i r haben nach der abgeände r t en Z i rkon ium-Al iza r inmethode F l u o r b e s t i m m u n g e n an P r o b e n angeste l l t , welche aus wenigen Milli- g r a m m e n (3 bis 50 mg) Na2SiF 6 u n d e twa zehnmal so viel Na2HPO ¢ zu- sammengese tz t waren.
Die P r o b e n wurden in e twa 50 c c m Wasser gelöst, welchem vorher e twa ~ 2 c c m einer 2 n-HNO~-Lösung zugegeben worden waren. Den
1) Diese Ztschrft. 76, 438 (1929). 2) Den Aluminiumnachweis mit tels des Farbstoffes Er iochromcyanin-R
in dem fluorhaltigen Sflberphosphatniederschlag kann man nur nach g ründ , lieher Entfernung des Fluorgehaltes ausführen, nicht nur weil man daa Aluminium zuerst mit tels Ammoniaks ausfällen muss und dies nur in Ab- wesenheit von Fluor ungestört gelingt, sondern auch aus dem Grunde, weil die Farbstoffreakt ion durch Fluor-Ionen ausserordentlich s tark gestört wird.
Wir führten den Aluminiumnachweis folgendermaßen aus: Einen (mit Fluor und Aluminium verunreinigten) Si lberphosphat .
niederschlag von etwa 100 m g lösten wir in einer Platinschale in wenig ver- d5nnter Salpetersäure auf, gaben etwa Õ,l g Borsäure und etwa t c c m konz. Schwefelsäure zur Lösung und dampften diese ein. Den Rücks tand erhi tzten wir gelinde bis zum Entweichen von SO3-DKmpfen, verdünnten darm die je tz t fluorfreie Lösung auf etwa 400--500 ccra und sät t igten sie heiss mi t Schwefelwasserstoff. Nach Abfil tr ieren des Silbersulfides engten wir die Lösung ein, neutral is ierten sie mi t Ammoniak und entfernten daraus die Phosphorsäure nach 1~. W o y [vergl. diese Ztschrft . 90, 165 (1932)]. Aus der so ven Fluor, Silber und Phosphorsgure befreiten Lösung schieden wir den Aluminiumgehalt in der in unserer früheren Arbei t angegebenen Weise [diese Ztschrft . 90, ~ 65 -- 166 (1932)] ab. Zur Identif izierung des so erhaltenen Aluminiumhydroxydniederschlages führten wir mi t d e m Niederschlag die Eriochr0meyaninreakt ion genau nach den Angaben von E. E e g r i w e durch.
lVian kann diese l~eaktion leicht ha lbquant i ta t iv ausfiihren. I n dieser Weise fanden wir in etwa t00 m g Silberphosphatniederschlag (ausgefällt aus einer Aluminium und Fluor enthal tende n Lösung) roh abgeschätzt ~t m g
Aluminium.

Über die Bestimmung von Silieiurn, Aluminium usw. 26~
Lösungen setzten wir 4 - -5 ccm einer 5%igen Sflbernitratlösung und dann tropfenweise so viel n-NaOH-LSsung hinzu, bis der zuerst mit gelber Farbe ausfallende Silberphosphatniederschlag von dem später ausfallenden Silberoxyd eben braun gefärbt wurde. Nach Absitzenlassen des Nieder- schlages filtrierten wir die Lösungen, wuschen den Niederschlag mir wenig Wasser gut nach und gaben dem Fil trat festes Natriumchlorid im Überschuss zu. Nach gründlichem Durehsehütteln der Flüssigkeit liessen wir den Silberchloridniederschlag über Nacht absitzen. (Nach kürzerem Stehen sind geringe Fluorverluste wahrzunehmen.) Sodann filtrierten wir die Lösung in eine Platinschale, wuschen den Nieder- schlag nach, fügten zu der Flüssigkeit i - - 2 Tropfen einer 2 n-NaOH- Lösung und dampften die schwach alkalische Flüssigkeit au f ein ganz kleines Volumen ein.
Diese Flüssigkeit wurde dann zur Fluorbestimmung benutzt, und zwar in derselben Weise, wie in Abschnitt 3 dieser Arbeit besehrieben.
Das Einwägen der Na2SiF«-Mengen geschah innerhalb der Fehler- grenzen von =~0,2 mg. Jede Bestimmung haben wir zweimal durchge- führt.
Oberhalb der Mengen von 20 n~g Na2SiF 6 haben wir zu den Bestimm- ungen Zirkoniumoxyehloridlösung ]3 und KF-Lösung B benutzt. Die erhaltenen Resultate haben wir in Tabelle I I I zusammengestellt:
Tabelle III, F l t l o r b e s t i m m u n g in N a t r i u m s i l i c o f l u o r i d be i A n w e s e n h e i t von
O r t h o p h o s p h o r s / ~ u r e n a c h der Z i r k o n i u m - A l i z a r i n m e % h o d e .
Fluorgehalt Fluorgehalt I ~it telwert I Fehler für Na2SiFs bestimmt in zwei der Fluor- 'den Mittelwert~ Einwage berechnet unabhängigen der Fluor-
~~Viessungen bestimmungen bestimmungen mg mg ,~g mg mg
3,0
5,0
t0,0
20,0
30,0
50,0
1 , 8 1 2,0 1,5
3,03 3,1 3,2
6,06 5,8 5,9
t2,t3 11,7 12,0
i
'18,20 18,4 18,1
30,32 30,6 L 29,9
~,8
3,2
5,9
ti,9
t8,3
30,3
-- 0,0
@ 0,2
~-- 0,2
- - 0 , 2
+o,~
= 0,0

262 Th. ~¢[illner und F. Kfinos:
7. Fluorbestimmung in geringen Mengen von I~'B3AIF 6 (Kryolith) nach der Zirkonium-Alizarinmethode bei Anwesenheit von 0rthophosphorsäure.
Wir führten Fluorbestimmungen nach der abgeänderten Zirkonium- Alizarinmethode an Proben aus, welche aus wenigen Mi]ligrammen (5--50 mg) Na3A1F « und etwa zehnmal so viel Na2HPO a zusammengesetzt waren. Dabei sind wir genau in der in Abschnitt 6 dieser Arbeit ange- gebenen Weise vorgegangen, mit der einzigen Erweiterung, dass wir das schon einmal gefällte Silberphosphat in verdünnter Salpetersäurc auf- gelöst und durch tropfenweises Zufügen von n-Natronlauge noch einmal ausgefällt haben. Die eigentliche Fluorbestimmung haben wir hernach in den vereinigten Fil tratcn der beiden Silberphosphatniedersehlgge vorgenommen,.
Das ]~inw~gen der NaaA1F«-lVfengen geschah auch hier innerhalb der ~ehlergrenzen von :J=0,2 mg. Oberhalb der menge von 20 mg NaaA1F « haben wir zu den Bestimmungen Zirkoniumoxychloridlösung B und KF-Lösung B benutzt. Die Resultate der Fluorbestimmungen haben wir in Tabelle IV zusammengestellt:
T a b e l l e IV. F l u o r b e s t i m m u n g in K r y o l i t h bei A n w e s e n h e i t v o n O r t h o -
p h o s p h o r s ä u r e n a c h de r Z i r k o n i u m - A l i z a r i n m e $ h o d e .
Na~AIFG Fehler (Kryolith) Fluorgehal~ Fluorgehalt
berechnet bestimmt der Fluor- Einwage bestimmtmg
mg mg ,mg mg
5,0 ~0,0 20,0 30,0 50,0
2,72 5,45
10,90 ~[6,35 27,25
2,4 5,4
~0,6 15,9 26,9
--0,3 ± 0,0 --0,3 --0,4 - - 0,4
Man sieht aus den Zahlen der Tabelle IV, dass trotz der zweimaligcn Abscheidung des Silberphosphates die Fluorwerte ein wenig zu niedrig ausfallen. Doch ist die Genauigkeit der Methode auch so noch für viele Zwecke ausreichend.
8. Fluorbestimmung in Gemengen von geringen Mengen Na2SiF 6 u n d
NaaAIF « (Kryolith) nach der Zirkonium-Alizarinmethode bei Anwesenheit von 0rthophosphorsäure.
Wir führten Fluorbestimmungen nach der abgeßnderten Zirkonium- Alizarinmethode an Proben aus, welche aus nur einigen Milligrammen iNa~SiF« und N~3A1F ~ und aus etwa zehnmal so viel 1Na~HPO4 zusammen- gesetzt wurden. Die Bestimmungen haben wir genau in der in Abschnitt 6
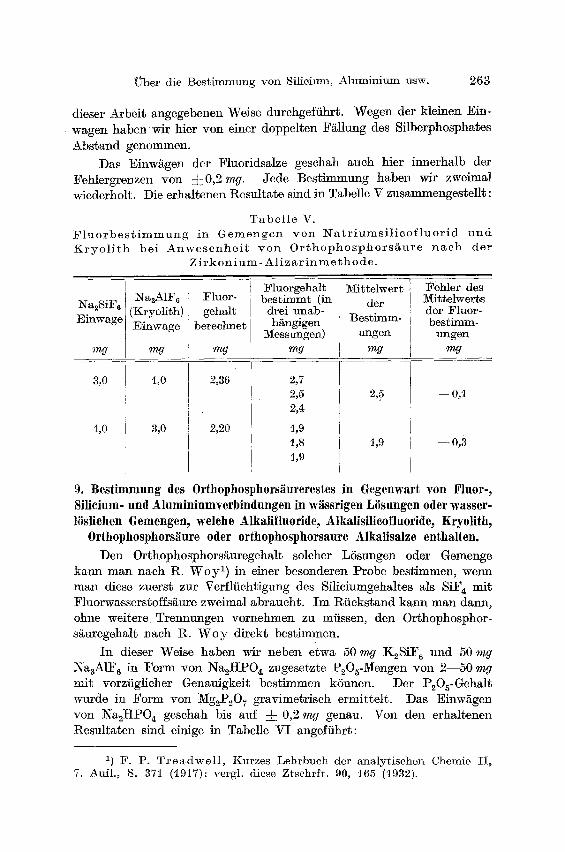
Über die Bestimmung von Silieium, Aluminium usw. 263
dieser Arbeit angegebenen Weise durchgeführt. Wegen der kleinen Ein- wagen haben wir hier von einer doppelten F~llung des Silberphosphates Abstand genommen.
Das Einw~gen der Fluoridsalze geschah auch hier innerhalb der Fehlergrenzen von =[=0,2 mg. Jede Bestimmung haben wir zweimal wiederholt. Die erhaltenen Resultate, sind in Tabelle V zusammengestellt:
T a b e l l e V. F l u o r b e s t i m m u n g in G e m e n g e n v o n N a t r i u m s i l i c o f l u o r i d u n d K r y o l i t h bei A n w e s e n h e i t von O r t h o p h o s p h o r s ä u r e n a c h der
Z i r k o n i u m - A]iz a r i n m e~h o de.
I Na2SiF6 ] NaaA1F«
(Kryolith) Einwage Einwage
mg nW
3,0
1,0
t,0
3,0
Fluor- gehMt
berechnet
mg
2,36
2,20
Fluorgehalt bestimmt (in
drei unab- hängigen
3/[essungen) mg
2,7 2,5 2,4
1,9 %8 ~,9
~/[ißtelwer t der
Bestimm- ungen
mg
2,5
t,9
Fehler des !VIittelwerts der Fluor- bestimm-
lmgen mg
@ 0,~l
--0,3
9. Bestimmung des 0rthophosphorsäurerestes in 6egenwart von Fluor-, Silieium- und Aluminiumverbindungen in wässrigen Lösungen oder wasser- löslichen Gemengen, welche Alkalifiuoride, Alkalisilico~luoride, Kryolith»
0rthophosphorsäure oder orthophosphorsaure Alkalisalze enthalten. Den Orthophosphors/~uregehalt solcher Lösungen oder Gemenge
kann man nach R. W o y 1) in einer besonderen Probe bestimmen, wenn man diese zuerst zur Verflüchtigung des Siliciumgehaltes als SiF a mit Fluorwasserstoffsäure zweimal abraucht. I m Rückstand kann man dann, ohne weitere Trennungen vornehmen zu müssen, den Ortho:phosphor- sä~uregehalt nach R. W o y direkt bestimmen.
In dieser Weise haben wir neben etwa 50 m g K2SiF 6 und 50 mg Na3AIF 6 in Form von Na2I-IPO 4 zugesetztc P205-Mengen von 2--50 mg mit vorzüglicher Genauigkeit bestimmen kölmen. Der P~Os-Gehalt wurde in Form von ~Vig2P207 gravimetrisch ermittelt. Das Einwägen von Na2HP04 geschah bis auf ± 0,2 mg genau. Von den erhaltenen Resultaten sind einige in Tabelle VI angeführt:
1) F. P. T r e a d w e l l , Kurzes Lehrbuch der analytischen Chemie II , 7. Aufl., S. 371 (1917); vergl, diese Ztsehrft. 90, 165 (1932).

264 Bericht: Allgemeine analytische Methoden usw.
Tabe l l e VI. B e s t i m m u n g von O r t h o p h o s p h o r s & u r e r e s t in G e g e n w a r t von F luor - , S i l ic ium- und A l u m i n i u m v e r b i n d u n g e n n a c h R. V¢oy.
K2SiF« Einwage
~ng
50,0 50,0 50,0 50,0
I~Ta3A]F6 (Kryolith) Einwage
mg
50,0 50,0 50,0 50,0
l~eO5 Einwage (In Form von P~O5
Na2HPOa bestimmt zugesetzt)
mg mg [
50,0 20,0 5,0 2,0
50,7 20,4 5,0 2A
Fehler der P2Os-
Bestimmung
mg
-~ 0,7 -[- 0,4 4- 0,0 -~ OA
Wie das mitgeteilte Zahlenmaterial zeigt, kann man nach den in unserer lrüheren und in der vorliegenden Arbeit beschriebenen Methoden Silicinm, Aluminium, Fluor und Orthophosphors~Lurerest auch bei Mengen von wenigen Milligramm in solchen wässrigen Lösungen oder wasser- löslichen Gemengen, welche Alkalifluoride, Alkalisilicofluoride, Kryolith, Orthophosphors~ure oder orthophosphorsaure Alkalisalze enthalten, zuverlässig quantitativ bestimmen.
Uj pcs t bei Budapeat, den 15. ~ebruar 1933.
Vereinigte Glühlampen- und Elektrizitäts A.-G. Tungsram.
Bericht über die Fortschritte der analytischen Chemie. I. Allgemeine analytische Methoden, analytische Operationen,
Apparate und Reagenzien. V o n
H. Brüekner. W. Forst.
Apparate. Die H e r s t e l l u n g e ines L a m p e n b a n k w i d e r s t a n d e s hat C, C. C o f f i n 1) beschriebcn. Der Widerstand besteht aus einem System parallel geschalteter Lampen» wobei der Kontakt durch eine stromführende Grundschiene und eine seitlich erhöht angebrachte, vom Bret t isolierte Seitenschiene gebildet wird. Der seitliche Kontakt der Lampe erh~lt einen Haken, der auf die seitliche Stromzuführung auf- gelegt wird, worauf die in horizontaler Richtung h~ngende Lampe mit der Spitze ihres Fusses auf der Grundschiene aufstösst. Je nach der gewünschten Stromstärke werden mehr oder weniger Lampen aufgelegt.
Unter dem I~Tamen W i l b e r f o r e e - K l a m m e r wird von d e m Hersteller C. B a k e r 2) eine Vorrichtung beschrieben, die geeignet ist, cylindrische Röhren sehr verschiedener Dicke festzuhalten. Sie besteht aus einem Stativarm, der einerseits ein aus vier zu je zwei rcchtwinklig
1) Jom'n. Americ. Chcm. Sec. 53, 2180 (t931). - - ~) Chem. Fabrik 3, 478 (1930).