fenomenos de superficies - …ecaths1.s3.amazonaws.com/quimicafisicafacena/1673928468.t_6... · de...
TRANSCRIPT

FENOMENOS DE
SUPERFICIESDra. Nelly Lidia Jorge
2017

BIBLIOGRAFIA
• Ball D. W. “Fisicoquímica”. E. Thomson. 2004
• S. Glasstone, “ Termodinámica para Químicos”,
Editorial Aguilar, Madrid, España, 1960.
• I. N. Levine, “Fisicoquímica”, Editorial McGraw
Hill, Vol. I. 1996.
• Maron y Prutton,”Fundamentos de
Fsicoquímica”, Editorial Limusa, Wiley S. A..
• Atkins, “Fisicoquímica”, Editorial
Panamericana.
• G. W. Castellan,” Fisicoquímica”, Editorial
Fondo Educativo Interamericano, S. A

La mayoría de los procesos fisicoquímicos naturales yartificiales ocurren en sistemas heterogéneos en dondelas diferentes fases que las componen están separadaspor una interfase, definida como la región del sistemamaterial cuyas propiedades fisicoquímicas se modifican.Las interfases pueden definirse según el tipo de estadode agregación de las fases que separa:
INTERFASES

INTERFASESEn termodinámica se define fase como una región del espaciocon propiedades intensivas constantes, como P, T, ci. Sitenemos dos fases en contacto, deben diferenciarse enalgunas de estas propiedades y por lo tanto debe existir unazona de transición donde las propiedades cambien desde suvalor en una fase hasta el valor que adquieren en otra.
Se denomina interfase a la región tridimensional decontacto entre dos fases α y β, en la que sus propiedadesvarían desde las correspondientes a la fase α hasta las de lafase β. Por ejemplo, si tenemos agua en contacto con suvapor, la propiedad concentración cambiará desde un valoralto en la fase líquida hasta un valor muy bajo en el vapor(tal y como se representa en la figura siguiente). Se trata portanto de una región no homogénea cuyo espesor se limita aunos pocos diámetros moleculares (normalmente de 3 a 4capas de moléculas).

¿Qué ocurre desde el punto de vista molecular? Todas las moléculascomprendidas por debajo del plano h1 tienen un mismo entorno yforman parte exclusivamente de la fase α. Del mismo modo, lasmoléculas situadas por encima del plano h2 tienen un mismo entorno yforman la fase β. Sin embargo, las moléculas situadas en la región h1h2
tienen un entorno molecular distinto al de las moléculas que están en elinterior de cada fase. En el caso de que las fases en contacto sean unlíquido y su vapor, la densidad que rodea a las moléculas de la interfaseno sería ni tan alta como en el interior de la fase líquida ni tan bajacomo en la fase gaseosa.

En la mayoría de los sistemas la fracción de moléculas en laregión interfacial es muy pequeña y la influencia sobre laspropiedades del sistema es despreciable. Existen, sin embargo,sistemas con una fracción significativa de moléculas en lasuperficie.
Los efectos de la interfase serán notables en sistemas conmucha superficie: coloides, sólidos porosos (como las zeolitas).También serán decisivas en aquellos procesos que tienen lugarúnicamente sobre superficies (corrosión, reacciones sobreelectrodos, membranas celulares…). Muchas aplicacionesquímicas en la industria se basan en fenómenos superficiales(adherencia, lubricación, detergencia…). Los fenómenos desuperficie implican al menos una fase condensada (sólido olíquido) ya que entre 2 gases las interacciones son tan débilesque las moléculas apenas notan cambio al pasar del interior deuna fase a una posición superficial. En este tema estudiamos losfenómenos de superficie con fases líquidas (liq-gas o liq-liq) yabordando luego las superficies sólidas.

TENSIÓN SUPERFICIAL En un fluido cada molécula interacciona con las que le rodean. El
radio de acción de las fuerzas moleculares es relativamente pequeño,abarca a las moléculas vecinas más cercanas. Vamos a determinar deforma cualitativa, la resultante de las fuerzas de interacción sobre unamolécula que se encuentra en A, el interior del líquido, B, en lasproximidades de la superficie C, en la superficie
Consideremos una molécula (en color rojo) en el seno de un líquido enequilibrio, alejada de la superficie libre tal como la A. Por simetría, laresultante de todas las fuerzas atractivas procedentes de lasmoléculas (en color azul) que la rodean, será nula.
En cambio, si la molécula se encuentra en B, por existir en valormedio menos moléculas arriba que abajo, la molécula en cuestiónestará sometida a una fuerza resultante dirigida hacia el interior dellíquido.

Si la molécula se encuentra en C, la resultante delas fuerzas de interacción es mayor que en el casoB. La fuerzas de interacción, hacen que lasmoléculas situadas en las proximidades de lasuperficie libre de un fluido experimenten unafuerza dirigida hacia el interior del líquido.Como todo sistema mecánico tiende a adoptarespontáneamente el estado de más baja energíapotencial, se comprende que los líquidos tengantendencia a presentar al exterior la superficie máspequeña posible.

COEFICIENTE DE TENSIÓN SUPERFICIAL
Imaginemos un alambre de un metal de pequeña sección como elesquema de la Figura 1, con una capa de líquido contenido dentro desu estructura: ABCD. El lado AB es móvil y se desplaza sinrozamiento con pequeños aros conectados sobre los lados AC y BD.
Se requiere una fuerza f para desplazar el alambre AB (llamadoémbolo superficial), aumentando la superficie de la película del líquidosin que ésta se rompa, fuerza que actúa contra la tensión superficial laque se opone a dicho desplazamiento.
Dc

Se define Tensión Superficial con el símbolo , como lafuerza que actúa sobre el émbolo AB, por unidad delongitud.El trabajo efectuado para desplazar el émbolo AB en un dx,trabajo que se realiza sobre el liquido.
Y la fuerza f es contrabalanceada por la de la Tensiónsuperficial, (dinas/cm) y puesto que hay dos (2)superficies (anverso y reverso ABCD) del film se tiene que:
w f dx (1)
lf 2y el trabajo diferencial:
siendo el producto del ancho l y la distancia infinitesimal dxigual al cambio infinitesimal en el área, dA, de la superficie(anverso y reverso), por tanto:
w l dx dA2
w
dA(2)

En conclusión es la fuerza expresada en dinas que actúa en 1 cm delongitud de superficie ( esta sobre la superficie) y de acuerdo a laexpresión anterior puede considerarse como EL TRABAJO EN ERGIOSNECESARIO PARA FORMAR UN CENTÍMETRO CUADRADO DE AREASUPERFICIAL, y se encuentra, por lo tanto, referido a la ENERGÍA LIBRESUPERFICIAL POR CENTIMETRO CUADRADO DE AREA.
La tensión superficial es una característica de un líquido que varía con latemperatura. Como se efectúa trabajo para modificar el área de unasuperficie, debemos poder relacionar este trabajo con una de lasfunciones de estado termodinámicas.
Vimos que la energía libre es igual a la máxima cantidad de trabajodiferente de pV que un proceso puede llevar a cabo. Puesto que cambiarel área de una superficie no es un trabajo del tipo pV, entonces el trabajodel tipo area-tensión- superficie debe relacionarse con la energía libre. Enel caso de un cambio reversible en el área de la superficie, que ocurre a Ty P ctes.
Esta ecuación implica tres cosas. Primero, podemos integrarla paraobtener:
dG W dA (3)
G W A (4)

Segundo es posible reordenar la ecuación (3) paradespejar la tensión superficial en términos de unaderivada parcial a T y P ctes.
Tercero si queremos tomar en cuenta la ecuación de lavariable natural para dG, en el caso de un sistema líquidocuya área superficial esta variando, debemos incluir elcambio en la energía de Gibbs debido a la variación delárea de la superficie.
Si tenemos un sistema de multicomponentes
,p T
G
A(5)
dG SdT VdP dA (6)
i i
i
dG SdT VdP dA dn (7)

MÉTODOS
Hay varios métodos para la determinación de y aquídesarrollaremos el de ASCENSO CAPILAR. En la figura 2,se esquematiza lo que sucede cuando un tubo (de vidrio)capilar se introduce en una cubeta que contiene un líquido,donde este puede ascender espontáneamente odescender, con respecto a la superficie del líquido. En estecaso (Fig.2) el tubo capilar abierto en ambos extremos,esta introducido en agua pura, observando un ascenso dellíquido, no obstante que la presión atmosférica lo trata de
impedir por su actuación sobre la rama abierta.

l
r

Como por efecto del fenómeno de la TensiónSuperficial, la superficie de interfase líquido-aire,tiene tendencia a disminuir, el agua se introduceen el tubo capilar como indican las flechas curvasy por ende hay una disminución de energía libre apresión y temperatura constante que estacompensada por la diferencial de energía libre delefecto gravitario de la pequeña masa del elementode volumen dV de altura dl y sección(siendo r el radio del capilar) y ubicado a unaaltura l de la superficie del líquido por tanto:

gravedadSup dGdG
De (3): dAdGSup
dlrdA 2 (superficie cilindrica)
dlrdGSup 2
La variación de la energía libre debido a lagravedad del peso de dV será igual al volumen xdensidad x aceleración de la gravedad:
(8)
dllgrdGgravedad2
(9)

dllgrdlr 22
Igualando (8) y (9)
lgr2
1De donde: (10)
La altura se mide con un catetómetro y el radio r por elmétodo de la gota u otro que permita determinarlo.En conclusión el líquido asciende espontáneamentemojando la superficie interior del capilar hasta que se llegaa un estado de equilibrio alcanzando una altura l que no semodificará en tanto y en cuanto no haya cambios detemperatura y presión atmosférica.
disminuye cuando aumenta la temperatura y se hace igual a cero en el estado crítico.

TENSIÓN INTERFACIAL Y LA
DISPERSIÓN DE LOS LIQUIDOS Cuando dos líquidos A y B son total o parcialmente
miscibles y se ponen en contacto, se encuentra que existe
una TENSION INTERFACIAL, en el límite de las dos
capas. Esta tensión AB es medible por métodos análogos a
los usados en la determinación de de líquidos puros. El
valor es intermedio del de A y B, en algunos casos es
inferior a los dos.Si una columna de un líquido puro de 1 cm2 de sección
transversal se divide, se crean dos superficies, cada una de
ellas con igual área. Como el trabajo necesario para crear
un área de superficie unitaria, como se expresó antes, es
igual a , el trabajo realizado en aquella división es
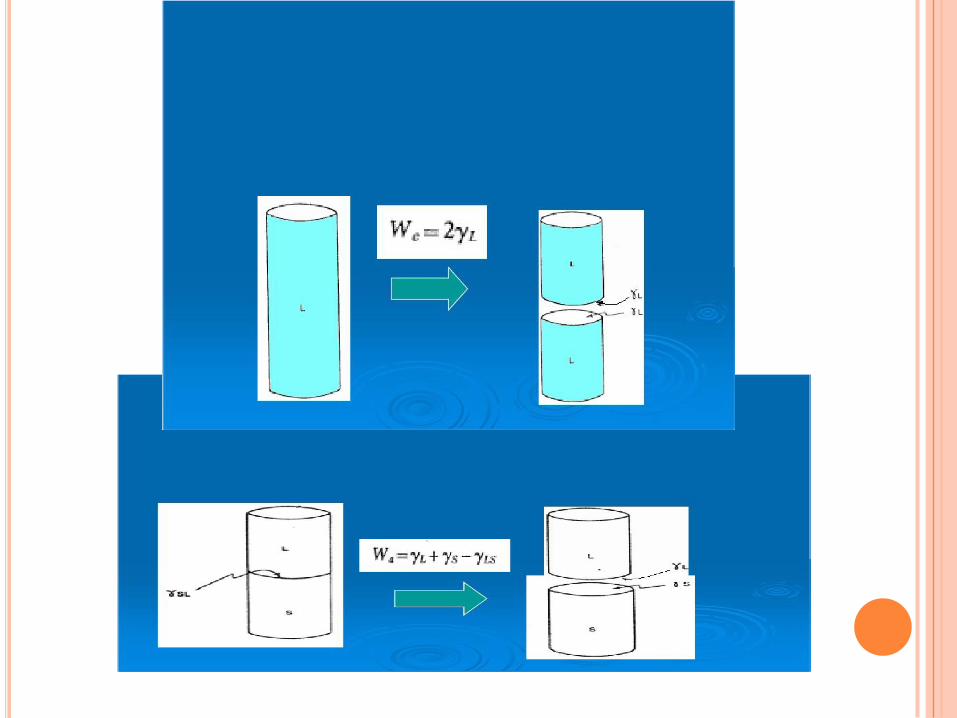
Denominándose WC: trabajo de cohesión del líquido.
CW 2 (11)

De forma análoga al caso de un líquido puro, si se considera
una columna de dos líquidos miscibles o parcialmente
miscibles, el trabajo para separarlos será el de adhesión
WA.
Estos dos tipos de trabajo conducen a una cantidad muy importantellamada coeficiente de dispersión. Si se quiere dispersar un líquidoB en la superficie A de otro líquido se llega al coeficiente dedispersión y esta dado por:
ABBAAW
BBA A CS W W
reemplazando
(14)
(12)
(13)
BBA A C A B AB B A B ABS W W 2
Un valor positivo de SBA significa que una pequeña cantidadde B en la superficie A, se esparcirá de igual forma como lohace el aceite en agua, si SBA < 0 no hay derrame y ellíquido adicionado permanece en forma de gotas sobre la
superficie.

TENSION SUPERFICIAL DE LAS
SOLUCIONES
El efecto de las sustancias disueltas en la tensión superficialdel solvente queda de manifiesto en los tres tipos de curvasde la Figura 3. En las soluciones de tipo I la adición de solutoconduce a un incremento de la tensión superficial, pero esteaumento no es grande. Tal comportamiento lo tienen loselectrolitos fuertes, sacarosa y el ácido aminobenzoico enagua, o la anilina en ciclohexano. Por otra parte, los noelectrolitos o los electrolitos débiles en el agua se comportansegún nos muestra la curva II. Esta conducta es muy comúny las soluciones presentan tensiones superficiales quedisminuyen con cierta regularidad cuando aumenta laconcentración de soluto.

(mN/m)
Concentración (M)
I
II
III
Figura 3. Efecto de Solutos sobre la
Tensión Superficial

El tipo III lo presentan ciertas soluciones acuosas de jabón,ácidos sulfónicos y sulfonatos, así como otros tipos decompuestos orgánicos. Estas sustancias, denominadasagentes activos superficiales tienen la capacidad de disminuirla tensión del agua a un valor incluso en concentracionespequeñas. Los solutos que conducen a un decrecimiento dela tensión superficial con el aumento de la concentración sedicen que presentan una actividad superficial positiva,mientras que los que aumentan dicha tensión tienen unaactividad superficial negativa.Gibbs demostró que la actividad superficial se debía a ladistribución desigual del soluto entre la superficie y el cuerpode la solución. Si un soluto se distribuye de manera que lasuperficie contiene un exceso q de moles de soluto por 1 cm2
de área al presente en el cuerpo de la solución , entonces enlas soluciones diluidas se cumple que:

C dq
RT dC(15)
Cuando la velocidad de variación de la tensiónsuperficial de la solución con la concentración, es positiva, esdecir cuando la tensión superficial de la solución aumentacon la concentración, q debe ser negativo, y el cuerpo de lasolución es mas rica en soluto que la superficie, como en elcaso de muchos electrolitos. Sin embargo, cuando la tensiónsuperficial de la solución disminuye con la concentración,es negativo , q es positivo y la superficie contiene unaconcentración mayor de soluto que en el seno de la solución.Esto último es lo que sucede con los agentes activossuperficiales. La ecuación se conoce como la de adsorciónde Gibbs. La deduciremos mas adelante.
ddC
ddC

SOLUTOS TIPO I
>
Aumentan
Solutos que son excluidos de la interfase
soluto : solvente
Na+/H2O (40 kJ/mol)solvente : solvente
H2O /H2O (20 kJ/mol)
Ejemplos NaCl, glicina, sales de amonio cuaternario

< solvente : solvente
H2O /H2O (20 kJ/mol)
SOLUTOS TIPO II y III
Disminuyen
Solutos que se concentran en la interfase
EjemplosII alcoholes y ácidos grasos
III jabones y detergentes
soluto : solvente
=CH2 /H2O (10 kJ/mol)

Suero 57-58
Líquido céfalo-raquídeo 62-65
Bilis 48
Líquido espermático 52-59
Humor acuoso 60
Orina 70-72
H2O 75
(mN m-1)
Tensión superficial de fluidos biológicos
La disminución de en fluidos biológicos (respecto de agua) es comparable al
efecto de 1-10 mM SDS.
El efecto está dado por ácidos grasos, prostaglandinas, sales biliares, que
favorecen el movimiento hidrodinámico de los fluidos

INTERFASES CURVASEl efecto de la tensión superficial es minimizar el áreainterfacial resultando en la formación de interfases curvas(los líquidos adoptan forma esférica en ausencia de otrasfuerzas). Esta curvatura provoca diferencias de presiónentre el interior y el exterior de la fase curvada, lo quetiene al menos dos consecuencias importantes: cambios dela presión de vapor y la capilaridad.

Ecuación de Young-Laplace
Esta ecuación, deducida independientemente por Youngy Laplace en 1805, describe la dependencia de la presiónde una fase con la curvatura de la superficie que lalimita.
Supongamos una fase formando una esfera de radio r(gota o burbuja) en el interior de una fase β. Si la esferase encuentra en situación estacionaria, las fuerzas queintentan comprimir la esfera (las debidas a la tensiónsuperficial y a la presión exterior) deben estarequilibradas por la fuerza que intenta expandirla (debidaa la presión interna). Las fuerzas que tienen su origen enla presión pueden escribirse como el producto de presiónpor el área, mientras que la debida a la tensiónsuperficial se puede escribir como el trabajo por unidadde longitud.

ecuación de Young-Laplace para
una interfase esférica: (16)
Dividiendo por
Dado que γ>0 y r>0, la presión en el interior de una superficiecurva es mayor que la presión en el exterior y aumenta amedida que r disminuye.

Si el sistema que se estudia es una burbuja:porque ambas superficies contribuirían a la
energía de la superficie, es decir a la tensión superficial.La ecuación (16) nos dice que cuanto menor sea la gota,mayor será la diferencia de presiones entre la parte interna yexterna de la interfase líquido-gas. Esto sugiere que lasgotas más pequeñas se evaporan más rápidamente.
Pr
4

INFLUENCIA DE LA TENSION DE VAPOR
CON EL RADIO DE CURVATURA
Aunque las propiedades superficiales de un líquido sondiferentes de la del interior del mismo, este efecto esdespreciable excepto en algunas ocasiones. Una de estases el en que el líquido está disperso en gotas pequeñas(mercurio sobre una superficie lisa sólida) y la superficiede las mismas corresponde a una fracción muyconsiderable de la cantidad total del líquido. Estasituación es similar cuando una sustancia sólida se divideen pequeños fragmentos.Si tenemos dn moles de un líquido en una superficie plana
y la transferimos a una gota esférica de radio inicial r y la
presión normal de vapor del líquido es P0 y la de la gota P,
teniendo en cuenta la ecuación: y para un
mol se tiene para el líquido plano:
Y para la gota:
atm
PRTgg
10 ln
001 PRTgg ln
PRTgg ln02

La variación de energía libre molar de transferir 1 mol dellíquido a P0 a la gota a P será:
y para un dn moles será:0
12P
PRTggg ln
0P
PRTdndGgdn ln (17)
En la figura 4 se puede apreciar este proceso termodinámico
Figura 4
oP dn
dn
drr

A su vez la variación de energía libre también se puede
calcular por el cambio de energía libre superficial que
resulta del aumento de superficie por la adición de dn
moles que conlleva a un aumento diferencial de
volumen: cc
donde M es el peso molecular y ρ la densidad, y que será
igual al aumento de volumen de la gota:
donde:
MdndV
drr 24
drrdnM 24
dnr
Mdr
24
De aquí: (18)
El aumento de energía libre superficial es el producto de
por el incremento de superficie que corresponde a un
aumento dr del radio de la gota:
dG dA r dr r r rdr dr r2 22 2 24 4 4 2

Resolviendo el cuadrado del binomio y despreciando el
término (dr)2 , se tiene que:
drrdG 8 (19)
Y sustituyendo dr por la ecuación (18) se tiene:
dnr
MrdG
248
dnr
MdG
2 (20)
Igualando la (17) y la (20)
TRr
M
P
P 2
0
ln (21)

ADSORCIÓN
Los procesos en las superficies gobiernan muchos aspectosde la vida cotidiana, incluyendo la vida misma. Aunque selimite la atención a las superficies sólidas, la importancia delos procesos apenas se reduce. La viabilidad de una industriaestá determinada bien constructivamente, como en lacatálisis, o bien destructivamente, como en la corrosión, porprocesos que tienen lugar en las superficies sólidas. Por otraparte, en los sistemas biológicos, la transferencia demateriales hacia el interior y hacia el exterior de las célulastiene lugar mediante adsorción sobre la membrana celular,penetración en la misma y desorción en la superficie opuestade la membrana. Se llama adsorción al fenómeno deacumulación de partículas sobre una superficie. La sustanciaque se adsorbe es el adsorbato y el material sobre el cual lohace es el adsorbente. El proceso inverso de la adsorción esla desorción.

PROCESOS DE ADSORCIÓN La adsorción es un proceso mediante el cual se extrae
materia de una fase y se concentra sobre la superficie deotra fase (generalmente sólida). Por ello se consideracomo un fenómeno superficial. La sustancia que seconcentra en la superficie o se adsorbe se llama"adsorbato" y la fase adsorbente se llama "adsorbente".
Por contra, la absorción es un proceso en el cual lasmoléculas o átomos de una fase interpenetran casiuniformemente en los de otra fase constituyéndose una"solución“ con esta segunda.
El proceso de cambio iónico supone un intercambio de unasustancia o ión por otra sobre la superficie del sólido.


El término sorción incluye la adsorción y la absorciónconjuntamente, siendo una expresión general para unproceso en el cual un componente se mueve desde una fasepara acumularse en otra, principalmente en los casos en quela segunda fase es sólida.La principal distinción entre sorción (adsorción y absorción) ycambio iónico es que las ecuaciones que describen la sorciónconsideran solamente una especie química, de manera que ladistribución del soluto entre la disolución y el sólido respondea una relación simple lineal o no. Las ecuaciones para elcambio iónico tienen en cuenta todos los iones que compitenpor los lugares de intercambio.

TIPOS DE ADSORCIÓN
Generalmente se reconocen dos tipos de adsorción, unaadsorción física o de Van der Waals y otra química oactivada.
ADSORCIÓN FÍSICA Hay una interacción de Van del Waals entre el adsorbato y el
adsorbente. En estos caso, la molécula adsorbida no está fija en unlugar específico de la superficie, sino más bien está libre de trasladarsedentro de la interfase.
Estas interacciones son de largo alcance pero son débiles.
Esta adsorción, en general, predomina a temperaturas bajas. Laenergía liberada es del orden de magnitud de la entalpia decondensación, numéricamente inferior a -40 kJ/ mol
No es específica.
Cuando la monocapa está completa, las interacciones intermolecularesentre moléculas adsorbidas en la monocapa y las mismas moléculas enel fluido pueden conducir a la formación de una segunda capa.
La pequeña energía puede ser absorbida como vibraciones de la red ydisipadas como movimiento térmico y una molécula botando por lasuperficie va perdiendo gradualmente su energía acomodación

ADSORCIÓN QUÍMICA
Las moléculas se adhieren a la superficie formando un enlacequímico y prefieren sitios que maximicen su número decoordinación con el sustrato.
Las energías de adsorción son elevadas, del orden de las de unenlace químico 200 kJ/ mol.
Cuando se forma una monocapa, no es posible que seproduzcan mas interacciones entre adsorbato y adsorbente.
Es irreversible
Es exotérmica. Un proceso espontáneo requiere que G<0.Puesto que la libertad de traslación del adsorbato se reducecuando se adsorbe, S es negativo. Por tanto H debe sernegativo

ISOTERMAS DE ADSORCIÓN
La relación entre la cantidad de sustancia adsorbida porun adsorbente y la presión o concentración de equilibrio auna temperatura constante se denomina isoterma deadsorción. Se han observado seis tipos de isotermas enla adsorción de gases en sólidos, que se muestra en lafigura siguiente. En los casos de quimiadsorción solo sepresentan isotermas del tipo I, mientras que en la físicatienen lugar los seis casos.
En las isotermas del tipo I, la cantidad de gas adsorbidopara una cantidad dada de adsorbente se incrementa conrapidez con la P y después más lentamente, conforme lasuperficie comienza a cubrirse con moléculas de gas.

TIPOS DE ISOTERMA DE ADSORCIÓN


ISOTERMA DE FREUNDLICH Para representar la variación de la cantidad de gas
adsorbido por unidad de área o de masa con la Presión,Freundlich propuso la ecuación.
Donde y es el peso o volumen de gas adsorbido porunidad de área o de masa de adsorbente, P es la presiónde equilibrio, k y n son constantes empíricas quedependen de la naturaleza del sólido y gas y de latemperatura.
nkPm
xy
1
Esta ecuación se puede verificar linealizándola mediante
logaritmo, resultando:
Si graficamos log y en función de log P, resulta una línea
recta, cuya pendiente es igual 1/ n y la ordenada al origen
log k
Pn
ky log1
loglog

ISOTERMA DE LANGMUIR El proceso de adsorción es
un proceso dinámico:
Se basa en tres suposiciones1.La adsorción no puede extenderse mas allá delrecubrimiento con una monocapa2.Todos los sitios son equivalentes y la superficie esuniforme3.La posibilidad de una molécula de adsorberse en unsitio dado es independiente de la ocupación de los sitiosvecinos, es decir no hay interacción intermolecular.
)superficie()superficie()( AMMgA

Considera que el proceso de adsorción consta de dos accionesopuestas, una de condensación de la molécula de la fase gaseosasobre la superficie y una de evaporación de las situadas en lasuperficie hacia el gas. Cuando se inicia la adsorción, cada moléculaque colisiona con la superficie puede condensarse en ella, pero alproseguir este proceso, cabe esperar que condensarán aquellasmoléculas que incidan en alguna parte de la superficie no cubiertatodavía. Así la velocidad inicial de condensación es elevada, perodisminuye a medida que disminuye la superficie libre disponible.
En cambio, una molécula adsorbida en la superficie, es capaz de
liberarse por la agitación térmica escapándose hacia el gas. La
velocidad de liberación dependerá a su vez de la superficie cubierta.
Estas dos velocidades, adsorción y desorción, alcanzan un momento
en que se hacen iguales, estableciéndose el equilibrio.
)superficie()superficie()( AMMgA

Si designamos por a la fracción de superficie total
cubierta por las moléculas adsorbidas en cualquier
instante, entonces la fracción de superficie disponible
para la adsorción será (1- ).
La teoría cinética me dice que la velocidad con que las
moléculas chocan con la unidad de superficie es
proporcional al gas, entonces la velocidad de
condensación debe quedar determinada por la P y la
fracción de superficie sin cubrir
Pkv óncondensaci 11
Donde k1 es una constante de proporcionalidad
Por otra parte la velocidad con que se evaporan las
moléculas desde la superficie será proporcional a la
fracción de superficie cubierta:
Para el equilibrio de adsorción estas dos velocidades se
igualan
2kv nevaporació
21 1 kPk

Si divido numerador y denominador por k1
La cantidad de gas adsorbido por unidad de área o por
unidad de masa de adsorbente, y, debe ser proporcional a
la fracción de superficie cubierta, y por lo tanto, si
consideramos que:
Pkk
PkkPkPkkPkPk
12
1211211
o
Pa
P
Pk
k
P
1
2
sup. la acubrir tod a ientecorrespond gas devolumen
adsorbido gas devolumen
mV
V
my
y

Esta ecuación es la isoterma de Langmuir. Las constantes
ym y a son características del sistema en consideración y se
evalúan a partir de datos experimentales.
mm
m
y
P
y
a
Y
Precíprocoeltomoy
PpormiembrosambosdividosiPa
P
y
y
:
yP
my1
my
a
P

ECUACIÓN DE GIBBS
Gibbs en 1879 e independientemente en 1888 Thompson,fueron los primeros en deducir la relación entre la tensiónsuperficial y la adsorción, se la conoce como ecuación deadsorción de Gibbs.
Cuando vimos la variación de energía libre no se tuvo encuenta la posibilidad de variaciones en la energíasuperficial, se suponía tácitamente que no habíaalteración en la magnitud de la superficie cuando elsistema sufría un cambio.
Con el objeto de tener en cuenta la posibilidad de unavariación en la energía libre como resultado de unaumento o disminución de la superficie expuesta, esnecesario incluir un término A.
Donde es la tensión superficial y A la magnitud de la
superficie.

La energía libre de un sistema de dos componentes a P y
T constantes:
Diferenciando se tiene:
Sabemos que si agregamos dA para un aumento de dA
en la superficie
Comparando los dos diferenciales resulta:
Imaginemos que el sistema se divide en dos partes, una
formada por toda la porción que esta bajo la influencia
de fuerzas superficiales y la otra el resto de la solución.
La primera se puede denominar fase de superficie y la
otra fase de volumen.
AnnG 2211
AddAdndndndndG 22221111
dAdndndG 2211
02211 Addndn

Dividiendo por el A se tiene
O sustituimos ni/A por i que son los excesos de lasconcentraciones en la superficie de los diversosconstituyentes del sistemas en este caso 1 y 2.
Si el sistema está en equilibrio, a temperatura, presión ysuperficie constantes, el potencial químico en la fase desuperficie de cualquier constituyente debe ser igual a supotencial químico en la fase de volumen, esto es, ladisolución. Por ello puedo escribir la ecuación de la forma
Recordar que y 1 y 2 dependen de la posiciónarbitraria elegida para la superficie geométrica. Nosconviene escoger la superficie de tal manera que 1, esdecir el exceso de conc en superficie del disolvente igual acero entonces.
022
11 dd
A
nd
A
n ss
ss
02211 ddd ss
02211 ddd
ss nn 21 y
022 ddT2
2

Sabemos que:
222022 aRTddaRT lnln
Sustituyendo
Son dos formas de la ecuación de Gibbs. Aunque en la
práctica se aplica al soluto, será igualmente válida para
ambos componentes de un sistema binario, si
prescindimos de los subíndices:
2
2
1
ad
d
RT ln 2
22
da
d
RT
a
da
d
RT
a

De la ecuación podemos deducir que si:
es negativa será positiva, la
concentración del soluto será, mayor en la fase de
superficie que en la fase de volumen.
es positiva será negativa, la
concentración será mayor en la fase de volumen
que en la fase de superficie.
dad
dad

COLOIDES
• Los coloides fueron por primera vez descritos por el químico inglés
Thomas Graham, en 1860.
• Un sistema coloidal esta compuesto por partículas cuyas dimensiones,
al menos en una dirección, se encuentra en el intervalo aproximado de
30 a 104 Å y por un medio en el que se encuentran dispersas estas
partículas.
• Las partículas se denominan partículas coloidales o fase dispersa; el
medio se denomina medio de dispersión o fase continua.
• Sustancias que no filtran a través de una membrana semipermeable
(proceso denominado DIALISIS)
• Sistemas con tamaño de partícula de soluto entre 10-9 m (10Å) y 10-6 m(1 )
• 1 = micrón= 1 x 10-4 cm
• 1 nm= nanómetro = 1 x 10-9 m
• 1Å = Amstrong = 1 x 10-10 m = 1 x 10-8 cm

Soluciones viscosas, lentamente difusibles, que
no atraviesan membranas semipermeables.
INCLUYEN
Emulsiones y Suspensiones
Agregados moleculares (micelas)
Soluciones de macromoléculas
Sistemas biológicos
PROPIEDADES CLÁSICAS
• No se ven fases diferentes al microscopio óptico común
• Presentan efecto Tyndall se pone de manifiesto la presencia de
partículas coloidales, al parecer, como puntos luminosos debido a la
luz que dispersan.
• Poseen carga eléctrica

En las dispersiones coloidales se distinguen dos partes :Fase dispersa : las llamadas micelas.Fase dispersante : en las que están dispersas las partículas coloidales.
Medio de dispersión Fase dispersa Nombre Ejemplos
GasLíquido
Sólido
Aerosol líquido
Aerosol sólido
Niebla, nubes,
polvo, humo.
Líquido
Gas
Líquido
Sólido
Espuma
Emulsión
Sol
Espumas (de jabón , cerveza, etc.), nata batida.
Leche, mayonesa.
Pinturas, tinta china, goma arábiga, jaleas
Sólido
Gas
Líquido
Sólido
Espuma sólida
Emulsión sólida
Sol sólido
Piedra pómez.
Mantequilla, queso.
Algunas aleaciones, piedras preciosas coloreadas
. Tipos de sistemas coloidales

La morfología de las micelas, en los sistemas coloidales, es variada, distinguimos tres tipos :Esféricas : cuyos coloide se llaman globulares, que son los más importantes, dentro de estos los de mayor importancia están formados por compuestos inorgánicos. Su grado de viscosidad es pequeño
.En forma de fibra : coloides fibrosos, formados por largas cadenas macromoleculares, de gran viscosidad.
Laminares : coloides laminares de viscosidad intermedia.

CLASIFICACIÓN. LAS PARTÍCULAS DE MUCHOS COLOIDES CONTIENEN
GRUPOS DE ÁTOMOS LOS CUALES SE DISOCIAN EN IONES, ESTOS GRUPOS
IONIZANTES HACEN QUE LA PARTÍCULA ESTE ELÉCTRICAMENTE CARGADA.
LAS PARTÍCULAS PUEDEN TAMBIÉN CARGARSE POR ADSORCIÓN DE IONES
DE LA SOLUCIÓN; ESTA CARGA ELÉCTRICA ES UNO DE LOS FACTORES DE
ESTABILIDAD, YA QUE LAS PARTÍCULAS CARGADAS POSITIVAMENTE SE
REPELEN ENTRE SÍ, AL IGUAL QUE, POR EJEMPLO, EN UN SOL
NEGATIVAMENTE CARGADO, SU ESTABILIDAD SE DEBE A LA REPULSIÓN
ELECTROSTÁTICA
Colóides liofóbico: en los soles liofóbicos no hay afinidad entre las
partículas y el solvente, la estabilidad de estos depende
principalmente de la carga de las partículas. Si el agua es el
solvente, se utiliza el nombre hidrófobo.
Coloides liofílico : en este tipo de coloides hay interacción entre las
partículas y el solvente. Este tipo de soles es mucho más estable que
los soles liofóbicos. Para el caso de los soles en agua se utilizara el
término hidrofílico. Las sustancias que forman estos coloides son de
naturaleza orgánica cuyas moléculas están constituidas por la larga
cadena hidrocarbonada con un grupo polar en uno de los extremos.

ESTABILIDAD DE COLOIDES LIÓFOBOS/HIDRÓFOBOS (1)
EMULSIÓN
Relación Superficie/volumen grande ( A/V)
Tendencia de las partículas a asociarse para reducir su área superficial
(G = A)
Flotación o
creaming
Sedimentación,
floculación,
coalescencia
Espontáneo Estabilización por
formulación

ESTABILIDAD DE COLOIDES LIÓFOBOS (2)
Velocidad de creaming o sedimentación = v
Ley de Stokes v = 2 g r2 ( 1 - 2)
9
r = radio de la partícula
( 1 - 2) = diferencia de densidad entre las dos fases
n = viscosidad de la fase dispersante
Movimiento de encuentros creaming o
partículas sedimentación

ASPECTOS ELÉCTRICOS DE LOS
FENÓMENOS DE SUPERFICIE
La fuente de estabilidad cinética de los coloides es laexistencia de una carga eléctrica sobre la superficie de laspartículas. A causa de esta carga eléctrica, los iones de cargaopuesta tienden a agruparse cerca y se forma una atmósferaiónica. Esta carga eléctrica puede tener diversos orígenes:a) Ionización en la superficie de la sustancia dispersadacuando ésta se encuentra en un medio acuoso.b) Adsorción preferente, en la superficie, de partículas deiones procedentes del medio dispersante (p. ej., la adsorciónde moléculas de tensioactivos iónicos sobre gotas de aceite,confiriéndoles una carga negativa o positiva a estaspartículas dispersadas en fase acuosa).

Imaginemos que las partículas adquieren carga negativadebido a alguno de los mecanismos mencionadosanteriormente. La superficie así cargada crea una asimetríade cargas dentro de la solución. Para mantener laneutralidad eléctrica debe disponerse en la proximidad desu superficie de un número de iones con carga positivatotal - contraiones - igual al número de cargas negativaspresentes en la superficie de la partícula.Las dos ordenaciones de carga positiva y negativa asícreadas se conocen como doble capa eléctrica.Idealmente, los contraiones deben disponerse de unaforma ordenada lo más cerca posible de la interfase. Sinembargo, en la práctica el ordenamiento es más complejo,como se muestra en la Figura, para sólidos cargadosnegativamente. Para una superficie con carga positiva, lafigura es similar pero las cargas son de signo contrario

Figura
Se distingue entonces dos regiones decarga. 1) hay una casi inmóvil de ionesque se adhieren a la superficie coloidal,el radio de la esfera que contiene estacapa fija se denomina radio de corte yes el principal factor determinante de lamovilidad de las partículas. El potencialeléctrico en el radio de corte relativo asu valor a una distancia en el solventese denomina potencial zeta, , opotencial electrocinético. 2) la unidadcargada atrae una atmósfera de cargaopuesta compuesta de iones móviles

La distribución de los contraiones en la disolución cerca de lapartícula cargada fue estudiada por Helmholtz (l879),Chapman (l904) y Stern (l924).
11
Modelos de la doble capa eléctrica:Modelos de la doble capa eléctrica:
+
+
+
+
+
+
–
–
–
–
–
–
–
–
–
–
–
–
+-
+
+
+
+
+
+
+
–
–
–
–
–
–
–
–
–
–
–
–
-
--+
+
+
+
-
+
+
+
+
+
+
+
–
–
–
–
–
–
–
–
–
–
–
–
-
-
-+
+ --
-
+
Helmholtz Gouy-Chapman Stern
(1879) (1904) (1924)

MODELO DE HELMHOLTZ PARA LA DOBLE CAPA
ELÉCTRICA:
Es el modelo más simple de la doble capa eléctrica. Esta teoríaconsidera a la doble capa eléctrica como un condensador deplacas paralelas donde º es el máximo potencial eléctricoexistente, y varía linealmente con la distancia a la superficie dela partícula. El exceso de iones en el lado de la solución de ladoble capa, se encuentra ubicado en un plano ("plano deHelmholtz") muy cercano a la superficie del electrodo. Enalgunas variantes de este modelo se plantea la existencia enrealidad de dos planos.En el "plano externo de Helmholtz" se encuentran los iones
solvatados y en el "plano interno de Helmholtz" se encuentran
iones que hayan perdido su capa de solvatación y por lo tanto
pueden aproximarse a la superficie del electrodo.

Este modelo sólo tiene validez cuando la carga total superficiales pequeña en magnitud absoluta o cuando la concentracióndel electrolito inerte es grande (compresión de la doble capa).
º
d x
(x)
+
+
+
+
+
+
-
-
-
-
-
-
d

DOBLE CAPA ELÉCTRICA (1)

DOBLE CAPA ELÉCTRICA (2)
HELMHOLTZ (1879): una capa de cargas en una
superficie sólida y una capa rígida de iones de carga
opuesta en la solución.
GOÜY-CHAPMAN (1910-1913): una capa de cargas en
una superficie sólida y una capa difusa de iones con
distribución estadística en la solución.
STERN (1924): una capa de cargas en una superficie
sólida y una capa de contraiones constituída por una
subcapa de contraiones absorbida en forma rígida y una
subcapa difusa de iones en solución.

DOBLE CAPA ELÉCTRICA (2)
•Potencial Z (ζ)
•Espesor de la doble capa (rd)

Si las partículas están
cargadas
FUERZAS
ELECTROSTÁTICAS
REPULSIVAS
Erep = cte r2 2 e-D/rd
R
•rd es el espesor de la
doble capa de iones.
• potencial zeta
•D =R-2 r
ENERGÍA DE REPULSIÓN
+r R
++
+
+
+
+
++
+
+
+
++
+
+
++
+
+
+
+ ++

a) Es el espesor de la doble capa
de iones.
b) Se calcula a partir de la teoría de
Debye-Huckel
rd = R T
2 F2 b I
c) Depende inversamente de la
fuerza iónica (I)
LA DOBLE CAPA SE CONTRAE A MEDIDA QUE
AUMENTA LA FUERZA IÓNICA
Donde rd :
0
1/2

ESPESOR DE LA DOBLE CAPA
Baja I,
rd
grande
Alta I,
rd
pequeño
(Goüy-Chapman)
Electrolito Espesor
(M) (nm)
0.00001 100
0.001 10
0.1 1
LA DOBLE CAPA SE CONTRAE A MEDIDA QUE AUMENTA LA FUERZA IÓNICA
+
++
++ +
++
+
+ +
+
++
+
+
+
++
++
++
++
++

13
El potencial de doble capa de los coloides
El tratamiento matemático del potencial de doble El tratamiento matemático del potencial de doble capa es similar al de un condensador eléctrico de capa es similar al de un condensador eléctrico de placas paralelasplacas paralelas
El potencial de doble capa también se conoce El potencial de doble capa también se conoce como potencial zeta y expresa la movilidad como potencial zeta y expresa la movilidad electroforética electroforética de los coloidesde los coloides
= 4 = 4 d / e Dd / e D
potencial zeta; d: distancia recorrida; e: potencial zeta; d: distancia recorrida; e: potencial aplicado; D: constante dieléctrica del potencial aplicado; D: constante dieléctrica del medio)medio)
Los valores del potencial zeta son del rango de 10 Los valores del potencial zeta son del rango de 10 a 75 mVa 75 mV

Es una medida de la estabilidad de una partícula e
indica el potencial que se requiere para penetrar la
capa de iones circundante en la partícula para
desestabilizarla. Por lo tanto, el potencial zeta es la
potencia electrostática que existe entre la separación
de las capas que rodean a la partícula.

FIJACIÓN DE LIGANDOS A
MACROMOLÉCULAS.
Las macromoléculas o biopolímeros, debido a su tamaño ycomplejidad estructural, pueden interaccionar de muydiversa manera con otras moléculas, mediante fuerzaselectrostáticas tales como coulómbicas, las de dispersión,pueden formar puentes de hidrógeno, etc. Si la moléculaque interactúa con el biopolímero, es pequeña desde elpunto de vista molecular, se la denomina ligando, y lainteracción resultante es la interacción ligando-macromolécula. Estas interacciones no covalentes son departicular importancia en todos los procesos biológicos,dentro de los cuales se pueden citar:

-catálisis enzimática, donde el sustrato es el ligando-neutralización de sustancias tóxicas extrañas alorganismo, por parte de los anticuerpos, en este caso elligando es el hapteno o antígeno.-estimulación de una actividad celular por parte dehormonas o neurotransmisores (ligandos), donde lamacromolécula es el receptor a nivel de membrana.Las interacciones ligando-macromolécula presentanlas siguientes propiedades:-reversibilidad-especificidad: es decir la macromolécula se une a unasustancia o familia de sustancias con determinadascaracterísticas estructurales, y en ciertos caso unirseselectivamente a un estereoisómero-competividad: la cantidad de ligando unido a lamacromolécula puede ser afectada por la presencia de otrasustancia (otro ligando), con características estructuralessimilares al ligando original.

-saturación: la macromolécula presenta un limitadonúmero de zonas de interacción (sitios) por lo que a altasconcentraciones de ligando se encontrarán ocupadas y unposterior incremento de dicha concentración no aumentarála cantidad de ligando unido (o fijado) a la macromolécula.Dado que la formación del complejo ligando-macromoléculaes el primer paso en la respuesta biológica a un efector esimportante dilucidar este fenómeno, lo que implicaconocer:A-¿Cuantos ligandos se unen a la macromolécula?B- ¿Con qué magnitud se produce la unión o fijación?C- Conocer la naturaleza de las fuerzas involucradas en lainteracción.Un análisis fisicoquímico basado en principiostermodinámicos proporciona el sustento para contestar lasdos primeras preguntas y sumandole informaciónmolecular, se puede conocer la última.

FIJACIÓN DE LIGANDOS A
MACROMOLÉCULAS CON UN SOLO SITIO
DE FIJACIÓN. PROPIEDADES.
En primer lugar abordaremos el desarrollo de las 2primeras cuestiones, teniendo en cuenta que cuando lasmacromoléculas se combinan reversiblemente conpequeñas moléculas o iones, dichos comportamientos songobernados por las leyes del equilibrio químicoEste es un caso particular en el cual una macromoléculapresenta un único sitio o zona de interacción a la cualpuede unirse el ligando.Consideremos, inicialmente, el caso en que un mol de A secombina con un mol de P
La constante de equilibrio para esta reacción de asociaciónes:
d
K
KP A PA
PAK
P A

Los Bioquímicos suelen referirse a una constante dedisociación que simplemente la recíproca.
Definamos por r el número de moles de A unidos a un molde P, bajo determinadas condiciones.Entonces:
A partir de la ec. (22)
Así que:
d
P AK
PA
concentración de A unido a P
concentración total de todas las formas de Pr
PAr
P PA
(22)
(23)
d
P APA
K
d
d
d
P AAK
r rP A K A
PK
(24)

Se observa que cuando r=0,5 (es decir P esta semisaturadocon A) [A]=Kd
La ecuación fundamental (24) puede ordenarse de distintasformas adecuadas para el tratamiento gráfico.Dos son las formas más comunes:
Gráficos de la doble recíprocaLa ecuación se reordena para dar:
Así la grafica de 1/r frente a 1/[A] da una linea recta dependiente Kd.
dK
r A
11
Pend=Kd
1/r
1/[A]-1/Kd

Gráficos de ScatchardLa ecuación se reordena
Al representar r/[A] frente a r se obtiene una recta cuyapendiente es –1/Kd.
d d
d d
r rrK r A A rK A r A
A K K
1
Pend=-1/Kd
r/[A]
r1
1/Kd

FIJACIÓN DE LIGANDOS A
MACROMOLÉCULAS CON SITIOS
MÚLTIPLES DE FIJACIÓN. PROPIEDADES
Si se considera el caso de una macromolécula P que puedeunir a n moléculas de un determinado ligando A, unenfoque del problema consiste en suponer quesucesivamente se establecen los siguientes equilibrios:
Cada etapa esta representada por una constante deafinidad, Ki, correspondiente a la fijación la iésima moléculade ligando, que se explicita en cada caso.
n n
P A PA
PA A PA
PA A PA
PA A PA
2
2 3
1

; ;....n
n
n
PA PA PAK K K
A P A PA A PA
2
1 2
1
Es importante puntualizar que las constantes de equilibriono son verdaderas constantes termodinámicas, ya que alexpresarlas en función de la concentración y noactividades, dependen del solvente, de la fuerza iónica delmedio, etc. Además son constantes macroscópicas, dadosque para su explicitación, no se hace presunción acerca dela interacción a nivel molecular y solamente se tiene encuenta la estequiometría múltiple de la unión. Paracuantificar los multiples equilibrios establecidos esnecesario determinar las concentraciones individuales decada especie, lo cual es dificultoso.
d
n Ar
K A(25)

La ecuación fundamental (25) puede ordenarse de distintasformas adecuadas para el tratamiento gráfico.Dos son las formas más comunes:
Gráficos de la doble recíprocaLa ecuación se reordena para dar:
Así la grafica de 1/r frente a 1/[A] da una linea recta dependiente Kd/n.
Gráficos de ScatchardLa ecuación se reordena
Al representar r/[A] frente a r se obtiene una recta cuyapendiente es –1/Kd y su intersección con el eje x es n.
[ ]
dK
r n n A
1 1
d d
r n r
A K K