synthesis, characterization, and cytotoxicity studies of cu (ii), zn (ii), and fe (iii) complexes of...
TRANSCRIPT

PAPER www.rsc.org/dalton | Dalton Transactions
Synthesis, characterization, and cytotoxicity of dinuclearplatinum-bisphosphonate complexes to be used as prodrugs in the localtreatment of bone tumours
Nicola Margiotta,a Rosa Ostuni,a Valentina Gandin,b Cristina Marzano,b Sara Piccinonnaa,c andGiovanni Natile*a
Received 22nd September 2009, Accepted 29th October 2009First published as an Advance Article on the web 16th November 2009DOI: 10.1039/b919721d
For over 30 years cisplatin has been one of the most active antitumour agents in clinical use,nevertheless research for overcoming cisplatin toxicity and resistance or for improving its efficacyhas never ceased. In this context we have recently proposed dinuclear Pt complexes with bridginggeminal bisphosphonates as novel Pt-prodrugs with potential activity at the bone surface afterembedment in inorganic matrices and implantation at the tumour site. In the present paper wereport the synthesis and full characterization of four new platinum complexes having a dinuclearstructure with a bisphosphonate (2-ammonium-1-hydroxyethane-1,1-diyl-bisphosphonate or3-ammonium-1-hydroxypropane-1,1-diyl-bisphosphonate, AHBP-H and PAM-H, respectively) actingas a bridging ligand between two platinum moieties (cis-[Pt(NH3)2]2+, directly related to cisplatin, and[Pt(cis-1,4-DACH)]2+, known to be able to overcome the cisplatin resistance). Moreover, as apreliminary investigation, the in vitro cytotoxicity of the new complexes has been evaluated on a panelof 13 human tumour cell lines including cisplatin- and multidrug-resistant sublines.
Introduction
Cisplatin is one of the most active antitumour agents in cur-rent clinical use;1-2 apart from being curative against testiculartumours,3 it is effective against several other cancers such asovarian, head and neck, cervical, and bladder.4-6 Despite itssuccess, treatment with cisplatin is limited by undesirable side ef-fects such as nephrotoxicity, nausea, vomiting, myelosuppression,ototoxicity and neurotoxicity.7,8 Moreover, tumour resistance tocisplatin (acquired during cycles of therapy or intrinsic in sometypes of cancers) is another factor that limits its use.9
Since the serendipitous discovery of the antitumoural activityof cisplatin,10,11 an intensive research has led to the synthesisof thousands of new platinum compounds with the aim ofovercoming cisplatin toxicity and resistance or to improve itsefficacy. The use of carrier ligands able to promote the specificaccumulation of the drug in target organs or cells has also beenexploited as a strategy to overcome the side effects of cisplatin. Forinstance, platinum complexes containing (aminoalkyl)phosphonicacids12,13 have been exploited by the group of Keppler14,15 for theirpotential use as selective platinum drugs for bone tumours. Thoseauthors were inspired by the analogy between these ligands andgeminal bisphosphonates (BPs), commercial drugs, which showaffinity for bones and other calcified tissues and are in clinical usefor the treatment of hypercalcaemia and skeletal metastases.16-19
aDipartimento Farmaco-Chimico, Universita degli Studi di Bari “A. Moro”,Via E. Orabona 4, 70125, Bari, Italy. E-mail: [email protected];Fax: +39-080-5442230bDipartimento di Scienze Farmaceutiche, Universita degli Studi di Padova,Via Marzolo 5, 35131, Padova, ItalycConsorzio Interuniversitario di Ricerca in Chimica dei Metalli nei SistemiBiologici (C.I.R.C.M.S.B.), Via C. Ulpiani 27, 70125, Bari, Italy
The combination of the bisphosphonic functionality with theplatinum moiety could promote the specific accumulation of theantitumour drug in the bone with consequent significant improve-ment of the biological effect and reduction of the systemic toxicity.Pharmacological investigations performed on Pt(II) complexeswith amino-bis/tris(methylenephosphonate) ligands were a proofof concept demonstrating their superior activity as compared tocisplatin in an orthotopically transplanted rat osteosarcoma modelclosely resembling the human osteosarcoma.20,21
A further lowering of the systemic side effects of a drugcould be obtained by local administration directly at the siteof the tumour and, in this context, we have recently proposeddinuclear Pt complexes with bridging geminal bisphosphonates(Chart 1) as novel Pt-prodrugs with potentiated activity at the
Chart 1 Sketches of already reported dinuclear Pt-bisphospho-nate complexes. AHBP-H = 2-ammonium-1-hyroxyethane-1,1-diyl-bisphosphonate; MED = medronate (methane-1,1-diyl-bisphosphonate);PHBP-H = 1-(pyridinium-4-yl)-1-hydroxy-methane-1,1-bisphosphonate.
10904 | Dalton Trans., 2009, 10904–10913 This journal is © The Royal Society of Chemistry 2009885

bone surface after embedment in inorganic matrices capable toactivate them.22-24
The dinuclear Pt-bisphosphonate complexes were loaded eitheronto silica xerogels, obtained with the sol–gel method, or ontosynthetic biomimetic hydroxyapatite (HA) nanocrystals in orderto obtain biocompatible inorganic composite materials capable toact as bone filler materials and to release an active antitumouralPt-species after local implant at the tumour site. The drug-releasing kinetic from the two inorganic composites was modu-lated by the presence of Ca2+ ions, which rendered the matricesable to retain the bisphosphonic ligand (this latter has a veryhigh affinity for calcium ions) so that only the antitumouralPt(en)2+ residue (en = ethylenediamine) was released from thesilica xerogels or from the biomimetic HA. Moreover, in a recentpaper the cytotoxicity of the Pt-complexes released from theHA were tested towards human cervix carcinoma cells and theyresulted to be more cytotoxic than the unmodified complexes25 andactive at a concentration very similar to that of pure [PtCl2(en)](the reference compound). It was also demonstrated that thereleased species from the matrices was [PtCl2(en)] or relatedsolvato-species.
The obtained results stimulated us to extend the investi-gation to the prototype platinum complex effectively used intherapy (cisplatin) and to another complex which is knownto be able to overcome the cisplatin-resistance ([PtCl2(cis-1,4-DACH)], DACH = diaminecyclohexane). Therefore, we havesynthesized four new dinuclear platinum(II) complexes (Chart 2)carrying the am(m)ine ligand(s) mentioned above and two typesof aminoalkyl-hydroxybisphosphonate bridging ligands (either2-ammonium-1-hydroxyethane-1,1-diyl-bisphosphonate (AHBP-H4) or 3-ammonium-1-hydroxy-propane-1,1-bisphosphonic acid(pamidronate, PAM-H4)).
Moreover, as a preliminary investigation, the in vitro cytotoxi-city of the new dinuclear complexes has been evaluated on a setof 13 human tumor cell lines including cisplatin- and multidrug-resistant (MDR) sublines.
Materials and methods
Chemicals
Commercial reagent grade chemicals and solvents were purchasedfrom Sigma-Aldrich Chemical Co. (St. Louis, MO, USA) and usedwithout further purification.
Instrumental measurements
Mass spectrometry: electrospray ionisation mass spectrometry(ESI-MS) was performed with an electrospray interface and anion trap mass spectrometer (1100 Series LC/MSD Trap systemAgilent, Palo Alto, CA) using H2O as solvent.
NMR spectra were recorded on Bruker Avance DPX instru-ments operating at 300 or 600 MHz (1H). Standard pulse sequenceswere used for 1H, 31P{1H} (121.5 MHz), and 195Pt{1H} (64.5 MHz)1D spectra. Chemical shifts are given in ppm. 1H chemical shiftswere referenced to internal sodium 3-(trimethylsilyl)propionate(TSP), 31P chemical shifts were referenced to external H3PO4
(85% w/w), and 195Pt chemical shifts were referenced to externalK2[PtCl4] in D2O fixed at -1628 ppm. Elemental analyses werecarried out with a Hewlett Packard 185 C, H, and N analyzer.
A Crison Micro-pH meter Model 2002 equipped with Crisonmicro-combination electrodes (5 and 3 diameter) and calibratedwith Crison standard buffer solution at pH 2.00, 4.01, 7.02, and9.26 was used for pH measurements. The pH readings for D2Osolution are indicated as pH* values and are uncorrected for theeffect of deuterium on glass electrodes.26
Synthesis of the bisphosphonic acids
Neutral 2-ammonium-1-hyroxyethane-1,1-diyl-bisphosphonicacid (AHBP-H4)22 and 3-ammonium-1-hydroxy-propane-1,1-bisphosphonic acid (pamidronate, PAM-H4)27 were preparedfollowing procedures reported in previous papers. The elementalanalyses and the spectroscopic characteristics of the synthesized
Chart 2 Sketches of the new dinuclear Pt-bisphosphonate complexes. AHBP-H = 2-ammonium-1-hyroxyethane-1,1-diyl-bisphosphonate; PAM-H =3-ammonium-1-hydroxy-propane-1,1-bisphosphonate (pamidronate).
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 10904–10913 | 10905886

geminal bisphosphonates were consistent with the data reportedin the literature.
Synthesis of the platinum compounds
[PtCl2(cis-1,4-DACH)],28 [Pt(OSO3)(cis-1,4-DACH)(OH2)],28 andcis-[PtI2(NH3)2]29 were prepared as already reported. The elemen-tal analyses and the spectroscopic features of the Pt precursorcomplexes were consistent with the data reported in the literature.
Preparation of cis-[Pt(OSO3)(NH3)2(OH2)]. Cis-[PtI2(NH3)2](0.550 g, 1.14 mmol) was suspended in water (20 mL) and treatedwith a solution of Ag2SO4 (0.356 g, 1.140 mmol in 5 mL H2O). Theobtained suspension was stirred for 24 h in the dark meanwhile ayellow solid (AgI) precipitated from the solution. The precipitatewas removed by filtration through Celite and the solution wasevaporated to dryness under reduced pressure at 50 ◦C. The yellowresidue was the desired cis-[Pt(OSO3)(NH3)2(OH2)] compound(0.344 g, 88% yield). Anal. calcd for cis-[Pt(OSO3)(NH3)2(OH2)](H8N2O5PtS): H 2.34%, N 8.24. Found: H 2.30%, N 8.16.
Preparation of [{cis-Pt(NH3)2}2(AHBP-H)]HSO4 (1A).2-Ammonium-1-hyroxyethane-1,1-diyl-bisphosphonic acid(AHBP-H4) (0.75 g, 0.339 mmol) was dissolved in H2O (15 mL)and the resulting solution was first partially neutralized withBa(OH)2·8H2O (0.315 g, 0.339 mmol) and then treated with asolution of cis-[Pt(OSO3)(NH3)2(OH2)] (0.236 g, 0.678 mmol, in15 mL of H2O). The resulting suspension was kept under stirringat room temperature for 40 h meanwhile a white solid (BaSO4)separated. After removal of the solid, the filtered solution wasconcentrated to ca. 2 mL and then treated with methanol whichcaused the precipitation of a yellow-greenish solid which wasthe desired product having HSO4
- as counterion. The precipitatewas left standing at 4 ◦C for 2 h and then it was isolated byfiltration of the solution, washed with methanol (2.0 mL), anddried under vacuum. Obtained 0.162 g (62% yield). Anal. calcdfor [{cis-Pt(NH3)2}2(AHBP-H)]HSO4·2H2O (C2H23N5O13P2Pt2S):C 2.97, H 2.86, N 8.65%. Found: C 3.00, H 2.98, N 8.53%.1H-NMR (D2O, pH* 7, labelling as in Fig. 1): 3.24 ppm (2 H, m,C(2)H2). 31P{1H}-NMR (D2O, pH* 7): 37.09 ppm. ESI-MS: calcdfor C2H22N5O9P2Pt2
+: 676.0. Found, m/z (% relative to the basepeak): 675.9 (100) [M]+.
Preparation of [{Pt(cis-1,4-DACH)}2(AHBP-H)]HSO4 (1B).AHBP-H4 (0.026 g, 0.118 mmol) was dissolved in H2O (15 mL)and treated first with Ba(OH)2·8H2O (0.037 g, 0.118 mmol)and then with a solution of [Pt(OSO3)(cis-1,4-DACH)(OH2)](0.100 g, 0.236 mmol in 15 mL of H2O). The resulting suspensionwas kept under stirring for 24 h and then left standing at4 ◦C for 1 h. After removal of the white solid (BaSO4), thefiltered solution was concentrated, under reduced pressure andat 45 ◦C, to a volume of ca. 3 mL. The pH of the resultingpale yellow solution was brought to 1 by addition of H2SO4 (1M) and then acetone was added to induce precipitation of thedesired product having HSO4
- as counter-ion. The product wasisolated by filtration of the solution and dried under vacuum.Obtained 0.0875 g (80% yield). Anal. calcd for [{Pt(cis-1,4-DACH)}2(AHBP-H)]HSO4·4H2O (C14H43N5O15P2Pt2S): C 16.72,H 4.31, N 6.96%. Found: C 16.41, H 4.27, N 6.85%. 1H-NMR(D2O, pH* 3, labelling as in Fig. 2): 5.38 ppm (4 H, br, cis-1,4DACH NH2), 5.02 ppm (4 H, br, cis-1,4 DACH NH2), 3.37 ppm
Fig. 1 1H and 31P{1H}-NMR spectra of compound 1A in D2O (pH* =7). The asterisk indicates the residual solvent peak. Numbering of protonsis reported in italics.
(2 H, t, C(2)H2), 3.13 ppm (4 H, s, C(a)H), 1.17 ppm (16 H,m, C(b)H2). 31P{1H}-NMR (D2O, pH* 3): 37.20 ppm. 195Pt{1H}-NMR (D2O, pH* 3): -1582 ppm (br) and -1595 ppm (br). ESI-MS:calcd for C14H34N5O7P2Pt2
+: 836.12. Found, m/z (% relative to thebase peak): 836.1 (100) [M]+.
Preparation of [{cis-Pt(NH3)2}2(PAM-H)]HSO4 (2A). 3-Ammonium-1-hydroxy-propane-1,1-bisphosphonic acid (PAM-H4) (0.109 g, 0.464 mmol) was dissolved in H2O (15 mL) andtreated first with Ba(OH)2·8H2O (0.146 g, 0.464 mmol) andthen with a solution of cis-[Pt(OSO3)(NH3)2(OH2)] (0.318 g,0.928 mmol in 15 mL of H2O). After stirring for 48 h at roomtemperature the obtained white solid (BaSO4) was removed andthe filtered solution was concentrated to a volume of ca. 2 mL. ThepH of the concentrated solution was lowered to 1 by addition ofH2SO4 (1 M) and then acetone was added to induce precipitationof a yellow-greenish solid which was left standing at 4 ◦C for2 h. The solid was isolated by filtration of the solution and driedunder vacuum. Obtained 0.143 g (40% yield). Anal. calcd for [{cis-Pt(NH3)2}2(PAM-H)]HSO4·4H2O (C3H29N5O15P2Pt2S): C 4.19, H3.40, N 8.15%. Found: C 4.56, H 3.03, N 7.69%. 1H-NMR (D2O,pH* 5, labelling as in Fig. 4): 4.35 ppm (12 H, br, ammine NH3),3.29 ppm (2 H, t, C(3)H2), 2.55–2.17 ppm (2 H, m, C(2)H2).31P{1H}-NMR (D2O, pH* 5): 39.81 ppm. ESI-MS: calcd forC3H20N5O7P2Pt2
+: 690.32. Found, m/z (% relative to the basepeak): 690.0 (100) [M]+.
10906 | Dalton Trans., 2009, 10904–10913 This journal is © The Royal Society of Chemistry 2009887
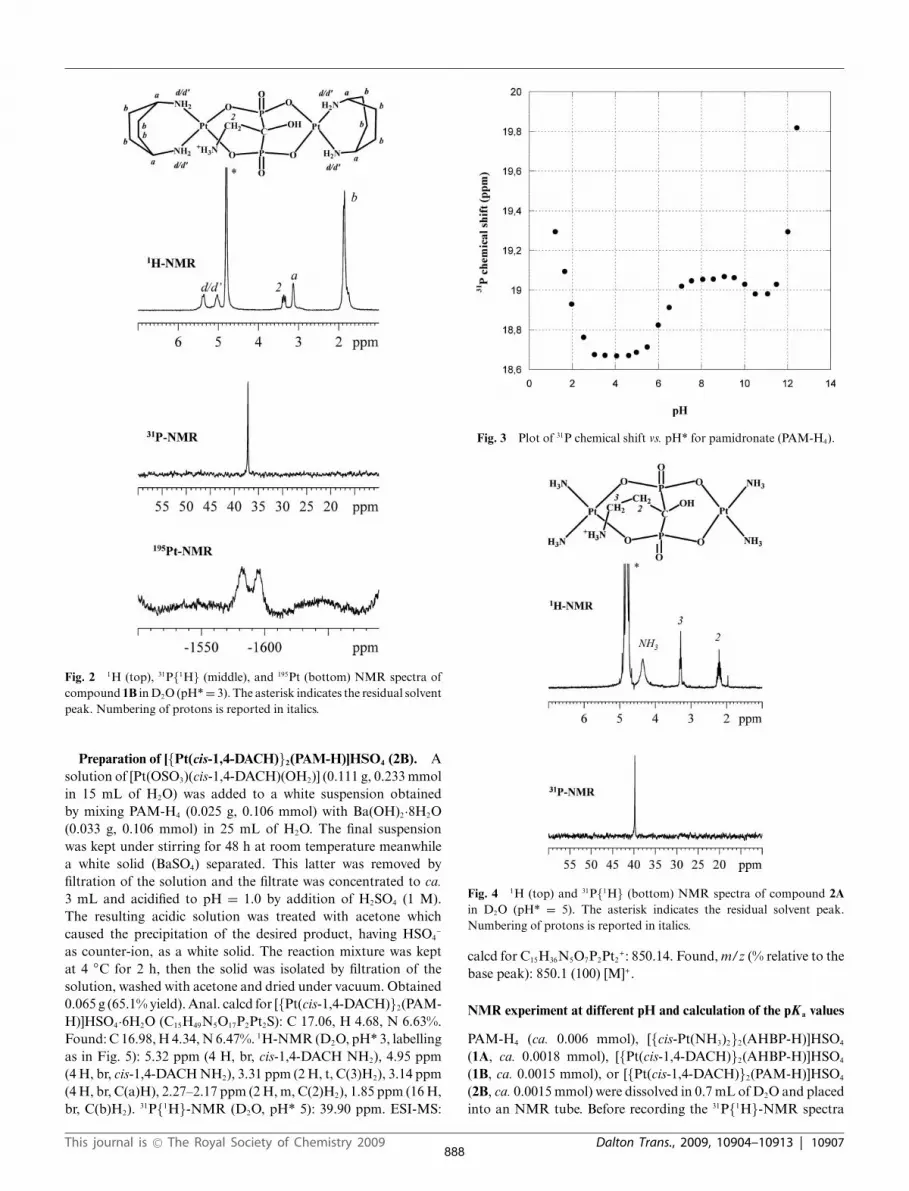
Fig. 2 1H (top), 31P{1H} (middle), and 195Pt (bottom) NMR spectra ofcompound 1B in D2O (pH* = 3). The asterisk indicates the residual solventpeak. Numbering of protons is reported in italics.
Preparation of [{Pt(cis-1,4-DACH)}2(PAM-H)]HSO4 (2B). Asolution of [Pt(OSO3)(cis-1,4-DACH)(OH2)] (0.111 g, 0.233 mmolin 15 mL of H2O) was added to a white suspension obtainedby mixing PAM-H4 (0.025 g, 0.106 mmol) with Ba(OH)2·8H2O(0.033 g, 0.106 mmol) in 25 mL of H2O. The final suspensionwas kept under stirring for 48 h at room temperature meanwhilea white solid (BaSO4) separated. This latter was removed byfiltration of the solution and the filtrate was concentrated to ca.3 mL and acidified to pH = 1.0 by addition of H2SO4 (1 M).The resulting acidic solution was treated with acetone whichcaused the precipitation of the desired product, having HSO4
-
as counter-ion, as a white solid. The reaction mixture was keptat 4 ◦C for 2 h, then the solid was isolated by filtration of thesolution, washed with acetone and dried under vacuum. Obtained0.065 g (65.1% yield). Anal. calcd for [{Pt(cis-1,4-DACH)}2(PAM-H)]HSO4·6H2O (C15H49N5O17P2Pt2S): C 17.06, H 4.68, N 6.63%.Found: C 16.98, H 4.34, N 6.47%. 1H-NMR (D2O, pH* 3, labellingas in Fig. 5): 5.32 ppm (4 H, br, cis-1,4-DACH NH2), 4.95 ppm(4 H, br, cis-1,4-DACH NH2), 3.31 ppm (2 H, t, C(3)H2), 3.14 ppm(4 H, br, C(a)H), 2.27–2.17 ppm (2 H, m, C(2)H2), 1.85 ppm (16 H,br, C(b)H2). 31P{1H}-NMR (D2O, pH* 5): 39.90 ppm. ESI-MS:
Fig. 3 Plot of 31P chemical shift vs. pH* for pamidronate (PAM-H4).
Fig. 4 1H (top) and 31P{1H} (bottom) NMR spectra of compound 2Ain D2O (pH* = 5). The asterisk indicates the residual solvent peak.Numbering of protons is reported in italics.
calcd for C15H36N5O7P2Pt2+: 850.14. Found, m/z (% relative to the
base peak): 850.1 (100) [M]+.
NMR experiment at different pH and calculation of the pKa values
PAM-H4 (ca. 0.006 mmol), [{cis-Pt(NH3)2}2(AHBP-H)]HSO4
(1A, ca. 0.0018 mmol), [{Pt(cis-1,4-DACH)}2(AHBP-H)]HSO4
(1B, ca. 0.0015 mmol), or [{Pt(cis-1,4-DACH)}2(PAM-H)]HSO4
(2B, ca. 0.0015 mmol) were dissolved in 0.7 mL of D2O and placedinto an NMR tube. Before recording the 31P{1H}-NMR spectra
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 10904–10913 | 10907888
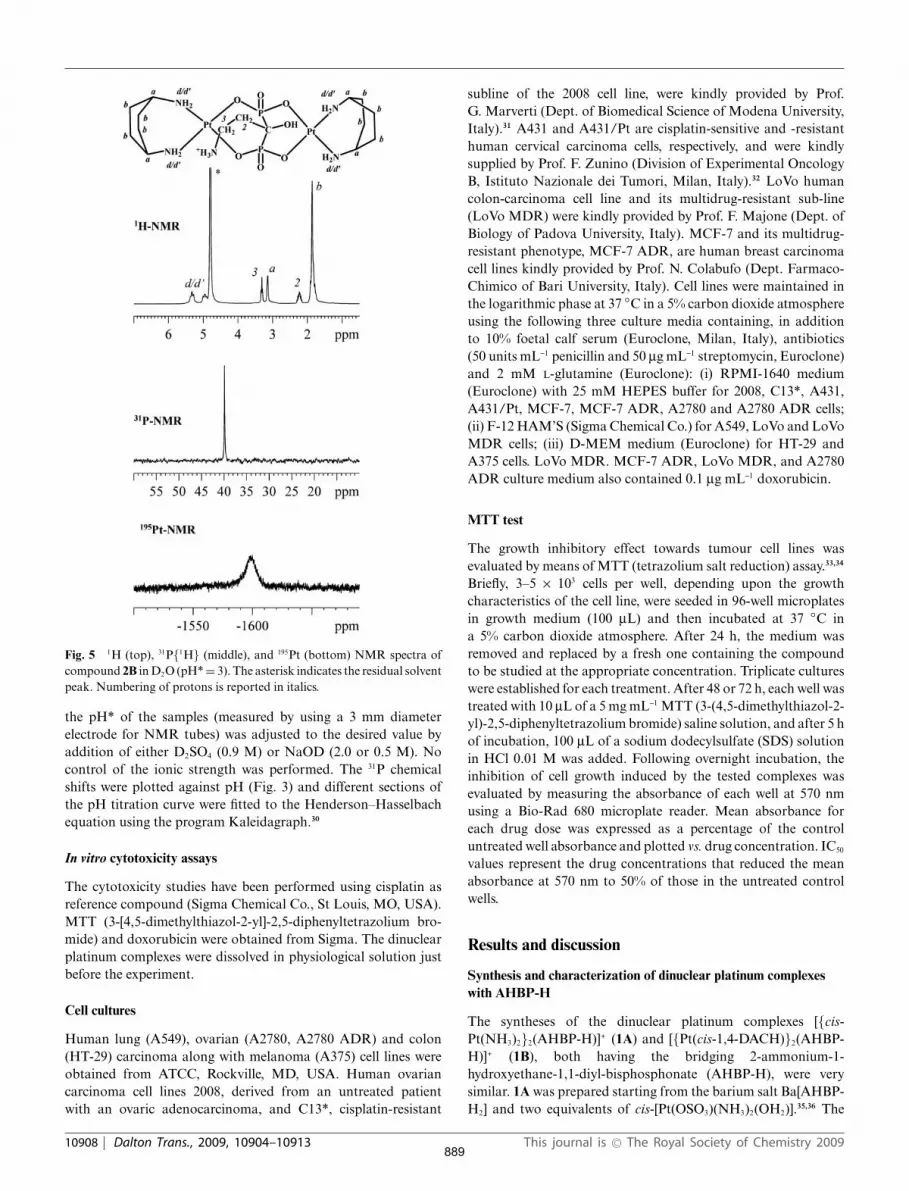
Fig. 5 1H (top), 31P{1H} (middle), and 195Pt (bottom) NMR spectra ofcompound 2B in D2O (pH* = 3). The asterisk indicates the residual solventpeak. Numbering of protons is reported in italics.
the pH* of the samples (measured by using a 3 mm diameterelectrode for NMR tubes) was adjusted to the desired value byaddition of either D2SO4 (0.9 M) or NaOD (2.0 or 0.5 M). Nocontrol of the ionic strength was performed. The 31P chemicalshifts were plotted against pH (Fig. 3) and different sections ofthe pH titration curve were fitted to the Henderson–Hasselbachequation using the program Kaleidagraph.30
In vitro cytotoxicity assays
The cytotoxicity studies have been performed using cisplatin asreference compound (Sigma Chemical Co., St Louis, MO, USA).MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bro-mide) and doxorubicin were obtained from Sigma. The dinuclearplatinum complexes were dissolved in physiological solution justbefore the experiment.
Cell cultures
Human lung (A549), ovarian (A2780, A2780 ADR) and colon(HT-29) carcinoma along with melanoma (A375) cell lines wereobtained from ATCC, Rockville, MD, USA. Human ovariancarcinoma cell lines 2008, derived from an untreated patientwith an ovaric adenocarcinoma, and C13*, cisplatin-resistant
subline of the 2008 cell line, were kindly provided by Prof.G. Marverti (Dept. of Biomedical Science of Modena University,Italy).31 A431 and A431/Pt are cisplatin-sensitive and -resistanthuman cervical carcinoma cells, respectively, and were kindlysupplied by Prof. F. Zunino (Division of Experimental OncologyB, Istituto Nazionale dei Tumori, Milan, Italy).32 LoVo humancolon-carcinoma cell line and its multidrug-resistant sub-line(LoVo MDR) were kindly provided by Prof. F. Majone (Dept. ofBiology of Padova University, Italy). MCF-7 and its multidrug-resistant phenotype, MCF-7 ADR, are human breast carcinomacell lines kindly provided by Prof. N. Colabufo (Dept. Farmaco-Chimico of Bari University, Italy). Cell lines were maintained inthe logarithmic phase at 37 ◦C in a 5% carbon dioxide atmosphereusing the following three culture media containing, in additionto 10% foetal calf serum (Euroclone, Milan, Italy), antibiotics(50 units mL-1 penicillin and 50 mg mL-1 streptomycin, Euroclone)and 2 mM L-glutamine (Euroclone): (i) RPMI-1640 medium(Euroclone) with 25 mM HEPES buffer for 2008, C13*, A431,A431/Pt, MCF-7, MCF-7 ADR, A2780 and A2780 ADR cells;(ii) F-12 HAM’S (Sigma Chemical Co.) for A549, LoVo and LoVoMDR cells; (iii) D-MEM medium (Euroclone) for HT-29 andA375 cells. LoVo MDR. MCF-7 ADR, LoVo MDR, and A2780ADR culture medium also contained 0.1 mg mL-1 doxorubicin.
MTT test
The growth inhibitory effect towards tumour cell lines wasevaluated by means of MTT (tetrazolium salt reduction) assay.33,34
Briefly, 3–5 ¥ 103 cells per well, depending upon the growthcharacteristics of the cell line, were seeded in 96-well microplatesin growth medium (100 mL) and then incubated at 37 ◦C ina 5% carbon dioxide atmosphere. After 24 h, the medium wasremoved and replaced by a fresh one containing the compoundto be studied at the appropriate concentration. Triplicate cultureswere established for each treatment. After 48 or 72 h, each well wastreated with 10 mL of a 5 mg mL-1 MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) saline solution, and after 5 hof incubation, 100 mL of a sodium dodecylsulfate (SDS) solutionin HCl 0.01 M was added. Following overnight incubation, theinhibition of cell growth induced by the tested complexes wasevaluated by measuring the absorbance of each well at 570 nmusing a Bio-Rad 680 microplate reader. Mean absorbance foreach drug dose was expressed as a percentage of the controluntreated well absorbance and plotted vs. drug concentration. IC50
values represent the drug concentrations that reduced the meanabsorbance at 570 nm to 50% of those in the untreated controlwells.
Results and discussion
Synthesis and characterization of dinuclear platinum complexeswith AHBP-H
The syntheses of the dinuclear platinum complexes [{cis-Pt(NH3)2}2(AHBP-H)]+ (1A) and [{Pt(cis-1,4-DACH)}2(AHBP-H)]+ (1B), both having the bridging 2-ammonium-1-hydroxyethane-1,1-diyl-bisphosphonate (AHBP-H), were verysimilar. 1A was prepared starting from the barium salt Ba[AHBP-H2] and two equivalents of cis-[Pt(OSO3)(NH3)2(OH2)].35,36 The
10908 | Dalton Trans., 2009, 10904–10913 This journal is © The Royal Society of Chemistry 2009889

elemental analysis of 1A was in accordance with the presenceof one aminobisphosphonate and two [cis-Pt(NH3)2]2+ residues.The 1H-NMR spectrum (Fig. 1) shows a multiplet centered at3.24 ppm assigned to the BP methylene protons. This resonanceis slightly downfield (Dd = 0.06 ppm) with respect to that ofthe free ligand and is the AA¢ portion of a AA¢XX¢ spin systemwhere AA¢ represent the methylenic protons and XX¢ are the twoP atoms. In our experimental conditions the determined couplingconstants were: 3JH–P = 3JH¢-P¢ = 20.28 Hz and 3JH–P¢ = 3JH¢-P =1.65 Hz.
The 31P{1H}-NMR spectrum (Fig. 1) shows a singlet withunresolved platinum satellites (platinum satellites usually broadenbeyond detection by chemical shift anisotropy relaxation of 195Ptespecially at high magnetic field strengths, as those used in ourexperiment)37 falling at 37.09 ppm assigned to the two phosphorusatoms of the bisphosphonic group, which are magnetically equiv-alent and shifted at lower field (Dd = 19.98 ppm) with respect tothat of the free AHBP ligand at the same pH*. Such a downfieldshift of the phosphorus nuclei is characteristic of phosphonategroups coordinated to a metal ion which have lost the acidicprotons.13,38
The acid dissociation constants of free AHBP-H4, determinedby 31P{1H}-NMR experiments at different pH, have already beenreported.22 31P{1H}-NMR experiments at different pH* were alsoperformed on 1A. The 31P chemical shift of 1A did not change inthe pH* range 0–9, supporting the involvement of all phosphonatehydroxyl oxygen atoms in coordination to platinum. The platinum-coordinated oxygen atoms cannot undergo neither protonationnor deprotonation steps by changes of pH* and therefore the31P chemical shift remains constant. Unfortunately, compound1A underwent structural changes at pH* higher than 9.0 withformation of new product(s) having non-equivalent phosphorusatoms. Probably, at high pH, the aminic group deprotonates andcoordinates to platinum by displacing one oxygen of the tetra-coordinated bisphosphonate. This rearrangement leads to a quitecomplex NMR spectrum, however no signals of free AHBP arepresent.
The synthesis of [{Pt(cis-1,4-DACH)}2(AHBP-H)]HSO4 (1B)was performed similarly, starting from the barium salt Ba[AHBP-H2] and two equivalents of [Pt(OSO3)(cis-1,4-DACH)(OH2)]. Theelemental analysis of 1B was in accordance with one aminobispho-sphonate per two [Pt(cis-1,4-DACH)]2+ moieties. The 1H-NMRspectrum (Fig. 2) shows two multiplets placed at 5.38 and 5.02 ppmassigned to the aminic protons of coordinated cis-1,4-DACH(geminal protons of each amine group are made non-equivalent bythe lack of symmetry with respect to the coordination plane). Theacidity conditions (pH* = 3) slow down the exchange of the aminicprotons with the deuterium of the solvent, allowing detection ofthe aminic protons in water solution.
The 1H-NMR spectrum shows also a triplet centered at3.37 ppm assigned to the methylenic protons of the bisphospho-nate. These protons are shifted at higher field (Dd = -0.08 ppm) ascompared to the free ligand at pH* = 3. The signals at 3.13 ppmand 1.77 ppm were assigned to the methynic and methylenicprotons of coordinated cis-1,4-DACH, respectively. The 31P{1H}-NMR spectrum shows a signal at 37.20 ppm assigned to the twophosphorus nuclei which are magnetically equivalent and shiftedat lower field (Dd = 20.74) with respect to the free AHBP ligandat the same pH*.
For a full characterization of complex 1B, the 195Pt-NMRspectrum was also recorded (Fig. 2). Two broad signals at -1582and -1595 ppm were observed. These signals are broadened bythe quadrupolar effects of 14N (99.6% abundance) from the amineligands and by chemical shift anisotropy relaxation of 195Pt. The195Pt signals are shifted at lower field with respect to the starting[Pt(cis-1,4-DACH)(H2O)2]2+ substrate. The same trend has alreadybeen observed in platinum complexes with a diamine and adicarboxylate ligand with respect to the species with a diamineand two aqua ligands.39,40,41
The presence of only one signal in the 31P{1H} spectrum andof two signals in the 195Pt spectrum fully supports the structurein which one bisphosphonate unit bridges two platinum atomsin a W conformation.36 The plane of symmetry passing throughthe two platinum atoms and perpendicular to the coordinationplane renders the two phosphorus atoms chemically equivalent.In contrast, the absence of a plane of symmetry passing throughthe P–C(1)–P atoms and perpendicular to the coordination plane(presence of two different substituents, –OH and –CH2NH3
+, onthe C(1) carbon atom), renders the two platinum atoms non-equivalent. Considering that the protonated amino group shouldshift at lower field the nearby Pt atom, the signal falling at-1595 ppm could be tentatively assigned to the Pt atom on the sideof the –OH group. The two aminic groups and the bifunctionalbisphosphonate saturate completely the coordination of eachplatinum center excluding the hydroxylic group from coordination.This is not the case for complexes with other metal ions in whichthe bisphosphonate can be tricoordinated contributing two oxygenatoms from the phosphonate groups and one from deprotonatedhydroxylic group to the same metal ion.42
Also for 1B we performed 31P{1H}-NMR experiments atdifferent pH*. As already observed for compound 1A, the 31Pchemical shift remains constant in the pH* range from 1 to 9,indicating that all the bisphosphonate hydroxyl oxygen atoms areinvolved in coordination to the platinum atom. As for complex1A, the titration was stopped at pH* 9 since 1B started torearrange. This is probably due to coordination to platinum ofthe deprotonated aminic group.
Synthesis and characterization of dinuclear platinum complexeswith PAM-H
Pamidronate (3-ammonium-1-hydroxy-propane-1,1-bisphos-phonic acid, PAM-H4) was synthesized according to a procedurealready reported by some of us.27 We have measured the pKa
values of the various deprotonation steps of pamidronate byrecording 31P{1H}-NMR spectra at different pH*. The plot of the31P chemical shift as a function of pH* is given in Fig. 3.
The titration curve shows four inflexion points corresponding tothe various deprotonation steps (Scheme 1). We did not determinethe constant related to the first deprotonation step (pKa1 inScheme 1) because this requires a hydrogen ion concentrationbeyond reach in our experimental conditions. As the pH* wasincreased from ca. 0 to 3 we observed a shift to higher field ofthe 31P chemical shift with an inflexion point (calculated by fittingthe points between pH* 0 and 3 to the Henderson–Hasselbachequation)30 falling at pH* 1.69. This pH value correspondsto the equilibrium constant of the second deprotonation step(pKa2, Scheme 1). As already found for the ligand AHBP-H4,22
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 10904–10913 | 10909890

Scheme 1
deprotonation of a phosphonate –OH shields the phosphorusnucleus directly attached to it and deshields the distal phosphonategroup present in the molecule. The former is called “direct effect”while the latter is called “remote effect”.13 Since we observed for thesecond deprotonation step a shielding of the 31P nuclei, the directeffect must prevail over the remote effect. By further increasingthe pH* from 3 to 7, we observed a deshielding of the phosphorusnuclei with an inflexion point falling at pH* 6.23. This valuecorresponds to pKa3 and represents the second deprotonationconstant of one phosphonate group. The deshielding of the 31Pnuclei indicates that in this case the remote effect prevails overthe direct effect. By further increasing the pH* from 7 to 14we observed first a shielding of the 31P nuclei with a inflexionpoint at pH* 10.1 (corresponding to pKa4, Scheme 1) and thena deshielding of the phosphorous nuclei with an inflexion pointoccurring at pH* 12.5 (corresponding to the deprotonation of theaminic group, pKa5, Scheme 1). As for AHBP-H4 and analogousaminobisphosphonates, the deprotonation of the aminic group ofPAM-H4, and hence the loss of the zwitterionic character, inducesa great deshielding of the phosphorous nuclei.43
Pamidronate was then used for the synthesis of the dinu-clear complexes [{cis-Pt(NH3)2}2(PAM-H)]+ (2A) and [{Pt(cis-1,4-DACH)}2(PAM-H)]+ (2B) in conditions very similar to those usedfor the syntheses of compounds 1A and 1B.
The complex [{cis-Pt(NH3)2}2(PAM-H)]+ (2A) was character-ized via elemental analysis and 1H and 31P{1H}-NMR. The1H-NMR spectrum (Fig. 4) shows a broad signal at 4.35 ppmassigned to the ammine protons of the platinum moieties. Thetriplet at 3.29 ppm, which is slightly shielded (Dd = -0.06) withrespect to the analogous signal of the free PAM at the samepH*, was assigned to the methylenic protons in position 3 ofcoordinated PAM-H. The multiplet falling in the range from 2.25to 2.17 ppm was assigned to the methylenic protons in position 2of the coordinated PAM-H.
The 31P{1H}-NMR spectrum (Fig. 4) shows a singlet withunresolved Pt satellites falling at 39.81 ppm and assigned to thephosphorous atoms of the two bisphosphonic groups which areshifted at lower field (Dd = 21.11) with respect to the analogous
signal of free PAM at the same pH*. Because of the low solubilityin water of this complex it has not been possible to record a 195Pt-NMR spectrum as well as to perform 31P{1H}-NMR experimentsat different pH*.
Also, the synthesis of [{Pt(cis-1,4-DACH)}2(PAM-H)]+ (2B)was performed starting from the barium salt Ba[PAM-H2] and twoequivalents of [Pt(OSO3)(cis-1,4-DACH)(H2O)]. The elementalanalysis of 2B confirmed the formation of a dinuclear speciescontaining one bisphosphonate per two [Pt(cis-1,4-DACH)]2+
residues. The characterization of 2B was performed by 1H,31P{1H}, and 195Pt-NMR. The 1H-NMR spectrum (Fig. 5) showstwo signals at 5.32 and 4.95 ppm assigned to the aminic protons ofcoordinated cis-1,4-DACH. The triplet at 3.31 ppm was assignedto the protons of the methylenic group in position 3 of coordinatedPAM-H. These protons are shifted at higher field with respect tothe free ligand (Dd = -0.04 ppm). The broad singlet at 3.14 ppmwas assigned to the methynic protons of coordinated cis-1,4-DACH. The multiplet (a triplet of triplets) falling in the range2.27–2.16 ppm was attributed to the methylenic protons in position2 of coordinated PAM-H. Finally, the broad signal falling at1.85 ppm was assigned to the methylenic protons of the cis-1,4-DACH ligands.
The 31P{1H}-NMR spectrum (Fig. 5) shows a signal at39.90 ppm assigned to the two P atoms of coordinated PAM-H,which are magnetically equivalent and shifted at lower field (Dd =21.2) with respect to the free ligand at the same pH*. The195Pt-NMR spectrum (Fig. 5) shows only one broad signal at-1599 ppm (a chemical shift characteristic of a Pt atom in a PtN2O2
coordination environment).40–41 Differently from complex 1B, 2Bshowed only one 195Pt signal, most probably because the longerchain of the aminic substituent prevents this group from exertingits deshielding effect on the proximal Pt atom.
For compound 2B, 31P{1H}-NMR spectra in the pH* rangefrom 0 to 13 were recorded. Differently from the case of free PAMligand, a constant 31P chemical shift (data not shown) was observedconfirming the involvement of all phosphonate hydroxyl oxygenatoms in coordination to platinum. Differently from complexes1A and 1B, compound 2B was found to be stable even at pH*
10910 | Dalton Trans., 2009, 10904–10913 This journal is © The Royal Society of Chemistry 2009891

values higher than 9. Most probably, the aminic group ofPAM-H, even after deprotonation, is unable to coordinate to plat-inum because of the long alkyl chain. This explanation is supportedby the observation of a single resonance for platinum while in theanalogous complex 1B, with a shorter alkyl chain, two distinctplatinum resonances were observed. Extending the 31P{1H}-NMRtitration above pH* 9, an inflexion point, corresponding to thedeprotonation of the aminic group of coordinated PAM-H, wasobserved at pH* ca. 11. It is to be noted that coordination ofPAM-H to platinum increases the acidity of the aminic groupeven if this latter group is not able to bind to the metal.
Cytotoxic activity
The in vitro cytotoxicity of the four new dinuclear complexeshas been evaluated in 13 human tumour cell lines of multipleorigins. The panel includes: LoVo and HT-29 (colorectal adeno-carcinoma cells), LoVo-MDR (Multi Drug Resistant colorectaladenocarcinoma cells), A549 (non small lung adenocarcinomacells), 2008 and A2780 (ovarian adenocarcinoma cells), A2780-ADR (Multi Drug Resistant ovarian adenocarcinoma cells),C13* (cisplatin resistant ovarian adenocarcinoma cells), MCF-7(mammary carcinoma cells), MCF-7-ADR (Multi Drug Resistantmammary adenocarcinoma cells), A375 (malignant melanomacells), A431 (cervical carcinoma cells), and A431/Pt (cisplatinresistant cervical carcinoma cells). Under the same experimentalconditions, cisplatin was also evaluated as reference metallodrug.IC50 values, calculated from the dose–survival curves obtainedafter 72 h of drug treatment from the MTT test, are reported inTable 1.
The less active complexes are those containing the [cis-Pt(NH3)2]2+ residues (1A and 2A), which show mean IC50 valuesof 50.34 (29.52–79.71) and 30.75 (14.83–49.83) mM, respectively,which are about 6 and 3.5 times higher than that of cisplatin(8.32 mM). In contrast, the two complexes containing [Pt(cis-1,4-DACH)]2+ (1B and 2B) show a better cytotoxic activity withmean IC50 values of 22.46 (6.25–66.65) and 13.57 (5.01–26.19) mM,respectively. In the panel, two pairs of cell lines have been selectedfor their sensitivity and resistance to cisplatin: A431 and A431/Ptand 2008 and C13* (first two pairs of cell lines in Table 1).Although cisplatin resistance is multifactorial, the main molecularmechanisms involved in Pt resistance of C13* and A431/Pt havebeen identified. In particular, in C13* cells resistance is correlatedto reduced cellular drug uptake, high cellular glutathione andthioredoxin reductase levels,44 and enhanced repair of DNAdamage.45 In A431/Pt cells, resistance is due to defect in druguptake and to decreased levels of proteins involved in DNAmismatch repair (MSH2), which causes an increased tolerance tocisplatin-induced DNA damage.46 By comparing the RF values(where RF is the resistance factor and is defined as the ratiobetween IC50 values calculated for the resistant cells and thosearising from the sensitive ones), it appears that all new compoundswith bisphosphonate ligands are able to overcome the cisplatin-resistance [mean RF values of 1.25 (1A and 2A), 0.92 (1B and 2B),and 1.73 (cisplatin) for A431 and A431/Pt cells, and of 1.12 (1Aand 2A), 0.93 (1B and 2B), and 10.46 (cisplatin) for 2008 and C13*cells]. The two complexes containing [Pt(cis-1,4-DACH)]2+ (1Band 2B) are slightly better than those containing [cis-Pt(NH3)2]2+
in overcoming the cisplatin resistance. This feature becomes much Tab
le1
Invi
tro
anti
tum
our
acti
vity
IC50
/mM
±S.
D.
Com
poun
dA
431
A43
1/P
t20
08C
13*
A37
5H
T-2
9A
549
LoV
oL
oVo
MD
RM
CF
-7M
CF
-7A
DR
A27
80A
2780
AD
R
1A39
.17
±0.
3851
.34
±1.
6451
.13
±2.
3259
.52
±1.
7351
.32
±1.
0579
.71
±1.
8459
.78
±2.
9046
.61
±0.
8239
.78
±0.
9553
.41
±1.
8551
.50
±1.
2129
.52
±1.
7341
.66
±2.
0(1
.31)
(1.1
6)(0
.86)
(0.9
6)(1
.41)
2A28
.43
±1.
0434
.19
±1.
1536
.26
±2.
2639
.19
±1.
9529
.26
±2.
1847
.03
±2.
0049
.83
±2.
2117
.43
±0.
7423
.09
±0.
9335
.64
±2.
1424
.72
±1.
2014
.83
±2.
2119
.91
±2.
0(1
.20)
(1.0
8)(1
.32)
(0.6
9)(1
.34)
1B19
.34
±1.
4912
.47
±0.
9213
.12
±1.
8713
.45
±1.
8835
.65
±2.
3466
.65
±2.
2731
.22
±3.
016.
25±
0.64
8.25
±1.
2419
.16
±2.
9337
.72
±1.
657.
22±
3.01
21.5
1±
1.11
(0.6
4)(1
.02)
(1.3
2)(1
.96)
(2.9
8)2B
10.2
5±
1.01
12.3
6±
2.53
11.8
0±
2.44
9.30
±2.
6021
.12
±1.
4526
.19
±2.
9117
.01
±1.
896.
25±
1.32
7.42
±1.
5110
.27
±0.
0823
.32
±0.
035.
01±
1.19
16.1
0±
0.33
(1.2
0)(0
.84)
(1.3
2)(2
.27)
(3.2
1)C
ispl
atin
1.67
±0.
392.
89±
0.64
2.12
±1.
6122
.18
±2.
011.
29±
2.05
21.2
9±
2.05
14.2
4±
1.37
6.31
±1.
156.
57±
0.39
7.60
±2.
498.
41±
1.22
6.54
±1.
177.
13±
2.4
(1.7
3)(1
0.46
)(1
.04)
(1.1
1)(1
.09)
Ant
itum
our
acti
vity
via
MT
Tte
st.I
C50
valu
esha
vebe
enev
alua
ted
bypr
obit
anal
ysis
(p<
0.05
,tes
tofc
2).
Cel
lsha
vebe
entr
eate
dfo
r72
hw
ith
incr
easi
ngco
ncen
trat
ion
ofte
sted
com
poun
ds.R
esis
tant
fact
or(R
F)
isde
fined
asIC
50re
sist
ant/
pare
ntlin
e.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 10904–10913 | 10911892

Table 2 Cisplatin cross-resistance profiles
IC50/mM ± S.D.
Compound A431 A431/Pt RF 2008 C13* RF
1A 53.23 ± 1.23 98.56 ± 2.38 1.85 87.41 ± 1.99 169.42 ± 1.59 1.932A 40.43 ± 1.04 71.31 ± 2.27 1.76 54.11 ± 1.46 98.12 ± 2.48 1.811B 32.13 ± 2.03 30.13 ± 1.49 0.96 29.24 ± 1.73 33.63 ±2.14 1.152B 15.62 ± 1.56 14.38 ± 1.42 0.85 16.34 ± 1.32 17.80 ± 1.37 1.08Cisplatin 22.06 ± 2.21 57.76 ± 3.94 2.61 5.23 ± 1.43 89.18 ± 2.08 17.05
Antitumour activity via MTT test. IC50 values have been evaluated by probit analysis (p < 0.05, test of c2). Cells have been treated for 48 h with increasingconcentration of tested compounds. RF (Resistant factor) is defined as IC50 resistant/parent line.
more evident by performing chemosensitivity tests at 48 h drugexposure (Table 2). Indeed, mean RF values for 1B and 2B appearroughly 3 and 15 times lower that those calculated for cisplatin incervical and ovarian carcinoma cells, respectively.
The human tumour cell line panel also includes three pairsof sensitive and multi-drug resistant cell lines (LoVo and LoVoMDR, MCF7 and MCF-7 ADR, and A2780 and A2780-ADR;the last three pairs in Table 1). In LoVo/MDR cells, resistance todoxorubicin is associated with an over expression of multispecificdrug transporters, such as the 170 kDa P-glycoprotein.47 Themulti-drug resistance of breast MCF-7 ADR and ovarian A2780ADR cancer cells is associated to an over expression of differentspecies of specific ATP-binding cassette transporters and mem-brane proteins.48,49 It appears that, while phosphonate compounds1A and 2A are able to overcome multi-drug resistance in all MDRsublines [mean RF values of 1.09 (LoVo and LoVo-MDR), 0.82(MCF7 and MCF7-ADR), and 1.37 (A2780 and A2780-ADR)],compounds 1B and 2B circumvent MDR only in the colorectalcell line pair, being the RF value 1.25 in LoVo/LoVo-MDR, 2.11in MCF7/MCF7-ADR, and 3.09 in A2780/A2780-ADR cells.This behaviour could be due to a different expression of MDR-related proteins in colorectal cancer in comparison with breast andovarian cancers, the latter notoriously possessing similar geneticpatterns.50
Furthermore, our preliminary pharmacological investigationindicates that the dinuclear complexes with pamidronate (secondand fourth row in Table 1) are more active than those with AHBP-H. This could be due to the greater stability of the PAM complexesat higher pH and the scarce tendency of the amine group in PAMto coordinate to platinum by displacing a phosphonate oxygen ora carrier amine ligand. The slightly greater activity of compoundsB, containing [Pt(cis-1,4-DACH)]2+ with respect to compoundsA containing [cis-Pt(NH3)2]2+, could also be explained with theinvolvement of the organic cation transporters (OCT) 1 and 2.These transporters have been recently invoked51 to explain thedifferent activity of cisplatin and oxaliplatin towards colo-rectalcancers. Oxaliplatin contains 1,2-diaminocyclohexane as non-leaving ligand and appears to be a good substrate for OCT1 andOCT2, while cisplatin is a poor substrate for these transporters.
Conclusion
Four new platinum complexes having a dinuclear structurewith a bisphosphonate (AHBP-H or PAM-H) bridging the twoplatinum moieties [cis-Pt(NH3)2]2+ or [Pt(cis-1,4-DACH)]2+) in aW conformation have been synthesized and fully characterized.
Although we have not been able to obtain crystals suitable forX-ray crystallography investigations for any of the four newcompounds to confirm their structure, all analytical and spec-troscopic measurements support the involvement in coordinationof all phosphonate hydroxyl oxygen atoms (each phosphonatecontributing one oxygen per platinum atom) resulting in aW-conformation of the type already reported and fully character-ized by X-ray diffraction for [{Pt(1R,2R-DACH)}2(medronate)].35
1H and 31P{1H}-NMR experiments at different pH values haveallowed the determination of the acidity constants for freepamidronate (those for free AHBP had already been reported)and an estimation of the stability of complexes 1A, 1B, and 2B (2Awas not sufficiently soluble in water). Complexes 1A and 1B arestable at pH values not exceeding 9.0, which was compatible withcarrying on the pharmacological investigations, while complex 2B(and presumably also 2A) is stable also at pH higher than 9. Thishas been found to be the result of the longer alkyl chain of PAM (ascompared to AHBP) holding the NH2 group farer from platinumand therefore less keen to coordination. This explanation was alsosupported by the equivalence of the platinum nuclei in 2B (oneresonance) but not in 1B (two separate resonances).
The in vitro cytotoxicity of the new complexes has been evalu-ated on a set of 13 human tumour cell lines comprising two pairsof cisplatin-sensitive and -resistant cell lines and 3 pairs of multi-drug-sensitive and -resistant cell lines. On the average, compoundsA (containing [cis-Pt(NH3)2]2+) are less active than B (containing[Pt(cis-1,4-DACH)]2+) and this could be due to an intrinsic featureof the [Pt(cis-1,4-DACH)]2+ moiety, which could have a betterpharmacokinetic and/or give more effective adducts with thetarget DNA. This latter hypothesis appears to be supported bythe better non-cross resistance of compounds B towards cisplatin-resistant cell lines. On the other hand compounds B appear to beless effective than compounds A against some multi-drug resistantcell lines. This latter aspect is worth of further investigation since,once more, platinum-DACH derivatives do not conform to thebehaviour of cisplatin analogues.
We also found that, on the average, compounds 2 are morecytotoxic than compounds 1. It is possible that this is the result ofa greater stability of compounds 2 with respect to compounds 1,particularly at high pH.
None of the four complexes has shown better activity thancisplatin. However, we believe that carboplatin, which unlikecisplatin but similarly to 1A, 1B, 2A, and 2B contains a O-donor chelate leaving ligand, could have been a better referencecompound. In fact preliminary data do indicate that several of thenew compounds possess similar or better activity than carboplatin.
10912 | Dalton Trans., 2009, 10904–10913 This journal is © The Royal Society of Chemistry 2009893

We want to recall that the new complexes are soluble in waterand are cationic under normal pH conditions, therefore theycould take advantage of the organic cation transporters and couldaccumulate more easily into the tumour cells.
The high solubility in water of complexes 1A, 1B and 2B rendersthese compounds ideal candidates for loading into inorganicmatrices to be used as bone-fillers and reservoir of Pt-based drugs.We have recently reported that the complex [{Pt(en)}2(AHBP-H)]+ 25 (Chart 1), embedded in inorganic composites (synthetichydroxyapatite nanocrystals or silica xerogels-based matrices), wasable, after activation, to release [PtCl2(en)] at a cytotoxic concen-tration. The new complexes reported in this work should havesimilar adsorption/release properties with respect to the inorganicmatrices but should be capable to release (after activation) theclinically used cisplatin or [PtCl2(cis-1,4-DACH)] (or their aquo-species), hence two Pt complexes with higher (or different) activitythan [PtCl2(en)]. We are currently investigating the release of thePt-complexes from inorganic matrices and their pharmacologicalproperties.
Acknowledgements
The authors thank the Universities of Bari and Padova, theItalian “Ministero dell’Universita e della Ricerca (MUR)”(PRIN 20078EWK9B), and the EC (COST Chemistry projectD39/0004/06), the Inter-University Consortium for Researchon the Chemistry of Metal Ions in Biological Systems(C.I.R.C.M.S.B.) for support.
References
1 Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug,ed. B. LippertVerlag Helvetica Chimica Acta, Zurich, 1999.
2 L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584.3 P. J. Loehrer and L. Einhorn, Ann. Intern. Med., 1984, 100, 704–713.4 R. C. Todd and S. J. Lippard, Metallomics, 2009, 1, 280–291, and
references therein.5 M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger, B. K.
Keppler, R. L. Reddick, M. Rozencweig, R. C. Young and F. M.Muggia, Dalton Trans., 2008, 183–194, and references therein.
6 D. D. Von Hoff, R. Schilsky, C. M. Reichert, R. L. Reddick, M.Rozencweig, R. C. Young and F. M. Muggia, Cancer Treat. Rep., 1979,63, 1527–1531.
7 I. H. Krakoff, in Platinum and Other Metal Coordination Compoundsin Cancer Chemotherapy: Clinical Applications of Platinum Complexes,ed. M. Nicolini, Martinus Nijhoff Publishing, Boston, 1988, 351.
8 P. J. Loehrer, S. D. Williams, Sr. and L. H. Einhorn, J. Natl. CancerInst., 1988, 80, 1373.
9 M. Kartalou and J. M. Essigmann, Mutat. Res., Fundam. Mol. Mech.Mutagen., 2001, 478, 23–43.
10 B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699.11 B. Rosenberg, L. Van Camp, J. E. Trosko and V. H. Mansour, Nature,
1969, 222, 385–386.12 T. G. Appleton, J. R. Hall and J. McMahon, Inorg. Chem., 1986, 25,
720–725.13 T. G. Appleton, J. R. Hall and J. McMahon, Inorg. Chem., 1986, 25,
726–734.14 M. Galanski, S. Slaby, M. A. Jakupec and B. Keppler, J. Med. Chem.,
2003, 46, 4946–4951.15 T. Klenner, P. Valenzuela-Paz, B. K. Keppler, G. Angres, H. R. Scherf,
F. Wingen, F. Amelung and D. Schmahl, Cancer Treat. Rev., 1990, 17,253–259.
16 J. M. Sanders, Y. Song, J. M. W. Chan, Y. Zhang, S. Jennings, T.Kostzowski, S. Odeh, R. Flessner, C. Schwerdtfeger, E. Kotsikorou,G. A. Meints, A. O. Gomez, D. Gonzalez-Pacanowska, A. M. Raker,H. Wang, E. R. van Beek, S. E. Papapoulos, C. T. Morita and E.Oldfield, J. Med. Chem., 2005, 48, 2957–2963.
17 E. Kotsikorou and E. Oldfield, J. Med. Chem., 2003, 46, 2932–2944.18 L. Widler, K. A. Jaeggi, M. Glatt, K. Muller, R. Bachmann, M. Bisping,
A.-R. Born, R. Cortesi, G. Guiglia, H. Jeker, R. Klein, U. Ramseier, J.Schmid, G. Schreiber, Y. Seltenmeyer and J. R. Green, J. Med. Chem.,2002, 45, 3721–3738.
19 J. H. Lin, Bone, 1996, 18, 75–85.20 T. Klenner, P. Valenzuela-Paz, B. K. Keppler and H. R. Scherf, J. Cancer
Res. Clin. Oncol., 1990, 116, 453–458.21 T. Klenner, F. Wingen, B. K. Keppler, B. Krempien and D. Schmahl,
J. Cancer Res. Clin. Oncol., 1990, 116, 341–350.22 N. Margiotta, R. Ostuni, D. Teoli, M. Morpurgo, N. Realdon, B.
Palazzo and G. Natile, Dalton Trans., 2007, 3131–3139.23 B. Palazzo, M. Iafisco, M. Laforgia, N. Margiotta, G. Natile, C. L.
Bianchi, D. Walsh, S. Mann and N. Roveri, Adv. Funct. Mater., 2007,17, 2180.
24 N. Margiotta, F. Capitelli, R. Ostuni and G. Natile, J. Inorg. Biochem.,2008, 102, 2078–2086.
25 M. Iafisco, B. Palazzo, M. Marchetti, N. Margiotta, R. Ostuni, G.Natile, M. Morpurgo, V. Gandin, C. Marzano and N. Roveri, J. Mater.Chem., 2009, 19, 8385–8392.
26 R. D. Feltham and R. G. Hayter, J. Chem. Soc., 1964, 4587–4591.27 M. Casolaro, I. Casolaro, A. Spreafico, C. Capperucci, B. Frediani, R.
Marcolongo, N. Margiotta, R. Ostuni, R. Mendichi, F. Samperi, T.Ishii and Y. Ito, Biomacromolecules, 2006, 7, 3417–3427.
28 R. Ranaldo, N. Margiotta, F. P. Intini, C. Pacifico and G. Natile, Inorg.Chem., 2008, 47, 2820–2830.
29 S. C. Dhara, Indian J. Chem., 1970, 8, 193–194.30 KaleidaGraph 3.5, Synergy Software, Reading, PA, USA, 2000.31 G. Marverti, P. A. Andrews, G. Piccinini, N. Ghiaroni and D. Barbieri,
Eur. J. Cancer, 1997, 33, 669–73.32 A. Castagna, P. Antonioli, H. Astner, M. Hamdan, S. C. Righettii, P.
Perego, F. Zumino and P. G. Righetti, Proteomics, 2004, 4, 3246–3267.33 D. T. Vistica, P. Skehan, D. A. Scudiero, A. Monks, A. Pittman and
M. R. Boyd, Cancer Res., 1991, 51, 2515–2520.34 M. C. Alley, D. A. Scudiero, A. Monks, M. L. Hursey, M. J. Czerwinski,
D. L. Fine, B. J. Abbott, J. G. Mayo, R. H. Shoemaker and M. R. Boyd,Cancer Res., 1988, 48, 589–601.
35 R. Bau, S. K. S. Huang, J.-A. Feng and C. E. McKenna, J. Am. Chem.Soc., 1988, 110, 7546–7547.
36 K. Libson, E. Deutsch and B. L. Barnett, J. Am. Chem. Soc., 1980, 102,2476–2478.
37 S. J. Berners-Price, L. Ronconi and P. J. Sadler, Prog. Nucl. Magn.Reson. Spectrosc., 2006, 49, 65–98.
38 D. G. Gorenstein, Prog. Nucl. Magn. Reson. Spectrosc., 1984, 16, 1–98.39 Y.-A. Lee, K. C. Young and S. S. Youn, J. Inorg. Biochem., 1997, 68,
289–294.40 F. D. Rochon and L. M. Gruia, Inorg. Chim. Acta, 2000, 306, 193–
204.41 F. D. Rochon and V. Buculei, Inorg. Chim. Acta, 2005, 358, 2040–2056.42 E. R. van Beek, C. W. G. M. Lowik, F. H. Ebetino and S. E. Papapoulos,
Bone, 1998, 23, 437–442.43 T. G. Appleton, J. R. Hall, A. D. Harris, H. A. Kimlin and I. J.
McMahon, Aust. J. Chem., 1984, 37, 1833–1840.44 C. Marzano, V. Gandin, A. Folda, G. Scutari, A. Bindoli and M. P.
Rigobello, Free Radical Biol. Med., 2007, 42, 872–81.45 E. L. Mamenta, E. E. Poma, W. K. Kaufmann, D. A. Delmastro, H. L.
Grady and S. G. Chaney, Cancer Res., 1994, 54, 3500–3505.46 C. Lanzi, P. Perego, R. Supino, S. Romanelli, T. Pensa, N. Carenino,
I. Viano, D. Colangelo, R. Leone, P. Apostoli, G. Cassinelli, R. A.Gambetta and F. Zunino, Biochem. Pharmacol., 1998, 55, 1247–54.
47 C. Wersinger, G. Rebel and I. H. Lelong-Rebel, Amino Acids, 2000, 19,667–685.
48 J. Sedlak, O. Sedlakova, P. Hlavcak, L. Hunakova, J. Bızik, M. Grofovaand B. Chorvath, Neoplasma, 1996, 43, 389–395.
49 U. Stein, W. Walther, M. Lemm, H. Naundorf and I. Fichtner,Int. J. Cancer, 1997, 72, 885–91.
50 M. E. Schaner, D. T. Ross, G. Ciaravino, T. Sorlie, O. Troyanskaya, M.Diehn, Y. C. Wang, G. E. Duran, T. L. Sikic, S. Caldeira, H. Skomedal,I. P. Tu, T. Hernandez-Boussard, S. W. Johnson, P. J. O’Dwyer, M. J.Fero, G. B. Kristensen, A. L. Borresen-Dale, T. Hastie, R. Tibshirani,M. van de Rijn, N. N. Teng, T. A. Longacre, D. Botstein, P. O. Brownand B. I. Sikic, Mol. Biol. Cell, 2003, 14, 4376–86.
51 K. S. Lovejoy, R. C. Todd, S- Zhang, M. S. McCornick, J. A. D’Aquino,J. T. Reardon, A. Sancar, K. M. Giacomini and S. J. Lippard, Proc. Natl.Acad. Sci. U. S. A., 2008, 105, 8902–8907.
This journal is © The Royal Society of Chemistry 2009 Dalton Trans., 2009, 10904–10913 | 10913894