endogenous endostatin inhibits choroidal neovascularization
TRANSCRIPT

The FASEB Journal • Research Communication
Endogenous endostatin inhibits choroidalneovascularization
Alexander G. Marneros,*,1 Haicheng She,† Hadi Zambarakji,† Hiroya Hashizume,‡
Edward J. Connolly,† Ivana Kim,† Evangelos S. Gragoudas,† Joan W. Miller,†
and Bjorn R. Olsen**Department of Developmental Biology, Harvard School of Dental Medicine, and Department ofCell Biology, Harvard Medical School, Boston, Massachusetts, USA; †Angiogenesis Laboratory andRetina Service, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts,USA; and ‡Division of Microscopic Anatomy and Bioimaging, Niigata University Postgraduate Schoolof Medical and Dental Sciences, Niigata City, Japan
ABSTRACT Endostatin, a fragment of the basementmembrane component collagen XVIII, exhibits antian-giogenic properties in vitro and in vivo when high dosesare administered. It is not known whether endogenousendostatin at physiological levels has a protective roleas an inhibitor of pathological angiogenesis, such aschoroidal neovascularization (CNV) in age-related mac-ular degeneration. Using a laser injury model, weinduced CNV in mice lacking collagen XVIII/endosta-tin and in control mice. CNV lesions in mutant micewere �3-fold larger than in control mice and showedincreased vascular leakage. These differences wereindependent of age-related changes at the choroid-retina interface. Ultrastructural analysis of the choroi-dal vasculature in mutant mice excluded morphologicalvascular abnormalities as a cause for the larger CNVlesions. When recombinant endostatin was adminis-tered to collagen XVIII/endostatin-deficient mice,CNV lesions were similar to those seen in control mice.In control mice treated with recombinant endostatin,CNV lesions were almost undetectable. These findingsdemonstrate that endogenous endostatin is an inhibitorof induced angiogenesis and that administration ofendostatin potently inhibits CNV growth and vascularleakage. Endostatin may have a regulatory role in thepathogenesis of CNV and could be used therapeuticallyto inhibit growth and leakage of CNV lesions.—Marn-eros, A. G., She, H., Zambarakji, H., Hashizume, H.,Connolly, E. J., Kim, I., Gragoudas, E. S., Miller, J. W.,Olsen, B. R. Endogenous endostatin inhibits choroidalneovascularization. FASEB J. 21, 3809–3818 (2007)
Key Words: angiogenesis � collagen XVIII � Bruch’s membrane� age-related macular degeneration
Choroidal neovascularization (cnv) is the hall-mark of the exudative form of age-related maculardegeneration (AMD), in which new vessels from thechoroidal vasculature penetrate Bruch’s membraneand extend into the subretinal space or into the planeof the retinal pigment epithelium (RPE). CNV maylead to RPE detachment, subretinal or intraretinal
hemorrhage, or fibrovascular scarring and accounts forthe majority of vision loss associated with AMD. It hasbeen proposed that CNV is induced by focally increasedlevels of proangiogenic factors at the RPE/Bruch’smembrane-choroid complex and/or by a decrease ofantiangiogenic factors. A major proangiogenic factorexpressed by RPE cells and macrophages is vascularendothelial growth factor (VEGF), which is also in-duced by hypoxia and up-regulated during the forma-tion of experimental CNV (1–7). Consequently, severalexperimental approaches to target CNV therapeuticallyhave utilized inhibitors of VEGF expression or activity,and these approaches have resulted in significant clin-ical benefit in patients with CNV (8–10). In contrast,hypoxia down-regulates some antiangiogenic factors,such as pigment epithelium-derived factor (PEDF), andthus may promote new vessel formation (11, 12).
The primary causes of such a proposed imbalance ofstimulators and inhibitors of angiogenesis in the patho-genesis of CNV remain largely unknown. Age-relatedchanges of Bruch’s membrane and abnormal sub-RPEdeposit formation may promote inflammation andcause abnormalities in RPE cell metabolism that lead toalterations in the transcriptional regulation of proan-giogenic or antiangiogenic factors.
In the process of choroidal neovascularization, thevascular overgrowth of choroidal vessels is coupled witha localized proteolytic degradation of Bruch’s mem-brane and penetration into the subretinal space. Thisproteolytic degradation of the basement membrane isbelieved to result in release of antiangiogenic frag-ments derived from basement membrane componentsthat may act to counterbalance excessive neovascular-ization locally (13). Thus, in addition to pro- andantiangiogenic factors expressed by RPE cells or inflam-matory cells at the site of CNV lesion formation, releaseof antiangiogenic proteolytic fragments from basement
1Correspondence: Department of Developmental Biology,Harvard School of Dental Medicine, 188 Longwood Ave., Bos-ton, MA 02115, USA. E-mail: [email protected]
doi: 10.1096/fj.07-8422com
38090892-6638/07/0021-3809 © FASEB

membrane components is thought to contribute to theextent of CNV formation as well.
A major constituent of Bruch’s membrane and thebasement membranes of the choroidal vessels is colla-gen XVIII (14). This collagen is a heparan sulfateproteoglycan of vascular and epithelial basement mem-branes. Although it is found in almost all basementmembranes, a particular requirement of collagenXVIII/endostatin for the eye has been shown by theidentification of ocular abnormalities in mice andhumans that either lack or have reduced expression ofcollagen XVIII (14–19). A proteolytic fragment of theC-terminal domain of collagen XVIII, termed en-dostatin, has been shown to possess antiangiogenicactivity in vitro and in vivo when administered at highdoses as a recombinant protein (20, 21). Based onthese findings, it has been suggested that collagenXVIII/endostatin may function as a physiologicalregulator of angiogenesis in vivo (17). However, arole for physiological levels of endostatin in regulat-ing angiogenesis in vivo has not yet been shown. Lackof physiological concentrations of this collagen or itsproteolytic fragment endostatin in Col18a1�/� micedoes not lead to obvious vascular defects during devel-opment or postnatal growth (15), indicating that colla-gen XVIII/endostatin does not function as a criticalnegative regulator of angiogenesis. This collagen maybe only one of many contributing factors involved inthe dynamic interaction of proangiogenic and antian-giogenic components that regulate the complex pro-cess of new vessel formation, in which other regulatorsof angiogenesis may compensate for its absence. How-ever, experiments with aortic explant cultures derivedfrom Col18a1�/� mice showed increased vascular out-growth compared with aortas derived from controllittermates (22). Thus, a sensitive assay that allows forprecise quantitation of neoangiogenesis in vivo mayreveal a role for physiological levels of collagen XVIII/endostatin in regulating induced vessel growth. There-fore, we induced CNV lesions with a diode laser photo-coagulator in Col18a1�/� mice and control littermates.Experimental CNV shows induced neoangiogenesis ina precise anatomic location with a clearly defined areaof vascular remodeling, allowing for a precise quantita-tion of vessel growth. Although CNV lesions are in-duced experimentally in this acute injury model, theyclosely resemble neovascular lesions in the chronicdisease AMD, and this acute laser injury model hasbeen established as a valid surrogate model for neovas-cular AMD that has helped reveal several aspects ofCNV pathogenesis.
Our data demonstrate a significant increase in thesize of experimental CNV lesions in Col18a1�/� mice,with lesions forming large confluent areas; in contrast,such lesions remained small and clearly circumscribedin control mice. Ultrastructural analysis of the choroi-dal vasculature in mutant mice excluded the possibilitythat morphological defects in these vessels caused theincrease in the size of CNV lesions. Furthermore, theincrease of CNV lesion size observed in mutant mice
was independent of age-related changes at the RPE-choroid interface. Notably, lack of collagen XVIII/endostatin also led to an increase in vascular perme-ability of CNV lesions, as assessed by fluoresceinangiography. Administration of recombinant endosta-tin to Col18a1�/� mice rescued the observed pheno-type, and CNV lesions were comparable to those seen incontrol mice. Notably, administration of recombinantendostatin to control mice resulted in CNV lesions thatwere hardly detectable.
These findings suggest that endogenous endostatin isa physiological inhibitor of induced angiogenesis invivo and may act as a regulator of CNV growth in AMD.Endostatin’s antiangiogenic properties could be usedtherapeutically to target CNV growth and vascularleakage from these lesions. Col18a1�/� mice are thefirst mouse model to show increased CNV formationdue to the absence of a single basement membranecomponent, and the data support the hypothesis thatreleased antiangiogenic proteolytic fragments frombasement membranes play a regulatory role duringpathological angiogenesis, such as in CNV formation.
MATERIALS AND METHODS
Animals
The generation and genotyping of Col18a1�/� mice havealready been described (15). The absence of collagen XVIIIexpression in Col18a1�/� mice was confirmed by a lack ofimmunostaining for collagen XVIII and endostatin usingpolyclonal antibodies, with positive staining in control eyes(14, 15). Uniformity of genetic backgrounds of control (wild-type, or WT) and Col18a1�/� (KO) mice was ensuredthrough backcrossing Col18a1�/� mice onto the C57BL/6Jbackground over 15 generations. Mice were fed a regularchow diet. For all procedures, anesthesia was achieved by i.p.injection of 100 mg/kg body weight ketamine hydrochloride(Abbott, North Chicago, IL, USA) and 10 mg/kg body weightxylazine (Bayer, Shawnee Mission, KS, USA); pupils weredilated with topical 0.5% tropicamide (Alcon, Humacao,Puerto Rico). All animal experiments were in accordancewith protocols reviewed and approved by the animal carecommittees of the Massachusetts Eye and Ear Infirmary andHarvard Medical School, and in accordance with the Associ-ation for Research in Vision and Ophthalmology Statementfor the Use of Animals in Ophthalmic and Vision Research.
Experimental CNV model
Eyes of age- and gender-matched Col18a1�/� mice andcontrol C57BL/6J littermates were exposed to laser photoco-agulation for induction of experimental CNV. Laser photo-coagulation was performed using a diode-pumped, frequency-doubled 532 nm laser (Oculight GLx Laser System, IRIDEXCorporation, Mountain View, CA, USA) attached to a slitlamp, and a coverslip was applied to the cornea to view theretina. Four lesions were induced using a power of 200 mW,a spot size of 50 �m, and a duration of 100 ms. The lesionswere located at the 3, 6, 9, and 12 o’clock meridians centeredon the optic nerve and located two or three disc diametersfrom the optic nerve. Laser-induced disruption of Bruch’smembrane was identified by the appearance of a bubble at
3810 Vol. 21 December 2007 MARNEROS ET AL.The FASEB Journal

the site of photocoagulation. Laser spots that did not result inthe formation of a bubble were excluded from the studies.
Fluorescein angiography
Two weeks after laser photocoagulation, fluorescein angiog-raphy (FA) was undertaken by an operator masked to thegenetic identity of the animals. FA was performed in anesthe-tized animals with dilated pupils using a digital funduscamera (Model TRC 50 IA; Topcon, Paramus, NJ, USA) andstandard fluorescein filters. Fluorescein injections were ad-ministered by i.p. injection (0.2 ml of 2% fluorescein sodium;Akorn Inc., Decatur, IL, USA) and the timer was started assoon as the fluorescein bolus was injected. A PMMA contactlens (base curve 1.65 mm, power 7.0 D, size 2.5 mm; UniconCorporation, Osaka, Japan) was placed on the mouse corneato improve visualization and prevent corneal drying. Twomasked retina specialists not involved in laser photocoagula-tion or angiography evaluated the fluorescein angiograms ata single sitting, and lesions were graded. Grade 0 lesions hadno hyperfluorescence. Grade 1 lesions exhibited hyperfluo-rescence without leakage. Grade 2A lesions exhibited hyper-fluorescence in the early or midtransit images and lateleakage. Grade 2B lesions showed bright hyperfluorescencein the transit images and late leakage beyond treated areas(grade 2B lesions were defined as clinically significant). Therelative distribution of FA grades for CNV lesions was deter-mined within each experimental group of mice. For conflu-ent CNV lesions, the same FA grade was assigned for alllesions within the confluent hyperfluorescent area.
Quantitation of the size of CNV lesions
Two weeks after laser injury, the size of CNV lesions wasmeasured using choroidal flat mounts. Mice were anesthe-tized and perfused through the left ventricle with a 30 gaugeblunt canula with 8 ml of lactated Ringer solution, followedby 5 ml of 5 mg/ml fluorescein-labeled dextran in 10%gelatin (2�106 MW; Sigma, St. Louis, MO, USA). The eyeswere enucleated and fixed in 4% formalin for 24 h. Theanterior segment and the lens were removed. The retina wasseparated from the underlying RPE and four relaxing radialincisions were made. The remaining RPE/choroid/scleracomplex was flat mounted (Immu-Mount Vectashield Mount-ing Medium; Vector Laboratories, Burlingame, CA, USA) andcoverslipped.
Choroidal flat mounts were analyzed by fluorescent micros-copy using a Leica DMR microscope (Leica Microsystems,Wetzlar, Germany) equipped with epifluorescence illumina-tion. Openlab software (Improvision, Boston, MA, USA) wasused to measure the magnitude of the hyperfluorescent areascorresponding to CNV.
Administration of recombinant endostatin
Human recombinant endostatin expressed in Pichia pastoriswas administered at a dose of 0.5 mg/kg/day via i.p. injec-tions for 14 days (U.S. Biological, Swampscott, MA, USA).Age- and gender-matched control mice received PBS instead.The first dose was given on the day of laser photocoagulation;the last dose was administered 14 days later, prior to prepa-ration of the choroidal flat mounts. This dose was calculatedto result in endostatin serum levels that approximate physio-logical serum levels (23).
Scanning electron microscopy
The fixative for scanning electron microscopy (SEM) con-tained 2% glutaraldehyde in 100 mmol/L phosphate buffer.
Eyes were enucleated and immersed in fixative for 2 days,then processed for SEM. Subsequently, eyes were treated with30% potassium hydroxide in distilled water for 8 min at 60°Cto remove the extracellular matrix around blood vessels. Thespecimens were stained with a solution of 2% tannic acid and1% osmium tetroxide, dehydrated with a graded series ofethanol, transferred to isoamyl acetate, and critical point-dried in liquid CO2. Some areas were cracked with fineforceps under a dissection microscope to expose blood vesselswithin the specimen. Dried specimens were put on aluminumstubs, coated with osmium tetroxide in an Osmium PlasmaCoater (Vacuum Device Corp, Japan), and examined with aHitachi S-4300N scanning electron microscope (24).
Immunohistochemistry
For immunohistochemical analysis, whole eyes were fixed in4% paraformaldehyde, followed by infiltration in 30% su-crose. Eyes were embedded in OCT (Tissue-Tek, Sakura,Japan) and 7 �m cryostat sections were made. Sections wereincubated with a monoclonal rat anti-mouse CD31 (BDPharMingen, San Diego, CA, USA) or a monoclonal ratantiperlecan (Lab Vision, Fremont, CA, USA) antibody. Sec-tions were incubated with FITC-conjugated secondary IgGantibodies (Vector Laboratories) and a mounting mediumcontaining DAPI was used (Vector Laboratories). For histo-logical analysis, sections were stained with hematoxylin andeosin according to standard procedures.
Electron and light microscopy
For morphological examination of CNV lesions, eyes wereenucleated and fixed for 24 h in 2.5% formaldehyde and1.5% glutaraldehyde in 0.1M cacodylate buffer (pH 7.4).After postfixation in 4% osmium tetroxide and dehydrationsteps, the eyes were embedded in TAAB epon (Marivac, Inc.,St. Laurent, Quebec, Canada) overnight. For light micros-copy, serial sections of 0.5 �m were stained with toluidin blueor azure II, and 85 nm-thin sections were used for standardtransmission electron microscopy.
Statistical analysis
A two-sample Student’s t test with unequal variance was usedfor statistical analyses of the quantitative CNV flat mount data.Statistical significance was set at P � 0.05. The hyperfluores-cent areas of CNV lesions in each eye were measured, and thetotal area of CNV lesions per eye was determined as their sum.For confluent lesions, the entire hyperfluorescent area wasmeasured and the number of initial CNV lesions withinconfluent lesions was counted.
RESULTS
Normal development and ultrastructural morphologyof choroidal vasculature in adult Col18a1�/� mice
Using immuno-EM labeling with polyclonal antibodiesto collagen XVIII (N-terminal noncollagenous domainNC11) and endostatin, we previously demonstratedthat collagen XVIII/endostatin is present in the cho-roidal endothelial basement membranes and in thebasement membrane of the RPE (14). Here we per-formed ultrastructural analysis of the choroidal vascu-lature by scanning EM, and found no significant differ-
3811ENDOSTATIN INHIBITS CHOROIDAL NEOVASCULARIZATION
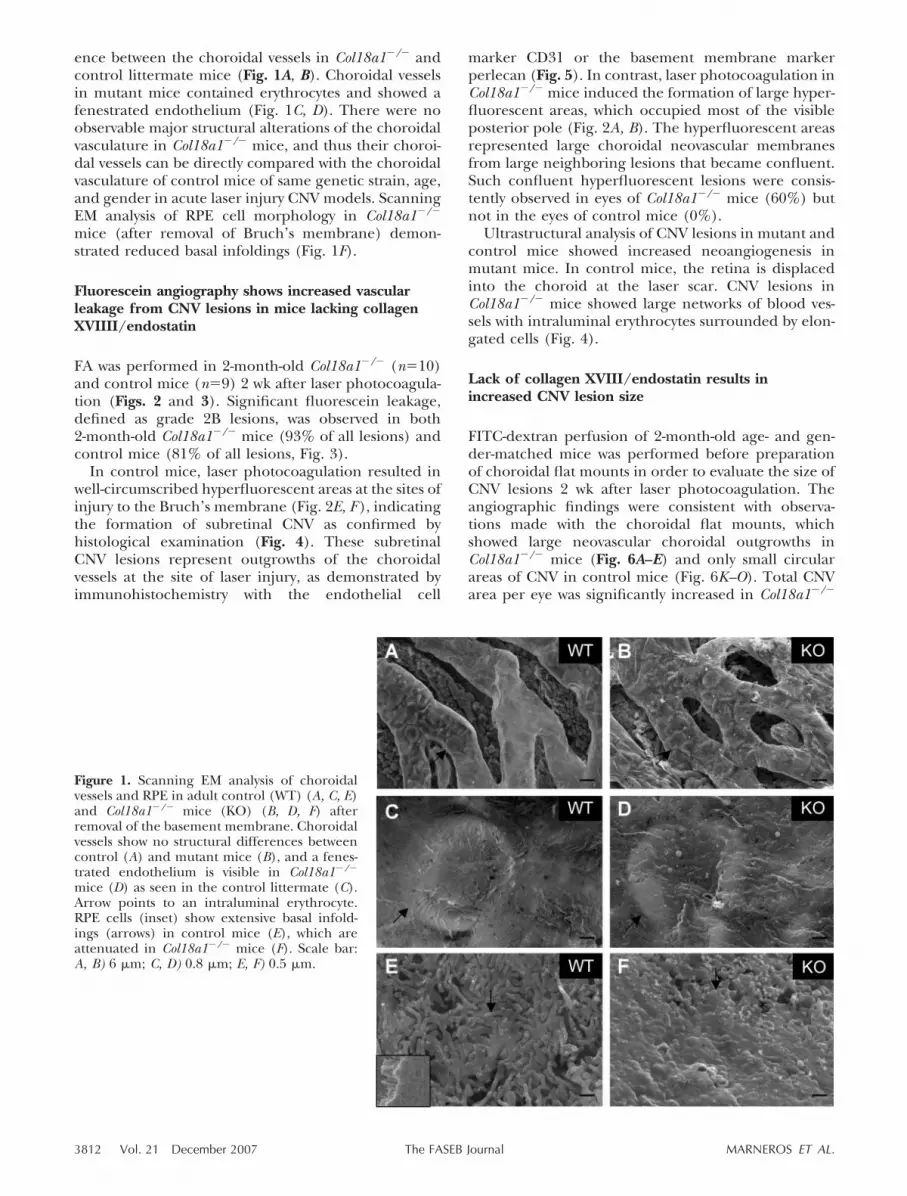
ence between the choroidal vessels in Col18a1�/� andcontrol littermate mice (Fig. 1A, B). Choroidal vesselsin mutant mice contained erythrocytes and showed afenestrated endothelium (Fig. 1C, D). There were noobservable major structural alterations of the choroidalvasculature in Col18a1�/� mice, and thus their choroi-dal vessels can be directly compared with the choroidalvasculature of control mice of same genetic strain, age,and gender in acute laser injury CNV models. ScanningEM analysis of RPE cell morphology in Col18a1�/�
mice (after removal of Bruch’s membrane) demon-strated reduced basal infoldings (Fig. 1F).
Fluorescein angiography shows increased vascularleakage from CNV lesions in mice lacking collagenXVIIII/endostatin
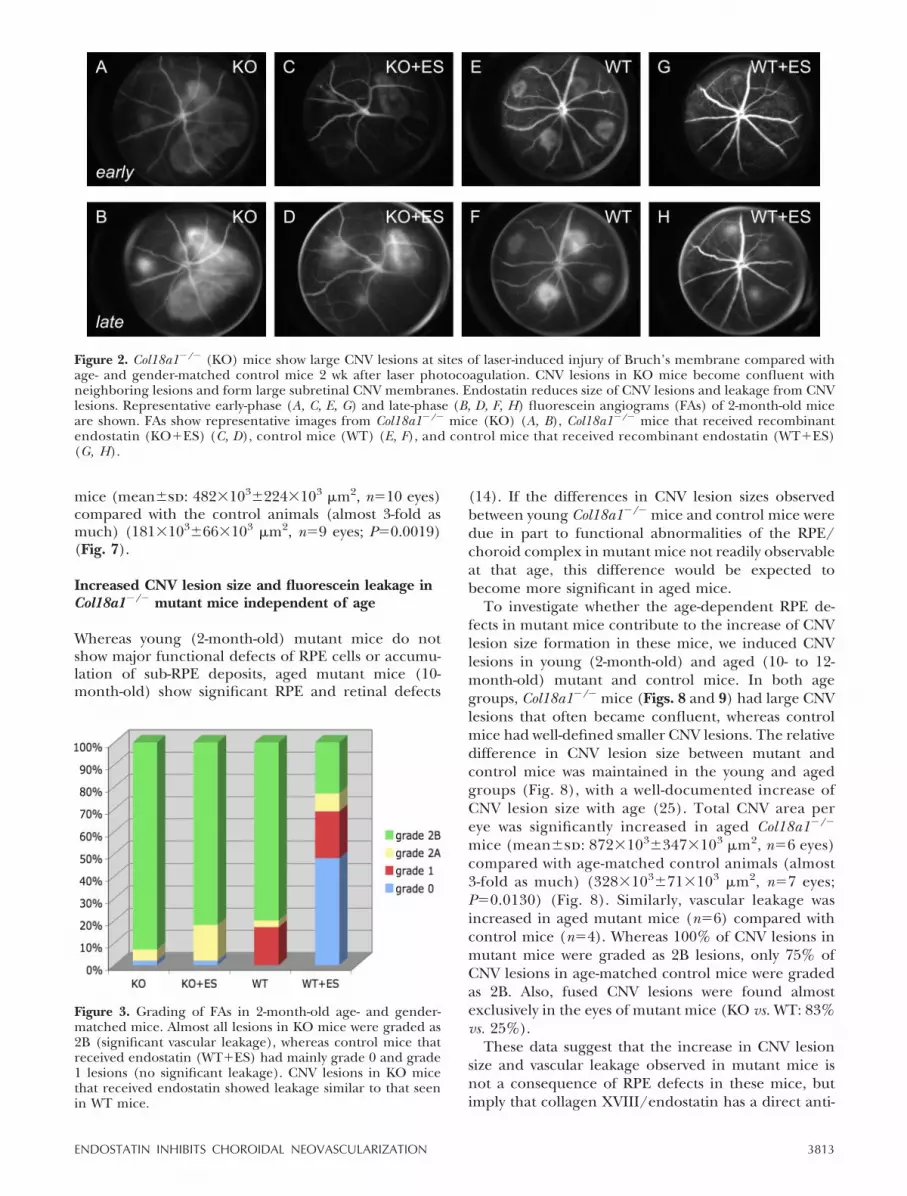
FA was performed in 2-month-old Col18a1�/� (n�10)and control mice (n�9) 2 wk after laser photocoagula-tion (Figs. 2 and 3). Significant fluorescein leakage,defined as grade 2B lesions, was observed in both2-month-old Col18a1�/� mice (93% of all lesions) andcontrol mice (81% of all lesions, Fig. 3).
In control mice, laser photocoagulation resulted inwell-circumscribed hyperfluorescent areas at the sites ofinjury to the Bruch’s membrane (Fig. 2E, F ), indicatingthe formation of subretinal CNV as confirmed byhistological examination (Fig. 4). These subretinalCNV lesions represent outgrowths of the choroidalvessels at the site of laser injury, as demonstrated byimmunohistochemistry with the endothelial cell
marker CD31 or the basement membrane markerperlecan (Fig. 5). In contrast, laser photocoagulation inCol18a1�/� mice induced the formation of large hyper-fluorescent areas, which occupied most of the visibleposterior pole (Fig. 2A, B). The hyperfluorescent areasrepresented large choroidal neovascular membranesfrom large neighboring lesions that became confluent.Such confluent hyperfluorescent lesions were consis-tently observed in eyes of Col18a1�/� mice (60%) butnot in the eyes of control mice (0%).
Ultrastructural analysis of CNV lesions in mutant andcontrol mice showed increased neoangiogenesis inmutant mice. In control mice, the retina is displacedinto the choroid at the laser scar. CNV lesions inCol18a1�/� mice showed large networks of blood ves-sels with intraluminal erythrocytes surrounded by elon-gated cells (Fig. 4).
Lack of collagen XVIII/endostatin results inincreased CNV lesion size
FITC-dextran perfusion of 2-month-old age- and gen-der-matched mice was performed before preparationof choroidal flat mounts in order to evaluate the size ofCNV lesions 2 wk after laser photocoagulation. Theangiographic findings were consistent with observa-tions made with the choroidal flat mounts, whichshowed large neovascular choroidal outgrowths inCol18a1�/� mice (Fig. 6A–E) and only small circularareas of CNV in control mice (Fig. 6K–O). Total CNVarea per eye was significantly increased in Col18a1�/�
Figure 1. Scanning EM analysis of choroidalvessels and RPE in adult control (WT) (A, C, E)and Col18a1�/� mice (KO) (B, D, F) afterremoval of the basement membrane. Choroidalvessels show no structural differences betweencontrol (A) and mutant mice (B), and a fenes-trated endothelium is visible in Col18a1�/�
mice (D) as seen in the control littermate (C).Arrow points to an intraluminal erythrocyte.RPE cells (inset) show extensive basal infold-ings (arrows) in control mice (E), which areattenuated in Col18a1�/� mice (F). Scale bar:A, B) 6 �m; C, D) 0.8 �m; E, F) 0.5 �m.
3812 Vol. 21 December 2007 MARNEROS ET AL.The FASEB Journal
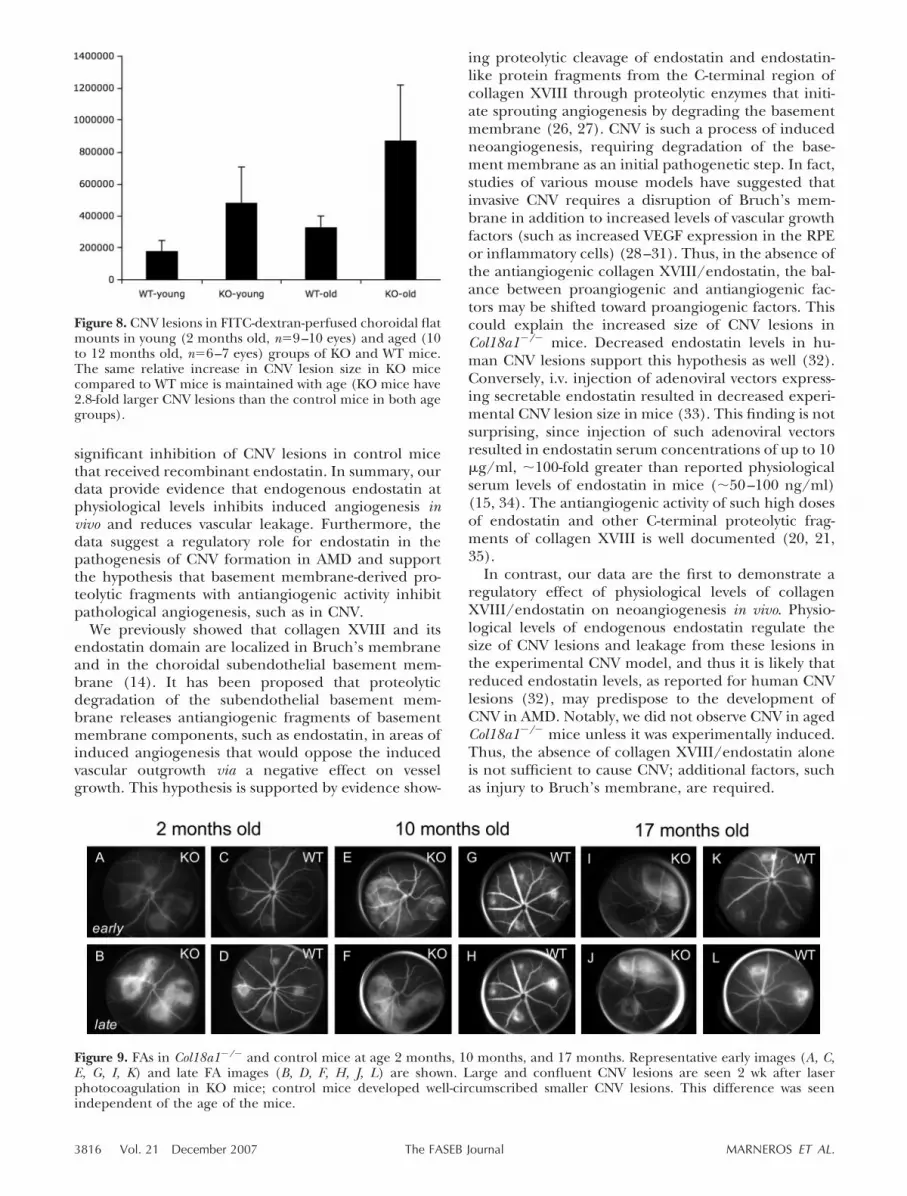
mice (mean�sd: 482�103�224�103 �m2, n�10 eyes)compared with the control animals (almost 3-fold asmuch) (181�103�66�103 �m2, n�9 eyes; P�0.0019)(Fig. 7).
Increased CNV lesion size and fluorescein leakage inCol18a1�/� mutant mice independent of age
Whereas young (2-month-old) mutant mice do notshow major functional defects of RPE cells or accumu-lation of sub-RPE deposits, aged mutant mice (10-month-old) show significant RPE and retinal defects
(14). If the differences in CNV lesion sizes observedbetween young Col18a1�/� mice and control mice weredue in part to functional abnormalities of the RPE/choroid complex in mutant mice not readily observableat that age, this difference would be expected tobecome more significant in aged mice.
To investigate whether the age-dependent RPE de-fects in mutant mice contribute to the increase of CNVlesion size formation in these mice, we induced CNVlesions in young (2-month-old) and aged (10- to 12-month-old) mutant and control mice. In both agegroups, Col18a1�/� mice (Figs. 8 and 9) had large CNVlesions that often became confluent, whereas controlmice had well-defined smaller CNV lesions. The relativedifference in CNV lesion size between mutant andcontrol mice was maintained in the young and agedgroups (Fig. 8), with a well-documented increase ofCNV lesion size with age (25). Total CNV area pereye was significantly increased in aged Col18a1�/�
mice (mean�sd: 872�103�347�103 �m2, n�6 eyes)compared with age-matched control animals (almost3-fold as much) (328�103�71�103 �m2, n�7 eyes;P�0.0130) (Fig. 8). Similarly, vascular leakage wasincreased in aged mutant mice (n�6) compared withcontrol mice (n�4). Whereas 100% of CNV lesions inmutant mice were graded as 2B lesions, only 75% ofCNV lesions in age-matched control mice were gradedas 2B. Also, fused CNV lesions were found almostexclusively in the eyes of mutant mice (KO vs. WT: 83%vs. 25%).
These data suggest that the increase in CNV lesionsize and vascular leakage observed in mutant mice isnot a consequence of RPE defects in these mice, butimply that collagen XVIII/endostatin has a direct anti-
Figure 2. Col18a1�/� (KO) mice show large CNV lesions at sites of laser-induced injury of Bruch’s membrane compared withage- and gender-matched control mice 2 wk after laser photocoagulation. CNV lesions in KO mice become confluent withneighboring lesions and form large subretinal CNV membranes. Endostatin reduces size of CNV lesions and leakage from CNVlesions. Representative early-phase (A, C, E, G) and late-phase (B, D, F, H) fluorescein angiograms (FAs) of 2-month-old miceare shown. FAs show representative images from Col18a1�/� mice (KO) (A, B), Col18a1�/� mice that received recombinantendostatin (KO�ES) (C, D), control mice (WT) (E, F), and control mice that received recombinant endostatin (WT�ES)(G, H).
Figure 3. Grading of FAs in 2-month-old age- and gender-matched mice. Almost all lesions in KO mice were graded as2B (significant vascular leakage), whereas control mice thatreceived endostatin (WT�ES) had mainly grade 0 and grade1 lesions (no significant leakage). CNV lesions in KO micethat received endostatin showed leakage similar to that seenin WT mice.
3813ENDOSTATIN INHIBITS CHOROIDAL NEOVASCULARIZATION

angiogenic activity that inhibits CNV lesion size andvascular leakage.
Endostatin inhibits CNV formationand fluorescein leakage
To determine whether the increased CNV lesions ob-served in mutant mice are the result of the lack ofcollagen XVIII as a structural basement membranecomponent or instead due to the lack of the antiangio-genic activity of its endostatin domain, we administeredrecombinant endostatin to mutant mice to achieveserum concentrations approximating physiological se-rum levels of endostatin. In this rescue experiment, thetotal CNV area per eye in Col18a1�/� mice that re-ceived endostatin was significantly smaller than inmutant mice, which received no endostatin (PBS) (KO:mean�sd: 482�103�224�103 �m2, n�10 eyes; KO�ES:
204�103�92�103 �m2, n�12 eyes; P�0.0031); thetotal CNV area per eye was almost the same size asseen in WT mice, with no detectable significantstatistical difference between the two groups(KO�ES: mean�sd: 203�103�92�103 �m2, n�12eyes; WT: 181�103�66�103 �m2, n�9 eyes;P�0.5231) (Fig. 7). Administration of recombinantendostatin to WT mice potently inhibited CNV lesionformation (WT: mean�sd: 181�103�66�103 �m2,n�10 eyes; WT�ES: 53�103�38�103 �m2, n�12eyes; P�0.0002) (Fig. 7). Thus, endostatin adminis-tration rescues the CNV phenotype in mutant mice.The data suggest that the antiangiogenic activity ofthe endostatin domain inhibits CNV lesion size.
Similarly, endostatin administration to mutant micereduced vascular leakage from CNV lesions to the amountseen in control mice. Furthermore, endostatin acted as apotent inhibitor of CNV leakage even when administered
Figure 4. CNV lesions in 2-month-old Col18a1�/� and control mice. A representative CNV lesion is shown in control (A, C, E,G) and Col18a1�/� (B, D, F, H) mice. A, B) Control mice have small well-circumscribed CNV lesions, but mutant mice formlarger CNV lesions (white arrows). C) In control mice the retina is displaced into the choroid at the laser scar (white arrows).CNV lesions in mutant mice show numerous vessels with intraluminal erythrocytes (white arrow in panel H), surrounded byelongated cells (F, H, black arrows; black arrows in panel D show RPE cell layer). EM images from the corresponding CNVlesions in panels A, B are shown in panels E–H. E, G) In control mice the CNV lesion shows at its borders abnormal pigmentedcells (black arrow) and, at its center, loss of photoreceptor outer segments. White arrows indicate the RPE cell layer. Blackarrowheads show endothelial cells of the choroid layer (E). F) Elongated endothelial cells (black arrows) are found between theRPE cell layer (white arrow) and the photoreceptor layer (*) in Col18a1�/� mice. *Inner segments of photoreceptors.Magnification: A, B) �20; C, D) �60. Bar in EM images: E) 10 �m; F, G) 5 �m; H) 2.5 �m.
Figure 5. Increased CNV lesion size in Col18a1�/� mice 2 wk after laser photocoagulation. A) A small CNV lesion (indicated bywhite arrowheads) in a choroidal flat mount of a 10-month-old control mouse visualized with FITC-dextran. B) A large confluentchoroidal neovascular membrane (indicated by white arrowheads) in a choroidal flat mount of a littermate Col18a1�/� mouse.Scale, 190 �m. Areas of hyperfluoresence in choroidal flat mounts correspond to subretinal neovascularization, as demonstratedby FITC immunostaining using monoclonal antibodies against perlecan (C) and CD31 (D) (indicated by white arrowheads). C,D) Original magnification, �10.
3814 Vol. 21 December 2007 MARNEROS ET AL.The FASEB Journal
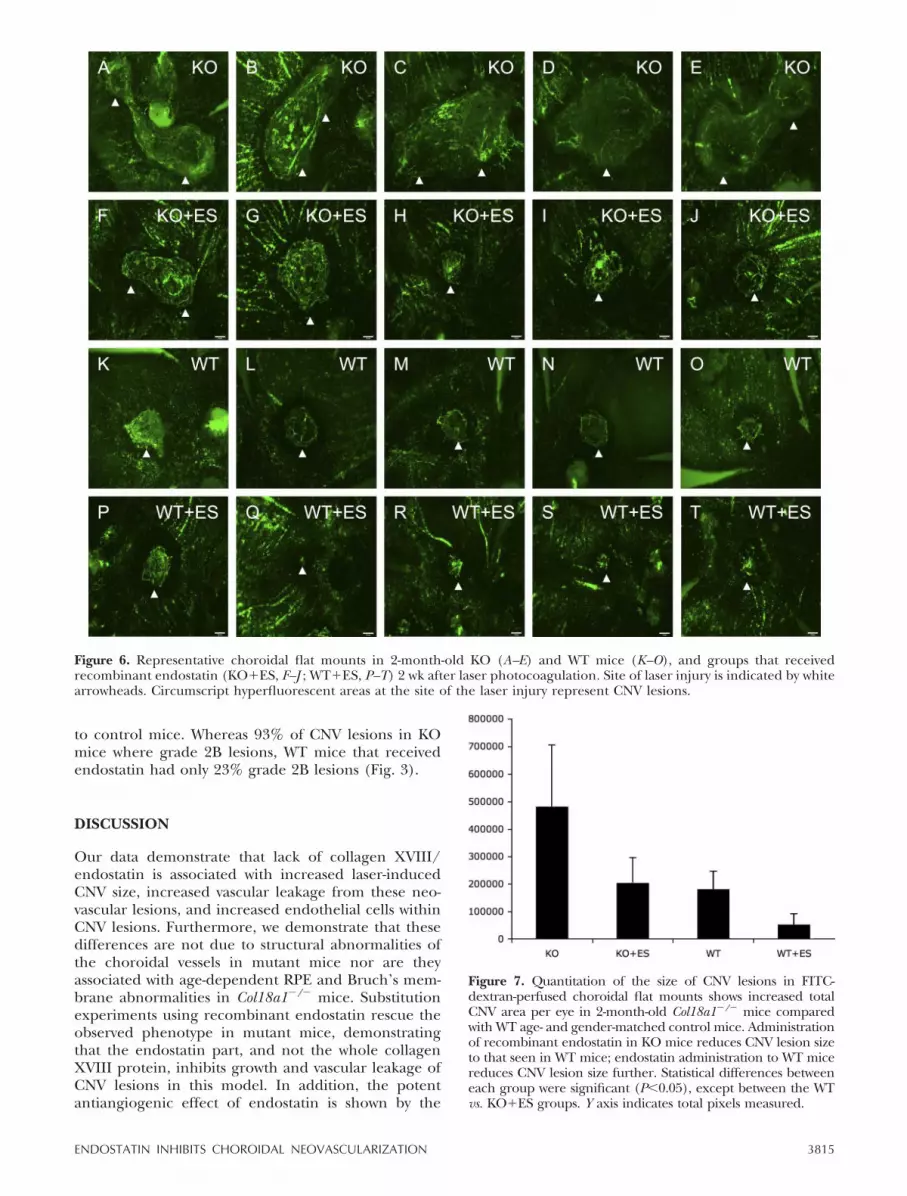
to control mice. Whereas 93% of CNV lesions in KOmice where grade 2B lesions, WT mice that receivedendostatin had only 23% grade 2B lesions (Fig. 3).
DISCUSSION
Our data demonstrate that lack of collagen XVIII/endostatin is associated with increased laser-inducedCNV size, increased vascular leakage from these neo-vascular lesions, and increased endothelial cells withinCNV lesions. Furthermore, we demonstrate that thesedifferences are not due to structural abnormalities ofthe choroidal vessels in mutant mice nor are theyassociated with age-dependent RPE and Bruch’s mem-brane abnormalities in Col18a1�/� mice. Substitutionexperiments using recombinant endostatin rescue theobserved phenotype in mutant mice, demonstratingthat the endostatin part, and not the whole collagenXVIII protein, inhibits growth and vascular leakage ofCNV lesions in this model. In addition, the potentantiangiogenic effect of endostatin is shown by the
Figure 6. Representative choroidal flat mounts in 2-month-old KO (A–E) and WT mice (K–O), and groups that receivedrecombinant endostatin (KO�ES, F–J ; WT�ES, P–T) 2 wk after laser photocoagulation. Site of laser injury is indicated by whitearrowheads. Circumscript hyperfluorescent areas at the site of the laser injury represent CNV lesions.
Figure 7. Quantitation of the size of CNV lesions in FITC-dextran-perfused choroidal flat mounts shows increased totalCNV area per eye in 2-month-old Col18a1�/� mice comparedwith WT age- and gender-matched control mice. Administrationof recombinant endostatin in KO mice reduces CNV lesion sizeto that seen in WT mice; endostatin administration to WT micereduces CNV lesion size further. Statistical differences betweeneach group were significant (P�0.05), except between the WTvs. KO�ES groups. Y axis indicates total pixels measured.
3815ENDOSTATIN INHIBITS CHOROIDAL NEOVASCULARIZATION
significant inhibition of CNV lesions in control micethat received recombinant endostatin. In summary, ourdata provide evidence that endogenous endostatin atphysiological levels inhibits induced angiogenesis invivo and reduces vascular leakage. Furthermore, thedata suggest a regulatory role for endostatin in thepathogenesis of CNV formation in AMD and supportthe hypothesis that basement membrane-derived pro-teolytic fragments with antiangiogenic activity inhibitpathological angiogenesis, such as in CNV.
We previously showed that collagen XVIII and itsendostatin domain are localized in Bruch’s membraneand in the choroidal subendothelial basement mem-brane (14). It has been proposed that proteolyticdegradation of the subendothelial basement mem-brane releases antiangiogenic fragments of basementmembrane components, such as endostatin, in areas ofinduced angiogenesis that would oppose the inducedvascular outgrowth via a negative effect on vesselgrowth. This hypothesis is supported by evidence show-
ing proteolytic cleavage of endostatin and endostatin-like protein fragments from the C-terminal region ofcollagen XVIII through proteolytic enzymes that initi-ate sprouting angiogenesis by degrading the basementmembrane (26, 27). CNV is such a process of inducedneoangiogenesis, requiring degradation of the base-ment membrane as an initial pathogenetic step. In fact,studies of various mouse models have suggested thatinvasive CNV requires a disruption of Bruch’s mem-brane in addition to increased levels of vascular growthfactors (such as increased VEGF expression in the RPEor inflammatory cells) (28–31). Thus, in the absence ofthe antiangiogenic collagen XVIII/endostatin, the bal-ance between proangiogenic and antiangiogenic fac-tors may be shifted toward proangiogenic factors. Thiscould explain the increased size of CNV lesions inCol18a1�/� mice. Decreased endostatin levels in hu-man CNV lesions support this hypothesis as well (32).Conversely, i.v. injection of adenoviral vectors express-ing secretable endostatin resulted in decreased experi-mental CNV lesion size in mice (33). This finding is notsurprising, since injection of such adenoviral vectorsresulted in endostatin serum concentrations of up to 10�g/ml, �100-fold greater than reported physiologicalserum levels of endostatin in mice (�50–100 ng/ml)(15, 34). The antiangiogenic activity of such high dosesof endostatin and other C-terminal proteolytic frag-ments of collagen XVIII is well documented (20, 21,35).
In contrast, our data are the first to demonstrate aregulatory effect of physiological levels of collagenXVIII/endostatin on neoangiogenesis in vivo. Physio-logical levels of endogenous endostatin regulate thesize of CNV lesions and leakage from these lesions inthe experimental CNV model, and thus it is likely thatreduced endostatin levels, as reported for human CNVlesions (32), may predispose to the development ofCNV in AMD. Notably, we did not observe CNV in agedCol18a1�/� mice unless it was experimentally induced.Thus, the absence of collagen XVIII/endostatin aloneis not sufficient to cause CNV; additional factors, suchas injury to Bruch’s membrane, are required.
Figure 8. CNV lesions in FITC-dextran-perfused choroidal flatmounts in young (2 months old, n�9–10 eyes) and aged (10to 12 months old, n�6–7 eyes) groups of KO and WT mice.The same relative increase in CNV lesion size in KO micecompared to WT mice is maintained with age (KO mice have2.8-fold larger CNV lesions than the control mice in both agegroups).
Figure 9. FAs in Col18a1�/� and control mice at age 2 months, 10 months, and 17 months. Representative early images (A, C,E, G, I, K) and late FA images (B, D, F, H, J, L) are shown. Large and confluent CNV lesions are seen 2 wk after laserphotocoagulation in KO mice; control mice developed well-circumscribed smaller CNV lesions. This difference was seenindependent of the age of the mice.
3816 Vol. 21 December 2007 MARNEROS ET AL.The FASEB Journal

In addition to the increased growth of CNV lesions inmutant mice, we also observed increased vascular leak-age from these lesions in Col18a1�/� mice, which wasinhibited by administration of recombinant endostatin.This finding suggests that endostatin inhibits vascularleakage directly and is consistent with previous obser-vations showing that endostatin inhibits VEGF-inducedretinal vascular permeability, possibly through an up-regulation of tight junction proteins (36, 37).
In vitro aortic explant cultures from Col18a1�/� andcontrol mice showed an increased vascular outgrowthin mutant mice, resembling in vitro the increasedangiogenesis and increased endothelial cells in the invivo CNV model (22). This difference was not observedafter the addition of low levels of endostatin to thecultures. While endothelial cells isolated fromCol18a1�/� mutant mice showed no difference in theirproliferation or growth factor-induced migration invitro, they showed an increased adhesion to fibronectin,a component of the extracellular matrix that binds toendothelial cells via integrins and cell surface proteo-glycans, which is highly expressed in CNV lesions aswell (22, 38–40). Endostatin can bind to cell surfaceproteoglycans and therefore may reduce fibronectinbinding sites on endothelial cells (22, 34, 35). Theantiangiogenic activity of endostatin thus may resultfrom a localized inhibition of endothelial cell/extracel-lular matrix interactions during vascular tube forma-tion at areas of neoangiogenesis, with subsequent de-stabilization and regression of vessels. Lack ofendostatin, in contrast, would result in an increasedangiogenic response through increased endothelialcell-extracellular matrix interactions, consistent withthe increased angiogenesis seen in vitro in the aorticexplant assay and in vivo in the CNV model inCol18a1�/� mice.
We thank Sofiya Plotkina and Elizabeth Benecchi fortechnical assistance. We are grateful to Anne Marie Lane foradvice on statistical analyses. This work was supported byNational Institutes of Health grant AR36820, by grants fromthe Ruth and Milton Steinbach Fund, Inc. and the Founda-tion Fighting Blindness, and by a core grant for visionresearch P30EY14104, T.F.C Frost Trust, and the Cripplegateand Dowsett Fellowship.
REFERENCES
1. Ishibashi, T., Hata, Y., Yoshikawa, H., Nakagawa, K., Sueishi, K.,and Inomata, H. (1997) Expression of vascular endothelialgrowth factor in experimental choroidal neovascularization.Graefes Arch. Clin. Exp. Ophthalmol. 235, 159–167
2. Yi, X., Ogata, N., Komada, M., Yamamoto, C., Takahashi, K.,Omori, K., and Uyama, M. (1997) Vascular endothelial growthfactor expression in choroidal neovascularization in rats. GraefesArch. Clin. Exp. Ophthalmol. 235, 313–319
3. Yi, X., Mai, L. C., Uyama, M., and Yew, D. T. (1998) Time-courseexpression of vascular endothelial growth factor as related tothe development of the retinochoroidal vasculature in rats. Exp.Brain Res. 118, 155–160
4. Gogat, K., Le Gat, L., Van Den Berghe, L., Marchant, D., Kobetz,A., Gadin, S., Gasser, B., Quere, I., Abitbol, M., and Menasche,M. (2004) VEGF and KDR gene expression during human
embryonic and fetal eye development. Invest. Ophthalmol. Vis.Sci. 45, 7–14
5. Adamis, A. P., Shima, D. T., Yeo, K. T., Yeo, T. K., Brown, L. F.,Berse, B., D’Amore, P. A., and Folkman, J. (1993) Synthesis andsecretion of vascular permeability factor/vascular endothelialgrowth factor by human retinal pigment epithelial cells. Biochem.Biophys. Res. Commun. 193, 631–638
6. Blaauwgeers, H. G., Holtkamp, G. M., Rutten, H., Witmer, A. N.,Koolwijk, P., Partanen, T. A., Alitalo, K., Kroon, M. E., Kijlstra,A., van Hinsbergh, V. W., and Schlingemann, R. O. (1999)Polarized vascular endothelial growth factor secretion by humanretinal pigment epithelium and localization of vascular endo-thelial growth factor receptors on the inner choriocapillaris.Evidence for a trophic paracrine relation. Am. J. Pathol. 155,421–428
7. Marneros, A. G., Fan, J., Yokoyama, Y., Gerber, H. P., Ferrara,N., Crouch, R. K., and Olsen, B. R. (2005) Vascular endothelialgrowth factor expression in the retinal pigment epithelium isessential for choriocapillaris development and visual function.Am. J. Pathol. 167, 1451–1459
8. Brown, D. M., Kaiser, P. K., Michels, M., Soubrane, G., Heier,J. S., Kim, R. Y., Sy, J. P., and Schneider, S. (2006) Ranibizumabversus verteporfin for neovascular age-related macular degener-ation. N. Engl. J. Med. 355, 1432–1444
9. Rosenfeld, P. J., Brown, D. M., Heier, J. S., Boyer, D. S., Kaiser,P. K., Chung, C. Y., and Kim, R. Y. (2006) Ranibizumab forneovascular age-related macular degeneration. N. Engl. J. Med.355, 1419–1431
10. Gragoudas, E. S., Adamis, A. P., Cunningham, E. T., Jr., Feinsod,M., and Guyer, D. R. (2004) Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 351, 2805–2816
11. Bhutto, I. A., McLeod, D. S., Hasegawa, T., Kim, S. Y., Merges,C., Tong, P., and Lutty, G. A. (2006) Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor(VEGF) in aged human choroid and eyes with age-relatedmacular degeneration. Exp. Eye Res. 82, 99–110
12. Gao, G., Li, Y., Zhang, D., Gee, S., Crosson, C., and Ma, J. (2001)Unbalanced expression of VEGF and PEDF in ischemia-inducedretinal neovascularization. FEBS Lett. 489, 270–276
13. Marneros, A. G., and Olsen, B. R. (2001) The role of collagen-derived proteolytic fragments in angiogenesis. Matrix Biol. 20,337–345
14. Marneros, A. G., Keene, D. R., Hansen, U., Fukai, N., Moulton,K., Goletz, P. L., Moiseyev, G., Pawlyk, B. S., Halfter, W., Dong,S., et al. (2004) Collagen XVIII/endostatin is essential for visionand retinal pigment epithelial function. EMBO J. 23, 89–99
15. Fukai, N., Eklund, L., Marneros, A. G., Oh, S. P., Keene, D. R.,Tamarkin, L., Niemela, M., Ilves, M., Li, E., Pihlajaniemi, T., andOlsen, B. R. (2002) Lack of collagen XVIII/endostatin results ineye abnormalities. EMBO J. 21, 1535–1544
16. Marneros, A. G., and Olsen, B. R. (2003) Age-dependent irisabnormalities in collagen XVIII/endostatin deficient mice withsimilarities to human pigment dispersion syndrome. Invest.Ophthalmol. Vis. Sci. 44, 2367–2372
17. Marneros, A. G., and Olsen, B. R. (2005) Physiological role ofcollagen XVIII and endostatin. FASEB J. 19, 716–728
18. Sertie, A. L., Sossi, V., Camargo, A. A., Zatz, M., Brahe, C., andPassos-Bueno, M. R. (2000) Collagen XVIII, containing anendogenous inhibitor of angiogenesis and tumor growth, playsa critical role in the maintenance of retinal structure and inneural tube closure (Knobloch syndrome). Hum. Mol. Genet. 9,2051–2058
19. Suzuki, O. T., Sertie, A. L., Der Kaloustian, V. M., Kok, F.,Carpenter, M., Murray, J., Czeizel, A. E., Kliemann, S. E.,Rosemberg, S., Monteiro, M., et al. (2002) Molecular analysis ofcollagen XVIII reveals novel mutations, presence of a thirdisoform, and possible genetic heterogeneity in Knobloch syn-drome. Am. J. Hum. Genet. 71, 1320–1329
20. O’Reilly, M. S., Boehm, T., Shang, Y., Fukai, N., Visions, G.,Lane, W. S., Flynn, E., Birched, J. R., Olsen, B. R., and Folkman,J. (1997) Endostatin: an endogenous inhibitor of angiogenesisand tumor growth. Cell. 88, 277–285
21. Yamaguchi, N., Anand-Apte, B., Lee, M., Sasaki, T., Fukai, N.,Shapiro, R., Que, I., Lowik, C., Timpl, R., and Olsen, B. R.(1999) Endostatin inhibits VEGF-induced endothelial cell mi-gration and tumor growth independent of zinc binding. EMBOJ. 18, 4414–4423
3817ENDOSTATIN INHIBITS CHOROIDAL NEOVASCULARIZATION

22. Li, Q., and Olsen, B. R. (2004) Increased angiogenic responsein aortic explants of collagen XVIII/endostatin-null mice. Am. J.Pathol. 165, 415–424
23. Kisker, O., Becker, C. M., Prox, D., Fannon, M., D’Amato, R.,Flynn, E., Fogler, W. E., Sim, B. K., Allred, E. N., Pirie-Shepherd,S. R., and Folkman, J. (2001) Continuous administration ofendostatin by intraperitoneally implanted osmotic pump im-proves the efficacy and potency of therapy in a mouse xenografttumor model. Cancer Res. 61, 7669–7674
24. Inai, T., Mancuso, M., Hashizume, H., Baffert, F., Haskell, A.,Baluk, P., Hu-Lowe, D. D., Shalinsky, D. R., Thurston, G.,Yancopoulos, G. D., and McDonald, D. M. (2004) Inhibition ofvascular endothelial growth factor (VEGF) signaling in cancercauses loss of endothelial fenestrations, regression of tumorvessels, and appearance of basement membrane ghosts. Am. J.Pathol. 165, 35–52
25. Espinosa-Heidmann, D. G., Suner, I., Hernandez, E. P., Frazier,W. D., Csaky, K. G., and Cousins, S. W. (2002) Age as anindependent risk factor for severity of experimental choroidalneovascularization. Invest. Ophthalmol. Vis. Sci. 43, 1567–1573
26. Felbor, U., Dreier, L., Bryant, R. A., Ploegh, H. L., Olsen, B. R.,and Mothes, W. (2000) Secreted cathepsin L generates endosta-tin from collagen XVIII. EMBO J. 19, 1187–1194
27. Ferreras, M., Felbor, U., Lenhard, T., Olsen, B. R., and Delaisse,J. (2000) Generation and degradation of human endostatinproteins by various proteinases. FEBS Lett. 486, 247–251
28. Schwesinger, C., Yee, C., Rohan, R. M., Joussen, A. M., Fernan-dez, A., Meyer, T. N., Poulaki, V., Ma, J. J., Redmond, T. M., Liu,S., Adamis, A. P., and D’Amato, R. J. (2001) Intrachoroidalneovascularization in transgenic mice overexpressing vascularendothelial growth factor in the retinal pigment epithelium.Am. J. Pathol. 158, 1161–1172
29. Baffi, J., Byrnes, G., Chan, C. C., and Csaky, K. G. (2000)Choroidal neovascularization in the rat induced by adenovirusmediated expression of vascular endothelial growth factor.Invest. Ophthalmol. Vis. Sci. 41, 3582–3589
30. Spilsbury, K., Garrett, K. L., Shen, W. Y., Constable, I. J., andRakoczy, P. E. (2000) Overexpression of vascular endothelialgrowth factor (VEGF) in the retinal pigment epithelium leads tothe development of choroidal neovascularization. Am. J. Pathol.157, 135–144
31. Wang, H., Ninomiya, Y., Sugino, I. K., and Zarbin, M. A. (2003)Retinal pigment epithelium wound healing in human Bruch’smembrane explants. Invest. Ophthalmol. Vis. Sci. 44, 2199–2210
32. Bhutto, I. A., Kim, S. Y., McLeod, D. S., Merges, C., Fukai, N.,Olsen, B. R., and Lutty, G. A. (2004) Localization of collagenXVIII and the endostatin portion of collagen XVIII in agedhuman control eyes and eyes with age-related macular degen-eration. Invest. Ophthalmol. Vis. Sci. 45, 1544–1552
33. Mori, K., Ando, A., Gehlbach, P., Nesbitt, D., Takahashi, K.,Goldsteen, D., Penn, M., Chen, C. T., Mori, K., Melia, M., et al.(2001) Inhibition of choroidal neovascularization by intrave-nous injection of adenoviral vectors expressing secretable en-dostatin. Am. J. Pathol. 159, 313–320
34. Sasaki, T., Fukai, N., Mann, K., Gohring, W., Olsen, B. R., andTimpl, R. (1998) Structure, function and tissue forms of theC-terminal globular domain of collagen XVIII containing theangiogenesis inhibitor endostatin. EMBO J. 17, 4249–4256
35. Sasaki, T., Larsson, H., Kreuger, J., Salmivirta, M., Claesson-Welsh, L., Lindahl, U., Hohenester, E., and Timpl, R. (1999)Structural basis and potential role of heparin/heparan sulfatebinding to the angiogenesis inhibitor endostatin. EMBO J. 18,6240–6248
36. Takahashi, K., Saishin, Y., Saishin, Y., Silva, R. L., Oshima, Y.,Oshima, S., Melia, M., Paszkiet, B., Zerby, D., Kadan, M. J., et al.(2003) Intraocular expression of endostatin reduces VEGF-induced retinal vascular permeability, neovascularization, andretinal detachment. FASEB J. 17, 896–898
37. Brankin, B., Campbell, M., Canning, P., Gardiner, T. A., andStitt, A. W. (2005) Endostatin modulates VEGF-mediated bar-rier dysfunction in the retinal microvascular endothelium. Exp.Eye Res. 81, 22–31
38. Bernfield, M., Banerjee, S. D., Koda, J. E., and Rapraeger, A. C.(1984) Remodelling of the basement membrane: morphogen-esis and maturation. Ciba Found. Symp. 108, 179–196
39. Hook, M., Couchman, J., Woods, A., Robinson, J., and Christ-ner, J. E. (1984) Proteoglycans in basement membranes. CibaFound. Symp. 108, 44–59
40. Nicolo, M., Biro, A., Cardillo-Piccolino, F., Castellani, P., Gio-vannini, A., Mariotti, C., Zingirian, M., Neri, D., and Zardi, L.(2003) Expression of extradomain-B-containing fibronectin insubretinal choroidal neovascular membranes. Am. J. Ophthalmol.135, 7–13
Received for publication April 14, 2007.Accepted for publication May 17, 2007.
3818 Vol. 21 December 2007 MARNEROS ET AL.The FASEB Journal