voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis
TRANSCRIPT

The FASEB Journal • Research Communication
Voltage-dependent anion channel 1 is involved inendostatin-induced endothelial cell apoptosis
Shaopeng Yuan, Yan Fu, Xiaofeng Wang, Hubing Shi, Yujie Huang, Xiaomin Song,Ling Li, Nan Song, and Yongzhang Luo1
Laboratory of Protein Chemistry, Beijing Key Laboratory for Protein Therapeutics, Department ofBiological Sciences and Biotechnology, Tsinghua University, Beijing, China
ABSTRACT Endostatin (ES) was reported to stimu-late apoptosis in endothelial cells, but the exact mech-anism remains controversial. In the present study, weelucidate the mechanism of ES-induced endothelial cellapoptosis. Our results indicate that ES induces cyto-chrome c release and caspase-9 activation in humanmicrovascular endothelial cells (HMECs) at the concen-tration of 1 �M for 24 h, which initiates the apoptosisprocess. Further, ATP production, mitochondrial mem-brane potential, and tubule formation assays showedthat ES promotes the mitochondrial permeability tran-sition pore (mPTP) opening via voltage-dependentanion channel 1 (VDAC1), a major component ofmitochondrial outer membrane. Knocking downVDAC1 by small interfering RNA attenuates ES-in-duced apoptosis, while overexpression of VDAC1 en-hances the sensitivity of endothelial cells to ES. More-over, we reveal that ES induces the reduction ofhexokinase 2 (HK2), which, in turn, promotes VDAC1phosphorylation and accumulation. Data from two-dimensional electrophoresis, immunoprecipitation,mPTP opening, and caspase-3 activation assays indicatethat two serine residues of VDAC1, Ser-12 and Ser-103,can modulate VDAC1 protein level and thus the sensi-tivity to apoptosis stimuli. On the basis of these find-ings, we conclude that VDAC1 plays a vital role inmodulating ES-induced endothelial cell apoptosis.—Yuan, S., Fu, Y., Wang, X., Shi, H., Huang, Y., Song, X.,Li, L., Song, N., Luo, Y. Voltage-dependent anionchannel 1 is involved in endostatin-induced endothelialcell apoptosis. FASEB J. 22, 2809–2820 (2008)
Key Words: angiogenesis � endothelium � hexokinase � mito-chondria � metabolism
Angiogenesis, the formation of new blood vesselsfrom preexisting vascular beds, is essential for tumorgrowth and survival (1). The angiogenesis process canbe regulated by a switch between concentrations ofinducers and inhibitors in vivo (2), and many angiogen-esis regulators have been developed as potential anti-tumor targets in clinical studies (3).
Endostatin (ES), the C-terminal globular domain ofcollagen XVIII, is one of the most powerful angiogen-esis inhibitors identified so far (4). ES specifically
targets angiogenic endothelial cells in the tumor re-gion, and its function has been related to severalsignaling events. Besides the inhibitory effect on endo-thelial cell migration and proliferation (5, 6), ES hasbeen proposed to induce endothelial cell apoptosis.Sukhatme and colleagues (7) reported that in cowpulmonary artery endothelial cells (C-PAECs), ESdown-regulates two major antiapoptotic proteins, bcl-2and bcl-xL, indicating that ES may induce apoptosis viathe intrinsic apoptotic pathway. In addition, othergroups reported that ES promotes the activation ofcaspase-8 in human dermal microvascular endothelialcells (HDMECs) and human umbilical vein endothelialcells (HUVECs), respectively, indicating that ES mayalso induce apoptosis via the extrinsic pathway (8, 9).Moreover, some other intracellular proteins are relatedto ES-induced apoptosis, such as Shb adaptor protein,AP-1, c-myc, mcl-1, and cyclin D1 (6, 10, 11). However,Skovseth and colleagues (12) reported that ES cannotinduce endothelial cell apoptosis at a low concentra-tion of 30–35 ng/ml in vivo. Conversely, ES was evenreported to enhance migration and proliferation inendothelial progenitor cells (13). Clearly, these studiesdemonstrate that the mechanism of ES-induced endo-thelial cell apoptosis remains controversial.
Mitochondria are major organelles involved in cellenergy conversion and apoptosis regulation (14). Inmitochondria-mediated cell apoptosis, opening of themitochondrial permeability transition pore (mPTP)plays a pivotal role (15). Decrease in mitochondrialmembrane potential, depletion of ATP, increase in freeCa2�, acidic and oxidative stresses are all signals thatpotentially enhance the probability of mPTP openingin cells (16). In addition, inhibitors targeting the mPTPopening, such as bongkrekic acid (BA) and cyclospor-ine A (CsA), appear to protect cells from the conse-quences of apoptosis stimuli (17, 18).
The mPTP is proposed to be a relatively nonspecificchannel spanning the inner and outer mitochondrialmembranes, which is composed of voltage-dependent
1 Correspondence: Laboratory of Protein Chemistry, Bei-jing Key Laboratory for Protein Therapeutics, Department ofBiological Sciences and Biotechnology, Tsinghua University,Beijing 100084, China. E-mail: [email protected]
doi: 10.1096/fj.08-107417
28090892-6638/08/0022-2809 © FASEB

anion channel (VDAC), adenine nucleotide transloca-tor (ANT), and cyclophilin D (CypD) (19–21). TheVDAC family proteins, including homologous isomersVDAC1, VDAC2, and VDAC3, locate on the mitochon-drial outer membrane. Under the regulation of Bcl-2family members, VDAC participates in the release ofcytochrome c and subsequent activation of caspase-9(22). In addition, caspase-independent apoptosis is alsorelated to VDAC (23). Among the isomers, VDAC1 isthe well recognized member involved in modulatingcell apoptosis (24). Up-regulation of VDAC1 promotesapoptosis in several cell lines (25–27).
Hexokinase 2 (HK2) is an important glycolytic pro-tein that prevents apoptosis through interacting withVDAC1 on the mitochondria (28). HK2 is up-regulatedin several tumor cells, which promotes endothelial cellangiogenesis (29). Recently, HK2 was determined to beregulated by hypoxia-inducible factor-1� (HIF-1�) inan angiogenesis modulating process (30). Since ES hasbeen reported to down-regulate HIF-1� in endothelialcells (11), it is plausible that ES regulates other glyco-lytic proteins, such as HK2, during the angiogenesisinhibition process.
In the present study, we show that ES induces up-regulation of VDAC1 and promotes the mPTP opening.We subsequently reveal that up-regulation of VDAC1results from ES-induced HK2 reduction. Our findingselucidate a novel mechanism of ES-induced endothelialcell apoptosis through mitochondria, providing insightinto the antiangiogenic mechanism of ES.
MATERIALS AND METHODS
Cell culture
HMECs, Hela, and HEK 293T cells are maintained at 37°C,5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM)(Hyclone, Logan, UT, USA) supplemented with 10% fetalcalf serum (FCS) (Hyclone), 100 �g/ml streptomycin, and100 U/ml penicillin. Endothelial cells were treated with 1 �MES for 24 h in the presence of 10 ng/ml basic fibroblastgrowth factor (bFGF) and 1% FCS unless indicated otherwise.
Chemicals, proteins, and antibodies
All chemicals were from Sigma-Aldrich (St. Louis, MO, USA)and all antibodies were from Santa Cruz Biotechnology(Santa Cruz, CA, USA) if not mentioned specifically. ES andbFGF were from Protgen Inc. (Beijing, China).
Monoclonal anti-VDAC1 antibody was from Calbiochem(Gibbstown, NJ, USA). MitoTracker-Red, Rhodamine 123(Rh123), and monoclonal anti-Cox IV antibody were fromMolecular Probes (Eugene, OR, USA).
TUNEL assay
The cultured cells were starved overnight, and then 1% FCSand 10 ng/ml bFGF were incubated with 1 �M ES. After 48 htreatment, the samples were prepared following the TACSTdT In Situ Apoptosis Detection Kit user’s manual (R&DSystems, Minneapolis, MN, USA) and were examined byfluorescence microscopy.
Annexin V/propidium iodide assay
Apoptosis was measured using the annexin V/propidiumiodide (PI) detection kit, according to the manufacturer’sinstruction (R&D Systems). Briefly, ES-treated cells weretrypsinized and resuspended in PBS supplemented with 0.1%BSA and labeled with annexin V-FITC and PI. Flow cytometricanalysis was then performed to monitor the green fluores-cence of annexin V and the red fluorescence of DNA-boundPI with excitation at 488 nm. All data were collected using aFACScan (Becton Dickinson, San Jose, CA, USA).
In vitro caspase activity assay
Caspase-3, -8, -9 activity was measured by the cleavage of theirrespective substrates, including DEVD-pNA, IETD-pNA, andLHED-pNA (BioVision, Mountain View, CA, USA). Briefly,1 � 106 cultured cells were lysed according to the user’smanual. Equal amounts of protein samples were transferredinto a 96-well microtiter plate with substrates. Then, theabsorbance was measured in a microplate reader at 405 nm(Bio-Rad Laboratories, Hercules, CA, USA). The activity ofcaspases was indicated by comparing the absorbance ofES-treated samples with an untreated control.
Cell fractionation assay
ES-treated cells were fractionated by differential centrifuga-tion. Briefly, cells were homogenized with a Dounce homog-enizer in a buffer containing 0.25 M sucrose and 10 mMHEPES (pH 7.4) supplemented with protease inhibitor cock-tail (Roche, Basel, Switzerland). The homogenate was thencentrifuged at 800 g for 10 min to remove nuclei andunbroken cells, and then the supernatant was centrifuged at12,000 g for 20 min to get the mitochondria. The cytosoliccytochrome c was thereby detected by immunoblotting assaywith cytochrome c monoclonal antibody.
For immunoblotting, proteins were separated by SDS-PAGE, transferred onto nitrocellulose membranes, and thenprobed with respective antibodies as indicated. Immunoreac-tive bands were visualized using enhanced chemilumines-cence (Pierce, Rockford, IL, USA).
Immunofluorescent and confocal microscopy
For detecting the localization of cytochrome c, ES-treatedcells were fixed by 4% paraformaldehyde and permeabilizedwith 0.2% Triton X-100. After washing twice with PBS, thecells were blocked with PBS containing 10% normal goatserum for 60 min and then incubated with monoclonalantibody against cytochrome c (1:200 dilution) and FITC-labeled goat anti-mouse secondary antibody (1:50 dilution) atroom temperature for 2 h. The mitochondria were directlystained with MitoTracker-Red, and the nuclei were stained byDAPI. Confocal fluorescence imaging was performed on anOlympus Fluoview laser scanning confocal imaging system(Olympus Inc., Tokyo, Japan).
Measurement of the cellular ATP levels
The ATP levels were determined by the luciferin-luciferasereaction. Briefly, 0.5 � 106 cultured cells were collected afterES treatment and then treated according to the manufactur-er’s instruction (BioVision, Mountain View, CA, USA). Theluminescence of 100 �l of lysate was measured immediatelyusing a luminometer.
2810 Vol. 22 August 2008 YUAN ET AL.The FASEB Journal

Measurement of mitochondrial membrane potential
ES-treated cells were trypsinized and resuspended in PBSsupplemented with 0.1% BSA and then incubated with Rh123for 30 min at 4°C. Flow cytometric analysis was then per-formed to monitor the green fluorescence of Rh123 withexcitation at 488 nm using FACScan.
mPTP opening assay
The mPTP opening was detected with the MitoProbe Transi-tion Pore Assay Kit according to the manufacturer’s instruc-tions (Molecular Probes). Briefly, cells were collected andstained with 2 �M calcein AM containing CoCl2 and ionomy-cin if required at 37°C for 15 min. Samples were then assessedfor fluorescence with excitation at 488 nm. Different intensi-ties of subgroup cells were respectively indicated in black(calcein AM only), green (calcein AM and CoCl2), and pink(calcein AM, CoCl2, and ionomycin). The mPTP opening wasindicated by the reduction of mitochondrial calcein signal(green subgroups).
Tubule formation assay
The tubule formation assay was carried out as describedpreviously. In brief, 24-well plates were coated with 100�l/well matrigel (Becton Dickinson Labware, Bedford, MA,USA). Endothelial cells were seeded on this matrix at 1 � 104
cells/well and incubated with 10 ng/ml bFGF. After 6 h, thewells were fixed and visualized. The tubule formation wasassessed in 3 randomly selected fields per well using aCoolSNAP charge-coupled device (CCD) digital camera(Roper Scientific, Tucson, AZ, USA).
Reactive oxygen species (ROS) release assay
The cell samples were prepared following the ROS assay kitmanufacturer’s instructions (Applygen Technologies Inc.,Beijing, China), and then detected by fluorescent micros-copy.
Phosphorylation of VDAC1 assay
ES-treated cells were lysed with 1% Chaps lysis buffer [10 mMHEPES (pH 7.4), 150 mM NaCl, 1 mM DTT, 20 mM NaF, 1mM Na3VO4, and 1% Chaps] containing protease inhibitors(Roche). The whole-cell lysates were immunoprecipitatedwith a rabbit polyclonal antibody against VDAC1 (Calbio-chem), electroblotted onto nitrocellulose membranes, andthen probed with an anti-phosphoserine monoclonal anti-body (Abcam plc, Cambridge, UK).
RNA interference
The sense sequences of double-strand siRNA were as follows:siVDAC1 (siVC1): 5� AGUGACGGGCAGUCUGGAATT 3�siHK2a: 5� GGAUAAGCUACAAAUCAAATT 3�siHK2b: 5� CGGGAAAGCAACUGUUUGATT 3�Scrambled siRNA, which serves as negative control, was
purchased from GenePharma (Shanghai, China). Transfec-tion of synthetic siRNA (25 nM) was performed with Lipo-fectamine 2000 (Invitrogen, Carlsbad, CA, USA) according tothe manufacturer’s instructions.
For RT-PCR assay, the primer sequences of HK2 andGAPDH were as follows:
HK2: Sense sequence: 5� TCAACCCCGGCAAGCAGAGG 3�Reverse sequence: 5� CCGCCGGGCCACCACAGT 3�
GAPDH: Sense sequence: 5� GAAGGTGAAGGTCGGAGTC 3�Reverse sequence: 5� GAAGATGGTGATGGGATTTC 3�
Statistical analysis
Data are expressed as means � sd. Multiple comparisonswere assessed by Student’s t test, and differences with P � 0.05were considered significant.
RESULTS
ES induces endothelial cell apoptosis viamitochondrial pathway
The mechanism of ES-induced endothelial cell apopto-sis has remained controversial. To clarify whether EScan indeed induce endothelial cell apoptosis, we firstperformed a series of apoptotic assays on HMECs toelucidate the proapoptotic function of ES. The TdT-mediated dUTP nick end labeling (TUNEL) assayshowed that in treated endothelial cells, ES promotednuclear DNA fragmentation, which is a hallmark ofapoptosis (Fig. 1A). Moreover, the results of the an-nexin V assay, which examined the exposed phospha-tidylserine (PS) on apoptotic endothelial cell mem-brane, showed that ES could induce HMEC apoptosisin a dose-dependent manner (data not shown).
To further characterize the ES-induced apoptosisprocess, we measured the activation of caspases, an-other hallmark of apoptosis. The DEVD-pNA cleavageactivity assay showed that caspase-3 was activated on EStreatment (Fig. 1B). Because different caspases partici-pate in different apoptotic pathways, we then set out toinvestigate which of the caspase-3 upstream modula-tors, caspase-8 and caspase-9, could be activated. Sepa-rate examination of the IETD-pNA and LHED-pNAcleavage activities indicated that only caspase-9 couldbe activated on ES treatment (Fig. 1B). ES-inducedactivation of caspase-3 and caspase-9 was further con-firmed by immunoblotting assay, which showed that theprecursors of these two caspases declined on ES treat-ment (Fig. 1C). Since activation of caspase-9 is normallytriggered by cytochrome c released to the cytoplasm(14), we thus examined released cytochrome c in thecytoplasm. Immunofluorescence and immunoblottingassays showed that ES significantly promoted the re-lease of cytochrome c to the cytoplasm (Fig. 1D). Inaddition, the release of cytochrome c was determined tooccur in an ES dose-dependent manner, which wasconsistent with the subsequent caspase-9 activation(Fig. 1E). Since cytochrome c release and caspase-9activation are both hallmarks of the mitochondria-mediated apoptosis process, these results thereforeindicate that the mitochondrial pathway is involved inthe ES-induced apoptotic process.
ES promotes endothelial cell mPTP opening
It is known that the efficiency of ATP synthesis onmitochondria is critical for the maintenance of func-
2811ENDOSTATIN INDUCES APOPTOSIS VIA VDAC1
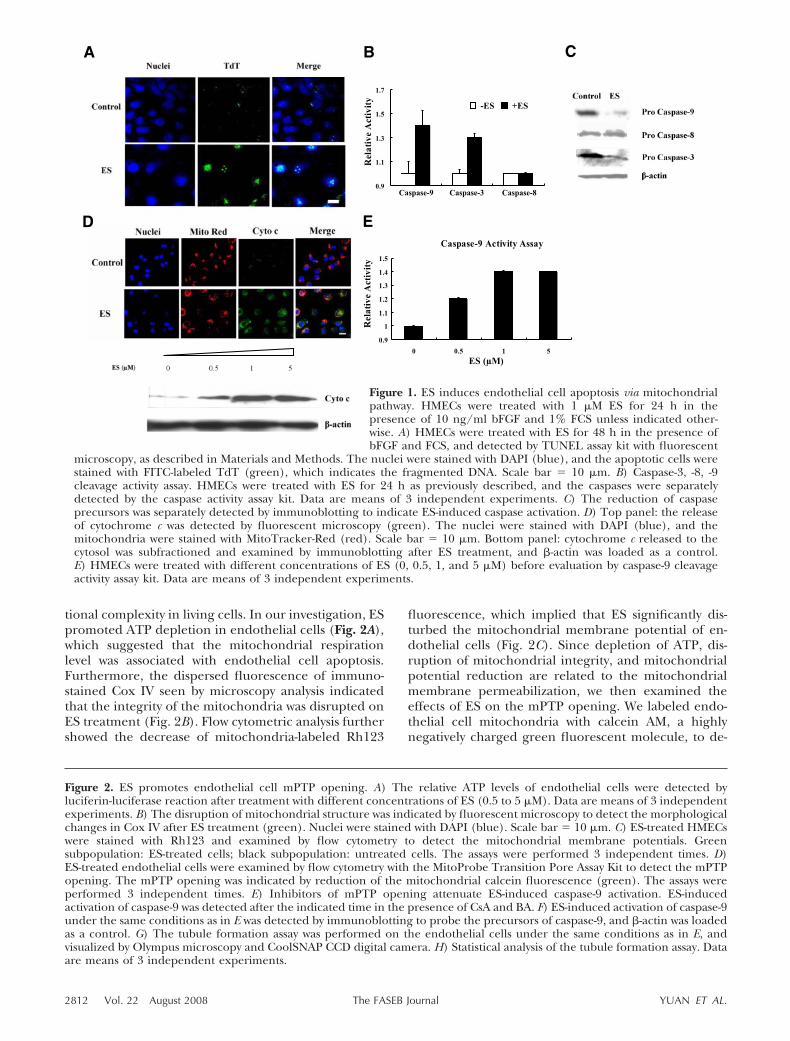
tional complexity in living cells. In our investigation, ESpromoted ATP depletion in endothelial cells (Fig. 2A),which suggested that the mitochondrial respirationlevel was associated with endothelial cell apoptosis.Furthermore, the dispersed fluorescence of immuno-stained Cox IV seen by microscopy analysis indicatedthat the integrity of the mitochondria was disrupted onES treatment (Fig. 2B). Flow cytometric analysis furthershowed the decrease of mitochondria-labeled Rh123
fluorescence, which implied that ES significantly dis-turbed the mitochondrial membrane potential of en-dothelial cells (Fig. 2C). Since depletion of ATP, dis-ruption of mitochondrial integrity, and mitochondrialpotential reduction are related to the mitochondrialmembrane permeabilization, we then examined theeffects of ES on the mPTP opening. We labeled endo-thelial cell mitochondria with calcein AM, a highlynegatively charged green fluorescent molecule, to de-
Figure 2. ES promotes endothelial cell mPTP opening. A) The relative ATP levels of endothelial cells were detected byluciferin-luciferase reaction after treatment with different concentrations of ES (0.5 to 5 �M). Data are means of 3 independentexperiments. B) The disruption of mitochondrial structure was indicated by fluorescent microscopy to detect the morphologicalchanges in Cox IV after ES treatment (green). Nuclei were stained with DAPI (blue). Scale bar � 10 �m. C) ES-treated HMECswere stained with Rh123 and examined by flow cytometry to detect the mitochondrial membrane potentials. Greensubpopulation: ES-treated cells; black subpopulation: untreated cells. The assays were performed 3 independent times. D)ES-treated endothelial cells were examined by flow cytometry with the MitoProbe Transition Pore Assay Kit to detect the mPTPopening. The mPTP opening was indicated by reduction of the mitochondrial calcein fluorescence (green). The assays wereperformed 3 independent times. E) Inhibitors of mPTP opening attenuate ES-induced caspase-9 activation. ES-inducedactivation of caspase-9 was detected after the indicated time in the presence of CsA and BA. F) ES-induced activation of caspase-9under the same conditions as in E was detected by immunoblotting to probe the precursors of caspase-9, and -actin was loadedas a control. G) The tubule formation assay was performed on the endothelial cells under the same conditions as in E, andvisualized by Olympus microscopy and CoolSNAP CCD digital camera. H) Statistical analysis of the tubule formation assay. Dataare means of 3 independent experiments.
Figure 1. ES induces endothelial cell apoptosis via mitochondrialpathway. HMECs were treated with 1 �M ES for 24 h in thepresence of 10 ng/ml bFGF and 1% FCS unless indicated other-wise. A) HMECs were treated with ES for 48 h in the presence ofbFGF and FCS, and detected by TUNEL assay kit with fluorescent
microscopy, as described in Materials and Methods. The nuclei were stained with DAPI (blue), and the apoptotic cells werestained with FITC-labeled TdT (green), which indicates the fragmented DNA. Scale bar � 10 �m. B) Caspase-3, -8, -9cleavage activity assay. HMECs were treated with ES for 24 h as previously described, and the caspases were separatelydetected by the caspase activity assay kit. Data are means of 3 independent experiments. C) The reduction of caspaseprecursors was separately detected by immunoblotting to indicate ES-induced caspase activation. D) Top panel: the releaseof cytochrome c was detected by fluorescent microscopy (green). The nuclei were stained with DAPI (blue), and themitochondria were stained with MitoTracker-Red (red). Scale bar � 10 �m. Bottom panel: cytochrome c released to thecytosol was subfractioned and examined by immunoblotting after ES treatment, and -actin was loaded as a control.E) HMECs were treated with different concentrations of ES (0, 0.5, 1, and 5 �M) before evaluation by caspase-9 cleavageactivity assay kit. Data are means of 3 independent experiments.
2812 Vol. 22 August 2008 YUAN ET AL.The FASEB Journal
2813ENDOSTATIN INDUCES APOPTOSIS VIA VDAC1
tect the effect in ES-treated living cells. As expected, EStreatment reduced the fluorescent signal of calcein onmitochondria, suggesting that ES promoted the mPTPopening (Fig. 2D). Furthermore, mPTP inhibitorscould attenuate ES-induced endothelial cell apoptosis.In our experiment, activation of caspase-9 was impairedby pretreatment with 5 �M BA and 4 �M CsA, respec-tively (Fig. 2E, F).
To investigate whether the opening of mPTP isinvolved in the antiangiogenic function of ES, weinvestigated the effects of BA and CsA on endothelialcell tubule formation. In the absence of BA and CsA,ES exhibited strong inhibition on the tubule forma-tion. However, this effect was attenuated in thepresence of BA and CsA (Fig. 2G, H). This resultdemonstrates that the inhibitory effect of ES onangiogenesis is dependent on the mPTP opening.Furthermore, it also implies that attenuation of en-
dothelial cell apoptosis can facilitate the angiogene-sis process.
ES induces VDAC1 up-regulation in endothelial cellsand modulates the apoptotic process
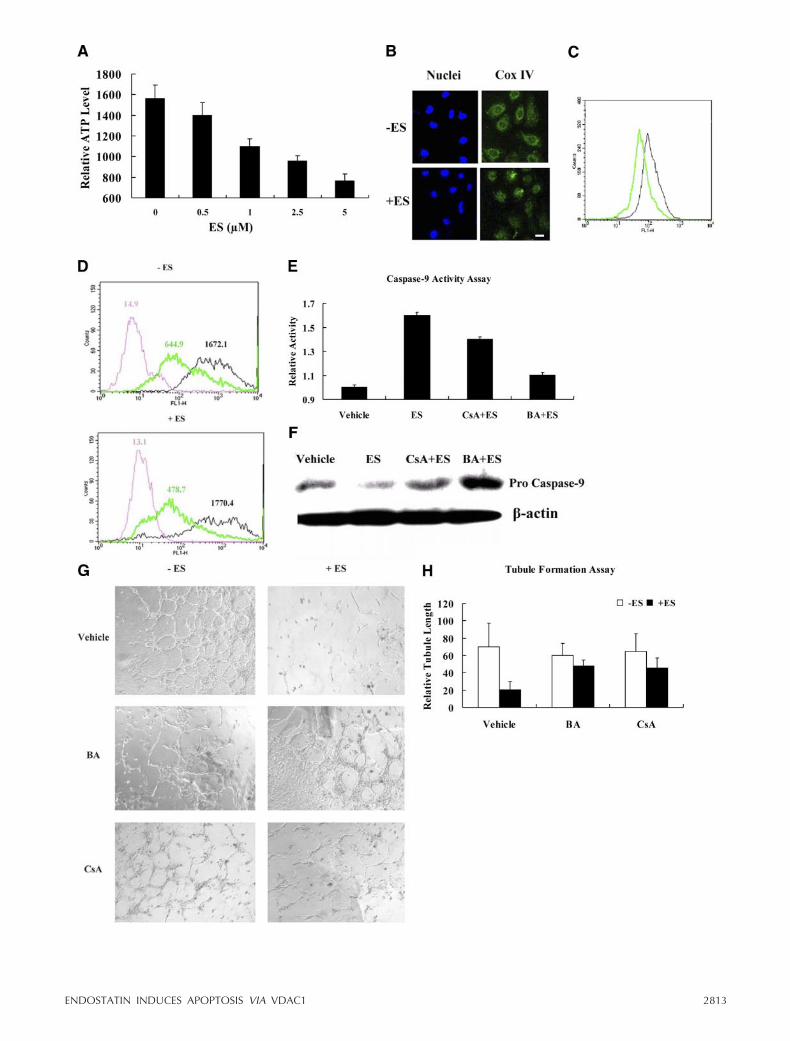
To further investigate the mechanism of ES-inducedmPTP opening, we focused on the component proteinsof mPTP and their roles in modulating apoptosis.Enhanced protein level of VDAC1, one of the essentialmPTP components, was observed in a dose-dependentmanner on ES treatment (Fig. 3A). Up-regulation ofVDAC1 was exclusively observed in endothelial cells,but not in nonendothelial cells such as Hela or HEK293T cells (data not shown). We therefore hypothe-sized that VDAC1 is a potential candidate in modulat-ing the ES-induced apoptotic process.
Figure 3. ES induces VDAC1 up-regulation in endothelial cells and modulates the apoptotic process. A) VDAC1 was probed withmonoclonal antibody after treatment with different concentrations of ES for 24 h, and -actin was loaded as a control. B) Theattenuation of ES-induced apoptosis was detected by annexin V/PI assay in the VDAC1-silenced endothelial cells. SyntheticVDAC1 siRNA (siVC1) was transfected in HMECs for 24 h before ES treatment; cells were collected and analyzed by flowcytometry. N.C. denotes negative control, which indicates scrambled siRNA. The assays were performed 3 independent times.C) The attenuation of caspase-9 activation was detected in ES-treated VDAC1-silenced endothelial cells under the sameconditions as in B. Data are means of 3 independent experiments. D) Immunoblotting to detect the attenuation of caspase-9activation in ES-treated VDAC1-silenced endothelial cells. E) pVC1 and GFP-VC1 indicate the vectors constructed with humanVDAC1 gene in the plasmid pcDNA3.1 and pEGFP-N1, respectively. HMECs were transiently transfected with pVC1 and emptyvectors and were incubated with or without 1 �M ES for 6 h before measuring the tubule formation. In another experiment,GFP-VC1 and pEGFP-N1 were transfected in HMECs, and the tubule formation was then measured by fluorescence microscopy(green). F) Statistical analysis of the tubule formation assay in E. Data are means of 3 independent experiments. G) Theactivation of caspase-9 was detected in pVC1 transfected HMECs incubated with or without ES. Data are means of 3 independentexperiments. H) pVC1 and empty vectors were transfected in HMECs, and then the ROS release was examined by fluorescencemicroscopy with probing dichlorofluorescin diacetate (DCFH-DA) fluorescence (green). Scale bar � 50 �m.
2814 Vol. 22 August 2008 YUAN ET AL.The FASEB Journal

To specify the function of VDAC1 in regulatingES-induced apoptosis, we knocked down VDAC1 inendothelial cells, using synthetic siRNA targetingVDAC1. As expected, the annexin V assay showed thatES-induced apoptosis was attenuated in VDAC1-si-lenced cells (Fig. 3B). In addition, the results of LHED-pNA cleavage activity and immunoblotting assay bothshowed that ES-induced caspase-9 activation was im-paired in VDAC1-silenced cells (Fig. 3C, D). Theseresults confirmed the important role of VDAC1 inmodulating ES-induced apoptosis.
To further examine the effect of VDAC1 up-regula-tion, we overexpressed VDAC1 and subsequently exam-ined the function on tubule formation. By fluorescentmicroscopy analysis, overexpression of VDAC1 was de-termined to directly inhibit endothelial cell tubuleformation (Fig. 3E, bottom panel), which enhanced thesensitivity of endothelial cells to ES (Fig. 3E, top panel;F). This inhibitory phenomenon might be the result ofoverexpression of VDAC1 promoting the death rate ofendothelial cells. Actually, overexpression of VDAC1 inendothelial cells did indeed enhance the activation ofcaspase-9 (Fig. 3G). Moreover, the production of ROS,another hallmark of mitochondria-mediated apoptosis,was elevated after overexpression of VDAC1 in endo-
thelial cells (Fig. 3H). These results demonstrate thatoverexpression of VDAC1 promotes the mitochondriadysfunction, which further confirms that ES inducesapoptosis via VDAC1 up-regulation and eventuallyleads to angiogenesis inhibition.
ES-induced reduction of HK2 results inVDAC1 up-regulation
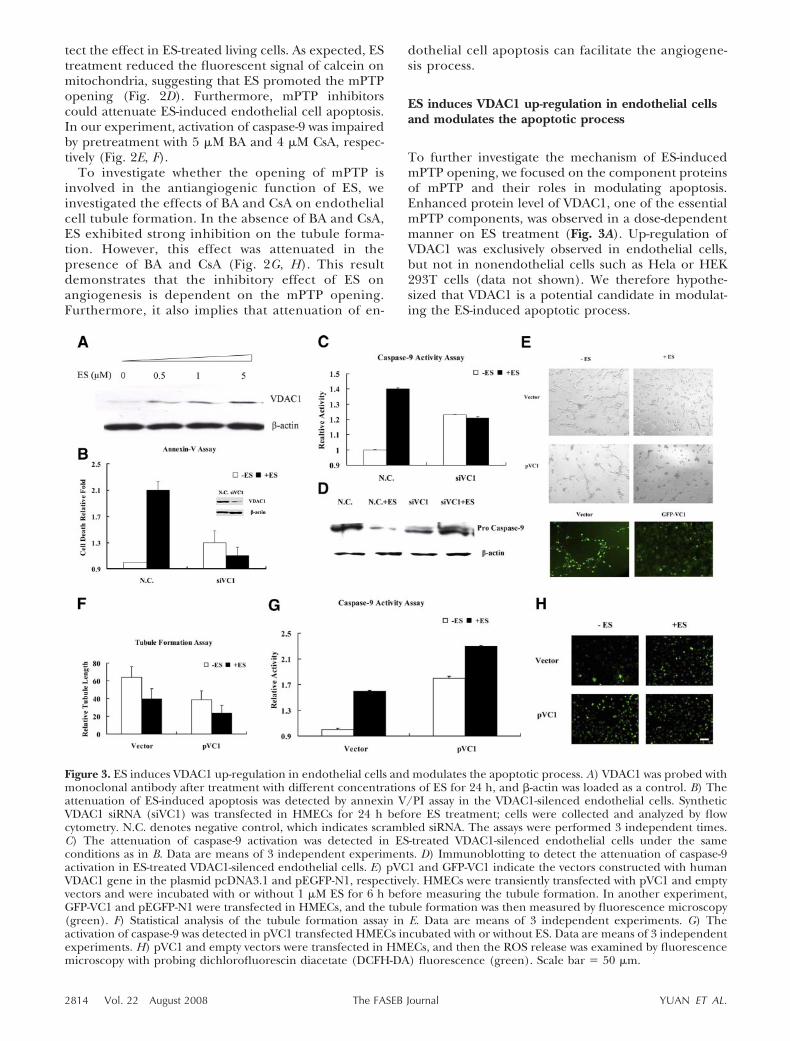
To further delineate the mechanism of how ES inducesapoptosis through VDAC1 up-regulation, we investi-gated VDAC1 binding proteins to screen the potentialmodulators. Among the VDAC1 binding proteins, HK2was reported to inhibit apoptosis (31). ES was observedto significantly reduce the protein level of HK2 after 4 hof treatment in endothelial cells (Fig. 4A). Consideringthe function of HK2 in apoptosis inhibition, the reduc-tion of HK2 predictably decreases its association withVDAC1. As expected, our results showed that ES dis-turbed the interaction between HK2 and VDAC1, andoverexpression of VDAC1 facilitated its dissociationfrom HK2 (Fig. 4B).
HK2 catalyzes the first committed step in glycolysis(28). Considering that endothelial cells are surrounded
Figure 4. ES-induced reduction of HK2 results in VDAC1 up-regulation.A) The protein levels of HK2 and VDAC1 were detected after EStreatment for different times (1, 4, and 12 h). B) pVC1 and the emptyvectors were transfected in HMECs and then were treated with ES ifrequired. After 24 h treatment, cells were collected and immunoprecipi-tation assay was performed. Different bands were separately detected withantibodies against HK2, VDAC1, and Bax. C) Two kinds of syntheticsiRNA targeting HK2 (siHK2a, siHK2b) were transfected in HMECs, andthe protein and mRNA levels of HK2 were separately measured byWestern blot analysis and RT-PCR after 24 h. -actin and GAPDH wereloaded as controls, respectively. D) Elevation of ES-induced caspase-9
activation was detected in HK2 silenced cells. Data are means of 3 independent experiments. E) After transfection ofsiRNA targeting HK2, released ROS (green) was examined in HK2-silenced HMECs in the presence of 10% FCS.Vehicle, nontransfected endothelial cells. Scale bar � 50 �m. F) The protein levels of HK2 and VDAC1 were analyzedin the HK2-silenced HMECs after ES treatment. -actin was loaded as a control.
2815ENDOSTATIN INDUCES APOPTOSIS VIA VDAC1

by a hypoxic microenvironment in vivo, we wonderedwhether HK2 was an important molecule in regulatingendothelial cell survival. To investigate the role of HK2,we examined the apoptotic effect of HK2 knockdownon endothelial cells. The HK2-specific siRNA efficientlysilenced its expression (Fig. 4C) and quenched theendothelial cell proliferation (see Supplemental Data).In addition, ES-induced apoptosis was elevated in HK2knockdown endothelial cells, concomitant with a signif-icantly enhanced activation of caspase-9 (Fig. 4D).These observations were further corroborated by in-creased ROS production in HK2-silenced endothelialcells (Fig. 4E). ROS elevation is an important signal topromote the mPTP opening. Because we have observedthat ROS elevated in VDAC1 overexpressed endothelialcells (Fig. 3G), we were curious whether VDAC1 wasaffected by HK2 reduction. Interestingly, we observedthat the protein level of VDAC1 increased after knock-ing down HK2 in endothelial cells (Fig. 4F).
Up-regulation of VDAC1 is due to its phosphorylation
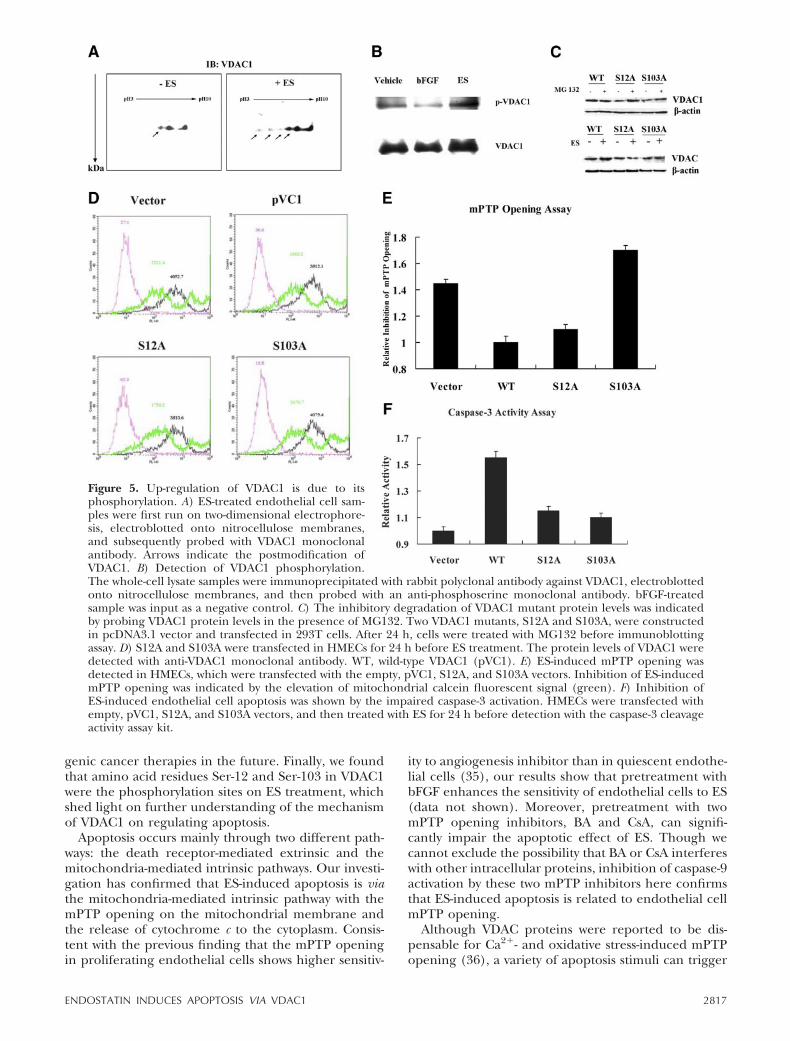
HK2 inhibits the translocation of Bax to the mitochon-dria (31). In our investigation, Bax was identified to beassociated with VDAC1 on ES treatment, and this kindof association was promoted in VDAC1-overexpressedendothelial cells (Fig. 4B). These results suggest thatHK2 and Bax probably compete to bind VDAC1 on theouter membrane of mitochondria. HK2 was reportedto modulate phosphorylation of VDAC, potentiallythrough glycogen synthase kinase 3 (GSK3) or pro-tein kinase C (PKC) (32, 33). In addition, the E72Qmutant of VDAC1, which lost the HK2 binding activity,was reported to impair the increased total protein levelof VDAC1 on exposure to RuR in 293T cells (24). Wethus wondered whether reduction of HK2 could lead tothe phosphorylation of VDAC1, which eventually re-sults in VDAC1 up-regulation. To answer this question,we performed two-dimensional electrophoresis to sep-arate VDAC1 in ES-treated endothelial cells and de-tected it with specific VDAC1 monoclonal antibody. Itwas clearly shown that VDAC1 was enriched in theacidic side of two-dimensional electrophoresis, suggest-ing the potential increase in VDAC1 phosphorylation(Fig. 5A). Since several serine residues were reported tobe the potential phosphorylation sites on VDAC1, wethus performed an immunoprecipitation assay to di-rectly detect ES-induced VDAC1 phosphorylation withmonoclonal antibody against phosphorylated serineresidues. It was observed that phosphorylation ofVDAC1 was enhanced after ES treatment (Fig. 5B).These results supported our hypothesis that ES couldpromote VDAC1 phosphorylation.
To further investigate the phosphorylated sites ofVDAC1, we constructed a series of VDAC1 mutants,which disturbed the potential serine phosphorylationon VDAC1. These mutants, including S12A, S103A, andS136A, were transfected into 293T cells, and theirprotein levels were detected by immunoblotting assay.Two VDAC1 mutants, S12A and S103A, could effec-
tively reduce the protein level of VDAC1 (Fig. 5C).These results indicate that dephosphorylation ofVDAC1 may impair its protein level elevation. In agree-ment with the findings of Zaid and his colleagues (24),there might be some degradation-related mechanismsto VDAC1 overexpression. Our results here showedthat reduction of S12A and S103A protein level wasinhibited by MG132, a well-known proteasome inhibi-tor (Fig. 5C). Because phosphorylation of a specificprotein could prevent the protease-mediated degrada-tion, our results indicate that phosphorylation ofVDAC1 probably impairs its degradation, which leadsto VDAC1 accumulation on ES treatment.
We then measured the function of S12A and S103Ain endothelial cells. The S12A and S103A mutantscould attenuate ES-induced up-regulation of VDAC1protein level in endothelial cells (Fig. 5D), whichimplied that ES-induced VDAC1 up-regulation mightresult from its phosphorylation. We further examinedthe effects of S12A and S103A on ES-induced apoptosisprocess. S12A and S103A were both observed to abolishES-induced mPTP opening (Fig. 5E) and caspase-3activation in endothelial cells (Fig. 5F), which furtherconfirmed our hypothesis that ES could affect VDAC1phosphorylation and accumulation.
Though the detailed mechanism of VDAC1 in mod-ulating ES-induced apoptosis needs further investiga-tion, our results here reveal a novel mechanism ofES-induced endothelial cell apoptosis through VDAC1up-regulation, and provide insight for further under-standing of the mechanisms of angiogenesis inhibitorssuch as ES.
DISCUSSION
As one of the most powerful endogenous angiogenesisinhibitors, ES has potent therapeutic effects on severaltypes of tumors in clinical studies (34). However, thedetailed molecular mechanism of ES-induced endothe-lial cell apoptosis is still controversial and needs furtherinvestigation. In the present study, we have elucidatedthe mechanism of ES-induced endothelial cell apopto-sis. First, we observed that certain mitochondria-relatedapoptotic signaling events occurred after ES treatment,which confirmed the previous reports that ES triggersapoptosis in endothelial cells. Second, we demon-strated that ES promoted the opening of mPTP andup-regulation of VDAC1. More specifically, ES-inducedVDAC1 up-regulation enhanced endothelial cell apo-ptosis, leading to tumor angiogenesis inhibition. This isthe first time that the antiangiogenesis function of ESlinks to the mitochondrial permeability in the apo-ptotic process. Third, ES was determined to affect anessential glycolytic enzyme, HK2, in endothelial cells,and up-regulation of VDAC1 was validated to be theresult of HK2 reduction, which promoted the phos-phorylation of VDAC1. These results elucidate the roleof glycolytic proteins in regulating angiogenesis, mayalso have implications in the treatment of antiangio-
2816 Vol. 22 August 2008 YUAN ET AL.The FASEB Journal
genic cancer therapies in the future. Finally, we foundthat amino acid residues Ser-12 and Ser-103 in VDAC1were the phosphorylation sites on ES treatment, whichshed light on further understanding of the mechanismof VDAC1 on regulating apoptosis.
Apoptosis occurs mainly through two different path-ways: the death receptor-mediated extrinsic and themitochondria-mediated intrinsic pathways. Our investi-gation has confirmed that ES-induced apoptosis is viathe mitochondria-mediated intrinsic pathway with themPTP opening on the mitochondrial membrane andthe release of cytochrome c to the cytoplasm. Consis-tent with the previous finding that the mPTP openingin proliferating endothelial cells shows higher sensitiv-
ity to angiogenesis inhibitor than in quiescent endothe-lial cells (35), our results show that pretreatment withbFGF enhances the sensitivity of endothelial cells to ES(data not shown). Moreover, pretreatment with twomPTP opening inhibitors, BA and CsA, can signifi-cantly impair the apoptotic effect of ES. Though wecannot exclude the possibility that BA or CsA interfereswith other intracellular proteins, inhibition of caspase-9activation by these two mPTP inhibitors here confirmsthat ES-induced apoptosis is related to endothelial cellmPTP opening.
Although VDAC proteins were reported to be dis-pensable for Ca2�- and oxidative stress-induced mPTPopening (36), a variety of apoptosis stimuli can trigger
Figure 5. Up-regulation of VDAC1 is due to itsphosphorylation. A) ES-treated endothelial cell sam-ples were first run on two-dimensional electrophore-sis, electroblotted onto nitrocellulose membranes,and subsequently probed with VDAC1 monoclonalantibody. Arrows indicate the postmodification ofVDAC1. B) Detection of VDAC1 phosphorylation.The whole-cell lysate samples were immunoprecipitated with rabbit polyclonal antibody against VDAC1, electroblottedonto nitrocellulose membranes, and then probed with an anti-phosphoserine monoclonal antibody. bFGF-treatedsample was input as a negative control. C) The inhibitory degradation of VDAC1 mutant protein levels was indicatedby probing VDAC1 protein levels in the presence of MG132. Two VDAC1 mutants, S12A and S103A, were constructedin pcDNA3.1 vector and transfected in 293T cells. After 24 h, cells were treated with MG132 before immunoblottingassay. D) S12A and S103A were transfected in HMECs for 24 h before ES treatment. The protein levels of VDAC1 weredetected with anti-VDAC1 monoclonal antibody. WT, wild-type VDAC1 (pVC1). E) ES-induced mPTP opening wasdetected in HMECs, which were transfected with the empty, pVC1, S12A, and S103A vectors. Inhibition of ES-inducedmPTP opening was indicated by the elevation of mitochondrial calcein fluorescent signal (green). F) Inhibition ofES-induced endothelial cell apoptosis was shown by the impaired caspase-3 activation. HMECs were transfected withempty, pVC1, S12A, and S103A vectors, and then treated with ES for 24 h before detection with the caspase-3 cleavageactivity assay kit.
2817ENDOSTATIN INDUCES APOPTOSIS VIA VDAC1

apoptosis by modulation of VDAC. For example, Chenand colleagues (26) reported that arsenic trioxide(As2O3) directly induced cytochrome c release fromisolated mouse liver mitochondria via mPTP openingand VDAC is the target. In addition, oxidative stress-induced apoptosis can be related to VDAC in smoothmuscle and endothelial cells (37). In the present study,we elucidate that VDAC1 is an indispensable protein,which modulates ES-induced apoptosis in endothelialcells. We determined that knocking down VDAC1 inendothelial cells impairs ES-induced apoptosis, whereasoverexpression of extraneous VDAC1 results in en-hanced sensitivity of endothelial cells to ES. To furtherinvestigate the mPTP opening in modulating ES-in-duced apoptosis, more studies may be performed onother mPTP component-deficient endothelial cells,such as CypD and ANT.
Bcl-2 family proteins, such as Bax and Bcl-xL, havebeen reported to form complexes with VDAC1 in theformation of mPTP (22). In our experiments, weobserved that ES promoted the translocation of Bax tothe mitochondria and enhanced the interaction withVDAC1, which possibly resulted in mPTP opening. Ourgroup recently reported that ES can be translocated toendothelial cell nuclei via nucleolin, inhibiting theproliferation of endothelial cells (38); it is thus reason-able that ES might regulate transcription factors innuclei to modulate the expression level of Bax, result-ing in its accumulation in the mitochondria and theassociation with VDAC1.
HK2 is another important VDAC1 binding proteinthat can inhibit apoptosis. Experiments with reconsti-tuted mPTPs in vitro indicate that the association ofHK2 with VDAC1 may mediate mPTP opening. Ourresults show that ES-induced HK2 reduction is corre-lated with endothelial cell apoptosis, because increasedapoptosis rate, caspase-9 activation, and ROS releasewere observed in HK2-silenced endothelial cells. Re-duction of HK2 could also explain how ES regulates theapoptotic process in endothelial cells, because silencingof HK2 elevates the expression level of VDAC1 andsensitizes endothelial cells to ES treatment.
It was reported that HIF-1� can promote tumorangiogenesis under the hypoxic microenvironment(39). HIF-1�, up-regulated by hypoxic stress, plays animportant role in regulating the transcription of theglycolytic enzymes in tumor and endothelial cells. SinceHK2 can be regulated by HIF-1� (30), and ES cansuppress the expression of HIF-1�, it is reasonable thatES possibly reduces the expression of HK2 throughHIF-1� by some unknown mechanism. Cobalt chloride(CoCl2) is known to stabilize HIF-1� by preventing theeffects of VHL protein (40). We observed that CoCl2could abolish ES-induced VDAC1 up-regulation (datanot shown). These results indicate that the reduction ofHK2 can regulate the protein level of VDAC1.
Phosphorylation is believed to play an important rolein modulating VDAC1 function. In our study, thetwo-dimensional gel electrophoresis and immunopre-cipitation studies indicate that ES enhances the phos-
phorylation of VDAC1. There were other reports thatphosphorylation of VDAC could modulate its function.For example, Sun et al. (33), reported that HK2 pro-motes the phosphorylation of VDAC although it was ona threonine instead of serine residue, consistent withour hypothesis that ES-induced HK2 reduction is re-lated to the phosphorylation of VDAC. VDAC can alsobe modulated by several other protein kinases withdifferent functions. Banerjee et al. (41) reported thatinteractions between VDAC with Bax and tBid areattenuated because of phosphorylation of VDAC bycyclic AMP-dependent protein kinase A (PKA). A sim-ilar result was reported for the inhibition of cyto-chrome c release from mitochondria by another kinase,c-Raf (42). Moreover, PKCε can phosphorylate VDACin vitro to inhibit the mPTP in cardiac mitochondria(43). On the contrary, Pastorin et al. (32) demonstratedthat the phosphorylation of VDAC by GSK3 in vitropromoted apoptosis by triggering HK2 disassociationfrom mitochondria.
Nevertheless, there have been few reports about thephosphorylation sites of VDAC until now. Hoppel andcolleagues (44) reported that two serine residues,Ser-12 and Ser-136, are phosphorylated in VDAC-1isolated from rat liver. Moreover, Ser-103 is anotherpotential phosphorylation site of VDAC1 (45). Thephosphorylated serine residues involved in our investi-gation were identified to be Ser-12 and Ser-103. Sincethe structure of VDAC1 is not resolved yet, thesephosphorylated amino acid residues are predicted tobe located on the cytosolic side of the mitochondrialouter membrane, accessible to protein kinases in thecytoplasm. For example, Ser-12 is a potential GSK3modulating site.
Because binding of HK2 to VDAC1 impairs VDAC1’sproapoptotic function, the interaction between HK2and VDAC1 might therefore bury the phosphorylationsites of VDAC1. We hypothesize that reduction of HK2probably promotes the exposure of the buried phos-phorylated sites to protein kinases, such as GSK3 orothers, which leads to VDAC1 phosphorylation. Phos-phorylation of VDAC1 may trigger the conformationalchanges of VDAC1 and inhibit its degradation, result-ing in VDAC1 accumulation. Phosphorylation ofVDAC1 might also facilitate Bax, tBid, or other mPTPcomponent proteins to interact with VDAC1, which, inturn, promotes mPTP opening and eventually inducescell death. In the present study, results that S12A andS103A can attenuate ES-induced VDAC1 up-regulation,endothelial cell mPTP opening, and apoptosis elicita-tion confirm our hypothesis.
Besides regulation of cell apoptosis, up-regulation ofVDAC1 can have other effects on endothelial cells.VDAC1 is a key determinant of Ca2� permeability at theER-mitochondria contact sites. VDAC1 up-regulationcan affect the Ca2� permeability at the ER-mitochon-dria contact sites, and facilitate the binding between ERand mitochondria and subsequently promote Ca2�
release from the ER (46). Unlike in other cell types,only less than 4% of mitochondria are close to the ER
2818 Vol. 22 August 2008 YUAN ET AL.The FASEB Journal

membrane surface in endothelial cells (16). VDAC1up-regulation in endothelial cells therefore might takeeffect on Ca2�-related proteins and signaling pathwaysin endothelial cells, such as calmodulin or calcium-dependent kinases. Actually, ES was reported to mod-ulate the cellular Ca2� concentration (47), which im-plies that ES possibly triggers the calcium-related eventsthrough VDAC1 up-regulation.
Taken together, results in the present study reveal anovel mechanism of ES-induced endothelial cell apo-ptosis through mitochondria dysfunction via VDAC1.These observations provide insight for further under-standing of the molecular mechanism of antiangio-genic function of ES. Moreover, elucidating the role ofVDAC1 in modulating ES-induced endothelial cell ap-optosis may also have implications in the future cancertherapies.
We gratefully acknowledge Quan Chen for his criticalreading of this manuscript and valuable suggestions. Thisstudy was supported by grants from the General Programs ofthe National Natural Science Foundation of China (30670419and 30771083), and the State Key Development Program forBasic Research of China (2006CB910305).
REFERENCES
1. Folkman, J. (1995) Angiogenesis in cancer, vascular, rheuma-toid and other disease. Nat. Med. 1, 27–31
2. Hanahan, D., and Folkman, J. (1996) Patterns and emergingmechanisms of the angiogenic switch during tumorigenesis. Cell86, 353–364
3. Ferrara, N., and Kerbel, R. S. (2005) Angiogenesis as a thera-peutic target. Nature 438, 967–974
4. O’Reilly, M. S., Boehm, T., Shing, Y., Fukai, N., Vasios, G., Lane,W. S., Flynn, E., Birkhead, J. R., Olsen, B. R., and Folkman, J.(1997) Endostatin: an endogenous inhibitor of angiogenesisand tumor growth. Cell 88, 277–285
5. Yamaguchi, N., Anand-Apte, B., Lee, M., Sasaki, T., Fukai, N.,Shapiro, R., Que, I., Lowik, C., Timpl, R., and Olsen, B. R.(1999) Endostatin inhibits VEGF-induced endothelial cell mi-gration and tumor growth independently of zinc binding.EMBO J. 18, 4414–4423
6. Shichiri, M., and Hirata, Y. (2001) Antiangiogenesis signals byendostatin. FASEB J. 15, 1044–1053
7. Dhanabal, M., Ramchandran, R., Waterman, M. J., Lu, H.,Knebelmann, B., Segal, M., and Sukhatme, V. P. (1999) En-dostatin induces endothelial cell apoptosis. J. Biol. Chem. 274,11721–11726
8. Addison, C. L., Nor, J. E., Zhao, H., Linn, S. A., Polverini, P. J.,and Delaney, C. E. (2005) The response of VEGF-stimulatedendothelial cells to angiostatic molecules is substrate-dependent[Online]. BMC Cell Biol. 6, 38
9. Kang, H. Y., Shim, D., Kang, S. S., Chang, S. I., and Kim, H. Y.(2006) Protein kinase B inhibits endostatin-induced apoptosisin HUVECs. J. Biochem. Mol. Biol. 39, 97–104
10. Dixelius, J., Larsson, H., Sasaki, T., Holmqvist, K., Lu, L.,Engstrom, A., Timpl, R., Welsh, M., and Claesson-Welsh, L.(2000) Endostatin-induced tyrosine kinase signaling throughthe Shb adaptor protein regulates endothelial cell apoptosis.Blood 95, 3403–3411
11. Abdollahi, A., Hahnfeldt, P., Maercker, C., Grone, H. J., Debus,J., Ansorge, W., Folkman, J., Hlatky, L., and Huber, P. E. (2004)Endostatin’s antiangiogenic signaling network. Mol. Cell 13,649–663
12. Skovseth, D. K., Veuger, M. J., Sorensen, D. R., De Angelis, P. M.,and Haraldsen, G. (2005) Endostatin dramatically inhibits en-dothelial cell migration, vascular morphogenesis, and perivas-cular cell recruitment in vivo. Blood 105, 1044–1051
13. Khan, Z. A., Melero-Martin, J. M., Wu, X., Paruchuri, S.,Boscolo, E., Mulliken, J. B., and Bischoff, J. (2006) Endothelialprogenitor cells from infantile hemangioma and umbilical cordblood display unique cellular responses to endostatin. Blood 108,915–921
14. Kroemer, G., and Reed, J. C. (2000) Mitochondrial control ofcell death. Nat. Med. 6, 513–519
15. Jacotot, E., Costantini, P., Laboureau, E., Zamzami, N., Susin,S. A., and Kroemer, G. (1999) Mitochondrial membrane per-meabilization during the apoptotic process. Ann. N. Y. Acad. Sci.887, 18–30
16. Davidson, S. M., and Duchen, M. R. (2007) Endothelial mito-chondria: contributing to vascular function and disease. Circ.Res. 100, 1128–1141
17. Marchetti, P., Castedo, M., Susin, S. A., Zamzami, N., Hirsch, T.,Macho, A., Haeffner, A., Hirsch, F., Geuskens, M., and Kroemer,G. (1996) Mitochondrial permeability transition is a centralcoordinating event of apoptosis. J. Exp. Med. 184, 1155–1160
18. Walter, D. H., Haendeler, J., Galle, J., Zeiher, A. M., andDimmeler, S. (1998) Cyclosporin A inhibits apoptosis of humanendothelial cells by preventing release of cytochrome C frommitochondria. Circulation 98, 1153–1157
19. Vander Heiden, M. G., Chandel, N. S., Li, X. X., Schumacker,P. T., Colombini, M., and Thompson, C. B. (2000) Outermitochondrial membrane permeability can regulate coupledrespiration and cell survival. Proc. Natl. Acad. Sci. U. S. A. 97,4666–4671
20. Crompton, M., Virji, S., and Ward, J. M. (1998) Cyclophilin Dbinds strongly to complexes of the voltage-dependent anionchannel and the adenine nucleotide translocase to form thepermeability transition pore. Eur. J. Biochem. 258, 729–735
21. Vieira, H. L., Haouzi, D., El Hamel, C., Jacotot, E., Belzacq, A. S.,Brenner, C., and Kroemer, G. (2000) Permeabilization of themitochondrial inner membrane during apoptosis: impact of theadenine nucleotide translocator. Cell Death Differ. 7, 1146–1154
22. Tsujimoto, Y., and Shimizu, S. (2000) VDAC regulation by theBcl-2 family of proteins. Cell Death Differ. 7, 1174–1181
23. Adachi, M., Higuchi, H., Miura, S., Azuma, T., Inokuchi, S.,Saito, H., Kato, S., and Ishii, H. (2004) Bax interacts with thevoltage-dependent anion channel and mediates ethanol-in-duced apoptosis in rat hepatocytes. Am. J. Physiol. Gastrointest.Liver Physiol. 287, G695–G705
24. Zaid, H., Abu-Hamad, S., Israelson, A., Nathan, I., and Shoshan-Barmatz, V. (2005) The voltage-dependent anion channel-1modulates apoptotic cell death. Cell Death Differ. 12, 751–760
25. Shimizu, S., Matsuoka, Y., Shinohara, Y., Yoneda, Y., and Tsuji-moto, Y. (2001) Essential role of voltage-dependent anionchannel in various forms of apoptosis in mammalian cells. J. CellBiol. 152, 237–250
26. Zheng, Y., Shi, Y., Tian, C., Jiang, C., Jin, H., Chen, J., Almasan,A., Tang, H., and Chen, Q. (2004) Essential role of thevoltage-dependent anion channel (VDAC) in mitochondrialpermeability transition pore opening and cytochrome c releaseinduced by arsenic trioxide. Oncogene 23, 1239–1247
27. Abu-Hamad, S., Sivan, S., and Shoshan-Barmatz, V. (2006) Theexpression level of the voltage-dependent anion channel con-trols life and death of the cell. Proc. Natl. Acad. Sci. U. S. A. 103,5787–5792
28. Pastorino, J. G., and Hoek, J. B. (2003) Hexokinase II: theintegration of energy metabolism and control of apoptosis. Curr.Med. Chem. 10, 1535–1551
29. Yasuda, S., Arii, S., Mori, A., Isobe, N., Yang, W., Oe, H.,Fujimoto, A., Yonenaga, Y., Sakashita, H., and Imamura, M.(2004) Hexokinase II and VEGF expression in liver tumors:correlation with hypoxia-inducible factor 1 alpha and its signif-icance. J. Hepatol. 40, 117–123
30. Kim, J. W., Gao, P., Liu, Y. C., Semenza, G. L., and Dang, C. V.(2007) Hypoxia-inducible factor 1 and dysregulated c-Myc co-operatively induce vascular endothelial growth factor and met-abolic switches hexokinase 2 and pyruvate dehydrogenase ki-nase 1. Mol. Cell. Biol. 27, 7381–7393
31. Pastorino, J. G., Shulga, N., and Hoek, J. B. (2002) Mitochon-drial binding of hexokinase II inhibits Bax-induced cytochromec release and apoptosis. J. Biol. Chem. 277, 7610–7618
32. Pastorino, J. G., Hoek, J. B., and Shulga, N. (2005) Activation ofglycogen synthase kinase 3beta disrupts the binding of hexoki-nase II to mitochondria by phosphorylating voltage-dependent
2819ENDOSTATIN INDUCES APOPTOSIS VIA VDAC1

anion channel and potentiates chemotherapy-induced cytotox-icity. Cancer Res. 65, 10545–10554
33. Sun L., Shukair S., Naik T. J., Moazed F., and Ardehali, H.(2008) Glucose phosphorylation and mitochondrial binding arerequired for the protective effects of hexokinase I and II. Mol.Cell. Biol. 28, 1007–1017.
34. Folkman, J. (2006) Antiangiogenesis in cancer therapy-endosta-tin and its mechanisms of action. Exp. Cell Res. 312, 594–607
35. Don, A. S., Kisker, O., Dilda, P., Donoghue, N., Zhao, X.,Decollogne, S., Creighton, B., Flynn, E., Folkman, J., and Hogg,P. J. (2003) A peptide trivalent arsenical inhibits tumor angio-genesis by perturbing mitochondrial function in angiogenicendothelial cells. Cancer Cell 3, 497–509
36. Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J., andMolkentin, J. D. (2007) Voltage-dependent anion channels aredispensable for mitochondrial-dependent cell death. Nat. CellBiol. 9, 487–489
37. Irani, K. (2000) Oxidant signaling in vascular cell growth, death,and survival: a review of the roles of reactive oxygen species insmooth muscle and endothelial cell mitogenic and apoptoticsignaling. Circ. Res. 87, 179–183
38. Shi, H., Huang, Y., Zhou, H., Song, X., Yuan, S., Fu, Y., and Luo,Y. (2007) Nucleolin is a receptor that mediates antiangiogenicand antitumor activity of endostatin. Blood 110, 2899–2906
39. Carmeliet, P., Dor, Y., Herbert, J. M., Fukumura, D., Brussel-mans, K., Dewerchin, M., Neeman, M., Bono, F., Abramovitch,R., Maxwell, P., Koch, C. J., Ratcliffe, P., Moons, L., Jain, R. K.,Collen, D., and Keshert, E. (1998) Role of HIF-1alpha inhypoxia-mediated apoptosis, cell proliferation and tumour an-giogenesis. Nature 394, 485–490
40. Chan, D. A., Sutphin, P. D., Denko, N. C., and Giaccia, A. J.(2002) Role of prolyl hydroxylation in oncogenically stabilizedhypoxia-inducible factor-1alpha. J. Biol. Chem. 277, 40112–40117
41. Banerjee, J., and Ghosh, S. (2006) Phosphorylation of rat brainmitochondrial voltage-dependent anion as a potential tool tocontrol leakage of cytochrome c. J. Neurochem. 98, 670–676
42. Le Mellay, V., Troppmair, J., Benz, R., and Rapp, U. R. (2002)Negative regulation of mitochondrial VDAC channels by C-Rafkinase (Online). BMC Cell Biol. 3, 14
43. Baines, C. P., Song, C. X., Zheng, Y. T., Wang, G. W., Zhang, J.,Wang, O. L., Guo, Y., Bolli, R., Cardwell, E. M., and Ping, P.(2003) Protein kinase C epsilon interacts with and inhibits thepermeability transition pore in cardiac mitochondria. Circ. Res.92, 873–880
44. Distler, A. M., Kerner, J., and Hoppel, C. L. (2007) Post-translational modifications of rat liver mitochondrial outermembrane proteins identified by mass spectrometry. Biochim.Biophys. Acta 1774, 628–636
45. Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C.,Mortensen, P., and Mann, M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127,635–648
46. Rapizzi, E., Pinton, P., Szabadkai, G., Wieckowski, M. R., Van-decasteele, G., Baird, G., Tuft, R. A., Fogarty, K. E., and Rizzuto,R. (2002) Recombinant expression of the voltage-dependentanion channel enhances the transfer of Ca2� microdomains tomitochondria. J. Cell Biol. 159, 613–624
47. Jiang, L., Jha, V., Dhanabal, M., Sukhatme, V. P., and Alper, S. L.(2001) Intracellular Ca(2�) signaling in endothelial cells by theangiogenesis inhibitors endostatin and angiostatin. Am. J.Physiol. Cell. Physiol. 280, C1140–C1150
Received for publication February 1, 2008.Accepted for publication March 6, 2008.
2820 Vol. 22 August 2008 YUAN ET AL.The FASEB Journal