preparation and in vitro evaluation of allopurinol-gelucire 50/13 solid dispersions
TRANSCRIPT

International Journal of Advances in Pharmaceutical Sciences
1 (2010) 60-67
http://www.arjournals.org/ijoaps.html
Research article ISSN:0976-1055
Preparation and in vitro evaluation of Allopurinol-Gelucire 50/13 solid dispersions
Jagdale S. C1*, Kuchekar B.S1, Chabukswar A.R1, Musale V.P1, Jadhao M.A2
*Corresponding author: 1MAEER’s Maharashtra
Institute of Pharmacy
S.No.124, MIT Campus,
Paud Road, Ex-Serviceman
Colony, Pune- 411038.
India
E-mail: [email protected]
2 Vidhyabharti College of
Pharmacy,
Amaravati. MH
India
Abstract The rate-limiting step to absorption of drugs from the gastrointestinal tract is often dissolution from the dosage form. Allopurinol is a commonly used drug in the treatment of chronic gout or hyperuricaemia associated with leukaemia, radiotherapy, anti-neoplastic agents. One of the major problems with allopurinol is that, it is practically insoluble in water, which results in poor bioavailability after oral administration. In the present study, solid dispersions of allopurinol were prepared by solvent evaporation method, kneading method, co-precipitation method, co-grinding method and closed melting method to increase its water solubility. In the present study amphiphilic carrier like gelucire 50/13 was used in the ratio of 1:1, 1:2 and 1:4. Prepared solid dispersions were characterized in the liquid state by phase solubility studies and in the solid state by Differential Scanning calorimetric analysis, Powder X-ray diffractometry and Fourier Transform Infrared spectroscopy. The aqueous solubility of allopurinol was preferential by the presence polymer with increasing concentration. Solid state characterizations indicated that allopurinol was present as an amorphous material and entrapped in polymer matrix. Mathematical modeling of in vitro dissolution data indicated the best fitting with Korsemeyer-Peppas model and the drug release kinetics primarily as Non-Fickian diffusion. Therefore, the current study showed that gelucire 50/13 has a significant solubilizing effect on allopurinol. Keywords: Allopurinol, gelucire 50/13, closed melting-method, co-grinding, dissolution study, powder X-ray diffraction analysis
Introduction Enhancement of the bioavailability of poorly water soluble drugs has been one of the main targets of drug development during the last decade and will continue in the upcoming years. Several techniques have been developed concerning enhancement of the dissolution
rate of these drugs, including particle size reduction [1], salt formation [2] and preparation of solid dispersions (SDs). Solid dispersions can be defined as molecular mixtures of poorly water soluble drugs in hydrophilic carriers, which present a drug release
doi:10.5138/ijaps.2010.0976.1055.01007 ©arjournals.org, All rights reserved.

Jagdale et al. International Journal of Advances in Pharmaceutical Sciences 1(2010) 60-67
61
profile that is driven by the polymer properties [3]. During the solid dispersion preparation, the aim is to disperse the drug homogenously within the carrier matrix and to encapsulate the hydrophobic drug to ensure complete wetting, fast carrier dissolution and improved drug stability. The most commonly used hydrophilic carriers for solid dispersions include polyvinylpyrrolidone [4,5], polyethylene glycols [6], and lipids, such as polyglycolized glycerides (Gelucire) [7], The solvent evaporation, melt adsorption, fusion, spray drying, spray freezing, spray congealing [8], and supercritical fluid precipitation [9], are the techniques reported for the preparation of solid dispersions. Recently, many workers reported solid dispersions using gelucires by fusion and solvent evaporation techniques [10]. Polyglycolized glycerides are available with a range of properties depending on their hydrophilic lipophilic balance (HLB) over the range of 1 to 18 and melting point between 33 ºC and 70 ºC. The carbamazepine solid dispersions with gelucire 50/13 have shown that crystallinity reduction and wetting with hydrophilic lipid are the main mechanisms responsible for increase in the dissolution rate [11]. Allopurinol (ALLO), selected in the current studies, is a poorly water-soluble drug known to demonstrate dissolution or solubility limited absorption. Based upon its aqueous solubility and various dissolution parameters, the drug bioavailability can unambiguously be regarded as limited solely to dissolution [12]. It is relatively insoluble in water, and freely soluble in alkaline aqueous solutions. Therefore, improvements in solubility or dissolution rate of poorly water-soluble drugs may be achieved through the formation of solid dispersions. In the present study, kneading (KN) [13], closed melting (CM) [14], co-precipitation (CP) [15], co-grinding (CG) [16], solvent evaporation (SE) [17] techniques has been used to prepare solid dispersions with lipid carrier i.e. gelucire 50/13. Gelucires are a group of glyceride-based excipients, composed of mixtures of mono, di and triglycerides with polyethylene glycol esters of fatty acid and are classified by two numbers, the first referring to the approximate melting point of the base and the second to the hydrophilic lypophilic balance (HLB) value. The nature and proportion of the components in the
gelucires determine the hydrophobicity and drug release properties of the corresponding dosage forms [18]. Solid dispersions in the form of dried powder were characterized in comparison with pure ALLO and corresponding physical mixtures in the same ratios by using Fourier Transform Infrared spectroscopy (FTIR), Differential Scanning calorimetric analysis (DSC), and Powder X-ray diffractometry (PXRD) techniques, and in vitro drug release. Material and Methods Material Allopurinol was kindly supplied by Nicolas piramal, (Pitampur, India) and gelucire 50/13 was obtained from Colorcon Asia Pvt. Ltd. Mumbai, India. All other chemicals and solvents were of analytical reagent grade. Methods Phase solubility studies Solubility measurements were performed in triplicate using the method reported by Higuchi and Connors [19]. An excess amount of ALLO was added to the aqueous solutions with increasing concentrations of gelucire 50/13 (i.e., 0, 1, 5, and 10 % w/v). Then the flasks were maintained at room temperature for 7 days with continuous starring by using magnetic stirrer. The saturated solution was sonicated for 20 minutes and then centrifuged; the supernatant were filtered through a Whatman filter paper No. 1. The filtrate was suitably diluted and analyzed spectrophotometrically at 250 nm on Varian carry 100, UV–vis Spectrophotometer, Australia [20]. Preparation of physical mixture and solid dispersions Physical mixtures (PM), for the sake of comparison physical mixtures of ALLO were prepared by mixing accurately weighed amounts of ALLO with gelucire 50/13 (ratio of drug: carrier was 1:1, 1:2 and 1:4) in mortar by simple trituration. Solvent evaporation method (SE), solid dispersions were prepared by dissolving accurately weighed amounts of ALLO and gelucire 50/13 in ethanol. After complete dissolution of ALLO and gelucire 50/13 in ethanol sonicated the solution for 20 minutes, and then solvent was evaporated under reduced pressure at

Jagdale et al. International Journal of Advances in Pharmaceutical Sciences 1(2010) 60-67
62
room temperature in a dessicator. Subsequently, the solid mass was ground and passed through sieve no 40. Co-grinding method (CG), ALLO was triturated with minimum quantity of ethanol in a glass mortar until it dissolved. The gelucire 50/13 was gradually added and prepared suspension was triturated rapidly at room temperature until the solvent evaporated. Solid mass was ground and passed through sieve no 40. Co-precipitation method (CP), solid dispersions were prepared by dissolving accurately weighed amounts of gelucire 50/13 in water and ALLO in ethanol. After complete dissolution, the aqueous solution of gelucire 50/13 was then poured into the ethanolic solution of the ALLO. The mixture of drug and carrier were then heated and evaporated under reduced pressure at room temperature in a dessicator. Subsequently, the solid dispersions were passed through sieve no 40 and stored in a dessicator until further evaluation. Kneading method (KN), a mixture of ALLO and gelucire 50/13 was wetted with water and kneaded thoroughly for 30 minutes in a glass mortar. The paste formed was dried under vacuum for 24 hrs. Dried powder was passed through sieve no. 40 and stored in a dessicator until further evaluation. Closed Melting method (CM), 1 gram of physical mixture was weighed in to glass ampoules (20 ml) and the glass ampoules were sealed, and then heated for 30 minutes in a water bath to prepare the solid dispersions. After slow cooling the ampoules caps were opened and collect the solid dispersions. All dispersions were pulverized with pestle and mortar, sieved through sieve no. 40 and dried in an oven at 60 ºC at least for 48 hrs. The physical mixtures and all dispersions were subsequently stored at room temperature in a screw capped glass vial until further evaluation. Evaluation and Characterization of solid dispersion All the formulations were evaluated by phase solubility studies and in-vitro dissolution studies. Selected samples of solid dispersions are characterized by UV, FTIR, DSC and PXRD analysis.
Fourier Transform Infrared spectroscopy, FTIR spectra of pure ALLO, gelucire 50/13, solid dispersions and physical mixture were recorded on 640 IR, Varian, Australia, using KBr pellet method. Triturate 10 mg of solid dispersions with 10-20 mg of dry KBr powder; fill the pellets with the same for scanning at 4000 to 400 cm-1. UV-Visible Spectroscopy, was performed by weighing solid dispersions accurately and dissolved in 0.1 Molar NaOH solution sufficiently diluting with distilled water to produce a final concentration of 10 mcg/ml. These solutions were scanned spectrophotometrically in the range of 240 nm to 300nm on Varian carry 100, UV–vis Spectrophotometer, Australia [21]. Differential Scanning Calorimetric analysis, A differential scanning calorimeter (DSC 823e, Mettler Toledo, Switzerland) was used to obtain the DSC curves of ALLO solid dispersions and its physical mixtures representing the rates of heat uptake. About 10 mg of sample was weighed in a standard open aluminum pans, were scanned from 20-450 °C, at a heating rate of 10 °C/minute while being purged with dry nitrogen. Powder X-ray diffractometry, Powder X-ray diffraction (PXRD) patterns were traced employing X-ray diffractometer (Philips PW 1729 Netherlands.) for the all samples, using Ni filter, CuK (α) radiation, a voltage of kV, a current of 20 mA and receiving slit of 0.2 in. The samples were analyzed over 2θ range of 5° to 60°, with scan step size of 0.020° (2θ) and scan step time of 1 second. In vitro Dissolution studies Release from the solid dispersions was determined in a calibrated 8-station USP-XXII apparatus (paddle method), TDT-08L Electrolab, Mumbai, India. Samples of drug, solid dispersions and physical mixture equivalent with 10 mg drug were packed in thin cloth thimble and then immersed in the dissolution medium consisted of 900 ml 0.1 N Hcl which was maintained at 37 ºC. The duration of the test and the rate of paddle stirring were 90 min and 75 rpm, respectively. At a time intervals of (5, 10, 15, 20, 30, 45, 60 and 90 minutes), 5 ml samples were withdrawn,
Jagdale et al. International Journal of Advances in Pharmaceutical Sciences 1(2010) 60-67
63
and replaced with 5 ml of fresh dissolution medium. The samples were filtered through a whatman filter paper No. 1 and analyzed spectrophotometrically at 250 nm on Varian carry 100, UV–vis Spectrophotometer, Australia. Cumulative percentages of the drug dissolved from the preparations were calculated [22]. Data analysis Phase-solubility studies The values of apparent stability constant Ks, between each drug–carrier combination were computed from the phase-solubility profiles, as described below:
Ks = Slope / Intercept (1- slope)
The values of Gibbs free energy of transfer, ∆Gotr of
ALLO from aqueous solution of the carriers were calculated according to the following relationship:
∆Gotr = - 2.303RT.log So/Ss
Where, So and Ss are the molar solubilities of ALLO in 1 % w/v aqueous solution of the carrier, respectively. The in-vitro drug release data was fitted into four models of data treatment for the solid dispersion formulations as follows:
1) Zero order: F = k X t 2) First order: ln F = k X t 3) Higuchi’s model: F= k√ t 4) Korsmeyer and Peppas model : F= ktn
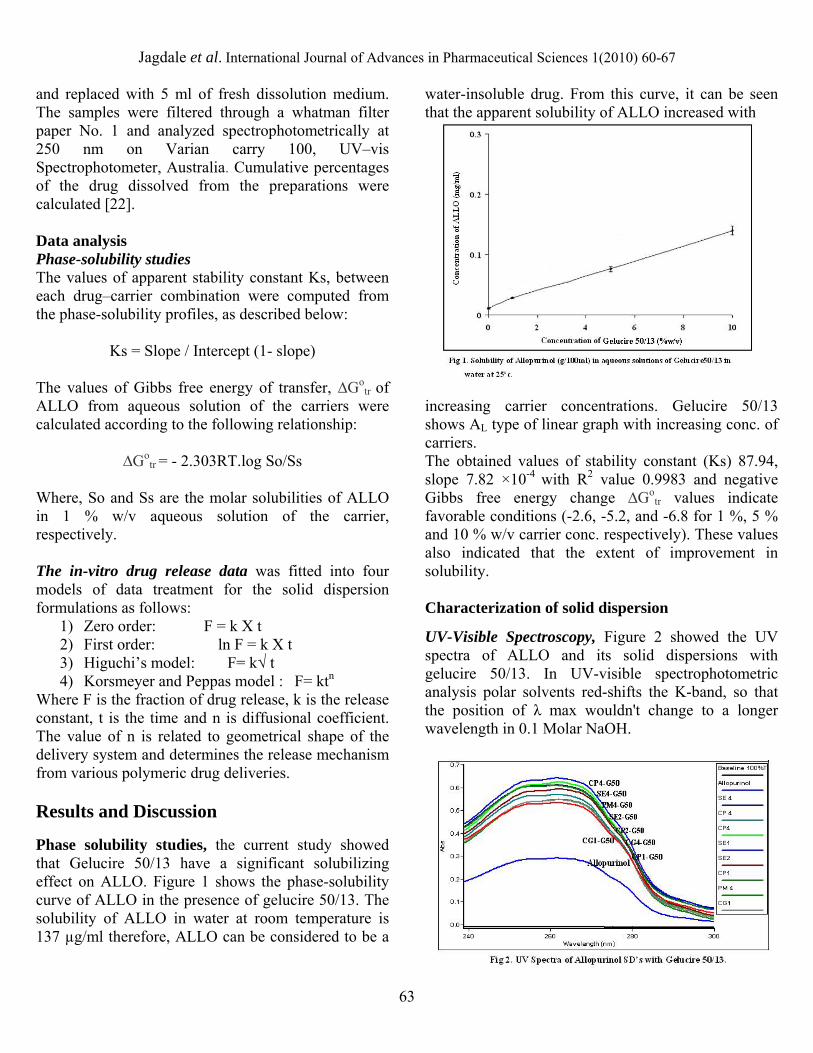
Where F is the fraction of drug release, k is the release constant, t is the time and n is diffusional coefficient. The value of n is related to geometrical shape of the delivery system and determines the release mechanism from various polymeric drug deliveries. Results and Discussion Phase solubility studies, the current study showed that Gelucire 50/13 have a significant solubilizing effect on ALLO. Figure 1 shows the phase-solubility curve of ALLO in the presence of gelucire 50/13. The solubility of ALLO in water at room temperature is 137 µg/ml therefore, ALLO can be considered to be a
water-insoluble drug. From this curve, it can be seen that the apparent solubility of ALLO increased with increasing carrier concentrations. Gelucire 50/13 shows AL type of linear graph with increasing conc. of carriers. The obtained values of stability constant (Ks) 87.94, slope 7.82 ×10-4 with R2 value 0.9983 and negative Gibbs free energy change ∆Go
tr values indicate favorable conditions (-2.6, -5.2, and -6.8 for 1 %, 5 % and 10 % w/v carrier conc. respectively). These values also indicated that the extent of improvement in solubility. Characterization of solid dispersion
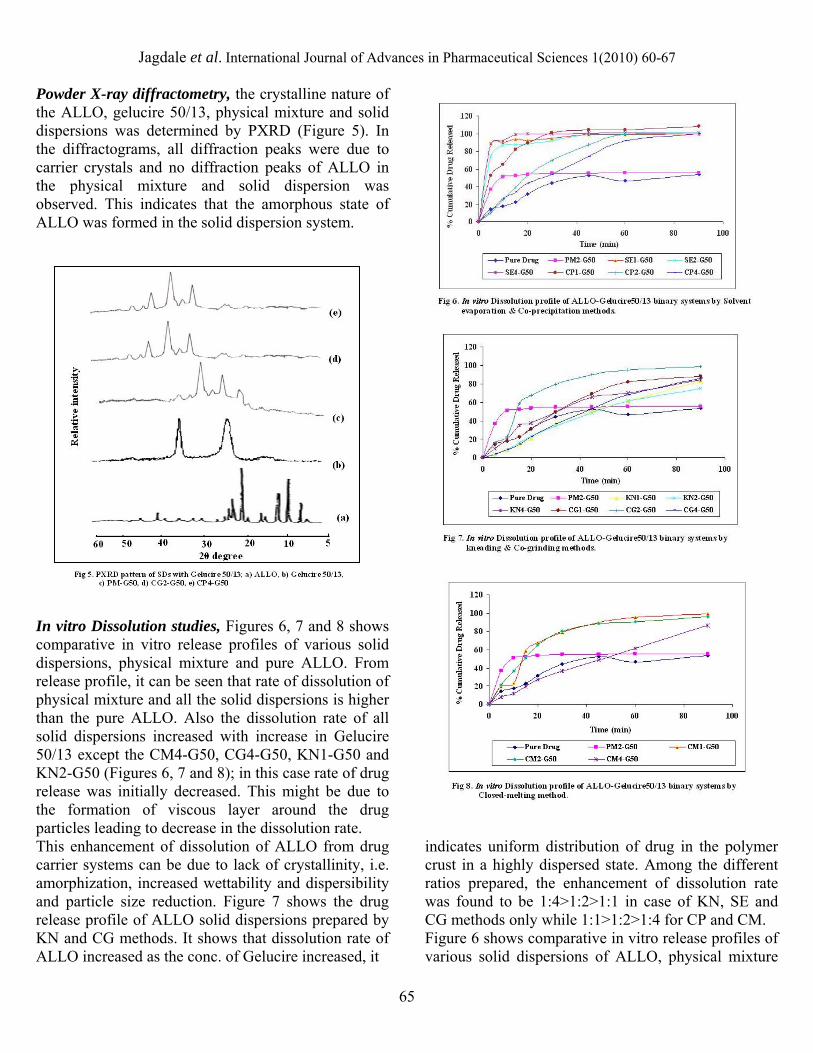
UV-Visible Spectroscopy, Figure 2 showed the UV spectra of ALLO and its solid dispersions with gelucire 50/13. In UV-visible spectrophotometric analysis polar solvents red-shifts the K-band, so that the position of λ max wouldn't change to a longer wavelength in 0.1 Molar NaOH.

Jagdale et al. International Journal of Advances in Pharmaceutical Sciences 1(2010) 60-67
64
In this case the solutions of physical mixture and solid dispersions showed UV spectra at same λ max i.e. 260 nm with increase in absorbance than ALLO. It was thought that the electrostatic interactions, including dipolar interactions and hydrogen bonds, would operate as intermolecular forces of drug-polymer. The hydrogen bonding is likely to contribute predominantly to the slight drug-polymer interaction. Fourier Transform Infrared spectroscopy, Figure 3 displays the FTIR spectra of pure ALLO, Gelucire 50/13, physical mixture and solid dispersions prepared by CP, SE and CG methods. The characteristic shoulders of ALLO are at 790, 811, 915, 1081, 1159, and 1232 cm−1, denoting CH in plane deformation; 1590 cm−1 representing ring vibration, 1700, 1763 cm−1 indicating CO stretching vibration of the -keto form of the 4-hydroxytautomer, 3060 cm−1 denoting CH stretching vibrations of pyrimidine ring and at 3400 cm−1 for NH stretching band. Gelucire 50/13 exhibited peaks of are the C-H stretching at 2890 cm−1 and the C-O (ether) stretching at 1125 cm−1, 450,710 and 1232 cm−1, denoting CH in plane deformation. The presence and absence of characteristic peaks associated with specific structural characteristics of the molecule was noted. The spectra of physical mixture
were equivalent to the addition spectrum of polymer and crystalline drug. This indicated that no interaction occurred with simple physical mixing of drug. Surprisingly, there was no interaction between the -O-H group of Gelucire and –N-H group of ALLO. In case of ALLO-gelucire solid dispersions, it was assumed that ALLO would also form the H-bond with Gelucire. During the amorphization ALLO itself from the intermolecular H-bonding between hydrogen of N-H and oxygen of O-H. Due to this the peak of allopurinol i.e. 1590 cm−1 (ring vibration) may becomes sharp and shifted to higher wavelength. Differential Scanning calorimetric analysis, the DSC curve of ALLO (Figure 4a) exhibited a sharp endothermic peak at 379.5 ºC due to fusion. Analogously, the thermal curve of Gelucire 50/13 (Figure 4b) showed a single endothermic effect with a peak at 50 ºC, corresponding to its melting point. The physical mixture showed an ALLO and Gelucire derived endothermic peaks with decreased intensity than pure ALLO. The thermo grams of both binary systems CP4-G50 (Figure 4d) and CP2-G50 (Figure 4e) showed no endothermic peak corresponding to ALLO. It is quite probable that ALLO might have dissolved in the molten carrier during DSC scan.
Jagdale et al. International Journal of Advances in Pharmaceutical Sciences 1(2010) 60-67
65
Powder X-ray diffractometry, the crystalline nature of the ALLO, gelucire 50/13, physical mixture and solid dispersions was determined by PXRD (Figure 5). In the diffractograms, all diffraction peaks were due to carrier crystals and no diffraction peaks of ALLO in the physical mixture and solid dispersion was observed. This indicates that the amorphous state of ALLO was formed in the solid dispersion system. In vitro Dissolution studies, Figures 6, 7 and 8 shows comparative in vitro release profiles of various solid dispersions, physical mixture and pure ALLO. From release profile, it can be seen that rate of dissolution of physical mixture and all the solid dispersions is higher than the pure ALLO. Also the dissolution rate of all solid dispersions increased with increase in Gelucire 50/13 except the CM4-G50, CG4-G50, KN1-G50 and KN2-G50 (Figures 6, 7 and 8); in this case rate of drug release was initially decreased. This might be due to the formation of viscous layer around the drug particles leading to decrease in the dissolution rate. This enhancement of dissolution of ALLO from drug carrier systems can be due to lack of crystallinity, i.e. amorphization, increased wettability and dispersibility and particle size reduction. Figure 7 shows the drug release profile of ALLO solid dispersions prepared by KN and CG methods. It shows that dissolution rate of ALLO increased as the conc. of Gelucire increased, it
indicates uniform distribution of drug in the polymer crust in a highly dispersed state. Among the different ratios prepared, the enhancement of dissolution rate was found to be 1:4>1:2>1:1 in case of KN, SE and CG methods only while 1:1>1:2>1:4 for CP and CM. Figure 6 shows comparative in vitro release profiles of various solid dispersions of ALLO, physical mixture

Jagdale et al. International Journal of Advances in Pharmaceutical Sciences 1(2010) 60-67
66
and pure ALLO prepared by the solvent evaporation and co-precipitation methods. The dissolution study showed that the release rates of ALLO from the solid dispersions by solvent evaporation were faster as compared with the solid dispersions by co-precipitation technique. This is because of ALLO was effectively transformed to amorphous. Also, the significant drug particle size reduction achieved, especially in the case of low drug loading, contributes to this improvement in dissolution rates. Hence, the present study showed the CP, SE and CG techniques are more efficient than, KN and CM techniques to increase the solubility/dissolution of allopurinol. Kinetic treatment of dissolution data To interpret the release kinetics and mechanism of drug release from solid dispersions. The coefficient of determination was considered as main parameter for interpreting the results. The n values of all formulations are as reported in Table 1. All formulations of solid dispersions were not fitting to a specific model. Formulations KN4-G50, CP2-G50, CP4-G50, CG2-50, CG4-G50 follows zero order kinetics with R2 values 0.9879, 0.9845, 0.9859, 0.9827 and 0.9839 respectively while only one formulation i.e. CM2-G50 followed first order kinetics with R2 value 0.9676. Formulations KN1-G50, KN2-G50, CM4-G50, CP4-G50, CG4-G50 follows Higuchi kinetics with R2 values 0.9894, 0.9943, 0.9874, 0.9835 and 0.9845 respectively. The release kinetic analyses of all formulations are as shown in Table 1. For formulations containing gelucire 50/13, KN1-G50 and KN2-G50, n values 1.0732 and 1.0602 respectively indicating that the release mechanism from these systems was Super case II Transport. And formulations KN4-G50, CM1-G50, CM2-G50, CM4-G50, CP2-G50, CP4-G50, CG2-G50 and CG4-G50, n values ranged from 0.5204 to 0.8025 indicating that the release mechanism from these systems was anomalous type (Non-Fickian transport), which refers to a combination of both diffusion and erosion controlled-drug release. Conclusion From the above study it was concluded that the solvent evaporation, co precipitation and co grinding are
powerful techniques for the preparation of rapidly dissolving formulations of allopurinol. All processes can potentially lead to better bioavailability of allopurinol drug products. The nature and the amount of the carrier used played an important role in the enhancement of the dissolution rate. The increased in the solubility and dissolution rate of allopurinol would provide the rapid onset of action. Acknowledgements The authors are grateful to Maharashtra Institute of pharmacy, Pune for providing essential laboratory conditions for present research work. Also an author acknowledges to Department of Physics, University of Pune for allowing the X-ray Diffraction and Differential Scanning Calorimetric studies. References 1. Merisko-Liversidge E, Liversidge GG, Cooper ER.
Nanosizing: a formulation approach for poorly-water-soluble compounds. Eur J Pharm Sci 2003; 18: 113.
2. Serajuddin ATM. Salt formation to improve drug solubility. Adv Drug Del Reviews 2007; 59: 603.
3. Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discovery Today 2007; 12: 1068-1075.
4. Ambike AA, Mahadik KR, Paradkar A. Stability study of amorphous valdecoxib. Int J Pharm 2004; 282: 151-162.
5. Paradkar A, Ambike AA, Jadhav BK. Characterization of curcumin-PVP solid dispersion obtained by spray drying. Int J Pharm 2004; 271: 281-286.
6. Doshi DH, Ravis WR, Betageri GV. Carbamazepine and polyethylene glycol solid dispersion preparation, in vitro dissolution, and characterization. Drug Dev Ind Pharm 1967; 23: 1167-1176.
7. Abdul-Fattah AM, Bhargava HN. Preparation and in vitro evaluation of solid dispersions of halfantrine. Int J Pharm 2002; 235: 17-33.

Jagdale et al. International Journal of Advances in Pharmaceutical Sciences 1(2010) 60-67
67
8. Fini A, Rodriguez C, Cavallari C. Ultrasound-compacted and spray-congealed indomethacin/polyethelyeneglycol systems. Int J Pharm 2002; 247: 11-22.
9. Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm 2004; 272: 1-10.
10. Pozzi F, Longo A, Lazzarini C. Formulations of Ubidecarenone with improved bioavailability. Eur J Pharm Biopharm 1991; 37: 243-246.
11. Passerini N, Perissutti B, Moneghini M. Characterization of cabamazepine-Gelucire 50/13 microparticles prepared by a spray congealing process using ultrasounds. J Pharm Sci 2002; 91: 699-707.
12. Ahuja N, Singh A, Singh B. Rofecoxib: an update on physicochemical, pharmaceutical, pharmacodynamic and pharmacokinetic aspects. J Pharm Pharmacol 2003; 55: 859-894.
13. Modi A, Tayade P. Enhancement of Dissolution Profile by Solid Dispersion (Kneading) Technique. AAPS PharmSciTech 2006; 7(3): 68.
14. Hasegawa S, Hamaura T, Furuyama N. Effects of water content in physical mixture and heating temperature on crystallinity of tropiglitazone- PVP K30 solid dispersions prepared by closed melting method. Int J Pharm 2005; 302: 103-112.
15. Ali N, Roya T, Hadi V. An Investigation on the Solid Dispersions of Chlordiazepoxide. Int J Biomed Sci 2007; 3(3): 211-217.
16. Dixit RP, Nagarsenkar MS. In vitro and In vivo advantage of Celecoxib surface solid dispersion and dosage form development. Indian J Pharm Sci 2007; 69(3): 370-377.
17. George Z, Bikiaris D, Karavas E. Effect of Physical State and Particle Size Distribution on Dissolution Enhancement of Nimodipine/PEG Solid Dispersions Prepared by Melt Mixing and Solvent Evaporation. AAPS PharmSciTech 2006; 8(4): E623-E631.
18. Craig DQM. The use of glycerides as controlled release matrices. In: Karsa DR, Stephenson RA (Eds.), Excipients and delivery systems for pharmaceutical formulations. Royal Society of Chemistry London; 1995. p. 148-173.
19. Higuchi T, Connors KA. Phase solubility techniques. 4th edi, Adv Anal Chem Instr 1965. p. 117-212.
20. Ahuja N, Katare O, Singh B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J of Pharm and Biopharm 2007; 65: 26–38.
21. Londhe VY, Nagarsenkar MS. Solid dispersion of hydroxypropyl β- cyclodextrin and carbamazepine: Study of complexation and in vitro dissolution profile. Indian drugs 1999; 36(1): 15-20.
22. Mahaparale PR, Gudsoorkar VR, Gajeli GB. Studies on Solid Dispersions of Meloxicam. Indian J Pharm Edu Res (Oct – Dec) 2006; 40(4): 241-245.
23. Vippagunta SR, Maul KA, Tallavajhala S. Solid-state characterization of nifedipine solid dispersions. Int J Pharm 2002; 236: 111–123.