long circulating lipid nanocapsules for drug detoxification
TRANSCRIPT

ARTICLE IN PRESS
0142-9612/$ - se
doi:10.1016/j.bi
�CorrespondE-mail addr
Biomaterials 28 (2007) 1248–1257
www.elsevier.com/locate/biomaterials
Long circulating lipid nanocapsules for drug detoxification
Anand Babu Dhanikulaa, Nabil Mohamed Khalida, Stephen D. Leeb, Rosanna Yeungb,Verica Risovicb, Kishor M. Wasanb, Jean-Christophe Lerouxa,�
aCanada Research Chair in Drug Delivery, Faculty of Pharmacy, C.P. 6128 Succ. Centre-ville, Montreal, QC, Canada H3C 3J7bFaculty of Pharmaceutical Sciences, University of British Columbia, 2146 East Mall Avenue, Vancouver, BC, Canada V6T 1Z3
Received 19 July 2006; accepted 26 October 2006
Abstract
Uncoated and poly(ethylene glycol) (PEG)-decorated lipid nanocapsules (NC) prepared from medium chain triglycerides were
investigated both in vitro and in vivo as parenteral detoxifying colloids for their ability to sequester haloperidol, docetaxel and paclitaxel.
In vitro studies showed that the uptake depended on the nature of the drug and the composition of NC core and shell. In the case of
haloperidol, maximal affinity was achieved upon incorporation of a complexing fatty acid. In plasma lipoprotein distribution studies, the
association of both haloperidol and docetaxel into triglyceride-rich lipoprotein fraction was significantly increased in the presence of NC.
The ability of the NC to lower the free drug concentrations in incubation medium was confirmed by cytotoxicity studies, where the
antiproliferative activity of docetaxel was significantly decreased in the presence of NC. Using docetaxel as drug model, the NC were
finally evaluated for their uptake potential in mice by one of the following administration sequences between the drug solution
(Taxoteres, DTX) and NC: NC-DTX, PEG(NC)-DTX and DTX-PEG(NC). Irrespective of the administration sequence, the NC
increased the blood levels of docetaxel due to the in situ sequestration of drug by the circulating carrier. These findings suggest that lipid
NC could be used as a non-specific mode to deal with the sequestration of molecules with high affinity for oils.
r 2006 Elsevier Ltd. All rights reserved.
Keywords: Nanocapsule; Detoxification; Uptake; Lipoprotein distribution; Haloperidol; Taxanes
1. Introduction
Overdoses of drugs either due to deliberate or accidentalreasons are not uncommon [1–5]. In the year 2000, theseincidents contributed to about a quarter of suicide deathsin the UK [6]. In Canada, approximately 8% ofhospitalizations were documented for detoxification treat-ment following substance abuse [7]. Deaths caused fromprescribed drug poisoning are on rise and are currentlyrecognized as a growing problem in the US [8,9].Intoxication resulting from chemical exposures can beaverted by the application of properly designed rescuemethods in the clinic [6,10]. Often, treatments are basedupon general modes of detoxification (gastric lavasion,dialysis, etc.) because the physician in the emergency wardis unaware of the nature of the toxic molecule. Each of the
e front matter r 2006 Elsevier Ltd. All rights reserved.
omaterials.2006.10.036
ing author. Tel.: +1514 343 6455; fax: +1 514 343 6871.
ess: [email protected] (J.-C. Leroux).
general methods of detoxification is associated withinherent limitations dictated by several factors such asconscious state of patient and extent of protein binding oftoxin. For instance, the oral administration of charcoal isnot feasible for an unconscious subject whereas dialysisand hemoperfusion are less effective methods for removalof highly protein bound drugs [11,12]. In this regard, aparenteral approach with rapid onset of action could be ofan invaluable aid for the physician while dealing withintoxicated patients. Recently, several alternative ap-proaches for sequestration of hydrophobic drugs havebeen investigated. Underhill et al. [13] prepared NC fromhexadecane core and cross-linked polysiloxane/silicate shelland observed 99% in vitro uptake of bupivacaine. Leeet al. [14] examined oligochitosan derivatives bearingelectron deficient aromatic rings to complex amitriptylineand concluded that hydrophobic interaction was the maindriving force for the molecular association. Fallonand Chauhan [15] observed a higher sequestration of

ARTICLE IN PRESSA. Babu Dhanikula et al. / Biomaterials 28 (2007) 1248–1257 1249
amitriptyline into liposomes bearing negative chargecompared to liposomes having no net charge. Thus,methods directed towards sequestration of lipophilicmolecules based on solvation, binding and/or complexa-tion seem to be gaining importance as promising detox-ification approaches.
Trigyceride oils (tricaprylin, soybean, safflower oils) arebiocompatible/biodegradable excipients [16] which, whenprocessed into emulsions, can present attractive featuresfor detoxification measures. Early attempts were based onthe use of orally administered multiple emulsions forremoval of uremia toxins [17], barbiturates and salicylates[18]. Morimoto et al. [19] demonstrated that paraffin oilmultiple emulsions co-administered orally with salicylicacid in rabbits reduced the blood peak concentration andthe area under concentration time curve, suggestingreduced drug exposure. The protective effects of lipidemulsions administered parenterally have also been re-ported. For example, the intravenous infusion of Intrali-pids emulsion attenuated the adverse haemodynamiceffects of bupivacaine [20]. More recently, it was shownthat the co-infusion of Intralipids resulted in significantlydecreased mortality of clomipramine [21] and verapamil[22] overdosed rats. However, despite these interestingfindings, the conventional emulsions, in general, faceinherent formulation difficulties arising from thermody-namic instability, which often leads to the coalescence of oildroplets over time [23]. In addition, the Intralipids
emulsions prepared from egg/soy lecithin and soybean/safflower oil having a droplet size range between 0.2 and0.6 mM, are also rapidly cleared from circulation due toadsorption of apolipoproteins [24]. Since the clearance rateof sequestration vectors is dependent on their size and thetype of surfactants used, vectors prepared from structuredtriglycerides and poly(ethylene glycol) (PEG)-derivedsurfactants could offer a new scope to replace conventionallipid emulsions intended for parenteral nutrition in thetreatment of drug poisoning.
Heurtault et al. [25] reported that nanocarriers(25–100 nm) i.e. lipid nanocapsules (NC), with uniqueinterfacial properties could be prepared from triglycerides.These NC had a rigid shell surrounding the oil core andwere physically stable for at least 18 months without fusionof the dispersed oily phase. In the present investigation,lipid NC were explored as an alternative to emulsions forthe treatment of drug intoxication. The underlyinghypothesis of this work is that administration of lipid-richNC would effectively sequester overdosed drugs in vivothereby reducing their systemic side-effects. For thispurpose, 3molecules namely, haloperidol, docetaxel andpaclitaxel were initially selected as model drugs for the invitro uptake studies. Haloperidol has a narrow thera-peutic window in humans [26–28], and cases involvingaccidental overdosing and poisoning have been reported inRefs. [29–31]. Alternatively, taxanes (paclitaxel and doc-etaxel) are useful hydrophobic model drugs due to thereoverlapping partition coefficient (Kp) values with the
commonly reported classes of drugs concerned with over-dose and intoxication, namely, antihistamines, anticholi-nergics, anticonvulsants and antidepressants. These feasi-bility experiments with taxanes, nevertheless, are con-fronted by the their low solubility in parenteral grade oils[32], their high affinity for blood components (albumin,lipoproteins) and the absence of electrostatic charge atphysiological pH. Plasma protein and lipoprotein distribu-tion and cell cytotoxicity studies were also performed tocomplement the in vitro data. Finally, the in vivo uptake ofdocetaxel by lipid NC and its distribution in mononuclearphagocyte system (MPS) organs (liver and spleen) wereinvestigated in mice.
2. Materials and methods
Haloperidol, bovine serum albumin (BSA), ethyl caprate, ethyl laurate,
ethyl oleate, lauric acid, tricaprylin, 3-(4,5-dimethylthiazol-2-yl)-2,5-
diphenyl-tetrazolium bromide (MTT) were purchased from Sigma
Chemical Co. (St. Louis, MO). Docetaxel was provided by Shanghai
Fudan Taxusal New Technology Co. (Shanghai, China). Paclitaxel was
purchased from Bioxel Pharma Inc. (Sainte-Foy, QC, Canada). 3H-
labelled haloperidol (12.8Ci/mmol), 14C-labelled cholesterol oleate (52Ci/
mol) and 3H-labelled cholesterol hexadecyl ether (51Ci/mmol) were from
Perkin Elmer (Woodbridge, ON, Canada). Taxoteres (DTX, 40mg/mL
of docetaxel in polysorbate 80, Aventis Pharma Ltd., Dagenham, UK)
was purchased from a retail pharmacy. 14C-labelled docetaxel (60Ci/mol)
and 3H-labelled paclitaxel (60Ci/mol) were purchased from American
Radiolabeled Chemicals Inc. (St. Louis, MO). Miglyols 810 was kindly
provided by Sasol GmbH (Arthur-Imhausen, Witten, Germany). Captexs
8000 and Captexs 810D were obtained from Abitec Corporation
(Columbus, OH). Labrafac CCs, Maisine 35–1s, Peceols were obtained
from Gattefosse S.A. (Saint-Priest, France). Corn, safflower, sesame
and soybean oils were provided by ICN Biomedicals (Aurora, OH).
Hydrogenated soy phosphatidylcholine (HSPC), 1,2-distearoyl-sn-glycero-
3-phosphatidylcholine (DSPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phos-
phatidylcholine (POPC), egg phosphatidylcholine (Egg PC), soy phos-
phatidylcholine (Soy PC), 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine
(DOPC) and 1,2-distearoyl-sn-glycero-3-phosphatidylethanolamine-N-
[methoxy(PEG)-2000] (DSPE-PEG2000) were purchased from Northern
Lipids Inc. (Vancouver, BC, Canada). Solutols HS15 (12-hydroxy stearic
acid ethoxylate) was provided by BASF (Ludwigshafen, Germany).
Sodium bromide and sodium chloride were obtained from Sigma Aldrich
(St. Louis, MO). Cholesterol standard and triglyceride standard were from
StanBio Laboratory (Boerne, TX) and Thermo Trace (Waltham, MA),
respectively. Cholesterol reagent and triglyceride reagent were purchased
from Roche Diagnostics (Laval, QC, Canada). The BCA Protein Assay
Kit used to characterize protein levels was obtained from Pierce
Biotechnology (Rockford, IL). Dulbecco’s modified Eagle’s medium, fetal
calf serum and antibiotic were from Invitrogen Corp. (Grand Island, NY).
Soluene 350s, Ultima Golds and Hionic-Fluors were obtained from
Perkin Elmer (Boston, MA) and Packard BioScience (Meriden, CT),
respectively. All other chemicals and solvents were of reagent grade. Water
was deionized with a MilliQ purification system (Millipore, Bedford, MA)
before use.
2.1. Partition coefficient
About 15 different oils were initially tested for their affinity towards
haloperidol based on the Kp values. These oils were selected so as to
include mono, di and triglycerides as well as ethyl esters. The triglycerides
consisted of both semi-processed (medium chain triglycerides) and natural
oils (long chain triglycerides). The Kp behaviour of haloperidol was also
investigated in safflower oil (long chain triglyceride), Miglyol 810 (medium

ARTICLE IN PRESSA. Babu Dhanikula et al. / Biomaterials 28 (2007) 1248–12571250
chain triglyceride) and Maisine 35-1 and Peceol (mixture of mono, di and
triglycerides) oils to check for the pH-dependent affinity of the drug based
on the polarity of oil. For the determination of Kp, 1 mg of the cold drug
doped with 0.045mCi of the radioactive drug was placed into a 2-mL
Eppendorf tube to which 750mL each of oil and oil-saturated phosphate
buffered saline (PBS, 150mM, pH 7.4), or pH 1 or 3 HCl solutions were
added. Then, the tubes were sealed and the contents were tumble-agitated
for 20 h at 25 1C. Following this equilibration time, the tubes were
centrifuged at 3900g for 10min. Each phase was separated and the drug
concentration was determined by scintillation counting (Packard-
Tri-Carb2100TR, Ramsey, MN). Kp was calculated from the ratio of
concentration of haloperidol in oil to buffer. Kp values were determined by
the same method for docetaxel and paclitaxel in PBS/Miglyol 810 and
PBS/tricaprylin phases.
2.2. Preparation of NC
NC were prepared by the phase-inversion method as described
elsewhere [25,33]. Briefly, the NC components namely (phospholipid,
Solutol HS15 and triglyceride) were weighed into a glass vial and heated to
about 70 1C to obtain a homogeneous mix. Then, an aqueous solution of
0.9% NaCl (1.9mL) was added and the lipid mixture was stirred to obtain
a homogeneous dispersion. This dispersion was subjected to three
consecutive phase inversion steps by repetitive heating above the phase-
inversion temperature (80–85 1C) and cooling to 60 1C. At the end of third
phase inversion step, the resulting formulation was rapidly cooled by
placing the vial in an ice bath. It was further diluted with 0.9% NaCl to
obtain the required NC concentration. PEGylated NC were obtained by
incubating the undiluted NC, prior to the dilution step, with a micellar
solution of DSPE-PEG2000 for 1.5 h period at 65 1C [34].
2.3. Size measurements
The mean diameter and size distribution of the NC were determined by
dynamic light scattering; 5–10mL of the formulation was diluted to 1mL
with filtered water and the size distribution was recorded on a Nano-ZS
Zetasizer (Malvern Instrument Ltd., Malvern, Worcestershire, UK) at
25 1C in back scattering mode. Atomic Force Microscopy (AFM) was
performed with Nanoscope IIIa, DimensionTM 3100 (Digital Instruments,
Santa Barbara, CA) in tapping mode. NC were deposited on a freshly
cleaved mica surface and allowed to partially dry-out at room
temperature. Subsequently, they were imaged in air at ambient conditions
using etched silicon probes with a tip radius of 5–10 nm and spring
constant in the range of 20–100N/m, oscillated at its fundamental
resonance frequency (200–400KHz).
2.4. In vitro drug uptake studies
The uptake of haloperidol, docetaxel and paclitaxel into lipid NC was
measured using jacketed Franz diffusion cells. The temperature was
maintained at 3770.2 1C using a circulating water bath and the cells were
continuously stirred at 600 rpm. All uptake studies were conducted at the
drug concentration of 40 ng/mL while the concentration of NC was
equivalent to 3mg/mL of glyceride oil. In the Franz cell, NC contained in
the lower compartment were physically separated from the sampling
(upper) compartment by a 50-nm pore diameter membrane filter. Prior to
setting-up the diffusion cells, both polycarbonate membrane and O-rings
were equilibrated in PBS containing cold drug along with its radioactive
counterpart. A second equilibration period was allowed after placing the
drug-buffer solution into the lower (4.8mL) and upper (0.8mL)
compartments of the diffusion cell. Then, 500mL of NC suspension was
introduced into the lower compartment through the side port. To estimate
the drug uptake, buffer samples were withdrawn from the sampling
compartment for radioactive counting. A control experiment run under
identical conditions with the replacement of NC with buffer showed no net
transfer of drug.
2.5. Plasma lipoprotein distribution studies
The objectives of plasma lipoprotein distribution studies were: (i) to
determine the equilibrium time by investigating plasma lipoprotein distribu-
tion of drug and NC in separate studies and (ii) to examine the effect of NC
concentration on drug distribution. The NC (tricaprylin/HSPC/Solutol: 52.3/
7.5/40.2% w/w) were labeled either with 14C-cholesterol oleate or 3H-
cholesterol hexadecyl ether. Male Sprague-Dawley rat plasma was added to
Beckman UltraclearTM ultracentrifuge tubes in 3-mL aliquots. A 20-mLaliquot of the appropriate drug (along with 3H-haloperidol or 14C-docetaxel)
stock solution was added and the tubes were mixed thoroughly using a vortex
mixer. The haloperidol and docetaxel concentrations in plasma were 0.7 and
5mg/mL, respectively. The plasma was incubated at 37 1C for 20, 40, 60, and
90min (n ¼ 5–6). An appropriate volume of the tricaprylin NC formulation
was then added to the ultracentrifuge tube to provide a final concentration of
0, 2, 3, 4, or 5mg/mL. The incubation was stopped with the addition of
sodium bromide (1.02 g) to adjust the density of the plasma and then the
samples were rapidly cooled on ice to 4 1C for a minimum of 2h.
Subsequently, the treated plasma samples were separated into four fractions:
triglyceride-rich lipoproteins (TRL), low-density lipoproteins (LDL), high-
density lipoproteins (HDL), and lipoprotein-deficient plasma (LPDP) by
density gradient ultracentrifugation as previously described [35–36]. Each
fraction was assayed for drug and NC content by scintillation counting.
2.6. Cytotoxicity studies
To assess the uptake potential of NC (tricaprylin/HSPC/Solutol: 52.3/
7.5/40.2% w/w), the reduction in the cytotoxicity of docetaxel was
measured by the MTT tetrazolium dye proliferation assay [37]. B16F10
melanoma cancer line was obtained from the American Type Culture
collection (ATCC Rockville, MA) and grown in Dulbecco’s modified
Eagle’s medium supplemented with 10% fetal calf serum and 1% (w/v)
penicillin–streptomycin in a humidified atmosphere at 37 1C with 5% CO2
in air. After the cells reached 85–90% confluence, they were trypsinized,
seeded at a density of 1000 cells/well in 96-well plates and were allowed to
adhere to the wells for 24 h. The medium was replaced with 100mLmedium containing increasing concentrations of docetaxel (0.1–100nM)
following which an equal volume of either medium or NC was added. The
plates were further incubated for 15 h at 37 1C. After treatment, the
content of each well was removed by aspiration followed by the addition
of 100mL of fresh medium, and the cells were allowed to grow for 48 h. At
the end of this period, the MTT reagent (10mL of 5mg/mL) was added
and the formazan produced was solubilized by the addition of 100 mL of
15% (w/v) SDS in 0.01 N HCl. Absorbance was measured at 470 nm with a
SafireTM plate reader (Tecan Austria GmbH, Salzburg, Austria).
2.7. In vivo drug uptake and biodistribution studies
To establish the proof-of-principle of drug uptake under in vivo
conditions, the pharmacokinetics and MPS organ-distribution of doc-
etaxel were evaluated in mice in the absence and presence of separately
injected NC. Two different experimental variables (effect of PEGylation
and administration sequence of the drug and NC on the sequestration
capabilities) were investigated with 5 mice/group. C57BL/6 mice (1 month
old, 18–20 g, Charles River Breeding Laboratories, Montreal, QC,
Canada) were conditioned in animal house under 12/12-h light/dark
cycle, and were allowed food and water ad libitum. All in vivo experiments
were carried out in accordance with current Canadian guidelines for the
care of laboratory animals and were approved by the animal ethical
committee of the University of Montreal.3H-labelled NC (tricaprylin/HSPC/Solutol: 52.3/7.5/40.2% w/w) with
or without PEG were prepared as described in Section 2.2. To follow the
time course of disposition, DTX and NC were, respectively, labelled
with 14C-docetaxel (3.1mCi/mg) and 3H-hexadecyl cholesteryl ether (0.09mCi/mg). In the first experiment, mice received non-PEGylated lipid NC followed
by DTX after a time lapse of 20min (NC-DTX). In the second experiment,
PEGylated lipid NC (PEG(NC)-DTX) were tested under the same

ARTICLE IN PRESSA. Babu Dhanikula et al. / Biomaterials 28 (2007) 1248–1257 1251
administration sequence while in the last case mice received DTX followed by
PEGylated lipid NC after a time lapse of 20min (DTX-PEG(NC)). Both
DTX and NC were administered under anaesthesia in isotonic saline (100mL)via the subclavian vein at docetaxel and triglyceride dose levels of 5 and
500mg/kg, respectively. A control with DTX (docetaxel dissolved in
polysorbate 80) was performed to compare the alterations in blood and
tissue distribution of drug caused by the NC. At 0.5, 1, 2, 4, 6, 12 and 24h
post-injection, blood was sampled under anaesthesia, and liver and spleen
were harvested. Blood and tissue samples were digested with Soluene-350s,
and decolourised with hydrogen peroxide. The samples were left to stand in
the dark overnight at 4 1C following the addition of Hionic Fluors
scintillation cocktail. Radioactivity was then measured using the scintillation
counter in dual mode (3H/14C).
2.8. Data treatment and statistical methods
Plasma concentration profiles were fitted to a non-compartment model
and all pharmacokinetic parameters were determined using the software
KINETICAs 4.1.1 (InnaPhase, Philadelphia, PA). For calculation of the
area under the blood concentration-time curve to the last sampling time-
point (AUClast), bootstrapping was performed. Data points were sampled
with replacement to generate 1000 different AUC curves for each study,
which were subsequently compared for any differences by statistical
analysis. The terminal disposition rate constant (lz) was determined from
the slope of terminal phase of log-linear blood concentration-time profile.
Clearance (Cl) was calculated by dividing the dose administered by the
observed AUC. Volume of distribution in terminal phase was calculated as
Vz ¼ Dose/(AUC� lz) and mean residence time (MRT) was obtained by
dividing the area under first moment curve by AUC. Data were compared
by ANOVA followed by a post hoc Tukey’s test for multiple comparisons.
Probability values of po0.05 were considered statistically significant.
3. Results and discussion
The current detoxification approach is based on thepremise that sequestration of free drug in the blood by
Table 1
Kp values for haloperidol and taxanes in various oils against PBS at room tem
OIL Kp Compositiona
Haloperidol Saturated fatty acid (%) Mono-
Captex 8000 9.9 (0.3) 100 (C8:0) —
Captex 810D 6.6 (1.0) 40 (C8:0+10:0) o5
Corn oil 4.6 (0.3) 12.7 (C14:0+16:0+18:0) 24.2
Ethyl caprate 14.1 (1.2) — —
Ethyl laurate 16.4 (1.2) — —
Ethyl oleate 11.2 (0.7) — —
Labrafac CC 9.2 (0.2) 100 (C8:0+10:0) —
Maisine 35-1 1.2 (0.2) 4–20 (C14:0+16:0+18:0) 10–35
Miglyol 810 11.9 (0.8) 100 (C8:0+C10:0) —
Peceol 2.4 (0.2) o16 (C14:0+16:0+18:0) 460
Safflower oil 8.6 (0.6) 9.6 (C14:0+16:0+18:0) 12.6
Sesame oil 4.8 (0.4) 14.2 (C14:0+16:0+18:0) 39.7
Soy bean oil 8.0 (0.1) 14.4 (C14:0+16:0+18:0) 23.3
Tricaprylin 11.9 (0.6) 100 (C8:0) —
Paclitaxel
Miglyol 810 15.4 (0.1) 100 (C8:0+10:0) —
Tricaprylin 15.7 (0.3) 100 (C8:0) —
Docetaxel
Miglyol 810 69.3 (4.8) 100 (C8:0+10:0) —
Tricaprylin 75.7 (3.3) 100 (C8:0) —
All values are mean (SD), n ¼ 4.aComposition as provided by manufacturer.
administration of long circulating lipid NC would serve tofavourably alter the pharmacokinetic parameters andconsequently reduce its acute toxicity [34,38,39]. In thepresent study, NC prepared from medium chain triglycer-ides have been investigated both in vitro and in vivo as apossible strategy to deal with drug overdosing. Mediumchain triglycerides have been reported to be non-toxic inacute toxicity studies across several animal species. Afterrepeated intramuscular administration of 0.5mL Miglyol812 (six times daily for 14 days) to rabbits, there were nogross clinical pathological manifestations [16]. The meanacute 50% kill lethal dose of tricaprylin was reported as3700mg/kg in mice by intravenous route [40]. Mediumchain triglycerides are also used for parenteral nutrition inhumans and depending on the needs, a single patient couldreceive between 200 and 600mL/day by intravenous route[16]. Thus, the safety profile of lipid NC fabricated frommedium chain triglycerides should not be limited by thetriglycerides for the detoxification approach.
3.1. Partitioning studies
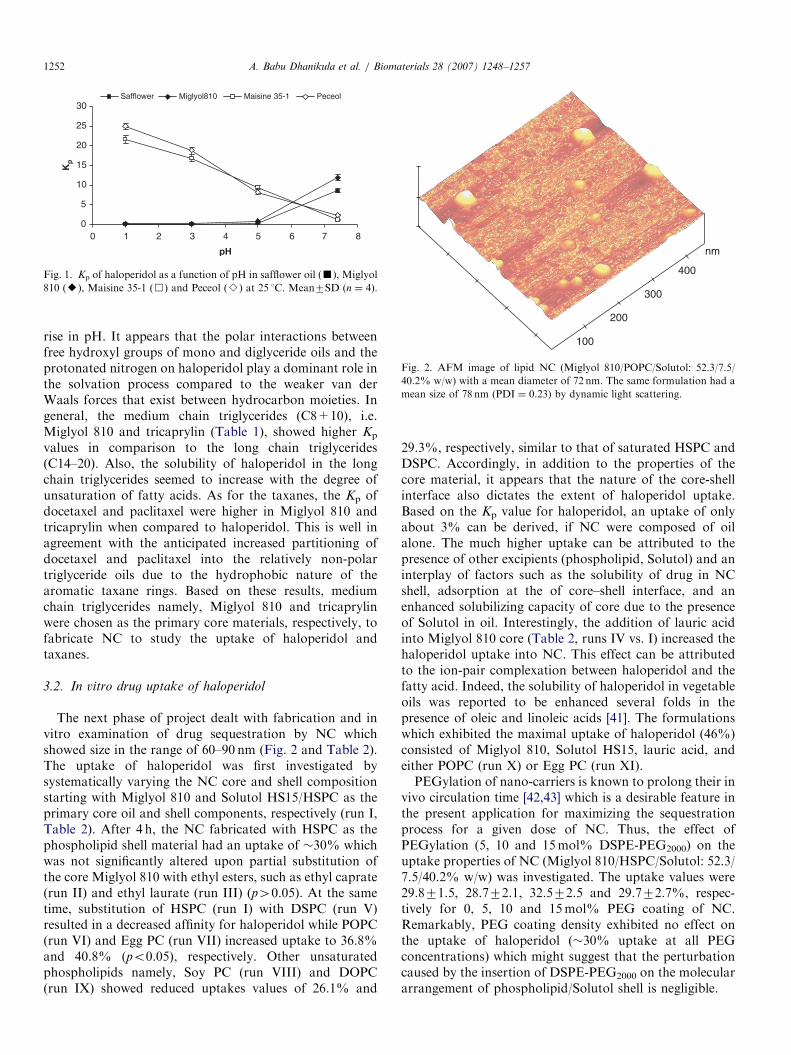
For the selection of the appropriate oil to make up thecore of lipid NC, Kp was used as a preliminary screeningcriterion (Table 1). The partitioning of haloperidol (base;pKa 8.3) into medium and long chain triglycerides waspH-dependent and the affinity for the oil increased with pH(Fig. 1). In contrast, the affinity of haloperidol for themono and diglycerides showed a trend opposite to that oftriglycerides. There was a decreased affinity of theprotonated haloperidol for Maisine 35-1 and Peceol with
perature
unsaturated fatty acid (C18:1) (%) Poly-unsaturated fatty acid (%)
—
40
59.5
—
—
—
—
450
—
o35
73.5
41.7
57.9
—
—
—
—
—
ARTICLE IN PRESS
0
5
10
15
20
25
30
0 1 2 3 4 65 7 8
pH
Kp
Safflower Miglyol810 Maisine 35-1 Peceol
Fig. 1. Kp of haloperidol as a function of pH in safflower oil (’), Miglyol
810 (E), Maisine 35-1 (&) and Peceol (B) at 25 1C. Mean7SD (n ¼ 4).
100
200
300
400
nm
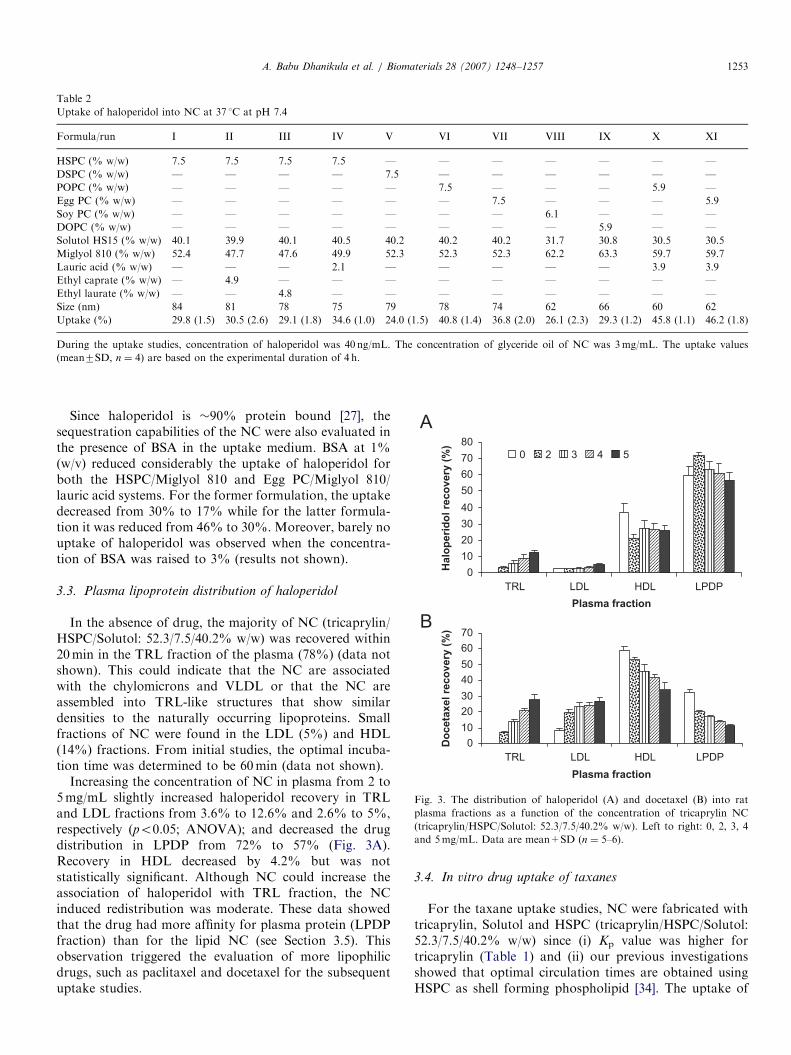
Fig. 2. AFM image of lipid NC (Miglyol 810/POPC/Solutol: 52.3/7.5/
40.2% w/w) with a mean diameter of 72 nm. The same formulation had a
mean size of 78 nm (PDI ¼ 0.23) by dynamic light scattering.
A. Babu Dhanikula et al. / Biomaterials 28 (2007) 1248–12571252
rise in pH. It appears that the polar interactions betweenfree hydroxyl groups of mono and diglyceride oils and theprotonated nitrogen on haloperidol play a dominant role inthe solvation process compared to the weaker van derWaals forces that exist between hydrocarbon moieties. Ingeneral, the medium chain triglycerides (C8+10), i.e.Miglyol 810 and tricaprylin (Table 1), showed higher Kp
values in comparison to the long chain triglycerides(C14–20). Also, the solubility of haloperidol in the longchain triglycerides seemed to increase with the degree ofunsaturation of fatty acids. As for the taxanes, the Kp ofdocetaxel and paclitaxel were higher in Miglyol 810 andtricaprylin when compared to haloperidol. This is well inagreement with the anticipated increased partitioning ofdocetaxel and paclitaxel into the relatively non-polartriglyceride oils due to the hydrophobic nature of thearomatic taxane rings. Based on these results, mediumchain triglycerides namely, Miglyol 810 and tricaprylinwere chosen as the primary core materials, respectively, tofabricate NC to study the uptake of haloperidol andtaxanes.
3.2. In vitro drug uptake of haloperidol
The next phase of project dealt with fabrication and invitro examination of drug sequestration by NC whichshowed size in the range of 60–90 nm (Fig. 2 and Table 2).The uptake of haloperidol was first investigated bysystematically varying the NC core and shell compositionstarting with Miglyol 810 and Solutol HS15/HSPC as theprimary core oil and shell components, respectively (run I,Table 2). After 4 h, the NC fabricated with HSPC as thephospholipid shell material had an uptake of �30% whichwas not significantly altered upon partial substitution ofthe core Miglyol 810 with ethyl esters, such as ethyl caprate(run II) and ethyl laurate (run III) (p40.05). At the sametime, substitution of HSPC (run I) with DSPC (run V)resulted in a decreased affinity for haloperidol while POPC(run VI) and Egg PC (run VII) increased uptake to 36.8%and 40.8% (po0.05), respectively. Other unsaturatedphospholipids namely, Soy PC (run VIII) and DOPC(run IX) showed reduced uptakes values of 26.1% and
29.3%, respectively, similar to that of saturated HSPC andDSPC. Accordingly, in addition to the properties of thecore material, it appears that the nature of the core-shellinterface also dictates the extent of haloperidol uptake.Based on the Kp value for haloperidol, an uptake of onlyabout 3% can be derived, if NC were composed of oilalone. The much higher uptake can be attributed to thepresence of other excipients (phospholipid, Solutol) and aninterplay of factors such as the solubility of drug in NCshell, adsorption at the of core–shell interface, and anenhanced solubilizing capacity of core due to the presenceof Solutol in oil. Interestingly, the addition of lauric acidinto Miglyol 810 core (Table 2, runs IV vs. I) increased thehaloperidol uptake into NC. This effect can be attributedto the ion-pair complexation between haloperidol and thefatty acid. Indeed, the solubility of haloperidol in vegetableoils was reported to be enhanced several folds in thepresence of oleic and linoleic acids [41]. The formulationswhich exhibited the maximal uptake of haloperidol (46%)consisted of Miglyol 810, Solutol HS15, lauric acid, andeither POPC (run X) or Egg PC (run XI).PEGylation of nano-carriers is known to prolong their in
vivo circulation time [42,43] which is a desirable feature inthe present application for maximizing the sequestrationprocess for a given dose of NC. Thus, the effect ofPEGylation (5, 10 and 15mol% DSPE-PEG2000) on theuptake properties of NC (Miglyol 810/HSPC/Solutol: 52.3/7.5/40.2% w/w) was investigated. The uptake values were29.871.5, 28.772.1, 32.572.5 and 29.772.7%, respec-tively for 0, 5, 10 and 15mol% PEG coating of NC.Remarkably, PEG coating density exhibited no effect onthe uptake of haloperidol (�30% uptake at all PEGconcentrations) which might suggest that the perturbationcaused by the insertion of DSPE-PEG2000 on the moleculararrangement of phospholipid/Solutol shell is negligible.
ARTICLE IN PRESS
0
10
20
30
40
50
60
70
80
A
B
TRL LDL HDL LPDP
Plasma fraction
Halo
peri
do
l re
co
very
(%
) 0 2 3 4 5
0
10
20
30
40
50
60
70
TRL LDL HDL LPDP
Plasma fraction
Do
ceta
xel re
co
very
(%
)
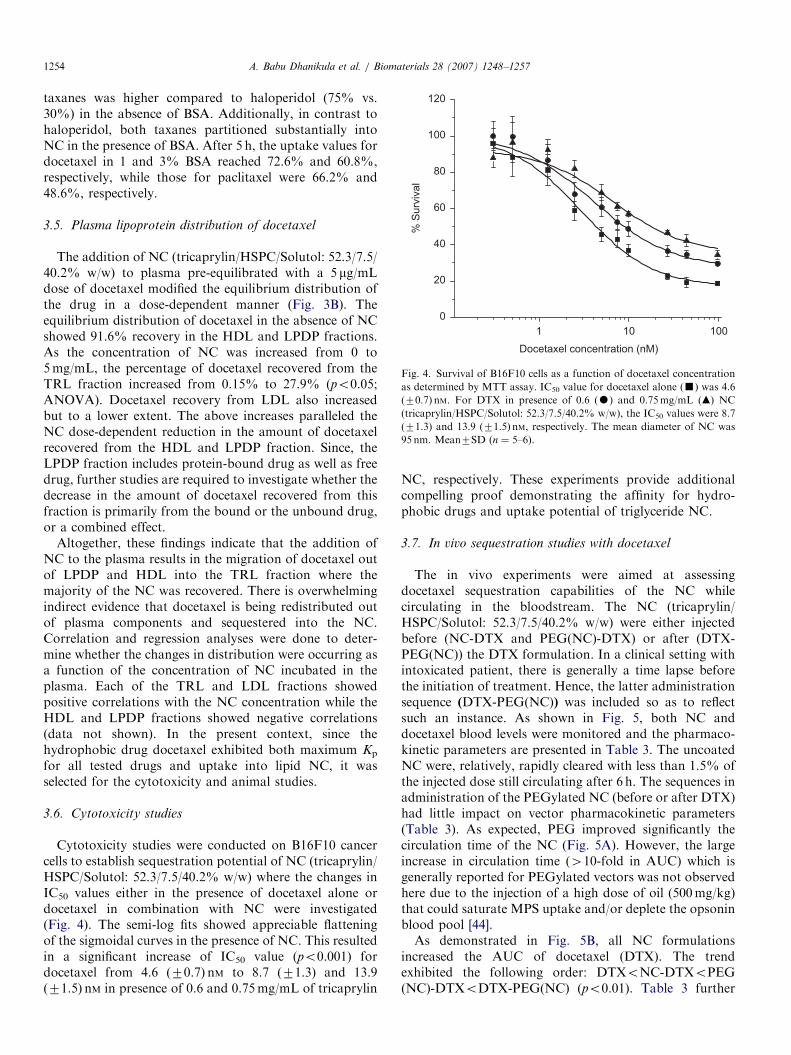
Fig. 3. The distribution of haloperidol (A) and docetaxel (B) into rat
plasma fractions as a function of the concentration of tricaprylin NC
(tricaprylin/HSPC/Solutol: 52.3/7.5/40.2% w/w). Left to right: 0, 2, 3, 4
and 5mg/mL. Data are mean+SD (n ¼ 5–6).
Table 2
Uptake of haloperidol into NC at 37 1C at pH 7.4
Formula/run I II III IV V VI VII VIII IX X XI
HSPC (% w/w) 7.5 7.5 7.5 7.5 — — — — — — —
DSPC (% w/w) — — — — 7.5 — — — — — —
POPC (% w/w) — — — — — 7.5 — — — 5.9 —
Egg PC (% w/w) — — — — — — 7.5 — — — 5.9
Soy PC (% w/w) — — — — — — — 6.1 — — —
DOPC (% w/w) — — — — — — — — 5.9 — —
Solutol HS15 (% w/w) 40.1 39.9 40.1 40.5 40.2 40.2 40.2 31.7 30.8 30.5 30.5
Miglyol 810 (% w/w) 52.4 47.7 47.6 49.9 52.3 52.3 52.3 62.2 63.3 59.7 59.7
Lauric acid (% w/w) — — — 2.1 — — — — — 3.9 3.9
Ethyl caprate (% w/w) — 4.9 — — — — — — — — —
Ethyl laurate (% w/w) — — 4.8 — — — — — — — —
Size (nm) 84 81 78 75 79 78 74 62 66 60 62
Uptake (%) 29.8 (1.5) 30.5 (2.6) 29.1 (1.8) 34.6 (1.0) 24.0 (1.5) 40.8 (1.4) 36.8 (2.0) 26.1 (2.3) 29.3 (1.2) 45.8 (1.1) 46.2 (1.8)
During the uptake studies, concentration of haloperidol was 40 ng/mL. The concentration of glyceride oil of NC was 3mg/mL. The uptake values
(mean7SD, n ¼ 4) are based on the experimental duration of 4 h.
A. Babu Dhanikula et al. / Biomaterials 28 (2007) 1248–1257 1253
Since haloperidol is �90% protein bound [27], thesequestration capabilities of the NC were also evaluated inthe presence of BSA in the uptake medium. BSA at 1%(w/v) reduced considerably the uptake of haloperidol forboth the HSPC/Miglyol 810 and Egg PC/Miglyol 810/lauric acid systems. For the former formulation, the uptakedecreased from 30% to 17% while for the latter formula-tion it was reduced from 46% to 30%. Moreover, barely nouptake of haloperidol was observed when the concentra-tion of BSA was raised to 3% (results not shown).
3.3. Plasma lipoprotein distribution of haloperidol
In the absence of drug, the majority of NC (tricaprylin/HSPC/Solutol: 52.3/7.5/40.2% w/w) was recovered within20min in the TRL fraction of the plasma (78%) (data notshown). This could indicate that the NC are associatedwith the chylomicrons and VLDL or that the NC areassembled into TRL-like structures that show similardensities to the naturally occurring lipoproteins. Smallfractions of NC were found in the LDL (5%) and HDL(14%) fractions. From initial studies, the optimal incuba-tion time was determined to be 60min (data not shown).
Increasing the concentration of NC in plasma from 2 to5mg/mL slightly increased haloperidol recovery in TRLand LDL fractions from 3.6% to 12.6% and 2.6% to 5%,respectively (po0.05; ANOVA); and decreased the drugdistribution in LPDP from 72% to 57% (Fig. 3A).Recovery in HDL decreased by 4.2% but was notstatistically significant. Although NC could increase theassociation of haloperidol with TRL fraction, the NCinduced redistribution was moderate. These data showedthat the drug had more affinity for plasma protein (LPDPfraction) than for the lipid NC (see Section 3.5). Thisobservation triggered the evaluation of more lipophilicdrugs, such as paclitaxel and docetaxel for the subsequentuptake studies.
3.4. In vitro drug uptake of taxanes
For the taxane uptake studies, NC were fabricated withtricaprylin, Solutol and HSPC (tricaprylin/HSPC/Solutol:52.3/7.5/40.2% w/w) since (i) Kp value was higher fortricaprylin (Table 1) and (ii) our previous investigationsshowed that optimal circulation times are obtained usingHSPC as shell forming phospholipid [34]. The uptake of
ARTICLE IN PRESS
1 10 100
0
20
40
60
80
100
120
% S
urv
ival
Docetaxel concentration (nM)
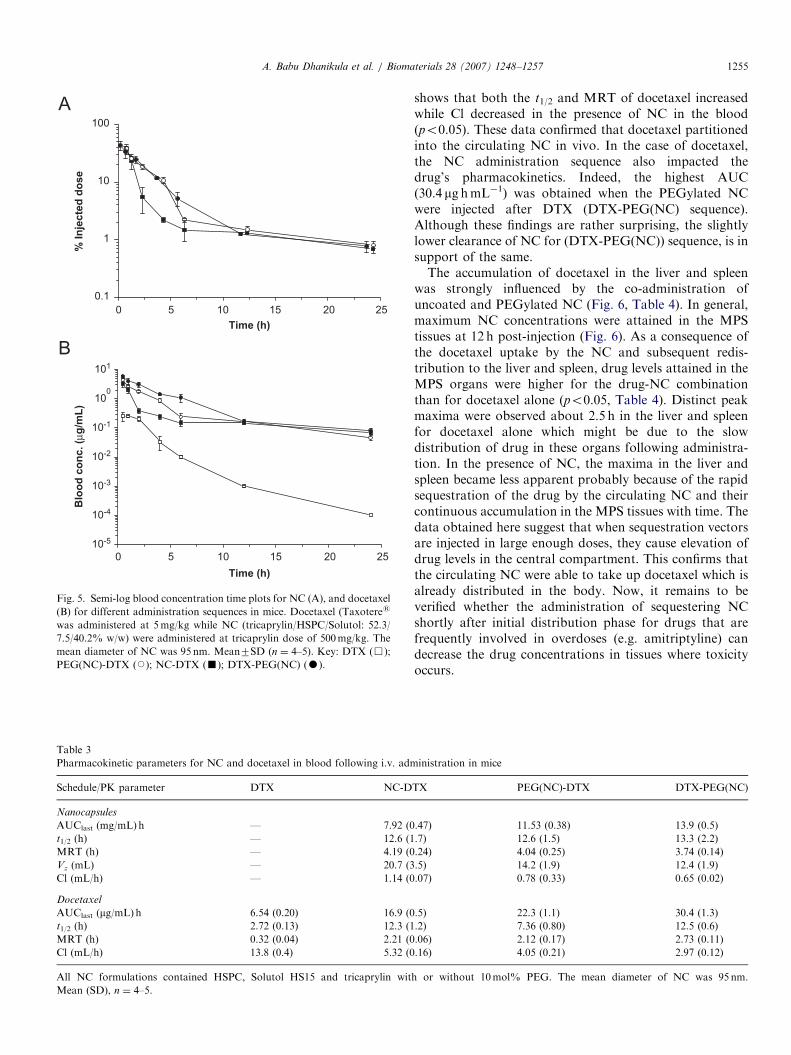
Fig. 4. Survival of B16F10 cells as a function of docetaxel concentration
as determined by MTT assay. IC50 value for docetaxel alone (’) was 4.6
(70.7) nM. For DTX in presence of 0.6 (K) and 0.75mg/mL (m) NC
(tricaprylin/HSPC/Solutol: 52.3/7.5/40.2% w/w), the IC50 values were 8.7
(71.3) and 13.9 (71.5) nM, respectively. The mean diameter of NC was
95 nm. Mean7SD (n ¼ 5–6).
A. Babu Dhanikula et al. / Biomaterials 28 (2007) 1248–12571254
taxanes was higher compared to haloperidol (75% vs.30%) in the absence of BSA. Additionally, in contrast tohaloperidol, both taxanes partitioned substantially intoNC in the presence of BSA. After 5 h, the uptake values fordocetaxel in 1 and 3% BSA reached 72.6% and 60.8%,respectively, while those for paclitaxel were 66.2% and48.6%, respectively.
3.5. Plasma lipoprotein distribution of docetaxel
The addition of NC (tricaprylin/HSPC/Solutol: 52.3/7.5/40.2% w/w) to plasma pre-equilibrated with a 5 mg/mLdose of docetaxel modified the equilibrium distribution ofthe drug in a dose-dependent manner (Fig. 3B). Theequilibrium distribution of docetaxel in the absence of NCshowed 91.6% recovery in the HDL and LPDP fractions.As the concentration of NC was increased from 0 to5mg/mL, the percentage of docetaxel recovered from theTRL fraction increased from 0.15% to 27.9% (po0.05;ANOVA). Docetaxel recovery from LDL also increasedbut to a lower extent. The above increases paralleled theNC dose-dependent reduction in the amount of docetaxelrecovered from the HDL and LPDP fraction. Since, theLPDP fraction includes protein-bound drug as well as freedrug, further studies are required to investigate whether thedecrease in the amount of docetaxel recovered from thisfraction is primarily from the bound or the unbound drug,or a combined effect.
Altogether, these findings indicate that the addition ofNC to the plasma results in the migration of docetaxel outof LPDP and HDL into the TRL fraction where themajority of the NC was recovered. There is overwhelmingindirect evidence that docetaxel is being redistributed outof plasma components and sequestered into the NC.Correlation and regression analyses were done to deter-mine whether the changes in distribution were occurring asa function of the concentration of NC incubated in theplasma. Each of the TRL and LDL fractions showedpositive correlations with the NC concentration while theHDL and LPDP fractions showed negative correlations(data not shown). In the present context, since thehydrophobic drug docetaxel exhibited both maximum Kp
for all tested drugs and uptake into lipid NC, it wasselected for the cytotoxicity and animal studies.
3.6. Cytotoxicity studies
Cytotoxicity studies were conducted on B16F10 cancercells to establish sequestration potential of NC (tricaprylin/HSPC/Solutol: 52.3/7.5/40.2% w/w) where the changes inIC50 values either in the presence of docetaxel alone ordocetaxel in combination with NC were investigated(Fig. 4). The semi-log fits showed appreciable flatteningof the sigmoidal curves in the presence of NC. This resultedin a significant increase of IC50 value (po0.001) fordocetaxel from 4.6 (70.7) nM to 8.7 (71.3) and 13.9(71.5) nM in presence of 0.6 and 0.75mg/mL of tricaprylin
NC, respectively. These experiments provide additionalcompelling proof demonstrating the affinity for hydro-phobic drugs and uptake potential of triglyceride NC.
3.7. In vivo sequestration studies with docetaxel
The in vivo experiments were aimed at assessingdocetaxel sequestration capabilities of the NC whilecirculating in the bloodstream. The NC (tricaprylin/HSPC/Solutol: 52.3/7.5/40.2% w/w) were either injectedbefore (NC-DTX and PEG(NC)-DTX) or after (DTX-PEG(NC)) the DTX formulation. In a clinical setting withintoxicated patient, there is generally a time lapse beforethe initiation of treatment. Hence, the latter administrationsequence (DTX-PEG(NC)) was included so as to reflectsuch an instance. As shown in Fig. 5, both NC anddocetaxel blood levels were monitored and the pharmaco-kinetic parameters are presented in Table 3. The uncoatedNC were, relatively, rapidly cleared with less than 1.5% ofthe injected dose still circulating after 6 h. The sequences inadministration of the PEGylated NC (before or after DTX)had little impact on vector pharmacokinetic parameters(Table 3). As expected, PEG improved significantly thecirculation time of the NC (Fig. 5A). However, the largeincrease in circulation time (410-fold in AUC) which isgenerally reported for PEGylated vectors was not observedhere due to the injection of a high dose of oil (500mg/kg)that could saturate MPS uptake and/or deplete the opsoninblood pool [44].As demonstrated in Fig. 5B, all NC formulations
increased the AUC of docetaxel (DTX). The trendexhibited the following order: DTXoNC-DTXoPEG(NC)-DTXoDTX-PEG(NC) (po0.01). Table 3 further
ARTICLE IN PRESS
0 5 10 15 20 25
0.1
1
10
100
0 5 10 15 20 25
10-5
10-4
10-3
10-2
10-1
100
101
Blo
od
co
nc. (µ
g/m
L)
% In
jecte
d d
ose
Time (h)
Time (h)
A
B
Fig. 5. Semi-log blood concentration time plots for NC (A), and docetaxel
(B) for different administration sequences in mice. Docetaxel (Taxoteres
was administered at 5mg/kg while NC (tricaprylin/HSPC/Solutol: 52.3/
7.5/40.2% w/w) were administered at tricaprylin dose of 500mg/kg. The
mean diameter of NC was 95 nm. Mean7SD (n ¼ 4–5). Key: DTX (&);
PEG(NC)-DTX (J); NC-DTX (’); DTX-PEG(NC) (K).
Table 3
Pharmacokinetic parameters for NC and docetaxel in blood following i.v. adm
Schedule/PK parameter DTX NC-D
Nanocapsules
AUClast (mg/mL) h — 7.92 (0
t1/2 (h) — 12.6 (1
MRT (h) — 4.19 (0
Vz (mL) — 20.7 (3
Cl (mL/h) — 1.14 (0
Docetaxel
AUClast (mg/mL) h 6.54 (0.20) 16.9 (0
t1/2 (h) 2.72 (0.13) 12.3 (1
MRT (h) 0.32 (0.04) 2.21 (0
Cl (mL/h) 13.8 (0.4) 5.32 (0
All NC formulations contained HSPC, Solutol HS15 and tricaprylin wit
Mean (SD), n ¼ 4–5.
A. Babu Dhanikula et al. / Biomaterials 28 (2007) 1248–1257 1255
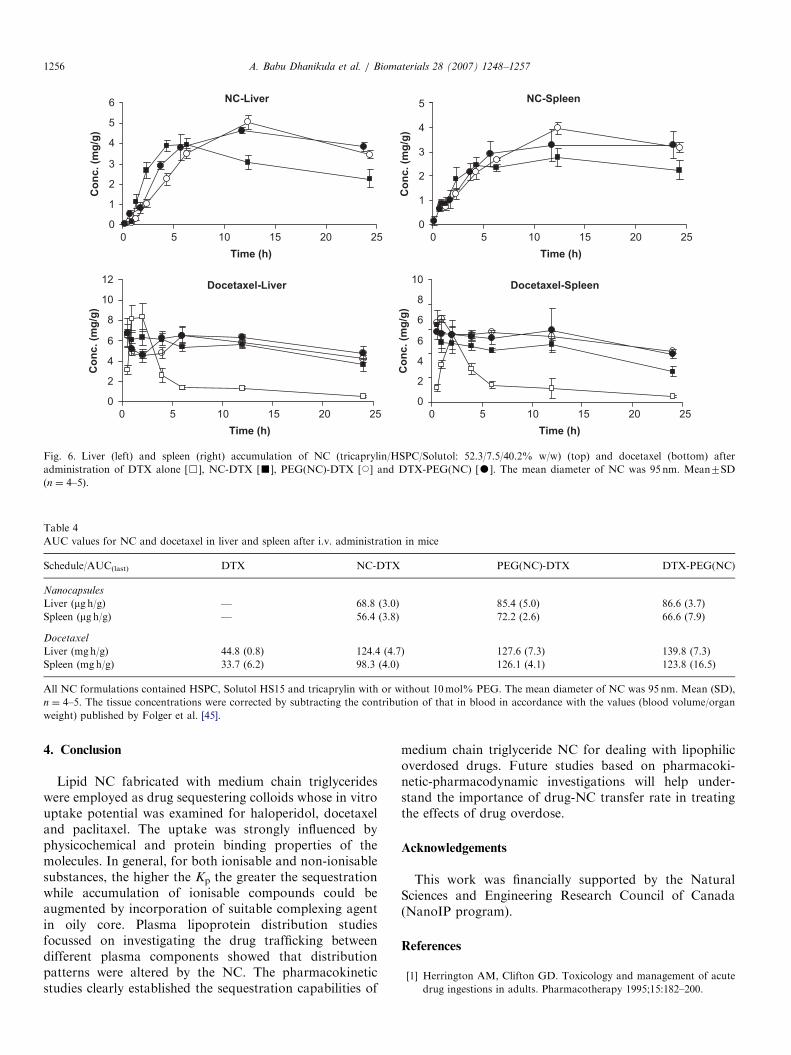
shows that both the t1/2 and MRT of docetaxel increasedwhile Cl decreased in the presence of NC in the blood(po0.05). These data confirmed that docetaxel partitionedinto the circulating NC in vivo. In the case of docetaxel,the NC administration sequence also impacted thedrug’s pharmacokinetics. Indeed, the highest AUC(30.4 mg hmL�1) was obtained when the PEGylated NCwere injected after DTX (DTX-PEG(NC) sequence).Although these findings are rather surprising, the slightlylower clearance of NC for (DTX-PEG(NC)) sequence, is insupport of the same.The accumulation of docetaxel in the liver and spleen
was strongly influenced by the co-administration ofuncoated and PEGylated NC (Fig. 6, Table 4). In general,maximum NC concentrations were attained in the MPStissues at 12 h post-injection (Fig. 6). As a consequence ofthe docetaxel uptake by the NC and subsequent redis-tribution to the liver and spleen, drug levels attained in theMPS organs were higher for the drug-NC combinationthan for docetaxel alone (po0.05, Table 4). Distinct peakmaxima were observed about 2.5 h in the liver and spleenfor docetaxel alone which might be due to the slowdistribution of drug in these organs following administra-tion. In the presence of NC, the maxima in the liver andspleen became less apparent probably because of the rapidsequestration of the drug by the circulating NC and theircontinuous accumulation in the MPS tissues with time. Thedata obtained here suggest that when sequestration vectorsare injected in large enough doses, they cause elevation ofdrug levels in the central compartment. This confirms thatthe circulating NC were able to take up docetaxel which isalready distributed in the body. Now, it remains to beverified whether the administration of sequestering NCshortly after initial distribution phase for drugs that arefrequently involved in overdoses (e.g. amitriptyline) candecrease the drug concentrations in tissues where toxicityoccurs.
inistration in mice
TX PEG(NC)-DTX DTX-PEG(NC)
.47) 11.53 (0.38) 13.9 (0.5)
.7) 12.6 (1.5) 13.3 (2.2)
.24) 4.04 (0.25) 3.74 (0.14)
.5) 14.2 (1.9) 12.4 (1.9)
.07) 0.78 (0.33) 0.65 (0.02)
.5) 22.3 (1.1) 30.4 (1.3)
.2) 7.36 (0.80) 12.5 (0.6)
.06) 2.12 (0.17) 2.73 (0.11)
.16) 4.05 (0.21) 2.97 (0.12)
h or without 10mol% PEG. The mean diameter of NC was 95 nm.
ARTICLE IN PRESS
Docetaxel-Liver Docetaxel-Spleen
NC-Liver NC-Spleen
0
1
2
3
4
5
6
0 5 10 15 20 25
Time (h)
Co
nc. (m
g/g
)
0
1
2
3
4
5
0 5 10 15 20 25
Time (h)
Co
nc. (m
g/g
)0
2
4
6
8
10
12
0 5 10 15 20 25
Time (h)
Co
nc. (m
g/g
)
0
2
4
6
6
8
10
0 5 10 15 20 25
Time (h)
Co
nc. (m
g/g
)
Fig. 6. Liver (left) and spleen (right) accumulation of NC (tricaprylin/HSPC/Solutol: 52.3/7.5/40.2% w/w) (top) and docetaxel (bottom) after
administration of DTX alone [&], NC-DTX [’], PEG(NC)-DTX [J] and DTX-PEG(NC) [K]. The mean diameter of NC was 95 nm. Mean7SD
(n ¼ 4–5).
Table 4
AUC values for NC and docetaxel in liver and spleen after i.v. administration in mice
Schedule/AUC(last) DTX NC-DTX PEG(NC)-DTX DTX-PEG(NC)
Nanocapsules
Liver (mg h/g) — 68.8 (3.0) 85.4 (5.0) 86.6 (3.7)
Spleen (mg h/g) — 56.4 (3.8) 72.2 (2.6) 66.6 (7.9)
Docetaxel
Liver (mgh/g) 44.8 (0.8) 124.4 (4.7) 127.6 (7.3) 139.8 (7.3)
Spleen (mgh/g) 33.7 (6.2) 98.3 (4.0) 126.1 (4.1) 123.8 (16.5)
All NC formulations contained HSPC, Solutol HS15 and tricaprylin with or without 10mol% PEG. The mean diameter of NC was 95 nm. Mean (SD),
n ¼ 4–5. The tissue concentrations were corrected by subtracting the contribution of that in blood in accordance with the values (blood volume/organ
weight) published by Folger et al. [45].
A. Babu Dhanikula et al. / Biomaterials 28 (2007) 1248–12571256
4. Conclusion
Lipid NC fabricated with medium chain triglycerideswere employed as drug sequestering colloids whose in vitrouptake potential was examined for haloperidol, docetaxeland paclitaxel. The uptake was strongly influenced byphysicochemical and protein binding properties of themolecules. In general, for both ionisable and non-ionisablesubstances, the higher the Kp the greater the sequestrationwhile accumulation of ionisable compounds could beaugmented by incorporation of suitable complexing agentin oily core. Plasma lipoprotein distribution studiesfocussed on investigating the drug trafficking betweendifferent plasma components showed that distributionpatterns were altered by the NC. The pharmacokineticstudies clearly established the sequestration capabilities of
medium chain triglyceride NC for dealing with lipophilicoverdosed drugs. Future studies based on pharmacoki-netic-pharmacodynamic investigations will help under-stand the importance of drug-NC transfer rate in treatingthe effects of drug overdose.
Acknowledgements
This work was financially supported by the NaturalSciences and Engineering Research Council of Canada(NanoIP program).
References
[1] Herrington AM, Clifton GD. Toxicology and management of acute
drug ingestions in adults. Pharmacotherapy 1995;15:182–200.

ARTICLE IN PRESSA. Babu Dhanikula et al. / Biomaterials 28 (2007) 1248–1257 1257
[2] Vernon DD, Gleich MC. Poisoning and drug overdose. Crit Care
Clin 1997;13:647–67.
[3] Mokhlesi B, Leikin JB, Murray P, Corbridge TC. Adult toxicology in
critical care: part I: general approach to the intoxicated patient. Chest
2003;123:577–92.
[4] Haydon E, Rehm J, Fischer B, Monga N, Adlaf E. Prescription drug
abuse in Canada and the diversion of prescription drugs into the illicit
drug market. Can J Public Health 2005;96:459–61.
[5] Greene SL, Dargan PI, Jones AL. Acute poisoning: understanding
90% of cases in a nutshell. Postgrad Med J 2005;81:204–16.
[6] Gunnell D, Ho D, Murray V. Medical management of deliberate
drug overdose: a neglected area for suicide prevention? Emerg Med J
2004;21:35–8.
[7] Single E, Robson L, Rehm J, Xie X. Morbidity and mortality
attributable to alcohol, tobacco and illicit drug use in Canada. Am J
Public Health 1999;89:385–90.
[8] Litovitz TL, Smilkstein M, Felberg L, Klein-Schwartz W, Berlin R,
Morgan J. 199 Annual report of the american association of poison
control centers toxic exposure surveillance system. Am J Emerg Med
1997;15:447–500.
[9] CDC. Increase in poisoning deaths caused by non-illicit drugs—
Utah, 1991-2003. MMWR 2005;293:1182–3.
[10] Deo N, Somasundaran T, Somasundaran P. Solution properties of
amitriptyline and its partitioning into lipid bilayers. Coll Surf B
Biointerfaces 2004;34:155–9.
[11] Chyka PA, Seger D. Position statement: single-dose activated
charcoal. American Academy of Clinical Toxicology; European
Association of Poisons Centres and Clinical Toxicologists. J Toxicol
Clin Toxicol 1997;35:721–41.
[12] Merigian KS, Blaho K. Diagnosis and management of the drug
overdose patient. Am J Ther 1997;4:99–113.
[13] Underhill R, Jovanovic A, Carino S, Varhney M, Shah D, Dennis D, et
al. Oil-filled silica nanocapsules for lipophilic drug uptake: implications
for drug detoxification therapy. Chem Mater 2002;14:4919–25.
[14] Lee D, Flint J, Morey T, Dennis D, Partch R, Baney R. Aromatic-
aromatic interaction of amitriptyline: implication of overdosed drug
detoxification. J Pharm Sci 2005;94:373–81.
[15] Fallon MS, Chauhan A. Sequestration of amitriptyline by liposomes.
J Colloid Interface Sci 2006;300:7–19.
[16] Traul KA, Driedger A, Ingle DL, Nakhasi D. Review of the
toxicologic properties of medium-chain triglycerides. Food Chem
Toxicol 2000;38:79–98.
[17] Asher WJ, Bovee KC, Frankenfeld JW, Hamilton RW, Henderson
LW, Holtzapple PG, et al. Liquid membrane system directed toward
chronic uremia. Kidney Int 1975;7(Suppl.):409–12.
[18] Frankenfeld JW, Fuller GC, Rhodes CT. Potential use of liquid
membranes for emergency treatment of drug overdose. Drug Dev
Commun 1976;2:405–19.
[19] Morimoto Y, Sugibayashi K, Yamaguchi Y, Kato Y. Detoxication
capacity of a multiple (w/o/w) emulsion for the treatment of drug
overdose: drug extraction into the emulsion in the gastro-intestinal
tract of rabbits. Chem Pharm Bull (Tokyo) 1979;27:3188–92.
[20] Weinberg G, Ripper R, Feinstein DL, Hoffman W. Lipid emulsion
infusion rescues dogs from bupivacaine-induced cardiac toxicity. Reg
Anesth Pain Med 2003;28:198–202.
[21] Yoav G, Odelia G, Shaltiel C. A lipid emulsion reduces mortality
from clomipramine overdose in rats. Vet Hum Toxicol 2002;44:30.
[22] Tebbutt S, Harvey M, Nicholson T, Cave G. Intralipid prolongs
survival in a rat model of verapamil toxicity. Acad Emerg Med
2006;13:134–9.
[23] Rollins CJ. Total nutrient admixtures: stability issues and their
impact on nursing practice. J Intraven Nurs 1997;20:299–304.
[24] Vilaro S, Llobera M. Uptake and metabolism of Intralipid by rat
liver: an electron-microscopic study. J Nutr 1988;118:932–40.
[25] Heurtault B, Saulnier P, Pech B, Proust JE, Benoit JP. A novel phase
inversion-based process for the preparation of lipid nanocarriers.
Pharm Res 2002;875:875–80.
[26] Froemming JS, Lam YW, Jann MW, Davis CM. Pharmacokinetics
of haloperidol. Clin Pharmacokinet 1989;17:396–423.
[27] Darby JK, Pasta DJ, Dabiri L, Clark L, Mosbacher D. Haloperidol
dose and blood level variability: toxicity and interindividual and
intraindividual variability in the nonresponder patient in the clinical
practice setting. J Clin Psychopharmacol 1995;15:334–40.
[28] Kudo S, Ishizaki T. Pharmacokinetics of haloperidol: an update. Clin
Pharmacokinet 1999;37:435–56.
[29] Levine BS, Wu SC, Goldberger BA, Caplan YH. Two fatalities
involving haloperidol. J Anal Toxicol 1991;15:282–4.
[30] Yoshida I, Sakaguchi Y, Matsuishi T, Yano E, Yamashita Y, Hayata
S, et al. Acute accidental overdosage of haloperidol in children. Acta
Paediatr 1993;82:877–80.
[31] Hansen LM, Megerian G, Donnenfeld AE. Haloperidol overdose
during pregnancy. Obstet Gynecol 1997;90:659–61.
[32] Kan P, Chen ZB, Lee CJ, Chu IM. Development of nonionic
surfactant/phospholipid o/w emulsion as a paclitaxel delivery system.
J Control Release 1999;58:271–8.
[33] Khalid MN, Simard P, Hoarau D, Dragomir A, Leroux JC. Long
circulating poly(ethylene glycol)-decorated lipid nanocapsules deliver
docetaxel to solid tumors. Pharm Res 2006;23:752–8.
[34] Hoarau D, Delmas P, David S, Roux E, Leroux JC. Novel long-
circulating lipid nanocapsules. Pharm Res 2004;21:1783–9.
[35] Wasan KM, Pritchard PH, Ramaswamy M, Wong W, Donnachie
EM, Brunner LJ. Differences in lipoprotein lipid concentration and
composition modify the plasma distribution of cyclosporine. Pharm
Res 1997;14:1613–20.
[36] Rossignol DP, Wasan KM, Choo E, Yau E, Wong N, Rose J, et al.
Safety, pharmacokinetics, pharmacodynamics, and plasma lipopro-
tein distribution of eritoran (E5564) during continuous intravenous
infusion into healthy volunteers. Antimicrob Agents Chemother
2004;48:3233–40.
[37] Mosmann T. Rapid colorimetric assay for cellular growth and
survival: application to proliferation and cytotoxicity assays.
J Immunol Methods 1983;65:55–63.
[38] Mosqueira VC, Legrand P, Morgat JL, Vert M, Mysiakine E, Gref
R, et al. Biodistribution of long-circulating PEG-grafted nanocap-
sules in mice: effects of PEG chain length and density. Pharm Res
2001;18:1411–9.
[39] Lamprecht A, Bouligand Y, Benoit JP. New lipid nanocapsules
exhibit sustained release properties for amiodarone. J Control
Release 2002;84:59–68.
[40] Wretlind A. The toxicity of low-molecular triglycerides. Acta Physiol
Scand 1957;40:338–43.
[41] Radd BL, Newman AC, Fegely BJ, Chrzanowski FA, Lichten JL,
Walkling WD. Development of haloperidol in oil injection formula-
tions. J Parenter Sci Technol 1985;39:48–51.
[42] Leroux JC, De Jaeghere F, Anner B, Doelker E, Gurny R. An
investigation on the role of plasma and serum opsonins on the
internalization of biodegradable poly(D,L-lactic acid) nanoparticles
by human monocytes. Life Sci 1995;57:695–703.
[43] Moghimi SM, Hunter AC. Capture of stealth nanoparticles by the
body’s defences. Crit Rev Ther Drug Carrier Syst 2001;18:527–50.
[44] Ueda K, Ishida M, Inoue T, Fujimoto M, Kawahara Y, Sakaeda T,
et al. Effect of injection volume on the pharmacokinetics of oil
particles and incorporated menatetrenone after intravenous injection
as O/W lipid emulsions in rats. J Drug Target 2001;9:353–60.
[45] Folger WE, Wade R, Brundish DE, Fidler IJ. Distribution and fate
of free and liposome-encapsulated [3H] Nor-muramyl dipeptide and
[3H] muramyl tripeptide phosphatidylethanolamine in mice.
J Immunol 1985;135:1372–7.