methamphetamine downregulates striatal glutamate receptors via diverse epigenetic mechanisms
TRANSCRIPT

ARCHIVAL REPORT
Methamphetamine Downregulates Striatal GlutamateReceptors via Diverse Epigenetic Mechanisms
Subramaniam Jayanthi, Michael T. McCoy, Billy Chen, Jonathan P. Britt, Saїd Kourrich,Hau-Jie Yau, Bruce Ladenheim, Irina N. Krasnova, Antonello Bonci, and Jean Lud CadetBackground: Chronic methamphetamine (METH) exposure causes neuroadaptations at glutamatergic synapses.
Methods: To identify the METH-induced epigenetic underpinnings of these neuroadaptations, we injected increasing METH doses torats for 2 weeks and measured striatal glutamate receptor expression. We then quantified the effects of METH exposure on histoneacetylation. We also measured METH-induced changes in DNA methylation and DNA hydroxymethylation.
Results: Chronic METH decreased transcript and protein expression of GluA1 and GluA2 alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and GluN1 N-methyl-D-aspartate receptor subunits. These changes were associated with alteredelectrophysiological glutamatergic responses in striatal neurons. Chromatin immunoprecipitation-polymerase chain reaction revealedthat METH decreased enrichment of acetylated histone H4 on GluA1, GluA2, and GluN1 promoters. Methamphetamine exposure alsoincreased repressor element-1 silencing transcription factor (REST) corepressor 1, methylated CpG binding protein 2, and histonedeacetylase 2 enrichment, but not of sirtuin 1 or sirtuin 2, onto GluA1 and GluA2 gene sequences. Moreover, METH caused interactionsof REST corepressor 1 and methylated CpG binding protein 2 with histone deacetylase 2 and of REST with histone deacetylase 1.Surprisingly, methylated DNA immunoprecipitation and hydroxymethylated DNA immunoprecipitation-polymerase chain reactionrevealed METH-induced decreased enrichment of 5-methylcytosine and 5-hydroxymethylcytosine at GluA1 and GluA2 promotersequences. Importantly, the histone deacetylase inhibitor, valproic acid, blocked METH-induced decreased expression of AMPAR andN-methyl-D-aspartate receptor subunits. Finally, valproic acid also attenuated METH-induced decrease H4K16Ac recruitment on AMPARgene sequences.
Conclusions: These observations suggest that histone H4 hypoacetylation may be the main determinant of METH-induced decreasedstriatal glutamate receptor expression.
Key Words: Addiction, AMPAR, CoREST, HDAC2, MeCP2, NMDAR,REST, valproic acid
Addictions are neuropsychiatric disorders that are secondary,in part, to altered synaptic plasticity in mesostriatal andcorticostriatal projection areas (1–3). The dorsal striatum is
important in the neural circuitry of addiction because the nidus ofcontrol for drug taking appears to shift from the ventral to the dorsalstriatum as drug taking becomes habitual (4–7). Repeated psychos-timulant injections can produce biochemical, molecular, and phys-iological alterations at striatal glutamatergic synapses (1,8).Specifically, cocaine administration is accompanied by changes inthe expression or trafficking of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs) in the mesolimbicsystem (1). Both contingent and noncontingent administration ofcocaine is associated with increased expression of AMPARs onneuronal membranes (9), increased expression of GluA2-lackingAMPARs (10,11), and physiological evidence of differential AMPARexpression (12) in the ventral striatum. Therefore, dynamic
From the Molecular Neuropsychiatry Research Branch (SJ, MTM, BL, INK,JLC) and Synaptic Plasticity Section (BC, JPB, SK, H-JY, AB), USDepartment of Health and Human Services/National Institutes ofHealth/National Institute on Drug Abuse/Intramural Research Program,Baltimore, MD.
Address correspondence to Jean Lud Cadet, M.D., Molecular Neuropsy-chiatry Research Branch, National Institute on Drug Abuse/NationalInstitutes of Health/US Department of Health and Human Services, 251Bayview Boulevard, Baltimore, MD 21224; E-mail: [email protected].
Received Jan 29, 2013; revised Sep 27, 2013; accepted Sep 30, 2013.
0006-3223/$36.00http://dx.doi.org/10.1016/j.biopsych.2013.09.034 Publis
alterations in AMPAR subunit composition might be involved inthe maintenance of drug seeking and/or in the occurrence ofrelapses (10,11). Parenthetically, very little is known about the effectsof methamphetamine (METH) on the expression of these receptors.In rodents, injections of increasing doses of METH (10–30 mg/kg) for7 consecutive days produced increased AMPA GluA2 proteinexpression in the dorsal striatum (13). Nevertheless, the transcrip-tional effects of METH on GluA1 or GluA2 or the epigenetic bases forany potential METH-induced changes in striatal AMPAR expressionare unknown.
Gene transcription is regulated by complex epigeneticchanges including posttranslational histone modifications andDNA methylation that regulate diverse genomic functions (14,15).Eukaryotic DNA is packaged into chromatin, whose basic unit, thenucleosome, contains four core histones that form an octamersurrounded by 147 base pair of DNA. The N-tails of histonespossess lysine residues that can be reversibly acetylated ordeacetylated by histone acetyltransferases or histone deacety-lases (HDACs), respectively (16). Because epigenetic phenomenaare involved in the clinical manifestations of neuropsychiatricdiseases, including addiction (17), we thought it is likely thatMETH could engender transcriptional and epigenetic changesthat are unique to this clinically devastating drug (18). Studies ofthe transcriptional effects of METH on AMPAR expression areimportant because its biochemical effects are different from thoseof cocaine. Specifically, METH interacts with vesicular monoaminetransporter and causes release of dopamine by reverse transport(18,19), whereas cocaine inhibits monoamine reuptake (20,21).The two main purposes of this study were to characterize theeffects of METH exposure on striatal AMPAR expression and toidentify potential epigenetic bases for any changes in receptorexpression.
BIOL PSYCHIATRY 2013;]:]]]–]]]hed by Elsevier Inc on behalf of Society of Biological Psychiatry

2 BIOL PSYCHIATRY 2013;]:]]]–]]] S. Jayanthi et al.
Methods and Materials
Animals and Drug TreatmentAll animal treatments and procedures were approved by the
National Institute on Drug Abuse Animal Care and Use Commit-tee and followed the Guide for the Care and Use of LaboratoryAnimals (ISBN 0-309-05377-3). Male Sprague-Dawley rats (CharlesRiver Labs, Wilmington, Massachusetts), weighing 250 g to 300 g,were housed in a humidity- and temperature-controlled (22.21C� .21C) room with free access to food and water. Followinghabituation, rats were assigned to two groups (eight rats each)and were injected daily for 2 weeks with either saline or METH, asshown in Table S1 in Supplement 1. The animals were euthan-ized 16 hours after the last saline or METH injection. This METHregimen was meant to mimic the patterns of METH abuse byhuman abusers who start at low to moderate doses (10–50 mg)and progressed to higher doses (22,23). This pattern of METHadministration to rats does not cause any striatal toxicity (24)(Figure S1 in Supplement 1).
For co-treatment with HDAC inhibitor, rats received intra-peritoneal sodium valproate (VPA) (300 mg/kg, dissolved in water;Sigma, Valencia, California) injections twice a day 30 minutesbefore either saline or METH injections. We chose VPA, a well-tolerated agent with extensive clinical use, recognizing its variedeffects on the brain (25). The VPA dose was based on thepublished literature (26). There were four groups for the co-treatment experiments: vehicle/saline (control); vehicle/METH(METH); VPA/saline (VPA); and VPA/METH (VPA � METH).
Quantitative Polymerase Chain Reaction Analysis ofMessenger RNA Levels
Total RNA was isolated from one striatal hemisphere usingRNeasy Mini kit (Santa Cruz Biotechnology, Santa Cruz, California)from eight rats per group. Quantitative polymerase chain reaction(PCR) was carried out essentially as described by us (27).
Subcellular FractionationSeparation of nuclear, cell membrane, and cytoplasmic frac-
tions from striatal tissues was performed by differential centrifu-gation at 4oC. Details are provided in Supplement 1.
Immunoblot AnalysisStriatal protein lysates (n ¼ 6) were separated by sodium dodecyl
sulfate polyacrylamide gel electrophoresis and electrophoreticallytransferred on polyvinylidene difluoride membranes, essentially asdescribed by us (see Supplement 1 for details). The membraneswere incubated overnight at 41C with specific antibodies againstGluA1, GluA2, GluN1/NR1, histone deacetylase 1 (HDAC1), andhistone deacetylase 2 (HDAC2) (Santa Cruz); H4K5ac, H4K12ac,and H4K16ac (Millipore, Billerica, California); and sirtuin 1 (SIRT1),sirtuin 2 (SIRT2), and methylated CpG binding protein 2 (MeCP2)(Cell Signaling, Danvers, Massachusetts).
Co-immunoprecipitationNuclear extracts were prepared from the striatum of saline-
and METH-treated rats according to Barrett et al. (28) with minormodifications. Details are included in Supplement 1.
Chromatin Immunoprecipitation AssaysStriatal tissue was processed for acetyl H4, repressor element 1
silencing transcription factor (REST), REST corepressor 1 (CoREST),HDAC1, HDAC2, and MeCP2 chromatin immunoprecipitation(ChIP) (29) or methylated DNA immunoprecipitation and
www.sobp.org/journal
hydroxymethylated DNA immunoprecipitation (30,31) accordingto published protocols. Details are provided in Supplement 1.
Enrichment of various proteins at GluA1, GluA2, and GluN1promoters were determined by quantitative real-time PCR usingspecific ChIP primers designed to amplify proximal or distalsequences from the transcription start site (TSS). Each PCRreaction was repeated at least twice. The specific primers usedare listed under Table S2 in Supplement 1.
ElectrophysiologyPerfused rat dorsal striatum was used in the electrophysiology
experiments. Details of the electrophysiological experiments wereessentially as described by Britt et al. (32) and are provided in theSupplement 1.
Statistical AnalysisAll the quantitative data are presented as mean � SEM. For
data comparing control and METH-treated groups, unpairedStudent t test was used (StatView version 4.02, St. Louis, Missouri).For the experiments involving VPA co-treatment, two-way analysisof variance (ANOVA) was used followed by Bonferroni post hoc.For electrophysiology, data were assessed using one-way ANOVAfor multiple group comparisons, with a Bonferroni post hoc. For allexperiments, the null hypothesis was rejected at p � .05.
Results
Chronic METH Administration Causes Decreased StriatalAMPAR Expression and Function
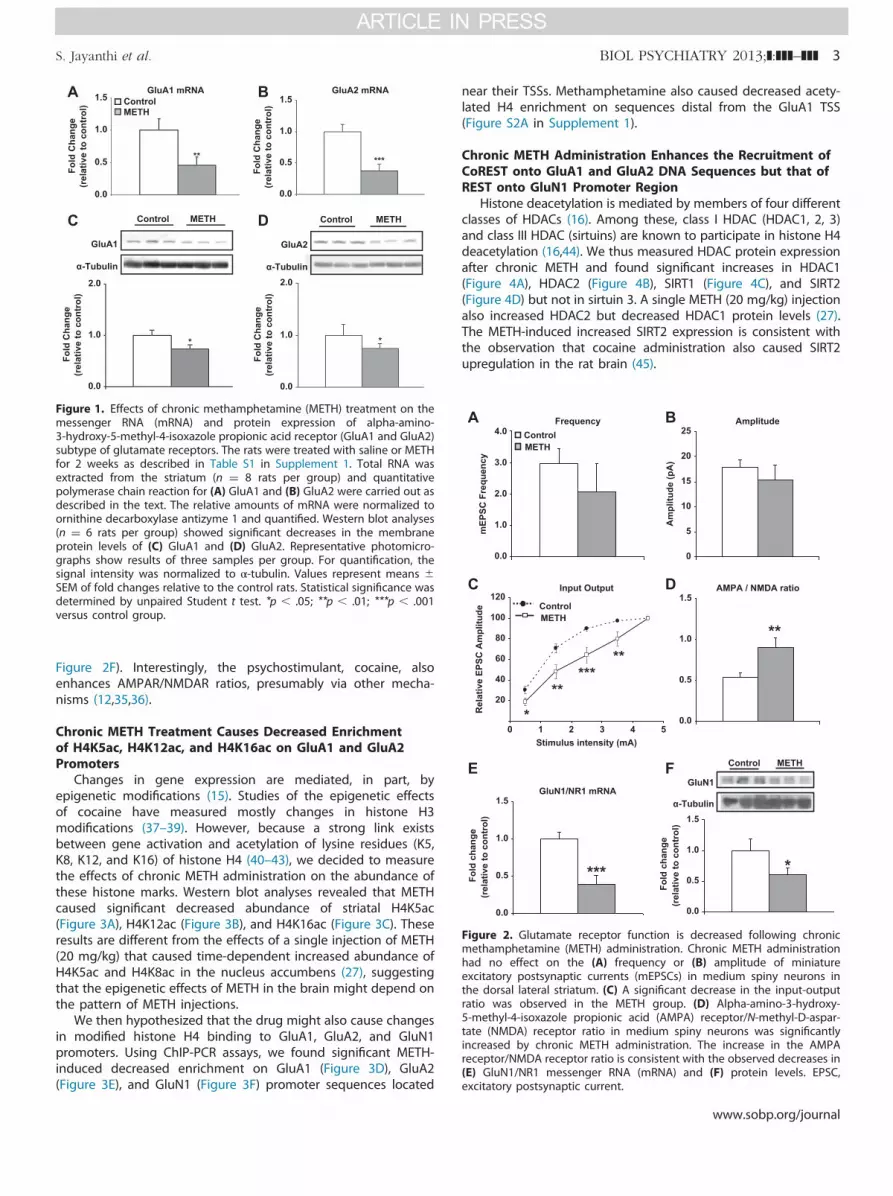
To identify the effects of METH on AMPAR expression, wetreated rats with either saline or increasing METH doses asdescribed above (Table S1 in Supplement 1). Chronic METHdecreased striatal messenger RNA (mRNA) expression of GluA1(Figure 1A) and GluA2 (Figure 1B). Methamphetamine alsocaused decreased GluA1 (Figure 1C) and GluA2 (Figure 1D)protein levels.
To determine whether these changes in AMPAR subtypes alterexcitatory synaptic transmission, ex vivo whole-cell patch clamprecordings were performed on striatal medium spiny neurons.Sixteen hours after the last METH or saline injection, rats weresacrificed and coronal slices containing the striatum wereobtained. Miniature excitatory postsynaptic currents on mediumspiny neurons (Figure 2) were measured blindly according toprevious descriptions (32). Unexpectedly, chronic METH did notcause significant changes in miniature excitatory postsynapticcurrent amplitude or frequency (Figure 2A,B), in contrast topublished observations with cocaine (12,33). We also increasedstimulus intensities and measured evoked excitatory postsynapticcurrents. We found that the input–output relationship betweenevoked excitatory postsynaptic currents and increasing stimulusintensities was significantly decreased in the METH group incomparison with control animals (Figure 2C). Surprisingly, wefound that the ratio of peak AMPAR- to peak N-methyl-D-aspartate receptor (NMDAR)-mediated evoked currents, a meas-ure of glutamate synaptic plasticity (34), was significantlyincreased in the chronic METH-treated group (Figure 2D). TheMETH-induced increases in AMPAR/NMDAR ratios appear to berelated, in part, to METH-induced decreased mRNA (Figure 2E)and protein (Figure 2F) levels of the obligatory N-methyl-D-aspartate (NMDA) receptor, GluN1/NR1, because the percentagedecrease in AMPA protein expression (�22% to 26%) was lessthan that of GluN1 (�45%) (compare Figures 1C and 1D with
Control METH
*
0.0
1.0
2.0
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
α-Tubulin
GluA1
Control METH
*
0.0
1.0
2.0
Fold
Cha
nge
(rel
ativ
e to
con
trol
)α-Tubulin
GluA2
GluA1 mRNA
0.0
1.0
1.5
**
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
0.5
0.0
1.0
1.5
0.5
GluA2 mRNA
***
Fold
Cha
nge
(rel
ativ
e to
con
trol
)ControlMETH
Figure 1. Effects of chronic methamphetamine (METH) treatment on themessenger RNA (mRNA) and protein expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (GluA1 and GluA2)subtype of glutamate receptors. The rats were treated with saline or METHfor 2 weeks as described in Table S1 in Supplement 1. Total RNA wasextracted from the striatum (n ¼ 8 rats per group) and quantitativepolymerase chain reaction for (A) GluA1 and (B) GluA2 were carried out asdescribed in the text. The relative amounts of mRNA were normalized toornithine decarboxylase antizyme 1 and quantified. Western blot analyses(n ¼ 6 rats per group) showed significant decreases in the membraneprotein levels of (C) GluA1 and (D) GluA2. Representative photomicro-graphs show results of three samples per group. For quantification, thesignal intensity was normalized to α-tubulin. Values represent means �SEM of fold changes relative to the control rats. Statistical significance wasdetermined by unpaired Student t test. *p � .05; **p � .01; ***p � .001versus control group.
AMPA / NMDA ratio
0.0
0.5
1.0
1.5
20
40
60
80
100
120Input Output
Rel
ativ
e EP
SC A
mpl
itude
ControlMETH
0 54321Stimulus intensity (mA)
mEP
SC F
requ
ency
0.0
3.0
2.0
1.0
4.0Frequency
Am
plitu
de (p
A)
0
15
10
5
20
25Amplitude
ControlMETH
GluN1/NR1 mRNA
Fold
cha
nge
(rel
ativ
e to
con
trol
)
0.0
0.5
1.0
1.5
Control METH
0.0
0.5
1.0
1.5
Fold
cha
nge
(rel
ativ
e to
con
trol
)
α-Tubulin
GluN1
Figure 2. Glutamate receptor function is decreased following chronicmethamphetamine (METH) administration. Chronic METH administrationhad no effect on the (A) frequency or (B) amplitude of miniatureexcitatory postsynaptic currents (mEPSCs) in medium spiny neurons inthe dorsal lateral striatum. (C) A significant decrease in the input-outputratio was observed in the METH group. (D) Alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor/N-methyl-D-aspar-tate (NMDA) receptor ratio in medium spiny neurons was significantlyincreased by chronic METH administration. The increase in the AMPAreceptor/NMDA receptor ratio is consistent with the observed decreases in(E) GluN1/NR1 messenger RNA (mRNA) and (F) protein levels. EPSC,excitatory postsynaptic current.
S. Jayanthi et al. BIOL PSYCHIATRY 2013;]:]]]–]]] 3
Figure 2F). Interestingly, the psychostimulant, cocaine, alsoenhances AMPAR/NMDAR ratios, presumably via other mecha-nisms (12,35,36).
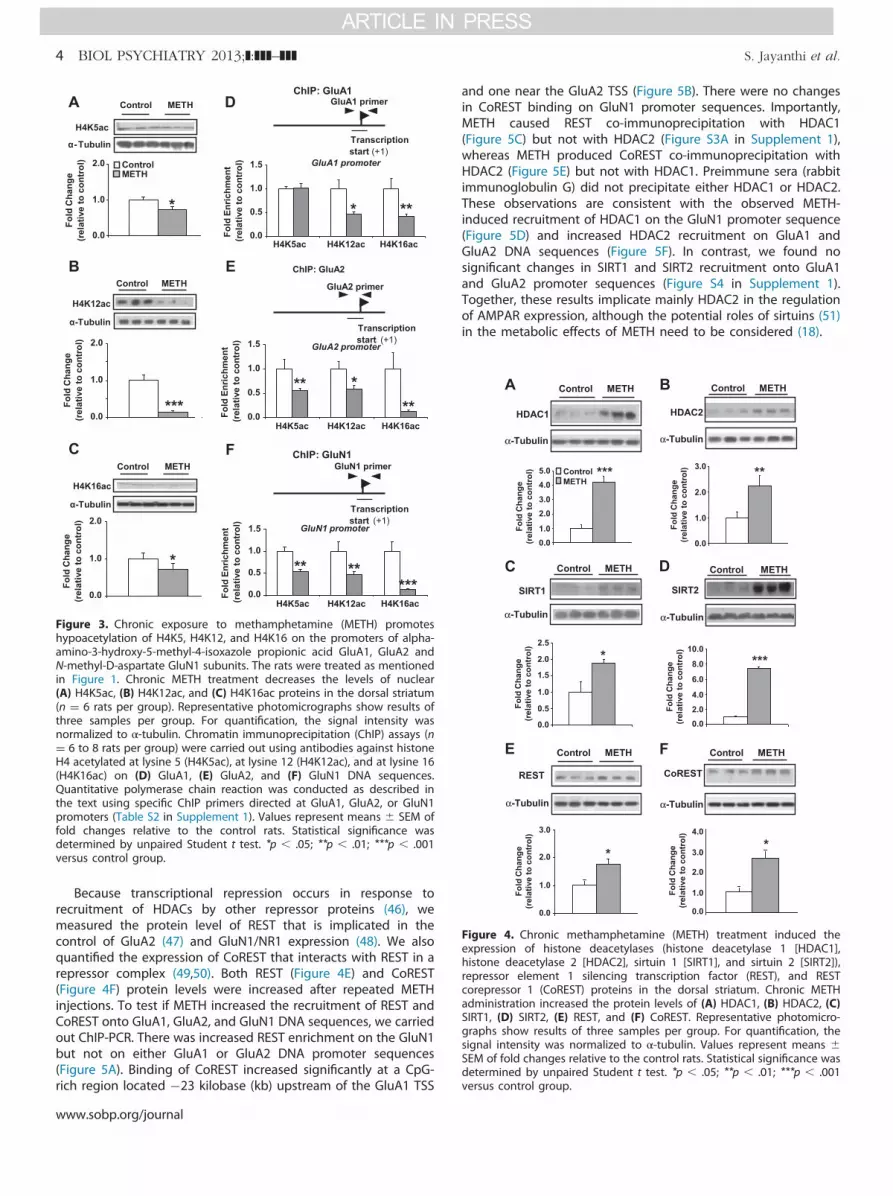
Chronic METH Treatment Causes Decreased Enrichmentof H4K5ac, H4K12ac, and H4K16ac on GluA1 and GluA2Promoters
Changes in gene expression are mediated, in part, byepigenetic modifications (15). Studies of the epigenetic effectsof cocaine have measured mostly changes in histone H3modifications (37–39). However, because a strong link existsbetween gene activation and acetylation of lysine residues (K5,K8, K12, and K16) of histone H4 (40–43), we decided to measurethe effects of chronic METH administration on the abundance ofthese histone marks. Western blot analyses revealed that METHcaused significant decreased abundance of striatal H4K5ac(Figure 3A), H4K12ac (Figure 3B), and H4K16ac (Figure 3C). Theseresults are different from the effects of a single injection of METH(20 mg/kg) that caused time-dependent increased abundance ofH4K5ac and H4K8ac in the nucleus accumbens (27), suggestingthat the epigenetic effects of METH in the brain might depend onthe pattern of METH injections.
We then hypothesized that the drug might also cause changesin modified histone H4 binding to GluA1, GluA2, and GluN1promoters. Using ChIP-PCR assays, we found significant METH-induced decreased enrichment on GluA1 (Figure 3D), GluA2(Figure 3E), and GluN1 (Figure 3F) promoter sequences located
near their TSSs. Methamphetamine also caused decreased acety-lated H4 enrichment on sequences distal from the GluA1 TSS(Figure S2A in Supplement 1).
Chronic METH Administration Enhances the Recruitment ofCoREST onto GluA1 and GluA2 DNA Sequences but that ofREST onto GluN1 Promoter Region
Histone deacetylation is mediated by members of four differentclasses of HDACs (16). Among these, class I HDAC (HDAC1, 2, 3)and class III HDAC (sirtuins) are known to participate in histone H4deacetylation (16,44). We thus measured HDAC protein expressionafter chronic METH and found significant increases in HDAC1(Figure 4A), HDAC2 (Figure 4B), SIRT1 (Figure 4C), and SIRT2(Figure 4D) but not in sirtuin 3. A single METH (20 mg/kg) injectionalso increased HDAC2 but decreased HDAC1 protein levels (27).The METH-induced increased SIRT2 expression is consistent withthe observation that cocaine administration also caused SIRT2upregulation in the rat brain (45).
www.sobp.org/journal
Control METH
0.0
1.0
2.0
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
H4K5ac
α-Tubulin
ChIP: GluA1
Transcriptionstart (+1)
GluA1 primer
0.0
0.5
1.0
1.5ControlMETH
Fold
Enr
ichm
ent
(rel
ativ
e to
con
trol
)H4K5ac H4K12ac H4K16ac
Fold
Enr
ichm
ent
(rel
ativ
e to
con
trol
)
H4K5ac H4K12ac H4K16ac
Transcriptionstart (+1)
GluA2 primer
GluA2 promoter
ChIP: GluA2
0.0
0.5
1.0
1.5
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
0.0
2.0
1.0
H4K12ac
α-Tubulin
Control METH
Transcriptionstart (+1)
GluN1 primerChIP: GluN1
Fold
Enr
ichm
ent
(rel
ativ
e to
con
trol
)
H4K5ac H4K12ac0.0
0.5
1.0
1.5
H4K16ac
α-Tubulin
Control METH
0.0
2.0
1.0
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
GluN1 promoter
GluA1 promoter
H4K16ac
Figure 3. Chronic exposure to methamphetamine (METH) promoteshypoacetylation of H4K5, H4K12, and H4K16 on the promoters of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid GluA1, GluA2 andN-methyl-D-aspartate GluN1 subunits. The rats were treated as mentionedin Figure 1. Chronic METH treatment decreases the levels of nuclear(A) H4K5ac, (B) H4K12ac, and (C) H4K16ac proteins in the dorsal striatum(n ¼ 6 rats per group). Representative photomicrographs show results ofthree samples per group. For quantification, the signal intensity wasnormalized to α-tubulin. Chromatin immunoprecipitation (ChIP) assays (n¼ 6 to 8 rats per group) were carried out using antibodies against histoneH4 acetylated at lysine 5 (H4K5ac), at lysine 12 (H4K12ac), and at lysine 16(H4K16ac) on (D) GluA1, (E) GluA2, and (F) GluN1 DNA sequences.Quantitative polymerase chain reaction was conducted as described inthe text using specific ChIP primers directed at GluA1, GluA2, or GluN1promoters (Table S2 in Supplement 1). Values represent means � SEM offold changes relative to the control rats. Statistical significance wasdetermined by unpaired Student t test. *p � .05; **p � .01; ***p � .001versus control group.
Control METH
0.01.0
2.03.0
4.05.0 ***
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
α-Tubulin
HDAC1
α-Tubulin
HDAC2
Control METH
Fold
Cha
nge
(rel
ativ
e to
con
trol
) **
0.0
1.0
2.0
3.0ControlMETH
0.0
1.0
2.0
4.0
3.0
Control METH
0.0
1.0
2.0
3.0
*
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
α-Tubulin
REST
Control METH
0.0
0.5
1.0
1.5
2.0
2.5
*
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
α-Tubulin
SIRT1
***
0.0
2.0
4.0
6.0
8.0
10.0
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
α-Tubulin
SIRT2
Control METH
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
α-Tubulin
CoREST
Control METH
*
Figure 4. Chronic methamphetamine (METH) treatment induced theexpression of histone deacetylases (histone deacetylase 1 [HDAC1],histone deacetylase 2 [HDAC2], sirtuin 1 [SIRT1], and sirtuin 2 [SIRT2]),repressor element 1 silencing transcription factor (REST), and RESTcorepressor 1 (CoREST) proteins in the dorsal striatum. Chronic METHadministration increased the protein levels of (A) HDAC1, (B) HDAC2, (C)SIRT1, (D) SIRT2, (E) REST, and (F) CoREST. Representative photomicro-graphs show results of three samples per group. For quantification, thesignal intensity was normalized to α-tubulin. Values represent means �SEM of fold changes relative to the control rats. Statistical significance wasdetermined by unpaired Student t test. *p � .05; **p � .01; ***p � .001versus control group.
4 BIOL PSYCHIATRY 2013;]:]]]–]]] S. Jayanthi et al.
Because transcriptional repression occurs in response torecruitment of HDACs by other repressor proteins (46), wemeasured the protein level of REST that is implicated in thecontrol of GluA2 (47) and GluN1/NR1 expression (48). We alsoquantified the expression of CoREST that interacts with REST in arepressor complex (49,50). Both REST (Figure 4E) and CoREST(Figure 4F) protein levels were increased after repeated METHinjections. To test if METH increased the recruitment of REST andCoREST onto GluA1, GluA2, and GluN1 DNA sequences, we carriedout ChIP-PCR. There was increased REST enrichment on the GluN1but not on either GluA1 or GluA2 DNA promoter sequences(Figure 5A). Binding of CoREST increased significantly at a CpG-rich region located �23 kilobase (kb) upstream of the GluA1 TSS
www.sobp.org/journal
and one near the GluA2 TSS (Figure 5B). There were no changesin CoREST binding on GluN1 promoter sequences. Importantly,METH caused REST co-immunoprecipitation with HDAC1(Figure 5C) but not with HDAC2 (Figure S3A in Supplement 1),whereas METH produced CoREST co-immunoprecipitation withHDAC2 (Figure 5E) but not with HDAC1. Preimmune sera (rabbitimmunoglobulin G) did not precipitate either HDAC1 or HDAC2.These observations are consistent with the observed METH-induced recruitment of HDAC1 on the GluN1 promoter sequence(Figure 5D) and increased HDAC2 recruitment on GluA1 andGluA2 DNA sequences (Figure 5F). In contrast, we found nosignificant changes in SIRT1 and SIRT2 recruitment onto GluA1and GluA2 promoter sequences (Figure S4 in Supplement 1).Together, these results implicate mainly HDAC2 in the regulationof AMPAR expression, although the potential roles of sirtuins (51)in the metabolic effects of METH need to be considered (18).

0.0
1.0
2.0
4.0
3.0
ChIP: REST
Fold
Enr
ichm
ent
(rel
ativ
e to
con
trol
)
GluA1 GluA20.0
1.0
4.0
2.0
3.0
5.0
GluN1
*ChIP: CoREST
0.0
1.0
4.0
2.0
3.0
Fold
Enr
ichm
ent
(rel
ativ
e to
con
trol
)
*
GluA1 GluA2
*
ControlMETH
GluN1
IP : CoRESTWB: HDAC2
HDAC2(60 kDa)
Inpu
t
IgGMETH Control Inpu
t
ChIP: HDAC2
Fold
Enr
ichm
ent
(rel
ativ
e to
con
trol
) ***
GluA1 GluA2 GluN10.0
1.0
2.0
4.0
3.0
IP : RESTWB: HDAC1
Inpu
t
Inpu
t
METH Control IgG
HDAC1
IgG(60 kDa)
Fold
Enr
ichm
ent
(rel
ativ
e to
con
trol
)
GluA1 GluA2 GluN1
*
ChIP: HDAC1
Figure 5. Chronic methamphetamine (METH) increases enrichment ofrepressor element 1 silencing transcription factor corepressor 1 (CoREST)on GluA1 and GluA2 gene promoters, whereas enrichment of repressorelement 1 silencing transcription factor (REST) was observed on GluN1promoter. Chromatin immunoprecipitation (ChIP) assays (n ¼ 8 rats pergroup) were performed on striata of control and METH-treated rats with(A) anti-REST and (B) anti-CoREST antibodies. Quantitative polymerasechain reaction was conducted as described in the text using specific ChIPprimers directed at GluA1 or GluA2 or GluN1 promoter (Table S2 inSupplement 1). Values represent means � SEM of fold changes relative tothe control rats. Statistical significance was determined by unpairedStudent t test. *p � .05; **p � .01 versus control group. Co-immunopre-cipitation assays of (C) REST and histone deacetylase 1 (HDAC1) and (E)CoREST and histone deacetylase 2 (HDAC2). Immunoprecipitates (IP) wereprepared from striatal nuclear extracts of control and METH-treated ratsusing antibody against anti-REST and anti-CoREST, and recovery of HDAC1and HDAC2 was determined by Western blot (WB) assay. The levels ofHDAC1 and HDAC2 from nonspecific immunoglobulin G (IgG) areindicated. Input levels (5%) of HDAC1 and HDAC2 are shown forcomparison. Chromatin immunoprecipitation assays (n ¼ 6 to 8 rats pergroup) were carried out using antibodies against HDAC1 (D) and HDAC2(F) on GluA1, GluA2, and GluN1 DNA sequences. Quantitative polymerasechain reaction was conducted as described in the text using specific ChIPprimers directed at GluA1, GluA2, or GluN1 promoters (Table S2 inSupplement 1). Values represent means � SEM of fold changes relativeto the control rats. Statistical significance was determined by unpairedStudent t test. *p � .05; **p � .01 versus control group.
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
αα-Tubulin
0.0
1.0
2.0
DNMT3A
Control METH
0.0
1.0
2.0
3.0
**
DNMT1
α-Tubulin
Control METH
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
0.0
1.0
2.0
α-Tubulin
DNMT3B
Control METH
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
IP : MeCP2WB : HDAC2
HDAC2(60 kDa)
IgG
Inpu
t
METHConInpu
t
ChIP: MeCP2
GluA1 GluA2 GluN1
*
*
Fold
Enr
ichm
ent
(rel
ativ
e to
con
trol
)
0.0
0.5
1.0
1.5
2.5
2.0α-Tubulin
*
0.0
2.0
1.0
MeCP2
Control METH
Fold
Cha
nge
(rel
ativ
e to
con
trol
)
ControlMETH
**
MeDIP
0.0
0.5
1.0
1.5
GluA1 GluA2
hMeDIP
0.0
0.5
1.0
1.5
GluA1 GluA2
* *
DN
A M
ethy
latio
n(r
elat
ive
to c
ontr
ol)
DN
Ahy
drox
ymet
hyla
tion
(rel
ativ
e to
con
trol
)
Figure 6. Chronic methamphetamine (METH) exposure causes down-regulation of GluA1 and GluA2 transcription by formation of a methylatedCpG binding protein 2 (MeCP2)-histone deacetylase 2 (HDAC2) complex.Western blot (WB) analysis of (A) MeCP2, (D) DNMT1, (E) DNMT3A, and (F)DNMT3B. Representative photomicrographs show results of three samplesper group. For quantification, the signal intensity was normalized toα-tubulin. Co-immunoprecipitation assays of (B) MeCP2 and HDAC2 showMETH-induced interactions of MeCP2 with HDAC2. The level of HDAC2from nonspecific immunoglobulin G (IgG) is indicated. Input levels (5%) ofHDAC2 are shown for comparison. Chromatin immunoprecipitation (ChIP)assays (n ¼ 6 to 8 rats per group) were carried out using antibodiesagainst MeCP2 (C). Quantitative polymerase chain reaction was conductedusing specific ChIP primers (Table S2 in Supplement 1). Denaturedgenomic DNA of �200 to 600 base pair (generated by sonication) wasincubated with an antibody directed against 5 mC (G) or 5 hmC (H), toisolate methylated or hydroxymethylated DNA by immunoprecipitation.Relative enrichment of 5 mC and 5 hmC in the bound over input fractionswas calculated by real-time polymerase chain reaction. Values representmeans � SEM of fold enrichment relative to the control rats. Statisticalsignificance was determined by unpaired Student t test. *p � .05; **p �.01 versus control group. Con, control; hMeDIP, hydroxymethylated DNAimmunoprecipitation; IP, immunoprecipitates; MeDIP, methylated DNAimmunoprecipitation.
S. Jayanthi et al. BIOL PSYCHIATRY 2013;]:]]]–]]] 5
Chronic METH-Induced Repression of AMPAR also InvolvesMeCP2 Recruitment
In addition to histone acetylation, decrease in gene expressioncan be regulated by DNA methylation (52). DNA methylation ismediated by three DNA methyl transferases (DNMTs), namely themaintenance enzyme, DNMT1 (53,54), and the de novo DNAmethylation enzymes, DNMT3a and DNMT3b (55). In associationwith DNA methylation, the multifunctional, methyl DNA-bindingtranscriptional regulator, MeCP2, can cause decreased geneexpression by recruiting HDACs (56). Methylated CpG bindingprotein 2 exists as a large complex (57), binds to DNMT1 (58), andinteracts with both methylated and unmethylated DNA (59–61).
To test the involvement of DNA methylation in METH-induceddecreased gene expression, we first measured MeCP2 proteinlevels because it participates in DNA methylation-related epige-netic phenomena (62). Chronic METH increased MeCP2 proteinlevels (Figure 6A). Because MeCP2 exerts its transcriptionalrepressing activities, in part, by recruiting HDAC complexes(56,63), we carried out co-immunoprecipitation studies and foundthat METH caused MeCP2 co-precipitation with HDAC2(Figure 6B) but not HDAC1 (Figure S3B in Supplement 1).Chromatin immunoprecipitation-PCR experiments also showedthat METH increased MeCP2 enrichment at sequences locatednear the GluA1 and GluA2 TSSs (Figure 6C). There were nochanges in MeCP2 binding on the GluN1 promoter (Figure 6C).Similar to the cases of histone H4 marks (Figure S2A inSupplement 1), METH also altered MeCP2 binding at the CpG-
www.sobp.org/journal
GluA2 mRNA
GluA1 mRNA
Fold
cha
nge
(rel
ativ
e to
veh
icle
/ sa
line) Saline
METH
GluN1 mRNA
0.0
0.5
1.5
1.0
*
Vehicle VPA
0.0
0.5
1.5
1.0
**
Vehicle VPA
Fold
cha
nge
(rel
ativ
e to
veh
icle
/ sa
line)
0.0
0.5
1.5
1.0
**
Vehicle VPA
Fold
cha
nge
(rel
ativ
e to
veh
icle
/ sa
line)
*
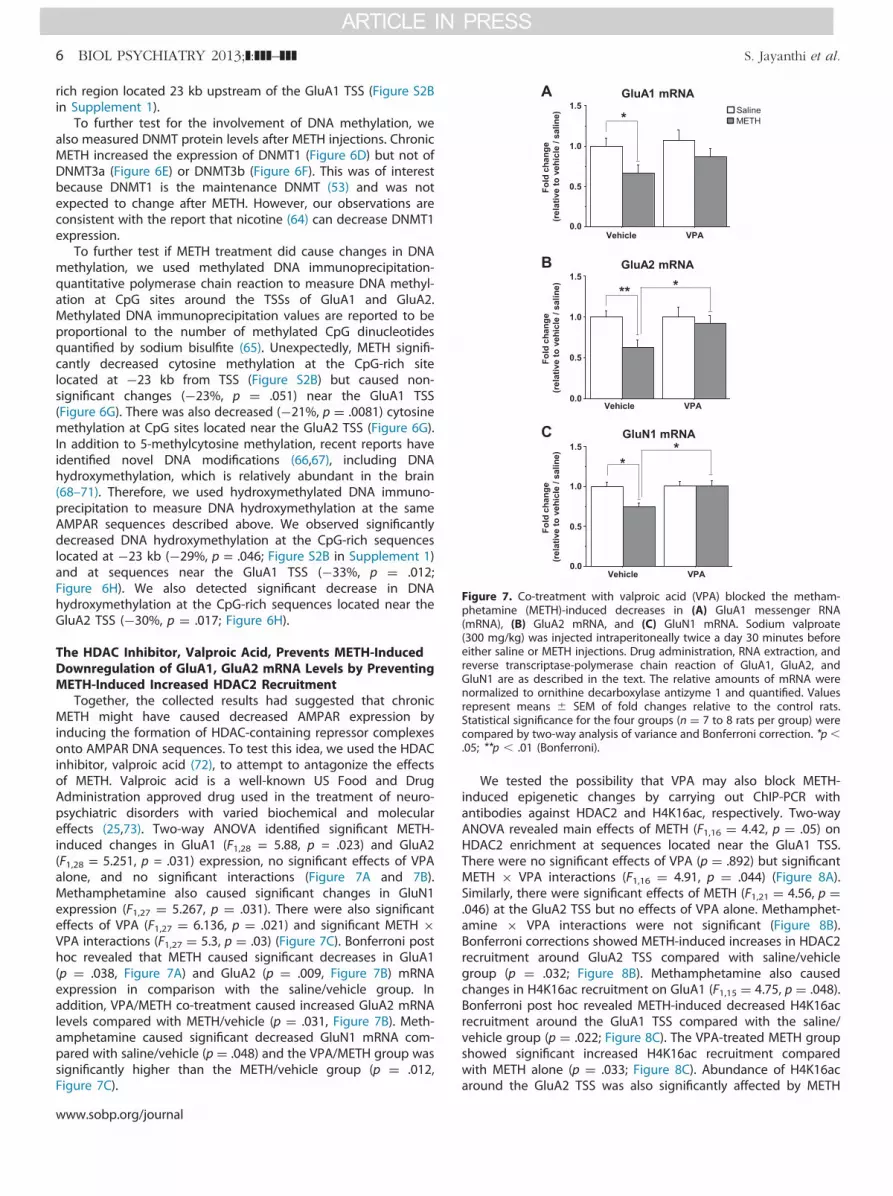
Figure 7. Co-treatment with valproic acid (VPA) blocked the metham-phetamine (METH)-induced decreases in (A) GluA1 messenger RNA(mRNA), (B) GluA2 mRNA, and (C) GluN1 mRNA. Sodium valproate(300 mg/kg) was injected intraperitoneally twice a day 30 minutes beforeeither saline or METH injections. Drug administration, RNA extraction, andreverse transcriptase-polymerase chain reaction of GluA1, GluA2, andGluN1 are as described in the text. The relative amounts of mRNA werenormalized to ornithine decarboxylase antizyme 1 and quantified. Valuesrepresent means � SEM of fold changes relative to the control rats.Statistical significance for the four groups (n ¼ 7 to 8 rats per group) werecompared by two-way analysis of variance and Bonferroni correction. *p �.05; **p � .01 (Bonferroni).
6 BIOL PSYCHIATRY 2013;]:]]]–]]] S. Jayanthi et al.
rich region located 23 kb upstream of the GluA1 TSS (Figure S2Bin Supplement 1).
To further test for the involvement of DNA methylation, wealso measured DNMT protein levels after METH injections. ChronicMETH increased the expression of DNMT1 (Figure 6D) but not ofDNMT3a (Figure 6E) or DNMT3b (Figure 6F). This was of interestbecause DNMT1 is the maintenance DNMT (53) and was notexpected to change after METH. However, our observations areconsistent with the report that nicotine (64) can decrease DNMT1expression.
To further test if METH treatment did cause changes in DNAmethylation, we used methylated DNA immunoprecipitation-quantitative polymerase chain reaction to measure DNA methyl-ation at CpG sites around the TSSs of GluA1 and GluA2.Methylated DNA immunoprecipitation values are reported to beproportional to the number of methylated CpG dinucleotidesquantified by sodium bisulfite (65). Unexpectedly, METH signifi-cantly decreased cytosine methylation at the CpG-rich sitelocated at �23 kb from TSS (Figure S2B) but caused non-significant changes (�23%, p ¼ .051) near the GluA1 TSS(Figure 6G). There was also decreased (�21%, p ¼ .0081) cytosinemethylation at CpG sites located near the GluA2 TSS (Figure 6G).In addition to 5-methylcytosine methylation, recent reports haveidentified novel DNA modifications (66,67), including DNAhydroxymethylation, which is relatively abundant in the brain(68–71). Therefore, we used hydroxymethylated DNA immuno-precipitation to measure DNA hydroxymethylation at the sameAMPAR sequences described above. We observed significantlydecreased DNA hydroxymethylation at the CpG-rich sequenceslocated at �23 kb (�29%, p = .046; Figure S2B in Supplement 1)and at sequences near the GluA1 TSS (�33%, p ¼ .012;Figure 6H). We also detected significant decrease in DNAhydroxymethylation at the CpG-rich sequences located near theGluA2 TSS (�30%, p ¼ .017; Figure 6H).
The HDAC Inhibitor, Valproic Acid, Prevents METH-InducedDownregulation of GluA1, GluA2 mRNA Levels by PreventingMETH-Induced Increased HDAC2 Recruitment
Together, the collected results had suggested that chronicMETH might have caused decreased AMPAR expression byinducing the formation of HDAC-containing repressor complexesonto AMPAR DNA sequences. To test this idea, we used the HDACinhibitor, valproic acid (72), to attempt to antagonize the effectsof METH. Valproic acid is a well-known US Food and DrugAdministration approved drug used in the treatment of neuro-psychiatric disorders with varied biochemical and moleculareffects (25,73). Two-way ANOVA identified significant METH-induced changes in GluA1 (F1,28 = 5.88, p = .023) and GluA2(F1,28 = 5.251, p = .031) expression, no significant effects of VPAalone, and no significant interactions (Figure 7A and 7B).Methamphetamine also caused significant changes in GluN1expression (F1,27 ¼ 5.267, p ¼ .031). There were also significanteffects of VPA (F1,27 ¼ 6.136, p ¼ .021) and significant METH �VPA interactions (F1,27 ¼ 5.3, p ¼ .03) (Figure 7C). Bonferroni posthoc revealed that METH caused significant decreases in GluA1(p ¼ .038, Figure 7A) and GluA2 (p ¼ .009, Figure 7B) mRNAexpression in comparison with the saline/vehicle group. Inaddition, VPA/METH co-treatment caused increased GluA2 mRNAlevels compared with METH/vehicle (p ¼ .031, Figure 7B). Meth-amphetamine caused significant decreased GluN1 mRNA com-pared with saline/vehicle (p ¼ .048) and the VPA/METH group wassignificantly higher than the METH/vehicle group (p ¼ .012,Figure 7C).
www.sobp.org/journal
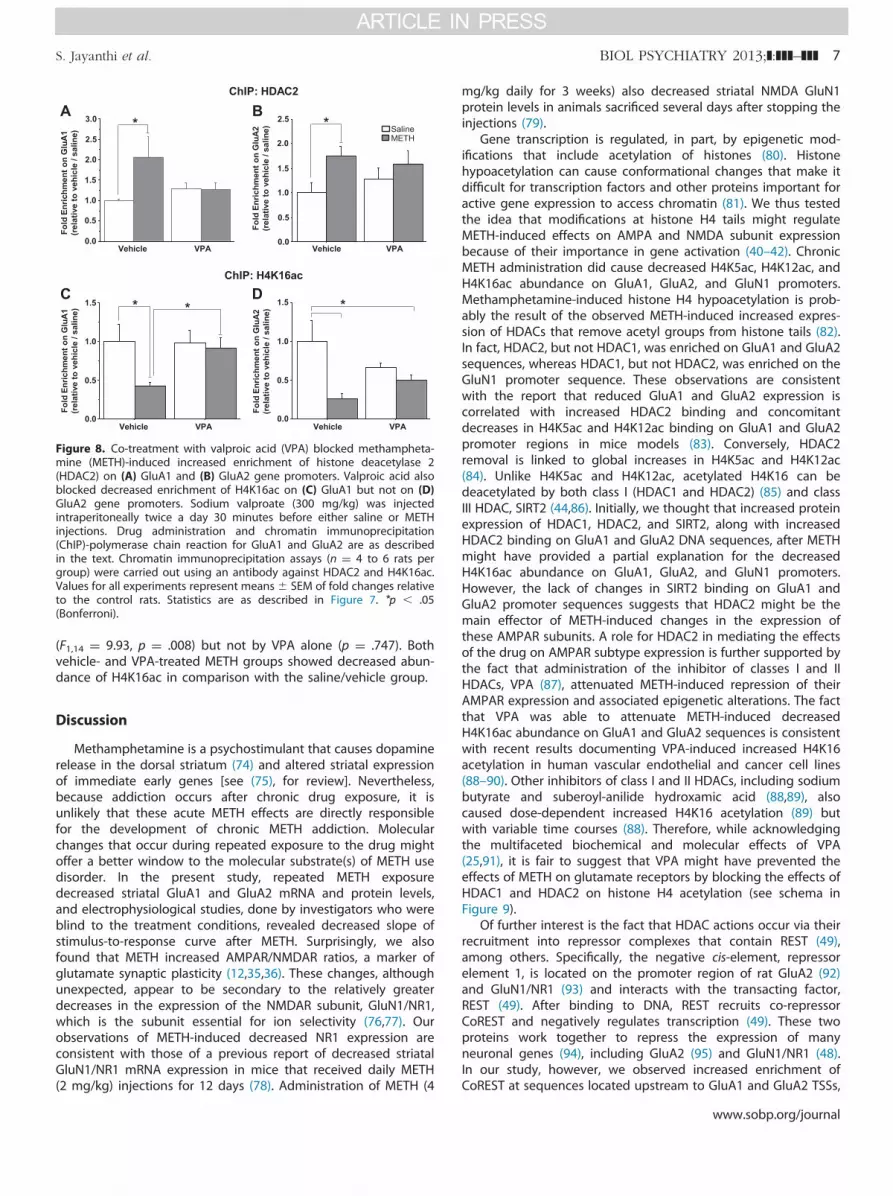
We tested the possibility that VPA may also block METH-induced epigenetic changes by carrying out ChIP-PCR withantibodies against HDAC2 and H4K16ac, respectively. Two-wayANOVA revealed main effects of METH (F1,16 ¼ 4.42, p ¼ .05) onHDAC2 enrichment at sequences located near the GluA1 TSS.There were no significant effects of VPA (p ¼ .892) but significantMETH � VPA interactions (F1,16 ¼ 4.91, p ¼ .044) (Figure 8A).Similarly, there were significant effects of METH (F1,21 ¼ 4.56, p ¼.046) at the GluA2 TSS but no effects of VPA alone. Methamphet-amine � VPA interactions were not significant (Figure 8B).Bonferroni corrections showed METH-induced increases in HDAC2recruitment around GluA2 TSS compared with saline/vehiclegroup (p ¼ .032; Figure 8B). Methamphetamine also causedchanges in H4K16ac recruitment on GluA1 (F1,15 ¼ 4.75, p ¼ .048).Bonferroni post hoc revealed METH-induced decreased H4K16acrecruitment around the GluA1 TSS compared with the saline/vehicle group (p ¼ .022; Figure 8C). The VPA-treated METH groupshowed significant increased H4K16ac recruitment comparedwith METH alone (p ¼ .033; Figure 8C). Abundance of H4K16acaround the GluA2 TSS was also significantly affected by METH
ChIP: HDAC2
ChIP: H4K16ac
0.0
0.5
1.5
2.5
1.0
2.0
Vehicle VPA
SalineMETH
Fold
Enr
ichm
ent o
n G
luA
1(r
elat
ive
to v
ehic
le /
salin
e)
0.0
0.5
1.5
1.0
2.0
Vehicle VPAFo
ld E
nric
hmen
t on
Glu
A2
(rel
ativ
e to
veh
icle
/ sa
line)
0.0
0.5
1.5
1.0
Vehicle VPA
Fold
Enr
ichm
ent o
n G
luA
1(r
elat
ive
to v
ehic
le /
salin
e)
0.0
0.5
1.5
1.0
Vehicle VPA
Fold
Enr
ichm
ent o
n G
luA
2(r
elat
ive
to v
ehic
le /
salin
e)
3.0
2.5
Figure 8. Co-treatment with valproic acid (VPA) blocked methampheta-mine (METH)-induced increased enrichment of histone deacetylase 2(HDAC2) on (A) GluA1 and (B) GluA2 gene promoters. Valproic acid alsoblocked decreased enrichment of H4K16ac on (C) GluA1 but not on (D)GluA2 gene promoters. Sodium valproate (300 mg/kg) was injectedintraperitoneally twice a day 30 minutes before either saline or METHinjections. Drug administration and chromatin immunoprecipitation(ChIP)-polymerase chain reaction for GluA1 and GluA2 are as describedin the text. Chromatin immunoprecipitation assays (n ¼ 4 to 6 rats pergroup) were carried out using an antibody against HDAC2 and H4K16ac.Values for all experiments represent means� SEM of fold changes relativeto the control rats. Statistics are as described in Figure 7. *p � .05(Bonferroni).
S. Jayanthi et al. BIOL PSYCHIATRY 2013;]:]]]–]]] 7
(F1,14 ¼ 9.93, p ¼ .008) but not by VPA alone (p ¼ .747). Bothvehicle- and VPA-treated METH groups showed decreased abun-dance of H4K16ac in comparison with the saline/vehicle group.
Discussion
Methamphetamine is a psychostimulant that causes dopaminerelease in the dorsal striatum (74) and altered striatal expressionof immediate early genes [see (75), for review]. Nevertheless,because addiction occurs after chronic drug exposure, it isunlikely that these acute METH effects are directly responsiblefor the development of chronic METH addiction. Molecularchanges that occur during repeated exposure to the drug mightoffer a better window to the molecular substrate(s) of METH usedisorder. In the present study, repeated METH exposuredecreased striatal GluA1 and GluA2 mRNA and protein levels,and electrophysiological studies, done by investigators who wereblind to the treatment conditions, revealed decreased slope ofstimulus-to-response curve after METH. Surprisingly, we alsofound that METH increased AMPAR/NMDAR ratios, a marker ofglutamate synaptic plasticity (12,35,36). These changes, althoughunexpected, appear to be secondary to the relatively greaterdecreases in the expression of the NMDAR subunit, GluN1/NR1,which is the subunit essential for ion selectivity (76,77). Ourobservations of METH-induced decreased NR1 expression areconsistent with those of a previous report of decreased striatalGluN1/NR1 mRNA expression in mice that received daily METH(2 mg/kg) injections for 12 days (78). Administration of METH (4
mg/kg daily for 3 weeks) also decreased striatal NMDA GluN1protein levels in animals sacrificed several days after stopping theinjections (79).
Gene transcription is regulated, in part, by epigenetic mod-ifications that include acetylation of histones (80). Histonehypoacetylation can cause conformational changes that make itdifficult for transcription factors and other proteins important foractive gene expression to access chromatin (81). We thus testedthe idea that modifications at histone H4 tails might regulateMETH-induced effects on AMPA and NMDA subunit expressionbecause of their importance in gene activation (40–42). ChronicMETH administration did cause decreased H4K5ac, H4K12ac, andH4K16ac abundance on GluA1, GluA2, and GluN1 promoters.Methamphetamine-induced histone H4 hypoacetylation is prob-ably the result of the observed METH-induced increased expres-sion of HDACs that remove acetyl groups from histone tails (82).In fact, HDAC2, but not HDAC1, was enriched on GluA1 and GluA2sequences, whereas HDAC1, but not HDAC2, was enriched on theGluN1 promoter sequence. These observations are consistentwith the report that reduced GluA1 and GluA2 expression iscorrelated with increased HDAC2 binding and concomitantdecreases in H4K5ac and H4K12ac binding on GluA1 and GluA2promoter regions in mice models (83). Conversely, HDAC2removal is linked to global increases in H4K5ac and H4K12ac(84). Unlike H4K5ac and H4K12ac, acetylated H4K16 can bedeacetylated by both class I (HDAC1 and HDAC2) (85) and classIII HDAC, SIRT2 (44,86). Initially, we thought that increased proteinexpression of HDAC1, HDAC2, and SIRT2, along with increasedHDAC2 binding on GluA1 and GluA2 DNA sequences, after METHmight have provided a partial explanation for the decreasedH4K16ac abundance on GluA1, GluA2, and GluN1 promoters.However, the lack of changes in SIRT2 binding on GluA1 andGluA2 promoter sequences suggests that HDAC2 might be themain effector of METH-induced changes in the expression ofthese AMPAR subunits. A role for HDAC2 in mediating the effectsof the drug on AMPAR subtype expression is further supported bythe fact that administration of the inhibitor of classes I and IIHDACs, VPA (87), attenuated METH-induced repression of theirAMPAR expression and associated epigenetic alterations. The factthat VPA was able to attenuate METH-induced decreasedH4K16ac abundance on GluA1 and GluA2 sequences is consistentwith recent results documenting VPA-induced increased H4K16acetylation in human vascular endothelial and cancer cell lines(88–90). Other inhibitors of class I and II HDACs, including sodiumbutyrate and suberoyl-anilide hydroxamic acid (88,89), alsocaused dose-dependent increased H4K16 acetylation (89) butwith variable time courses (88). Therefore, while acknowledgingthe multifaceted biochemical and molecular effects of VPA(25,91), it is fair to suggest that VPA might have prevented theeffects of METH on glutamate receptors by blocking the effects ofHDAC1 and HDAC2 on histone H4 acetylation (see schema inFigure 9).
Of further interest is the fact that HDAC actions occur via theirrecruitment into repressor complexes that contain REST (49),among others. Specifically, the negative cis-element, repressorelement 1, is located on the promoter region of rat GluA2 (92)and GluN1/NR1 (93) and interacts with the transacting factor,REST (49). After binding to DNA, REST recruits co-repressorCoREST and negatively regulates transcription (49). These twoproteins work together to repress the expression of manyneuronal genes (94), including GluA2 (95) and GluN1/NR1 (48).In our study, however, we observed increased enrichment ofCoREST at sequences located upstream to GluA1 and GluA2 TSSs,
www.sobp.org/journal

GluA1
GluA2
GluN1
His
tone
H4
hypo
acet
ylat
ion
GluA1GluA2GluN1
Chronic METH
Figure 9. Schematic models showing chronic methamphetamine (METH)-induced epigenetic modifications in the dorsal striatum. Under controlcondition, there exists a balance between histone acetylases and histonedeacetylases that regulate the histone acetylation/deacetylation statusthat maintain the baseline transcription levels. However, chronic METHexposure leads to formation of protein repressor complexes methylatedCpG binding protein 2 (MeCP2)-repressor element 1 silencing transcrip-tion factor corepressor 1 (CoREST)-histone deacetylase 2 (HDAC2) thatcause H4K5, H4K12, and H4K16 hypoacetylation at enhancer or promotersequences of GluA1 and GuA2 genes. This then leads to decreasedexpression of these receptors. Rats chronically exposed to METH also showformation of a protein repressor complex that contains repressor element1 silencing transcription factor (REST)-histone deacetylase 1 (HDAC1) thatproduces hypoacetylation of H4K5, H4K12, and H4K16 at the promoterregion of GluN1 and subsequent decreased GluN1 (NR1) expression in thedorsal striatum. Co-treatment of METH-treated animals with valproic acid(VPA) that inhibits HDAC1 and HDAC2 blocked METH-mediated hypoace-tylation and METH-induced decreased expression of these glutamatereceptors. Ac, acetyl moiety; RE1, repressor element 1; TSS, transcriptionstart site.
8 BIOL PSYCHIATRY 2013;]:]]]–]]] S. Jayanthi et al.
while there was increased enrichment of REST, but not of CoREST,on the GluN1 promoter. These observations are consistent withrecent reports that CoREST and REST can function independently(50). For example, specific CoREST target genes that do notcontain the repressor element 1 motif to which REST binds arestill targeted by CoREST for transcriptional repression (96). Theobserved repression was shown to result from interactions ofCoREST with other repressor proteins (94,97), including HDACs(see above discussion). In addition to a role for CoREST, ourfindings indicate that MeCP2, an important protein that isinvolved in Rett syndrome (98), also participates in METH-induced repression of AMPAR expression. The METH-inducedincreased MeCP2 recruitment at distal sites to the GluA1TSS isconsistent with a potential effect on enhancer sequences that areknown to interact with promoter sequences to regulate tran-scription (99,100). The observation of METH-induced increasedMeCP2 binding had suggested that METH might have also causedincreased DNA methylation, a suggestion that we thought wassupported by our demonstration of increased striatal expressionof the maintenance DNMT, DNMT1 (101). However, our failure toobserve any METH-induced increased DNA methylation in GluApromoter or enhancer regions argues against the involvement ofDNA modifications in the regulation of AMPAR expression in thepresent model. The present observations also suggest that DNAmethylation might not be obligatory for the gene suppressiveeffects of MeCP2 that binds to both methylated and unmethy-lated DNA (60,61,102,103). Finally, the observations that the HDACinhibitor, VPA, also attenuated the METH-induced decreased
www.sobp.org/journal
glutamate receptor expression, HDAC2 binding to GluA1 andGluA2 promoters, and decreased H4K16Ac recruitment to thesereceptor promoter sequences also argue for a preferential role forhistone hypoacetylation for METH-induced decreased AMPARexpression.
In summary, our study provides direct evidence for epigeneticregulation of transcriptional effects of chronic METH exposure onglutamate receptors. Figure 9 provides a schematic representa-tion that describes potential roles of REST, CoREST, MeCP2,HDAC1, and HDAC2 in mediating METH-induced downregulationof GluA1, GluA2, and GluN1 mRNA levels. The scheme suggeststwo distinct regulatory mechanisms: one for GluA expression thatinvolves MeCP2 and CoREST recruitment of HDAC2 onto thechromatin, with resulting H4K5, K12, and K16 deacetylation anddecreased H4K5ac, K12ac, and K16ac binding onto GluA1 andGluA2 DNA sequences, and another one for the regulation of NR1expression that is mediated by REST and HDAC1 with decreasedH4K5ac, K12ac, and K16ac binding onto GluN1 promoter region.Because the clinically effective US Food and Drug Administrationapproved neuropsychiatric medication, VPA, was able to blockMETH-induced decreases in AMPAR and NMDAR expression, it istempting to speculate that this medication or similar epigeneticagents could be used to mitigate some neuropsychiatric mani-festations of chronic METH exposure in humans.
This work was supported by funds of the Intramural ResearchProgram of the US Department of Health and Human Services/National Institutes of Health/National Institute on Drug Abuse.
We thank Dr. Genevieve Beauvais, Dr. Ingrid Tulloch, and TraceyMartin for their help with animal injections and dissection andDr. Benita Gonzalez with help with statistical analyses. We alsothank the reviewers whose constructive criticisms helped to improvethe content, presentation, and discussion of our results.
The authors report no biomedical financial interests or potentialconflicts of interest.
Supplementary material cited in this article is available online athttp://dx.doi.org/10.1016/j.biopsych.2013.09.034.
1. Bowers MS, Chen BT, Bonci A (2010): AMPA receptor synaptic plas-ticity induced by psychostimulants: The past, present, and thera-peutic future. Neuron 67:11–24.
2. Chen BT, Hopf FW, Bonci A (2010): Synaptic plasticity in the meso-limbic system: Therapeutic implications for substance abuse. Ann N YAcad Sci 1187:129–139.
3. Luscher C, Malenka RC (2011): Drug-evoked synaptic plasticity inaddiction: From molecular changes to circuit remodeling. Neuron 69:650–663.
4. Belin D, Everitt BJ (2008): Cocaine seeking habits depend upondopamine-dependent serial connectivity linking the ventral with thedorsal striatum. Neuron 57:432–441.
5. Doyon J, Bellec P, Amsel R, Penhune V, Monchi O, Carrier J, et al.(2009): Contributions of the basal ganglia and functionally relatedbrain structures to motor learning. Behav Brain Res 199:61–75.
6. Everitt BJ, Robbins TW (2013): From the ventral to the dorsalstriatum: Devolving views of their roles in drug addiction [publishedonline ahead of print February 21]. Neurosci Biobehav Rev. doi:10.1016/j.neubiorev.2013.02.010.
7. Wise RA (2009): Roles for nigrostriatal—not just mesocorticolimbic—dopamine in reward and addiction. Trends Neurosci 32:517–524.
8. Kalivas PW, Volkow ND (2011): New medications for drug addictionhiding in glutamatergic neuroplasticity. Mol Psychiatry 16:974–986.
9. Lu L, Dempsey J, Shaham Y, Hope BT (2005): Differential long-termneuroadaptations of glutamate receptors in the basolateral andcentral amygdala after withdrawal from cocaine self-administrationin rats. J Neurochem 94:161–168.

S. Jayanthi et al. BIOL PSYCHIATRY 2013;]:]]]–]]] 9
10. Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y,et al. (2008): Formation of accumbens GluR2-lacking AMPA receptorsmediates incubation of cocaine craving. Nature 454:118–121.
11. Wolf ME, Ferrario CR (2010): AMPA receptor plasticity in the nucleusaccumbens after repeated exposure to cocaine. Neurosci BiobehavRev 35:185–211.
12. Kourrich S, Rothwell PE, Klug JR, Thomas MJ (2007): Cocaineexperience controls bidirectional synaptic plasticity in the nucleusaccumbens. J Neurosci 27:7921–7928.
13. Simoes PF, Silva AP, Pereira FC, Marques E, Milhazes N, Borges F,et al. (2008): Methamphetamine changes NMDA and AMPA gluta-mate receptor subunit levels in the rat striatum and frontal cortex.Ann N Y Acad Sci 1139:232–241.
14. Martin C, Zhang Y (2007): Mechanisms of epigenetic inheritance.Curr Opin Cell Biol 19:266–272.
15. Murr R (2010): Interplay between different epigenetic modificationsand mechanisms. Adv Genet 70:101–141.
16. de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB(2003): Histone deacetylases (HDACs): Characterization of the clas-sical HDAC family. Biochem J 370:737–749.
17. Robison AJ, Nestler EJ (2011): Transcriptional and epigenetic mech-anisms of addiction. Nat Rev Neurosci 12:623–637.
18. Krasnova IN, Cadet JL (2009): Methamphetamine toxicity andmessengers of death. Brain Res Rev 60:379–407.
19. Sulzer D (2011): How addictive drugs disrupt presynaptic dopamineneurotransmission. Neuron 69:628–649.
20. Kuhar MJ, Ritz MC, Boja JW (1991): The dopamine hypothesis of thereinforcing properties of cocaine. Trends Neurosci 14:299–302.
21. Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ (1987): Cocaine receptorson dopamine transporters are related to self-administration ofcocaine. Science 237:1219–1223.
22. Cho AK, Melega WP (2002): Patterns of methamphetamine abuseand their consequences. J Addict Dis 21:21–34.
23. Kramer JC, Fischman VS, Littlefield DC (1967): Amphetamine abuse.Pattern and effects of high doses taken intravenously. JAMA 201:305–309.
24. Cadet JL, McCoy MT, Cai NS, Krasnova IN, Ladenheim B, Beauvais G,et al. (2009): Methamphetamine preconditioning alters midbraintranscriptional responses to methamphetamine-induced injury inthe rat striatum. PLoS One 4:e7812.
25. Rosenberg G (2007): The mechanisms of action of valproate inneuropsychiatric disorders: Can we see the forest for the trees? CellMol Life Sci 64:2090–2103.
26. Rowley HL, Marsden CA, Martin KF (1995): Differential effects ofphenytoin and sodium valproate on seizure-induced changes ingamma-aminobutyric acid and glutamate release in vivo. Eur JPharmacol 294:541–546.
27. Martin TA, Jayanthi S, McCoy MT, Brannock C, Ladenheim B, GarrettT, et al. (2012): Methamphetamine causes differential alterations ingene expression and patterns of histone acetylation/hypoacetylationin the rat nucleus accumbens. PLoS One 7:e34236.
28. Barrett LE, Van Bockstaele EJ, Sul JY, Takano H, Haydon PG, Eberwine JH(2006): Elk-1 associates with the mitochondrial permeability transitionpore complex in neurons. Proc Natl Acad Sci U S A 103:5155–5160.
29. Tsankova NM, Kumar A, Nestler EJ (2004): Histone modifications atgene promoter regions in rat hippocampus after acute and chronicelectroconvulsive seizures. J Neurosci 24:5603–5610.
30. Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL,Schübeler D (2005): Chromosome-wide and promoter-specific anal-yses identify sites of differential DNA methylation in normal andtransformed human cells. Nat Genet 37:853–862.
31. Vucetic Z, Kimmel J, Totoki K, Hollenbeck E, Reyes TM (2010):Maternal high-fat diet alters methylation and gene expression ofdopamine and opioid-related genes. Endocrinology 151:4756–4764.
32. Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A(2012): Synaptic and behavioral profile of multiple glutamatergicinputs to the nucleus accumbens. Neuron 76:790–803.
33. Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001): Long-termdepression in the nucleus accumbens: A neural correlate ofbehavioral sensitization to cocaine. Nat Neurosci 4:1217–1223.
34. Hsia AY, Malenka RC, Nicoll RA (1998): Development of excitatorycircuitry in the hippocampus. J Neurophysiol 79:2013–2024.
35. Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM,et al. (2008): Cocaine but not natural reward self-administration nor
passive cocaine infusion produces persistent LTP in the VTA. Neuron59:288–297.
36. Ungless MA, Whistler JL, Malenka RC, Bonci A (2001): Single cocaineexposure in vivo induces long-term potentiation in dopamineneurons. Nature 411:583–587.
37. Covington HE 3rd, Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY,et al. (2011): A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron 71:656–670.
38. Kennedy PJ, Feng J, Robison AJ, Maze I, Badimon A, Mouzon E, et al.(2013): Class I HDAC inhibition blocks cocaine-induced plasticityby targeted changes in histone methylation. Nat Neurosci 16:434–440.
39. Rogge GA, Wood MA (2013): The role of histone acetylation incocaine-induced neural plasticity and behavior. Neuropsychophar-macology 38:94–110.
40. Dion MF, Altschuler SJ, Wu LF, Rando OJ (2005): Genomic character-ization reveals a simple histone H4 acetylation code. Proc Natl AcadSci U S A 102:5501–5506.
41. Henriksen P, Wagner SA, Weinert BT, Sharma S, Bacinskaja G,Rehman M, et al. (2012): Proteome-wide analysis of lysine acetylationsuggests its broad regulatory scope in Saccharomyces cerevisiae.Mol Cell Proteomics 11:1510–1522.
42. Rundlett SE, Carmen AA, Suka N, Turner BM, Grunstein M (1998):Transcriptional repression by UME6 involves deacetylation of lysine5 of histone H4 by RPD3. Nature 392:831–835.
43. Vettese-Dadey M, Grant PA, Hebbes TR, Crane- Robinson C, Allis CD,Workman JL (1996): Acetylation of histone H4 plays a primary role inenhancing transcription factor binding to nucleosomal DNA in vitro.EMBO J 15:2508–2518.
44. Vaquero A, Sternglanz R, Reinberg D (2007): NAD�-dependentdeacetylation of H4 lysine 16 by class III HDACs. Oncogene 26:5505–5520.
45. Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE 3rd, Maze I,et al. (2009): Genome-wide analysis of chromatin regulation bycocaine reveals a role for sirtuins. Neuron 62:335–348.
46. Bithell A (2011): REST: Transcriptional and epigenetic regulator.Epigenomics 3:47–58.
47. Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, et al.(2003): Ischemic insults derepress the gene silencer REST in neuronsdestined to die. J Neurosci 23:2112–2121.
48. Bai G, Zhuang Z, Liu A, Chai Y, Hoffman PW (2003): The role of theRE1 element in activation of the NR1 promoter during neuronaldifferentiation. J Neurochem 86:992–1005.
49. Andres ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME,Grimes J, et al. (1999): CoREST: A functional corepressor required forregulation of neural-specific gene expression. Proc Natl Acad Sci U S A96:9873–9878.
50. Abrajano JJ, Qureshi IA, Gokhan S, Zheng D, Bergman A, Mehler MF(2009): REST and CoREST modulate neuronal subtype specification,maturation and maintenance. PLoS One 4:e7936.
51. Zhang F, Wang S, Gan L, Vosler PS, Gao Y, Zigmond MJ, Chen J(2011): Protective effects and mechanisms of sirtuins in the nervoussystem. Prog Neurobiol 95:373–395.
52. Suzuki MM, Bird A (2008): DNA methylation landscapes: Provocativeinsights from epigenomics. Nat Rev Genet 9:465–476.
53. Denis H, Ndlovu MN, Fuks F (2011): Regulation of mammalian DNAmethyltransferases: A route to new mechanisms. EMBO Rep 12:647–656.
54. Kar S, Deb M, Sengupta D, Shilpi A, Parbin S, Torrisani J, et al. (2012): Aninsight into the various regulatory mechanisms modulating humanDNA methyltransferase 1 stability and function. Epigenetics 7:994–1007.
55. Chedin F (2011): The DNMT3 family of mammalian de novo DNAmethyltransferases. Prog Mol Biol Transl Sci 101:255–285.
56. Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N,et al. (1998): Methylated DNA and MeCP2 recruit histone deacetylaseto repress transcription. Nat Genet 19:187–191.
57. Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF,et al. (2005): Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2.Proc Natl Acad Sci U S A 102:17551–17558.
58. Kimura H, Shiota K (2003): Methyl-CpG-binding protein, MeCP2, is atarget molecule for maintenance DNA methyltransferase, Dnmt1. JBiol Chem 278:4806–4812.
www.sobp.org/journal

10 BIOL PSYCHIATRY 2013;]:]]]–]]] S. Jayanthi et al.
59. Adams VH, McBryant SJ, Wade PA, Woodcock CL, Hansen JC (2007):Intrinsic disorder and autonomous domain function in the multi-functional nuclear protein, MeCP2. J Biol Chem 282:15057–15064.
60. Hansen JC, Ghosh RP, Woodcock CL (2010): Binding of the Rettsyndrome protein, MeCP2, to methylated and unmethylated DNAand chromatin. IUBMB Life 62:732–738.
61. Meehan RR, Lewis JD, Bird AP (1992): Characterization of MeCP2, avertebrate DNA binding protein with affinity for methylated DNA.Nucleic Acids Res 20:5085–5092.
62. Feng J, Fan G (2009): The role of DNA methylation in the central nervoussystem and neuropsychiatric disorders. Int Rev Neurobiol 89:67–84.
63. Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, BirdA (1998): Transcriptional repression by the methyl-CpG-binding proteinMeCP2 involves a histone deacetylase complex. Nature 393:386–389.
64. Satta R, Maloku E, Zhubi A, Pibiri F, Hajos M, Costa E, Guidotti A(2008): Nicotine decreases DNA methyltransferase 1 expression andglutamic acid decarboxylase 67 promoter methylation in GABAergicinterneurons. Proc Natl Acad Sci U S A 105:16356–16361.
65. Dong E, Nelson M, Grayson DR, Costa E, Guidotti A (2008): Clozapineand sulpiride but not haloperidol or olanzapine activate brain DNAdemethylation. Proc Natl Acad Sci U S A 105:13614–13619.
66. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. (2011): Tetproteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333:1300–1303.
67. Nabel CS, Manning SA, Kohli RM (2012): The curious chemicalbiology of cytosine: Deamination, methylation, and oxidation asmodulators of genomic potential. ACS Chem Biol 7:20–30.
68. Guo JU, Su Y, Zhong C, Ming GL, Song H (2011): Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in theadult brain. Cell 145:423–434.
69. Kriaucionis S, Heintz N (2009): The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain.Science 324:929–930.
70. Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, et al. (2011): 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopmentand aging. Nat Neurosci 14:1607–1616.
71. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y,et al. (2009): Conversion of 5-methylcytosine to 5-hydroxy-methylcytosine in mammalian DNA by MLL partner TET1. Science324:930–935.
72. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS(2001): Histone deacetylase is a direct target of valproic acid, apotent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem276:36734–36741.
73. Nalivaeva NN, Belyaev ND, Turner AJ (2009): Sodium valproate: Anold drug with new roles. Trends Pharmacol Sci 30:509–514.
74. Kazahaya Y, Akimoto K, Otsuki S (1989): Subchronic methamphet-amine treatment enhances methamphetamine- or cocaine-induceddopamine efflux in vivo. Biol Psychiatry 25:903–912.
75. Cadet JL, Jayanthi S, McCoy MT, Beauvais G, Cai NS (2010):Dopamine D1 receptors, regulation of gene expression in the brain,and neurodegeneration. CNS Neurol Disord Drug Targets 9:526–538.
76. Cull-Candy SG, Leszkiewicz DN (2004): Role of distinct NMDAreceptor subtypes at central synapses. Sci STKE 2004:re16.
77. Madden K (2002): NMDA receptor antagonists and glycine siteNMDA antagonists. Curr Med Res Opin 18(suppl 2):s27–s31.
78. Motawaj M, Arrang JM (2011): Ciproxifan, a histamine H-receptorantagonist / inverse agonist, modulates methamphetamine-inducedsensitization in mice. Eur J Neurosci 33:1197–1204.
79. Yamamoto H, Kitamura N, Lin XH, Ikeuchi Y, Hashimoto T, ShirakawaO, Maeda K (1999): Differential changes in glutamatergic trans-mission via N-methyl-D-aspartate receptors in the hippocampus andstriatum of rats behaviourally sensitized to methamphetamine. Int JNeuropsychopharmacol 2:155–163.
80. Jenuwein T, Allis CD (2001): Translating the histone code. Science293:1074–1080.
81. Braunstein M, Sobel RE, Allis CD, Turner BM, Broach JR (1996): Efficienttranscriptional silencing in Saccharomyces cerevisiae requires a hetero-chromatin histone acetylation pattern. Mol Cell Biol 16:4349–4356.
82. Wolffe AP (1996): Histone deacetylase: A regulator of transcription.Science 272:371–372.
www.sobp.org/journal
83. Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, et al. (2012): Anepigenetic blockade of cognitive functions in the neurodegenerat-ing brain. Nature 483:222–226.
84. Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J,et al. (2009): HDAC2 negatively regulates memory formation andsynaptic plasticity. Nature 459:55–60.
85. Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, JacksonSP (2010): Human HDAC1 and HDAC2 function in the DNA-damageresponse to promote DNA nonhomologous end-joining. Nat StructMol Biol 17:1144–1151.
86. Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, et al.(2006): SirT2 is a histone deacetylase with preference for histone H4Lys 16 during mitosis. Genes Dev 20:1256–1261.
87. Kramer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J,Peters MA, et al. (2003): The histone deacetylase inhibitor valproicacid selectively induces proteasomal degradation of HDAC2. EMBO J22:3411–3420.
88. Barbetti V, Gozzini A, Cheloni G, Marzi I, Fabiani E, Santini V, et al.(2013): Time- and residue-specific differences in histone acetylationinduced by VPA and SAHA in AML1/ETO-positive leukemia cells.Epigenetics 8:210–219.
89. Bartels M, Geest CR, Bierings M, Buitenhuis M, Coffer PJ (2010):Histone deacetylase inhibition modulates cell fate decisions duringmyeloid differentiation. Haematologica 95:1052–1060.
90. Larsson P, Ulfhammer E, Magnusson M, Bergh N, Lunke S, El-Osta A,et al. (2012): Role of histone acetylation in the stimulatory effect ofvalproic acid on vascular endothelial tissue-type plasminogenactivator expression. PLoS One 7:e31573.
91. Monti B, Polazzi E, Contestabile A (2009): Biochemical, molecular andepigenetic mechanisms of valproic acid neuroprotection. Curr MolPharmacol 2:95–109.
92. Myers SJ, Peters J, Huang Y, Comer MB, Barthel F, Dingledine R(1998): Transcriptional regulation of the GluR2 gene: Neural-specificexpression, multiple promoters, and regulatory elements. J Neurosci18:6723–6739.
93. Bai G, Norton DD, Prenger MS, Kusiak JW (1998): Single-strandedDNA-binding proteins and neuron-restrictive silencer factor partic-ipate in cell-specific transcriptional control of the NMDAR1 gene. JBiol Chem 273:1086–1091.
94. Lunyak VV, Burgess R, Prefontaine GG, Nelson C, Sze SH, ChenowethJ, et al. (2002): Corepressor-dependent silencing of chromosomalregions encoding neuronal genes. Science 298:1747–1752.
95. Huang Y, Doherty JJ, Dingledine R (2002): Altered histone acetylationat glutamate receptor 2 and brain-derived neurotrophic factor genesis an early event triggered by status epilepticus. J Neurosci 22:8422–8428.
96. Abrajano JJ, Qureshi IA, Gokhan S, Molero AE, Zheng D, Bergman A,Mehler MF (2010): Corepressor for element-1-silencing transcriptionfactor preferentially mediates gene networks underlying neural stemcell fate decisions. Proc Natl Acad Sci U S A 107:16685–16690.
97. Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G (2005): REST and itscorepressors mediate plasticity of neuronal gene chromatinthroughout neurogenesis. Cell 121:645–657.
98. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY(1999): Rett syndrome is caused by mutations in X-linked MECP2,encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188.
99. Marsman J, Horsfield JA (2012): Long distance relationships:Enhancer-promoter communication and dynamic gene transcription.Biochim Biophys Acta 1819:1217–1227.
100. Ong CT, Corces VG (2011): Enhancer function: New insights into theregulation of tissue-specific gene expression. Nat Rev Genet 12:283–293.
101. Bird A (2002): DNA methylation patterns and epigenetic memory.Genes Dev 16:6–21.
102. Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA,Hansen JC (2003): Chromatin compaction by human MeCP2. Assem-bly of novel secondary chromatin structures in the absence of DNAmethylation. J Biol Chem 278:32181–32188.
103. Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP,et al. (2007): Integrasted epigenomic analyses of neuronal MeCP2reveal a role for long-range interaction with active genes. Proc NatlAcad Sci U S A 104:19416–19421.