o d o b r e n o - halmed - agencija za lijekove i medicinske · pdf file ·...
TRANSCRIPT

1
SAŽETAK OPISA SVOJSTAVA LIJEKA 1. NAZIV GOTOVOG LIJEKA Janumet 50 mg/850 mg filmom obložene tablete Janumet 50 mg/1000 mg filmom obložene tablete 2. KVALITATIVNI I KVANTITATIVNI SASTAV Jedna Janumet 50 mg/850 mg filmom obložena tableta sadrži 50 mg sitagliptina u obliku sitagliptinfosfat hidrata i 850 mg metforminklorida. Jedna Janumet 50 mg/1000 mg filmom obložena tableta sadrži 50 mg sitagliptina u obliku sitagliptinfosfat hidrata i 1000 mg metforminklorida. Za cjeloviti popis pomoćnih tvari vidjeti dio 6.1. 3. FARMACEUTSKI OBLIK Filmom obložena tableta (tableta). Janumet 50 mg/850 mg: ružičasta filmom obložena tableta u obliku kapsule, s utisnutom oznakom "515" s jedne strane. Janumet 50 mg/1000 mg: crvena filmom obložena tableta u obliku kapsule, s utisnutom oznakom "577" s jedne strane. 4. KLINIČKI PODACI 4.1 Terapijske indikacije Za bolesnike s dijabetesom melitus tipa 2: Janumet je indiciran kao dopuna dijeti i tjelovježbi za bolju regulaciju glikemije u bolesnika u kojih ista nije dovoljno dobro regulirana primjenom najveće podnošljive doze metformina ili u bolesnika koji se već liječe kombinacijom sitagliptina i metformina. Janumet je indiciran u kombinaciji sa sulfonilurejom (tj. kao dio trojne kombinirane terapije) kao dopuna dijeti i tjelovježbi u bolesnika u kojih glikemija nije dovoljno dobro regulirana primjenom najveće podnošljive doze metformina i sulfonilureje. Janumet je indiciran kao dio trojne terapije u kombinaciji s agonistom receptora za aktivator proliferacije peroksisoma-gama (engl. peroxisome proliferator-activated receptor gamma, PPAR) (tj. tiazolidindionom) kao dopuna dijeti i tjelovježbi u bolesnika u kojih glikemija nije dovoljno dobro regulirana primjenom najveće podnošljive doze metformina i agonista PPAR. Janumet je također indiciran kao dodatak inzulinu (tj. kao dio trojne kombinirane terapije), a kao dopuna dijeti i tjelovježbi, za poboljšanje regulacije glikemije u bolesnika u kojih ista nije dovoljno dobro regulirana postojanom dozom inzulina i metforminom.
H A L M E D
27 - 09 - 2012
O D O B R E N O

2
4.2 Doziranje i način primjene Doziranje Dozu lijeka Janumet u antidijabetičkoj terapiji treba prilagoditi svakom pojedinom bolesniku prema postojećoj terapiji, učinkovitosti i podnošljivosti lijekova, ne prelazeći pritom najveću preporučenu dozu sitagliptina od 100 mg na dan. Za bolesnike u kojih glikemija nije dobro regulirana monoterapijom najvećom podnošljivom dozom metformina U bolesnika u kojih glikemija nije dobro regulirana samo metforminom, uobičajena početna doza lijeka Janumet mora biti 50 mg sitagliptina dvaput na dan (ukupna dnevna doza 100 mg) plus dosadašnja doza metformina. Za bolesnike koji su do sada uzimali sitagliptin i metformin i prelaze na Janumet U bolesnika koji su do sada uzimali sitagliptin i metformin, liječenje lijekom Janumet mora započeti dosadašnjim dozama sitagliptina i metformina. Za bolesnike u kojih glikemija nije dobro regulirana dvojnom kombiniranom terapijom najvećim podnošljivim dozama metformina i sulfonilureje Doza lijeka Janumet mora biti 50 mg sitagliptina dvaput na dan (ukupna dnevna doza 100 mg) uz dozu metformina koja odgovara dosadašnjoj. Kad se Janumet uzima u kombinaciji sa sulfonilurejom, možda će biti potrebno sniziti dozu sulfonilureje kako bi se smanjio rizik od hipoglikemije (vidjeti dio 4.4). Za bolesnike u kojih glikemija nije dobro regulirana dvojnom kombiniranom terapijom najvećim podnošljivim dozama metformina i agonista PPAR Doza lijeka Janumet mora biti 50 mg sitagliptina dvaput na dan (ukupna dnevna doza 100 mg) uz dozu metformina koja odgovara dosadašnjoj. Za bolesnike u kojih glikemija nije dobro regulirana dvojnom kombiniranom terapijom inzulinom i najvećom podnošljivom dozom metformina Doza lijeka Janumet mora biti 50 mg sitagliptina dvaput na dan (ukupna dnevna doza 100 mg) uz dozu metformina koja odgovara dosadašnjoj. Kad se Janumet uzima u kombinaciji s inzulinom, možda će biti potrebno sniziti dozu inzulina kako bi se smanjio rizik od hipoglikemije (vidjeti dio 4.4). Za različite doze metformina, Janumet je dostupan u dozama od 50 mg sitagliptina i 850 mg metforminklorida ili 1000 mg metforminklorida. Svi bolesnici moraju se i dalje pridržavati odgovarajućeg načina prehrane i raspodijeliti unos ugljikohidrata kroz dan. Bolesnici s prekomjernom tjelesnom težinom trebaju nastaviti s niskokalorijskom prehranom. Posebne skupine bolesnika Bolesnici s oštećenjem bubrega Janumet se ne smije primjenjivati u bolesnika s umjerenim ili teškim oštećenjem bubrega (klirens kreatinina < 60 ml/min) (vidjeti djelove 4.3 i 4.4). Bolesnici s oštećenjem jetre Janumet se ne smije primjenjivati u bolesnika s oštećenjem jetre (vidjeti djelove 4.3 i 5.2). Starije osobe Budući da se metformin i sitagliptin izlučuju putem bubrega, s povećanjem životne dobi Janumet treba primjenjivati uz oprez. Potrebno je kontrolirati funkciju bubrega radi sprječavanja laktacidoze povezane s metforminom, pogotovo u starijih osoba (vidjeti djelove 4.3 i 4.4). Podaci o sigurnosti primjene sitagliptina u osoba starijih od 75 godina su ograničeni pa se lijek mora primjenjivati uz oprez.
H A L M E D
27 - 09 - 2012
O D O B R E N O

3
Pedijatrijska populacija Janumet se ne preporučuje u djece mlađe od 18 godina jer nema podataka o sigurnosti primjene i djelotvornosti lijeka u toj dobnoj skupini. Način primjene Janumet se mora uzimati dvaput na dan s hranom kako bi se ublažile gastrointestinalne nuspojave povezane s metforminom. 4.3 Kontraindikacije Janumet je kontraindiciran u bolesnika: - preosjetljivih na neku od djelatnih ili pomoćnih tvari lijeka (vidjeti djelove 4.4 i 4.8); - s dijabetičkom ketoacidozom, dijabetičkom pretkomom; - s umjerenim i teškim oštećenjem bubrega (klirens kreatinina < 60 ml/min) (vidjeti dio 4.4); - s akutnim stanjima koja mogu utjecati na funkciju bubrega kao što su:
- dehidracija, - teška infekcija, - šok, - intravaskularna primjena kontrastnog sredstva koje sadrži jod (vidjeti dio 4.4);
- s akutnom ili kroničnom bolešću koja može izazvati hipoksiju tkiva kao što su: - zatajenje srca ili pluća, - nedavni infarkt miokarda, - šok;
- s oštećenjem jetre; - s akutnim trovanjem alkoholom, alkoholičara; - za vrijeme dojenja. 4.4 Posebna upozorenja i mjere opreza pri uporabi Općenito Janumet se ne smije primjenjivati u bolesnika s dijabetesom tipa 1 niti za liječenje dijabetičke ketoacidoze. Pankreatitis U razdoblju nakon stavljanja lijeka na tržište među spontano prijavljenim nuspojavama zabilježen je akutni pankreatitis. Bolesnike treba poučiti o karakterističnim simptomima akutnog pankreatitisa: jakoj boli u trbuhu koja ne prestaje. Nakon povlačenja sitagliptina (sa ili bez drugih lijekova) iz terapije opaženo je povlačenje pankreatitisa; međutim, zabilježeni su vrlo rijetki slučajevi nekrotizirajućeg ili hemoragičnog pankreatitisa i/ili smrtni ishod. Ako se sumnja na pankreatitis, treba prekinuti primjenu lijeka Janumet i drugih lijekova za koje se sumnja da bi ga mogli izazvati. Laktacidoza Laktacidoza je vrlo rijetka, ali ozbiljna metabolička komplikacija (s visokom smrtnošću ako se pravodobno ne liječi) koja može nastupiti uslijed nakupljanja metformina. Među bolesnicima liječenima metforminom, laktacidoza je prijavljena uglavnom u dijabetičara sa značajnim zatajenjem bubrega. Učestalost laktacidoze može se i mora smanjiti tako da se u obzir uzmu i ostali čimbenici rizika poput loše regulacije dijabetesa, ketoze, dugotrajnog gladovanja, prekomjerne konzumacije alkohola, insuficijencije jetre i bilo kojeg stanja povezanog s hipoksijom. Dijagnoza Značajke laktacidoze su acidozna zaduha, bol u trbuhu i hipotermija nakon koje slijedi koma. Laboratorijski nalazi su sniženi pH krvi, razina laktata u plazmi iznad 5 mmol/l te povišen anionski procijep i omjer laktata/piruvata. Posumnja li se na metaboličku acidozu, treba odmah prekinuti primjenu lijeka i hospitalizirati bolesnika (vidjeti dio 4.9).
H A L M E D
27 - 09 - 2012
O D O B R E N O

4
Funkcija bubrega Poznato je da se metformin i sitagliptin značajno izlučuju putem bubrega. Laktacidoza povezana s metforminom pojačava se što je veći stupanj oštećenja bubrežne funkcije; stoga treba redovito mjeriti koncentraciju kreatinina u serumu: - najmanje jedanput godišnje u bolesnika s normalnom funkcijom bubrega - najmanje 2 do 4 puta godišnje u bolesnika u kojih su razine kreatinina na gornjoj granici
normalnih vrijednosti ili više, te u starijih osoba. U starijih osoba smanjena bubrežna funkcija je česta i nema simptoma. Posebna pozornost potrebna je u okolnostima kada može doći do oštećenja bubrežne funkcije; npr. kada se uvodi liječenje antihipertenzivima, diureticima ili nesteroidnim protuupalnim lijekom. Hipoglikemija Bolesnici koji Janumet uzimaju u kombinaciji sa sulfonilurejom ili s inzulinom mogu biti izloženi riziku od hipoglikemije. Stoga može biti neophodno sniziti dozu sulfonilureje ili inzulina. Reakcije preosjetljivosti Prijavljene su ozbiljne reakcije preosjetljivosti u bolesnika liječenih sitagliptinom nakon stavljanja lijeka na tržište. One obuhvaćaju anafilaksiju, angioedem i eksfolijativne kožne bolesti, uključujući Stevens-Johnsonov sindrom. Ove su se reakcije javile u prva tri mjeseca od početka primjene sitagliptina, a u nekim slučajevima prijavljene su nakon primjene prve doze. Pri sumnji na reakciju preosjetljivosti, treba prekinuti liječenje lijekom Janumet, procijeniti koji su drugi mogući uzroci te započeti liječenje drugim antidijabetikom (vidjeti dio 4.8). Operativni zahvat Budući da Janumet sadrži metforminklorid, njegovu se primjenu mora prekinuti 48 sati prije elektivnog operativnog zahvata pod općom, spinalnom ili epiduralnom anestezijom. Janumet se obično ponovno uvodi tek nakon što prođe 48 sati od operacije i to samo onda kada se utvrdi da je funkcija bubrega normalna. Primjena kontrastnog sredstva koje sadrži jod U bolesnika koji uzimaju metformin, intravaskularna primjena kontrastnog sredstva koje sadrži jod u radiološkim pretragama može dovesti do zatajenja bubrega povezanog s laktacidozom. Zbog toga treba prekinuti uzimanje lijeka Janumet prije ili za vrijeme pretrage, a liječenje se može nastaviti 48 sati nakon pretrage i to samo onda kada se utvrdi da je funkcija bubrega normalna (vidjeti dio 4.5). Promjene u kliničkom statusu bolesnika s prethodno reguliranim dijabetesom tipa 2 Ako se utvrde abnormalni laboratorijski nalazi ili klinička bolest (posebno ako je slabo izražena) u bolesnika s dijabetesom tipa 2 u kojeg je glikemija do tada bila dobro regulirana lijekom Janumet, treba odmah potražiti znakove ketoacidoze ili laktacidoze. Laboratorijske pretrage trebaju obuhvatiti određivanje vrijednosti elektrolita i ketona u serumu, razine glukoze u krvi te, prema indikaciji, vrijednosti pH krvi, laktata, piruvata i metformina. Utvrdi li se ketoacidoza ili laktacidoza, mora se odmah prekinuti primjena lijeka Janumet i uvesti odgovarajuće korektivne mjere. 4.5 Interakcije s drugim lijekovima i drugi oblici interakcija Istodobno uzimanje višekratnih doza sitagliptina (50 mg dvaput na dan) i metformina (1000 mg dvaput na dan) nisu u bolesnika s dijabetesom tipa 2 značajno utjecali na farmakokinetiku sitagliptina ni metformina. Do sada nisu provedena farmakokinetička istraživanja interakcije drugih lijekova s lijekom Janumet; međutim, takva su istraživanja provedena s djelatnim tvarima lijeka Janumet, sitagliptinom i metforminom, pojedinačno. Pri akutnom trovanju alkoholom (osobito nakon gladovanja, u slučaju neishranjenosti ili insuficijencije jetre) povećan je rizik od razvoja laktacidoze zbog metformina kao djelatne tvari u
H A L M E D
27 - 09 - 2012
O D O B R E N O

5
lijeku Janumet (vidjeti dio 4.4). Zbog toga se mora izbjegavati uzimanje alkohola i lijekova koji sadrže alkohol. Moguća je interakcija između metformina i kationskih lijekova koji se izlučuju putem bubrežnih tubula (npr. cimetidin), budući da se natječu za zajedničke sustave transporta bubrežnim tubulima. Ispitivanje provedeno na sedam zdravih dobrovoljaca pokazalo je da cimetidin u dozi od 400 mg dvaput na dan povisuje sistemsku izloženost metforminu (AUC) za 50%, a vršne koncentracije (Cmax) za 81%. Stoga, kad se istodobno daju kationski lijekovi koji se izlučuju bubrežnim tubulima, treba razmotriti strogi nadzor nad regulacijom glikemije, prilagodbu doze u okviru preporučenog doziranja i promjene u režimu liječenja dijabetesa. Intravaskularna primjena kontrastnog sredstva koje sadrži jod u radiološkim pretragama može dovesti do zatajivanja bubrega, a time i do nakupljanja metformina i rizika od laktacidoze. Zbog toga treba prekinuti uzimanje lijeka Janumet prije ili za vrijeme pretrage, a liječenje se može nastaviti 48 sati nakon pretrage i to samo kada se utvrdi da je funkcija bubrega normalna (vidjeti dio 4.4). Kombinacije kod kojih je potreban oprez Glukokortikoidi (sistemski i lokalni), beta-2 agonisti i diuretici sami po sebi djeluju hiperglikemijski. S time treba upoznati bolesnika i češće kontrolirati glukozu u krvi, a osobito na početku liječenja ovim lijekovima. Ako je potrebno, dozu antidijabetika treba prilagoditi u vrijeme ili nakon prekida liječenja drugim lijekovima. ACE inhibitori mogu sniziti razinu glukoze u krvi. Ako je potrebno, dozu antidijabetika treba prilagoditi u vrijeme ili nakon prekida liječenja drugim lijekovima. Djelovanje drugih lijekova na sitagliptin Podaci iz kliničkih ispitivanja ukazuju da je malen rizik od klinički značajnih interakcija pri istodobnoj primijeni s drugim lijekovima. Ciklosporin: Provedeno je ispitivanje djelovanja ciklosporina, snažnog inhibitora p-glikoproteina, na farmakokinetiku sitagliptina. Pri istodobnoj primjeni jednokratne oralne doze sitagliptina od 100 mg i jednokratne oralne doze ciklosporina od 600 mg AUC sitagliptina povećan je za oko 29%, a Cmax za oko 68%. Ove promjene u farmakokinetici sitagliptina ne smatraju se klinički značajnima. Izlučivanje sitagliptina putem bubrega nije značajno promijenjeno. Stoga se ne očekuju značajne interakcije s ostalim inhibitorima p-glikoproteina. Istraživanja in vitro ukazuju na to da je za ograničeni metabolizam sitagliptina ponajviše odgovoran enzim CYP3A4, a pridonosi mu i CYP2C8. U bolesnika s normalnom bubrežnom funkcijom, metabolizam, uključujući onaj putem CYP3A4, ima tek manju ulogu u izlučivanju sitagliptina. Metabolizam može imati značajniju ulogu u eliminaciji sitagliptina u bolesnika s teškim oštećenjem bubrega ili terminalnom fazom bolesti bubrega. Stoga je moguće da snažni inhibitori CYP3A4 (npr. ketokonazol, itrakonazol, ritonavir, klaritromicin) promijene farmakokinetiku sitagliptina u bolesnika s teškim oštećenjem bubrega ili terminalnom fazom bolesti bubrega. Djelovanje snažnih inhibitora CYP3A4 u bolesnika s insuficijencijom bubrega još nije ispitano u kliničkom ispitivanju. Istraživanja prijenosa in vitro pokazala su da je sitagliptin supstrat p-glikoproteina i organskog anionskog prijenosnika 3 (OAT3). Probenecid je inhibirao prijenos sitagliptina in vitro putem OAT3, ali se rizik od klinički značajnih interakcija smatra malim. Nije ispitana istodobna primjena sitagliptina s inhibitorima OAT3 in vivo.
H A L M E D
27 - 09 - 2012
O D O B R E N O

6
Djelovanje sitagliptina na druge lijekove Podaci iz istraživanja in vitro ukazuju na to da sitagliptin ne inhibira niti inducira izoenzime CYP450. U kliničkim ispitivanjima sitagliptin nije značajno utjecao na farmakokinetiku metformina, gliburida, simvastatina, roziglitazona, varfarina ili oralnih kontraceptiva te je u istraživanjima in vivo iskazao neznatnu sklonost interakciji sa supstratima CYP3A4, CYP2C8, CYP2C9 i organskim kationskim prijenosnikom (OCT). Također je u istraživanjima in vivo sitagliptin pokazao neznatno djelovanje na koncentracije digoksina u plazmi i blagu inhibiciju p-glikoproteina. Digoksin: Sitagliptin je tek neznatno djelovao na koncentracije digoksina u plazmi. Nakon istodobne primjene digoksina u dozi od 0,25 mg i sitagliptina u dozi od 100 mg na dan u trajanju od 10 dana, AUC digoksina porastao je u prosjeku za 11%, a njegova vršna koncentracija (Cmax) u plazmi za 18%. Stoga nije potrebno prilagođavati dozu digoksina. Ipak, kad se sitagliptin i digoksin istodobno primjenjuju, treba nadzirati bolesnike s rizikom od toksičnoga djelovanja digoksina. 4.6 Trudnoća i dojenje Nema dovoljno podataka o primjeni sitagliptina u trudnica. Istraživanja na životinjama pokazala su reproduktivnu toksičnost sitagliptina u visokim dozama (vidjeti dio 5.3). Ograničeni podaci ukazuju na to da primjena metformina u trudnica nije povezana s povećanim rizikom od urođenih malformacija. Istraživanja na životinjama s metforminom nisu pokazala štetno djelovanje na trudnoću, razvoj embrija i fetusa, porod i postnatalni razvoj (vidjeti i dio 5.3). Janumet se ne smije uzimati u trudnoći. Ako bolesnica planira trudnoću ili zatrudni, treba prekinuti liječenje Janumet tabletama i što prije započeti liječenje inzulinom. Do sada nisu provedena istraživanja djelovanja kombinacije djelatnih tvari lijeka Janumet na životinjama u laktaciji. Istraživanja provedena s djelatnim tvarima zasebno pokazala su da se i sitagliptin i metformin izlučuju u mlijeko štakorica. Metformin se u malim količinama izlučuje u majčino mlijeko. Nije poznato izlučuje li se sitagliptin u majčino mlijeko. Stoga dojilje ne smiju uzimati Janumet (vidjeti dio 4.3). 4.7 Utjecaj na sposobnost upravljanja vozilima i rada na strojevima Janumet nema poznatog utjecaja na sposobnost upravljanja vozilima i rada na strojevima. Ipak, kod upravljanja vozilom ili rada na strojevima treba uzeti u obzir da su pri primjeni sitagliptina prijavljene omaglica i pospanost. Bolesnike također treba upozoriti da postoji rizik od hipoglikemije uzima li se Janumet u kombinaciji sa sulfonilurejom ili s inzulinom. 4.8 Nuspojave Nisu provedena klinička ispitivanja s Janumet tabletama; međutim, utvrđena je bioekvivalencija lijeka Janumet i kombinacije sitagliptina i metformina (vidjeti dio 5.2). Sitagliptin i metformin Niže su navedene nuspojave za koje se smatra da su povezane s lijekom, a koje su u dvostruko slijepim kliničkim ispitivanjima prijavljivane češće u bolesnika liječenih sitagliptinom u kombinaciji s metforminom nego u bolesnika koji su uzimali placebo (> 0,2 % uz razliku > 1 bolesnika). Nuspojave su klasificirane prema organskim sustavima i apsolutnoj učestalosti (Tablica 1), uz korištenje preporučene terminologije MedDRA. Učestalost pojavljivanja definirana je kao: vrlo često (≥1/10), često (≥1/100 i <1/10), manje često (≥1/1000 i <1/100), rijetko (≥1/10.000 i <1/1000), vrlo rijetko (<1/10.000).
H A L M E D
27 - 09 - 2012
O D O B R E N O

7
Tablica 1. Učestalost nuspojava prijavljenih u placebom kontroliranim kliničkim ispitivanjima Nuspojava Učestalost nuspojave prema režimu liječenja
Sitagliptin s metforminom 1
Sitagliptin s metforminom i sulfonilurejom 2
Sitagliptin s metforminom i
agonistom PPARγ
(roziglitazon)3
Sitagliptin s metforminom i
inzulinom4
Vrijeme 24 tjedna 24 tjedna 18 tjedana 24 tjedna Poremećaji metabolizma i prehrane hipoglikemija* vrlo često često vrlo često Poremećaji živčanog sustava glavobolja često manje često somnolencija manje često Poremećaji probavnog sustava proljev manje često često mučnina često opstipacija često bol u gornjem dijelu abdomena
manje često
povraćanje često suha usta manje često Opći poremećaji i reakcije na mjestu primjeneperiferni edem često† Pretrage sniženje razine glukoze u krvi
manje često
* U kliničkim ispitivanjima sa sitagliptinom kao monoterapijom ili u kombinaciji s metforminom ili metforminom i agonistom PPARγ učestalost hipoglikemije prijavljene u bolesnika koji su uzimali sitagliptin bila je slična onoj u bolesnika koji su uzimali placebo. † Zabilježeno u analizi nakon 54. tjedna ispitivanja. 1 U ovom placebom kontroliranom 24-tjednom kliničkom ispitivanju primjene sitagliptina u dozi od
100 mg jedanput na dan kao dodatka dotadašnjem liječenju metforminom, učestalost nuspojava za koje se smatralo da su povezane s primjenom lijeka iznosila je 9,3% u bolesnika koji su uz dotadašnje liječenje metforminom uzimali sitagliptin, u odnosu na 10,1% u bolesnika koji su uz metformin uzimali placebo.
U dodatnom jednogodišnjem kliničkom ispitivanju primjene sitagliptina u dozi od 100 mg jedanput na dan kao dodatka dotadašnjem liječenju metforminom, učestalost nuspojava za koje se smatralo da su povezane s primjenom lijeka iznosila je 14,5% u bolesnika koji su uz dotadašnje liječenje metforminom uzimali sitagliptin, u odnosu na 30,3% u bolesnika koji su uz metformin uzimali sulfonilureju.
Prema zbirnim podacima iz kliničkih ispitivanja koja su trajala do godine dana i u kojima se uspoređivala primjena sitagliptina kao dodatka dotadašnjem liječenju metforminom s primjenom lijeka iz skupine sulfonilureja kao dodatka dotadašnjem liječenju metforminom, nuspojave za koje se smatralo da su povezane s primjenom lijeka i koje su u bolesnika koji su uzimali sitagliptin 100 mg bile češće (> 0,2% uz razliku > 1 bolesnika) nego u bolesnika koji su uzimali sulfonilureju
H A L M E D
27 - 09 - 2012
O D O B R E N O

8
bile su: anoreksija (Poremećaji metabolizma i prehrane; učestalost: manje često) i gubitak tjelesne težine (Pretrage; učestalost: manje često).
2 U ovom 24-tjednom placebom kontroliranom kliničkom ispitivanju primjene sitagliptina u dozi od
100 mg jedanput na dan kao dodatka dotadašnjem kombiniranom liječenju glimepiridom i metforminom, ukupna učestalost nuspojava za koje se smatralo da su povezane s primjenom lijeka iznosila je 18,1% u bolesnika koji su kao dodatak dotadašnjem liječenju glimepiridom i metforminom uzimali sitagliptin, u odnosu na 7,1% u bolesnika koji su uz glimepirid i metformin uzimali placebo.
3 U ovom kliničkom ispitivanju sitagliptina u dozi od 100 mg jedanput na dan u kombinaciji s
roziglitazonom i metforminom, koje je trajalo 54 tjedna, učestalost nuspojava za koje se smatralo da su povezane s primjenom lijeka u bolesnika koji su kao dio kombinacije uzimali sitagliptin iznosila je 15,3%, u odnosu na 10,9% u bolesnika koji su uz roziglitazon i metformin uzimali placebo. Ostale nuspojave povezane s primjenom lijeka zabilježene u analizi podataka nakon 54. tjedna (učestalost: često) u bolesnika liječenih kombiniranim liječenjem sa sitagliptinom, a koje su se javljale češće (> 0,2% uz razliku > 1 bolesnika) nego u bolesnika koji su kao dio kombinacije primali placebo bile su: glavobolja, kašalj, povraćanje, hipoglikemija, gljivične infekcije kože i infekcije gornjih dišnih putova.
4 U ovom 24-tjednom placebom kontroliranom kliničkom ispitivanju primjene sitagliptina u dozi od 100 mg jedanput na dan kao dodatka postojećem liječenju inzulinom i metforminom, učestalost nuspojava za koje se smatralo da su povezane s primjenom lijeka u bolesnika koji su uzimali sitagliptin u kombinaciji s inzulinom i metforminom iznosila je 16,2% u odnosu na 9,0% u bolesnika koji su uz inzulin i metformin uzimali placebo.
U 24-tjednom kliničkom ispitivanju početnog kombiniranog liječenja sitagliptinom i metforminom dvaput na dan (sitagliptin 50 mg plus metformin 500 mg, odnosno sitagliptin 50 mg plus metformin 1000 mg) ukupna učestalost nuspojava za koje se smatra da su povezane s primjenom lijeka bila je 14,0% u bolesnika liječenih kombinacijom sitagliptina i metformina u odnosu na 9,7% u bolesnika koji su uzimali placebo. Ukupna učestalost nuspojava za koje se smatra da su povezane s primjenom lijeka u bolesnika koji su uzimali kombinaciju sitagliptina i metformina bila je podjednaka onoj u bolesnika koji su uzimali samo metformin (14,0%), a veća nego u bolesnika koji su uzimali samo sitagliptin (6,7%), s tim da se ta razlika uglavnom odnosila na nuspojave u probavnom sustavu. Dodatni podaci o pojedinačnim djelatnim tvarima koje se nalaze u fiksnoj kombinaciji Sitagliptin U kliničkim ispitivanjima koja su trajala do 24 tjedna u kojima se uzimao samo sitagliptin u dozi od 100 mg jedanput na dan, nuspojave za koje se smatra da su povezane s primjenom lijeka prijavljene češće u bolesnika koji su uzimali sitagliptin (>2% uz razliku >1 bolesnika) nego u bolesnika koji su uzimali placebo bile su: glavobolja, hipoglikemija, opstipacija i omaglica. Osim navedenih nuspojava povezanih s primjenom lijeka, neželjeni događaji (bez obzira jesu li povezani s primjenom lijeka ili ne) zabilježeni u najmanje 5% bolesnika, a češće u bolesnika liječenih sitagliptinom, obuhvaćali su infekciju gornjih dišnih putova i nazofaringitis. Ostali neželjeni događaji zabilježeni češće u bolesnika liječenih sitagliptinom (koji nisu dosegli razinu učestalosti od 5%, ali im je učestalost sa sitagliptinom veća za 0,5% nego u kontrolnoj skupini) uključuju osteoartritis i bol u udovima. U kliničkim ispitivanjima opažen je mali porast broja leukocita (razlika od oko 200 stanica po mikrolitri u odnosu na placebo; uz srednji početni broj leukocita od oko 6600 stanica po mikrolitri) uslijed porasta broja neutrofila. To je zamijećeno u većini, ali ne i u svim kliničkim ispitivanjima. Ova promjena u laboratorijskim nalazima ne smatra se klinički značajnom. Pri primjeni sitagliptina nisu opažene klinički značajne promjene u vitalnim znakovima niti u EKG-u (uključujući promjene QT-intervala).
H A L M E D
27 - 09 - 2012
O D O B R E N O

9
Podaci nakon stavljanja lijeka na tržište Nakon stavljanja na tržište lijeka Janumet, odnosno sitagliptina, jedne od djelatnih tvari ovog lijeka, prijavljene su dodatne nuspojave (učestalost nepoznata). Te su nuspojave prijavljene kad su se Janumet, odnosno sitagliptin, primjenjivali samostalno i/ili u kombinaciji s drugim antidijabeticima, a obuhvaćaju: reakcije preosjetljivosti uključujući anafilaksiju, angioedem, osip, urtikariju, kožni vaskulitis i eksfolijativne kožne bolesti, uključujući Stevens-Johnsonov sindrom (vidjeti dio 4.4); akutni pankreatitis, uključujući hemoragični i nekrotizirajući pankreatitis sa ili bez smrtnog ishoda (vidjeti dio 4.4); poremećaj bubrežne funkcije, uključujući akutno zatajenje bubrega (koje u nekim slučajevima zahtijeva dijalizu); povraćanje. Metformin Podaci iz kliničkih ispitivanja i nakon stavljanja lijeka na tržište U Tablici 2 prikazane su nuspojave prema organskim sustavima i učestalosti. Kategorije učestalosti temelje se na podacima dobivenima iz Sažetka opisa svojstava lijeka metformina s tržišta Europske Unije. Tablica 2. Učestalost nuspojava metformina zabilježenih u kliničkim ispitivanjima i nakon stavljanja lijeka na tržište Nuspojava Učestalost Poremećaji metabolizma i prehrane laktacidoza vrlo rijetko nedostatak vitamina B12a vrlo rijetko Poremećaji živčanog sustava metalni okus često Poremećaji probavnog sustava probavne smetnjeb vrlo često Poremećaji jetre i žuči poremećaj funkcije jetre, hepatitis vrlo rijetko Poremećaji kože i potkožnog tkiva urtikarija, eritem, svrbež vrlo rijetko aDugotrajno liječenje metforminom povezano je sa smanjenom resorpcijom vitamina B12, što u vrlo rijetkim slučajevima može dovesti do klinički značajnoga nedostatka (deficijencije) vitamina B12 (npr. megaloblastične anemije). bSimptomi u probavnom sustavu poput mučnine, povraćanja, proljeva, boli u trbuhu i gubitka teka najčešće se javljaju na početku liječenja i u većini se slučajeva sami povuku. 4.9 Predoziranje Nema podatka o predoziranju lijekom Janumet. Zdravi ispitanici su u kontroliranim kliničkim ispitivanjima općenito dobro podnosili jednokratne doze do najviše 800 mg sitagliptina. U jednom ispitivanju pri dozi od 800 mg sitagliptina opaženo je minimalno povećanje QTc-intervala koje nije bilo klinički značajno. Nema iskustava s primjenom doza viših od 800 mg u kliničkim ispitivanjima. U kliničkim ispitivanjima faze I s višekratnim dozama
H A L M E D
27 - 09 - 2012
O D O B R E N O

10
nisu zamijećene o dozi ovisne klinički značajne nuspojave sitagliptina kada se davao u dozama do najviše 600 mg na dan tijekom najdulje 10 dana ili u dozi od 400 mg na dan tijekom najdulje 28 dana. Predoziranje visokim dozama metformina (ili već postojeći rizik od laktacidoze) može dovesti do laktacidoze koja zahtijeva hitnu bolničku intervenciju. Najdjelotvorniji način uklanjanja laktata i metformina jest hemodijaliza. Sitagliptin se manjim dijelom može ukloniti hemodijalizom. U kliničkim je ispitivanjima hemodijalizom u trajanju od 3-4 sata uklonjeno oko 13,5% doze lijeka. Ako je to klinički opravdano, može se razmotriti produljenje hemodijalize. Nije poznato je li moguće ukloniti sitagliptin peritonejskom dijalizom. U slučaju predoziranja, treba primijeniti uobičajene suportivne mjere, npr. ukloniti neresorbirani lijek iz probavnog sustava, uvesti klinički nadzor (uključujući i EKG) i po potrebi započeti suportivno liječenje. 5. FARMAKOLOŠKA SVOJSTVA 5.1 Farmakodinamička svojstva Farmakoterapijska skupina: Lijekovi za liječenje šećerne bolesti; Kombinacije oralnih antidijabetika, ATK oznaka: A10BD07 Janumet sadrži kombinaciju dvaju antidijabetika s komplementarnim mehanizmima djelovanja koji poboljšavaju regulaciju glikemije u bolesnika s dijabetesom tipa 2: sitagliptinfosfat, inhibitor dipeptidil peptidaze 4 (DPP-4), i metforminklorid, lijek iz skupine bigvanida. Sitagliptin Sitagliptinfosfat je snažan i vrlo selektivan peroralni inhibitor enzima dipeptidil peptidaze 4 (DPP-4), namijenjen liječenju dijabetesa tipa 2. Inhibitori enzima DPP-4 pripadaju skupini lijekova koji pojačavaju djelovanje inkretina. Inhibicijom enzima DPP-4 sitagliptin povisuje razine dvaju poznatih aktivnih inkretinskih hormona, peptida-1 nalik glukagonu (engl. glucagon-like peptide-1, GLP-1) i inzulinotropnog polipeptida ovisnog o glukozi (engl. glucose-dependent insulinotropic polypeptide, GIP). Inkretini su dio endogenog sustava koji sudjeluje u fiziološkoj regulaciji homeostaze glukoze. Kad je koncentracija glukoze u krvi normalna ili povišena, GLP-1 i GIP povećavaju sintezu inzulina i njegovo izlučivanje iz beta stanica gušterače. Uz to, GLP-1 smanjuje i izlučivanje glukagona iz alfa stanica gušterače, čime se smanjuje proizvodnja glukoze u jetri. Pri niskim razinama glukoze u krvi ne dolazi do poticanja izlučivanja inzulina niti do smanjenja lučenja glukagona. Sitagliptin je snažan i vrlo selektivan inhibitor enzima DPP-4 te pri terapijskim koncentracijama ne inhibira njemu vrlo srodne enzime DPP-8 i DPP-9. Po svojoj kemijskoj strukturi i farmakološkom djelovanju, sitagliptin se razlikuje od analoga GLP-1, inzulina, sufonilureja ili meglitinida, bigvanida, agonista receptora za aktivator proliferacije peroksisoma-gama (engl. proliferator-activated receptor gamma, PPAR), inhibitora alfa-glukozidaze i analoga amilina. U dvodnevnom ispitivanju u zdravih osoba, sitagliptin sam povisio je koncentraciju aktivnog GLP-1 dok je metformin sam povisio koncentraciju i aktivnog i ukupnog GLP-1 do sličnih vrijednosti. Istodobna primjena sitagliptina i metformina imala je aditivan učinak na koncentraciju aktivnog GLP-1. Sitagliptin je, ali ne i metformin, povisio koncentraciju aktivnog GIP. Sveukupno, sitagliptin je poboljšao regulaciju glikemije kada se uzimao kao monoterapija ili kao dio kombiniranog liječenja. U kliničkim je ispitivanjima sitagliptin, primijenjen kao monoterapija, poboljšao regulaciju glikemije i značajno snizio hemoglobin A1c (HbA1c) te glukozu natašte i nakon obroka. Sniženje razine glukoze u plazmi natašte (engl. fasting plasma glucose, FPG) opaženo je pri prvoj vremenskoj točki mjerenja, tj.
H A L M E D
27 - 09 - 2012
O D O B R E N O

11
nakon tri tjedna liječenja. Zamijećena učestalost hipoglikemije u bolesnika liječenih sitagliptinom bila je slična onoj u skupini koja je uzimala placebo. Liječenje sitagliptinom nije dovelo do porasta tjelesne težine u odnosu na početnu. Opažena su poboljšanja zamjenskih pokazatelja funkcije beta stanica, uključujući procjenu modela homeostaze beta (engl. Homeostasis Model Assessment-ß, HOMA-ß), omjer proinzulina i inzulina te rezultate odgovora beta stanica na temelju testa podnošljivosti često uzorkovanih obroka (engl. frequently-sampled meal tolerance test). Klinička ispitivanja primjene sitagliptina u kombinaciji s metforminom U 24-tjednom placebom kontroliranom kliničkom ispitivanju djelotvornosti i sigurnosti primjene dodavanja sitagliptina u dozi od 100 mg jedanput na dan postojećem liječenju metforminom, sitagliptin je značajno poboljšao glikemijske parametre u odnosu na placebo. Promjena tjelesna težina u odnosu na početnu bila je slična u bolesnika koji su uzimali sitagliptin i onih koji su uzimali placebo. U ovoj je studiji također u obje skupine bolesnika bila slična učestalost hipoglikemije. U drugom 24-tjednom placebom kontroliranom faktorijelnom kliničkom ispitivanju početnog liječenja, sitagliptin u dozi od 50 mg dvaput na dan u kombinaciji s metforminom (u dozama od 500 mg ili 1000 mg dvaput na dan) doveo je do značajnog poboljšanja glikemijskih parametara u odnosu na oba lijeka primijenjena u monoterapiji. Gubitak tjelesne težine u skupini koja je uzimala kombinaciju sitagliptina i metformina bio je sličan onome opaženom u skupini koja je uzimala samo metformin ili placebo; u bolesnika koji su uzimali samo sitagliptin nije bilo promjene u odnosu na početne vrijednosti. Učestalost hipoglikemije bila je podjednaka u svim ispitanim skupinama. Kliničko ispitivanje primjene sitagliptina u kombinaciji s metforminom i sulfonilurejom U 24-tjednom placebom kontroliranom kliničkom ispitivanju cilj je bio ocijeniti djelotvornost i sigurnost primjene sitagliptina (100 mg jedanput na dan) kao dodatka liječenju glimepiridom (samom ili u kombinaciji s metforminom). Dodatak sitagliptina glimepiridu i metforminu značajno je poboljšao glikemijske parametre. U bolesnika liječenih sitagliptinom došlo je do blagog porasta tjelesne težine (+1,1 kg) u odnosu na bolesnike koji su uzimali placebo. Kliničko ispitivanje primjene sitagliptina u kombinaciji s metforminom i agonistom PPAR U 54-tjednom placebom kontroliranom kliničkom ispitivanju cilj je bio ocijeniti djelotvornost i sigurnost primjene sitagliptina (100 mg jedanput na dan) kao dodatka liječenju roziglitazonom u kombinaciji s metforminom. Dodatak sitagliptina roziglitazonu i metforminu značajno je poboljšao glikemijske parametre u primarnoj kontrolnoj vremenskoj točki nakon 18 tjedana, a poboljšanje se održalo do završetka ispitivanja. Porast tjelesne težine bio je sličan u bolesnika liječenih sitagliptinom i onih koji su uzimali placebo (1,9 kg naprama 1,3 kg). Kliničko ispitivanje primjene sitagliptina u kombinaciji s metforminom i inzulinom U 24-tjednom placebom kontroliranom kliničkom ispitivanju cilj je bio ocijeniti djelotvornost i sigurnost primjene sitagliptina (100 mg jedanput na dan) kao dodatka liječenju inzulinom (u stabilnoj dozi tijekom najmanje 10 tjedana) sa ili bez primjene metformina (najmanje 1500 mg). U bolesnika koji su primali predmiješani inzulin, prosječna dnevna doza bila je 70,9 U/dan. U bolesnika koji su primali nemiješani inzulin (srednjedugog ili dugog djelovanja), prosječna dnevna doza bila je 44,3 U/dan. Podaci za 73% bolesnika koji su uzimali i metformin prikazani su u Tablici 3. Dodatak sitagliptina inzulinu značajno je poboljšao glikemijske parametre. Niti u jednoj skupini nije bilo značajne promjene u porastu tjelesne težine u odnosu na početne vrijednosti. Tablica 3: Vrijednosti HbA1c u placebom kontroliranim kliničkim ispitivanjima kombiniranog liječenja sitagliptinom i metforminom*
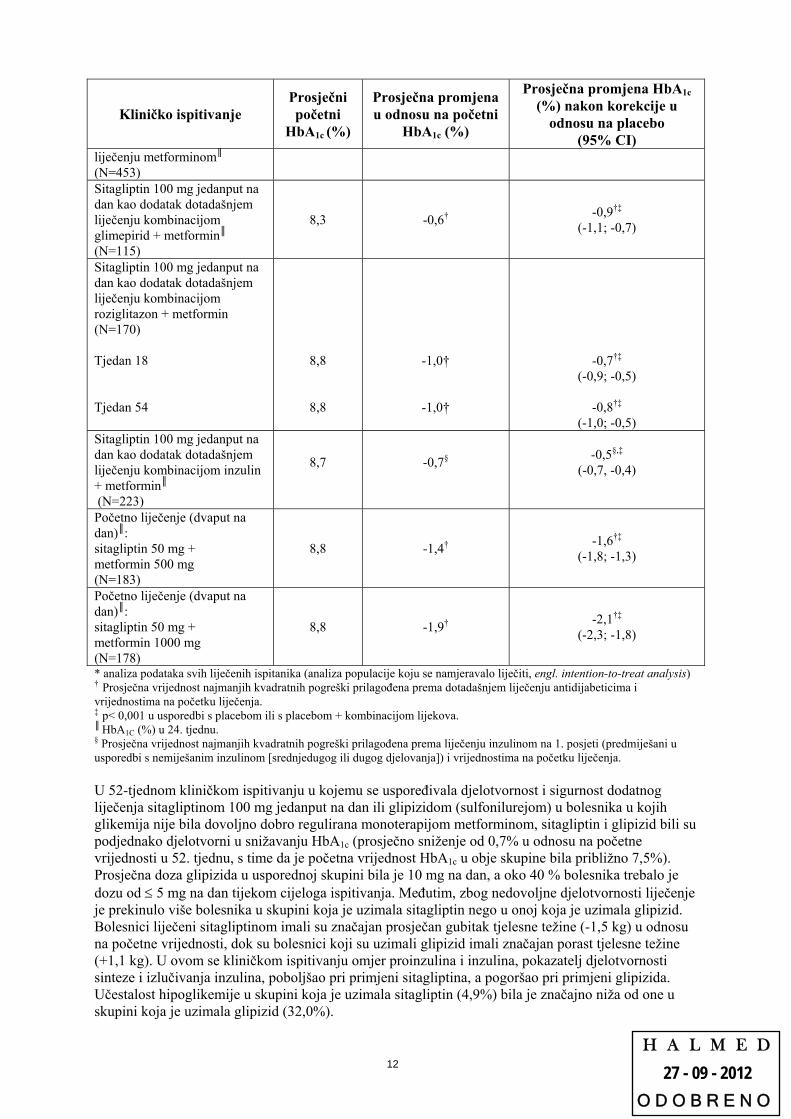
Kliničko ispitivanje Prosječni početni
HbA1c (%)
Prosječna promjena u odnosu na početni
HbA1c (%)
Prosječna promjena HbA1c (%) nakon korekcije u
odnosu na placebo
(95% CI) Sitagliptin 100 mg jedanput na dan kao dodatak dotadašnjem
8,0 -0,7† -0,7†‡
(-0,8; -0,5)
H A L M E D
27 - 09 - 2012
O D O B R E N O
12
Kliničko ispitivanje Prosječni početni
HbA1c (%)
Prosječna promjena u odnosu na početni
HbA1c (%)
Prosječna promjena HbA1c (%) nakon korekcije u
odnosu na placebo
(95% CI) liječenju metforminom║ (N=453) Sitagliptin 100 mg jedanput na dan kao dodatak dotadašnjem liječenju kombinacijom glimepirid + metformin║ (N=115)
8,3 -0,6† -0,9†‡
(-1,1; -0,7)
Sitagliptin 100 mg jedanput na dan kao dodatak dotadašnjem liječenju kombinacijom roziglitazon + metformin (N=170) Tjedan 18 Tjedan 54
8,8
8,8
-1,0†
-1,0†
-0,7†‡
(-0,9; -0,5)
-0,8†‡
(-1,0; -0,5) Sitagliptin 100 mg jedanput na dan kao dodatak dotadašnjem liječenju kombinacijom inzulin + metformin║ (N=223)
8,7
-0,7§
-0,5§,‡
(-0,7, -0,4)
Početno liječenje (dvaput na dan)║: sitagliptin 50 mg + metformin 500 mg (N=183)
8,8 -1,4† -1,6†‡
(-1,8; -1,3)
Početno liječenje (dvaput na dan)║: sitagliptin 50 mg + metformin 1000 mg (N=178)
8,8 -1,9† -2,1†‡
(-2,3; -1,8)
* analiza podataka svih liječenih ispitanika (analiza populacije koju se namjeravalo liječiti, engl. intention-to-treat analysis) † Prosječna vrijednost najmanjih kvadratnih pogreški prilagođena prema dotadašnjem liječenju antidijabeticima i vrijednostima na početku liječenja. ‡ p< 0,001 u usporedbi s placebom ili s placebom + kombinacijom lijekova. ║ HbA1C (%) u 24. tjednu. § Prosječna vrijednost najmanjih kvadratnih pogreški prilagođena prema liječenju inzulinom na 1. posjeti (predmiješani u usporedbi s nemiješanim inzulinom [srednjedugog ili dugog djelovanja]) i vrijednostima na početku liječenja. U 52-tjednom kliničkom ispitivanju u kojemu se uspoređivala djelotvornost i sigurnost dodatnog liječenja sitagliptinom 100 mg jedanput na dan ili glipizidom (sulfonilurejom) u bolesnika u kojih glikemija nije bila dovoljno dobro regulirana monoterapijom metforminom, sitagliptin i glipizid bili su podjednako djelotvorni u snižavanju HbA1c (prosječno sniženje od 0,7% u odnosu na početne vrijednosti u 52. tjednu, s time da je početna vrijednost HbA1c u obje skupine bila približno 7,5%). Prosječna doza glipizida u usporednoj skupini bila je 10 mg na dan, a oko 40 % bolesnika trebalo je dozu od 5 mg na dan tijekom cijeloga ispitivanja. Međutim, zbog nedovoljne djelotvornosti liječenje je prekinulo više bolesnika u skupini koja je uzimala sitagliptin nego u onoj koja je uzimala glipizid. Bolesnici liječeni sitagliptinom imali su značajan prosječan gubitak tjelesne težine (-1,5 kg) u odnosu na početne vrijednosti, dok su bolesnici koji su uzimali glipizid imali značajan porast tjelesne težine (+1,1 kg). U ovom se kliničkom ispitivanju omjer proinzulina i inzulina, pokazatelj djelotvornosti sinteze i izlučivanja inzulina, poboljšao pri primjeni sitagliptina, a pogoršao pri primjeni glipizida. Učestalost hipoglikemije u skupini koja je uzimala sitagliptin (4,9%) bila je značajno niža od one u skupini koja je uzimala glipizid (32,0%).
H A L M E D
27 - 09 - 2012
O D O B R E N O

13
Metformin Metformin je bigvanid koji djeluje antihiperglikemijski, snižavajući bazalne i postprandijalne vrijednosti glukoze u krvi. Ne stimulira lučenje inzulina te stoga ne izaziva hipoglikemiju. Metformin može djelovati putem tri mehanizma: - smanjujući proizvodnju glukoze u jetri time što inhibira glukoneogenezu i glikogenolizu - u mišićima, umjereno povećavajući osjetljivost na inzulin, čime se poboljšava pohrana i
iskorištavanje glukoze u perifernim tkivima - usporavajući resorpciju glukoze u crijevima. Metformin potiče unutarstaničnu sintezu glikogena time što djeluje na glikogen sintazu. Metformin povećava transportni kapacitet pojedinih tipova membranskih transportera glukoze (GLUT-1 i GLUT-4). U ljudi, neovisno o djelovanju na glukozu, metformin ima povoljan učinak na metabolizam lipida. Kontrolirana srednjeduga i dugotrajna klinička ispitivanja pokazala su da metformin pri terapijskim dozama snižava razine ukupnog i LDL kolesterola te triglicerida. U prospektivnom randomiziranom kliničkom ispitivanju (pod nazivom UKPDS) utvrđeni su dugoročni povoljni učinci intenzivne regulacije glikemije u bolesnika s dijabetesom tipa 2. Analiza rezultata u bolesnika s prekomjernom tjelesnom težinom liječenih metforminom nakon neuspjelog liječenja samo dijetom pokazala je sljedeće: - značajno smanjenje apsolutnog rizika od bilo koje komplikacije povezane s dijabetesom u
skupini liječenoj metforminom (29,8 događaja na 1000 bolesnik-godina) u odnosu na skupinu koja je bila samo na dijeti (43,3 događaja na 1000 bolesnik-godina), p=0,0023, i u odnosu na objedinjene podatke skupina koje su uzimale samo sulfonilureju ili primale samo inzulin (40,1 događaja na 1000 bolesnik-godina), p=0,0034
- značajno smanjenje apsolutnog rizika od smrtnosti povezane s dijabetesom - metformin: 7,5 događaja na 1000 bolesnik-godina; skupina samo na dijeti: 12,7 događaja na 1000 bolesnik-godina, p=0,017
- značajno smanjenje apsolutnog rizika od ukupne smrtnosti: 13,5 događaja na 1000 bolesnik-godina u bolesnika na metforminu naprama 20,6 događaja na 1000 bolesnik-godina za skupinu samo na dijeti (p=0,011), odnosno 18,9 događaja na 1000 bolesnik-godina za objedinjene podatke skupina koje su uzimale samo sulfonilureju ili primale samo inzulin (p=0,021)
- značajno smanjenje apsolutnog rizika od infarkta miokarda - metformin: 11 događaja na 1000 bolesnik-godina, skupina samo na dijeti: 18 događaja na 1000 bolesnik-godina (p=0,01).
Europska agencija za lijekove je izuzela obvezu podnošenja rezultata ispitivanja lijeka Janumet u svim podskupinama pedijatrijske populacije u dijabetesu mellitu tipa 2 (vidjeti dio 4.2 za informacije o pedijatrijskoj primjeni). 5.2 Farmakokinetička svojstva Janumet Ispitivanje bioekvivalencije u zdravih osoba pokazalo je da su Janumet kombinirane tablete (sitagliptin/metforminklorid) bioekvivalentne istodobnoj primjeni pojedinačnih tableta sitagliptinfosfata i tableta metforminklorida. Sljedeći podaci odnose se na farmakokinetička svojstva pojedinačnih djelatnih tvari u lijeku Janumet .
H A L M E D
27 - 09 - 2012
O D O B R E N O

14
Sitagliptin Apsorpcija Nakon peroralne primjene doze od 100 mg u zdravih osoba, sitagliptin se brzo resorbira, a vršna koncentracija u plazmi (medijan Tmax) zabilježena je 1 do 4 sata nakon primjene doze; srednji AUC sitagliptina u plazmi iznosi 8,52 µM•hr, a Cmax 950 nM. Apsolutna bioraspoloživost sitagliptina je približno 87%. Budući da istodobno uzimanje s punomasnim obrokom ne utječe na farmakokinetiku lijeka, sitagliptin se može uzimati s hranom ili bez nje. AUC sitagliptina u plazmi raste proporcionalno dozi. Proporcionalnost dozi nije utvrđena za Cmax i C24hr (porast Cmax je bio veći, a C24hr manji od porasta proporcionalnog dozi). Distribucija Srednji volumen raspodjele sitagliptina u stanju dinamičke ravnoteže nakon intravenske primjene doze od 100 mg u zdravih osoba iznosi oko 198 litara. Udio sitagliptina koji se reverzibilno veže za bjelančevine u plazmi je nizak (38%). Biotransformacija Sitagliptin se prvenstveno eliminira mokraćom u neizmijenjenom obliku, a tek manji dio se razgrađuje. Mokraćom se u neizmijenjenom obliku izluči oko 79% sitagliptina. Nakon primjene peroralne doze [14C]sitagliptina približno 16% radioaktivnosti izlučilo se u obliku metabolita lijeka. Otkriveno je šest metabolita u tragovima i ne očekuje se da oni pridonose inhibicijskom djelovanju sitagliptina u plazmi na DPP-4. Istraživanja in vitro ukazuju na to da je za ograničeni metabolizam sitagliptina u najvećoj mjeri odgovoran enzim CYP3A4, uz doprinos CYP2C8. In vitro istraživanja pokazala su da sitagliptin ne inhibira CYP izoenzime CYP3A4, 2C8, 2C9, 2D6, 1A2, 2C19 ili 2B6 te da ne inducira CYP3A4 i CYP1A2. Eliminacija Nakon primjene peroralne doze [14C]sitagliptina u zdravih osoba, približno 100% radioaktivnosti izluči se fecesom (13%) ili mokraćom (87%) unutar tjedan dana od primjene. Prividno terminalno t1/2 sitagliptina nakon peroralne primjene doze od 100 mg iznosi oko 12,4 sata. Nakupljanje sitagliptina pri višekratnim dozama je minimalno. Renalni klirens sitagliptina iznosi približno 350 ml/min. Sitagliptin se najvećim dijelom eliminira izlučivanjem bubrezima putem aktivne tubularne sekrecije. Sitagliptin je supstrat humanog organskog anionskog prijenosnika-3 (engl. human organic anion transporter-3, hOAT-3) koji bi mogao biti uključen u eliminaciju sitagliptina putem bubrega. Dosad nije utvrđena klinička važnost hOAT-3 u prijenosu sitagliptina. Sitagliptin je također i supstrat p-glikoproteina koji bi također mogao posredovati u eliminaciji sitagliptina bubrezima. Međutim, ciklosporin kao inhibitor p-glikoproteina, nije smanjio renalni klirens sitagliptina. Sitagliptin nije supstrat prijenosnika OCT2, OAT1 ni PEPT1/2. In vitro, sitagliptin ne inhibira prijenos posredovan OAT3 (IC50=160 M) ili p-glikoproteinom (do 250 M) pri terapijski značajnim koncentracijama u plazmi. U jednom je kliničkom ispitivanju sitagliptin imao mali učinak na koncentracije digoksina u plazmi, što ukazuje da bi sitagliptin mogao biti blagi inhibitor p-glikoproteina. Osobitosti lijeka u bolesnika Farmakokinetika sitagliptina općenito je podjednaka u zdravih osoba i u bolesnika s dijabetesom tipa 2.
H A L M E D
27 - 09 - 2012
O D O B R E N O

15
Oštećenje bubrega U otvorenom kliničkom ispitivanju jednokratne doze utvrđivala se farmakokinetika snižene doze sitagliptina (50 mg) u bolesnika s kroničnim oštećenjem bubrega različitoga stupnja u odnosu na zdrave kontrolne ispitanike. U ispitivanje su uključeni bolesnici s oštećenjem bubrega klasificiranim na temelju klirensa kreatinina na blago (50 do <80 ml/min), umjereno (30 do <50 ml/min) i teško (<30 ml/min), kao i bolesnici na hemodijalizi s bolešću bubrega u terminalnoj fazi (ESRD). Bolesnici s blagim oštećenjem bubrega nisu imali klinički značajno povišenje koncentracije sitagliptina u plazmi u odnosu na zdrave kontrolne ispitanike. Otprilike dvostruki porast AUC-a sitagliptina u plazmi zamijećen je u bolesnika s umjerenim oštećenjem bubrega, dok je u bolesnika s teškim oštećenjem i onih s ESRD-om na dijalizi porast bio četiri puta veći u odnosu na zdrave kontrolne ispitanike. Sitagliptin se umjereno uklanja hemodijalizom (13,5% nakon dijalize u trajanju 3-4 sata započete 4 sata nakon primjene lijeka). Budući da je iskustvo vrlo ograničeno, ne preporučuje se primjena sitagliptina u bolesnika s umjerenim ili teškim oštećenjem bubrega, uključujući one s ESRD-om (vidjeti dio 4.2). Oštećenje jetre U bolesnika s blagim ili umjerenim oštećenjem jetre (rezultat 9 po Child Pugh klasifikaciji) nije potrebno prilagođavati dozu sitagliptina. Nema kliničkih podataka o bolesnicima s teškim oštećenjem jetre (rezultat >9 po Child Pugh klasifikaciji). Međutim, budući da se sitagliptin najvećim dijelom uklanja putem bubrega, ne očekuje se da bi teško oštećenje jetre moglo utjecati na farmakokinetiku lijeka. Starije osobe Nije potrebno prilagođavati dozu s obzirom na dob. Prema analizi podataka populacijske farmakokinetike iz kliničkih ispitivanja faze I i II, dob nema klinički značajan utjecaj na farmakokinetiku sitagliptina. Starije osobe (65-80 godina) imale su oko 19% više koncentracije sitagliptina u plazmi u usporedbi s mlađim bolesnicima. Pedijatrijska populacija Nisu provedena klinička ispitivanja sitagliptina u pedijatrijskih bolesnika. Ostale osobitosti u bolesnika Nije potrebno prilagođavati dozu s obzirom na spol, rasu ili indeks tjelesne mase (BMI). Prema zbirnoj analizi farmakokinetičkih podataka iz kliničkih ispitivanja faze I kao i podataka o populacijskoj farmakokinetici iz kliničkih ispitivanja faze I i II, ove osobitosti bolesnika nemaju klinički značajan utjecaj na farmakokinetiku sitagliptina. Metformin Apsorpcija Nakon primjene peroralne doze metformina, tmax se postiže za 2,5 h. Apsolutna bioraspoloživost tablete metformina od 500 mg u zdravih osoba iznosi oko 50-60%. Nakon primjene peroralne doze, neapsorbirani dio koji se izluči fecesom iznosi 20-30%. Nakon peroralne primjene, apsorpcija metformina je nepotpuna jer dolazi do zasićenja. Pretpostavlja se da farmakokinetika apsorpcije metformina nije linearna. Pri uobičajenim dozama i režimu primjene metformina, koncentracije u stanju dinamičke ravnoteže u plazmi postižu se za 24 do 48 sati i obično su niže od 1 µg/ml. U kontroliranim kliničkim ispitivanjima, vršne razine metformina u plazmi (Cmax) nisu čak ni pri najvećim dozama prelazile prag od 4 µg/ml. Hrana smanjuje i neznatno usporava apsorpciju metformina. Nakon primjene doze od 850 mg, opažena je 40% niža vršna koncentracija lijeka u plazmi, smanjenje AUC-a za 25% te produljenje vremena do postizanja vršne koncentracije u plazmi za 35 min. Nije poznato jesu li ovi podaci klinički značajni.
H A L M E D
27 - 09 - 2012
O D O B R E N O

16
Distribucija Vezanje za bjelančevine u plazmi je zanemarivo. Metformin se raspodjeljuje u eritrocite. Vršna koncentracija u krvi je niža nego u plazmi, a postiže se gotovo istovremeno. Eritrociti su vrlo vjerojatno sekundarni prostor raspodjele. Srednji volumen raspodjele (Vd) kreće se između 63 l i 276 l. Biotransformacija Metformin se izlučuje neizmijenjen mokraćom. U ljudi nisu utvrđeni njegovi metaboliti. Eliminacija Brzina bubrežnog klirensa metformina iznosi >400 ml/min, što ukazuje na to da se eliminira glomerularnom filtracijom i tubularnom sekrecijom. Nakon peroralne doze prividno terminalno poluvrijeme eliminacije iznosi oko 6,5 h. Kad je oštećena funkcija bubrega, bubrežni se klirens smanjuje proporcionalno klirensu kreatinina te je poluvrijeme eliminacije produljeno, što dovodi do povišenih razina metformina u plazmi. 5.3 Neklinički podaci o sigurnosti primjene Nisu provedena istraživanja lijeka Janumet na životinjama. U ispitivanjima na psima u trajanju od 16 tjedana, kombinacija metformina i sitagliptina nije pokazala dodatnu toksičnost u odnosu na primjenu samog metformina. U ovim je istraživanjima izloženost sitagliptinu koja nije dovela do štetnih učinaka (tzv. NOEL) bila šest, a kod metformina 2,5 puta viša od izloženosti u ljudi. Sljedeći se podaci odnose na rezultate ispitivanja sitagliptina ili metformina pojedinačno. Sitagliptin Toksično djelovanje na bubrege i jetru opaženo je kod glodavaca pri 58 puta većoj sistemskoj izloženosti lijeku od one u ljudi, dok je prag netoksičnog djelovanja utvrđen pri 19 puta većoj izloženosti nego u ljudi. U štakora su zamijećene abnormalnosti na sjekutićima pri razinama izloženosti 67 puta višima od kliničke; u 14- tjednom ispitivanju na štakorima utvrđeno je da je prag netoksičnog djelovanja, s obzirom na ovaj nalaz, izloženost 58 puta viša od kliničke razine. Nije poznat značaj ovih saznanja za ljude. Prolazni fizikalni znakovi povezani s liječenjem, od kojih neki upućuju na neurotoksičnost, poput disanja otvorenih usta, jačeg slinjenja, povraćanja bijele pjene, ataksije, drhtavice, smanjene aktivnosti i/ili pogrbljenog držanja tijela zamijećeni su kod pasa pri izloženosti otprilike 23 puta većoj od kliničke. Histološki je, osim toga, opažena neznatna ili vrlo mala degeneracija skeletnih mišića pri dozama koje su dovele do sistemske izloženosti oko 23 puta veće od one u ljudi. Prag netoksičnog djelovanja za ove učinke bio je pri razini izloženosti 6 puta većoj od kliničkih razina. Sitagliptin se u nekliničkim istraživanjima nije pokazao genotoksičnim. Kod miševa se nije pokazao kancerogenim. Kod štakora je opažena povećana učestalost adenoma i karcinoma jetre pri sistemskoj izloženosti 58 puta većoj od razina izloženosti u ljudi. Budući da je utvrđena korelacija između hepatotoksičnosti lijeka i nastanka neoplazija u jetri štakora, pri tako visokoj dozi vjerojatno je povećana učestalost tumora jetre posljedica kroničnog toksičnog djelovanja na jetru. Budući da su sigurnosne granice visoke (19 puta više od koncentracije bez učinka), ove se neoplazije ne smatraju relevantnima za primjenu lijeka u ljudi. Kod štakora i štakorica koji su sitagliptin primali prije i tijekom parenja nisu zamijećeni učinci na plodnost povezani s primjenom lijeka. Sitagliptin nije pokazao štetno djelovanje na razvoj fetusa i mladunčadi štakora. Ispitivanja reproduktivne toksičnosti ukazala su na neznatno veću učestalost prenatalnih malformacija rebara povezanih s primjenom lijeka (nedostatak, nerazvijenost i "valovita" rebra) u mladunčadi štakora pri sistemskoj izloženosti 29 puta većoj od izloženosti koja se postiže u ljudi. Toksičnost za
H A L M E D
27 - 09 - 2012
O D O B R E N O

17
majku opažena je kod zečica pri izloženosti 29 puta većoj od izloženosti koja se postiže u ljudi. Budući da je granica sigurnosti visoka, ova saznanja ne upućuju na to da postoji značajan rizik za reprodukciju u ljudi. Sitagliptin se u znatnim količinama izlučuje u mlijeko štakorica (omjer mlijeko/plazma je 4:1). Metformin Neklinički podaci ne ukazuju na poseban rizik za ljude na temelju konvencionalnih ispitivanja sigurnosne farmakologije, toksičnosti ponovljenih doza, genotoksičnosti, kancerogenosti i reproduktivne toksičnosti. 6. FARMACEUTSKI PODACI 6.1 Popis pomoćnih tvari Jezgra tablete: celuloza, mikrokristalična (E460) povidon K29/32 (E1201) natrijev laurilsulfat natrijev stearilfumarat Ovojnica tablete: Polivinilni alkohol makrogol 3350 talk (E553b) titanijev dioksid (E171) željezov oksid, crveni (E172) željezov oksid, crni (E172) 6.2 Inkompatibilnosti Nije primjenjivo. 6.3 Rok valjanosti 24 mjeseca. 6.4 Posebne mjere pri čuvanju lijeka Čuvati na temperaturi ispod 30°C. 6.5 Vrsta i sadržaj unutarnjeg pakovanja (spremnika) Neprozirni PVC/PE/PVDC–aluminijsko blister pakovanje sa 28 (2x14) ili 56 (4x14) tableta, u kartonskoj kutiji. Na tržištu se ne moraju nalaziti sve veličine pakovanja. 6.6 Upute za uporabu i rukovanje i posebne mjere za uklanjanje neiskorištenog lijeka ili
otpadnih materijala koji potječu od lijeka Sav neiskorišteni lijek ili otpad potrebno je zbrinuti u skladu s propisima za zbrinjavanje opasnog otpada.
H A L M E D
27 - 09 - 2012
O D O B R E N O

18
7. NAZIV I ADRESA NOSITELJA ODOBRENJA ZA STAVLJANJE GOTOVOG LIJEKA U PROMET
MERCK SHARP & DOHME d.o.o. Heinzelova 62a, 10 000 Zagreb HRVATSKA 8. KLASA RJEŠENJA O ODOBRENJU ZA STAVLJANJE LIJEKA U PROMET
UP/I- 530-09/08-01/377 UP/I- 530-09/08-01/378 UP/I- 530-09/08-01/379 UP/I- 530-09/08-01/380
9. DATUM PRVOG ODOBRENJA ZA STAVLJANJE LIJEKA U PROMET/ DATUM OBNOVE ODOBRENJA ZA STAVLJANJE LIJEKA U PROMET
Datum prvog odobrenja: 9. srpnja 2009./ 10. DATUM REVIZIJE SAŽETKA OPISA SVOJSTAVA LIJEKA Rujan, 2012.
H A L M E D
27 - 09 - 2012
O D O B R E N O