progress in enzyme-based biosensors using optical transducers
TRANSCRIPT

Microchim. Acta 148, 107–132 (2004)
DOI 10.1007/s00604-004-0273-8
Fundamental Review
Progress in Enzyme-Based Biosensors Using Optical Transducers
Martin M. F. Choi�
Department of Chemistry, Hong Kong Baptist University, 224 Waterloo Road, Kowloon Tong, Hong Kong SAR, P.R. China
Received July 6, 2004; accepted August 24, 2004; published online October 21, 2004
# Springer-Verlag 2004
Abstract. Enzyme-based biosensors are well devel-
oped and relatively mature technique in the biosensing
field. Biosensors that utilise enzymes as the recogni-
tion elements represent the most extensively studied
area. The organisation of an enzyme-based biosensor
requires the integration of the biocatalyst with the sup-
port or immobilised materials to the extent that the
biocatalytic transformation is either optically or elec-
tronically transduced. Any optical or electrical changes
at the support material as a result of the biocatalytic
process, that is, depletion of the reactant or formation of
the product, provide routes for the opto=electronic
transduction of the biological process occurring at
the sensing surface. This review focuses on the dis-
cussion of some enzyme immobilisation techniques
including physical and chemical immobilisation.
Enzyme-based biosensors using various optical detec-
tion methods such as absorptiometry, luminometry,
chemiluminescence, evanescent wave, and surface
plasmon resonance are also included. Finally, different
types of enzyme-based optical biosensors for ascorbic
acid, bilirubin, cholesterol, choline, ethanol, glucose,
glutamate=glutamine, lactate, penicillin, urea, and uric
acid determinations are presented.
Key words: Optical transducers; enzyme; biosensors.
Biosensors and their associated techniques are a fast
and rapid growing field which combine biochemistry,
biology, chemistry, physics, electronics and computer
science, etc. The term ‘‘biosensor’’ began to appear in
the scientific literature in the late 1970s. The first bio-
sensor, known as the ‘‘enzyme electrode’’, was pres-
ented by Clark in 1956 [1], and Clark and Lyons in
1962 [2] when the enzyme glucose oxidase (GOx) was
coupled with an amperometric oxygen (O2) electrode.
The change in the dissolved O2 was sensed by the elec-
trode and shown to be proportional to the concentration
of glucose in the sample. In the years that followed,
enzyme electrodes for a variety of other clinically
important substances were also demonstrated by incor-
porating the relevant enzymes to electrochemical sen-
sors. In 1977, Rechnitz et al. [3] designed a selective
electrode for arginine by immobilising living microor-
ganisms on the surface of an ammonia gas-sensing elec-
trode and they described it as a ‘‘bio-selective sensor’’.
Bio-selective sensor was later shortened to ‘‘biosensor’’
and has remained as the popular term for detection
devices which combines a sensing material of biological
origin and a physical transducer. Since then, the designs
and applications of biosensors in various fields of ana-
lytical science have continued to grow tremendously.
IUPAC [4, 5] recently defines a biosensor as a spe-
cific type of chemical sensor comprising a biological or
biologically derived recognition element either inte-
grated within or intimately associated with a physico-
chemical transducer. The biological element is capable
E-mail: [email protected]�On sabbatical leave at The University of North Carolina at
Chapel Hill in July 2004–July 2005

of recognising the presence, activity or concentration of
a specific analyte in solution. The recognition can be a
binding process for example, an affinity-based biosen-
sor consisting of an antibody, nucleic acid, receptor
protein or cell receptor, and synthetic receptor. The
other class of recognition elements is based on biocata-
lytic reactions, for instances, biosensors derived from
enzymes, microorganisms, tissue slices, and biomimetic
catalysts. The interaction of the recognition element
with a target analyte results in a measurable change in
a solution property, such as depletion of a reactant or
formation of a product. The transducer converts the
change in solution property into a quantifiable electrical
signal. The mode of transduction may be one of several
approaches, including electrochemical, optical, piezo-
electric, magnetic or thermometric transducers.
The fundamental and most important feature of a
biosensor is the construction of the biological recog-
nition element or more specifically the biological
recognition site for the interaction with the target ana-
lyte. These biological materials can be proteins, that is
enzymes [6, 7], receptors [6, 8], antibodies or antigens
[9–11], oligonucleotides or deoxyribonucleic acid
(DNA) fragments [12], or low molecular weight
molecules exhibiting affinity interactions with other
biomaterials such as cofactors, namely, nicotinamide
adenine dinucleotide (NAD) [13], biotin [14–18], or
for example, saccharides exhibiting affinity interac-
tions with lectins [19]. For biosensing, the interactions
between the biological recognition elements and the
analytes should also induce some kinds of signals
which can be interpreted and picked up by signal
transducers based on optical spectroscopy (e.g. ab-
sorption, reflectance, luminescence, optical rotation,
evanescent wave, and surface plasmon resonance),
and electrical transduction (e.g. current, potential,
capacitance, impedance, piezoelectric transduction,
and field effect transistor transduction).
Biosensors that utilise enzymes as the recognition
elements represent the most extensively studied area.
The high specificity of enzyme-substrate interactions,
and the usually high turnover rates of biocatalysts,
open the way to tailor-made sensitive and specific
enzyme-based biosensor devices [20–22]. The orga-
nisation of an enzyme-based biosensor requires the
integration of the biocatalyst with the support or
immobilised materials to the extent that the biocata-
lytic transformation is either optically or electroni-
cally transduced. Any optical or electrical changes
at the support material as a result of the biocatalytic
process, that is, depletion of the reactant or formation
of the product, provide routes for the opto=electronic
transduction of the biological process occurring at the
sensing surface as displayed in Fig. 1.
For example, the biocatalytic conversion of glucose
to gluconic acid at the GOx-immobilised materials
results in biocatalysed O2 depletion, hydrogen perox-
ide (H2O2) and Hþ formation.
D-glucose þ O2 þ H2O
���������!glucose oxidaseD-gluconic acid þ H2O2
Therefore, amperometric and chemiluminescence
detection of O2 and H2O2 were often used in the
development of biosensor devices.
The tailoring of biosensing systems requires the
assembly of biomaterials on solid supports and the
design of the appropriate signal transduction between
the biological matrices and the support material. The
chemical means to support the biomaterials include
the immobilisation of enzymes on solid surfaces by
means of polymers [23], membranes [24], carbon
paste [25–28], and sol-gel matrices [29, 30], etc.
Functionalisation of solid surfaces with monolayers
of organic compounds and biomaterials for the attach-
ment of biological recognition elements has been a
subject of extensive research [31]. It is one of the
crucial procedures to produce highly active enzyme-
immobilised materials for biosensing which in fact
can determine the success of a biosensor. Since the
main aim of this article is to review the current prog-
ress of the optical transducers for the enzyme-based
biosensors, the enzyme immobilisation methods can
only be briefly discussed in the later sections. Readers
interested in this topic can go through an excellent
review article [32] to obtain more information.
Optical Enzyme-Based Biosensors
Extensive studies in the field of sensors, and, in par-
ticular, in optical biosensors have been performed
Fig. 1. General scheme of biosensing technique
108 M. M. F. Choi

during the last few decades. The studies were basi-
cally driven by the vital practical needs of industry,
medicine, and environmental control and monitoring,
etc. for such analytical systems and devices. These
needs continue to grow at a rapid pace. Enzyme-based
biosensors using optical transducers have been devel-
oped over nearly half-century. Perhaps the earliest
example of an optical biosensor for clinical applica-
tion is a test strip for glucose in urine commercialised
in 1957 [33]. Glucose oxidase and peroxidase were
coimmobilised onto a cellulose pad. The H2O2 pro-
duced from the enzymatic oxidation of glucose reacts
with o-tolidine in the presence of peroxidase to form a
dye. The colour intensity is proportional to the glu-
cose concentration and determined visually by the
eye. It represents a semiquantitative measurement of
glucose. Later, the colour intensity was determined
more accurately by the reflectance meters.
The success of enzyme-based biosensors is based
on mainly two factors: The first one is the immobili-
sation or fixation of enzymes on solid substrates or
platforms. This step is extremely important as the
immobilisation method can enhance the working life-
time and shelf-time of the biosensors. It also affects
the sensitivity and limit of detection of the biosensors
to analytes. The second part is the optical transducers
which can affect the sensitivity, selectivity and limit
of detection of the biosensors. The sensitivity of a
biosensor depends on the affinity or catalytic proper-
ties of the biological component and the sensitivity of
the physico-chemical transducer. In this article, the
enzyme immobilisation methods are briefly reviewed,
and then followed by various techniques ofoptical trans-
ducers. Finally, some examples of optical enzyme-based
biosensors are presented.
This review covers approximately the period up to
June 2004. Consequently, there is a possibility that
some of the articles will be missed by the time this
review is published. The author has tried to fish out
articles from a computer search of SciFinder Scholar
(CAS). The author apologises for any omissions
which have not caught his eyes. There is certainly
some excellent work on optical enzyme-based biosen-
sors research going on that is not reported here. As
stated above, this review does not claim to be com-
prehensive. Rather, it will offer some guidance in
identifying interesting development in the current
enzyme-based optical biosensors research.
Techniques of Enzyme Immobilisation
The main reason for enzyme immobilisation is to
allow the re-use of immobilised-enzyme over an
extended period of time, hence leading to cost sav-
ings. Furthermore, it allows easier biosensor manipu-
lation and operation. Numerous methods have been
utilised for the immobilisation of enzymes in optical
biosensors and generally the techniques comprise
either chemical or physical methods. Both methods
intend to keep the enzymes on the solid support with-
out being washed out by solutions. Physical immo-
bilisation of enzyme includes adsorption on solid
supports=polymeric gels, entrapment in inorganic=organic polymeric gels, and confinement within semi-
permeable membranes as portrayed in Fig. 2 [34].
Chemical immobilisation of enzymes can be achieved
by covalently bonding the enzymes to functionalised
solid materials or intermolecular cross-linking of the
biomolecules as illustrated in Fig. 3 [34]. In general,
chemical immobilisation can provide longer shelf-life
and stable biosensors; however, the enzymatic activity
often is not as good as in case of immobilised-
enzymes prepared by physical immobilisation.
Physical Immobilisation
In the early stage of development of enzyme-based
optical biosensors, solid enzyme powders or enzyme
solution were deposited onto a solid support and then
kept in position by a semi-permeable membrane. The
semi-permeable membrane allows the free passage of
the substrate and product but confines the enzyme in a
Fig. 2. Physical immobilisation of enzymes
(black circles) by (a) confinement, (b) adsorp-
tion, and (c) entrapment
Progress in Enzyme-Based Biosensors Using Optical Transducers 109

fixed cavity between the membrane and optical trans-
ducer. The selectivity can be maintained by control-
ling the porosity and chemical properties of the
membrane. The simplest and most common of these
semi-permeable membranes are the dialysis mem-
branes, cellulose-based membranes, and ultra-filtra-
tion membranes.
Another type of simple and easy method of physical
immobilisation is the adsorption of enzymes onto solid
supports. The enzyme is dissolved in solution and the
solid material is in contact with the enzyme solution
for a fixed period of time. The unadsorded enzyme is
then removed by washing with buffer. The adsorption
mechanisms are governed by electrostatic attraction,
hydrophobic interaction and van der Waal’s forces. This
method is simple, mild and causes little or no enzyme
inactivation; hence, the enzyme activity is maintained
and the sensitivity of the biosensor will be higher.
Unfortunately, the immobilised enzymes are loosely
bound and tend to desorb from the solid materials after
repeated use. Consequently, this is not the method of
choice for most workers. In order to circumvent this
drawback, various enzymes have been entrapped within
cross-linked organic or inorganic polymers. The prep-
aration is done by cross-linking of the polymer in the
presence of the enzyme; thus, the enzyme can be phy-
sically entrapped in the micro-pores of the polymer.
The size of the pores is small, and this can prevent
larger enzyme from leaching but allows small analytes
to pass through to react with the entrapped enzyme
[34]. Sol-gel methods were popular with the enzyme
adding to the sol-gel solution. After the sol-gel has been
gelled, the enzyme will be entrapped within the poly-
meric network of the porous gel [35].
Chemical Immobilisation
The most popular chemical immobilisation for
enzyme-based biosensors is the covalent bonding of
enzymes to solid supports. The binding of the
enzymes to the solid support is achieved by activating
the surface of the support and following with coupling
to the activated surface. The excess and unreacted
enzymes are removed by washing with buffer. This
technique provides permanent and stable attachment
of enzyme molecules to the support; however, some
of the enzymes are prone to be deactivated in the
coupling reaction. The following section summarises
some common ways of activating the solid surfaces
for the covalent attachment of enzymes.
Cyanuryl chloride (2,4,6-trichlorotriazine) is a
versatile reagent for the activation of surfaces con-
taining hydroxy groups and subsequent immobi-
lisation of enzyme layers [36–38]. Solid surfaces
functionalised by hydroxy groups react with 2,4,6-
trichlorotriazine to oxy-substituted mono- and di-
chlorotriazine layers as shown in Fig. 4 [32]. These
Fig. 3. Chemical immobilisation of enzy-
mes (black circles) by (a) surface covalent
bonding, and (b) cross-linking
Fig. 4. Attachment of enzyme via 2,4,6-trichlorotriazine to solid
surfaces
110 M. M. F. Choi

residues react with the amino acid residues of the
enzymes to form a monolayer of enzyme on the solid
surface.
Similarly, solid surfaces can be functionalised to
carboxylic acid with subsequent attachment with
enzymes. The carboxylic acid residues are converted
to an active ester using carbodimide reagents [39], to
acyl halides using thionyl chloride [40], or to a mixed
anhydride by the reaction with an anhydride [41] prior
to the coupling of the enzymes on the solid support as
displayed in Fig. 5 [32]. Likewise, solid surfaces can
be functionalised to amino groups with subsequent
attachment with enzymes. The enzyme is coupled
with the amino residues surface by glutaraldehyde
[39] as shown in Fig. 6 [32].
Alkoxy- or halosilanes are reactive substances for
the derivatisation of hydroxy-functionalised supports,
and their hydrolysis groups yields siloxane mono-
layers or multilayers on the solid surfaces (Fig. 7
[32]). In essence, the covalently linkage of enzyme
to solid surfaces can be achieved by a wide range of
chemical reactions. Fig. 8a and b [32] lists a series
of functionalities used for the modification of solid
surfaces and applied to the secondary coupling
of enzymes. The amino functional groups of lysine
residues on the enzymes can be linked to surface
carboxy groups generating amide bonds or to surface
aldehyde functional groups to yield imine bonds
[42–44]. Alternative techniques for attaching enzymes
to solid surfaces include nucleophilic substitution of
Fig. 5. Covalent linkage of enzyme layers to solid sur-
faces by coupling of carboxy functionalities
Fig. 6. Covalent linkage of enzyme layers to
solid surfaces by coupling of amino function-
alities
Progress in Enzyme-Based Biosensors Using Optical Transducers 111

surfaces funtionalised with cyanuric chloride [45–52],
incorporation of the isothiocyanate functionalities
[53, 54], and Michael additions to quinone function-
alities linked to solid surfaces [55]. Aromatic amines
linked to surfaces enable the generation of the sur-
face-bound diazonium salt, which reacts with tyro-
sine or histidine residues to form enzymes linked
to the solid support through azo groups [56]. The
photochemical activation of a phenylazide monolayer
yields the respective nitrene, which reacts with the
amino group of lysine residues of the enzyme [57].
Enzymes containing cysteine residues can be cova-
lently linked to surfaces functionalised by p-mercury-
benzoate [58], iodoacetamide [59] or maleimide
[60–62].
The immobilisation of enzymes by cross-linking
with bifunctional reagents provides another means
for incorporating enzymes onto solid surfaces. The
enzyme molecules can be either cross-linked with
each other or in the presence of a functionally
protein such as bovine serum albumin. The latter
reagent is added in order to increase the number of
cross-linking sites and to provide physical strength.
Of the considerable number of cross-linking agents
available, glutaraldehyde has found widespread use
in enzyme-based optical biosensors. The glutaralde-
hyde cross-linking procedures have been attractive
due to their simplicity and the strong chemical bond-
ing achieved by the biomolecules. The main draw-
back has been the possibility of activity losses due to
the distortion of the active enzyme conformation and
the chemical alterations of the active site during
cross-linking [32].
Optical Transducers
The transducer is an important component in a bio-
sensor through which the measurement of the target
analyte(s) is achieved by selective transformation of
a biomolecule-analyte interaction into a quantifiable
electrical or optical signal. A wide range of optical
and electrochemical instruments have been em-
ployed in conjunction with biosensing. In general the
optical transducers of most common enzyme biosen-
sors is based on optical techniques such as absorp-
tion, reflectance, luminescence, chemiluminescence,
evanescent wave, surface plasmon resonance, and
interferometry.
Absorbance=Reflectance-Based Transducers
The optical detection principle of absorbance trans-
ducers is fundamentally based on the Beer-Lambert
law:
log ðIo=IÞ ¼ A ¼ "Cl
where Io is the intensity of incident light; I is the
intensity of transmitted light; A is absorbance; " is
the molar absorbance of the analyte at a specific
wavelength; C is the concentration of analyte; and l
is the path length of light through solution.
Fig. 7. Assembly of functionalised, layered siloxane assem-
blies on solid surfaces for the immobilisation of enzymes. Z
is a complementary functional group for X
112 M. M. F. Choi

Fig. 8. (a) Various monolayer surface functionalities for the covalent linkage of enzymes. (b) Various monolayer surface functionalities for
the covalent linkage of enzymes
Progress in Enzyme-Based Biosensors Using Optical Transducers 113

Some biosensing methods employed this photo-
metric properties at which the analyte or product
involve absorbance changes at specific wavelengths.
The common absorbance transducers use a single fibre
or fibre bundle brings light to the analyte-sensitive
reagent phase and the transmitted or reflected light is
Fig. 8 (continued)
114 M. M. F. Choi

returned to a measurement instrument or detector via
fibre(s) as displayed in Fig. 9a. The extent of the ab-
sorption depends on the absorption cross-section of the
transducing molecule, the optical path length and the
illumination wavelength. Changes in chemical envi-
ronment can modify the absorption of the biorecogni-
tion element, and this modification is monitored as a
change in transmitted intensity within the biosensor.
When dealing with non-transparent measuring
environment, it becomes difficult to measure trans-
mitted light satisfactorily and in these cases the inten-
sity of the reflected light may be used as a measure of
the colour of the recognition element, analyte, or prod-
uct as shown in Fig. 9b. The Kubelka-Munk equation
provides an effective approach for quantitatively relat-
ing the observed signal to the sample concentration
for diffuse reflectance measurements. The Kubelka-
Munk function, f(R), is defined as:
f ðRÞ ¼ ð1 � RÞ2=2R
where R is the absolute diffuse reflectance. This func-
tion is directly related to the concentration of an
absorbing sample species, C, by
f ðRÞ ¼ "C=S
where " is the molar absorbance of the sample species
and S is the scattering coefficient of the sample
surface.
The best known example of biosensor is the involve-
ment of NADþ=NADH in the biosensing reactions:
pyruvateþNADHþHþ�����������������!lactate
dehydrogenaseL-lactateþNADþ
NADH has a strong absorbance at absorption peak
maximum 340 nm, but NADþ has no absorbance at
this wavelength. NADH also gives fluorescence at
�450 nm. The absorbance-based transducer can moni-
tor the absorption change at 340–360 nm and the sig-
nal is then related to the concentration of pyruvate or
L-lactate.
Luminescence Transducers
Luminescence transducers combine high selectivity
and sensitivity to monitor changes of various param-
eters in the analytical system such as concentration
of the analytes and products, concentration of quench-
ers, protolytic and complexation reactions, membrane
potential, and hydrophobicity or hydrophilicity, etc.
Luminescence transducers provide a large and im-
portant basis for the development of enzyme-based
biosensors. Main approaches in luminescence sens-
ing utilise either luminescence intensity or lifetime
measurement.
The relationship between luminescence intensity
and analyte concentration under weakly absorbing
solutions can be described by the Parker’s equation:
L ¼ 2:31Io�"Clk
where L is the luminescence intensity; Io is the inten-
sity of excitation light; � is the luminescence quantum
yield; " is the molar absorbance of the analyte at the
excitation wavelength; C is the concentration of ana-
lyte; l is the path length of light through solution; and
k is an instrumental constant.
Fig. 9. Absorbance-based optical transducer with (a) absorption configuration, and (b) reflectance configuration
Progress in Enzyme-Based Biosensors Using Optical Transducers 115

The construction of luminescence-based trans-
ducer is similar to that of an absorbance-based
transducer. Excitation light is guided to the recogni-
tion element which is exposed to the analyte. The
fluorescence is then collected by the detection sys-
tem as depicted in Fig. 10. Any change in lumines-
cence intensity, phase or lifetime can be related to
the interaction of recognition element, analyte, and
product.
In addition, the principle of luminescence quench-
ing is also commonly employed in luminescence-
based transducer. According to Stern and Volmer,
the relationship between intensities or lifetimes in
the absence and presence of quencher is given by:
Io=I ¼ 1 þ KSV ½Q� ¼ �o=�
where Io and I are the luminescence intensities in the
absence and presence of quencher Q, respectively;
KSV is the Stern-Volmer constant; [Q] is the quench-
er concentration; �o and � are the luminescence life-
times in the absence and presence of quencher Q,
respectively.
A typical example for this type of transducer is
represented by the oxygen sensors based on lumines-
cence quenching of long-lived ruthenium complexes
[63–67]. These oxygen sensors have been applied
successfully to some enzyme-based optical biosensors
which are described in subsequent sections.
Chemiluminescence Transducers
The emission of light due to the chemiluminescence
reaction between reagent and analyte has been em-
ployed in enzyme-based optical biosensors. For
example, Freeman and Seitz [68] developed a H2O2
biosensor based on the peroxidase-mediated oxidation
of luminol by H2O2 at alkaline conditions. In general,
the chemiluminescence reaction can be represented as:
A þ B����!catalystC�
C� ! C þ h�
Chemiluminescence occurs as a result of the
oxidation of certain substances, usually with O2 or
H2O2, to produce light in the cold and in the absence
of any exciting illumination. The optical transducer
is configured to pick up the emitted light and trans-
mitted to an optical detector for recording as shown
Fig. 10. Biosensor with luminescence-based optical transducer. (a) 90�, and (b) 180� arrangement
116 M. M. F. Choi

in Fig. 11. The main disadvantages of this type of
optical transducer are finite lifetime due to reagent
consumption and steady-state mass transfer required
to get a constant signal.
Evanescent Wave Transducer
An interesting approach of optical transducer applied
to enzyme-based biosensors has been proposed. For
example, a biorecognition element is immobilised on
the surface of an optical fibre to sense analyte when it
contacts the fibre surface. At each internal reflection
in the optical fibre, interference between the incident
and reflected internal beam creates a non-propagating
standing wave in the medium, perpendicular to the
reflecting surface. The energy associated with this
wave tails out to the surroundings, where it can inter-
act with analytes in the environment. This tailing phe-
nomenon is known as the evanescent wave. In a rough
estimation, the depth of penetration of the field typi-
cally is in the order of the wavelength of the light
used. Analytes interacted with the recognition element
on the lateral optical fibre surface change the light
propagation characteristics of the optical fibre by their
absorbance, dielectric or reflective index behaviour.
The amount of light leaking into the environment is
easily affected by these changes, and such alterations
can be measured at the distal end of the optical fibre
as depicted in Fig. 12. The major field of evanescent
wave transducers appears to be in immunodetection
with labelled antigen=antibody couples.
Surface Plasmon Resonance Transducer
Another interesting approach for optical transducer
is based on the surface plasmon resonance (SPR)
effect on metal surface. A thin metallic film is
deposited on a polished surface (e.g. a prism) which
acts as the support for the surface plasmon. A high-
ly refractive dielectric biorecognition layer is then
deposited on the metal film to monitor the interac-
tion of biorecognition element and analyte. A plane-
polarised light beam is directed onto the prism
placed on a rotation stage. Reflectance is measured
as a function of angle. The sample solution passes on
the metal surface as shown in Fig. 13. The interac-
tion is highly dependent on the surface refractive
index of the analyte and is detected by the change
in the resonance angle of the exciting plane po-
larised light. Again, the major field of application
of SPR transducer is in the immunosensing, because
the binding of, for example, an antigen on the metal
surface to an antibody in solution causes the reso-
nance angle to change significantly.
Fig. 11. Biosensor with chemiluminescence-based optical transducer
Fig. 12. Biosensor with evanescent wave transducer
Progress in Enzyme-Based Biosensors Using Optical Transducers 117

Other Optical Transducers
Scientific knowledge will always advance and new opti-
cal techniques will be employed in the enzyme-based
optical biosensors. To date many optical techniques
including interferometry, scattering spectrometry, pho-
toacoustic spectrometry, ellipsometry, polarimetry and
diffraction, etc. have been explored and used as optical
transducers in enzyme-based biosensors. So far their
popularities are not as good as the above-mentioned
optical transducers since they often require complex
and expensive apparatus for construction. Thus, these
optical transducers are not discussed in this review.
However, readers interested with these topics can con-
sult a book edited by Wolfbeis [69].
Examples of Enzyme-Based
Optical Biosensors
Numerous review articles [32, 35, 70–81] have fo-
cused on techniques and theories of enyzme-based
optical and electrochemical biosensors. This review
gives typical and useful examples of some common
enzyme-based optical biosensors. From a practical
and commercial point of view, four typical biosensors
have been widely used. Glucose biosensors are ap-
plied in diagnosis and treatment of diabetes, food
science and biotechnology. Lactate biosensors are
used in sports medicine, critical care, food science
and biotechnology. Urea biosensors are employed in
clinical applications. Glutamate=glutamine biosensors
are utilised in food science and biotechnology. In
addition, other developed enzyme-based optical bio-
sensors such as ascorbic acid, bilirubin, cholesterol,
choline, ethanol, glutamate=glutamine, penicillin, and
uric acid are also presented.
Ascorbic Acid Optical Biosensors
Ascorbic acid is an essential part of healthy diet and
thus it is important to quantify the amount of this
biochemical in food substances. Ascorbic acid is a
reducing agent which can involve in many chemical
reactions and these can form the basis for its detec-
tion. Unfortunately, only very few enzyme-based opti-
cal biosensors for ascorbic acid have been reported.
For instance, an ascorbic acid optical biosensor was
fabricated by immobilising ascorbic acid oxidase on
an oxygen-sensitive sensor membrane which mea-
sured the O2 consumption in the enzymatic oxidation
of ascorbic acid [82]. The detection principle was
simply based on the O2 fluorescence quenching on
an O2-sensitive sensor membrane.
Fig. 13. Biosensor with surface plasmon resonance transducer: (a) experimental arrangement, and (b) attenuated reflective curves for SPR
(1) before and, (2) after exposure to analyte
118 M. M. F. Choi

Bilirubin Optical Biosensors
Bile determination is very useful to diagnose many
gastric pathologies. The determination of bilirubin in
clinical analysis becomes an important issue. Biliru-
bin was oxidised to biliverdin by bilirubin oxidase,
and was quantitatively determined by measuring the
fluorescence quenching of a luminescence dye caused
by O2 consumption in the enzymatic reaction [83, 84].
Alternatively, coupling with immobilized GOx=haemoglobin and glucose, bilirubin was oxidised to
biliverdin by H2O2. The decrease in its absorption at
450 nm was monitored and its concentration was then
determined [85].
Cholesterol Optical Biosensors
The determination of cholesterol levels is of particular
importance in the clinical diagnosis of diseases such
as coronary heart disease, myocardial infarction and
arteriosclerosis. To date most of the enzyme-based
cholesterol biosensors utilise cholesterol oxidase
(ChOx) to catalyse the oxidation of cholesterol by
molecular O2 to 4-cholesten-3-one and H2O2. As a
result, cholesterol can be determined by either mon-
itoring the O2 or H2O2 level optically or ampero-
metrically. For instances, Wu and Choi [86] employed
the hydrogel network matrices to entrap ChOx and
octadecylsilica for optical biosensing cholesterol
in hydrophobic organic or aqueous micelle solvents
in conjunction with an optical O2 transducer. This
organic-phase cholesterol biosensor was successfully
applied to determine the free cholesterol content in
commercial butter samples.
Similarly, other cholesterol biosensors based on
O2 transducer were proposed. Cholesterol oxidase was
entrapped=immobilised in, a cellulose acetate mem-
brane coupled with an O2-sensitive membrane [87], a
graphite powder layer deposited onto an O2-sensitive
silicone film [88], and covalently on a nylon mem-
brane in conjunction with an O2-sensitive membrane
[89].
Alternatively, cholesterol may be determined by
chemiluminescence. Marquette et al. [90] exploited
the luminol electrochemiluminescence for the
development of fibre-optic biosensors for cholesterol.
Zhang and Ma [91] fabricated a fibre-optic biosensor
responding to both of cholesterol esters and free cho-
lesterol by covalently coupling cholesterol esterase,
ChOx and horseradish peroxidase (HRP) to bovine
serum albumin via glutaraldehyde. This chemilumi-
nescence biosensor was based on H2O2-luminol-
HRP to produce the analytical signal.
Choline Optical Biosensors
Choline is an important neurotransmitter in mammals.
Due to the growing needs for on-site clinical monitor-
ing of choline, many efforts have been devoted to
develop choline biosensors. Choline oxidase (CLOx)
and an O2-sensitive dye was dispersed in a Nafion
film. Choline oxidase catalysed the oxidation of cho-
line to betaine and H2O2 while consuming O2.
choline þ 2O2 þ H2O���!CLOxbetaine þ H2O2
The fluorescence intensity of the O2-sensitive dye was
then related to the choline concentration [92]. A simi-
lar approach to determine choline-containing phos-
pholipids in serum with a fibre-optic biosensor was
also described. Choline oxidase was immobilised on
a nylon membrane and positioned onto an O2-sensi-
tive silicone membrane. Phospholipids were hydro-
lysed by the enzyme phospholipase-D to choline
which was subsequently analysed by the O2 trans-
ducer [93]. Furthermore, a fibre-optic biosensor based
on luminol electrochemiluminescence integrated in a
flow injection analysis (FIA) system was developed
for the detection of choline. Choline oxidase was ini-
tially immobilised covalently on solid supports. The
electrochemiluminescence of luminol was generated
by a glassy carbon electrode polarised at þ425 mV vs.
a platinum pseudo-reference electrode to detect
choline [94].
Ethanol Optical Biosensors
Ethanol optical biosensors based on alcohol oxidase
(AOx) and alcohol dehydrogenase (ADH) are com-
monly employed as the ethanol and alcohols recogni-
tion elements. Alcohol oxidase involves the catalytic
oxidation of short-chain aliphatic alcohols whereas
ADH possesses low selectivity for catalytic oxidation
of both aromatic and aliphatic alcohols [95]. Thus,
optical fluorometric biosensors for ethanol was devel-
oped, which was based on the enzymatic oxidation of
ethanol. Sensors layer contained O2-sensitive indi-
cators which showed the decrease in the local O2 par-
tial pressure as the result of the enzymatic oxidation
of ethanol [96, 97]. Other types of optical ethanol
biosensors employed ADH to convert NADþ into
Progress in Enzyme-Based Biosensors Using Optical Transducers 119

NADH in the presence of ethanol. The rate of produc-
tion of NADH was monitored fluorometrically and
related to the ethanol concentration [98, 99]. Fibre-
optic chemiluminescent biosensors for monitoring
aqueous ethanol also have been fabricated. The bio-
sensors operation schemes were based upon the AOx
enzymatic oxidation of ethanol to produce H2O2,
which then reacted with luminol to produce light.
The intensity of the emitted light was determined
and served as a measure for ethanol concentration
[100–102].
Glucose Optical Biosensors
Continuous monitoring of blood glucose is essential to
avoid the long-term consequences of elevated blood
glucose. A wide variety of optical sensing methods
has been proposed and includes colorimetry [103, 104],
fluorimetry [105–109], absorptiometry [110], near in-
frared spectroscopy [111, 112] and optical rotation
[113, 114]. The most commonly used technology for
blood glucose determination is an enzyme-based
method. Most of these biosensors utilise GOx as the
biological recognition element [115–130]. In the pres-
ence of O2, the enzyme converts glucose into gluco-
nolactaone and H2O2. These biosensors determine the
glucose concentration by monitoring amperometri-
cally the formation of H2O2 or the depletion of O2.
The fabrication of such sensors heavily focuses on
ways to eliminate interferences by, e.g., ascorbate,
urate, and acetaminophen.
For glucose optical biosensors, these interferences
do not exist to a large extent as the optical transducer
does not suffer from these interferents. Instead, the
determination of glucose is done by monitoring opti-
cally either the consumption of O2 or the formation
of H2O2. As O2 is an excellent quencher for most
luminescent compounds, most of the enzyme-based
optical glucose biosensor developed to date is based
on O2 luminescence quenching. Figure 14 displays
the general scheme of this type of glucose optical
biosensors.
In 1984, Uwira et al. [131] reported the first fluori-
metric determination of glucose by measurement of
the depletion in the dissolved O2 in the analyte sample.
The optical transducer was fabricated from an O2-sen-
sitive dye with which the fluorescence intensity was
monitored. Other fibre-optic glucose biosensors have
been prepared by covalently immobilising GOx on
the surface of S-layer ultrafiltration membrane [132],
incorporating GOx in a bilayer lipid film [133],
cross-linking GOx with glutaraldehye [134, 135] and
adsorpting and crosslinking GOx [136] on O2 sensors.
The optical transducers of these biosensors were based
on the luminescence quenching effect of O2 on ruthe-
nium(II) complexes. Other luminescence O2-sensitive
dyes such as decacyclene [137] have also been em-
ployed in O2 transducers for biosensing glucose.
Sol-gel matrices have been proved to be very useful
solid supports for the immobilisation of enzymes as
they can retain the enzyme activity and are considered
to be the best way of immobilising GOx to date [138].
Sol-gel based glucose biosensors employing optical O2
transducers have been developed [35, 139, 140]. The
encapsulated GOx exhibited excellent characteristics
in terms of activity, operational lifetime and optical
transparency. They enabled continuous monitoring of
the glucose concentrations in real samples. Again,
the optical transducers were based on luminescence
quenching effect of O2 on ruthenium(II) complexes.
Eggshell membrane biomaterial has been recently
reported to be an ideal enzyme immobilisation plat-
form for enzymes and the lifetimes of the immobilised
enzymes were much extended. An optical glucose
biosensor was then fabricated from a GOx-immobi-
lised eggshell membrane and an O2-sensitive ruthe-
nium complex sensor membrane [141].
A micrometer-sized fibre-optic fluorescence biosen-
sor for glucose was recently reported [142]. A lumines-
cent O2-sensitive ruthenium complex and GOx were
incorporated into an acrylamide polymer that was
attached covalently to a silanised optical fibre tip sur-
face by photocontrolled polymerisation. The perfor-
mance of this biosensor was comparable with that of
larger glucose biosensors but with the additional advan-
tages of miniaturisation. A disadvantage of this biosen-
sor was that the lifetime was short due to the leaching of
dye and enzyme from the fibre tip after repeating use.
Recently Xu et al. [143] stepped forward to reduce
the size of glucose optical biosensors to nano-scale forFig. 14. General design of glucose optical biosensors
120 M. M. F. Choi

intracellular glucose imaging. These optical nanosen-
sors were fabricated from polyacrylamide PEBBLEs
(Probes Encapsulated By Biologically Localised
Embedding) containing GOx, an O2-sensitive fluores-
cent ruthenium complex, and an O2-insensitive fluo-
rescent reference dye, Oregon Green 488-dextran or
Texas Red-dextran, for the purpose of ratiometric
intensity measurements. The enzymatic oxidation of
glucose to gluconic acid resulted in the local depletion
of O2, which was measured by the O2-sensitive ruthe-
nium dye. The PEBBLES matrix protected the en-
zyme and fluorescence dyes from interference by
proteins in cells. As the size of these nanosensors is
very small, they show potential for intracellular glu-
cose assay.
In addition, Papkovsky et al. [144] described an
immunosensor based on GOx using a luminescence
decay lifetime-based O2 sensor with phase measure-
ments. Although this immunosensor was not intended
to determine glucose, it could be applied to detect
human lactate dehydrogenase isoenzymes. The optical
transducer was operated under luminescence decay
time using the phase measurement technique. Based
on this similar technique, an enzyme-based biosensor
utilising GOx immobilised on a microporous O2 sen-
sor membrane was fabricated to determine glucose by
the phase shift measurement of the luminescent plati-
num complex of octaethylporphine-ketone [145].
The intrinsic fluorescence property of GOx has also
been employed as the optical transducer for glucose
biosensing. The fluorescence from the redox active
site flavine adenine dinucleotides (FADs) of GOx
was monitored on exposure to various glucose con-
centrations [146]. Similarly, Hartnett et al. [147]
focused on the study of the behaviour of GOx seques-
tered within sol-gel derived glasses. The steady-state
and time-resolved fluorescence from FADs of GOx
were monitored when in contact with glucose solu-
tions. The fluorescence intensity decreased with the
increase in glucose concentration. The results demon-
strated that a simple biosensing platform based on the
intrinsic fluorescence from the FAD residues within
GOx could be employed to quantify glucose.
Besides glucose biosensors based on O2 optical
transducers, an optical pH transducer has also been
employed to quantify glucose [148, 149]. Glucose
oxidase was physically immobilised on an optical
fibre. When GOx catalysed the oxidation of glucose
to produce gluconic acid, which, in turn, lowered the
pH in the microenvironment of the biosensor and this
change was then picked up by a pH-sensitive dye
incorporated into the sensing layer.
Finally, Wang et al. [150] reported the electro-
chemiluminescent detection of H2O2 which could be
applied to determine glucose by coimmobilisation of
polymeric luminol, iron(II) tris(5-aminophenanthro-
line) and GOx on an indium tin oxide glass. Unfor-
tunately it was an irreversible sensing scheme as
luminol was consumed during the reactions. Recently
Wolfbeis et al. [151] reported the use of a reversible
H2O2 sensor to quantify glucose by incorporating with
the GOx enzymatic reaction to generate H2O2.
In conclusion, glucose biosensors are, by far, the
most widely employed and therefore continue to drive
research toward better biosensors. The analytical per-
formance of most GOx-based optical biosensors to
date is thus summarised in Table 1 for the ease of
comparison.
Glutamate=Glutamine Optical Biosensors
The amino acid glutamate is the major excitatory neu-
rotransmitter used in the nervous system for interneuro-
nal communication. It is used throughout the brain by
various neuronal pathways including those involved in
learning and memory, locomotion, and sensory percep-
tion. Since glutamate is released from neurons on a
millisecond time scale into sub-micrometer spaces,
the development of a glutamate biosensor with high
temporal and spatial resolution is of great interest for
the study of neurological function and disease. Several
optical biosensors have been developed to monitor glu-
tamate. Glutamate dehydrogenase (GDH) was encap-
sulated in a silica sol-gel film on the tip of an optical
fibre whereas it catalysed the oxidative deamination
of glutamate to �-ketoglutarate and the simultaneous
reduction of NADþ to NADH, whose fluorescence
formed the basis of the detection [152]. Wang and
Arnold [153] employed a dual-enzyme of immobilised
GDH and glutamate-pyruvate transaminase (GPT) to
produce NADH at the tip of a fibre-optic probe. NADH
luminescence was monitored through this probe and
the measured fluorescence intensity was related to
the concentration of glutamate. Glutamate dehydroge-
nase catalyses the formation of NADH, and GPT drives
the GDH reaction by removing the reaction product
and regenerating glutamate. Cordek et al. [154] used
similar methods to immobilise GDH on a submicro-
metre optical fibre and studied the subcellular level
neurophysiological responses of glutamate.
Progress in Enzyme-Based Biosensors Using Optical Transducers 121

Table
1.
An
aly
tica
lp
erfo
rman
ceo
fvar
iou
sG
Ox
-bas
edo
pti
cal
bio
sen
sors
Bio
sen
sor
des
crip
tio
nT
ran
sdu
cer
Sta
bil
ity
Dy
nam
ic=
lin
ear
ran
ge
Det
ecti
on
lim
it
Res
po
nse
tim
e
Inte
rfer
ence
sR
ef.
En
zym
em
emb
ran
es
wit
h2
-Dcr
yst
alli
ne
stru
ctu
re
O2
lum
ines
cen
ce
qu
ench
ing
20
0h
(co
nti
nu
ou
s
op
erat
ion
)
1–
80
mM
(flow
mo
de)
0.5
–1
5m
M
(ste
ady
-sta
tem
od
e)
?1
00
s?
13
2
Bil
ayer
lip
iden
zym
e
film
O2
lum
ines
cen
ce
qu
ench
ing
?2
–5
mM
??
?1
33
GO
xcr
oss
lin
kin
g
enzy
me
lay
er
O2
lum
ines
cen
ce
qu
ench
ing
40
0h
(co
nti
nu
ou
s
op
erat
ion
)
0.1
–5
00
mM
(FIA
,zo
ne
sam
pli
ng
)
0.1
–1
0m
M
(FIA
,d
irec
tin
ject
ion
)
??
?1
34
GO
x-g
luta
rald
ehy
de
cro
ssli
nk
ing
enzy
me
lay
er
O2
lum
ines
cen
ce
qu
ench
ing
4w
eek
s
(use
dd
aily
)
0.0
6–
1m
M?
6m
in?
13
5
GO
xad
sorp
tio
n
and
cro
ssli
nk
ing
enzy
me
lay
er
O2
lum
ines
cen
ce
qu
ench
ing
6m
on
ths
0–
2m
M?
12
s?
13
6
GO
xim
mo
bil
ised
on
ny
lon
6–
6m
emb
ran
e
O2
lum
ines
cen
ce
qu
ench
ing
5m
on
ths
0.1
–2
0m
M0
.05
–0
.1m
M1
50
–2
10
s?
13
7
En
trap
ped
GO
x
sol-
gel
pow
der
O2
lum
ines
cen
ce
qu
ench
ing
28
0d
ays
0.0
6–
30
mM
6mM
5–
8m
inas
corb
ate,
caff
ein
e
(sli
gh
tin
terf
eren
ce)
13
9
San
dw
ich
edG
Ox
0.1
–1
5m
M2
50
–6
00
s
GO
xen
trap
ped
sol-
gel
O2
lum
ines
cen
ce
qu
ench
ing
>4
mo
nth
s0
.1–
8m
M?
50
–1
00
s?
35
En
trap
ped
GO
x
sol-
gel
pow
der
0.1
–4
mM
15
0–
25
0s
En
trap
ped
-GO
x
insi
lica
texer
og
el
hy
bri
dis
ed
wit
hh
yd
rox
yet
hy
l
carb
ox
ym
eth
yl
cell
ulo
se
O2
lum
ines
cen
ce
qu
ench
ing
3y
ears
0.0
09
–1
00
mM
9mM
6–
9m
inn
14
0
GO
x-i
mm
ob
ilis
ed
egg
shel
lm
emb
ran
e
O2
lum
ines
cen
ce
qu
ench
ing
>5
mo
nth
s0
.3–
2m
M0
.3m
M5
min
asco
rbat
e,u
rate
,
sucr
ose
(sli
gh
tin
terf
eren
ce)
14
1
Mic
rom
eter
-siz
e
op
tica
lfi
bre
glu
cose
bio
sen
sor
O2
lum
ines
cen
ce
qu
ench
ing
4–
6d
ays
0.7
–1
0m
M1
fmo
l;
0.7
5m
M
2s
?1
42
PE
BB
LE
sn
ano
sen
sor
O2
lum
ines
cen
ce
qu
ench
ing
?�
0.3
–8
mM
?�
10
0–
20
0s
n1
43
GO
xo
nm
icro
po
rou
s
lig
ht-
scat
teri
ng
sup
po
rtm
ater
ial
O2
lum
ines
cen
ce
qu
ench
ing
>3
mo
nth
s0
.2–
20
mM
?fe
wse
con
ds
?1
45
122 M. M. F. Choi

Intr
insi
cfl
uo
resc
ence
of
GO
x
FA
Dfl
uo
resc
ence
?1
.5–
2m
M?
2–
30
min
?1
46
GO
xw
ith
inso
l-g
el
der
ived
gla
ss
O2
lum
ines
cen
ce
qu
ench
ing
>6
mo
nth
s�
2–
7m
M?
1–
3m
in?
14
7
Ph
ysi
call
yim
mo
bil
ised
GO
xo
no
pti
cal
fib
re
pH
lum
ines
cen
ce
inte
nsi
ty
?0
.1–
2m
M?
8–
12
min
?1
48
Ph
ysi
call
yim
mo
bil
ised
GO
xo
no
pti
cal
fib
re
pH
lum
ines
cen
ce
inte
nsi
ty
?0
.1–
2m
M?
??
14
9
Po
lym
eric
lum
ino
l,
Fe(
ph
en-N
H2) 3
2þ
,
GO
xse
nso
r
H2O
2el
ectr
och
emi-
lum
ines
cen
ce
?1
0mM
–1
mM
50mM
?as
corb
ate
15
0
GO
xim
mo
bil
ised
on
H2O
2-s
ensi
tiv
e
hy
dro
gel
mem
bra
ne
H2O
2lu
min
esce
nce
tran
sdu
cer
?0
.1–
5m
M�
0.2
mM
>1
0m
inC
u2þ
,F
e3þ
,
citr
ate,
ph
osp
hat
e
15
1
nN
o,?:
no
tav
aila
ble
or
un
clea
r.
Progress in Enzyme-Based Biosensors Using Optical Transducers 123

Besides GDH, enzymes such as glutamate oxidase
(GLOx) and glutamate decarboxylase (GDC) have
also been used. Kar and Arnold [155] immobilised
GLOx at the tip of an ammonia sensor to produce a
glutamate biosensor whereas Dremel et al. [156] pro-
posed two kinds of fibre-optic glutamate biosensors.
In the first type, an O2 sensor was covered with a
membrane onto which was immobilised GLOx. The
decrease in O2 partial pressure in the presence of glu-
tamate as a result of enzymatic reaction was deter-
mined via dynamic quenching of the fluorescence of
the O2-sensitive indicator. In the second type, a carbon
dioxide-sensitive sensor was covered with a mem-
brane of immobilised GDC. The production of carbon
dioxide in the presence of substrate was determined
via the changes in the pH of a carbon dioxide sensor
consisting of a membrane-covered pH-sensitive fluo-
rescent pH indicator entrapped in a hydrogen car-
bonate buffer. The application of both biosensors to
determine glutamate in food and pharmaceutical sam-
ples was demonstrated.
Glutamine is a major source of nitrogen and carbon
in cell culture media. Thus, glutamine monitoring is
important in bioprocess control. Cattaneo et al.
[157, 158] developed chemiluminescence fibre-optic
biosensor systems for the determination of glutamine
in mammalian cell cultures. Glutaminase (GLA) and
GLOx were immobilised onto porous aminopropyl
glass beads by glutaraldehyde activation and packed
to form an enzyme column. These two enzymes acted
in sequence on glutamine to produce H2O2, which was
then reacted with luminol in the presence of ferricya-
nide to produce a chemiluminescent light signal that
was detected and quantified with a fibre-optic system.
In a similar fashion, glutamate and glutamine were
determined by luminol chemiluminescence with FIA
based on immobilised GLOx and GLA coupled with
Arthromyces ramosus peroxidase. The H2O2 produced
in the enzymatic reactions was determined by luminol
chemiluminescence catalysed by the peroxidase [159].
Lactate Optical Biosensors
Determination of lactate is essential in clinical analy-
sis for the diagnosis of lactate acidosis as a result of
metabolic, respiratory, or haemodynamic disturbance.
Typical fields of application for lactate biosensor are
in sports medicine for exercise control and in food
industry for the control of dairy products. Lactate
oxidase (LOx) is widely employed in biosensors to
determine lactate concentration based on the follow-
ing enzymatic reaction:
CH3CHðOHÞCO2�
ðL-lactateÞþO2���!
LOxCH3COCO2
�
ðpyruvateÞþH2O2
In most lactate biosensors, the consumption of O2 or
the production of H2O2 is monitored amperometri-
cally or optically and the lactate concentration is then
calculated. Other enzymes such as lactate dehy-
drogenase (LDH) and lactate monooxygenase (LMO)
have also been applied in the lactate biosensors:
CH3CHðOHÞCO2� þ NADþ ���!LDH
CH3COCO2�
þ NADH þ Hþ
CH3CHðOHÞCO2�þO2���!
LMOCH3COO�þCO2þH2O
Amperometric lactate biosensors have been studied
extensively as their configuration and design are very
similar to amperometric glucose biosensors. However,
the development of lactate optical biosensor is relative-
ly very scarce. For examples, lactate monooxygenase
was immobilised covalently on nylon membranes,
and the consumption of O2 was measured by follow-
ing, via a fibre-optic bundle, the changes in the fluo-
rescence of an O2-sensitive dye when the biosensor
was in contact with lactate [160]. Another biosensor
for the determination of L-lactic acid based on a fibre-
optic O2 sensor immobilised with LOx was developed.
The consumption of O2 was determined by dynamic
quenching of a fluorescence dye by molecular O2. For
the detection of L-lactic acid in milk products, sample
pretreatment proved necessary for protection of the bio-
sensor. Continuous L-lactate analyses could be carried
out with this enzyme-based biosensor for �2 days
[161]. Similarly, a fibre-optic biosensor based on bacte-
rial cytoplasmicmembranesas the biological recognition
element and an O2-sensitive dye layer as the transducer
has been described for the detection of lactate. Bacteria
with an induced LOx system were adsorbed onto a cel-
lulose disk. The disk was fixed mechanically over the
O2-sensitive siloxane layer on the distal end of an optical
fibre. This system detected lactate with no interference
from glucose, fructose or glutamic acid [162]. Lactate
biosensor based on measurement of phosphorescence
decay time of platinum(II) and palladium(II) complexes
of porphyrins was reviewed by Papkovsky et al. [163].
A fibre-optic biosensor based on the intrinsic fluo-
rescence of immobilised flavoproteins of LMO has been
employed to detect lactate. This method was based on
124 M. M. F. Choi

the change in fluorescence intensity of FAD as a pros-
thetic group of LMO during its interaction with lactate.
The fluorescence was monitored via fibre-optic light
guides at wavelengths >500 nm with fluorescence
excitation at around 410–450 nm. A characteristic fea-
ture of this biosensor is the narrow dynamic range that
usually does not exceed 3 mM [164].
An optical biosensor for the determination of L-
lactate based on immobilised HRP and LOx has been
described [165]. The fluorescence intensity of the
dimeric product of the enzyme catalysed oxidation
of homovanillic acid was utilised to determine H2O2
which was generated from the enzymatic oxidation of
L-lactate in the presence of O2. The biosensor has
been applied to the determination of lactate in control
serum.
Sol-gel encapsulation of LDH and its cofactor has
been employed as a disposable biosensor for L-lactate.
This biosensor utilised the changes in fluorescence
from the reduced cofactor NADH upon exposure
to L-lactate [166, 167]. Although problems such as
diminished enzymatic activity and=or leaching of
enzyme from the sol-gel matrix occurred, the sol-gel
process was sufficiently mild to permit retention of
enzymatic activity. The biosensor had a linear dynamic
range over the normal physiological L-lactate level and
had a long-term storage stability of at least 3 weeks
[149]. An ultrasensitive optical fibre lactate sensor with
rapid response time and 50mm size was developed.
Lactate dehydrogenase was covalently immobilised
onto an optical fibre probe surface and was applied
to determine the lactate content in food samples [168].
A bioluminescent fibre-optic sensor for the analysis
of L-lactate was developed using three different
enzymes, luciferase, NAD(P)H:FMN oxidoreductase
and LDH, immobilised on polyamide membranes.
Two kinds of sensing layer were studied, one consisting
of only one membrane on which the enzymes were
randomly coimmobilised, the other being a compart-
mentalised system obtained by stacking a luciferase=
oxidoreductase membrane on a LDH membrane.
After optimisation, the performances of the biosensor
in terms of sensitivity, detection limit and dynamic lin-
ear range of measurements were strongly improved by
compartmentalisation of the sensing layer compared
with those obtained using the coimmobilised system.
A fivefold increase in biosensor sensitivity was obtained
and the detection limit was 0.2mM lactate [169].
The luminol electrochemiluminescence has been
exploited for the development of several biosensors
allowing the detection of H2O2 and of substrates of
H2O2-producing oxidases. A glassy carbon electrode
was polarised at a fixed potential. Luminol was then
electrochemically oxidised and reacted in the pres-
ence of H2O2 to produce light. Lactate oxidase was
chemically immobilised on collagen or polyamide
membranes. Electro-optical flow injection analysis
of lactate was made possible [90, 170, 171].
Penicillin Optical Biosensors
Determination of penicillin is an important stage in
the control of biotechnological production processes
as well as in the quality control antibiotics prepara-
tion. In most of the developed enzyme-based optical
penicillin biosensors, penicillinase was used to catal-
yse the reaction of hydrolysis of the �-lactam ring in
penicillin to penicilloic acid [172] and the change in
pH was picked up by various pH optical sensors
[173, 174]. For instances, Polster et al. [175] em-
ployed phenol red as the basis of pH measurement.
Healey et al. [176, 177] immobilised penicillinase
and a pH-sensitive dye, fluorescein, on optical im-
aging fibres for fluorometric determination of peni-
cillin. Xie et al. [178] coimmobilised fluorescein
isothiocyanate and penicillinase on a preactivated
biodyne B membrane attached to the end of a bifur-
cated optical fibre for sensing penicillin. In addition,
Carlyon et al. [179] designed a single mode fibre-
optic evanescent penicillin biosensor. Penicillin G
was monitored at 633 nm by the decoloration of the
starch-iodine reagent when Bacillus cereus penicil-
linase was immobilised over the exposed core of a
monomode fibre.
Urea Optical Biosensors
The first enzyme-based urea optical biosensor was
developed by Goldfinch and Lowe [180] in 1984. A
pH-sensitive dye, bromothymol blue, and enzyme
urease were covalently immobilised on a glutathione
membrane. Enzymatic hydrolysis of urea resulted in
an increase in the pH of the medium,
ðNH2Þ2CO þ H2O���!urease2NH3 þ CO2
thus causing the colour change of the pH probe.
Most of the urea optical biosensors developed after-
wards fundamentally employed this enzymatic reaction
and a pH=NH3 optical transducer for determination of
urea. For instances, cellulose acetate membranes with
Progress in Enzyme-Based Biosensors Using Optical Transducers 125

brilliant yellow [181] or bromothymol blue [182], and
triacetylcellulose membranes with immobilised pH
indicator, neutral red, and urease [183] have been used
as absorption-based transducers for determination of
urea concentration whereas Koncki et al. [184, 185]
immobilised the pH-sensitive polymer polypyrrol=Prussian Blue, and urease on transparent film for spec-
trophotometric measurement of urea.
Another urea optical biosensor relied on the urease-
catalysed hydrolysis product, NH4þ which was
detected with an ion-selective sensing membrane
containing nonactin as ion-selective ionophore and
ETH 5294 chromoionophore in a 1-mm plasticised
poly(vinyl chloride) film. The transducer membrane
and the enzyme containing reaction layer were
sandwich-cast with spin coating onto the surface of
the sensing slide. The attenuation of the laser light
propagating inside the glass waveguide was used as
signal for urea analysis [186].
Wolfbeis and Li [187], and Chen and Wang [188]
designed fluorometric urea biosensors based on an
NH4þ-selective membrane. A urease enzyme layer
was immobilised on the NH4þ-selective polymer
membrane. The NH4þ-selective membrane utilised
dichlorofluorescein octadecyl ester as the anionic
chromophore and nonactin as the neutral ionophore.
Enzymatic hydrolysis of urea produced NH4þ ion
which was extracted into the polymer film to form
complexes with nonactin. Hþ was released which
resulted in a fluorescence change of the sensor mem-
brane due to the charge neutrality principle. This bio-
sensor was applied to determine urea in diluted serum
samples.
De Marcos et al. [189] directly photoimmobilised
urease with polyacrylamide onto a polypyrrole (PPy)
film. This PPy film showed an absorbance spectrum in
the near infrared range which was pH dependent. The
variation of absorbance was thus directly related to the
change of pH caused during the enzymatic reaction,
which was also dependent on the urea concentration.
The main advantage of this biosensor was that no pH-
sensitive indicator was needed.
Covalent immobilisation of amphiphilic mono-
layers containing urease onto optical fibres for fluoro-
metric detection of urea has been proposed [190].
Bifunctional 11 or 12 carbon chain length amphi-
philes with a triethoxychlorosilane group at one
terminus and an amine functionality at the other
terminus were covalently immobilised onto planar
quartz wafers and optical fibres. A small amount of
the fluorescent probe nitrobenzaoxadiazole dipalmi-
toylphosphatidylethanolamine (NBD-PE) was par-
titioned into the membranes from an aqueous
suspension. For coated wafers or fibres placed into
aqueous solutions, alterations of pH change the phys-
ical and electrostatic structure of the membranes,
which in turn altered the emission intensity of the
NBD-PE owing to changes in self-quenching. The
fluorescence intensity decreased as the degree of ioni-
sation of headgroups within the membrane decreased,
consistent with an increase in self-quenching. Urease
was covalently linked onto the functional groups at
the surface of these membranes. Addition of urea to
this system produced NH3 and carbonic acid and
results in changes in fluorescence intensity from the
immobilised layer owing to alteration of surface
charge at the membrane.
Furthermore, Rhines and Arnold [191] employed
two fluoresceins to probe the pH change after the
enzymatic hydrolysis of urea while Xie et al. [192]
developed fibre-optic urea biosensor based on immo-
bilised urease coupled to a fluorescence pH indicator
trisodium 8-hydroxypyrene-1,3,6-trisulphonate. The
enzymatically generated NH3 diffused through the
membrane into a solution of trisodium 8-hydroxy-
pyrene-1,3,6-trisulphonate resulting in the change in
fluorescence intensity. Instead of using absorption and
fluorescence spectroscopies, Gauglitz and Reichert
[193] also proposed the use of reflectance spectros-
copy and a pH-sensitive dye for determination of
urea.
Uric Acid Optical Biosensors
Uric acid and other oxypurines are the principal final
products of purine metabolism in the human body.
Abnormal levels of uric acid can cause gout, hyper-
uricemia, and Lesch-Nyan disease [194]. As such, it
is important to determine the concentration of uric
acid in human urine and=or blood to diagnose these
diseases. A fibre-optic biosensor for uric acid based
on immobilised uricase has been reported [195].
Uricase and HRP were coimmobilised on the inert
matrix bovine albumin via glutaraldehyde. Enzymat-
ic oxidation of uric acid resulted in the production
of H2O2 which subsequently reacted with thiamine
in the presence of HRP to produce the fluorescent
product thiochrome. The fluorescence intensity was
related to the uric acid concentration. Alternative-
ly, a chemiluminescent technique [196] was also
126 M. M. F. Choi
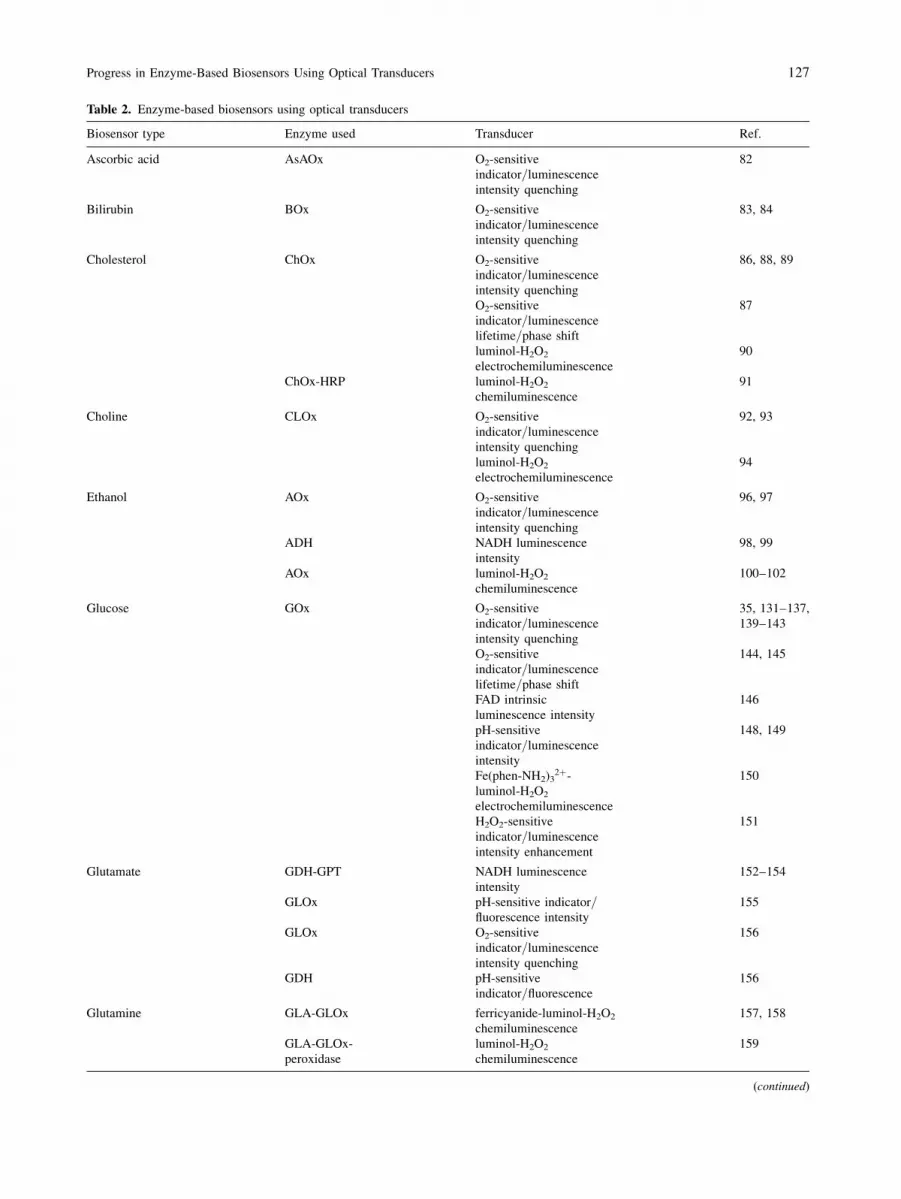
Table 2. Enzyme-based biosensors using optical transducers
Biosensor type Enzyme used Transducer Ref.
Ascorbic acid AsAOx O2-sensitive
indicator=luminescence
intensity quenching
82
Bilirubin BOx O2-sensitive
indicator=luminescence
intensity quenching
83, 84
Cholesterol ChOx O2-sensitive
indicator=luminescence
intensity quenching
86, 88, 89
O2-sensitive
indicator=luminescence
lifetime=phase shift
87
luminol-H2O2
electrochemiluminescence
90
ChOx-HRP luminol-H2O2
chemiluminescence
91
Choline CLOx O2-sensitive
indicator=luminescence
intensity quenching
92, 93
luminol-H2O2
electrochemiluminescence
94
Ethanol AOx O2-sensitive
indicator=luminescence
intensity quenching
96, 97
ADH NADH luminescence
intensity
98, 99
AOx luminol-H2O2
chemiluminescence
100–102
Glucose GOx O2-sensitive
indicator=luminescence
intensity quenching
35, 131–137,
139–143
O2-sensitive
indicator=luminescence
lifetime=phase shift
144, 145
FAD intrinsic
luminescence intensity
146
pH-sensitive
indicator=luminescence
intensity
148, 149
Fe(phen-NH2)32þ-
luminol-H2O2
electrochemiluminescence
150
H2O2-sensitive
indicator=luminescence
intensity enhancement
151
Glutamate GDH-GPT NADH luminescence
intensity
152–154
GLOx pH-sensitive indicator=fluorescence intensity
155
GLOx O2-sensitive
indicator=luminescence
intensity quenching
156
GDH pH-sensitive
indicator=fluorescence
156
Glutamine GLA-GLOx ferricyanide-luminol-H2O2
chemiluminescence
157, 158
GLA-GLOx-
peroxidase
luminol-H2O2
chemiluminescence
159
(continued)
Progress in Enzyme-Based Biosensors Using Optical Transducers 127

employed to fabricate an uric acid biosensor. Uri-
case and HRP were coimmobilised on a solid sup-
port. Uricase catalysed the oxidation of uric acid to
produce H2O2, which then reacted with luminol
under the catalysis of HRP to produce chemilumi-
nescence. The light intensity was related to the con-
centration of uric acid in the sample solution.
Finally, Table 2 summarises all the above-mentioned
enzyme-based biosensors using various enzymes and
optical transducers.
Table 2 (continued)
Biosensor type Enzyme used Transducer Ref.
Lactate LOx O2-sensitive
indicator=luminescence
intensity quenching
161, 162
LOx O2-sensitive
indicator=luminescence
lifetime=phase shift
163
LOx luminol-H2O2
electrochemiluminescence
90, 170, 171
LOx-HRP H2O2-sensitive
indicator=luminescence
intensity
165
LMO O2-sensitive
indicator=luminescence
intensity quenching
160
LMO FAD intrinsic
luminescence intensity
164
LDH NADH luminescence
intensity
149, 166–168
LDH, luciferase,
NAD(P)H:FMN
oxidoreductase
bioluminescence 169
Penicillin PC pH-sensitive
indicator=absorption
175
pH-sensitive
indicator=fluorescence
intensity
176–178
pH-sensitive
indicator=evanescent
wave absorption
179
Urea UA pH-sensitive
indicator=absorption
180–185
polypyrrole
film=absorption
189
pH-sensitive
indicator=reflectance
193
pH-sensitive
indicator=fluorescence
intensity
191, 192
NH4þ-sensitive
film=absorption
186
NH4þ-sensitive
film=fluorescence
intensity
187, 188, 190
Uric acid UC fluorescence intensity 195
luminol-H2O2
chemiluminescence
196
ADH Alcohol dehydrogenase; AOx alcohol oxidase; AsAOx ascorbic acid oxidase; BOx bilirubin oxidase; ChOx cholesterol oxidase; CLOx
choline oxidase; GDC glutamate decarboxylase; GLA glutaminase; GOx glucose oxidase; GDH glutamate dehydrogenase; GLOx glutamate
oxidase; GPT glutamate-pyruvate transaminase; HRP horseradish peroxidase; LDH lactate dehydrogenase; LMO lactate monooxygenase;
LOx lactate oxidase; NADH reduced nicotinamide adenine dinucleotide; PC penicillinase; UA urease; and UC uricase.
128 M. M. F. Choi

Techniques of Background Correction
for Enzyme-Based Optical Biosensors
The success in fabrication of enzyme-based optical
biosensors has been addressed in the previous sec-
tions. Unfortunately, it is often observed that the vari-
able background levels of factors such as dissolved
O2 and pH in real samples can affect the results of
enzyme-based biosensing especially when O2, pH,
NH3, or CO2 sensors are employed as the optical
transducers. Currently several strategies have been
used to overcome this problem [34]. The first method
is to employ a two-sensor technique [35, 148]. For
example, Wolfbeis et al. [35] utilised this technique
to determine the glucose concentration with the first
sensor only sensitive to the dissolved O2 and the other
to both O2 and glucose in the sample solution. The
second strategy is to make use of two different dyes
for ratiometric measurement. For instance, the first
dye serving as a reference indicator is insensitive to
the biosensing process while another indicator is sen-
sitive to the analyte of interest. The ratio of the two
signals is then correlated to the analyte concentration
[143, 197]. Finally the commonly used technique is to
keep the dissolved O2 content and pH of the standards
and samples almost constant by using air-saturated
buffered solutions and performing the analysis in a
relatively closed environment such as continuous-flow
or flow-injection systems [82, 134, 135, 137, 139,
140, 156].
Conclusion and Perspectives
The key elements in the integration of enzymes with
optical transducers include the physical and chemical
deposition of the enzymes on the solid supports, the
electrical=optical communication between the enzyme-
immobilised layers and the transducers, and the elec-
tronic transduction of the biocatalysed transformation
that occurs at the immobilised enzyme=transducer
interfaces. Monolayer and multilayer arrays of en-
zymes can be organised on transducers using co-
valent bonds, affinity interactions, or hydrophobic=hydrophilic interactions. Clearly, the architecture of
enzyme-immobilised layers on optical transducer ele-
ments is envisaged to offer exciting perspectives at the
frontiers of chemistry, biology, physics, medicine, and
material science. Biosensors are one of the most fruit-
ful, exciting, and interdisciplinary areas of research in
analytical science. The concepts of selectivity, revers-
ibility, detection limits, ruggedness, and shelf-life
remain extremely important characteristics of any bio-
sensors and always should be borne in mind. Despite
the complexity of sample matrix and the problems
associated with real sample measurements, significant
progress has already been made in improving reliabil-
ity and extending capabilities to higher sensitivity and
selectivity, and faster response time of biosensors. The
author believes that the biosensor field continues to
advance at an accelerated pace. Looking forward, the
largest market for biosensors is still in clinical and
environmental analysis.
Finally, the current trend of reducing costs of
biological recognition elements and instrumentation
will open new and cost-effective analytical horizons
and broaden existing areas such as clinical and
environmental analysis. Enzyme-based optical bio-
sensors provide novel ways of performing the rapid,
remote, in-line determination of a wide variety of
analytes in a range of application fields. Enzyme-
based biosensors either using optical or electrochem-
ical transducers are at the heart of the development
of compact, self-contained devices for diversified
chemical or biological analysis, which play an
increasingly important role in our modern society.
Their importance will certainly increase in the years
ahead.
References
[1] Clark L C Jr (1956) Trans Am Soc Artif Intern Organs 2:
41–57
[2] Clark L C Jr, Lyons C (1962) Ann N Y Acad Sci 102: 29–45
[3] Rechnitz G A, Kobos R K, Riechel S J, Gebauer C R (1977)
Anal Chim Acta 94: 357–365
[4] Turner A P F (2000) Science 290: 1315–1317
[5] Thevenot D R, Toth K, Durst R A, Wilson G S (2001) Biosens
Bioelectron 16: 121–131
[6] G€oopel W (1995) Biosens Bioelectron 10: 853–883
[7] Schmidt H-L, Schuhmann W (1996) Biosens Bioelectron 11:
127–135
[8] Zhong C-J, Porter M D (1995) Anal Chem 67: 709A–715A
[9] Bourdillon C, Demaille C, Gueris J, Moirous J, Sav�eeant J-M
(1993) J Am Chem Soc 115: 12264–12269
[10] Bourdillon C, Demaille C, Moirous J, Sav�eeant J-M (1994)
J Am Chem Soc 116: 10328–10329
[11] Bourdillon C, Demaille C, Moirous J, Sav�eeant J-M (1996)
Acc Chem Res 29: 529–535
[12] Yang M, McGovern M E, Thompson M (1997) Anal Chim
Acta 346: 259–275
[13] Bardea A, Katz E, B€uuckmann A F, Willner I (1997) J Am
Chem Soc 119: 9114–9119
[14] Pantano P, Morton T H, Kuhr W G (1991) J Am Chem Soc
113: 1832–1833
[15] Hoshi T, Takeshita H, Anzai J, Osa T (1995) Anal Sci 11:
311–312
Progress in Enzyme-Based Biosensors Using Optical Transducers 129

[16] Du X, Anzai J, Osa T, Motohashi R (1996) Electroanalysis 8:
813–816
[17] Chen Q, Kobayashi Y, Takeshita H, Hoshi T, Anzai J (1998)
Electroanalysis 10: 94–97
[18] Anzai J, Takeshita H, Kobayashi Y, Osa T (1998) Anal Chem
70: 811–817
[19] Willner I, Rubin S, Cohen Y (1993) J Am Chem Soc 115:
4937–4938
[20] Katz E, Heleg-Shabtai V, Willner B, Willner I, B€uuckmann A
F (1997) Bioelectrochem Bioenerg 42: 95–104
[21] Willner I, Katz E, Willner B, Blonder R, Heleg-Shabtai V,
B€uuckmann A F (1997) Biosens Bioelectron 12: 337–356
[22] Krull U J, Heimlich M S, Kallury K M R, Piunno P A E,
Brennan J D, Brown R S, Nikolelis D P (1995) Can J Chem
73: 1239–1250
[23] Curulli A, Carelli I, Trischitta O, Palleschi G (1997) Biosens
Bioelectron 12: 1043–1055
[24] Scheller F W, Schubert F, Neumann B, Pfeiffer D, Hintsche
R, Dransfeld I, Wollenberger U, Renneberg R, Warsinke A,
Johansson G, Skoog M, Yang X, Bogdanovskaya V,
B€uuckmann A, Zaitsev S Y (1991) Biosens Bioelectron 6:
245–253
[25] Dicks J M, Aston W J, Davis G, Turner A P F (1986) Anal
Chim Acta 182: 103–112
[26] Gorton L, Karan H I, Hall P D, Inagaki T, Okamoto Y,
Skotheim T A (1990) Anal Chim Acta 228: 23–30
[27] Wring S A, Hart J P (1992) Analyst 117: 1215–1229
[28] Rosen-Margalit I, Rishpon J (1993) Biosens Bioelectron 8:
315–323
[29] Glezer V, Lev O (1993) J Am Chem Soc 115: 2533–2534
[30] Avnir D, Braun S, Lev O, Ottolenghi M (1994) Chem Mater
6: 1605–1614
[31] Swalen J D, Allara D L, Andrade J D, Chandross E A, Caroff
S, Israelshvili J, McCarthy T J, Murray R, Pease R F, Rabolt
J F, Wynne K J, Yu H (1987) Langmuir 3: 932–950
[32] Willner I, Kata E (2000) Angew Chem Int Ed 39: 1180–1218
[33] Free A H (1977) Ann Clin Lab Sci 7: 479–485
[34] Kuswandi B, Andres R, Narayanaswamy R (2001) Analyst
126: 1469–1491
[35] Wolfbeis O S, Oehme I, Papkovskaya N, Klimant I (2000)
Biosens Bioelectron 15: 69–76
[36] Lin A W C, Yeh P, Yacynych A M, Kuwana T (1977)
J Electroanal Chem 84: 411–419
[37] Yacynych A M, Kuwana T (1978) Anal Chem 50:
640–645
[38] Ianniello R M, Wieck H J, Yacynych A M (1983) Anal Chem
55: 2067–2070
[39] Kamin R A, Wilson G S (1980) Anal Chem 52: 1198–1285
[40] Koval C A, Anson F C (1978) Anal Chem 50: 223–229
[41] Rocklin R D, Murray R W (1979) J Electroanal Chem 100:
271–282
[42] Bourdillon C, Bourgeois J P, Thomas D (1979) Biotechnol
Bioeng 21: 1877–1879
[43] Bourdillon C, Bourgeois J P Thomas D (1980) J Am Chem
Soc 102: 4231–4235
[44] Kuznetsov B A, Byzova N A, Shumakovich G P (1994)
J Electroanal Chem 371: 85–92
[45] Ianniello R M, Yacynych A M (1981) Anal Chem 53:
2090–2095
[46] Ianniello R M, Yacynych A M (1981) Anal Chim Acta 131:
123–132
[47] Ianniello R M, Lindsay T J, Yacynych A M (1982) Anal
Chem 54: 1098–1101
[48] Ianniello R M, Yacynych A M (1983) Anal Chim Acta 146:
249–253
[49] Razumas V J, Jasaitis J J, Kulys J J (1983) Bioelectrochem
Bioenerg 10: 427–439
[50] Jonsson G, Gorton L (1985) Biosensors 1: 355–368
[51] Osborn J A, Yacynych A M, Roberts D C (1986) Anal Chim
Acta 183: 287–292
[52] Antrim R F, Yacynych A M, Wieck H J, Lutter G W (1988)
Analyst 113: 341–344
[53] Katz E, Riklin A, Willner I (1993) J Electroanal Chem 354:
129–144
[54] Willner I, Riklin A, Shoham B, Rivenzon D, Katz E (1993)
Adv Mater 5: 912–915
[55] Mann-Buxbaum E, Pittner F, Schalkhammer T, Jachimowicz
A, Jobst G, Olcaytug F, Urban G (1990) Sens Actuators B 1:
518–522
[56] Williams R A, Blanch H W (1994) Biosens Bioelectron 9:
159–167
[57] Miyasaka T, Koyama K, Watanabe T (1990) Chem Lett,
pp 627–630
[58] Solov’ev A A, Katz E, Shuvalov V A, Erokhin Y E (1991)
Bioelectrochem Bioenerg 26: 29–41
[59] Katz E (1994) J Electroanal Chem 365: 157–164
[60] Hong H-G, Bohn P W, Sligar S G (1993) Anal Chem 65:
1635–1638
[61] Hong H-G, Jiang M, Sligar S G, Bohn P W (1994) Langmuir
10: 153–158
[62] Firestone M A, Shank M L, Sligar S G, Bohn P W (1996)
J Am Chem Soc 118: 9033–9041
[63] Wolfbeis O S, Leiner M J P, Posch H E (1986) Mikrochim
Acta III: 359–366
[64] Bacon J R, Demas J N (1987) Anal Chem 59: 2780–2785
[65] Lippitsch M E, Pusterhofer J, Leiner M J P, Wolfbeis O S
(1988) Anal Chim Acta 205: 1–6
[66] Carraway E R, Demas J N, DeGraff B A, Bacon J R (1991)
Anal Chem 63: 337–342
[67] Klimant I, Belser P, Wolfbeis O S (1994) Talanta 41: 985–991
[68] Freeman T M, Seitz W R (1978) Anal Chem 50: 1242–1246
[69] Wolfbeis O S (1991) Fiber optic chemical sensors and
biosensors, vol. 1. CRC Press, Boca Raton, FL, USA
[70] Owen V M (1985) Ann Clim Biochem 22: 559–564
[71] Hall E A (1986) Enzyme Microb Technol 8: 651–658
[72] Scheller F W, Hintsche R, Pfeiffer D, Schubert F, Riedel K,
Kindervater R (1991) Sens Actuators B 4: 197–200
[73] Wagner G, Guilbault G G (1994) Food biosensor analysis,
Marcel Dekker, New York, USA
[74] Turner A P F (1994) Curr Opin Biotech 5: 49–53
[75] Mascini M (1995) Sens Actuators B 29: 121–125
[76] Aboul-Enein H Y, Stefan R-I, van Staden J F (1999) Crit Rev
Anal Chem 29: 323–331
[77] Wang J (1999) Anal Chem 71: 328R–332R
[78] Wolfbeis O S (2000) Anal Chem 72: 81R–90R
[79] Wilson G S, Hu Y (2000) Chem Rev 100: 2693–2704
[80] D’Orazio P (2003) Clin Chim Acta 334: 41–69
[81] Wolfbeis O S (2004) Anal Chem 76: 3269–3284
[82] Schaffar B P H, Dremel B A A, Schmid R D (1989) GBF
Monographs 13: 229–232
[83] Li X, Fortuney A, Guilbault G G, Suleiman A A (1996) Anal
Lett 29: 171–180
[84] Li X, Rosenzweig Z (1997) Anal Chim Acta 353: 263–273
[85] Vidal M M, Delgadillo I, Gil M H, Alonso-Chamarro J
(1996) Biosens Bioelectron 11: 347–354
[86] Wu X J, Choi M M F (2003) Anal Chem 75: 4019–4027
[87] Yan Y, Xu Y, Li S (2002) Shengwu Yixue Gongchengxue
Zazhi 19: 101–102
[88] Carazuela M D, Cuesta B, Moreno-Bondi M C, Quejido A
(1997) Biosens Bioelectron 12: 233–240
130 M. M. F. Choi

[89] Trettnak W, Wolfbeis O S (1989) Anal Biochem 184: 124–127
[90] Marquette C A, Leca B D, Blum L J (2001) Luminescence
16: 159–165
[91] Zhang Z, Ma W (1993) Gaodeng Xuexiao Huaxue Xuebao
14: 1366–1369
[92] Wang H, Cao X, Jia K, Chai X, Lu H, Lu Z (2001) Proc SPIE-
Int Soc Opt Eng 4601: 115–118
[93] Marazuela M D, Moreno-Bondi M C (1998) Anal Chim Acta
374: 19–29
[94] Tsafack V C, Marquette C A, Leca B, Blum L J (2000)
Analyst 125: 151–155
[95] Hasunuma T, Kuwabata S, Fukusaki E-I, Kobayashi A
(2004) Anal Chem 76: 1500–1506
[96] Wolfbeis O S, Posch H E (1988) Fresen Z Anal Chem 332:
255–257
[97] Lau R C W, Choi M M F, Lu J (1999) Talanta 48: 321–331
[98] Walters B S, Nielsen T J, Arnold M A (1988) Talanta 35:
151–155
[99] Gautier S M, Blum L J, Coulet P R (1990) J Bioluminesc
Chemiluminesc 5: 57–63
[100] Xie X, Suleiman A A, Guilbault G G, Yang Z, Sun Z (1992)
Anal Chim Acta 266: 325–329
[101] Marshall R W, Gibson T D (1992) Anal Chim Acta 266:
309–315
[102] Verostko C E, Atwater J E, Akse J R, Dehart J L, Wheeler R R
(1998) US Pat 5792621
[103] Schier G M, Moses R G, Gan I E T, Blair S C (1988) Diabetes
Res Clin Pract 4: 177–181
[104] Clarke W, Becker D J, Cox D, Santiago J V, White N H,
Betschart J, Eckenrode K, Levandoski L A, Prusinki E A,
Simineiro L M, Snyder A L, Tideman A M, Yaegar T (1988)
Diabetes Res Clin Pract 4: 209–213
[105] Meadows D, Schultz J S (1988) Talanta 35: 145–150
[106] Trettnak W, Wolfbeis O S (1989) Anal Chim Acta 221:
195–203
[107] Tolosa L, Malak H, Rao G, Lakowicz J R (1997) Sens
Actuators B 45: 93–99
[108] Tolosa L, Gryczynski I, Eichorn L R, Dattelbaum J D,
Castellano F N, Rao G, Lakowicz J R (1999) Anal Biochem
267: 114–120
[109] D’Auria S, Dicesare N, Gryczynski Z, Gryczynski I, Rossi
M, Lakowicz J R (2000) Biochem Biophys Res Commun
274: 727–731
[110] Aslan K, Lakowicz J R, Geddes C D (2004) Anal Chim Acta
517: 139–144
[111] Robinson M R, Eaton R P, Haaland D M, Koepp G W,
Thomas E V, Stallard B R, Robinson P L (1992) Clin Chem
38: 1618–1622
[112] Heise H M, Marbach R, Koschinsky T H, Gries F A (1994)
Ann Occup Hyg 18: 439–447
[113] March W F, Rabinovitch B, Adams R, Wise J R, Melton M
(1982) Trans Am Soc Artif Intern Organs 28: 232–235
[114] Rabinovitch B, March W F, Adams R L (1982) Diabetes Care
5: 254–265
[115] Moscone D, Pasini M (1992) Talanta 39: 1039–1044
[116] Tatsu Y, Yamashita K, Yamaguchi M, Yamamura S,
Yamamoto H, Yoshikawa S (1992) Chem Lett, pp 1615–1618
[117] Narang U, Prasad P N, Bright F V (1994) Anal Chem 66:
3139–3144
[118] Male K B, Gartu P O, Kamen A A, Luong J H T (1997)
Biotechnol Bioeng 55: 497–504
[119] Wang B, Li B, Deng Q, Dong S (1998) Anal Chem 70:
3170–3174
[120] Chen T, Friedman K A, Lei I, Heller A (2000) Anal Chem 72:
3757–3763
[121] Sapre A G, Bedekar A, Deshpande A V, Lali A M (2000)
Biotechnol Lett 22: 569–573
[122] Chen X, Matsumoto N, Hu Y, Wilson G S (2002) Anal Chem
74: 368–372
[123] Karyakin A A, Kotel’nikova E A, Lukachova L V, Karyakina
E E, Wang J (2002) Anal Chem 74: 1597–1603
[124] Matsumoto R, Mochizuki M, Kano K, Ikeda T (2002) Anal
Chem 74: 3297–3303
[125] Wei X, Cruz J, Gorski W (2002) Anal Chem 74: 5039–5046
[126] Palmisano F, Zambonin P G, Centonze D, Quinto M (2002)
Anal Chem 74: 5913–5918
[127] Gouda M D, Kumar M A, Thakur M S, Karanth N G (2002)
Biosens Bioelectron 7: 503–507
[128] Haouz A, Stieg S (2002) Enzyme Microb Technol 30:
129–133
[129] Zen J-M, Kumar A S, Chung C-R (2003) Anal Chem 75:
2703–2709
[130] Hrapovic S, Luong J H T (2003) Anal Chem 75: 3308–3315
[131] Uwira N, Opitz N, Lubbers D W (1984) Adv Exp Med Biol
169: 915–921
[132] Neubauer A, Pum D, Sleytr U B, Klimant I, Wolfbeis O S
(1996) Anal Chim Acta 11: 317–325
[133] Dremel B A A, Kalisz H, Schaffar B P H, Draxler S,
Lippitsch M E, Schmid R D, Wolfbeis O S (1992) In
GBF monographs: biosensors, fundamentals, technologies
and applications, vol. 17. In: Scheller F, Schmid R D (eds)
VHC, Weinheim, Germany, p 259–262
[134] Dremel B A A, Schaffar B P H, Schmid R D (1989) Anal
Chim Acta 225: 293–301
[135] Moreno-Bondi M C, Wolfbeis O S, Leiner M J P, Schaffar
B P H (1990) Anal Chem 62: 2377–2380
[136] Schaffar B P H, Wolfbeis O S (1990) Biosens Bioelectron 6:
137–148
[137] Trettnak W, Leiner M J P, Wolfbeis O S (1988) Analyst 113:
1519–1523
[138] Chen Q, Kenausis G L, Heller A (1998) J Am Chem Soc 120:
4582–4585
[139] Wu X, Choi M M F, Xiao D (2000) Analyst 125: 157–162
[140] Wu X J, Choi M M F (2004) Anal Chim Acta 514:
219–226
[141] Choi M M F, Pang W S H, Xiao D, Wu X (2001) Analyst 126:
1558–1563
[142] Rosenzweig Z, Kopelman R (1996) Anal Chem 68: 1408–1413
[143] Xu H, Aylott J W, Kopelman R (2002) Analyst 127: 1471–1477
[144] Papkovsky D B, O’Riordan T C, Guilbault G G (1999) Anal
Chem 71: 1568–1573
[145] Papkovsky D B, Ovchinnikov A N, Ogurtsov V I, Ponomarev
G V, Korpela T (1998) Sens Actuators B 51: 137–145
[146] Trettnak W, Wolfbeis O S (1989) Anal Chim Acta 221:
195–203
[147] Hartnett A M, Ingersoll C M, Baker G A, Bright F V (1999)
Anal Chem 71: 1215–1224
[148] Trettnak W, Leiner M J P, Wolfbeis O S (1988) Biosensors 4:
15–26
[149] Li C-I, Lin Y-H, Shih C-L, Tsaur J-P, Chau L-K (2002)
Biosens Bioelectron 17: 323–330
[150] Wang C H, Chen S M, Wang C M (2002) Analyst 127:
1507–1511
[151] Wolfbeis O S, Sch€aaferling M, D€uurkop A (2003) Microchim
Acta 143: 221–227
[152] Rickus J L, Tobin A J, Zink J I, Dunn B (2002) Materials
Research Society Symposium Proceedings 723: 155–160
[153] Wang A J, Arnold M A (1992) Anal Chem 64: 1051–1055
[154] Cordek J, Wang X, Tan W (1999) Anal Chem 71: 1529–1533
[155] Kar S, Arnold M A (1992) Anal Chem 64: 2438–2443
Progress in Enzyme-Based Biosensors Using Optical Transducers 131

[156] Dremel B A A, Schmid R D, Wolfbeis O S (1991) Anal Chim
Acta 248: 351–359
[157] Cattaneo M V, Male K B, Luong J H T (1992) Biosens
Bioelectron 7: 569–574
[158] Cattaneo M V, Luong J H T (1993) Biotechnol Bioeng 41:
659–665
[159] Blankenstein G, Preuschoff F, Spohn U, Mohr K-H, Kula
M-R (1993) Anal Chim Acta 271: 231–237
[160] Trettnak W, Wolfbeis O S (1989) Anal Lett 22: 2191–2197
[161] Dremel B A A, Yang W, Schmid R D (1990) Anal Chim Acta
234: 107–112
[162] Ignatov S G, Ferguson J A, Walt D R (2001) Biosens
Bioelectron 16: 109–113
[163] Papkovsky D B, O’Riordan T, Soini A (2000) Biochem Soc T
28: 74–77
[164] Trettnak W, Wolfbeis O S (1989) Fresen Z Anal Chem 334:
427–430
[165] Schubert F, Wang F, Rinneberg H (1995) Mikrochim Acta
121: 237–247
[166] Arnold M A (1988) Proc SPIE-Int Soc Opt Eng 906: 128–133
[167] Wangsa J, Arnold M A (1988) Anal Chem 60: 1080–1082
[168] Liu X, Tan W (1999) Mikrochim Acta 131: 129–135
[169] Michel P E, Gautier S M, Blum L J (1996) Anal Lett 29:
1139–1155
[170] Marquette C A, Blum L J (1999) Anal Chim Acta 381: 1–10
[171] Marquette C A, Degiuli A, Blum L J (2000) Appl Biochem
Biotech 89: 107–115
[172] Koncki R, Leszczy�nnska E, Cybulska A, Glab S (1996) Anal
Chim Acta 321: 27–34
[173] Mueller C, Hitzmann B, Schubert F, Scheper T (1995) Proc
SPIE-Int Soc Opt Eng 2388: 558–567
[174] Mueller C, Hitzmann B, Schubert F, Scheper T (1997) Sens
Actuators B 340: 71–77
[175] Polster J, Prestel G, Wollenweber M, Kraus G, Gauglitz G
(1995) Talanta 42: 2065–2072
[176] Healey B G, Walt D R (1995) Anal Chem 67: 4471–4476
[177] Healey B G, Li L, Walt D R (1997) Biosens Bioelectron 12:
521–529
[178] Xie X, Suleiman A A, Guilbault G G (1992) Biotechnol
Bioeng 39: 1147–1150
[179] Carlyon E E, Lowe C R, Reid D, Bennion I (1992) Biosens
Bioelectron 7: 141–146
[180] Goldfinch M J, Lowe C R (1984) Anal Biochem 138: 430–436
[181] Sansubrino A, Mascini M (1994) Biosens Bioelectron 9:
207–216
[182] Xie X, Suleiman A A, Guilbault G G (1990) Anal Lett 23:
2143–2153
[183] Krysteva M, Al Hallak M (2003) The Scientific World 3:
585–592
[184] Koncki R, Lenarczuk T, Radomska A, Glab S (2001) Analyst
126: 1080–1085
[185] Radomska A, Glab S, Koncki R (2001) Analyst 126:
1564–1567
[186] Kovacs B, Nagy G, Dombi R, Toth K (2003) Biosens
Bioelectron 18: 111–118
[187] Wolfbeis O S, Li H (1993) Biosens Bioelectron 8: 161–166
[188] Chen H, Wang E (2000) Anal Lett 33: 997–1011
[189] De Marcos S, Hortiguela R, Galban J, Castillo J R, Wolfbeis
O S (1999) Microchim Acta 130: 267–272
[190] Brennan J D, Brown R S, Della Manna A, Kallury K M R,
Piunno P A, Krull U J (1993) Sens Actuators B 11:
109–119
[191] Rhines T D, Arnold M A (1989) Anal Chim Acta 227:
387–396
[192] Xie X, Suleiman A A, Guilbault G G (1991) Talanta 38:
1197–1200
[193] Gauglitz G, Reichert M (1992) Sens Actuators B 6: 83–86
[194] Popa E, Kubota Y, Tryk D A, Fujishima A (2000) Anal Chem
72: 1724–1727
[195] Gong Z, Zhang Z (1996) Anal Lett 29: 695–709
[196] Zhang Z, Ma W, Yang M (1992) Fenxi Huaxue 20: 1048–1051
[197] Ge X, Tolosa L, Rao G (2004) Anal Chem 76: 1403–1410
132 Progress in Enzyme-Based Biosensors Using Optical Transducers