morphological and biochemical studies on aging and autophagy
TRANSCRIPT

R
M
RA
a
ARRAA
KAAMB
1
oiotgbwaadgitqc
mi(oomr
1d
Ageing Research Reviews 11 (2012) 10– 31
Contents lists available at SciVerse ScienceDirect
Ageing Research Reviews
jo u r n al hom epage: www.elsev ier .com/ locate /ar r
eview
orphological and biochemical studies on aging and autophagy
ita Rezzani ∗, Alessandra Stacchiotti, Luigi Fabrizio Rodellanatomy Section, Department of Biomedical Sciences and Biotechnology, University of Brescia, Viale Europa 11, 25123 Brescia, Italy
r t i c l e i n f o
rticle history:eceived 14 July 2011eceived in revised form 5 September 2011ccepted 8 September 2011vailable online 16 September 2011
a b s t r a c t
To maintain health in the elderly is a crucial objective for modern medicine that involves both basicand clinical researches. Autophagy is a fundamental auto-cannibalizing process that preserves cellularhomeostasis and, if altered, either by excess or defect, greatly changes cell fate and can result in incapaci-tating human diseases. Efficient autophagy may prolong lifespan, but unfortunately this process becomesless efficient with age.
eywords:utophagygingorphological markers
iochemical markers
The present review is focused on the close relationship between autophagy and age-related disorders indifferent tissues/organs and in transgenic animal models. In particular, it comments on the up to date liter-ature on mechanisms responsible for age-related impairment of autophagy. Moreover, before discussingabout these mechanisms, it is necessary to describe the metabolic autophagic regulation of autophagyand the proteins involved in this process. At the end, these data would summarize the autophagic linkwith aging process, as important tools in the future biogerontology scenario.
. Introduction
One of the major challenges in the 21st century is the agingf the population and the associated medical social and economicmplications. Life expectancy exceeds 80 years in several devel-ped countries and is projected to continue to increase duringhe next half century (Kirkwood, 2008). Since the elderly peopleradually increases, it becomes more important to understand theiological bases (i.e., processes and their mechanisms) of aging asell as morphological and molecular aspects underlining various
ge-related diseases. This knowledge may lead to future geneticnd pharmacological interventions that can delay many aspects ofecline occurring at advanced ages, prolonging the age of people inood health (Vellai et al., 2009). In the last two decades, the biolog-cal basis of aging shed light on many aging mysteries describinghe age-related changes in organs of organisms. However, manyuestions are unresolved since the aging shows many aspects andauses (Rajawat et al., 2009; Troen, 2003).
Aging is a process characterized by an accumulation of aberrantacromolecules and organelles during post-development period
nducing a decrease of time survival and an increase of death riskRajawat and Bossis, 2008). During aging process there is a failuref maintenance and repair pathways and this induces the origin
f age-related diseases and eventual death (Rattan, 2006). Accu-ulation of worn-out organelles and different cellular structureseduces molecular and cellular efficiency of biological processes
∗ Corresponding author. Tel.: +39 0303717483; fax: +39 0303717486.E-mail address: [email protected] (R. Rezzani).
568-1637/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.arr.2011.09.001
© 2011 Elsevier B.V. All rights reserved.
that are important for organ homeostasis and survival. Since mod-ifications of biological processes are the main causes of agingor senescence, they are the final manifestations of unsuccessfulhomeostasis or failure of homeodynamics (Holliday, 2007). In par-ticular, it is possible to report that the lifespan of an organismis the sum of deleterious changes and maintenance mechanismsresponding to the damage. In particular, it is known that the lifes-pan is determined by the balance between metabolism, whichleads to the accumulation of damage (thus causing aging), andcompensatory responses (Fig. 1). The changes are due to theaccumulation of oxidized, misfolded, cross-linked or aggregatedmacromolecules that are morphologically not normal and so, theycannot properly function. Moreover, it is possible that these macro-molecules interfere with other molecules and organelles, or theiraggregates compromising cellular functions sending also erroneoussignals. Thus, the cells need to eliminate them for survival. It isknown also that the cellular damage is related to aging-pathologies,including cancer, neurodegeneration, infection and muscle atrophy(Kirkwood, 2008; Hekimi and Guarente, 2003).
Oxidation of DNA induces, for example, a single-or double strandbreaks leading to possible mutation and determining cancer, andother diseases (Rubinsztein, 2006; Pinkston et al., 2006; Matheuet al., 2007). Nevertheless, the not defined or not known cellu-lar and biochemical markers of aging make useless the efforts foridentifying the primary and secondary mechanisms responsibleto damages. Many theories regarding aging have been proposed
(Weinert and Timiras, 2003) and they have been divided in twocategories (Troen, 2003): stochastic and genetic theories. The firsttheory considers the alterations occurring during the entire lifes-pan of cells as above reported, while the genetic theories suggest
R. Rezzani et al. / Ageing Research
tl(
fgIatltCo2“aihrla2
bioc
msmfitiimaa
2
ms2pm(t
Fig. 1. Lifespan and metabolism.
hat aging is part of the genetically programmed and controlledifespan including development, maturation, senescence and deathWeinert and Timiras, 2003).
These theories are not mutually exclusive, when considering theree radical/mitochondrial DNA theory of aging, the influence ofenes is reduced whereas that of stochastic events is increased.ncreased evidence suggested that cellular senescence and agingre manifestations of evolutionary pressures to prevent malignantransformation, i.e. cancer (Troen, 2003). It is now clear that cel-ular senescence is a crucial anticancer mechanism that preventshe growth of cells at risk for neoplastic transformation (Rodier andampisi, 2011). It is important to note that the term “senescence” isbtained by human diploid fibroblasts in culture (Shay and Wright,000) and indicates the “irreversible state” of cell cycle respect toreversible” quiescent state coming down to a stress-responsivelteration of phenotype (Young and Narita, 2010). However, evenf the senescence can be considered a state of cell cycle arrest, itas an active metabolism (Young et al., 2009), and a delayed stressesponse involving multiple mechanisms such as epigenetic regu-ation (Adams, 2007), DNA damage response (Mallette et al., 2007)nd a senescence-associated secretion phenotype (Acosta et al.,008).
Although many aging and age-related disease theories haveeen postulated, there are more points no defined. Now, many stud-
es indicated that autophagy is directly involved in the progressionf diseases linked to aging even if the autophagic pathways are veryomplex and they still remain a big puzzle.
Here, we highlight: (1) the contribution of autophagy to theaintenance of cellular homeostasis, that reduces the progres-
ive post-maturational deterioration of tissues and organs and itsetabolic regulation in physiological conditions; (2) the recent
ndings linking the autophagic system to lifespan extension andhe proteins involved in this pathway; and (3) the main proteinsnvolved in the autophagy and age-related cell deterioration lead-ng to associated diseases. We suggest that if these proteins, likely to
arkers are identified, it should be possible to predict the onset ofge-related diseases, as well as individual life expectancies (Simmnd Johnson, 2010).
. Aging and cellular mechanism
Aging progressively affects cellular functions such as oxidativeetabolism, and important organelles such as mitochondria, lyso-
omes and the endoplasmic reticulum (ER) (Chandel and Budinger,007; Rajawat et al., 2009). Eukaryotic cells have two major
roteolytic systems for degrading abnormal proteins: the lysoso-al pathway (Dice, 2000) and the ubiquitin-proteosome pathwayUPS). UPS is linked to ubiquitin, that is a small 76 aminoacids pro-ein responsible for the demolition of many cytosolic, nuclear and
Reviews 11 (2012) 10– 31 11
endoplasmic proteins (Ciechanover, 2005). Moreover, UPS, presentin the cytoplasm and in the nucleus, is able to recognize anddestroy oxidatively damaged and ubiquinated proteins losing theirfunction with the aging (Carrard et al., 2002; Ward, 2002). Theproteolytic system is supported by molecular chaperones, whichdetermines the fate of the damaged proteins. In detail, in nor-mal young cells, molecular chaperones recognize damaged proteinsand attempt to repair them or, if it is not possible, target themfor degradation by intracellular proteolytic systems. In old cells,the major proteolytic systems are defective leading to intracellularaccumulation of damaged/unfolded or misfolded proteins. Proteinaggregates slowly accumulate in all cells throughout life, but theirformation can accelerate under particular cellular conditions or incertain pathologies (Fig. 2). Damaged proteins are first recognizedby molecular proteins and, if their alterations are too extensive,they will be degraded. The protein degradation is essential also dur-ing embriogenesis and morphogenesis and also as protective actionagainst harmful agents and pathogens (Martinez-Vicente et al.,2005). At this regard, the lysosome activity is required for removingdamaged proteins and the main alteration linked to lysosome-aging-related is the accumulation of lipofuscins, that are inverselylinked to age and no degradable. Although growing cells are ableto destroy lipofuscins by dilution in each mitotic cycle, postmi-totic cells such as neurons are no able to do this. It is known thatAlzheimer disease is associated with enhanced lipofuscin depositsand reduced efficiency of lysosomal protein degradation (Nixonet al., 2001). Moreover, accumulation of ubiquinated proteins islinked to other neurodegenerative disorders, such as Parkinsondisease (Ciechanover et al., 2000).
There are many other organelles accumulating during aging, likemitochondria (Mammucari and Rizzuto, 2010) and peroxisomes,organelles that are greatly involved in the generation of reactiveoxygen species (ROS) (Terlecky et al., 2006). The “free radical the-ory” of aging proposed by Harman in 1956 indicated that ROS,produced during metabolic pathway, induce a progressive loss incellular functions, leading to the aging phenotype (Harman, 1956).Since ROS are mainly produced in mitochondria during oxidativerespiration, they are very important targets of aging and longevitystudies (Chan, 2006; Pérez et al., 2009). The mitochondrial DNA(mtDNA) is strongly subject to ROS exposure and this induces inmitochondria an easy replace of normal mtDNA in senescent tissues(Ozawa, 1997). These mitochondrial dysfunctions are associatedwith hypoxia in many tissues like the brain (Roberts et al., 1997),heart (Pepe, 2000) and kidney (Nangaku et al., 2008). A usefuland well known mechanism for prolonging lifespan and attenuat-ing mtDNA dysfunction is caloric restriction (Cavallini et al., 2008;Guarente, 2008); this treatment is able to increase mitochondrialfunction without decreasing the respiration rate.
Finally, aging, a natural decline in the biological pathways ofan organism, is in part due to decrease of system function, suchas molecular chaperones, proteasome and lysosome-mediatedautophagy, that are important for cellular homeostasis and for pro-tection against premature aging (Vellai, 2009).
3. Autophagy and self-eating
Autophagy means (auto)-eating (phagy). Three primary types ofautophagy have been defined: macroautophagy, microautophagy,and chaperone-mediated autophagy (Fig. 3).
Macroautophagy is due to the formation of a cup-shapeddouble membrane structure called the phagophore sequestrat-
ing a portion of cytoplasm. This phagophore expands and sealsto form an autophagosome. Then, autophagosome fuses with alysosome, forming an autolysosome, that is responsible to thedegradation of intra-autophagosomal components by lysosomal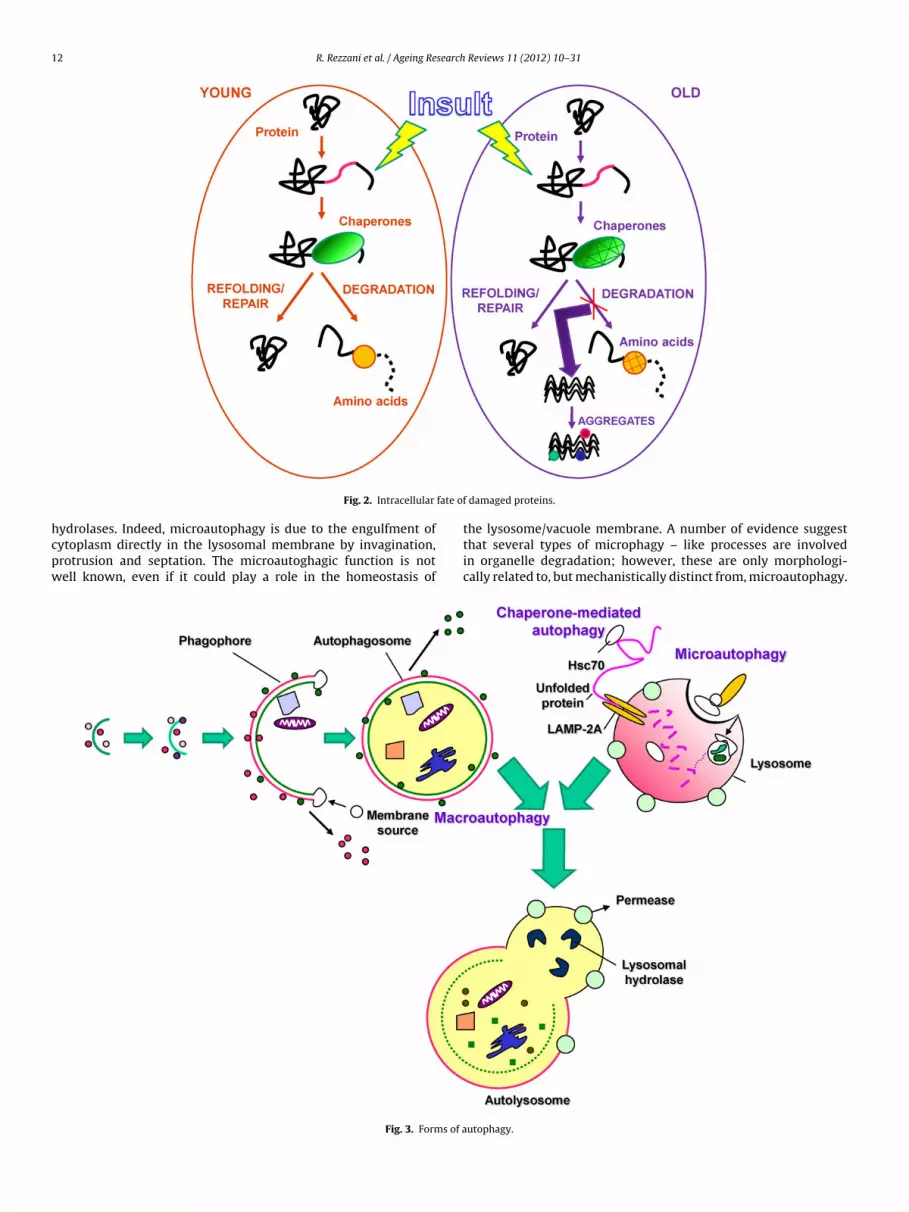
12 R. Rezzani et al. / Ageing Research Reviews 11 (2012) 10– 31
fate of
hcpw
Fig. 2. Intracellular
ydrolases. Indeed, microautophagy is due to the engulfment ofytoplasm directly in the lysosomal membrane by invagination,rotrusion and septation. The microautoghagic function is notell known, even if it could play a role in the homeostasis of
Fig. 3. Forms of a
damaged proteins.
the lysosome/vacuole membrane. A number of evidence suggestthat several types of microphagy – like processes are involvedin organelle degradation; however, these are only morphologi-cally related to, but mechanistically distinct from, microautophagy.
utophagy.

search
Cphsaa
cd1icalh(ts1
bti
pctiiMmwteio
3
mctbialo
adtbe(twmtptdsdic
R. Rezzani et al. / Ageing Re
haperone-mediated autophagy (CMA) is a process of direct trans-ort of unfolded proteins via the cytosolic and lysosomal chaperonesc70 and lysosomal receptor protein type 2A (LAMP-2A), a lyso-omal membrane receptor. If one considers these three types ofutophagy, macroautophagy is best characterized and well knownnd it is normally indicated with the term “autophagy”.
At the beginning of the scientific history, autophagy is normallyonsidered to be involved in alterations of cellular cytoarchitectureuring differentiation and development. Successively, in the late960s, autophagy was found to be present in various organs includ-
ng newborn kidney. In 1970, Pfeifer showed that the breakdown ofytoplasmic components is functionally related to gluconeogenesisnd Sybers and colleagues suggested that the cellular injury stimu-ated the endoplasmic reticulum to enclose the altered componentselping the lysosomal digestion without damaging the entire cellPfeifer, 1978; Sybers et al., 1976). Later, autophagy was found par-icularly efficient in the removing cytoplasmic components ando, it plays an important role in intracellular components (Pfeifer,978).
Since the lysosomal compartment is useful for autophagy, thisiological pathway exists in all eukaryotic cell types and it is impor-ant for long-lived protein degradation as well as organelle turnovern order to control the quality or health of the cytoplasm.
Autophagy can be also considered as a cellular response tohysiological challenges including nutrient starvation, cell growthontrol, intracellular proteins and organelle turnover, cell death,umor suppression and inherent immunity. A role for autophagyn cell survival is well recognized but it has been also suggestedts involvement in cell death from excessive cellular consumption.
oreover, it has been also indicated that autophagy is deter-ined by cytotoxic events making very complicated to concludehether autophagy is a death or survival pathway. Furthermore,
he autophagy has been observed to play a role in certain dis-ases including cancer, renal and cardiovascular disorders becauset can, selectively or not selectively, engulfs various cytoplasmicrganelles (Periyasamy-Thandavan et al., 2009).
.1. Autophagic dogma
Normally, autophagy can be considered on the basis of howacromolecules and organelles are brought into the lysosomal
ompartment and on the types of substrates carried for degrada-ion. In light of these considerations, macro-microautophagy haseen found in all eukaryotes, whereas CMA has been detected only
n mammals (Cuervo, 2004). It is important to underline that UPSnd macroautophagy mainly act in the degradation of short- or longived proteins, respectively and also in the degradation of a subsetf proteins (Korolchuk et al., 2009; Lamark and Johansen, 2010).
The stone of the macroautophagy is the formation of theutophagosome. The origin of the autophagosomal membrane isebated and many morphological studies report that it is linkedo the ER (Ylä-Anttila et al., 2009; Hayashi-Nishino et al., 2010),ut also derived from membrane of trans-Golgi compartment (Yent al., 2010), mitochondria (Hailey et al., 2010) or plasma membraneLongatti et al., 2010). Moreover, Ravikumar et al. (2010) suggestedhat plasma membrane is involved for producing autophagosome,hen a large amount of membrane need. In this case the plasmaembrane is an important tank of membranes during stress condi-
ions limiting the lipid synthesis that can occur. However, this lastoint was not fully explored and other studies will need to evaluatehe model reported above. Recently, many data indicated that theifferent types of cells use several membranes to form autophago-
omes or that different inducers of autophagy involve the use ofifferent donor membranes. The plasticity of the autophagy makest an extremely adaptable pathway functioning in a wide range ofonditions, that are very important for survival.
Reviews 11 (2012) 10– 31 13
In yeast, it has been found that a single phagophore assemblysite (PAS) is involved only in autophagosome formation (Suzukiet al., 2001). Since the autophagic mechanism is similar from yeastto mammals, it is possible that PAS is a site upstream of thephagophore. The increase in genetic knowledge of yeasts allowedto well clarify the molecular mechanisms of autophagy identifyingmore than 30 autophagy-related genes (Atg) and half of them havebeen also found in mammalian as homologues (Xie and Klionsky,2007; Yang and Klionsky, 2009).
CMA is an inducible mechanism activated by oxidative damageand nutritional stress and it also participates in antigen presenta-tion. It targets particular cytosolic proteins crossing the lysosomalmembrane without vesicles or membrane deformation (Arias andCuervo, 2010). About one-third of cellular protein are degraded byCMA and this is possible through the interaction of a cytosolic chap-erone, the heat shock-cognate sequence of 70 kDa protein, hsc70.The originated complex (substrate-hsc70 chaperone) is recognizedby a LAMP-2A, and translocated into lysosome lumen where it isdegraded. LAMP-2A is an important receptor and component forthe CMA translocation complex. These results are also reportedby Bandyopadhyay et al. (2008) and they established that CMAbinds LAMP-2A only in its monomeric form. After its binding, thecomplex changes its molecular weight (700 kDa) for obtaining thesubstrate translocation (Bandyopadhyay et al., 2008). The final pro-cess depends from two lysosomal proteins, lys-hsc70 and hsc-90.Although these data, the pathways responsible for translocationacross the lysosomal membrane via CMA are still poorly character-ized. Recently, Ana Maria Cuervo’s group demonstrated that glialfibrillary acid protein (GFAP), a cytoplasmic intermediate filamentprotein, involved in the formation of the intracellular cytoarchi-tecture, participates in the modulation of CMA and it is regulatedby GTP (Bandyopadhyay et al., 2010). They show that, in culturedcells, GFAP interacts with LAMP-2A in the lysosomal membranein response to starvation and oxidative stress, that are two potentCMA activators. They suggest that a fine-tuned regulatory mech-anism for substrate binding and translocation via CMA, based onLAMP-2A assembly into a multimeric complex and on the stabilityof the translocation complex in lysosomal membrane, exists.
As reported above, the different forms of autophagy are notindependent from each other, but they act together or there isalso the possibility that it is induced first one type of autophagyand then another, depending from the stimuli or specific cellularrequirement (Kaushik et al., 2008). Indeed, it is known that there isa strict link between CMA and macroautophagy and an upregula-tion of some autophagic pathways after the damage of a particularform of autophagy could be a basis for therapeutic strategies in sev-eral diseases, i.e. cancer and neurodegenerative disorders. Finally,Wong and Cuervo (2010) identified several forms of autophagywhich are different from the canonical forms seen during basalor starvation-induced autophagy. It has become evident that dif-ferent mechanisms can lead to the formation of autophagosomes,whereas some molecular components once thought to be essentialfor macroautophagy can be dispensable, and the use of alternativevariants to compensate for the defective ones could represent atherapeutic alternative still unexplored.
3.2. Formation of autophagosomes and markers or proteinsinvolved in this process
During autophagy, the cytoplasm containing the material thatmust be degraded is surrounded by a double membrane formingan autophagosome. This latter fuses with endocytic compartment
forming an amphisome and with lysosomes by a mechanismGTPase Rab7 and LAMP-2 dependent. In this process, the outerautophagosomal membrane fuses with lysosomal membranewhile the inner autophagosomal membrane is released into the
1 search
lrosTtfBiAc3tpwliolmt(sbdtstw
(
(
(
4
sl2retcmstait2t2
4 R. Rezzani et al. / Ageing Re
ysosome interior. The vesicle is then degraded and the mate-ial return to the cytosol using specific permeases. The formationf autophagosomes, consisting of nucleation, membrane expan-ion, and vesicle closure, is the rate-limiting step in autophagy.his step requires 18 Atg that have been identified in yeasts buthey have the same counterparts in mammalians. In particular,or the beginning of autophagosomes, the Atg1–Atg 6 (beclin-1,ECN-1)–Atg13–Atg17 complex plays an important role in the
nduction; the phosphatylinositol 3-kinase complex (PI3K) and thetg5–Atg12–Atg16 complex, that is important in the process ofonjugation of Atg8 (microtubule-associated protein 1 light chain
(LC3-I) in mammalian cells with phosphatidylethanolamine (PE)o form Atg8-PE (LC3II in mammalian cells), need for autophagicathway. ATg8-PE is present on autophagosome membranes andhile, when autophagosome has formed, Atg5–Atg12 complex is
ost, Atg8-PE (LC3II) remains on autophagosome membranes. Thenner membrane is degraded by lysosomal enzymes after the fusionf autophagosomes with lysosomes and the outer membrane LC3IIost Atg4 and returns to the cytosol. LC3II is considered a useful
arker of autophagy even if it can be present on protein aggregates,hat have been formed in a manner independent of autophagyTanida et al., 2005; Shvets and Elazar, 2008). For many years, theources used for autophagosome formation have been discussed,ut now it is accepted that for example the lipids for the membraneserived from ER (Axe et al., 2008). There are many components ofhe lysosomal membranes considered responsible of autophago-ome formation; the major components of these membranes arehe homologus type I transmembrane proteins LAMP1 and LAMP2ith 37% identity in humans (Saftig et al., 2008).
So, we can summarize the autophagy process in three steps:
1) Induction: following external/internal stimuli (e.g. nutrient-poor conditions) mammalian target of rapamycin (mTOR) isinhibited leading to induction of autophagy.
2) Autophagosome formation: cytosolic proteins and organellesare sequestrated by a double membrane vesicle, arising fromER. Formation of this vesicle is co-ordinated by complexesof Atg proteins, in particular Atg1–Atg6–Atg13–Atg17 com-plex. While this complex is responsible to the induction ofautophagic process, Atg5–Atg12–Atg17 complex is responsi-ble to recruitment of Atg8 (LC3). This step is important for theformation of autophagosomes.
3) Docking and fusion with lysosomes with LAMP protein pres-ence.
. Autophagy in physiological processes and its regulation
Physiological role of autophagy in the mammalian system washown in vivo animal models (Hur et al., 2010) and its failureeads to alterations of cavitation during embryogenesis (Qu et al.,007), or accumulation of abnormal organelles such as endoplasmiceticulum or mitochondria in several tissues of animals (Komatsut al., 2005; Masiero et al., 2009). Moreover, it is involved in cer-ain tissue-specific functions and in the maintenance of cellularytoarchitecture: – intracellular biogenesis of surfactant in pneu-ocytes II (Meijer and Codogno, 2004); – elimination of organelles,
uch as ribosomes and mitochondria during erythrocyte matura-ion (Meijer and Codogno, 2004); – regulation of structure and massnd functions of pancreatic �-cells (Hur et al., 2010). Moreover, it ismportant to report that amino acids inhibit autophagy by activa-
ion of mTOR-dependent signaling in concert with insulin (Meijer,008); – glucagon and catecholamine, i.e. norepinephrine regulatehis process respectively for example in liver and in heart (Yin et al.,008; Aránguiz-Urroz et al., 2011). In the following section theReviews 11 (2012) 10– 31
metabolic regulation of autophagy regarding insulin, aminoacids,glucagon, and catecholamine will be discussed.
4.1. Role of autophagy in different organs and its regulation
Jung et al. (2008) and Ebato et al. (2008) studied the phys-iological role of constitutive autophagy in pancreatic �-cells byproducing mice with �-cell specific disruption of Atg7, anotheressential gene in autophagy. These mice had decreased prolifer-ation of �-cells with the reduction in the �-cell mass. Obviously,these alterations produced a pancreatic insulin content decreaseand a reduction of insulin granule number. Morphological analysisof �-cells showed an increase of ubiquitinated proteins, damagedmitochondria and dilated ER. As a consequence these animals hadhypoinsulinemia and hyperglycemia. So, these data indicated thatautophagy plays a role in the maintenance of pancreatic cytoarchi-tecture, that is important for survival and function of the organsand in particular of �-cells. Moreover, it is possible to indicate thatautophagy deficiency regulates insulin content (Jung et al., 2008;Ebato et al., 2008).
Instead, it is accepted that insulin in synergy with amino acidsare able to inhibit the autophagic processes (Meijer and Codogno,2009) via mTOR signaling pathways. This pathway is controlled bytwo steps: (1) insulin linking to its receptors (insulin receptor sub-strates 1 and 2, IRS1 and IRS2), involves class I phosphatidylinositol3-kinase (PI3K) inducing the formation of protein kinase B (PKB).This step of the pathway is involved in regulation of glucose trans-port in muscle and adypocytes; (2) this part of the insulin-signalingpathway involves components such as eukaryotic initiation fac-tor 4E binding protein (4E-BP), ribosomal protein S6, that play animportant role in regulating protein synthesis. The activity of mTORis inhibited by the tuberculous sclerosis complex, TSC1/TSC2, whichacts as a GTP-ase activating a protein complex for the small G pro-tein Rheb. This latter binds and activates mTOR, which is presentin a complex TORC1 (target of rapamycin complex 1) acting as ascaffold for mTOR phosphorylation substrates, the proteins G�L,and PRAS40. Moreover, mTOR could be present as TORC2 (targetof rapamycin complex 2) and its role is responsible of kinase Bphosphorylation. It is important to remember that TORC1 activityis not determined by insulin alone but also by low concentrations ofamino acids (Dann and Thomas, 2006). Instead, high concentrationsof amino acids alone can activate mTOR without insulin presenceas reported by Hara et al. (1998). Amino acids do not stimulate classI PI3K or protein kinase B. Thus, it indicates that their mechanism isdifferent from that of insulin (Avruch et al., 2009). Moreover, otherauthors demonstrated also that inhibitors of class I PI3K interferewith amino acid-dependent signaling without insulin, suggestingthat basal activity of PI3K is enough for mTOR activity in thesesituations (Tang et al., 2003).
We must report also that the activation of mTOR activity inducedby amino acids not only stimulates protein synthesis but alsoinhibits autophagy and rapamycin stimulates autophagy in pres-ence of amino acids. Since it has been demonstrated that rapamycinstimulates autophagy also in yeasts (Prick et al., 2006), and plants(Bassham, 2007), its role is maintained during evolution.
In hepatocytes, low concentrations of amino acids, insulin stim-ulates mTOR signaling inhibiting autophagy, whereas glucagon hasopposite effects (Meijer, 2008). This is consistent with the rolethat these hormones have on the liver and this is important fromthe point of view of metabolic regulation (Droge and Kinscherf,2008).
The mechanisms by which TOR acts has been demonstrated in
yeast and this indicated that inhibition of TOR by starvation orrapamicin treatment is accompanied by defosphorylation of Atg1and Atg13 and binding of Atg1 to Atg 13 and Atg 17 inducing anincreased activity of Atg 1 protein (Kawamata et al., 2008).
search
ticemptvptiltit
5
mcmcUasisDiBwi2
dstaedgit2prKppecta2
ta
tatbi
R. Rezzani et al. / Ageing Re
Regarding catecholamine and autophagy, recent data suggestedhat adrenergic stimulation regulate cardiac fibroblast autophagy,nducing an increase collagen degradation, thus playing a cru-ial role in the maintenance matrix homeostasis (Aránguiz-Urrozt al., 2011). These data, observed via transmission electronicroscopy, fluorescent microscopy studies and endogenous LC3
rocessing/lipidation assayed by immunowesternblot, indicatedhat pro-autophagic effects of beta-adrenergic receptor (�-AR) acti-ation led to an increased degradation of collagen-1, an effectreviously unexplored in the contribution of catecholamines tohe regulation of cardiac collagen homeostasis. Moreover, it ismportant to report that adrenergic stimulation in cardiac fibrob-asts triggers cell proliferation (Colombo et al., 2003), increaseshe production of nitric oxide (Gustafsson and Brunton, 2000),ncreases DNA synthesis (Kim et al., 2002) reducing collagen secre-ion (Ostrom et al., 2003).
. Aging and autophagy: a challenge for the future
Aging is an inevitable process due to progressive accu-ulation of abnormal macromolecules and organelles in the
ytosol. To destroy short-lived cytosolic proteins, there areany non-lysosomal proteolytic pathways, like calpain, cytosolic
alcium-dependent cysteine proteases (Sorimachi et al., 1997) andPS (Wójcik et al., 2004). Moreover, long-lived cytosolic proteinsnd damaged organelles are removed by the autophagic/lysosomalystem (Martinez-Vicente et al., 2005; Kim et al., 2008). This systems greatly damaged and compromised in old age, whereas the otherhows minor or no substantial age-related changes (Cuervo andice, 2000; Cuervo et al., 2005). The high involvement of autophagy
n aging is due to reduced detoxification ability of aging cells. Ettoreergamini’s group showed that the autophagic mechanism is notorking well in different aging models in the liver in vitro and
n vivo (Donati et al., 2001; Del Roso et al., 2003; Bergamini et al.,004).
Autophagy, as reported above, is considered to be the majoregradative process of eukaryotic cells with two fundamentalurvival functions: to provide an energy source during starva-ion, and to eliminate affected or superfluous macromoleculesnd organelles, thus contributing to subcellular clearance (Moreaut al., 2010). The increased presence of autophagosomes inying/dead cells allowed to define this process as type II pro-rammed cell death (Schweichel and Merker, 1973); this concepts true also for selective targeting of key cell survival elements or ashe results of excessive self-digestion (Yu et al., 2006; Scarlatti et al.,009). Indeed, when the cells are stressed, the autophagy plays arotective role because Atg gene knockdown/knockout acceleratesather than delays cell death (Levine and Yuan, 2005; Levine androemer, 2008). It is a relevant issue whether autophagy is only arotective response or can also be a detrimental process, but at thisurpose it is accepted that it represents a self-limited survival strat-gy that, if left unchecked, can lead to adverse effects in cells underertain pathological conditions. Therefore, the prevailing idea ishat autophagy is a necessary process, but one that must be bal-nced by positive and negative regulators (Heymann, 2006; Liang,010).
As reported above the ability of cells to remove damaged pro-eins or organelles decrease with aging and this concept has beenlso reported in hepatic cells of old rats (Yin et al., 2008).
Droge (2004) proposed that autophagy has three functionshat are relevant to the aging process: regulation of the amino
cid balance, damaged protein accumulation control, and selec-ive organelle turnover (Droge, 2004). In old age, autophagy maye compromised by altered absorption of amino acids or defects innsulin-receptor signaling. In contrast, intensification of autophagy
Reviews 11 (2012) 10– 31 15
by caloric restriction is a well-known practice that is associatedwith increased longevity. In lower eukaryotes such as yeast and Dic-tyostelium discoideum, autophagy is essential for survival in timesof limited nutrient availability (Klionsky and Emr, 2000). Moreover,the cleaning of damaged cellular components and old organellessuch as mitochondria by macroautophagy is crucial for the mainte-nance of a healthy cell. Indeed, normal metabolism often resultsin oxidative damage to cells, and if altered organelles are notremoved by autophagy, they may themselves cause increased freeradical production. As the mitochondrial permeability transitioninitiates autophagy, there is a strong possibility that mitochon-dria may be particular targets for this process. For this reason,efficient autophagy is assumed to protect cells against death asso-ciated with defective or damaged mitochondria. Finally, autophagystrictly regulates protein turnover, and if altered, can lead to certainproteinopathies and neurodegeneration typical of aging (Wong andCuervo, 2010).
5.1. Longevity pathways and autophagy
Aging is a complicated process involving the integrationbetween environmental/extracellular factors with longevity path-ways strictly dependent by autophagy: these include the insulinreceptor, amino acid-dependent signaling (Droge, 2004) and ROSaccumulation (Scherz-Shouval and Elazar, 2007). During agingall of these signals are compromised by changes in conditionsaffecting amino acid delivery and the insulin receptor that ledto dysregulation, and contribute to the mechanism of aging(Rajawat et al., 2009). During starvation, constitutive autophagyis strongly induced whereas mTOR, a key governor of cell growthand metabolism is inhibited. So, it is a master negative regulator ofautophagy (Dennis et al., 2001; Wullschleger et al., 2006).
The inhibition of TOR as part of autophagy induction occurs inall eukaryotic cells, whereas other regulatory mechanisms suchas extracellular regulated protein kinase- 1,2 (ERK1,2) and p38-mitogen-activated protein kinase (p38-MAPK) signaling are lessuniversal (Neufeld, 2010). Indeed, the nutrient sensing TOR path-way is also emerging as a key regulator of aging (Hands et al., 2009).In particular, the insulin and insulin-like growth factor 1 (IGF-1)receptor constitutes a common signaling axis that controls agingfrom yeast to mice (Kaeberlein et al., 2005; Harrison et al., 2009).
mTOR is able to act under nutrient-rich but also under starva-tion conditions. This protein kinase interacts with other proteinsto form two main complexes, mTOR complexes 1 and 2 (mTORC1and mTORC2) (Blagosklonny and Hall, 2009). mTORC1 is activatedby hormones, mitogens and growth factors, and amino acids, butis negatively regulated in the absence of nutrients or by stress-ful conditions, such as decreased energy (ATP) availability (Hall,2008; Wang and Proud, 2006). Indeed, mTORC1 regulates a range ofessential cellular functions, the best understood of these being pro-tein synthesis (mRNA translation), which is positively regulated bymTORC1. Conversely, mTORC1 signaling inhibits autophagy (Wangand Proud, 2006). It has also been suggested that mTOR modu-lates autophagy through the regulation of the phosphatase PP2A(Yorimitsu et al., 2009), via p70S6 and its transcriptional targets,or signaling through Akt. For example, using RNAi knockdown incell lines, p70S6 kinase has recently been shown to be requiredfor the starvation-induced autophagic response (Armour et al.,2009). The important role of macroautophagy, as nonselective func-tions, in removing, for example, oxidized proteins, increased inaged cells under normal growth conditions as well as selectiveautophagy can occur to remove damaged or old organelles. There
is also accumulating evidence for selective autophagic processesin response to ROS and oxidative stress associated with aging. Inparticular, mitophagy, the selective degradation of mitochondria,can be induced by several stimuli and it is known to decrease the
1 search
paptce
tfioooacmdtigrocf
tyrpnp2pi2affK
diafi
TAa
6 R. Rezzani et al. / Ageing Re
otential oxidative damage from defective mitochondria (Essicknd Sam, 2010). We must underline that autophagy, in the earlyhase of oxidative stress, plays a protective role against it, and thathe loss of autophagic function in aged cells is the reason why theseells have a lower tolerance to oxidative-induced injury (Vittorinit al., 1999).
The recent challenges in the research on aging are two-fold:o find new factors contributing to aging, and to translate thesendings into therapeutic strategies for improving the health-spanf the aging human population. At this regards, the decreasef autophagy could be a useful point explaining why the olderrganisms are unable to fight off cell abnormalities, and whyutophagy may improve cell function delaying the aging-associatedhanges and the onset of aging-related disorders. Moreover, bothacroautophagy (Donati, 2006) and CMA (Cuervo and Dice, 2000)
ecrease in aging and in several age-related disorders together withhe malfunctioning of different proteolytic systems, and it makesmpossible to activate compensatory mechanisms. Autophagyenetically interacts with many pathways to mediate the longevityesponse and may be placed at the base of the signaling networkf aging (Madeo et al., 2010). Whether age-associated changes inritical autophagy-related proteins contribute to macroautophagyailure is, thus, the subject of many studies.
It is accepted that the basal autophagy is related to the selec-ive removal of old, damaged and not necessary organelles ineast and mammals, but it is also important to consider that theequire of autophagy is different among cell types; for exam-le, autophagy plays a crucial role in non-dividing cells such aseurons, glomerular podocytes and myocytes, where the overex-ression of autophagy genes prolongs lifespan (Simonsen et al.,008; Hartleben et al., 2010). There is a large evidence that lifes-an extension is dependent on autophagy action, which declines
n aging resulting in impaired protein homeostasis (Koga et al.,011). Although the mechanism responsible for age-associatedutophagic impairment is unknown, a considerable part may deriverom the inability of lipofuscin-loaded secondary lysosomes touse with autophagosomes and form autolysosomes (Levine androemer, 2008).
Recent studies by the Cuervo’s group determined a number ofefects that lead to decreased activity of CMA with age, suggest-
ng the way to correct and improve cellular function (Orensteinnd Cuervo, 2010; Cuervo, 2010). The studies of many groups areocusing on the breakdown of autophagic pathways, and if restor-ng them would lead to normal cellular activity. Using a transgenic
able 1 list of the phenotypes resulting from tissue-specific autophagy disruption in mice genllele in each murine model; “promoter” indicates the promoter controlling Cre recombin
Tissue Targeted gene Promoter Phenotype
Liver Atg7 Mx1(I); AIb Hepatomegaly, acSkeletal muscle Atg5 Atg7 HSA MyLc1 Type I fiber atroph
accumulation of pCardiac muscle Atg5 Atg5 A-MyHC (I)
MyLC2vCardiac hypertropmortality, age-relmitochondria
Pancreas �-cells Atg7 Rip Impaired glucose
Pancreas acinar cells Atg5 EL-1 Resistance to acutSmall intestine Atg5, Atg7 Villin Paneth cell dysfunT cells Atg5, Atg7 Lck Lymphopenia (T cB cells Atg5 CD19 Inefficient B cells
Dendritic cells Atg5 CD11c Defective MHCII-dHematopoietic cells Atg7 Vav Severe anemia, lyPodocytes Atg5 Podocin Age-related glomeAdipose tissue Atg7 Ap2 Resistance to obes
reduced white adiNeural cells Atg5, Atg7 FIP200 Nestin Nestin Neurodegeneratio
increased mortaliPurkinje cells Atg5, Atg7 Pcp2 Axonal degenerat
Reviews 11 (2012) 10– 31
mouse model with an inducible exogenous copy of LAMP-2A (Tet-off-LAMP-2A), they found that the activation of the transgene inliver of 22 to 26 months old mice (the equivalent of octogenari-ans in human years), was enough to restore levels of CMA closeto those observed in young mice. In contrast, the livers of normalmice in a control group began to fail. Remarkably, reduced levels ofLAMP-2A are not due to transcriptional downregulation or prob-lems in lysosomal targeting of this protein, but instead originatefrom age-related changes in the lysosomal membrane that affectlysosomal distribution and turnover of LAMP-2A and render thisprotein unstable (Kiffin et al., 2007). The lower levels of oxidizedand aggregated proteins observed in the old transgenic mice, alongwith their improved response to hepatotoxic stressors and betteroverall liver function, further support the participation of CMA incellular homeostasis and the stress response, and underscore thecontribution of reduced CMA activity to the functional decline inaging organisms (Kon and Cuervo, 2010).
Autophagy dysregulation is also associated with the mainsenescence-associated diseases including those associated withmetabolic disorders, neurodegeneration, inflammation, circulatorysyndromes and cancer (summarized in Table 1). In aging, thelipid peroxidation-related impairment of lysosomal function maycontribute to retinal pigment epithelium (RPE) cell dysfunctionvia intracellular processes that are directly affected by reducedlysosomal activity, such as lipofuscinogenesis and autophagy.Excessive lipofuscinogenesis secondary to intralysosomal accumu-lation of undegraded material, is detrimental to RPE cells due toits phototoxic properties (Schütt et al., 2000). Moreover, decreasedlysosomal capacity will impair autophagy. Particularly in long-lived, non-dividing cells like the postmitotic RPE, autophagy is vitalfor cellular homeostasis, and reduced autophagy activity results inaccelerated cell aging and cellular degeneration (Kaarniranta et al.,2009; Terman et al., 2007).
The decrease of both macroautophagy and CMA with age canbe prevented in animals subjected to caloric restriction, which isthe only intervention known to slow down aging. In the case ofmacroautophagy, caloric restriction maintains the ability of thelysosomal system to respond to nutritional and hormonal changeseven at advanced age. Although the mechanism by which caloricrestriction guarantees proper functioning of CMA in old rodents is
not clear, it is interesting to point out that in these animals CMAis constitutively activated to values up to the ones detected afterstarvation in young rats. This constitutive activation of CMA couldcontribute to the continuous removal of damaged proteins, thuserated using the Cre–Lox targeting system. “targeted gene” represents the floxedase expression in each model.
cumulation of ubiquitin-positive inclusions, lipotoxic degenerationy, accumulation of p62-ubiquitin inclusions; General muscolar atrophy,62-ubiquitin inclusions and damaged mitochondriahy, ventricular dilatation, contractile dysfunction; Cardiac failure and increased
ated cardiomyopathy, disorganized sarcomere structure and collapsed
tolerance, increased cell death in response to high fat diete pancreatitisctionells), apoptosis, accumulation of damaged mitochondriadevelopmentependent antigen presentation, viral infection
mphopenia, increased mortalityrulopathy, accumulation of p62-ubiquitin inclusions, ER stress, proteinuriaity, accumulation of p62-ubiquitin inclusions and damaged mitochondria,pose tissue and increased brown adiposen, ataxia, accumulation of p62-ubiquitin inclusions and damaged mitochondria,
ty (more in the Atg5, Atg7 neural knockout)ion, progressive late-onset ataxia, accumulation of p62-ubiquitin inclusions

search
pcs
5
mmomi2w(dFlAleatilhrnLiilH2vap
5p
m2sannbea
1
2
3
4
iWti
R. Rezzani et al. / Ageing Re
reventing their accumulation inside cells. However, the long-termonsequences of this continuous CMA activation will need furthertudy.
.2. Autophagy in progeria models: a paradoxical finding
In the last few years, studies on accelerated aging in animalodels have underlined and indicated that the maintenance orodulation of autophagy contributes to extend longevity. On the
ther hand, Lopez-Otin’s group has reported that a progeroidurine model exhibits extensive autophagy and its decrease dur-
ng normal aging (Marino and Lopez-Otìn, 2008; Marino et al., 2008,010). In humans, progeroid syndromes are dramatic diseases inhich certain features of human aging are prematurely developed
Navarro et al., 2006). Lopez-Otin’s group studied Zmpste24-eficient progeroid mice, lacking the Zmpste24 protein (called alsoACE-1), a zinc-metalloproteinase involved in the maturation ofamin A, an essential component of the nuclear envelope. Also lamin-deficient mice exhibited nuclear abnormalities and histopatho-
ogical defects remembering an accelerated aging process andlevated autophagy. They demonstrated that these mice showedn autophagy increase due to mTOR inhibition and to upregula-ion of AMP-activated protein kinase (AMPK) activity, a decreasen blood glucose levels and an increase of circulating adiponectinevels. These results indicated a deregulation of glucose and lipidomeostasis. This paradoxical autophagy might indicate a tempo-ary adaptation to reduce metabolic activity and cell division rateormally present in different progeroid syndromes (Marino andopez-Otìn, 2008). On the contrary, although autophagy activations useful for adaptation to stress, this pathway may lead to cell deathf chronically activated, and also contribute to progressive muscu-ar and cardiac wasting observed in progeria murine models andutchinson–Gilford progeria syndrome patients (Merideth et al.,008). At the end, the authors indicated that this issue is no wellerified and clarified, so future studies need to better evaluated ifutophagy could be a possible clinical marker for improving therognosis of progeria patients.
.3. Proteins involved in aging (biomarkers) and autophagyathways
During aging, the ability to activate stress response genes andarker proteins decrease and it is also compromised (Verbeke et al.,
001). The concept of “biomarkers of aging” was defined at leastince the 1980s when major efforts were undertaken to separateging and age-related diseases (Baker and Sprott, 1988). Unfortu-ately, most of the markers actually under discussion are relatedot only to age but also to diseases; thus, is difficult to find a “pure”iomarker of aging (Simm et al., 2008). However, the American Fed-ration for Aging Research has proposed the following criteria for
biomarker of aging (Johnson, 2006; Sprott, 2010):
. It must predict the rate of aging. In other words, it must be abetter predictor of lifespan than chronological age alone.
. It must monitor a basic process that underlies the aging process,not the effects of disease.
. It must be able to be tested repeatedly without harming theperson, for example, a blood test or an imaging technique.
. It must be something that works in humans and in laboratoryanimals, such as mice.
The importance for identifying the proteins involved in aging
s the possibility to evaluate their involvement in cell senescence.ith the term “senescence”, one considers a different cellular sta-us from aging because the cell is unable to proliferate, becomesnsensitive to growth factors and other signals able to induce cell
Reviews 11 (2012) 10– 31 17
proliferation, and is associated with DNA damage (Sikora et al.,2011).
In our opinion, the senescence is the intermediate state of cel-lular life since this indicate the clock of replicative pathway andgrowth arrest that is essentially permanent and cannot be reversedby physiological stimuli. It is surprising that there are four faces ofsenescence (tumor suppression, tumor promotion, aging and tissuerepair), some of which have apparently opposing effects. In light ofthese considerations, it is evident that we agree with a recent paperpublished by Rodier and Campisi (2011) in which these authorsreferred that the senescent cells are the orchestrators of tumor sup-pression, cancer, wound healing, and aging (Rodier and Campisi,2011). This explains why cellular senescence has a rich history,marked by unexpected complexity and why some aspects of itsphysiological significance remain conjecture, and several aspectsof its regulation remain enigmatic.
Cellular senescence has been identified in vivo and in vitroin different species, such as in human, baboon and mouse skin,human and rodent vascular endothelial and smooth muscle cellsin animal skeletal muscle, fat tissue and liver (Jeyapalan andSedivy, 2008). There are different types of senescence: replica-tive senescence depending on telomere erosion, stress-inducedpremature and also oncogene-induced senescence telomerase-independent. The senescence is correlated with altered cellularmorphology and the appearance of specific proteins, such assenescence-associated-�-galactosidase (SA-�-GAL), DNA damageresponse (DDR), promyelocytic leukaemia nuclear bodies (PMLNBs) (Sikora et al., 2011) (Fig. 4). In particular, based on histochem-istry, it has been observed increased beta galactosidase at pH 6.0in cultured cells under replicative or induced senescence, and isstrictly associated with lysosomes but no present in proliferatingcells (Lee et al., 2006). When the cells are stopped in senescencegrowth, there are different pathways supporting it, like cyclin-dependent kinase inhibitors (CDKIs) p21 and p16, that are regulatedby the tumor suppressor p53 (Campisi and d’Adda di Fagagna,2007). Several stimuli that generate a DNA damage response inducesenescence mainly through p53 signaling that is regulated at mul-tiple points by proteins such as alternate-reading frame (ARF), andthe ubiquitin-protein ligase (MDM2 in mice). It has been demon-strated that the expression of ARF and its coregulator, p16 INK4aincreased in senescent 25-month-old mice. In detail, this overex-pression has been seen in several organs, such as kidney, ovarystroma, pancreas, spleen and uterus. Moreover, the caloric restric-tion induced a decrease of these markers and an improvement oforgan damages (Krishnamurthy et al., 2004).
One of the first suggestions of a strict link between autophagyand aging was reported by Meléndez et al. (2003) showing that inCaenorhabditis elegans, RNAi-mediated depletion of Atg-6 results inincomplete morphogenesis and reduced lifespan. In contrast caloricrestriction produces lifespan extension and autophagy inductionwith a loss of cytosolic p53 (Tasdemir et al., 2008a). Interestingly, itwas demonstrated that decreased p53 was determined by variousautophagy inducers whereas inhibition of p53 prevents autophagy.These results have been well clarified by Tasdemir et al. (2008a)showing a double role of autophagy on the basis of its localization.If p53 is present in the nucleus, it can induce autophagy throughtranscriptional effects, but, if it is present in the cytosol, it acts asrepressor of autophagy (Tasdemir et al., 2008b).
Another important markers activated by caloric restriction aresirtuins, a number of NAD+-dependent histone/protein deacety-lases, observed in organisms ranging from yeasts to mammals,regulating cellular metabolism through deacetylation of several
transcription factors, such as FoxO proteins, p53, and the p65component of the NF-kB complex. In particular, sirtuin-1 (SIRT-1)antagonizes cellular senescence in human diploid fibroblasts andprotects mouse cardiac muscle against oxidative stress (Salminen
18 R. Rezzani et al. / Ageing Research Reviews 11 (2012) 10– 31
SStress
�
�
�
�
�
Different signalling
pathways involved:Accumulation of
senescent cells:Cell cycle
arrest
SA-β- GAL
Changed
morpholo gy
Chromatin
modifica tion
DNA da mag e
Instability of
chromos omes
Secretome
Obesity and
dia betes
Neurodeg ene ration
Atheroscler osis
Cancer
teins
athdtmeoo2t(plthEae
r
Fig. 4. Biochemical pro
nd Kaarniranta, 2009; Huang et al., 2008; Ghosh and Sil, 2008). Ifhe main function of autophagic degradation seems to be cellularousekeeping, probably SIRT-1 and autophagy are strictly depen-ent (Morselli et al., 2010). Indeed, Lee et al. (2008) demonstratedhat SIRT-1 deacetylates specific Atg proteins in a NAD+-dependent
anner, whereas the absence of this protein in cultured cells and inmbryonic and neonatal tissues increases their acetylation. More-ver, SIRT-1, through FoxO3 deacetylation, stimulates autophagy ofrganelles such as mitochondria in aged mice kidney (Kume et al.,010). Recent studies point out different activators of SIRT-1 withherapeutic potency in several metabolic and age-related diseasesOpie and Lecour, 2007). In particular, red wine polyphenolic com-ounds, e.g., resveratrol, can increase SIRT-1 activity and extend
ifespan by mimicking the effects of caloric restriction. It is par-icularly interesting that resveratrol, besides SIRT-1, triggers theeat shock response increasing HSP70 chaperone expression andR stress; this effect may compensate for aging changes, and medi-
te resistance and lifespan extension (Hashimoto et al., 2001; Steint al., 1999).Other important marker regulating metabolism and stressesistance in mammals is FoxO (forkhead box O) a family of
Fig. 5. Sirtuin1 and
involved in autophagy.
transcription factors, homologs of the DAF-16 protein in C. ele-gans (De Gaetano et al., 2002). Recently, FoxO3 protein was able tomodulate the autophagic pathways along with the UPS in skeletalmuscle, whereas in non-muscle cell types, the same marker stim-ulates autophagy without proteasomal involvement. mTOR andalso p53, if it is present in the cytosol, as reported above, are themain inhibitors of autophagy. So, they have inhibitory effects onlongevity. Indeed, SIRT-1, stimulated by caloric restriction, resver-atrol and cofactor nicotinamide adenine dinucleotide (NAD+) havepositive effects on autophagy and on longevity. Moreover, SIRT-1 positively regulates FoxO transcription factors and, in particular,FoxO3, which, in its turn, is able to activate autophagy and promotethe longevity (Fig. 5).
The Sestrins (Sesns), a family of highly inducible conservedstress-responsive proteins, which have been suggested to adjuststress and metabolic responses in different organisms fromDrosophila to mammals are also important markers (Budanov,
2011). Remarkably, Sesns induce autophagy by inhibition of TORC1and can also reduce ROS production by increasing the efficiency ofmitochondrial respiration, so they retard the appearance of age-associated cardiac pathologies (Lee et al., 2011). The question is:autophagy.

search
mot
6a
ittomscat
6
gtctptpaaoicIsa
haipscscta1atabsoAvra
imuaol
R. Rezzani et al. / Ageing Re
ay hypoxic preconditioning induce Sesns and stimulate the levelf autophagy, thus preventing cardiac and neurological attacks? So,his hypothesis must still be evaluated.
. Morphological and biochemical studies on autophagy inging and aging-dependent diseases
Morphological studies can provide invaluable insight in thenterpretation of autophagy linked to aging. Moreover, the iden-ification of autophagic proteins, that are mainly common alsoo aging, plays a primary role in the actual biomedical researchn longevity, where morphological approaches must be comple-entary to the molecular or proteomic findings. Hereafter, we
ummarize recent studies dealing with morphological and bio-hemical studies of autophagy in several tissues/organs in agingnd aging-dependent diseases in humans and experimental sys-ems.
.1. Cartilage
Aging commonly affects joints and articular cartilage by the pro-ressive accumulation of damaged macromolecules and organelles,hus reducing normal function and cellular survival. The reducedellularity and degradation of extracellular matrix are major his-ological features of osteoarthritis (OA), the most common jointathology in senescence (Taniguchi et al., 2009). In particular, thisissue is characterized by a very low rate of cell turnover, and, thus,hysiological mechanisms that favor the repair of cellular dam-ge are very critical for the maintenance of chondrocyte survivalnd function. However, until now little was known about the rolef autophagy in articular cartilage. Roach et al. (2004) describedn chondrocytes a peculiar variant of apoptotic cell death, calledhondroptosis, a term that also includes an autophagy component.n a rat OA model, the progression of cartilage degeneration in theuperficial zone and partial middle zone is driven by chondrocyteutophagy in addition to cell death (Roach et al., 2004).
The mechanistic aspects of autophagy in OA pathogenesisave not been clarified, but hypoxia inducible factor 1 (HIF-1)ppears to actively function in this process. When HIF-1 expressions silenced in chondrocytes, BECN-1 and microtubule-associatedrotein 1 light chain 3 (MAP1LC3) expression are significantlyuppressed. Because both BECN-1 and LC3 are required to exe-ute the autophagy program, this finding suggests that HIF-1erves to promote autophagy (Bohensky et al., 2007). On theontrary, HIF-2 seems to function as a negative regulator ofhe autophagic pathway because HIF-2 silencing elicits a strongutophagic response in chondrocytes, and an upregulation of HIF-
expression. Accordingly, in HIF-2�−/− mice, elevated autophagicctivity was observed in growth plates. HIF-2 expression seemso diminish in OA chondrocytes, implying deregulation of theutophagic pathway (Bohensky et al., 2009). As discussed above,oth HIF-1� and HIF-2� are small ubiquitin-like modifier (SUMO)ubstrates. Therefore, SUMO regulation of HIF may influence notnly the hypoxic response, but also autophagy in chondrocytes.utophagy also intertwines with cellular senescence and death inarious contexts, which further complicates its currently obscureole in chondrocyte biology (Vellai et al., 2009). Again, HIF-1 mayct as a node, which interconnects these pathways.
Recent studies carried out by Carames et al. (2010) usingmmunohistochemical analysis showed the presence of three
arkers mainly involved in induction (Ser/Thr kinase ULK1), reg-
lation (BECN-1) and execution (LC3) of autophagy in the normalnd OA human cartilage and in knee joints of mice with OA. Allf the above markers are constitutively expressed in normal carti-age where basal autophagy may be a protective and homeostaticReviews 11 (2012) 10– 31 19
mechanism, but in aging- or surgically induced OA there isa significant reduction or loss of their staining, related toincreased apoptosis detected by PARP/p85 overexpression. Thus,the autophagic pathways are associated with the onset or develop-ment of OA, due to the accumulation of damaged macromolecules.
Another interesting morphological study by Couve andSchmachtenberg (2011) recently outlined in vitro the deep rela-tion between autophagy and aging in odontoblasts. Indeed,these long-lived postmitotic cells are responsible for secretionand maintenance of dentin. Odontoblasts express both LC3 andLAMP-2, autophagosomal and lysosomal markers, in an age-dependent pattern, and display progressive lipofuscin depositionduring aging. The prevalence of lipofuscins and the reduction ofthe autophagy–lysosomal system might be responsible for thereduced dentinogenic secretory ability in this cell type (Couve andSchmachtenberg, 2011).
In light of the data reported above, it is clear that there areconflictual results about autophagy changes in OA and aging chon-drocyte.
6.2. Liver
The intracellular storage and utilization of lipids are critical tomaintain hepatocyte energy homeostasis. During nutrient depri-vation, cellular lipids stored as triglycerides in lipid droplets arehydrolyzed into fatty acids for energy. A second cellular responseto starvation is the induction of autophagy, which delivers intra-cellular proteins and organelles sequestered in double-membranevesicles (autophagosomes) to lysosomes for degradation and useas an energy source. Lipolysis and autophagy share similarities inregulation and function (Shih et al., 2010). Decreased autophagyin the liver with aging may contribute to hepatic lipid accumula-tion that occurs along with an increased incidence of the metabolicsyndrome in aged humans. The ability of increased lipid content toimpair autophagy also indicates that lipid accumulation could con-tribute to the decrease in autophagic function with aging. It existsa parallelism between autophagy and lipolysis. They are regulatedhormonally by insulin and glucagon and are increased during star-vation. Except for the processing of endocytosed lipoproteins, nodirect involvement of the lysosomal degradation pathway in lipidmetabolism has been established. The regulatory and functionalsimilarities between autophagy and lipolysis, along with the capa-bility of lysosomes to degrade lipids, indicated that autophagy maycontribute to lipid droplets and triglyceride breakdown. Therapeu-tic strategies to increase autophagic function may therefore providea new approach to prevent the metabolic syndrome and its associ-ated pathologies. At this regards, it is useful to report that dietarycaloric restrictions and the antilipolytic agents have been provento efficiently stimulate autophagy in old rodents (Cuervo et al.,2005) indicating that an antiaging strategy exists that exploits thisphenomenon.
The reduction or loss of autophagy results in severe injuryassociated with large amounts of cytoplasmic ubiquitin-positiveproteinaceous inclusions, typically present in steatohepatitis,alcoholic hepatitis, and �1-antitrypsin deficiency (Stumptneret al., 2002). Komatsu et al. (2007) using immunofluorescencedemonstrated that in the liver of transgenic GFP-LC3 mice theautophagosomal signal colocalizes with p62, and that a similarfinding is also evident in GFP-LC3 transgenic mice starved for1 day. p62 is a unique protein largely conserved in metazoansand plants, but not in yeasts, which can bind other proteins tomediate diverse signaling pathways, and is also associated with
liver inclusions in autophagy-deficient hepatocytes. Indeed, in p62Knockout mice and flies, its suppression determines the appearanceof ubiquitin-positive protein aggregates, indicating the importanceof this protein in the formation of inclusions. So, the p62-impaired
2 search
tltvchbaaceadaa
wtiAlit
6
fooagmmemnrfmdtUorp
rssmmtoslmpam(mUudt
0 R. Rezzani et al. / Ageing Re
urnover is a major cause of the pathogenic changes seen in theiver of autophagy-deficient mice (Komatsu et al., 2010). Many ofhese changes are strictly corroborated by morphological obser-ations such as hepatocyte hypertrophy, narrowing of sinusoidalapillaries, and abnormal bile ducts, all signs of cholestasis and/oremostasis in the liver. The novel autophagy-stimulating drug car-amazepine has been recently demonstrated to be very usefulgainst �1-antitrypsin deficiency where it reduced liver fibrosis in
mouse model of the disease (Hidvegi et al., 2010). Moreover, inhronic primary biliary cirrhosis, autophagy may be involved in bilepithelial cells lesions; LC3 immunostaining was detected by Sasakind Nakanuma (2010), where it may mediate the process of biliaryucts senescence. Indeed, senescent markers such as p21 and p16re coexpressed with LC3 in damaged epithelial biliary cells (Sasakind Nakanuma, 2010).
Overall, autophagy seems to be a pro-survival, protective path-ay in the liver, and its role in liver injury, if any, remains
o be elucidated (Rautou et al., 2010). In the liver, autophagys a prominent survival mechanism under nutrient deprivation.ggregation-prone mutant �1-antitrypsin is also cleared from the
iver by autophagy. Mice deficient for Atg7 in the liver demonstratempaired adaptation to starvation, organelle turnover, accumula-ion of aggregated proteins and organelles, and hepatomegaly.
.3. Skeletal muscle system
Muscle aging is characterized by a decline in functional per-ormance and restriction of adaptability, due to progressive lossf muscle tissue coupled with a decrease in strength and forceutput. Together with selective activation of apoptotic pathways,
hallmark of age-related muscle loss or sarcopenia is the pro-ressive incapacity of regeneration machinery to replace damageduscle. These characteristics are shared by pathologies involvinguscle wasting, such as muscular dystrophies or amyotrophic lat-
ral sclerosis, cancer and AIDS, all characterized by alterations inetabolic and physiological parameters, and progressive weak-
ess in specific muscle groups. Modulation of extracellular agonists,eceptors, protein kinases, intermediate molecules, transcriptionactors and tissue-specific gene expression collectively compro-
ise the functionality of skeletal muscle tissue, leading to muscleegeneration and persistent protein degradation through activa-ion of proteolytic systems, such as those involving calpains, thePS and caspases (Vinciguerra et al., 2010). A better understandingf the mechanisms underlying the pathogenesis of muscle atrophyepresents an important first step for the development of thera-eutic approaches to rescue muscle atrophy in aging and disease.
The role of autophagy in regulating muscle mass has been onlyecently addressed with regard to organelles (Sandri, 2010). Themall size of autophagosomes in skeletal tissue made it harder totudy autophagy morphologically before the era of transgenic ani-als. Transgenic GFP-LC3 mice expressing LC3 represent a usefulodel to study activation of autophagy during fasting in skele-
al muscle. This animal model was adopted to compare the sizef autophagosomes in skeletal muscle relative to other tissues,uch as liver, heart, and pancreas where the autophagosomes arearger (Mizushima et al., 2004). However, if excessive autophagy
ay be dangerous to muscle cells due to higher wasting of cyto-lasmic proteins (Dobrowolny et al., 2008) inhibition of autophagylso plays a role in many myopathies with inclusions or abnor-al mitochondria (Temiz et al., 2009). Recently, Masiero et al.
2009) showed, by morphological analysis, a significant loss ofuscle mass associated with compensatory upregulation of the
PS and apoptosis in muscle-specific Atg7 knockout mice. Usingltrastructural studies, the authors detected swollen mitochondria,ilated sarcoplasmic reticulum and atypical inclusions similar tohose observed in Atg7-deficient livers and Atg5-deficient heartsReviews 11 (2012) 10– 31
(Nakai et al., 2007). By succinate dehydrogenase histochemistryand p62 immunostaining performed in both glycolytic and oxida-tive muscles in a tamoxifen-inducible specific adult Atg7 knockoutmodel, they further demonstrated the accumulation of abnormalmitochondria, and hypothesized that autophagy inhibition, con-firmed by undetectable LC3, triggers muscle wasting and maycontribute to aging sarcopenia. In this genetic model of autophagy,three weeks of autophagy inhibition is sufficient to promote muscleatrophy and weakness associated with impaired force transmis-sion. Ultrastructural analysis clearly outlines altered sarcomericdisposition, and myofibrils assemble associated with centrallynucleated fibers, whose number increase with age (Masiero andSandri, 2010). Thus, the maintenance of autophagy in skeletal mus-cles may represent an important tool to rejuvenate organelles andto prevent dysfunctional inclusions.
6.4. Cardiovascular system
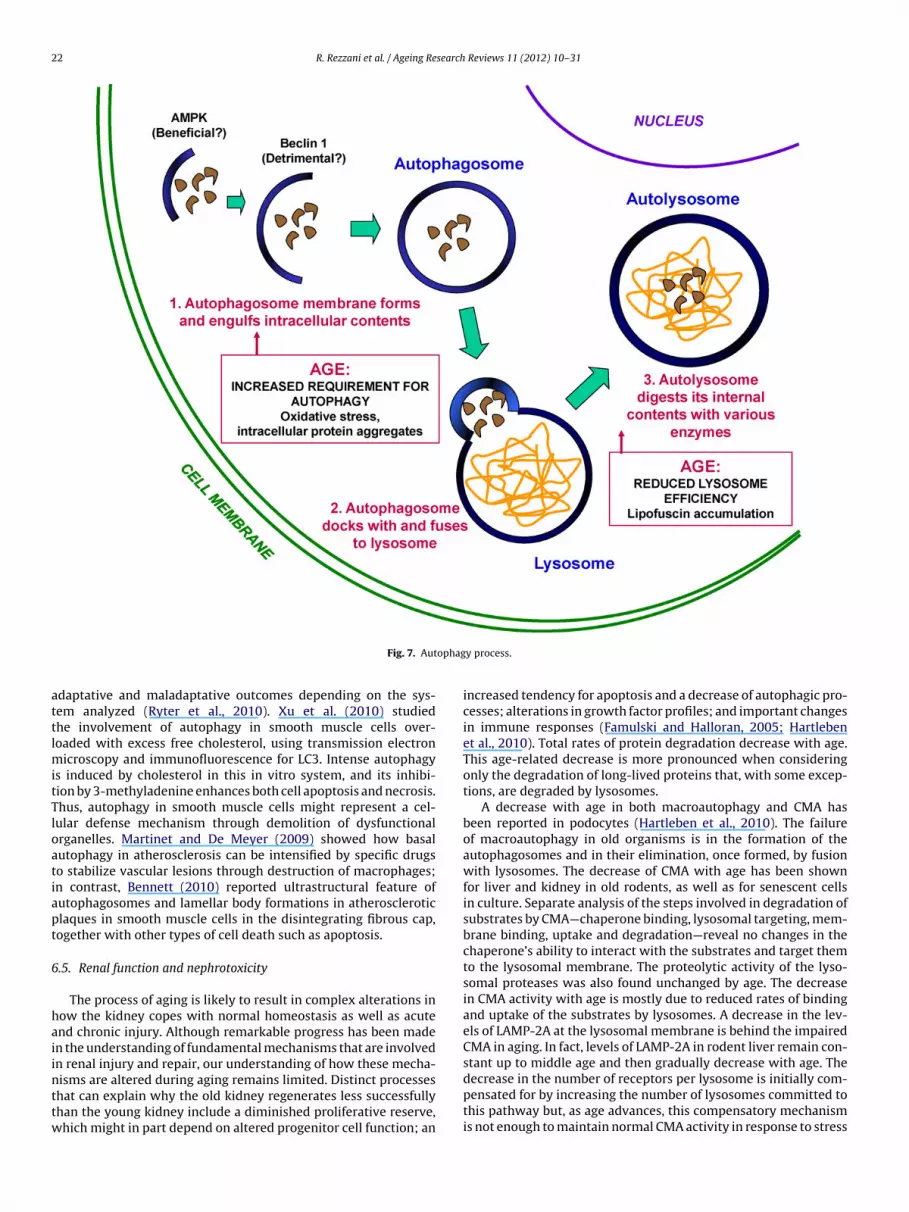
Elderly patients are more likely than young patients to experi-ence a myocardial infarction (MI) and are more likely to developheart failure following MI. The old people shows increase in stressexposure and shifts in signaling pathways determining changes inthe biology of cardiomyocytes. In fact, aging is characterized bydecreased cardiomyocyte renewal, possibly through the impair-ment of both stem cell differentiation into cardiomyoctyes andcardiomyocyte division. Additionally, changes in cardiomyocytecytoarchitecture and metabolic pathways increase the cell’s apo-ptosis. When the damaged proteins and organelles increase incardiomyoctyes block the autophagy pathways (Shih et al., 2010).In fact, during aging, cardiomyocyte renewal decreases throughthe impairment of both stem cell differentiation and cellular divi-sion. Moreover, progressive accumulation of damaged proteins andorganelles in aged cardiomyocytes blocks autophagy, leading toapoptosis of the cell. Compensatory neurohormonal signals inducethe remaining cardiomyocytes to undergo hypertrophy, increasingtheir metabolic demand (Fig. 6).
Autophagy is essential for cardiomyocytes because the longevityof cardiomyocytes and their high energy requirements requireshigh levels of cumulative oxidative damage and stress. Mean-while, many cardiomyocytes do not divide, the cellular wastethrough mitosis is not an option. Effective autophagy in cardiomy-ocytes is therefore necessary to continue normal metabolism and tomaintain the cellular survival. The failure of autophagy causes car-diomyopathy in experimental and clinical models and one markerresponsible of these problems is the gene Atg5. In fact, an accu-mulation of damaged proteins, due to disruption of Atg5 gene,leads to hypertrophy, subsequent dilated cardiomyopathy, andheart failure. Although functional autophagy is crucial for normalfunctioning of cardiomyocytes, the effects of myocardial infarc-tion (MI) on autophagy, and conversely the effects of autophagyon the heart following MI, remain incompletely described. Inhearts subjected to reperfused MI, autophagy is activated firstby an AMP activated protein kinase (AMPK)-dependent mecha-nism during the ischemic period, followed by BECN-1-dependentautophagy during reperfusion (Matsui et al., 2007). It is importantto report that autophagy promotes the survival of cardiomy-ocytes during the AMPK-dependent (ischemic) phase, but becomesdetrimental during the BECN-1-dependent (reperfusion) phase(Fig. 7). Cardiomyocyte requirement for autophagy increases withage. Damage from ROS impairs functional organelles is especiallypronounced with aging. The accumulation of oxidative damageproduces mitochondrial damages with changes in their cytoarchi-
tecture. These changes make the organelles disables for energyproduction and prone to ROS leakage (Brunk and Terman, 2002).Moreover, mutation accumulation and protein aggregation, impairmitochondrial fission (Terman et al., 2003) which otherwise serves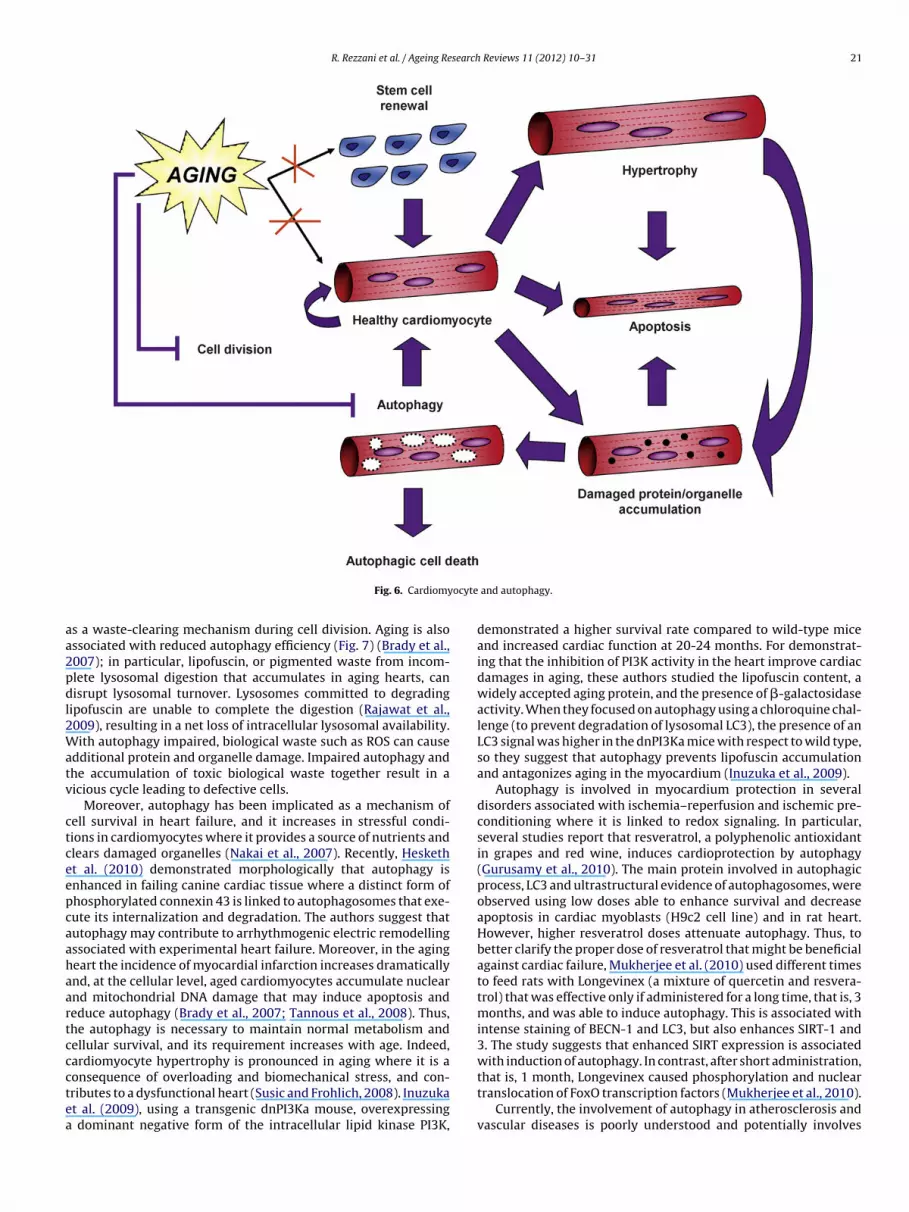
R. Rezzani et al. / Ageing Research Reviews 11 (2012) 10– 31 21
ocyte
aa2pdl2Watv
ctceepcaahaartccctea
Fig. 6. Cardiomy
s a waste-clearing mechanism during cell division. Aging is alsossociated with reduced autophagy efficiency (Fig. 7) (Brady et al.,007); in particular, lipofuscin, or pigmented waste from incom-lete lysosomal digestion that accumulates in aging hearts, canisrupt lysosomal turnover. Lysosomes committed to degrading
ipofuscin are unable to complete the digestion (Rajawat et al.,009), resulting in a net loss of intracellular lysosomal availability.ith autophagy impaired, biological waste such as ROS can cause
dditional protein and organelle damage. Impaired autophagy andhe accumulation of toxic biological waste together result in aicious cycle leading to defective cells.
Moreover, autophagy has been implicated as a mechanism ofell survival in heart failure, and it increases in stressful condi-ions in cardiomyocytes where it provides a source of nutrients andlears damaged organelles (Nakai et al., 2007). Recently, Hesketht al. (2010) demonstrated morphologically that autophagy isnhanced in failing canine cardiac tissue where a distinct form ofhosphorylated connexin 43 is linked to autophagosomes that exe-ute its internalization and degradation. The authors suggest thatutophagy may contribute to arrhythmogenic electric remodellingssociated with experimental heart failure. Moreover, in the agingeart the incidence of myocardial infarction increases dramaticallynd, at the cellular level, aged cardiomyocytes accumulate nuclearnd mitochondrial DNA damage that may induce apoptosis andeduce autophagy (Brady et al., 2007; Tannous et al., 2008). Thus,he autophagy is necessary to maintain normal metabolism andellular survival, and its requirement increases with age. Indeed,ardiomyocyte hypertrophy is pronounced in aging where it is a
onsequence of overloading and biomechanical stress, and con-ributes to a dysfunctional heart (Susic and Frohlich, 2008). Inuzukat al. (2009), using a transgenic dnPI3Ka mouse, overexpressingdominant negative form of the intracellular lipid kinase PI3K,
and autophagy.
demonstrated a higher survival rate compared to wild-type miceand increased cardiac function at 20-24 months. For demonstrat-ing that the inhibition of PI3K activity in the heart improve cardiacdamages in aging, these authors studied the lipofuscin content, awidely accepted aging protein, and the presence of �-galactosidaseactivity. When they focused on autophagy using a chloroquine chal-lenge (to prevent degradation of lysosomal LC3), the presence of anLC3 signal was higher in the dnPI3Ka mice with respect to wild type,so they suggest that autophagy prevents lipofuscin accumulationand antagonizes aging in the myocardium (Inuzuka et al., 2009).
Autophagy is involved in myocardium protection in severaldisorders associated with ischemia–reperfusion and ischemic pre-conditioning where it is linked to redox signaling. In particular,several studies report that resveratrol, a polyphenolic antioxidantin grapes and red wine, induces cardioprotection by autophagy(Gurusamy et al., 2010). The main protein involved in autophagicprocess, LC3 and ultrastructural evidence of autophagosomes, wereobserved using low doses able to enhance survival and decreaseapoptosis in cardiac myoblasts (H9c2 cell line) and in rat heart.However, higher resveratrol doses attenuate autophagy. Thus, tobetter clarify the proper dose of resveratrol that might be beneficialagainst cardiac failure, Mukherjee et al. (2010) used different timesto feed rats with Longevinex (a mixture of quercetin and resvera-trol) that was effective only if administered for a long time, that is, 3months, and was able to induce autophagy. This is associated withintense staining of BECN-1 and LC3, but also enhances SIRT-1 and3. The study suggests that enhanced SIRT expression is associatedwith induction of autophagy. In contrast, after short administration,
that is, 1 month, Longevinex caused phosphorylation and nucleartranslocation of FoxO transcription factors (Mukherjee et al., 2010).Currently, the involvement of autophagy in atherosclerosis andvascular diseases is poorly understood and potentially involves
22 R. Rezzani et al. / Ageing Research Reviews 11 (2012) 10– 31
ophag
attlmitTloatiapt
6
haiinttw
Fig. 7. Aut
daptative and maladaptative outcomes depending on the sys-em analyzed (Ryter et al., 2010). Xu et al. (2010) studiedhe involvement of autophagy in smooth muscle cells over-oaded with excess free cholesterol, using transmission electron
icroscopy and immunofluorescence for LC3. Intense autophagys induced by cholesterol in this in vitro system, and its inhibi-ion by 3-methyladenine enhances both cell apoptosis and necrosis.hus, autophagy in smooth muscle cells might represent a cel-ular defense mechanism through demolition of dysfunctionalrganelles. Martinet and De Meyer (2009) showed how basalutophagy in atherosclerosis can be intensified by specific drugso stabilize vascular lesions through destruction of macrophages;n contrast, Bennett (2010) reported ultrastructural feature ofutophagosomes and lamellar body formations in atheroscleroticlaques in smooth muscle cells in the disintegrating fibrous cap,ogether with other types of cell death such as apoptosis.
.5. Renal function and nephrotoxicity
The process of aging is likely to result in complex alterations inow the kidney copes with normal homeostasis as well as acutend chronic injury. Although remarkable progress has been maden the understanding of fundamental mechanisms that are involvedn renal injury and repair, our understanding of how these mecha-
isms are altered during aging remains limited. Distinct processeshat can explain why the old kidney regenerates less successfullyhan the young kidney include a diminished proliferative reserve,hich might in part depend on altered progenitor cell function; any process.
increased tendency for apoptosis and a decrease of autophagic pro-cesses; alterations in growth factor profiles; and important changesin immune responses (Famulski and Halloran, 2005; Hartlebenet al., 2010). Total rates of protein degradation decrease with age.This age-related decrease is more pronounced when consideringonly the degradation of long-lived proteins that, with some excep-tions, are degraded by lysosomes.
A decrease with age in both macroautophagy and CMA hasbeen reported in podocytes (Hartleben et al., 2010). The failureof macroautophagy in old organisms is in the formation of theautophagosomes and in their elimination, once formed, by fusionwith lysosomes. The decrease of CMA with age has been shownfor liver and kidney in old rodents, as well as for senescent cellsin culture. Separate analysis of the steps involved in degradation ofsubstrates by CMA—chaperone binding, lysosomal targeting, mem-brane binding, uptake and degradation—reveal no changes in thechaperone’s ability to interact with the substrates and target themto the lysosomal membrane. The proteolytic activity of the lyso-somal proteases was also found unchanged by age. The decreasein CMA activity with age is mostly due to reduced rates of bindingand uptake of the substrates by lysosomes. A decrease in the lev-els of LAMP-2A at the lysosomal membrane is behind the impairedCMA in aging. In fact, levels of LAMP-2A in rodent liver remain con-stant up to middle age and then gradually decrease with age. The
decrease in the number of receptors per lysosome is initially com-pensated for by increasing the number of lysosomes committed tothis pathway but, as age advances, this compensatory mechanismis not enough to maintain normal CMA activity in response to stress
search
(L
fbeoiHacao
pPaektlblmbnMpdpmtaatcpiiteo
naratattpftmEsoacmswf
r
R. Rezzani et al. / Ageing Re
Cuervo and Dice, 2000). What causes the age-related decrease inAMP-2A levels remains unknown.
Growing evidence supports intercommunication among the dif-erent proteolytic systems, which are likely to be in a continuousalance. For example, the same protein can be degraded by differ-nt proteases, the same components are shared by different formsf autophagy, and the blockage of one of the autophagic pathwayss compensated, at least temporarily, by activation of the others.owever, since both macroautophagy and CMA are impaired withge, compensation is probably no longer possible, and this failureould contribute to the decrease in the ability to remove dam-ged intracellular components and to adapt to stress, as seen inld organisms.
Glomerular function and integrity are strictly associated withodocyte integrity, and its maintenance in aging (Wiggins, 2007).odocytes are terminally differentiated cells, like neurons, thatdopt autophagy to prevent the progression of glomerular dis-ase (Hartleben et al., 2010). Unfortunately, very little has beennown about the molecular nature of the reparative mechanismshat defend the podocyte against environmental stress and cel-ular aging. The understanding of this missing link could be theasis for successful therapeutic prevention of irreversible glomeru-
osclerosis. Our data now indicate that autophagy is an importantechanism for podocyte homeostasis. Autophagy has previously
een shown to be an essential cell repair and turnover mecha-ism for postmitotic cells such as neurons (Cuervo et al., 2005;izushima et al., 2008). Strikingly, in the kidney, glomerular
odocytes most prominently display autophagic activity. Atg5-eficient podocytes that are incapable of performing autophagy,rogressively accumulate biological “garbage” such as damageditochondria and ubiquitinated protein aggregates, underlining
he fact that the removal of this cellular waste depends onutophagy in this cell type. The compromised clearance of oldnd/or damaged mitochondria by autophagy coupled with reducedurnover of long-lived proteins probably contributes to the intra-ellular accumulation of oxidized proteins seen in Atg5-deficientodocytes. Podocytes are particularly susceptible to oxidative
njury. Both ER stress and oxidative stress ultimately lead torreversible podocyte injury and podocyte loss. This clearly dis-inguishes podocytes from other glomerular structures that arefficiently renewed due to proliferation, providing a dilution ofxidatively or otherwise damaged proteins and cell organelles.
The efficiency of autophagy seems to be a direct determi-ant of cellular aging for long-lived glomerular podocytes. Indeed,utophagy-deficient podocytes perfectly phenocopy many age-elated cellular alterations. The most prominent signs of thisccelerated aging in Atg5-deficient podocytes are the accumula-ion of lipofuscin, the formation of ubiquitinated protein aggregatesnd aggresomes, the occurrence of damaged mitochondria, andhe increase in the load of oxidized proteins. To our knowledge,his is the first genetic mouse model clearly featuring an agingodocyte. Aging in humans and rodents progressively impairs renalunction. The age-related structural kidney changes are charac-erized by loss of podocytes and glomerulosclerosis. The exact
echanisms underlying age-dependent renal injury are unknown.ven in the absence of known risk factors such as hyperten-ion or diabetes, otherwise healthy individuals (65 years old orlder) frequently present glomerulosclerosis (Wiggins, 2007). Theutophagy–lysosome system undergoes striking changes in agingells. Aging in general leads to a reduction in autophagosome for-ation and autophagosome–lysosome fusion. It is intriguing to
peculate that a gradual decrease of glomerular autophagic activity
ith age could play a major role in the functional decline of renalunction in human aging kidneys.In aging mice, ultrastructural analysis and immunofluorescence
eveal that ablation of Atg5 in podocytes results in podocyte
Reviews 11 (2012) 10– 31 23
loss and glomerulosclerosis. Indeed autophagy-deficient podocytesshare characteristics with aging neurons such as accumulation ofoxidized proteins, lipofuscins, and mitochondria abnormalities. Allthese findings demonstrate the importance of basal autophagy inmaintaining glomerular function.
Another recent study dealing with the protective role ofautophagy in a relevant kidney disease, ischemic damage, wasreported by Jiang et al. (2010) in vitro and in vivo. Nevertheless,if autophagy might be protective or detrimental for the kidney isnot clear at the moment, and its role in cisplatin nephrotoxicity iscontroversial (Inoue et al., 2010; Jiang and Dong, 2010).
6.6. Central nervous system (CNS) and neurodegeneration
Several neurodegenerative diseases, such as dementia, andAlzheimer and Parkinson diseases are a key risk factor aging andso they are an important issue to take in consideration in aging.To study the anatomical changes observed in aging and for dis-tinguishing these variations from dementia, it is necessary toidentify them from different point of view. These changes includealterations in receptors, loss of dendrites and spines, and myelindystrophy, as well as alterations in synaptic transmission. Patholog-ical processes such as the abnormal aggregation of specific proteinsare seen in many neurodegenerative diseases. Together, these mul-tiple factors likely constitute the substrate for age-related loss ofcognitive function. Cognitive aging can significantly affect produc-tivity and quality of life, and effective symptomatic or preventativetreatments could provide many benefits to individuals and society(Shineman et al., 2010). So, understanding the complexity, that isresponsible to distinguish the syndrome of cognitive aging fromnormal and diseases will allow the researchers to develop drugsimproving cognitive performance and to better known the biolog-ical mechanisms leading to brain damages.
Autophagy is essential for neuronal homeostasis, and itsdysfunction has been directly linked to a growing number ofneurodegenerative disorders. The problems inducing autophagicfailure in nervous system can be different because there are manymolecular players involved in the steps determining autophagicprocesses. It is very important to know the different step(s)involved and altered in the autophagic processes to better know thecourse of the neurological pathologies. Moreover, this will be essen-tial to developing targeted therapeutic approaches for each diseasebased on modulation of autophagy (Wong and Cuervo, 2010).
The nervous system is more sensible to decrease of autophagyduring acute starvation and it is more damaged when the damagedorganelles and misfolded proteins accumulated inside neuronsunless they are successfully removed. This is possible because thecellular division is mostly limited to developmental stages, andmature neurons have a limited or null potential of proliferation,and the accumulation of organelles and misfolded proteins cannotbe redistributed among daughter cells (Marino et al., 2011). Hence,the maintenance of a proper intracellular quality control is probablyone of the most demanding goals that neuronal cells must obtain(Nixon et al., 2008).
Normally, the balance between the formation and degradationof cellular proteins is required for cell survival. The damaged pro-teins are degraded by the UPS and autophagy (Ciechanover, 2005;Rubinsztein, 2006) and if these systems do not work correctly thedegeneration, as seen in Parkinson, Alzheimer, and Huntington dis-ease, and amyotrophic lateral sclerosis, and other related proteinconformation disorders are possible.
Nowadays, the protective role of autophagy in CNS has been
well established through the characterization of a CNS-specificautophagy-deficient animal model. The knockout models areavailable for prolonged neurodegeneration, including abnormalreflexes, progressive motor deficits, ataxia, growth retardation and,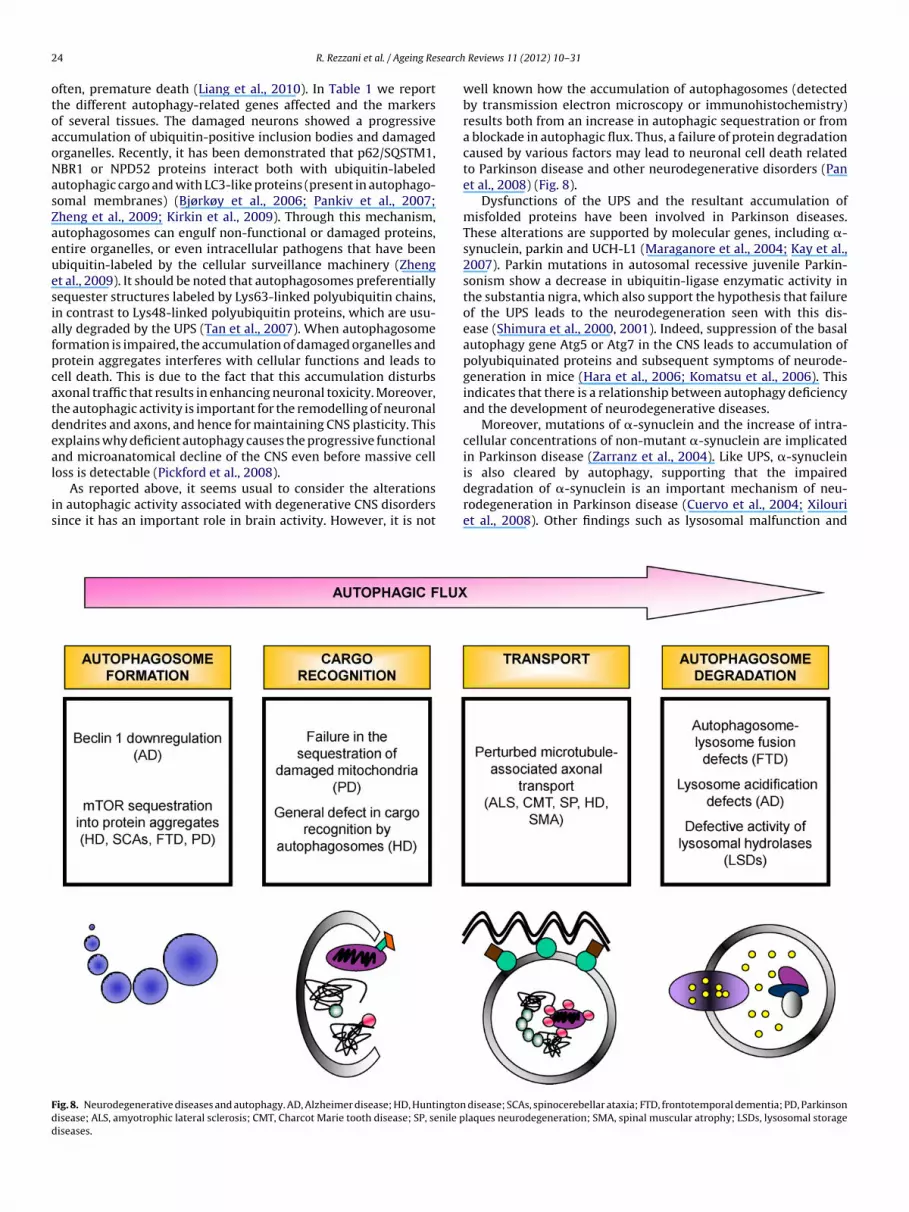
2 search
otoaoNasZaeuesiafpcatdeal
is
Fdd
4 R. Rezzani et al. / Ageing Re
ften, premature death (Liang et al., 2010). In Table 1 we reporthe different autophagy-related genes affected and the markersf several tissues. The damaged neurons showed a progressiveccumulation of ubiquitin-positive inclusion bodies and damagedrganelles. Recently, it has been demonstrated that p62/SQSTM1,BR1 or NPD52 proteins interact both with ubiquitin-labeledutophagic cargo and with LC3-like proteins (present in autophago-omal membranes) (Bjørkøy et al., 2006; Pankiv et al., 2007;heng et al., 2009; Kirkin et al., 2009). Through this mechanism,utophagosomes can engulf non-functional or damaged proteins,ntire organelles, or even intracellular pathogens that have beenbiquitin-labeled by the cellular surveillance machinery (Zhengt al., 2009). It should be noted that autophagosomes preferentiallyequester structures labeled by Lys63-linked polyubiquitin chains,n contrast to Lys48-linked polyubiquitin proteins, which are usu-lly degraded by the UPS (Tan et al., 2007). When autophagosomeormation is impaired, the accumulation of damaged organelles androtein aggregates interferes with cellular functions and leads toell death. This is due to the fact that this accumulation disturbsxonal traffic that results in enhancing neuronal toxicity. Moreover,he autophagic activity is important for the remodelling of neuronalendrites and axons, and hence for maintaining CNS plasticity. Thisxplains why deficient autophagy causes the progressive functionalnd microanatomical decline of the CNS even before massive celloss is detectable (Pickford et al., 2008).
As reported above, it seems usual to consider the alterationsn autophagic activity associated with degenerative CNS disordersince it has an important role in brain activity. However, it is not
ig. 8. Neurodegenerative diseases and autophagy. AD, Alzheimer disease; HD, Huntingtonisease; ALS, amyotrophic lateral sclerosis; CMT, Charcot Marie tooth disease; SP, senile piseases.
Reviews 11 (2012) 10– 31
well known how the accumulation of autophagosomes (detectedby transmission electron microscopy or immunohistochemistry)results both from an increase in autophagic sequestration or froma blockade in autophagic flux. Thus, a failure of protein degradationcaused by various factors may lead to neuronal cell death relatedto Parkinson disease and other neurodegenerative disorders (Panet al., 2008) (Fig. 8).
Dysfunctions of the UPS and the resultant accumulation ofmisfolded proteins have been involved in Parkinson diseases.These alterations are supported by molecular genes, including �-synuclein, parkin and UCH-L1 (Maraganore et al., 2004; Kay et al.,2007). Parkin mutations in autosomal recessive juvenile Parkin-sonism show a decrease in ubiquitin-ligase enzymatic activity inthe substantia nigra, which also support the hypothesis that failureof the UPS leads to the neurodegeneration seen with this dis-ease (Shimura et al., 2000, 2001). Indeed, suppression of the basalautophagy gene Atg5 or Atg7 in the CNS leads to accumulation ofpolyubiquinated proteins and subsequent symptoms of neurode-generation in mice (Hara et al., 2006; Komatsu et al., 2006). Thisindicates that there is a relationship between autophagy deficiencyand the development of neurodegenerative diseases.
Moreover, mutations of �-synuclein and the increase of intra-cellular concentrations of non-mutant �-synuclein are implicatedin Parkinson disease (Zarranz et al., 2004). Like UPS, �-synucleinis also cleared by autophagy, supporting that the impaired
degradation of �-synuclein is an important mechanism of neu-rodegeneration in Parkinson disease (Cuervo et al., 2004; Xilouriet al., 2008). Other findings such as lysosomal malfunction anddisease; SCAs, spinocerebellar ataxia; FTD, frontotemporal dementia; PD, Parkinsonlaques neurodegeneration; SMA, spinal muscular atrophy; LSDs, lysosomal storage

R. Rezzani et al. / Ageing Research Reviews 11 (2012) 10– 31 25
Genet ic fa ctors:
Mutations in DJ-1, PINK1 and LR RK2
Enviro nmental toxins Aging
Mitochondri al dysfunction
Mutations in α-synuc lein ATP Free radicals
Mutations in Pa rkin , UCHL1 α-synuc lein aggregation Mutation in ATP13A2
UPS dysfunctionAutopha gy
dysfunction
Fail to deg rade mis folded proteins
Dopaminergic neu rons death
y fact
msin
attaudta
vattDdcbhltw(
t2w1
Fig. 9. Parkinson disease and man
utations in ATP13A2, a lysosomal ATPase, accompanies �-ynuclein aggregation in a mouse model (Meredith et al., 2002) andndicate that autophagy dysfunction is an important mechanism ofeurodegeneration (Di Fonzo et al., 2007) (Fig. 9).
In addition to the primary causes of autophagy dysfunctionlready discussed, aging may be another important factor relatedo the dysfunction of protein control systems, since it is known thathe activities of both the UPS and autophagy are decreased in almostll old organisms (Kiffin et al., 2007). Moreover, it is important tonderline that age is one of the chief risk factors for Parkinsonisease (Nagatsu and Sawada, 2006), and it is possible to suggesthat the aging brain is particularly vulnerable to dysfunction ofutophagy.
Since autophagy is maintained at low level with nutrient star-ation (Kiffin et al., 2007), and they have also the ability to induceutophagy in response to factors other than nutrient limitation,hus, autophagy enhancement might be a novel strategy for thereatment of neurodegenerative disorders (Menzies et al., 2006).uring cerebral ischemia, necrosis is accompanied by mitochon-rial swelling, dilation of ER and extensive vacuolation of theytoplasm in the ischemic core. In the ischemic penumbra, instead,esides hybrid forms of cell death and apoptosis, biochemicalallmarks of apoptosis may be involved in the execution of morpho-
ogical aspects of autophagy, pointing to a major cross talk betweenhe two lethal subroutines. This new kind of apoptotic cell deathith a concomitant upregulation of BECN-1 is called “apophagy”
Fig. 10).It has been demonstrated that BECN-1, a Bcl-2-interacting pro-
ein, is the mammalian homolog of yeast Atg6/Vps30 (Liang et al.,001; Erlich et al., 2006) and it promotes autophagy associatedith inhibition of cellular proliferation and tumorigenesis. BECN-
is a component of the phosphatidylinositol 3-kinase complex
ors responsible of this pathology.
that is required for autophagy (Kihara et al., 2001). More recently,it has been demonstrated that heterozygous disruption of BECN-1 induces spontaneous tumors, and cells with reduced BECN-1expression exhibit reduced autophagic activity. Erlich et al.’s (2006)studies demonstrated an increase of BECN-1 levels near the braininjury in mice. Interestingly, BECN-1 increase is visible at earlystages post-injury in neurons and at later stages in astrocytes.Neuronal cells, but not astrocytes overexpressing BECN-1, havedamaged DNA, but without changes in nuclear morphology. Theseobservations indicated that not all the BECN1 overexpressing cellswill die. The elevation of BECN-1 at the site of injury may representenhanced autophagy as a mechanism to destroy injured cells andreduce damage to cells by disposing of injured components (Erlichet al., 2006).
In this regard, Rami (2008) showed a dramatic elevation inBECN-1 1 levels and LC3 in the penumbra of rats challenged bycerebral ischemia. LC3, the mammalian ortholog of yeast Atg8, wasoriginally identified as the light chain 3 of microtubule-associatedproteins (MAPs) (Shimizu et al., 2004). An increased autophagicactivity is reflected by the enhanced conversion of LC3-I (cytoso-lic) to LC3-II (lipidated), together with recruitment of fluorescentprotein-tagged LC3 (GFP-LC3) to autophagosomes indicated bythe increased number of GFP-LC3 puncta. Moreover, Rami (2008)found also that a subpopulation of BECN-1-upregulating cells alsoexpresses the active form of caspase-3, and that all BECN1 upreg-ulating cells display dense staining of LC3. Neuronal cells thatoverexpress BECN1 showed damaged DNA but without changesin nuclear morphology, which indicates that not all the BECN1-
upregulating cells are predestined to die. Upon cerebral ischemia,BECN-1 levels in the neurons of cortex and striatum are increased.Additionally, the BECN-1-upregualtion was also observed in somecaspase-3-positive cells, indicating that BECN-1 may have either a
26 R. Rezzani et al. / Ageing Research Reviews 11 (2012) 10– 31
Fig. 10. Cerebral ischemia. Glut, glutamate; NO, nitric oxide; �-calpain, micro calpain.
Insulin rece ptor PTEN
Ambra1 Class I PI3K Wortmannin , LY29400 2
Beclin 1 Akt/PKB Nutrient tra nspor ters
(GLUT1)
Metabolic
substr ates
Class III PtdIns3K mTOR Mitochondr iaWortmannin,
LY29400 2
ATP
IMPase Autophagy Gluco se, gro wth fa ctor,
amino acids
Mood-stab ilizing drugs, IMPase
inhibitors , trehalose
3-MA, Baf1 Rapamycin, hypox ia
Fig. 11. Autophagy’s regulation.

search
rbti
thia2roO(tdsRtbui
mltpilcdgp
7
itelnurekpop
daa
A
EMi
R
A
R. Rezzani et al. / Ageing Re
epair function via autophagy or unidentified functions upon cere-ral ischemia. Since release of materials from mitochondria triggershe intrinsic pathway of apoptosis, sequestration of mitochondrian autophagic vacuoles protects cells against apoptosis (Fig. 11).
Other studies suggest that cystatin C (CysC) is increased inhe brain of animal models with degenerative diseases and inuman epileptic patients, and they have a neuroprotective role
n response to oxidative stress. CysC enhances autophagic clear-nce of aggregates via mTOR-dependent pathway (Essick and Sam,010). On the contrary, BECN1 siRNA or 3-MA inhibits the neu-oprotective role of CysC (Tizon et al., 2010). The mechanism ofxidative stress-induced autophagy in neural cells involves alsoxi-�, a neuroprotective protein identified in dopamine neurons
Yoo et al., 2004). Oxi-� is downregulated during oxidative stress,hus rendering neurons susceptible to oxidative stress-inducedeath. Under normal conditions, Oxi-� activates mTOR and thusuppresses autophagosome formation. However, increased level ofOS downregulates Oxi-� and leads to decreased mTOR activity,hereby increasing autophagosome formation. Furthermore, inhi-ition of autophagy with 3-MA protects against neuronal deathnder oxidative stress, confirming the involvement of autophagy
n cell death in neurodegenerative diseases (Choi et al., 2010).Recent studies show that Snapin is a protein acting as a dynein
otor adaptor, and that it coordinates retrograde transport andate endosomal-lysosomal trafficking (Cai and Sheng, 2011) main-aining efficient autophagy–lysosomal function in neurons. Thisathway is possible because Snapin−/− neurons display damage
n the retrograde transport, aberrant accumulation of immatureysosomes and impaired clearance of autolysosomes. Snapin defi-iency leads to reduced neuron viability, neurodegeneration andevelopmental effects in the CNS. Reintroducing the Snapin trans-ene rescues these phenotypes, so, Snapin is a candidate molecularrotein for autophagy–lysosomal regulation.
. Conclusions
Accumulating evidence has suggested that lifespan extensions dependent on efficient autophagy, but the fundamental ques-ion is: how does autophagy promote longevity? Recently, Madeot al. (2010) outlined the role of longevity-promoting regimens,ike caloric restriction but also inhibition of mTOR, together withatural compounds, like spermidine, a natural polyamine that stim-lates autophagy. So, removing cellular damage by autophagy mayepresent a promising common denominator of many lifespan-xtending manipulations. In this review we summarize the currentnowledge regarding the main proteins involved in the autophagicrocesses and the changes of their expression during aging. More-ver, we report the metabolic regulation of autophagic process inhysiological conditions.
The nature of the autophagic defect, the cellular response to thatefect, and elapsed time into the progression of the disease shouldll be taken into account during the implementation of therapeuticpproaches, since they can affect the quality of life.
cknowledgements
The authors thank Dr Favero Gaia, Dr Rossini Claudia and Dr Foglioleonora for their excellent work in helping to prepare the chapter.oreover, the authors thank Dr Foglio Eleonora for her contribution
n realizing the figures.
eferences
costa, J.C., O’Loghlen, A., Banito, A., Guijarro, M.V., Augert, A., Raguz, S., Fumagalli,M., Da Costa, M., Brown, C., Popov, N., Takatsu, Y., Melamed, J., d‘Adda di Fagagna,F., Bernard, D., Hernando, E., Gil, J., 2008. Chemokine signaling via the CXCR2receptor reinforces senescence. Cell 133, 1006–1018.
Reviews 11 (2012) 10– 31 27
Adams, P.D., 2007. Remodeling of chromatin structure in senescent cells and itspotential impact on tumor suppression and aging. Gene 397, 84–93.
Arias, E., Cuervo, A.M., 2010. Chaperone-mediated autophagy in protein qualitycontrol. Curr. Opin. Cell Biol. 23, 1–6.
Armour, S.M., Baur, J.A., Hsieh, S.N., Land-Bracha, A., Thomas, S.M., Sinclair, D.A.,2009. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy.Aging (Albany NY) 1, 515–528.
Aránguiz-Urroz, P., Canales, J., Copaja, M., Troncoso, R., Vicencio, J.M., Carrillo, C., Lara,H., Lavandero, S., Díaz-Araya, G., 2011. Beta(2)-adrenergic receptor regulatescardiac fibroblast autophagy and collagen degradation. Biochim. Biophys. Acta1812, 23–31.
Avruch, J., Long, X., Ortiz-Vega, S., Rapley, J., Papageorgiou, A., Dai, N., 2009.Amino acid regulation of TOR complex. Am. J. Physiol. Endocrinol. Metab. 296,E592–E602.
Axe, E.L., Walker, S.A., Manifava, M., Chandra, P., Roderick, H.L., Habermann, A.,Griffiths, G., Ktistakis, N.T., 2008. Autophagosome formation from membranecompartments enriched in phosphatidylinositol 3-phosphate and dynamicallyconnected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701.
Baker III, G.T., Sprott, R.L., 1988. Biomarkers of aging. Exp. Gerontol. 23, 223–239.Bandyopadhyay, U., Kaushik, S., Varticovski, L., Cuervo, A.M., 2008. The chaperone-
mediated autophagy receptor organizes in dynamic protein complexes at thelysosomal membrane. Mol. Cell. Biol. 28, 5747–5763.
Bandyopadhyay, U., Sridar, S., Kaushik, S., Kiffin, R., Cuervo, A.M., 2010. Identificationof regulators of chaperone-mediated autophagy. Mol. Cell 39, 535–547.
Bassham, D.C., 2007. Plant autophagy—more than a starvationresponse. Curr. Opin.Plant Biol. 10, 587–593.
Bennett, M.R., 2010. Life and death in the atherosclerotic plaque. Curr. Opin. Lipidol.21, 422–426.
Bergamini, E., Cavallini, G., Donati, A., Gori, Z., 2004. The role of macroautophagy inthe ageing process, anti-ageing intervention and age-associated diseases. Int. J.Biochem. Cell Biol. 36, 2392–2404.
Bjørkøy, G., Lamark, T., Johansen, T., 2006. p62/SQSTM1: a missing link betweenprotein aggregates and the autophagy machinery. Autophagy 2, 138–139.
Blagosklonny, M.V., Hall, M.N., 2009. Growth and aging: a common molecular mech-anism. Aging 1, 357–362.
Bohensky, J., Shapiro, I.M., Leshinsky, S., Terkhorn, S.P., Adams, C.S., Srinivas, V.,2007. HIF-1 regulation of chondrocyte apoptosis: induction of the autophagicpathway. Autophagy 3, 207–214.
Bohensky, J., Terkhorn, S.P., Freeman, T.A., Adams, C.S., Garcia, J.A., Shapiro, I.M.,Srinivas, V., 2009. Regulation of autophagy in human and murine cartilage:hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Rheum.60, 1406–1415.
Brady, N.R., Hamacher-Brady, A., Yuan, H., Gottlieb, R.A., 2007. The autophagicresponse to nutrient deprivation in the hl-1 cardiac myocyte is modu-lated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J. 274,3184–3197.
Brunk, U.T., Terman, A., 2002. The mitochondrial–lysosomal axis theory of aging:accumulation of damaged mitochondria as a result of imperfect autophagocy-tosis. Eur. J. Biochem. 269, 1996–2002.
Budanov, A.V., 2011. Stress-responsive Sestrins link p53 with redox regulation andmTOR signaling. Antioxid. Redox Signal. 15, 1679–1690.
Cai, Q., Sheng, Z.H., 2011. Uncovering the role of Snapin in regulatingautophagy–lysosomal function. Autophagy 7, 445–447.
Campisi, J., d’Adda di Fagagna, F., 2007. Cellular senescence: when bad things happento good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740.
Carames, B., Taniguchi, N., Otsuki, S., Blanco, F.J., Lotz, M., 2010. Autophagy is a pro-tective mechanism in normal cartilage, and its aging-related loss is linked withcell death and osteoarthritis. Arthritis Rheum. 62, 791–801.
Carrard, G., Bulteau, A.L., Petropoulos, I., Friguet, B., 2002. Impairment of proteasomestructure and function in aging. Int. J. Biochem. Cell Biol. 34, 1461–1474.
Cavallini, G., Donati, A., Gori, Z., Bergamini, E., 2008. Towards an understanding ofthe anti-aging mechanism of caloric restriction. Curr. Aging Sci. 1, 4–9.
Chan, D.C., 2006. Mitochondria: dynamic organelles in disease, aging, and develop-ment. Cell 125, 1241–1252.
Chandel, N.S., Budinger, G.R., 2007. The cellular basis for diverse responses to oxygen.Free Radic. Biol. Med. 42, 165–174.
Choi, K.C., Kim, S.H., Ha, J.Y., Kim, S.T., Son, H.T., 2010. A novel mTOR activat-ing protein protects dopamine neurons against oxidative stress by repressingautophagy related cell death. J. Neurochem. 112, 366–376.
Ciechanover, A., 2005. Proteolysis: from the lysosome to ubiquitin and the protea-some. Nat. Rev. Mol. Cell Biol. 6, 79–87.
Ciechanover, A., Orian, A., Schwartz, A.L., 2000. The ubiquitin-mediated proteolyticpathway: mode of action and clinical implications. J. Cell. Biochem. Suppl. 34,40–51.
Colombo, F., Gosselin, H., El-Helou, V., Calderone, A., 2003. Beta-adrenergic receptor-mediated DNA synthesis in neonatal rat cardiac fibroblasts proceeds via aphosphatidylinositol 3-kinase dependent pathway refractory to the antiprolif-erative action of cyclic AMP. J. Cell. Physiol. 195, 322–330.
Couve, E., Schmachtenberg, O., 2011. Autophagic activity and aging in human odon-toblasts. J. Dent. Res. 90, 523–528.
Cuervo, A.M., 2004. Autophagy: many paths to the same end. Mol. Cell. Biochem.
263, 55–72.Cuervo, A.M., 2010. Chaperone-mediated autophagy: selectivity pays off. TrendsEndocrinol. Metab. 21, 142–150.
Cuervo, A.M., Dice, J.F., 2000. Age-related decline in chaperone-mediated autophagy.J. Biol. Chem. 275, 31505–31513.

2 search
C
C
D
D
D
D
D
D
D
D
D
D
D
E
E
E
F
G
G
G
G
H
HH
H
H
H
H
H
8 R. Rezzani et al. / Ageing Re
uervo, A.M., Bergamini, E., Brunk, U.T., Dröge, W., Ffrench, M., Terman, A., 2005.Autophagy and Aging—the importance of maintaining clean cells. Autophagy 1,131–140.
uervo, A.M., Stefanis, L., Fredenburg, R., Lansbury, P.T., Sulzer, D., 2004. Impaireddegradation of mutant alpha-synuclein by chaperone-mediated autophagy. Sci-ence 305, 1292–1295.
ann, S.G., Thomas, G., 2006. The amino acid sensitive TOR pathway from yeast tomammals. FEBS Lett. 580, 2821–2829.
e Gaetano, G., De Curtis, A., di Castelnuovo, A., Donati, M.B., Iacoviello, L., Rotondo,S., 2002. Antithrombotic effect of polyphenols in experimental models: a mech-anism of reduced vascular risk by moderate wine consumption. Ann. N.Y. Acad.Sci. 957, 174–188.
el Roso, A., Vittorini, S., Cavallini, G., Donati, A., Gori, Z., Masini, M., Pollera, M.,Bergamini, E., 2003. Ageing-related changes in the in vivo function of rat livermacroautophagy and proteolysis. Exp. Gerontol. 38, 519–527.
ennis, P.B., Jaeschke, A., Saitoh, M., Fowler, B., Kozma, S.C., Thomas, G., 2001. Mam-malian TOR: a homeostatic ATP sensor. Science 294, 1102–1105.
ice, J., 2000. Lysosomal Pathways of Protein Degradation. Landes Bioscience, Austin,TX.
i Fonzo, A., Chien, H.F., Socal, M., Giraudo, S., Tassorelli, C., Iliceto, G., Fabbrini,G., Marconi, R., Fincati, E., Abbruzzese, G., Marini, P., Squitieri, F., Horstink,M.W., Montagna, P., Libera, A.D., Stocchi, F., Goldwurm, S., Ferreira, J.J., Meco,G., Martignoni, E., Lopiano, L., Jardim, L.B., Oostra, B.A., Barbosa, E.R., Bonifati,V., Italian Parkinson Genetics Network, 2007. ATP13A2 missense mutationsin juvenile Parkinsonism and young onset Parkinson disease. Neurology 68,1557–1562.
obrowolny, G., Aucello, M., Rizzuto, E., Beccafico, S., Mammucari, C., Boncompagni,S., Belia, S., Wannenes, F., Nicoletti, C., Del Prete, Z., Rosenthal, N., Molinaro, M.,Protasi, F., Fanò, G., Sandri, M., Musarò, A., 2008. Skeletal muscle is a primarytarget of SOD1G93A-mediated toxicity. Cell Metab. 8, 425–436.
onati, A., 2006. The involvement of macroautophagy in aging and anti-aging inter-ventions. Mol. Aspects Med. 27, 455–470.
onati, A., Cavallini, G., Paradiso, C., Vittorini, S., Pollera, M., Gori, Z., Bergamini,E., 2001. Age-related changes in the regulation of autophagic proteolysis in ratisolated hepatocytes. J. Gerontol. A Biol. Sci. Med. Sci. 56, 288–293.
roge, W., 2004. Autophagy and aging – importance of amino acid levels. Mech.Ageing Dev. 125, 161–168.
roge, W., Kinscherf, R., 2008. Aberrant insulin receptor signalling and amino acidhomeostasis as a major cause of oxidative stress in aging. Antioxid. Redox Signal.10, 661–678.
bato, C., Uchida, T., Arakawa, M., Komatsu, M., Ueno, T., Komiya, K., Azuma, K.,Hirose, T., Tanaka, K., Kominami, E., Kawamori, R., Fujitani, Y., Watada, H., 2008.Autophagy is important in islet homeostasis and compensatory increase of betacell mass in response to high-fat diet. Cell Metab. 8, 325–332.
rlich, S., Shohami, E., Pinkas-Kramarski, R., 2006. Neurodegeneration inducesupregulation of Beclin 1. Autophagy 2, 49–51.
ssick, E.E., Sam, F., 2010. Oxidative stress and autophagy in cardiac disease, neuro-logical disorders, aging and cancer. Oxid. Med. Cell. Longev. 3, 168–177.
amulski, K.S., Halloran, P.F., 2005. Molecular events in kidney ageing. Curr. Opin.Nephrol. Hypertens. 14, 243–248.
hosh, A., Sil, P.C., 2008. A protein from Cajanus indicus Spreng protects liver andkidney against mercuric chloride-induced oxidative stress. Biol. Pharm. Bull. 31,1651–1658.
uarente, L., 2008. Mitochondria – a nexus for aging, calorie restriction, and sirtuins?Cell 132, 171–176.
urusamy, N., Lekli, I., Mukherjee, S., Ray, D., Ahsan, M.K., Gherghiceanu, M.,Popescu, L.M., Das, D.K., 2010. Cardioprotection by resveratrol: a novel mech-anism via autophagy involving the mTORC2 pathway. Cardiovasc. Res. 86,103–112.
ustafsson, A.B., Brunton, L.L., 2000. Beta-adrenergic stimulation of rat cardiacfibroblasts enhances induction of nitric-oxide synthase by interleukin-1beta viamessage stabilization. Mol. Pharmacol. 58, 1470–1478.
ailey, D.W., Rambold, A.S., Satpute-Krishnan, P., Mitra, K., Sougrat, R., Kim, P.K.,Sougrat, R., Kim, P.K., Lippincott-Schwartz, J., 2010. Mitochondria supply mem-branes for autophagosome biogenesis during starvation. Cell 141, 656–667.
all, M.N., 2008. m-TOR-what does it do? Transplant. Proc. 40, 5–8.ands, S.L., Proud, C., Wyttenbach, A., 2009. mTOR’s role in ageing: protein synthesis
or autophagy? Aging 1, 586–597.ara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R.,
Yokoyama, M., Mishima, K., Saito, I., Okano, H., Mizushima, N., 2006. Suppressionof basal autophagy in neural cells causes neurodegenerative disease in mice.Nature 441, 885–889.
ara, K., Yonezawa, K., Weng, Q.P., Kozlowski, M.T., Belham, C., Avruch, J., 1998.Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 througha common effector mechanism. J. Biol. Chem. 273, 14484–14494.
arman, D., 1956. Aging: a theory based on free radical and radiation chemistry.J. Gerontol. 11, 298–300.
arrison, D.E., Strong, R., Sharp, Z.D., Nelson, J.F., Astle, C.M., Flurkey, K., Nadon,N.L., Wilkinson, J.E., Frenkel, K., Carter, C.S., Pahor, M., Javors, M.A., Fernandez,E., Miller, R.A., 2009. Rapamycin fed late in life extends lifespan in geneticallyheterogeneous mice. Nature 460, 392–395.
artleben, B., Godel, M., Meyer-Schwesinger, C., Liu, S., Ulrich, T., Köbler, S., Wiech,T., Grahammer, F., Arnold, S.J., Lindenmeyer, M.T., Cohen, C.D., Pavenstädt, H.,Kerjaschki, D., Mizushima, N., Shaw, A.S., Walz, G., Huber, T.B., 2010. Autophagyinfluences glomerular disease susceptibility and maintains podocyte homeosta-sis in aging mice. J. Clin. Invest. 120, 1084–1096.
Reviews 11 (2012) 10– 31
Hashimoto, M., Kim, S., Eto, M., Iijima, K., Ako, J., Yoshizumi, M., Akishita, M., Kondo,K., Itakura, H., Hosoda, K., Toba, K., Ouchi, Y., 2001. Effect of acute intake of redwine on flow-mediated vasodilatation of the brachial artery. Am. J. Cardiol. 88,1457–1460.
Hayashi-Nishino, M., Fujita, N., Noda, T., Yamaguchi, A., Yoshimori, T., Yamamoto, A.,2010. Electron tomography reveals the endoplasmic reticulum as a membranesource for autophagosome formation. Autophagy 6, 301–303.
Hekimi, S., Guarente, L., 2003. Genetics and the specificity of the aging process.Science 299, 1351–1354.
Hesketh, G., Shah, M., Halperin, V., Cooke, C., Akar, F., Yen, T., Kass, D.A., Machamer,C.E., Van Eyk, J.E., Tomaselli, G.F., 2010. Ultrastructure and regulation of lateral-ized connexin43 in the failing heart. Circ. Res. 106, 1153–1163.
Heymann, D., 2006. Autophagy: a protective mechanism in response to stress andinflammation. Curr. Opin. Invest. Drugs 7, 443–450.
Hidvegi, T., Ewing, M., Hale, P., Dippold, C., Beckett, C., Kemp, C., Maurice, N., Mukher-jee, A., Goldbach, C., Watkins, S., Michalopoulos, G., Perlmutter, D.H., 2010. Anautophagy-enhancing drug promotes degradation of mutant �1-Antitrypsin Zand reduces hepatic fibrosis. Science 329, 229–232.
Holliday, R., 2007. Ageing: The Paradox of Life. Springer, Dordrecht, The Netherlands.Huang, J., Gan, Q., Han, L., Li, J., Zhang, H., Sun, Y., Zhang, Z., Tong, T., 2008. SIRT1 over-
expression antagonizes cellular senescence with activated ERK/S6k1 signalingin human diploid fibroblasts. PLoS ONE 3, 1710.
Hur, K.Y., Jung, H.S., Lee, M.S., 2010. Role of autophagy in �-cell function and mass.Diabetes Obes. Metab. 12 (Suppl. 2), 20–26.
Inoue, K., Kuwana, H., Shimamura, Y., Ogata, K., Taniguchi, Y., Kagawa, T., Horino, T.,Takao, T., Morita, T., Sasaki, S., Mizushima, N., Terada, Y., 2010. Cisplatin-inducedmacroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin. Exp.Nephrol. 14, 112–122.
Inuzuka, Y., Okuda, J., Kawashima, T., Kato, T., Niizuma, S., Tamaki, Y., Iwanaga, Y.,Yoshida, Y., Kosugi, R., Watanabe-Maeda, K., Machida, Y., Tsuji, S., Aburatani,H., Izumi, T., Kita, T., Shioi, T., 2009. Suppression of phosphoinositide 3-kinaseprevents cardiac aging in mice. Circulation 120, 1695–1703.
Jeyapalan, J.C., Sedivy, J.M., 2008. Cellular senescence and organismal aging. Mech.Ageing Dev. 129, 467–474.
Jiang, M., Dong, Z., 2010. Self-eating for death or survival during cisplatin nephro-toxicity? Clin. Exp. Nephrol. 14, 516–517.
Jiang, M., Liu, K., Luo, J., Dong, Z., 2010. Autophagy is a renoprotective mechanismduring in vitro hypoxia and in vivo ischemia–reperfusion injury. Am. J. Pathol.176, 1181–1192.
Johnson, T.E., 2006. Recent results: biomarkers of aging. Exp. Gerontol. 41,1243–1246.
Jung, H.S., Chung, K.W., Won Kim, J., Kim, J., Komatsu, M., Tanaka, K., Nguyen, Y.H.,Kang, T.M., Yoon, K.H., Kim, J.W., Jeong, Y.T., Han, M.S., Lee, M.K., Kim, K.W., Shin,J., Lee, M.S., 2008. Loss of autophagy diminishes pancreatic beta cell mass andfunction with resultant hyperglycemia. Cell Metab. 8, 318–324.
Kaarniranta, K., Salminen, A., Eskelinen, E.L., Kopitz, J., 2009. Heat shock proteinsas gatekeepers of proteolytic pathways—implications for age-related maculardegeneration (AMD). Ageing Res. Rev. 8, 128–139.
Kaeberlein, M., Powers III, R.W., Steffen, K.K., Westman, E.A., Hu, D., Dang, N., Kerr,E.O., Kirkland, K.T., Fields, S., Kennedy, B.K., 2005. Regulation of yeast replicativelife span by TOR and Sch9 in response to nutrients. Science 310, 1193–1196.
Kaushik, S., Massey, A.C., Mizushima, N., Cuervo, A.M., 2008. Constitutive activationof chaperone-mediated autophagy in cells with impaired macroautophagy. Mol.Biol. Cell 19, 2179–2192.
Kay, D.M., Moran, D., Moses, L., Poorkaj, P., Zabetian, C.P., Nutt, J., Factor, S.A., Yu, C.E.,Montimurro, J.S., Keefe, R.G., Schellenberg, G.D., Payami, H., 2007. Heterozygousparkin point mutations are as common in control subjects as in Parkinson’spatients. Ann. Neurol. 61, 47–54.
Kawamata, T., Kamada, Y., Kabeya, Y., Sekito, T., Ohsumi, Y., 2008. Organization ofthe pre-autophagosomal structure responsible for autophagosome formation.Mol. Biol. Cell 19, 2039–2050.
Kiffin, R., Kaushik, S., Zeng, M., Bandyopadhyay, U., Zhang, C., Massey, A.C., Martinez-Vicente, M., Cuervo, A.M., 2007. Altered dynamics of the lysosomal receptor forchaperone-mediated autophagy with age. J. Cell Sci. 120, 782–791.
Kihara, A., Kabeya, Y., Ohsumi, Y., Yoshimori, T., 2001. Beclin-phosphatidylinositol3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2,330–335.
Kim, J., Eckhart, A.D., Eguchi, S., Koch, W.J., 2002. Beta-adrenergic receptor-mediatedDNA synthesis in cardiac fibroblasts is dependent on transactivation of theepidermal growth factor receptor and subsequent activation of extracellularsignal-regulated kinases. J. Biol. Chem. 277, 32116–32123.
Kim, P.K., Hailey, D.W., Mullen, R.T., Lippincott-Schwartz, J., 2008. Ubiquitin signalsautophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad.Sci. U.S.A. 105, 20567–20574.
Kirkin, V., Lamark, T., Sou, Y.S., Bjørkøy, G., Nunn, J.L., Bruun, J.A., Shvets, E., McEwan,D.G., Clausen, T.H., Wild, P., Bilusic, I., Theurillat, J.P., Øvervatn, A., Ishii, T., Elazar,Z., Komatsu, M., Dikic, I., Johansen, T., 2009. A role for NBR1 in autophagosomaldegradation of ubiquitinated substrates. Mol. Cell 33, 505–516.
Kirkwood, T.B., 2008. A systematic look at an old problem. Nature 451, 644–647.Klionsky, D., Emr, S., 2000. Autophagy as a regulated pathway of cellular degradation.
Science 290, 1717–1721.
Koga, H., Kaushik, S., Cuervo, A.M., 2011. Protein homeostasis and aging: the impor-tance of exquisite quality control. Ageing Res. Rev. 10, 205–215.Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., Sou,
Y.S., Ueno, I., Sakamoto, A., Tong, K.I., Kim, M., Nishito, Y., Iemura, S., Natsume,T., Ueno, T., Kominami, E., Motohashi, H., Tanaka, K., Yamamoto, M., 2010. The

search
K
K
K
K
K
K
K
L
L
L
L
L
L
LL
L
L
M
M
M
M
M
M
M
M
M
M
M
M
M
R. Rezzani et al. / Ageing Re
selective autophagy substrate p62 activates the stress responsive transcriptionfactor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–226.
omatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., Ueno, T.,Koike, M., Uchiyama, Y., Kominami, E., Tanaka, K., 2006. Loss of autophagyin the central nervous system causes neurodegeneration in mice. Nature 441,880–884.
omatsu, M., Waguri, S., Koike, M., Sou, Y., Ueno, T., Hara, T., Mizushima, N., Iwata, J.,Ezaki, J., Murata, S., Hamazaki, J., Nishito, Y., Iemura, S., Natsume, T., Yanagawa,T., Uwayama, J., Warabi, E., Yoshida, H., Ishii, T., Kobayashi, A., Yamamoto, M.,Yue, Z., Uchiyama, Y., Kominami, E., Tanaka, K., 2007. Homeostatic levels of p62control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell131, 1149–1163.
omatsu, M., Waguri, S., Ueno, T., Iwata, J., Murata, S., Tanida, I., Ezaki, J., Mizushima,N., Ohsumi, Y., Uchiyama, Y., Kominami, E., Tanaka, K., Chiba, T., 2005. Impair-ment of starvation-induced and constitutive autophagy in Atg7-deficient mice.J. Cell Biol. 169, 425–434.
on, M., Cuervo, A.M., 2010. Chaperone-mediated autophagy in health and disease.FEBS Lett. 584, 1399–1404.
orolchuk, V.I., Mansilla, A., Menzies, F.M., Rubinsztein, D.C., 2009. Autophagy inhi-bition compromises degradation of ubiquitin-proteasome pathway substrates.Mol. Cell 33, 517–527.
rishnamurthy, J., Torrice, C., Ramsey, M.R., Kovalev, G.I., Al-Regaiey, K., Su, L., Sharp-less, N.E., 2004. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 114,1299–1307.
ume, S., Uzu, T., Horiike, K., Chin-Kanasaki, M., Isshiki, K., Araki, S., Sugimoto, T.,Haneda, M., Kashiwagi, A., Koya, D., 2010. Calorie restriction enhances cell adap-tation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouseaged kidney. J. Clin. Invest. 120, 1043–1055.
amark, T., Johansen, T., 2010. Autophagy: links with the proteasome. Curr. Opin.Cell Biol. 22, 192–198.
ee, B.Y., Han, J.A., Im, J.S., Morrone, A., Johung, K., Goodwin, E.C., Kleijer, W.J., DiMaio,D., Hwang, E.S., 2006. Senescence-associated beta-galactosidase is lysosomalbeta-galactosidase. Aging Cell 5, 187–195.
ee, H.M., Shin, D.M., Yuk, J.M., Shi, G., Choi, D.K., Lee, S.H., Huang, S.M., Kim, J.M.,Kim, C.D., Lee, J.H., Jo, E.K., 2011. Autophagy negatively regulates keratinocyteinflammatory responses via scaffolding protein p62/SQSTM1. J. Immunol. 186,1248–1258.
ee, I.H., Cao, L., Mostoslavsky, R., Lombard, D.B., Liu, J., Bruns, N.E., Tsokos, M., Alt,F.W., Finkel, T., 2008. A role for the NAD-dependent deacetylase Sirt1 in theregulation of autophagy. Proc. Natl. Acad. Sci. U.S.A. 105, 3374–3379.
evine, B., Kroemer, G., 2008. Autophagy in the pathogenesis of disease. Cell 132,27–42.
evine, B., Yuan, J., 2005. Autophagy in cell death: an innocent convict? J. Clin. Invest.115, 2679–2688.
iang, C., 2010. Negative regulation of autophagy. Cell Death Differ. 17, 1807–1815.iang, C.C., Wang, C., Peng, X., Gan, B., Guan, J.L., 2010. Neural-specific deletion of
FIP200 leads to cerebellar degeneration caused by increased neuronal death andaxon degeneration. J. Biol. Chem. 285, 3499–3509.
iang, X.H., Yu, J., Brown, K., Levine, B., 2001. Beclin 1 contains a leucine-rich nuclearexport signal that is required for its autophagy and tumor suppressor function.Cancer Res. 61, 3443–3449.
ongatti, A., Orsi, A., Tooze, S.A., 2010. Autophagosome formation: not necessarilyan inside job. Cell Res. 20, 1181–1184.
adeo, F., Tavernarakis, N., Kroemer, G., 2010. Can autophagy promote longevity?Nat. Cell Biol. 12, 842–846.
allette, F.A., Gaumont-Leclerc, M.F., Ferbeyre, G., 2007. The DNA damage signallingpathway is a critical mediator of oncogene-induced senescence. Genes Dev. 21,43–48.
ammucari, C., Rizzuto, R., 2010. Signaling pathways in mitochondrial dysfunctionand aging. Mech. Ageing Dev. 131, 536–543.
araganore, D.M., Lesnick, T.G., Elbaz, A., Chartier-Harlin, M.C., Gasser, T., Krüger, R.,Hattori, N., Mellick, G.D., Quattrone, A., Satoh, J., Toda, T., Wang, J., Ioannidis, J.P.,de Andrade, M., Rocca, W.A., UCHL1 Global Genetics Consortium, 2004. UCHL1is a Parkinson’s disease susceptibility gene. Ann. Neurol. 55, 512–521.
arino, G., Lopez-Otìn, C., 2008. Autophagy and aging. New lessons from progeroidmice. Autophagy 4, 807–809.
arino, G., Ugalde, A.P., Salvador-Montoliu, N., Varela, I., Quiròs, P.M., Cadinanos,J., van der Pluijm, I., Freije, J.M., López-Otín, C., 2008. Premature aging in miceactivates a systemic metabolic response involving autophagy induction. Hum.Mol. Genet. 17, 2196–2211.
arino, G., Fernàndez, A.F., López-Otín, C., 2010. Autophagy and aging: lessons fromprogeria models. Adv. Exp. Med. Biol. 694, 61–68.
arino, G., Madeo, F., Kroemer, G., 2011. Autophagy for tissue homeostasis andneuroprotection. Curr. Opin. Cell Biol. 23, 198–206.
artinet, W., De Meyer, G.R., 2009. Autophagy in atherosclerosis: a cell survival anddeath phenomenon with therapeutic potential. Circ. Res. 104, 304–317.
artinez-Vicente, M., Sovak, G., Cuervo, A.M., 2005. Protein degradation and aging.Exp. Gerontol. 40, 622–633.
asiero, E., Sandri, M., 2010. Autophagy inhibition induces atrophy and myopathyin adult skeletal muscles. Autophagy 6, 307–309.
asiero, E., Agatea, L., Mammucari, C., Blaaw, B., Loro, E., Komatsu, M., Metzger, D.,
Reggiani, C., Schiaffino, S., Sandri, M., 2009. Autophagy is required to maintainmuscle mass. Cell Metab. 10, 507–515.atheu, A., Maraver, A., Klatt, P., Flores, I., Garcia-Cao, I., Borras, C., Flores, J.M., Vina,J., Blasco, M.A., Serrano, M., 2007. Delayed ageing through damage protectionby the Arf/p53 pathway. Nature 448, 375–379.
Reviews 11 (2012) 10– 31 29
Matsui, Y., Takagi, H., Qu, X., Abdellatif, M., Sakoda, H., Asano, T., Levine, B.,Sadoshima, J., 2007. Distinct roles of autophagy in the heart during ischemiaand reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediatingautophagy. Circ. Res. 100, 914–922.
Meijer, A.J., 2008. Amino acid regulation of autophagosome formation. Methods Mol.Biol. 445, 89–109.
Meijer, A.J., Codogno, P., 2009. Autophagy: regulation and role in disease. Crit. Rev.Clin. Lab. Sci. 46, 210–240.
Meijer, A.J., Codogno, P., 2004. Regulation and role of autophagy in mammalian cells.Int. J. Biochem. Cell Biol. 36, 2445–2462.
Meléndez, A., Tallóczy, Z., Seaman, M., Eskelinen, E.L., Hall, D.H., Levine, B., 2003.Autophagy genes are essential for Dauer development and life-span extensionin C. elegans. Science 301, 1387–1391.
Menzies, F.M., Ravikumar, B., Rubinsztein, D.C., 2006. Protective roles for inductionof autophagy in multiple proteinopathies. Autophagy 2, 224–225.
Meredith, G.E., Totterdell, S., Petroske, E., Santa Cruz, K., Callison Jr., R.C., Lau, Y.S.,2002. Lysosomal malfunction accompanies alpha-synuclein aggregation in aprogressive mouse model of Parkinson’s disease. Brain Res. 956, 156–165.
Merideth, M.A., Gordon, L.B., Clauss, S., Sachdev, V., Smith, A.C., Perry, M.B., Brewer,C.C., Zalewski, C., Kim, H.J., Solomon, B., Brooks, B.P., Gerber, L.H., Turner,M.L., Domingo, D.L., Hart, T.C., Graf, J., Reynolds, J.C., Gropman, A., Yanovski,J.A., Gerhard-Herman, M., Collins, F.S., Nabel, E.G., Cannon III, R.O., Gahl, W.A.,Introne, W.J., 2008. Phenotype and course of Hutchinson–Gilford progeria syn-drome. N. Engl. J. Med. 358, 592–604.
Mizushima, N., Levine, B., Cuervo, A.M., Klionsky, D.J., 2008. Autophagy fights diseasethrough cellular self-digestion. Nature 451, 1069–1075.
Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T., Ohsumi, Y., 2004. In vivoanalysis of autophagy in response to nutrient starvation using transgenic miceexpressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111.
Moreau, K., Luo, S., Rubinsztein, D.C., 2010. Cytoprotective roles for autophagy. Curr.Opin. Cell Biol. 22, 206–211.
Morselli, E., Maiuri, M.C., Markaki, M., Megalou, E., Pasparaki, A., Palikaras, K., Criollo,A., Galluzzi, L., Malik, S.A., Vitale, I., Michaud, M., Madeo, F., Tavernarakis, N.,Kroemer, G., 2010. The life span-prolonging effect of sirtuin-1 is mediated byautophagy. Autophagy 6, 186–188.
Mukherjee, S., Ray, D., Lekli, I., Bak, I., Tosaki, A., Das, D., 2010. Effects of Longivinex(modified resveratrol) on cardioprotection and its mechanisms of actions. Can.J. Physiol. Pharmacol. 88, 1017–1025.
Nagatsu, T., Sawada, M., 2006. Cellular and molecular mechanisms of Parkinson’sdisease: neurotoxins, causative genes, and inflammatory cytokines. Cell. Mol.Neurobiol. 26, 781–802.
Nakai, A., Yamaguchi, O., Takeda, T., Higuchi, Y., Hikoso, S., Taniike, M., Omiya, S.,Mizote, I., Matsumura, Y., Asahi, M., Nishida, K., Hori, M., Mizushima, N., Otsu, K.,2007. The role of autophagy in cardiomyocytes in the basal state and in responseto haemodynamic stress. Nat. Med. 13, 619–624.
Nangaku, M., Inagi, R., Miyata, T., Fujita, T., 2008. Hypoxia and hypoxia-induciblefactor in renal disease. Nephron. Exp. Nephrol 110, e1–e7.
Navarro, C.L., Cau, P., Lévy, N., 2006. Molecular bases of progeroid syndromes. Hum.Mol. Genet. 15, 151–161.
Neufeld, T.P., 2010. TOR-dependent control of autophagy: biting the hand that feeds.Curr. Opin. Cell Biol. 22, 157–168.
Nixon, R.A., Mathews, P.M., Cataldo, A.M., 2001. The neuronal endosomal-lysosomalsystem in Alzheimer’s disease. J. Alzheimers Dis. 3, 97–107.
Nixon, R.A., Yang, D.S., Lee, J.H., 2008. Neurodegenerative lysosomal disorders: acontinuum from development to late age. Autophagy 4, 590–599.
Opie, L.H., Lecour, S., 2007. The red wine hypothesis: from concepts to protectivesignalling molecules. Eur. Heart J. 28, 1683–1693.
Orenstein, S.J., Cuervo, A.M., 2010. Chaperone-mediated autophagy: molecularmechanisms and physiological relevance. Semin. Cell Dev. Biol. 21, 719–726.
Ostrom, R.S., Naugle, J.E., Hase, M., Gregorian, C., Swaney, J.S., Insel, P.A., Brunton,L.L., Meszaros, J.G., 2003. Angiotensin II enhances adenylyl cyclase signalingvia Ca2+/calmodulin. Gq-Gs cross-talk regulates collagen production in cardiacfibroblasts. J. Biol. Chem. 278, 24461–24468.
Ozawa, T., 1997. Genetic and functional changes in mitochondria associated withaging. Physiol. Rev. 77, 425–464.
Pan, T., Kondo, S., Le, W., Jankovic, J., 2008. The role of autophagy–lysosomepathway in neurodegeneration associated with Parkinson’s disease. Brain 131,1969–1978.
Pankiv, S., Clausen, T.H., Lamark, T., Brech, A., Bruun, J.A., Outzen, H., Øvervatn, A.,Bjørkøy, G., Johansen, T., 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facili-tate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem.282, 24131–24145.
Pepe, S., 2000. Mitochondrial function in ischemia and reperfusion of the ageingheart. Clin. Exp. Pharmacol. Physiol. 27, 745–750.
Pérez, V., Bokov, A., Van Remmen, H., Mele, J., Ran, Q., Ikeno, Y., Richardson, A.,2009. Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 1790,1005–1014.
Periyasamy-Thandavan, S., Jiang, M., Schoenlein, P., Dong, Z., 2009. Autophagy:molecular machinery, regulation, and implications for renal pathophysiology.Am. J. Physiol. Renal Physiol. 297, F244–F256.
Pfeifer, U., 1978. Inhibition by insulin of the formation of autophagic vacuoles in rat
liver. A morphometric approach to the kinetics of intracellular degradation byautophagy. J. Cell Biol. 78, 152–167.Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P.A.,Small, S., Spencer, B., Rockenstein, E., Levine, B., Wyss-Coray, T., 2008. Theautophagy-related protein beclin 1 shows reduced expression in early

3 search
P
P
Q
R
R
R
R
R
R
R
R
R
R
R
S
SS
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
S
0 R. Rezzani et al. / Ageing Re
Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin.Invest. 118, 2190–2199.
inkston, J.M., Garigan, D., Hansen, M., Kenyon, C., 2006. Mutations that increase thelife span of C. elegans inhibit tumor growth. Science 313, 971–975.
rick, T., Thumm, M., Kohrer, K., Häussinger, D., vom Dahl, S., 2006. In yeast, loss ofHog1 leads to osmosensitivity of autophagy. Biochem. J. 394, 153–161.
u, X., Zou, Z., Sun, Q., Luby-Phelps, K., Cheng, P., Hogan, R.N., Gilpin, C., Levine, B.,2007. Autophagy gene-dependent clearance of apoptotic cells during embryonicdevelopment. Cell 128, 931–946.
ajawat, Y.S., Bossis, I., 2008. Autophagy in aging and in neurodegenerative disor-ders. Hormones 7, 46–61.
ajawat, Y.S., Hilioti, Z., Bossis, I., 2009. Aging: central role for autophagy and thelysosomal degradative system. Ageing Res. Rev. 8, 199–213.
ami, A., 2008. Upregulation of Beclin 1 in the ischemic penumbra. Autophagy 4,227–229.
attan, S.I., 2006. Theories of biological aging: genes, proteins, and free radicals. FreeRadic. Res. 40, 1230–1238.
autou, P.E., Mansouri, A., Lebrec, D., Durand, F., Valla, D., Moreau, R., 2010.Autophagy in liver diseases. J. Hepatol. 53, 1123–1134.
avikumar, B., Moreau, K., Jahreiss, L., Puri, C., Rubinsztein, D.C., 2010. Plasma mem-brane contributes to the formation of pre-autophagosomal structures. Nat. CellBiol. 12, 747–757.
oach, H.I., Aigner, T., Kouri, J.B., 2004. Chondroptosis: a variant of apoptotic celldeath in chondrocytes? Apoptosis 9, 265–277.
oberts Jr., E.L., Chih, C.P., Rosenthal, M., 1997. Age-related changes in brainmetabolism and vulnerability to anoxia. Adv. Exp. Med. Biol. 411, 83–89.
odier, F., Campisi, J., 2011. Four faces of cellular senescence. J. Cell Biol. 192,547–556.
ubinsztein, D.C., 2006. The roles of intracellular protein-degradation pathways inneurodegeneration. Nature 443, 780–786.
yter, S., Lee, S., Smith, A., Choi, A., 2010. Autophagy in vascular disease. Proc. Am.Thorac. Soc. 7, 40–47.
aftig, P., Beertsen, W., Eskelinen, E.L., 2008. LAMP-2: a control step for phagosomeand autophagosome maturation. Autophagy 4, 510–512.
andri, M., 2010. Autophagy in skeletal muscle. FEBS Lett. 584, 1411–1416.asaki, M., Nakanuma, Y., 2010. Biliary epithelial apoptosis, autophagy, and senes-
cence in primary biliary cirrhosis. Hepat. Res. Treat. 2010, 205128.alminen, A., Kaarniranta, K., 2009. SIRT1: regulation of longevity via autophagy.
Cell. Signal. 21, 1356–1360.carlatti, F., Granata, R., Meijer, A.J., Codogno, P., 2009. Does autophagy have a license
to kill mammalian cells? Cell Death Differ. 16, 12–20.cherz-Shouval, R., Elazar, Z., 2007. ROS, mitochondria and the regulation of
autophagy. Trends Cell Biol. 17, 422–427.chütt, F., Davies, S., Kopitz, J., Holz, F.G., Boulton, M.E., 2000. Photodamage to human
RPE cells by A2-E, a retinoid component of lipofuscin. Invest. Ophthalmol. Vis.Sci. 41, 2303–2308.
chweichel, J.U., Merker, H.J., 1973. The morphology of various types of cell deathin prenatal tissues. Teratology 7, 253–266.
hay, J.W., Wright, W.E., 2000. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol.Cell Biol. 1, 72–76.
hih, H., Lee, B., Lee, R.J., Boyle, A.J., 2010. The aging heart and post-infarction leftventricular remodeling. J. Am. Coll. Cardiol. 57, 9–17.
himizu, S., Kanaseki, T., Mizushima, N., Mizuta, T., Arakawa-Kobayashi, S., Thomp-son, C.B., Tsujimoto, Y., 2004. Role of Bcl-2 family proteins in a non-apoptoticprogrammed cell death dependent on autophagy genes. Nat. Cell Biol. 6,1221–1228.
himura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., Shimizu, N.,Iwai, K., Chiba, T., Tanaka, K., Suzuki, T., 2000. Familial Parkinson disease geneproduct, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305.
himura, H., Schlossmacher, M.G., Hattori, N., Frosch, M.P., Trockenbacher, A.,Schneider, R., Mizuno, Y., Kosik, K.S., Selkoe, D.J., 2001. Ubiquitination of a newform of alpha-synuclein by parkin from human brain: implications for Parkin-son’s disease. Science 293, 263–269.
hineman, D.W., Salthouse, T.A., Launer, L.J., Hof, P.R., Bartzokis, G., Kleiman, R.,Luine, V., Buccafusco, J.J., Small, G.W., Aisen, P.S., Lowe, D.A., Fillit, H.M., 2010.Therapeutics for cognitive aging. Ann. N.Y. Acad. Sci. 1191 (S1), 1–15.
hvets, E., Elazar, Z., 2008. Autophagy-independent incorporation of GFP-LC3 intoprotein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy4, 1054–1056.
ikora, E., Arendt, T., Bennett, M., Narita, M., 2011. Impact of cellular senescencesignature on ageing research. Ageing Res. Rev. 10, 146–152.
imm, A., Johnson, T.E., 2010. Biomarkers of ageing: a challenge for the future. Exp.Gerontol. 45, 731–732.
imm, A., Nass, N., Bartling, B., Hofmann, B., Silber, R.E., Navarrete Santos, A., 2008.Potential biomarkers of ageing. Biol. Chem. 389, 257–265.
imonsen, A., Cumming, R.C., Brech, A., Isakson, P., Schubert, D.R., Finley, K., 2008.Promoting basal levels of autophagy in the nervous system enhances longevityand oxidant resistance in adult Drosophila. Autophagy 4, 176–184.
orimachi, H., Ishiura, S., Suzuki, K., 1997. Structure and physiological function ofcalpains. Biochem. J. 328, 721–732.
prott, R.L., 2010. Biomarkers of aging and disease: introduction and definitions.
Exp. Gerontol. 45, 2–4.tein, J.H., Keevil, J.G., Wiebe, D.A., Aeschlimann, S., Folts, J.D., 1999. Purple grapejuice improves endothelial function and reduces the susceptibility of LDL choles-terol to oxidation in patients with coronary artery disease. Circulation 100,1050–1055.
Reviews 11 (2012) 10– 31
Stumptner, C., Fuchsbichler, A., Heid, H., Zatloukal, K., Denk, H., 2002. Mallory body– a disease-associated type of sequestosome. Hepatology 35, 1053–1062.
Susic, D., Frohlich, E.D., 2008. The aging hypertensive heart: a brief update. Nat. Clin.Pract. Cardiovasc. Med. 5, 104–110.
Suzuki, K., Kirisako, T., Kamada, Y., Mizushima, N., Noda, T., Oshumi, Y., 2001. Thepre-autophagosomal structure organized by concerted functions of APG genesis essential for autophagosome formation. EMBO J. 20, 5971–5981.
Sybers, H.D., Ingwall, J., DeLuca, M., 1976. Autophagy in cardiac myocytes. RecentAdv. Stud. Cardiac Struct. Metab. 12, 453–463.
Tan, J.M., Wong, E.S., Dawson, V.L., Dawson, T.M., Lim, K.L., 2007. Lysine 63-linkedpolyubiquitin potentially partners with p62 to promote the clearance of proteininclusions by autophagy. Autophagy 4, 251–253.
Tang, X., Wang, L., Proud, C.G., Downes, C.P., 2003. Muscarinic receptor-mediatedactivation of p70 S6 kinase 1 (S6K1) in 1321N1 astrocytoma cells: permissiverole of phosphoinositide 3-kinase. Biochem. J. 374, 137–143.
Tanida, I., Minematsu-Ikeguchi, N., Ueno, T., Kominami, E., 2005. Lysosomal turnover,but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy1, 84–91.
Taniguchi, N., Caramés, B., Ronfani, L., Ulmer, U., Komiya, S., Bianchi, M.E., Lotz, M.,2009. Aging-related loss of the chromatin protein HMGB2 in articular cartilageis linked to reduced cellularity and osteoarthritis. Proc. Natl. Acad. Sci. U.S.A.106, 1181–1186.
Tannous, P., Zhu, H., Nemchencko, A., Berry, J.M., Johnstone, J.L., Shelton, J.M.,Miller Jr., F.J., Rothermel, B.A., Hill, J.A., 2008. Intracellular protein aggre-gation is a proximal trigger of cardiomyocyte autophagy. Circulation 117,3070–3078.
Tasdemir, E., Maiuri, M.C., Galluzzi, L., Vitale, I., Djavaheri-Mergny, M., D‘Amelio, M.,Criollo, A., Morselli, E., Zhu, C., Harper, F., Nannmark, U., Samara, C., Pinton, P.,Vicencio, J.M., Carnuccio, R., Moll, U.M., Madeo, F., Paterlini-Brechot, P., Rizzuto,R., Szabadkai, G., Pierron, G., Blomgren, K., Tavernarakis, N., Codogno, P., Cecconi,F., Kroemer, G., 2008a. Regulation of autophagy by cytoplasmic p53. Nat. CellBiol. 10, 676–687.
Tasdemir, E., Chiara Maiuri, M., Morselli, E., Criollo, A., D’Amelio, M., Djavaheri-Mergny, M., Cecconi, F., Tavernarakis, N., Kroemer, G., 2008b. A dual role of p53in the control of autophagy. Autophagy 4, 810–814.
Temiz, P., Weihl, C.C., Pestronk, A., 2009. Inflammatory myopathies with mitochon-drial pathology and protein aggregates. J. Neurol. Sci. 278, 25–29.
Terlecky, S.R., Koepke, J.I., Walton, P.A., 2006. Peroxisomes and aging. Biochim.Biophys. Acta 1763, 1749–1754.
Terman, A., Dalen, H., Eaton, J.W., Neuzil, J., Brunk, U.T., 2003. Mitochondrial recyclingand aging of cardiac myocytes: the role of autophagocytosis. Exp. Gerontol. 38,863–876.
Terman, A., Gustafsson, B., Brunk, U.T., 2007. Autophagy, organelles and ageing.J. Pathol. 211, 134–143.
Tizon, B., Sahoo, S., Yu, H., Gauthier, S., Kumar, A.R., Mohan, P., Figliola, M.,Pawlik, M., Grubb, A., Uchiyama, Y., Bandyopadhyay, U., Cuervo, A.M.,Nixon, R.A., Levy, E., 2010. Induction of autophagy by cystatin C: a mecha-nism that protects murine primary cortical neurons and neuronal cell lines.PLoS ONE 5, e9819.
Troen, B.R., 2003. The biology of aging. Mount Sinai J. Med. 70, 3–22.Vellai, T., 2009. Autophagy genes and ageing. Cell Death Differ. 16, 94–102.Vellai, T., Takács-Vellai, K., Sass, M., Klionsky, D.J., 2009. The regulation of aging: does
autophagy underlie longevity? Trends Cell Biol. 19, 487–494.Verbeke, P., Fonager, J., Clark, B.F., Rattan, S.I., 2001. Heat shock response and ageing:
mechanisms and applications. Cell Biol. Int. 25, 845–857.Vinciguerra, M., Musaro, A., Rosenthal, N., 2010. Regulation of muscle atrophy in
aging and disease. Adv. Exp. Med. Biol. 694, 211–233.Vittorini, S., Paradiso, C., Donati, A., Cavallini, G., Masini, M., Gori, Z., Pollera, M.,
Bergamini, E., 1999. The age-related accumulation of protein carbonyl in rat livercorrelates with the age-related decline in liver proteolytic activities. J. Gerontol.A Biol. Sci. Med. Sci. 54, 318–323.
Wang, X., Proud, C.G., 2006. The mTOR pathway in the control of protein synthesis.Physiology 21, 362–369.
Ward, W.F., 2002. Protein degradation in the aging organism. Prog. Mol. Subcell. Biol.29, 35–42.
Weinert, B.T., Timiras, P.S., 2003. Invited review: theories of aging. J. Appl. Physiol.95, 1706–1716.
Wiggins, R.C., 2007. The spectrum of podocytopathies: a unifying view of glomerulardiseases. Kidney Int. 71, 1205–1214.
Wójcik, C., Yano, M., DeMartino, G.N., 2004. RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked toubiquitin/proteasome-dependent proteolysis. J. Cell Sci. 117, 281–292.
Wong, E., Cuervo, A.M., 2010. Autophagy gone awry in neurodegenerative diseases.Nat. Neurosci. 13, 805–811.
Wullschleger, S., Loewith, R., Hall, M.N., 2006. TOR signalling in growth andmetabolism. Cell 124, 471–484.
Xie, Z., Klionsky, D.J., 2007. Autophagosome formation: core machinery and adap-tations. Nat. Cell Biol. 9, 1102–1109.
Xilouri, M., Vogiatzi, T., Vekrellis, K., Stefanis, L., 2008. Alpha-synuclein degradationby autophagic pathways: a potential key to Parkinson’s disease pathogenesis.Autophagy 4, 917–919.
Xu, K., Yang, Y., Yan, M., Zhan, J., Fu, X., Zheng, X., 2010. Autophagy plays a protectiverole in free cholesterol overload-induced death of smooth muscle cells. J. LipidRes. 51, 2581–2590.
Yang, Z., Klionsky, D.J., 2009. An overview of the molecular mechanism of autophagy.Curr. Top. Microbiol. Immunol. 335, 1–32.

search
Y
Y
Y
Y
Y
R. Rezzani et al. / Ageing Re
en, W.L., Shintani, T., Nair, U., Cao, Y., Richardson, B.C., Li, Z., Hughson, F.M., Baba,M., Klionsky, D.J., 2010. The conserved oligomeric Golgi complex is involvedin double-membrane vesicle formation during autophagy. J. Cell Biol. 188,101–114.
in, X.M., Ding, W.X., Gao, W., 2008. Autophagy in the liver. Hepatology 47,1773–1785.
lä-Anttila, P., Vihinen, H., Jokitalo, E., Eskelinen, E.L., 2009. 3D tomography revealsconnections between the phagophore and endoplasmic reticulum. Autophagy5, 1180–1185.
oo, M.S., Kawamata, H., Kim, D.J., Chun, H.S., Son, J.H., 2004. Experimental strategy
to identify genes susceptible to oxidative stress in nigral dopaminergic neurons.Neurochem. Res. 29, 1223–1234.orimitsu, T., He, C., Wang, K., Klionsky, D.J., 2009. Tap42-associated protein phos-phatase type 2A negatively regulates induction of autophagy. Autophagy 5,616–624.
Reviews 11 (2012) 10– 31 31
Young, A., Narita, M., 2010. Connecting autophagy to senescence in pathophysiology.Curr. Opin. Cell Biol. 22, 234–240.
Young, A., Narita, M., Ferreira, M., Kirschner, K., Sadaie, M., Darot, J., Tavaré, S.,Arakawa, S., Shimizu, S., Watt, F.M., Narita, M., 2009. Autophagy mediates themitotic senescence transition. Genes Dev. 23, 798–803.
Yu, L., Wan, F., Dutta, S., Welsh, S., Liu, Z., Freundt, E., Baehrecke, E.H., Lenardo,M., 2006. Autophagic programmed cell death by selective catalase degradation.Proc. Natl. Acad. Sci. U.S.A. 103, 4952–4957.
Zarranz, J.J., Alegre, J., Gómez-Esteban, J.C., Lezcano, E., Ros, R., Ampuero, I., Vidal, L.,Hoenicka, J., Rodriguez, O., Atarés, B., Llorens, V., Gomez Tortosa, E., del Ser, T.,
Munoz, D.G., de Yebenes, J.G., 2004. The new mutation, E46K, of alpha-synucleincauses Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173.Zheng, L., Kågedal, K., Dehvari, N., Benedikz, E., Cowburn, R., Marcusson, J., Terman,A., 2009. Oxidative stress induces macroautophagy of amyloid beta-protein andensuing apoptosis. Free Radic. Biol. Med. 46, 422–429.