oxidative stress in toxicology - hindawi.com
TRANSCRIPT

Oxidative Stress in Toxicology
Journal of Toxicology

Oxidative Stress in Toxicology

Journal of Toxicology
Oxidative Stress in Toxicology

Copyright © 2011 Hindawi Publishing Corporation. All rights reserved.
This is a focus issue published in volume 2011 of “Journal of Toxicology.” All articles are open access articles distributed under theCreative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided theoriginal work is properly cited.

Editorial Board
Syed F. Ali, USAMichael Aschner, USAThomas Burbacher, USASteven J. Bursian, USAJames Bus, USALucio Guido Costa, USAEdmond Edmond Creppy, FranceKevin Crofton, USAMichael L. Cunningham, USAAnthony DeCaprio, USADavid Doolittle, USAPaul R. Ebert, AustraliaLaurence D. Fechter, USAM. Teresa Colomina Fosch, SpainM. Ian Gilmour, USABhaskar Gollapudi, USA
Hisato Iwata, JapanMargaret James, USAYujian James Kang, USAMary Kanz, USAM. Firoze Khan, USAPaul Kostyniak, USARobert Krieger, USAKannan Krishnan, CanadaB. L. Lasley, USAPamela Lein, USARobert Luebke, USAMichael R. Moore, AustraliaJack Ng, AustraliaP. J. O’Brien, CanadaCurtis Omiecinski, USAOrish Ebere Orisakwe, Nigeria
Gary H. Perdew, USACinta Porte, SpainRobert H. Rice, USARudy Richardson, USAArleen Rifkind, USAJeanClare Seagrave, USAJames Sikarskie, USAJ. J. Stegeman, USASusan Sumner, USARobert Tanguay, USAKenneth Turteltaub, USABrad Upham, USAWilliam Valentine, USAJ. T. Zelikoff, USAWei Zheng, USA

Contents
Laboratory and Field Testing of an Automated Atmospheric Particle-Bound Reactive Oxygen SpeciesSampling-Analysis System, Yungang Wang, Philip K. Hopke, Liping Sun, David C. Chalupa,and Mark J. UtellVolume 2011, Article ID 419476, 9 pages
Behavioral Characterization of GCLM-Knockout Mice, a Model for Enhanced Susceptibility to OxidativeStress, Toby B. Cole, Gennaro Giordano, Aila L. Co, Isaac Mohar, Terrance J. Kavanagh, and Lucio G. CostaVolume 2011, Article ID 157687, 7 pages
Protective Action of Neurotrophic Factors and Estrogen against Oxidative Stress-MediatedNeurodegeneration, Tadahiro Numakawa, Tomoya Matsumoto, Yumiko Numakawa, Misty Richards,Shigeto Yamawaki, and Hiroshi KunugiVolume 2011, Article ID 405194, 12 pages
24-Epibrassinolide, a Phytosterol from the Brassinosteroid Family, Protects Dopaminergic Cells againstMPP+-Induced Oxidative Stress and Apoptosis, Julie Carange, Fanny Longpre, Benoit Daoust,and Maria-Grazia MartinoliVolume 2011, Article ID 392859, 13 pages
Evidence for a Role of Oxidative Stress in the Carcinogenicity of Ochratoxin A, M. Marin-Kuan,V. Ehrlich, T. Delatour, C. Cavin, and B. SchilterVolume 2011, Article ID 645361, 15 pages
Assessment of Protective Effect of Some Modern Agrochemicals against Ozone-Induced Stress inSensitive Clover and Tobacco Cultivars, Oleg Blum, Nataliya Didyk, Nataliya Pavluchenko,and Barbara GodzikVolume 2011, Article ID 308598, 4 pages
Research Strategies in the Study of the Pro-Oxidant Nature of Polyphenol Nutraceuticals, Harvey Babich,Alyssa G. Schuck, Jeffrey H. Weisburg, and Harriet L. ZuckerbraunVolume 2011, Article ID 467305, 12 pages
Estimation of the Postmortem Duration of Mouse Tissue by Electron Spin Resonance Spectroscopy,Shinobu Ito, Tomohisa Mori, Hideko Kanazawa, and Toshiko SawaguchiVolume 2011, Article ID 973172, 11 pages
Oxidative Toxicity in Neurodegenerative Diseases: Role of Mitochondrial Dysfunction and TherapeuticStrategies, Katie Facecchia, Lee-Anne Fochesato, Sidhartha D. Ray, Sidney J. Stohs, and Siyaram PandeyVolume 2011, Article ID 683728, 12 pages
Oxidative Stress and Air Pollution Exposure, Maura Lodovici and Elisabetta BigagliVolume 2011, Article ID 487074, 9 pages
Liposomal Antioxidants for Protection against Oxidant-Induced Damage, Zacharias E. SuntresVolume 2011, Article ID 152474, 16 pages

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 419476, 9 pagesdoi:10.1155/2011/419476
Research Article
Laboratory and Field Testing of an AutomatedAtmospheric Particle-Bound Reactive OxygenSpecies Sampling-Analysis System
Yungang Wang,1 Philip K. Hopke,1 Liping Sun,1 David C. Chalupa,2 and Mark J. Utell2
1 Center for Air Resource Engineering and Science, Clarkson University, Potsdam, NY 13699-5708, USA2 Department of Environmental Medicine, University of Rochester Medical Center, Rochester, NY 14627, USA
Correspondence should be addressed to Philip K. Hopke, [email protected]
Received 20 October 2010; Accepted 20 January 2011
Academic Editor: M. Ian Gilmour
Copyright © 2011 Yungang Wang et al. This is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
In this study, various laboratory and field tests were performed to develop an effective automated particle-bound ROS sampling-analysis system. The system uses 2′ 7′-dichlorofluorescin (DCFH) fluorescence method as a nonspecific, general indicator ofthe particle-bound ROS. A sharp-cut cyclone and a particle-into-liquid sampler (PILS) were used to collect PM2.5 atmosphericparticles into slurry produced by a DCFH-HRP solution. The laboratory results show that the DCFH and H2O2 standard solutionscould be kept at room temperature for at least three and eight days, respectively. The field test in Rochester, NY, shows that theaverage ROS concentration was 8.3±2.2 nmol of equivalent H2O2 m−3 of air. The ROS concentrations were observed to be greaterafter foggy conditions. This study demonstrates the first practical automated sampling-analysis system to measure this ambientparticle component.
1. Introduction
Substantial efforts are being made to elucidate the mecha-nisms of adverse human health effects by airborne particulatematter (PM). Fine particles (PM2.5) have been found to becorrelated with cardiopulmonary morbidity and mortality[1]. Ultrafine particles (UFPs, Dp < 100 nm) have beenassociated with effects in animals [2, 3] and humans [4, 5].However, the chemical components of the particles that drivethe mechanisms resulting in health effects are not yet wellunderstood. Since oxidative stress is thought to be a criticalfactor in driving health effects [1], it is essential to identifyand link specific oxidative particulate components, such asreactive oxygen species (ROS).
ROS include oxygen-containing compounds with strongoxidative capacity. Molecules like H2O2, organic peroxides,and nitrite peroxides, ions like hypochlorite ion (OCl−)peroxide anion (O2
−), and radicals like hydroxyl (•OH) andsuperoxide radicals (•O2
−), and organic peroxyl (ROO•)are all grouped as “reactive oxygen species”. ROS can be
generated endogenously during the cell metabolism throughreaction of the inhaled PM components such as metals (Fe,Cu, and Zn) and polycyclic aromatic hydrocarbon (PAH)[6, 7]. The excess oxidative stress from the ROS leads tolipid peroxidation, DNA damage, and protein oxidation,and has been implicated in the increased incidence ofcardiopulmonary disease, asthma, and chronic obstructivepulmonary disease [8–11]. Recently, ROS was found to bepresent in PM, especially in the UFPs component [12, 13].These particle-bound ROS are believed to induce effects onhuman health analogous to that of endogenous ROS.
The major sources of particle-bound ROS in the atmo-sphere are reaction between volatile organic compounds(VOC) and oxidants such as ozone (O3) or hydroxyl radicals(OH). For example, the oxidation products of biogenic VOCand O3 have low vapor pressure and can easily condense onthe surface of existing PM or nucleate to form secondaryorganic aerosols (SOA). These components also includeperoxides and radical species that constitute some of theparticle-bound ROS [14, 15]. In principle, photochemical

2 Journal of Toxicology
reactions generate the majority of free radical species inthe atmosphere during the daytime. Without sunlight,the particle-bound ROS formation mechanism is largelyinfluenced by the NO3 radical [16] and the OH radical,the latter of which was formed from the ozone and alkenereactions [17]. The specific route through which atmosphericparticle-bound ROS are formed remains unclear.
Efforts have been made to characterize the ambi-ent particle-bound ROS. The photochemical intensity wasa major factor affecting ROS concentrations in smallerparticles, especially in UFPs [18]. The concentration of tro-pospheric hydroxyl radicals can be described by a lineardependence on solar ultraviolet radiation [19]. Hydroper-oxides were simultaneously measured in both gas andaerosol phases, and about 40% of particle-bound H2O2
were associated with PM2.5 [20]. Concentration data onatmospheric ROS in the particle phase are limited andreported in the unit of nmol of equivalent H2O2 m−3 of air[12, 13, 18, 21, 22].
In prior studies, filters were commonly used to manuallycollect particle-bound ROS. ROS was then extracted fromthe filters and analyzed using the 2′ 7′-dichlorofluorescin(DCFH) fluorescence technique in the laboratory. Thismethod might underestimate ROS concentrations becausethe short lived species may be more chemically active thanthe components measured days or weeks later. The methodis quite labor intensive [23]. The lack of suitable methods toroutinely sample and immediately analyze ROS in the fieldhas restricted the evaluation of the health effects of particle-bound ROS.
A continuous, automated particle-bound ROS systemwas previously developed [23]. DCFH was employed asa general, nonspecific indicator of particle-bound ROS con-centration. A sharp cut cyclone and a particle-into-liquid-sampler (PILS) were used to collect PM2.5 into aqueousslurry that contained the DCFH solution. The fluorescentintensity (FI) was then measured with a flow-through flu-orescence detector. Quantification was obtained by relatingthe sample’s FI to that of an equivalent concentrationof H2O2. This initial laboratory system was not deployedbecause of uncertainties in its operation in the field. Issuesof concern included the stability of the reagent solutionsunder field conditions and the complexity of the design. Thecurrent study presents the results from the laboratory testingof a modified system and measurement of the solutionstabilities leading to field measurements of atmosphericparticle-bound ROS concentrations in Rochester, NY.
2. Experimental
2.1. Instruments. A schematic diagram of the automatedsampling-analysis system is shown in Figure 1. The detaileddesign and construction of the system were introduced in theprevious study [23]. During the optimization and laboratorytesting of the system, the membrane reactor and superser-pentine reactor were found not to significantly improve thereaction among the DCFH, horseradish peroxidase (HRP)and ROS. Therefore, they were removed from the system andthe HRP was directly dissolved into the DCFH solution.
The current system included a PM2.5 sharp-cut cyclone,a manganese dioxide (MnO2) denuder to remove gas phaseoxidants, and a particle-into-liquid-sampler (PILS, MetrohmInc.) as the inlet system. The solutions are circulated usingan 8-channel peristaltic pump through a selection valve, anda fluorescence detector (FP2020, Jasco Inc.). The sampleand blank cycles were run for 3 minutes and 7 minutes,respectively, via the selection valve to eliminate effects ofone cycle on the next. To minimize variability arising byvisible and long-wavelength UV radiation, as well as toprevent photo-oxidation of the DCFH, the flow lines werecovered with aluminum foil. The sampling flow rate was16.7 L/min.
2.2. Reagents. Two solutions, DCFH with HRP and H2O2
standards, were prepared in a dark environment beforethe measurements. DCFH is a nonfluorescent reagent thatbecomes fluorescent upon reaction with ROS. Glass contain-ers were wrapped with aluminum foil to prevent exposureto light. All solutions were prepared with high purity water(resistivity: 18.2 MΩ·cm at 25◦C, Millipore Corp.).
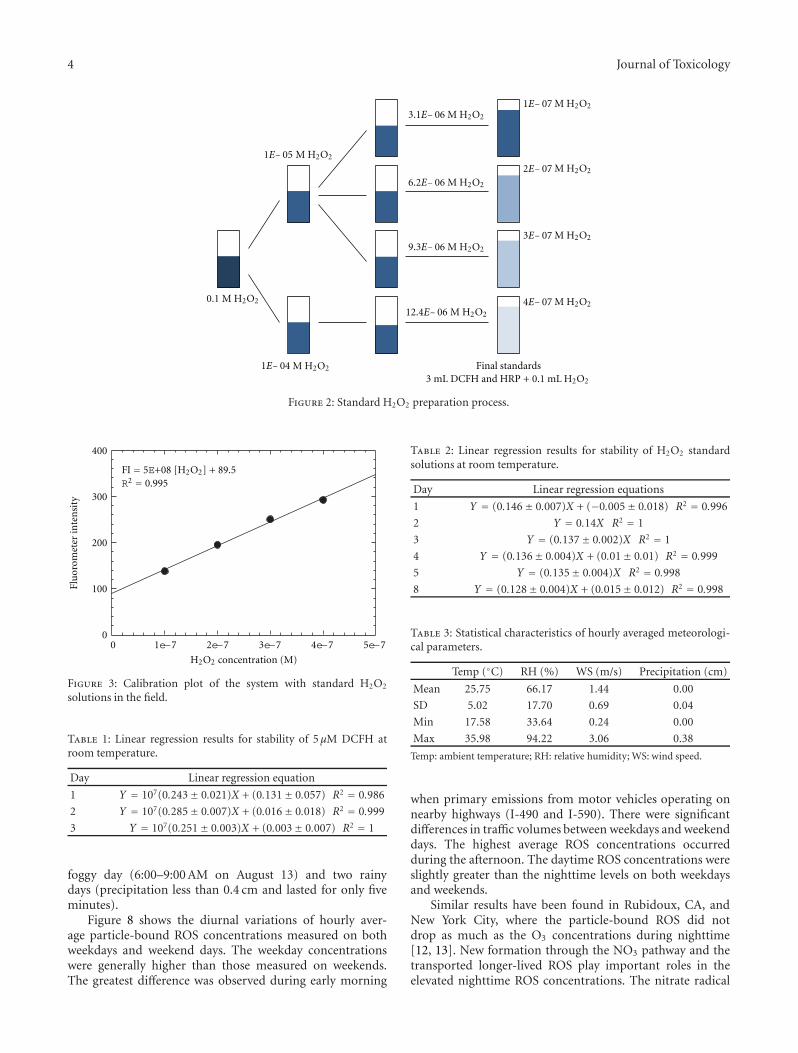
The DCFH and HRP solutions were prepared at 5 μMand 0.5 units/mL, respectively, as described in Appendix A.An standard H2O2 solution was used to develop the cal-ibration curve. The specific preparation process of H2O2
standards through a series of dilutions of 30% H2O2 isshown in Figure 2. Final H2O2 concentrations of 1 × 10−7,2 × 10−7, 3 × 10−7, 4 × 10−7 M were made by mixing0.1 mL of intermediate H2O2 solutions of 3.1×10−6 M, 6.2×10−6 M, 9.3 × 10−6 M, and 12.4 × 10−6 M with 3 mL DCFHsolution prepared with HRP. Standard curves were developedfrom measuring the FI of these final four concentrations ofH2O2.
2.3. Procedure. The standard operation procedure for run-ning the automated ROS system is given in Appendix B.Calibration of the system was performed with standard H2O2
solutions of concentrations ranging from 100 to 400 nM,prepared by serial dilutions of a 30% stock solution ofH2O2, with MilliQ water serving as a blank. A HEPAfilter was placed in front of the system during calibra-tion running. Figure 3 shows the blank-subtracted linearcalibration curve obtained in the field. The system waslinear (R2 = 0.995) over the range of H2O2 concentrationsby least-squares analysis. The relationship between H2O2
concentration and FI is expressed as the equation in thefigure.
2.4. Sampling Location. The particle-bound ROS concen-trations, O3 concentrations and meteorological parameters(ambient temperature, relative humidity, wind direction andspeed) were continuously measured during the period ofAugust 12 to 18, 2009 at the New York State Departmentof Environmental Conservation (NYSDEC) site in Rochester,NY. The site is located at 43◦08′46′′ N, 77◦32′53′′ W, adjacentto Interstate Highway I-490 and I-590, as well as NY Route96, a major route carrying traffic traveling to and fromdowntown Rochester (see Figure 4).

Journal of Toxicology 3
Vacuumpump
Fluorescencedetector
Denuder
Particle size selector
PM2.5
Gas removal
PILS
Debubbler
Sampling
selectionvalve
Waste
Peristaltic pump
MilliQwater
Air dryer
DCFH+ HRP
123
54
678
13
52
GasWaste waterMilliQ waterDCFH + HRP
Figure 1: Schematic diagram of the particle-bound ROS automated system.
3. Results and Discussion
3.1. Stabilities of the DCFH and H2O2 Solutions. The stabilityof the chemical reagents is important for a practical systemthat can be maintained in the field with a reasonable levelof effort. Therefore, the stabilities of DCFH and H2O2
standards were examined. The experimental stability resultsfor 5 μM DCFH stored at room temperature are presentedin Figure 5 and Table 1. It can be seen that 5 μM DCFHwas stable for three days at room temperature. The stabilityof the H2O2 standards is shown in Figure 6 and Table 2.The solutions can be kept at room temperature for up toeight days. These results provide the feasibility in the fielddeployment of the automated sampling-analysis system sincethe unit does not require daily solution preparation.
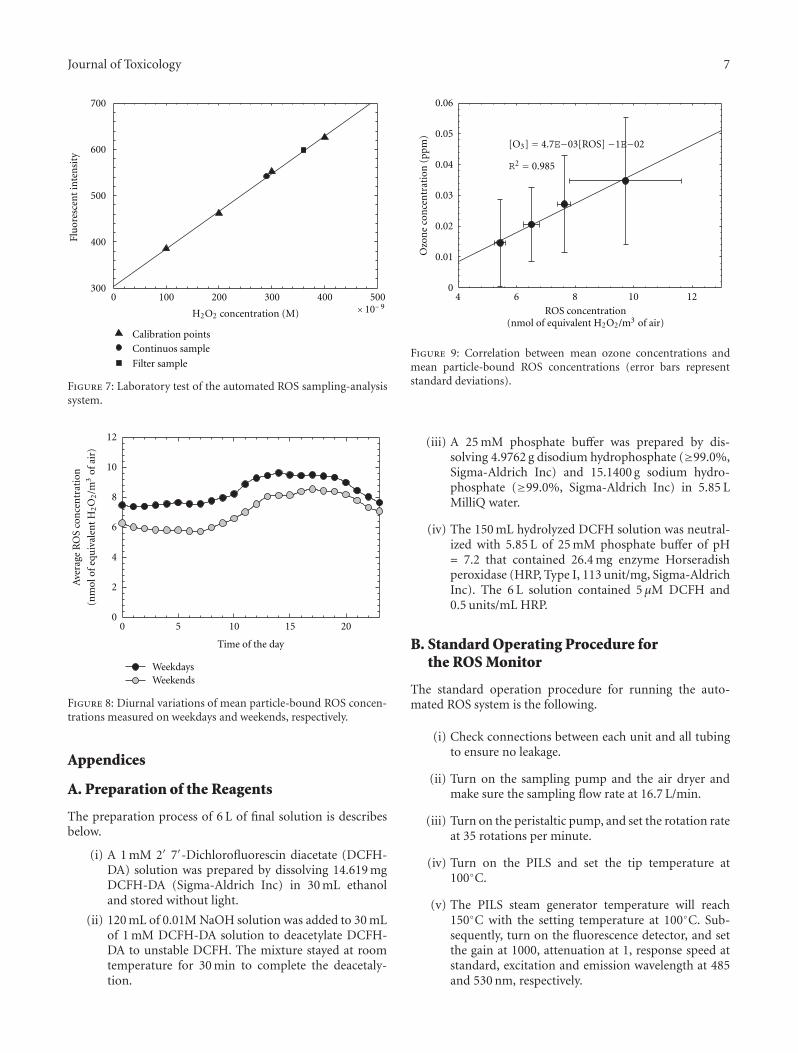
3.2. Laboratory Testing of the System. Laboratory tests wereperformed by sampling particle-bound ROS from an α-pinene-ozone generator [24] for 30 minutes at a flow rateof 16.7 L min−1. The continuous sample and filter samplewere compared with H2O2 standard solutions (see Figure 7).During a 30-minute sampling period, the FI was constant.The filter point represents sample taken on a baked quartzfilter for 15 minute intervals. This sample duration limitsthe loss of short lifetime ROS. 50 mL of 5 μM DCFH wasadded to the filter sample and the filter was then sonicated
for another 15 minutes. The FI of the filter particle-boundROS was comparable to that measured with the continuoussystem. The FI results of filter and continuous samples wereplotted in the standard calibration curve shown in Figure 7.
A somewhat higher FI was obtained from the filtersample, which contradicts the assumption that the filtersampling method may result in the loss of short lifetime ROS,leading to lower FI in filter sample than from continuoussystem sample [23]. The 15-minute extraction of the filtersample probably increased the extent of DCFH oxidizationrather than decreased short lifetime ROS. Another possiblereason was that the extraction volume of DCFH solutionwas 50 mL, which was larger than the volume used for thecontinuous system sample (10 mL). Therefore, higher FI forfilter particle-bound ROS was produced. After the chemicalreagents stability check and laboratory performance testing,the automated particle-bound ROS sampling-analysis systemwas ready for field testing.
3.3. Field Testing of the System. Table 3 summarizes statisticsof meteorological parameters. Persistently sunny and humidweather (average ambient temperature: 25.75◦C, averagerelative humidity: 66.17%) was given by Ontario Lake seatedto the north. The prevailing winds during this period weremainly from the southwest with an average wind speed of1.44 m/s. During the seven days of study, there was one
4 Journal of Toxicology
1E− 07
2E− 07
3E− 07
4E− 07
Final standards3 mL DCFH and HRP + 0.1 mL
1E− 05
0.1
1E− 04
3.1E− 06
6.2E− 06
9.3E− 06
12.4E− 06
M H2O2
M H2O2
M H2O2
M H2O2
M H2O2
M H2O2
M H2O2
M H2O2
M H2O2
M H2O2
M H2O2
H2O2
Figure 2: Standard H2O2 preparation process.
0 1e−7 2e−7 3e−7 4e−7 5e−7
Flu
orom
eter
inte
nsi
ty
0
100
200
300
400
H2O2 concentration (M)
FI = 5E+08 [H2O2] + 89.5R2 = 0.995
Figure 3: Calibration plot of the system with standard H2O2
solutions in the field.
Table 1: Linear regression results for stability of 5 μM DCFH atroom temperature.
Day Linear regression equation
1 Y = 107(0.243± 0.021)X + (0.131± 0.057) R2 = 0.986
2 Y = 107(0.285± 0.007)X + (0.016± 0.018) R2 = 0.999
3 Y = 107(0.251± 0.003)X + (0.003± 0.007) R2 = 1
foggy day (6:00–9:00 AM on August 13) and two rainydays (precipitation less than 0.4 cm and lasted for only fiveminutes).
Figure 8 shows the diurnal variations of hourly aver-age particle-bound ROS concentrations measured on bothweekdays and weekend days. The weekday concentrationswere generally higher than those measured on weekends.The greatest difference was observed during early morning
Table 2: Linear regression results for stability of H2O2 standardsolutions at room temperature.
Day Linear regression equations
1 Y = (0.146± 0.007)X + (−0.005± 0.018) R2 = 0.996
2 Y = 0.14X R2 = 1
3 Y = (0.137± 0.002)X R2 = 1
4 Y = (0.136± 0.004)X + (0.01± 0.01) R2 = 0.999
5 Y = (0.135± 0.004)X R2 = 0.998
8 Y = (0.128± 0.004)X + (0.015± 0.012) R2 = 0.998
Table 3: Statistical characteristics of hourly averaged meteorologi-cal parameters.
Temp (◦C) RH (%) WS (m/s) Precipitation (cm)
Mean 25.75 66.17 1.44 0.00
SD 5.02 17.70 0.69 0.04
Min 17.58 33.64 0.24 0.00
Max 35.98 94.22 3.06 0.38
Temp: ambient temperature; RH: relative humidity; WS: wind speed.
when primary emissions from motor vehicles operating onnearby highways (I-490 and I-590). There were significantdifferences in traffic volumes between weekdays and weekenddays. The highest average ROS concentrations occurredduring the afternoon. The daytime ROS concentrations wereslightly greater than the nighttime levels on both weekdaysand weekends.
Similar results have been found in Rubidoux, CA, andNew York City, where the particle-bound ROS did notdrop as much as the O3 concentrations during nighttime[12, 13]. New formation through the NO3 pathway and thetransported longer-lived ROS play important roles in theelevated nighttime ROS concentrations. The nitrate radical

Journal of Toxicology 5
N
S
W E
2 km
Prevailingwind direction
NYS DEC site
Figure 4: Location of the sampling site in Rochester, NY.
Table 4: Summary of previous particle-bound ROS studies.
Location Concentration (nmol H2O2/m3-air) Period Reference
Flushing, NY, USA 0.87 ± 0.18 Jan-Feb 2004 [13]
Singapore traffic 15.10 ± 0.10 Dec 2005 [21]
Singapore ambient 5.71 ± 2.30 Dec 2005 (10 am–1 pm) [21]
Taipei, Taiwan 0.54 ± 0.40 Jul–Dec 2000 [18]
Rubidoux, CA, USA 5.90 ± 1.70 July 2003 [12, 22]
Rochester, NY, USA 8.30 ± 2.19 Aug 2009 This study
reactions along with the oxidation of alkenes by the residualozone led to ROS concentrations that were only slightlylower than the daytime concentrations [22]. These diurnalpatterns suggest that photochemical reactions and vehicularemissions are the main sources of the atmospheric particle-bound ROS in urban areas.
Table 4 compares the particle-bound ROS concentra-tions measured in different urban locations with filtercollection and extraction methods. Except for the flushing,NY study, all of the studies were conducted during thesummer. The overall average ROS concentration from allthe studies was 6.1 nmol m−3. The lowest ROS concentration(0.54 nmol m−3) was measured in Taipei, Taiwan, which wasan order of magnitude lower than the ROS concentrationsin the other studies. Short-lived ROS with lifetime less than3-hr cannot be estimated, since 3-hr samples were collectedin that study [22]. Thus, concentrations of ROS with lifetimeless than 3-hr might be greatly underestimated. The averageparticle-bound ROS concentration of 8.3 ± 2.2 nmol m−3
measured in this study is among the typical values reportedfor the urban sites of U.S. and Asia.
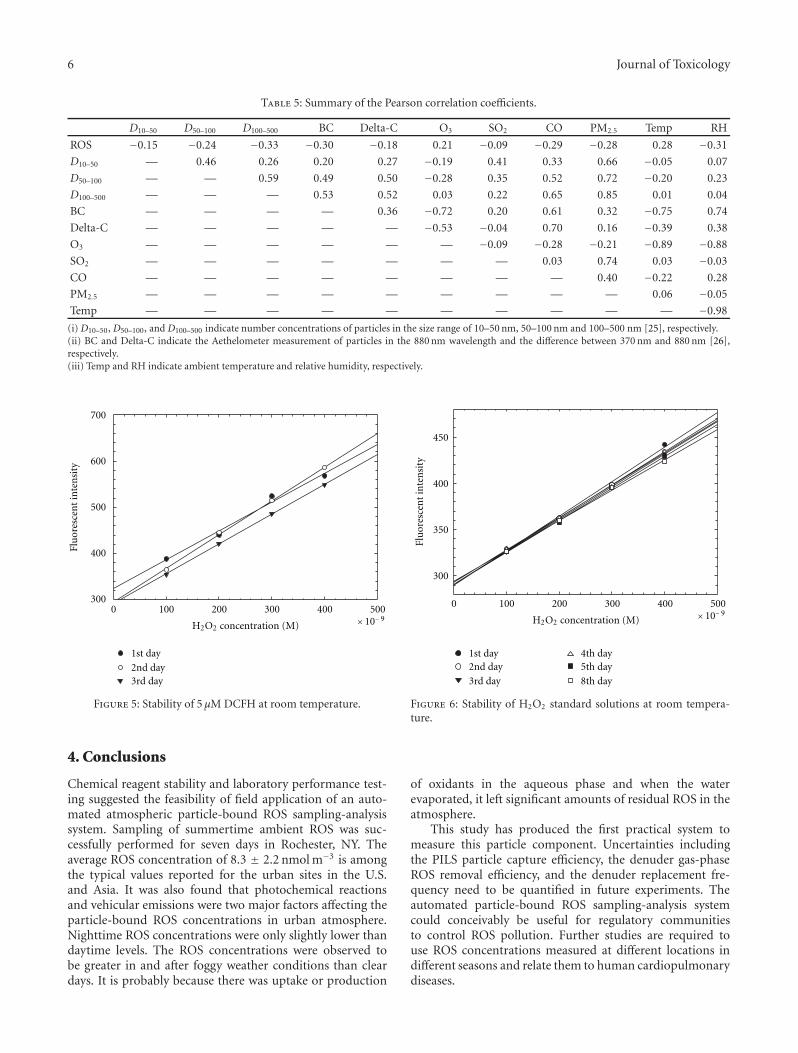
Table 5 summarizes the Pearson correlation coefficientsbetween the hourly averaged ROS values, the other measured
pollutants, and the meteorological variables. All the variableswere measured at the same site and were averaged to hourlyvalues. Details of the measurements are described elsewhere[25–27]. The scatter plot of the average ozone concentrationsand the corresponding ROS concentrations for the entiresampling period is shown in Figure 9. The ozone concentra-tions, measured as a potential indicator of the intensity ofphotochemical reactions [18], were obtained from standardphotometric ozone monitors maintained by the NYSDEC atthis location. The statistically high correlation (r2 = 0.985)between ozone and ROS concentrations indicates that theformation of ROS is strongly influenced by photochemicalactivity, consistent with the previous studies [13, 18, 22].
The largest standard deviation of ROS concentration wasfound for the highest level shown as the top point. It was dueto the higher ROS concentrations measured on the August13, a foggy morning, with an average ROS concentration of12.31 nmol m−3. This event may have resulted from rapiduptake of water-soluble oxidants into the aqueous phaseleading to high residual ROS concentration. In addition, theyields of H2O2 and other complex peroxides were observedto increase substantially in the presence of water vapor in theair from another recent study [28].
6 Journal of Toxicology
Table 5: Summary of the Pearson correlation coefficients.
D10–50 D50–100 D100–500 BC Delta-C O3 SO2 CO PM2.5 Temp RH
ROS −0.15 −0.24 −0.33 −0.30 −0.18 0.21 −0.09 −0.29 −0.28 0.28 −0.31
D10–50 — 0.46 0.26 0.20 0.27 −0.19 0.41 0.33 0.66 −0.05 0.07
D50–100 — — 0.59 0.49 0.50 −0.28 0.35 0.52 0.72 −0.20 0.23
D100–500 — — — 0.53 0.52 0.03 0.22 0.65 0.85 0.01 0.04
BC — — — — 0.36 −0.72 0.20 0.61 0.32 −0.75 0.74
Delta-C — — — — — −0.53 −0.04 0.70 0.16 −0.39 0.38
O3 — — — — — — −0.09 −0.28 −0.21 −0.89 −0.88
SO2 — — — — — — — 0.03 0.74 0.03 −0.03
CO — — — — — — — — 0.40 −0.22 0.28
PM2.5 — — — — — — — — — 0.06 −0.05
Temp — — — — — — — — — — −0.98
(i) D10–50, D50–100, and D100–500 indicate number concentrations of particles in the size range of 10–50 nm, 50–100 nm and 100–500 nm [25], respectively.(ii) BC and Delta-C indicate the Aethelometer measurement of particles in the 880 nm wavelength and the difference between 370 nm and 880 nm [26],respectively.(iii) Temp and RH indicate ambient temperature and relative humidity, respectively.
H2O2 concentration (M) × 10− 90 100 200
300300
400
400
500
500
600
700
Flu
ores
cen
tin
ten
sity
1st day
2nd day3rd day
Figure 5: Stability of 5 μM DCFH at room temperature.
4. Conclusions
Chemical reagent stability and laboratory performance test-ing suggested the feasibility of field application of an auto-mated atmospheric particle-bound ROS sampling-analysissystem. Sampling of summertime ambient ROS was suc-cessfully performed for seven days in Rochester, NY. Theaverage ROS concentration of 8.3 ± 2.2 nmol m−3 is amongthe typical values reported for the urban sites in the U.S.and Asia. It was also found that photochemical reactionsand vehicular emissions were two major factors affecting theparticle-bound ROS concentrations in urban atmosphere.Nighttime ROS concentrations were only slightly lower thandaytime levels. The ROS concentrations were observed tobe greater in and after foggy weather conditions than cleardays. It is probably because there was uptake or production
H2O2 concentration (M) × 10− 90 100 200 300 400 500
1st day2nd day
3rd day
4th day5th day
8th day
300
350
400
450
Flu
ores
cen
tin
ten
sity
Figure 6: Stability of H2O2 standard solutions at room tempera-ture.
of oxidants in the aqueous phase and when the waterevaporated, it left significant amounts of residual ROS in theatmosphere.
This study has produced the first practical system tomeasure this particle component. Uncertainties includingthe PILS particle capture efficiency, the denuder gas-phaseROS removal efficiency, and the denuder replacement fre-quency need to be quantified in future experiments. Theautomated particle-bound ROS sampling-analysis systemcould conceivably be useful for regulatory communitiesto control ROS pollution. Further studies are required touse ROS concentrations measured at different locations indifferent seasons and relate them to human cardiopulmonarydiseases.
Journal of Toxicology 7
H2O2 concentration (M) × 10− 90 100 200
300300
400
400
500
500
600
700
Calibration points
Continuos sample
Filter sample
Flu
ores
cen
tin
ten
sity
Figure 7: Laboratory test of the automated ROS sampling-analysissystem.
0 5 10 15 200
2
4
6
8
10
12
Time of the day
WeekdaysWeekends
Ave
rage
RO
Sco
nce
ntr
atio
n
(nm
olof
equ
ival
ent
H2O
2/m
3of
air)
Figure 8: Diurnal variations of mean particle-bound ROS concen-trations measured on weekdays and weekends, respectively.
Appendices
A. Preparation of the Reagents
The preparation process of 6 L of final solution is describesbelow.
(i) A 1 mM 2′ 7′-Dichlorofluorescin diacetate (DCFH-DA) solution was prepared by dissolving 14.619 mgDCFH-DA (Sigma-Aldrich Inc) in 30 mL ethanoland stored without light.
(ii) 120 mL of 0.01M NaOH solution was added to 30 mLof 1 mM DCFH-DA solution to deacetylate DCFH-DA to unstable DCFH. The mixture stayed at roomtemperature for 30 min to complete the deacetaly-tion.
0
0.01
0.02
0.03
0.04
0.05
0.06
Ozo
ne
con
cen
trat
ion
(ppm
)
[O3] = 4.7E−03[ROS] −1E−02
= 0.985
4 6 8 10 12ROS concentration
(nmol of equivalent H2O2/m3 of air)
R2
Figure 9: Correlation between mean ozone concentrations andmean particle-bound ROS concentrations (error bars representstandard deviations).
(iii) A 25 mM phosphate buffer was prepared by dis-solving 4.9762 g disodium hydrophosphate (≥99.0%,Sigma-Aldrich Inc) and 15.1400 g sodium hydro-phosphate (≥99.0%, Sigma-Aldrich Inc) in 5.85 LMilliQ water.
(iv) The 150 mL hydrolyzed DCFH solution was neutral-ized with 5.85 L of 25 mM phosphate buffer of pH= 7.2 that contained 26.4 mg enzyme Horseradishperoxidase (HRP, Type I, 113 unit/mg, Sigma-AldrichInc). The 6 L solution contained 5 μM DCFH and0.5 units/mL HRP.
B. Standard Operating Procedure forthe ROS Monitor
The standard operation procedure for running the auto-mated ROS system is the following.
(i) Check connections between each unit and all tubingto ensure no leakage.
(ii) Turn on the sampling pump and the air dryer andmake sure the sampling flow rate at 16.7 L/min.
(iii) Turn on the peristaltic pump, and set the rotation rateat 35 rotations per minute.
(iv) Turn on the PILS and set the tip temperature at100◦C.
(v) The PILS steam generator temperature will reach150◦C with the setting temperature at 100◦C. Sub-sequently, turn on the fluorescence detector, and setthe gain at 1000, attenuation at 1, response speed atstandard, excitation and emission wavelength at 485and 530 nm, respectively.

8 Journal of Toxicology
(vi) Start the computer, set the sampling period of 3minutes and rinsing period of 7 minutes.
(vii) Start the “Logger Lite” software (version 1.3.2,Vernier Software & Technology) and build a file tosave the data.
Acknowledgments
This work was supported by the United States EnvironmentalProtection Agency (EPA) through Science to Achieve Results(STAR) Grant no. RD83107801, a Syracuse Center of Excel-lence CARTI Project Award, which is supported by a grantfrom the U.S. Environmental Protection Agency [Award no.X-83232501-0], the Electric Power Research Institute underAgreement no. W06325, and the New York State EnergyResearch and Development Authority (NYSERDA) throughContracts nos. 8650 and 10604. Although the researchdescribed in this paper has been funded wholly or in part bythe EPA, it has not been subjected to the Agency’s peer andpolicy review and, therefore, does not necessarily reflect theviews of the Agency and no official endorsement should beinferred.
References
[1] C. A. Pope and D. W. Dockery, “Health effects of fine partic-ulate air pollution: lines that connect,” Journal of the Air andWaste Management Association, vol. 56, no. 6, pp. 709–742,2006.
[2] W. G. Kreyling, M. Semmler-Behnke, and W. Moller, “Healthimplications of nanoparticles,” Journal of NanoparticleResearch, vol. 8, no. 5, pp. 543–562, 2006.
[3] G. Oberdorster and M. J. Utell, “Ultrafine particles in theurban air: to the respiratory tract—and beyond?” Environ-mental Health Perspectives, vol. 110, no. 8, pp. A440–A441,2002.
[4] A. Peters, S. von Klot, M. Heier et al., “Exposure to traffic andthe onset of myocardial infarction,” The New England Journalof Medicine, vol. 351, no. 17, pp. 1721–1730, 2004.
[5] C. Sioutas, R. J. Delfino, and M. Singh, “Exposure assessmentfor atmospheric ultrafine particles (UFPs) and implicationsin epidemiologic research,” Environmental Health Perspectives,vol. 113, no. 8, pp. 947–955, 2005.
[6] G. L. Squadrito, R. Cueto, B. Dellinger, and W. A. Pryor,“Quinoid redox cycling as a mechanism for sustained freeradical generation by inhaled airborne particulate matter,”Free Radical Biology and Medicine, vol. 31, no. 9, pp. 1132–1138, 2001.
[7] S. J. Stohs, D. Bagchi, and M. Bagchi, “Toxicity of traceelements in tobacco smoke,” Inhalation Toxicology, vol. 9, no.9, pp. 867–890, 1997.
[8] J. Ciencewicki, S. Trivedi, and S. R. Kleeberger, “Oxidantsand the pathogenesis of lung diseases,” Journal of Allergy andClinical Immunology, vol. 122, no. 3, pp. 456–468, 2008.
[9] R. Kelishadi, M. Hashemi, N. Mohammadifard, S. Asgary,and N. Khavarian, “Association of changes in oxidative andproinflammatory states with changes in vascular function aftera lifestyle modification trial among obese children,” ClinicalChemistry, vol. 54, no. 1, pp. 147–153, 2008.
[10] P. Kirkham and I. Rahman, “Oxidative stress in asthma andCOPD: antioxidants as a therapeutic strategy,” Pharmacologyand Therapeutics, vol. 111, no. 2, pp. 476–494, 2006.
[11] W. Yang and S. T. Omaye, “Air pollutants, oxidative stressand human health,” Mutation Research/Genetic Toxicology andEnvironmental Mutagenesis, vol. 674, no. 1-2, pp. 45–54, 2009.
[12] P. Venkatachari, P. K. Hopke, B. D. Grover, and D. J.Eatough, “Erratum: ”Measurement of particle-bound reactiveoxygen species in Rubidoux Aerosols”,” Journal of AtmosphericChemistry, vol. 52, no. 3, pp. 325–326, 2005.
[13] P. Venkatachari, P. K. Hopke, W. H. Brune et al., “Characteri-zation of wintertime reactive oxygen species concentrations inFlushing, New York,” Aerosol Science and Technology, vol. 41,no. 2, pp. 97–111, 2007.
[14] J. L. Mauderly and J. C. Chow, “Health effects of organicaerosols,” Inhalation Toxicology, vol. 20, no. 3, pp. 257–288,2008.
[15] T. Sakulyanontvittaya, T. Duhl, C. Wiedinmyer et al., “Monot-erpene and sesquiterpene emission estimates for the UnitedStates,” Environmental Science and Technology, vol. 42, no. 5,pp. 1623–1629, 2008.
[16] R. P. Wayne, I. Barnes, P. Biggs et al., “The nitrate radical:physics, chemistry, and the atmosphere,” Atmospheric Environ-ment Part A, vol. 25, no. 1, pp. 1–203, 1991.
[17] S. E. Paulson and J. J. Orlando, “The reactions of ozone withalkenes: an important source of HOx in the boundary layer,”Geophysical Research Letters, vol. 23, no. 25, pp. 3727–3730,1996.
[18] H. F. Hung and C. S. Wang, “Experimental determination ofreactive oxygen species in Taipei aerosols,” Journal of AerosolScience, vol. 32, no. 10, pp. 1201–1211, 2001.
[19] F. Rohrer and H. Berresheim, “Strong correlation betweenlevels of tropospheric hydroxyl radicals and solar ultravioletradiation,” Nature, vol. 442, no. 7099, pp. 184–187, 2006.
[20] A. S. Hasson and S. E. Paulson, “An investigation of therelationship between gas-phase and aerosol-borne hydroper-oxides in urban air,” Journal of Aerosol Science, vol. 34, no. 4,pp. 459–468, 2003.
[21] S. W. See, Y. H. Wang, and R. Balasubramanian, “Contrastingreactive oxygen species and transition metal concentrations incombustion aerosols,” Environmental Research, vol. 103, no. 3,pp. 317–324, 2007.
[22] P. Venkatachari, P. K. Hopke, B. D. Grover, and D. J. Eatough,“Measurement of particle-bound reactive oxygen species inrubidoux aerosols,” Journal of Atmospheric Chemistry, vol. 50,no. 1, pp. 49–58, 2005.
[23] P. Venkatachari and P. K. Hopke, “Development and labora-tory testing of an automated monitor for the measurement ofatmospheric particle-bound reactive oxygen species (ROS),”Aerosol Science and Technology, vol. 42, no. 8, pp. 629–635,2008.
[24] P. Venkatachari and P. K. Hopke, “Development and evalu-ation of a particle-bound reactive oxygen species generator,”Journal of Aerosol Science, vol. 39, no. 2, pp. 168–174, 2008.
[25] Y. Wang, P. K. Hopke, D. C. Chalupa, and M. J. Utell, “Long-term study of urban ultrafine particles and other pollutants,”Atmospheric Environment. In press.
[26] Y. Wang, P. K. Hopke, O. V. Rattigan, and Y. Zhu, “Charac-terization of ambient black carbon and wood burning par-ticles in urban areas,” submitted to Journal of EnvironmentalMonitoring.

Journal of Toxicology 9
[27] Y. Wang, J. Huang, T. J. Zananski, P. K. Hopke, and T. M.Holsen, “Impacts of the Canadian forest fires on atmosphericmercury and carbonaceous particles in northern New York,”Environmental Science and Technology, vol. 44, no. 22, pp.8435–8440, 2010.
[28] C. E. Reeves and S. A. Penkett, “Measurements of peroxidesand what they tell us,” Chemical Reviews, vol. 103, no. 12, pp.5199–5218, 2003.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 157687, 7 pagesdoi:10.1155/2011/157687
Research Article
Behavioral Characterization of GCLM -Knockout Mice, a Modelfor Enhanced Susceptibility to Oxidative Stress
Toby B. Cole,1, 2, 3 Gennaro Giordano,1 Aila L. Co,2, 3 Isaac Mohar,1 Terrance J. Kavanagh,1
and Lucio G. Costa1, 4
1 Department of Environmental and Occupational Health Sciences, University of Washington, Seattle, WA 98195, USA2 Division of Medical Genetics, University of Washington, Seattle, WA 98195, USA3 Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA4 Department of Human Anatomy, Pharmacology and Forensic Science, University of Parma Medical School, 43121 Parma, Italy
Correspondence should be addressed to Lucio G. Costa, [email protected]
Received 20 November 2010; Accepted 25 February 2011
Academic Editor: M. Teresa Colomina Fosch
Copyright © 2011 Toby B. Cole et al. This is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Glutathione (GSH) is a major player in cellular defense against oxidative stress. Deletion of the modifier subunit of glutamatecysteine ligase (GCLM), the first and the rate-limiting enzyme in the synthesis of GSH, leads to significantly lower GSH levelsin all tissues including the brain. GCLM-knockout (Gclm−/−) mice may thus represent a model for compromised response tooxidative stress amenable to in vitro and in vivo investigations. In order to determine whether the diminished GSH content wouldby itself cause behavioral alterations, a series of behavioral tests were carried out comparing young adult Gclm−/− with wild-typemice. Tests included the rotarod, acoustic startle reflex and prepulse inhibition of the startle reflex, open field behavior, and theplatform reversal variant of the Morris Water Maze. Results showed no differences between Gclm−/− and wild-type mice in any ofthe neurobehavioral tests. However, more subtle alterations, or changes which may appear as animals age, cannot be excluded.
1. Introduction
Oxidative stress refers to the cytotoxic consequences of reac-tive oxygen species (ROS), which are generated as byproductsof normal and aberrant metabolic processes that use molec-ular oxygen. The tripeptide glutathione (GSH; γ-glutamyl-cysteinyl-glycine) is one of the most abundant cellular thiols.GSH is a major player in cellular defense against ROS,because it nonenzymatically scavenges both singlet oxygenand hydroxyl radicals, and is used by glutathione peroxidasesand glutathione transferases to limit the levels of certainreactive aldehydes and peroxides within the cell [1, 2]. WhenROS production exceeds the antioxidant defense capacityof the cell, oxidative stress ensues, leading to the damageof DNA, proteins, and membrane lipids. The first and therate-limiting step in the synthesis of GSH is carried out byglutamate-cysteine ligase (GCL; [1]). The enzyme consists oftwo subunits, a larger (73 kD) catalytic subunit (GCLC) anda smaller (31 kD) modifier, or regulatory, subunit (GCLM),which are coded by separate genes [3]. GCLC alone provides
catalytic activity and is the site of GSH feedback inhibition.By lowering the Km of GCL for glutamate and raisingthe Ki for GSH, GCLM, although enzymatically inactive,plays an important regulatory function, as the holoenzyme(GCLholo) has higher catalytic efficiency than GCLC [3, 4].While disruption of the Gclc gene in mice is embryolethal[5], no overt phenotype is observed upon disruption of theGclm gene in mice [4, 6, 7]. In the absence of GCLM, theability of GCLC to synthesize GSH is drastically reduced [8].In tissues from Gclm−/− mice, GSH levels are only 10–30%of those found in Gclm+/+ animals [6, 7, 9]. In brain tissueand cells, GSH levels in Gclm−/− mice are 17–35% of thosepresent in wild-type mice [6, 10, 11].
Gclm−/− mice are more sensitive to the hepatotoxicityof acetaminophen [7], and neurons and astrocytes isolatedfrom the brain of Gclm−/− mice have been shown tobe particularly susceptible to the toxicity of agents thatincrease oxidative stress, such as domoic acid [6], certainorganophosphorus insecticides [12], methylmercury andPCBs [13], and polybrominated diphenyl ethers [10].

2 Journal of Toxicology
A relatively common C588T polymorphism has beendiscovered in the 5′-flanking region of the human GCLMgene [14]. Individuals carrying the T allele have a lowerpromoter activity in a luciferase reporter gene assay inresponse to oxidants and significantly lower plasma GSHlevels [14]. These individuals are also at risk for myocardialinfarction and present with impairments in nitric oxide-mediated coronary vasomotor function [14, 15]. An associa-tion between GCLM polymorphisms and schizophrenia hasalso been suggested [16] but is still controversial [17, 18].
Nevertheless, individuals with GCLM polymorphismsleading to lower GSH levels would be expected to displayan enhanced sensitivity to the adverse effects of oxidativestress. The Gclm−/− mouse thus represents a useful model forsuch GCLM polymorphisms, amenable for in vitro, as wellas in vivo studies. In order to extend in vitro observationsto an in vivo situation, an initial behavioral characterizationof Gclm−/− mice was carried out, to determine whetherthe genetically determined diminished GSH level would byitself affect behavioral outcomes. Indeed, glutathione dys-regulation is associated with the etiology and progression ofseveral diseases, including neurotoxic and neurodegenerativedisorders [19, 20].
2. Materials and Methods
2.1. Generation of Gclm-Null Mice and Genotyping. All ani-mal use protocols were approved by the Institutional AnimalCare and Use Committee at the University of Washington,and experiments were carried out in accordance with theNational Research Council Guide for the Care and Use ofLaboratory Animals, as adopted by the National Institutesof Health. Wild-type and Gclm-null (Gclm−/−) mice ofbackcrossed C57Bl/6J (B6.129) strain background [6, 7] werehoused in a centralized, AAALAC-accredited, and specificpathogen-free facility at the University of Washington. Micewere maintained in a 12 h light-dark cycle with ad libitumaccess to food (standard mouse chow) and water. Male andfemale mice hemizygous for the Gclm deletion (Gclm-Hz)were intercrossed, generating wild-type, Gclm−/−, and Gclm-Hz mice, in the expected Mendelian ratios [21].
To genotype pups, genomic DNA was isolated fromear punch tissue using a Qiagen DNeasy kit, and micewere genotyped by PCR amplification of the wild-typeand disrupted Gclm alleles (i.e., amplification of β-geo), aspreviously described [6, 7].
As seen previously, all pups developed normally andexhibited no differences in phenotypic landmarks comparedto wild-type littermates. At weaning, mice were transferredto the neurobehavioral testing facility and housed two to fourper cage for the duration of testing.
2.2. Neurobehavioral Assessment. One wild-type (total = 12)or Gclm−/− (total = 13) male mouse, each taken from adifferent litter, was used for neurobehavioral testing. Testswere chosen to investigate possible differences between thetwo mouse genotypes in sensory functions, motor activityand coordination, and learning and memory.
Auditory startle and prepulse inhibition of startle weretested at 12 weeks of age using an automated auditorystartle chamber (San Diego Instruments). During a 15-minute test session, mice were placed in the startle chamberand presented with 30 stimuli at randomized intervals. Thestimuli consisted of a 120 dB tone, a 120 dB tone precededby a 70 dB prepulse, or a “null” stimulus involving no tone.Each type of stimulus was presented 10 times. The order ofstimulus presentation was first determined using a randomnumber table, after which each mouse received the stimuliin the same order. The startle chambers used a piezoelectricsensor to measure the maximum amplitude (Vmax) of thestartle response after each stimulus and the latency to themaximum startle response (Tmax). Prepulse inhibition ofstartle was calculated as the percent inhibition of the auditorystartle response by the 70 dB prepulse, after subtracting thestartle response to the null stimulus [22, 23].
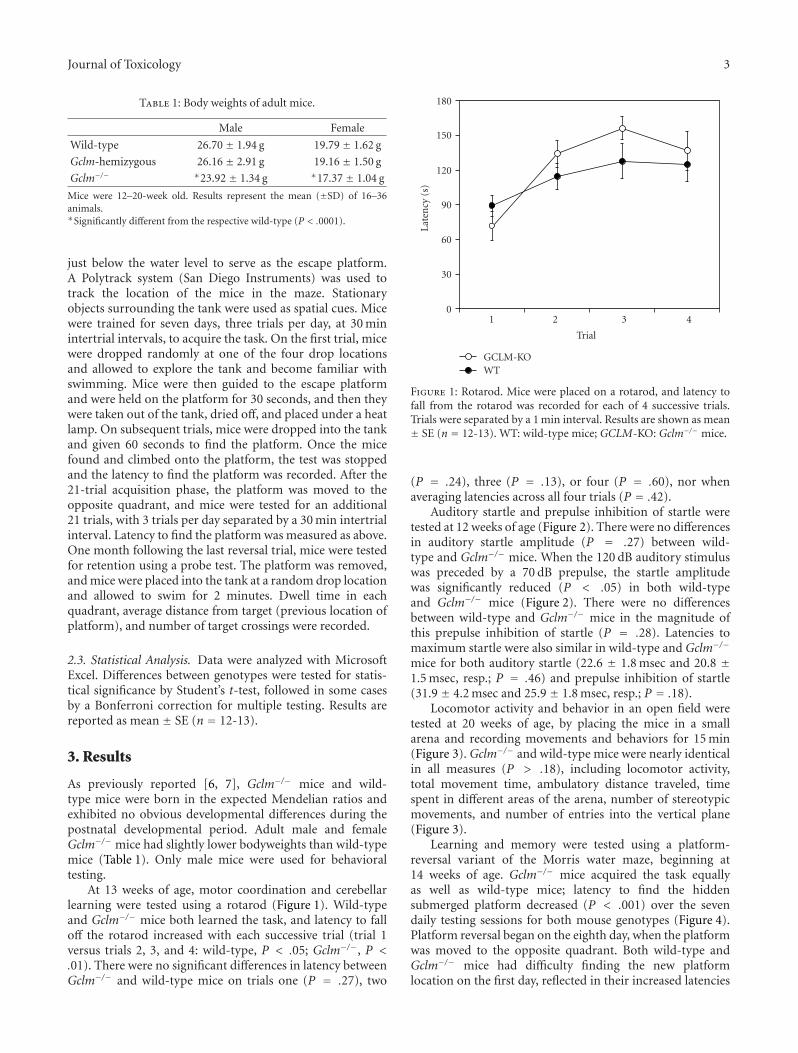
A rotarod (Coulbourn Instruments) was used to testmotor coordination and cerebellar learning [24, 25] at 13weeks of age. Mice were placed on the rotarod cylinder, whichaccelerated to 5 rpm/min from a baseline rate of 3 rpm.Latency to fall off the cylinder was recorded for each of foursuccessive trials, with a 5 min intertrial interval.
Open-field behavior and locomotor activity were testedat 20 weeks of age using a Tru-Scan photo beam trackingsystem (Coulbourn Instruments, Whitehall PA). Mice wereplaced in an open-field arena that was 25.4 cm wide, 25.4 cmdeep, and 40.64 cm high, and movements and behaviorswere recorded for 15 min using dual sensor rings to measureinfrared beam breaks in the horizontal or vertical plane.Beams were spaced 1.52 cm apart, providing 0.76 cm spatialresolution. Data were collected in 30 sec bins, and totals overthe 15 min testing period were calculated as the sum of the30 individual values. Data were also analyzed individuallyfor the first 5 min period, the second 5-min period, andthe third 5 min period. Specific measures included totalnumber of movements, total movement time, total rest time,ambulatory move time, latency to first movement, latencyto first ambulatory movement, total movement distance,ambulatory distance, mean velocity, ambulatory velocity,distance traveled in arena margin and arena center, timespent in arena margin and arena center, number of entriesinto arena center, time spent in back half and front half ofarena, number of entries into back half and front half ofarena, number of entries into vertical plane, time spent invertical plane, number of jumps from floor plane, numberof movements in vertical plane, number of stereotypicmovements, number of stereotypic episodes, total timeof stereotypic behavior, and number of counterclockwiseand clockwise center point rotations. Data were collectedautomatically using Tru-Scan 2.0 software, and raw data wereexported for analysis by Microsoft Excel.
A platform-reversal variant of the Morris water maze wasused to test learning and memory [26–28] beginning at 14weeks of age. This test utilized the polytrack system above.The maze consisted of a 165-gallon (624.6 liter), circular,galvanized stock tank, 4 ft (1.22 meter) in diameter and 2 ft(0.61 meter) in height, filled with room temperature water.A 10 cm square plexiglass stand was placed in the tank
Journal of Toxicology 3
Table 1: Body weights of adult mice.
Male Female
Wild-type 26.70 ± 1.94 g 19.79 ± 1.62 g
Gclm-hemizygous 26.16 ± 2.91 g 19.16 ± 1.50 g
Gclm−/− ∗23.92 ± 1.34 g ∗17.37 ± 1.04 g
Mice were 12–20-week old. Results represent the mean (±SD) of 16–36animals.∗Significantly different from the respective wild-type (P < .0001).
just below the water level to serve as the escape platform.A Polytrack system (San Diego Instruments) was used totrack the location of the mice in the maze. Stationaryobjects surrounding the tank were used as spatial cues. Micewere trained for seven days, three trials per day, at 30 minintertrial intervals, to acquire the task. On the first trial, micewere dropped randomly at one of the four drop locationsand allowed to explore the tank and become familiar withswimming. Mice were then guided to the escape platformand were held on the platform for 30 seconds, and then theywere taken out of the tank, dried off, and placed under a heatlamp. On subsequent trials, mice were dropped into the tankand given 60 seconds to find the platform. Once the micefound and climbed onto the platform, the test was stoppedand the latency to find the platform was recorded. After the21-trial acquisition phase, the platform was moved to theopposite quadrant, and mice were tested for an additional21 trials, with 3 trials per day separated by a 30 min intertrialinterval. Latency to find the platform was measured as above.One month following the last reversal trial, mice were testedfor retention using a probe test. The platform was removed,and mice were placed into the tank at a random drop locationand allowed to swim for 2 minutes. Dwell time in eachquadrant, average distance from target (previous location ofplatform), and number of target crossings were recorded.
2.3. Statistical Analysis. Data were analyzed with MicrosoftExcel. Differences between genotypes were tested for statis-tical significance by Student’s t-test, followed in some casesby a Bonferroni correction for multiple testing. Results arereported as mean ± SE (n = 12-13).
3. Results
As previously reported [6, 7], Gclm−/− mice and wild-type mice were born in the expected Mendelian ratios andexhibited no obvious developmental differences during thepostnatal developmental period. Adult male and femaleGclm−/− mice had slightly lower bodyweights than wild-typemice (Table 1). Only male mice were used for behavioraltesting.
At 13 weeks of age, motor coordination and cerebellarlearning were tested using a rotarod (Figure 1). Wild-typeand Gclm−/− mice both learned the task, and latency to falloff the rotarod increased with each successive trial (trial 1versus trials 2, 3, and 4: wild-type, P < .05; Gclm−/−, P <.01). There were no significant differences in latency betweenGclm−/− and wild-type mice on trials one (P = .27), two
0
30
60
90
120
150
180
Late
ncy
(s)
1 2 3 4
Trial
GCLM-KOWT
Figure 1: Rotarod. Mice were placed on a rotarod, and latency tofall from the rotarod was recorded for each of 4 successive trials.Trials were separated by a 1 min interval. Results are shown as mean± SE (n = 12-13). WT: wild-type mice; GCLM-KO: Gclm−/− mice.
(P = .24), three (P = .13), or four (P = .60), nor whenaveraging latencies across all four trials (P = .42).
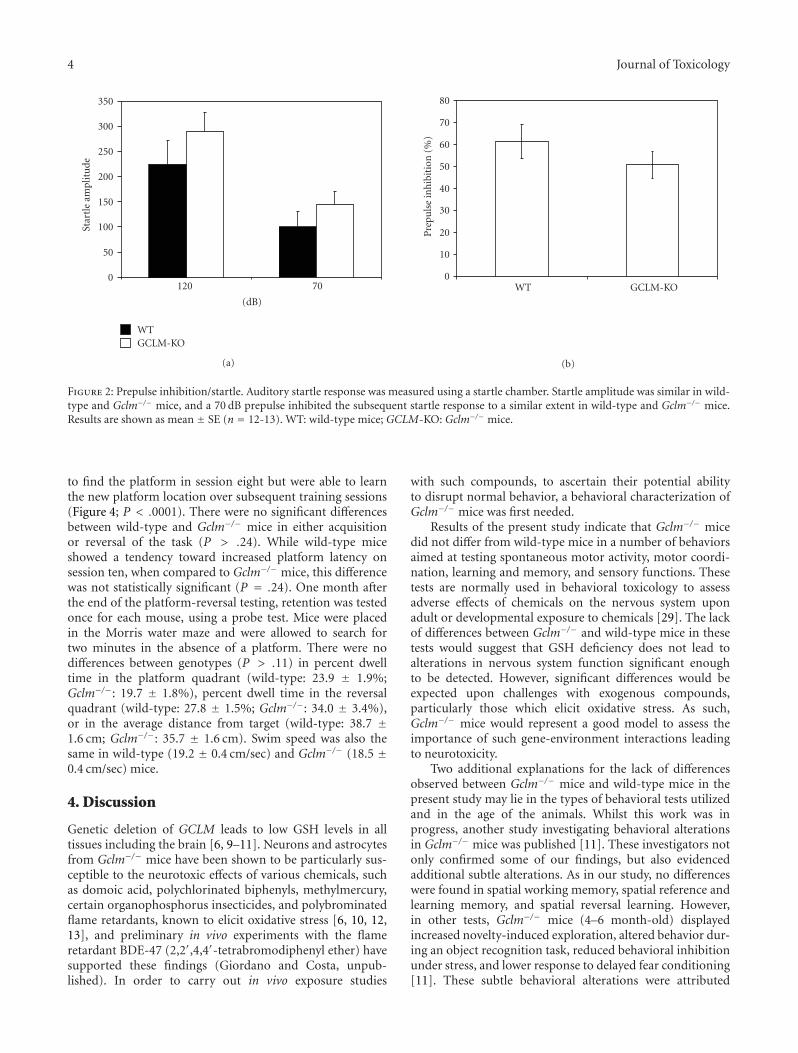
Auditory startle and prepulse inhibition of startle weretested at 12 weeks of age (Figure 2). There were no differencesin auditory startle amplitude (P = .27) between wild-type and Gclm−/− mice. When the 120 dB auditory stimuluswas preceded by a 70 dB prepulse, the startle amplitudewas significantly reduced (P < .05) in both wild-typeand Gclm−/− mice (Figure 2). There were no differencesbetween wild-type and Gclm−/− mice in the magnitude ofthis prepulse inhibition of startle (P = .28). Latencies tomaximum startle were also similar in wild-type and Gclm−/−
mice for both auditory startle (22.6 ± 1.8 msec and 20.8 ±1.5 msec, resp.; P = .46) and prepulse inhibition of startle(31.9 ± 4.2 msec and 25.9 ± 1.8 msec, resp.; P = .18).
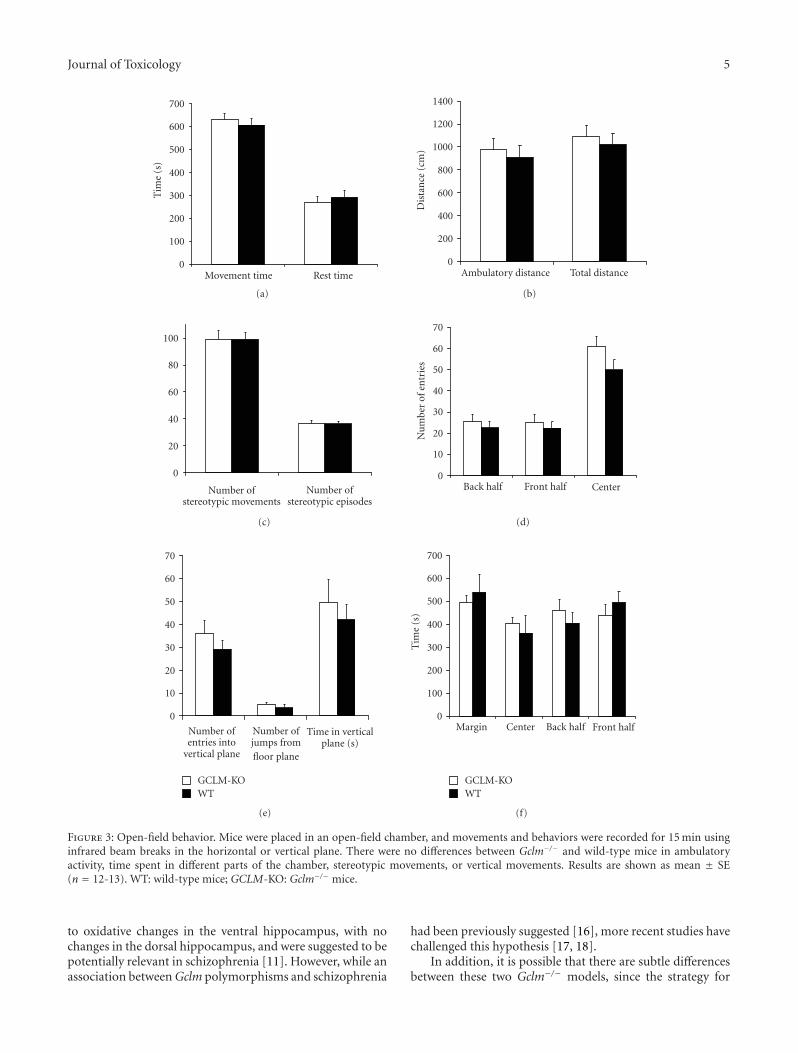
Locomotor activity and behavior in an open field weretested at 20 weeks of age, by placing the mice in a smallarena and recording movements and behaviors for 15 min(Figure 3). Gclm−/− and wild-type mice were nearly identicalin all measures (P > .18), including locomotor activity,total movement time, ambulatory distance traveled, timespent in different areas of the arena, number of stereotypicmovements, and number of entries into the vertical plane(Figure 3).
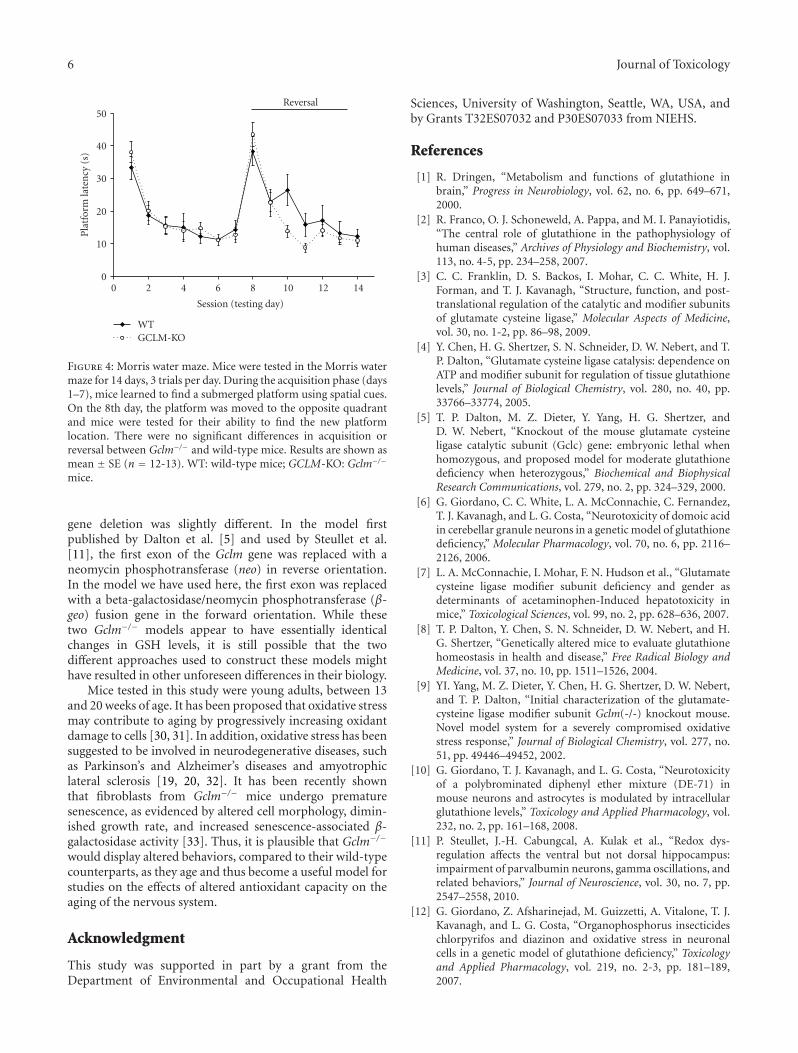
Learning and memory were tested using a platform-reversal variant of the Morris water maze, beginning at14 weeks of age. Gclm−/− mice acquired the task equallyas well as wild-type mice; latency to find the hiddensubmerged platform decreased (P < .001) over the sevendaily testing sessions for both mouse genotypes (Figure 4).Platform reversal began on the eighth day, when the platformwas moved to the opposite quadrant. Both wild-type andGclm−/− mice had difficulty finding the new platformlocation on the first day, reflected in their increased latencies
4 Journal of Toxicology
0
50
100
150
200
250
300
350
Star
tle
ampl
itu
de
120 70
(dB)
GCLM-KOWT
(a)
0
10
20
30
40
50
60
70
80
Pre
puls
ein
hib
itio
n(%
)
WT GCLM-KO
(b)
Figure 2: Prepulse inhibition/startle. Auditory startle response was measured using a startle chamber. Startle amplitude was similar in wild-type and Gclm−/− mice, and a 70 dB prepulse inhibited the subsequent startle response to a similar extent in wild-type and Gclm−/− mice.Results are shown as mean ± SE (n = 12-13). WT: wild-type mice; GCLM-KO: Gclm−/− mice.
to find the platform in session eight but were able to learnthe new platform location over subsequent training sessions(Figure 4; P < .0001). There were no significant differencesbetween wild-type and Gclm−/− mice in either acquisitionor reversal of the task (P > .24). While wild-type miceshowed a tendency toward increased platform latency onsession ten, when compared to Gclm−/− mice, this differencewas not statistically significant (P = .24). One month afterthe end of the platform-reversal testing, retention was testedonce for each mouse, using a probe test. Mice were placedin the Morris water maze and were allowed to search fortwo minutes in the absence of a platform. There were nodifferences between genotypes (P > .11) in percent dwelltime in the platform quadrant (wild-type: 23.9 ± 1.9%;Gclm−/−: 19.7 ± 1.8%), percent dwell time in the reversalquadrant (wild-type: 27.8 ± 1.5%; Gclm−/−: 34.0 ± 3.4%),or in the average distance from target (wild-type: 38.7 ±1.6 cm; Gclm−/−: 35.7 ± 1.6 cm). Swim speed was also thesame in wild-type (19.2 ± 0.4 cm/sec) and Gclm−/− (18.5 ±0.4 cm/sec) mice.
4. Discussion
Genetic deletion of GCLM leads to low GSH levels in alltissues including the brain [6, 9–11]. Neurons and astrocytesfrom Gclm−/− mice have been shown to be particularly sus-ceptible to the neurotoxic effects of various chemicals, suchas domoic acid, polychlorinated biphenyls, methylmercury,certain organophosphorus insecticides, and polybrominatedflame retardants, known to elicit oxidative stress [6, 10, 12,13], and preliminary in vivo experiments with the flameretardant BDE-47 (2,2′,4,4′-tetrabromodiphenyl ether) havesupported these findings (Giordano and Costa, unpub-lished). In order to carry out in vivo exposure studies
with such compounds, to ascertain their potential abilityto disrupt normal behavior, a behavioral characterization ofGclm−/− mice was first needed.
Results of the present study indicate that Gclm−/− micedid not differ from wild-type mice in a number of behaviorsaimed at testing spontaneous motor activity, motor coordi-nation, learning and memory, and sensory functions. Thesetests are normally used in behavioral toxicology to assessadverse effects of chemicals on the nervous system uponadult or developmental exposure to chemicals [29]. The lackof differences between Gclm−/− and wild-type mice in thesetests would suggest that GSH deficiency does not lead toalterations in nervous system function significant enoughto be detected. However, significant differences would beexpected upon challenges with exogenous compounds,particularly those which elicit oxidative stress. As such,Gclm−/− mice would represent a good model to assess theimportance of such gene-environment interactions leadingto neurotoxicity.
Two additional explanations for the lack of differencesobserved between Gclm−/− mice and wild-type mice in thepresent study may lie in the types of behavioral tests utilizedand in the age of the animals. Whilst this work was inprogress, another study investigating behavioral alterationsin Gclm−/− mice was published [11]. These investigators notonly confirmed some of our findings, but also evidencedadditional subtle alterations. As in our study, no differenceswere found in spatial working memory, spatial reference andlearning memory, and spatial reversal learning. However,in other tests, Gclm−/− mice (4–6 month-old) displayedincreased novelty-induced exploration, altered behavior dur-ing an object recognition task, reduced behavioral inhibitionunder stress, and lower response to delayed fear conditioning[11]. These subtle behavioral alterations were attributed
Journal of Toxicology 5
0
100
200
300
400
500
600
700
Tim
e(s
)
Movement time Rest time
(a)
0
200
400
600
800
1000
1200
1400
Dis
tan
ce(c
m)
Ambulatory distance Total distance
(b)
0
20
40
60
80
100
Number ofstereotypic movements
Number ofstereotypic episodes
(c)
0
10
20
30
40
50
60
70
Nu
mbe
rof
entr
ies
Back half Front half Center
(d)
0
10
20
30
40
50
60
70
Number ofentries into
vertical plane
Number ofjumps fromfloor plane
Time in verticalplane (s)
GCLM-KOWT
(e)
0
100
200
300
400
500
600
700
Tim
e(s
)
Margin Center Back half Front half
GCLM-KOWT
(f)
Figure 3: Open-field behavior. Mice were placed in an open-field chamber, and movements and behaviors were recorded for 15 min usinginfrared beam breaks in the horizontal or vertical plane. There were no differences between Gclm−/− and wild-type mice in ambulatoryactivity, time spent in different parts of the chamber, stereotypic movements, or vertical movements. Results are shown as mean ± SE(n = 12-13). WT: wild-type mice; GCLM-KO: Gclm−/− mice.
to oxidative changes in the ventral hippocampus, with nochanges in the dorsal hippocampus, and were suggested to bepotentially relevant in schizophrenia [11]. However, while anassociation between Gclm polymorphisms and schizophrenia
had been previously suggested [16], more recent studies havechallenged this hypothesis [17, 18].
In addition, it is possible that there are subtle differencesbetween these two Gclm−/− models, since the strategy for
6 Journal of Toxicology
0
10
20
30
40
50
Pla
tfor
mla
ten
cy(s
)
0 2 4 6 8 10 12 14
Session (testing day)
GCLM-KOWT
Reversal
Figure 4: Morris water maze. Mice were tested in the Morris watermaze for 14 days, 3 trials per day. During the acquisition phase (days1–7), mice learned to find a submerged platform using spatial cues.On the 8th day, the platform was moved to the opposite quadrantand mice were tested for their ability to find the new platformlocation. There were no significant differences in acquisition orreversal between Gclm−/− and wild-type mice. Results are shown asmean ± SE (n = 12-13). WT: wild-type mice; GCLM-KO: Gclm−/−
mice.
gene deletion was slightly different. In the model firstpublished by Dalton et al. [5] and used by Steullet et al.[11], the first exon of the Gclm gene was replaced with aneomycin phosphotransferase (neo) in reverse orientation.In the model we have used here, the first exon was replacedwith a beta-galactosidase/neomycin phosphotransferase (β-geo) fusion gene in the forward orientation. While thesetwo Gclm−/− models appear to have essentially identicalchanges in GSH levels, it is still possible that the twodifferent approaches used to construct these models mighthave resulted in other unforeseen differences in their biology.
Mice tested in this study were young adults, between 13and 20 weeks of age. It has been proposed that oxidative stressmay contribute to aging by progressively increasing oxidantdamage to cells [30, 31]. In addition, oxidative stress has beensuggested to be involved in neurodegenerative diseases, suchas Parkinson’s and Alzheimer’s diseases and amyotrophiclateral sclerosis [19, 20, 32]. It has been recently shownthat fibroblasts from Gclm−/− mice undergo prematuresenescence, as evidenced by altered cell morphology, dimin-ished growth rate, and increased senescence-associated β-galactosidase activity [33]. Thus, it is plausible that Gclm−/−
would display altered behaviors, compared to their wild-typecounterparts, as they age and thus become a useful model forstudies on the effects of altered antioxidant capacity on theaging of the nervous system.
Acknowledgment
This study was supported in part by a grant from theDepartment of Environmental and Occupational Health
Sciences, University of Washington, Seattle, WA, USA, andby Grants T32ES07032 and P30ES07033 from NIEHS.
References
[1] R. Dringen, “Metabolism and functions of glutathione inbrain,” Progress in Neurobiology, vol. 62, no. 6, pp. 649–671,2000.
[2] R. Franco, O. J. Schoneweld, A. Pappa, and M. I. Panayiotidis,“The central role of glutathione in the pathophysiology ofhuman diseases,” Archives of Physiology and Biochemistry, vol.113, no. 4-5, pp. 234–258, 2007.
[3] C. C. Franklin, D. S. Backos, I. Mohar, C. C. White, H. J.Forman, and T. J. Kavanagh, “Structure, function, and post-translational regulation of the catalytic and modifier subunitsof glutamate cysteine ligase,” Molecular Aspects of Medicine,vol. 30, no. 1-2, pp. 86–98, 2009.
[4] Y. Chen, H. G. Shertzer, S. N. Schneider, D. W. Nebert, and T.P. Dalton, “Glutamate cysteine ligase catalysis: dependence onATP and modifier subunit for regulation of tissue glutathionelevels,” Journal of Biological Chemistry, vol. 280, no. 40, pp.33766–33774, 2005.
[5] T. P. Dalton, M. Z. Dieter, Y. Yang, H. G. Shertzer, andD. W. Nebert, “Knockout of the mouse glutamate cysteineligase catalytic subunit (Gclc) gene: embryonic lethal whenhomozygous, and proposed model for moderate glutathionedeficiency when heterozygous,” Biochemical and BiophysicalResearch Communications, vol. 279, no. 2, pp. 324–329, 2000.
[6] G. Giordano, C. C. White, L. A. McConnachie, C. Fernandez,T. J. Kavanagh, and L. G. Costa, “Neurotoxicity of domoic acidin cerebellar granule neurons in a genetic model of glutathionedeficiency,” Molecular Pharmacology, vol. 70, no. 6, pp. 2116–2126, 2006.
[7] L. A. McConnachie, I. Mohar, F. N. Hudson et al., “Glutamatecysteine ligase modifier subunit deficiency and gender asdeterminants of acetaminophen-Induced hepatotoxicity inmice,” Toxicological Sciences, vol. 99, no. 2, pp. 628–636, 2007.
[8] T. P. Dalton, Y. Chen, S. N. Schneider, D. W. Nebert, and H.G. Shertzer, “Genetically altered mice to evaluate glutathionehomeostasis in health and disease,” Free Radical Biology andMedicine, vol. 37, no. 10, pp. 1511–1526, 2004.
[9] YI. Yang, M. Z. Dieter, Y. Chen, H. G. Shertzer, D. W. Nebert,and T. P. Dalton, “Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(-/-) knockout mouse.Novel model system for a severely compromised oxidativestress response,” Journal of Biological Chemistry, vol. 277, no.51, pp. 49446–49452, 2002.
[10] G. Giordano, T. J. Kavanagh, and L. G. Costa, “Neurotoxicityof a polybrominated diphenyl ether mixture (DE-71) inmouse neurons and astrocytes is modulated by intracellularglutathione levels,” Toxicology and Applied Pharmacology, vol.232, no. 2, pp. 161–168, 2008.
[11] P. Steullet, J.-H. Cabungcal, A. Kulak et al., “Redox dys-regulation affects the ventral but not dorsal hippocampus:impairment of parvalbumin neurons, gamma oscillations, andrelated behaviors,” Journal of Neuroscience, vol. 30, no. 7, pp.2547–2558, 2010.
[12] G. Giordano, Z. Afsharinejad, M. Guizzetti, A. Vitalone, T. J.Kavanagh, and L. G. Costa, “Organophosphorus insecticideschlorpyrifos and diazinon and oxidative stress in neuronalcells in a genetic model of glutathione deficiency,” Toxicologyand Applied Pharmacology, vol. 219, no. 2-3, pp. 181–189,2007.

Journal of Toxicology 7
[13] L. G. Costa, V. Fattori, G. Giordano, and A. Vitalone, “Anin vitro approach to assess the toxicity of certain food con-taminants: methylmercury and polychlorinated biphenyls,”Toxicology, vol. 237, no. 1—3, pp. 65–76, 2007.
[14] S.-I. Nakamura, K. Kugiyama, S. Sugiyama et al., “Polymor-phism in the 5′-flanking region of human glutamate-cysteineligase modifier subunit gene is associated with myocardialinfarction,” Circulation, vol. 105, no. 25, pp. 2968–2973, 2002.
[15] S. I. Nakamura, S. Sugiyama, D. Fujioka, K. I. Kawabata, H.Ogawa, and K. Kugiyama, “Polymorphism in glutamate—cysteine ligase modifier subunit gene is associated withimpairment of nitric oxide—mediated coronary vasomotorfunction,” Circulation, vol. 108, no. 12, pp. 1425–1427, 2003.
[16] M. Tosic, J. Ott, S. Barral, P. Bovet, P. Deppen, F. Gheorghitaet al., “Schizophrenia and oxidative stress: glutamate cysteineligase modifier as a susceptibility gene,” American Journal ofHuman Genetics, vol. 79, no. 3, pp. 586–592, 2006.
[17] C. Butticaz, T. Werge, J. S. Beckmann, M. Cuenod, K. Q. Do,and C. Rivolta, “Mutation screening of the glutamate cysteineligase modifier (GCLM) gene in patients with schizophrenia,”Psychiatric Genetics, vol. 19, no. 4, pp. 201–208, 2009.
[18] J. Ma, D. M. Li, R. Zhang et al., “Genetic analysis of glutamatecysteine ligase modifier (GCLM) gene and schizophrenia inHan Chinese,” Schizophrenia Research, vol. 119, no. 1—3, pp.273–274, 2010.
[19] L. M. Sayre, G. Perry, and M. A. Smith, “Oxidative stress andneurotoxicity,” Chemical Research in Toxicology, vol. 21, no. 1,pp. 172–188, 2008.
[20] N. Ballatori, S. M. Krance, S. Notenboom, S. Shi, K. Tieu, andC. L. Hammond, “Glutathione dysregulation and the etiologyand progression of human diseases,” Biological Chemistry, vol.390, no. 3, pp. 191–214, 2009.
[21] D. Botta, C. C. White, P. Vliet-Gregg et al., “Modulating GSHsynthesis using glutamate cysteine ligase transgenic and gene-targeted mice,” Drug Metabolism Reviews, vol. 40, no. 3, pp.465–477, 2008.
[22] S. F. Logue, E. H. Owen, D. L. Rasmussen, and J. M. Wehner,“Assessment of locomotor activity, acoustic and tactile startle,and prepulse inhibition of stratle in inbred mouse strains andF1 hybrids: implications of genetic background for single geneand quantitative trait loci analyses,” Neuroscience, vol. 80, no.4, pp. 1075–1086, 1997.
[23] R. Paylor and J. N. Crawley, “Inbred strain differences inprepulse inhibition of the mouse startle response,” Psy-chopharmacology, vol. 132, no. 2, pp. 169–180, 1997.
[24] J. Altman and K. Sudarshan, “Postnatal development oflocomotion in the laboratory rat,” Animal Behaviour, vol. 23,no. 4, pp. 896–920, 1975.
[25] V. J. Moser, “Neurobehavioral screening in rodents,” inCurrent Protocols in Toxicology, L. G. Costa, E. Hodgson, andD. J. Reed, Eds., John Wiley & Sons, Inc., 1999.
[26] R. Morris, “Developments of a water-maze procedure forstudying spatial learning in the rat,” Journal of NeuroscienceMethods, vol. 11, no. 1, pp. 47–60, 1984.
[27] G. L. Wenk, “Assessment of spatial memory,” in CurrentProtocols in Toxicology, L. G. Costa, E. Hodgson, and D. J. Reed,Eds., pp. 11.3.1–11.3.18, John Wiley & Sons, Inc., 1999.
[28] V. Voikar, S. Koks, E. Vasar, and H. Rauvala, “Strain and genderdifferences in the behavior of mouse lines commonly used intransgenic studies,” Physiology and Behavior, vol. 72, no. 1-2,pp. 271–281, 2001.
[29] K. C. Raffaele and W. P. Weisenburgen, “Neurotoxicology test-ing,” in Toxicological Testing Handbook. Principles, Applicationsand Data Interpretation, D. Jacobson-Kram and K. A. Keller,Eds., chapter 4, pp. 357–390, Informa Healthcare, New York,NY, USA, 2006.
[30] T. Finkel and N. J. Holbrook, “Oxidants, oxidative stress andthe biology of ageing,” Nature, vol. 408, no. 6809, pp. 239–247,2000.
[31] R. S. Sohal, R. J. Mockett, and W. C. Orr, “Mechanisms ofaging: an appraisal of the oxidative stress hypothesis,” FreeRadical Biology and Medicine, vol. 33, no. 5, pp. 575–586, 2002.
[32] M. Lee, T. Cho, N. Jantaratnotai, Y. T. Wang, E. McGeer, and P.L. McGeer, “Depletion of GSH in glia cells induces neurotoxic-ity: relevance to aging and degenerative neurological diseases,”FASEB Journal, vol. 24, no. 7-8, pp. 2533–2545, 2010.
[33] Y. Chen, E. Johansson, Y. Fan et al., “Early onset senescenceoccurs when fibroblasts lack the glutamate-cyteine ligasemodifier subunit,” Free Radical Biology and Medicine, vol. 47,no. 4, pp. 410–418, 2009.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 405194, 12 pagesdoi:10.1155/2011/405194
Review Article
Protective Action of Neurotrophic Factors and Estrogen againstOxidative Stress-Mediated Neurodegeneration
Tadahiro Numakawa,1, 2 Tomoya Matsumoto,2, 3 Yumiko Numakawa,4 Misty Richards,1, 5
Shigeto Yamawaki,2, 3 and Hiroshi Kunugi1, 2
1 Department of Mental Disorder Research, National Institute of Neuroscience, National Center of Neurology and Psychiatry,Tokyo 187-8502, Japan
2 Core Research for Evolutional Science and Technology Program (CREST), Japan Science and Technology Agency (JST),Saitama 332-0012, Japan
3 Department of Psychiatry and Neurosciences, Division of Frontier Medical Science, Graduate School of Biomedical Sciences,Hiroshima University, 1-2-3 Kasumi, Minami-ku, Hiroshima 734-8551, Japan
4 Peptide-prima Co., Ltd., 1-25-81, Nuyamazu, Kumamoto 861-2102, Japan5 The Center for Neuropharmacology and Neuroscience, Albany Medical College, Albany, NY 12208, USA
Correspondence should be addressed to Tadahiro Numakawa, [email protected]
Received 11 January 2011; Revised 28 February 2011; Accepted 29 March 2011
Academic Editor: Laurence D. Fechter
Copyright © 2011 Tadahiro Numakawa et al. This is an open access article distributed under the Creative Commons AttributionLicense, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properlycited.
Oxidative stress is involved in the pathogenesis of neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease,and Huntington’s disease. Low levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) are important formaintenance of neuronal function, though elevated levels lead to neuronal cell death. A complex series of events includingexcitotoxicity, Ca2+ overload, and mitochondrial dysfunction contributes to oxidative stress-mediated neurodegeneration. Asexpected, many antioxidants like phytochemicals and vitamins are known to reduce oxidative toxicity. Additionally, growingevidence indicates that neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and estrogens significantly preventneuronal damage caused by oxidative stress. Here, we review and discuss recent studies addressing the protective mechanisms ofneurotrophic factors and estrogen within this system.
1. Introduction
It is well established that the brain consumes a large quantityof oxygen and glucose [1–5]. Brain neurons utilize suchnutrients, requiring a consistent and steady supply in orderto function appropriately. Not surprisingly, brain neuronsare vulnerable to oxidative stress [6], which threatens theoverall functionality of the brain. Though various systemsprotecting against oxidative toxicity exist in the brain atcellular and molecular levels, a disruption of the defensivesystem may be involved in neurological deficits observedin neurodegenerative diseases. Indeed, many studies suggestthat oxidative toxicity is related to Alzheimer’s disease (AD),Parkinson’s disease (PD), and Huntington’s disease (HD)[7]. In addition, a correlation between an accumulation ofoxidative stress and aging has also been established [8]. Thus,
it is important to clarify the detailed relationship betweenoxidative stress and cellular damage in neurodegenerativediseases and the aging process. In the cellular and molecularmechanisms underlying oxidative stress-induced cell death,it is well known that excitotoxicity, Ca2+ overload, mito-chondrial dysfunction, and the stimulation of intracellularsignaling cascades play a role [9]. As expected, antioxidantsincluding many phytochemicals and vitamins have beenfound to support the survival of neurons under oxidativestress.
Brain-derived neurotrophic factor (BDNF), a member ofthe neurotrophin family, is known to be a strong survival-promoting factor against various neuronal insults. As aresult, the molecular mechanisms underlying neurotrophin-dependent survival promotion when exposed to oxidativestress have been extensively studied. BDNF plays a critical

2 Journal of Toxicology
role in cell proliferation, cell differentiation, neuronal pro-tection, and the regulation of synaptic function in the centralnervous system (CNS) via stimulating key intracellular sig-naling cascades [10, 11]. In addition to BDNF, glial cell line-derived neurotrophic factor (GDNF) and hepatocyte growthfactor (HGF) are also effective for neuronal survival [12, 13].Furthermore, estrogens, which regulate synaptic plasticity inaddition to sex differentiation of the brain [14–16], are foundto exert protective actions against toxic conditions such asoxidative stress [17]. Here, we review the current issuesconcerning protective functions of neurotrophic factors andestrogen on neurons under oxidative stress.
2. The Role of Oxidative Stress inNeurodegenerative Diseases
Low levels of ROS and RNS have a physiological effecton cellular functions including neuronal plasticity [18].However, in excess, ROS/RNS cause oxidation/nitrosylationof lipids, proteins, and nucleic acids, resulting in neuronalcell death (Figure 1). Such damage occurs as a result ofeither overproduction of ROS/RNS or reduced activityof enzymatic and nonenzymatic antioxidants. Thus, thedelicate balance between pro- and antioxidant reactions iscritical for maintaining normal neuronal function.
Oxidative stress-mediated toxicity may be closely relatedto the pathogenesis of neurodegenerative diseases such asAD, PD, and HD [7]. For example, in AD brains, mark-ers for protein oxidation (protein carbonyls and 3-nitro-tyrosine (3-NT)), lipid oxidation (4-hydroxy-2′-nonenal (4-HNE)), and DNA oxidation (8-hydroxy-2-deoxyoguanine(8-OHdG)) are elevated [19]. Indeed, the accumulationof amyloid beta (Aβ), a hallmark of AD, produces ROSincluding hydrogen peroxide (H2O2) in the presence ofFe3+ or Cu2+ [20–22], but see [23]. In PD brains, inwhich a selective and progressive loss of dopamine (DA)neurons in the substantia nigra pars compacta occurs, 4-HNE, protein carbonyls, 3-NT, and 8-OHdG are all increasedwhile glutathione (GSH, a major intracellular antioxidant)is decreased [24]. Interestingly, 4-HNE covalently binds toalpha-synuclein (α-Syn), a central protein in PD patho-genesis, resulting in neurotoxic effects on DAergic andGABAergic neuronal cultures [25]. Similarly, HD brains(where significant neuronal loss in the striatum and cor-tex is observed) demonstrate elevated 3-NT, lipofuscin (aproduct of unsaturated fatty acid peroxidation), malondi-aldehyde (a marker for lipid oxidation), and 8-OHdG [26].Reduced levels of GSH were also confirmed in culturedneurons from mice expressing mutant Huntingtin protein(Htt140Q/140Q) [27].
Oxidative toxicity is also involved in cerebral ischemia/reperfusion injury. Brain regions and types of neurons thatare vulnerable to ischemia are limited. It may be becausecerebral blood flow is highly spatiotemporally modulated[2], and this view could also be important to understandwhy specific types of neurons in different brain regionsare affected in each neurodegenerative disease. In addition,a large body of evidence suggests that accumulation of
oxidative stress-dependent damage occurs during normalaging, which may cause a noticeable decline in cognitivefunction [8, 28]. Considering that cognitive deficits areobserved in neurodegenerative diseases such as AD as well,a common mechanism underlying oxidative stress-mediatedneuronal cell death may exist. In the following section,we summarize the current knowledge concerning oxidativestress-mediated neuronal cell death.
3. Oxidative Stress-Mediated NeuronalCell Death
3.1. Mitochondrial Dysfunction, Ca2+ Overload and Excito-toxicity. Apoptosis, a prototypic form of programmed celldeath, is a major mode of cell death in neurodegenerativediseases. Various mechanisms including excitotoxicity, Ca2+
overload, mitochondrial dysfunction, endoplasmic reticu-lum stress, and oxidative stress have been found to contributeto apoptosis [9] (Figure 1). Mitochondria produce low levelsof ROS in a process known as cellular respiration throughthe electron transport chain (ETC). The ETC consists offive protein complexes (I–V), and a disruption of thiselectron transport system leads to excess generation of ROS[29]. Importantly, a number of studies reported possibleinvolvement of mitochondrial dysfunction, including alteredactivity of the ETC, in patients and animal models for AD[30], PD [31], HD [32], and stroke [33]. Some reportssuggest that patients with psychiatric disorders, such asschizophrenia [34], depression [35], and bipolar disorder[36], also display mitochondrial dysfunction.
In addition, mitochondria regulate/impact/affect Ca2+
homeostasis by sequestering excess cytosolic Ca2+ into theirmatrix (named Ca2+ loading). However, an uncontrolledCa2+ loading may be involved in neurodegeneration. In astudy investigating striatal mitochondria of Hdh150 knock-in HD mice, a disrupted Ca2+ homeostasis was found[37]. Another study discovered that a deficiency of phos-phatase and tensin homolog deleted on chromosome 10(PTEN)-induced putative kinase 1 (PINK1, a mitochondrialkinase linked to familial PD) results in mitochondrialCa2+ accumulation in cultured neurons [38]. Endoplasmicreticulum also regulates intracellular Ca2+ concentrationthrough inositol-1,4,5-triphosphate receptors (InsP3Rs) andryanodine receptors (RyRs). Interestingly, presenilin (PS) 1and 2, genes involved in the pathogenesis of AD, acted asa passive endoplasmic reticulum Ca2+ channel to maintainsteady-state Ca2+ levels, which was disrupted by mutant PS1-M146V and PS2-N141I [39, 40]. These PS mutants enhancedthe gating activity of InsP3Rs, leading to Aβ generation [41].Furthermore, it was shown that Aβ-containing senile plaquescause Ca2+ overload [42]. Taken together, it seems likely thatmutant PSs and Aβ contribute to the disruption of Ca2+
homeostasis, which may cause mitochondrial dysfunctionleading to neuronal degeneration [30].
Remarkably, nicotinamide adenine dinucleotide phos-phate (NADPH) oxidase (Nox) may generate ROS ina mitochondria-independent manner. In cultured corti-cal neurons lacking p47(phox), a cytosolic subunit of

Journal of Toxicology 3
Endoplasmicreticulum stress
Excitotoxicity
Mitochondrialdysfunction
Ca2+ overload
Accumulation ofoxidative stress
Death signalingp53, MAPK (JNK, p38, ERK) etc.
Mitochondrial signaling
Bax, PUMA, cytochrome c
Caspase 9/caspase 3
Apoptosis
Apoptosis-inducing factor
Oxidation ofproteins
lipidsDNA
Ubiquitin-proteasomesystem
dysfunction
Protein aggregation
PhytochemicalsVitamin EEstrogenNeurotrophic factors(BDNF, GDNF, HGF etc.)
Figure 1: Mechanisms underlying oxidative stress-mediated neuronal apoptosis. Accumulation of oxidative stress is involved in thedevelopment/progression of neurodegenerative diseases. A number of events including excitotoxicity, mitochondrial dysfunction, Ca2+
overload, and endoplasmic reticulum stress are associated with excess reactive oxygen species (ROS) and reactive nitrogen species (RNS)generation. High levels of ROS/RNS lead to oxidation of proteins, lipids, and DNA. Oxidized lipids induce damage of the ubiquitin-proteasome system (UPS). The UPS dysfunction and oxidation of proteins result in aggregation of proteins, recognized as a hallmarkof several neurodegenerative diseases. Under oxidative stress, death signaling pathways (p53, mitogen-activated protein kinase (MAPK),etc.) are activated. Activation of p53 leads to induction of proapoptotic proteins such as Bax and p53-upregulated modulator of apoptosis(PUMA), followed by translocation of these proteins into mitochondria. Finally, mitochondrial cytochrome c is released, which thenstimulates the activation of caspase 9/caspase 3. Alternatively, mitochondria secrete apoptosis-inducing factor (AIF), leading to caspase-independent apoptosis. As shown, recent studies suggest antioxidant effects of phytochemicals, vitamin E, estrogen, and neurotrophicfactors including brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), and hepatocyte growthfactor (HGF), leading to increased preservation of neuronal function.
Nox, extensive N-methyl-D-aspartic acid (NMDA) receptoractivation failed to produce ROS, while H2O2 or themitochondrial complex III inhibitor (antimycin) increasedROS [43]. Furthermore, ROS production and oxidativedamage in the hippocampal CA1 neurons after ischemiawere dramatically attenuated in mice either treated withNox inhibitor or lacking gp91(phox), another Nox subunit[44]. Considering the fact that overactivation of NMDAreceptors occurs in ischemia [45], it is possible thatNMDA-mediated excitotoxicity may cause mitochondria-independent, but Nox-dependent, ROS production in cere-bral ischemia/reperfusion injury.
3.2. Signaling Pathways in Apoptosis. p53, a transcriptionfactor, is activated by ROS, and induces the upregulationof mitochondrial proapoptotic proteins including B-celllymphoma-2-associated X protein (Bax) and members of theB-cell lymphoma-2-homology 3 (BH3) family consisting ofBH3 interacting death agonist (Bid), Nox activator 1 (Noxa),and p53-upregulated modulator of apoptosis (PUMA) [33].Indeed, oxidative stressors including H2O2 increased Noxa,Bim, and PUMA (but not Bid) in cultured cortical neurons
[46]. Importantly, PUMA, but not Noxa or Bim, wasinvolved in Bax-dependent apoptosis [46]. The contributionof p53/PUMA to delayed cell death of hippocampal neuronsafter stroke was also reported [47]. These studies suggestthat p53-mediated PUMA expression may be a key event inneuronal apoptosis (Figure 1).
As the final step of apoptosis, cytochrome c is releasedfrom mitochondria via the permeability transition pore(PTP), which consists of the mitochondrial inner and outermembrane proteins including B-cell lymphoma-2 (Bcl-2)and Bax (Figure 1). Cytosolic cytochrome c participates inthe formation of the apoptosome, a multiprotein complexincluding apoptosis protease-activating factor 1 (Apaf-1) andcaspase-9, which activates caspase-3, an executioner in celldeath [48]. On the other hand, apoptosis-inducing factor(AIF) is involved in mitochondria-mediated, but caspase-independent, apoptosis [49] (Figure 1).
3.3. Antioxidative Factors. Considering that oxidative stressmay be associated with the pathogenesis of neurodegen-erative diseases, a key therapeutic intervention would beto block or delay accumulating oxidative stress levels via

4 Journal of Toxicology
increasing the function of endogenous antioxidants and/orsuppressing ROS production (Figure 1). Well-known antiox-idants include glutathione precursor [50, 51], polyphe-nols [52–54], catechins [55], flavonoids [56], and sulfatedpolysaccharides [57]. As the toxicity of phytochemicals islow, these substances offer a new therapeutic approachagainst neurodegenerative diseases [58]. On the other hand,whether oxidative stress is a cause or consequence ofneurodegenerative disease remains to be elucidated [7]. Agrowing body of evidence suggests that oxidative stressdirectly initiates and progresses to neuronal cell death.However, it is possible that accumulation of oxidative stressis easily induced in neurons weakened by other insults.Indeed, in the apoptotic process, many cellular eventsincluding mitochondrial dysfunction, Ca2+ overload, andexcitotoxicity activate death signaling cascades (Figure 1).Such negative feedback loops may influence cell viability.These events probably occur in parallel and have an additiveor synergic effect in the induction of cell death. Therefore,in addition to blocking accumulation of oxidative stress,inhibiting death-signaling cascades and activating survivalsignaling would also be effective. In the following section,we focus specifically on neurotrophic factors and steroidhormones that may exert a beneficial influence.
4. Neurotrophins and Oxidative Stress inNeurodegenerative Diseases
As mentioned above, oxidative stress may be involvedin the onset of HD, AD, PD, and amyotrophic lateralsclerosis (ALS) [7, 9]. Interestingly, neurotrophic factors,including neurotrophins, may also be associated with thepathology of these neurodegenerative diseases. For example,both mRNA and protein levels of BDNF are decreased inpatients and animal models of HD [59–61]. In addition,the level of TrkB (tropomyosin-related kinase B), a highaffinity receptor for BDNF, is also reduced in knockinHD striatal cells, in which mutant huntingtin with 111glutamines (7 glutamines in normal) is expressed [62].Following TrkB activation stimulated by BDNF, the mitogen-activated protein kinase/extracellular signal-regulated pro-tein kinase (MAPK/ERK), phospholipase Cγ (PLCγ), andphosphatidylinositol 3-kinase (PI3K) pathways are primarilytriggered [10]. In the knock-in HD striatal cells, a down-regulation of ERK signaling occurred, while PI3K/Akt andPLCγ pathways were intact. Such a decrease in ERK signalingin these striatal cells resulted in an increase in the cell deathcaused by H2O2 [63]. As expected, it was revealed that BDNF,neurotrophin-3 (NT-3), and NT-4/5 prevent neuronal celldeath in an animal model of HD [64].
Recent reports suggest that the upregulation of BDNFexpression/function plays a role in neuroprotection withinAD models. Counts and Mufson showed that noradrenaline(NA) is neuroprotective against Aβ-dependent toxicity inhuman NTera-2N (hNT) neurons and rat hippocampalneurons [65]. NA prevented an increase in ROS causedby Aβ. Notably, coapplication with functional blockingantibodies for BDNF or nerve growth factor (NGF) signifi-cantly inhibited the NA-dependent protective effect against
Aβ toxicity [65]. As AD is well known as an age-relatedneurodegenerative illness, the senescence-accelerated mouseprone 8 (SAMP8) mice, which show age-related impairmentof cognitive function, is a useful model of AD [66]. Usingthe SAMP8 mice, Zhao et al. investigated the effect ofginsenoside, a component of ginseng, on memory [67]. Theyreported that chronic treatment with ginsenoside preventedloss of memory in aged SAMP8 mice. Such a treatmentwith ginsenoside decreased the Aβ and, in turn, increasedantioxidation and synaptic plasticity-related proteins such asBDNF [67].
Oxidative stress may damage nigral DA neurons, result-ing in the onset of PD. Under oxidative stress, hemeoxygenase-1 (HO-1) increases and exerts a positive effecton nigral DA neurons. Overexpression of HO-1 in ratsubstantia nigra rescued DA neurons from cell death causedby 1-methyl-4-phenylpyridinium (MPP(+)), which is aninhibitor for mitochondrial complex I and is well known toproduce PD symptoms. After HO-1 overexpression, GDNF,in addition to BDNF, was upregulated [68]. Additionally, itwas reported that bilirubin, a downstream product of HO-1, increased GDNF and BDNF expression through ERK andPI3K/Akt pathways [69]. These results suggest that HO-1 protects neurons through increasing these neurotrophicfactors. A role of the novel DA D3 receptor agonist D-264 in neuroprotection was reported [70]. In the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP, an inhibitorof mitochondrial complex I)-induced neurodegenerationmouse model for PD, D-264 treatment improved behavioralperformance and reduced neuronal loss. Remarkably, the D-264 treatment induced an upregulation of BDNF and GDNFin MPTP-treated animals [70]. Finally, using an in vitrosystem, L-theanine (a glutamate analog) was shown to pro-mote SH-SY5Y cell survival and inhibited downregulationof both BDNF and GDNF under neurotoxicant (rotenoneand dieldrin) application [71]. Generally, GDNF and BDNFare important for survival/morphological change of DAneurons, and both have a recovery effect on PD-like behavior[12, 72, 73]. Taken together, it is possible that upregulation ofgrowth factors including BDNF and GDNF is necessary forthe prevention of DA neuronal damage.
5. BDNF and Oxidative Stress-InducedCell Death
BDNF exerts protective effects against neuronal cell deathby activating intracellular signaling cascades via TrkB [10,11, 74]. Interestingly, trypanosome trans-sialidase (TS,sialic acid-transferring enzyme) mimics neurotrophins.Woronowicz et al. showed that TS induced phosphorylationof TrkB in rat pheochromocytoma (PC12) cells express-ing TrkB and promoted cell survival under H2O2 stress[75]. The PI3K pathway was important for TS-mediatedsurvival promotion. On the other hand, BDNF protectscultured cortical neurons from NMDA- or H2O2-inducedcell death via suppressing the MAPK pathway [76]. Onceexposed to NMDA or H2O2, retinoblastoma protein andE2F1 transcription factor, which are cell cycle regulators,

Journal of Toxicology 5
were stimulated. BDNF inhibited such activation of cellcycle regulators, suggesting that the prevention of cell cyclereentry is involved in BDNF function during oxidativestress [76]. Moreover, the activation of cyclic adenosine3′,5′-monophosphate (cAMP)-responsive element-bindingprotein (CREB) is involved in BDNF neuroprotection.Transgenic mice expressing A-CREB, a dominant negativeform of CREB, showed a significant increase in vulnerabilityto seizure activity. The A-CREB mice demonstrated increasedROS levels and decreased neuroprotection by BDNF applica-tion, suggesting that CREB is an essential upstream effectorof neuroprotection against oxidative toxicity [77]. Impor-tantly, CREB also regulates the transcriptional productionof BDNF [78]. The BDNF gene consists of nine exons, andexon IX corresponds to the common open reading frame ofthe protein. The remaining exons have distinct promoters,respectively. Thus, the transcript of BDNF consists of one ofeight 5′ untranslated exons (exon I∼VIII) and 3′ exon IX[79]. Interestingly, the action of CREB via promoter IV iscritical for experience-dependent production of BDNF [80].Therefore, positive-feedback mechanisms may be involved inBDNF-mediated neuroprotection.
As mentioned, BDNF seems to be beneficial in thetherapeutic approach to neurodegenerative diseases. How-ever, previous clinical trials have revealed numerous sideeffects of neurotrophins as well as their poor penetrationthrough the blood brain barrier, making it very difficult touse these proteins as a drug [81]. Therefore, many studieshave been performed in an effort to find a drug thatupregulates BDNF. In SH-SY5Y cells after H2O2 application,tripterygium regelii extract (TRE), a traditional herbalmedicine, increased tyrosine hydroxylase, a dopaminergicmarker, and BDNF [82]. TRE was shown to repress theupregulation of proapoptotic proteins Bax and caspase-3, while inhibiting downregulation of antiapoptotic Bcl-2 under H2O2 application [82]. Sonic hedgehog (SHH)protein, a member of the Hedgehog family of signalingmolecules [83], is putatively involved as a neuroprotectiveagent in oxidative stress-related neurodegenerative diseaseand ischemia. After H2O2 exposure, the SHH pathway wasstimulated in cultured cortical neurons, and the increase inSHH pathway activation was noticeably protective againstcell death caused by H2O2 [84]. In that in vitro system,exogenous SHH increased levels of vascular endothelialgrowth factor (VEGF) and BDNF, as well as activity ofsuperoxide dismutase (SOD) and Bcl-2 expression [84].
Positive effects of the antioxidant vitamin E on oxidativestress-mediated toxicity in vitro [85–87] and in vivo [88,89] have been reported. Vitamin E has also been shownto exert beneficial effects against neurodegenerative diseases[90, 91]. Our research demonstrates that pretreatment withvitamin E analogs including α- and γ-tocopherol (αT andγT, respectively) and α- and γ-tocotrienol (αT3 and γT3)protected cultured cortical neurons against H2O2-mediatedneuronal cell death [92]. In our cultures, αT stimulatedthe activation of both the ERK and PI3K pathways andcaused the upregulation of Bcl-2. Importantly, αT-mediatedsurvival and Bcl-2 upregulation disappeared in the presenceof inhibitors for ERK and PI3K signaling, suggesting the
involvement of both pathways in neuroprotection by vitaminE analogs. However, the neuroprotection was not via BDNFsignaling, as αT unchanged TrkB activation and BDNFexpression [92]. It would be interesting to examine possiblecontributions from other neurotrophic factors.
It is now critical to further investigate the mechanismsunderlying the upregulation of BDNF and/or other effectivegrowth factors in order to discover more efficacious medi-cations. In general, BDNF levels are regulated by neuronalactivity. In addition to the influx of Ca2+, neuronal activ-ity, including glutamatergic regulation, contributes to theproduction and secretion of BDNF [93–98]. Change in theproduction and secretion of BDNF is thought to be involvedin the activity-dependent synaptic plasticity in the CNS [99,100]. Interestingly, two recent studies have demonstratedthe role of synaptic activity in neuroprotection. In culturedhippocampal neurons, action potential bursting reduced thelevels of p53, PUMA and Apaf1 [101]. Furthermore, NMDAreceptor stimulation inhibited PUMA-mediated apoptosisvia reducing levels of Apaf1 and procaspase-9 [102]. Insupport of these current studies, a previous study demon-strated that transcranial magnetic stimulation, which is wellknown to potentiate neuronal activity, inhibited toxic effectsof 3-nitropropionic acid (3-NPA) (protein/lipid oxidations,reduction in activities of catalase, GSH peroxidase andsuccinate dehydrogenase, and GSH deficiency) and rescuedthe striatal neuronal loss in rats [103]. It is necessary toinvestigate whether or not such neuronal activity-mediatedprotection occurs via the upregulation of BDNF. Addition-ally, future studies investigating the role of neuronal activityin the expression of neurotrophic factors that are influencedby molecules that cross the blood brain barrier are needed.
Transplantation of growth factor-secreting cells mayserve as an alternative method to treat neurodegenerativediseases. Indeed, the grafting of neurotrophin-secreting celllines has been shown to protect neurons against quinolinate-induced cell death in an animal model of HD [64]. In addi-tion, it was shown that erythropoietin-transduced humanmesenchymal stromal cells (EPO-MSCs) played a neuropro-tective role in the rat model for ischemic stroke [104]. Inthe EPO-MSCs, neurotrophic factors including BDNF andHGF were upregulated. The implantation of EPO-MSCs intoischemic rats reversed impairment in neurological functionand infarct volumes [104]. Finally, a gene transfer approachmay be a potentially effective strategy as well. In an in vivocognitive dysfunction model induced by Aβ injection, HGFgene transfer improved impairment of cognitive behavior.It was suggested that BDNF upregulation was involved inthe positive action of HGF gene transfer [105]. Furtherinvestigation on the possible mechanisms underlying theBDNF upregulation is interesting.
6. Estrogen Signaling and Oxidative Stress
Estrogen, one of the sex steroids, has various roles in sexdifferentiation, neuroprotection, and synaptic plasticity [14–16, 106]. Furthermore, estrogenic protection from toxicityincluding excitotoxicity and oxidative stress is well studied[107–109]. Importantly, the maintenance of mitochondrial
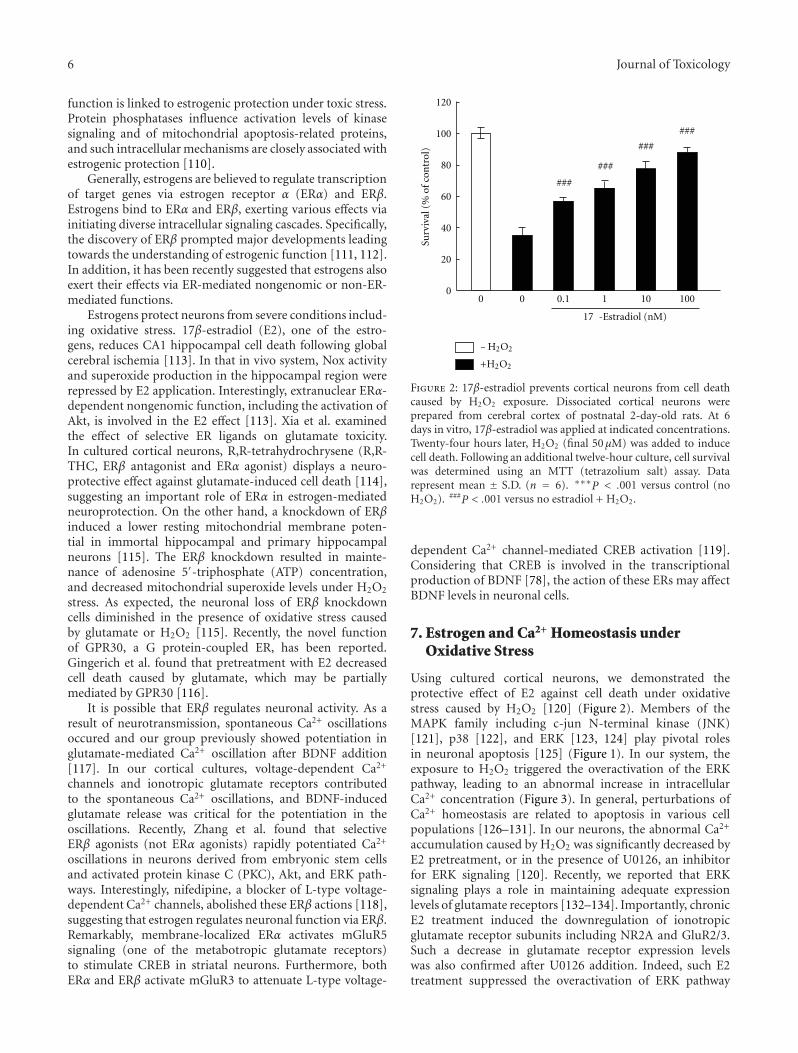
6 Journal of Toxicology
function is linked to estrogenic protection under toxic stress.Protein phosphatases influence activation levels of kinasesignaling and of mitochondrial apoptosis-related proteins,and such intracellular mechanisms are closely associated withestrogenic protection [110].
Generally, estrogens are believed to regulate transcriptionof target genes via estrogen receptor α (ERα) and ERβ.Estrogens bind to ERα and ERβ, exerting various effects viainitiating diverse intracellular signaling cascades. Specifically,the discovery of ERβ prompted major developments leadingtowards the understanding of estrogenic function [111, 112].In addition, it has been recently suggested that estrogens alsoexert their effects via ER-mediated nongenomic or non-ER-mediated functions.
Estrogens protect neurons from severe conditions includ-ing oxidative stress. 17β-estradiol (E2), one of the estro-gens, reduces CA1 hippocampal cell death following globalcerebral ischemia [113]. In that in vivo system, Nox activityand superoxide production in the hippocampal region wererepressed by E2 application. Interestingly, extranuclear ERα-dependent nongenomic function, including the activation ofAkt, is involved in the E2 effect [113]. Xia et al. examinedthe effect of selective ER ligands on glutamate toxicity.In cultured cortical neurons, R,R-tetrahydrochrysene (R,R-THC, ERβ antagonist and ERα agonist) displays a neuro-protective effect against glutamate-induced cell death [114],suggesting an important role of ERα in estrogen-mediatedneuroprotection. On the other hand, a knockdown of ERβinduced a lower resting mitochondrial membrane poten-tial in immortal hippocampal and primary hippocampalneurons [115]. The ERβ knockdown resulted in mainte-nance of adenosine 5′-triphosphate (ATP) concentration,and decreased mitochondrial superoxide levels under H2O2
stress. As expected, the neuronal loss of ERβ knockdowncells diminished in the presence of oxidative stress causedby glutamate or H2O2 [115]. Recently, the novel functionof GPR30, a G protein-coupled ER, has been reported.Gingerich et al. found that pretreatment with E2 decreasedcell death caused by glutamate, which may be partiallymediated by GPR30 [116].
It is possible that ERβ regulates neuronal activity. As aresult of neurotransmission, spontaneous Ca2+ oscillationsoccured and our group previously showed potentiation inglutamate-mediated Ca2+ oscillation after BDNF addition[117]. In our cortical cultures, voltage-dependent Ca2+
channels and ionotropic glutamate receptors contributedto the spontaneous Ca2+ oscillations, and BDNF-inducedglutamate release was critical for the potentiation in theoscillations. Recently, Zhang et al. found that selectiveERβ agonists (not ERα agonists) rapidly potentiated Ca2+
oscillations in neurons derived from embryonic stem cellsand activated protein kinase C (PKC), Akt, and ERK path-ways. Interestingly, nifedipine, a blocker of L-type voltage-dependent Ca2+ channels, abolished these ERβ actions [118],suggesting that estrogen regulates neuronal function via ERβ.Remarkably, membrane-localized ERα activates mGluR5signaling (one of the metabotropic glutamate receptors)to stimulate CREB in striatal neurons. Furthermore, bothERα and ERβ activate mGluR3 to attenuate L-type voltage-
Surv
ival
(%of
con
trol
)
120
100
80
60
40
20
0
17 -Estradiol (nM)
0 0 0.1 1 10 100
−H2O2
+H2O2
###
###
###
###
Figure 2: 17β-estradiol prevents cortical neurons from cell deathcaused by H2O2 exposure. Dissociated cortical neurons wereprepared from cerebral cortex of postnatal 2-day-old rats. At 6days in vitro, 17β-estradiol was applied at indicated concentrations.Twenty-four hours later, H2O2 (final 50 μM) was added to inducecell death. Following an additional twelve-hour culture, cell survivalwas determined using an MTT (tetrazolium salt) assay. Datarepresent mean ± S.D. (n = 6). ∗∗∗P < .001 versus control (noH2O2). ###P < .001 versus no estradiol + H2O2.
dependent Ca2+ channel-mediated CREB activation [119].Considering that CREB is involved in the transcriptionalproduction of BDNF [78], the action of these ERs may affectBDNF levels in neuronal cells.
7. Estrogen and Ca2+ Homeostasis underOxidative Stress
Using cultured cortical neurons, we demonstrated theprotective effect of E2 against cell death under oxidativestress caused by H2O2 [120] (Figure 2). Members of theMAPK family including c-jun N-terminal kinase (JNK)[121], p38 [122], and ERK [123, 124] play pivotal rolesin neuronal apoptosis [125] (Figure 1). In our system, theexposure to H2O2 triggered the overactivation of the ERKpathway, leading to an abnormal increase in intracellularCa2+ concentration (Figure 3). In general, perturbations ofCa2+ homeostasis are related to apoptosis in various cellpopulations [126–131]. In our neurons, the abnormal Ca2+
accumulation caused by H2O2 was significantly decreased byE2 pretreatment, or in the presence of U0126, an inhibitorfor ERK signaling [120]. Recently, we reported that ERKsignaling plays a role in maintaining adequate expressionlevels of glutamate receptors [132–134]. Importantly, chronicE2 treatment induced the downregulation of ionotropicglutamate receptor subunits including NR2A and GluR2/3.Such a decrease in glutamate receptor expression levelswas also confirmed after U0126 addition. Indeed, such E2treatment suppressed the overactivation of ERK pathway

Journal of Toxicology 7
Oxidative stress
ERK
pERK
Ca2+
Ca2+
+17 -Estradiol
Death signaling
ERK
Oxidative stress
Figure 3: 17β-estradiol inhibits neuronal cell death under oxidativestress via reducing the series of events evoked by exposure toH2O2, including overactivation of the ERK signaling and overloadof Ca2+. Upper: After H2O2 addition, marked phosphorylated(activated) ERK (pERK) and resultant increase in intracellular Ca2+
concentration were observed, resulting in cell death. Lower: Pre-treatment with 17β-estradiol induced downregulation of ionotropicglutamate receptors via decreasing ERK activation, while alsoserving to decrease levels of Ca2+ influx triggered by H2O2. Sucha decrease in glutamate receptor expression and intracellular Ca2+
was also confirmed in the presence of U0126, an inhibitor ofERK signaling. As expected, chronic 17β-estradiol reduced levelsof pERK stimulated by H2O2. A blockade of glutamate receptorsrescued cortical cells from H2O2-dependent death. Therefore, itis possible that 17β-estradiol promotes survival via suppressingglutamate receptor-mediated Ca2+ influx, due to downregulation ofionotropic glutamate receptors [120].
stimulated by H2O2. Furthermore, inhibitors of ionotropicglutamate receptors blocked cell death caused by H2O2.Taken together, it is possible that E2 exerts survival-promoting effects through repressing glutamate receptor-mediated Ca2+ influx [120] (Figure 3). As described, ERKsignaling is essential for maintenance of glutamate receptorlevels, making it interesting to investigate how estrogensinfluence ERK signaling.
p66Shc also generates mitochondrial ROS (H2O2), causesimpairment in Ca2+ homeostasis, and is associated withapoptosis [135, 136]. Almeida et al. found that H2O2
stimulates PKCβ/p66Shc/NF-κB signaling to apoptosis inosteoblastic cells, and that E2 prevents H2O2-dependent acti-vation of p66Shc and NF-κB via repressing phosphorylationof PKCβ, resulting in protection from cell death [137].
8. Estrogen In Vivo Approach
In 6-hydroxydopamine (6-OHDA, a PD mimetic)-lesionedrats, a neuroprotective effect of silymarin (SM, one offlavonolignans) was shown [138]. SM administration pro-tected neurons of the substantia nigra pars compacta from 6-OHDA toxicity, while fulvestrant, an ER antagonist, partiallyblocked the effects of SM. Additionally, the effect of oralestrogen on ROS generation was reported. Wing et al.demonstrated a beneficial effect of chronic oral estrogentreatment on oxidative stress and atherosclerosis in apoE-deficient mice [139]. Using ovariectomized apoE-deficientmice, it was revealed that atherosclerosis was reduced whentreated with E2 (6 μg/day) for 12 weeks. Importantly, afterE2 treatment, superoxide anion and expression of Noxdecreased, while Cu/ZnSOD and MnSOD increased [139].Last, Schwann cells (SC) play a critical role in spinal cordinjury repair, though SC survival after transplantation isvery difficult. Current research is focused on discovering ifE2 pretreatment protects SC, in an effort to generate moresuccessful spinal cord transplantation procedures [140]. Inprimary SC cultures, strong expression of ERα and ERβ,and overall E2-dependent survival mechanisms against H2O2
exposure were confirmed, though ICI182780 (an ER antag-onist) had no influence on E2 effects. These findings suggestthat genomic signaling via ERs is not involved. Importantly,in spinal cord injury, sustained E2 administration was foundto be an effective treatment improving SC transplantation[140].
9. Conclusion
An increase in neuronal damage at the cellular and molecularlevel may be involved in the pathogenesis of brain illness,including neurodegenerative disease. It is possible thatoxidative stress leads to neuronal cell death via increasingglutamate-mediated excitotoxicity, intracellular Ca2+ con-centration, mitochondrial dysfunction, activation of death-signaling cascades, and decreasing overall survival signaling.Several drug candidates, which were found to attenuatedeleterious symptoms in various models of neurodegener-ative disease, are reported to upregulate the expression ofneurotrophic factors including BDNF. Considering this, itseems pertinent to further investigate the possible mech-anisms underlying such neurotrophic factor upregulation.On the other hand, estrogenic survival promotion is alsowell studied, though further investigation addressing howeach ER contributes to neuronal protection against oxidativetoxicity is needed. Finally, as a close relationship betweensteroid hormones and BDNF in various neuronal functionsincluding cell survival is known [141], detailed studiesconcerning how estrogen and BDNF interact with each otherin CNS neurons under oxidative stress are important.

8 Journal of Toxicology
Acknowledgments
This work was supported by a grant from the Core Researchfor Evolutional Science and Technology Program (CREST)Japan Science and Technology Agency (JST) (T. N., T. M.,S. Y. and H. K.), the Takeda Science Foundation (T. N.),the Japan Health Sciences Foundation (Research on HealthSciences focusing on Drug Innovation) (H. K.), Health andLabor Sciences Research Grants (Comprehensive Researchon Disability, Health, and Welfare) (H. K.), IntramuralResearch Grants (20-3, 21-9) for Neurological and Psychi-atric Disorders of NCNP (H. K.), and Grants-in-Aid forScientific Research (B) (grant number 20390318) (H. K.) andYoung Scientists (A) (21680034) (T. N.) from the Ministryof Education, Culture, Sports, Science, and Technology ofJapan.
References
[1] O. Sakurada, C. Kennedy, J. Jehle, J. D. Brown, G. L. Carbin,and L. Sokoloff, “Measurement of local cerebral bloodflow with iodo [14C] antipyrine,” The American Journal ofPhysiology, vol. 234, no. 1, pp. H59–H66, 1978.
[2] J. E. Cremer and M. P. Seville, “Regional brain blood flow,blood volume, and haematocrit values in the adult rat,”Journal of Cerebral Blood Flow and Metabolism, vol. 3, no. 2,pp. 254–256, 1983.
[3] O. U. Scremin, R. R. Sonnenschein, and E. H. Rubinstein,“Cholinergic cerebral vasodilatation: lack of involvement ofcranial parasympathetic nerves,” Journal of Cerebral BloodFlow and Metabolism, vol. 3, no. 3, pp. 362–368, 1983.
[4] L. Sokoloff, M. Reivich, C. Kennedy et al., “The[C]deoxyglucose method for the measurement of localcerebral glucose utilization: theory, procedure, and normalvalues in the conscious and anesthetized albino rat,” Journalof Neurochemistry, vol. 28, no. 5, pp. 897–916, 1977.
[5] L. Sokoloff, “Localization of functional activity in the centralnervous system by measurement of glucose utilization withradioactive deoxyglucose,” Journal of Cerebral Blood Flow andMetabolism, vol. 1, no. 1, pp. 7–36, 1981.
[6] T. Satoh, Y. Enokido, T. Kubo, M. Yamada, and H. Hatanaka,“Oxygen toxicity induces apoptosis in neuronal cells,” Cellu-lar and Molecular Neurobiology, vol. 18, no. 6, pp. 649–666,1998.
[7] J. K. Andersen, “Oxidative stress in neurodegeneration: causeor consequence?” Nature Medicine, vol. 10, pp. S18–S25,2004.
[8] D. Harman, “Free radical theory of aging: an update—increasing the functional life span,” Annals of the New YorkAcademy of Sciences, vol. 1067, no. 1, pp. 10–21, 2006.
[9] E. Bossy-Wetzel, R. Schwarzenbacher, and S. A. Lip-ton, “Molecular pathways to neurodegeneration,” NatureMedicine, vol. 10, no. 7, pp. S2–S9, 2004.
[10] E. J. Huang and L. F. Reichardt, “Trk receptors: roles in neu-ronal signal transduction,” Annual Review of Biochemistry,vol. 72, pp. 609–642, 2003.
[11] T. Numakawa, S. Suzuki, E. Kumamaru, N. Adachi, M.Richards, and H. Kunugi, “BDNF function and intracellularsignaling in neurons,” Histology and Histopathology, vol. 25,no. 2, pp. 237–258, 2010.
[12] S. S. Gill, N. K. Patel, G. R. Hotton et al., “Direct braininfusion of glial cell line-derived neurotrophic factor in
Parkinson disease,” Nature Medicine, vol. 9, no. 5, pp. 589–595, 2003.
[13] F. Maina and R. Klein, “Hepatocyte growth factor, a versatilesignal for developing neurons,” Nature Neuroscience, vol. 2,no. 3, pp. 213–217, 1999.
[14] R. D. Brinton, “Estrogen-induced plasticity from cells tocircuits: predictions for cognitive function,” Trends in Phar-macological Sciences, vol. 30, no. 4, pp. 212–222, 2009.
[15] A. W. Lee and D. W. Pfaff, “Hormone effects on specific andglobal brain functions,” Journal of Physiological Sciences, vol.58, no. 4, pp. 213–220, 2008.
[16] S. Tobet, J. G. Knoll, C. Hartshorn et al., “Brain sexdifferences and hormone influences: a moving experience?”Journal of Neuroendocrinology, vol. 21, no. 4, pp. 387–392,2009.
[17] J. W. Simpkins, K. D. Yi, S. H. Yang, and J. A. Dykens,“Mitochondrial mechanisms of estrogen neuroprotection,”Biochimica et Biophysica Acta, vol. 1800, no. 10, pp. 1113–1120, 2010.
[18] K. T. Kishida and E. Klann, “Sources and targets of reactiveoxygen species in synaptic plasticity and memory,” Antioxi-dants and Redox Signaling, vol. 9, no. 2, pp. 233–244, 2007.
[19] D. A. Butterfield, T. Reed, S. F. Newman, and R. Sul-tana, “Roles of amyloid β-peptide-associated oxidative stressand brain protein modifications in the pathogenesis ofAlzheimer’s disease and mild cognitive impairment,” FreeRadical Biology and Medicine, vol. 43, no. 5, pp. 658–677,2007.
[20] X. Huang, C. S. Atwood, M. A. Hartshorn et al., “The Aβpeptide of Alzheimer’s disease directly produces hydrogenperoxide through metal ion reduction,” Biochemistry, vol. 38,no. 24, pp. 7609–7616, 1999.
[21] X. Huang, M. P. Cuajungco, C. S. Atwood et al., “Cu(II)potentiation of Alzheimer Aβ neurotoxicity. Correlationwith cell-free hydrogen peroxide production and metalreduction,” Journal of Biological Chemistry, vol. 274, no. 52,pp. 37111–37116, 1999.
[22] D. Jiang, L. Men, J. Wang et al., “Redox reactions of coppercomplexes formed with different β-amyloid peptides andtheir neuropathalogical relevance,” Biochemistry, vol. 46, no.32, pp. 9270–9282, 2007.
[23] R. C. Nadal, S. E. J. Rigby, and J. H. Viles, “Amyloid β-Cu2+
complexes in both monomeric and fibrillar forms do notgenerate H2O2 catalytically but quench hydroxyl radicals,”Biochemistry, vol. 47, no. 44, pp. 11653–11664, 2008.
[24] C. Zhou, Y. Huang, and S. Przedborski, “Oxidative stressin Parkinson’s disease: a mechanism of pathogenic andtherapeutic significance,” Annals of the New York Academy ofSciences, vol. 1147, pp. 93–104, 2008.
[25] Z. Qin, D. Hu, S. Han, S. H. Reaney, D. A. Di Monte, and A.L. Fink, “Effect of 4-hydroxy-2-nonenal modification on α-synuclein aggregation,” Journal of Biological Chemistry, vol.282, no. 8, pp. 5862–5870, 2007.
[26] E. C. Stack, W. R. Matson, and R. J. Ferrante, “Evidence ofoxidant damage in Huntington’s disease: translational strate-gies using antioxidants,” Annals of the New York Academy ofSciences, vol. 1147, pp. 79–92, 2008.
[27] X. Li, A. Valencia, E. Sapp et al., “Aberrant rab 11-dependenttrafficking of the neuronal glutamate transporter EAAClcauses oxidative stress and cell death in huntington’s disease,”Journal of Neuroscience, vol. 30, no. 13, pp. 4552–4561, 2010.
[28] N. A. Bishop, T. Lu, and B. A. Yankner, “Neural mechanismsof ageing and cognitive decline,” Nature, vol. 464, no. 7288,pp. 529–535, 2010.

Journal of Toxicology 9
[29] D. G. Nicholls, “Oxidative stress and energy crises inneuronal dysfunction,” Annals of the New York Academy ofSciences, vol. 1147, pp. 53–60, 2008.
[30] V. Adam-Vizi and A. A. Starkov, “Calcium and mitochondrialreactive oxygen species generation: how to read the facts,”Journal of Alzheimer’s Disease, vol. 20, supplment 2, pp. S413–S426, 2010.
[31] C. Henchcliffe and F. M. Beal, “Mitochondrial biology andoxidative stress in Parkinson disease pathogenesis,” NatureClinical Practice Neurology, vol. 4, no. 11, pp. 600–609, 2008.
[32] J. M. Gil and A. C. Rego, “Mechanisms of neurodegenerationin Huntington’s disease,” European Journal of Neuroscience,vol. 27, no. 11, pp. 2803–2820, 2008.
[33] K. Niizuma, H. Endo, and P. H. Chan, “Oxidative stressand mitochondrial dysfunction as determinants of ischemicneuronal death and survival,” Journal of Neurochemistry, vol.109, no. 1, pp. 133–138, 2009.
[34] D. Ben-Shachar and D. Laifenfeld, “Mitochondria, synapticplasticity, and schizophrenia,” International Review of Neuro-biology, vol. 59, pp. 273–296, 2004.
[35] A. Gardner and R. G. Boles, “Beyond the serotonin hypoth-esis: mitochondria, inflammation and neurodegeneration inmajor depression and affective spectrum disorders,” Progressin Neuro-Psychopharmacology & Biological Psychiatry, vol. 35,no. 3, pp. 730–743, 2011.
[36] T. Kato, “Role of mitochondrial DNA in calcium signalingabnormality in bipolar disorder,” Cell Calcium, vol. 44, no. 1,pp. 92–102, 2008.
[37] J. M. A. Oliveira, M. B. Jekabsons, S. Chen et al., “Mitochon-drial dysfunction in Huntington’s disease: the bioenergeticsof isolated and in situ mitochondria from transgenic mice,”Journal of Neurochemistry, vol. 101, no. 1, pp. 241–249, 2007.
[38] S. Gandhi, A. Wood-Kaczmar, Z. Yao et al., “PINK1-associated Parkinson’s disease is caused by neuronal vulnera-bility to calcium-induced cell death,” Molecular Cell, vol. 33,no. 5, pp. 627–638, 2009.
[39] H. Tu, O. Nelson, A. Bezprozvanny et al., “Presenilins formER Ca2+ leak channels, a function disrupted by familialAlzheimer’s disease-linked mutations,” Cell, vol. 126, no. 5,pp. 981–993, 2006.
[40] H. Zhang, S. Sun, A. Herreman, B. De Strooper, and I.Bezprozvanny, “Role of presenilins in neuronal calciumhomeostasis,” Journal of Neuroscience, vol. 30, no. 25, pp.8566–8580, 2010.
[41] K. H. Cheung, D. Shineman, M. Muller et al., “Mechanismof Ca2+ disruption in Alzheimer’s disease by presenilinregulation of InsP3 receptor channel gating,” Neuron, vol. 58,no. 6, pp. 871–883, 2008.
[42] K. V. Kuchibhotla, S. T. Goldman, C. R. Lattarulo, H. Y. Wu,B. T. Hyman, and B. J. Bacskai, “Aβ plaques lead to aberrantregulation of calcium homeostasis in vivo resulting instructural and functional disruption of neuronal networks,”Neuron, vol. 59, no. 2, pp. 214–225, 2008.
[43] A. M. Brennan, S. W. Suh, S. J. Won et al., “NADPH oxidase isthe primary source of superoxide induced by NMDA receptoractivation,” Nature Neuroscience, vol. 12, no. 7, pp. 857–863,2009.
[44] H. Chen, Y. S. Song, and P. H. Chan, “Inhibition of NADPHoxidase is neuroprotective after ischemia-reperfusion,” Jour-nal of Cerebral Blood Flow and Metabolism, vol. 29, no. 7, pp.1262–1272, 2009.
[45] D. W. Choi, “Neurodegeneration: cellular defences de-stroyed,” Nature, vol. 433, no. 7027, pp. 696–698, 2005.
[46] D. Steckley, M. Karajgikar, L. B. Dale et al., “Puma is adominant regulator of oxidative stress induced bax activationand neuronal apoptosis,” Journal of Neuroscience, vol. 27, no.47, pp. 12989–12999, 2007.
[47] K. Niizuma, H. Endo, C. Nito, D. J. Myer, and P. H. Chan,“Potential role of PUMA in delayed death of hippocampalCA1 neurons after transient global cerebral ischemia,” Stroke,vol. 40, no. 2, pp. 618–625, 2009.
[48] S. J. Riedl and G. S. Salvesen, “The apoptosome: signallingplatform of cell death,” Nature Reviews Molecular CellBiology, vol. 8, no. 5, pp. 405–413, 2007.
[49] C. Cande, F. Cecconi, P. Dessen, and G. Kroemer, “Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death?” Journal of Cell Science,vol. 115, no. 24, pp. 4727–4734, 2002.
[50] K. Aoyama, W. S. Sang, A. M. Hamby et al., “Neuronal glu-tathione deficiency and age-dependent neurodegeneration inthe EAAC1 deficient mouse,” Nature Neuroscience, vol. 9, no.1, pp. 119–126, 2006.
[51] A. E. Berman, W. Y. Chan, A. M. Brennan et al., “N-acetylcysteine prevents loss of dopaminergic neurons in theEAAC1(-/-) mouse,” Annals of Neurology, vol. 69, no. 3, pp.509–520, 2011.
[52] M. Kairisalo, A. Bonomo, A. Hyrskyluoto et al., “Resver-atrol reduces oxidative stress and cell death and increasesmitochondrial antioxidants and XIAP in PC6.3-cells,” Neu-roscience Letters, vol. 488, no. 3, pp. 263–266, 2011.
[53] S. Fallarini, G. Miglio, T. Paoletti et al., “Clovamide androsmarinic acid induce neuroprotective effects in in vitromodels of neuronal death,” British Journal of Pharmacology,vol. 157, no. 6, pp. 1072–1084, 2009.
[54] Y. Shimojo, K. Kosaka, Y. Noda, T. Shimizu, and T. Shirasawa,“Effect of rosmarinic acid in motor dysfunction and life spanin a mouse model of familial amyotrophic lateral sclerosis,”Journal of Neuroscience Research, vol. 88, no. 4, pp. 896–904,2010.
[55] J. Chao, W. K. W. Lau, M. J. Huie et al., “A pro-drug of thegreen tea polyphenol (-)-epigallocatechin-3-gallate (EGCG)prevents differentiated SH-SY5Y cells from toxicity inducedby 6-hydroxydopamine,” Neuroscience Letters, vol. 469, no. 3,pp. 360–364, 2010.
[56] R. Lagoa, C. Lopez-Sanchez, A. K. Samhan-Arias, C.M. Ganan, V. Garcia-Martinez, and C. Gutierrez-Merino,“Kaempferol protects against rat striatal degenerationinduced by 3-nitropropionic acid,” Journal of Neurochem-istry, vol. 111, no. 2, pp. 473–487, 2009.
[57] D. Luo, Q. Zhang, H. Wang et al., “Fucoidan protects againstdopaminergic neuron death in vivo and in vitro,” EuropeanJournal of Pharmacology, vol. 617, no. 1–3, pp. 33–40, 2009.
[58] J. Kim, H. J. Lee, and K. W. Lee, “Naturally occurringphytochemicals for the prevention of Alzheimer’s disease,”Journal of Neurochemistry, vol. 112, no. 6, pp. 1415–1430,2010.
[59] W. Duan, Z. Guo, H. Jiang, M. Ware, X. J. Li, and M. P.Mattson, “Dietary restriction normalizes glucose metabolismand BDNF levels, slows disease progression, and increasessurvival in huntingtin mutant mice,” Proceedings of theNational Academy of Sciences of the United States of America,vol. 100, no. 5, pp. 2911–2916, 2003.
[60] I. Ferrer, E. Goutan, C. Marın, M. J. Rey, and T. Ribalta,“Brain-derived neurotrophic factor in Huntington disease,”Brain Research, vol. 866, no. 1-2, pp. 257–261, 2000.
[61] S. Gines, I. S. Seong, E. Fossale et al., “Specific progressivecAMP reduction implicates energy deficit in presymptomatic

10 Journal of Toxicology
Huntington’s disease knock-in mice,” Human MolecularGenetics, vol. 12, no. 5, pp. 497–508, 2003.
[62] S. Gines, M. Bosch, S. Marco et al., “Reduced expression ofthe TrkB receptor in Huntington’s disease mouse models andin human brain,” European Journal of Neuroscience, vol. 23,no. 3, pp. 649–658, 2006.
[63] S. Gines, P. Paoletti, and J. Alberch, “Impaired TrkB-mediated ERK1/2 activation in huntington disease knock-in striatal cells involves reduced p52/p46 Shc expression,”Journal of Biological Chemistry, vol. 285, no. 28, pp. 21537–21548, 2010.
[64] E. Perez-Navarro, A. M. Canudas, P. Akerud, J. Alberch,and E. Arenas, “Brain-derived neurotrophic factor,neurotrophin-3, and neurotrophin-4/5 prevent thedeath of striatal projection neurons in a rodent modelof Huntington’s disease,” Journal of Neurochemistry, vol. 75,no. 5, pp. 2190–2199, 2000.
[65] S. E. Counts and E. J. Mufson, “Noradrenaline activationof neurotrophic pathways protects against neuronal amyloidtoxicity,” Journal of Neurochemistry, vol. 113, no. 3, pp. 649–660, 2010.
[66] H. Cheng, J. Yu, Z. Jiang et al., “Acupuncture improvescognitive deficits and regulates the brain cell proliferation ofSAMP8 mice,” Neuroscience Letters, vol. 432, no. 2, pp. 111–116, 2008.
[67] H. Zhao, Q. Li, Z. Zhang, X. Pei, J. Wang, and Y. Li, “Long-term ginsenoside consumption prevents memory loss inaged SAMP8 mice by decreasing oxidative stress and up-regulating the plasticity-related proteins in hippocampus,”Brain Research, vol. 1256, no. C, pp. 111–122, 2009.
[68] S. Y. Hung, H. C. Liou, K. H. Kang, R. M. Wu, C. C.Wen, and W. M. Fu, “Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity,” Molecular Phar-macology, vol. 74, no. 6, pp. 1564–1575, 2008.
[69] S. Y. Hung, H. C. Liou, and W. M. Fu, “The mechanismof heme oxygenase-1 action involved in the enhancement ofneurotrophic factor expression,” Neuropharmacology, vol. 58,no. 2, pp. 321–329, 2010.
[70] C. Li, S. Biswas, X. Li, A. K. Dutta, and W. Le,“Novel D3 dopamine receptor-preferring agonist D-264:evidence of neuroprotective property in Parkinson’s dis-ease animal models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and lactacystin,” Journal of NeuroscienceResearch, vol. 88, no. 11, pp. 2513–2523, 2010.
[71] H. S. Cho, S. Kim, S. Y. Lee, J. A. Park, S. J. Kim, andH. S. Chun, “Protective effect of the green tea component,l-theanine on environmental toxins-induced neuronal celldeath,” NeuroToxicology, vol. 29, no. 4, pp. 656–662, 2008.
[72] S. Takai, M. Yamada, T. Araki, H. Koshimizu, H. Nawa,and H. Hatanaka, “Shp-2 positively regulates brain-derivedneurotrophic factor-promoted survival of cultured ven-tral mesencephalic dopaminergic neurons through a brainimmunoglobulin-like molecule with tyrosine-based activa-tion motifs/Shp substrate-1,” Journal of Neurochemistry, vol.82, no. 2, pp. 353–364, 2002.
[73] S. Love, P. Plaha, N. K. Patel, G. R. Hotton, D. J. Brooks, andS. S. Gill, “Glial cell line-derived neurotrophic factor inducesneuronal sprouting in human brain,” Nature Medicine, vol.11, no. 7, pp. 703–704, 2005.
[74] T. Numakawa, E. Kumamaru, N. Adachi, Y. Yagasaki, A.Izumi, and H. Kunugi, “Glucocorticoid receptor interactionwith TrkB promotes BDNF-triggered PLC-γ signaling forglutamate release via a glutamate transporter,” Proceedings
of the National Academy of Sciences of the United States ofAmerica, vol. 106, no. 2, pp. 647–652, 2009.
[75] A. Woronowicz, S. R. Amith, V. W. Davis et al., “Try-panosome trans-sialidase mediates neuroprotection againstoxidative stress, serum/glucose deprivation, and hypoxia-induced neurite retraction in Trk-expressing PC12 cells,”Glycobiology, vol. 17, no. 7, pp. 725–734, 2007.
[76] N. Boutahar, E. Reynaud, F. Lassabliere, and J. Borg, “Brain-derived neurotrophic factor inhibits cell cycle reentry butnot endoplasmic reticulum stress in cultured neurons fol-lowing oxidative or excitotoxic stress,” Journal of NeuroscienceResearch, vol. 88, no. 10, pp. 2263–2271, 2010.
[77] B. Lee, R. Cao, Y. S. Choi et al., “The CREB/CRE transcrip-tional pathway: protection against oxidative stress-mediatedneuronal cell death,” Journal of Neurochemistry, vol. 108, no.5, pp. 1251–1265, 2009.
[78] B. E. Lonze and D. D. Ginty, “Function and regulation ofCREB family transcription factors in the nervous system,”Neuron, vol. 35, no. 4, pp. 605–623, 2002.
[79] T. Aid, A. Kazantseva, M. Piirsoo, K. Palm, and T. Timmusk,“Mouse and rat BDNF gene structure and expression revis-ited,” Journal of Neuroscience Research, vol. 85, no. 3, pp. 525–535, 2007.
[80] E. J. Hong, A. E. McCord, and M. E. Greenberg, “A biologicalfunction for the neuronal activity-dependent component ofBdnf transcription in the development of cortical inhibition,”Neuron, vol. 60, no. 4, pp. 610–624, 2008.
[81] S. J. Allen and D. Dawbarn, “Clinical relevance of theneurotrophins and their receptors,” Clinical Science, vol. 110,no. 2, pp. 175–191, 2006.
[82] B. S. Choi, K. Sapkota, S. Kim, H. J. Lee, H. S. Choi, andS. J. Kim, “Antioxidant activity and protective effects oftripterygium regelii extract on hydrogen peroxide-inducedinjury in human dopaminergic cells, SH-SY5Y,” Neurochem-ical Research, vol. 35, no. 8, pp. 1269–1280, 2010.
[83] J. E. Hooper and M. P. Scott, “Communicating with hedge-hogs,” Nature Reviews Molecular Cell Biology, vol. 6, no. 4, pp.306–317, 2005.
[84] R. L. Dai, S. Y. Zhu, Y. P. Xia et al., “Sonic hedgehog protectscortical neurons against oxidative stress,” NeurochemicalResearch, vol. 36, no. 1, pp. 67–75, 2011.
[85] S. Khanna, S. Roy, H. Ryu et al., “Molecular basis ofvitamin E action: tocotrienol modulates 12-lipoxygenase,a key mediator of glutamate-induced neurodegeneration,”Journal of Biological Chemistry, vol. 278, no. 44, pp. 43508–43515, 2003.
[86] T. B. Sherer, R. Betarbet, C. M. Testa et al., “Mechanism oftoxicity in rotenone models of Parkinson’s disease,” Journalof Neuroscience, vol. 23, no. 34, pp. 10756–10764, 2003.
[87] F. Osakada, A. Hashino, T. Kume, H. Katsuki, S. Kaneko,and A. Akaike, “α-tocotrienol provides the most potentneuroprotection among vitamin E analogs on culturedstriatal neurons,” Neuropharmacology, vol. 47, no. 6, pp. 904–915, 2004.
[88] V. Conte, K. Uryu, S. Fujimoto et al., “Vitamin E reducesamyloidosis and improves cognitive function in Tg2576mice following repetitive concussive brain injury,” Journal ofNeurochemistry, vol. 90, no. 3, pp. 758–764, 2004.
[89] H. Nakashima, T. Ishihara, O. Yokota et al., “Effects of α-tocopherol on an animal model of tauopathies,” Free RadicalBiology and Medicine, vol. 37, no. 2, pp. 176–186, 2004.
[90] A. Kontush and S. Schekatolina, “Vitamin E in neurodegen-erative disorders: Alzheimer’s disease,” Annals of the New YorkAcademy of Sciences, vol. 1031, pp. 249–262, 2004.

Journal of Toxicology 11
[91] D. Pratico, “Evidence of oxidative stress in Alzheimer’sdisease brain and antioxidant therapy: lights and shadows,”Annals of the New York Academy of Sciences, vol. 1147, pp.70–78, 2008.
[92] Y. Numakawa, T. Numakawa, T. Matsumoto et al., “VitaminE protected cultured cortical neurons from oxidative stress-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase,”Journal of Neurochemistry, vol. 97, no. 4, pp. 1191–1202,2006.
[93] P. B. Shieh, S. C. Hu, K. Bobb, T. Timmusk, and A. Ghosh,“Identification of a signaling pathway involved in calciumregulation of BDNF expression,” Neuron, vol. 20, no. 4, pp.727–740, 1998.
[94] X. Tao, S. Finkbeiner, D. B. Arnold, A. J. Shaywitz, and M.E. Greenberg, “Ca2+ influx regulates BDNF transcription bya CREB family transcription factor-dependent mechanism,”Neuron, vol. 20, no. 4, pp. 709–726, 1998.
[95] B. J. Gwag and J. E. Springer, “Activation of NMDA receptorsincreases brain-derived neurotrophic factor (BDNF) mRNAexpression in the hippocampal formation,” NeuroReport, vol.5, no. 2, pp. 125–128, 1993.
[96] M. Hartmann, R. Heumann, and V. Lessmann, “Synapticsecretion of BDNF after high-frequency stimulation ofglutamatergic synapses,” EMBO Journal, vol. 20, no. 21, pp.5887–5897, 2001.
[97] A. Balkowiec and D. M. Katz, “Activity-dependent release ofendogenous brain-derived neurotrophic factor from primarysensory neurons detected by ELISA in situ,” Journal ofNeuroscience, vol. 20, no. 19, pp. 7417–7423, 2000.
[98] A. Balkowiec and D. M. Katz, “Cellular mechanisms reg-ulating activity-dependent release of native brain-derivedneurotrophic factor from hippocampal neurons,” Journal ofNeuroscience, vol. 22, no. 23, pp. 10399–10407, 2002.
[99] M. E. Greenberg, B. Xu, B. Lu, and B. L. Hempstead,“New insights in the biology of BDNF synthesis and release:implications in CNS function,” Journal of Neuroscience, vol.29, no. 41, pp. 12764–12767, 2009.
[100] S. W. Flavell and M. E. Greenberg, “Signaling mechanismslinking neuronal activity to gene expression and plasticity ofthe nervous system,” Annual Review of Neuroscience, vol. 31,pp. 563–590, 2008.
[101] D. Lau and H. Bading, “Synaptic activity-mediated sup-pression of p53 and induction of nuclear calcium-regulatedneuroprotective genes promote survival through inhibitionof mitochondrial permeability transition,” Journal of Neuro-science, vol. 29, no. 14, pp. 4420–4429, 2009.
[102] F. Leveille, S. Papadia, M. Fricker et al., “Suppression of theintrinsic apoptosis pathway by synaptic activity,” Journal ofNeuroscience, vol. 30, no. 7, pp. 2623–2635, 2010.
[103] I. Tunez, R. Drucker-Colın, I. Jimena et al., “Transcranialmagnetic stimulation attenuates cell loss and oxidativedamage in the striatum induced in the 3-nitropropionicmodel of Huntington’s disease,” Journal of Neurochemistry,vol. 97, no. 3, pp. 619–630, 2006.
[104] G. W. Cho, S. H. Koh, M. H. Kim et al., “The neuroprotectiveeffect of erythropoietin-transduced human mesenchymalstromal cells in an animal model of ischemic stroke,” BrainResearch, vol. 1353, pp. 1–13, 2010.
[105] D. Takeuchi, N. Sato, M. Shimamura et al., “Alleviation ofAβ-induced cognitive impairment by ultrasound-mediatedgene transfer of HGF in a mouse model,” Gene Therapy, vol.15, no. 8, pp. 561–571, 2008.
[106] R. D. Brinton, “Cellular and molecular mechanisms of estro-gen regulation of memory function and neuroprotectionagainst Alzheimer’s disease: recent insights and remainingchallenges,” Learning and Memory, vol. 8, no. 3, pp. 121–133,2001.
[107] K. Kurata, M. Takebayashi, S. Morinobu, and S. Yamawaki,“β-estradiol, dehydroepiandrosterone, and dehydroepian-drosterone sulfate protect against N-methyl-D-aspartate-induced neurotoxicity in rat hippocampal neurons by differ-ent mechanisms,” Journal of Pharmacology and ExperimentalTherapeutics, vol. 311, no. 1, pp. 237–245, 2004.
[108] J. W. Simpkins and J. A. Dykens, “Mitochondrial mechanismsof estrogen neuroprotection,” Brain Research Reviews, vol. 57,no. 2, pp. 421–430, 2008.
[109] S. Liu and F. Mauvais-Jarvis, “Minireview: estrogenic pro-tection of β-cell failure in metabolic diseases,” Endocrinology,vol. 151, no. 3, pp. 859–864, 2010.
[110] J. W. Simpkins, K. D. Yi, and S. H. Yang, “Role of pro-tein phosphatases and mitochondria in the neuroprotectiveeffects of estrogens,” Frontiers in Neuroendocrinology, vol. 30,no. 2, pp. 93–105, 2009.
[111] G. G. J. M. Kuiper, E. Enmark, M. Pelto-Huikko, S. Nilsson,and J. A. Gustafsson, “Cloning of a novel estrogen receptorexpressed in rat prostate and ovary,” Proceedings of theNational Academy of Sciences of the United States of America,vol. 93, no. 12, pp. 5925–5930, 1996.
[112] C. Zhao, K. Dahlman-Wright, and J. A. Gustafsson, “Estro-gen signaling via estrogen receptor β,” The Journal ofBiological Chemistry, vol. 285, no. 51, pp. 39575–39579, 2010.
[113] Q. G. Zhang, L. Raz, R. Wang et al., “Estrogen attenuatesischemic oxidative damage via an estrogen receptor α-mediated inhibition of NADPH oxidase activation,” Journalof Neuroscience, vol. 29, no. 44, pp. 13823–13836, 2009.
[114] Y. Xia, J. Z. Xing, and T. L. Krukoff, “Neuroprotectiveeffects of R,R-tetrahydrochrysene against glutamate-inducedcell death through anti-excitotoxic and antioxidant actionsinvolving estrogen receptor-dependent and -independentpathways,” Neuroscience, vol. 162, no. 2, pp. 292–306, 2009.
[115] S. H. Yang, S. N. Sarkar, R. Liu et al., “Estrogen receptor βas a mitochondrial vulnerability factor,” Journal of BiologicalChemistry, vol. 284, no. 14, pp. 9540–9548, 2009.
[116] S. Gingerich, G. L. Kim, J. A. Chalmers et al., “Estrogenreceptor alpha and G-protein coupled receptor 30 mediatethe neuroprotective effects of 17β-estradiol in novel murinehippocampal cell models,” Neuroscience, vol. 170, no. 1, pp.54–66, 2010.
[117] T. Numakawa, S. Yamagishi, N. Adachi et al., “Brain-derivedneurotrophic factor-induced potentiation of Ca2+ oscilla-tions in developing cortical neurons,” Journal of BiologicalChemistry, vol. 277, no. 8, pp. 6520–6529, 2002.
[118] L. Zhang, B. E. Blackman, M. D. Schonemann et al.,“Estrogen receptor β-selective agonists stimulate calciumoscillations in human and mouse embryonic stem cell-derived neurons,” PLoS ONE, vol. 5, no. 7, article e11791,2010.
[119] D. Grove-Strawser, M. I. Boulware, and P. G. Mermelstein,“Membrane estrogen receptors activate the metabotropicglutamate receptors mGluR5 and mGluR3 to bidirectionallyregulate CREB phosphorylation in female rat striatal neu-rons,” Neuroscience, vol. 170, no. 4, pp. 1045–1055, 2010.
[120] Y. Numakawa, T. Matsumoto, D. Yokomaku et al., “17β-estradiol protects cortical neurons against oxidative stress-induced cell death through reduction in the activity of

12 Journal of Toxicology
mitogen-activated protein kinase and in the accumulation ofintracellular calcium,” Endocrinology, vol. 148, no. 2, pp. 627–637, 2007.
[121] N. Inamura, Y. Enokido, and H. Hatanaka, “Involvement ofc-Jun N-terminal kinase and caspase 3-like protease in DNAdamage-induced, p53-mediated apoptosis of cultured mousecerebellar granule neurons,” Brain Research, vol. 904, no. 2,pp. 270–278, 2001.
[122] S. L. Valles, C. Borras, J. Gambini et al., “Oestradiol orgenistein rescues neurons from amyloid beta-induced celldeath by inhibiting activation of p38,” Aging Cell, vol. 7, no.1, pp. 112–118, 2008.
[123] Y. Ishikawa, E. Kusaka, Y. Enokido, T. Ikeuchi, and H.Hatanaka, “Regulation of Bax translocation through phos-phorylation at Ser-70 of Bcl-2 by MAP kinase in NO-inducedneuronal apoptosis,” Molecular and Cellular Neuroscience,vol. 24, no. 2, pp. 451–459, 2003.
[124] S. Yamagishi, T. Matsumoto, T. Numakawa et al., “ERK1/2are involved in low potassium-induced apoptotic signalingdownstream of ASK1-p38 MAPK pathway in cultured cere-bellar granule neurons,” Brain Research, vol. 1038, no. 2, pp.223–230, 2005.
[125] E. K. Kim and E. J. Choi, “Pathological roles of MAPK sig-naling pathways in human diseases,” Biochimica et BiophysicaActa, vol. 1802, no. 4, pp. 396–405, 2010.
[126] M. P. Mattson, “Apoptosis in neurodegenerative disorders,”Nature Reviews Molecular Cell Biology, vol. 1, no. 2, pp. 120–129, 2000.
[127] M. Stanciu, Y. Wang, R. Kentor et al., “Persistent activationof ERK contributes to glutamate-induced oxidative toxicityin a neuronal cell line and primary cortical neuron cultures,”Journal of Biological Chemistry, vol. 275, no. 16, pp. 12200–12206, 2000.
[128] A. J. Crossthwaite, S. Hasan, and R. J. Williams, “Hydrogenperoxide-mediated phosphorylation of ERK1/2, AKt/PKBand JNK in cortical neurones: dependence on Ca2+ and PI3-kinase,” Journal of Neurochemistry, vol. 80, no. 1, pp. 24–35,2002.
[129] Y. Matsumoto, S. Yamamoto, Y. Suzuki et al., “Na+/H+
exchanger inhibitor, SM-20220, is protective against excito-toxicity in cultured cortical neurons,” Stroke, vol. 35, no. 1,pp. 185–190, 2004.
[130] D. M. Hartley, M. C. Kurth, L. Bjerkness, J. H. Weiss, and D.W. Choi, “Glutamate receptor-induced 45Ca2+ accumulationin cortical cell culture correlates with subsequent neuronaldegeneration,” Journal of Neuroscience, vol. 13, no. 5, pp.1993–2000, 1993.
[131] J. H. Weiss, D. M. Hartley, J. Koh, and D. W. Choi, “Thecalcium channel blocker nifedipine attenuates slow excitatoryamino acid neurotoxicity,” Science, vol. 247, no. 4949 I, pp.1474–1477, 1990.
[132] T. Matsumoto, T. Numakawa, D. Yokomaku et al., “Brain-derived neurotrophic factor-induced potentiation of gluta-mate and GABA release: different dependency on signalingpathways and neuronal activity,” Molecular and CellularNeuroscience, vol. 31, no. 1, pp. 70–84, 2006.
[133] T. Tuerxun, T. Numakawa, N. Adachi et al., “SA4503, asigma-1 receptor agonist, prevents cultured cortical neuronsfrom oxidative stress-induced cell death via suppression ofMAPK pathway activation and glutamate receptor expres-sion,” Neuroscience Letters, vol. 469, no. 3, pp. 303–308, 2010.
[134] H. Kawashima, T. Numakawa, E. Kumamaru et al., “Glu-cocorticoid attenuates brain-derived neurotrophic factor-dependent upregulation of glutamate receptors via the
suppression of microRNA-132 expression,” Neuroscience, vol.165, no. 4, pp. 1301–1311, 2010.
[135] M. Giorgio, E. Migliaccio, F. Orsini et al., “Electron transferbetween cytochrome c and p66Shc generates reactive oxygenspecies that trigger mitochondrial apoptosis,” Cell, vol. 122,no. 2, pp. 221–233, 2005.
[136] M. Pellegrini, F. Finetti, V. Petronilli et al., “p66Shc promotesT cell apoptosis by inducing mitochondrial dysfunction andimpaired Ca2+ homeostasis,” Cell Death and Differentiation,vol. 14, no. 2, pp. 338–347, 2007.
[137] M. Almeida, L. Han, E. Ambrogini, S. M. Bartell, and S.C. Manolagas, “Oxidative stress stimulates apoptosis andactivates NF-κB in osteoblastic cells via a PKCβ/p66Shc signal-ing cascade: counter regulation by estrogens or androgens,”Molecular Endocrinology, vol. 24, no. 10, pp. 2030–2037,2010.
[138] T. Baluchnejadmojarad, M. Roghani, and M. Mafakheri,“Neuroprotective effect of silymarin in 6-hydroxydopaminehemi-parkinsonian rat: involvement of estrogen receptorsand oxidative stress,” Neuroscience Letters, vol. 480, no. 3, pp.206–210, 2010.
[139] L. Y. C. Wing, Y. C. Chen, Y. Y. Shih, J. C. Cheng, Y. J. Lin,and M. J. Jiang, “Effects of oral estrogen on aortic ROS-generating and -scavenging enzymes and atherosclerosis inapoE-deficient mice,” Experimental Biology and Medicine,vol. 234, no. 9, pp. 1037–1046, 2009.
[140] A. Siriphorn, S. Chompoopong, and C. L. Floyd, “17β-estradiol protects Schwann cells against H2O2-induced cyto-toxicity and increases transplanted Schwann cell survival in acervical hemicontusion spinal cord injury model,” Journal ofNeurochemistry, vol. 115, no. 4, pp. 864–872, 2010.
[141] T. Numakawa, D. Yokomaku, M. Richards, H. Hori, N.Adachi, and H. Kunugi, “Functional interactions betweensteroid hormones and neurotrophin BDNF,” World Journalof Biological Chemistry, vol. 1, no. 5, pp. 133–143, 2010.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 392859, 13 pagesdoi:10.1155/2011/392859
Research Article
24-Epibrassinolide, a Phytosterol fromthe Brassinosteroid Family, Protects Dopaminergic Cellsagainst MPP+-Induced Oxidative Stress and Apoptosis
Julie Carange,1 Fanny Longpre,1 Benoit Daoust,1 and Maria-Grazia Martinoli1, 2
1 Department of Biochemistry, Neurosciences Research Group, Universite du Quebec a Trois-Rivieres, Trois-Rivieres,QC, Canada G9A 5H7
2 Neuroscience Research Unit, Centre de recherche, Universite Laval, Ste-Foy, QC, Canada G1V 4G2
Correspondence should be addressed to Maria-Grazia Martinoli, [email protected]
Received 15 December 2010; Revised 7 March 2011; Accepted 28 March 2011
Academic Editor: William Valentine
Copyright © 2011 Julie Carange et al. This is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Oxidative stress and apoptosis are frequently cited to explain neuronal cell damage in various neurodegenerative disorders,such as Parkinson’ s disease. Brassinosteroids (BRs) are phytosterols recognized to promote stress tolerance of vegetables viamodulation of the antioxidative enzyme cascade. However, their antioxidative effects on mammalian neuronal cells have neverbeen examined so far. We analyzed the ability of 24-epibrassinolide (24-Epi), a natural BR, to protect neuronal PC12 cells from1-methyl-4-phenylpyridinium- (MPP+-) induced oxidative stress and consequent apoptosis in dopaminergic neurons. Our resultsdemonstrate that 24-Epi reduces the levels of intracellular reactive oxygen species and modulates superoxide dismutase, catalase,and glutathione peroxidase activities. Finally, we determined that the antioxidative properties of 24-Epi lead to the inhibitionof MPP+-induced apoptosis by reducing DNA fragmentation as well as the Bax/Bcl-2 protein ratio and cleaved caspase-3. Thisis the first time that the potent antioxidant and neuroprotective role of 24-Epi has been shown in a mammalian neuronal cellline.
1. Introduction
Parkinson’s disease (PD) is characterized by the selectivedegeneration of nigrostriatal dopaminergic (DAergic) neu-rons, resulting in dopamine (DA) depletion [1]. While theetiology of PD is not completely clear, several pathologystudies have demonstrated that, in postmortem samples ofsubstantia nigra pars compacta (SNpc), DAergic neuronsexhibit markers of oxidative stress, such as lipid peroxidation,DNA oxidative damage, and carbonyl modifications ofsoluble proteins [2, 3]. The PD brain is also character-ized by oxidative damage and functionally impaired andmisassembled mitochondrial complex I, which affirm theinvolvement of oxidative stress in the pathophysiology ofPD [4]. Indeed, recent data have shown that Mn-dependentsuperoxide dismutase (SOD) level and activity are increasedin PD brains [3, 5].
Further evidence implicating oxidative stress in PDcomes from studies with the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which has been widelyused as a DAergic neurotoxin because it causes a severe PD-like syndrome in humans as well as in monkeys and mice[6]. Its administration to C57BL mice leads to a declineof striatal DA and tyrosine hydroxylase (TH) levels in theSNpc [7, 8] as well as the death of DAergic neurons [1].MPTP crosses the blood-brain barrier and is converted intoits toxic metabolite 1-methyl-4-phenylpyridinium (MPP+)in astrocytes by the enzyme monoamine oxidase B. MPP+ isthen selectively taken up by DAergic neurons via the high-affinity DA transporter (DAT) and is actively transportedinto mitochondria where it interferes with mitochondrialrespiration through complex I inhibition [9–11], elevatingreactive oxygen species (ROS) levels and increasing SOD,catalase (CAT), and glutathione peroxidase (GPx) activities

2 Journal of Toxicology
in MPTP-treated mice and MPP+-treated neuronal cells [12–14]. Also, MPTP and its active metabolite MPP+ induce THnitration, that is associated with the degeneration of DAergicneurons [15]. On the other hand, H2O2, O2
•− and •OH havebeen detected after complex I inhibition with MPP+ [16]. Inaddition, the decrease of complex I and subsequent oxidativestress evoked by MPP+ administration elicit neuronal celldeath by apoptosis [17, 18].
Brassinosteroids (BRs) are steroidal plant growth regula-tors found in several vegetables, including Vicia faba (broadbean) seed and pollen [19–21]. BRs, considered to be anew group of plant hormones, are currently being studiedintensively to understand their role in plant metabolism[22]. Their main physiological effects in plants include theregulation of hormonal balance, activation of protein andnucleic acid synthesis, enzyme activity, growth promotion,increased size and quantity of fruits, and, most interestingly,augmented resistance to unfavorable environmental factors,stress, and disease (for review see [23]). It has also beendemonstrated that the exogenous application of natural BRsto other vegetables has a specific antioxidative effect. Thenatural BR 24-epibrassinolide (24-Epi) occurring in Viciafaba [20] increases the enzymatic antioxidant activities ofSOD, CAT, and GPx in Lycopersicon esculentum (tomato)leaves and Brassica juncea L. (Indian mustard) plants [24,25]. 24-Epi also reduces lipid peroxidation in Oryza sativa L.(rice) and Indian mustard plants [25, 26].
The antioxidative properties of BRs, clearly apparent invegetables, strongly suggest that these compounds exert anantioxidant and neuroprotective role in mammals by curbingapoptosis, as reported recently for other natural molecules(for review see [13, 27–34]). Indeed, in mammals, the effectsof BRs are just starting to be elucidated. BRs are known toexert anticancer and antiproliferative activities on human celllines [35–38]. Antiviral activity has also been identified innatural BRs and synthetic analogs [39–41].
Moreover, beans from Vicia faba also contain L-3,4-dihydroxyphenylalanine (L-dopa) [42, 43], the amino acidprecursor of DA, which is nowadays the most effectivesymptomatic treatment of PD [44]. Clinical reports indicatethat the consumption of Vicia faba has a beneficial outcomein PD patients [45, 46]. However, L-dopa concentrations inVicia faba are not sufficient to explain the magnitude of theresponses observed in PD patients and raise the possibilitythat other compounds from Vicia faba, such as BRs,may complement the L-dopa effect by their antioxidativeactivities.
The aim of our study was to examine, in detail, theinfluence of 24-Epi, a BR present in Vicia faba, on MPP+-induced oxidative stress in a well-known model of PD, nervegrowth factor- (NGF-) differentiated PC12 cells [47, 48]. Weshowed that 24-Epi reduces apoptotic cellular death as well asprotein markers of apoptosis, modulates SOD, CAT, and GPxactivities, and decreases intracellular ROS concentrations.Overall, our findings clearly demonstrate that 24-Epi is anew, efficient, protective molecule against MPP+-inducedoxidative stress and might thus be regarded as a novel agentin complementary and/or preventive therapies of neuro-degenerative diseases.
2. Materials and Methods
All reagents were purchased from Sigma (St. Louis, MO)unless stated otherwise.
2.1. Cell Culture and Treatments. PC12 cells, obtained fromthe American Type Culture Collection (Rockville, MD), weremaintained in a controlled environment at 37◦C and in 5%CO2 atmosphere. They were grown in RMPI-1640 mediumsupplemented with 5% fetal bovine serum (FBS), 10% horseserum, and gentamicin (50 μg/mL). The culture medium waschanged every 2 days and the cells were seeded at a densityof 30,000 cells/cm2. Neuronal differentiation was inducedfor 4 days with 50 ng/mL NGF in RPMI-1640 mediumsupplemented with 1% FBS. To examine the effects of 24-Epi on MPP+-induced cellular death and oxidative stress,neuronal PC12 cells were pretreated with 24-Epi (10−9 M)or vehicle (culture medium) for 3 h and then exposed toMPP+ 5 mM for 1, 3, 15, or 24 h [49, 50] (Figure 1). Inapoptosis experiments, we used 500 μM of MPP+ for 24 h, asreported elsewhere [49–51]. After kinetics and dose-responsestudies [49, 50], the final concentration of 10−9 M 24-Epi(see Figure 2 for chemical structure) was chosen as the lowestdose capable of rescuing cells from MPP+-induced cellulardeath (data not included). All experiments were performedin phenol red-free medium and charcoal-stripped serum toremove steroids from the medium.
2.2. Cytotoxicity. Cytotoxicity was evaluated by colorimetricassay based on the measurement of lactate dehydrogenase(LDH) activity released from damaged cells into the super-natant [52]. LDH, a stable cytoplasmic enzyme present inall cells, is rapidly released into the cell culture supernatantupon plasma membrane injury. The amount of enzymeactivity detected in the culture supernatant correlates withthe proportion of lysed cells [53, 54].
NGF-differentiated PC12 cells were grown and treated incollagen-coated 96-well plates. Then, 100 μL of LDH sub-strate mixture was added to 50 μL of cell-free supernatant,as described elsewhere [49]. Absorbance was measured ata wavelength of 490 nm in a microplate reader (ThermolabSystem, Franklin, MA). Total cellular LDH was determinedby lysing the cells with 1% Triton X-100 (high control); theassay medium served as a low control and was subtractedfrom all absorbance measurements:
Cytotoxicity (%)
=(Experimental value− Low control
)
(High control− Low control
) × 100.(1)
2.3. ROS Detection. The antioxidative effect of 24-Epi againstMPP+-induced ROS was evaluated by dihydrorhodamine(DHR) 123 assay and MitoSOX Red (Invitrogen, Toronto,ON, Canada), according to a previously-described method[13, 55]. Briefly, to detect OH•, NO2
•, CO3•−, H2O2, HOCl,
and ONOO− by DHR [56–59], NGF-differentiated PC12cells were grown and treated on collagen-coated slides in24-well plates. A stock solution of DHR was prepared in
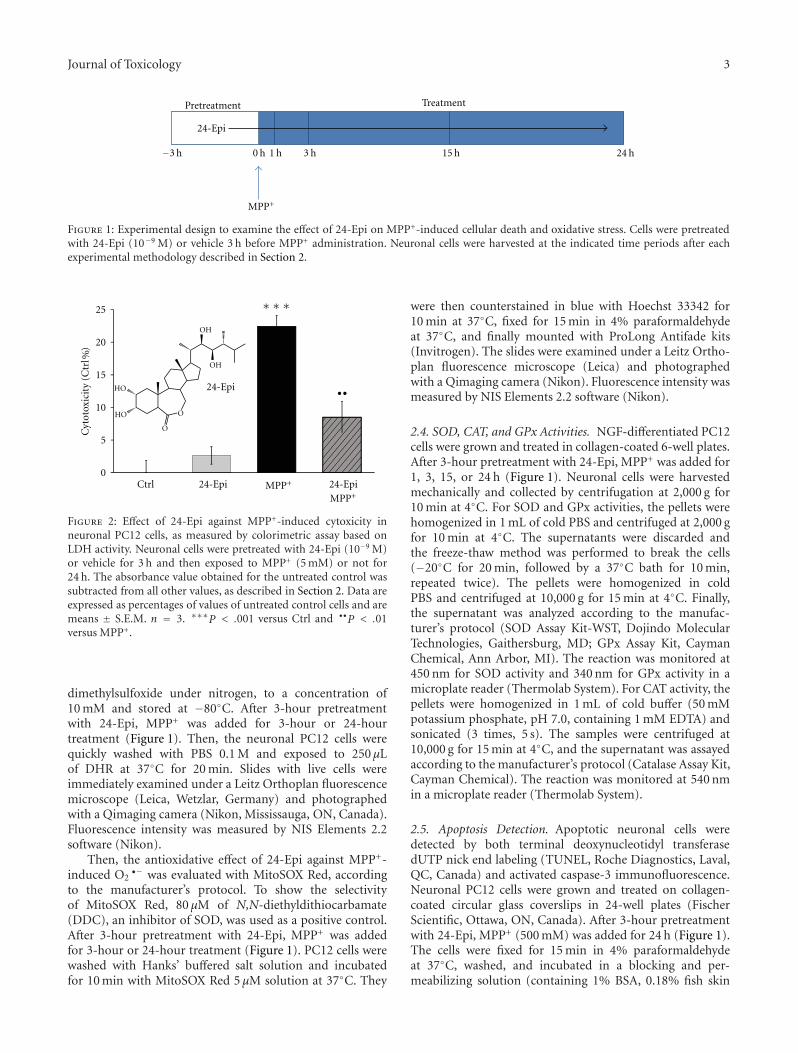
Journal of Toxicology 3
24-Epi
MPP+
Pretreatment
0 h 24 h15 h3 h1 h
Treatment
−3 h
Figure 1: Experimental design to examine the effect of 24-Epi on MPP+-induced cellular death and oxidative stress. Cells were pretreatedwith 24-Epi (10−9 M) or vehicle 3 h before MPP+ administration. Neuronal cells were harvested at the indicated time periods after eachexperimental methodology described in Section 2.
0
5
10
15
20
25
Ctrl 24-Epi MPP+ 24-Epi
MPP+
∗ ∗ ∗
••24-Epi
Cyt
otox
icit
y(C
trl
)%
HO
HO
OH
OH
O
O
Figure 2: Effect of 24-Epi against MPP+-induced cytoxicity inneuronal PC12 cells, as measured by colorimetric assay based onLDH activity. Neuronal cells were pretreated with 24-Epi (10−9 M)or vehicle for 3 h and then exposed to MPP+ (5 mM) or not for24 h. The absorbance value obtained for the untreated control wassubtracted from all other values, as described in Section 2. Data areexpressed as percentages of values of untreated control cells and aremeans ± S.E.M. n = 3. ∗∗∗P < .001 versus Ctrl and ••P < .01versus MPP+.
dimethylsulfoxide under nitrogen, to a concentration of10 mM and stored at −80◦C. After 3-hour pretreatmentwith 24-Epi, MPP+ was added for 3-hour or 24-hourtreatment (Figure 1). Then, the neuronal PC12 cells werequickly washed with PBS 0.1 M and exposed to 250 μLof DHR at 37◦C for 20 min. Slides with live cells wereimmediately examined under a Leitz Orthoplan fluorescencemicroscope (Leica, Wetzlar, Germany) and photographedwith a Qimaging camera (Nikon, Mississauga, ON, Canada).Fluorescence intensity was measured by NIS Elements 2.2software (Nikon).
Then, the antioxidative effect of 24-Epi against MPP+-induced O2
•− was evaluated with MitoSOX Red, accordingto the manufacturer’s protocol. To show the selectivityof MitoSOX Red, 80 μM of N,N-diethyldithiocarbamate(DDC), an inhibitor of SOD, was used as a positive control.After 3-hour pretreatment with 24-Epi, MPP+ was addedfor 3-hour or 24-hour treatment (Figure 1). PC12 cells werewashed with Hanks’ buffered salt solution and incubatedfor 10 min with MitoSOX Red 5 μM solution at 37◦C. They
were then counterstained in blue with Hoechst 33342 for10 min at 37◦C, fixed for 15 min in 4% paraformaldehydeat 37◦C, and finally mounted with ProLong Antifade kits(Invitrogen). The slides were examined under a Leitz Ortho-plan fluorescence microscope (Leica) and photographedwith a Qimaging camera (Nikon). Fluorescence intensity wasmeasured by NIS Elements 2.2 software (Nikon).
2.4. SOD, CAT, and GPx Activities. NGF-differentiated PC12cells were grown and treated in collagen-coated 6-well plates.After 3-hour pretreatment with 24-Epi, MPP+ was added for1, 3, 15, or 24 h (Figure 1). Neuronal cells were harvestedmechanically and collected by centrifugation at 2,000 g for10 min at 4◦C. For SOD and GPx activities, the pellets werehomogenized in 1 mL of cold PBS and centrifuged at 2,000 gfor 10 min at 4◦C. The supernatants were discarded andthe freeze-thaw method was performed to break the cells(−20◦C for 20 min, followed by a 37◦C bath for 10 min,repeated twice). The pellets were homogenized in coldPBS and centrifuged at 10,000 g for 15 min at 4◦C. Finally,the supernatant was analyzed according to the manufac-turer’s protocol (SOD Assay Kit-WST, Dojindo MolecularTechnologies, Gaithersburg, MD; GPx Assay Kit, CaymanChemical, Ann Arbor, MI). The reaction was monitored at450 nm for SOD activity and 340 nm for GPx activity in amicroplate reader (Thermolab System). For CAT activity, thepellets were homogenized in 1 mL of cold buffer (50 mMpotassium phosphate, pH 7.0, containing 1 mM EDTA) andsonicated (3 times, 5 s). The samples were centrifuged at10,000 g for 15 min at 4◦C, and the supernatant was assayedaccording to the manufacturer’s protocol (Catalase Assay Kit,Cayman Chemical). The reaction was monitored at 540 nmin a microplate reader (Thermolab System).
2.5. Apoptosis Detection. Apoptotic neuronal cells weredetected by both terminal deoxynucleotidyl transferasedUTP nick end labeling (TUNEL, Roche Diagnostics, Laval,QC, Canada) and activated caspase-3 immunofluorescence.Neuronal PC12 cells were grown and treated on collagen-coated circular glass coverslips in 24-well plates (FischerScientific, Ottawa, ON, Canada). After 3-hour pretreatmentwith 24-Epi, MPP+ (500 mM) was added for 24 h (Figure 1).The cells were fixed for 15 min in 4% paraformaldehydeat 37◦C, washed, and incubated in a blocking and per-meabilizing solution (containing 1% BSA, 0.18% fish skin
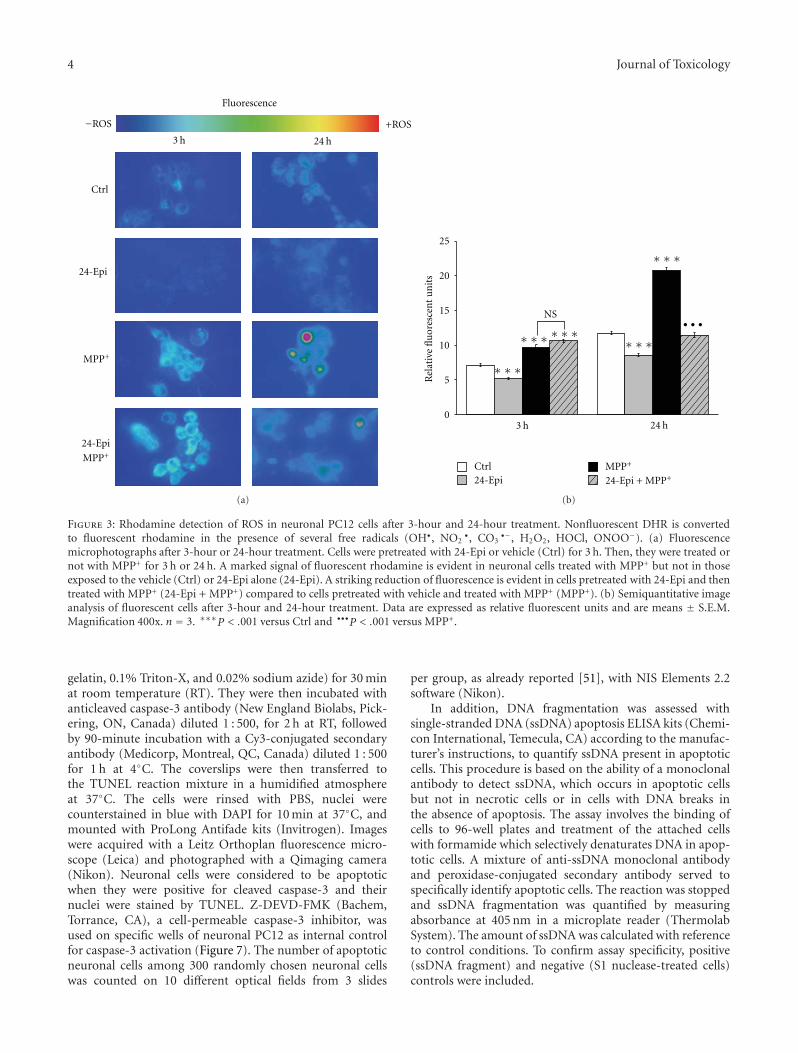
4 Journal of Toxicology
3 h 24 h
+ROS
Fluorescence
Ctrl
24-Epi
MPP+
24-Epi
MPP+
ROS−
(a)
0
5
10
15
20
25
3 h 24 hR
elat
ive
flu
ores
cen
tu
nit
s
Ctrl24-Epi
MPP+
24-Epi + MPP+
∗ ∗ ∗
∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗ ∗
∗ ∗ ∗
NS• • •
(b)
Figure 3: Rhodamine detection of ROS in neuronal PC12 cells after 3-hour and 24-hour treatment. Nonfluorescent DHR is convertedto fluorescent rhodamine in the presence of several free radicals (OH•, NO2
•, CO3•−, H2O2, HOCl, ONOO−). (a) Fluorescence
microphotographs after 3-hour or 24-hour treatment. Cells were pretreated with 24-Epi or vehicle (Ctrl) for 3 h. Then, they were treated ornot with MPP+ for 3 h or 24 h. A marked signal of fluorescent rhodamine is evident in neuronal cells treated with MPP+ but not in thoseexposed to the vehicle (Ctrl) or 24-Epi alone (24-Epi). A striking reduction of fluorescence is evident in cells pretreated with 24-Epi and thentreated with MPP+ (24-Epi + MPP+) compared to cells pretreated with vehicle and treated with MPP+ (MPP+). (b) Semiquantitative imageanalysis of fluorescent cells after 3-hour and 24-hour treatment. Data are expressed as relative fluorescent units and are means ± S.E.M.Magnification 400x. n = 3. ∗∗∗P < .001 versus Ctrl and •••P < .001 versus MPP+.
gelatin, 0.1% Triton-X, and 0.02% sodium azide) for 30 minat room temperature (RT). They were then incubated withanticleaved caspase-3 antibody (New England Biolabs, Pick-ering, ON, Canada) diluted 1 : 500, for 2 h at RT, followedby 90-minute incubation with a Cy3-conjugated secondaryantibody (Medicorp, Montreal, QC, Canada) diluted 1 : 500for 1 h at 4◦C. The coverslips were then transferred tothe TUNEL reaction mixture in a humidified atmosphereat 37◦C. The cells were rinsed with PBS, nuclei werecounterstained in blue with DAPI for 10 min at 37◦C, andmounted with ProLong Antifade kits (Invitrogen). Imageswere acquired with a Leitz Orthoplan fluorescence micro-scope (Leica) and photographed with a Qimaging camera(Nikon). Neuronal cells were considered to be apoptoticwhen they were positive for cleaved caspase-3 and theirnuclei were stained by TUNEL. Z-DEVD-FMK (Bachem,Torrance, CA), a cell-permeable caspase-3 inhibitor, wasused on specific wells of neuronal PC12 as internal controlfor caspase-3 activation (Figure 7). The number of apoptoticneuronal cells among 300 randomly chosen neuronal cellswas counted on 10 different optical fields from 3 slides
per group, as already reported [51], with NIS Elements 2.2software (Nikon).
In addition, DNA fragmentation was assessed withsingle-stranded DNA (ssDNA) apoptosis ELISA kits (Chemi-con International, Temecula, CA) according to the manufac-turer’s instructions, to quantify ssDNA present in apoptoticcells. This procedure is based on the ability of a monoclonalantibody to detect ssDNA, which occurs in apoptotic cellsbut not in necrotic cells or in cells with DNA breaks inthe absence of apoptosis. The assay involves the binding ofcells to 96-well plates and treatment of the attached cellswith formamide which selectively denaturates DNA in apop-totic cells. A mixture of anti-ssDNA monoclonal antibodyand peroxidase-conjugated secondary antibody served tospecifically identify apoptotic cells. The reaction was stoppedand ssDNA fragmentation was quantified by measuringabsorbance at 405 nm in a microplate reader (ThermolabSystem). The amount of ssDNA was calculated with referenceto control conditions. To confirm assay specificity, positive(ssDNA fragment) and negative (S1 nuclease-treated cells)controls were included.
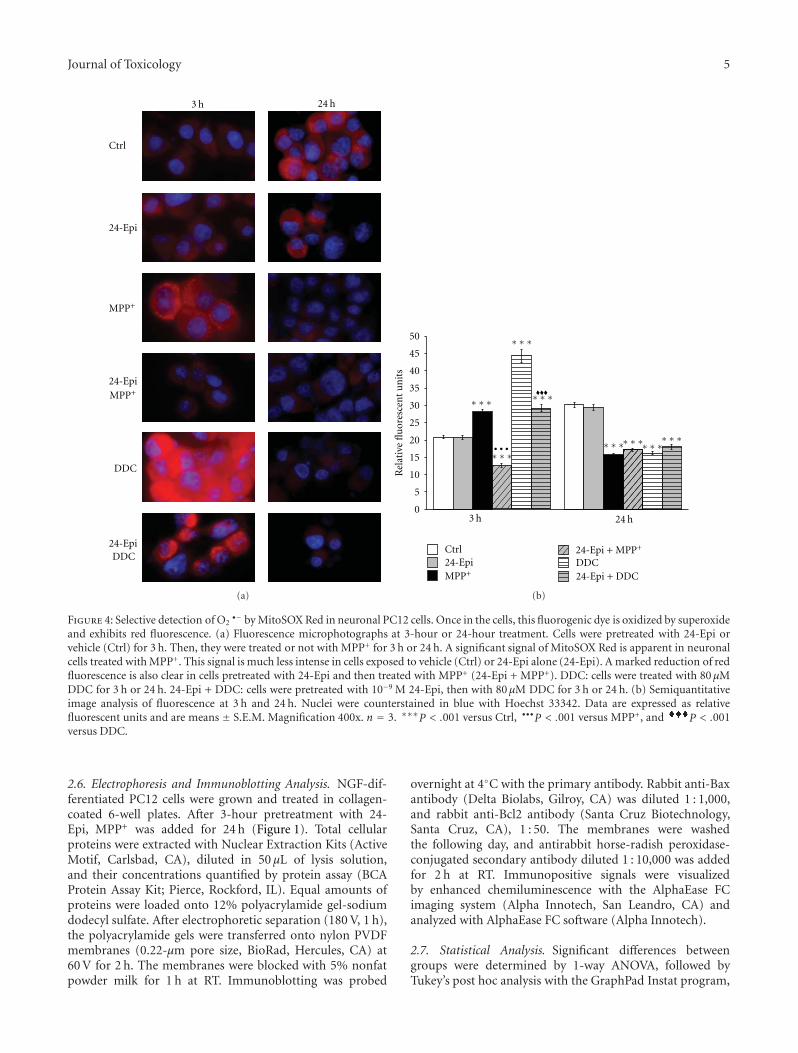
Journal of Toxicology 5
3 h 24 h
Ctrl
24-Epi
MPP+
24-Epi
MPP+
24-Epi
DDC
DDC
(a)
0
5
10
15
20
25
30
35
40
45
50
3 h 24 h
Rel
ativ
efl
uor
esce
nt
un
its
Ctrl24-EpiMPP+
24-Epi + MPP+
DDC24-Epi + DDC
∗ ∗ ∗
∗ ∗ ∗
∗ ∗ ∗
∗ ∗ ∗∗ ∗ ∗∗ ∗ ∗∗ ∗ ∗
∗ ∗ ∗• • •
(b)
Figure 4: Selective detection of O2•− by MitoSOX Red in neuronal PC12 cells. Once in the cells, this fluorogenic dye is oxidized by superoxide
and exhibits red fluorescence. (a) Fluorescence microphotographs at 3-hour or 24-hour treatment. Cells were pretreated with 24-Epi orvehicle (Ctrl) for 3 h. Then, they were treated or not with MPP+ for 3 h or 24 h. A significant signal of MitoSOX Red is apparent in neuronalcells treated with MPP+. This signal is much less intense in cells exposed to vehicle (Ctrl) or 24-Epi alone (24-Epi). A marked reduction of redfluorescence is also clear in cells pretreated with 24-Epi and then treated with MPP+ (24-Epi + MPP+). DDC: cells were treated with 80 μMDDC for 3 h or 24 h. 24-Epi + DDC: cells were pretreated with 10−9 M 24-Epi, then with 80 μM DDC for 3 h or 24 h. (b) Semiquantitativeimage analysis of fluorescence at 3 h and 24 h. Nuclei were counterstained in blue with Hoechst 33342. Data are expressed as relativefluorescent units and are means ± S.E.M. Magnification 400x. n = 3. ∗∗∗P < .001 versus Ctrl, •••P < .001 versus MPP+, and P < .001versus DDC.
2.6. Electrophoresis and Immunoblotting Analysis. NGF-dif-ferentiated PC12 cells were grown and treated in collagen-coated 6-well plates. After 3-hour pretreatment with 24-Epi, MPP+ was added for 24 h (Figure 1). Total cellularproteins were extracted with Nuclear Extraction Kits (ActiveMotif, Carlsbad, CA), diluted in 50 μL of lysis solution,and their concentrations quantified by protein assay (BCAProtein Assay Kit; Pierce, Rockford, IL). Equal amounts ofproteins were loaded onto 12% polyacrylamide gel-sodiumdodecyl sulfate. After electrophoretic separation (180 V, 1 h),the polyacrylamide gels were transferred onto nylon PVDFmembranes (0.22-μm pore size, BioRad, Hercules, CA) at60 V for 2 h. The membranes were blocked with 5% nonfatpowder milk for 1 h at RT. Immunoblotting was probed
overnight at 4◦C with the primary antibody. Rabbit anti-Baxantibody (Delta Biolabs, Gilroy, CA) was diluted 1 : 1,000,and rabbit anti-Bcl2 antibody (Santa Cruz Biotechnology,Santa Cruz, CA), 1 : 50. The membranes were washedthe following day, and antirabbit horse-radish peroxidase-conjugated secondary antibody diluted 1 : 10,000 was addedfor 2 h at RT. Immunopositive signals were visualizedby enhanced chemiluminescence with the AlphaEase FCimaging system (Alpha Innotech, San Leandro, CA) andanalyzed with AlphaEase FC software (Alpha Innotech).
2.7. Statistical Analysis. Significant differences betweengroups were determined by 1-way ANOVA, followed byTukey’s post hoc analysis with the GraphPad Instat program,
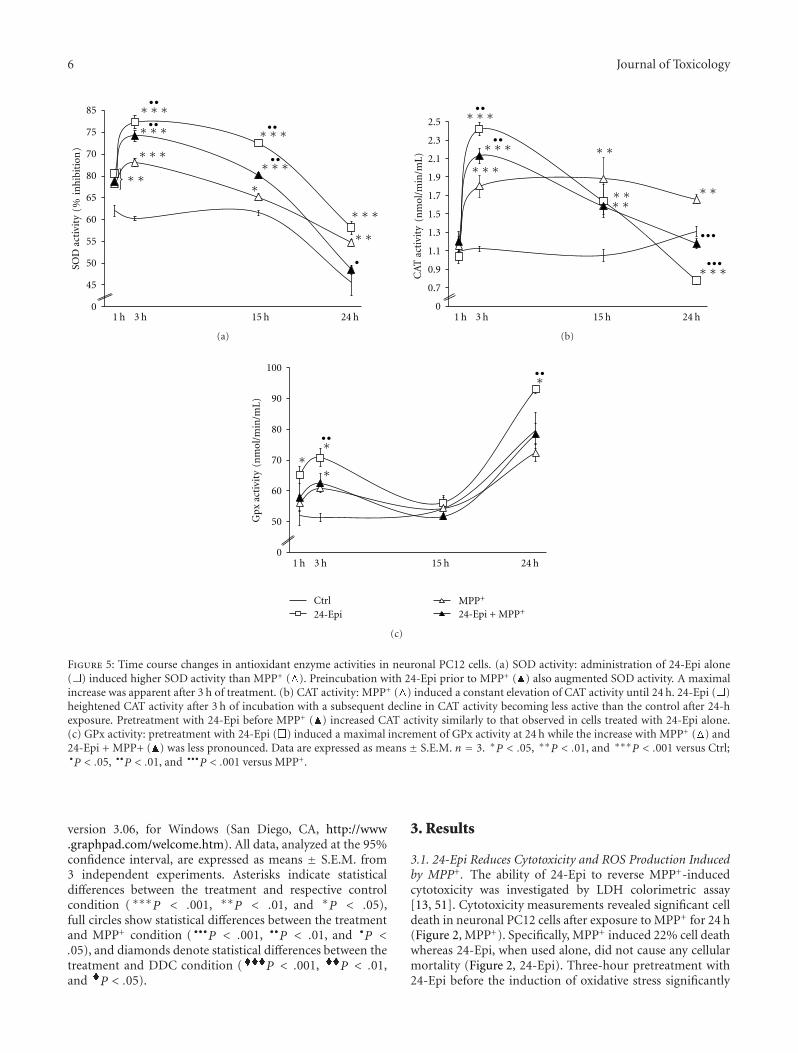
6 Journal of Toxicology
0
45
50
55
60
65
70
75
80
85
SOD
acti
vity
(in
hib
itio
n)
∗ ∗
∗ ∗ ∗∗ ∗ ∗
∗ ∗ ∗
••
•• ••
••
∗ ∗ ∗
∗ ∗ ∗∗
∗ ∗ ∗
∗ ∗
•
1 h 3 h 15 h 24 h
%
(a)
1 h 3 h 15 h 24 h0
0.7
0.9
1.1
1.3
1.5
1.7
1.9
2.1
2.3
2.5
CA
Tac
tivi
ty(n
mol
/min
/mL
)
∗ ∗ ∗
∗ ∗ ∗
∗ ∗ ∗
∗ ∗
∗ ∗ ∗ ∗∗ ∗
••
•••
•••
∗ ∗ ∗••
(b)
0
50
60
70
80
90
100
Gpx
acti
vity
(nm
ol/m
in/m
L)
∗∗
∗
∗
••
••
1 h 3 h 15 h 24 h
Ctrl24-Epi
MPP+
24-Epi + MPP+
(c)
Figure 5: Time course changes in antioxidant enzyme activities in neuronal PC12 cells. (a) SOD activity: administration of 24-Epi alone( ) induced higher SOD activity than MPP+ ( ). Preincubation with 24-Epi prior to MPP+ ( ) also augmented SOD activity. A maximalincrease was apparent after 3 h of treatment. (b) CAT activity: MPP+ ( ) induced a constant elevation of CAT activity until 24 h. 24-Epi ( )heightened CAT activity after 3 h of incubation with a subsequent decline in CAT activity becoming less active than the control after 24-hexposure. Pretreatment with 24-Epi before MPP+ ( ) increased CAT activity similarly to that observed in cells treated with 24-Epi alone.(c) GPx activity: pretreatment with 24-Epi ( ) induced a maximal increment of GPx activity at 24 h while the increase with MPP+ ( ) and24-Epi + MPP+ ( ) was less pronounced. Data are expressed as means ± S.E.M. n = 3. ∗P < .05, ∗∗P < .01, and ∗∗∗P < .001 versus Ctrl;•P < .05, ••P < .01, and •••P < .001 versus MPP+.
version 3.06, for Windows (San Diego, CA, http://www.graphpad.com/welcome.htm). All data, analyzed at the 95%confidence interval, are expressed as means ± S.E.M. from3 independent experiments. Asterisks indicate statisticaldifferences between the treatment and respective controlcondition (∗∗∗P < .001, ∗∗P < .01, and ∗P < .05),full circles show statistical differences between the treatmentand MPP+ condition ( •••P < .001, ••P < .01, and •P <.05), and diamonds denote statistical differences between thetreatment and DDC condition ( P < .001, P < .01,and P < .05).
3. Results
3.1. 24-Epi Reduces Cytotoxicity and ROS Production Inducedby MPP+. The ability of 24-Epi to reverse MPP+-inducedcytotoxicity was investigated by LDH colorimetric assay[13, 51]. Cytotoxicity measurements revealed significant celldeath in neuronal PC12 cells after exposure to MPP+ for 24 h(Figure 2, MPP+). Specifically, MPP+ induced 22% cell deathwhereas 24-Epi, when used alone, did not cause any cellularmortality (Figure 2, 24-Epi). Three-hour pretreatment with24-Epi before the induction of oxidative stress significantly
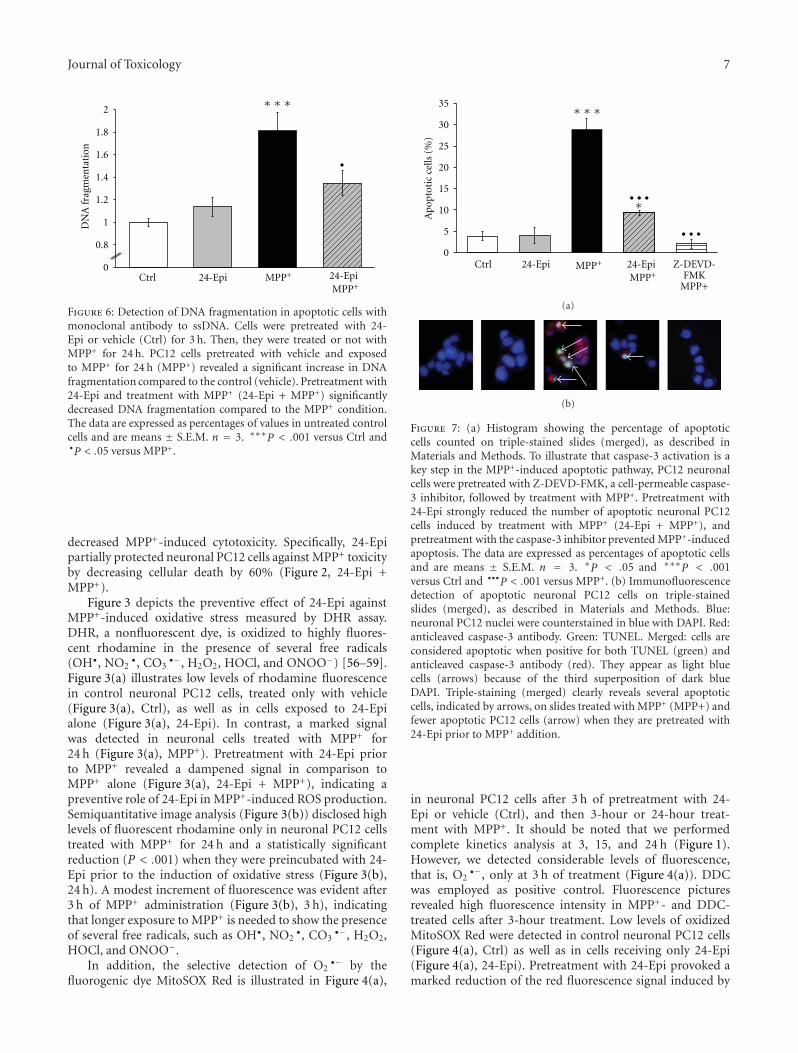
Journal of Toxicology 7
0
0.8
1
1.2
1.4
1.6
1.8
2
Ctrl 24-Epi MPP+ 24-EpiMPP+
DN
Afr
agm
enta
tion
∗ ∗ ∗
•
Figure 6: Detection of DNA fragmentation in apoptotic cells withmonoclonal antibody to ssDNA. Cells were pretreated with 24-Epi or vehicle (Ctrl) for 3 h. Then, they were treated or not withMPP+ for 24 h. PC12 cells pretreated with vehicle and exposedto MPP+ for 24 h (MPP+) revealed a significant increase in DNAfragmentation compared to the control (vehicle). Pretreatment with24-Epi and treatment with MPP+ (24-Epi + MPP+) significantlydecreased DNA fragmentation compared to the MPP+ condition.The data are expressed as percentages of values in untreated controlcells and are means ± S.E.M. n = 3. ∗∗∗P < .001 versus Ctrl and•P < .05 versus MPP+.
decreased MPP+-induced cytotoxicity. Specifically, 24-Epipartially protected neuronal PC12 cells against MPP+ toxicityby decreasing cellular death by 60% (Figure 2, 24-Epi +MPP+).
Figure 3 depicts the preventive effect of 24-Epi againstMPP+-induced oxidative stress measured by DHR assay.DHR, a nonfluorescent dye, is oxidized to highly fluores-cent rhodamine in the presence of several free radicals(OH•, NO2
•, CO3•−, H2O2, HOCl, and ONOO−) [56–59].
Figure 3(a) illustrates low levels of rhodamine fluorescencein control neuronal PC12 cells, treated only with vehicle(Figure 3(a), Ctrl), as well as in cells exposed to 24-Epialone (Figure 3(a), 24-Epi). In contrast, a marked signalwas detected in neuronal cells treated with MPP+ for24 h (Figure 3(a), MPP+). Pretreatment with 24-Epi priorto MPP+ revealed a dampened signal in comparison toMPP+ alone (Figure 3(a), 24-Epi + MPP+), indicating apreventive role of 24-Epi in MPP+-induced ROS production.Semiquantitative image analysis (Figure 3(b)) disclosed highlevels of fluorescent rhodamine only in neuronal PC12 cellstreated with MPP+ for 24 h and a statistically significantreduction (P < .001) when they were preincubated with 24-Epi prior to the induction of oxidative stress (Figure 3(b),24 h). A modest increment of fluorescence was evident after3 h of MPP+ administration (Figure 3(b), 3 h), indicatingthat longer exposure to MPP+ is needed to show the presenceof several free radicals, such as OH•, NO2
•, CO3•−, H2O2,
HOCl, and ONOO−.In addition, the selective detection of O2
•− by thefluorogenic dye MitoSOX Red is illustrated in Figure 4(a),
0
5
10
15
20
25
30
35
Ctrl MPP+ Z-DEVD-FMK
MPP+
Apo
ptot
icce
lls(%
)
∗ ∗ ∗
∗• • •
• • •
24-Epi 24-EpiMPP+
(a)
(b)
Figure 7: (a) Histogram showing the percentage of apoptoticcells counted on triple-stained slides (merged), as described inMaterials and Methods. To illustrate that caspase-3 activation is akey step in the MPP+-induced apoptotic pathway, PC12 neuronalcells were pretreated with Z-DEVD-FMK, a cell-permeable caspase-3 inhibitor, followed by treatment with MPP+. Pretreatment with24-Epi strongly reduced the number of apoptotic neuronal PC12cells induced by treatment with MPP+ (24-Epi + MPP+), andpretreatment with the caspase-3 inhibitor prevented MPP+-inducedapoptosis. The data are expressed as percentages of apoptotic cellsand are means ± S.E.M. n = 3. ∗P < .05 and ∗∗∗P < .001versus Ctrl and •••P < .001 versus MPP+. (b) Immunofluorescencedetection of apoptotic neuronal PC12 cells on triple-stainedslides (merged), as described in Materials and Methods. Blue:neuronal PC12 nuclei were counterstained in blue with DAPI. Red:anticleaved caspase-3 antibody. Green: TUNEL. Merged: cells areconsidered apoptotic when positive for both TUNEL (green) andanticleaved caspase-3 antibody (red). They appear as light bluecells (arrows) because of the third superposition of dark blueDAPI. Triple-staining (merged) clearly reveals several apoptoticcells, indicated by arrows, on slides treated with MPP+ (MPP+) andfewer apoptotic PC12 cells (arrow) when they are pretreated with24-Epi prior to MPP+ addition.
in neuronal PC12 cells after 3 h of pretreatment with 24-Epi or vehicle (Ctrl), and then 3-hour or 24-hour treat-ment with MPP+. It should be noted that we performedcomplete kinetics analysis at 3, 15, and 24 h (Figure 1).However, we detected considerable levels of fluorescence,that is, O2
•−, only at 3 h of treatment (Figure 4(a)). DDCwas employed as positive control. Fluorescence picturesrevealed high fluorescence intensity in MPP+- and DDC-treated cells after 3-hour treatment. Low levels of oxidizedMitoSOX Red were detected in control neuronal PC12 cells(Figure 4(a), Ctrl) as well as in cells receiving only 24-Epi(Figure 4(a), 24-Epi). Pretreatment with 24-Epi provoked amarked reduction of the red fluorescence signal induced by
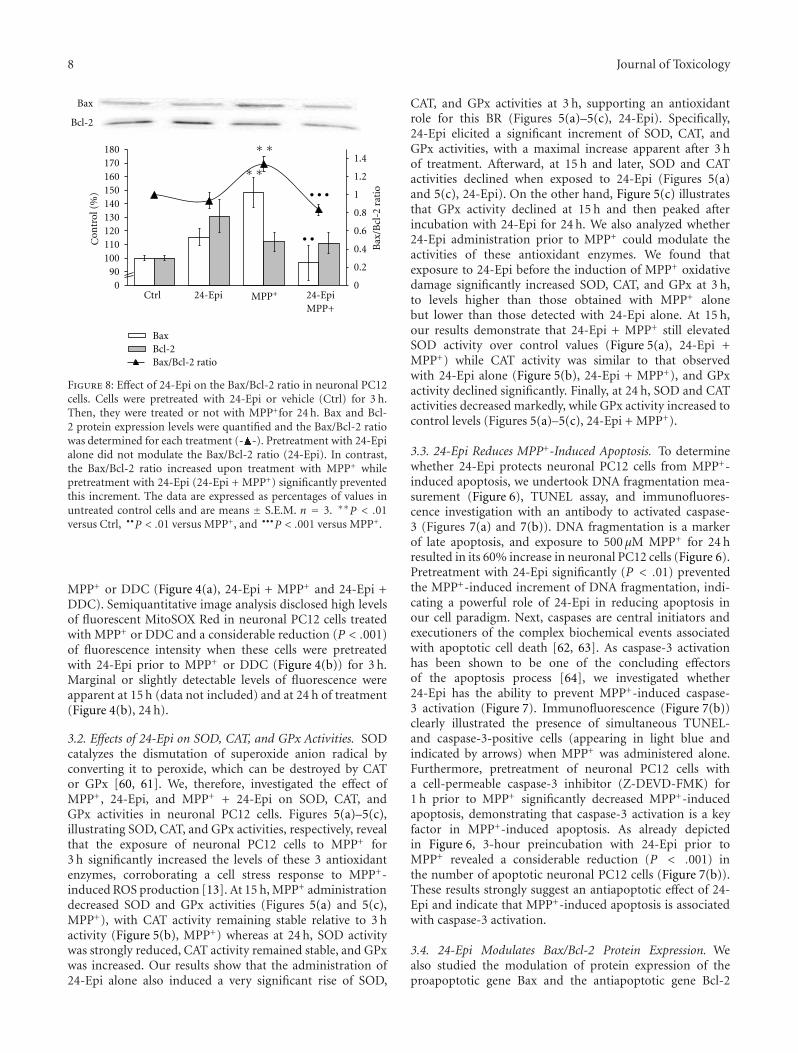
8 Journal of Toxicology
0
0.2
0.4
0.6
0.8
1
1.2
1.4
090
100110120130140150160170180
Ctrl 24-Epi MPP+ 24-EpiMPP+
Bax
/Bcl
-2ra
tio
Bax
Bcl-2
BaxBcl-2Bax/Bcl-2 ratio
∗ ∗
• • •
• •
∗ ∗
ontr
ol(%
)C
Figure 8: Effect of 24-Epi on the Bax/Bcl-2 ratio in neuronal PC12cells. Cells were pretreated with 24-Epi or vehicle (Ctrl) for 3 h.Then, they were treated or not with MPP+for 24 h. Bax and Bcl-2 protein expression levels were quantified and the Bax/Bcl-2 ratiowas determined for each treatment (- -). Pretreatment with 24-Epialone did not modulate the Bax/Bcl-2 ratio (24-Epi). In contrast,the Bax/Bcl-2 ratio increased upon treatment with MPP+ whilepretreatment with 24-Epi (24-Epi + MPP+) significantly preventedthis increment. The data are expressed as percentages of values inuntreated control cells and are means ± S.E.M. n = 3. ∗∗P < .01versus Ctrl, ••P < .01 versus MPP+, and •••P < .001 versus MPP+.
MPP+ or DDC (Figure 4(a), 24-Epi + MPP+ and 24-Epi +DDC). Semiquantitative image analysis disclosed high levelsof fluorescent MitoSOX Red in neuronal PC12 cells treatedwith MPP+ or DDC and a considerable reduction (P < .001)of fluorescence intensity when these cells were pretreatedwith 24-Epi prior to MPP+ or DDC (Figure 4(b)) for 3 h.Marginal or slightly detectable levels of fluorescence wereapparent at 15 h (data not included) and at 24 h of treatment(Figure 4(b), 24 h).
3.2. Effects of 24-Epi on SOD, CAT, and GPx Activities. SODcatalyzes the dismutation of superoxide anion radical byconverting it to peroxide, which can be destroyed by CATor GPx [60, 61]. We, therefore, investigated the effect ofMPP+, 24-Epi, and MPP+ + 24-Epi on SOD, CAT, andGPx activities in neuronal PC12 cells. Figures 5(a)–5(c),illustrating SOD, CAT, and GPx activities, respectively, revealthat the exposure of neuronal PC12 cells to MPP+ for3 h significantly increased the levels of these 3 antioxidantenzymes, corroborating a cell stress response to MPP+-induced ROS production [13]. At 15 h, MPP+ administrationdecreased SOD and GPx activities (Figures 5(a) and 5(c),MPP+), with CAT activity remaining stable relative to 3 hactivity (Figure 5(b), MPP+) whereas at 24 h, SOD activitywas strongly reduced, CAT activity remained stable, and GPxwas increased. Our results show that the administration of24-Epi alone also induced a very significant rise of SOD,
CAT, and GPx activities at 3 h, supporting an antioxidantrole for this BR (Figures 5(a)–5(c), 24-Epi). Specifically,24-Epi elicited a significant increment of SOD, CAT, andGPx activities, with a maximal increase apparent after 3 hof treatment. Afterward, at 15 h and later, SOD and CATactivities declined when exposed to 24-Epi (Figures 5(a)and 5(c), 24-Epi). On the other hand, Figure 5(c) illustratesthat GPx activity declined at 15 h and then peaked afterincubation with 24-Epi for 24 h. We also analyzed whether24-Epi administration prior to MPP+ could modulate theactivities of these antioxidant enzymes. We found thatexposure to 24-Epi before the induction of MPP+ oxidativedamage significantly increased SOD, CAT, and GPx at 3 h,to levels higher than those obtained with MPP+ alonebut lower than those detected with 24-Epi alone. At 15 h,our results demonstrate that 24-Epi + MPP+ still elevatedSOD activity over control values (Figure 5(a), 24-Epi +MPP+) while CAT activity was similar to that observedwith 24-Epi alone (Figure 5(b), 24-Epi + MPP+), and GPxactivity declined significantly. Finally, at 24 h, SOD and CATactivities decreased markedly, while GPx activity increased tocontrol levels (Figures 5(a)–5(c), 24-Epi + MPP+).
3.3. 24-Epi Reduces MPP+-Induced Apoptosis. To determinewhether 24-Epi protects neuronal PC12 cells from MPP+-induced apoptosis, we undertook DNA fragmentation mea-surement (Figure 6), TUNEL assay, and immunofluores-cence investigation with an antibody to activated caspase-3 (Figures 7(a) and 7(b)). DNA fragmentation is a markerof late apoptosis, and exposure to 500 μM MPP+ for 24 hresulted in its 60% increase in neuronal PC12 cells (Figure 6).Pretreatment with 24-Epi significantly (P < .01) preventedthe MPP+-induced increment of DNA fragmentation, indi-cating a powerful role of 24-Epi in reducing apoptosis inour cell paradigm. Next, caspases are central initiators andexecutioners of the complex biochemical events associatedwith apoptotic cell death [62, 63]. As caspase-3 activationhas been shown to be one of the concluding effectorsof the apoptosis process [64], we investigated whether24-Epi has the ability to prevent MPP+-induced caspase-3 activation (Figure 7). Immunofluorescence (Figure 7(b))clearly illustrated the presence of simultaneous TUNEL-and caspase-3-positive cells (appearing in light blue andindicated by arrows) when MPP+ was administered alone.Furthermore, pretreatment of neuronal PC12 cells witha cell-permeable caspase-3 inhibitor (Z-DEVD-FMK) for1 h prior to MPP+ significantly decreased MPP+-inducedapoptosis, demonstrating that caspase-3 activation is a keyfactor in MPP+-induced apoptosis. As already depictedin Figure 6, 3-hour preincubation with 24-Epi prior toMPP+ revealed a considerable reduction (P < .001) inthe number of apoptotic neuronal PC12 cells (Figure 7(b)).These results strongly suggest an antiapoptotic effect of 24-Epi and indicate that MPP+-induced apoptosis is associatedwith caspase-3 activation.
3.4. 24-Epi Modulates Bax/Bcl-2 Protein Expression. Wealso studied the modulation of protein expression of theproapoptotic gene Bax and the antiapoptotic gene Bcl-2

Journal of Toxicology 9
by 24-Epi. The ratio of proapoptotic Bax to antiapoptoticBcl-2 (Bax/Bcl-2) has been reported to be correlated withapoptosis [51, 65]. Our results reveal that the administrationof 24-Epi alone did not significantly modulate the Bax/Bcl-2 ratio (Figure 8, triangles on a continuous line). Treatmentwith MPP+ alone significantly increased the Bax/Bcl-2 ratio,indicating that MPP+-induced apoptosis of PC12 cells maybe mediated by the mitochondrial pathway. The MPP+-induced increase of the Bax/Bcl-2 ratio was considerablyattenuated in cells pretreated with 24-Epi (Figure 8, 24-Epi +MPP+) to control levels, suggesting, for the first time, thatthe BR 24-Epi is a strong modulator of proapoptotic andantiapoptotic gene expression.
4. Discussion
In this paper, we demonstrated, for the first time, that 24-Epi,a BR found in a variety of vegetables as well as in Vicia faba,can exert antioxidative and consequent antiapoptotic actionsin mammalian neural cells. In particular, we studied PC12cells, a known, reliable, and efficient model for the investiga-tion of oxidative stress and neuroprotection of DA neurons[49, 66]. After NGF administration, PC12 cells differentiateinto a neuronal-like phenotype that secretes high DA levelsand expresses TH, DAT, neurofilaments as well as estrogenreceptor-alpha and -beta (ERα and ERβ) [49, 66–68].
Recent studies have reported the powerful propertiesof various natural polyphenols against oxidative stress inseveral cellular and in vivo paradigms of neurodegenerativedisease [30–34]. Currently, many natural polyphenols areunder intense investigation for their antioxidative effectsand their possible use as complementary and/or preventivetherapies of diseases [33, 34]. Our aim was to demonstratethat BRs, contained in a wide variety of vegetables, indeedexert antioxidative as well as neuroprotective properties inneuronal PC12 cells, a cellular model of PD [47, 48].
At present, phytosterols are recognized antioxidants [69],and some of them possess antioxidative properties associatedwith neuroprotective effects [70, 71]. Others, such as β-sitosterol, modulate SOD, GPx, and CAT activities [72], andthe ginsenoside Rg1, a phytosterol derived from ginseng, isalso reported to be antiapoptotic in neuronal PC12 cells afteroxidative stress [73, 74]. However, BRs, in particular, aremuch less studied, even if the recent literature is pointingto their interesting potential in mammalian systems, suchas antiviral [40], anticancer, and antiproliferative activities[35–37]. At present, no data on a possible antioxidativeand antiapoptotic role of BRs are available in mammalianneurons in vitro or in vivo. As such, in this study, weexamined, for the first time, the neuroprotective, antioxi-dant, and antiapoptotic consequences of low-dose 24-Epi(10−9 M), a common BR, against oxidative damage inducedby treatments with MPP+, the active metabolite of MPTP,a known Parkinsonian toxin. The positive outcomes wereported in DAergic neuronal culture, using nanomolardoses of 24-Epi, on parameters of neuroprotection, oxidativemetabolism, and apoptosis, are supported by the fact thatBRs may be considered the plant equivalent of steroidhormones in vertebrates, sharing similar metabolic pathways
[75]. Thus, BRs could easily pass through the blood-brainbarrier and are likely to accumulate in the brain andserum, as demonstrated for plant sterol and stanol estersin Watanabe rabbits [76]. In particular, we established that24-Epi can protect DA neuronal cells from MPP+-inducedcellular death by reducing intracellular ROS production.Indeed, nonfluorescent DHR has the capacity to enter cellsand, once inside them, it is oxidized by oxygen species(superoxide anion, peroxynitrite) to fluorescent rhodamine[57]. Accordingly, our results show increased rhodaminefluorescence after MPP+ treatment and reduced fluorescencewhen 24-Epi is administered to neuronal PC12 cells priorto MPP+. DHR has been deployed extensively to measureintracellular ROS, but it does not quantify O2
•− production.MitoSOX Red is a selective indicator of mitochondrial O2
•−
production and becomes highly fluorescent when oxidizedby this ROS but not by other oxidants. With MitoSOX Red,we illustrated an increase of fluorescence when MPP+ wasadministered alone for 3 h, and a substantial reduction with24-Epi treatment given prior to MPP+ for 3 h, suggestinga potent scavenging role for 24-Epi. As O2
•− is a highlyreactive ROS, we could barely detect its presence by MitoSOXRed at 15 or 24 h of treatment. However, at the cellular level,since antioxidant enzymes are the primary defense mecha-nisms of protection against ROS damage, SOD, CAT, andGPx are pivotal in preventing cellular injury and apoptosis.In our study, augmented SOD activity demonstrated that24-Epi may enhance the ability to eliminate ROS duringvarious oxidative stresses and may indicate a protectiverole in pretreatment experiments. Besides, several othernatural and synthetic molecules are reported to heightenSOD activity in various cellular systems [13, 77–79]. MPP+
augmented SOD activity in our experiments, as describedin recent literature in vitro [13] and in vivo, where MPTPincreased SOD activity by generating superoxide ions [14].This apparent contrasting result should be analyzed bycomparing it with those obtained by fluorescent rhodamineand MitoSOX Red. Indeed, low ROS levels, illustrated bylow rhodamine and low MitoSOX Red fluorescence (24-Epi and 24-Epi + MPP+, Figures 3 and 4), sustain theability of 24-Epi to induce SOD activity, as demonstratedby our data. When MPP+ was administered, rhodamineand MitoSOX Red fluorescence indicated high ROS levelsand, consequently, the cells responded by augmenting SODactivity, as already reported [13, 14]. Our findings clearlyshow that SOD activity may be induced by 2 different mech-anisms, a protective mechanism (24-Epi) and a response-to-stress mechanism (MPP+). More importantly, pretreatmentwith 24-Epi prior to MPP+ administration indicates lowROS levels, as revealed by rhodamine and MitoSOX Redfluorescence, suggesting that the relatively low SOD activityinduced by 24-Epi pretreatment may have already scavengedMPP+-generated ROS before 24 h.
CAT activity is another parameter of oxidative stress.Our results point out that at 15 h and 24 h, 24-Epipretreatment reduces the MPP+-induced increase in CATactivity, confirming a scavenging role for 24-Epi in thepretreatment experimental condition, as already reported foranother natural antioxidant molecule, sesamin [13]. GPx

10 Journal of Toxicology
is a selenium-dependent enzyme involved in antioxidantdefense and intracellular redox regulation and modulation.Cardiovascular and neuroprotective effects of the traceelement selenium have been observed, although long-termsupplementation has a “ying-yang” effect [80]. The glu-tathione response after MPP+ treatments has already beendescribed in a DAergic cell line and is in accordance withour results, demonstrating an increase in GPx activity at24 h after toxin administration [81]. Our data and otherfindings suggest a change in glutathione regulatory enzymeactivities during the kinetics of MPP+ administration [81].More interestingly, our data show a significant increase inGPx activity after 24-Epi pretreatment, indicating that thismolecule may augment stock of the antioxidant enzymeabove control levels.
In addition, our results demonstrate a clear neuropro-tective and antiapoptotic role of 24-Epi against cellulardeath induced by MPP+ administration. We also documentthat 24-Epi is a potent modulator of apoptosis, opposingMPP+-induced DNA fragmentation and decreasing MPP+-evoked apoptotic/antiapoptotic protein expression, namely,the Bax/Bcl-2 ratio. Altogether, our data establish thatoxidative stress-induced apoptosis in DAergic cells can bereversed by preadministration of 24-Epi. Thus, 24-Epi maybe accounted for by another natural molecule interactingwith intracellular apoptotic pathways [51, 82]. Recent studieshave reported the anticancer and antiproliferative activitiesof 2 BRs, 28-homocastasterone and 24-Epi [35], supportingtheir cytotoxic and apoptotic role. This is not the firsttime that natural neuroprotective and antioxidant moleculesappear to act as “double agents” on apoptotic parameters,depending on the cell lines studied and the concentrationstested. First, it should be noted that to demonstrate theanticancer and antiproliferative activities of BRs, theseauthors used micromolar concentrations, and toxicity wasapparent at 10 μM and higher dose levels [35]. In ourstudy, we tested nanomolar concentrations of 24-Epi sincemicromolar levels would likely be difficult to sustain invivo in the human brain. On the other hand, neuronalPC12 cells are differentiated cells expressing a neuronalphenotype as well as DAT, neurofilament proteins, and ERαand ERβ, in contrast to native mitotic PC12 cells, where 17-β estradiol or several polyphenols do not counteract MPP+-induced cellular death [50, 55]. It is certainly importantin future work to study in vivo models of PD to betterunderstand the role of plant steroids in mammalian neuronalsystems.
Finally, this is the first investigation to highlight 24-Epi’s powerful function in parameters of neuronal celldistress, apoptosis, and cellular death. Other studies shouldbe performed to elucidate the possible modulation of 24-Epion the intrinsic parameters of DAergic neurotransmission.In addition, we cannot completely exclude a role of 24-Epion MPP+ uptake, in particular via the modulation of DATand VMAT expression, that could mimic downstream events.Though, altogether these results can open the way to furtherdocument, in an animal model of PD, the importance of BRsas natural molecules in preventive or/and complementarystrategies to control neurodegeneration.
Acknowledgments
This paper was supported by a grant from Le FondsQuebecois de la Recherche sur la Nature et les Technologies(FQRNT), Quebec, Canada. The editorial assistance of OvidDa Silva is acknowledged.
References
[1] W. Dauer and S. Przedborski, “Parkinson’ s disease: mecha-nisms and models,” Neuron, vol. 39, no. 6, pp. 889–909, 2003.
[2] C. W. Olanow, “The pathogenesis of cell death in Parkinson’sdisease–2007,” Movement Disorders, vol. 22, 17, pp. S335–S342, 2007.
[3] C. Zhou, Y. Huang, and S. Przedborski, “Oxidative stressin parkinson’ s disease: a mechanism of pathogenic andtherapeutic significance,” Annals of the New York Academy ofSciences, vol. 1147, pp. 93–104, 2008.
[4] P. M. Keeney, J. Xie, R. A. Capaldi et al., “Parkinson’ s diseasebrain mitochondrial complex i has oxidatively damagedsubunits and is functionally impaired and misassembled,” TheJournal of Neuroscience, vol. 26, no. 19, pp. 5256–5264, 2006.
[5] A. Navarro, A. Boveris, M. J. Bandez et al., “Human braincortex: mitochondrial oxidative damage and adaptive responsein parkinson disease and in dementia with Lewy bodies,” FreeRadical Biology and Medicine, vol. 46, no. 12, pp. 1574–1580,2009.
[6] F. Calon, N. Lavertu, A. M. Lemieux et al., “Effect ofMPTP-induced denervation on basal ganglia GABA receptors:correlation with dopamine concentrations and dopaminetransporter,” Synapse, vol. 40, no. 3, pp. 225–234, 2001.
[7] M. W. Jakowec, K. Nixon, E. Hogg et al., “Tyrosine hydroxylaseand dopamine transporter expression following 1-Methyl–4-Phenyl-1,2,3,6-tetrahydropyridine-induced neurodegenera-tion of the mouse nigrostriatal pathway,” Journal of Neuro-science Research, vol. 76, no. 4, pp. 539–550, 2004.
[8] J. Blanchet, F. Longpre, G. Bureau et al., “Resveratrol, a redwine polyphenol, protects dopaminergic neurons in MPTP-treated mice,” Progress in Neuro-Psychopharmacology andBiological Psychiatry, vol. 32, no. 5, pp. 1243–1250, 2008.
[9] Y. Mizuno, N. Sone, and T. Saitoh, “Effects of 1-methyl–4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl–4-phenylpyridinium ion on activities of the enzymes inthe electron transport system in mouse brain,” Journal ofNeurochemistry, vol. 48, no. 6, pp. 1787–1793, 1987.
[10] A. Schober, “Classic toxin-induced animal models of Parkin-son’s disease: 6-OHDA and MPTP,” Cell and Tissue Research,vol. 318, no. 1, pp. 215–224, 2004.
[11] S. Przedborski, K. Tieu, C. Perier, and M. Vila, “MPTP asa mitochondrial neurotoxic model of parkinson’s disease,”Journal of Bioenergetics and Biomembranes, vol. 36, no. 4, pp.375–379, 2004.
[12] D. S. Cassarino, C. P. Fall, R. H. Swerdlow et al., “Elevatedreactive oxygen species and antioxidant enzyme activities inanimal and cellular models of Parkinson’ s disease,” BiochimicaEt Biophysica Acta, vol. 1362, no. 1, pp. 77–86, 1997.
[13] V. Lahaie-Collins, J. Bournival, M. Plouffe, J. Carange, andM. G. Martinoli, “Sesamin modulates tyrosine hydroxylase,superoxide dismutase, catalase, inducible NO synthase andinterleukin-6 expression in dopaminergic cells under MPP-induced oxidative stress,” Oxidative Medicine and CellularLongevity, vol. 1, no. 1, pp. 54–62, 2008.

Journal of Toxicology 11
[14] B. Thomas, K. S. Saravanan, and K. P. Mohanakumar, “In vitroand in vivo evidences that antioxidant action contributes tothe neuroprotective effects of the neuronal nitric oxide syn-thase and monoamine oxidase-B inhibitor, 7-nitroindazole,”Neurochemistry International, vol. 52, no. 6, pp. 990–1001,2008.
[15] J. Ara, S. Przedborski, A. B. Naini et al., “Inactivation oftyrosine hydroxylase by nitration following exposure to per-oxynitrite and 1-methyl–4-phenyl-1,2,3,6-tetrahydropyridine(MPTP),” Proceedings of the National Academy of Sciences ofthe United States of America, vol. 95, no. 13, pp. 7659–7663,1998.
[16] J. D. Adams Jr., L. K. Klaidman, and A. C. Leung, “MPP+andMPDP+induced oxygen radical formation with mitochondrialenzymes,” Free Radical Biology and Medicine, vol. 15, no. 2, pp.181–186, 1993.
[17] T. Shang, S. Kotamraju, S. V. Kalivendi, C. J. Hillard, andB. Kalyanaraman, “1-Methyl–4-phenylpyridinium-inducedapoptosis in cerebellar granule neurons is mediated bytransferrin receptor iron-dependent depletion of tetrahydro-biopterin and neuronal nitric-oxide synthase-derived super-oxide,” Journal of Biological Chemistry, vol. 279, no. 18, pp.19099–19112, 2004.
[18] A. Hartley, J. M. Stone, C. Heron, J. M. Cooper, and A.H. Schapira, “Complex i inhibitors induce dose-dependentapoptosis in PC12 cells: relevance to parkinson’ s disease,”Journal of Neurochemistry, vol. 63, no. 5, pp. 1987–1990, 1994.
[19] K. H. Park, T. Yokota, A. Sakurai, and N. Takahashi,“Occurence of castasterone, brassinolide and methyl 4-chloroindole-3-acetate in immature Vicia faba seeds,” Agricul-tural and Biological Chemistry, vol. 51, no. 11, pp. 3081–3086,1987.
[20] N. Ikekawa, F. Nishiyama, and Y. Fujimoto, “Identification of24-epibrassinolide in bee pollen of the broad bean, Vicia fabaL.,” Chemical and Pharmaceutical Pulletin, vol. 36, no. 1, pp.405–407, 1988.
[21] K. Gamoh, K. Omote, N. Okamoto, and S. Takatsuto, “High-performance liquid chromatography of brassinosteroids inplants with derivatization using 9-phenanthreneboronic acid,”Journal of Chromatography, vol. 469, pp. 424–428, 1989.
[22] V. A. Khripach, V. N. Zhabinskii, and A. E. de Groot, inBrassinosteroids: A New Class of Plant Hormones, AcademicPress, San Diego, Calif, USA, 1999.
[23] V. Khripach, V. Zhabinsk, and A. De Groot, “Twenty yearsof brassinosteroids: steroidal plant hormones warrant bettercrops for the XXI century,” Annals of Botany, vol. 86, no. 3, pp.441–447, 2000.
[24] L. M. Mazorra, M. Nunez, M. Hechavarria, F. Coll, and M. J.Sanchez-Blanco, “Influence of brassinosteroids on antioxidantenzymes activity in tomato under different temperatures,”Biologia Plantarum, vol. 45, no. 4, pp. 593–596, 2002.
[25] B. Ali, S. Hayat, Q. Fariduddin, and A. Ahmad, “24-Epibrassinolide protects against the stress generated by salinityand nickel in Brassica juncea,” Chemosphere, vol. 72, no. 9, pp.1387–1392, 2008.
[26] F. Ozdemir, M. Bor, T. Demiral et al., “Effects of 24-epibrassinolide on seed germination, seedling growth, lipidperoxidation, proline content and antioxidative system ofrice(Oryza sativa L.)under salinity stress,” Plant GrowthRegulation, vol. 42, no. 3, pp. 203–211, 2004.
[27] V. Calabrese, D. A. Butterfield, and A. M. Stella, “Nutritionalantioxidants and the heme oxygenase pathway of stresstolerance: novel targets for neuroprotection in Alzheimer’ sdisease,” The Italian Journal of Biochemistry, vol. 52, no. 4, pp.177–181, 2003.
[28] T. S. Anekonda, “Resveratrol-a boon for treating Alzheimer’ sdisease?” Brain Research Reviews, vol. 52, no. 2, pp. 316–326,2006.
[29] J. A. Baur and D. A. Sinclair, “Therapeutic potential of resvera-trol: the in vivo evidence,” Nature Reviews Drug Discovery, vol.5, no. 6, pp. 493–506, 2006.
[30] M. Singh, M. Arseneault, T. Sanderson et al., “Challenges forresearch on polyphenols from foods in Alzheimer’ s disease:bioavailability, metabolism, and cellular and molecular mech-anisms,” Journal of Agricultural and Food Chemistry, vol. 56,no. 13, pp. 4855–4873, 2008.
[31] F. J. Alcain and J. M. Villalba, “Sirtuin activators,” ExpertOpinion on Therapeutic Patents, vol. 19, no. 4, pp. 403–414,2009.
[32] K. B. Pandey and S. I. Rizvi, “Plant polyphenols as dietaryantioxidants in human health and disease,” Oxidative Medicineand Cellular Longevity, vol. 2, no. 5, pp. 270–278, 2009.
[33] J. A. Baur, “Resveratrol, sirtuins, and the promise of a DRmimetic,” Mechanisms of Ageing and Development, vol. 131,no. 4, pp. 261–269, 2010.
[34] M. Iriti, S. Vitalini, G. Fico, and F. Faoro, “Neuroprotectiveherbs and foods from different traditional medicines anddiets,” Molecules, vol. 15, no. 5, pp. 3517–3555, 2010.
[35] J. Malikova, J. Swaczynova, Z. Kolar, and M. Strnad, “Anti-cancer and antiproliferative activity of natural brassinos-teroids,” Phytochemistry, vol. 69, no. 2, pp. 418–426, 2008.
[36] Y. D. Wu and Y. J. Lou, “Brassinolide, a plant sterol from pollenof Brassica napus L., induces apoptosis in human prostatecancer PC-3 cells,” Pharmazie, vol. 62, no. 5, pp. 392–395,2007.
[37] B. Slavikova, L. Kohout, M. Budesinsky, J. Swaczynova, and A.Kasal, “Brassinosteroids: synthesis and activity of some fluoroanalogues,” Journal of Medicinal Chemistry, vol. 51, no. 13, pp.3979–3984, 2008.
[38] A. H. Hamdy, E. A. Aboutabl, S. Sameer et al., “3-Keto-22-epi-28-nor-cathasterone, a brassinosteroid-related metabolitefrom Cystoseira myrica,” Steroids, vol. 74, no. 12, pp. 927–930,2009.
[39] J. A. Ramirez, O. M. Teme Centurin, E. G. Gros, andL. R. Galagovsky, “Synthesis and bioactivity evaluation ofbrassinosteroid analogs,” Steroids, vol. 65, no. 6, pp. 329–337,2000.
[40] F. M. Michelini, J. A. Ramirez, A. Berra, L. R. Galagovsky, andL. E. Alche, “In vitro and in vivo antiherpetic activity of threenew synthetic brassinosteroid analogues,” Steroids, vol. 69, no.11-12, pp. 713–720, 2004.
[41] M. B. Wachsman, J. A. Ramirez, L. B. Talarico, L. R.Galagovsky, and C. E. Coto, “Antiviral activity of natural andsynthetic brassinosteroids,” Current Medicinal Chemistry, vol.3, pp. 163–179, 2004.
[42] E. Farina, P. Piu, and L. Strinna, “Extraction of L-DOPA fromvicia faba L. and other plants of the leguminous genera,”Bollettino della Societa italiana di biologia sperimentale, vol. 50,no. 8, pp. 508–511, 1974.
[43] J. M. Rabey, Y. Vered, H. Shabtai, E. Graff, A. Harsat, andA. D. Korczyn, “Broad bean(Vicia faba)consumption andParkinson’s disease,” Advances in Neurology, vol. 60, pp. 681–684, 1993.
[44] P. A. LeWitt, “Levodopa therapeutics for Parkinson’s disease:new developments,” Parkinsonism and Related Disorders, vol.15, supplement 1, pp. S31–S34, 2009.
[45] H. Apaydin, S. Ertan, and S. Ozekmekci, “Broad bean(Viciafaba)- a natural source of L-dopa-prolongs “on” periodsin patients with Parkinson’ s disease who have “on-off”

12 Journal of Toxicology
fluctuations,” Movement Disorders, vol. 15, no. 1, pp. 164–166,2000.
[46] L. Raguthu, S. Varanese, L. Flancbaum, E. Tayler, and A. DiRocco, “Fava beans and Parkinson’s disease: useful ’naturalsupplement’ or useless risk?” European Journal of Neurology,vol. 16, no. 10, p. e171, 2009.
[47] L. A. Greene and A. S. Tischler, “Establishment of a noradren-ergic clonal line of rat adrenal pheochromocytoma cells whichrespond to nerve growth factor,” Proceedings of the NationalAcademy of Sciences of the United States of America, vol. 73, no.7, pp. 2424–2428, 1976.
[48] E. J. Ryu, J. M. Angelastro, and L. A. Greene, “Analysis of geneexpression changes in a cellular model of Parkinson disease,”Neurobiology of Disease, vol. 18, no. 1, pp. 54–74, 2005.
[49] S. Gelinas and M. G. Martinoli, “Neuroprotective effect ofestradiol and phytoestrogens on MPP+-induced cytotoxicityin neuronal PC12 cells,” Journal of Neuroscience Research, vol.70, no. 1, pp. 90–96, 2002.
[50] B. Gagne, S. Gelinas, G. Bureau et al., “Effects of estradiol,phytoestrogens, and Ginkgo biloba extracts against 1-methyl-4-phenyl-pyridine-induced oxidative stress,” Endocrine, vol.21, no. 1, pp. 89–95, 2003.
[51] J. Bournival, P. Quessy, and M. G. Martinoli, “Protective effectsof resveratrol and quercetin against MPP+-induced oxidativestress act by modulating markers of apoptotic death indopaminergic neurons,” Cellular and Molecular Neurobiology,vol. 29, no. 8, pp. 1169–1180, 2009.
[52] C. Korzeniewski and D. M. Callewaert, “An enzyme-releaseassay for natural cytotoxicity,” Journal of Immunological Meth-ods, vol. 64, no. 3, pp. 313–320, 1983.
[53] A. Martin and M. Clynes, “Acid phosphatase: endpoint forin vitro toxicity tests,” In Vitro Cellular and DevelopmentalBiology, vol. 27A, no. 3, part 1, pp. 183–184, 1991.
[54] T. Decker and M. L. Lohmann-Matthes, “A quick and simplemethod for the quantitation of lactate dehydrogenase releasein measurements of cellular cytotoxicity and tumor necrosisfactor(TNF)activity,” Journal of Immunological Methods, vol.115, no. 1, pp. 61–69, 1988.
[55] S. Gelinas, G. Bureau, B. Valastro et al., “Alpha and betaestradiol protect neuronal but not native PC12 cells fromparaquat-induced oxidative stress,” Neurotoxicity Research,vol. 6, no. 2, pp. 141–148, 2004.
[56] L. M. Henderson and J. B. Chappell, “Dihydrorhodamine123: a fluorescent probe for superoxide generation?” EuropeanJournal of Biochemistry, vol. 217, no. 3, pp. 973–980, 1993.
[57] N. W. Kooy, J. A. Royall, H. Ischiropoulos et al., “Peroxynitrite-mediated oxidation of dihydrorhodamine 123,” Free RadicalBiology and Medicine, vol. 16, no. 2, pp. 149–156, 1994.
[58] P. Wardman, “Fluorescent and luminescent probes for mea-surement of oxidative and nitrosative species in cells andtissues: progress, pitfalls, and prospects,” Free Radical Biologyand Medicine, vol. 43, no. 7, pp. 995–1022, 2007.
[59] M. Wrona, K. Patel, and P. Wardman, “Reactivity of 2’ , 7’-dichlorodihydrofluorescein and dihydrorhodamine 123 andtheir oxidized forms toward carbonate, nitrogen dioxide, andhydroxyl radicals,” Free Radical Biology and Medicine, vol. 38,no. 2, pp. 262–270, 2005.
[60] J. M. Mates and F. Sanchez-Jimenez, “Antioxidant enzymesand their implications in pathophysiologic processes,” Fron-tiers in Bioscience, vol. 4, pp. D339–D345, 1999.
[61] J. A. Klein and S. L. Ackerman, “Oxidative stress, cell cycle, andneurodegeneration,” Journal of Clinical Investigation, vol. 111,no. 6, pp. 785–793, 2003.
[62] I. Chowdhury, B. Tharakan, and G. K. Bhat, “Caspases-anupdate,” Comparative Biochemistry and Physiology B, vol. 151,no. 1, pp. 10–27, 2008.
[63] B. Mignotte and J. L. Vayssiere, “Mitochondria and apoptosis,”European Journal of Biochemistry, vol. 252, no. 1, pp. 1–15,1998.
[64] A. Hartmann, S. Hunot, P. P. Michel et al., “Caspase-3: avulnerability factor and final effector in apoptotic death ofdopaminergic neurons in Parkinson’ s disease,” Proceedingsof the National Academy of Sciences of the United States ofAmerica, vol. 97, no. 6, pp. 2875–2880, 2000.
[65] Y. Wu, Y. Shang, S. G. Sun et al., “Protective effect of ery-thropoietin against 1-methyl-4-phenylpyridinium- inducedneurodegenaration in PC12 cells,” Neuroscience Bulletin, vol.23, no. 3, pp. 156–164, 2007.
[66] K. Chiasson, V. Lahaie-Collins, J. Bournival, B. Delapierre, S.Gelinas, and M.-G. Martinoli, “Oxidative stress and 17-α- and17-β-estradiol modulate neurofilaments differently,” Journal ofMolecular Neuroscience, vol. 30, no. 3, pp. 297–310, 2006.
[67] T. Kadota, T. Yamaai, Y. Saito et al., “Expression of dopaminetransporter at the tips of growing neurites of PC12 cells,”Journal of Histochemistry and Cytochemistry, vol. 44, no. 9, pp.989–996, 1996.
[68] J. Nilsen, G. Mor, and F. Naftolin, “Raloxifene inducesneurite outgrowth in estrogen receptor positive PC12 cells,”Menopause, vol. 5, no. 4, pp. 211–216, 1998.
[69] Y. Yasukazu and N. Etsuo, “Antioxidant effects of phytosteroland its components,” Journal of Nutritional Science andVitaminology, vol. 49, no. 4, pp. 277–280, 2003.
[70] H. B. Zhao, S. Z. Wang, QI. H. He et al., “Ganoderma totalsterol(GS)and GS1 protect rat cerebral cortical neurons fromhypoxia/reoxygenation injury,” Life Sciences, vol. 76, no. 9, pp.1027–1037, 2005.
[71] X. C. Chen, Y. C. Zhou, Y. Chen, Y.-G. Zhu, F. Fang,and L.-M. Chen, “Ginsenoside Rg1 reduces MPTP-inducedsubstantia nigra neuron loss by suppressing oxidative stress,”Acta Pharmacologica Sinica, vol. 26, no. 1, pp. 56–62, 2005.
[72] M. Vivancos and J. J. Moreno, “β-sitosterol modulates antiox-idant enzyme response in RAW 264.7 macrophages,” FreeRadical Biology and Medicine, vol. 39, no. 1, pp. 91–97, 2005.
[73] X. C. Chen, Y. G. Zhu, L. A. Zhu et al., “Ginsenoside Rg1attenuates dopamine-induced apoptosis in PC12 cells by sup-pressing oxidative stress,” European Journal of Pharmacology,vol. 473, no. 1, pp. 1–7, 2003.
[74] X. C. Chen, Y. G. Zhu, X. Z. Wang, L.-A. Zhu, and C. Huang,“Protective effect of ginsenoside Rg1 on dopamine-inducedapoptosis in PC12 cells,” Acta Pharmacologica Sinica, vol. 22,no. 8, pp. 673–678, 2001.
[75] F. Rosati, G. Danza, A. Guarna et al., “New evidence of simi-larity between human and plant steroid metabolism: 5alpha-reductase activity in Solanum malacoxylon,” Endocrinology,vol. 144, no. 1, pp. 220–229, 2003.
[76] C. B. Fricke, M. Schroder, M. Poulsen et al., “Increased plantsterol and stanol levels in brain of Watanabe rabbits fedrapeseed oil derived plant sterol or stanol esters,” The BritishJournal of Nutrition, vol. 98, no. 5, pp. 890–899, 2007.
[77] L. J. An, S. Guan, G. F. Shi et al., “Protocatechuic acid fromAlpinia oxyphylla against MPP+-induced neurotoxicity inPC12 cells,” Food and Chemical Toxicology, vol. 44, no. 3, pp.436–443, 2006.
[78] R. C. Hou, C. C. Wu, C. H. Yang, and K.-C. G. Jeng, “Protectiveeffects of sesamin and sesamolin on murine BV-2 microgliacell line under hypoxia,” Neuroscience Letters, vol. 367, no. 1,pp. 10–13, 2004.

Journal of Toxicology 13
[79] T. W. Jung, J. Y. Lee, W. S. Shim et al., “Rosiglitazone protectshuman neuroblastoma SH-SY5Y cells against acetaldehyde-induced cytotoxicity,” Biochemical and Biophysical ResearchCommunications, vol. 340, no. 1, pp. 221–227, 2006.
[80] H. Steinbrenner and H. Sies, “Protection against reactiveoxygen species by selenoproteins,” Biochimica et BiophysicaActa, vol. 1790, no. 11, pp. 1478–1485, 2009.
[81] D. A. Drechsel, L. P. Liang, and M. Patel, “1-Methyl–4-phenylpyridinium-induced alterations of glutathione statusin immortalized rat dopaminergic neurons,” Toxicology andApplied Pharmacology, vol. 220, no. 3, pp. 341–348, 2007.
[82] G. Bureau, F. Longpre, and M. G. Martinoli, “Resveratroland quercetin, two natural polyphenols, reduce apoptoticneuronal cell death induced by neuroinflammation,” Journalof Neuroscience Research, vol. 86, no. 2, pp. 403–410, 2008.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 645361, 15 pagesdoi:10.1155/2011/645361
Review Article
Evidence for a Role of Oxidative Stress inthe Carcinogenicity of Ochratoxin A
M. Marin-Kuan, V. Ehrlich, T. Delatour, C. Cavin, and B. Schilter
Chemical Food Safety Group, Quality & Safety Department, Nestle Research Center, P.O. Box 44, Vers-chez-les-Blanc,1000 Lausanne 26, Switzerland
Correspondence should be addressed to M. Marin-Kuan, [email protected]
Received 30 September 2010; Accepted 20 April 2011
Academic Editor: Brad Upham
Copyright © 2011 M. Marin-Kuan et al. This is an open access article distributed under the Creative Commons AttributionLicense, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properlycited.
The in vitro and in vivo evidence compatible with a role for oxidative stress in OTA carcinogenicity has been collected and described.Several potential oxido-reduction mechanisms have been identified in the past. More recently, the possibility of a reduction ofcellular antioxidant defense has been raised as an indirect source of oxidative stress. Consequences resulting from the productionof oxidative stress are observed at different levels. First, OTA exposure has been associated with increased levels of oxidative DNA,lipid, and protein damage. Second, various biological processes known to be mobilized under oxidative stress were shown tobe altered by OTA. These effects have been observed in both in vitro and in vivo test systems. In vivo, active doses were oftenwithin doses documented to induce renal tumors in rats. In conclusion, the evidence for the induction of an oxidative stressresponse resulting from OTA exposure can be considered strong. Because the contribution of the oxidative stress response in thedevelopment of cancers is well established, a role in OTA carcinogenicity is plausible. Altogether, the data reviewed above supportthe application of a threshold-based approach to establish safe level of dietary human exposure to OTA.
1. Introduction
Ochratoxin A (OTA) is a mycotoxin produced by severalfood-borne species of Aspergillus and Penicillium fungi. OTAoccurs in various food materials and therefore humansare continuously exposed to relatively small amounts of it.Because of its wide occurrence and consequent exposure,together with a potent carcinogenic potential in animalmodels, OTA has attracted significant public health attentionover the last few years. Several national and internationalfood safety organizations and expert groups have conducteda thorough review of the situation as well as risk assessmentsin order to provide an insight on the health significance ofOTA in food.
Human epidemiology has been inconclusive: a numberof studies have suggested a correlation between exposure toOTA and Balkan Endemic Nephropathy (BEN) and mortalityfrom urinary tract tumors [1, 2]. Epidemiological datawere recently reviewed by several expert groups [3–6].All concluded that causality between intake of OTA and
human nephropathy could not be established. Therefore,the IARC statement that there is inadequate evidence forcarcinogenicity in humans (group 2B) [7] appears still valid.Recently, other nephrotoxic agents have been put forward asthe primary cause of BEN [6, 8, 9].
In absence of adequate human data, risk assessmentshave relied on animal data. Kidney has been considered as thekey target organ of OTA toxicity. In all animal species studied,OTA was found to produce renal toxicity, while in rodentsrenal carcinogenicity was clearly established. Recently, OTArenal and hepatic carcinogenicity was also observed in chicks[10]. Using a LOAEL of 8 mcg/kg bw/day based on earlymarkers of renal toxicity in pig (the most sensitive animalspecies) and applying an uncertainty factor of 450, EFSA [4]allocated a Tolerable Weekly Intake (TWI) of 120 ng/kg bw.Analysis of dietary exposure throughout Europe revealed thatthe current average OTA exposure (50–60 ng/kg bw/week)is well within the TWI [4]. The joint FAO/WHO ExpertCommittee on Food Additives (JECFA) first evaluated OTAat their 37th meeting [11]. Based on the LOAEL in pig, and

2 Journal of Toxicology
applying an uncertainty factor of 500, JECFA allocated aProvisional Tolerable Weekly Intake (PTWI) of 112 ng/kg bw[11]. This value was rounded to 100 ng/kg bw/week andconfirmed in several subsequent meetings [12, 13].
In 2008, JECFA applied a benchmark dose (BMD) mod-eling approach using carcinogenicity data [14]. The BMDis the dose estimated to cause a predefined increase (e.g.,10% for the BMD10) in tumor incidence over background.The BMDL is the lower limit of a one-sided 95% confidenceinterval of the BMD. The occurrence of combined adenomasand carcinomas in the kidneys of male rats was considered byJECFA to be the most appropriate data for modeling. Valuesof 18–33 mcg/kg bw/d and 15–25 mcg/kg bw/day were calcu-lated for, respectively, the BMD10 and BMDL10. Because theBMD approach did not provide a lower point of departurethan the LOAEL in pig, JECFA decided to reconfirm thePTWI of 100 ng/kg bw/day [14].
A recent health risk assessment, performed by HealthCanada [15] recommends to regulate OTA as a nonthresholdcarcinogen, because of the uncertainties regarding the modeof action. The authors defined a negligible cancer risk intake(NCRI, risk level 1 : 100 000) using the tumorigenic dose atwhich 5% of rats are likely to develop tumors (TD05, derivedthrough modeling) as point of departure. Importantly, thereis considerable convergence between the NCRI established inthis assessment and the TDI derived by EFSA.
In the risk assessment of carcinogenic substances, con-sideration of the mode of action (MOA) is essential, deter-mining the method to be applied in order to define levelsof exposure below which a low safety concern is expected.The key events analysis framework of the MOA has not yetbeen formally applied to OTA. However, the approach usedby most expert groups (EFSA, JECFA, ILSI) to establish thesafe level of exposure of OTA (based on uncertainty factors)implies the consideration of key events compatible with athreshold effect. For these groups, amongst the mechanismsof action highlighted as possible, oxidative stress has beenpresented as one of the most probable [5, 14].
1.1. Scope of the Paper. Over the last decades, studiesaimed at elucidating the modes of action implicated inOTA toxicity and carcinogenicity have been published [16].There has been considerable debate for many years over thegenotoxicity of OTA and its actual role in carcinogenicity [3–5, 11–15, 17, 18].
Although genotoxicity is likely to play a role in OTAcarcinogenicity [3, 4], the actual molecular mechanisminvolved, either through covalent adduct formation, throughother indirect modifications, or both is still unclear. Thepotential of OTA to form covalent DNA adducts has beensubjected to debate due to conflicting data in the literature.Using 32P-postlabelling analysis, large numbers of putativeOTA-derived DNA adducts have been reported to be presentin a wide range of tissues from OTA-treated rats, mice as wellas pigs [2, 17, 19–22]. However, so far, these adducts havenever been observed by any other highly specific techniquessuch as radioactivity measurements using 3H-labelled OTA(3H-OTA) [23], accelerator mass spectrometry (AMS) [24],
or liquid chromatography-tandem mass spectrometry (LC-MS/MS) [25].
The present paper is not intended to provide a thoroughreview of the complex and controversial scientific literatureon DNA-adduct formation. However, it is important to keepin mind that DNA adducts are increasingly considered asmarkers of exposures and not only of effects [26, 27] and thatDNA-covalent binding does not necessarily determine theshape of the dose-response at low level of exposure [28, 29].According to Mantle and coworkers [21, 30], experimentaldose-response data for OTA’s renal carcinogenesis makes acompelling case for OTA being a thresholded carcinogen inmale rat. In this context, it appears important to also considerother potential modes of action, which could potentiallycontribute to OTA carcinogenicity. In the near future, theapplication of the mode of action framework [28] willlikely help to understand the individual contribution of allmechanisms described up to now for OTA.
The focus of the present short paper was to collect andhighlight the evidence associated with a role for oxidativestress as a plausible mechanism to consider for OTA. A list ofthe studies used to illustrate the main messages of the presentpaper is provided in Table 1. Although not exhaustive, the listshows that over the last two decades, numerous investigatorshave documented the generation of oxidative stress as a resultof OTA treatment in both in vitro and in vivo model systems.
2. Sources of OTA-MediatedOxygen-Species Generation
Production of reactive oxygen species (ROS) leading tooxidative stress and macromolecular damage is known tocontribute to the pathogenesis of age-related as well aschronic diseases including cancer [31–35]. A number ofstudies are available documenting that OTA is associatedwith the production of reactive oxygen species and resultingoxidative stress through various direct and indirect mecha-nisms.
2.1. Oxido-Reduction Mechanisms. Several oxido-reductionmechanisms elicited by OTA have been proposed. In areconstituted system consisting of phospholipid vesicles, theflavoprotein NADPH-cytochrome P450 reductase and Fe3+,OTA was found to chelate ferric ions (Fe3+), facilitatingtheir reduction to ferrous ions (Fe2+), which in the presenceof oxygen, provided the active species initiating lipid per-oxidation [36]. Results indicated that the hydroxyl radicalwas not involved in the process. A role for cytochromeP450 in this reaction was also suggested [36]. In contrast,others found that OTA induced oxidative damage throughthe generation of hydroxyl radicals. This reaction conductedwith microsomes, in presence of NADPH and O2 did notrequire exogenous Fe [20]. Structure-activity studies havealso suggested that the toxicity of OTA may be attributableto its isocoumarin moiety and that the lactone carbonylgroup may be involved in its toxicity. Using a Bacillusbrevis model, Hoehler’s et al. showed that OTA behaved asa cell pro-oxidant through mobilization of Fe2+ and Ca2+
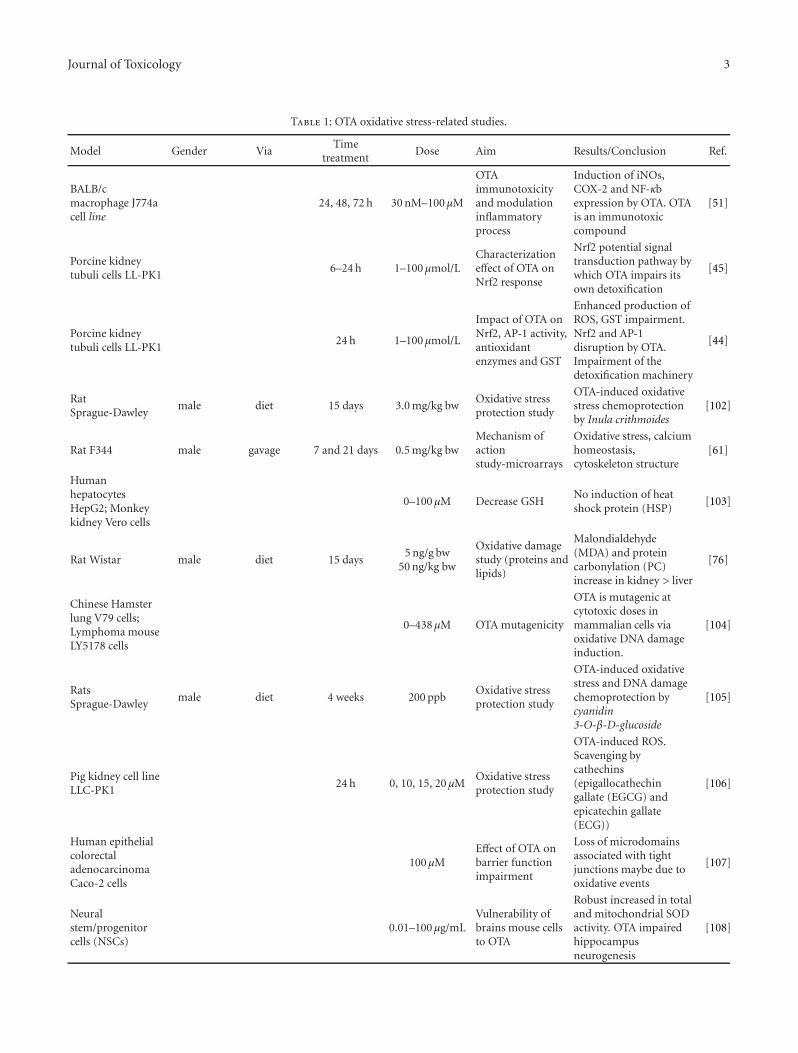
Journal of Toxicology 3
Table 1: OTA oxidative stress-related studies.
Model Gender ViaTime
treatmentDose Aim Results/Conclusion Ref.
BALB/cmacrophage J774acell line
24, 48, 72 h 30 nM–100 μM
OTAimmunotoxicityand modulationinflammatoryprocess
Induction of iNOs,COX-2 and NF-κbexpression by OTA. OTAis an immunotoxiccompound
[51]
Porcine kidneytubuli cells LL-PK1
6–24 h 1–100 μmol/LCharacterizationeffect of OTA onNrf2 response
Nrf2 potential signaltransduction pathway bywhich OTA impairs itsown detoxification
[45]
Porcine kidneytubuli cells LL-PK1
24 h 1–100 μmol/L
Impact of OTA onNrf2, AP-1 activity,antioxidantenzymes and GST
Enhanced production ofROS, GST impairment.Nrf2 and AP-1disruption by OTA.Impairment of thedetoxification machinery
[44]
RatSprague-Dawley
male diet 15 days 3.0 mg/kg bwOxidative stressprotection study
OTA-induced oxidativestress chemoprotectionby Inula crithmoides
[102]
Rat F344 male gavage 7 and 21 days 0.5 mg/kg bwMechanism ofactionstudy-microarrays
Oxidative stress, calciumhomeostasis,cytoskeleton structure
[61]
HumanhepatocytesHepG2; Monkeykidney Vero cells
0–100 μM Decrease GSHNo induction of heatshock protein (HSP)
[103]
Rat Wistar male diet 15 days5 ng/g bw
50 ng/kg bw
Oxidative damagestudy (proteins andlipids)
Malondialdehyde(MDA) and proteincarbonylation (PC)increase in kidney > liver
[76]
Chinese Hamsterlung V79 cells;Lymphoma mouseLY5178 cells
0–438 μM OTA mutagenicity
OTA is mutagenic atcytotoxic doses inmammalian cells viaoxidative DNA damageinduction.
[104]
RatsSprague-Dawley
male diet 4 weeks 200 ppbOxidative stressprotection study
OTA-induced oxidativestress and DNA damagechemoprotection bycyanidin3-O-β-D-glucoside
[105]
Pig kidney cell lineLLC-PK1
24 h 0, 10, 15, 20 μMOxidative stressprotection study
OTA-induced ROS.Scavenging bycathechins(epigallocathechingallate (EGCG) andepicatechin gallate(ECG))
[106]
Human epithelialcolorectaladenocarcinomaCaco-2 cells
100 μMEffect of OTA onbarrier functionimpairment
Loss of microdomainsassociated with tightjunctions maybe due tooxidative events
[107]
Neuralstem/progenitorcells (NSCs)
0.01–100 μg/mLVulnerability ofbrains mouse cellsto OTA
Robust increased in totaland mitochondrial SODactivity. OTA impairedhippocampusneurogenesis
[108]
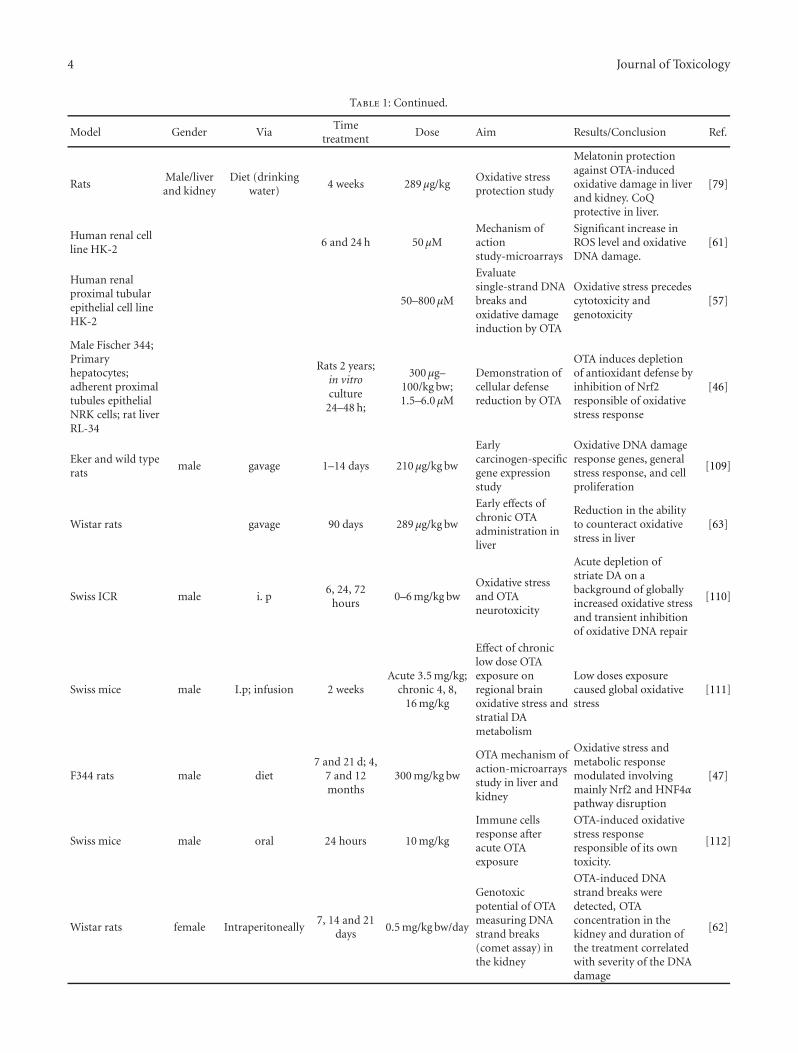
4 Journal of Toxicology
Table 1: Continued.
Model Gender ViaTime
treatmentDose Aim Results/Conclusion Ref.
RatsMale/liverand kidney
Diet (drinkingwater)
4 weeks 289 μg/kgOxidative stressprotection study
Melatonin protectionagainst OTA-inducedoxidative damage in liverand kidney. CoQprotective in liver.
[79]
Human renal cellline HK-2
6 and 24 h 50 μMMechanism ofactionstudy-microarrays
Significant increase inROS level and oxidativeDNA damage.
[61]
Human renalproximal tubularepithelial cell lineHK-2
50–800 μM
Evaluatesingle-strand DNAbreaks andoxidative damageinduction by OTA
Oxidative stress precedescytotoxicity andgenotoxicity
[57]
Male Fischer 344;Primaryhepatocytes;adherent proximaltubules epithelialNRK cells; rat liverRL-34
Rats 2 years;in vitroculture
24–48 h;
300 μg–100/kg bw;1.5–6.0 μM
Demonstration ofcellular defensereduction by OTA
OTA induces depletionof antioxidant defense byinhibition of Nrf2responsible of oxidativestress response
[46]
Eker and wild typerats
male gavage 1–14 days 210 μg/kg bw
Earlycarcinogen-specificgene expressionstudy
Oxidative DNA damageresponse genes, generalstress response, and cellproliferation
[109]
Wistar rats gavage 90 days 289 μg/kg bw
Early effects ofchronic OTAadministration inliver
Reduction in the abilityto counteract oxidativestress in liver
[63]
Swiss ICR male i. p6, 24, 72
hours0–6 mg/kg bw
Oxidative stressand OTAneurotoxicity
Acute depletion ofstriate DA on abackground of globallyincreased oxidative stressand transient inhibitionof oxidative DNA repair
[110]
Swiss mice male I.p; infusion 2 weeksAcute 3.5 mg/kg;
chronic 4, 8,16 mg/kg
Effect of chroniclow dose OTAexposure onregional brainoxidative stress andstratial DAmetabolism
Low doses exposurecaused global oxidativestress
[111]
F344 rats male diet7 and 21 d; 4,
7 and 12months
300 mg/kg bw
OTA mechanism ofaction-microarraysstudy in liver andkidney
Oxidative stress andmetabolic responsemodulated involvingmainly Nrf2 and HNF4αpathway disruption
[47]
Swiss mice male oral 24 hours 10 mg/kg
Immune cellsresponse afteracute OTAexposure
OTA-induced oxidativestress responseresponsible of its owntoxicity.
[112]
Wistar rats female Intraperitoneally7, 14 and 21
days0.5 mg/kg bw/day
Genotoxicpotential of OTAmeasuring DNAstrand breaks(comet assay) inthe kidney
OTA-induced DNAstrand breaks weredetected, OTAconcentration in thekidney and duration ofthe treatment correlatedwith severity of the DNAdamage
[62]
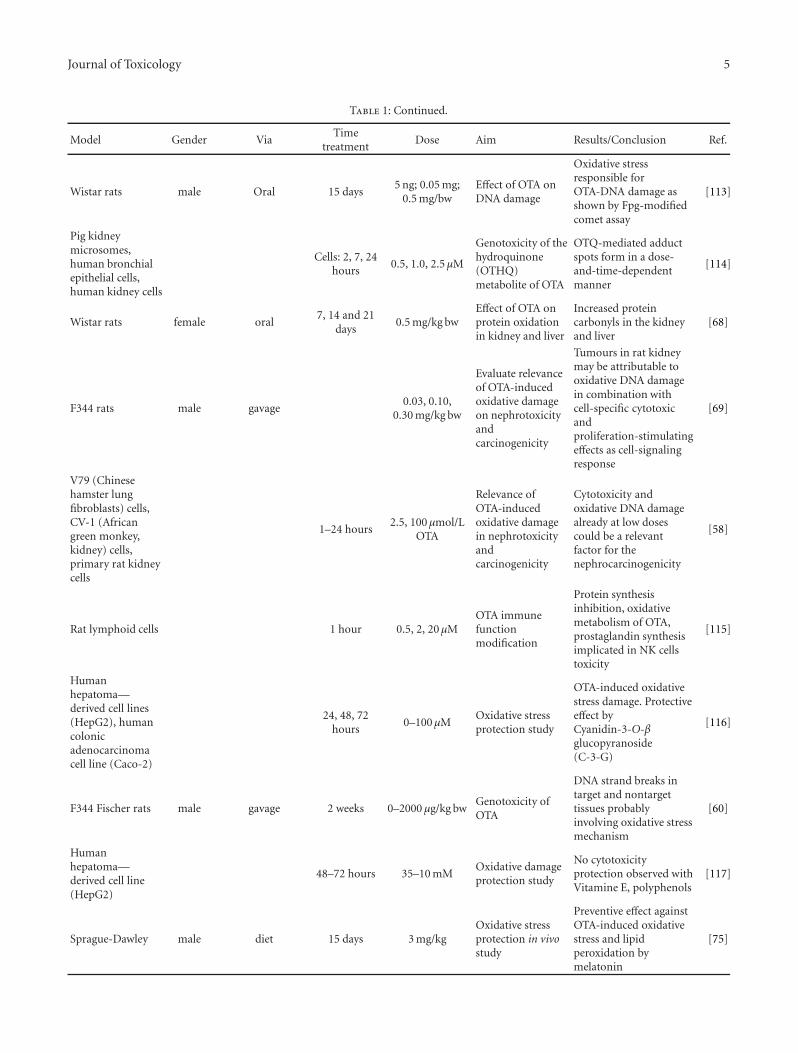
Journal of Toxicology 5
Table 1: Continued.
Model Gender ViaTime
treatmentDose Aim Results/Conclusion Ref.
Wistar rats male Oral 15 days5 ng; 0.05 mg;
0.5 mg/bwEffect of OTA onDNA damage
Oxidative stressresponsible forOTA-DNA damage asshown by Fpg-modifiedcomet assay
[113]
Pig kidneymicrosomes,human bronchialepithelial cells,human kidney cells
Cells: 2, 7, 24hours
0.5, 1.0, 2.5 μM
Genotoxicity of thehydroquinone(OTHQ)metabolite of OTA
OTQ-mediated adductspots form in a dose-and-time-dependentmanner
[114]
Wistar rats female oral7, 14 and 21
days0.5 mg/kg bw
Effect of OTA onprotein oxidationin kidney and liver
Increased proteincarbonyls in the kidneyand liver
[68]
F344 rats male gavage0.03, 0.10,
0.30 mg/kg bw
Evaluate relevanceof OTA-inducedoxidative damageon nephrotoxicityandcarcinogenicity
Tumours in rat kidneymay be attributable tooxidative DNA damagein combination withcell-specific cytotoxicandproliferation-stimulatingeffects as cell-signalingresponse
[69]
V79 (Chinesehamster lungfibroblasts) cells,CV-1 (Africangreen monkey,kidney) cells,primary rat kidneycells
1–24 hours2.5, 100 μmol/L
OTA
Relevance ofOTA-inducedoxidative damagein nephrotoxicityandcarcinogenicity
Cytotoxicity andoxidative DNA damagealready at low dosescould be a relevantfactor for thenephrocarcinogenicity
[58]
Rat lymphoid cells 1 hour 0.5, 2, 20 μMOTA immunefunctionmodification
Protein synthesisinhibition, oxidativemetabolism of OTA,prostaglandin synthesisimplicated in NK cellstoxicity
[115]
Humanhepatoma—derived cell lines(HepG2), humancolonicadenocarcinomacell line (Caco-2)
24, 48, 72hours
0–100 μMOxidative stressprotection study
OTA-induced oxidativestress damage. Protectiveeffect byCyanidin-3-O-βglucopyranoside(C-3-G)
[116]
F344 Fischer rats male gavage 2 weeks 0–2000 μg/kg bwGenotoxicity ofOTA
DNA strand breaks intarget and nontargettissues probablyinvolving oxidative stressmechanism
[60]
Humanhepatoma—derived cell line(HepG2)
48–72 hours 35–10 mMOxidative damageprotection study
No cytotoxicityprotection observed withVitamine E, polyphenols
[117]
Sprague-Dawley male diet 15 days 3 mg/kgOxidative stressprotection in vivostudy
Preventive effect againstOTA-induced oxidativestress and lipidperoxidation bymelatonin
[75]
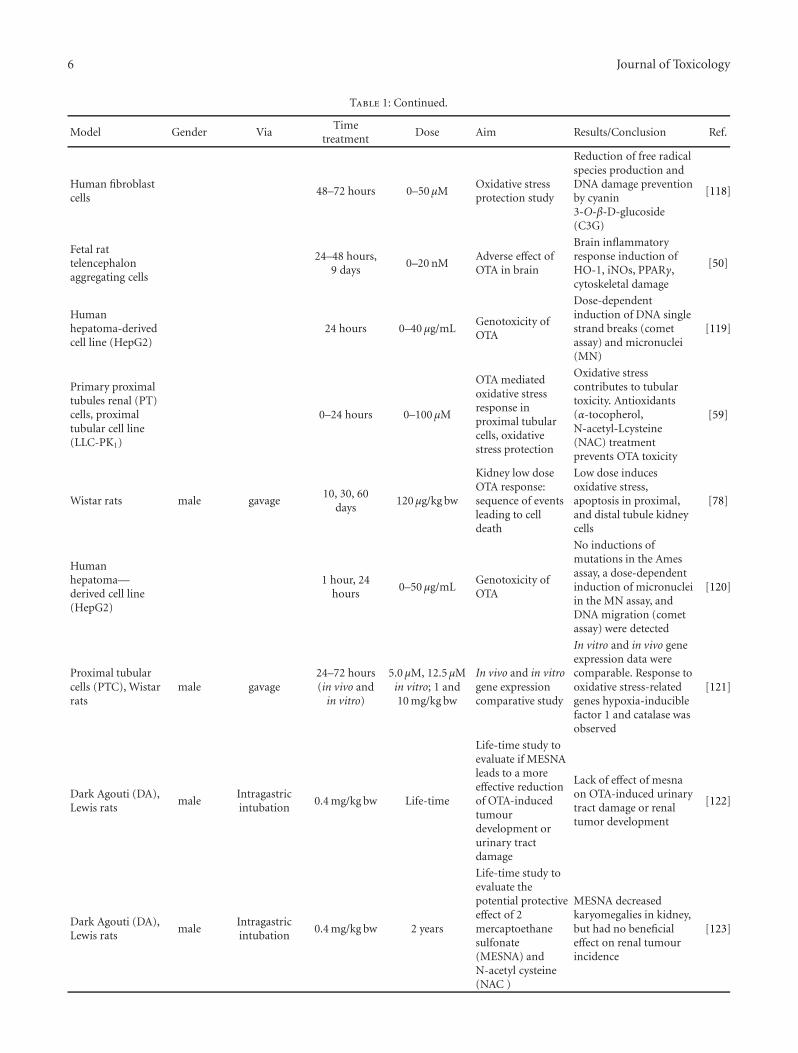
6 Journal of Toxicology
Table 1: Continued.
Model Gender ViaTime
treatmentDose Aim Results/Conclusion Ref.
Human fibroblastcells
48–72 hours 0–50 μMOxidative stressprotection study
Reduction of free radicalspecies production andDNA damage preventionby cyanin3-O-β-D-glucoside(C3G)
[118]
Fetal rattelencephalonaggregating cells
24–48 hours,9 days
0–20 nMAdverse effect ofOTA in brain
Brain inflammatoryresponse induction ofHO-1, iNOs, PPARγ,cytoskeletal damage
[50]
Humanhepatoma-derivedcell line (HepG2)
24 hours 0–40 μg/mLGenotoxicity ofOTA
Dose-dependentinduction of DNA singlestrand breaks (cometassay) and micronuclei(MN)
[119]
Primary proximaltubules renal (PT)cells, proximaltubular cell line(LLC-PK1)
0–24 hours 0–100 μM
OTA mediatedoxidative stressresponse inproximal tubularcells, oxidativestress protection
Oxidative stresscontributes to tubulartoxicity. Antioxidants(α-tocopherol,N-acetyl-Lcysteine(NAC) treatmentprevents OTA toxicity
[59]
Wistar rats male gavage10, 30, 60
days120 μg/kg bw
Kidney low doseOTA response:sequence of eventsleading to celldeath
Low dose inducesoxidative stress,apoptosis in proximal,and distal tubule kidneycells
[78]
Humanhepatoma—derived cell line(HepG2)
1 hour, 24hours
0–50 μg/mLGenotoxicity ofOTA
No inductions ofmutations in the Amesassay, a dose-dependentinduction of micronucleiin the MN assay, andDNA migration (cometassay) were detected
[120]
Proximal tubularcells (PTC), Wistarrats
male gavage24–72 hours(in vivo and
in vitro)
5.0 μM, 12.5 μMin vitro; 1 and10 mg/kg bw
In vivo and in vitrogene expressioncomparative study
In vitro and in vivo geneexpression data werecomparable. Response tooxidative stress-relatedgenes hypoxia-induciblefactor 1 and catalase wasobserved
[121]
Dark Agouti (DA),Lewis rats
maleIntragastricintubation
0.4 mg/kg bw Life-time
Life-time study toevaluate if MESNAleads to a moreeffective reductionof OTA-inducedtumourdevelopment orurinary tractdamage
Lack of effect of mesnaon OTA-induced urinarytract damage or renaltumor development
[122]
Dark Agouti (DA),Lewis rats
maleIntragastricintubation
0.4 mg/kg bw 2 years
Life-time study toevaluate thepotential protectiveeffect of 2mercaptoethanesulfonate(MESNA) andN-acetyl cysteine(NAC )
MESNA decreasedkaryomegalies in kidney,but had no beneficialeffect on renal tumourincidence
[123]
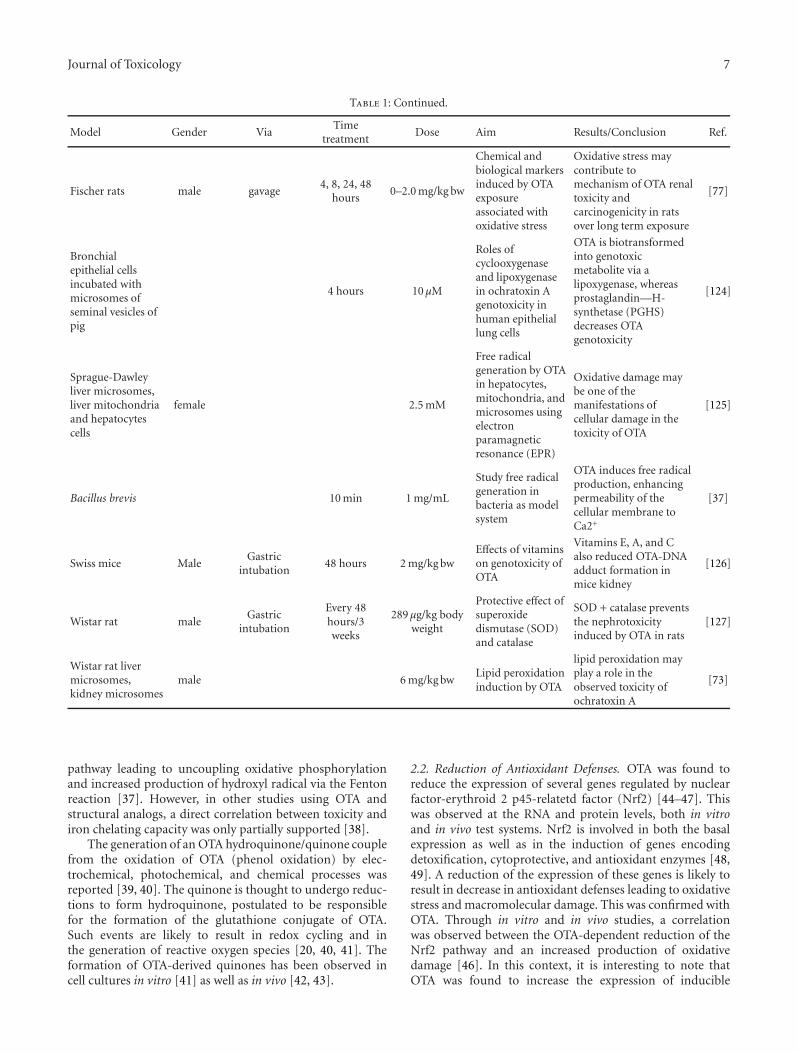
Journal of Toxicology 7
Table 1: Continued.
Model Gender ViaTime
treatmentDose Aim Results/Conclusion Ref.
Fischer rats male gavage4, 8, 24, 48
hours0–2.0 mg/kg bw
Chemical andbiological markersinduced by OTAexposureassociated withoxidative stress
Oxidative stress maycontribute tomechanism of OTA renaltoxicity andcarcinogenicity in ratsover long term exposure
[77]
Bronchialepithelial cellsincubated withmicrosomes ofseminal vesicles ofpig
4 hours 10 μM
Roles ofcyclooxygenaseand lipoxygenasein ochratoxin Agenotoxicity inhuman epitheliallung cells
OTA is biotransformedinto genotoxicmetabolite via alipoxygenase, whereasprostaglandin—H-synthetase (PGHS)decreases OTAgenotoxicity
[124]
Sprague-Dawleyliver microsomes,liver mitochondriaand hepatocytescells
female 2.5 mM
Free radicalgeneration by OTAin hepatocytes,mitochondria, andmicrosomes usingelectronparamagneticresonance (EPR)
Oxidative damage maybe one of themanifestations ofcellular damage in thetoxicity of OTA
[125]
Bacillus brevis 10 min 1 mg/mL
Study free radicalgeneration inbacteria as modelsystem
OTA induces free radicalproduction, enhancingpermeability of thecellular membrane toCa2+
[37]
Swiss mice MaleGastric
intubation48 hours 2 mg/kg bw
Effects of vitaminson genotoxicity ofOTA
Vitamins E, A, and Calso reduced OTA-DNAadduct formation inmice kidney
[126]
Wistar rat maleGastric
intubation
Every 48hours/3weeks
289 μg/kg bodyweight
Protective effect ofsuperoxidedismutase (SOD)and catalase
SOD + catalase preventsthe nephrotoxicityinduced by OTA in rats
[127]
Wistar rat livermicrosomes,kidney microsomes
male 6 mg/kg bwLipid peroxidationinduction by OTA
lipid peroxidation mayplay a role in theobserved toxicity ofochratoxin A
[73]
pathway leading to uncoupling oxidative phosphorylationand increased production of hydroxyl radical via the Fentonreaction [37]. However, in other studies using OTA andstructural analogs, a direct correlation between toxicity andiron chelating capacity was only partially supported [38].
The generation of an OTA hydroquinone/quinone couplefrom the oxidation of OTA (phenol oxidation) by elec-trochemical, photochemical, and chemical processes wasreported [39, 40]. The quinone is thought to undergo reduc-tions to form hydroquinone, postulated to be responsiblefor the formation of the glutathione conjugate of OTA.Such events are likely to result in redox cycling and inthe generation of reactive oxygen species [20, 40, 41]. Theformation of OTA-derived quinones has been observed incell cultures in vitro [41] as well as in vivo [42, 43].
2.2. Reduction of Antioxidant Defenses. OTA was found toreduce the expression of several genes regulated by nuclearfactor-erythroid 2 p45-relatetd factor (Nrf2) [44–47]. Thiswas observed at the RNA and protein levels, both in vitroand in vivo test systems. Nrf2 is involved in both the basalexpression as well as in the induction of genes encodingdetoxification, cytoprotective, and antioxidant enzymes [48,49]. A reduction of the expression of these genes is likely toresult in decrease in antioxidant defenses leading to oxidativestress and macromolecular damage. This was confirmed withOTA. Through in vitro and in vivo studies, a correlationwas observed between the OTA-dependent reduction of theNrf2 pathway and an increased production of oxidativedamage [46]. In this context, it is interesting to note thatOTA was found to increase the expression of inducible

8 Journal of Toxicology
nitric oxide synthase (iNOs) [50–52], an enzyme responsiblefor the production of nitric oxide (NO). An associationbetween iNOs expression and the development of cancerswas suggested in humans and animals in vivo [53]. In excess,NO may behave as a toxic radical producing nitrosativestress. NO is known to react with oxygen anion radicalsuperoxide to form the pro-oxidant peroxynitrite. Underphysiological conditions, peroxynitrite rapidly decomposesto generate a nitro radical intermediate leading to proteinand DNA nitration. OTA was shown to stimulate proteinand possibly DNA nitration [52, 53] indicating that OTAexposure may be considered as a source of both oxygen andalso reactive nitrogen radicals/species (RNS).
3. OTA-Mediated Oxidative Damage
3.1. DNA-Damage. ROS, such as hydroxyl radicals and nitricoxide, are capable of forming oxidized DNA bases thatdirectly produce diverse types of DNA damage [54–56].The oxidized DNA bases appear to be capable of inducingmutations that are commonly observed in neoplasia [33]. Asillustrated in Table 1, diverse biomarkers have been analyzedshowing that OTA induces DNA damage. In cell cultures,the OTA-dependent production of ROS was correlated withan increased formation of 8-oxoguanine [57–59]. Moreover,OTA was shown to induce DNA strand breaks as assessedby comet assay in liver, kidney, and spleen of F344 ratsgiven 0, 0.25, 0.50, 1.0, and 2.0 mg/kg bw/day by gavagefor 2 weeks [60]. In liver and kidney, the extent of DNAdamage analyzed by comet assay was further enhanced inthe presence of formamidopyrimidine glycosylase (Fpg), anenzyme involved in excision repair of oxidized DNA bases[57, 58, 61]. Another study [62] detected DNA-strand breaks(using the comet assay) in the kidney of female Wistar ratstreated intraperitoneally with OTA (0.5 mg/kg bw/day for 7,14, and 21 days; n = 5 per group). The severity of theDNA lesions in the kidney increased according to the OTAdose and was at maximum after 21 days of treatment. Otherauthors have observed oxidative DNA-damage in varioustissues of animals treated with a wide range of OTA dosesand treatment durations [46, 60, 63, 64].
3.2. Protein Damage. Carbonylation of proteins occursthrough a variety of oxidative pathways [54, 65–67]. Car-bonylation is an important protein modification associatedwith alterations of protein (enzymes) function, proteinmisfolding, protein fate, and proteolysis. An increase inprotein carbonyl content of tissues has been associated witha number of pathological disorders. Due to their abundancein mammalian cells, cytoskeletal proteins like actin arecommon targets for a variety of ROS and low-molecularweight reactive carbonyl species [67]. Inconsistent data havebeen reported on the potential impact of OTA on proteincarbonylation [52, 59, 63, 68, 69]. No increase in proteinoxidation was observed in liver and kidney of F344 ratstreated with OTA at 0.3 mg/kg bw per day for 4 weeks [69].The same finding was reported in liver of Wistar rats treatedwith OTA at 289 μg/kg bw for 90 days [63]. In contrast, in
another in vivo study measuring protein oxidation in Wistarrats treated with OTA (0.5 mg/kg bw/day) for 24 h, 7, 14and 21 days, a significant increase in protein carbonyls wasfound after 14 and 21 days of treatment in, respectively, thekidney and the liver [68]. Oxidative protein modification wasobserved in vitro [52].
3.3. Lipid Damage. Lipid peroxidation is among the mostextensively investigated processes induced by free radicals.Of these, the by-products, 4-hydroxy-2-nonenal (HNE), thetautomer malondialdehyde (MDA), acrolein and crotonalde-hyde have been widely studied. The ability of these reactiveelectrophiles to modify DNA bases, yielding promutageniclesions, is considered to contribute to the mutagenic andcarcinogenic effects associated with oxidative stress-inducedlipid peroxidation. HNE and MDA have increasingly beenimplicated in carcinogenesis [33, 70–72]. OTA has beenreported to increase MDA formation. Initially, Rahimtula’sgroup [73] observed that OTA was able to stimulate lipidperoxidation when added to liver or kidney microsomesor when administered to rats in vivo. Stimulation of lipidperoxidation by OTA-iron complex facilitating the reductionof iron was further reported [36]. Moreover, additionalstudies [74] indicated that OTA induced lipid peroxidationaccompanied by leakage of calcium from calcium-loadedmicrosomes. Increased formation of MDA was observedin animal models treated orally with different doses ofOTA [75–79]. HNE-protein adducts were measured in cellcultures treated with OTA [52].
4. OTA-Mediated Biological Response
It is widely acknowledged that reactive oxygen and ni-trosative species can trigger biological responses such asstimulation or inhibition of signal transduction and geneexpression. Such biological responses are considered tocontribute to the expression of the carcinogenic potentialof the reactive chemicals. A number of in vitro and in vivostudies are consistent with a role of oxidative and probablynitrosative stress as messengers involved in the adversebiological effects of OTA (Table 1).
5. Cell Signaling
ROS induces release of calcium from intracellular stores,resulting in the activation of kinases, such as protein kinaseC (PKC). ROS species play also a critical role in the selectivemobilization of other cell signaling responses. Cell signal-ing phosphoproteins of mitogen-activated protein (MAP)kinases including ERK, c-Jun N-terminal kinases (JNK), andp38 kinases are involved in proliferation, differentiation, andapoptosis. Activation of these molecules has been observedin response to changes in the cellular redox balance andare considered as vectors of ROS biological effects [33]. Invitro experiments provided evidence of an effect of OTAon intracellular calcium (Ca2+) homeostasis [74, 80–82].Ca2+ homeostasis modulation by OTA was also observedin vivo [83]. An increased rate of microsomal Ca2+ uptake

Journal of Toxicology 9
was observed after OTA administration in vivo and in vitro[84]. Gene profiling analysis suggested a modulation ofgenes involved in calcium homeostasis by OTA [47, 85].These data indicate that Ca2+ dependent signal transductionpathways may be affected by OTA treatment. Interferenceswere observed with other cell signaling pathways. OTA wasshown to stimulate phosphorylation of ERK1/2, SAP/JNK,and p38 using in vitro models [82, 86, 87]. Using an invivo model ERK1/2 a specific signaling response was alsoobserved with mobilization of the atypical protein kinase C(PKCζ) and the insulin-like growth factor-1 (IGF-1) system[88]. Gene profiling data supported a potential role of the(IGF)-PI3K-PKB pathway in OTA-mediated renal toxicity inmale rats [89].
5.1. Redox Modulation of Transcription Factors. Numerousreports have characterized interactions of ROS and RNSwith activity of transcription factors [90–92]. Transcriptionfactors contain a conserved redox sensitive cysteine residue;the oxidation of this residue inactivates the DNA-bindingdomain of the factor. Several studies have observed aninactivation of transcription factors resulting from increasedconcentrations of ROS [93–95]. For example, it was shownthat the complex AP-1 is a basic leucine zipper protein,which is highly sensitive to changes in redox environmentdue to a cysteine residue in the DNA-binding domain[96]. Gene expression profile studies performed using invivo models have shown that OTA impairs the antioxi-dant defense system regulated by Nrf2 [47]. This effectwas supported by further studies [44–46]. Using in vitromodels together with electrophoretic mobility shift assay,an inhibition of Nrf2 and AP-1 activity was shown as aresult of OTA treatment [46]. Interestingly, Nrf2 binding siterepresents a bZip domain that interacts with the AntioxidantResponse Element (ARE) DNA-binding site triggering thetranscription of Nrf2 regulated genes. The ARE motif (5′-A/GTGAC/TnnnGCA/G-3′) shares structural similarity withAP-1 binding site (5′-TGACTCA) involving both cysteinerich residues, the target of ROS oxidation [97, 98]. These datastrongly suggest a correlation between the generation of ROSby OTA and the subsequent inactivation of Nrf2 as previouslydescribed for AP-1. This molecular mechanism appears as alikely molecular component explaining the reduction of thedefense response observed under OTA treatment.
In vivo experiments provided direct evidence that S-nitrosylation can interfere with transcription [91, 92]. Nitricoxide (NO) induces the nitrosylation of cysteine residues(thiol groups) within or near the DNA-binding domainand/or insertion of the zinc finger, which is a DNA-bindingmotif, resulting in the inhibition of the DNA-binding activityof transcription factors [99–101]. This is illustrated by theexample of the suppression of P450 gene expression byNO. NO-donors were found to suppress CYP2D6 promoteractivity through inhibition of the transcription factor calledhepatocyte nuclear factor 4 (HNF4) [94, 95]. Interestingly,in a gene expression study performed by our group, OTAwas associated with a significant reduction of the expressionof genes regulated by HNF4 suggesting an indirect evidence
of the role oxidative stress and the transcription factorsregulation by OTA [47].
5.2. Alterations of Gap-Junction Intercellular Communication(GJIC). A strong correlation between the ability of a com-pound to block cell-to-cell communication in cultured cellsand its ability to induce rodent tumors through nongeno-toxic mechanisms has been documented [33, 128–131].Disruption of gap junction intercellular communication wasspecifically reported in human renal cancer cell lines [132].For example, Connexin 32 (Cx32) was discovered to begenerally downregulated in human renal cell carcinoma(RCC) cell lines and in cancerous regions of the kidney[133]. ROS such as H2O2, an established tumor promoter,is known to modulate cell-to-cell communication. Likewise,certain chemicals inducing ROS were shown to inhibitintercellular communication in a variety of cells in culturesystems [33]. Data on the potential effects of OTA on GJICare inconsistent. Kidney epithelial cells treated with OTAresulted in modulation of gap junction-mediated intercel-lular communication, through a reduced expression of thegap-junction protein CX43 [134]. In addition, OTA stronglyreduced the expression of other gap-junction proteins, CX26,CX32, and CX43, in liver of rats treated with OTA [63]. Inanother study, even though a strong reduction of CX43 wasfound in renal cells in vitro, OTA inhibited GJIC only in liverbut not in kidney [135].
6. Prevention of OTA-Induced Oxidative Stress
6.1. Counteracting OTA-Mediated Reduction of Nrf2 Activity.As mentioned earlier, OTA was found to reduce the expres-sion of antioxidant enzymes through inhibition of Nrf2activity [46]. This reduction in antioxidant gene expressionwas correlated with increased oxidative damage of proteinand DNA [46]. To further confirm the actual role of thereduction of defense mechanisms in the induction of cellularoxidative damage, inducers of Nrf2 were applied in cellcultures in vitro. All reported OTA effects at the levels of Nrf2activity, Nrf2-regulated gene expression, and DNA damagewere prevented in cell pretreated with Nrf2 inducers [46],strongly indicating a causal relationship between Nrf2 effectsand oxidative damage.
6.2. Application of Radical Scavengers. Several studies havebeen performed to try to counteract the adverse effects ofoxygen radicals generated under OTA-treatment. A numberof molecules with various antioxidant properties were tested,including inula crithmoides, cyanidin 3-O-b-D-glucoside,catechins, melatonin, superoxide dismutase, catalase, and N-acetyl-L-cysteine (NAC), using in vivo or in vitro models.Protection against OTA-induced DNA damage, lipid peroxi-dation as well as cytotoxicity was observed [75, 79, 102, 105,106, 116, 118, 127] further confirming the link between OTAexposure and oxidative macromolecular damage. However,up to now, the application of chemicals known to possessantioxidant properties failed to prevent the development ofOTA-induced tumors in animal models. Authors reported

10 Journal of Toxicology
Ochratoxin A
Oxidative stress
HNF4
Nrf2/ARE
ROSRNSRCS
Mutation, apoptosis, cytotoxicity
Tumors
DNAdamage
Lipiddamage
Ca2+ cellsignaling
Proteindamage
Cell transformation/proliferation
Intercellularcommunication
disruption
Gap juntionalintercellular
communication
Defensesystem
impairment
Transcriptionfactors
inactivation
Cytoskeleton(actin)
Figure 1: Scheme to illustrate the oxidative stress-mediated mode of action proposed for OTA. Increased production of ROS, RNS,and RCS is likely to originate either from direct redox reactions involving OTA or through the inhibition of cellular defenses such asthrough the inhibition of transcription factors as Nrf2 which regulates enzymes with antioxidant properties. The generation of radicalswill induce macromolecular oxidative damage such as oxidized DNA bases which may be converted into mutation resulting into generationof transformed cells. In addition, radicals will trigger biological responses which may impair intercellular communication and induce cellproliferation as well as reduction in cellular defense in oxidative stress. This last effect is likely to amplify the oxidative stress-mediated effectsof OTA. Altogether, these molecular mechanisms will result in cancer development.
protection against nephrotoxicity but not carcinogenicityinduced by ochratoxin A, implicating two separate pathways[122, 123].
6.3. Application of Peroxidase Inhibitors. Indomethacin andaspirin were found to prevent OTA genotoxicity in theurinary bladder and kidney of mice [136]. These datasuggested the possible co-oxidation of OTA by enzymesinvolved in arachidonic acid biotransformation, espe-cially prostaglandin-H-synthase (PGHS) and/or lipoxyge-nase [124]. Such reactions thought to produce activated OTAmetabolites are also known to generate ROS which then mayinduce oxidation [124].
7. Conclusion
The carcinogenic mycotoxin OTA has been reviewed bya number of expert groups [3–5, 14, 137]. These expertgroups identified the production of oxidative stress as animportant event in the mode of action of OTA-inducednephrocarcinogenicity. In the present paper, the actualevidence available supporting such hypothesis was collectedand reviewed.
It has been clearly shown that OTA generates oxida-tive stress predominantly in kidney through potentiallydirect (redox cycling) and indirect (reduction of cellularantioxidant defenses) mechanisms. Interestingly, these twomechanisms may interact with each other. The reductionof defense may amplify the impact of the direct productionof radicals. Consequences resulting from the production ofoxidative stress were observed at different levels. High kidneysusceptibility to oxidative stress conditions may explain thetarget-specific toxicity of OTA. Oxidative stress has beenincriminated in a number of kidney pathological pathways[138–140].
As depicted in Figure 1, first, OTA exposure was asso-ciated with increased levels of oxidative DNA, lipid, andprotein damage. Second, various biological pathways knownto be mobilized under oxidative stress were shown to bealtered by OTA. Importantly, these effects were observed inboth in vitro and in vivo test systems. Active in vivo doseswere within doses known to induce tumors in kidney. Thesemechanisms are likely to be relevant for humans.
In conclusion, the evidence for the induction of anoxidative stress response resulting from OTA exposure canbe considered strong. Because the contribution of theoxidative stress response in the development of cancers is

Journal of Toxicology 11
well established, a role in OTA carcinogenicity is plausible.Altogether, the data reviewed above support the applicationof a threshold-based approach to establish safe level ofdietary human exposure to OTA.
References
[1] C. A. Tatu, D. Drugarin, V. Paunescu, D. I. Stanescu, and F.Schneider, “Balkan endemic nephropathy, the haematopoi-etic system and the environmental connection,” Food andChemical Toxicology, vol. 36, no. 3, pp. 245–247, 1998.
[2] A. Pfohl-Leszkowicz and R. A. Manderville, “Ochratoxin A:an overview on toxicity and carcinogenicity in animals andhumans,” Molecular Nutrition and Food Research, vol. 51, no.1, pp. 61–99, 2007.
[3] J. Fink-Gremmels, “Conclusions from the workshops onOchratoxin A in Food: recent developments and significance,organized by ILSI Europe in Baden (Austria), 29 June-1 July2005,” Food Additives and Contaminants, vol. 22, supplement1, pp. 1–5, 2005.
[4] EFSA, “Opinion of the Scientific Panel on Contaminants inthe food chain on a request from the commission related toOchratoxin A in Food,” 365, 1-56, 2006.
[5] JECFA, “Ochratoxin A,” WHO Technical Report Series 947.IPCS, WHO, Geneva, Switzerland, pp. 169–180. 2007.
[6] Symposium on Recent Advances in Endemic Nephropathy:the role of toxins in an environmental disease, Croatia, 2006.
[7] IARC, Some Naturally Occuring Substances: Food Items andConstituents, Hetrocyclic Aromatic Amines and Mycotoxins,IARC Monographs on the Evaluation of Carcinogenic Riskto Humans, IARC, Lyon, France, 1993.
[8] A. P. Grollman and B. Jelakovic, “Role of environmentaltoxins in endemic (Balkan) nephropathy,” Journal of theAmerican Society of Nephrology, vol. 18, no. 11, pp. 2817–2823, 2007.
[9] A. P. Grollman, S. Shibutani, M. Moriya et al., “Aristolochicacid and the etiology of endemic (Balkan) nephropathy,”Proceedings of the National Academy of Sciences of the UnitedStates of America, vol. 104, no. 29, pp. 12129–12134, 2007.
[10] S. D. Stoev, “Studies on some feed additives and materialsgiving partial protection against the suppressive effect ofochratoxin A on egg production of laying hens,” Research inVeterinary Science, vol. 88, pp. 486–491, 2010.
[11] JECFA, “Evaluation of certain food additives and contam-inants: ochratoxin A,” WHO Technical Report Series 806,WHO, Geneva, Switzerland, 1991.
[12] JECFA, “Evaluation of certain food additives and contam-inants: ochratoxin A,” WHO Technical Report Series 859,WHO, Geneva, Switzerland, 1995.
[13] JECFA, “Evaluation of certain mycotoxins in food: ochra-toxin A,” WHO Technical Report Series 906, WHO, Geneva,Switzerland, 2002.
[14] JECFA, “Ochratoxin A (addendum),” WHO Food AdditiveSeries 59: Safety Evaluations of certain food additives andcontaminants. IPCS, WHO, Geneva, Switzerland, 2008.
[15] T. Kuiper-Goodman, C. Hilts, S. M. Billiard, Y. Kiparissis, I.D. K. Richard, and S. Hayward, “Health risk assessment ofochratoxin a for all age-sex strata in a market economy,” FoodAdditives and Contaminants A, vol. 27, no. 2, pp. 212–240,2010.
[16] B. Schilter, M. Marin-Kuan, T. Delatour, S. Nestler, P.Mantle, and C. Cavin, “Ochratoxin A: potential epigenetic
mechanisms of toxicity and carcinogenicity,” Food Additivesand Contaminants, vol. 22, supplement 1, pp. 88–93, 2005.
[17] R. A. Manderville, “A case for the genotoxicity of ochratoxina by bioactivation and covalent DNA adduction,” ChemicalResearch in Toxicology, vol. 18, no. 7, pp. 1091–1097, 2005.
[18] R. J. Turesky, “Perspective: ochratoxin A is not a genotoxiccarcinogen,” Chemical Research in Toxicology, vol. 18, no. 7,pp. 1082–1090, 2005.
[19] V. Faucet, A. Pfohl-Leszkowicz, J. Dai, M. Castegnaro, andR. A. Manderville, “Evidence for covalent DNA adduction byochratoxin A following chronic exposure to rat and subacuteexposure to pig,” Chemical Research in Toxicology, vol. 17, no.9, pp. 1289–1296, 2004.
[20] R. A. Manderville and A. Pfohl-Leszkowicz, “Chapter 4genotoxicity of chlorophenols and ochratoxin A,” Advancesin Molecular Toxicology, vol. 1, pp. 85–138, 2006.
[21] P. G. Mantle, V. Faucet-Marquis, R. A. Manderville, B.Squillaci, and A. Pfohl-Leszkowicz, “Structures of covalentadducts between DNA and ochratoxin a: a new factor indebate about genotoxicity and human risk assessment,”Chemical Research in Toxicology, vol. 23, no. 1, pp. 89–98,2010.
[22] A. Pfohl-Leszkowicz and M. Castegnaro, “Further argumentsin favour of direct covalent binding of Ochratoxin A(OTA) after metabolic biotransformation,” Food Additivesand Contaminants, vol. 22, supplement 1, pp. 75–87, 2005.
[23] C. Schlatter, J. Studer-Rohr, and T. Rasonyi, “Carcinogenicityand kinetic aspects of ochratoxin A,” Food Additives andContaminants, vol. 13, supplement, pp. 43–44, 1996.
[24] A. Mally, H. Zepnik, P. Wanek et al., “Ochratoxin A: lack offormation of covalent DNA adducts,” Chemical Research inToxicology, vol. 17, no. 2, pp. 234–242, 2004.
[25] T. Delatour, A. Mally, J. Richoz et al., “Absence of 2′-deoxyguanosine-carbon 8-bound ochratoxin A adduct in ratkidney DNA monitored by isotope dilution LC-MS/MS,”Molecular Nutrition and Food Research, vol. 52, no. 4, pp.472–482, 2008.
[26] A. M. Jarabek, L. H. Pottenger, L. S. Andrews et al.,“Creating context for the use of DNA adduct data in cancerrisk assessment: I. Data organization,” Critical Reviews inToxicology, vol. 39, no. 8, pp. 659–678, 2009.
[27] J. A. Swenberg, E. Fryar-Tita, Y. C. Jeong et al., “Biomarkersin toxicology and risk assessment: informing critical dose-response relationships,” Chemical Research in Toxicology, vol.21, no. 1, pp. 253–265, 2008.
[28] A. R. Boobis, G. P. Daston, R. J. Preston, and S. S. Olin,“Application of key events analysis to chemical carcinogensand noncarcinogens,” Critical Reviews in Food Science andNutrition, vol. 49, no. 8, pp. 690–707, 2009.
[29] G. E. Johnson, S. H. Doak, S. M. Griffiths et al., “Non-lineardose-response of DNA-reactive genotoxins: recommenda-tions for data analysis,” Mutation Research, vol. 678, no. 2,pp. 95–100, 2009.
[30] P. G. Mantle, “Minimum tolerable exposure period andmaximum threshold dietary intake of ochratoxin A forcausing renal cancer in male Dark Agouti rats,” Food andChemical Toxicology, vol. 47, no. 10, pp. 2419–2424, 2009.
[31] S. Loft and H. E. Poulsen, “Cancer risk and oxidative DNAdamage in man,” Journal of Molecular Medicine, vol. 74, no.6, pp. 297–312, 1996.
[32] J. E. Klaunig, Y. Xu, J. S. Isenberg et al., “The role ofoxidative stress in chemical carcinogenesis,” EnvironmentalHealth Perspectives, vol. 106, supplement 1, pp. 289–295,1998.

12 Journal of Toxicology
[33] J. E. Klaunig and L. M. Kamendulis, “The role of oxidativestress in carcinogenesis,” Annual Review of Pharmacology andToxicology, vol. 44, pp. 239–267, 2004.
[34] M. Valko, C. J. Rhodes, J. Moncol, M. Izakovic, and M.Mazur, “Free radicals, metals and antioxidants in oxidativestress-induced cancer,” Chemico-Biological Interactions, vol.160, no. 1, pp. 1–40, 2006.
[35] M. E. Goetz and A. Luch, “Reactive species: a cell damagingrout assisting to chemical carcinogens,” Cancer Letters, vol.266, no. 1, pp. 73–83, 2008.
[36] R. F. Omar, B. B. Hasinoff, F. Mejilla, and A. D. Rahimtula,“Mechanism of ochratoxin a stimulated lipid peroxidation,”Biochemical Pharmacology, vol. 40, no. 6, pp. 1183–1191,1990.
[37] D. Hoehler, R. R. Marquardt, A. R. Mclntosh, and H. Xiao,“Free radical generation as induced by ochratoxin A andits analogs in bacteria (Bacillus brevis),” Journal of BiologicalChemistry, vol. 271, no. 44, pp. 27388–27394, 1996.
[38] H. Xiao, S. Madhyastha, R. R. Marquardt et al., “Toxicityof ochratoxin A, its opened lactone form and several ofits analogs: structure-activity relationships,” Toxicology andApplied Pharmacology, vol. 137, no. 2, pp. 182–192, 1996.
[39] J. Dai, G. Park, M. W. Wright, M. Adams, S. A. Akman,and R. A. Manderville, “Detection and characterization of aglutathione conjugate of ochratoxin A,” Chemical Research inToxicology, vol. 15, no. 12, pp. 1581–1588, 2002.
[40] I. G. Gillman, T. N. Clark, and R. A. Manderville, “Oxidationof ochratoxin A by an Fe-porphyrin system: model forenzymatic activation and DNA cleavage,” Chemical Researchin Toxicology, vol. 12, no. 11, pp. 1066–1076, 1999.
[41] V. Faucet-Marquis, F. Pont, F. C. Størmer, T. Rizk, M.Castegnaro, and A. Pfohl-Leszkowicz, “Evidence of a newdechlorinated ochratoxin A derivative formed in opossumkidney cell cultures after pretreatment by modulators of glu-tathione pathways: correlation with DNA-adduct formation,”Molecular Nutrition and Food Research, vol. 50, no. 6, pp.530–542, 2006.
[42] R. A. Manderville, “Pfohl-Leszkowicz bioactivation andDNA adduction as a rationale for ochratoxin A carcinogene-sis,” World Mycotoxin Journal, vol. 1, pp. 357–367, 2008.
[43] A. Pfohl-Leszkowicz, “Ochratoxin A and aristolochic acidinvolvement in nephropathies and associated urothelial tracttumours,” Arhiv za Higijenu Rada i Toksikologiju, vol. 60, no.4, pp. 465–483, 2009.
[44] C. Boesch-Saadatmandi, A. Loboda, A. Jozkowicz et al.,“Effect of ochratoxin A on redox-regulated transcription fac-tors, antioxidant enzymes and glutathione-S-transferase incultured kidney tubulus cells,” Food and Chemical Toxicology,vol. 46, no. 8, pp. 2665–2671, 2008.
[45] C. Boesch-Saadatmandi, A. E. Wagner, A. C. Graeser, C.Hundhausen, S. Wolffram, and G. Rimbach, “Ochratoxin Aimpairs Nrf2-dependent gene expression in porcine kidneytubulus cells,” Journal of Animal Physiology and AnimalNutrition, vol. 93, no. 5, pp. 547–554, 2009.
[46] C. Cavin, T. Delatour, M. Marin-Kuan et al., “Reduction inantioxidant defenses may contribute to ochratoxin A toxicityand carcinogenicity,” Toxicological Sciences, vol. 96, no. 1, pp.30–39, 2007.
[47] M. Marin-Kuan, S. Nestler, C. Verguet et al., “A toxicoge-nomics approach to identify new plausible epigenetic mech-anisms of ochratoxin a carcinogenicity in rat,” ToxicologicalSciences, vol. 89, no. 1, pp. 120–134, 2006.
[48] J. D. Hayes, S. A. Chanas, C. J. Henderson et al., “The Nrf2transcription factor contributes both to the basal expression
of glutathione S-transferases in mouse liver and to theirinduction by the chemopreventive synthetic antioxidants,butylated hydroxyanisole and ethoxyquin,” Biochemical Soci-ety Transactions, vol. 28, no. 2, pp. 33–41, 2000.
[49] T. W. Kensler, N. Wakabayashi, and S. Biswal, “Cell survivalresponses to environmental stresses via the Keap1-Nrf2-AREpathway,” Annual Review of Pharmacology and Toxicology,vol. 47, pp. 89–116, 2007.
[50] M. G. Zurich, S. Lengacher, O. Braissant, F. Monnet-Tschudi,L. Pellerin, and P. Honegger, “Unusual astrocyte reactivitycaused by the food mycotoxin ochratoxin a in aggregating ratbrain cell cultures,” Neuroscience, vol. 134, no. 3, pp. 771–782,2005.
[51] M. C. Ferrante, G. M. Raso, M. Bilancione, E. Esposito, A.Iacono, and R. Meli, “Differential modification of inflam-matory enzymes in J774A.1 macrophages by ochratoxin Aalone or in combination with lipopolysaccharide,” ToxicologyLetters, vol. 181, no. 1, pp. 40–46, 2008.
[52] C. Cavin, T. Delatour, M. Marin-Kuan et al., “OchratoxinA—mediated DNA and protein damage: roles of nitrosativeand oxidative stresses,” Toxicological Sciences, vol. 110, no. 1,pp. 84–94, 2009.
[53] J. A. Crowell, V. E. Steele, C. C. Sigman, and J. R. Fay, “Isinducible nitric oxide synthase a target for chemopreven-tion?” Molecular Cancer Therapeutics, vol. 2, no. 8, pp. 815–823, 2003.
[54] R. De Bont and N. van Larebeke, “Endogenous DNA damagein humans: a review of quantitative data,” Mutagenesis, vol.19, no. 3, pp. 169–185, 2004.
[55] Y. Hiraku and S. Kawanishi, “Immunohistochemical analysisof 8-nitroguanine, a nitrative DNA lesion, in relation toinflammation-associated carcinogenesis,” Methods in Molec-ular Biology, vol. 512, pp. 3–13, 2009.
[56] L. I. Shukla, A. Adhikary, R. Pazdro, D. Becker, and M.D. Sevilla, “Formation of 8-oxo-7,8-dihydroguanine-radicalsin γ-irradiated DNA by multiple one-electron oxidations,”Nucleic Acids Research, vol. 32, no. 22, pp. 6565–6574, 2004.
[57] L. Arbillaga, A. Azqueta, O. Ezpeleta, and A. L. De Cerain,“Oxidative DNA damage induced by ochratoxin A in the HK-2 human kidney cell line: evidence of the relationship withcytotoxicity,” Mutagenesis, vol. 22, no. 1, pp. 35–42, 2007.
[58] H. G. Kamp, G. Eisenbrand, J. Schlatter, K. Wurth, andC. Janzowski, “Ochratoxin A: induction of (oxidative) DNAdamage, cytotoxicity and apoptosis in mammalian cell linesand primary cells,” Toxicology, vol. 206, no. 3, pp. 413–425,2005.
[59] G. J. Schaaf, S. M. Nijmeijer, R. F. M. Maas, P. Roestenberg, E.M. De Groene, and J. Fink-Gremmels, “The role of oxidativestress in the ochratoxin A-mediated toxicity in proximaltubular cells,” Biochimica et Biophysica Acta, vol. 1588, no. 2,pp. 149–158, 2002.
[60] A. Mally, G. Pepe, S. Ravoori et al., “Ochratoxin A causesDNA damage and cytogenetic effects but no DNA adductsin rats,” Chemical Research in Toxicology, vol. 18, no. 8, pp.1253–1261, 2005.
[61] L. Arbillaga, A. Azqueta, J. H. M. van Delft, and A. Lopezde Cerain, “In vitro gene expression data supporting aDNA non-reactive genotoxic mechanism for ochratoxin A,”Toxicology and Applied Pharmacology, vol. 220, no. 2, pp.216–224, 2007.
[62] D. Zeljezic, A. M. Domijan, and M. Peraica, “DNA damageby ochratoxin A in rat kidney assessed by the alkaline cometassay,” Brazilian Journal of Medical and Biological Research,vol. 39, no. 12, pp. 1563–1568, 2006.

Journal of Toxicology 13
[63] N. Gagliano, I. D. Donne, C. Torri et al., “Early cytotoxiceffects of ochratoxin A in rat liver: a morphological, bio-chemical and molecular study,” Toxicology, vol. 225, no. 2-3,pp. 214–224, 2006.
[64] H. G. Kamp, G. Eisenbrand, C. Janzowski et al., “OchratoxinA induces oxidative DNA damage in liver and kidney afteroral dosing to rats,” Molecular Nutrition and Food Research,vol. 49, no. 12, pp. 1160–1167, 2005.
[65] I. Dalle-Donne, R. Rossi, D. Giustarini, A. Milzani, andR. Colombo, “Protein carbonyl groups as biomarkers ofoxidative stress,” Clinica Chimica Acta, vol. 329, no. 1-2, pp.23–38, 2003.
[66] I. Dalle-Donne, D. Giustarini, R. Colombo, R. Rossi, and A.Milzani, “Protein carbonylation in human diseases,” Trendsin Molecular Medicine, vol. 9, no. 4, pp. 169–176, 2003.
[67] I. Dalle-Donne, R. Rossi, R. Colombo, D. Giustarini, and A.Milzani, “Biomarkers of oxidative damage in human disease,”Clinical Chemistry, vol. 52, no. 4, pp. 601–623, 2006.
[68] A. M. Domijan, K. Rudes, and M. Peraica, “The effect ofochratoxin A on the concentration of protein carbonyls inrats,” Arhiv za Higijenu Rada i Toksikologiju, vol. 56, no. 4,pp. 311–315, 2005.
[69] H. G. Kamp, G. Eisenbrand, C. Janzowski et al., “OchratoxinA induces oxidative DNA damage in liver and kidney afteroral dosing to rats,” Molecular Nutrition and Food Research,vol. 49, no. 12, pp. 1160–1167, 2005.
[70] L. J. Marnett, “Lipid peroxidation—DNA damage by malon-dialdehyde,” Mutation Research, vol. 424, no. 1-2, pp. 83–95,1999.
[71] L. J. Marnett, “Oxy radicals, lipid peroxidation and DNAdamage,” Toxicology, vol. 181-182, pp. 219–222, 2002.
[72] U. Nair, H. Bartsch, and J. Nair, “Lipid peroxidation-inducedDNA damage in cancer-prone inflammatory diseases: areview of published adduct types and levels in humans,” FreeRadical Biology and Medicine, vol. 43, no. 8, pp. 1109–1120,2007.
[73] A. D. Rahimtula, J. C. Bereziat, V. Bussacchini-Griot, and H.Bartsch, “Lipid peroxidation as a possible cause of ochratoxina toxicity,” Biochemical Pharmacology, vol. 37, no. 23, pp.4469–4477, 1988.
[74] S. Khan, M. Martin, H. Bartsch, and A. D. Rahimtula,“Perturbation of liver microsomal calcium homeostasis byochratoxin A,” Biochemical Pharmacology, vol. 38, no. 1, pp.67–72, 1989.
[75] M. A. Abdel-Wahhab, M. M. Abdel-Galil, and M. El-Lithey, “Melatonin counteracts oxidative stress in rats fed anochratoxin A contaminated diet,” Journal of Pineal Research,vol. 38, no. 2, pp. 130–135, 2005.
[76] A. M. Domijan, M. Peraica, A. L. Vrdoljak, B. Radic, V.Zlender, and R. Fuchs, “The involvement of oxidative stressin ochratoxin A and fumonisin B toxicity in rats,” MolecularNutrition and Food Research, vol. 51, no. 9, pp. 1147–1151,2007.
[77] J. C. Gautier, D. Holzhaeuser, J. Markovic, E. Gremaud, B.Schilter, and R. J. Turesky, “Oxidative damage and stressresponse from ochratoxin A exposure in rats,” Free RadicalBiology and Medicine, vol. 30, no. 10, pp. 1089–1098, 2001.
[78] J. Petrik, T. Zanic-Grubisic, K. Barisic et al., “Apoptosis andoxidative stress induced by ochratoxin A in rat kidney,”Archives of Toxicology, vol. 77, no. 12, pp. 685–693, 2003.
[79] E. Sutken, E. Aral, F. Ozdemir, S. Uslu, O. Alatas, and O.Colak, “Protective role of melatonin and coenzyme Q inochratoxin A toxicity in rat liver and kidney,” InternationalJournal of Toxicology, vol. 26, no. 1, pp. 81–87, 2007.
[80] A. Benesic, S. Mildenberger, and M. Gekle, “Nephrito-genic ochratoxin A interferes with hormonal signalling inimmortalized human kidney epithelial cells,” Pflugers ArchivEuropean Journal of Physiology, vol. 439, no. 3, pp. 278–287,2000.
[81] X. Chong and A. D. Rahimtula, “Alterations in ATP-dependent calcium uptake by rat renal cortex microsomesfollowing ochratoxin A administration in vivo or addition invitro,” Biochemical Pharmacology, vol. 44, no. 7, pp. 1401–1409, 1992.
[82] M. Gekle, C. Sauvant, and G. Schwerdt, “Ochratoxin A atnanomolar concentrations: a signal modulator in renal cells,”Molecular Nutrition and Food Research, vol. 49, no. 2, pp.118–130, 2005.
[83] E. Rached, G. C. Hard, K. Blumbach et al., “Ochratoxin A:13-week oral toxicity and cell proliferation in male F344/ Nrats,” Toxicological Sciences, vol. 97, no. 2, pp. 288–298, 2007.
[84] A. D. Rahimtula and X. Chong, “Alterations in calciumhomeostasis as a possible cause of ochratoxin A nephrotoxic-ity,” IARC Scientific Publications, no. 115, pp. 207–214, 1991.
[85] L. Arbillaga, A. Vettorazzi, A. G. Gil, J. H. van Delft, J.A. Garcıa-Jalon, and A. Lopez de Cerain, “Gene expressionchanges induced by ochratoxin A in renal and hepatictissues of male F344 rat after oral repeated administration,”Toxicology and Applied Pharmacology, vol. 230, no. 2, pp.197–207, 2008.
[86] M. Gekle, G. Schwerdt, R. Freudinger et al., “OchratoxinA induces JNK activation and apoptosis in MDCK-C7 cellsat nanomolar concentrations,” Journal of Pharmacology andExperimental Therapeutics, vol. 293, no. 3, pp. 837–844, 2000.
[87] C. Sauvant, H. Holzinger, and M. Gekle, “Proximal tubulartoxicity of ochratoxin A is amplified by simultaneous inhibi-tion of the extracellular signal-regulated kinases 1/2,” Journalof Pharmacology and Experimental Therapeutics, vol. 313, no.1, pp. 234–241, 2005.
[88] M. Marin-Kuan, S. Nestler, C. Verguet et al., “MAPK-ERKactivation in kidney of male rats chronically fed ochratoxin Aat a dose causing a significant incidence of renal carcinoma,”Toxicology and Applied Pharmacology, vol. 224, no. 2, pp.174–181, 2007.
[89] K. Stemmer, H. Ellinger-Ziegelbauer, H. J. Ahr, and D. R.Dietrich, “Carcinogen-specific gene expression profiles inshort-term treated Eker and wild-type rats indicative ofpathways involved in renal tumorigenesis,” Cancer Research,vol. 67, no. 9, pp. 4052–4068, 2007.
[90] A. P. Arrigo, “Gene expression and the thiol redox state,” FreeRadical Biology and Medicine, vol. 27, no. 9-10, pp. 936–944,1999.
[91] H. E. Marshall, K. Merchant, and J. S. Stamler, “Nitrosationand oxidation in the regulation of gene expression,” FASEBJournal, vol. 14, no. 13, pp. 1889–1900, 2000.
[92] C. Vossen and M. Erard, “Down-regulation of nuclearreceptor DNA-binding activity by nitric oxide—HNF4 as amodel system,” Medical Science Monitor, vol. 8, no. 10, pp.RA217–RA220, 2002.
[93] G. I. Giles, “The redox regulation of thiol dependent signal-ing pathways in cancer,” Current Pharmaceutical Design, vol.12, no. 34, pp. 4427–4443, 2006.
[94] H. Hara and T. Adachi, “Contribution of hepatocyte nuclearfactor-4 to down-regulation of CYP2D6 gene expression bynitric oxide,” Molecular Pharmacology, vol. 61, no. 1, pp. 194–200, 2002.

14 Journal of Toxicology
[95] R. Vuppugalla and R. Mehvar, “Hepatic disposition andeffects of nitric oxide donors: rapid and concentration-dependent reduction in the cytochrome P450-mediateddrug metabolism in isolated perfused rat livers,” Journal ofPharmacology and Experimental Therapeutics, vol. 310, no. 2,pp. 718–727, 2004.
[96] S. Karimpour, J. Lou, L. L. Lin et al., “Thioredoxin reductaseregulates AP-1 activity as well as thioredoxin nuclear local-ization via active cysteines in response to ionizing radiation,”Oncogene, vol. 21, no. 41, pp. 6317–6327, 2002.
[97] K. Iwasaki, E. L. MacKenzie, K. Hailemariam, K. Sakamoto,and Y. Tsuji, “Hemin-mediated regulation of an antioxidant-responsive element of the human ferritin H gene and roleof ref-1 during erythroid differentiation of K562 cells,”Molecular and Cellular Biology, vol. 26, no. 7, pp. 2845–2856,2006.
[98] Y. Tsuji, “JunD activates transcription of the human ferritinH gene through an antioxidant response element duringoxidative stress,” Oncogene, vol. 24, no. 51, pp. 7567–7578,2005.
[99] K. D. Kroncke, L. O. Klotz, C. V. Suschek, and H. Sies,“Comparing nitrosative versus oxidative stress toward zincfinger-dependent transcription. Unique role for NO,” Journalof Biological Chemistry, vol. 277, no. 15, pp. 13294–13301,2002.
[100] K. D. Kroncke, “Nitrosative stress and transcription,” Biolog-ical Chemistry, vol. 384, no. 10-11, pp. 1365–1377, 2003.
[101] D. Nikitovic, A. Holmgren, and G. Spyrou, “Inhibition ofAP-1 DNA binding by nitric oxide involving conservedcysteine residues in Jun and Fos,” Biochemical and BiophysicalResearch Communications, vol. 242, no. 1, pp. 109–112, 1998.
[102] M. A. Abdel-Wahhab, S. H. Abdel-Azim, and A. A. El-Nekeety, “Inula crithmoides extract protects against ochra-toxin A-induced oxidative stress, clastogenic and mutagenicalterations in male rats,” Toxicon, vol. 52, no. 4, pp. 566–573,2008.
[103] W. Hassen, I. Ayed-Boussema, A. Bouslimi, and H. Bacha,“Heat shock proteins (Hsp 70) response is not systematic tocell stress. Case of the mycotoxin ochratoxin A,” Toxicology,vol. 242, no. 1–3, pp. 63–70, 2007.
[104] N. Palma, S. Cinelli, O. Sapora, S. H. Wilson, and E.Dogliotti, “Ochratoxin A-induced mutagenesis in mam-malian cells is consistent with the production of oxidativestress,” Chemical Research in Toxicology, vol. 20, no. 7, pp.1031–1037, 2007.
[105] C. Di Giacomo, R. Acquaviva, A. Piva et al., “Protective effectof cyanidin 3-O-β-d-glucoside on ochratoxin A-mediateddamage in the rat,” British Journal of Nutrition, vol. 98, no.5, pp. 937–943, 2007.
[106] S. Costa, A. Utan, R. Cervellati, E. Speroni, and M. C.Guerra, “Catechins: natural free-radical scavengers againstochratoxin A-induced cell damage in a pig kidney cell line(LLC-PK1),” Food and Chemical Toxicology, vol. 45, no. 10,pp. 1910–1917, 2007.
[107] D. Lambert, P. J. Padfield, J. McLaughlin, S. Cannell,and C. A. O’Neill, “Ochratoxin A displaces claudins fromdetergent resistant membrane microdomains,” Biochemicaland Biophysical Research Communications, vol. 358, no. 2, pp.632–636, 2007.
[108] V. Sava, A. Velasquez, S. Song, and J. Sanchez-Ramos,“Adult hippocampal neural stem/progenitor cells in vitroare vulnerable to the mycotoxin ochratoxin-A,” ToxicologicalSciences, vol. 98, no. 1, pp. 187–197, 2007.
[109] K. Stemmer, H. Ellinger-Ziegelbauer, H. J. Ahr, and D. R.Dietrich, “Carcinogen-specific gene expression profiles inshort-term treated Eker and wild-type rats indicative ofpathways involved in renal tumorigenesis,” Cancer Research,vol. 67, no. 9, pp. 4052–4068, 2007.
[110] V. Sava, O. Reunova, A. Velasquez, R. Harbison, and J.Sanchez-Ramos, “Acute neurotoxic effects of the fungalmetabolite ochratoxin-A,” NeuroToxicology, vol. 27, no. 1, pp.82–92, 2006.
[111] V. Sava, O. Reunova, A. Velasquez, and J. Sanchez-Ramos,“Can low level exposure to ochratoxin-A cause parkinson-ism?” Journal of the Neurological Sciences, vol. 249, no. 1, pp.68–75, 2006.
[112] M. C. Ferrante, M. Bilancione, G. M. Raso et al., “Expressionof COX-2 and hsp72 in peritoneal macrophages after an acuteochratoxin A treatment in mice,” Life Sciences, vol. 79, no. 13,pp. 1242–1247, 2006.
[113] A. M. Domijan, D. Zeljezic, N. Kopjar, and M. Peraica,“Standard and Fpg-modified comet assay in kidney cells ofochratoxin A- and fumonisin B-treated rats,” Toxicology, vol.222, no. 1-2, pp. 53–59, 2006.
[114] M. Tozlovanu, V. Faucet-Marquis, A. Pfohl-Leszkowicz,and R. A. Manderville, “Genotoxicity of the hydroquinonemetabolite of ochratoxin A: structure-activity relationshipsfor covalent DNA adduction,” Chemical Research in Toxicol-ogy, vol. 19, no. 9, pp. 1241–1247, 2006.
[115] L. Alvarez-Erviti, C. Leache, E. Gonzalez-Penas, and A. LopezDe Cerain, “Alterations induced in vitro by ochratoxin A inrat lymphoid cells,” Human and Experimental Toxicology, vol.24, no. 9, pp. 459–466, 2005.
[116] M. C. Guerra, F. Galvano, L. Bonsi et al., “Cyanidin-3-O-β-glucopyranoside, a natural fee-radical scavenger againstaflatoxin B1-and ochratoxin A-induced cell damage in ahuman hepatoma cell line (Hep G2) and a human colonicadenocarcinoma cell line (CaCo-2),” British Journal of Nutri-tion, vol. 94, no. 2, pp. 211–220, 2005.
[117] C. Hundhausen, C. Bosch-Saadatmandi, K. Augustin, R.Blank, S. Wolffram, and G. Rimbach, “Effect of vitamin E andpolyphenols on ochratoxin A-induced cytotoxicity in liver(HepG2) cells,” Journal of Plant Physiology, vol. 162, no. 7,pp. 818–822, 2005.
[118] A. Russo, L. La Fauci, R. Acquaviva et al., “OchratoxinA-induced DNA damage in human fibroblast: protectiveeffect of cyanidin 3-O-β-D-glucoside,” Journal of NutritionalBiochemistry, vol. 16, no. 1, pp. 31–37, 2005.
[119] S. Knasmuller, C. Cavin, A. Chakraborty et al., “Structurallyrelated mycotoxins ochratoxin A, ochratoxin B, and citrinindiffer in their genotoxic activities and in their mode of actionin human-derived liver (HepG2) cells: implications for riskassessment,” Nutrition and Cancer, vol. 50, no. 2, pp. 190–197, 2004.
[120] V. Ehrlich, F. Darroudi, M. Uhl et al., “Genotoxic effects ofochratoxin A in human-derived hepatoma (HepG2) cells,”Food and Chemical Toxicology, vol. 40, no. 8, pp. 1085–1090,2002.
[121] A. Luhe, H. Hildebrand, U. Bach, T. Dingermann, and H.J. Ahr, “A new approach to studying ochratoxin A (OTA)-induced nephrotoxicity: expression profiling in vivo and invitro employing cDNA microarrays,” Toxicological Sciences,vol. 73, no. 2, pp. 315–328, 2003.
[122] W. C. Son, K. Kamino, Y. S. Lee, and K. S. Kang, “Lack ofeffects of sodium 2-mercaptoethane sulfonate (mesna) on

Journal of Toxicology 15
Ochratoxin A induced renal tumorigenicity following life-time oral administration of Ochratoxin A in DA and Lewisrats,” Toxicology Letters, vol. 142, no. 1-2, pp. 19–27, 2003.
[123] A. Pfohl-Leszkowicz, H. Bartsch, B. Azemar, U. Mohr, J.Esteve, and M. Castegnaro, “MESNA protects rats againstnephrotoxicity but not carcinogenicity induced by ochra-toxin A, implicating two separate pathways,” Facta Universi-tatis Series Medicine and Biology, vol. 9, pp. 57–63, 2002.
[124] E. Pinelli, C. El Adlouni, B. Pipy, F. Quartulli, and A. Pfohl-Leszkowicz, “Roles of cyclooxygenase and lipoxygenases inochratoxin A genotoxicity in human epithelial lung cells,”Environmental Toxicology and Pharmacology, vol. 7, no. 2, pp.95–107, 1999.
[125] D. Hoehler, R. R. Marquardt, A. R. McIntosh, and G. M.Hatch, “Induction of free radicals in hepatocytes, mitochon-dria and microsomes of rats by ochratoxin A and its analogs,”Biochimica et Biophysica Acta, vol. 1357, no. 2, pp. 225–233,1997.
[126] Y. Grosse, L. Chekir-Ghedira, A. Huc et al., “Retinol, ascorbicacid and α-tocopherol prevent DNA adduct formationin mice treated with the mycotoxins ochratoxin A andzearalenone,” Cancer Letters, vol. 114, no. 1-2, pp. 225–229,1997.
[127] I. Baudrimont, A. M. Betbeder, A. Gharbi, A. Pfohl-Leszkowicz, G. Dirheimer, and E. E. Creppy, “Effect ofsuperoxide dismutase and catalase on the nephrotoxicityinduced by subchronical administration of ochratoxin A inrats,” Toxicology, vol. 89, no. 2, pp. 101–111, 1994.
[128] J. E. Klaunig, L. M. Kamendulis, and Y. Xu, “Epigeneticmechanisms of chemical carcinogenesis,” Human and Exper-imental Toxicology, vol. 19, pp. 543–555, 2000.
[129] J. E. Trosko and R. J. Ruch, “Cell-cell communication incarcinogenesis,” Frontiers in Bioscience, vol. 3, pp. d208–d236,1998.
[130] J. E. Trosko, C. C. Chang, B. Upham, and M. Wilson, “Epige-netic toxicology as toxicant-induced changes in intracellularsignalling leading to altered gap junctional intercellularcommunication,” Toxicology Letters, vol. 102-103, pp. 71–78,1998.
[131] H. Yamasaki, V. Krutovskikh, M. Mesnil, A. Columbano, H.Tsuda, and N. Ito, “Gap junctional intercellular communi-cation and cell proliferation during rat liver carcinogenesis,”Environmental Health Perspectives, vol. 101, supplement 5,pp. 191–197, 1993.
[132] M. Noguchi, K. Nomata, J. I. Watanabe, H. Sato, H. Kanetake,and Y. Saito, “Disruption of gap junctional intercellularcommunication in human renal cancer cell lines,” Urology,vol. 53, no. 1, pp. 218–222, 1999.
[133] H. Sato, H. Hagiwara, Y. Ohde, H. Senba, N. Virgona, and T.Yano, “Regulation of renal cell carcinoma cell proliferation,invasion and metastasis by connexin 32 gene,” Journal ofMembrane Biology, vol. 216, no. 1, pp. 17–21, 2007.
[134] A. Mally, M. Decker, M. Bekteshi, and W. Dekant, “Ochra-toxin A alters cell adhesion and gap junction intercellularcommunication in MDCK cells,” Toxicology, vol. 223, no. 1-2,pp. 15–25, 2006.
[135] A. Horvath, B. L. Upham, V. Ganev, and J. E. Trosko,“Determination of the epigenetic effects of ochratoxin in ahuman kidney and a rat liver epithelial cell line,” Toxicon, vol.40, no. 3, pp. 273–282, 2002.
[136] S. Obrecht-Pflumio, Y. Grosse, A. Pfohl-Leszkowicz, and G.Dirheimer, “Protection by indomethacin and aspirin againstgenotoxicity of ochratoxin A, particularly in the urinary
bladder and kidney,” Archives of Toxicology, vol. 70, no. 3-4,pp. 244–248, 1996.
[137] Centre for Food Safety (CFS), “Ochratoxin A in Food,” Foodand Environmental Hygiene Department. Risk AssessmentStudies Report no. 23. The Government of the Hong KongSpecial Administrative Region, Hong Kong, 2006.
[138] P. J. Beisswenger, K. S. Drummond, R. G. Nelson, S. K.Howell, B. S. Szwergold, and M. Mauer, “Susceptibility todiabetic nephropathy is related to dicarbonyl and oxidativestress,” Diabetes, vol. 54, no. 11, pp. 3274–3281, 2005.
[139] C. Coskun, A. Kural, Y. Doventas et al., “Hemodialysis andprotein oxidation products,” Annals of the New York Academyof Sciences, vol. 1100, pp. 404–408, 2007.
[140] B. Descamps-Latscha and V. Witko-Sarsat, “Importanceof oxidatively modified proteins in chronic renal failure,”Kidney International, Supplement, vol. 59, no. 78, pp. S108–S113, 2001.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 308598, 4 pagesdoi:10.1155/2011/308598
Research Article
Assessment of Protective Effect of Some ModernAgrochemicals against Ozone-Induced Stress in SensitiveClover and Tobacco Cultivars
Oleg Blum,1 Nataliya Didyk,1 Nataliya Pavluchenko,1 and Barbara Godzik2
1 M. M. Gryshko National Botanical Garden, National Academy of Sciences of Ukraine, Timiryazevs’ka St., 1, 01014 Kyiv, Ukraine2 W. Szafer Institute of Botany, Polish Academy of Sciences, Lubicz 46, 31-512 Krakow, Poland
Correspondence should be addressed to Nataliya Didyk, natasha [email protected]
Received 9 January 2011; Revised 30 March 2011; Accepted 28 April 2011
Academic Editor: Cinta Porte
Copyright © 2011 Oleg Blum et al. This is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Some modern agrochemicals with antioxidant potential were tested for their protective effect against ozone injury using cloverand tobacco ozone-sensitive cultivars as model plants subjected to ambient ozone at two sites (Kyiv city in Ukraine and Szarowvillage in Poland). All used agrochemicals showed partial protective effects against ozone injury on clover and tobacco. Conductedstudies confirmed the effectiveness of modern fungicides belonging to strobilurin group as protectants of sensitive crops againstozone damage. The effectiveness of new growth regulators “Emistym C” and “Agrostymulin” was showed for the first time. Outof the studied agrochemicals, fungicide “Strobi” and natural growth regulator “Emistym C” demonstrated the best protectiveeffects. These agrochemicals present promise for further studies of their possible utilization for enhancement of ozone toleranceof sensitive crops.
1. Introduction
Ozone is one of the major toxic to vegetation and humanhealth gaseous air pollutants. It contributes to crop losses andforest decline. It was estimated that the present day globalrelative yield losses range between 7% and 12% for wheat,between 6% and 16% for soybean, between 3% and 4% forrice, and between 3% and 5% for maize (range resulting fromdifferent metrics used) [1]. For the year 2000 global cropyield loss due to ambient ozone was estimated to be worth$14–26 billion. About 40% of this damage is occurring inChina and India [1]. Crop yield loss due to ambient ozonein the U.S.A. was estimated to be worth $3–5 billion annually[2].
The ozone-induced crop losses could be mitigated by twoways: (i) selection of O3-tolerant cultivars; (ii) applicationof chemical protectants. So far there has been only limitedsuccess in producing genetically transformed plants withincreased tolerance to elevated ozone concentrations becauseplant resistance to ozone is a very complicated phenomenoninvolving multiple signaling pathways and defense responses[3, 4].
The phytotoxicity of ozone results primarily from theoxidative stress imposed by the pollutant on sensitivecomponents of the plasmalemma [3, 5]. In connection tothis, application of antioxidants for protection of crops fromozone injury has been extensively studied over the last fourdecades [6, 7]. A large number of antioxidants (fungicides,insecticides, herbicides, growth regulators, etc.) have beenevaluated [6]. Among them the systemic antioxidant ethy-lene diurea—N-[2-(2-oxo-1-imidazolidinyl) ethyl]-N′ phe-nylurea (EDU) was found to be the most efficient [6]. EDU,applied as a foliar spray, soil drench, or stem infusion agent,was shown to prevent acute ozone injury and inhibit plantsenescence [6, 8]. Physiological effects of EDU associatedwith its protective properties are still unclear. Yet, there issome evidence that EDU may confer tolerance to ozonethrough the induction of enzyme systems involved in theelimination of activated oxygen species and free radicals [9].
Presently, modern agrochemicals containing antioxidantcompounds, such as fungicides belonging to strobilurinsand triazoles (azoxystrobin, epoxiconazole, penconazole,etc.), are gaining increasing attention as possible ozone

2 Journal of Toxicology
protectants [10]. The above-mentioned fungicides, especiallystrobilurins, which were developed on the basis of naturalsubstances extracted from the fungus Strobilurus tenacellus(Pers.) Singer, have low toxicity for human health and envi-ronment. Therefore they are more preferable for applicationin agriculture as protectants, than other synthetic agroche-micals such as EDU. In spite of the above-said a ratherlimited number of studies have been devoted to the protec-tive effects of strobilurins against oxidative stress from theenvironment. Yet it was shown that application of strobil-urins to plants caused marked enhancement of antioxidativeenzymes and enhanced scavenging of potentially harmfulreactive oxygen species.
Another promising group of agrochemicals is growthregulators containing phytohormones or/and others antio-xidants. Apart from their effects on plant development, somephytohormones, that is, cytokinins, gibberellins, salicylicacid and their synthetic analogs were shown to enhance tol-erance of crops to abiotic stresses including ambient ozone[6, 7]. The aim of this study was to assess protective prop-erties of some modern agrochemicals with antioxidantpotential including three fungicides belonging to strobilurinsand some new growth regulators in comparison with EDU.
2. Materials and Methods
The experiments were conducted simultaneously on twomonitoring stations located in semiurban (National BotanicGarden in Kyiv city, Ukraine) and rural (Field Station ofInstitute of Botany Polish Academy of Sciences in Szarow,30 km from Krakow, Poland) sites. In Kyiv plants of subter-ranean clover (Trifolium subterraneum L., O3-sensitive cv.Geraldton) and tobacco (Nicotiana tabacum L.) cvs. Bel-W3 (O3-sensitive) and Bel-B (O3-resistant) were used. InSzarow only subterranean clover was used. Prior to exposureto ozone, tobacco and clover were cultivated in ozone freeair until they reach 4 leaves. In Kyiv plants were cultivatedin the special chamber, made from organic glass, withthe volume of 1.1 m3, at 25–28◦C temperature, 65–70%RH, light illumination of 66 μmol m−2·s−1 PAR and 14 : 10(L : D) h photoperiod. The air was pumped up throughcharcoal filter with the rate of 0.3 m3 per min. In Szarowplants were cultivated in the special ozone free greenhouseat temperature of 25 ± 2◦C with a relative humidity of75% under natural photoperiod. Afterwards, the plants weresprayed with distilled water (control) or solutions of one ofthe following agrochemicals: (1) EDU (ethylenediurea), inconcentrations of 150 mg L−1; (2) “Emistym C”, a naturalgrowth regulator containing exometabolites of micorhizalfungi (flavonoids, phytohormones and organic acids), inconcentration of 30 and 150 mg L−1; (3) “Agrostimulin,” con-taining synthetic growth regulator—ivin, in concentrationof 30 and 150 mg L−1; modern fungicides belonging to thegroup of strobilurins such as (4) “Quadris” (containing 25%of azoxystrobin ) in concentration of 150 and 300 mg L−1; (5)“Flint” (containing 50% of trifloxystrobin) in concentrationof 150 and 300 mg L−1; (6) “Strobi” (containing 50% ofkresoxim-methyl) in concentration of 150 and 300 mg L−1.
0
10
20
30
40
50
60
70
80
Inju
red
leav
es(%
)
Tobacco
1 1 2 3 4 5 6 7Bel-B Bel-W3
Treatment
LSD0.05 = 4.6
0
5
10
15
20
25
30
35
1 2 3 4 5 6 7 1 2 3 4 5 6 7
Treatment
Kyiv SzarowClover
LSD0.05 = 3.2 LSD0.05 = 2.8
Figure 1: Ozone-induced visible injuries on leaves of cloverand tobacco sprayed with distilled water (1), solutions of EDU,150 mg L−1 (2), “Emistym C”, 150 mg L−1 in Kyiv and 30 mg L−1
in Szarow (3), “Agrostymulin”, 150 mg L−1 in Kyiv and 30 mg L−1
in Szarow (4), “Quadris”, 300 mg L−1 in Kyiv and 150 mg L−1 inSzarow (5), “Flint”, 300 mg L−1 in Kyiv and 150 mg L−1 in Szarow(6), “Strobi”, 300 mg L−1 in Kyiv and 150 mg L−1 in Szarow (7) onthe last day of exposure to ambient ozone. Vertical bars representstandard error. LSD0,05: least significant difference at P < 0.05.
Ozone protective effect of “Emistym C” and “Agrostimulin”has been studied for the first time.
Two modes of agrochemical application (except forEDU) were studied. In Kyiv higher concentrations of thechemicals were applied only once (one day before exposure).In Sharow lower concentrations of agrochemicals wereapplied twice (one day before exposure and on 16th day ofexposure). We used EDU as a standard ozone protectant,in concentration, whose effectiveness is well established onmany plant species [6, 8].
The test plants were exposed into the field on thefollowing day after application of agrochemicals. The dura-tion of exposure of the test plants to ambient ozone was2 and 4 weeks for tobacco and clover, respectively. Onthe last day of exposure ozone-induced foliar injury (%)and content of photosynthetic pigments (chlorophylls aand b, carotenoids) in leaves of test plants were evaluated.Photosynthetic pigments were extracted by 100% acetoneand determined spectrophotometrically using “Spekol 11”(Carl zeiss, Jena, Germany) [11]. Dry biomass of test plants
Journal of Toxicology 3
0
20
40
60
80
100
120
140
Con
ten
tof
phot
osyn
thet
icpi
gmen
ts,m
gp
er10
0g
ofle
affr
esh
wei
ght
1 2 3 4 5 6 7
Treatment
2 2
1
1 2
Chlorophyll a
Chlorophyll b
Carotenoids
LSD0.05 =
LSD0.05 =
LSD0.05 =
3.
.
.
(a)
0
0
5
10
15
20
25
30
35
40
1 2 3 4 5 6 7
Treatment
1
Bel-B Bel-W3
2Chlorophyll a
Chlorophyll b
Carotenoids
Con
ten
tof
phot
osyn
thet
icpi
gmen
ts,m
gp
er10
0g
ofle
affr
esh
wei
ght
LSD0.05 =
LSD0.05 =
LSD0.05 =
3
.
2.
1.8
(b)
Figure 2: Content of photosynthetic pigments in leaves of clover(a) and tobacco (b) sprayed with distilled water (1), solutions ofEDU, 150 mg L−1 (2), “Emistym C”, 150 mg L−1 (3), “Agrostymulin”150 mg L−1 (4), “Quadris”, 300 mg L−1 (5), “Flint”, 300 mg L−1 (6),“Strobi”, 300 mg L−1 (7) on the last day of exposure to ambientozone in Kyiv. Vertical bars represent standard error. LSD0.05: leastsignificant difference at P < 0.05 for ∗chlorophyll a; ∗∗chlorophyllb; ∗∗∗carotenoids.
was also determined. For clover eight replications of tenplants each per treatment were used in both locations. Thus,values reported are the means of 80 plants. For tobacco tenreplications of one plant each per treatment were used. Thus,values reported are the means of 10 plants.
During the period of the experiments ambient ozoneconcentrations were continuously monitored at the bothlocations. In Kyiv and Szarow ozone monitoring was con-ducted using the Thermo Environmental UV Photometric
1.4
1.2
1
0.8
0.6
0.4
0.2
Toba
cco,
dry
wei
ght
(gp
erpl
ant)
Bel-B Bel-W3
Kyiv
Treatment
1 2 3 4 5 6 7
LSD0.05 = 2.1
Treatment
1 2 3 4 5 6 7 1 2 3 4 5 6 7
SzarowKyiv
0
2
4
6
8
10
12
14
Clo
ver,
dry
wei
ght
(mg
per
pot)
LSD0.05 = 1.1LSD0.05 = 1.4
Figure 3: Mean dry weight of clover and tobacco sprayed withdistilled water (control) (1), solutions of EDU, 150 mg L−1 (2),“Emistym C”, 150 mg L−1 in Kyiv and 30 mg L−1 in Szarow (3),“Agrostymulin”, 150 mg L−1 in Kyiv and 30 mg L−1 in Szarow (4),“Quadris”, 300 mg L−1 in Kyiv and 150 mg L−1 in Szarow (5),“Flint”, 300 mg L−1 in Kyiv and 150 mg L−1 in Szarow (6), “Strobi”,300 mg L−1 in Kyiv and 150 mg L−1 in Szarow (7) on the last day ofexposure to ambient ozone. Vertical bars represent standard error.LSD0.05— least significant difference at P < 0.05.
Ozone Analyzer Model 49-003 and Model 49 C, correspond-ingly. Average daily ozone and temperature concentrations aswell as the doses of ozone, which test plants received duringthe period of experimentation were calculated using index ofAOT 40 (accumulated exposure over a threshold of 40 ppb).
Statistical analysis of the results obtained was conductedwith the usage of descriptive statistics and analysis ofvariance. Treatment means were compared using ANOVAand LSD-test (Statistica 6.0 software) [12].
3. Results and Discussion
For the most part of the periods of observations the averagedaily ambient ozone concentrations exceeded the thresholdof subterranean clover and tobacco Bel-W3 cv. sensitivityto ozone of 25 ppb [13, 14] in both experimental sites.The dose of ozone (calculated using index of AOT 40),which test plants received during the exposition periods,

4 Journal of Toxicology
was 828 ppb·h for clover and 700 ppb·h for tobacco inKyiv, and 1662 ppb·h for clover in Sharow. Despite the factthat in Szarow ambient ozone concentrations during theperiod of experimentation were higher than in Kyiv, cloverplants in control (without application of agrochemicals)displayed similar degree of visible foliar injury (about 28%)in both sites (Figure 1). However, dry phytomass of cloverplants in Sharow was almost two times lower than thatin Kyiv. O3-sensitive tobacco plants demonstrated highersensitivity to ozone than clover in terms of visible foliarinjuries. All the applied agrochemicals (with the exceptionof “Quadris”, which have no significant effect on foliarinjuries and biomass, accumulation in tobacco) showedpartial protective effect to O3-sensitive plants. The plantstreated with the solutions of the mentioned agrochemicalshad less foliar injuries, higher biomass and higher content ofphotosynthetic pigments (chlorophylls a and b) in leaves ascompared to control (sprayed with distil water) (Figures 1, 2,and 3). Tobacco plants treated with solutions of “EmistymC” had even higher biomass than plants of O3-resistantBel-B cv. treated with distill water, which was evidentlydue to the growth promoting effect of this agrochemicalrather than its ozone protective effect. Among the studiedagrochemicals the most effective as protectant against ozone-induced injuries was fungicide “Strobi”. Natural growthregulator “Emistym C” was slightly less effective. Fungicide“Quadris” demonstrated the lowest effectiveness.
In terms of visible foliar injuries and biomass accumula-tion, all the applied agrochemicals more effectively protectedclover plants when they were applied two times (one daybefore exposure and on 16th day of exposure) in lowerconcentration (in Szarow) than when they were applied once(one day before exposure) in higher concentration (in Kyiv).For biomass accumulation, this tendency was less defined.It could be supposed that repeated application of the testedagrochemicals even in lower concentrations is more effectiveas it allows protection of newly emerged leaves. Though, thedifferences in ozone doses, which plants received in bothlocations, could also contribute to the observed effects. Thedegree of protective effect for all tested agrochemicals washigher in clover plants as compared to tobacco. Evidentlyit was a consequence of higher degree of ozone injuries intobacco as compared to clover.
Thus, conducted studies confirmed the effectiveness ofozone protective effect of EDU and fungicides belongingto the group of strobilurins [6, 8, 10]. The protectiveeffect of growth regulators “Emistym C” and “Agrostimulin”against ozone damage of plants has been shown for the firsttime. Out of the studied agrochemicals fungicide “Strobi”and natural growth regulator “Emistym C” demonstratedthe best protective effects. These agrochemicals presentpromise for further studies of their possible utilization forenhancement of ozone tolerance of sensitive crops.
Acknowledgments
This study was funded by STCU (Project N3894) and bythe “M. M. Gryshko” National Botanical Garden of the
National Academy of Sciences of Ukraine and by the W.Szafer Institute of Botany of Polish Academy of Sciences.
References
[1] R. Van Dingenen, F. J. Dentener, F. Raes, M. C. Krol, L.Emberson, and J. Cofala, “The global impact of ozone onagricultural crop yields under current and future air qualitylegislation,” Atmospheric Environment, vol. 43, no. 3, pp. 604–618, 2009.
[2] E. L. Fiscus, F. L. Booker, and K. O. Burkey, “Crop responsesto ozone: uptake, modes of action, carbon assimilation andpartitioning,” Plant, Cell and Environment, vol. 28, no. 8, pp.997–1011, 2005.
[3] Z. Chen and D. R. Gallie, “Increasing tolerance to ozone byelevating foliar ascorbic acid confers greater protection againstozone than increasing avoidance,” Plant Physiology, vol. 138,no. 3, pp. 1673–1689, 2005.
[4] N. Tosti, S. Pasqualini, A. Borgogni et al., “Gene expressionprofiles of O3-treated Arabidopsis plants,” Plant, Cell andEnvironment, vol. 29, no. 9, pp. 1686–1702, 2006.
[5] M. C. Puckette, Y. Tang, and R. Mahalingam, “Transcriptomicchanges induced by acute ozone in resistant and sensitiveMedicago truncatula accessions,” BMC Plant Biology, vol. 8,article 46, 2008.
[6] D. Archambault, D. J. Slaski, and J. J. Li, “Ozone protection inplants. The potential use of chemical protectants to measureoxidant damage in Alberta crops,” Report prepared for theAir Research Users Group, Alberta Environment, Edmonton,Canada, 2000.
[7] N. P. Didyk and O. B. Blum, “Natural antioxidants of plant ori-gin against ozone damage of sensitive crops,” Acta PhysiologiaePlantarum, vol. 33, no. 1, pp. 25–34, 2011.
[8] B. Godzik and W. J. Manning, “Relative effectiveness of eth-ylenediurea, and constituent amounts of urea and phenylureain ethylenediurea, in prevention of ozone injury to tobacco,”Environmental Pollution, vol. 103, no. 1, pp. 1–6, 1998.
[9] B. D. Whitaker, E. H. Lee, and R. A. Rowland, “EDU and ozoneprotection: foliar glycerolipids and steryl lipids in snapbeanexposed to O3,” Physiologia Plantarum, vol. 80, no. 2, pp. 286–293, 1990.
[10] Y. X. Wu and A. Von Tiedemann, “Impact of fungicides onactive oxygen species and antioxidant enzymes in spring barley(Hordeum vulgare L.) exposed to ozone,” EnvironmentalPollution, vol. 116, no. 1, pp. 37–47, 2002.
[11] M. M. Musiyenko, T. B. Parshikova, and P. C. Slavnyi, Spec-trophotometric Methods in the Practice of Physiology, Biochem-istry and Ecology of Plants, Phytosociotsentr, Kyiv, Ukraine,2001.
[12] G. N. Zaytsev, Mathematical Statistics in Experimental Botany,Nauka, Moscow, Russia, 1984.
[13] G. P. Karlsson, H. Pleijel, E. Sild et al., “Clover Sweden—anational three-year study of the effects of tropospheric ozoneon Trifolium subterraneum, L.,” Water, Air, and Soil Pollution,vol. 85, no. 3, pp. 1503–1508, 1995.
[14] H. E. Heggestad, “Origin of Bel-W3, Bel-C and Bel-B tobaccovarieties and their use as indicators of ozone,” EnvironmentalPollution, vol. 74, no. 4, pp. 264–291, 1991.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 467305, 12 pagesdoi:10.1155/2011/467305
Review Article
Research Strategies in the Study of the Pro-Oxidant Nature ofPolyphenol Nutraceuticals
Harvey Babich, Alyssa G. Schuck, Jeffrey H. Weisburg, and Harriet L. Zuckerbraun
Department of Biology, Stern College for Women, Yeshiva University, 245 Lexington Avenue, New York, NY 10016, USA
Correspondence should be addressed to Harvey Babich, [email protected]
Received 10 January 2011; Accepted 12 April 2011
Academic Editor: P. J. O’Brien
Copyright © 2011 Harvey Babich et al. This is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Polyphenols of phytochemicals are thought to exhibit chemopreventive effects against cancer. These plant-derived antioxidantpolyphenols have a dual nature, also acting as pro-oxidants, generating reactive oxygen species (ROS), and causing oxidative stress.When studying the overall cytotoxicity of polyphenols, research strategies need to distinguish the cytotoxic component derivedfrom the polyphenol per se from that derived from the generated ROS. Such strategies include (a) identifying hallmarks of oxidativedamage, such as depletion of intracellular glutathione and lipid peroxidation, (b) classical manipulations, such as polyphenolexposures in the absence and presence of antioxidant enzymes (i.e., catalase and superoxide dismutase) and of antioxidants(e.g., glutathione and N-acetylcysteine) and cotreatments with glutathione depleters, and (c) more recent manipulations, suchas divalent cobalt and pyruvate to scavenge ROS. Attention also must be directed to the influence of iron and copper ions and tothe level of polyphenols, which mediate oxidative stress.
1. Introduction
Concerns to reduce the risk of chronic diseases have ledto much research to discern those lifestyle choices that po-tentially lessen developing chronic pathologies, cancer inparticular. Epidemiological evidence has shown that thedietary consumption of fruits and vegetables can mediatethe risk of developing malignancies. Studies with laboratoryanimal models and in vitro research with cells in culturehave confirmed the anticarcinogenic effects of natural phy-tochemicals. In particular, the polyphenol components ofphytochemicals have been identified as anticarcinogens. Themost studied polyphenol is (−)-epigallocatechin-3-gallate(EGCG), the major constituent in green tea. Such nonnu-tritive polyphenol phytochemicals are termed nutraceuticals,and their ready bioavailability makes their consumptionas potential cancer chemopreventive agents a meaningfullifestyle choice [1].
Polyphenols, a heterogeneous class of phytochemicalswith a wide range of pharmacological properties, are mostknown for their antioxidant properties and their abilitiesto act as scavengers of reactive oxygen species (ROS).ROS include hydrogen peroxide (H2O2), superoxide anion
(O2·−), and hydroxyl radical (OH·). ROS are formed as
by-products of mitochondrial respiration or by certainoxidases, such as nicotine adenine dinucleotide phosphate(NADPH) oxidase. ROS are involved in many cellular events,including as second messengers in the activation of severalsignaling pathways leading to the activation of transcriptionfactors, mitogenesis, gene expression, and the induction ofapoptosis, or programmed cell death [2–4]. Overproductionof ROS, as indicated by a change in the redox state of thecell, may lead to oxidative damage of proteins, lipids, andDNA. To prevent oxidative stress, neutralization of excessiveROS is accomplished by antioxidant enzymes, includingsuperoxide dismutase (SOD) to detoxify O2
·− and catalaseand glutathione peroxidase to detoxify H2O2. In addition, thetripeptide, glutathione (γ-glutamylcysteinylglycine; GSH),plays a major role in maintaining intracellular redox balanceand in alleviating ROS-induced oxidative stress. Synthesizedenzymatically by γ-glutamylcysteine synthetase and glu-tathione synthetase, a prime function of GSH is to scavengeROS and thereby to prevent oxidative damage [5]. Becauseoxidative stress has been implicated with cancer, as wellas with other chronic diseases and pathologies, includingatherosclerosis, neurodegenerative diseases, and aging, much
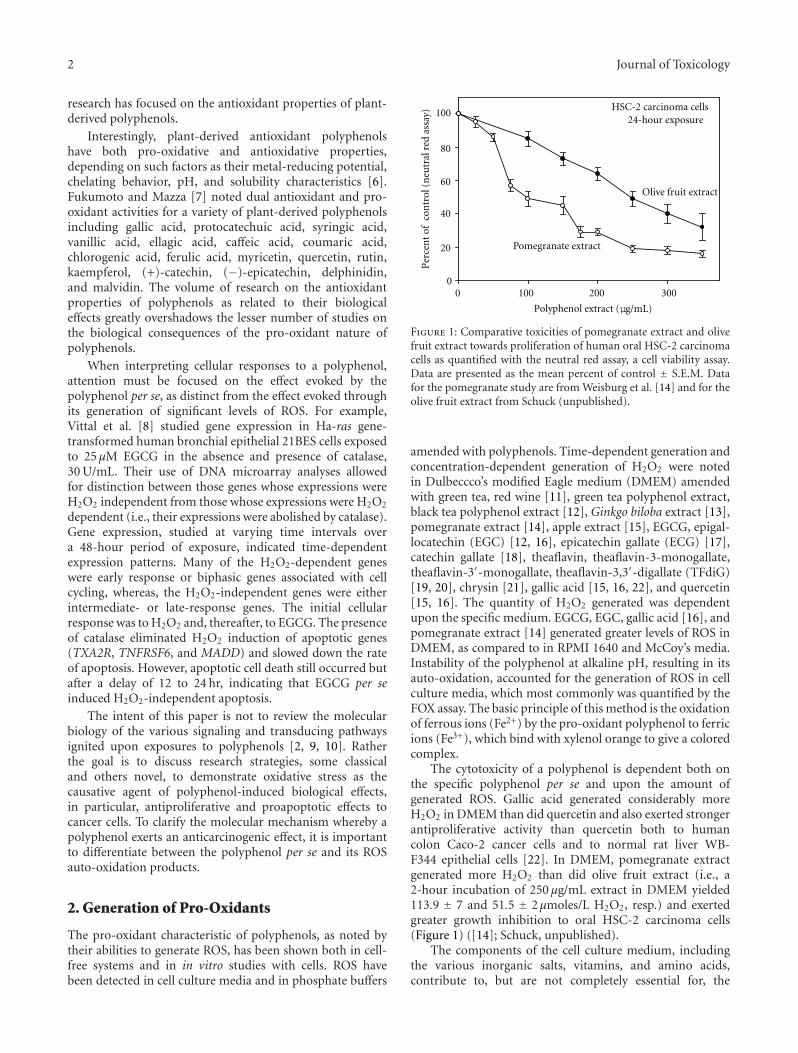
2 Journal of Toxicology
research has focused on the antioxidant properties of plant-derived polyphenols.
Interestingly, plant-derived antioxidant polyphenolshave both pro-oxidative and antioxidative properties,depending on such factors as their metal-reducing potential,chelating behavior, pH, and solubility characteristics [6].Fukumoto and Mazza [7] noted dual antioxidant and pro-oxidant activities for a variety of plant-derived polyphenolsincluding gallic acid, protocatechuic acid, syringic acid,vanillic acid, ellagic acid, caffeic acid, coumaric acid,chlorogenic acid, ferulic acid, myricetin, quercetin, rutin,kaempferol, (+)-catechin, (−)-epicatechin, delphinidin,and malvidin. The volume of research on the antioxidantproperties of polyphenols as related to their biologicaleffects greatly overshadows the lesser number of studies onthe biological consequences of the pro-oxidant nature ofpolyphenols.
When interpreting cellular responses to a polyphenol,attention must be focused on the effect evoked by thepolyphenol per se, as distinct from the effect evoked throughits generation of significant levels of ROS. For example,Vittal et al. [8] studied gene expression in Ha-ras gene-transformed human bronchial epithelial 21BES cells exposedto 25 μM EGCG in the absence and presence of catalase,30 U/mL. Their use of DNA microarray analyses allowedfor distinction between those genes whose expressions wereH2O2 independent from those whose expressions were H2O2
dependent (i.e., their expressions were abolished by catalase).Gene expression, studied at varying time intervals overa 48-hour period of exposure, indicated time-dependentexpression patterns. Many of the H2O2-dependent geneswere early response or biphasic genes associated with cellcycling, whereas, the H2O2-independent genes were eitherintermediate- or late-response genes. The initial cellularresponse was to H2O2 and, thereafter, to EGCG. The presenceof catalase eliminated H2O2 induction of apoptotic genes(TXA2R, TNFRSF6, and MADD) and slowed down the rateof apoptosis. However, apoptotic cell death still occurred butafter a delay of 12 to 24 hr, indicating that EGCG per seinduced H2O2-independent apoptosis.
The intent of this paper is not to review the molecularbiology of the various signaling and transducing pathwaysignited upon exposures to polyphenols [2, 9, 10]. Ratherthe goal is to discuss research strategies, some classicaland others novel, to demonstrate oxidative stress as thecausative agent of polyphenol-induced biological effects,in particular, antiproliferative and proapoptotic effects tocancer cells. To clarify the molecular mechanism whereby apolyphenol exerts an anticarcinogenic effect, it is importantto differentiate between the polyphenol per se and its ROSauto-oxidation products.
2. Generation of Pro-Oxidants
The pro-oxidant characteristic of polyphenols, as noted bytheir abilities to generate ROS, has been shown both in cell-free systems and in in vitro studies with cells. ROS havebeen detected in cell culture media and in phosphate buffers
Polyphenol extract (
0 100 200 300
Per
cen
t of
con
trol
(n
eutr
al r
ed a
ssay
)
0
20
40
60
80
10024-hour exposure
Pomegranate extract
Olive fruit extract
HSC-2 carcinoma cells
μg/mL)
Figure 1: Comparative toxicities of pomegranate extract and olivefruit extract towards proliferation of human oral HSC-2 carcinomacells as quantified with the neutral red assay, a cell viability assay.Data are presented as the mean percent of control ± S.E.M. Datafor the pomegranate study are from Weisburg et al. [14] and for theolive fruit extract from Schuck (unpublished).
amended with polyphenols. Time-dependent generation andconcentration-dependent generation of H2O2 were notedin Dulbeccco’s modified Eagle medium (DMEM) amendedwith green tea, red wine [11], green tea polyphenol extract,black tea polyphenol extract [12], Ginkgo biloba extract [13],pomegranate extract [14], apple extract [15], EGCG, epigal-locatechin (EGC) [12, 16], epicatechin gallate (ECG) [17],catechin gallate [18], theaflavin, theaflavin-3-monogallate,theaflavin-3′-monogallate, theaflavin-3,3′-digallate (TFdiG)[19, 20], chrysin [21], gallic acid [15, 16, 22], and quercetin[15, 16]. The quantity of H2O2 generated was dependentupon the specific medium. EGCG, EGC, gallic acid [16], andpomegranate extract [14] generated greater levels of ROS inDMEM, as compared to in RPMI 1640 and McCoy’s media.Instability of the polyphenol at alkaline pH, resulting in itsauto-oxidation, accounted for the generation of ROS in cellculture media, which most commonly was quantified by theFOX assay. The basic principle of this method is the oxidationof ferrous ions (Fe2+) by the pro-oxidant polyphenol to ferricions (Fe3+), which bind with xylenol orange to give a coloredcomplex.
The cytotoxicity of a polyphenol is dependent both onthe specific polyphenol per se and upon the amount ofgenerated ROS. Gallic acid generated considerably moreH2O2 in DMEM than did quercetin and also exerted strongerantiproliferative activity than quercetin both to humancolon Caco-2 cancer cells and to normal rat liver WB-F344 epithelial cells [22]. In DMEM, pomegranate extractgenerated more H2O2 than did olive fruit extract (i.e., a2-hour incubation of 250 μg/mL extract in DMEM yielded113.9 ± 7 and 51.5 ± 2μmoles/L H2O2, resp.) and exertedgreater growth inhibition to oral HSC-2 carcinoma cells(Figure 1) ([14]; Schuck, unpublished).
The components of the cell culture medium, includingthe various inorganic salts, vitamins, and amino acids,contribute to, but are not completely essential for, the
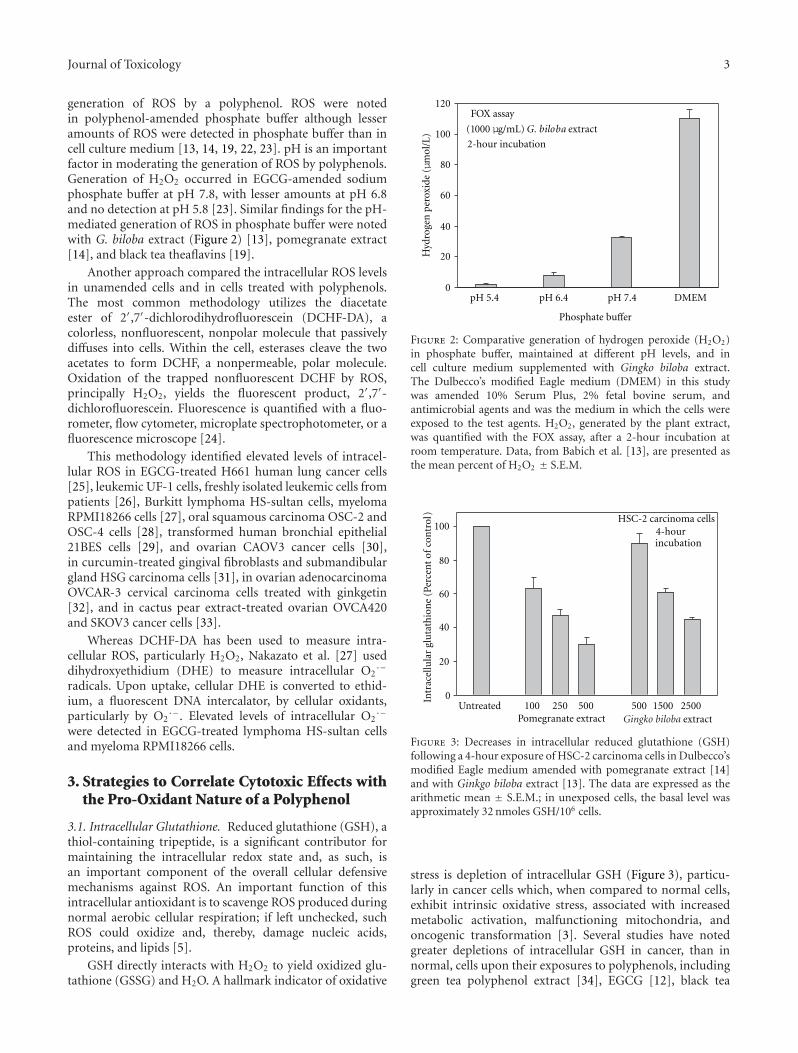
Journal of Toxicology 3
generation of ROS by a polyphenol. ROS were notedin polyphenol-amended phosphate buffer although lesseramounts of ROS were detected in phosphate buffer than incell culture medium [13, 14, 19, 22, 23]. pH is an importantfactor in moderating the generation of ROS by polyphenols.Generation of H2O2 occurred in EGCG-amended sodiumphosphate buffer at pH 7.8, with lesser amounts at pH 6.8and no detection at pH 5.8 [23]. Similar findings for the pH-mediated generation of ROS in phosphate buffer were notedwith G. biloba extract (Figure 2) [13], pomegranate extract[14], and black tea theaflavins [19].
Another approach compared the intracellular ROS levelsin unamended cells and in cells treated with polyphenols.The most common methodology utilizes the diacetateester of 2′,7′-dichlorodihydrofluorescein (DCHF-DA), acolorless, nonfluorescent, nonpolar molecule that passivelydiffuses into cells. Within the cell, esterases cleave the twoacetates to form DCHF, a nonpermeable, polar molecule.Oxidation of the trapped nonfluorescent DCHF by ROS,principally H2O2, yields the fluorescent product, 2′,7′-dichlorofluorescein. Fluorescence is quantified with a fluo-rometer, flow cytometer, microplate spectrophotometer, or afluorescence microscope [24].
This methodology identified elevated levels of intracel-lular ROS in EGCG-treated H661 human lung cancer cells[25], leukemic UF-1 cells, freshly isolated leukemic cells frompatients [26], Burkitt lymphoma HS-sultan cells, myelomaRPMI18266 cells [27], oral squamous carcinoma OSC-2 andOSC-4 cells [28], transformed human bronchial epithelial21BES cells [29], and ovarian CAOV3 cancer cells [30],in curcumin-treated gingival fibroblasts and submandibulargland HSG carcinoma cells [31], in ovarian adenocarcinomaOVCAR-3 cervical carcinoma cells treated with ginkgetin[32], and in cactus pear extract-treated ovarian OVCA420and SKOV3 cancer cells [33].
Whereas DCHF-DA has been used to measure intra-cellular ROS, particularly H2O2, Nakazato et al. [27] useddihydroxyethidium (DHE) to measure intracellular O2
·−
radicals. Upon uptake, cellular DHE is converted to ethid-ium, a fluorescent DNA intercalator, by cellular oxidants,particularly by O2
·−. Elevated levels of intracellular O2·−
were detected in EGCG-treated lymphoma HS-sultan cellsand myeloma RPMI18266 cells.
3. Strategies to Correlate Cytotoxic Effects withthe Pro-Oxidant Nature of a Polyphenol
3.1. Intracellular Glutathione. Reduced glutathione (GSH), athiol-containing tripeptide, is a significant contributor formaintaining the intracellular redox state and, as such, isan important component of the overall cellular defensivemechanisms against ROS. An important function of thisintracellular antioxidant is to scavenge ROS produced duringnormal aerobic cellular respiration; if left unchecked, suchROS could oxidize and, thereby, damage nucleic acids,proteins, and lipids [5].
GSH directly interacts with H2O2 to yield oxidized glu-tathione (GSSG) and H2O. A hallmark indicator of oxidative
Hyd
roge
np
erox
ide
(μm
ol/L
)
0
20
40
60
80
100
120
pH 5.4 pH 6.4 pH 7.4
Phosphate buffer
DMEM
FOX assay
(1000 μg/mL) G. biloba extract
2-hour incubation
Figure 2: Comparative generation of hydrogen peroxide (H2O2)in phosphate buffer, maintained at different pH levels, and incell culture medium supplemented with Gingko biloba extract.The Dulbecco’s modified Eagle medium (DMEM) in this studywas amended 10% Serum Plus, 2% fetal bovine serum, andantimicrobial agents and was the medium in which the cells wereexposed to the test agents. H2O2, generated by the plant extract,was quantified with the FOX assay, after a 2-hour incubation atroom temperature. Data, from Babich et al. [13], are presented asthe mean percent of H2O2 ± S.E.M.
0
20
40
60
80
100
Intr
acel
lula
rgl
uta
thio
ne
()
Untreated 100 250 500 500 1500 2500Pomegranate extract extract
HSC-2 carcinoma cells
Perc
ent
of c
ontr
ol
Gingko biloba
4-hourincubation
Figure 3: Decreases in intracellular reduced glutathione (GSH)following a 4-hour exposure of HSC-2 carcinoma cells in Dulbecco’smodified Eagle medium amended with pomegranate extract [14]and with Ginkgo biloba extract [13]. The data are expressed as thearithmetic mean ± S.E.M.; in unexposed cells, the basal level wasapproximately 32 nmoles GSH/106 cells.
stress is depletion of intracellular GSH (Figure 3), particu-larly in cancer cells which, when compared to normal cells,exhibit intrinsic oxidative stress, associated with increasedmetabolic activation, malfunctioning mitochondria, andoncogenic transformation [3]. Several studies have notedgreater depletions of intracellular GSH in cancer, than innormal, cells upon their exposures to polyphenols, includinggreen tea polyphenol extract [34], EGCG [12], black tea
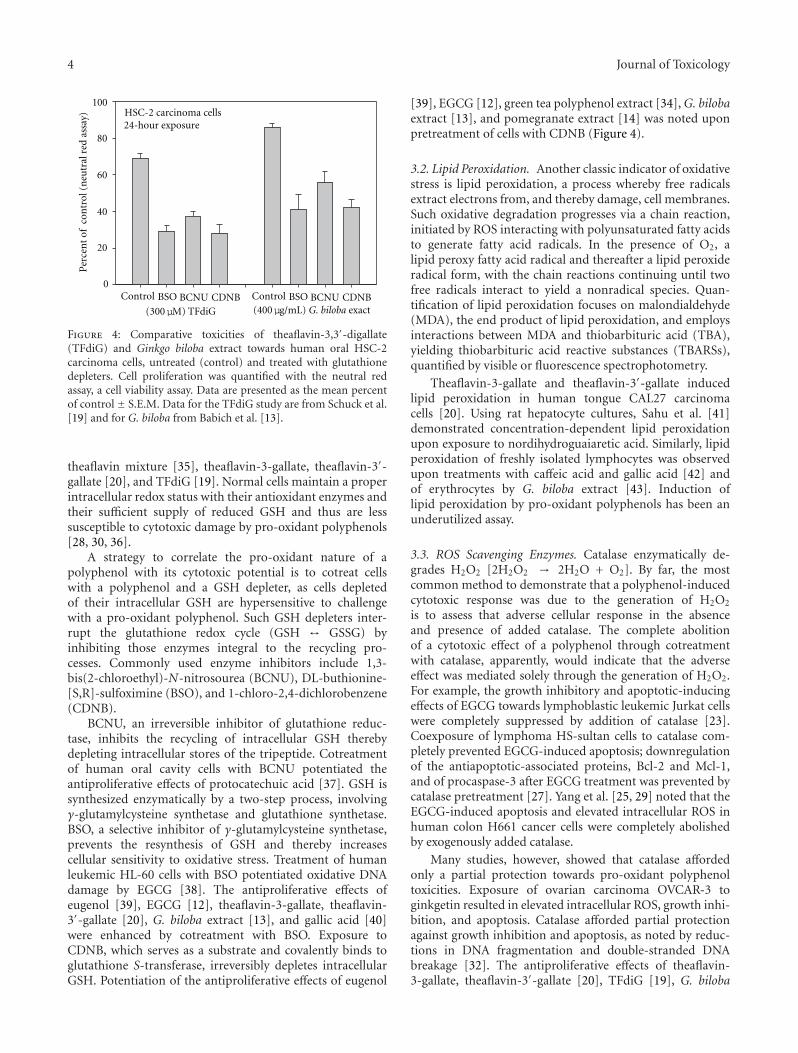
4 Journal of Toxicology
HSC-2 carcinoma cells
Control BSO BCNU CDNB Control BSO BCNU CDNB(300μM) TFdiG (400μg/mL) G. biloba exact
0
20
40
60
80
100
24-hour exposure
Per
cen
t of
con
trol
(n
eutr
al r
ed a
ssay
)
Figure 4: Comparative toxicities of theaflavin-3,3′-digallate(TFdiG) and Ginkgo biloba extract towards human oral HSC-2carcinoma cells, untreated (control) and treated with glutathionedepleters. Cell proliferation was quantified with the neutral redassay, a cell viability assay. Data are presented as the mean percentof control ± S.E.M. Data for the TFdiG study are from Schuck et al.[19] and for G. biloba from Babich et al. [13].
theaflavin mixture [35], theaflavin-3-gallate, theaflavin-3′-gallate [20], and TFdiG [19]. Normal cells maintain a properintracellular redox status with their antioxidant enzymes andtheir sufficient supply of reduced GSH and thus are lesssusceptible to cytotoxic damage by pro-oxidant polyphenols[28, 30, 36].
A strategy to correlate the pro-oxidant nature of apolyphenol with its cytotoxic potential is to cotreat cellswith a polyphenol and a GSH depleter, as cells depletedof their intracellular GSH are hypersensitive to challengewith a pro-oxidant polyphenol. Such GSH depleters inter-rupt the glutathione redox cycle (GSH ↔ GSSG) byinhibiting those enzymes integral to the recycling pro-cesses. Commonly used enzyme inhibitors include 1,3-bis(2-chloroethyl)-N-nitrosourea (BCNU), DL-buthionine-[S,R]-sulfoximine (BSO), and 1-chloro-2,4-dichlorobenzene(CDNB).
BCNU, an irreversible inhibitor of glutathione reduc-tase, inhibits the recycling of intracellular GSH therebydepleting intracellular stores of the tripeptide. Cotreatmentof human oral cavity cells with BCNU potentiated theantiproliferative effects of protocatechuic acid [37]. GSH issynthesized enzymatically by a two-step process, involvingγ-glutamylcysteine synthetase and glutathione synthetase.BSO, a selective inhibitor of γ-glutamylcysteine synthetase,prevents the resynthesis of GSH and thereby increasescellular sensitivity to oxidative stress. Treatment of humanleukemic HL-60 cells with BSO potentiated oxidative DNAdamage by EGCG [38]. The antiproliferative effects ofeugenol [39], EGCG [12], theaflavin-3-gallate, theaflavin-3′-gallate [20], G. biloba extract [13], and gallic acid [40]were enhanced by cotreatment with BSO. Exposure toCDNB, which serves as a substrate and covalently binds toglutathione S-transferase, irreversibly depletes intracellularGSH. Potentiation of the antiproliferative effects of eugenol
[39], EGCG [12], green tea polyphenol extract [34], G. bilobaextract [13], and pomegranate extract [14] was noted uponpretreatment of cells with CDNB (Figure 4).
3.2. Lipid Peroxidation. Another classic indicator of oxidativestress is lipid peroxidation, a process whereby free radicalsextract electrons from, and thereby damage, cell membranes.Such oxidative degradation progresses via a chain reaction,initiated by ROS interacting with polyunsaturated fatty acidsto generate fatty acid radicals. In the presence of O2, alipid peroxy fatty acid radical and thereafter a lipid peroxideradical form, with the chain reactions continuing until twofree radicals interact to yield a nonradical species. Quan-tification of lipid peroxidation focuses on malondialdehyde(MDA), the end product of lipid peroxidation, and employsinteractions between MDA and thiobarbituric acid (TBA),yielding thiobarbituric acid reactive substances (TBARSs),quantified by visible or fluorescence spectrophotometry.
Theaflavin-3-gallate and theaflavin-3′-gallate inducedlipid peroxidation in human tongue CAL27 carcinomacells [20]. Using rat hepatocyte cultures, Sahu et al. [41]demonstrated concentration-dependent lipid peroxidationupon exposure to nordihydroguaiaretic acid. Similarly, lipidperoxidation of freshly isolated lymphocytes was observedupon treatments with caffeic acid and gallic acid [42] andof erythrocytes by G. biloba extract [43]. Induction oflipid peroxidation by pro-oxidant polyphenols has been anunderutilized assay.
3.3. ROS Scavenging Enzymes. Catalase enzymatically de-grades H2O2 [2H2O2 → 2H2O + O2]. By far, the mostcommon method to demonstrate that a polyphenol-inducedcytotoxic response was due to the generation of H2O2
is to assess that adverse cellular response in the absenceand presence of added catalase. The complete abolitionof a cytotoxic effect of a polyphenol through cotreatmentwith catalase, apparently, would indicate that the adverseeffect was mediated solely through the generation of H2O2.For example, the growth inhibitory and apoptotic-inducingeffects of EGCG towards lymphoblastic leukemic Jurkat cellswere completely suppressed by addition of catalase [23].Coexposure of lymphoma HS-sultan cells to catalase com-pletely prevented EGCG-induced apoptosis; downregulationof the antiapoptotic-associated proteins, Bcl-2 and Mcl-1,and of procaspase-3 after EGCG treatment was prevented bycatalase pretreatment [27]. Yang et al. [25, 29] noted that theEGCG-induced apoptosis and elevated intracellular ROS inhuman colon H661 cancer cells were completely abolishedby exogenously added catalase.
Many studies, however, showed that catalase affordedonly a partial protection towards pro-oxidant polyphenoltoxicities. Exposure of ovarian carcinoma OVCAR-3 toginkgetin resulted in elevated intracellular ROS, growth inhi-bition, and apoptosis. Catalase afforded partial protectionagainst growth inhibition and apoptosis, as noted by reduc-tions in DNA fragmentation and double-stranded DNAbreakage [32]. The antiproliferative effects of theaflavin-3-gallate, theaflavin-3′-gallate [20], TFdiG [19], G. biloba
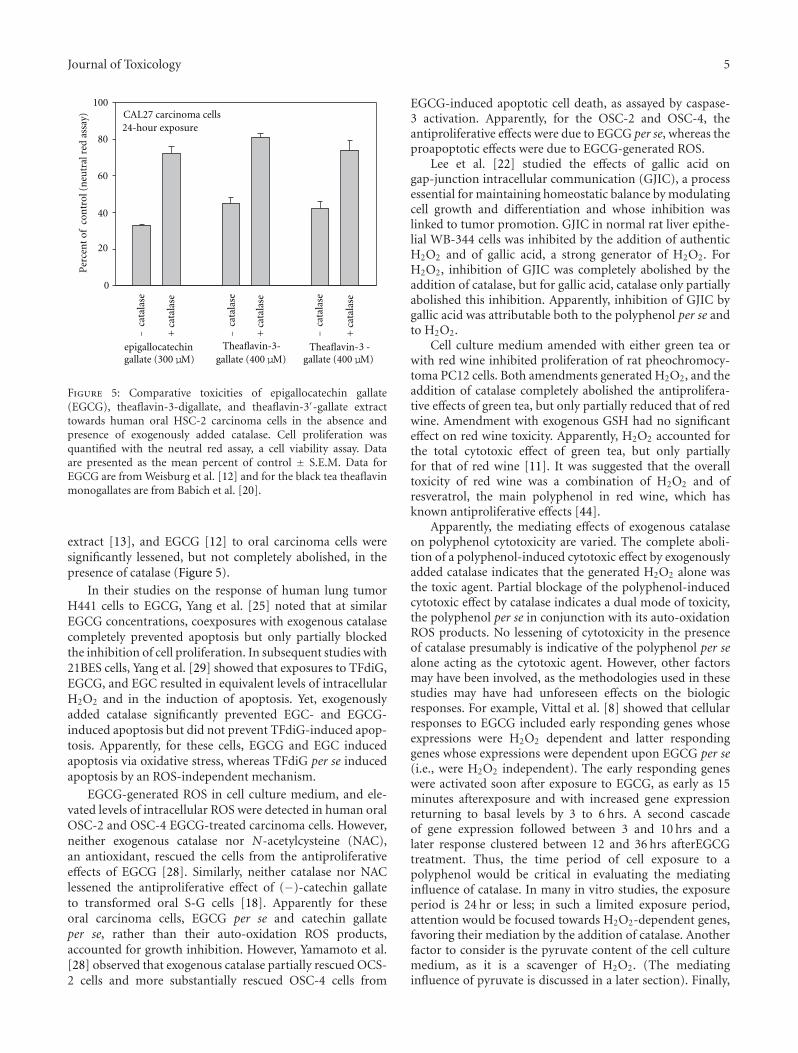
Journal of Toxicology 5
epigallocatechingallate (300 μM)
Theaflavin Theaflavin-3-gallate (400 μM)
- -gallate (400 μM)
CAL27 carcinoma cells
0
20
40
60
80
100
+ca
tala
se
+ca
tala
se
+ca
tala
se
−ca
tala
se
−ca
tala
se
−ca
tala
se
3
24-hour exposure
Per
cen
t of
con
trol
(n
eutr
al r
ed a
ssay
)
Figure 5: Comparative toxicities of epigallocatechin gallate(EGCG), theaflavin-3-digallate, and theaflavin-3′-gallate extracttowards human oral HSC-2 carcinoma cells in the absence andpresence of exogenously added catalase. Cell proliferation wasquantified with the neutral red assay, a cell viability assay. Dataare presented as the mean percent of control ± S.E.M. Data forEGCG are from Weisburg et al. [12] and for the black tea theaflavinmonogallates are from Babich et al. [20].
extract [13], and EGCG [12] to oral carcinoma cells weresignificantly lessened, but not completely abolished, in thepresence of catalase (Figure 5).
In their studies on the response of human lung tumorH441 cells to EGCG, Yang et al. [25] noted that at similarEGCG concentrations, coexposures with exogenous catalasecompletely prevented apoptosis but only partially blockedthe inhibition of cell proliferation. In subsequent studies with21BES cells, Yang et al. [29] showed that exposures to TFdiG,EGCG, and EGC resulted in equivalent levels of intracellularH2O2 and in the induction of apoptosis. Yet, exogenouslyadded catalase significantly prevented EGC- and EGCG-induced apoptosis but did not prevent TFdiG-induced apop-tosis. Apparently, for these cells, EGCG and EGC inducedapoptosis via oxidative stress, whereas TFdiG per se inducedapoptosis by an ROS-independent mechanism.
EGCG-generated ROS in cell culture medium, and ele-vated levels of intracellular ROS were detected in human oralOSC-2 and OSC-4 EGCG-treated carcinoma cells. However,neither exogenous catalase nor N-acetylcysteine (NAC),an antioxidant, rescued the cells from the antiproliferativeeffects of EGCG [28]. Similarly, neither catalase nor NAClessened the antiproliferative effect of (−)-catechin gallateto transformed oral S-G cells [18]. Apparently for theseoral carcinoma cells, EGCG per se and catechin gallateper se, rather than their auto-oxidation ROS products,accounted for growth inhibition. However, Yamamoto et al.[28] observed that exogenous catalase partially rescued OCS-2 cells and more substantially rescued OSC-4 cells from
EGCG-induced apoptotic cell death, as assayed by caspase-3 activation. Apparently, for the OSC-2 and OSC-4, theantiproliferative effects were due to EGCG per se, whereas theproapoptotic effects were due to EGCG-generated ROS.
Lee et al. [22] studied the effects of gallic acid ongap-junction intracellular communication (GJIC), a processessential for maintaining homeostatic balance by modulatingcell growth and differentiation and whose inhibition waslinked to tumor promotion. GJIC in normal rat liver epithe-lial WB-344 cells was inhibited by the addition of authenticH2O2 and of gallic acid, a strong generator of H2O2. ForH2O2, inhibition of GJIC was completely abolished by theaddition of catalase, but for gallic acid, catalase only partiallyabolished this inhibition. Apparently, inhibition of GJIC bygallic acid was attributable both to the polyphenol per se andto H2O2.
Cell culture medium amended with either green tea orwith red wine inhibited proliferation of rat pheochromocy-toma PC12 cells. Both amendments generated H2O2, and theaddition of catalase completely abolished the antiprolifera-tive effects of green tea, but only partially reduced that of redwine. Amendment with exogenous GSH had no significanteffect on red wine toxicity. Apparently, H2O2 accounted forthe total cytotoxic effect of green tea, but only partiallyfor that of red wine [11]. It was suggested that the overalltoxicity of red wine was a combination of H2O2 and ofresveratrol, the main polyphenol in red wine, which hasknown antiproliferative effects [44].
Apparently, the mediating effects of exogenous catalaseon polyphenol cytotoxicity are varied. The complete aboli-tion of a polyphenol-induced cytotoxic effect by exogenouslyadded catalase indicates that the generated H2O2 alone wasthe toxic agent. Partial blockage of the polyphenol-inducedcytotoxic effect by catalase indicates a dual mode of toxicity,the polyphenol per se in conjunction with its auto-oxidationROS products. No lessening of cytotoxicity in the presenceof catalase presumably is indicative of the polyphenol per sealone acting as the cytotoxic agent. However, other factorsmay have been involved, as the methodologies used in thesestudies may have had unforeseen effects on the biologicresponses. For example, Vittal et al. [8] showed that cellularresponses to EGCG included early responding genes whoseexpressions were H2O2 dependent and latter respondinggenes whose expressions were dependent upon EGCG per se(i.e., were H2O2 independent). The early responding geneswere activated soon after exposure to EGCG, as early as 15minutes afterexposure and with increased gene expressionreturning to basal levels by 3 to 6 hrs. A second cascadeof gene expression followed between 3 and 10 hrs and alater response clustered between 12 and 36 hrs afterEGCGtreatment. Thus, the time period of cell exposure to apolyphenol would be critical in evaluating the mediatinginfluence of catalase. In many in vitro studies, the exposureperiod is 24 hr or less; in such a limited exposure period,attention would be focused towards H2O2-dependent genes,favoring their mediation by the addition of catalase. Anotherfactor to consider is the pyruvate content of the cell culturemedium, as it is a scavenger of H2O2. (The mediatinginfluence of pyruvate is discussed in a later section). Finally,

6 Journal of Toxicology
the toxicity of a polyphenol may be dependent upon ROSother than H2O2, in particular upon O2
·−, whose activityis unaffected by catalase, but rather by SOD. As such, torule out ROS as mediators of a polyphenol-induced cytotoxiceffect, cell exposures to the polyphenol need to be done in theabsence of antioxidant enzymes and in presence of catalase,of SOD, and of a combination of catalase and SOD.
As compared to catalase, fewer studies have been directedtowards the effects of exogenous SOD on the toxic potencyof pro-oxidant polyphenols. Superoxide radical is convertedto H2O2, either spontaneously or enzymatically via SOD.Under physiological conditions, SOD, which requires metalcofactors, catalyzes the dismutation of the superoxide radical[2O2
·−+ 2H+ → O2 + H2O2]. Elevation of intracellular O2·−
was observed in EGCG-treated lymphoma and myeloma cells[27]. Treatment of HL-60 leukemia cells with EGCG resultedin elevated intracellular ROS, with a concomitant increase inDNA damage, as detected in the alkaline comet assay. Coex-posures either with SOD or with catalase largely preventedEGCG-mediated DNA damage and greatly reduced ROSformation; combinations of SOD and catalase completelyinhibited intracellular ROS formation and DNA damage[45].
The generation of O2·− may be a major component of
EGCG- [46] and of theaflavin-mediated toxicity, as Yen et al.[47] noted that DNA damage to EGCG-, EGC-, andtheaflavin-treated Chang liver cells was associated with thegeneration of O2
·−. In a cell-free system, SOD lessened theEGCG-mediated generation of H2O2 in sodium phosphatebuffer (pH 7.8) and in RPMI medium, as well as suppressingthe cytotoxicity of EGCG to Jurkat cells [23]. DNA breakage,as quantified in the comet assay, in human peripheral lym-phocytes treated with resveratrol was inhibited by treatmentwith SOD [48].
3.4. Antioxidants. N-acetylcysteine (NAC), a derivative ofthe amino acid, L-cysteine with an acetyl group attachedto the N atom, is a precursor in the synthesis of glu-tathione. NAC and GSH are typically used as exogenouslyadded antioxidants to lessen the potency of pro-oxidantpolyphenols. Using oral cancer cells as the bioindicators andinhibition of cell proliferation as the cytotoxicity endpoint,the potencies of EGCG [49] and of G. biloba extract [13] werelessened by NAC, of curcumin by NAC and GSH [50], andof green tea polyphenol extract by GSH [34]. Treatment ofmyeloid leukemic UF-1 cells with EGCG resulted in elevatedintracellular ROS and in the induction of apoptosis, bothof which were blocked upon coexposure with NAC [26].Curcumin-induced onset of early apoptosis, as detected bythe externalization of phosphatidylserine on cell surfaces ofnormal human gingival fibroblasts and human submandibu-lar gland HSG carcinoma cells, and of the generation ofintracellular H2O2, were lessened upon coexposures withexogenous NAC and GSH [31]. EGCG-induced activationof mitogen-activated protein kinases (MAPKs), release ofcytochrome c, and apoptotic cell death of human colorectalHT-29 cells were significantly blocked in the presence of GSHand NAC. Interestingly, pretreatment with catalase failed to
lessen MAPK activation and apoptotic cell death, indicatingthe involvement of ROS other than H2O2 [51].
3.5. Pyruvate. Pyruvate, amended to cell culture medium assodium pyruvate, is a scavenger of H2O2; it nonenzymaticallyparticipates in a direct oxidative decarboxylation with H2O2
to yield acetate, carbon dioxide, and water [CH3COCOO− +H2O2 → CH3COO− + CO2 + H2O] [52–54]. As such, pyr-uvate affords protection against pro-oxidant polyphenols.Exposure of human ovarian SKOV3 and CAOV3 cancer cellsto EGCG resulted in growth inhibition, accompanied byincreased intracellular levels of H2O2. Addition of pyruvateto the culture medium neutralized the cytotoxicity of EGCG[30].
The antiproliferative toxicities of TFdiG [19], G. bilobaextract [13], and pomegranate extract [14] to oral carcinomaHSC-2 cells were lessened in the presence of exogenous pyru-vate. Studies with normal gingival fibroblasts also noted thatpyruvate lessened the antiproliferative toxicity of caffeic acid,EGCG, TFdiG, black tea theaflavin mixture, and G. bilobaextract [55]. Caspase-mediated cleavage of poly(ADP-ribose)polymerase (PARP), an indicator of apoptosis, occurredupon exposure of HSC-2 cells to 200 and 250 μg/mLpomegranate extract. In cells cotreated with pomegranateextract and pyruvate, PARP cleavage was greatly reduced,indicating that pomegranate extract-induced apoptosis wasa function of the induction of oxidative stress [14].
Of the various mediators of pro-oxidant polyphenolcytotoxicity, pyruvate may have the most far reaching effects,as it is a component in some, but not in all, commerciallyavailable formulations of cell culture media. Furthermore,even within one particular type of medium, there maybe different formulations, some with and some withoutpyruvate. Thus, RPMI, McCoy, Medium 199, and MEMtypically lack pyruvate, whereas MEMα contains pyruvate.DMEM, the most commonly used cell culture medium,has various formulations, the majority of which containpyruvate and others, much lesser in number, lack pyruvate.Figure 6 shows the comparative cytotoxicities to HSC-2cells of pomegranate extract and olive fruit extract in twocommercially available formulations of DMEM, one withand the other without pyruvate. The mediating effect ofpyruvate on H2O2 toxicity presumably is not well known, asthe pyruvate content of the cell culture medium is seldomreported in the experimental design. Thus, cell responsesto a pro-oxidant polyphenol may vary among laboratories,depending on the pyruvate content of the exposure media,which should be noted in publications.
In addition to directly scavenging exogenous H2O2, pyr-uvate has other properties to mediate oxidative stress. Pyr-uvate readily penetrates cells by a specific H+-monocar-boxylic cotransporter [56] and is a readily oxidized fuel,enhancing the cytosolic energy state to maintain cellularfunctioning in the face of metabolic challenge [57]. Pyruvatedampens the mitochondrial generation of ROS, stabilizesmitochondrial ATP production compromised by oxidativestress, maintains the mitochondrial membrane potentialunder oxidative stress [58], and upregulates the expression

Journal of Toxicology 7
of glutathione peroxidase, which is concerned with mito-chondrial scavenging of H2O2 and in maintaining GSH levels[59]. The pyruvate content of the cell culture medium isan amendment that should be reckoned in studies of pro-oxidant polyphenols.
3.6. Cobalt. Divalent cobalt (Co2+) catalyzes the decompo-sition of H2O2 to H2O and O2, without an accompany-ing change in its valence [60]. Additions of mixtures ofblack tea polyphenols or of green tea polyphenols to cellculture medium generated significant amounts of H2O2,which progressively decreased in the presence of increasingconcentrations of added Co2+, as CoCl2. As a scavengerof H2O2, amendments of cell culture medium with Co2+
afforded protection to transformed oral S-G cells from greenand black tea polyphenol mixtures [61]. The antiproliferativeeffects of EGCG to oral HCS-2 and HSG carcinoma cells[49] and to normal gingival fibroblasts [62], of a black teatheaflavin mixture to transformed gingival GT1 fibroblastsand tongue carcinoma CAL27 cells [35], and of Gingkobiloba extract [13] and of pomegranate extract [14] to HSC-2 cells were substantially lessened in the presence of CoCl2.Activation of caspase-3, an indicator of apoptosis, was notedin HSC-2 cells treated with pomegranate extract; whencotreated with pomegranate extract and CoCl2, activation ofcaspase-3 was greatly lessened [14]. In many of these studies[13, 14, 35], Co2+ afforded almost complete protectionagainst polyphenol-induced growth inhibition (Figure 7).At the concentrations used in these studies, Co2+ usuallyprovided greater protection from oxidative damage thancatalase.
3.7. Iron. Nakagawa et al. [63] noted that o-phenanthroline,a Fe2+-chelating agent, suppressed EGCG-induced cell deathin cultured osteoclastic cells. In a cell-free system, it wasfurther shown that the reduction of Fe3+ to Fe2+ by EGCGtriggered a Fenton reaction to form a highly reactivehydroxyl radical from the EGCG-generated H2O2 [H2O2 +Fe2+ → OH· + OH− + Fe3+]. Subsequent studies byNakagawa et al. [23] noted that the growth inhibitory andapoptotic-inducing effects of EGCG towards Jurkat cells werepartially suppressed by o-phenanthroline. These investiga-tors suggested that the mode of cytotoxicity of EGCG wasthrough the generation of H2O2, which triggered the Fe2+-dependent Fenton reaction, thereby generating highly toxicOH· radicals to inhibit growth and to induce apoptotic celldeath.
Lipid peroxidation in transformed oral S-G cells exposedto protocatechuic acid was enhanced in the presence of Fe2+
[37]. Similarly, Fe2+ potentiated the EGCG-lipid peroxida-tion towards oral cancerous and normal gingival fibroblasts[12]. In both studies, the polyphenol per se had little effecton lipid peroxidation, thus, indicating the involvement of aFenton reaction.
3.8. Copper. Several studies have shown that plant polyphe-nols in the presence of metal ions cause oxidative damage to
DMEM
200 μg/mL PE
250 μg/mL PE
250 μg/mL OFE
300 μg/mL OFE
HSC-2 carcinoma cells
0
20
40
60
80
100
−py
r
−py
r
−py
r
+py
r
+py
r
+py
r
−py
r
+py
r
Per
cen
t of
con
trol
(n
eutr
al r
ed a
ssay
)
Figure 6: Comparative toxicities of pomegranate extract (PE) andolive fruit extract (OFE) towards human oral HSC-2 carcinomacells, in Dulbecco’s modified Eagle medium (DMEM) commerciallyformulated without pyruvate (−pyr) and with pyruvate (+pyr).Cell proliferation was quantified with the neutral red assay, a cellviability assay. Data are presented as the mean percent of control ±S.E.M. Data for pomegranate extract are from Weisburg et al. [14]and for olive fruit extract from Schuck (unpublished).
0
20
40
60
80
100−
CoC
l 2
+C
oCl 2
−C
oCl 2
+C
oCl 2
−C
oCl 2
+C
oCl 2
HSC-2 carcinoma cells
(400 μM) theaflavin- (1mg/mL) (250 μg/mL)pomegrante3, -digallate extract extract
Gingko
biloba3
24-hour exposure
Per
cen
t of
con
trol
(n
eutr
al r
ed a
ssay
)
Figure 7: Comparative toxicities of theaflavin-3,3′-digallate,Ginkgo biloba extract, and pomegranate extract towards humanoral HSC-2 carcinoma cells, untreated (control) and treated withcobalt chloride. Cell proliferation was quantified with the neutralred assay, a cell viability assay. Data are presented as the meanpercent of control ± S.E.M. Data for the theaflavin-3,3′-gallate arefrom Schuck et al. [19], for G. biloba extract from Babich et al. [13],and for pomegranate extract from Weisburg et al. [14].
DNA. Divalent copper (Cu2+), one of the most redox sensi-tive metal ions in cells, is closely associated with chromatin.In a study by Bhat et al. [42], freshly isolated peripherallymphocytes were treated with caffeic acid, and DNA damagewas evaluated with the comet assay. DNA breakage, observedwith 200 μM caffeic acid, was progressively lessened in the

8 Journal of Toxicology
presence of increasing concentrations of neocuproine, aCu1+-specific chelating agent. Apparently, DNA breakageby caffeic acid involved endogenous copper, with Cu1+ anintermediate in the mechanistic pathway leading to DNAcleavage. Treatments with catalase and SOD reduced caffeicacid-mediated DNA damage; also, caffeic acid-mediated lipidperoxidation was lessened in the presence of neocuproine.Apparently, both Cu1+ and ROS were involved in oxidativedamage by caffeic acid. To explain the mechanism forDNA damage by caffeic acid, it was postulated that thepolyphenol bound both DNA and Cu2+ to form a ternarycomplex, leading to the reduction of Cu2+ to Cu1+, whichsubsequently interacted with polyphenol-generated H2O2
to afford OH· radicals via a Fenton reaction. Similarly,damage of cellular DNA in lymphocytes was noted uponexposures to EGCG, gallic acid [42], and quercetin [48],with neocuproine sequestering DNA damage. Others havenoted interactions between pro-oxidant polyphenols, ROS,and Cu2+ → Cu1+ to cause oxidative DNA damage althoughother mechanistic pathways, not involving a ternary complexbut rather direct interactions between the polyphenol andCu2+, were suggested [38, 64, 65].
4. Cellular Responses as Mediated byPro-Oxidant Polyphenol Concentration
Three distinct cellular responses appear to result fromexposure to polyphenols, with each response dependentupon the concentration and pro-oxidant nature of thepolyphenols. (a) A mild exposure causes mild oxidative stressand thereby ignites cellular antioxidant defense systems. (b)An intermediate to high exposure gradually overwhelms theantioxidant defense systems and induces apoptotic cell death.(c) A very high exposure quickly overwhelms the cellularantioxidant defenses and causes oxidative damage leading tocell death by necrosis. In designing experiments to identifyoxidative stress as the causative mechanism of cytotoxicity, acareful balance is needed between the generated ROS and theexperimental variable (i.e., scavenger or potentiator of ROStoxicity) mediating oxidative stress.
As noted, glutathione, the major contributor for main-taining the redox state of the cell, exists in both areduced (GSH) and an oxidized form (GSSG). Maintainingsuitable levels of GSH is crucial to counteract oxida-tive stress and involves the transactivation of phase IIdetoxification/antioxidant genes encoding enzymes for GSHsynthesis. Several gene response elements are involved intranscriptional regulation of GSH metabolism, including theantioxidant/electrophile response elements (AREs/EpREs),which have promoters with a specific consensus sequence ofnucleotides that respond to molecules with antioxidant prop-erties. Thus, plant-derived polyphenols, as antioxidants, reg-ulate AREs/EpREs, leading to the synthesis of GSH. Althoughthe specific mechanism is unclear, one approach suggestedthat the pro-oxidant nature of the polyphenol was the criticalfactor, in that auto-oxidation of the polyphenol generatedROS, which lessened the concentration of GSH, therebyto ignite transcriptional activation of γ-glutamylcysteine
synthetase [5]. Exposure of COS-1 cells, an immortalizedAfrican green monkey kidney cell line, to noncytotoxic con-centrations of quercetin enhanced synthesis of GSH throughupregulation of γ-glutamylcysteine synthetase. Exposure ofthese cells to high concentrations of polyphenols led toelevated levels of ROS, which quickly depleted GSH storesand thereby increased cellular susceptibility to oxidative freeradical attack, resulting in cell death by either apoptosisand/or necrosis [66].
Studies with gingival fibroblasts showed that a 4-hourexposure to a nontoxic concentration of TFdiG [19] orof EGCG [12] stimulated the resynthesis of GSH, oftento levels exceeding baseline. Early studies by Arrick et al.[67] noted that after a brief exposure to GSH depleters,cells rapidly resynthesized GSH, often overshooting normallevels. It has been suggested that a potential health benefitfor the consumption of polyphenols at dietary levels is togenerate low levels of ROS, so as to induce mild oxidativestress and thereby boost antioxidant defense systems tocounteract potential challenge by elevated levels of ROS,perhaps through mitochondrial respiration [5, 9].
Raza and John [68], using PC cells, derived from apheochromocytoma of the rat adrenal medulla, showedthat the level of EGCG was critical in evoking either adefensive mechanism or cell death by apoptosis. Low levels ofEGCG (e.g., 50 μM) apparently induced mild oxidative stress,igniting cellular antioxidant defenses, such as the stimulationof GSH synthesis and increased activity of glutathione-S-transferase. A higher level EGCG (e.g., 400 μM) potenti-ated oxidative stress, as indicated by a persistent elevatedintracellular level of ROS that overwhelmed and maskedthe antioxidant defensive mechanisms, leading to disruptionof the intracellular GSH pool and an increase in lipidperoxidation. Also noted at 400 μM EGCG was an increasedexpression of the enzyme, cytochrome P450 2E1, a memberof the cytochrome P450 mixed-function oxidase systeminvolved in the metabolism of ROS and a participant incellular oxidative stress-related toxicity.
In an interesting study by Hsuuw and Chan [69] withhuman breast cancer MCF-7 cells, responses were comparedafter exposures to moderate (20–50 μM) and to high levels(100–400 μM) of EGCG. Moderate levels of EGCG-inducedapoptosis and elevated levels of EGCG induced cell necrosis(cell lysis). At moderate levels of EGCG, cell viability waslessened, and apoptosis was induced, and both correlatedwith increased oxidative stress, as indicated by intracellulargeneration of ROS, a loss of mitochondrial membranepotential, activation of caspase-3, caspase-9, and c-Jun-terminal kinase (JNK), and an increased expression level ofBax protein and a decreased level of Bcl-2 protein, therebyshifting the Bax-Bcl-2 ratio to favor apoptosis. Pretreatmentof the MCF-7 cells with the antioxidants, NAC or α-tocopherol, attenuated intracellular ROS generation and res-cued the cells from apoptotic death. Exposure to 25–50 μMEGCG did not adversely affect ATP production; pretreatmentwith antimycin A, an inhibitor of mitochondrial respiratoryactivity, rescued the cells from apoptotic death, indicatingthat high levels of ATP were required to induce apoptosis.Conversely, at the high concentrations of EGCG, lesser levels

Journal of Toxicology 9
of intracellular ROS and of apoptotic cells and only minoreffects on caspase and JNK activations, on the Bax-Bcl-2ratio, and on mitochondrial membrane potential were noted.Instead, cell death correlated with leakage of LDH, a sign ofnecrosis. Lowered ATP levels were observed at the high levelsof EGCG. It was suggested that the switch from apoptoticdeath to necrosis was controlled by the intracellular level ofATP, with high ATP levels favoring apoptosis and decreasedlevels favoring necrosis.
5. Studies on the Pro-OxidantEffects of Polyphenols in LaboratoryAnimals and Humans
The limited in vitro studies on the pro-oxidant nature ofnatural photochemical dwarfs the minuscule research withlaboratory animal model systems. Li et al. [70] cited theirstudy as “the first demonstration that EGCG induces ROSformation and consequently causes DNA oxidative damagein tumor cells in animals.” NCr nu/nu mice bearing humanlung cancer H1299 xenograft tumors were maintained ona diet supplemented with 0.1, 0.3, and 0.5% EGCG. Atthe end of the 45-day experimental period, tumor growthwas found to be dose dependently inhibited by EGCG. Tocorrelate tumor inhibition with EGCG-induced oxidativestress, immunohistochemistry staining of the xenografttumors was performed. The parameters evaluated included(a) the formation of the oxidative DNA-product, 8-hydroxyl-2′-deoxyguanosine (8-OHdG), a commonly used markerof oxidative stress, (b) the formation of phosphorylatedhistone 2A variant X (γ-H2AX), a cellular marker for thepresence of double-strand DNA breaks, which can be causedby ROS, and (c) apoptotic activity, as measured by theinduction of caspase-3. Dose-dependent induction of allthree biochemical parameters was observed in the xenografttumors.
Animal studies using elevated doses of EGCG havereported hepatotoxicity linked to oxidative damage. Treat-ment of CF mice with a single high oral dose of 1,500 mg/kgEGCG or two once-daily doses of 750 mg/kg EGCG causedliver necrosis, associated with induction of apoptosis andincreased lipid peroxidation and γ-histone 2AX proteinexpression. Plasma levels of 8-isoprostane, a nonenzymaticmarker of arachidonic acid oxidation, were also increasedupon EGCG treatment [71].
Apparently, there are no other laboratory animal studieson the pro-oxidant effects of polyphenol nutraceuticals.
It has been noted that oral consumption of tea protectedagainst carcinomas in the human oral cavity. Li et al. [72]showed that oral and topical administration of a mixedgreen tea preparation significantly reduced the size of oralprecancerous lesions and the incidence of micronucleatedoral mucosa cells in leukoplakia patients. Halder et al. [73]noted similar results in patients with oral leukoplakia whowere administered a regimen of black tea. These studies didnot evaluate whether the pro-oxidative nature of the teas wasinvolved in chemoprevention. However, studies by Yang et al.[74], Lee et al. [75], and Lambert et al. [76] observed that
holding tea solutions in the oral cavity or chewing tea leavesgenerated high salivary levels of catechins and theaflavins,accompanied with high levels of salivary H2O2. Catechin-and theaflavin-generated salivary H2O2 may have played arole in controlling the precancerous lesions in the studies byLi et al. [72] and Halder et al. [73].
6. Concluding Remarks
Cell response to a polyphenol challenge reflects a dualityof toxicities, that of the polyphenol per se and that of thegenerated ROS, both of which modulate chemical signalingpathways leading to antiproliferative and apoptotic effects.To distinguish between the two chemical challenges, studiesin the absence and in the presence of ROS scavengers arewarranted. Some of the research strategies employed to eluci-date oxidative stress are well established, for example, the useof glutathione depleters and ROS scavenging enzymes; otherstrategies are less known, that is, use of divalent cobalt andpyruvate to scavenge ROS. In studies in our laboratory, cobalt(usually, at 250 μM CoCl2) was a more efficient scavengerof H2O2 than was catalase (usually, at 100 Units/mL). Mostimportant for studies of oxidative stress is acknowledgementof the ROS scavenging property of pyruvate, as it isincorporated in formulations of some commercially availablemedia and not in others. DMEM is probably the mostutilized cell culture medium, yet its pyruvate is seldom notedin the experimental design. Of the numerous formulationsof DMEM, relatively few lack pyruvate. As noted in ourlaboratory [55], the magnitude of the cellular response to apolyphenol differed significantly if the study was performedin DMEM without pyruvate as compared to using DMEMwith pyruvate. As the pyruvate status of the medium isseldom incorporated into the description of the experimentaldesign, it can be presumed that the ROS scavenging propertyof pyruvate is not well known.
Acknowledgment
Appreciation is expressed to Stern College for Women ofYeshiva University for kindly providing funding for theresearch reviewed in this paper.
References
[1] R. Beliveau and D. Gingras, “Role of nutrition in preventingcancer,” Canadian Family Physician, vol. 53, no. 11, pp. 1905–1911, 2007.
[2] S. Nair, W. Li, and A. N. T. Kong, “Natural dietary anti-cancer chemopreventive compounds: redox-mediated differ-ential signaling mechanisms in cytoprotection of normal cellsversus cytotoxicity in tumor cells,” Acta Pharmacologica Sinica,vol. 28, no. 4, pp. 459–472, 2007.
[3] H. Pelicano, D. Carney, and P. Huang, “ROS stress in cancercells and therapeutic implications,” Drug Resistance Updates,vol. 7, no. 2, pp. 97–110, 2004.
[4] H. U. Simon, A. Haj-Yehia, and F. Levi-Schaffer, “Roleof reactive oxygen species (ROS) in apoptosis induction,”Apoptosis, vol. 5, no. 5, pp. 415–418, 2000.

10 Journal of Toxicology
[5] J. O. Moskaug, H. Carlsen, M. C. Myhrstad, and R. Blomhoff,“Polyphenols and glutathione synthesis regulation,” TheAmerican Journal of Clinical Nutrition, vol. 71, supplement, pp.16985–17025, 2005.
[6] E. A. Decker, “Phenolics: prooxidants or antioxidants?” Nutri-tion Reviews, vol. 55, no. 11, pp. 396–398, 1997.
[7] L. R. Fukumoto and G. Mazza, “Assessing antioxidant andprooxidant activities of phenolic compounds,” Journal ofAgricultural and Food Chemistry, vol. 48, no. 8, pp. 3597–3604,2000.
[8] R. Vittal, Z. E. Selvanayagam, Y. Sun et al., “Gene expressionchanges induced by green tea polyphenol (−)-epigallocat-echin-3-gallate in human bronchial epithelial 21BES cellsanalyzed by DNA microarray,” Molecular Cancer Therapeutics,vol. 3, no. 9, pp. 1091–1099, 2004.
[9] J. D. Lambert and R. J. Elias, “The antioxidant and pro-oxidant activities of green tea polyphenols: a role in cancerprevention,” Archives of Biochemistry and Biophysics, vol. 501,no. 1, pp. 65–72, 2010.
[10] C. S. Yang, J. D. Lambert, Z. Hou, J. Ju, G. Lu, and X. Hao,“Molecular targets for the cancer preventive activity of teapolyphenols,” Molecular Carcinogenesis, vol. 45, no. 6, pp. 431–435, 2006.
[11] P. C. Chai, L. H. Long, and B. Halliwell, “Contribution ofhydrogen peroxide to the cytotoxicity of green tea and redwines,” Biochemical and Biophysical Research Communications,vol. 304, no. 4, pp. 650–654, 2003.
[12] J. H. Weisburg, D. B. Weissman, T. Sedaghat, and H. Babich,“In vitro cytotoxicity of epigallocatechin gallate and teaextracts to cancerous and normal cells from the human oralcavity,” Basic and Clinical Pharmacology and Toxicology, vol.95, no. 4, pp. 191–200, 2004.
[13] H. Babich, N. J. Ackerman, F. Burekhovich, H. L. Zucker-braun, and A. G. Schuck, “Gingko biloba leaf extract inducesoxidative stress in carcinoma HSC-2 cells,” Toxicology in Vitro,vol. 23, no. 6, pp. 992–999, 2009.
[14] J. H. Weisburg, A. G. Schuck, M. S. Silverman et al., “Pome-granate extract, A prooxidant with antiproliferative andproapoptotic activities preferentially towards carcinoma cells,”Anti-Cancer Agents in Medicinal Chemistry, vol. 10, no. 8, pp.634–643, 2010.
[15] T. Lapidot, M. D. Walker, and J. Kanner, “Can apple antiox-idants inhibit tumor cell proliferation? Generation of H2O2
during interaction of phenolic compounds with cell culturemedia,” Journal of Agricultural and Food Chemistry, vol. 50, no.11, pp. 3156–3160, 2002.
[16] L. H. Long, M. V. Clement, and B. Halliwell, “Artifacts in cellculture: rapid generation of hydrogen peroxide on addition of(−)-epigallocatechin, (−)-epigallocatechin gallate, (+)-cate-chin, and quercetin to commonly used cell culture media,”Biochemical and Biophysical Research Communications, vol.273, no. 1, pp. 50–53, 2000.
[17] H. Babich, M. E. Krupka, H. A. Nissim, and H. L. Zucker-braun, “Differential in vitro cytotoxicity of (−)-epicatechingallate (ECG) to cancer and normal cells from the human oralcavity,” Toxicology in Vitro, vol. 19, no. 2, pp. 231–242, 2005.
[18] H. Babich, H. L. Zuckerbraun, and S. M. Weinerman, “Invitro cytotoxicity of (−)-catechin gallate, a minor polyphenolin green tea,” Toxicology Letters, vol. 171, no. 3, pp. 171–180,2007.
[19] A. G. Schuck, M. B. Ausubel, H. L. Zuckerbraun, and H.Babich, “Theaflavin-3,3′-digallate, a component of black tea:an inducer of oxidative stress and apoptosis,” Toxicology inVitro, vol. 22, no. 3, pp. 598–609, 2008.
[20] H. Babich, R. T. Gottesman, E. J. Liebling, and A. G. Schuck,“Theaflavin-3-gallate and theaflavin-3′-gallate, polyphenolsin black tea with prooxidant properties,” Basic and ClinicalPharmacology and Toxicology, vol. 103, no. 1, pp. 66–74, 2008.
[21] T. Lapidot, M. D. Walker, and J. Kanner, “Antioxidant andprooxidant effects of phenolics on pancreatic β-cells in vitro,”Journal of Agricultural and Food Chemistry, vol. 50, no. 25, pp.7220–7225, 2002.
[22] K. W. Lee, H. J. Hur, H. J. Lee, and C. Y. Lee, “Antiproliferativeeffects of dietary phenolic substances and hydrogen peroxide,”Journal of Agricultural and Food Chemistry, vol. 53, no. 6, pp.1990–1995, 2005.
[23] H. Nakagawa, K. Hasumi, J. T. Woo, K. Nagai, and M. Wachi,“Generation of hydrogen peroxide primarily contributes tothe induction of Fe(II)-dependent apoptosis in Jurkat cells by(−)-epigallocatechin gallate,” Carcinogenesis, vol. 25, no. 9, pp.1567–1574, 2004.
[24] S. G. Rhee, T. S. Chang, W. Jeong, and D. Kang, “Methods fordetection and measurement of hydrogen peroxide inside andoutside of cells,” Molecules and Cells, vol. 29, no. 6, pp. 539–549, 2010.
[25] G. Y. Yang, J. Liao, K. Kim, E. J. Yurkow, and C. S. Yang,“Inhibition of growth and induction of apoptosis in humancancer cell lines by tea polyphenols,” Carcinogenesis, vol. 19,no. 4, pp. 611–616, 1998.
[26] T. Nakazato, K. Ito, Y. Miyakawa et al., “Catechin, a green teacomponent, rapidly induces apoptosis of myeloid leukemiccells via modulation of reactive oxygen species production invitro and inhibits tumor growth in vivo,” Haematologica, vol.90, no. 3, pp. 317–325, 2005.
[27] T. Nakazato, K. Ito, Y. Ikeda, and M. Kizaki, “Green teacomponent, catechin, induces apoptosis of human malignantB cells via production of reactive oxygen species,” ClinicalCancer Research, vol. 11, no. 16, pp. 6040–6049, 2005.
[28] T. Yamamoto, J. Lewis, J. Wataha et al., “Roles of catalase andhydrogen peroxide in green tea polyphenol-induced chemo-preventive effects,” Journal of Pharmacology and ExperimentalTherapeutics, vol. 308, no. 1, pp. 317–323, 2004.
[29] G. Y. Yang, J. Liao, C. Li et al., “Effect of black and green teapolyphenols on c-jun phosphorylation and H2O2 productionin transformed and non-transformed human bronchial celllines: possible mechanisms of cell growth inhibition andapoptosis induction,” Carcinogenesis, vol. 21, no. 11, pp. 2035–2039, 2000.
[30] M. M. Chan, K. J. Soprano, K. Weinstein, and D. Fong,“Epigallocatechin-3-gallate delivers hydrogen peroxide toinduce death of ovarian cancer cells and enhances theircisplatin susceptibility,” Journal of Cellular Physiology, vol. 207,no. 2, pp. 389–396, 2006.
[31] T. Atsumi, K. Tonosaki, and S. Fujisawa, “Induction of earlyapoptosis and ROS-generation activity in human gingivalfibroblasts (HGF) and human submandibular gland carci-noma (HSG) cells treated with curcumin,” Archives of OralBiology, vol. 51, no. 10, pp. 913–921, 2006.
[32] S. Yeu, C. M. Sun, H. H. Chuang, and P. T. Chang, “Studieson the cytotoxic mechanisms of ginkgetin in a human ovarianadenocarcinoma cell line,” Naunyn-Schmiedeberg’s, vol. 362,no. 1, pp. 82–90, 2000.
[33] J. M. Feugang, F. Ye, D. Y. Zhang et al., “Cactus pear extractsinduce reactive oxygen species production and apoptosis inovarian cancer cells,” Nutrition and Cancer, vol. 62, no. 5, pp.692–699, 2010.
[34] H. Babich, A. R. Selevan, and E. R. Ravkin, “Glutathione as amediator of the in vitro cytotoxicity of a green tea polyphenol

Journal of Toxicology 11
extract,” Toxicology Mechanisms and Methods, vol. 17, no. 6,pp. 357–369, 2007.
[35] H. Babich, S. M. Pinsky, E. T. Muskin, and H. L. Zuckerbraun,“In vitro cytotoxicity of a theaflavin mixture from black tea tomalignant, immortalized, and normal cells from the humanoral cavity,” Toxicology in Vitro, vol. 20, no. 5, pp. 677–688,2006.
[36] J. Lu, C. T. Ho, G. Ghai, and . Kuan Yu Chen, “Differentialeffects of theaflavin monogallates on cell growth, apoptosis,and Cox-2 gene expression in cancerous versus normal cells,”Cancer Research, vol. 60, no. 22, pp. 6465–6471, 2000.
[37] H. Babich, A. Sedletcaia, and B. Kenigsberg, “In vitro cyto-toxicity of protocatechuic acid to cultured human cells fromoral tissue: involvement in oxidative stress,” Pharmacology andToxicology, vol. 91, no. 5, pp. 245–253, 2002.
[38] A. Furukawa, S. Oikawa, M. Murata, Y. Hiraku, and S. Kawan-ishi, “(−)-Epigallocatechin gallate causes oxidative damage toisolated and cellular DNA,” Biochemical Pharmacology, vol. 66,no. 9, pp. 1769–1778, 2003.
[39] H. Babich, A. Stern, and E. Borenfreund, “Eugenol cytotoxicityevaluated with continuous cell lines,” Toxicology in Vitro, vol.7, no. 2, pp. 105–109, 1993.
[40] B. R. You and W. H. Park, “Gallic acid-induced lung cancercell death is related to glutathione depletion as well as reactiveoxygen species increase,” Toxicology in Vitro, vol. 24, no. 5, pp.1356–1362, 2010.
[41] S. C. Sahu, D. I. Ruggles, and M. W. O’Donnell, “Prooxidantactivity and toxicity of nordihydroguaiaretic acid in clone-9rat hepatocyte cultures,” Food and Chemical Toxicology, vol. 44,no. 10, pp. 1751–1757, 2006.
[42] S. H. Bhat, A. S. Azmi, and S. M. Hadi, “Prooxidant DNAbreakage induced by caffeic acid in human peripheral lym-phocytes: involvement of endogenous copper and a putativemechanism for anticancer properties,” Toxicology and AppliedPharmacology, vol. 218, no. 3, pp. 249–255, 2007.
[43] S. B. Sarikcioglu, G. Oner, and E. Tercan, “Antioxidant effectof EGb 761 on hydrogen peroxide-induced lipoperoxidationof G-6-PD deficient erythrocytes,” Phytotherapy Research, vol.18, no. 10, pp. 837–840, 2004.
[44] H. Babich, A. G. Reisbaum, and H. L. Zuckerbraun, “In vitroresponse of human gingival epithelial S-G cells to resveratrol,”Toxicology Letters, vol. 114, no. 1–3, pp. 143–153, 2000.
[45] L. Elbling, R. M. Weiss, O. Teufelhofer et al., “Green tea extractand (−)-epigallocatechin-3-gallate, the major tea catechin,exert oxidant but lack antioxidant activities,” FASEB Journal,vol. 19, no. 7, pp. 807–809, 2005.
[46] Z. Hou, S. Sang, H. You et al., “Mechanism of actionof (−)-epigallocatechin-3-gallate: auto-oxidation- dependentinactivation of epidermal growth factor receptor and directeffects on growth inhibition in human esophageal cancerKYSE 150 cells,” Cancer Research, vol. 65, no. 17, pp. 8049–8056, 2005.
[47] G. C. Yen, J. W. Ju, and C. H. Wu, “Modulation of tea andtea polyphenols on benzo(a)pyrene-induced DNA damage inChang liver cells,” Free Radical Research, vol. 38, no. 2, pp. 193–200, 2004.
[48] S. M. Hadi, S. H. Bhat, A. S. Azmi, S. Hanif, U. Shamim,and M. F. Ullah, “Oxidative breakage of cellular DNA by plantpolyphenols: a putative mechanism for anticancer properties,”Seminars in Cancer Biology, vol. 17, no. 5, pp. 370–376, 2007.
[49] A. Ishino, S. Mita, S. Watanabe, and H. Sakagami, “Effect ofanticancer drugs, metals and antioxidants on cytotoxic activityof epigallocatechin gallate,” Anticancer Research, vol. 19, no. 5,pp. 4343–4348, 1999.
[50] S. Fujisawa, T. Atsumi, M. Ishihara, and Y. Kadoma, “Cytotox-icity, ROS-generation activity and radical-scavenging activityof curcumin and related compounds,” Anticancer Research,vol. 24, no. 2, pp. 563–569, 2004.
[51] C. Chen, G. Shen, V. Hebbar, R. Hu, E. D. Owuor, and A.N. T. Kong, “Epigallocatechin-3-gallate-induced stress signalsin HT-29 human colon adenocarcinoma cells,” Carcinogenesis,vol. 24, no. 8, pp. 1369–1378, 2003.
[52] K. A. Nath, H. Enright, L. Nutter, M. Fischereder, J. N. Zou,and R. P. Hebbel, “Effect of pyruvate on oxidant injury toisolated and cellular DNA,” Kidney International, vol. 45, no.1, pp. 166–176, 1994.
[53] R. T. Mallet, “Pyruvate: metabolic protector of cardiac perfor-mance,” Proceedings of the Society for Experimental Biology andMedicine, vol. 223, no. 2, pp. 136–148, 2000.
[54] A. Shostak, L. Gotloib, R. Kushnier, and V. Wajsbrot, “Protec-tive effect of pyruvate upon cultured mesothelial cells exposedto 2 mM hydrogen peroxide,” Nephron, vol. 84, no. 4, pp. 362–366, 2000.
[55] H. Babich, E. J. Liebling, R. F. Burger, H. L. Zuckerbraun,and A. G. Schuck, “Choice of DMEM, formulated with orwithout pyruvate, plays an important role in assessing the invitro cytotoxicity of oxidants and prooxidant nutraceuticals,”In vitro Cellular & Developmental Biology, vol. 45, no. 5-6, pp.226–233, 2009.
[56] C. K. Garcia, J. L. Goldstein, R. K. Pathak, R. G. W. Anderson,and M. S. Brown, “Molecular characterization of a membranetransporter for lactate, pyruvate, and other monocarboxylates:implications for the Cori cycle,” Cell, vol. 76, no. 5, pp. 865–873, 1994.
[57] R. Bunger, R. T. Mallet, and D. A. Hartman, “Pyruvate-enhanced phosphorylation potential and inotropism innormoxic and postischemic isolated working heart. Near-complete prevention of reperfusion contractile failure,” Euro-pean Journal of Biochemistry, vol. 180, no. 1, pp. 221–233,1989.
[58] X. Wang, E. Perez, R. Liu, L. J. Yan, R. T. Mallet, and S. H.Yang, “Pyruvate protects mitochondria from oxidative stressin human neuroblastoma SK-N-SH cells,” Brain Research, vol.1132, no. 1, pp. 1–9, 2007.
[59] F. M. Maiorino, R. Brigelius-Flohe, K. D. Aumann, A. Roveri,D. Schomburg, and L. Flohe, “[5] Diversity of glutathioneperoxidases,” Methods in Enzymology, vol. 252, pp. 38–48,1995.
[60] M. K. Eberhardt, C. Santos, and M. A. Soto, “Formation ofhydroxyl radicals and Co3+ in the reaction of Co2+-EDTAwith hydrogen peroxide. Catalytic effect of Fe3+,” Biochimicaet Biophysica Acta, vol. 1157, no. 1, pp. 102–106, 1993.
[61] H. Babich, T. Gold, and R. Gold, “Mediation of the in vitrocytotoxicity of green and black tea polyphenols by cobaltchloride,” Toxicology Letters, vol. 155, no. 1, pp. 195–205, 2005.
[62] T. Sakagami, K. Satoh, M. Ishihara et al., “Effect of cobaltion on radical intensity and cytotoxic activity of antioxidants,”Anticancer Research, vol. 20, no. 5, pp. 3143–3150, 2000.
[63] H. Nakagawa, M. Wachi, J. T. Woo et al., “Fenton reaction isprimarily involved in a mechanism of (3)-epigallocatechin-3-gallate to induce osteoclastic cell death,” Biochemical andBiophysical Research Communications, vol. 292, no. 1, pp. 94–101, 2002.
[64] S. Inoue, K. Ito, K. Yamamoto, and S. Kawanishi, “Caffeic acidcauses metal-dependent damage to cellular and isolated DNAthrough H2O2 formation,” Carcinogenesis, vol. 13, no. 9, pp.1497–1502, 1992.

12 Journal of Toxicology
[65] S. Oikawa, A. Furukawa, H. Asada, K. Hirakawa, and S.Kawanishi, “Catechins induce oxidative damage to cellularand isolated DNA through the generation of reactive oxygenspecies,” Free Radical Research, vol. 37, no. 8, pp. 881–890,2003.
[66] M. C. W. Myhrstad, H. Carlsen, O. Nordstrom, R. Blomhoff,and J. Ø. Moskaug, “Flavonoids increase the intracellularglutathione level by transactivation of the γ-glutamylcysteinesynthetase catalytical subunit promoter,” Free Radical Biologyand Medicine, vol. 32, no. 5, pp. 386–393, 2002.
[67] B. A. Arrick, C. F. Nathan, O. W. Griffith, and Z. A. Cohn,“Glutathione depletion sensitizes tumor cells to oxidativecytolysis,” The Journal of Biological Chemistry, vol. 257, no. 3,pp. 1231–1237, 1982.
[68] H. Raza and A. John, “Green tea polyphenol epigallocatechin-3-gallate differentially modulates oxidative stress in PC12 cellcompartments,” Toxicology and Applied Pharmacology, vol.207, no. 3, pp. 212–220, 2005.
[69] Y. D. Hsuuw and W. H. Chan, “Epigallocatechin gallate dose-dependently induces apoptosis or necrosis in human MCF-7cells,” Annals of the New York Academy of Sciences, vol. 1095,pp. 428–440, 2007.
[70] G. X. Li, Y. K. Chen, Z. Hou et al., “Pro-oxidative activities anddose-response relationship of (−)-epigallocatechin-3-gallatein the inhibition of lung cancer cell growth: a comparativestudy in vivo and in vitro,” Carcinogenesis, vol. 31, no. 5, pp.902–910, 2010.
[71] J. D. Lambert, M. J. Kennett, S. Sang, K. R. Reuhl, J. Ju, and C.S. Yang, “Hepatotoxicity of high oral dose (−)-epigallocate-chin-3-gallate in mice,” Food and Chemical Toxicology, vol. 48,no. 1, pp. 409–416, 2010.
[72] N. Li, Z. Sun, C. Han, and J. Chen, “The chemopreven-tive effects of tea on human oral precancerous mucosalesions,” Proceedings of the Society for Experimental Biology andMedicine, vol. 220, no. 4, pp. 218–224, 1999.
[73] A. Halder, R. Raychowdhury, A. Ghosh, and M. De, “Blacktea (Camellia sinensis) as a chemopreventive agent in oralprecancerous lesions,” Journal of Environmental Pathology,Toxicology and Oncology, vol. 24, no. 2, pp. 141–144, 2005.
[74] C. S. Yang, M. J. Lee, and L. Chen, “Human salivary teacatechin levels and catechin esterase activities: implicationin human cancer prevention studies,” Cancer EpidemiologyBiomarkers and Prevention, vol. 8, no. 1, pp. 83–89, 1999.
[75] M. J. Lee, J. D. Lambert, S. Prabhu et al., “Delivery of teapolyphenols to the oral cavity by green tea leaves and blacktea extract,” Cancer Epidemiology Biomarkers and Prevention,vol. 13, no. 1, pp. 132–137, 2004.
[76] J. D. Lambert, S. J. Kwon, J. Hong, and C. S. Yang, “Salivaryhydrogen peroxide produced by holding or chewing green teain the oral cavity,” Free Radical Research, vol. 41, no. 7, pp. 850–853, 2007.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 973172, 11 pagesdoi:10.1155/2011/973172
Research Article
Estimation of the Postmortem Duration of Mouse Tissue byElectron Spin Resonance Spectroscopy
Shinobu Ito,1, 2 Tomohisa Mori,3 Hideko Kanazawa,2 and Toshiko Sawaguchi4
1 I.T.O. Provitamin Research Center, 1-6-7-3F Nakamachi, Musashino, Tokyo, Japan2 Faculty of Pharmacy, Keio University, 1-5-30 Shibakoen, Minato-ku, Tokyo 105-8512, Japan3 Department of Pharmacology and Experimental Neuroscience, College of Medicine, University of Nebraska at Omaha,Omaha, NE 68182, USA
4 Department of Occupational Therapy, Faculty of Regional Health Therapy, Teikyo Heisei University, 4-1 Uruido-minami,Ichihara, Chiba, Japan
Correspondence should be addressed to Toshiko Sawaguchi, [email protected]
Received 14 January 2011; Revised 29 March 2011; Accepted 12 April 2011
Academic Editor: Lucio Guido Costa
Copyright © 2011 Shinobu Ito et al. This is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Electron spin resonance (ESR) method is a simple method for detecting various free radicals simultaneously and directly. However,ESR spin trap method is unsuited to analyze weak ESR signals in organs because of water-induced dielectric loss (WIDL).To minimize WIDL occurring in biotissues and to improve detection sensitivity to free radicals in tissues, ESR cuvette wasmodified and used with 5,5-dimethtyl-1-pyrroline N-oxide (DMPO). The tissue samples were mouse brain, hart, lung, liver,kidney, pancreas, muscle, skin, and whole blood, where various ESR spin adduct signals including DMPO-ascorbyl radical (AsA∗),DMPO-superoxide anion radical (OOH), and DMPO-hydrogen radical (H) signal were detected. Postmortem changes in DMPO-AsA∗ and DMPO-OOH were observed in various tissues of mouse. The signal peak of spin adduct was monitored until the 205thday postmortem. DMPO-AsA∗ in liver (y = 113.8–40.7 log (day), R1 = −0.779, R2 = 0.6, P < .001) was found to linearly decreasewith the logarithm of postmortem duration days. Therefore, DMPO-AsA∗ signal may be suitable for detecting an oxidation stresstracer from tissue in comparison with other spin adduct signal on ESR spin trap method.
1. Introduction
Electron spin resonance (ESR) or electron paramagneticresonance (EPR) is now widely used to analyze free radicalspecies in living body and materials. Possibility of applicationof ESR is studied in a forensic science area. It can bepotentially used for estimating postmortem duration in thecause of death. Pashinian and Proshut [1], who suggestedthe potential of using ESR in forensic medicine, attempted todetermine the time of the occurrence of mechanical traumaby measuring the ESR signals of bone marrow. Several studieshave analyzed blood by ESR, because blood contains iron-containing proteins such as hemoglobin. Uzeneva [2], forexample, studied on the ESR signals of posttraumatic blood.Mil’ et al. [3] reported that the ESR signal intensity of bloodof patients exposed to radiation at the Chernobyl nuclearaccident is higher than that of healthy people. Nakamura
et al. [4] reported on ESR signals induced by ionizingradiation in teeth. Quarino and kobilinsky. [5] used ESRto detect human hemoglobin from bloodstains. Turkes etal. [6] analyzed blood stored under blood bank conditionsusing ESR. They reported that the intensity of ESR signalsfrom methemoglobin, nonheme irons, and organic radicalsin dried human blood increase with time. Fujita et al. [7]showed that (1) ESR signals from bloodstains are effectivein estimating the age of human and (2) ESR signals regularlychange over time within the period of 432 days. In these ESRstudies, measurements were performed at low temperature(140◦K) for detecting the ESR signal of protein-bonded ions.As described above, ESR is now widely used to analyzeliving body and material in forensic medicine, and it can bepotentially used to estimate the age of human from blood-stains. In those cases, ESR measurements were performedat room temperature unless otherwise mentioned. However,

2 Journal of Toxicology
(I) (II) (III–VI) (VII–X) (XI) (XII)
(I ) (II ) (III ) (VI )
(a) DMPO-OOH signal generated by hypoxantin-xantine reaction
(I) (II)
(b) DMPO-AsA∗ signal generated by ascorbic acid-hemoglobin reaction
(I) (II)(III)
(IV)
(V)
(VI)
(VII)
(VIII) (IX)
(c) DMPO-H signal generated by hematoporphyrin-ultraviolet rays reaction
(I)
(II) (III)
(IV)
(d) DMPO-OH signal generated by Fe(II)-H2O2 reaction
Figure 1: Standard ESR signals of DMPO- and DPPMPO-adducts.
with the exception of blood, few studies have examinedthe postmortem changes in ESR signals found in organsand tissues. In this study, we investigated the origins ofESR signals in postmortem tissues and the time courses ofchanges in the signals.
ESR spin trapping and probing is a method that hasrecently attracted attention and is used to analyze the freeradicals of tissues. ESR spin trapping method is performedby a conventional X-band ESR analysis system [8, 9], whichdetects individual radical types as spin adducts and identifyand quantify reactive oxygen species (ROS) types based onthe signal patterns. ESR spin probing method has recentlybeen applied to three-dimensional ESR imaging for livingbody [10–14], but the method has to analyze relatively weaksignals from living body [15, 16]. Ascorbic acid (AsA) is asuperior scavenger; it reacts with hydroxyl radicals strongly,the rate of reaction is 7.0 × 109–1.1 × 1010 M−1S−1 [17],and Ascorbyl radical (AsA∗) is generated after the reaction.
The detection of ESR signals of AsA∗ is straightforword,because the spin trap adduct signal of AsA∗ is simple. AsA∗
has a possibility as an important indicator for oxidationstress in tissue. Previous study of AsA∗ spin adduct signalwas limited to tissues having strong oxidative stresses orAsA administration mouse having a high AsA∗ level. Adoublet peak spectrum was found to obtain following AsAinjection in mouse, and the signals were confirmed indifferent ways due to AsA∗ [18]. It was reported that (1)tissue constantly suffers from the oxidation of AsA andiron proteins [19] and (2) the oxidation reaction couldproceed by the reaction of AsA by these recycle Fentonreactions [20]. AsA∗ was detected in those tissue sufferingfrom oxidative stress. DMPO-(5,5-dimethtyl-1-pyrroline N-oxide) AsA∗ was detected in oxidative stress mouse skininduced by X-ray irradiation [21]. A method to detectDMPO-AsA∗ signal with a high sensitivity from a brain wasreported recently by Masumizu et al. [22]. Since ESR signals
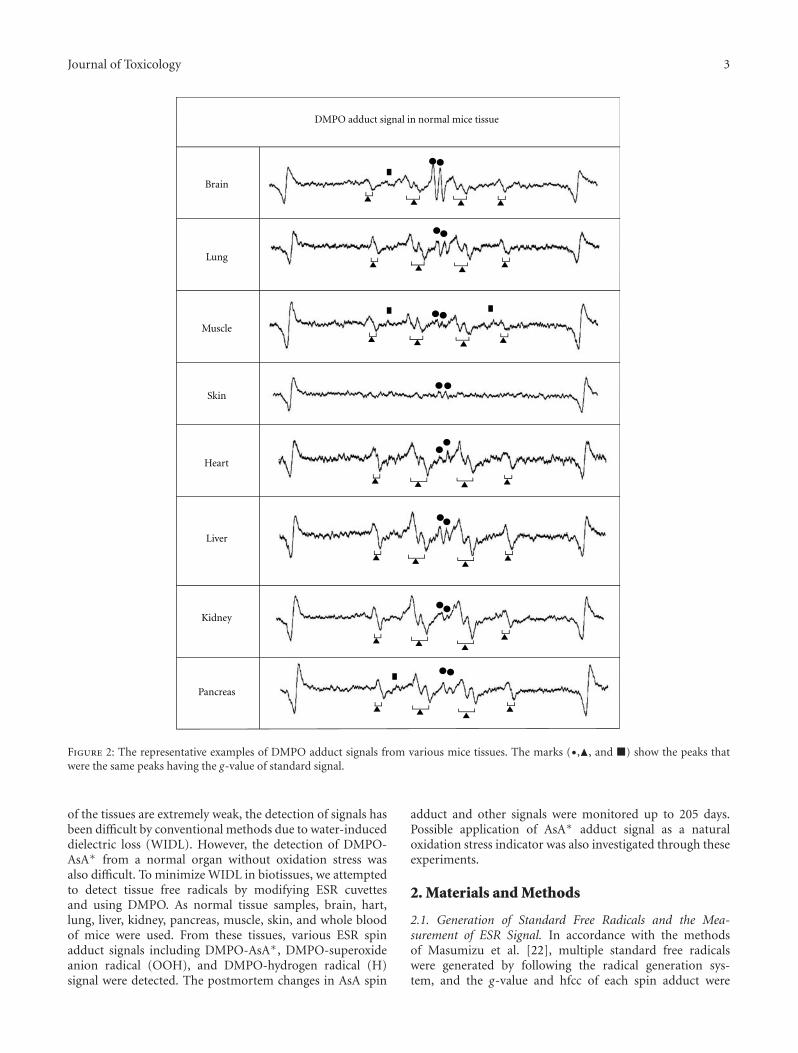
Journal of Toxicology 3
Brain
Lung
Muscle
Skin
Heart
Liver
Kidney
Pancreas
DMPO adduct signal in normal mice tissue
Figure 2: The representative examples of DMPO adduct signals from various mice tissues. The marks (•,�, and �) show the peaks thatwere the same peaks having the g-value of standard signal.
of the tissues are extremely weak, the detection of signals hasbeen difficult by conventional methods due to water-induceddielectric loss (WIDL). However, the detection of DMPO-AsA∗ from a normal organ without oxidation stress wasalso difficult. To minimize WIDL in biotissues, we attemptedto detect tissue free radicals by modifying ESR cuvettesand using DMPO. As normal tissue samples, brain, hart,lung, liver, kidney, pancreas, muscle, skin, and whole bloodof mice were used. From these tissues, various ESR spinadduct signals including DMPO-AsA∗, DMPO-superoxideanion radical (OOH), and DMPO-hydrogen radical (H)signal were detected. The postmortem changes in AsA spin
adduct and other signals were monitored up to 205 days.Possible application of AsA∗ adduct signal as a naturaloxidation stress indicator was also investigated through theseexperiments.
2. Materials and Methods
2.1. Generation of Standard Free Radicals and the Mea-surement of ESR Signal. In accordance with the methodsof Masumizu et al. [22], multiple standard free radicalswere generated by following the radical generation sys-tem, and the g-value and hfcc of each spin adduct were
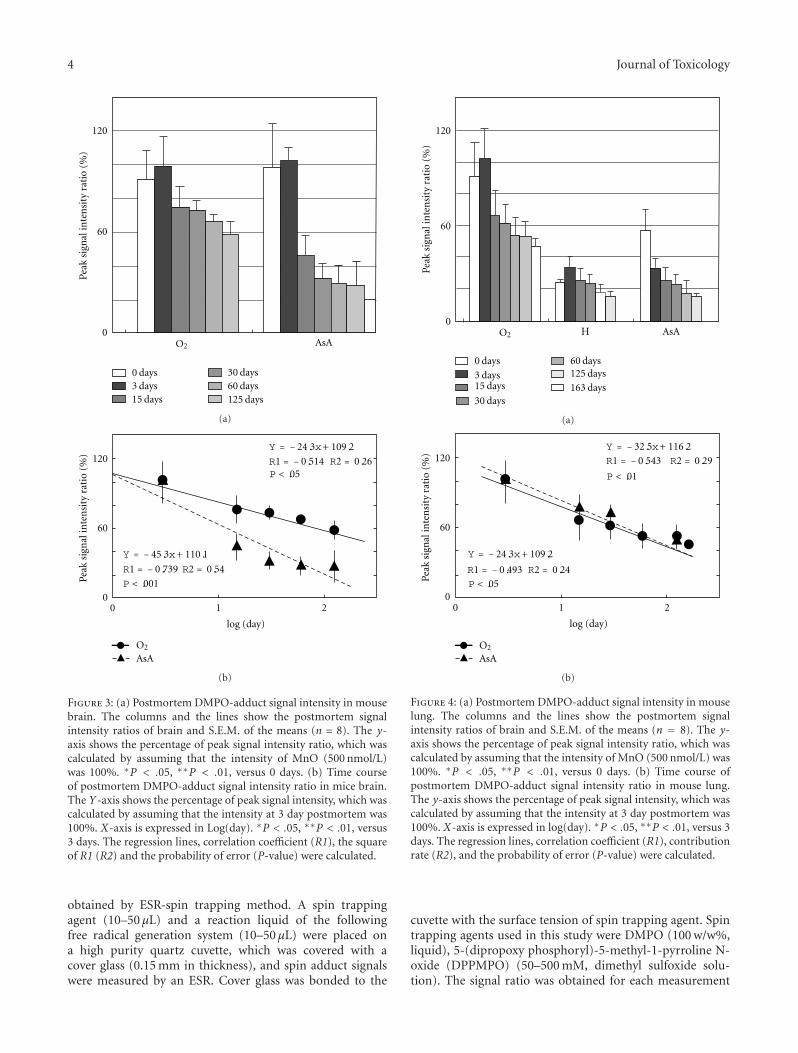
4 Journal of Toxicology
120
60
0AsA
0 days3 days15 days
30 days60 days125 days
Peak
sign
alin
ten
sity
rati
o(%
)
O2
(a)
O2
120
60
0
Peak
sign
alin
ten
sity
rati
o(%
)
Y = − 24.3x+ 109.2
Y = − 45.3x+ 110.1
0 1 2
log (day)
AsA
R1 = − 0.514 R2 = 0.26
R1 = − 0.739 R2 = 0.54
P < .05
P < .001
(b)
Figure 3: (a) Postmortem DMPO-adduct signal intensity in mousebrain. The columns and the lines show the postmortem signalintensity ratios of brain and S.E.M. of the means (n = 8). The y-axis shows the percentage of peak signal intensity ratio, which wascalculated by assuming that the intensity of MnO (500 nmol/L)was 100%. ∗P < .05, ∗∗P < .01, versus 0 days. (b) Time courseof postmortem DMPO-adduct signal intensity ratio in mice brain.The Y-axis shows the percentage of peak signal intensity, which wascalculated by assuming that the intensity at 3 day postmortem was100%. X-axis is expressed in Log(day). ∗P < .05, ∗∗P < .01, versus3 days. The regression lines, correlation coefficient (R1), the squareof R1 (R2) and the probability of error (P-value) were calculated.
obtained by ESR-spin trapping method. A spin trappingagent (10–50 μL) and a reaction liquid of the followingfree radical generation system (10–50 μL) were placed ona high purity quartz cuvette, which was covered with acover glass (0.15 mm in thickness), and spin adduct signalswere measured by an ESR. Cover glass was bonded to the
120
60
0H AsA
0 days
3 days15 days
30 days
60 days125 days
163 days
Peak
sign
alin
ten
sity
rati
o(%
)
O2
(a)
Y = − 32.5x+ 116.2
Y = − 24.3x+ 109.2
120
60
0
Peak
sign
alin
ten
sity
rati
o(%
)
0 1 2
log (day)
AsA
R1 = − 0.543 R2 = 0.29
R1 = − 0.493 R2 = 0.24
O2
P < .01
P < .05
(b)
Figure 4: (a) Postmortem DMPO-adduct signal intensity in mouselung. The columns and the lines show the postmortem signalintensity ratios of brain and S.E.M. of the means (n = 8). The y-axis shows the percentage of peak signal intensity ratio, which wascalculated by assuming that the intensity of MnO (500 nmol/L) was100%. ∗P < .05, ∗∗P < .01, versus 0 days. (b) Time course ofpostmortem DMPO-adduct signal intensity ratio in mouse lung.The y-axis shows the percentage of peak signal intensity, which wascalculated by assuming that the intensity at 3 day postmortem was100%. X-axis is expressed in log(day). ∗P < .05, ∗∗P < .01, versus 3days. The regression lines, correlation coefficient (R1), contributionrate (R2), and the probability of error (P-value) were calculated.
cuvette with the surface tension of spin trapping agent. Spintrapping agents used in this study were DMPO (100 w/w%,liquid), 5-(dipropoxy phosphoryl)-5-methyl-1-pyrroline N-oxide (DPPMPO) (50–500 mM, dimethyl sulfoxide solu-tion). The signal ratio was obtained for each measurement
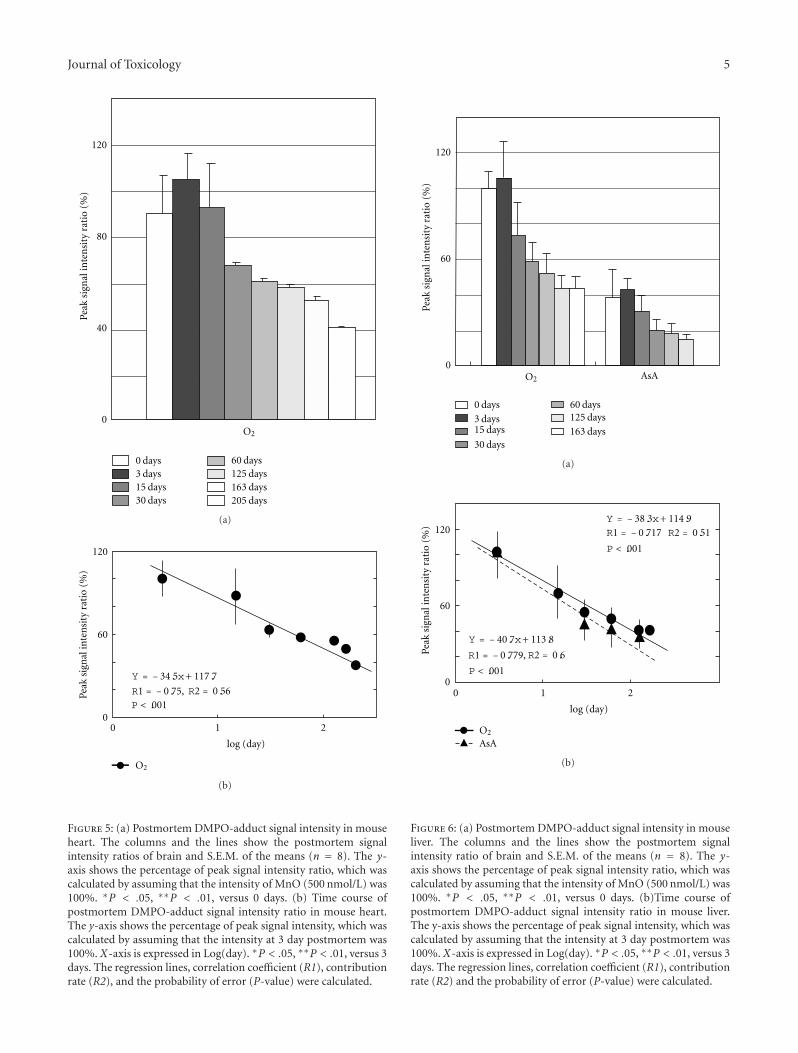
Journal of Toxicology 5
120
80
40
0
0 days3 days15 days30 days
60 days125 days163 days205 days
Peak
sign
alin
ten
sity
rati
o(%
)
O2
(a)
120
60
0
Peak
sign
alin
ten
sity
rati
o(%
)
0 1 2
log (day)
Y = − 34.5x+ 117.7
O2
R2 = 0.56P < .001R1 = − 0.75,
(b)
Figure 5: (a) Postmortem DMPO-adduct signal intensity in mouseheart. The columns and the lines show the postmortem signalintensity ratios of brain and S.E.M. of the means (n = 8). The y-axis shows the percentage of peak signal intensity ratio, which wascalculated by assuming that the intensity of MnO (500 nmol/L) was100%. ∗P < .05, ∗∗P < .01, versus 0 days. (b) Time course ofpostmortem DMPO-adduct signal intensity ratio in mouse heart.The y-axis shows the percentage of peak signal intensity, which wascalculated by assuming that the intensity at 3 day postmortem was100%. X-axis is expressed in Log(day). ∗P < .05, ∗∗P < .01, versus 3days. The regression lines, correlation coefficient (R1), contributionrate (R2), and the probability of error (P-value) were calculated.
0 days
3 days15 days
30 days
60 days125 days
163 days
120
60
0
Peak
sign
alin
ten
sity
rati
o(%
)
AsAO2
(a)
0 1 2
log (day)
AsA
Y = − 38.3x+ 114.9
Y = − 40.7x+ 113.8
120
60
0
Peak
sign
alin
ten
sity
rati
o(%
)
R1 = − 0.717 R2 = 0.51
R2 = 0.6
O2
P < .001
P < .001
R1 = − 0.779,
(b)
Figure 6: (a) Postmortem DMPO-adduct signal intensity in mouseliver. The columns and the lines show the postmortem signalintensity ratio of brain and S.E.M. of the means (n = 8). The y-axis shows the percentage of peak signal intensity ratio, which wascalculated by assuming that the intensity of MnO (500 nmol/L) was100%. ∗P < .05, ∗∗P < .01, versus 0 days. (b)Time course ofpostmortem DMPO-adduct signal intensity ratio in mouse liver.The y-axis shows the percentage of peak signal intensity, which wascalculated by assuming that the intensity at 3 day postmortem was100%. X-axis is expressed in Log(day). ∗P < .05, ∗∗P < .01, versus 3days. The regression lines, correlation coefficient (R1), contributionrate (R2) and the probability of error (P-value) were calculated.

6 Journal of Toxicology
using the signal of MnO, an internal standard substance, as astandard.
2.2. Standard Free Radical Adduct Signal. Standard ESRsignals of various free radicals were provided by the followingsystems.
2.2.1. Superoxide Anion Radical Generation System. Superox-ide was generated with a hypoxanthin-xanthine oxidase reac-tion system. Superoxide dismutase (SOD) solution (30 μL)(0.1 phosphate buffer/saline, pH 7.8, 200 U/mL) was addedto it, and an appeared peak is assigned to DMPO-OOH(superoxide radical) or DPPMPO-OOH.
2.2.2. Hydroxyl Radical Generation System. Hydroxyl radicalswere generated from the reaction of 10 mmol/L FeSO4 and20 mmol/L H2O2 (Fenton reaction). Hydroxyl radical wereconfirmed by adding 30 μL AsA solution (0.1 mol/L AsA),and tan appeared peak is assigned to DMPO-OH (hydroxylradical) or DPPMPO-OH.
2.2.3. AsA∗ Generation System. AsA∗ was generated byreacting hemoglobin (0.1 w/w%) and AsA (1 mmol/L). AsA∗
was also generated by adding 10 μL L-ascorbic acid solution(10 mmol/L) to the 10 μL hydroxyl radical generation systemdescribed above.
2.2.4. Hydrogen Radical. Hydrogen radical was generated byhematoporphyrin (1 w/w%) with UV irradiation at 365 nm(the intensity: 5 mW/cm2) (Ushio Optical Modulex, SX-UI500MQQ)(Ushio, Tokyo, Japan). Hydrogen radical is alsogenerated by electrolyzing 0.01 w/w% NaCl solution withTI-8000 (Nihon Trim, Osaka, Japan). For confirming thegeneration of hydrogen radicals, DBNBS was added to thehydrogen radical solution and the color of the solution wasobserved to be orange (P2002-350420A). The g-value of freeradical signal obtained and identifyied and the signal wasidentified by the calculation of both frequency and magnetfield of the ESR signal. For correcting internal cavity forquantitative analysis, manganese oxide (MnO) was used asthe internal standard of ESR cavity. DMPO signals wererecorded between 3rd and 4th MnO signals. The relativeintensity of radicals was calculated by comparison with the3rd MnO signal intensity. The g-value and the distance(mT) between the peaks for hfcc were measured by softwarecoming with ESR device. ESR equipment and its conditionused in this study were followings. The measurements ofg-value and hfcc were calculated by analysis software (A-System vl.40 ISAJ, FA-manager vl.20, JES, Tokyo, Japan)accompanying with ESR spectrometer. Numerical valuewas measured more than three times, and the numericalmaximum dispersion range is shown in ± number.
2.3. Equipment. Electron spin resonance (ESR) spectrometer(JEOL, JES-FA200 spectrometer, Tokyo). ESR spectrometryconditions used to estimate each radical with spin-trappingreagent were as follows: microwave frequency: 9414.499 ±5.000 MHz, microwave power: 4.00 mW, field center: 335.32
± 0.5 mT, sweep width: ±5.00 mT, modulation frequency:100.00 kHz, modulation width +/−: 0.1 mT, sweep time: 0.5–5 min, amplitude: 1.500–2.500, and time constant: 0.03–0.5 s, at room temperature. ESR universal cavity (JEOL,ES-UCX2 : TE011 mode cavity) with an X-band microwaveunit (8.750–9.650 GHz). ESR standard marker: manganeseoxide (MnO) powder (JEOL DATUM, MO7-FB-4) aqueoussample cell (JEOL, ES-LC12), sample volume: 20–100 μL. Atissue-type quartz cell (Labotec, Tokyo) with home-madecover glass (Size: 40 × 5 × 0.5 mm in thickness).
2.4. ESR Signal Measurement in Animal Tissue
2.4.1. Animals. For postmortem change experiments, maleddY mice (Nihon SLC, Shizuoka, Japan) weighing 20.1–25.7 g (6 to 8 weeks old) were used. The animals were housedat a room temperature of 20.2–25.3◦C under a 12-h light-dark cycle (lights on at 7:00 a.m.). Food and water wereavailable ad libitum. All of the following procedures wereconducted in accordance with the guiding principles forthe care and use of laboratory animals promulgated by theJapanese Pharmacological Society and with the guidelines foranimal care in our laboratories, as approved by the TokyoWomen’s Medical University Committee on animal care anduse. Food was withdrawn 24 h before experiments.
2.4.2. Removal of Tissue and ESR Analysis. Mice weresacrificed by dislocating their cervical spine. The tissueswere immediately removed and placed on an ice-cold plateafter being rinsed with ice-cold buffer (0.1 mol/L phosphatebuffer/saline, pH 7.8). The tissues were sliced into 0.2–0.3 mm in thickness using a microtome (KN3150465) (Kenis,Osaka, Japan). Slice weight was measured for normalizingESR signal of each radical. Brain tissues were removed fromthe cerebral hemisphere. Hart tissues were removed from thelower tip of the atrium. Tissues of lung, liver, kidney, andpancreas were collected. Muscle tissues were removed fromthe thigh muscle of right legs. Skin tissues were removedfrom the tip of ears. Whole blood was sampled from theheart. DMPO (10–50 μL) was added to the tissue samples(10–50 mg) or the blood (10–50 μL) immediately after beingweighed, and at precisely five minutes after remove, ESRsignals were measured. To identify obtained peaks, the signalsmeasured were analyzed by specialized analysis software,installed in the ESR device, for determining the g-value andhfcc calculated from the distance between peaks. After theadduct signals of superoxide were confirmed, the decaying ofthe peak was monitored by adding SOD solution to cuvettescontaining samples.
2.5. Postmortem Change. Mice were sacrificed by dislo-cating their cervical spine. Their tissues were collected,and their ESR spin adduct signals were detected by theprocedures described above. For observing postmortemchange of ESR signal, the sliced tissues were stored at4◦C and for 3–125, 163, 205 days after being sealed withpolyvinylidene chloride film to prevent water evaporation forcreating fixed decomposition conditions. The sliced mouse

Journal of Toxicology 7
tissues were ESR-analyzed on the 3, 15, 30, 60, 125, 163,and 205 days postmortem by the procedures describedabove.
2.6. Chemicals. The chemicals used in the present studywere 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) (Labotec,Tokyo, Japan), 5-(dipropoxy phosphoryl)-5-methyl-1-pyrroline N-oxide (DPPMPO), (Dojin chemicals, Kumamo-to, Japan), xanthine oxidase (MP Biomedicals, Ohio,USA), hypoxanthine (Wako Pure Chemical Industries,Osaka, Japan), methanol, (USP grade), dimethyl sulfoxide,sequencing (DMSO) (Pierce Biotechnology, Ill, USA),hydrogen peroxide (Wako Pure Chemical,) ferrous sulfate,(USP grade), superoxide dismutase, from bovine eryth-rocytes (Cu/Zn Type) (Wako Pure Chemical), L(+)-ascorbic acid (Wako Pure Chemical), and 3,5-dibromo-4-nitrosobenzenesulfonic acid sodium salt (MP Biomedicals,Inc., Ohio, USA). All other chemicals were of analyticalgrade.
2.7. Data Analysis. Standard ESR signals were recordedon the ESR computer system, the position and heightof peaks were recorded together with the height ofthe internal standard. g-value and hfcc were calcu-lated automatically after measurements with ESR com-puter software. The ESR signals of samples were identi-fied by determining the g-value and hfcc of measurablepeaks and compared with the peak values of standardradicals.
2.8. Statistical Analysis. Data are expressed as the mean withS.E.M. One-way ANOVA followed by Dunnett’s multiplecomparison test was used for evaluating the significance ofdifference. A P less than <.05 was considered significant.In the analysis of an ESR signal of postmortem change,regression line, correlation coefficient (R1), contributionrate (R2), and the significant difference calibration werecalculated by a method of Pearson. A correlation coefficient(R1) � 0.7 and a contribution rate � 0.5 were consideredsignificant.
3. Results
Signals A, B, C, and D shown in Figure 1 were originatedfrom DMPO-OOH (superoxide adduct signal), DMPO-AsA∗ (AsA radical adduct signal), DMPO-H (hydrogenradical adduct signal), and DMPO-OH (hydroxyl radicaladduct signal), respectively. The hfcc of DMPO-OOH,DMPO-AsA∗, and DMPO-H was in agreement to threedigits after decimal point within the max range of ±0.0009,and this value was almost the same as the previous study[20]. With regard to DMPO-OOH, when peaks decayed,adjacent peaks were merged, making it difficult to distinguishtheir g-values, because aHγ values (0.132) was quite small.Therefore, the g-values of combined adjacent peaks weredetermined by the standard peaks in advance, and theg-values and the distance between the combined peakswere used for identification. The following g-values were
determined from the standard waveform and used to identifythe peaks detected in the organs when peaks decayed intissue. For example, in Figure 1(a), (I′) was the centerpoint g-value between the g-value of the highest point ofpeak (I) and the g-value of the lowest point of peak (II)(2.0173). (II′) was the center point the g-value betweenthe g-value of the highest point of peak (III) and the g-value of the lowest point of peak (VI) (2.0096). Furthertests for identification showed that the peak of DMPO-OH was decayed with the addition of AsA solution, whilethe peak of DMPO-OOH was decayed with the additionof SOD solution. The existence of hydrogen radicals wasconfirmed by observing orange color in the DDTMK-addedhydrogen-radical solution. These results confirmed that thepeaks of the spin adducts artificially generated were adequateas standard peaks. The g-value and hfcc of DMPO-AsA∗
signal were 2.0045 and 0.187, respectively. These values werealmost the same as the previous study [22]. The tissuesamples were the brain, hart, lung, liver, kidney, pancreas,muscle, and skin of ddy mice. Various ESR spin adductsignals including AsA signal were detected in these samples.These results showed that DMPO-AsA∗, DMPO-OOH, andDMPO-H were detectable and identifiable in the mousetissue.
Figure 2 shows ESR signals of DMPO adduct of normalmice from brain, lung, muscle from femoral, skin from ear,heart, liver, kidney, and pancreas. The marks (•, �, and�) were added to the peak that accords with the g-valueof standard signal in Figures 1 and 2. Measurement of thestandard waveform and hyperfine structure constant of freeradicals was performed . Figure 1 shows the spectrum ofDMPO standard adduct signals of various free radicals thatwere artificially generated. Postmortem changes in DMPO-AsA∗ and DMPO-OOH signals were observed at 4 ◦C invarious tissues of the mouse. Figure 3(a) shows the changeof signal from brain; Figure 4(a), lung; Figure 5(a), heart;Figure 6(a), liver. The brain was measured up to 125 daysbefore tissue destruction, the lungs and the liver were ableto be measured up to 163 days, and the heart, a strongtissue, was measured up to 205 days. The signal peaks of thespin adducts identified were monitored at the 125, 163, and205th day after postmortem. The increase of signal intensityratio was observed from 0 to 3 days posthumously but nosignificant difference.
All peak intensity ratios decreased postmortem. Thedecrease rate was found to be straight on the logarithmicalscale of postmortem period. The regression analysis ofthe signal intensity ratio and the postmortem period wasperformed for each tissue. The results of regression analysisof brain were shown in Figure 3(b); lung, Figure 4(b);heart, Figure 5(b); liver, Figure 6(b). Furthermore, corre-lation coefficient (R1), the square of R1 (R2), and theprobability of error (P) were also calculated. The linearity ofDMPO-AsA∗ was found to be better than that of DMPO-OOH from the former’s higher correlation coefficient. Thelinearity of DMPO-AsA∗ in the liver tissue (y = −40.7x +113.8, x = log(day), R1 = −0.779, (P < .001)) and brain(y = − 45.3x + 110.1, x = log(day), R1 = − 0.739, P < .001)was meaningful.

8 Journal of Toxicology
4. Discussion
The significant amount of AsA is found in the body and isone of redox molecules first consumed by oxidative stress[23]. AsA is particularly an essential factor in eliminatingROS (reactive oxygen species) in which hydroxyl radicalsare the most highly toxic. The lifetime of AsA∗ is extremelyshort, being measured in microseconds, which makes themextremely difficult to be detected. To date, research hasbeen carried out via an ESR spin drum (trapping) method.However, X-band electromagnetic waves, which are emittedfrom whole tissues using the spin trapping method, areattenuated due to the body moisture of WIEL. Therefore,detecting the signals with this conventional method isdifficult and is limited to be applied to a tissue such as brainemitting comparatively strong signals.
In this experiment, a high purity quart cuvette wasimproved (modified), and by a new spin trap agent, DMPO,with a high permeability to brain tissues, the detectionsensitivity of signal of AsA∗ in brain tissues was improvedsuccessfully by modifying the method described as follows.The permeability of X-band waves was improved by a thintissue sample that was half of that used. WIDL was alsominimized by drastically reducing the overall fluid volume(including a large quantity of moisture) added to ESR cavityfor each organ from 150 μL to 20–50 μL. The attenuation inelectromagnetic waves caused by cover glass was also reducedby making cover glass thinner (0.15 μm). The ratio of the spintrap solution and the mass of each tissue slice was changed to1.2 : 1 to improve its sensitivity. While the Masumizu methodused grease on the edge of the cover glass to fix specimen, theweight of cover glass in our method was very light and itssurface tension was sufficient to attach sample tissue withoutgrease, ignoring the spectral and chemical changes inducedby the grease itself. The new procedure was able to measureESR signal from sample with high sensitivity.
It is demonstrated that a substantial improvement inthe sensitivity of detecting DMPO-AsA∗ and DMPO-OOHsignals occurred and that the obvious peaks of DMPO-AsA∗
and DMPO-OOH were detected with ESR even withoutoxidative stress. In a condition without oxidative stress,DMPO-OOH signals were high in heart, liver, and kidneyamong postmortem tissues while DMPO-AsA∗ signals weredetected especially high in the brain and lungs samples. Inthe previous study [20], only the traces of AsA∗ adductswere detected in tissue with a low AsA concentration andit was difficult to measure them except brain. However, thisstudy was able to show the spin adduct signals of AsA∗
even in tissue with a low AsA concentration without addingoxidative stress. In other words, AsA∗ adducts can now bedetected in almost all tissue including brain, lung, heart, liver,kidney, pancreas, muscle, and skin. These results indicatethat oxidative stress can be easily detected in almost all tissueas long as DMPO-AsA∗ adducts are used as indicators foroxidative stress. AsA∗ adducts are observed, when AsA reactswith hydroxyl radicals or ROS such as superoxide and alsowhen AsA removes an electron in the regeneration process oftocopheryl radicals. On that time, these AsA∗ react with spintrap agents resulting in DMPO-AsA∗. Reacting especially
with hydroxyl radicals, AsA is known as a hydroxyl radicalscavenger in the body. It is highly possible that the spinadducts of AsA∗ are the byproducts of these radical reactions.The peaks of DMPO-AsA∗ were composed of twin peaksof the same height, with g-values of 2.0057 and 2.0045 anda hfcc of 0.187 mT. The signals of DMPO-AsA∗ agree wellwith those values reported by Masumizu et al. [22], thatis, the g-value and hfcc (aH) of a doublet were 2.0048 and0.187 mT. Regarding the hydrogen radical, the g-values andthose of its nine characteristic peaks were measured (Figures1 and 2). Although all nine peaks were unable to be observedalways, the g-values of peaks observed in tissue were ableto be measured and compared with the standard peaks. Inmouse tissue in this study, DMPO detected an AsA radicalsadduct signal, superoxide adduct signal, and several otheradduct signals with the same g-values as a hydrogen radicalsadduct signal.
ESR signals from sample tissue were identified as DMPO-OOH, DMPO-AsA∗, and DMPO-H by comparing their hfccwith the reference values. Regarding DMPO-OH, a peak ofequal g-values was detected only at trace level from tissue.Although the peak height of spin adducts in the spectrumvaries depending on the concentration of spin trap agents,by thin-sliced tissue and the conditioning of spelling Q-DIP,DMPO-AsA∗ clearly showed better sensitivity than DMPO-OOH or DMPO-H which are detected at the same time.Since the signal of DMPO-AsA∗ was stable in comparisonwith DMPO-OOH or DMPO-H, DMPO-AsA∗ might bemore useful in detecting intracellular oxidative stress thanDMPO-OOH or DMPO-H.
As one disadvantage, DMPO-OOH is overlapped bybackground signals, especially in heart and other muscles,thus making it difficult to distinguish. Conversely, DMPO-AsA∗ can yield clear signals, although the g-value overlapsthat of DMPO-OOH. Therefore, DMPO is considered auseful spin trap agent, especially for detecting intracellularAsA∗. The signals of DMPO-AsA∗ was detected in mousebrain and lung more clearly than another tissue in this ESRmeasurement. The signals of DMPO-OOH were detectedin mouse heart and liver more clearly than other tissues.Intracellular AsA concentration in tissues (brain, lung,liver, kidney, and pancreas) is higher than extracellularAsA concentration (like in blood), because superoxide wasspeculated to have an extracellular source. In regards toAsA∗, for DMPO-AsA∗, the detection peaks for both of thesewere, in order from the highest to the lowest, brain > lung> liver > kidney > heart > muscle. Brain and lungs tissueshave an antioxidation stress system, because both tissues areable to receive oxidation stress easily [24–27]. As for the highdetection levels of AsA∗ in brain and lung, the concentrationof AsA in organ is brain > lung > liver > kidney > heart >muscle [28]. Brain and lung are known to have an especiallyhigh AsA concentration. Therefore, there is a possibility thatthe peak heights of DPPMPO AsA reflect AsA concentrationwithin respective tissue. The signal intensity ratio of DMPO-OOH was high in heart and liver tissues.
The heights of respective peaks of DMPO-OOH at0 days postmortem were, except blood, in order, heart(50%) and liver (50%) > brain (45%) and lungs (45%);

Journal of Toxicology 9
this appeared to have matched the ranking of respectiveiron concentrations within each organ. The peak heightsof DMPO-OOH were found to be dependent on the ironconcentrations of hemoglobin being the main representativeamong components within each tissue. It is the experi-mentally found results. In a previous study, regarding ironconcentrations in each organ, the blood was reported to havethe highest concentrations; for a mouse of age 100 days,iron concentrations in heart, liver, kidney, and brain were298, 254, 245, and 89 ng Fe/mg dry wt, respectively [29].In an organism, 70% Fe exists in blood hemoglobin, while20% to 25% exists in water-soluble ferritin and insolublehemosiderin in the liver, spleen, bone marrow, and so forth.In blood serum, Fe exists in transferrin. Numerous reportsdescribe multiple generation systems producing superoxidefrom blood. For example, a system generating superoxideis activated by phagocytes. Especially, the present studywas able to continue to detect superoxide and AsA radicaladduct signals in heart over 200 days postmortem (Figure 5).Further, at around 200 days, heart tissue color was foundto change from black to a yellowish brown, and as anorgan dries and hardens, the most of cells in the organ arepresumed to die. Nevertheless, even from such tissue, thetrace amounts of superoxide continued to be detected. Thelinearity of DMPO-AsA∗ was found to be better than that ofDMPO-OOH from the correlation coefficients. The linearityof DMPO-AsA∗ liver (y = 40.7x + 113.8, x = log(day),R1 = −0.779) was found to be the best (Figure 6), becauseit was thought that (1) the liver AsA level at death time wascomparatively high and (2) the configuration of liver tissuewas stable posthumously.
As a possible system for generating superoxide over a longpostmortem duration, the best candidate was thought to bethe oxidation process of iron in hemoglobin.
Hemoglobin contains four hemes; when heme iron isFe(II), it reversibly binds with oxygen. Hemoglobin withoxygen (oxyhemoglobin) oxidizes postmortem and becomesmethemoglobin containing Fe(III). At the reaction step, elec-trons are released and superoxide and H2O2 are generated(Haber-Weiss reaction) [30]:
Fe(II) + O2 −→ Fe(III) + (O2•)− (1)
It is a well-known fact that hydrogen peroxide isproduced from the reaction of superoxide dismutase andsuperoxide [31]. It has recently become clear that Fe(II)generates hydrogen peroxide (H2O2) [32, 33] due to thereaction of superoxide and H+(hydrogen ion) and further,that due to the reaction of (3), a hydroxyl radical is produced[34]:
Fe(II) + (O2•)− + 2H+ −→ Fe(III) + H2O2 (2)
The iron in these reactions may be dissolved or surfacebound as these reactions can occur in solution or onpyrite surface [35]. The hydrogen peroxide is generated withreaction (2) due to the Fenton reaction with Fe(II) andproduces a hydroxyl radical:
Fe(II) + H2O2 −→ •O3H + OH− + Fe(III) (3)
It is thought that the large amounts of generated superox-ide cause further hemoglobin oxidation and that theypromote further the production of methemoglobin. Ito etal., reported that iron oxidation reaction proceeds in ironprotein and AsA [20].
Furthermore, AsA reduces iron as the following reactionof iron-proteins and AsA; this reaction would be recycled.Possible reactions proceed in the following sequence:
Fe(II) Protein + O2 −→ Fe(III) Protein +(O•
2
)(4)
Fe(II) Protein + (O2•)− + 2H+−→Fe(III) Protein + H2O2
(5)
Fe(II) Protein + H2O2 −→ •O3H + OH− + Fe(III) Protein
+OH− + Fe(III)(6)
Fe(III) Protein + AsA −→ Fe(II) Protein + AsA• (7)
Iron-protein recycling reaction with AsA suggested thatthe reactions would potentially continue for long time.
Hochstein and collaborators [36, 37] reported that theoxidation of myoglobin into ferrylmyoglobin (MbIV) isa critical event in tissue damage associated with cardiacischaemic reperfusion states. Also, superoxide extricates freeFe from Fe-binding proteins such as ferritin, thereby assistingin the oxidation of Fe [38]. Further, in this study, DMPO-OOH originating from the blood was confirmed to besuppressed (inhibited) by the addition of citric acid, aniron chelator. It is because the content is an experimentalresult. In this way, in the postmortem observations in ourexperiments, over a long term, DMPO-OOH from tissuesamples was detected, because the superoxide generationsystem became the main source for generating superoxide intissue during postmortem. The linear decrease of superoxideindicated the reduction of superoxide generation in theoxidation process of Fe(II) to Fe(III).
However, in the tissue, numerous other O2 genera-tion sources were observed in addition to iron oxidation.For example, in several days postmortem, phagocytes inblood such as neutrophils, eosinophils, monocytes, andmacrophages, and so forth, were thought to produce super-oxide by NADPH and NADPH oxidase reactions from thestimuli of bacterial proliferation, protein degeneration, andand so forth [39]. From ESR signal of liver, the peak wherethe g-values of DMPO-OOH, DMPO-AsA∗, and DMPO-Hwere recognized. In the liver, mitochondria in hepatocytesare active occurred , and superoxide is produced by drugmetabolism [40, 41]. Superoxide apparently appears morein the liver, which is an organ most easily exposed tosuperoxide, because of the extremely high level of superoxidedismutase (SOD) activity reported in the liver [41]. SODactivity is also high in the liver, kidneys, and heart [42]. Themain sources of superoxide in the liver are reportedly micro-some P450 (P450IIE1) and NADH during the metabolism

10 Journal of Toxicology
of substances like alcohol [43]. In a previous study, itwas reported that the majority of superoxides originatingfrom tissue are metabolic byproducts from mitochondria,respiration, and microsomes [40]. In the brain, superoxideis reportedly produced, when the nervous system is directlyexposed to hemoglobin, which releases a large amount ofiron [44]. In nerve cells, superoxide is produced by theoxidizing system of dopamine and catecholamine [45]. Thegeneration of superoxide in the brain gives neuronal death,which is considered as a cause of damage to the nerve cells,as manifested in diseases including multiple sclerosis, thedeterioration of cognitive function with aging, dementia,amyotrophic lateral sclerosis (ALS), and Alzheimer’s andParkinson’s diseases [46].
The results of our experiments showed a tendency forincreasing the peak height of DMPO-OOH, a spin adductfor superoxide, from immediately to several days after death.DMPO-OOH occurring from the superoxide productionsystem of hepatocytes as mentioned above is also consideredas the part of this increase. All mean values on three daysafter death were slightly above the line of the superoxidedecay curve; it may be the indication of other superoxidesources than Fe. However, the marginal differences in thesevalues imply that the postmortem occurrence of superoxidestill remains a major source for generating superoxide threedays after death regardless of the origin of control. For severaldays postmortem, cells in tissue samples remain alive andthe tissues are under ischemic condition. The increase ofDMPO-OOH during these postmortem days is speculated tobe due to the occurrence of superoxide caused by reactionwith ischemia from hypoxic condition.
In this study, the sensitivity of detecting DMPO adductsignals in the tissue was improved using an X-band ESRand the spin trap method. For reducing the decay causedby WIDL of X-band, sample cuvette in ESR instrumentwas also modified and DMPO was examined as a new spintrap agent. By these improvements, spin adduct signals weredetected from brain, lungs, heart, liver, kidneys, pancreas,muscles, and skin tissues and the signals were confirmedto be the genuine adduct signals of superoxide and AsA∗
from their g-values and hfcc-values of standard signal. Inthe postmortem follow up, DMPO-OOH, DMPO-AsA∗, andDMPO-H were detected not only in the fresh tissue butalso in the tissue of a mouse that had been stored morethan 200 days after death in 4◦C. DMPO-AsA∗ in liverwas found to linearly decrease to logarithm of postmortem.It was related linearly to the logarithm of duration andnot linearly to postmortem duration. Therefore, DMPO-AsA∗ signals were found to be a useful indicator estimatingpostmortem duration and oxidative damages in varioustissue.
References
[1] G. A. Pashinian and V. L. Proshut, “Determination of the timeof occurrence of mechanical trauma by the EPR spectra of thebone marrow,” Sudebno-Meditsinskaia Ekspertiza, vol. 31, no.4, pp. 9–11, 1988.
[2] R. V. Uzeneva, “The diagnosis of intravital mechanicaltrauma by the blood EPR-spectral parameters,” Sudebno-Meditsinskaya Ekspertisa, vol. 32, no. 4, pp. 16–18, 1989.
[3] E. M. Mil’, V. V. Kasparov, V. I. Biniukov, N. V. Tabatchikova,and O. A. Borisova, “Changes in the EPR spectra of the nitrosylcomplexes of blood proteins in the low-intensity whole-bodyirradiation of mice,” Radiatsionnaia, Biologiia, Radioecologiia,vol. 40, no. 3, pp. 305–309, 2000.
[4] N. Nakamura, H. M. Cullings, Y. Kodama et al., “A methodto differentiate between the levels of ESR signals induced bysunlight and by ionizing radiation in teeth from atomic bombsurvivors,” Radiation Research, vol. 165, no. 3, pp. 359–364,2006.
[5] L. Quarino and L. Kobilinsky, “Development of a radioim-munoassay technique for the detection of human hemoglobinin dried bloodstains,” Journal of Forensic Sciences, vol. 33, no.6, pp. 1369–1378, 1988.
[6] S. Turkes, O. Korkmaz, and M. Korkmaz, “Time course of theage-related alterations in stored blood,” Biophysical Chemistry,vol. 105, no. 1, pp. 143–150, 2003.
[7] Y. Fujita, K. Tsuchiya, S. Abe, Y. Takiguchi, S. I. Kubo, andH. Sakurai, “Estimation of the age of human bloodstainsby electron paramagnetic resonance spectroscopy: long-termcontrolled experiment on the effects of environmental fac-tors,” Forensic Science International, vol. 152, no. 1, pp. 39–43,2005.
[8] M. Culcasi, S. Pietri, and P. J. Cozzone, “Use of 3,3,5,5-tetramethyl-1-pyrroline-1-oxide spin trap for the continuousflow ESR monitoring of hydroxyl radical generation inthe ischemic and reperfused myocardium,” Biochemical andBiophysical Research Communications, vol. 164, no. 3, pp.1274–1280, 1989.
[9] S. M. Musser, Y. C. Fann, R. J. Gurbiel, B. M. Hoffman,and S. I. Chan, “Q-band electron nuclear double resonance(ENDOR) and X-band EPR of the sulfobetaine 12 heat-treated cytochrome c oxidase complex,” Journal of BiologicalChemistry, vol. 272, no. 1, pp. 203–209, 1997.
[10] P. Kuppusamy, S. T. Ohnishi, Y. Numagami, T. Ohnishi,and J. L. Zweier, “Three-dimensional imaging of nitric oxideproduction in the rat brain subjected to ischemia-hypoxia,”Journal of Cerebral Blood Flow and Metabolism, vol. 15, no. 6,pp. 899–903, 1995.
[11] H. Yokoyama, Y. Lin, O. Itoh et al., “EPR imaging forin vivo analysis of the half-life of a nitroxide radical inthe hippocampus and cerebral cortex of rats after epilepticseizures,” Free Radical Biology and Medicine, vol. 27, no. 3-4,pp. 442–448, 1999.
[12] H. Togashi, T. Matsuo, H. Shinzawa et al., “Ex vivo mea-surement of tissue distribution of a nitroxide radical afterintravenous injection and its in vivo imaging using a rapidscan ESR-CT system,” Magnetic Resonance Imaging, vol. 18,no. 2, pp. 151–156, 2000.
[13] A. Hirayama, S. Nagase, A. Ueda et al., “In vivo imag-ing of oxidative stress in ischemia-reperfusion renal injuryusing electron paramagnetic resonance,” American Journal ofPhysiology—Renal Physiology, vol. 288, no. 3, pp. F597–F603,2005.
[14] K. Matsumoto, S. Kawai, C. F. Chignell, and H. Utsumi,“Location of anthralin radical generation in mouse skinby UV-A irradiation: an estimation using microscopic EPRspectral-spatial imaging,” Magnetic Resonance in Medicine, vol.55, no. 4, pp. 738–742, 2006.
[15] K. Takeshita, H. Utsumi, and A. Hamada, “ESR measurementof radical clearance in lung of whole mouse,” Biochemical and

Journal of Toxicology 11
Biophysical Research Communications, vol. 177, no. 2, pp. 874–880, 1991.
[16] H. Utsumi, K. Ichikawa, K. Takeshita et al., “In vivo ESRmeasurements of free radical reactions in living mice,” Journalof Toxicological Sciences, vol. 21, no. 5, pp. 293–295, 1996.
[17] J. Bielski, A. Sawinska, and J. Pianowska, “Disorders of thebioelectric activity of the brain in workers exposed to theelectromagnetic fields of different frequency,” Polski TygodnikLekarski, vol. 37, no. 26, pp. 769–771, 1982.
[18] X. Wang, J. Liu, I. Yokoi, M. Kohno, and A. Mori, “Directdetection of circulating free radicals in the rat using elec-tron spin resonance spectrometry,” Free Radical Biology andMedicine, vol. 12, no. 2, pp. 121–126, 1992.
[19] S. Ito, K. Itoga, M. Yamato, H. Akamatsu, and T. Okano, “Theco-application effects of fullerene and ascorbic acid on UV-Birradiated mouse skin,” Toxicology, vol. 267, no. 1–3, pp. 27–38, 2010.
[20] S. Ito, T. Mori, H. Kanazawa, and T. Sawaguchi, “Differ-ential effects of the ascorbyl and tocopheryl derivative onthe methamphetamine-induced toxic behavior and toxicity,”Toxicology, vol. 240, no. 1-2, pp. 96–110, 2007.
[21] C. Chi, R. Tanaka, Y. Okuda et al., “Quantitative measure-ments of oxidative stress in mouse skin induced by X-rayirradiation,” Chemical and Pharmaceutical Bulletin, vol. 53, no.11, pp. 1411–1415, 2005.
[22] T. Masumizu, Y. Noda, A. Mori, and L. Packer, “Electronspin resonance assay of ascorbyl radical generation in mousehippocampal slices during and after kainate-induced seizures,”Brain Research Protocols, vol. 16, no. 1–3, pp. 65–69, 2005.
[23] B. Frei, R. Stocker, and B. N. Ames, “Antioxidant defenses andlipid peroxidation in human blood plasma,” Proceedings of theNational Academy of Sciences of the United States of America,vol. 85, no. 24, pp. 9748–9752, 1988.
[24] G. W. Hunninghake and R. G. Crystal, “Cigarette smoking andlung destruction. Accumulation of neutrophils in the lungsof cigarette smokers,” American Review of Respiratory Disease,vol. 128, no. 5, pp. 833–838, 1983.
[25] J. C. Hogg, “Neutrophil kinetics and lung injury,” PhysiologicalReviews, vol. 67, no. 4, pp. 1249–1295, 1987.
[26] C. W. Olanow, “Oxidation reactions in Parkinson’s disease,”Neurology, vol. 40, no. 10, supplement 3, pp. 37–39, 1990.
[27] C. E. Cross, A. van der Vliet, C. A. O’Neill, and J. P. Eiserich,“Reactive oxygen species and the lung,” Lancet, vol. 344, no.8927, pp. 930–933, 1994.
[28] K. Tanaka, T. Hashimoto, S. Tokumaru, H. Iguchi, and S. Kojo,“Interactions between vitamin C and vitamin E are observedin tissues of inherently scorbutic rats,” Journal of Nutrition, vol.127, no. 10, pp. 2060–2064, 1997.
[29] H. R. Massie, V. R. Aiello, and V. Banziger, “Iron accumulationand lipid peroxidation in aging C57BL/6J mice,” ExperimentalGerontology, vol. 18, no. 4, pp. 277–285, 1983.
[30] C. A. Cohn, R. Laffers, and M. A. Schoonen, “Using yeastRNA as a probe for generation of hydroxyl radicals by earthmaterials,” Environmental Science and Technology, vol. 40, no.8, pp. 2838–2843, 2006.
[31] B. M. Babior, “Phagocytes and oxidative stress,” AmericanJournal of Medicine, vol. 109, no. 1, pp. 33–44, 2000.
[32] M. J. Borda, A. R. Elsetinow, M. A. Schoonen, and D. R.Strongin, “Pyrite-induced hydrogen peroxide formation as adriving force in the evolution of photosynthetic organisms onan early earth,” Astrobiology, vol. 1, no. 3, pp. 283–288, 2001.
[33] C. A. Cohn, A. Pak, M. A. Schoonen, and D. R. Strongin,“Quantifying hydrogen peroxide in iron-containing solutions
using leuco crystal violet,” Journal of Pharmacological andToxicological Methods, vol. 50, no. 3, pp. 47–52, 2005.
[34] C. A. Cohn, R. Laffers, S. R. Simon, T. O’Riordan, and M. A.Schoonen, “Role of pyrite in formation of hydroxyl radicalsin coal: possible implications for human health,” Particle andFibre Toxicology, vol. 3, no. 16, Article ID 16, pp. 1–10, 2006.
[35] T. O’riordan and M. A. Schoonen, “Role of pyrite in formationof hydroxyl radicals in coal: possible implications for humanhealth,” Particle and Fibre Toxicology, vol. 19, no. 3, p. 16, 2006.
[36] A. Arduini, L. Eddy, and P. Hochstein, “The reduction offerryl myoglobin by ergothioneine: a novel function forergothioneine,” Archives of Biochemistry and Biophysics, vol.281, no. 1, pp. 41–43, 1990.
[37] D. Galaris, L. Eddy, A. Arduini, E. Cadenas, and P. Hochstein,“Mechanisms of reoxygenation injury in myocardial infarc-tion: implications of a myoglobin redox cycle,” Biochemicaland Biophysical Research Communications, vol. 160, no. 3, pp.1162–1168, 1989.
[38] P. Biemond, A. J. Swaak, H. G. van Eijk, and J. F. Koster,“Superoxide dependent iron release from ferritin in inflam-matory diseases,” Free Radical Biology and Medicine, vol. 4, no.3, pp. 185–198, 1988.
[39] J. M. McCord, “The evolution of free radicals and oxidativestress,” American Journal of Medicine, vol. 108, no. 8, pp. 652–659, 2000.
[40] B. Chance, H. Sies, and A. Boveris, “Hydroperoxidemetabolism in mammalian organs,” Physiological Reviews, vol.59, no. 3, pp. 527–605, 1979.
[41] M. W. J. Cleeter, J. M. Cooper, and A. H. Schapira, “Irre-versible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridinium: evidence for free radical involvement,”Journal of Neurochemistry, vol. 58, no. 2, pp. 786–789, 1992.
[42] M. Phillips, J. Camakaris, and D. M. Danks, “Comparisons ofcopper deficiency states in the murine mutants blotchy andbrindled. Changes in copper-dependent enzyme activity in 13-day-old mice,” Biochemical Journal, vol. 238, no. 1, pp. 177–183, 1986.
[43] F. Cristiano, J. B. de Haan, R. C. Iannello, and I. Kola,“Changes in the levels of enzymes which modulate theantioxidant balance occur during aging and correlate withcellular damage,” Mechanisms of Ageing and Development, vol.12, no. 80, pp. 93–105, 1995.
[44] J. M. Braughler and E. D. Hall, “Central nervous systemtrauma and stroke. I. Biochemical considerations for oxygenradical formation and lipid peroxidation,” Free Radical Biologyand Medicine, vol. 6, no. 3, pp. 289–301, 1989.
[45] D. G. Graham, “On the origin and significance of neurome-lanin,” Archives of Pathology and Laboratory Medicine, vol. 103,no. 7, pp. 359–362, 1979.
[46] J. N. Keller and M. P. Mattson, “Roles of lipid peroxidationin modulation of cellular signaling pathways, cell dysfunction,and death in the nervous system,” Reviews in the Neurosciences,vol. 9, no. 2, pp. 105–116, 1998.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 683728, 12 pagesdoi:10.1155/2011/683728
Review Article
Oxidative Toxicity in Neurodegenerative Diseases:Role of Mitochondrial Dysfunction and Therapeutic Strategies
Katie Facecchia,1 Lee-Anne Fochesato,1 Sidhartha D. Ray,2
Sidney J. Stohs,3 and Siyaram Pandey1
1 Department of Chemistry & Biochemistry, University of Windsor, 277-1 Essex Hall, 401 Sunset Avenue,Windsor, ON, Canada N9B 3P4
2 College of Pharmacy and Toxicology, Long Island University, Brooklyn, NY 11436-1331, USA3 School of Pharmacy and Health Professions, Creighton University Medical Center, Omaha, NE 68178, USA
Correspondence should be addressed to Siyaram Pandey, [email protected]
Received 31 January 2011; Accepted 8 May 2011
Academic Editor: M. Firoze Khan
Copyright © 2011 Katie Facecchia et al. This is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Besides fluorine, oxygen is the most electronegative element with the highest reduction potential in biological systems. Metabolicpathways in mammalian cells utilize oxygen as the ultimate oxidizing agent to harvest free energy. They are very efficient, butnot without risk of generating various oxygen radicals. These cells have good antioxidative defense mechanisms to neutralizethese radicals and prevent oxidative stress. However, increased oxidative stress results in oxidative modifications in lipid, protein,and nucleic acids, leading to mitochondrial dysfunction and cell death. Oxidative stress and mitochondrial dysfunction havebeen implicated in many neurodegenerative disorders including Alzheimer’s disease, Parkinson’s disease, and stroke-related braindamage. Research has indicated mitochondria play a central role in cell suicide. An increase in oxidative stress causes mitochondrialdysfunction, leading to more production of reactive oxygen species and eventually mitochondrial membrane permeabilization.Once the mitochondria are destabilized, cells are destined to commit suicide. Therefore, antioxidative agents alone are not sufficientto protect neuronal loss in many neurodegenerative diseases. Combinatorial treatment with antioxidative agents could stabilizemitochondria and may be the most suitable strategy to prevent neuronal loss. This review discusses recent work related to oxidativetoxicity in the central nervous system and strategies to treat neurodegenerative diseases.
1. Sensitivity of Neurons to Oxidative Stress
Neuronal cells in the brain are highly sensitive to oxidativestress due to their large dependence on oxidative phospho-rylation for energy as compared to other cells. The demandfor oxygen consumption is extremely high with 1-2% ofthe oxygen being converted into superoxide anion radicals(O2
•−) and hydrogen peroxide, leading to oxidative stress[1]. Oxidative stress exists when there is an imbalance ofreactive oxygen species (ROS) production and antioxidantactivity. Since there are high levels of metals such as ironin the brain, metal toxicity is also a problem leading tooxidative stress. One way that the brain combats stressis by employing the aerobic isoenzymatic form of lactatedehydrogenase when glucose is metabolized [2]. Previousreports have indicated that these isoenzymatic forms oflactate dehydrogenase play a significant role in the metabolic
functions of neurons [3]. Since neurons in the brain alsostrongly depend on mitochondrial driven aerobic respira-tion, when the mitochondria become dysfunctional, theseneurons become much more susceptible to oxidative stress.Mitochondria already have a high level of oxidative stressand, therefore, any increase in internal or external reactiveoxygen species (ROS) leads to dysfunctional mitochondria,which in turn produces more ROS leading to a vicious anddetrimental cycle (Figure 1).
Mitochondria have their own enzyme for combating ROSproduction by converting superoxide radicals to hydrogenperoxide by manganese superoxide dismutase (MnSOD),which are further broken down into water by peroxidases[4]. With heightened levels of oxidative stress, however, thesecombatants are not enough, and as we age, our defensesagainst oxidative stress decrease.

2 Journal of Toxicology
III II I
IV
V
ROS
Environmentaltoxins:MPP+
ParaquatRotenone
Lipid peroxidationProtein oxidationDNA oxidation
MMP
VDACmPTP
Apoptogenic factors:AIF
Cytochrome c
ATP
O2
Figure 1: Environmental toxins cause the production of ROS by inhibiting complex I of the electron transport chain (ETC) and decreasethe production of ATP. This ROS contributes to a loss in the mitochondrial membrane potential and well as disruption of mitochondrialpermeability transition pores and voltage-dependant anion channels contributing to apoptosis. ROS also moves to the cytosol where itoxidizes proteins, DNA, and lipids.
1.1. Factors Leading to Oxidative Stress. Generation of ROSat complex I, coined “complex I syndrome,” in the mito-chondrial electron transport chain (ETC) has been linked toage-associated modifications in the central nervous system[4, 5]. When mitochondrial DNA is the target of oxidation, itcan lead to mutations, rearrangements, and transcriptionalerrors that impair important mitochondrial componentsleading to more oxidative stress and eventual cell death.This has been shown to be more sensitive in cerebellargranule neuronal cells due to their deficiency in repairingmitochondrial DNA damaged by oxidative stress [4].
Lipid peroxidation causes a collapse of plasma andmitochondrial membranes, releasing cytochrome c, andinducing apoptosis. The brain is most affected by lipidperoxidation because of its high oxidizable lipid and metalcontent in comparison with other tissue [5].
Superoxide radicals and hydrogen peroxide can alsocreate further oxidative stress by metal-catalyzed reactions[6]. Under oxidative stress, superoxide radicals can oxidizeiron molecules. The released iron then takes part in theFenton reaction and generates hydroxyl [6]. It has beenshown that inactivation of mitochondrial aconitase (anenzyme involved in the citric acid cycle) by ROS contributesto the release of free iron and hydrogen peroxide leading toneuronal cell [7].
As a result of the reactions mentioned above, thereare increased levels of oxidized glutathione (GSSG) witha concomitant decrease in reduced glutathione (GSH),oxidized protein, and increased lipid peroxidation, all ofwhich are commonly used as markers of oxidative stress andthe extent of damage caused by it. We have shown a decreasein GSH levels when rats are challenged with the herbicide
paraquat, known to cause neurotoxicity and depletion ofsubstantia nigra neurons due to oxidative stress (Figure 2).
Direct oxidative stress by hydrogen peroxide has beenshown to induce inflammation by NF-κB activation andinterleukins and is involved in the stress activated proteinkinase (JNK) pathway [8]. Recent studies on chronic expo-sure of neuronal cells to hydrogen peroxide elicit dynamicresponses, including changes in cytoskeletal structure, energymetabolic shifts (aerobic to glycolysis), and transmembranereceptor activity [9]. In other studies, chronic exposure tohydrogen peroxide has been shown to have a protective roleby inducing the upregulation of antioxidant enzymes such ascatalase and superoxide dismutase [10, 11].
1.2. Oxidative Stress in Neurodegenerative Diseases Inducedby Environmental Toxins. Oxidative stress has been linkedto aging and two of the most common neurodegenerativediseases, namely, Alzheimer’s disease (AD) and Parkinson’sdisease (PD). AD is characterized by the loss of neurons,synapses, and neurotransmitters throughout the brain, butespecially in the hippocampus and cerebral cortex. Mito-chondrial dysfunction may be the underlying reason forthe loss, marked by an increase in ROS, lipid peroxidation,and protein oxidation, which are found in AD brains,thus contributing to oxidative stress [28, 29]. Amyloid beta(Aβ), one of the hallmarks of AD, is also involved inoxidative stress and mitochondrial dysfunction by reducingthe mitochondrial membrane potential. As an age-relateddisease, this reduction is intensified in the brain of aginganimal models compared to younger animals [28].
In PD, the oxidative stress is most damaging and selectiveto the mitochondria, specifically in the substantia nigra

Journal of Toxicology 3
SHSH
Oxidative stress
ARE
Keap1Keap1Nrf2Nrf2
SS
Nrf2P
Nrf2P
MAPKPK CPIK3
ActinActin
Figure 2: Oxidative stress induces Nrf2 dissociation from Keap1. Nrf2 is activated by phosphorylation and translocated into the nucleuswhere it may act as a transcription factor for antioxidant response genes.
region of the midbrain. Susceptibility to this disease canbe due to genetics, environmental toxins (including mostpesticides and herbicides), or a combination of the two,which can cause mitochondrial damage leading to oxidativestress [30].
Many cell culture models have been used to establishthe role of oxidative stress in PD in hopes of translatingthe observed results to an in vivo model. For example,glutamate excitotoxicity on mixed neuronal-glial cell culturesalong with hypoxia-induced neuronal cell death decreasedATP production and increased ROS [31]. Direct hydrogenperoxide insult has been shown to induce all the samenegative factors mentioned above in conjunction with PD[32] and is also associated with the proapoptotic protein Bax[33].
One well-established in vivo model for studying PDis the use of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine(MPTP), a byproduct found in synthetic heroine. MPTPcrosses the blood-brain barrier and is metabolized into1-methyl-4-phenylpyridinium (MPP+) where it has beenshown to block complex I of the ETC and increase ROS, lipidperoxidation, and protein oxidation [13, 34]. As mentionedpreviously, genetics and environmental toxins can providenegative synergistic effects contributing to PD. Recently,it has been found that DJ-1 deficient mice challengedwith MPTP have an increase in both oxidative stress anddopaminergic neuronal cell death [35]. DJ-1 is a geneassociated with an early onset form of PD.
Exposure to paraquat, which is structurally similar toMPP+, has been correlated with the risk of PD in multiplestudies [36–38]. Paraquat treatment on human neuronal cellculture causes mitochondrial permeabilization and oxidativestress [39]. It is believed to induce symptoms of PD byreacting in its reduced form with oxygen to produce asuperoxide radical [40, 41]. Although banned in Europe in2007, this herbicide is still the most used worldwide [42, 43].In addition, rotenone, a pesticide and complex I inhibitor,induces the symptoms of PD similar to paraquat and MPP+.
Exposure to paraquat has also been established as an invivo model of PD. Both paraquat-injected rats and mice show
parkinsonian symptoms, oxidative stress, and dopaminergicneural loss in the substantia nigra region of the midbrain [13,44, 45]. These symptoms could also be attenuated by water-soluble coenzyme Q10, an ETC component and antioxidant[13].
It was recently shown that the mechanism in whichparaquat and rotenone induce dopaminergic cell deathmight be through the JNK pathway. This is believed to bedue to increased phosphorylation of JNK as demonstrated inprimary cultured dopaminergic neurons under paraquat androtenone insult [46, 47] and caspase-3-dependent apoptosis[48]. Another mechanistic pathway of dopaminergic celldeath by both paraquat and MPP+ is believed to be throughthe activation of NADPH oxidase-1 (a superoxide-generatingenzyme complex), [49, 50].
It was also observed that rotenone induced dopamin-ergic neurodegeneration in an animal model by meansof microglial activation, causing NADPH oxidase-derivedsuperoxides to be formed [51]. However, recent studies showthat human cell line microglia, although activated, onlyproduce extracellular ROS and, therefore, do not contributeto neurodegeneration when exposed to chronic, low doses ofrotenone [52]. Paraquat has been shown to induce oxidativestress in the cytosol, whereas MPP+ and rotenone induceoxidative stress in the mitochondria [48]. Although all thesechemicals induce symptoms of PD, their mechanism ofneuronal cell death varies, therefore, providing multipleapproaches to not only study the mechanisms associated withPD but also to develop innovative therapeutic interventionsfor combating this disease.
1.3. Role of Stress-Responsive Transcription Factor Nrf2 (NF-E2-Related Factor 2) in Protection against Oxidative Toxicity.Role of Nrf2, an important stress-responsive transcriptionfactor of the “cap-and-collar” β-leucine zipper family, isnow considered instrumental to several neurodegenerativedisorders [53–55]. Expression of a number of Phase IIenzymes (e.g., NQO1, GSTs) and antioxidant proteins (e.g.,GCL, HO-1, thioredoxin) are regulated by this gene. It isbelieved that this process is driven by the association of
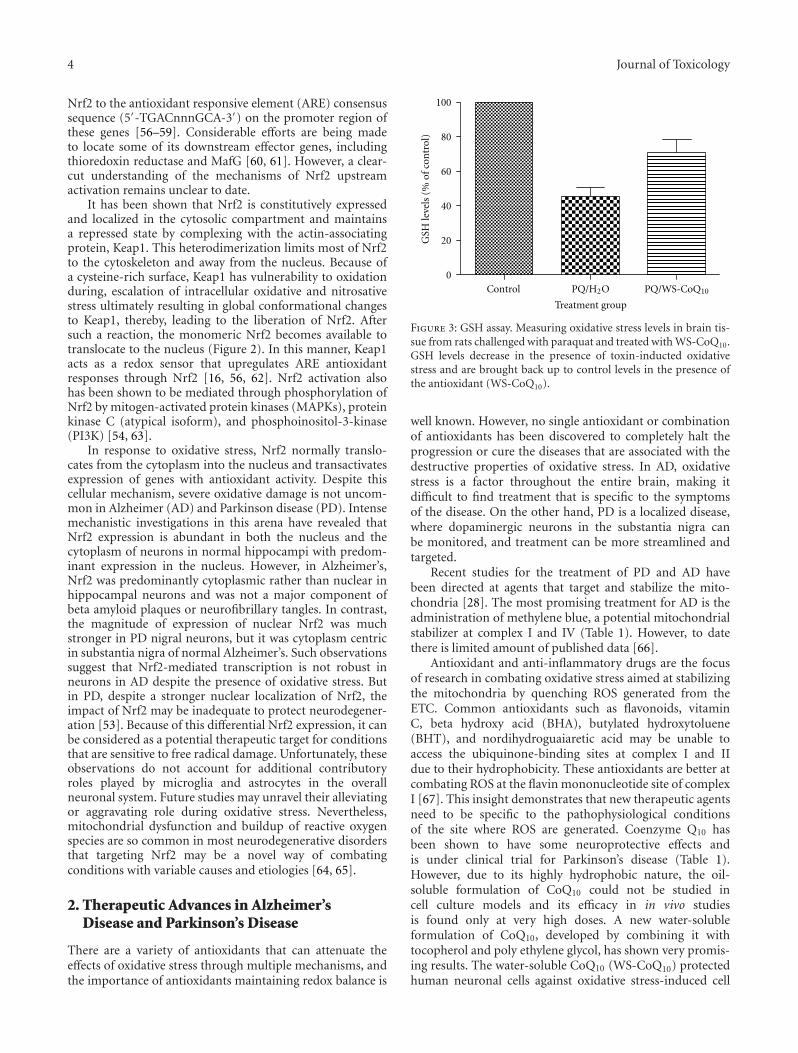
4 Journal of Toxicology
Nrf2 to the antioxidant responsive element (ARE) consensussequence (5′-TGACnnnGCA-3′) on the promoter region ofthese genes [56–59]. Considerable efforts are being madeto locate some of its downstream effector genes, includingthioredoxin reductase and MafG [60, 61]. However, a clear-cut understanding of the mechanisms of Nrf2 upstreamactivation remains unclear to date.
It has been shown that Nrf2 is constitutively expressedand localized in the cytosolic compartment and maintainsa repressed state by complexing with the actin-associatingprotein, Keap1. This heterodimerization limits most of Nrf2to the cytoskeleton and away from the nucleus. Because ofa cysteine-rich surface, Keap1 has vulnerability to oxidationduring, escalation of intracellular oxidative and nitrosativestress ultimately resulting in global conformational changesto Keap1, thereby, leading to the liberation of Nrf2. Aftersuch a reaction, the monomeric Nrf2 becomes available totranslocate to the nucleus (Figure 2). In this manner, Keap1acts as a redox sensor that upregulates ARE antioxidantresponses through Nrf2 [16, 56, 62]. Nrf2 activation alsohas been shown to be mediated through phosphorylation ofNrf2 by mitogen-activated protein kinases (MAPKs), proteinkinase C (atypical isoform), and phosphoinositol-3-kinase(PI3K) [54, 63].
In response to oxidative stress, Nrf2 normally translo-cates from the cytoplasm into the nucleus and transactivatesexpression of genes with antioxidant activity. Despite thiscellular mechanism, severe oxidative damage is not uncom-mon in Alzheimer (AD) and Parkinson disease (PD). Intensemechanistic investigations in this arena have revealed thatNrf2 expression is abundant in both the nucleus and thecytoplasm of neurons in normal hippocampi with predom-inant expression in the nucleus. However, in Alzheimer’s,Nrf2 was predominantly cytoplasmic rather than nuclear inhippocampal neurons and was not a major component ofbeta amyloid plaques or neurofibrillary tangles. In contrast,the magnitude of expression of nuclear Nrf2 was muchstronger in PD nigral neurons, but it was cytoplasm centricin substantia nigra of normal Alzheimer’s. Such observationssuggest that Nrf2-mediated transcription is not robust inneurons in AD despite the presence of oxidative stress. Butin PD, despite a stronger nuclear localization of Nrf2, theimpact of Nrf2 may be inadequate to protect neurodegener-ation [53]. Because of this differential Nrf2 expression, it canbe considered as a potential therapeutic target for conditionsthat are sensitive to free radical damage. Unfortunately, theseobservations do not account for additional contributoryroles played by microglia and astrocytes in the overallneuronal system. Future studies may unravel their alleviatingor aggravating role during oxidative stress. Nevertheless,mitochondrial dysfunction and buildup of reactive oxygenspecies are so common in most neurodegenerative disordersthat targeting Nrf2 may be a novel way of combatingconditions with variable causes and etiologies [64, 65].
2. Therapeutic Advances in Alzheimer’sDisease and Parkinson’s Disease
There are a variety of antioxidants that can attenuate theeffects of oxidative stress through multiple mechanisms, andthe importance of antioxidants maintaining redox balance is
PQ/WS-CoQ10Control0
20
40
60
80
100
Treatment group
GSH
leve
ls(%
ofco
ntr
ol)
PQ/H2O
Figure 3: GSH assay. Measuring oxidative stress levels in brain tis-sue from rats challenged with paraquat and treated with WS-CoQ10.GSH levels decrease in the presence of toxin-inducted oxidativestress and are brought back up to control levels in the presence ofthe antioxidant (WS-CoQ10).
well known. However, no single antioxidant or combinationof antioxidants has been discovered to completely halt theprogression or cure the diseases that are associated with thedestructive properties of oxidative stress. In AD, oxidativestress is a factor throughout the entire brain, making itdifficult to find treatment that is specific to the symptomsof the disease. On the other hand, PD is a localized disease,where dopaminergic neurons in the substantia nigra canbe monitored, and treatment can be more streamlined andtargeted.
Recent studies for the treatment of PD and AD havebeen directed at agents that target and stabilize the mito-chondria [28]. The most promising treatment for AD is theadministration of methylene blue, a potential mitochondrialstabilizer at complex I and IV (Table 1). However, to datethere is limited amount of published data [66].
Antioxidant and anti-inflammatory drugs are the focusof research in combating oxidative stress aimed at stabilizingthe mitochondria by quenching ROS generated from theETC. Common antioxidants such as flavonoids, vitaminC, beta hydroxy acid (BHA), butylated hydroxytoluene(BHT), and nordihydroguaiaretic acid may be unable toaccess the ubiquinone-binding sites at complex I and IIdue to their hydrophobicity. These antioxidants are better atcombating ROS at the flavin mononucleotide site of complexI [67]. This insight demonstrates that new therapeutic agentsneed to be specific to the pathophysiological conditionsof the site where ROS are generated. Coenzyme Q10 hasbeen shown to have some neuroprotective effects andis under clinical trial for Parkinson’s disease (Table 1).However, due to its highly hydrophobic nature, the oil-soluble formulation of CoQ10 could not be studied incell culture models and its efficacy in in vivo studiesis found only at very high doses. A new water-solubleformulation of CoQ10, developed by combining it withtocopherol and poly ethylene glycol, has shown very promis-ing results. The water-soluble CoQ10 (WS-CoQ10) protectedhuman neuronal cells against oxidative stress-induced cell
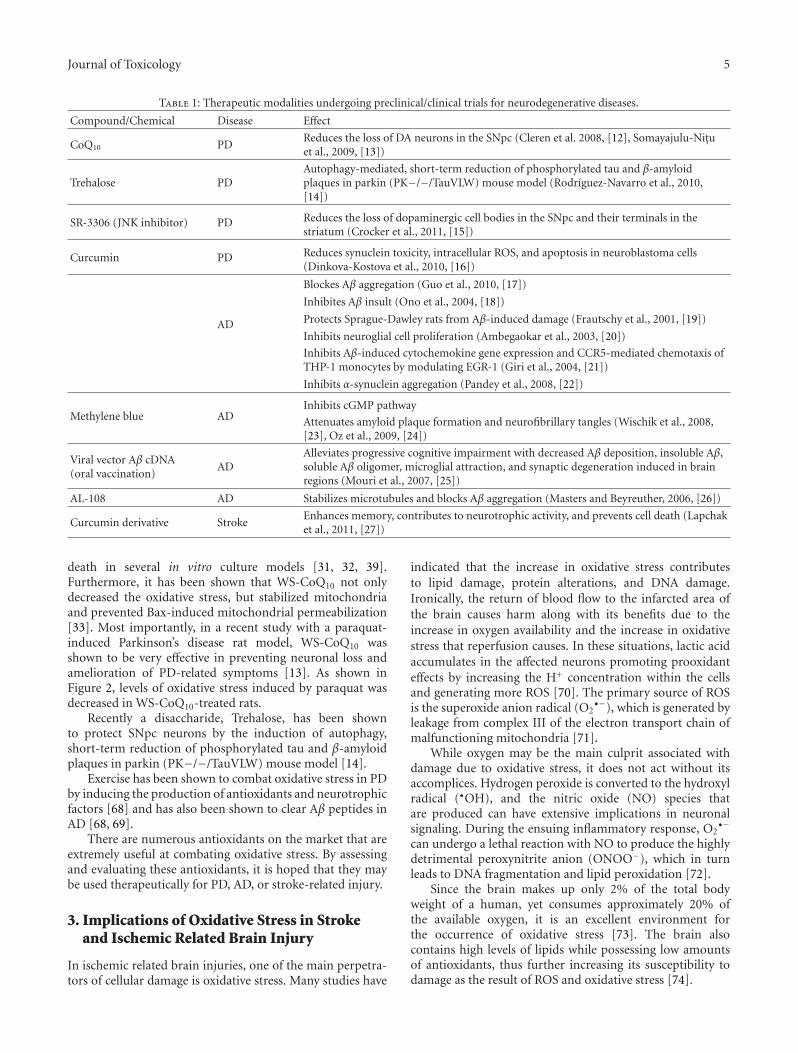
Journal of Toxicology 5
Table 1: Therapeutic modalities undergoing preclinical/clinical trials for neurodegenerative diseases.
Compound/Chemical Disease Effect
CoQ10 PDReduces the loss of DA neurons in the SNpc (Cleren et al. 2008, [12], Somayajulu-Nituet al., 2009, [13])
Trehalose PDAutophagy-mediated, short-term reduction of phosphorylated tau and β-amyloidplaques in parkin (PK−/−/TauVLW) mouse model (Rodrıguez-Navarro et al., 2010,[14])
SR-3306 (JNK inhibitor) PD Reduces the loss of dopaminergic cell bodies in the SNpc and their terminals in thestriatum (Crocker et al., 2011, [15])
Curcumin PD Reduces synuclein toxicity, intracellular ROS, and apoptosis in neuroblastoma cells(Dinkova-Kostova et al., 2010, [16])
AD
Blockes Aβ aggregation (Guo et al., 2010, [17])
Inhibites Aβ insult (Ono et al., 2004, [18])
Protects Sprague-Dawley rats from Aβ-induced damage (Frautschy et al., 2001, [19])
Inhibits neuroglial cell proliferation (Ambegaokar et al., 2003, [20])
Inhibits Aβ-induced cytochemokine gene expression and CCR5-mediated chemotaxis ofTHP-1 monocytes by modulating EGR-1 (Giri et al., 2004, [21])
Inhibits α-synuclein aggregation (Pandey et al., 2008, [22])
Methylene blue ADInhibits cGMP pathway
Attenuates amyloid plaque formation and neurofibrillary tangles (Wischik et al., 2008,[23], Oz et al., 2009, [24])
Viral vector Aβ cDNA(oral vaccination)
ADAlleviates progressive cognitive impairment with decreased Aβ deposition, insoluble Aβ,soluble Aβ oligomer, microglial attraction, and synaptic degeneration induced in brainregions (Mouri et al., 2007, [25])
AL-108 AD Stabilizes microtubules and blocks Aβ aggregation (Masters and Beyreuther, 2006, [26])
Curcumin derivative StrokeEnhances memory, contributes to neurotrophic activity, and prevents cell death (Lapchaket al., 2011, [27])
death in several in vitro culture models [31, 32, 39].Furthermore, it has been shown that WS-CoQ10 not onlydecreased the oxidative stress, but stabilized mitochondriaand prevented Bax-induced mitochondrial permeabilization[33]. Most importantly, in a recent study with a paraquat-induced Parkinson’s disease rat model, WS-CoQ10 wasshown to be very effective in preventing neuronal loss andamelioration of PD-related symptoms [13]. As shown inFigure 2, levels of oxidative stress induced by paraquat wasdecreased in WS-CoQ10-treated rats.
Recently a disaccharide, Trehalose, has been shownto protect SNpc neurons by the induction of autophagy,short-term reduction of phosphorylated tau and β-amyloidplaques in parkin (PK−/−/TauVLW) mouse model [14].
Exercise has been shown to combat oxidative stress in PDby inducing the production of antioxidants and neurotrophicfactors [68] and has also been shown to clear Aβ peptides inAD [68, 69].
There are numerous antioxidants on the market that areextremely useful at combating oxidative stress. By assessingand evaluating these antioxidants, it is hoped that they maybe used therapeutically for PD, AD, or stroke-related injury.
3. Implications of Oxidative Stress in Strokeand Ischemic Related Brain Injury
In ischemic related brain injuries, one of the main perpetra-tors of cellular damage is oxidative stress. Many studies have
indicated that the increase in oxidative stress contributesto lipid damage, protein alterations, and DNA damage.Ironically, the return of blood flow to the infarcted area ofthe brain causes harm along with its benefits due to theincrease in oxygen availability and the increase in oxidativestress that reperfusion causes. In these situations, lactic acidaccumulates in the affected neurons promoting prooxidanteffects by increasing the H+ concentration within the cellsand generating more ROS [70]. The primary source of ROSis the superoxide anion radical (O2
•−), which is generated byleakage from complex III of the electron transport chain ofmalfunctioning mitochondria [71].
While oxygen may be the main culprit associated withdamage due to oxidative stress, it does not act without itsaccomplices. Hydrogen peroxide is converted to the hydroxylradical (•OH), and the nitric oxide (NO) species thatare produced can have extensive implications in neuronalsignaling. During the ensuing inflammatory response, O2
•−
can undergo a lethal reaction with NO to produce the highlydetrimental peroxynitrite anion (ONOO−), which in turnleads to DNA fragmentation and lipid peroxidation [72].
Since the brain makes up only 2% of the total bodyweight of a human, yet consumes approximately 20% ofthe available oxygen, it is an excellent environment forthe occurrence of oxidative stress [73]. The brain alsocontains high levels of lipids while possessing low amountsof antioxidants, thus further increasing its susceptibility todamage as the result of ROS and oxidative stress [74].

6 Journal of Toxicology
Stroke is a leading cause of death and long-term disabilityin industrialized nations [75, 76] and is a condition thatregularly leaves its victims in a state of impaired cognitionand motor deficits, with nearly 40% of patients not expectedto make a full recovery [70]. The damage and detrimentaleffects of stroke are heavily influenced by oxidative stress andthe production of free radicals.
Two types of stroke can occur, hemorrhagic stroke, andthe more common, ischemic stroke. In hemorrhagic stroke,rupture of an artery results in uncontrolled bleeding to theaffected area of the brain. In ischemic stroke, there is ablockage of blood flow to the brain due to the formationof a blood clot. This deprivation of oxygenated bloodresults in the formation of the ischemic core where cells dierather quickly and irreversibly due to necrosis. The onsetof lipolysis, protein degradation, and the breakdown of ionhomeostasis are some of the events responsible for the rapiddeath of these cells [77].
In the area between the unaffected brain and the ischemiccore lies a region where the struggle between the life anddeath of neurons ensues. This region of the brain is knownas the penumbra. It is here that the brain is composed ofdamaged and malfunctioning, yet salvageable tissue. Cellsin this region are susceptible to a programmed form of celldeath known as apoptosis. These cells can remain viable andfor several days following the onset of stroke [78].
Here in the penumbra region is where a host of eventsrelated to oxidative stress take place. Ironically enough,reperfusion acts as a double-edged sword. While reperfusionis essential to save the cells affected by ischemia, it alsobrings along with it its own threat. When reperfusion occurs,there is a large and rapid influx of oxygenated blood to theinfarct region. While this delivers the necessary blood, it alsobrings with it the elements necessary for producing ROS thatcontribute to the oxidative stress placed upon the alreadydamaged brain tissue.
When platelets are exposed to conditions of reperfusion,they generate additional ROS in the form of O2
•− and •OH.Furthermore, the ROS that are produced during reperfusionare responsible for the activation and transcription of manyproteins. For example, ROS stimulate the production of JNKand mitogen-activated protein kinase phosphorylation (p38MAPK) in the affected neurons of the penumbra. JNK-1 isfavored in the nucleus of neurons during reperfusion, andactivator protein-1 (AP-1) binding is also enhanced [78].The activation of AP-1 is necessary for the induction ofapoptosis to occur [79]. This action, along with the activa-tion of caspase-3, are several examples of how reperfusion isresponsible for initiating cell death within the neurons of thepenumbra by controlling the expression of certain genes.
Along with their role in effecting the transcription ofvarious proteins, ROS generated by reperfusion can itselfcause direct cellular stress. Reperfusion causes such a largeinflux of oxygen that all of it cannot be used by themitochondria, and normal radical scavenging mechanismssuch as superoxide dismutase (SOD), glutathione peroxidase,and catalase are overwhelmed and cannot adequately quenchthe multitude of free radicals that are produced and leakfrom the system [80]. The cells of the penumbra are already
vulnerable to damage, and the generation of ROS exacerbatesthe damage that may have already occurred to these cellsby lipid peroxidation. In particular, phospholipid membranedegradation is a major concern. The brain is especiallysusceptible to such damage due to the large amount oflipids that compose its structure. Lipid peroxidation targetsthe polyunsaturated fatty acids (PUFAs) in the brain, thus,decreasing the membrane integrity. The decrease in mem-brane stability is especially important because the membranecontains receptor proteins and ion channel entities. Alongwith its own deleterious effects, lipid peroxidation is alsoresponsible for the inhibition of lipid repair enzymes such aslysophosphatidylcholine acyltransferase and fatty acyl CoA-synthase [81].
While cells in the infarct region die instantly via necrosis,cells of the penumbra are likely to die by means of apoptosis.Apoptosis is a programmed form of cell death where the cellexpends energy towards its own demise. It is controlled bya complex interconnection of proteins and is often triggeredby oxidative stress and the release of cytochrome c from themitochondria [82]. The increased level of ROS is involvedin generating the signal that causes permeabilization ofthe mitochondrial membrane, and, thus, the release ofcytochrome c into the cytosol. Once this occurs, the initiationof the cascade of caspases occurs. Activation of caspases 3, 8,and 9 will eventually lead to the death of the cell and othersurrounding cells [83].
3.1. Stroke-Induced Inflammation. Inflammation is a bio-logical response to harmful stimuli and often occurs as aresult of stroke. One of the key contributors to the inflam-matory response are glial cells, more specifically microglia,that secrete proinflammatory cytokines and chemokinesthat contribute to the damage done to the penumbra.The most common contributors to the damage due toinflammation are tumour necrosis factor alpha (TNF-alpha)and interlenukin-B (IL-Beta), among others [84]. Thesecytokines are responsible for the increased expression ofcellular adhesion molecules (CAMs) that in combinationwith platelets, adhere to vessel walls causing a “no-flow”constriction [85] and the release of more proinflammatorymolecules. Ultimately, the inflammatory response results indecreased blood brain barrier function, increased cerebraledema, and cell death [84].
3.2. The Role of Proapoptotic Proteins in Stroke. As previouslymentioned, apoptosis is controlled by a wide range ofproteins. Oxidative stress can cause the activation of p53-tumor suppressor gene which in turn is responsible for theincreased transcription of Bcl-2 associated X protein (Bax)and the p53 upregulated modulator of apoptosis (PUMA)[86]. PUMA has been shown to be able to interact withthe Bcl-2 family of proteins to assist in initiating apoptosis.It has been shown that PUMA is able to associate withthe mitochondrial membrane along with Bax to promotethe release of cytochrome c [87]. Studies involving the PD-associated DJ-1 gene indicate that this gene protects the cellsagainst excitotoxicity and the effects of ischemia. DJ-1 wasfound to decrease the presence of oxidative stress markers,

Journal of Toxicology 7
and, thus, protect the cell due to the alleviation of the effectsof oxidative stress [88].
A large majority of the proteins responsible for thebalance between death and survival belong to the Bcl-2family of proteins that protect cells from apoptosis inducedby a wide variety of stimuli. One of these proteins is theproapoptotic protein Bax which exists in the cytosol as aharmless 24 kDa monomer. However, in cases of increasedoxidative stress, the protein product of the p53 tumorsuppressor gene causes increased transcription of Bax tooccur [89]. In response to this increased amount of Baxdue to oxidative stress, Bax undergoes dimerization eitherwith itself or other members of the Bcl-2 family (e.g.,tBID) through interactions of alpha helix 2 with the BH3domain [90]. This dimerized form of Bax then begins itsmigration towards the mitochondria. Once in range of themitochondria, the dimerized form of Bax may associate withthe protein transition pore (PTP) of the voltage-dependentanion channel (VDAC) of the mitochondrial membrane.This action allows for the uncontrolled flow of cytotoxicfactors, such as cytochrome c, to be released from themitochondria into the cytosol, and inevitably, the demise ofthe mitochondria [91].
Studies have demonstrated that Bax channel activity isnecessary for apoptosis to occur since cell death was haltedwith the use of Bax channel blockers [92]. Since Bax isan essential protein in the regulation of apoptosis, it isan excellent target candidate for therapeutic approaches.Not only does its extensive involvement in the process ofcell death make Bax a good therapeutic target, its positionin the apoptosis cascade does as well. While focusingon antioxidants may be a valid point of investigation,bolstering of the antioxidant defense machinery still resultsin permeabilized mitochondria. Blocking the initiation ofapoptosis further up the chain by inhibiting Bax functionmay save the mitochondria and halt apoptosis.
3.3. Experimental Models of Stroke. In the cellular modelof stroke, an excellent way to mimic ischemic assault isby inducing hypoxia. Hypoxia is the deprivation of anadequate amount of oxygen to tissues, usually accompaniedby detrimental effects. To accomplish this in a cellular model,cells can be placed in an oxygen-free chamber for a periodof time before being removed [93]. It was found that whenconditions of hypoxia exist, hypoxia-inducible factor alpha(HIF-1α) expression increases [94] and, therefore, is impli-cated in hypoxia-induced apoptosis. HIF-1α participates bystabilizing the structure of the tumor suppressor gene p53,which leads to the expression of apoptosis-related genes[94, 95].
HIF-1α has also been shown to have antiapoptotic effectsbecause those cells with increased levels of HIF-1α showresistance to hypoxia-induced apoptosis [96]. The decidingfactor of whether HIF-1α is protective or harmful to a cellseems to depend on the level of hypoxia. If conditions ofmild hypoxia exist, HIF-1α is phosphorylated and associatedwith HIF-1β, and the transcription of p53 is low withanti-apoptotic genes being activated [97, 98]. However, inhigh hypoxic conditions, the reverse is the case. HIF-1α is
dephosphorylated and p53 levels are unregulated, eventuallyleading to the activation of pro-apoptotic proteins such asBax [97, 99, 100]. This is similar to conditions of ischemicstroke where up to 24 hours after ischemia, most pro-apoptotic genes are upregulated, whereas 48 hours to 8 daysafter ischemia anti-apoptotic genes are the majority of thoseinduced [94].
There are a variety of different in vivo models of strokeranging from middle cerebral artery occlusion (MCAO)[101] and four vessel carotid artery occlusion [102]. Onemodel that our laboratory has employed to investigate theeffects of stroke in a rat model is the bilateral carotid arteryocclusion and two vessel occlusion hypovolemic hypotension(2VO/HT) model [103]. In this model, global forebrainischemia is induced by occluding the 2 carotid arteries andremoving a certain volume of blood from the animal tomaintain a mean arterial pressure of 50 mm Hg. This resultsin an interruption of blood flow to the brain, successfully cre-ating an infarct region similar to stroke and the surroundingpenumbra. This model can be used to accurately investigatethe effects of various therapeutic reagents and their abilitiesto protect neurons under conditions similar to stroke.
3.4. Therapeutic Approaches for Stroke. At this time, the onlyknown treatment for victims of stroke is the use of throm-bolytics, most commonly, tissue plasminogen activator. Thedownfall to this avenue of treatment is that it must beadministered within 3 hours of the onset of stroke. This isrelatively hard to accomplish, as most stroke victims do notarrive at hospital to receive treatment within this timeframe.Also, thrombolytics can lead to an increased likelihood ofhemorrhages occurring within the brain [85]. Hypothermiahas also been investigated as a possible treatment for stroke.It has been found that lowering the body temperature of astroke victim may improve the neurological outcome [104].However this technique remains highly experimental as thetemperature, duration, and onset of cooling still remains tobe accurately determined.
An emerging field of study for treatment of ischemiaincludes the use of bone marrow stromal cells (BMSC).These cells can differentiate into neural and glial cells, both invivo and in vitro, after being transplanted into animal modelbrains following neurological insult such as intracerebralhemorrhage (ICH) [105]. These neural stem cells migrateto the area of the brain that is injured in order to replacethe neuron deficit that was lost due to hemorrhagic stroke.Recent studies have found that these BMSCs are able togenerate functional recovery in Wistar rats following ICH[106].
Another interesting avenue of exploration into potentialtherapeutics for stroke is the use of curcumin derivatives.Curcumin has been shown to prevent Alzheimer’s markersin animal models of the disease [107] and has also beenshown to be effective in reducing the deficits of middlecerebral artery occlusion in a rat [108, 109]. Recent studieshave shown that when used as a treatment in a modelof stroke, a pyrazole derivative of curcumin was able toenhance memory, contribute to neurotrophic activity, and,most importantly, prevent cell death [110].

8 Journal of Toxicology
Research has focused on blocking pro-apoptotic proteinsthat are responsible for causing cell death. Recently, advancesin treatments for stoke have been made by the use of lowmolecular weight compounds that inhibit proteins (suchas Bax) that are critical in the apoptosis cascade. Thisis a critical stage for the inhibition of apoptosis due tothe fact that Bax channel formation is required for thedestabilization of the mitochondria, and subsequent releaseof cytotoxic factors [92]. These inhibitory compounds weremodeled after single-domain antibodies that were able tobind specifically to Bax [93]. They are small enough tohave the potential to cross the blood brain barrier and arenot susceptible to proteolysis. Recent research completedin our laboratory indicates that these compounds show ahigh specificity towards the pro-apoptotic protein Bax andare able to block its function and save the neurons of thepenumbra from apoptosis [111]. These compounds are ableto competitively bind to Bax even when in the presenceof single-domain antibodies that are specific to Bax. Bybinding in some manner to Bax, these compounds preventthe association of Bax with the mitochondria and preventmitochondrial destabilization, thus, limiting the influx ofcytotoxic factors into the cytosol. It is hoped that thesecompounds will not need-to be administered in such a short-time frame following stroke as is the case for thrombolytics,nor pose the risk of hemorrhage that thrombolytics do. Withmore investigation, it is likely that the use of low molecularweight compounds will become valid treatment options forstroke patients.
4. Conclusion
It is now well established that oxidative stress and mitochon-drial dysfunction are the early and key biochemical mecha-nisms leading to Alzheimer’s disease, Parkinson’s disease, andstroke-related pathologies. Mitochondria are greatly involvedin neuronal cell death due to the vicious cycle of oxidativetoxicity, which causes mitochondrial dysfunction that leadsto more ROS and the potential collapse of the mitochondrialmembrane. Environmental toxins, amyloid-beta toxicity,and ischemia/hyper-perfusion-related toxicity all lead tooxidative toxicity directly or indirectly by mitochondrialdestabilization. Significant progress has been made to inhibitneuronal cell death by using anti-oxidants or blockers of pro-apoptotic proteins, but a combinatorial treatment to reduceoxidative stress and stabilize mitochondria to halt neuronalloss needs to be explored.
Acknowledgments
The authors gratefully acknowledge the funding fromNSERC and CIHR for this research project. K. Facecchia andL.-A. Fochesato contributed equally to this paper.
References
[1] M. B. Moura, L. S. Dos Santos, and B. Van Houten,“Mitochondrial dysfunction in neurodegenerative diseases
and cancer,” Environmental and Molecular Mutagenesis, vol.51, no. 5, pp. 391–405, 2010.
[2] P. G. Bittar, Y. Charnay, L. Pellerin, C. Bouras, and P. J.Magistretti, “Selective distribution of lactate dehydrogenaseisoenzymes in neurons and astrocytes of human brain,”Journal of Cerebral Blood Flow and Metabolism, vol. 16, no.6, pp. 1079–1089, 1996.
[3] G. Tholey, B. F. Roth-Schechter, and P. Mandel, “Activity andisoenzyme pattern of lactate dehydrogenase in neurons andastroblasts cultured from brains of chick embryos,” Journalof Neurochemistry, vol. 36, no. 1, pp. 77–81, 1981.
[4] J. F. Harrison, S. B. Hollensworth, D. R. Spitz, W. C.Copeland, G. L. Wilson, and S. P. LeDoux, “Oxidative stress-induced apoptosis in neurons correlates with mitochondrialDNA base excision repair pathway imbalance,” Nucleic AcidsResearch, vol. 33, no. 14, pp. 4660–4671, 2005.
[5] H. V. Nobre Jr., M. M.D.F. Fonteles, and R. M.D. De Freitas,“Acute seizure activity promotes lipid peroxidation, increasednitrite levels and adaptive pathways against oxidative stressin the frontal cortex and striatum,” Oxidative Medicine andCellular Longevity, vol. 2, no. 3, pp. 130–137, 2009.
[6] K. Jomova, D. Vondrakova, M. Lawson, and M. Valko,“Metals, oxidative stress and neurodegenerative disorders,”Molecular and Cellular Biochemistry, vol. 345, no. 1-2, pp. 91–104, 2010.
[7] D. Cantu, J. Schaack, and M. Patel, “Oxidative inactivation ofmitochondrial aconitase results in iron and H2O2-mediatedneurotoxicity in rat primary mesencephalic cultures,” PLoSONE, vol. 4, no. 9, article e7095, 2009.
[8] W. -C. Wu, D. -N. Hu, H. -X. Gao et al., “Subtoxic levelshydrogen peroxide-induced production of interleukin-6 byretinal pigment epithelial cells,” Molecular Vision, vol. 16, pp.1864–1873, 2010.
[9] W. Chadwick, Y. Zhou, S. -S. Park et al., “Minimal peroxideexposure of neuronal cells induces multifaceted adaptiveresponses,” PLoS ONE, vol. 5, no. 12, article e14352, 2010.
[10] P. Sen, P. K. Chakraborty, and S. Raha, “p38 Mitogen-activated protein kinase (p38MAPK) upregulates catalaselevels in response to low dose H2O2 treatment throughenhancement of mRNA stability,” FEBS Letters, vol. 579, no.20, pp. 4402–4406, 2005.
[11] M. C. Gomez-Cabrera, E. Domenech, and J. Vina, “Moderateexercise is an antioxidant: upregulation of antioxidant genesby training,” Free Radical Biology and Medicine, vol. 44, no. 2,pp. 126–131, 2008.
[12] C. Cleren, L. Yang, B. Lorenzo et al., “Therapeutic effectsof coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTPmodel of Parkinsonism,” Journal of Neurochemistry, vol. 104,no. 6, pp. 1613–1621, 2008.
[13] M. Somayajulu-Nitu, J. K. Sandhu, J. Cohen et al., “Paraquatinduces oxidative stress, neuronal loss in substantia nigraregion and Parkinsonism in adult rats: neuroprotection andamelioration of symptoms by water-soluble formulation ofcoenzyme Q10,” BMC Neuroscience, vol. 10, article88, 2009.
[14] J. A. Rodrıguez-Navarro, L. Rodrıguez, M. J. Casarejos et al.,“Trehalose ameliorates dopaminergic and tau pathology inparkin deleted/tau overexpressing mice through autophagyactivation,” Neurobiology of Disease, vol. 39, no. 3, pp. 423–438, 2010.
[15] C. E. Crocker, S. Khan, M. D. Cameron, H. A. Robertson,G. S. Robertson, and P. Lograsso, “JNK inhibition protectsdopamine neurons and provides behavioral improvement ina rat 6-hydroxydopamine model of Parkinson’s disease,” ACSChemical Neuroscience, vol. 2, no. 4, pp. 207–212, 2011.

Journal of Toxicology 9
[16] A. T. Dinkova-Kostova, P. Talalay, J. Sharkey et al., “Anexceptionally potent inducer of cytoprotective enzymes:elucidation of the structural features that determine inducerpotency and reactivity with Keap1,” Journal of BiologicalChemistry, vol. 285, no. 44, pp. 33747–33755, 2010.
[17] J. -P. Guo, S. Yu, and P. L. McGeer, “Simple in vitro assaysto identify amyloid-β aggregation blockers for Alzheimer’sdisease therapy,” Journal of Alzheimer’s Disease, vol. 19, no.4, pp. 1359–1370, 2010.
[18] K. Ono, K. Hasegawa, H. Naiki, and M. Yamada, “Curcuminhas potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro,” Journal of Neuroscience Research, vol.75, no. 6, pp. 742–750, 2004.
[19] S. A. Frautschy, W. Hu, P. Kim et al., “Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitivedeficits and neuropathology,” Neurobiology of Aging, vol. 22,no. 6, pp. 993–1005, 2001.
[20] S. S. Ambegaokar, L. Wu, K. Alamshahi et al., “Curcumininhibits dose-dependently and time-dependently neuroglialcell proliferation and growth,” Neuroendocrinology Letters,vol. 24, no. 6, pp. 469–473, 2003.
[21] R. K. Giri, V. Rajagopal, and V. K. Kalra, “Curcumin, theactive constituent of turmeric, inhibits amyloid peptide-induced cytochemokine gene expression and CCR5-mediated chemotaxis of THP-1 monocytes by modulatingearly growth response-1 transcription factor,” Journal ofNeurochemistry, vol. 91, no. 5, pp. 1199–1210, 2004.
[22] N. Pandey, J. Strider, W. C. Nolan, S. X. Yan, and J. E.Galvin, “Curcumin inhibits aggregation of alpha-synuclein,”Acta Neuropathologica, vol. 115, no. 4, pp. 479–489, 2008.
[23] C. M. Wischik, P. Bentham, D. J. Wischik, and K. M. Seng,“Tau aggregation inhibitor (TAI) therapy with rememberTMarrests disease progression in mild and moderate Alzheimer’sdisease over 50 weeks,” Alzheimer’s Dementia, vol. 4, p. T167,2008.
[24] M. Oz, D. E. Lorke, and G. A. Petroianu, “Methylene blue andAlzheimer’s disease,” Biochemical Pharmacology, vol. 78, no.8, pp. 927–932, 2009.
[25] A. Mouri, Y. Noda, H. Hara, H. Mizoguchi, T. Tabira,and T. Nabeshima, “Oral vaccination with a viral vectorcontaining Aβ cDNA attenuates age-related Aβ accumulationand memory deficits without causing inflammation in amouse Alzheimer model,” FASEB Journal, vol. 21, no. 9, pp.2135–2148, 2007.
[26] C. L. Masters and K. Beyreuther, “Alzheimer’s centenniallegacy: prospects for rational therapeutic intervention tar-geting the Aβ amyloid pathway,” Brain, vol. 129, no. 11, pp.2823–2839, 2006.
[27] P. A. Lapchak, D. R. Schubert, and P. A. Maher, “Delayedtreatment with a novel neurotrophic compound reducesbehavioral deficits in rabbit ischemic stroke,” Journal ofNeurochemistry, vol. 116, no. 1, pp. 122–131, 2011.
[28] W. E. Muller, A. Eckert, C. Kurz, G. P. Eckert, and K.Leuner, “Mitochondrial dysfunction: common final pathwayin brain aging and Alzheimer’s disease-therapeutic aspects,”Molecular Neurobiology, vol. 41, no. 2-3, pp. 159–171, 2010.
[29] A. Atlante, A. Bobba, V. A. Petragallo, and E. Marra,“Alzheimer’s proteins, oxidative stress, and mitochondrialdysfunction interplay in a neuronal model of Alzheimer’sdisease,” International Journal of Alzheimer’s Disease, vol.2010, Article ID 621870, 11 pages, 2010.
[30] T. P. Brown, P. C. Rumsby, A. C. Capleton, L. Rushton, and L.S. Levy, “Pesticides and Parkinson’s disease—is there a link?”
Environmental Health Perspectives, vol. 114, no. 2, pp. 156–164, 2006.
[31] J. K. Sandhu, S. Pandey, M. Ribecco-Lutkiewicz et al.,“Molecular mechanisms of glutamate neurotoxicity in mixedcultures of NT2-derived neurons and astrocytes: protectiveeffects of coenzyme Q10,” Journal of Neuroscience Research,vol. 72, no. 6, pp. 691–703, 2003.
[32] M. Somayajulu, S. McCarthy, M. Hung, M. Sikorska, H.Borowy-Borowski, and S. Pandey, “Role of mitochondria inneuronal cell death induced by oxidative stress; neuroprotec-tion by Coenzyme Q10,” Neurobiology of Disease, vol. 18, no.3, pp. 618–627, 2005.
[33] J. Naderi, M. Somayajulu-Nitu, A. Mukerji et al., “Water-soluble formulation of Coenzyme QM10 inhibits Bax-induced destabilization of mitochondria in mammaliancells,” Apoptosis, vol. 11, no. 8, pp. 1359–1369, 2006.
[34] R. J. Smeyne and V. Jackson-Lewis, “The MPTP model ofParkinson’s disease,” Molecular Brain Research, vol. 134, no.1, pp. 57–66, 2005.
[35] R. H. Kim, P. D. Smith, H. Aleyasin et al., “Hypersensitivity ofDJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6- tetrahy-dropyrindine (MPTP) and oxidative stress,” Proceedings of theNational Academy of Sciences of the United States of America,vol. 102, no. 14, pp. 5215–5220, 2005.
[36] H. H. Liou, M. C. Tsai, C. J. Chen et al., “Environmentalrisk factors and Parkinson’s disease: a case-control study inTaiwan,” Neurology, vol. 48, no. 6, pp. 1583–1588, 1997.
[37] B. Ritz and F. Yu, “Parkinson’s disease mortality and pesticideexposure in California 1984–1994,” International Journal ofEpidemiology, vol. 29, no. 2, pp. 323–329, 2000.
[38] A. S. Dhillon, G. L. Tarbutton, J. L. Levin et al., “Pesti-cide/environmental exposures and Parkinson’s disease in EastTexas,” Journal of Agromedicine, vol. 13, no. 1, pp. 37–48,2008.
[39] S. McCarthy, M. Somayajulu, M. Sikorska, H. Borowy-Borowski, and S. Pandey, “Paraquat induces oxidative stressand neuronal cell death; neuroprotection by water-solubleCoenzyme Q10,” Toxicology and Applied Pharmacology, vol.201, no. 1, pp. 21–31, 2004.
[40] M. Somayajulu-Nitu, D. Domazet-Damjanov, A. Matei,E. Schwartzenberger, J. Cohen, and S. Pandey, “Role ofenvironmental and inflammatory toxicity in neuronal celldeath,” The Open Toxicology Journal, vol. 2, pp. 26–42, 2008.
[41] F. F. Neves, R. B. Sousa, A. Pazin-Filho, P. Cupo, J. Elias Jr.,and M. H. Nogueira-Barbosa, “Severe paraquat poisoning:clinical and radiological findings in a survivor,” JornalBrasileiro de Pneumologia, vol. 36, no. 4, pp. 513–516, 2010.
[42] T. Reuters, “EU court reimposes ban on paraquat weedkiller,”2007, http://uk.reuters.com/article/idUKL11666800200707-11.
[43] Syngenta Crop Protection AG, “Paraquat InformationCenter,” 2011, http://www.paraquat.com/english/paraquat-information-center.
[44] A. L. McCormack, M. Thiruchelvam, A. B. Manning-Boget al., “Environmental risk factors and Parkinson’s dis-ease: selective degeneration of nigral dopaminergic neuronscaused by the herbicide paraquat,” Neurobiology of Disease,vol. 10, no. 2, pp. 119–127, 2002.
[45] A. L. McCormack, J. G. Atienza, L. C. Johnston, J. K.Andersen, S. Vu, and D. A. Di Monte, “Role of oxidativestress in paraquat-induced dopaminergic cell degeneration,”Journal of Neurochemistry, vol. 93, no. 4, pp. 1030–1037,2005.

10 Journal of Toxicology
[46] W. S. Choi, G. Abel, H. Klintworth, R. A. Flavell, andZ. Xia, “JNK3 mediates paraquat-and rotenone-induceddopaminergic neuron death,” Journal of Neuropathology andExperimental Neurology, vol. 69, no. 5, pp. 511–520, 2010.
[47] J. Peng, X. O. Mao, F. F. Stevenson, M. Hsu, and J. K.Andersen, “The herbicide paraquat induces dopaminergicnigral apoptosis through sustained activation of the JNKpathway,” Journal of Biological Chemistry, vol. 279, no. 31, pp.32626–32632, 2004.
[48] S. Ramachandiran, J. M. Hansen, D. P. Jones, J. R. Richard-son, and G. W. Miller, “Divergent mechanisms of paraquat,MPP+, and rotenone toxicity: oxidation of thioredoxin andcaspase-3 activation,” Toxicological Sciences, vol. 95, no. 1, pp.163–171, 2007.
[49] A. C. Cristovao, D. H. Choi, G. Baltazar, M. F. Beal,and Y. S. Kim, “The role of NADPH oxidase 1-derivedreactive oxygen species in paraquat-mediated dopaminergiccell death,” Antioxidants and Redox Signaling, vol. 11, no. 9,pp. 2105–2118, 2009.
[50] V. Anantharam, S. Kaul, C. Song, A. Kanthasamy, andA. G. Kanthasamy, “Pharmacological inhibition ofneuronal NADPH oxidase protects against 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress andapoptosis in mesencephalic dopaminergic neuronal cells,”NeuroToxicology, vol. 28, no. 5, pp. 988–997, 2007.
[51] H. M. Gao, B. Liu, and J. S. Hong, “Critical role formicroglial NADPH oxidase in rotenone-induced degenera-tion of dopaminergic neurons,” Journal of Neuroscience, vol.23, no. 15, pp. 6181–6187, 2003.
[52] S. B. Shaikh and L. F. B. Nicholson, “Effects of chroniclow dose rotenone treatment on human microglial cells,”Molecular Neurodegeneration, vol. 4, no. 1, article 55, 2009.
[53] C. P. Ramsey, C. A. Glass, M. B. Montgomery et al.,“Expression of Nrf2 in neurodegenerative diseases,” Journalof Neuropathology and Experimental Neurology, vol. 66, no. 1,pp. 75–85, 2007.
[54] M. J. Calkins, D. A. Johnson, J. A. Townsend et al., “TheNrf2/ARE pathway as a potential therapeutic target in neu-rodegenerative disease,” Antioxidants and Redox Signaling,vol. 11, no. 3, pp. 497–508, 2009.
[55] M. R. Vargas, D. A. Johnson, D. W. Sirkis, A. Messing, andJ. A. Johnson, “Nrf2 activation in astrocytes protects againstneurodegeneration in mouse models of familial amyotrophiclateral sclerosis,” Journal of Neuroscience, vol. 28, no. 50, pp.13574–13581, 2008.
[56] K. Itoh, K. I. Tong, and M. Yamamoto, “Molecular mecha-nism activating Nrf2-Keap1 pathway in regulation of adap-tive response to electrophiles,” Free Radical Biology andMedicine, vol. 36, no. 10, pp. 1208–1213, 2004.
[57] J. M. Lee and J. A. Johnson, “An important role of Nrf2-ARE pathway in the cellular defense mechanism,” Journal ofBiochemistry and Molecular Biology, vol. 37, no. 2, pp. 139–143, 2004.
[58] S. Numazawa and T. Yoshida, “Nrf2-dependent gene expres-sions: a molecular toxicological aspect,” Journal of Toxicolog-ical Sciences, vol. 29, no. 2, pp. 81–89, 2004.
[59] H. K. Ho, C. C. White, C. Fernandez et al., “Nrf2 acti-vation involves an oxidative-stress independent pathway intetrafluoroethylcysteine-induced cytotoxicity,” ToxicologicalSciences, vol. 86, no. 2, pp. 354–364, 2005.
[60] F. Katsuoka, H. Motohashi, J. D. Engel, and M. Yamamoto,“Nrf2 transcriptionally activates the mafG gene through anantioxidant response element,” Journal of Biological Chem-istry, vol. 280, no. 6, pp. 4483–4490, 2005.
[61] A. Sakurai, M. Nishimoto, S. Himeno et al., “Transcriptionalregulation of thioredoxin reductase 1 expression by cadmiumin vascular endothelial cells: role of NF-E2-related factor-2,”Journal of Cellular Physiology, vol. 203, no. 3, pp. 529–537,2005.
[62] M. I. Kang, A. Kobayashi, N. Wakabayashi, S. G. Kim,and M. Yamamoto, “Scaffolding of Keap1 to the actincytoskeleton controls the function of Nrf2 as key regulatorof cytoprotective phase 2 genes,” Proceedings of the NationalAcademy of Sciences of the United States of America, vol. 101,no. 7, pp. 2046–2051, 2004.
[63] K. Nakaso, H. Yano, Y. Fukuhara, T. Takeshima, K. Wada-Isoe, and K. Nakashima, “PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin inhuman neuroblastoma cells,” FEBS Letters, vol. 546, no. 2-3,pp. 181–184, 2003.
[64] J. A. Johnson, D. A. Johnson, A. D. Kraft et al., “The Nrf2-ARE pathway: an indicator and modulator of oxidative stressin neurodegeneration,” Annals of the New York Academy ofSciences, vol. 1147, pp. 61–69, 2008.
[65] T. Greco and G. Fiskum, “Neuroprotection through stim-ulation of mitochondrial antioxidant protein expression,”Journal of Alzheimer’s Disease, vol. 20, supplement 2, pp.S427–S437, 2010.
[66] T. Gura, “Hope in Alzheimer’s fight emerges from unex-pected places,” Nature Medicine, vol. 14, no. 9, p. 894, 2008.
[67] Y. Liu and D. R. Schubert, “The specificity of neuroprotectionby antioxidants,” Journal of Biomedical Science, vol. 16, no. 1,article 98, 2009.
[68] R. J. Bloomer, B. K. Schilling, R. E. Karlage, M. S. Ledoux,R. F. Pfeiffer, and J. Callegari, “Effect of resistance trainingon blood oxidative stress in Parkinson disease,” Medicine andScience in Sports and Exercise, vol. 40, no. 8, pp. 1385–1389,2008.
[69] E. T. Ang, Y. K. Tai, S. Q. Lo, R. Seet, and T. W. Soong,“Neurodegerative diseases: exercising toward neurogensisand neuoregeneration,” Frontiers in Aging Neuroscience, vol.25, p. 2, 2010.
[70] C. L. Allen and U. Bayraktutan, “Oxidative stress and its rolein the pathogenesis of ischaemic stroke,” International journalof stroke, vol. 4, no. 6, pp. 461–470, 2009.
[71] J. F. Turrens, A. Alexandre, and A. L. Lehninger,“Ubisemiquinone is the electron donor for superoxideformation by complex III of heart mitochondria,” Archivesof Biochemistry and Biophysics, vol. 237, no. 2, pp. 408–414,1985.
[72] L. Bergendi, L. Benes, Z. Durackova, and M. Ferencik,“Chemistry, physiology and pathology of free radicals,” LifeSciences, vol. 65, no. 18-19, pp. 1865–1874, 1999.
[73] D. D. Clarke and L. Sokoloff, “Circulation and energymetabolism of the brain,” in Baisic Neurochemistry: Molec-ular, Cellular and Medical Aspects, pp. 637–669, Lippincott-Raven, Philadelphia, Pa, USA, 1999.
[74] S. A. Saeed, K. F. Shad, T. Saleem, F. Javed, and M. U.Khan, “Some new prospects in the understanding of themolecular basis of the pathogenesis of stroke,” ExperimentalBrain Research, vol. 182, no. 1, pp. 1–10, 2007.
[75] M. L. Alexandrova and P. G. Bochev, “Oxidative stress duringthe chronic phase after stroke,” Free Radical Biology andMedicine, vol. 39, no. 3, pp. 297–316, 2005.
[76] M. A. Moro, A. Almeida, J. P. Bolanos, and I. Lizasoain,“Mitochondrial respiratory chain and free radical generationin stroke,” Free Radical Biology and Medicine, vol. 39, no. 10,pp. 1291–1304, 2005.

Journal of Toxicology 11
[77] R. Brouns and P. P. De Deyn, “The complexity of neurobio-logical processes in acute ischemic stroke,” Clinical Neurologyand Neurosurgery, vol. 111, no. 6, pp. 483–495, 2009.
[78] B. Schaller and R. Graf, “Cerebral ischemia and reperfusion:the pathophysiologic concept as a basis for clinical therapy,”Journal of Cerebral Blood Flow and Metabolism, vol. 24, no. 4,pp. 351–371, 2004.
[79] Y. C. Chen, S. H. Tsai, S. Y. Lin-Shiau, and J. K. Lin,“Elevation of apoptotic potential by anoxia hyperoxia shift inNIH3T3 cells,” Molecular and Cellular Biochemistry, vol. 197,no. 1-2, pp. 147–159, 1999.
[80] C. Li, M. Wright, and R. Jackson, “Reactive species-mediatedlung epithelia cells death after hypoxia and reoxygenation,”Experimental Lung Research, vol. 28, pp. 373–389, 2002.
[81] B. Schaller and R. Graf, “Cerebral ischemia and reperfusion:the pathophysiologic concept as a basis for clinical therapy,”Journal of Cerebral Blood Flow and Metabolism, vol. 24, no. 4,pp. 351–371, 2004.
[82] M. E. Soriano and L. Scorrano, “The interplay between BCL-2 family proteins and mitochondrial morphology in theregulation of apoptosis,” Advances in Experimental Medicineand Biology, vol. 687, pp. 97–114, 2010.
[83] K. L. Moffitt, S. L. Martin, and B. Walker, “From sentencingto execution—the processes of apoptosis,” Journal of Phar-macy and Pharmacology, vol. 62, no. 5, pp. 547–562, 2010.
[84] S. E. Lakhan, A. Kirchgessner, and M. Hofer, “Inflammatorymechanisms in ischemic stroke: therapeutic approaches,”Journal of Translational Medicine, vol. 7, article 97, 2009.
[85] G. J. del Zoppo, “Acute anti-inflammatory approaches toischemic stroke,” Annals of the New York Academy of Sciences,vol. 1207, pp. 143–148, 2010.
[86] K. Nakano and K. H. Vousden, “PUMA, a novel proapoptoticgene, is induced by p53,” Molecular Cell, vol. 7, no. 3, pp. 683–694, 2001.
[87] S. Zinkel, A. Gross, and E. Yang, “BCL2 family in DNAdamage and cell cycle control,” Cell Death and Differentiation,vol. 13, no. 8, pp. 1351–1359, 2006.
[88] H. Aleyasin, M. W. C. Rousseaux, M. Phillips et al., “TheParkinson’s disease gene DJ-1 is also a key regulator of stroke-induced damage,” Proceedings of the National Academy ofSciences of the United States of America, vol. 104, no. 47, pp.18748–18753, 2007.
[89] C. Culmsee and M. P. Mattson, “p53 in neuronal apoptosis,”Biochemical and Biophysical Research Communications, vol.331, no. 3, pp. 761–777, 2005.
[90] X. M. Yin, Z. N. Oltvai, and S. J. Korsmeyer, “BH1 and BH2domains of Bcl-2 are required for inhibition of apoptosis andheterodimerization with Bax,” Nature, vol. 369, no. 6478, pp.321–323, 1994.
[91] A. Scharstuhl, H. A. M. Mutsaers, S. W. C. Pennings, F. G. M.Russel, and F. A. D. T. G. Wagener, “Involvement of VDAC,Bax and ceramides in the efflux of AIF from mitochondriaduring curcumin-induced apoptosis,” PLoS ONE, vol. 4, no.8, article e6688, 2009.
[92] C. Hetz, P. A. Vitte, A. Bombrun et al., “Bax channelinhibitors prevent mitochondrion-mediated apoptosis andprotect neurons in a model of global brain ischemia,” Journalof Biological Chemistry, vol. 280, no. 52, pp. 42960–42970,2005.
[93] D. Gueorguieva, S. Li, N. Walsh, A. Mukerji, J. Tanha,and S. Pandey, “Identification of single-domain, Bax-specificintrabodies that confer resistance to mammalian cells againstoxidative-stress-induced apoptosis,” The FASEB Journal, vol.20, no. 14, pp. 2636–2638, 2006.
[94] L. Li, YI. Qu, J. Li, Y. Xiong, M. Mao, and D. Mu, “Rela-tionship between HIF-1α expression and neuronal apoptosisin neonatal rats with hypoxia-ischemia brain injury,” BrainResearch, vol. 1180, no. 1, pp. 133–139, 2007.
[95] X. Fan, C. J. Heijnen, M. A. van der Kooij, F. Groenendaal,and F. van Bel, “The role and regulation of hypoxia-induciblefactor-1α expression in brain development and neonatalhypoxic-ischemic brain injury,” Brain Research Reviews, vol.62, no. 1, pp. 99–108, 2009.
[96] D. Chen, M. Li, J. Luo, and W. Gu, “Direct interactionsbetween HIF-1α and Mdm2 modulate p53 function,” Journalof Biological Chemistry, vol. 278, no. 16, pp. 13595–13598,2003.
[97] A. E. Greijer, P. van der Groep, D. Kemming et al., “Up-regualtion of gene expression by hypoxia is mediated pre-dominantly by hypoxia-inducible factor I (HIF-I),” Journalof Pathology, vol. 206, no. 3, pp. 291–304, 2005.
[98] M. W. Halterman, C. C. Miller, and H. J. Federoff, “Hypoxia-inducible factor-1α mediates hypoxia-induced delayed neu-ronal death that involves p53,” Journal of Neuroscience, vol.19, no. 16, pp. 6818–6824, 1999.
[99] H. Suzuki, A. Tomida, and T. Tsuruo, “Dephosphorylatedhypoxia-inducible factor 1α as a mediator of p53-dependentapoptosis during hypoxia,” Oncogene, vol. 20, no. 41, pp.5779–5788, 2001.
[100] J. P. Piret, D. Mottet, M. Raes, and C. Michiels, “IsHIF-1α a pro- or an anti-apoptotic protein?” BiochemicalPharmacology, vol. 64, no. 5-6, pp. 889–892, 2002.
[101] J. W. Calvert, J. Cahill, M. Yamaguchi-Okada, and J. H.Zhang, “Oxygen treatment after experimental hypoxia-ischemia in neonatal rats alters the expression of HIF-1α andits downstream target genes,” Journal of Applied Physiology,vol. 101, no. 3, pp. 853–865, 2006.
[102] J. Koizumi, Y. Yoshida, T. Nakazawa, and G. Ooneda,“Experimental studies of ischemic brain edema. 1. A newexperimental model of cerebral embolism in rats in whichrecirculation can be introduced in the ischemic area,”Japanese Journal of Stroke, vol. 8, pp. 1–8, 1986.
[103] W. A. Pulsinelli and J. B. Brierley, “A new model of bilateralhemispheric ischemia in the unanesthetized rat,” Stroke, vol.10, no. 3, pp. 267–272, 1979.
[104] G. G. Roberts, M. J. Di Loreto, M. Marshall, J. Wang, andD. J. DeGracia, “Hippocampal cellular stress responses afterglobal brain ischemia and reperfusion,” Antioxidants andRedox Signaling, vol. 9, no. 12, pp. 2265–2275, 2007.
[105] M. A. Yenari and T. M. Hemmen, “Therapeutic hypothermiafor brain ischemia: where have we come and where do wego?” Stroke, vol. 41, no. 10 SUPPL. 1, pp. S72–S74, 2010.
[106] D. Woodbury, E. J. Schwarz, D. J. Prockop, and I. B.Black, “Adult rat and human bone marrow stromal cellsdifferentiate into neurons,” Journal of Neuroscience Research,vol. 61, no. 4, pp. 364–370, 2000.
[107] L. Otero, M. Zurita, C. Bonilla et al., “Late transplantationof allogeneic bone marrow stromal cells improves neurologicdeficits subsequent to intracerebral hemorrhage,” Cytother-apy, vol. 13, no. 5, pp. 562–571, 2011.
[108] S. A. Frautschy, W. Hu, P. Kim et al., “Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitivedeficits and neuropathology,” Neurobiology of Aging, vol. 22,no. 6, pp. 993–1005, 2001.
[109] M. Thiyagarajan and S. S. Sharma, “Neuroprotective effect ofcurcumin in middle cerebral artery occlusion induced focalcerebral ischemia in rats,” Life Sciences, vol. 74, no. 8, pp. 969–985, 2004.

12 Journal of Toxicology
[110] P. Dohare, P. Garg, V. Jain, C. Nath, and M. Ray, “Dosedependence and therapeutic window for the neuroprotectiveeffects of curcumin in thromboembolic model of rat,”Behavioural Brain Research, vol. 193, no. 2, pp. 289–297,2008.
[111] K. McGonigal, J. Tanha, E. Palazov, S. Li, D. Gueorguieva-Owens, and S. Pandey, “Isolation and functional character-ization of single domain antibody modulators of caspase-3and apoptosis,” Applied Biochemistry and Biotechnology, vol.157, no. 2, pp. 226–236, 2009.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 487074, 9 pagesdoi:10.1155/2011/487074
Review Article
Oxidative Stress and Air Pollution Exposure
Maura Lodovici and Elisabetta Bigagli
Department of Pharmacology and Toxicology, University of Florence, Viale Pieraccini 6, 50139 Florence, Italy
Correspondence should be addressed to Maura Lodovici, [email protected]
Received 15 December 2010; Revised 10 May 2011; Accepted 30 June 2011
Academic Editor: Susan Sumner
Copyright © 2011 M. Lodovici and E. Bigagli. This is an open access article distributed under the Creative Commons AttributionLicense, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properlycited.
Air pollution is associated with increased cardiovascular and pulmonary morbidity and mortality. The mechanisms of airpollution-induced health effects involve oxidative stress and inflammation. As a matter of fact, particulate matter (PM), especiallyfine (PM2.5, PM< 2.5 μm) and ultrafine (PM0.1, PM< 0.1 μm) particles, ozone, nitrogen oxides, and transition metals, are potentoxidants or able to generate reactive oxygen species (ROS). Oxidative stress can trigger redox-sensitive pathways that lead todifferent biological processes such as inflammation and cell death. However, it does appear that the susceptibility of targetorgan to oxidative injury also depends upon its ability to upregulate protective scavenging systems. As vehicular traffic is knownto importantly contribute to PM exposure, its intensity and quality must be strongly relevant determinants of the qualitativecharacteristics of PM spread in the atmosphere. Change in the composition of this PM is likely to modify its health impact.
1. Introduction
Numerous epidemiological studies have shown an increasedmorbidity and mortality due to environmental air pollution[1, 2]. Environmental air does contain a complex mixture oftoxics, including particulate matter (PM), irritant gases, andbenzene. The chemical composition of particles does varygreatly and depends on numerous geographical, meteorolog-ical, and source-specific variables. Generally, environmentalparticles include inorganic components (sulfates, nitrates,ammonium, chloride, and trace metals), elemental andorganic carbon, biological components (bacteria, spores,and pollens), and adsorbed volatile and semivolatile organiccompounds [3]. In addition, environmental particles, whenmixed with atmospheric gases (ozone, sulfur nitric oxides,and carbon monoxide) can generate environmental aerosols.Particles are usually defined as PM10 and PM2.5 with diam-eter less than 10 and 2.5 μm, respectively. Any fraction mayhave different effects; that is, PM with aerodynamic diameterless than 10 to 2.5 μm does generate a bigger amount ofhydroxyl radical due to the heavy metals adsorbed on thepores and surfaces of the particles, whereas particles of largersize (PM10) deposit mainly in the upper airways and can becleared by the mucociliary system [4, 5]. Recently, however,interest has also focused on the ultrafine particles (UFPs)
with a diameter less than 100 nm; UFPs are consideredimportant with respect to health effects because of their veryhigh alveolar deposition fraction, large surface area, chemicalcomposition, and ability to enter into the circulation andinduce inflammation. Vehicle emissions, in particular relatedto diesel engines, diesel exhaust particles (DEPs), are a majorsource of environmental UFPs, which in the presence of poorventilation may penetrate indoor, where additional sourcesincluding environmental tobacco smoke, cooking, burningof candles, and chemical reactions are present [6–10]. Long-term exposure to high levels of such particles can increaserisk of cancer, respiratory diseases, and arteriosclerosis,whereas short-term exposure peaks can cause exacerbationof bronchitis, asthma, and other respiratory diseases aswell as changes in heart-rate variability [2, 11–13]. Thegeneral consensus does indicate that the mechanism of airpollution-induced health effects involves an inflammation-related cascade and oxidation stress both in lung, vascular,and heart tissue [14–19]. Inflammation is initially a pro-tective mechanism which removes the injurious stimuli andproduces reactive oxygen species (ROS) able to induce cellkilling. In the early phase of inflammation, oxidant stressdoes not directly cause cell damage and can induce the tran-scription of stress defense genes including antioxidant genes.This preconditioning effect of ROS enhances the resistance

2 Journal of Toxicology
against future inflammatory oxidant stress and promotes theinitiation of tissue repair processes. The additional release ofcell contents amplifies the inflammatory process and conse-quently can induce tissue injury [20]. Oxidation damage hasbeen implicated in many degenerative and nondegenerativediseases, including cardiovascular and pulmonary diseases,diabetes, and Alzheimer disease. Oxidation stress derivedfrom an unbalance between ROS formation and individualantioxidant activity potentially does lead to damage of lipids,proteins, and macromolecules such as DNA and RNA [21].This paper will focus on the mechanisms of oxidative stressinduction and cellular damage by air pollution exposure onpulmonary and cardiovascular systems.
2. Possible Mechanisms of Oxidative StressInduced by Air Pollution Exposure
In the last decades, great attention has been paid to air pollu-tion exposure due to vehicular traffic and other combustionprocesses. PM and gas pollutants are considered to be themost important factors in urban areas, and several mecha-nisms have been hypothesized to explain the adverse healtheffects in humans, especially in the cardiopulmonary system[22]. Although each air pollutant can exert its own specifictoxicity in the respiratory and cardiovascular systems, ozone,oxides of nitrogen, and suspended particulates all share acommon property of being potent oxidants, either throughdirect effects on lipids and proteins or indirectly through theactivation of intracellular oxidant pathways [23–25].
ROS can be generated from the surface of particles wherepolycyclic aromatic hydrocarbons (PAH) and nitro-PAHare adsorbed, other than transition metals (iron, copper,chromium, and vanadium) that catalyzing Fenton’s reaction(Fe2+ + H2O2 + H+ → Fe3++ OH• + H2O) generate thehighly reactive hydroxyl radical able to induce oxidativeDNA damage [26, 27]. Several studies have shown thatiron and other transition metals leaching from particlesor by their presence on particle surfaces play a role inthe generation of ROS in biological systems [28]. Particlesbound benzo(a)pyrene has been shown to be bioavailableand can induce oxidative DNA damage in systemic targetorgans, including lung and kidney [29, 30]. Moreover,it should be noted that ozone and nitrogen dioxide areusually present together with particles in environmental air.They are also oxidants with potential effects in terms ofoxidative DNA damage. Similarly, volatile compounds, suchas benzene, in urban air pollution can induce DNA oxidation[31, 32]. In addition, photochemical oxidants (ozone andperoxyacetyl nitrate), secondary pollutants formed by theaction of sunlight on an atmosphere that does containreactive hydrocarbons and NOx, contribute to increaseoxidation stress [33]. Then, in the presence of high ROS for-mation, mitochondrial damage with induction of NADPH-oxidase isoform 4 (NOX4) does occur, together with anactivation of inflammatory cells (neutrophils, eosinophils,and monocytes) and increased numbers of macrophagescapable of ROS and reactive nitrogen species generation[34–36]. Initially, when oxidative stress is relatively low,various transcription factors, such as the nuclear factor
erythroid-2 (Nrf2), induce a series of antioxidant and detox-ification enzymes (e.g., catalase, superoxide dismutase, andglutathione S-transferase) that counteract ROS formationprotecting from adverse biological outcomes [37, 38]. Inthe second phase, if the protective antioxidant response failsor is inadequate to deal with increasing ROS production,the result is a proinflammatory situation with variouscytotoxic effects [39]. These effects are mediated by theredox-sensitive mitogen-activated protein kinase (MAPK)and NF-κB cascades that are responsible for the expressionof cytokines, chemokines, and adhesion molecules, which areinvolved in inflammatory processes [39].
3. Atmospheric Gases
Gaseous pollutants contribute to a great extent in compo-sition variations of the atmosphere and are mainly due tocombustion of fossil fuels and to emission of motor vehicles[40].
Ozone is a strong oxidizing agent formed in the tropo-sphere through a complex series of reactions involving theaction of sunlight in nitrogen dioxide and hydrocarbons.Ozone initiate intracellular oxidative stress through ozonideand hydroperoxide formation. This mechanism of oxidativedamage involves the activation of Nrf2, heat shock protein70, NF-κB, increased expression of a range of proinflamma-tory cytokines (TNFα and interleukin 1β), chemokines (e.g.,interleukin 8), and adhesion genes; ozone is also an activatorof protein-1 fos and c-jun onco genes [41, 42]. The majorsource of anthropogenic emissions of nitrogen oxides intothe atmosphere is the combustion of fossil fuels derivingfrom stationary sources (heating, power generation) andmotor vehicles. In environmental conditions, nitric oxide israpidly transformed into nitrogen dioxide by atmosphericoxidants such as ozone [43].
Various antioxidants, like ascorbic acid, uric acid, andthiols, act as powerful scavengers of O3 and NO•
2 radicalin body fluids, likely protecting lung lining fluids againstinhaled oxidizing air pollutants [44]. When such defensemechanisms are overwhelmed, O3 may injure the underlyingcells by inducing lipid peroxidation and activating inflam-matory gene expression [45]. In vitro and in vivo studies,both in animals and human beings, confirm the capacityof nitrogen dioxide to activate oxidant pathways althoughless potently than ozone [46]. Volatile organic compoundsare a class of compounds which includes chemical speciesof organic nature such as benzene, but the majority ofgaseous pollutants are inhaled and, therefore. mainly affectthe respiratory and cardiovascular systems. Among gaseouspollutants, carbon monoxide (CO) has been described as oneof the main pollutants responsible for the development ofcardiovascular diseases [47], while benzene can also inducehaematological problems and cancer [48].
Benzene is a commonly used industrial chemical and aconstituent of gasoline [31]. Inhalation is the most importantroute of absorption during occupation-related exposure.Benzene toxicity is attributed to its metabolism, whichdoes lead to the formation of reactive metabolites such ashydroquinone and its oxidized form benzoquinone which

Journal of Toxicology 3
are highly reactive molecules and, by means of redox cycling,produce ROS [49]. Furthermore, the addition of antioxidantenzymes has been shown to block oxidative damage inducedby the above-mentioned metabolites confirming the roleof ROS production and oxidative stress in hydroquinoneand benzoquinone cytotoxicity [50]. Uzma et al. [31]demonstrated that occupation-related exposure to benzenecauses oxidative stress, immune suppression, and inducingthe expression of tumour-suppressing gene p53 in gasolinefilling workers. These authors hypothesized that the increasein the p53 expression may block the cell cycle at G1 phaseand go on to repair DNA damage, which is the initial step intumour suppression.
4. Oxidative Stress from Organic Fraction
Ambient PM, does consist of complex and various mixturesof particles suspended in the breathing air [50]. Majorsources of PM are factories, power plants, refuse inciner-ators, motor vehicles, building activity, fires, and naturalwindblown dust. The size of the particles vary, and there isstrong evidence supporting that ultrafine and fine particlesare more hazardous than larger ones in terms of mortalityand cardiovascular and respiratory effects [51].
Results from various surveys have demonstrated thatoxidative potential of fine and ultrafine particles is theresult of significant amounts of organic carbon compounds,such as quinones and PAHs. In the organic fraction orig-inating in the air from incomplete combustion processes,the major reactive and toxic compounds are substituted(e.g., methyl naphthalene) and unsubstituted PAH, nitro-PAH (1-nitropyrene and 3-nitro-fluoranthene), dinitro-PAH(dinitro pyrene) and peroxyacetyl nitrate [52, 53]. Moreover,reactive intermediates in the oxidation of mixtures of volatileorganic compounds (VOCs), oxides of nitrogen (NOx),hydroxy radical, and ozone are shown to play a central rolein the formation and fate of airborne toxic chemicals, PAH,and fine particles [52]. The main pathways of metabolicactivation of PAHs are generation of diol epoxides catalyzedby cytochrome P450 (CYP450), leading to DNA adductformation, formation of radical cations catalyzed by CYP450peroxidases, and formation of redox-active quinones [54].Valavanidis et al. [55] demonstrated that redox-active transi-tion metals, redox cycling quinoids and PAH act synergicallyto produce ROS. J. Y. C. Ma and J. K. H. Ma [56] reportedthat organic fraction of DEP, mainly constituent of PAH andquinones, does undergo to metabolic activation in the lungand liver of exposed animals, is able to induce CYP4501A1isoform expression that generates ROS and reactive PAH-quinones. In addition, PM initiates inflammatory damageupregulating proinflammatory cytokines and chemokines;in vitro observations have shown that PM exposure maycause expression of nuclear factor NF-κB-related genes andoxidant-dependent NF-κB activation [57, 58]. To defendagainst oxidative damage, cells increase the production ofantioxidant enzymes through the activation of the Nrf2,[37] and PM appears to inhibit protective enzymes involvedin oxidative stress responses leading to the activation ofadditional intracellular signaling cascades that regulate the
expression of cytokine and chemokine genes [59]. Manyrecent observations have shown that DEPs, because of theirfine and ultrafine composition, play an important role onoxidative cellular damage through ROS generation causinglipid peroxidation and oxidative DNA damage. Some DEPsconsist of a carbon core or organic droplets with adsorbedorganic compounds, such as PAH, quinines, and redox-activemetals. The capacity of DEPs to induce oxidative stress islargely related to these adsorbed components [60, 61].
5. Oxidative Stress Induced by Transition Metals
Transition metals such as iron, lead, mercury, cadmium,silver, nickel, vanadium, chromium, manganese, and copperare detectable in PM2.5 and UFPs adsorbed on their surfaceand are capable of ROS formation by Fenton’s reaction [35].As critical constituents of PM, transition metals were postu-lated to be involved in a number of pathological processes ofthe respiratory system through free radical-mediated damage[62]. They are natural components of the earth’s crust andenter into the environment through a wide variety of sources,including combustion, waste water discharges, and manufac-turing facilities. Iron is a well-known soot suppressant thatmight be emitted into the atmosphere in the form of ultrafineparticles [63]. Zinc is a major metal element detected intraffic derived PM2.5, deriving from waste oil samples [64].Copper is a component of car brake pads, however, ceramicbrake pads contain 10%–20% copper by mass, while themetallic brake pads contained about 70% iron with verylittle copper. This metal in PM has also been linked toroad traffic sources associated to PM2.5 [64]. Soluble metalsin inhaled particles, such as Fe, Ni, V, Co, Cu, and Cr,were associated with increased ROS production, followed bycellular oxidative stress in airway epithelial cells [65].
6. Air Pollution Induced-OxidativeDamage in Target Organs: Cardiovascularand Pulmonary Systems
6.1. Cardiovascular System. Diesel and gasoline vehicleemissions in the urban areas have dominant contributionsto environmental particles, especially those located in theultrafine range. Because of their small size and large surfacearea, UFPs have demonstrated unique biochemical charac-teristics, such as enhanced ability to adsorb or absorb organicmolecules and to penetrate into cellular targets in the humanpulmonary and cardiovascular systems [66, 67]. UFPs maybe directly transported to the cardiac vasculature, wherethey can induce arrhythmias, reduce myocyte contractility,and decrease coronary blood flow [10, 68]. Studies byBrook et al. [69] demonstrated that fine particulate airpollution and ozone cause acute arterial vasoconstriction inhealthy humans, while Urch et al. [70] reported that fineparticles exposure pollution raise blood pressure and impairvascular function. In addition, UFP exposure depressesmyocardial contractile response and coronary flow in bothspontaneously hypertensive and wild-type rats [71], the sameobservation was found by Simkhovich et al. [72] in young

4 Journal of Toxicology
and old rat hearts. Long-term exposure to low concentra-tions of PM2.5 has been shown to alter vasomotor tone,lead to vascular inflammation, and potentiate atherosclerosisinduced by highly fat-containing chow in susceptible mice[73]. In addition, Suwa et al. [74] reported that exposureto PM10 cause progression of atherosclerotic lesions towardsa more advanced phenotype hyperlipidemic rabbits. More-over, atherosclerotic lesions of thoracic aorta were reportedto be significantly increased with pronounced macrophageinfiltration and lipid deposition in Apolipoprotein E (−/−)ApoE (−/−) mice exposed to PM2.5 through NADPH oxidasedependent pathways [75]. ApoE (−/−) mice exposed toozone showed increased oxidative stress and mitochondrialDNA damage, decreased vascular endothelial nitric oxidesynthase, and significantly increased atherogenesis comparedto filtered air exposed controls [76]. Recently, Cherng et al.[77] reported that DEP exposure enhances vasoconstrictionand diminishes acetylcholine-induced dilatation in coronaryarteries of animals in a nitric oxide synthase-dependentmanner. Baccarelli et al. [78] showed that air pollution isassociated with changes in the global coagulation function,after short-term exposure to air pollution in normal subjectsresident in Lombardia Region, Italy. Road traffic-relatedpollutants may increase a heart-rate-corrected QT intervalamong people with diabetes, obesity and nonsmoking elderlyindividuals and the number of genetic variants relatedto oxidative stress does increase this effect [79]. On thecontrary, Mordukhovich et al. [80], despite the positiveassociations between blood pressure and black carbon,found no effects on gene variants related to antioxidativedefense. Increases in black carbon and PM2.5 were associatedwith increases in blood pressure, heart-rate, endothelin-1, vascular endothelial growth factor, and oxidative stressmarkers and with a decrease in brachial artery diameterin nonsmoking seniors [81]. More recently, Kooter et al.[82] showed that diesel engine exhaust exposure induces apulmonary antioxidant response, with an increased activityof the anti-oxidant enzymes glutathione peroxidase, super-oxide dismutase, heme oxygenase-1 protein, heme oxygenaseactivity, and uric acid which precedes the inflammatoryresponse (an increase in IL-6 and TNF-α) in rats. Inaddition, since the authors found that increased plasmathrombogenicity and antioxidant defense gene expressionin aorta tissue shortly after the exposure does occur, theyhypothesized a direct translocation of diesel engine exhaustcomponents to the vasculature even if the mediation by otherpathways cannot be excluded [82].
6.2. Pulmonary. A strong correlation has been foundbetween PM concentration of redox-active compounds anddamage in macrophages and bronchial epithelial cells [83–85]. Moreover, in human airway epithelial cells, organiccompounds adsorbed on particle surfaces does promoteinflammation through CYP1A1-mediated ROS generationand release of cytokines after activation of transductionpathways involving MAPK and the transcription factor NF-kappaB [86]. Recently, Andersson et al. [26] reported that 1-nitropyrene, one of the most abundant nitro-PAHs in dieselexhausts, induces DNA damage by ROS formation in human
endothelial cells, and this effect was mainly mediated bymetabolites mainly generating by reduction of nitro group,as it has been previously reported by Topinka et al. [87] in rathepatocytes. Increased production of ROS after PM exposureis suggested by the finding that many of the proinflammatorygenes (TNF-α and IL-8, among others) induced uponexposure to PM are regulated by redox sensitive transcriptionfactors such as NF-κB, activator protein 1 (AP-1) andCAATT/enhancer binding protein (C/EBP). Activation ofthese transcription factors and increased transcription ofdownstream genes has been reported in human alveolar andbronchial epithelial cells in response to PM exposure [88–92]. Several studies have demonstrated that air pollutionparticles induce inflammatory mediator release and oxidativestress in lung epithelial cells and alveolar macrophages. Whenreaching the bone marrow [93], cytokines and chemokinesreleased from the lung stimulate migration of neutrophilsand their precursors into the circulation. In the short-term, there is acute tissue damage with activation of theepidermal-growth-factor receptor pathway, and evidence fororgan-repair responses [94]. Vanadium pentoxide (V2O5)is a component of PM derived from fuel combustionas well as a source of occupation-related exposure inhumans [95]. Sørensen et al. [95] indicate that vanadiumand chromium (VI) detectable in PM(2.5) have an effecton oxidative DNA damage in human lymphocytes, afterreduction to chromium (III) in the cells. Since, outdoorPM and urinary 1-hydroxypyrene (PAH exposure marker)were synergistically associated with urinary MDA levels ofschoolchildren, Bae et al. [96] concluded that exposure toPM air pollution and PAH can induce oxidative stress inschoolchildren. In addition, these authors found that urinaryMDA levels are also associated with some metals boundto PM10 and PM2.5 suggesting that metals bound to PMare responsible, at least in part, for the oxidative stress[96]. The oxidized species arising from the reaction betweenozone and lining fluid are involved in the signaling cascadeof inflammatory cells into the lung and contribute to theacute bronchoconstrictor response and hyperresponsivenessobserved in asthma on exposure to this pollutant [97, 98].Furthermore, has been reported that ozone is able to induceapoptosis, DNA damage, and cytotoxicity on human alveolarepithelial type I-like cells and in mice exposed to ozonefor 6 weeks [99, 100]. While, Ferecatu et al. [84] reportedan antiapoptotic effect of PAH adsorbed on PM2.5 that inaddition to the well-documented inflammatory responsemay explain the persistence of a prolonged inflammationstate induced after pollution exposure and might delay repairprocesses of injured tissues in primary cultures of humanbronchial epithelial cells. Chirino et al. [101] found ROSgeneration and decreased glutathione and the activity ofthe antioxidant enzymes, such as superoxide dismutase andglutathione reductase, in a human lung epithelial cell lineexposed to PM10. Recently, it has been found that bus driversexposed to PAH and volatile compounds displayed a higherlevel of DNA instability and oxidative damage than thecontrols and the incidence of oxidized lesions in lymphocyteDNA correlated with exposure to benzene. Moreover, thoseof the drivers with at least one variant of 8-oxoguanine

Journal of Toxicology 5
glycosylate 1 (hOGG1) (Cys/Cys or Ser/Cys) allele tendedto have higher oxidative DNA damage in lymphocyte thanthose with the wild genotype [102]. In addition, in the sameyear Delfino et al. [103] reported that PM (ranged from 0.25to 2.5 μm) and O3 were positively associated with exhalednitrogen monoxide and that PM0.25, CO, and NO werepositively associated with IL-6, while ROS were associatedwith both outcomes in elderly subjects enrolled.
7. Defense Mechanisms against ROS Formation
Antioxidants in the lung are the first line of defense againstROS [104]. The composition and quantity of antioxidantsin respiratory tract lining fluids may represent an importantdeterminant of individual responsiveness to air pollutants,but it should be thought of as a dynamic equilibriumwith the antioxidant defenses within the epithelium and amore remote plasma pool [105]. Interestingly, the resultsobtained by Osburn and Kensler [106] demonstrated thatthe activation of transcriptional factor Nrf2 determinesan upregulation of antioxidant enzymes that representsan adaptive response to face the exposure to oxidantpollutants providing a pivotal defense mechanism againstenvironmental hazards, including various air pollutants.Successively, Rubio et al. [107] observed that Nrf2 doesprotect against benzene metabolites in human lung cells,and knockdown of Nrf2 greatly does enhance cytotoxicityand cell death associated with reduced glutathione levels andloss of inducibility of antioxidant response elements (ARE-driven) genes.
Although the interrelation among antioxidant levels inthe respiratory tract, cellular and plasma levels are not wellunderstood, it appears that the susceptibility of the lung tooxidative injury depends largely on its ability to upregulateprotective scavenging systems. A recent review by Rubioet al. [108] indicates that air pollutants are Nrf2 pathwayinductors which regulate the expression of cytoprotectiveand detoxifying enzymes as well as antioxidants having animportant role in the defense against atmospheric pollutant-induced toxicity.
8. Conclusions
In conclusion, several experimental and epidemiologicalstudies have proved exposure to air pollution to be an impor-tant determinant of overall pulmonary and cardiovascularrisk damage and possibly have an influence on traditionalrisk factors. Although each environmental pollutant has itsown mechanism of toxicity, most pollutants, like UFP, PM2.5,ozone, nitrogen oxides, and transition metals, are potentoxidants or capable of ROS production. Consequently, thepromotion of oxidative stress has been identified as oneof the most important mechanisms responsible for toxicair pollutant effects. Oxidative stress can trigger redox-sensitive pathways that lead to different biological processeslike inflammation and cell death. Recently, EnvironmentalPollution Agency (EPA) revised the level of the 24-h PM2.5
standard to 35 μg/m3, moved the 24-h PM10 standard from
75 at 150 μg/m3, and revoked the annual standard, becauseavailable evidence generally did not suggest a link betweenlong-term exposure to current ambient levels of coarseparticles and health or welfare effects [109]. However, avast number of data indicate that in general, smaller sizefraction, containing higher concentration of PAH, transitionmetal, and semiquinones, has a higher ROS capacity andconsequently should be capable to induce severe toxico-logical effects. Thus, change in the composition of thisPM are likely to modify its health impact. Road traffic isknown to vastly contribute to PM exposure. Traffic intensityand quality should then be important determinants of thequalitative characteristics of PM spread in the atmosphere.In addition, although the interrelation between antioxidantlevels in respiratory and cardiovascular systems, cellular andplasma levels is not yet well understood; it appears thatthe susceptibility of target organs to oxidative injury largelydepends on cell ability to upregulate protective scavengingsystems such as Nrf2. This transcription factor does regulatethe expression of numerous cytoprotective genes that detox-ify reactive species playing an important role in the defenseagainst atmospheric pollutant-induced toxicity.
However, many questions remain unanswered, but inthe future, rapid developments in molecular biology, pro-teomics, and genomics will help to completely clarify thebiological mechanisms involved in pulmonary and cardio-vascular injuries caused by air pollution.
Acknowledgment
This work is supported by a fund of University of Florence.
References
[1] J. Schwartz, “Air pollution and daily mortality: a review andmeta analysis,” Environmental Research, vol. 64, no. 1, pp. 36–52, 1994.
[2] D. W. Dockery, A. C. Pope III, X. Xu et al., “An associationbetween air pollution and mortality in six U.S. cities,” NewEngland Journal of Medicine, vol. 329, no. 94, pp. 1753–1759,1993.
[3] R. M. Harrison and J. Yin, “Particulate matter in theatmosphere: which particle properties are important for itseffects on health?” Science of the Total Environment, vol. 249,no. 1–3, pp. 85–101, 2000.
[4] Environmental Protection Agency, Air Quality Criteria forParticulate Matter, vol. III of EPA/600/P-95/001CF, NationalCenter for Environmental Assessment, Research TrianglePark, NC, USA, 1996.
[5] J. Ferin, G. Oberdorster, and D. P. Penney, “Pulmonaryretention of ultrafine and fine particles in rats,” AmericanJournal of Respiratory Cell and Molecular Biology, vol. 6, no.5, pp. 535–542, 1992.
[6] J. Ferin, “Pulmonary retention and clearance of particles,”Toxicology Letters, vol. 72, no. 1–3, pp. 121–125, 1994.
[7] L. Kliucininkas, D. Martuzevicius, E. Krugly et al., “Indoorand outdoor concentrations of fine particles, particle-boundPAHs and volatile organic compounds in Kaunas, Lithuania,”Journal of Environmental Monitoring, vol. 13, no. 1, pp. 182–191, 2011.

6 Journal of Toxicology
[8] M. Dennekamp, S. Howarth, C. A. J. Dick, J. W. Cherrie, K.Donaldson, and A. Seaton, “Ultrafine particles and nitrogenoxides generated by gas and electric cooking,” Occupationaland Environmental Medicine, vol. 58, no. 8, pp. 511–516,2001.
[9] J. I. Levy, T. Dumyahn, and J. D. Spengler, “Particulatematter and polycyclic aromatic hydrocarbon concentrationsin indoor and outdoor microenvironments in Boston, Mas-sachusetts,” Journal of Exposure Analysis and EnvironmentalEpidemiology, vol. 12, no. 2, pp. 104–114, 2002.
[10] U. Franck, O. Herbarth, B. Wehner, A. Wiedensohler, and M.Manjarrez, “How do the indoor size distributions of airbornesubmicron and ultrafine particles in the absence of significantindoor sources depend on outdoor distributions?” IndoorAir, vol. 13, no. 2, pp. 174–181, 2003.
[11] B. Brunekreef and S. T. Holgate, “Air pollution and health,”The Lancet, vol. 360, no. 9341, pp. 1233–1242, 2002.
[12] A. Peters, D. W. Dockery, J. E. Muller, and M. A. Mittleman,“Increased particulate air pollution and the triggering ofmyocardial infarction,” Circulation, vol. 103, no. 23, pp.2810–2815, 2001.
[13] Y. J. Li, H. Takizawa, and T. Kawada, “Role of oxidativestresses induced by diesel exhaust particles in airway inflam-mation, allergy and asthma: their potential as a target ofchemoprevention,” Inflammation and Allergy, vol. 9, no. 4,pp. 300–305, 2010.
[14] A. J. Ghio, C. Kim, and R. B. Devlin, “Concentrated ambientair particles induce mild pulmonary inflammation in healthyhuman volunteers,” American Journal of Respiratory andCritical Care Medicine, vol. 162, no. 3 I, pp. 981–988, 2001.
[15] K. Donaldson and V. Stone, “Current hypotheses onthe mechanisms of toxicity of ultrafine particles,” Annalidell’Istituto Superiore di Sanita, vol. 39, no. 3, pp. 405–410,2003.
[16] X. Y. Li, D. Brown, S. Smith, W. MacNee, and K. Donaldson,“Short-term inflammatory responses following intratrachealinstillation of fine and ultrafine carbon black in rats,”Inhalation Toxicology, vol. 11, no. 8, pp. 709–731, 1999.
[17] Y. Bai, A. K. Suzuki, and M. Sagai, “The cytotoxic effectsof diesel exhaust particles on human pulmonary arteryendothelial cells in vitro: role of active oxygen species,” FreeRadical Biology and Medicine, vol. 30, no. 5, pp. 555–562,2001.
[18] S. Hirano, A. Furuyama, E. Koike, and T. Kobayashi,“Oxidative-stress potency of organic extracts of dieselexhaust and urban fine particles in rat heart microvesselendothelial cells,” Toxicology, vol. 187, no. 2-3, pp. 161–170,2003.
[19] L. E. Wold, B. Z. Simkhovich, M. T. Kleinman et al., “In vivoand in vitro models to test the hypothesis of particle-inducedeffects on cardiac function and arrhythmias,” CardiovascularToxicology, vol. 6, no. 1, pp. 69–78, 2006.
[20] H. Jaeschke, “Reactive oxygen and mechanisms of inflamma-tory liver injury: present concepts,” Journal of Gastroenterol-ogy and Hepatology, vol. 26, no. 1, pp. 173–179, 2011.
[21] L. Risom, P. Møller, and S. Loft, “Oxidative stress-induced DNA damage by particulate air pollution,” MutationResearch, vol. 592, no. 1-2, pp. 119–137, 2005.
[22] J. Schnelle-Kreis, U. Kupper, M. Sklorz et al., “Daily measure-ment of organic compounds in ambient particulate matterin Augsburg, Germany: new aspects on aerosol sources andaerosol related health effects,” Biomarkers, vol. 14, no. 1, pp.39–44, 2009.
[23] P. MØller and S. Loft, “Oxidative damage to DNA and lipidsas biomarkers of exposure to air pollution,” EnvironmentalHealth Perspectives, vol. 118, no. 8, pp. 1126–1136, 2010.
[24] A. Valavanidis, K. Fiotakis, and T. Vlachogianni, “Airborneparticulate matter and human health: toxicological assess-ment and importance of size and composition of particles foroxidative damage and carcinogenic mechanisms,” Journal ofEnvironmental Science and Health C, vol. 26, no. 4, pp. 339–362, 2008.
[25] H. A. Jeng, “Chemical composition of ambient particulatematter and redox activity,” American Journal of PhysiologyLung Cell Molecular Physiology, vol. 293, pp. 170–181, 2007.
[26] H. Andersson, E. Piras, J. Demma, and B. Hellman, “Lowlevels of the air pollutant 1-nitropyrene induce DNA damage,increased levels of reactive oxygen species and endoplasmicreticulum stress in human endothelial cells,” Toxicology, vol.262, no. 1, pp. 57–64, 2009.
[27] J. H. Park, A. B. Troxel, R. G. Harvey, and T. M. Penning,“Polycyclic aromatic hydrocarbon (PAH) o-quinones pro-duced by the Aldo-Keto-Reductases (AKRs) generate abasicsites, oxidized pyrimidines, and 8-Oxo-dGuo via reactiveoxygen species,” Chemical Research in Toxicology, vol. 19, no.5, pp. 719–728, 2006.
[28] A. J. Ghio, J. H. Richards, J. D. Carter, and M. C. Madden,“Accumulation of iron in the rat lung after tracheal instilla-tion of diesel particles,” Toxicologic Pathology, vol. 28, no. 4,pp. 619–627, 2000.
[29] P. Gerde, B. A. Muggenburg, M. Lundborg, Y. Tesfaigzi, andA. R. Dahl, “Respiratory epithelial penetration and clearanceof particle-borne benzo[a]pyrene,” Research Report, vol. 101,pp. 5–27, 2001.
[30] K. B. Kim and B. M. Lee, “Oxidative stress to DNA,protein, and antioxidant enzymes (superoxide dismutase andcatalase) in rats treated with benzo(a)pyrene,” Cancer Letters,vol. 113, no. 1-2, pp. 205–212, 1997.
[31] N. Uzma, S. S. Kumar, and M. A. H. Hazari, “Exposure tobenzene induces oxidative stress, alters the immune responseand expression of p53 in gasoline filling workers,” AmericanJournal of Industrial Medicine, vol. 53, no. 12, pp. 1264–1270,2010.
[32] M. Sørensen, H. Skov, H. Autrup, O. Hertel, and S. Loft,“Urban benzene exposure and oxidative DNA damage,”Science of the Total Environment, vol. 309, no. 1–3, pp. 69–80,2003.
[33] H. H. Liu, Y. C. Wu, and H. L. Chen, “Production ofozone and reactive oxygen species after welding,” Archives ofEnvironmental Contamination and Toxicology, vol. 53, no. 4,pp. 513–518, 2007.
[34] A. Baulig, M. Garlatti, V. Bonvallot et al., “Involvement ofreactive oxygen species in the metabolic pathways triggeredby diesel exhaust particles in human airway epithelial cells,”American Journal of Physiology, vol. 285, no. 3, pp. L671–L679, 2003.
[35] N. Li, C. Sioutas, A. Cho et al., “Ultrafine particulate pol-lutants induce oxidative stress and mitochondrial damage,”Environmental Health Perspectives, vol. 111, no. 4, pp. 455–460, 2003.
[36] N. Amara, R. Bachoual, M. Desmard et al., “Dieselexhaust particles induce matrix metalloprotease-1 in humanlung epithelial cells via a NADP(H) oxidase/NOX4 redox-dependent mechanism,” American Journal of Physiology, vol.293, no. 1, pp. L170–L181, 2007.

Journal of Toxicology 7
[37] K. W. Kang, S. J. Lee, and S. G. Kim, “Molecular mechanismof Nrf2 activation by oxidative stress,” Antioxidants andRedox Signaling, vol. 7, no. 11-12, pp. 1664–1673, 2005.
[38] J. D. Hayes and M. McMahon, “NRF2 and KEAP1 mutations:permanent activation of an adaptive response in cancer,”Trends in Biochemical Sciences, vol. 34, no. 4, pp. 176–188,2009.
[39] A. H. Sprague and R. A. Khalil, “Inflammatory cytokinesin vascular dysfunction and vascular disease,” BiochemicalPharmacology, vol. 78, no. 6, pp. 539–552, 2009.
[40] K. Katsouyanni, “Ambient air pollution and health,” BritishMedical Bulletin, vol. 68, pp. 143–156, 2003.
[41] B. G. Nichols, J. S. Woods, D. L. Luchtel, J. Corral, and J.Q. Koenig, “Effects of ozone exposure on nuclear factor-κBactivation and tumor necrosis factro-α expression in humannasal epithelial cells,” Toxicological Sciences, vol. 60, no. 2, pp.356–362, 2001.
[42] D. Bassett, C. Elbon-Copp, S. Otterbein, H. Barraclough-Mitchell, M. DeLorme, and H. Yang, “Inflammatory cellavailability affects ozone-induced lung damage,” Journal ofToxicology and Environmental Health A, vol. 64, no. 7, pp.547–565, 2001.
[43] WHO, “Air quality guidelines: for Europe,” World HealthOrganization Regional Publications, no. 91, pp. 1–287, 2001.
[44] W. Yang and S. T. Omaye, “Air pollutants, oxidative stress andhuman health,” Mutation Research, vol. 674, no. 1-2, pp. 45–54, 2009.
[45] B. Kosmider, J. E. Loader, R. C. Murphy, and R. J. Mason,“Apoptosis induced by ozone and oxysterols in humanalveolar epithelial cells,” Free Radical Biology and Medicine,vol. 48, no. 11, pp. 1513–1524, 2010.
[46] H. Bayram, R. J. Sapsford, M. M. Abdelaziz, and O. A.Khair, “Effect of ozone and nitrogen dioxide on the releaseof proinflammatory mediators from bronchial epithelial cellsof nonatopic nonasthmatic subjects and atopic asthmaticpatients in vitro,” Journal of Allergy and Clinical Immunology,vol. 107, no. 2, pp. 287–294, 2001.
[47] M. L. Bell, R. D. Peng, F. Dominici, and J. M. Samet,“Emergency hospital admissions for cardiovascular diseasesand ambient levels of carbon monoxide results for 126 unitedstates urban counties,” Circulation, vol. 120, no. 11, pp. 949–955, 2009.
[48] G. Barreto, D. Madureira, F. Capani, L. Aon-Bertolino, E.Saraceno, and L. D. Alvarez-Giraldez, “The role of catecholsand free radicals in benzene toxicity: an oxidative DNAdamage pathway,” Environmental and Molecular Mutagenesis,vol. 50, no. 9, pp. 771–780, 2009.
[49] J. L. Bolton, M. A. Trush, T. M. Penning, G. Dryhurst, andT. J. Monks, “Role of quinones in toxicology,” ChemicalResearch in Toxicology, vol. 13, no. 3, pp. 135–160, 2000.
[50] U. Poschl, “Atmospheric aerosols: composition, transforma-tion, climate and health effects,” Angewandte Chemie, vol. 44,no. 46, pp. 7520–7540, 2005.
[51] E. Boldo, C. Linares, J. Lumbreras et al., “Health impactassessment of a reduction in ambient PM(2.5) levels inSpain,” Environment International, vol. 37, no. 2, pp. 342–348, 2011.
[52] G. L. Squadrito, R. Cueto, B. Dellinger, and W. A. Pryor,“Quinoid redox cycling as a mechanism for sustained freeradical generation by inhaled airborne particulate matter,”Free Radical Biology and Medicine, vol. 31, no. 9, pp. 1132–1138, 2001.
[53] B. J. Finlayson-Pitts Jr. and J. N. Pitts, “Troposphericair pollution: ozone, airborne toxics, polycyclic aromatichydrocarbons, and particles,” Science, vol. 276, no. 5315, pp.1045–1052, 1997.
[54] V. Bonvallot, A. Baeza-Squiban, A. Baulig et al., “Organiccompounds from diesel exhaust particles elicit a proin-flammatory response in human airway epithelial cells andinduce cytochrome p450 1A1 expression,” American Journalof Respiratory Cell and Molecular Biology, vol. 25, no. 4, pp.515–521, 2001.
[55] A. Valavanidis, K. Fiotakis, E. Bakeas, and T. Vlahogianni,“Electron paramagnetic resonance study of the generationof reactive oxygen species catalysed by transition metalsand quinoid redox cycling by inhalable ambient particulatematter,” Redox Report, vol. 10, no. 1, pp. 37–51, 2005.
[56] J. Y. C. Ma and J. K. H. Ma, “The dual effect of the particulateand organic components of diesel exhaust particles on thealteration of pulmonary immune/inflammatory responsesand metabolic enzymes,” Journal of Environmental Scienceand Health C, vol. 20, no. 2, pp. 117–147, 2002.
[57] A. Shukla, C. Timblin, K. BeruBe et al., “Inhaled particulatematter causes expression of nuclear factor (NF)-κB-relatedgenes and oxidant-dependent NF-κB activation in vitro,”American Journal of Respiratory Cell and Molecular Biology,vol. 23, no. 2, pp. 182–187, 2000.
[58] R. Li, Z. Ning, R. Majumdar et al., “Ultrafine particlesfrom diesel vehicle emissions at different driving cyclesinduce differential vascular pro-inflammatory responses:implication of chemical components and NF-κB signaling,”Particle and Fibre Toxicology, vol. 7, no. 1, pp. 6–18, 2010.
[59] R. Li, Z. Ning, J. Cui et al., “Ultrafine particles from dieselengines induce vascular oxidative stress via JNK activation,”Free Radical Biology and Medicine, vol. 15, no. 46, pp. 775–782, 2009.
[60] K. S. Kouassi, S. Billet, G. Garcon et al., “Oxidative damageinduced in A549 cells by physically and chemically charac-terized air particulate matter (PM2.5) collected in Abidjan,Cote d’Ivoire,” Journal of Applied Toxicology, vol. 30, no. 4,pp. 310–320, 2010.
[61] A. Saxon and D. Diaz-Sanchez, “Air pollution and allergy:you are what you breathe,” Nature Immunology, vol. 6, no.3, pp. 223–226, 2005.
[62] E. Vidrio, H. Jung, and C. Anastasio, “Generation of hydroxylradicals from dissolved transition metals in surrogate lungfluid solutions,” Atmospheric Environment, vol. 42, no. 18, pp.4369–4379, 2008.
[63] P. J. Schaumann, J. Borm, A. Herbrich et al., “Metal-rich ambient particles (particulate matter2.5) cause airwayinflammation in healthy subjects,” American Journal ofRespiratory and Critical Care Medicine, vol. 170, no. 8, pp.898–903, 2004.
[64] M. E. Gerlofs-Nijland, J. A. M. A. Dormans, H. J. T. Bloemenet al., “Toxicity of coarse and fine particulate matter fromsites with contrasting traffic profiles,” Inhalation Toxicology,vol. 19, no. 13, pp. 1055–1069, 2007.
[65] A. M. Knaapen, T. Shi, P. J. A. Borm, and R. P. F. Schins,“Soluble metals as well as the insoluble particle fraction areinvolved in cellular DNA damage induced by particulatematter,” Molecular and Cellular Biochemistry, vol. 234-235,pp. 317–326, 2002.
[66] A. Peters, H. E. Wichmann, T. Tuch, J. Heinrich, and J.Heyder, “Respiratory effects are associated with the numberof ultrafine particles,” American Journal of Respiratory andCritical Care Medicine, vol. 155, no. 4, pp. 1376–1383, 1997.

8 Journal of Toxicology
[67] C. A. Pope, J. B. Muhlestein, H. T. May, D. G. Renlund,J. L. Anderson, and B. D. Horne, “Ischemic heart diseaseevents triggered by short-term exposure to fine particulateair pollution,” Circulation, vol. 114, no. 23, pp. 2443–2448,2006.
[68] R. J. Delfino, C. Sioutas, and S. Malik, “Potential role ofultrafine particles in associations between airborne particlemass and cardiovascular health,” Environmental Health Per-spectives, vol. 113, no. 8, pp. 934–946, 2005.
[69] R. D. Brook, J. R. Brook, B. Urch, R. Vincent, S. Rajagopalan,and F. Silverman, “Inhalation of fine particulate air pollutionand ozone causes acute arterial vasoconstriction in healthyadults,” Circulation, vol. 105, no. 13, pp. 1534–1536, 2002.
[70] B. Urch, F. Silverman, P. Corey et al., “Acute blood pressureresponses in healthy adults during controlled air pollutionexposures,” Environmental Health Perspectives, vol. 113, no.8, pp. 1052–1055, 2005.
[71] H. Hwang, R. A. Kloner, M. T. Kleinman, and B. Z.Simkhovich, “Direct and acute cardiotoxic effects of ultrafineair pollutants in spontaneously hypertensive rats and Wistar-Kyoto rats,” Journal of Cardiovascular Pharmacology andTherapeutics, vol. 13, no. 3, pp. 189–198, 2008.
[72] B. Z. Simkhovich, P. Marjoram, M. T. Kleinman, and R.A. Kloner, “Direct and acute cardiotoxicity of ultrafineparticles in young adult and old rat hearts,” Basic Researchin Cardiology, vol. 102, no. 6, pp. 467–475, 2006.
[73] Q. Sun, A. Wang, X. Jin et al., “Long-term air pollutionexposure and acceleration of atherosclerosis and vascularinflammation in an animal model,” Journal of the AmericanMedical Association, vol. 294, no. 23, pp. 3003–3010, 2005.
[74] T. Suwa, J. C. Hogg, K. B. Quinlan, A. Ohgami, R. Vincent,and S. F. Van Eeden, “Particulate air pollution inducesprogression of atherosclerosis,” Journal of the AmericanCollege of Cardiology, vol. 39, no. 6, pp. 935–942, 2002.
[75] G. C. Chuang, Z. Yang, D. G. Westbrook et al., “Pulmonaryozone exposure induces vascular dysfunction, mitochondrialdamage, and atherogenesis,” American Journal of Physiology,vol. 297, no. 2, pp. L209–L216, 2009.
[76] M. J. Campen, A. K. Lund, T. L. Knuckles et al., “Inhaleddiesel emissions alter atherosclerotic plaque composition inApoE(-/-) mice,” Toxicology and Applied Pharmacology, vol.242, no. 3, pp. 310–317, 2010.
[77] T. W. Cherng, M. L. Paffett, O. Jackson-Weaver, M. J.Campen, B. R. Walker, and N. L. Kanagy, “Mechanisms ofdiesel-induced endothelial nitric oxide synthase dysfunctionin coronary arterioles,” Environmental Health Perspectives,vol. 119, no. 1, pp. 98–103, 2011.
[78] A. Baccarelli, A. Zanobetti, I. Martinelli et al., “Effects ofexposure to air pollution on blood coagulation,” Journal ofThrombosis and Haemostasis, vol. 5, no. 2, pp. 252–260, 2007.
[79] E. S. Baja, J. D. Schwartz, G. A. Wellenius et al., “Traffic-related air pollution and QT interval: modification bydiabetes, obesity, and oxidative stress gene polymorphismsin the normative aging study,” Environmental Health Perspec-tives, vol. 118, no. 6, pp. 840–846, 2010.
[80] I. Mordukhovich, E. Wilker, H. Suh et al., “Black carbonexposure, oxidative stress genes, and blood pressure in arepeated-measures study,” Environmental Health Perspectives,vol. 117, no. 11, pp. 1767–1772, 2009.
[81] L. Liu, T. Ruddy, M. Dalipaj et al., “Effects of indoor, outdoor,and personal exposure to particulate air pollution on car-diovascular physiology and systemic mediators in seniors,”Journal of Occupational and Environmental Medicine, vol. 51,no. 9, pp. 1088–1098, 2009.
[82] I. M. Kooter, M. E. Gerlofs-Nijland, A. J. Boere et al.,“Diesel engine exhaust initiates a sequence of pulmonaryand cardiovascular effects in rats,” Environmental HealthPerspectives, vol. 118, no. 8, pp. 1126–1136, 2010.
[83] L. J. den Hartigh, M. W. Lame, W. Ham, M. J. Kleeman, F.Tablin, and D. W. Wilson, “Endotoxin and polycyclic aro-matic hydrocarbons in ambient fine particulate matter fromFresno, California initiate human monocyte inflammatoryresponses mediated by reactive oxygen species,” Toxicology inVitro, vol. 24, no. 7, pp. 1993–2002, 2010.
[84] I. Ferecatu, M. C. Borot, C. Bossard et al., “Polycyclicaromatic hydrocarbon components contribute to themitochondria-antiapoptotic effect of fine particulate matteron human bronchial epithelial cells via the aryl hydrocarbonreceptor,” Particle and Fibre Toxicology, vol. 7, no. 1, pp.18–32, 2010.
[85] A. Baulig, M. Garlatti, V. Bonvallot et al., “Involvement ofreactive oxygen species in the metabolic pathways triggeredby diesel exhaust particles in human airway epithelial cells,”American Journal of Physiology, vol. 285, no. 3, pp. L671–L679, 2003.
[86] A. Churg, C. Xie, X. Wang, R. Vincent, and R. D. Wang, “Airpollution particles activate NF-κB on contact with airwayepithelial cell surfaces,” Toxicology and Applied Pharmacology,vol. 208, no. 1, pp. 37–45, 2005.
[87] J. Topinka, L. R. Schwarz, F. Kiefer et al., “DNA adductformation in mammalian cell cultures by polycyclic aromatichydrocarbons (PAH) and nitro-PAH in coke oven emissionextract,” Mutation Research, vol. 419, no. 1–3, pp. 91–105,1998.
[88] L. A. Jimenez, J. Thompson, D. A. Brown et al., “Activationof NF-κB by PM10 occurs via an iron-mediated mechanismin the absence of IκB degradation,” Toxicology and AppliedPharmacology, vol. 15, no. 166, pp. 101–110, 2000.
[89] S. Becker, L. A. Dailey, J. M. Soukup, S. C. Grambow, R.B. Devlin, and Y. C. T. Huang, “Seasonal variations in airpollution particle-induced inflammatory mediator releaseand oxidative stress,” Environmental Health Perspectives, vol.113, no. 8, pp. 1032–1038, 2005.
[90] S. Becker, S. Mundandhara, R. B. Devlin, and M. Madden,“Regulation of cytokine production in human alveolarmacrophages and airway epithelial cells in response toambient air pollution particles: further mechanistic studies,”Toxicology and Applied Pharmacology, vol. 207, no. 2, supple-ment, pp. S269–S275, 2005.
[91] S. S. Salvi, C. Nordenhall, A. Blomberg et al., “Acute exposureto diesel exhaust increases IL-8 and GRO-α production inhealthy human airways,” American Journal of Respiratory andCritical Care Medicine, vol. 161, no. 2 I, pp. 550–557, 2000.
[92] E. D. Karoly, Z. Li, X. Hyseni, and Y. C. T. Huang, “Up-regulation of tissue factor in human pulmonary arteryendothelial cells after ultrafine particle exposure,” Environ-mental Health Perspectives, vol. 115, no. 4, pp. 535–540, 2007.
[93] H. Ishii, S. Hayashi, J. C. Hogg et al., “Alveolar macrophage-epithelial cell interaction following exposure to atmosphericparticles induces the release of mediators involved in mono-cyte mobilization and recruitment,” Respiratory Research, vol.6, pp. 87–99, 2005.
[94] G. Oberdorster, “Significance of particle parameters inthe evaluation of exposure-dose-response relationships ofinhaled particles,” Inhalation Toxicology, vol. 8, pp. 73–89,1996.

Journal of Toxicology 9
[95] M. Sørensen, R. P. F. Schins, O. Hertel, and S. Loft, “Transi-tion metals in personal samples of PM2.5 and oxidative stressin human volunteers,” Cancer Epidemiology Biomarkers andPrevention, vol. 14, no. 5, pp. 1340–1343, 2005.
[96] S. Bae, X. C. Pan, S. Y. Kim et al., “Exposures to particulatematter and polycyclic aromatic hydrocarbons and oxidativestress in schoolchildren,” Environmental Health Perspectives,vol. 118, no. 4, pp. 579–583, 2010.
[97] H. Song, W. Tan, and X. Zhang, “Ozone induces inflamma-tion in bronchial epithelial cells,” Journal of Asthma, vol. 48,no. 1, pp. 79–83, 2011.
[98] I. S. Mudway and F. J. Kelly, “Ozone and the lung: a sensitiveissue,” Molecular Aspects of Medicine, vol. 21, no. 1–2, pp. 1–48, 2000.
[99] B. Kosmider, J. E. Loader, R. C. Murphy, and R. J. Mason,“Apoptosis induced by ozone and oxysterols in humanalveolar epithelial cells,” Free Radical Biology and Medicine,vol. 48, no. 11, pp. 1513–1524, 2010.
[100] K. Triantaphyllopoulos, F. Hussain, M. Pinart et al., “Amodel of chronic inflammation and pulmonary emphysemaafter multiple ozone exposures in mice,” American Journal ofPhysiology, vol. 300, no. 5, pp. L691–L700, 2011.
[101] Y. I. Chirino, Y. Sanchez-Perez, A. R. Osornio-Vargas etal., “PM10 impairs the antioxidant defense system andexacerbates oxidative stress driven cell death,” ToxicologyLetters, vol. 193, no. 3, pp. 209–216, 2010.
[102] Y. Bagryantseva, B. Novotna, P. Rossner et al., “Oxidativedamage to biological macromolecules in Prague bus driversand garagemen: impact of air pollution and genetic polymor-phisms,” Toxicology Letters, vol. 199, no. 1, pp. 60–68, 2010.
[103] R. J. Delfino, N. Staimer, T. Tjoa et al., “Associationsof primary and secondary organic aerosols with airwayand systemic inflammation in an elderly panel cohort,”Epidemiology, vol. 21, no. 6, pp. 892–902, 2010.
[104] F. J. Kelly and T. D. Tetley, “Nitrogen dioxide depletes uricacid and ascorbic acid but not glutathione from lung liningfluid,” Biochemical Journal, vol. 325, no. 1, pp. 95–99, 1997.
[105] A. F. Behndig, I. S. Mudway, J. L. Brown et al., “Airwayantioxidant and inflammatory responses to diesel exhaustexposure in healthy humans,” European Respiratory Journal,vol. 27, no. 2, pp. 359–365, 2006.
[106] W. O. Osburn and T. W. Kensler, “Nrf2 signaling: an adaptiveresponse pathway for protection against environmental toxicinsults,” Mutation Research, vol. 659, no. 1-2, pp. 31–39, 2008.
[107] V. Rubio, J. Zhang, M. Valverde, E. Rojas, and Z. Z. Shi,“Essential role of Nrf2 in protection against hydroquinone-and benzoquinone-induced cytotoxicity,” Toxicology in Vitro,vol. 25, no. 2, pp. 521–529, 2010.
[108] V. Rubio, M. Valverde, and E. Rojas, “Effects of atmosphericpollutants on the Nrf2 survival pathway,” EnvironmentalScience and Pollution Research, vol. 17, no. 2, pp. 369–382,2010.
[109] EPA, “Integrated science assessment for particulate matter,”Provisional Draft (Personal communication), 2008.

Hindawi Publishing CorporationJournal of ToxicologyVolume 2011, Article ID 152474, 16 pagesdoi:10.1155/2011/152474
Review Article
Liposomal Antioxidants for Protection againstOxidant-Induced Damage
Zacharias E. Suntres
Medical Sciences Division, Northern Ontario School of Medicine, Lakehead University, 955 Oliver Road, Thunder Bay,ON, Canada P7B 5E1
Correspondence should be addressed to Zacharias E. Suntres, [email protected]
Received 7 December 2010; Revised 13 April 2011; Accepted 24 May 2011
Academic Editor: JeanClare Seagrave
Copyright © 2011 Zacharias E. Suntres. This is an open access article distributed under the Creative Commons AttributionLicense, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properlycited.
Reactive oxygen species (ROS), including superoxide anion, hydrogen peroxide, and hydroxyl radical, can be formed as normalproducts of aerobic metabolism and can be produced at elevated rates under pathophysiological conditions. Overproductionand/or insufficient removal of ROS result in significant damage to cell structure and functions. In vitro studies showed thatantioxidants, when applied directly and at relatively high concentrations to cellular systems, are effective in conferring protectionagainst the damaging actions of ROS, but results from animal and human studies showed that several antioxidants provide onlymodest benefit and even possible harm. Antioxidants have yet to be rendered into reliable and safe therapies because of theirpoor solubility, inability to cross membrane barriers, extensive first-pass metabolism, and rapid clearance from cells. There isconsiderable interest towards the development of drug-delivery systems that would result in the selective delivery of antioxidants totissues in sufficient concentrations to ameliorate oxidant-induced tissue injuries. Liposomes are biocompatible, biodegradable, andnontoxic artificial phospholipid vesicles that offer the possibility of carrying hydrophilic, hydrophobic, and amphiphilic molecules.This paper focus on the use of liposomes for the delivery of antioxidants in the prevention or treatment of pathological conditionsrelated to oxidative stress.
1. Introduction
Oxidative stress (OS) is defined as an imbalance betweenthe production of reactive oxygen species and antioxidantdefenses that can lead to cellular and tissue damage [1–8]. Apotential pharmacological strategy in preventing or treatingoxidant-induced cellular and tissue damage involves theuse of appropriate antioxidants. Antioxidants are substanceswhich are able to prevent, delay, or remove oxidative damageto a molecule [4, 9–11]. Yet, their efficacy is hindered withchallenges such as poor solubility, inability to cross cell-membrane barriers, extensive first-pass metabolism, andrapid clearance of antioxidants from cells [12, 13]. Toimprove the pharmacological and pharmacokinetic prop-erties of antioxidants, diverse systems such as antioxidantchemical modifications and coupling to affinity carriers,micelles, and liposomes are being developed [4, 13–18].This paper focus on the use of liposomes for the deliv-ery of antioxidants in the prevention or treatment of
several pathological conditions linked to oxidative stress.Liposomes are artificial vesicles consisting of an aqueouscore enclosed in one or more phospholipid layers; water-soluble compounds can be encapsulated in the aqueous core,while lipid-soluble compounds can be incorporated intothe lipid bilayer of the liposome (Figure 1) [19–21]. Encap-sulation of enzymatic antioxidants (superoxide dismutaseand catalase) or nonenzymatic antioxidants (glutathione, N-acetylcysteine, CoQ10, curcumin, resveratrol, α-tocopherol,and γ-tocopherol) in liposomes improves their therapeuticpotential against oxidant-induced tissue injuries, becauseliposomes apparently facilitate intracellular delivery and pro-long the retention time of entrapped agents inside the cell.
2. Oxidative Stress and the AntioxidantDefense System
The involvement of oxidants in several pathological dis-orders, including cancer, diabetes, cardiovascular diseases,

2 Journal of Toxicology
chronic inflammatory disease, postischaemic organ injury,neurodegenerative disorders, and xenobiotic/drug toxicityhas been widely accepted, but whether such oxidants arethe major cause of tissue injury in human disease orsimply produced during the development of injury is stillunder debate [1–8]. Regardless, the interaction of reactiveoxygen species (ROS) with macromolecules, organelles, andtissues results in damage. For example, membrane lipidoxidation may result in an irreversible injury to the plasmamembrane [22]. Protein oxidation may lead to a loss ofcritical sulphydryl groups in addition to modifications ofamino acids leading to the formation of carbonyl and otheroxidized moieties [23]. Attack of ROS on nucleic acids cangenerate various modified bases in DNA which play a criticalrole in carcinogenesis and aging [1].
In general, oxidative stress describes the result of anincreased ROS production and/or a decrease in their elim-ination [24]. Such ROS include, but certainly not limitedto, superoxide anion, hydrogen peroxide, hydroxyl radical,and lipid peroxides [25, 26]. Some of these species, suchas superoxide or hydroxyl radicals, are extremely unstable,whereas others, like hydrogen peroxide, are freely diffusibleand relatively long lived. The sources of ROS may be exoge-nous or endogenous [2, 11]. During their metabolism, agentssuch as paraquat or doxorubicin generate ROS resultingin pneumotoxicity or cardiotoxicity, respectively [4, 27].Intrinsically generated oxidants may be derived from theelectron transport chain in the mitochondria, with the mostimportant source of damaging oxidants being the phagocyticcells (residential macrophages and recruited neutrophils)that can generate ROS and reactive nitrogen species fromthe NADPH oxidase assembly located on their cell surfaces[2, 28, 29]. Other pathways that may participate in theformation of ROS include xanthine oxidase in ischemia-reperfusion and cytochrome P-450-dependent activation ofxenobiotics [11, 26].
Sequential reduction of molecular oxygen leads to theformation of superoxide anion, hydrogen peroxide, andhydroxyl radical. The univalent reduction of molecularoxygen forms superoxide anion in a reaction mediated byenzymes such as xanthine oxidase and NAD(P)H oxidaseor nonenzymatically by redox-reactive compounds suchas the semiubiquinone compound of the mitochondrialelectron transport chain [11, 26, 30]. Superoxide dismutasesconvert the superoxide anion into hydrogen peroxide whichin the presence of reduced transition metals (e.g., ferrousor cuprous ions) is metabolized to the highly reactivehydroxyl radical [30]. Other reactive species include nitricoxide, lipid radicals, peroxynitrite, and hypochlorous acid[11, 29]. Nitric oxide is synthesized from the guanidinegroup of L-arginine by a family of enzymes termed nitricoxide synthases. Simultaneous generation of nitric oxide andsuperoxide anion favors the production of a strong oxidant,the peroxynitrite anion, which may account for some of thedeleterious effects associated with nitric oxide production[31]. All these reactive oxygen species are potentially veryreactive molecules and, at high enough concentration, resultin damage to critical cellular constituents (such as proteins,
Hydrophobic
space(shaded area)
Hydrophilic
space
N-acetylcysteine
-tocopherol
Figure 1: Schematic representation of a liposome containingantioxidants. Liposomes are artificially prepared vesicles madeof lipid bilayer. Water-soluble compounds (e.g., N-acetylcysteine(NAC)) can be encapsulated in the aqueous phase, while lipid-soluble compounds (e.g., α-tocopherol) can be incorporated intothe lipid bilayer of the liposome.
nucleic acids, carbohydrates, and lipids) resulting in cellnecrosis [11, 26, 28, 29].
At physiological low levels, ROS function as “redoxmessengers” in intracellular signaling and regulation. Redoxsignaling is a well-recognized stress response that leadsto a variety of downstream effects including increasedexpression of protective and repair enzymes [32, 33]. Thereis also mounting evidence that redox signaling is part ofnormal metabolism in nonstressed cells. In this situation,endogenously generated oxidants act as second messengersfor receptor agonists such as growth factors and hormones,signaling the proliferative or metabolic changes associatedwith these ligands [32, 33]. Oxidants can activate andinactivate transcription factors, membrane channels, andmetabolic enzymes, and modulate calcium-dependent andphosphorylation signaling pathways. These processes incor-porate the major regulatory networks of cells, giving redoxsignals the capacity to stimulate and adjust most aspects ofcell physiology [34].
Exposure to reactive oxygen species from a variety ofsources has led organisms to develop a series of defensemechanisms [9, 35]. The harmful effects of ROS are coun-terbalanced by the antioxidant action of both antioxidantenzymes and nonenzymatic antioxidants [30]. Antioxidantshave been defined as substances that are able to prevent,delay, or remove oxidative damage to a molecule [36] andmay be categorized in the following groups: (i) those aimedat preventing the generation and distribution of reactiveoxygen and nitrogen species (e.g., the effective controlof iron distribution and the destruction of peroxides bycatalase or by glutathione peroxidase), (ii) those aimed atreactive metabolite scavenging including the maintenance ofeffective levels of antioxidants, such as vitamin E, vitamin C,β-carotene, and glutathione as well as the enzyme superoxide

Journal of Toxicology 3
dismutase, and (iii) those aimed at free radical repair,particularly the maintenance of effective levels of glutathione[4, 10, 11, 36].
The antioxidant enzymes found in living cells includethe superoxide dismutase, catalase, glutathione peroxidase,peroxiredoxins, and thioredoxin reductase [4, 10, 11, 13,25, 36–38]. These antioxidant enzymes are not consumedduring their catalytic actions, and they have high affinityand rate of reaction with ROS consequently allowing moreeffective protection against acute massive oxidative insults.Superoxide dismutases (SODs) are metalloenzymes thatcatalyze the conversion of superoxide anion to hydrogenperoxide and include the cytosolic copper and zinc con-taining form (Cu-Zn-SOD), the mitochondrial manganese-containing SOD (MnSOD), and an extracellular form (EC-SOD) [39]. Catalase, which catalyzes the detoxication ofhydrogen peroxide to water and oxygen, occurs abundantlyin the body, with the highest activity in the liver, followed byerythrocytes, then the lungs [13, 25]. Glutathione peroxidaseplays a major role in the detoxication of hydrogen peroxide,other hydroperoxides, and lipid peroxides via the glutathioneredox cycle [11, 25]. The thioredoxin system, consistingof thioredoxin reductase in conjunction with thioredoxinand NADPH, is a ubiquitous oxidoreductase system withantioxidant and redox regulatory roles; mammalian thiore-doxin reductase has a highly reactive active site selenocys-teine residue resulting in a profound reductive capacity,reducing several substrates in addition to thioredoxin [37].Peroxiredoxins are ubiquitous thiol-dependent peroxidasesthat accomplish the same function as other antioxidantenzymes such as catalase and glutathione peroxidase, buttheir catalytic activity is lower than that of these enzymes.Peroxiredoxin 6 is a bifunctional protein with both GSH per-oxidase and phospholipase A2 activities that play importantphysiological roles in antioxidant defense and lung surfactantmetabolism [38, 40].
Nonenzymatic antioxidative systems are not as specificas enzymatic ones, but they serve as the first line of defenseand are, therefore, of high importance in cellular responseto oxidative stress. The major nonenzymatic antioxidantsinclude vitamin C (ascorbic acid), vitamin E (tocopherolsand tocotrienols), glutathione, carotenoids, flavonoids, andother micronutrients. Vitamin C, a water-soluble vitamin,is effective in scavenging ROS, including hydroxyl radical,aqueous peroxyl radicals, and superoxide anion [10]. Vita-min E, the principal antioxidant in the body, is composedof four tocopherols and four tocotrienols, and, due toits lipophilicity, it is present in all cellular membranes[41]. The antioxidant functions of vitamin E include thetermination of chain reactions in polyunsaturated fatty acidsin cell membranes from peroxidation by scavenging peroxylradicals in membranes and neutralizing the highly reactivesinglet oxygen molecules [42]. Glutathione (GSH) is themost abundant nonprotein thiol in living organisms andcan exert its protective effects by acting as a nucleophile toform conjugates with many xenobiotic compounds and/ortheir metabolites and serve as a reductant in the metabolismof hydrogen peroxide and other organic hydroperoxides,a reaction catalyzed by glutathione peroxidases found in
cytosols and mitochondria of various cells [43, 44]. Inhumans, a number of neurodegenerative and neuropsy-chiatric conditions are associated with disturbances inglutathione [45]. Repletion and maintenance of neuronalGSH is, therefore, important to cell health and viability andcould provide therapeutic benefit in situations when GSHis deficient. Carotenoids are a class of natural fat-solublepigments found principally in plants, the most abundantin the diet being beta-carotene, lycopene, lutein, beta-cryptoxanthin, zeaxanthin, and astaxanthin. The antioxidantactions of carotenoids are based on their singlet oxygenquenching properties and their ability to trap peroxyl radicals[46, 47]. Flavonoids (i.e., quercetin) and phenolic acids (i.e.,resveratrol) are polyphenolic substances that have chemicalstructures supporting the scavenging of free radicals and thechelation of redox-active metals [48].
3. Antioxidant Therapy
Antioxidant therapy has been defined as any treatment thatprevents or decreases the adverse effects of oxidants. One ofthe strategies for pharmacological modification of oxidant-mediated tissue injury focuses on increasing the antioxidantcapacity of cells or preventing the generation of ROS [12, 13].The effectiveness of exogenous antioxidants to protect tissuesfrom oxidant stress in vivo depends on the antioxidant used,its physicochemical and biopharmaceutical properties, itsavailability at the site of action, and, the nature of the oxidantstress [12, 13].
Only a few available drugs have been documented topossess antioxidant properties that might contribute to theirefficacy. For example, N-acetylcysteine is used as a mucolyticagent and in the treatment of acetaminophen poisoning [86],deferoxamine is used as a chelating agent in iron overload[87], and probucol, calcium antagonists, β-adrenoreceptorantagonists, antiarrhythmic drugs, and statins are used inthe treatment of cardiovascular diseases [88, 89]. Most ofthe studies examining the prophylactic or therapeutic effectsof antioxidants against several diseases and disorders haveresulted from epidemiological studies in ethnic groups whohave different lifestyles and have been exposed to differentenvironmental factors [13]. For example, the use of redwine in the diet of French people is associated with lowerincidences of cardiovascular disease [13]. Also, the highlevel of antioxidants in the Mediterranean diet has alsobeen associated with lower incidences of cardiovasculardisease and morbidity and mortality [13]. Other studieshave focused on the use of antioxidants by patients asdietary supplements in the hope of maintaining health,preventing disease, or reducing the toxicity of chemotherapyand radiotherapy, but the results so far are not conclusive[12, 13, 90].
The benefit of dietary antioxidants has been exploredin both in vitro and in vivo systems. Most antioxidants,when applied directly and at relatively high concentrations tocellular systems in vitro, are effective in conferring protectionagainst oxidant insults. On the other hand, results fromstudies in animals and humans have revealed that severalantioxidants provide only modest benefit and have yet to

4 Journal of Toxicology
Neutral lipids
Dissolved inorganic solvent
Evaporatedunder heat
Dry lipid film
Sonication
NAC andbuffer added
Multi- and uni-lamellar vesicles
Figure 2: Preparation of liposomal-N-acetylcysteine (L-NAC). L-NAC is prepared from a mixture of DPPC and NAC in a 1 : 1 molarratio by a dehydration-rehydration method. The lipids are dissolvedin chloroform in a round-bottomed flask and dried at 45◦C with arotary evaporator. The lipid film is dried with nitrogen to eliminatetraces of chloroform and hydrated with a solution of NAC andsubsequently sonicated. Sonication is a simple method for reducingthe size of liposomes. Upon rehydration, free NAC is separatedby high-speed centrifugation (24400 g at 4◦C, for 30 min), a stepperformed twice. At the end of this procedure, the liposomal vesiclesize is usually below 200 nm mean diameter with an encapsulationefficiency of 35% NAC.
be rendered into reliable and safe antioxidant therapies. Forexample, exposure of animals to the antioxidant enzymessuperoxide dismutase and catalase has been encounteredwith antigenicity and immunogenicity problems [91, 92],and both of these enzymes are poorly absorbed from andrapidly degraded in the gastrointestinal tract [13]. Otherstudies have shown that flavonoids possess potent antiox-idant activity in vitro, but their antioxidant action in vivohas been challenged by studies examining the bioavailabilityof flavonoids, which indicate that they reach only very lowconcentrations in human plasma after the consumptionof flavonoid-rich foods [93]. In addition, most flavonoidsare extensively metabolized in vivo, which can affect theirantioxidant capacity [93]. It is obvious that the failure ofantioxidants to seriously modify the injurious actions ofoxidants is primarily attributed to their physicochemicalcharacteristics and/or pharmacokinetic properties withoutexcluding other possibilities such as some antioxidants alsohave prooxidant activities, particularly in the presence oftransitional metals [94–97]. Experimental evidence, there-fore, supports the need for the development of formulationsthat would enhance the delivery and retention of antioxi-dants in the tissues. Strategies to improve the effectivenessof antioxidants are focused on chemical modifications ofantioxidants (e.g., attachment of masking pegylated (PEG)-groups), coupling of antioxidants to affinity carriers (e.g.,antibodies against cellular adhesion molecules) and drug-delivery systems such as micelles and liposomes [4, 13–15, 17]. In this paper, we will address the role of liposomesas a delivery system for antioxidants.
4. Liposomes
Liposomes are nanosized artificial vesicles of spherical shapethat can be produced from natural or synthetic phospho-lipids. The polar head groups are located at the surface of themembranes, in contact with the medium, whereas the fatty
acid chains form the hydrophobic core of the membranes,shielded from the water. Polar molecules can be encapsulatedin the aqueous core, while hydrophobic molecules dissolvein the bilayers of liposomes [19]. Lipid film hydration isthe simplest method for the encapsulation of water-solubledrugs; a lipid film is hydrated with an aqueous solution con-taining the drug (Figure 2). Entrapment efficiency may varydepending on: (i) the compound itself (charge, size, solubil-ity, etc.), (ii) the lipid variations (a single type or a mixtureof lipids, composition, charge, etc.), and (iii) the prepara-tion methods. The manipulation and design of liposomesendowed with the ability for targeting specific cell sites (oralternately, the temporary avoidance of these sites), resultsin long circulation, increased biodistribution, and favourablepharmacodynamics [98–100]. Currently, a number of lipo-some formulations are in clinical use to combat cancer andinfectious diseases, while others await clinical trial outcomes.For example, while DaunoXome, Doxil, and Ambisomeare currently clinically approved, CPX-1 and LE-SN38 areexamples of liposomal-based drugs that encapsulate a topoi-somerase I inhibitor and are currently in Phase-II clinicaltrials for the treatment of colon or colorectal cancer [100].Some of the setbacks and disadvantages of liposomes includetime-consuming preparation techniques, low entrapmentvolumes, and toxicity concerns due to the presence ofresidual toxic organic compounds during preparation [98].
The lipids used for the preparation of liposomesare predominantly phospholipids or surfactants whichform bilayers similar to those found in biological mem-branes. The surfactants dimyristoyl-phosphatidyl-glycerol(DMPG), dimyristoyl-phosphatidylcholine (DMPC), dipal-mitoyl-phosphatidylcholine (DPPC), and desaturated-phos-phatidylcholine (DSPC) are naturally occurring but can beproduced synthetically as well. Extensive testing of thesephospholipids has revealed them to be remarkably safefor pharmaceutical use. For example, phosphatidylcholine(i.e., DPPC) (a phospholipid that carries no net charges, isthe major constituent of cell membranes, and it providesa structural framework for the membrane and maintainsthe permeability barrier) is well tolerated in animal andhuman studies. Inhaled nondrug containing liposomes (15and 150 mg of lipid DPPC/mL) for 1 hour on pulmonaryfunction and on oximetry in healthy nonsmoking volunteersshowed that liposome inhalation is well tolerated, and nooxygen desaturation, decrements in pulmonary function, orside effects were noted [101]. Other investigators have uti-lized fluorescein-labelled liposomes to examine the clearanceof aerosolized liposomes from the lungs of human volunteersand reported no untoward side effects [102]. Intravenousadministration of DPPC (5 to 50 mg of lipid) did not induceimmediate or delayed toxicity in mice and did not produceany changes in body weight and weight of major organs 2weeks after administration. It has been reported that thetoxicity of intravenously administered liposomes composedof DPPC is so low that accurate assessment of an LD50 valueis difficult and has been estimated to be of the order of10 g/kg in mice [103]. However, it is important to note thataddition of other constituents to liposomes in order to alter

Journal of Toxicology 5
stability or kinetics can result in an increase in toxic potential,particularly on parenteral administration of liposomes [104].
A potential problem with conventional liposomes, par-ticularly when delivered intravenously, is their rapid removalfrom circulation by cells of the reticuloendothelial system(RES) particularly in the liver and spleen [14, 105]. Tocircumvent the phagocytic cells of the immune system andhence enhance their half-life in the circulation, “stealthliposomes” have been designed [14, 105]. Stealth liposomesare created by coating the liposomes with a layer ofpolyethylene glycol-phosphatidylethanolamine (PEG lipo-somes). PEGylation is the process of covalent attachment ofpolyethylene glycol polymer to another molecule maskingthe agent from the host’s immune and metabolic systemsand creating a shield around the pegylated agent due to itslarge hydrodynamic volume, thus protecting it from renalclearance and consequently prolonging its circulatory time[14, 105, 106].
5. Liposomes as an Antioxidant Delivery System
Liposomes have been considered to be excellent models ofcell membranes and have been used for the evaluation of theantioxidant properties of several lipophilic and hydrophilicantioxidants against oxidant insults. As a drug deliverysystem, liposomes have been used for the transport of water-soluble and lipid-soluble antioxidants as well as antioxidantenzymes to different organs and tissues for the treatmentof oxidative stress-induced damage. Up-to-date, delivery ofmolecules with antioxidant properties to different organsand tissues include the lipophilic antioxidants α-tocopherol[107, 108] and CoQ10 [13, 109, 110], the hydrophilicantioxidants glutathione, N-acetylcysteine [64, 111, 112],and quercetin [13, 83, 113], and the antioxidant enzymessuperoxide dismutase and catalase [4, 14] (Table 1). Lipo-somes can facilitate intracellular delivery of several thera-peutic agents via fusion with the plasma membrane lipids,receptor-mediated endocytosis, and phagocytosis [114–116].
5.1. Liposomal Superoxide Dismutase (SOD) and/or Catalase(CAT). The effectiveness of the antioxidant enzymes, SODand CAT, in the treatment of oxidant-induced injuriesis limited because of their unfavorable physicochemicalproperties [13, 91, 92, 117, 118]. SOD is unable to cross cellmembranes due to its high molecular mass (which preventsintracellular transport) or its charge (which prevents itsadherence to targets) and possess immunogenic properties[15, 16, 91]. To enhance their effectiveness, polyethyleneglycol was attached to antioxidant enzymes (pegylated SODand/or pegylated CAT), an approach shown to increasetheir in vivo half-lives, cellular uptake by endothelial cells,and effectiveness in preventing pulmonary oxygen toxicityin rats [119, 120]. However, when compared to liposomalentrapped SOD and CAT, PEG antioxidant enzymes are lessprotective against lung injury from continuous hyperoxia;endothelial cells treated with liposomal entrapped SOD and
CAT increase the activity of these enzymes by 44-fold within2 hours [119, 121]. Another strategy involves the conjugationof SOD and CAT with an antibody to platelet-endothelial celladhesion molecule-1 (PECAM-1) which bind to endothelialcells and alleviate oxidative stress in cell culture models aswell as a mouse model of vascular oxidative stress [122–124].Such validated strategies may be used effectively to developfuture liposomal therapeutics by conjugating antibodies andother molecules to liposomes in order to provide specifictargeting to endothelial cells or any other cell type [125, 126].
The pharmacokinetic profile of the antioxidant enzymesalso provides hindrance in their effective use in the treatmentof oxidant injuries. Intravenously administered SOD showsa biological half-life of a few minutes, and enteral admin-istration is ineffective due to biodegradation of the enzymein the gastrointestinal system [15, 16, 118]. On the otherhand, systemic administration of liposomally encapsulatedsuperoxide dismutase and catalase prolongs their circulatinghalf-life and enhances their protection in animal models ofoxidant-induced organ injuries [49, 57, 58]. For example,the short half-lives of native SOD and catalase in circulatingblood and their inability to cross the blood-brain barrierlimit their therapeutic usefulness in treating ischemic braininjury. Intravenous administration of Cu, ZnSOD liposomesfacilitates the delivery of SOD into the brain, not only inthe infarct but also in the noninfarcted subcortical area,resulting in significantly elevated levels of SOD activityand protection against cerebral ischemia/reperfusion injury[53–55]. Also, the addition of polyethylene glycol to thesurface of the liposomes gives the liposomes a hydrophilic“sterically stabilized” surface, a property that contributes to alower affinity of macrophages of the mononuclear phagocytesystem (MPS) for the circulating liposome particles and con-sequently to an even extended prolonged blood circulation(more than 5-fold prolongation of liposome circulation timein blood) [127]. Systemic administration of PEG-liposomesfor targeting SOD to arthritic sites with that of non-PEG-liposomes containing stearylamine in rats with adjuvantarthritis demonstrate that PEG-liposomes are superior withrespect to circulation time and extent of localization atarthritic sites [50].
Intratracheally administered surfactant liposomes, en-capsulating CuZn-superoxide dismutase and catalase, in-creases the alveolar type II cell antioxidant activity andprotects cells against oxidant stress in the lungs of adult,premature or newborn animals [59–61, 65]) or againstbleomycin-induced lung injury [62, 63]. The coinstillationwith 2-chloroethyl ethyl sulfide (CEES) of liposomes con-taining pegylated- (PEG-) catalase (CAT), PEG-superoxidedismutase (SOD), or the combination, greatly attenuatedthe development of lung injury [64]. Results from a limitednumber of clinical studies suggest that liposomal superoxidedismutase might protect against radiation-induced fibrosis[51, 52]. The biological half-life of recombinant humanCu/Zn SOD (rhSOD) within the systemic circulation byliposomal encapsulation and aerosolization into the lungs ofpigs leads to long-term and uniform uptake into systemiccirculation without acute deleterious effects on respiratorytract suggesting that aerosolization of liposomal rhSOD
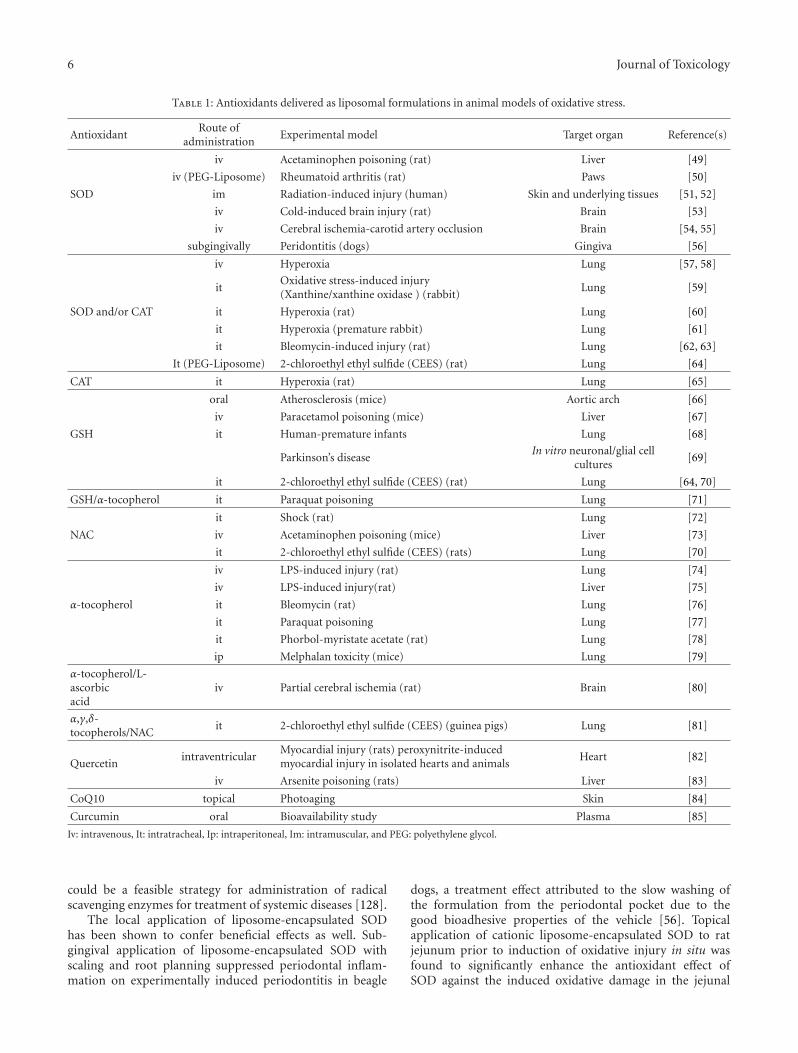
6 Journal of Toxicology
Table 1: Antioxidants delivered as liposomal formulations in animal models of oxidative stress.
AntioxidantRoute of
administrationExperimental model Target organ Reference(s)
iv Acetaminophen poisoning (rat) Liver [49]
iv (PEG-Liposome) Rheumatoid arthritis (rat) Paws [50]
SOD im Radiation-induced injury (human) Skin and underlying tissues [51, 52]
iv Cold-induced brain injury (rat) Brain [53]
iv Cerebral ischemia-carotid artery occlusion Brain [54, 55]
subgingivally Peridontitis (dogs) Gingiva [56]
iv Hyperoxia Lung [57, 58]
itOxidative stress-induced injury(Xanthine/xanthine oxidase ) (rabbit)
Lung [59]
SOD and/or CAT it Hyperoxia (rat) Lung [60]
it Hyperoxia (premature rabbit) Lung [61]
it Bleomycin-induced injury (rat) Lung [62, 63]
It (PEG-Liposome) 2-chloroethyl ethyl sulfide (CEES) (rat) Lung [64]
CAT it Hyperoxia (rat) Lung [65]
oral Atherosclerosis (mice) Aortic arch [66]
iv Paracetamol poisoning (mice) Liver [67]
GSH it Human-premature infants Lung [68]
Parkinson’s diseaseIn vitro neuronal/glial cell
cultures[69]
it 2-chloroethyl ethyl sulfide (CEES) (rat) Lung [64, 70]
GSH/α-tocopherol it Paraquat poisoning Lung [71]
it Shock (rat) Lung [72]
NAC iv Acetaminophen poisoning (mice) Liver [73]
it 2-chloroethyl ethyl sulfide (CEES) (rats) Lung [70]
iv LPS-induced injury (rat) Lung [74]
iv LPS-induced injury(rat) Liver [75]
α-tocopherol it Bleomycin (rat) Lung [76]
it Paraquat poisoning Lung [77]
it Phorbol-myristate acetate (rat) Lung [78]
ip Melphalan toxicity (mice) Lung [79]
α-tocopherol/L-ascorbicacid
iv Partial cerebral ischemia (rat) Brain [80]
α,γ,δ-tocopherols/NAC
it 2-chloroethyl ethyl sulfide (CEES) (guinea pigs) Lung [81]
Quercetinintraventricular
Myocardial injury (rats) peroxynitrite-inducedmyocardial injury in isolated hearts and animals
Heart [82]
iv Arsenite poisoning (rats) Liver [83]
CoQ10 topical Photoaging Skin [84]
Curcumin oral Bioavailability study Plasma [85]
Iv: intravenous, It: intratracheal, Ip: intraperitoneal, Im: intramuscular, and PEG: polyethylene glycol.
could be a feasible strategy for administration of radicalscavenging enzymes for treatment of systemic diseases [128].
The local application of liposome-encapsulated SODhas been shown to confer beneficial effects as well. Sub-gingival application of liposome-encapsulated SOD withscaling and root planning suppressed periodontal inflam-mation on experimentally induced periodontitis in beagle
dogs, a treatment effect attributed to the slow washing ofthe formulation from the periodontal pocket due to thegood bioadhesive properties of the vehicle [56]. Topicalapplication of cationic liposome-encapsulated SOD to ratjejunum prior to induction of oxidative injury in situ wasfound to significantly enhance the antioxidant effect ofSOD against the induced oxidative damage in the jejunal

Journal of Toxicology 7
mucosa, compared with their free forms; this effect wascaused by the increased mucosal adhesion of the liposomeswhich potentially provide protection for the SOD againstproteolysis and premature scavenging by mucin components[129]. In another study, topical application of liposomal-encapsulated SOD reduced postburn wound size and edemaformation for the reason that liposomes enhanced theantioxidant effect of SOD against the neutrophil-mediatedoxidant injury by promoting the controlled sustained releaseof SOD at the site of injury [130].
5.2. Liposomal Glutathione. Glutathione in its reducedform (GSH) is the most powerful intracellular antioxidant.Decreases in circulating and intracellular concentrations ofGSH levels can result in a lowered cellular redox potentialinfluencing the translocation of the transcription factor NFkB which regulates the synthesis of cytokines and adhesionmolecules [131]. One possibility to protect cells fromdamage caused by reactive oxygen species is to restore theintracellular glutathione levels. Cellular GSH concentrationcan be influenced by exogenous administration of GSH (asintravenous infusion or as aerosol) or glutathione esters[131]. The systemic bioavailability of orally administeredGSH and glutathione monoethyl ester (GSHE) in the rat islow without affecting the circulating concentrations of GSHand cysteine, while intravenous administration of GSH or itsester results in rapid elimination albeit slower for the GSHE[132, 133]. Dietary glutathione is not possible to increasecirculating glutathione to a clinically beneficial extent by theoral administration of a single dose of glutathione (3 grams)because of hydrolysis of glutathione by intestinal and hepaticgamma-glutamyltransferase [134]. However, consumptionof liposomal glutathione (50 mg/kg/day) by atheroscle-rotic apolipoprotein E-deficient (E0) mice (which developatherosclerotic lesions with many features common withhuman lesions) for 2 months significantly reduced serumand macrophage oxidative stress, macrophage cholesterolmass, and as a result of these effects, significantly attenuatedatherosclerosis development when compared to mice thatconsumed similar concentration of control liposomes [66].In another study, glutathione injected intravenously 2 hoursbefore acetaminophen administration as a liposomal formu-lation was more effective than soluble GSH in protectingagainst drug-induced liver necrosis lipid peroxidation, andhepatic glutathione depletion [67].
In animal studies where GSH is administered intratra-cheally, only 1%-2% of the dose administered remained inthe lung 24 hours after treatment, while liposome encapsu-lation improved the pulmonary retention of GSH, with 18%and 10% of the dose administered remaining in the lung 24 hand 48 h after treatment [135, 136]. Accordingly, Suntres andShek [71] showed that intratracheal instillation of liposome-entrapped GSH yielded a better protection than free GSHagainst paraquat-induced lung injury. The improved pro-tection conferred by the liposomal GSH formulation wasattributed to the extended retention of liposomes in the lung,thus allowing a slow release of its GSH content [71]. Inanother study, Smith et al. [136], showed that intratrachealinstillation of liposomal GSH was better than free GSH
in protecting against hyperoxia-induced lung injury [136].Intratracheal liposomal glutathione instillation in ventilatedpreterm infants raised the pulmonary glutathione and signif-icantly reduced the levels of lipid peroxidation products [68].
It has been shown that liposomal GSH can be utilized forrepletion and maintenance of intracellular GSH in neuronalcells and that liposomal GSH can provide significant protec-tion to neurons in a model system relevant to Parkinson’sdisease [69]. Neurons, like most other cells, do not possesstransport mechanisms for GSH and elevation of extracellularGSH may pose potential toxicity problems that increase neu-ronal vulnerability during ischemia, while the encapsulationof GSH into lipid vesicles may avoid the potential toxicityto neurons associated with extracellular GSH elevation andmay facilitate drug delivery to cells as has been shown forother liposomal preparations [137, 138]. The concentrationneeded for half maximal repletion in mixed mesencephaliccultures containing approximately 70% neurons and 30%glia was 100-fold less when GSH was encapsulated into lipo-somal vesicles (4.75 μM for liposomal GSH versus 533 μM fornonliposomal fully reduced GSH) [69].
6. Liposomal N-Acetylcysteine
N-acetylcysteine (NAC), a thiol-containing compound,when administered in its conventional form did not protectagainst the prolonged shock-induced acute lung injury butwhen administered as a liposomal formulation directly tothe lungs of animals protected against the lung injury[72]. The protective effect conferred by the liposomal NACoccurred at low NAC doses [72], since liposomes areknown to prolong the retention of the antioxidant in thelungs. L-NAC was also shown to have a prophylactic effectagainst both LPS-induced lung injuries [112] and LPS- oracetaminophen-induced hepatotoxicity in animals [73, 111].N-Acetylcysteine is a drug used in the clinic for the treatmentof acetaminophen-induced hepatotoxicity and as a mucolyticagent. It possesses free radical-scavenging properties [4,139] which are attributed to the nucleophilicity and redoxinteractions of its thiol group [139, 140]. Additionally, NACis a source of cysteine, often the limiting precursor of denovo GSH synthesis [139, 141, 142]. Furthermore, NAC hasbeen shown to influence redox-sensitive cell-signaling andtranscription pathways, such as NF-κB (which regulates pro-inflammatory genes), and the p38, ERK1/2, SAPK/JNK, c-Jun, and c-Fos pathways, among others, in a wide varietyof different systems [139, 143]. N-Acetylcysteine has beenshown to promote cell growth and survival by activating theMAPK pathway in response to ROS-induced injuries (whichnormally lead to growth arrest and apoptosis) and is ableto limit inflammatory processes, such as the release of pro-inflammatory cytokines [144].
The use of L-NAC was also investigated in half sulfurmustard-induced acute lung injury in rats [64, 70] andguinea pigs [108]. McClintock et al. [64] and Hoesel etal. [70] found that the effects of airway instillation of 2-chloroethyl ethyl sulfide (CEES), which has been shownto induce acute lung injury (assessed by the leakage ofplasma albumin into the lung) in rats, were attenuated

8 Journal of Toxicology
with immediate and 1-hour-delayed instillation of lipo-somes containing reducing agents (i.e., NAC, GSH, orresveratrol) or bifunctional liposomes (containing NAC andGSH). Additionally, Hoesel et al. [70] found that airwayinstillation of L-NAC was protective when administered4 hours following CEES application in rats, as well as 3weeks after CEES with bifunctional α-tocopherol and NACliposomes, though not with L-NAC alone after 3 weeks. Also,the intratracheal administration of liposomal antioxidantformulation containing alpha/gamma-tocopherol alone orwith NAC immediately after instillation of CEES attenuatedthe short-term as well as long-term (fibrotic) effects of CEES-induced lung injury [64].
Irrespective of the route of administration, liposomalNAC was far superior to the conventional NAC. It has beenshown that intravenous administration of NAC alone at adose of 25 mg/kg was not effective in conferring any signifi-cant protection against LPS-induced hepatotoxicity while anequivalent dose of NAC delivered as a liposomal formulationconferred protection. This is not surprising, because moststudies have shown that intravenous administration ofconventional NAC preparations are demonstrated to beeffective against animal models of sepsis or endotoxemiaat doses greater than 50 mg/kg, iv [145–148]. Results frompharmacokinetic studies have shown that following intra-venous administration, NAC undergoes rapid and extensivemetabolism in the liver resulting in bioavailability of about10% [149, 150]. This finding further supports the role ofliposomes in their use as a drug delivery system because oftheir capability of delivering hepatoprotective agents to theliver, an ideal approach to increase local concentration ofthe agent, to reduce adverse effects, and to achieve maximaltherapeutic efficiency. In a separate study examining thesafety and pharmacokinetics of L-NAC in control rats, ithas been demonstrated that the half-life of intravenouslyadministered L-NAC is significantly increased by 4-fold from6 min to 30 min (unpublished observation). Similarly, resultspresented by other investigators have shown that intravenousadministration of drugs as liposomal formulations prolongtheir circulation time in blood and increase their distributionto major organs, including the lung [4, 13].
6.1. Liposomal Tocopherols. α-Tocopherol, the major com-ponent of Vitamin E, is a lipid-soluble hydrocarbon com-pound that partitions into lipid storage organelles and cellmembranes. It is an efficient scavenger of lipid peroxylradicals and, hence, it is able to break peroxyl chain prop-agation reactions in cellular membranes preventing lipidperoxidation [151, 152]. In studies with model membranesystems, α-tocopherol intercalates into phospholipid bilayerswith the long axis of the molecule oriented parallel to thelipid hydrocarbon chains, and it is able to rotate aboutits long axis and diffuse laterally within fluid lipid bilayers[152, 153]. Because of its membrane stabilizing effect, α-tocopherol has been used in the preparation of liposomesfor the delivery of several drugs [84, 154]. The maximumamount of α-tocopherol that can be contained in eggphosphatidylcholine or phosphatidylcholine liposomes isapproximately 33% α-tocopherol. α-Tocopherol alters the
membrane characteristics of liposomes by making themmore stable and less permeable to aqueous solutes and highlyresistant to protein-induced disruption [155]. The suppres-sion of protein-induced disruption is more pronouncedwith α-tocopherol than with cholesterol (used in liposomalpreparation to increase the physical stability of liposomesparticularly in the presence of biological fluids such asplasma), even at lower molar ratios. Also, α-tocopherol inliposomes can undergo spontaneous intermembrane transferto an acceptor membrane without the fusion of the withα-tocopherol liposomes [156]. Thus, liposomes containingα-tocopherol (15 to 30 mol%) may be useful for deliveringphysiological quantities of this vitamin component or otherdrugs to cells in culture or to tissues in vivo.
Many studies have clearly demonstrated the impor-tant role of α-tocopherol in modulating oxidant-inducedcellular injury, permitting cells with high levels of theantioxidant to become more resistant to oxidative insults.It has been shown that the administration of vitamin E,prior to an oxidative challenge, reduces the level of lipidperoxidation in several tissues and improves survival [152,157–159]. However, in contrast to our studies [4, 74–78,160], where the intratracheal instillation of α-tocopherolliposomes conferred a significant protective effect againstthe acute lung injuries and inflammation induced byseveral chemicals including paraquat, bleomycin, phorbolmyristate acetate, and lipopolysaccharide (LPS), the oral orparenteral administration of conventional α-tocopherol toanimals offered limited or no protection against paraquator lipopolysaccharide-induced lung damage [161–165]. Inour studies, the intratracheal instillation of α-tocopherolliposomes achieved a substantially higher antioxidant levelin the lung, approximately 1 mg/g lung weight [166], whilethe amount of antioxidant recovered from the lungs ofanimals after oral or parenteral administration of vitaminE was most likely less than 40 μg/g lung tissue [167, 168].Vitamin E, used as oral supplements, is often in the formof tocopheryl esters which are highly stable to oxidationbut are absorbed only after they have been unesterified bythe intestinal esterases. The active form of α-tocopherol ishighly viscous oil, practically insoluble in water and readilyoxidized by atmospheric oxygen. In our laboratory, attemptswere made to deliver α-tocopherol to the lungs of animalsby other means, but results from these studies showed thatorganic solvents, such as ethanol and dimethylsulphoxide,used to solubilize the viscous free α-tocopherol, were toxicto the lung, and emulsifying agents and detergents, such aspolyethylene glycol and Tween 80, caused respiratory failure,possibly by disrupting surface tension of the lung [166]. Anintravenous form of vitamin E, E-ferol, that contained dl-(α)-tocopherol and polysorbate 80 was first clinically used,albeit without detailed safety tests, in the early 1980s in low-birth-weight and premature infants for the treatment of theretrolental fibroplasias and resulted in severe adverse effects,including hepatomegaly and renal failure most likely due tothe excipient polysorbate 80 [169].
The effectiveness of α-tocopherol-loaded liposomes hasbeen examined in tissue injuries other than the lung as

Journal of Toxicology 9
well. Sinha et al. 2001 [80] showed that α-tocopherol-loaded liposomes were protective against an experimental invivo rat model of global cerebral ischemia controlling moreeffectively the conjugated diene increment in the brain ofrats after ischemia and reperfusion of rats [80]. Liposomalα-tocopherol formulations have been proven to be effectivein tissue injuries induced by alkylating agents such asmelphalan, 2-chloroethyl ethyl sulfide (CEES), [79, 108].
7. Other Liposomal Antioxidants
7.1. Liposomal Coenzyme Q. Coenzyme Q (CoQ) is anaturally occurring compound also known as ubiquinone.It belongs to a homologous series of compounds thatshare a common benzoquinone ring structure but differin the length of the isoprenoid side chain; in humansand a few other mammalian species, the side chain iscomprised of 10 isoprene units, hence it is called coenzymeQ10 (CoQ10). CoQ10 has a fundamental role in cellularbioenergetics as a cofactor in the mitochondrial electrontransport chain (respiratory chain) and is, therefore, essentialfor the production of ATP. CoQ10 in its reduced form asthe hydroquinone (called ubiquinol) is a potent lipophilicantioxidant and is capable of recycling and regeneratingother antioxidants such as tocopherol and ascorbate [170,171]. Data from several laboratories have demonstratedthat CoQ10 pretreatment protects the myocardium fromischemia-reperfusion injury via both antioxidant and bioen-ergetic pathways [110]. However, the high molecular weightand lipophilicity of CoQ10 makes it poorly water solubleand consequently leads to low systemic availability [18].Following oral administration of CoQ10 either as a powderor as an oil suspension elicit practically no response inhuman subjects and it takes days to increase the CoQ10blood and possibly other tissue [109, 170]. The intravenousadministration of CoQ10 as a liposomal formulation raisedthe serum and myocardial levels of CoQ10 and significantlyimproved the function and efficiency of myocardial tissueand reduced oxidant injury observed following ischemia-reperfusion [109]. Several advancements have been madeto enhance the bioavailability of CoQ10 using variousapproaches like size reduction, solubility enhancement (bysolid dispersion, prodrug, complexation, and ionization) anduse of novel drug carriers such as liposomes, microspheres,nanoparticles, nanoemulsions, and self-emulsifying system(see review by Beg et al. 2010 [18]).
Liposomal CoQ10 is a promising candidate for thetopical application of CoQ10. CoQ10 encapsulated in N-trimethyl chitosan- (TMC-) coated liposomes is being exam-ined as potential ophthalmic drug delivery system. TMCexhibits an excellent absorption-enhancing effect by openingthe tight junctions between adjacent cells of epithelialcell monolayers [172] while Q10 ameliorates the oxidativestress in the lenses, the most common damaging factorfor the development of cataract [173]. Also, cosmeticallyapplied CoQ10 has the ability to reduce photoaging, witha corresponding decrease in wrinkle depth [84], but itsbioavailability in skin is poor. A liposomal formulationcomposed of soybean phosphatidylcholine (SPC) and alpha-
tocopherol (Vit E) used to encapsulate CoQ10 for topicalapplication significantly enhanced its accumulation (at leasttwofold) in rat skin, compared with an unencapsulatedsuspension. Prolonging the treatment time and increasingthe content of CoQ10 in the formulation both raised theamount of CoQ10 in rat skin [84].
7.2. Liposomal Curcumin. Curcumin, a hydrophobic poly-phenolic compound derived from dietary spice turmeric,possesses diverse pharmacologic effects including anti-inflammatory, antioxidant, antiproliferative, and antiangio-genic activities. Curcumin has been used extensively intraditional medicine since ancient times as a householdremedy against various diseases, including hepatic disorders,cough, sinusitis, rheumatism, and biliary disorders. Inthe past few decades, a number of studies have beencarried out to confirm curcumin’s potential role in treatinginflammatory disorders, cardiovascular disease, cancer,AIDS, and neurological disorders [174–176]. The maindrawback associated with the therapeutic potential ofcurcumin is its poor aqueous solubility and stability ingastrointestinal fluids, which leads to poor bioavailability[175, 176]. Several novel drug-delivery approaches, includ-ing microemulsions, nanoemulsions, liposomes, solid lip-id nanoparticles, microspheres, solid dispersion, poly-meric nanoparticles, and self-microemulsifying drug-delivery systems have been used to enhance the bio-availability and tissue-targeting ability of curcumin in vitroand in vivo [175–177]. Oral administration of liposomalcurcumin increased its bioavailability and antioxidantactivity in plasma [85]. A recent study examining theantitumor activity of liposomal curcumin against humanpancreatic carcinoma cells demonstrated that liposomalcurcumin inhibits pancreatic carcinoma growth and, inaddition, exhibits antiangiogenic effects [178].
7.3. Liposomal Resveratrol. Resveratrol, the main activepolyphenol in red wine, has been implicated as a possi-ble contributor to the cardiovascular protection conferredby red wine consumption, since this compound can befound in grapes among other fruits [179, 180]. The car-diovascular effect of resveratrol can, in turn, be relatedwith its proved capacity to act as a modulator on themetabolism of lipoproteins inhibiting the oxidation of low-density lipoproteins (LDLs), and inhibiting either plateletaggregation or proatherogenic eicosanoids production byhuman platelets and neutrophils. These potential therapeuticand prophylactic applications are, however, restricted by thelow bioavailability caused by its physical properties [179,180]. Resveratrol has low water solubility and stability aswell as its is rapidly and extensively metabolized making itsclinical use challenging [181]. To our knowledge, there havebeen only a few studies comparing the antioxidant propertiesof liposomal resveratrol with that of free resveratrol andreported that the liposomal formulation is more effectivein ameliorating the oxidant-induced cellular injury inducedfrom UV-B irradiation on human-derived renal epithelialcells [182].

10 Journal of Toxicology
7.4. Liposomal Quercetin. Quercetin is a polyphenolicflavonoid, commonly found in apples, onions, berries, andred wine, with strong antioxidant and antiinflammatoryproperties. Its use is limited by its low water solubility andhigh rate of metabolism [183]. Pretreatment of animalswith the flavonoid quercetin was ineffective in protectingagainst carbon tetrachloride-induced hepatotoxicity. Carbontetrachloride is known to mediate its liver toxicity via free-radical mechanisms including oxidative stress. Pretreatmentof animals with mannosylated liposomal quercetin signifi-cantly lowered the hepatotoxic effect of carbon tetrachloride[83]. In another study, it was demonstrated that quercetin-filled liposomes improved the protective effects of theantioxidant against peroxynitrite-induced myocardial injuryin isolated cardiac tissues and anesthetized animals [82].Pegylated liposomal quercetin significantly improved thesolubility and bioavailability of quercetin and enhanced itsantitumour activity in immunocompetent C57BL/6N micebearing LL/2 Lewis lung cancer and in BALB/c mice bearingCT26 colon adenocarcinoma and H22 hepatoma [113].
7.5. Liposomal Astaxanthin. Astaxanthin, is a red-orangelipid-soluble carotenoid pigment with powerful antioxi-dant and antinflammatory properties. As measured by afluorometric assay based on visible-absorbing fluorescentprobes that belong to the BODIPY class of dyes (BODIPY581/591 C11 and BODIPY 665/676) to measure antioxidantactivities of antioxidants in a lipid environment, astaxanthinpossess an antioxidative capability 10-times stronger thanzeaxanthin, lutein, canthaxanthin, and β-carotene [184].However, its use is often limited due to its poor water sol-ubility and extremely low bioavailability when administeredorally. The transport and protective effects of astaxanthinin Hep3B and HepG2 cell lines challenged with gammaradiation was far superior when the antioxidant was deliv-ered with egg-yolk phosphatidylcholine liposomes [185].Incorporation of astaxanthin into phosphatidylcholine mul-tilamellar liposomes strongly reduced lipid damage when thelipoperoxidation promoters—H2O2, tert-butyl hydroperox-ide (t-ButOOH) or ascorbate—and Fe2+:EDTA were addedsimultaneously to the liposomes [186] but also liposomalencapsulation is also effective in slowing the oxidation ofastaxanthin [185].
8. Liposomes Containing More thanOne Antioxidant
It has been shown that the administration of liposomescontaining more than one antioxidant is more advantageousin ameliorating oxidant-induced tissue injuries [57, 71, 80].The antioxidant effect of liposomal formulations containingthe enzymes SOD and catalase is more effective than thosecontaining the individual antioxidant enzyme [57, 58]. Thetherapeutic efficacy of an antioxidant liposome formulation,containing a lipophilic antioxidant, α-tocopherol, can beimproved by encapsulating another antioxidant, such as GSHor L-ascorbic acid, in the same liposome preparation. Theseliposomes, containing both α-tocopherol and GSH wereshown to be more effective in protecting against oxidant-
induced lung injuries and lipid peroxidation than thosecontaining either α-tocopherol or GSH alone presumablyby α-tocopherol scavenging free radicals and stabilizingbiological membranes, while GSH, in addition to its abilityto act as a free radical scavenger, can also regenerate α-tocopherol from its oxidized form [71]. Similarly, liposomescontaining both α-tocopherol and L-ascorbic acid were moreeffective in preventing the generation of conjugated diene incerebral tissues by the induction of global cerebral ischemiaand reperfusion than the those containing L-ascorbic acidthrough a synergistic action [80]. In a more recent study,it was shown that liposomes containing α-tocopherol, γ-tocopherol, δ-tocopherol, and NAC were more effectivethan liposomes containing only NAC or GSH in blockingthe CEES-induced inflammatory response and lung injuries[81, 108]. Liposomal delivery of curcumin and resveratrolreduced cancer in prostate-specific PTEN knockout mice andthis treatment effect was far superior to that seen followingadministration of each agent alone either in their free orliposomal form [187].
9. Concluding Remarks
Antioxidant liposomes hold great promise in the treatmentof many diseases in which oxidative stress plays a signif-icant role. Antioxidant enzymes and many of the otherantioxidants do not easily penetrate the plasma membraneof cells and some have poor stability and short half-life inplasma when administered through conventional deliverymodes. Liposomes are highly efficient in terms of facilitatingantioxidant delivery and achieving prophylactic and ther-apeutic efficacies against oxidative stress-induced damage.Liposomes can be prepared from natural phospholipids thatare biocompatible, biodegradable, and nonimmunogenicand practically do not cause toxic effects or antigenicreactions. Although successful clinical applications in thefield of drug delivery and treatment of cancer and systemicor local fungal infections have demonstrated the potentialof the technology, clinical applications for the utilizationof liposomal antioxidant formulations have been limited.Controversy on the efficacy and safety of antioxidants stillexists due to lack of well-controlled clinical trials. There is aneed for large multicentre prospective randomized controltrials to assess the effects of different types and doses ofantioxidant in selected groups of patients in various chronicand acute oxidative stress-related diseases. Perhaps, it is timeto vigorously seek alternative approaches and not simplycontinue to study the use of antioxidants by patients asdietary supplements in the hope of maintaining health andpreventing disease. The ability for precise adjustments ofliposome parameters such as size, charge, lipid composition,and the conjugation of ligands coupled with the moreefficient and safer loading techniques offers the flexibilityto make liposomes an effective platform for the delivery ofantioxidants.
Conflict of Interests
The author declares that there is no conflict of interests.

Journal of Toxicology 11
Acknowledgments
This work was supported by a grant from the Natural Sci-ences and Engineering Research Council of Canada (NSERCno. 312533-2008). Certain parts of this paper are reprintedfrom Chapter 11, “The role of liposomal antioxidants inoxidative stress” by Z. Suntres and A. Omri, in: NanocarrierTechnologies: frontiers of nanotherapy (ed. RM Mozafari),2006 [188], with kind permission from Springer Science +Business Media B.V.
References
[1] D. Ziech, R. Franco, A. G. Georgakilas et al., “The role ofreactive oxygen species and oxidative stress in environmen-tal carcinogenesis and biomarker development,” Chemico-Biological Interactions, vol. 188, no. 2, pp. 334–339, 2010.
[2] P. A. Ward, “Oxidative stress: acute and progressive lunginjury,” Annals of the New York Academy of Sciences, vol. 1203,pp. 53–59, 2010.
[3] E. Hopps, D. Noto, G. Caimi, and M. R. Averna, “A novelcomponent of the metabolic syndrome: the oxidative stress,”Nutrition, Metabolism and Cardiovascular Diseases, vol. 20,no. 1, pp. 72–77, 2010.
[4] Z. E. Suntres, “Role of antioxidants in paraquat toxicity,”Toxicology, vol. 180, no. 1, pp. 65–77, 2002.
[5] K. A. Jellinger, “Basic mechanisms of neurodegeneration: acritical update,” Journal of Cellular and Molecular Medicine,vol. 14, no. 3, pp. 457–487, 2010.
[6] M. Keel and O. Trentz, “Pathophysiology of polytrauma,”Injury, vol. 36, no. 6, pp. 691–709, 2005.
[7] C. M. Bergamini, S. Gambetti, A. Dondi, and C. Cervellati,“Oxygen, reactive oxygen species and tissue damage,” CurrentPharmaceutical Design, vol. 10, no. 14, pp. 1611–1626, 2004.
[8] J. M. McCord, “The evolution of free radicals and oxidativestress,” American Journal of Medicine, vol. 108, no. 8, pp. 652–659, 2000.
[9] I. F. F. Benzie, “Evolution of antioxidant defence mecha-nisms,” European Journal of Nutrition, vol. 39, no. 2, pp. 53–61, 2000.
[10] P. Evans and B. Halliwell, “Micronutrients: oxidant/anti-oxidant status,” British Journal of Nutrition, vol. 85, no. 2, pp.S67–S74, 2001.
[11] H. Sies, “Oxidative stress: oxidants and antioxidants,” Exper-imental Physiology, vol. 82, no. 2, pp. 291–295, 1997.
[12] S. R. Steinhubl, “Why have antioxidants failed in clinicaltrials?” American Journal of Cardiology, vol. 101, no. 10, pp.S14–S19, 2008.
[13] D. V. Ratnam, D. D. Ankola, V. Bhardwaj, D. K. Sahana, andM. N. V. R. Kumar, “Role of antioxidants in prophylaxis andtherapy: a pharmaceutical perspective,” Journal of ControlledRelease, vol. 113, no. 3, pp. 189–207, 2006.
[14] W. L. Stone and M. Smith, “Therapeutic uses of antioxidantliposomes,” Applied Biochemistry and Biotechnology, vol. 27,no. 3, pp. 217–230, 2004.
[15] V. R. Muzykantov, “Targeting of superoxide dismutase andcatalase to vascular endothelium,” Journal of ControlledRelease, vol. 71, no. 1, pp. 1–21, 2001.
[16] V. R. Muzykantov, “Delivery of antioxidant enzyme proteinsto the lung,” Antioxidants and Redox Signaling, vol. 3, no. 1,pp. 39–62, 2001.
[17] R. Carnemolla, V. V. Shuvaev, and V. R. Muzykantov,“Targeting antioxidant and antithrombotic biotherapeutics
to endothelium,” Seminars in Thrombosis and Hemostasis,vol. 36, no. 3, pp. 332–342, 2010.
[18] S. Beg, S. Javed, and K. Kohli, “Bioavailability enhancementof coenzyme q10: an extensive review of patents,” RecentPatents on Drug Delivery and Formulation, vol. 4, no. 3, pp.245–255, 2010.
[19] F. Schuber, A. Kichler, C. Boeckler, and B. Frisch, “Lipo-somes: from membrane models to gene therapy,” Pure andApplied Chemistry, vol. 70, no. 1, pp. 89–96, 1998.
[20] A. Samad, Y. Sultana, and M. Aqil, “Liposomal drug deliverysystems: an update review,” Current Drug Delivery, vol. 4, no.4, pp. 297–305, 2007.
[21] D. S. Pisal, M. P. Kosloski, and S. V. Balu-Iyer, “Delivery oftherapeutic proteins,” Journal of Pharmaceutical Sciences, vol.99, no. 6, pp. 2557–2575, 2010.
[22] G. Stark, “Functional consequences of oxidative membranedamage,” Journal of Membrane Biology, vol. 205, no. 1, pp.1–16, 2005.
[23] M. Comporti, “Three models of free radical-induced cellinjury,” Chemico-Biological Interactions, vol. 72, no. 1-2, pp.1–56, 1989.
[24] A. Azzi, “Oxidative stress: a dead end or a laboratory hypoth-esis?” Biochemical and Biophysical Research Communications,vol. 362, no. 2, pp. 230–232, 2007.
[25] Z. Durackova, “Some current insights into oxidative stress,”Physiological Research, vol. 59, no. 4, pp. 459–469, 2010.
[26] V. B. Djordjevic, “Free radicals in cell biology,” in Inter-national Review of Cytology, W. J. Kwang, Ed., pp. 57–89,Academic Press, New York, NY, USA, 2004.
[27] A. L. A. Ferreira, L. S. Matsubara, and B. B. Matsubara,“Anthracycline-induced cardiotoxicity,” Cardiovascular andHematological Agents in Medicinal Chemistry, vol. 6, no. 4,pp. 278–281, 2008.
[28] F. Di Virgilio, “New pathways for reactive oxygen species gen-eration in inflammation and potential novel pharmacologicaltargets,” Current Pharmaceutical Design, vol. 10, no. 14, pp.1647–1652, 2004.
[29] H. J. Forman and M. Torres, “Signaling by the respiratoryburst in macrophages,” IUBMB Life, vol. 51, no. 6, pp. 365–371, 2001.
[30] W. Droge, “Free radicals in the physiological control of cellfunction,” Physiological Reviews, vol. 82, no. 1, pp. 47–95,2002.
[31] G. Ferrer-Sueta and R. Radi, “Chemical biology of peroxyni-trite: kinetics, diffusion, and radicals,” ACS Chemical Biology,vol. 4, no. 3, pp. 161–177, 2009.
[32] M. L. Circu and T. Y. Aw, “Reactive oxygen species, cellularredox systems, and apoptosis,” Free Radical Biology andMedicine, vol. 48, no. 6, pp. 749–762, 2010.
[33] K. T. Turpaev, “Reactive oxygen species and regulation ofgene expression,” Biochemistry, vol. 67, no. 3, pp. 281–292,2002.
[34] C. C. Winterbourn and M. B. Hampton, “Thiol chemistryand specificity in redox signaling,” Free Radical Biology andMedicine, vol. 45, no. 5, pp. 549–561, 2008.
[35] M. Valko, D. Leibfritz, J. Moncol, M. T. D. Cronin, M. Mazur,and J. Telser, “Free radicals and antioxidants in normalphysiological functions and human disease,” InternationalJournal of Biochemistry and Cell Biology, vol. 39, no. 1, pp.44–84, 2007.
[36] B. Harwell, “Biochemistry of oxidative stress,” BiochemicalSociety Transactions, vol. 35, no. 5, pp. 1147–1150, 2007.
[37] J. Nordberg and E. S. J. Arner, “Reactive oxygen species,antioxidants, and the mammalian thioredoxin system,” Free

12 Journal of Toxicology
Radical Biology and Medicine, vol. 31, no. 11, pp. 1287–1312,2001.
[38] Y. Manevich and A. B. Fisher, “Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense andlung phospholipid metabolism,” Free Radical Biology andMedicine, vol. 38, no. 11, pp. 1422–1432, 2005.
[39] I. N. Zelko, T. J. Mariani, and R. J. Folz, “Superoxidedismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) genestructures, evolution, and expression,” Free Radical Biologyand Medicine, vol. 33, no. 3, pp. 337–349, 2002.
[40] Y. Wang, S. I. Feinstein, and A. B. Fisher, “Peroxiredoxin 6 asan antioxidant enzyme: protection of lung alveolar epithelialtype II cells from H2O2-induced oxidative stress,” Journal ofCellular Biochemistry, vol. 104, no. 4, pp. 1274–1285, 2008.
[41] C. K. Chow, “Biological functions and metabolic fate ofvitamin E revisited,” Journal of Biomedical Science, vol. 11, no.3, pp. 295–302, 2004.
[42] M. G. Traber and J. Atkinson, “Vitamin E, antioxidant andnothing more,” Free Radical Biology and Medicine, vol. 43, no.1, pp. 4–15, 2007.
[43] M. E. Anderson and J. L. I. Luo, “Glutathione therapy: fromprodrugs to genes,” Seminars in Liver Disease, vol. 18, no. 4,pp. 415–424, 1998.
[44] M. C. Fotia and R. Amorati, “Non-phenolic radical-trappingantioxidants,” Journal of Pharmacy and Pharmacology, vol.61, no. 11, pp. 1435–1448, 2009.
[45] J. S. Bains and C. A. Shaw, “Neurodegenerative disorders inhumans: the role of glutathione in oxidative stress-mediatedneuronal death,” Brain Research Reviews, vol. 25, no. 3, pp.335–358, 1997.
[46] S. A. R. Paiva, R. M. Russell, and S. K. Dutta, “β-carotene andother carotenoids as antioxidants,” Journal of the AmericanCollege of Nutrition, vol. 18, no. 5, pp. 426–433, 1999.
[47] N. I. Krinsky and E. J. Johnson, “Carotenoid actions and theirrelation to health and disease,” Molecular Aspects of Medicine,vol. 26, no. 6, pp. 459–516, 2005.
[48] K. D. Croft, “The chemistry and biological effects offlavonoids and phenolic acids,” Annals of the New YorkAcademy of Sciences, vol. 854, pp. 435–442, 1998.
[49] D. Nakae, H. Yoshiji, K. Yamamoto et al., “Influence of tim-ing of administration of liposome-encapsulated superoxidedismutase on its prevention of acetaminophen-induced livercell necrosis in rats,” Acta Pathologica Japonica, vol. 40, no. 8,pp. 568–573, 1990.
[50] M. L. Corvo, O. C. Boerman, W. J. G. Oyen et al., “Intra-venous administration of superoxide dismutase entrapped inlong circulating liposomesII. In vivo fate in a rat model ofadjuvant arthritis,” Biochimica et Biophysica Acta, vol. 1419,no. 2, pp. 325–334, 1999.
[51] S. Delanian, “Successful treatment of radiation-inducedfibrosis using liposomal Cu/Zn superoxide dismutase: clin-ical trial,” Radiotherapy and Oncology, vol. 32, no. 1, pp. 12–20, 1994.
[52] F. Baillet, M. Housset, A. M. Michelson, and K. Puget,“Treatment of radiofibrosis with liposomal superoxide dis-mutase. Preliminary results of 50 cases,” Free Radical ResearchCommunications, vol. 1, no. 6, pp. 387–394, 1986.
[53] P. H. Chan, S. Longar, and R. A. Fishman, “Protective effectsof liposome-entrapped superoxide dismutase on posttrau-matic brain edema,” Annals of Neurology, vol. 21, no. 6, pp.540–547, 1987.
[54] S. Imaizumi, V. Woolworth, R. A. Fishman, and P. H. Chan,“Liposome-entrapped superoxide dismutase reduces cerebral
infarction in cerebral ischemia in rats,” Stroke, vol. 21, no. 9,pp. 1312–1317, 1990.
[55] S. Imaizumi, V. Woolworth, H. Kinouchi, S. F. Chen, R. A.Fishman, and P. H. Chan, “Liposome-entrapped superox-ide dismutase ameliorates infarct volume in focal cerebralischaemia,” Acta Neurochirurgica, Supplement, vol. 51, pp.236–238, 1990.
[56] M. Petelin, Z. Pavlica, T. Ivanusa, M. Sentjurc, and U. Ska-leric, “Local delivery of liposome-encapsulated superoxidedismutase and catalase suppress periodontal inflammation inbeagles,” Journal of Clinical Periodontology, vol. 27, no. 12, pp.918–925, 2000.
[57] B. A. Freeman, J. F. Turrens, and Z. Mirza, “Modulation ofoxidant lung injury by using liposome-entrapped superoxidedismutase and catalase,” Federation Proceedings, vol. 44, no.10, pp. 2591–2595, 1985.
[58] J. F. Turrens, J. D. Crapo, and B. A. Freeman, “Protectionagainst oxygen toxicity by intravenous injection of liposome-entrapped catalase and superoxide dismutase,” The Journal ofClinical Investigation, vol. 73, no. 1, pp. 87–95, 1984.
[59] M. L. Barnard, R. R. Baker, and S. Matalon, “Mitigationof oxidant injury to lung microvasculature by intratrachealinstillation of antioxidant enzymes,” American Journal ofPhysiology, vol. 265, no. 4, part 1, pp. L340–L345, 1993.
[60] R. V. Padmanabhan, R. Gudapaty, and I. E. Liener, “Protec-tion against pulmonary oxygen toxicity in rats by the intra-tracheal administration of liposome-encapsulated superox-ide dismutase or catalase,” American Review of RespiratoryDisease, vol. 132, no. 1, pp. 164–167, 1985.
[61] F. J. Walther, R. David-Cu, and S. L. Lopez, “Antioxidant-surfactant liposomes mitigate hyperoxic lung injury inpremature rabbits,” American Journal of Physiology, vol. 269,no. 5, part 1, pp. L613–L617, 1995.
[62] A. Ledwozyw, “Protective effect of liposome-entrappedsuperoxide dismutase and catalase on bleomycin-inducedlung injury in rats. I. Antioxidant enzyme activities and lipidperoxidation,” Acta Veterinaria Hungarica, vol. 39, no. 3-4,pp. 215–224, 1991.
[63] A. Ledwozyw, “Protective effect of liposome-entrappedsuperoxide dismutase and catalase on bleomycin-inducedlung injury in rats. II. Phospholipids of the lung surfactant,”Acta Physiologica Hungarica, vol. 78, no. 2, pp. 157–162, 1991.
[64] S. D. McClintock, L. M. Hoesel, S. K. Das et al., “Attenuationof half sulfur mustard gas-induced acute lung injury in rats,”Journal of Applied Toxicology, vol. 26, no. 2, pp. 126–131,2006.
[65] D. W. Thibeault, M. Rezaiekhaligh, S. Mabry, and T. Beringer,“Prevention of chronic pulmonary oxygen toxicity in youngrats with liposome-encapsulated catalase administered intra-tracheally,” Pediatric Pulmonology, vol. 11, no. 4, pp. 318–327,1991.
[66] M. Rosenblat, N. Volkova, R. Coleman, and M. Aviram,“Anti-oxidant and anti-atherogenic properties of liposomalglutathione: studies in vitro, and in the atheroscleroticapolipoprotein E-deficient mice,” Atherosclerosis, vol. 195, no.2, pp. e61–e68, 2007.
[67] A. Wendel, H. Jaeschke, and M. Gloger, “Drug-induced lipidperoxidation in mice—II. Protection against paracetamol-induced liver necrosis by intravenous liposomally entrappedglutathione,” Biochemical Pharmacology, vol. 31, no. 22, pp.3601–3605, 1982.
[68] R. W. I. Cooke and J. A. Drury, “Reduction of oxidative stressmarker in lung fluid of preterm infants after administration

Journal of Toxicology 13
of intra-tracheal liposomal glutathione,” Biology of theNeonate, vol. 87, no. 3, pp. 178–180, 2005.
[69] G. D. Zeevalk, L. P. Bernard, and F. T. Guilford, “Liposomal-glutathione provides maintenance of intracellular glu-tathione and neuroprotection in mesencephalic neuronalcells,” Neurochemical Research, vol. 35, no. 10, pp. 1575–1587,2010.
[70] L. M. Hoesel, M. A. Flierl, A. D. Niederbichler et al., “Abilityof antioxidant liposomes to prevent acute and progressivepulmonary injury,” Antioxidants and Redox Signaling, vol. 10,no. 5, pp. 973–981, 2008.
[71] Z. E. Suntres and P. N. Shek, “Alleviation of paraquat-induced lung injury by pretreatment with bifunctionalliposomes containing α-tocopherol and glutathione,” Bio-chemical Pharmacology, vol. 52, no. 10, pp. 1515–1520, 1996.
[72] J. Fan, P. N. Shek, Z. E. Suntres, Y. H. Li, G. D. Oreopou-los, and O. D. Rotstein, “Liposomal antioxidants provideprolonged protection against acute respiratory distress syn-drome,” Surgery, vol. 128, no. 2, pp. 332–338, 2000.
[73] L. Khairy, G. E. Isom, and D. O. Kildsig, “Reversalof acetaminophen intoxication with an N-acetylcysteine-liposome preparation,” Research Communications in Chem-ical Pathology and Pharmacology, vol. 42, no. 1, pp. 153–156,1983.
[74] Z. E. Suntres and P. N. Shek, “Prophylaxis againstlipopolysaccharide-induced acute lung injury by α- toco-pherol liposomes,” Critical Care Medicine, vol. 26, no. 4, pp.723–729, 1998.
[75] Z. E. Suntres and P. N. Shek, “Treatment of LPS-inducedtissue injury: role of liposomal antioxidants,” Shock, vol. 6,no. 6, pp. S57–S64, 1996.
[76] Z. E. Suntres and P. N. Shek, “Protective effect of liposomalalpha-tocopherol against bleomycin-induced lung injury,”Biomedical and Environmental Sciences, vol. 10, supplement1, pp. 47–59, 1997.
[77] Z. E. Suntres and P. N. Shek, “Liposomal α-tocopherolalleviates the progression of paraquat-induced lung damage,”Journal of Drug Targeting, vol. 2, no. 6, pp. 493–500, 1995.
[78] Z. E. Suntres and P. N. Shek, “Prevention of phorbolmyristate acetate-induced acute lung injury by α-tocopherolliposomes,” Journal of Drug Targeting, vol. 3, no. 3, pp. 201–208, 1995.
[79] E. Wigenstam, D. Rocksn, B. Ekstrand-Hammarstrm, andA. Bucht, “Treatment with dexamethasone or liposome-encapsuled vitamin e provides beneficial effects afterchemical-induced lung injury,” Inhalation Toxicology, vol. 21,no. 11, pp. 958–964, 2009.
[80] J. Sinha, N. Das, and M. K. Basu, “Liposomal antioxidantsin combating ischemia-reperfusion injury in rat brain,”Biomedicine and Pharmacotherapy, vol. 55, no. 5, pp. 264–271, 2001.
[81] S. Mukhopadhyay, S. Mukherjee, B. K. Ray, A. Ray, W.L. Stone, and S. K. Das, “Antioxidant liposomes protectagainst CEES-induced lung injury by decreasing SAF-1/MAZ-mediated inflammation in the guinea pig lung,”Journal of Biochemical and Molecular Toxicology, vol. 24, no.3, pp. 187–194, 2010.
[82] A. Soloviev, A. Stefanov, A. Parshikov et al., “Arrhythmogenicperoxynitrite-induced alterations in mammalian heart con-tractility and its prevention with quercetin-filled liposomes,”Cardiovascular Toxicology, vol. 2, no. 2, pp. 129–139, 2002.
[83] A. K. Mandal, S. Das, M. K. Basu, R. N. Chakrabarti, andN. Das, “Hepatoprotective activity of liposomal flavonoid
against arsenite-induced liver fibrosis,” Journal of Pharmacol-ogy and Experimental Therapeutics, vol. 320, no. 3, pp. 994–1001, 2007.
[84] W. C. Lee and T. H. Tsai, “Preparation and characterizationof liposomal coenzyme Q10 for in vivo topical application,”International Journal of Pharmaceutics, vol. 395, no. 1-2, pp.78–83, 2010.
[85] M. Takahashi, S. Uechi, K. Takara, Y. Asikin, and K. Wada,“Evaluation of an oral carrier system in rats: bioavailabilityand antioxidant properties of liposome-encapsulated cur-cumin,” Journal of Agricultural and Food Chemistry, vol. 57,no. 19, pp. 9141–9146, 2009.
[86] P. J. Millea, “N-acetylcysteine: multiple clinical applications,”American Family Physician, vol. 80, no. 3, pp. 265–269, 2009.
[87] R. S. Britton, K. L. Leicester, and B. R. Bacon, “Iron toxicityand chelation therapy,” International Journal of Hematology,vol. 76, no. 3, pp. 219–228, 2002.
[88] A. R. Weseler and A. Bast, “Oxidative stress and vascularfunction: implications for pharmacologic treatments,” Cur-rent Hypertension Reports, vol. 12, no. 3, pp. 154–161, 2010.
[89] S. Yamashita and Y. Matsuzawa, “Where are we with probu-col: a new life for an old drug?” Atherosclerosis, vol. 207, no.1, pp. 16–23, 2009.
[90] G. M. D’Andrea, “Use of antioxidants during chemotherapyand radiotherapy should be avoided,” CA Cancer Journal forClinicians, vol. 55, no. 5, pp. 319–321, 2005.
[91] M. L. Nucci, J. Olejarczyk, and A. Abuchowski, “Immuno-genicity of polyethylene glycol-modified superoxide dismu-tase and catalase,” Journal of Free Radicals in Biology andMedicine, vol. 2, no. 5-6, pp. 321–325, 1986.
[92] A. Arslantas, “Development of functional models for a SOD,”Metal-Based Drugs, vol. 9, no. 1-2, pp. 9–18, 2002.
[93] S. B. Lotito and B. Frei, “Consumption of flavonoid-richfoods and increased plasma antioxidant capacity in humans:cause, consequence, or epiphenomenon?” Free Radical Biol-ogy and Medicine, vol. 41, no. 12, pp. 1727–1746, 2006.
[94] M. Maiorino, A. Zamburlini, A. Roveri, and F. Ursini,“Prooxidant role of vitamin E in copper induced lipid perox-idation,” The FEBS Letters, vol. 330, no. 2, pp. 174–176, 1993.
[95] Z. E. Suntres and E. M. K. Lui, “Age-related differences iniron-nitrilotriacetate hepatotoxicity in the guinea pig: roleof copper metallothionein,” Journal of Pharmacology andExperimental Therapeutics, vol. 258, no. 3, pp. 797–806, 1991.
[96] Z. E. Suntres and E. M. K. Lui, “Prooxidative effect ofcopper-metallothionein in the acute cytotoxicity of hydrogenperoxide in Ehrlich ascites tumour cells,” Toxicology, vol.217, no. 2-3, pp. 155–168, 2006.
[97] N. Abudu, J. J. Miller, M. Attaelmannan, and S. S. Levinson,“Vitamins in human arteriosclerosis with emphasis onvitamin C and vitamin E,” Clinica Chimica Acta, vol. 339, no.1-2, pp. 11–25, 2004.
[98] Z. Drulis-Kawa and A. Dorotkiewicz-Jach, “Liposomes asdelivery systems for antibiotics,” International Journal ofPharmaceutics, vol. 387, no. 1-2, pp. 187–198, 2010.
[99] P. Goyal, K. Goyal, S. G. V. Kumar, A. Singh, O. P. Katare, andD. N. Mishra, “Liposomal drug delivery systems—clinicalapplications,” Acta Pharmaceutica, vol. 55, no. 1, pp. 1–25,2005.
[100] N. A. Kshirsagar, S. K. Pandya, B. G. Kirodian, and S. Sanath,“Liposomal drug delivery system from laboratory to clinic,”Journal of Postgraduate Medicine, vol. 51, no. 5, supplement1, pp. S5–S15, 2005.

14 Journal of Toxicology
[101] D. A. Thomas, M. A. Myers, B. Wichert, H. Schreier, andR. J. Gonzalez-Rothi, “Acute effects of liposome aerosolinhalation on pulmonary function in healthy humanvolunteers,” Chest, vol. 99, no. 5, pp. 1268–1270, 1991.
[102] H. G. Eichler, J. Senior, A. Stadler, S. Gasic, P. Pfundner,and G. Gregoriadis, “Kinetics and disposition of fluorescein-labelled liposomes in healthy human subjects,” EuropeanJournal of Clinical Pharmacology, vol. 34, no. 5, pp. 475–479,1988.
[103] G. Storm, H. P. Wilms, and D. J. A. Crommelin, “Liposomesand biotherapeutics,” Biotherapy, vol. 3, no. 1, pp. 25–42,1991.
[104] M. J. Parnham and H. Wetzig, “Toxicity screening ofliposomes,” Chemistry and Physics of Lipids, vol. 64, no. 1–3,pp. 263–274, 1993.
[105] P. Srinath, M. G. Chary, S. P. Vyas, and P. V. Diwan, “Long-circulating liposomes of indomethacin in arthritic rats—abiodisposition study,” Pharmaceutica Acta Helvetiae, vol. 74,no. 4, pp. 399–404, 2000.
[106] L. Cattel, M. Ceruti, and F. Dosio, “From conventional tostealth liposomes a new frontier in cancer chemotherapy,”Tumori, vol. 89, no. 3, pp. 237–249, 2003.
[107] T. Minko, A. Stefanov, and V. Pozharov, “Selected contri-bution: lung hypoxia: antioxidant and antiapoptotic effectsof liposomal α-tocopherol,” Journal of Applied Physiology,vol. 93, no. 4, pp. 1550–1560, 2002.
[108] S. Mukherjee, W. L. Stone, H. Yang, M. G. Smith, and S. K.Das, “Protection of half sulfur mustard gas-induced lunginjury in guinea pigs by antioxidant liposomes,” Journal ofBiochemical and Molecular Toxicology, vol. 23, no. 2, pp.143–153, 2009.
[109] K. Niibori, H. Yokoyama, J. A. Crestanello, and G. J. R.Whitman, “Acute administration of liposomal coenzymeQ10 increases myocardial tissue levels and improves toler-ance to ischemia reperfusion injury,” Journal of SurgicalResearch, vol. 79, no. 2, pp. 141–145, 1998.
[110] H. Yokoyama, D. M. Lingle, J. A. Crestanello et al.,“Coenzyme Q10 protects coronary endothelial functionfrom ischemia reperfusion injury via an antioxidant effect,”Surgery, vol. 120, no. 2, pp. 189–196, 1996.
[111] M. Alipour, A. Omri, M. G. Smith, and Z. E. Suntres,“Prophylactic effect of liposomal N-acetylcysteine againstLPS-induced liver injuries,” Journal of Endotoxin Research,vol. 13, no. 5, pp. 297–304, 2007.
[112] P. Mitsopoulos, A. Omri, M. Alipour, N. Vermeulen, M. G.Smith, and Z. E. Suntres, “Effectiveness of liposomal-N-acetylcysteine against LPS-induced lung injuries in rodents,”International Journal of Pharmaceutics, vol. 363, no. 1-2, pp.106–111, 2008.
[113] Z. P. Yuan, L. J. Chen, L. Y. Fan et al., “Liposomal quercetinefficiently suppresses growth of solid tumors in murinemodels,” Clinical Cancer Research, vol. 12, no. 10, pp.3193–3199, 2006.
[114] T. M. Allen, “Liposomal drug formulations: rationale fordevelopment and what we can expect for the future,” Drugs,vol. 56, no. 5, pp. 747–756, 1998.
[115] G. Gregoriadis, “Overview of liposomes,” Journal of Anti-microbial Chemotherapy, vol. 28, supplement B, pp. 39–48,1991.
[116] V. P. Torchilin, “Recent approaches to intracellular deliveryof drugs and DNA and organelle targeting,” Annual Reviewof Biomedical Engineering, vol. 8, pp. 343–375, 2006.
[117] C. Regnault, M. Soursac, M. Roch-Arveiller, E. Postaire, andG. Hazebroucq, “Pharmacokinetics of superoxide dismutasein rats after oral administration,” Biopharmaceutics and DrugDisposition, vol. 17, no. 2, pp. 165–174, 1996.
[118] T. Fujita, M. Nishikawa, C. Tamaki, Y. Takakura, M. Hashida,and H. Sezaki, “Targeted delivery of human recombinantsuperoxide dismutase by chemical modification with mono-and polysaccharide derivatives,” Journal of Pharmacology andExperimental Therapeutics, vol. 263, no. 3, pp. 971–978, 1992.
[119] C. W. White, J. H. Jackson, A. Abuchowski et al., “Poly-ethylene glycol-attached antioxidant enzymes decrease pul-monary oxygen toxicity in rats,” Journal of Applied Physiology,vol. 66, no. 2, pp. 584–590, 1989.
[120] G. Tang, J. E. White, R. J. Gordon, P. D. Lumb, and M. F.Tsan, “Polyethylene glycol-conjugated superoxide dismutaseprotects rats against oxygen toxicity,” Journal of AppliedPhysiology, vol. 74, no. 3, pp. 1425–1431, 1993.
[121] J. S. Beckman, R. L. Minor Jr., and B. A. Freeman, “Aug-mentation of antioxidant enzymes in vascular endothelium,”Journal of Free Radicals in Biology and Medicine, vol. 2, no.5-6, pp. 359–365, 1986.
[122] V. V. Shuvaev, M. Christofidou-Solomidou, F. Bhora et al.,“Targeted detoxification of selected reactive oxygen speciesin the vascular endothelium,” Journal of Pharmacology andExperimental Therapeutics, vol. 331, no. 2, pp. 404–411, 2009.
[123] V. V. Shuvaev, J. Han, K. J. Yu et al., “PECAM-targeteddelivery of SOD inhibits endothelial inflammatory response,”The FASEB Journal, vol. 25, no. 1, pp. 348–357, 2011.
[124] V. V. Shuvaev, S. Tliba, M. Nakada, S. M. Albelda, and V. R.Muzykantov, “Platelet-endothelial cell adhesion molecule-1-directed endothelial targeting of superoxide dismutasealleviates oxidative stress caused by either extracellularor intracellular superoxide,” Journal of Pharmacology andExperimental Therapeutics, vol. 323, no. 2, pp. 450–457, 2007.
[125] S. Muro and V. R. Muzykantov, “Targeting of antioxidantand anti-thrombotic drugs to endothelial cell adhesionmolecules,” Current Pharmaceutical Design, vol. 11, no. 18,pp. 2383–2401, 2005.
[126] J. W. Park, C. C. Benz, and F. J. Martin, “Future directions ofliposome- and immunoliposome-based cancer therapeutics,”Seminars in Oncology, vol. 31, no. 6, supplement 13, pp.196–205, 2004.
[127] D. Papahadjopoulos, T. M. Allen, A. Gabizon et al., “Stericallystabilized liposomes: improvements in pharmacokineticsand antitumor therapeutic efficacy,” Proceedings of theNational Academy of Sciences of the United States of America,vol. 88, no. 24, pp. 11460–11464, 1991.
[128] M. Kaipel, A. Wagner, E. Wassermann et al., “Increasedbiological half-life of aerosolized liposomal recombinanthuman Cu/Zn superoxide dismutase in pigs,” Journal ofAerosol Medicine and Pulmonary Drug Delivery, vol. 21, no.3, pp. 281–290, 2008.
[129] T. T. Jubeh, S. Antler, S. Haupt, Y. Barenholz, and A. Rubin-stein, “Local prevention of oxidative stress in the intestinalepithelium of the rat by adhesive liposomes of superoxidedismutase and tempamine,” Molecular Pharmaceutics, vol. 2,no. 1, pp. 2–11, 2005.
[130] K. Vorauer-Uhl, E. Furnschlief, A. Wagner, B. Ferko, andH. Katinger, “Topically applied liposome encapsulatedsuperoxide dismutase reduces postburn wound size andedema formation,” European Journal of PharmaceuticalSciences, vol. 14, no. 1, pp. 63–67, 2001.

Journal of Toxicology 15
[131] R. Exne, B. Wessner, N. Manhart, and E. Roth, “Therapeuticpotential of glutathione,” Wiener Klinische Wochenschrift,vol. 112, no. 14, pp. 610–616, 2000.
[132] I. Grattagliano, P. Wieland, C. Schranz, and B. H. Lauterburg,“Effect of oral glutathione monoethyl ester and glutathioneon circulating and hepatic sulfydrils in the rat,” Pharmacologyand Toxicology, vol. 75, no. 6, pp. 343–347, 1994.
[133] I. Grattagliano, P. Wieland, C. Schranz, and B. H. Lauterburg,“Disposition of glutathione monoethyl ester in the rat:glutathione ester is a slow release form of extracellularglutathione,” Journal of Pharmacology and ExperimentalTherapeutics, vol. 272, no. 2, pp. 484–488, 1995.
[134] A. Witschi, S. Reddy, B. Stofer, and B. H. Lauterburg, “Thesystemic availability of oral glutathione,” European Journal ofClinical Pharmacology, vol. 43, no. 6, pp. 667–669, 1992.
[135] Z. E. Suntres and P. N. Shek, “Incorporation of α-tocopherolin liposomes promotes the retention of liposome-encap-sulated glutathione in the rat lung,” Journal of Pharmacy andPharmacology, vol. 46, no. 1, pp. 23–28, 1994.
[136] L. J. Smith, J. Anderson, and M. Shamsuddin, “Glutathionelocalization and distribution after intratracheal instillation:implications for treatment,” American Review of RespiratoryDisease, vol. 145, no. 1, pp. 153–159, 1992.
[137] A. Chonn and P. R. Cullis, “Recent advances in liposomaldrug-delivery systems,” Current Opinion in Biotechnology,vol. 6, no. 6, pp. 698–708, 1995.
[138] R. E. Pagano and J. N. Weinstein, “Interactions of liposomeswith mammalian cells,” Annual Review of Biophysics andBioengineering, vol. 7, pp. 435–468, 1978.
[139] A. M. Sadowska, B. Manuel-y-Keenoy, and W. A. De Backer,“Antioxidant and anti-inflammatory efficacy of NAC in thetreatment of COPD: discordant in vitro and in vivo dose-effects: a review,” Pulmonary Pharmacology and Therapeutics,vol. 20, no. 1, pp. 9–22, 2007.
[140] K. R. Atkuri, J. J. Mantovani, L. A. Herzenberg, and L. A.Herzenberg, “N-Acetylcysteine-a safe antidote for cysteine/glutathione deficiency,” Current Opinion in Pharmacology,vol. 7, no. 4, pp. 355–359, 2007.
[141] G. S. Kelly, “Clinical applications of N-acetylcysteine,”Alternative Medicine Review, vol. 3, no. 2, pp. 114–127, 1998.
[142] M. C. G. van de Poll, C. H. C. Dejong, and P. B. Soeters,“Adequate range for sulfur-containing amino acids andbiomarkers for their excess: lessons from enteral andparenteral nutrition,” Journal of Nutrition, vol. 136, no. 6,pp. 1694S–1700S, 2006.
[143] F. Pajonk, K. Riess, A. Sommer, and W. H. McBride,“N-acetyl-L-cysteine inhibits 26S proteasome function:implications for effects on NF-κB activation,” Free RadicalBiology and Medicine, vol. 32, no. 6, pp. 536–543, 2002.
[144] M. Zafarullah, W. Q. Li, J. Sylvester, and M. Ahmad, “Molec-ular mechanisms of N-acetylcysteine actions,” Cellular andMolecular Life Sciences, vol. 60, no. 1, pp. 6–20, 2003.
[145] S. Atis, A. Nayci, A. Ozge, U. Comelekoglu, S. Gunes, andO. Bagdatoglu, “N-acetylcysteine protects the rats againstphrenic nerve dysfunction in sepsis,” Shock, vol. 25, no. 1,pp. 30–35, 2006.
[146] M. Caglikulekci, M. Dirlik, C. Pata et al., “Effect of N-Acetylcysteine on blood and tissue lipid peroxidation inlipopolysaccharide-induced obstructive jaundice,” Journal ofInvestigative Surgery, vol. 19, no. 3, pp. 175–184, 2006.
[147] B. G. Hsu, R. P. Lee, F. L. Yang, H. J. Harn, and H. I.Chen, “Post-treatment with N-acetylcysteine ameliorates
endotoxin shock-induced organ damage in conscious rats,”Life Sciences, vol. 79, no. 21, pp. 2010–2016, 2006.
[148] V. Smyrniotis, N. Arkadopoulos, G. Kostopanagiotou etal., “Attenuation of ischemic injury by N-acetylcysteinepreconditioning of the liver,” Journal of Surgical Research,vol. 129, no. 1, pp. 31–37, 2005.
[149] S. Fishbane, J. H. Durham, K. Marzo, and M. Rudnick, “N-acetylcysteine in the prevention of radiocontrast-inducednephropathy,” Journal of the American Society of Nephrology,vol. 15, no. 2, pp. 251–260, 2004.
[150] A. Gillissen and D. Nowak, “Characterization of N-acetylcysteine and ambroxol in anti-oxidant therapy,”Respiratory Medicine, vol. 92, no. 4, pp. 609–623, 1998.
[151] G. W. Burton, “Vitamin E: molecular and biologicalfunction,” Proceedings of the Nutrition Society, vol. 53, no. 2,pp. 251–262, 1994.
[152] X. Wang and P. J. Quinn, “Vitamin E and its function inmembranes,” Progress in Lipid Research, vol. 38, no. 4, pp.309–336, 1999.
[153] R. Meyer, A. F. P. Sonnen, and W. M. Nau, “Phase-dependentlateral diffusion of α-tocopherol in DPPC liposomesmonitored by fluorescence quenching,” Langmuir, vol. 26,no. 18, pp. 14723–14729, 2010.
[154] C. M. S. Cereda, G. R. Tofoli, R. B. De Brito Junior etal., “Stability and local toxicity evaluation of a liposomalprilocaine formulation,” Journal of Liposome Research, vol.18, no. 4, pp. 329–339, 2008.
[155] S. Urano, M. Kitahara, Y. Kato, Y. Hasegawa, and M. Matsuo,“Membrane stabilizing effect of vitamin E: existence of ahydrogen bond between α-tocopherol and phospholipidsin bilayer liposomes,” Journal of Nutritional Science andVitaminology, vol. 36, no. 6, pp. 513–519, 1990.
[156] C. P. Verdon and J. B. Blumberg, “Influence of dietaryvitamin E on the intermembrane transfer of α-tocopherol asmediated by an α-tocopherol binding protein,” Proceedingsof the Society for Experimental Biology and Medicine, vol. 189,no. 1, pp. 52–60, 1988.
[157] S. Banudevi, G. Krishnamoorthy, P. Venkataraman, C.Vignesh, M. M. Aruldhas, and J. Arunakaran, “Role ofα-tocopherol on antioxidant status in liver, lung and kidneyof PCB exposed male albino rats,” Food and ChemicalToxicology, vol. 44, no. 12, pp. 2040–2046, 2006.
[158] A. A. Shvedova, E. R. Kisin, A. R. Murray et al., “Vitamin Edeficiency enhances pulmonary inflammatory response andoxidative stress induced by single-walled carbon nanotubesin C57BL/6 mice,” Toxicology and Applied Pharmacology, vol.221, no. 3, pp. 339–348, 2007.
[159] A. M. Terrasa, M. H. Guajardo, C. A. Marra, and G. Zapata,“α-Tocopherol protects against oxidative damage to lipidsof the rod outer segments of the equine retina,” VeterinaryJournal, vol. 182, no. 3, pp. 463–468, 2009.
[160] Z. E. Suntres and P. N. Shek, “Intratracheally administeredliposomal alpha-tocopherol protects the lung against long-term toxic effects of paraquat,” Biomedical and EnvironmentalSciences, vol. 8, no. 4, pp. 289–300, 1995.
[161] L. J. Ramazzotto and R. Engstrom, “Dietary vitamin Eand the effects of inhaled nitrogen dioxide on rat lungs,”Environmental Physiology & Biochemistry, vol. 5, no. 4, pp.226–234, 1975.
[162] H. M. Redetzki, C. D. Wood, and W. D. Grafton, “Vitamin Eand paraquat poisoning,” Veterinary and Human Toxicology,vol. 22, no. 6, pp. 395–397, 1980.

16 Journal of Toxicology
[163] R. J. Stephens, D. J. Buntman, and D. S. Negi, “Tissue levelsof vitamin E in the lung and the cellular response to injuryresulting from oxidant gas exposure,” Chest, vol. 83, no. 5,supplement 1, pp. 37S–39S, 1983.
[164] D. L. Warren, D. M. Hyde, and J. A. Last, “Synergisticinteraction of ozone and respirable aerosols on rat lungs.IV. Protection by quenchers of reactive oxygen species,”Toxicology, vol. 53, no. 1, pp. 113–133, 1988.
[165] T. Yasaka, K. Okudaira, and H. Fujito, “Further studies oflipid peroxidation in human paraquat poisoning,” Archivesof Internal Medicine, vol. 146, no. 4, pp. 681–685, 1986.
[166] Z. E. Suntres, S. R. Hepworth, and P. N. Shek, “Pulmonaryuptake of liposome-associated α-tocopherol followingintratracheal instillation in rats,” Journal of Pharmacy andPharmacology, vol. 45, no. 6, pp. 514–520, 1993.
[167] M. E. Knight and R. J. Roberts, “Tissue vitamin E levelsin newborn rabbits after pharmacologic dosing. Influenceof dose, dosage form, and route of administration,”Developmental Pharmacology and Therapeutics, vol. 8, no. 2,pp. 96–106, 1985.
[168] N. Hidiroglou, L. F. Laflamme, and L. R. McDowell, “Bloodplasma and tissue concentrations of vitamin E in beef cattleas influenced by supplementation of various tocopherolcompounds,” Journal of Animal Science, vol. 66, no. 12, pp.3227–3234, 1988.
[169] P. P. Constantinides, J. Han, and S. S. Davis, “Advances inthe use of tocols as drug delivery vehicles,” PharmaceuticalResearch, vol. 23, no. 2, pp. 243–255, 2006.
[170] H. N. Bhagavan and R. K. Chopra, “Coenzyme Q10: absorp-tion, tissue uptake, metabolism and pharmacokinetics,” FreeRadical Research, vol. 40, no. 5, pp. 445–453, 2006.
[171] F. L. Crane, “Biochemical functions of coenzyme Q10,”Journal of the American College of Nutrition, vol. 20, no. 6,pp. 591–598, 2001.
[172] Y. Zambito, C. Zaino, and G. Di Colo, “Effects ofN-trimethylchitosan on transcellular and paracellulartranscorneal drug transport,” European Journal of Pharma-ceutics and Biopharmaceutics, vol. 64, no. 1, pp. 16–25, 2006.
[173] S. Wang, J. Zhang, T. Jiang et al., “Protective effect ofCoenzyme Q10 against oxidative damage in human lensepithelial cells by novel ocular drug carriers,” InternationalJournal of Pharmaceutics, vol. 403, no. 1-2, pp. 219–229, 2011.
[174] A. S. Strimpakos and R. A. Sharma, “Curcumin: preventiveand therapeutic properties in laboratory studies and clinicaltrials,” Antioxidants and Redox Signaling, vol. 10, no. 3, pp.511–545, 2008.
[175] P. Anand, S. G. Thomas, A. B. Kunnumakkara et al.,“Biological activities of curcumin and its analogues(Congeners) made by man and Mother Nature,” BiochemicalPharmacology, vol. 76, no. 11, pp. 1590–1611, 2008.
[176] A. Kumar, A. Ahuja, J. Ali, and S. Baboota, “Conundrum andtherapeutic potential of curcumin in drug delivery,” CriticalReviews in Therapeutic Drug Carrier Systems, vol. 27, no. 4,pp. 279–312, 2010.
[177] S. Padhye, D. Chavan, S. Pandey, J. Deshpande, K. V. Swamy,and F. H. Sarkar, “Perspectives on chemopreventive andtherapeutic potential of curcumin analogs in medicinalchemistry,” Mini Reviews in Medicinal Chemistry, vol. 10, no.5, pp. 372–387, 2010.
[178] C. M. Mach, L. Mathew, S. A. Mosley, R. Kurzrock, andJ. A. Smith, “Determination of minimum effective doseand optimal dosing schedule for liposomal curcumin in
a xenograft human pancreatic cancer model,” AnticancerResearch, vol. 29, no. 6, pp. 1895–1899, 2009.
[179] J. Brittes, M. Lucio, C. Nunes, J. L. F. C. Lima, and S. Reis,“Effects of resveratrol on membrane biophysical properties:relevance for its pharmacological effects,” Chemistry andPhysics of Lipids, vol. 163, no. 8, pp. 747–754, 2010.
[180] S. Sadruddin and R. Arora, “Resveratrol: biologic andtherapeutic implications,” Journal of the CardioMetabolicSyndrome, vol. 4, no. 2, pp. 102–106, 2009.
[181] C. H. Cottart, V. Nivet-Antoine, C. Laguillier-Morizot, andJ. L. Beaudeux, “Resveratrol bioavailability and toxicity inhumans,” Molecular Nutrition and Food Research, vol. 54, no.1, pp. 7–16, 2010.
[182] J. Kristl, K. Teskac, C. Caddeo, Z. Abramovic, and M.Sentjurc, “Improvements of cellular stress response onresveratrol in liposomes,” European Journal of Pharmaceuticsand Biopharmaceutics, vol. 73, no. 2, pp. 253–259, 2009.
[183] B. A. Graf, W. Mullen, S. T. Caldwell et al., “Disposition andmetabolism of [2-14C]quercetin-4′- glucoside in rats,” DrugMetabolism and Disposition, vol. 33, no. 7, pp. 1036–1043,2005.
[184] Y. M. A. Naguib, “Antioxidant activities of astaxanthinand related carotenoids,” Journal of Agricultural and FoodChemistry, vol. 48, no. 4, pp. 1150–1154, 2000.
[185] C. H. Peng, C. H. Chang, R. Y. Peng, and C. C. Chyau,“Improved membrane transport of astaxanthine byliposomal encapsulation,” European Journal of Pharmaceuticsand Biopharmaceutics, vol. 75, no. 2, pp. 154–161, 2010.
[186] M. P. Barros, E. Pinto, P. Colepicolo, and M. Pedersen,“Astaxanthin and peridinin inhibit oxidative damage inFe2+-loaded liposomes: scavenging oxyradicals or changingmembrane permeability?” Biochemical and BiophysicalResearch Communications, vol. 288, no. 1, pp. 225–232, 2001.
[187] N. K. Narayanan, D. Nargi, C. Randolph, and B. A.Narayanan, “Liposome encapsulation of curcumin andresveratrol in combination reduces prostate cancer incidencein PTEN knockout mice,” International Journal of Cancer,vol. 125, no. 1, pp. 1–8, 2009.
[188] Z. E. Suntres and A. Omri, “The role of liposomalantioxidants in oxidative stress,” in Nanocarrier Technologies,M. R. Mozafari, Ed., pp. 191–205, Springer, Amsterdam, TheNetherlands, 2006.