involvement of cytochrome p450 1a in sanguinarine detoxication
TRANSCRIPT

Toxicology Letters 151 (2004) 375–387
Involvement of cytochrome P450 1Ain sanguinarine detoxication
Jirı Vrba1, Pavel Kosina, Jitka Ulrichová, Martin Modrianský∗
Institute of Medical Chemistry and Biochemistry, Faculty of Medicine, Palacký University,Hnevotınská 3, 775 15 Olomouc, Czech Republic
Received 1 December 2003; received in revised form 27 February 2004; accepted 8 March 2004
Available online 17 April 2004
Abstract
Sanguinarine (SA), a member of the benzo[c]phenanthridine alkaloids, is a potent anti-microbial agent with anti-inflammatoryand anti-neoplastic properties. However, toxicity of the alkaloid severely limits its medical applications. Recent report byWilliams et al. [Vet. Hum. Toxicol. 42 (2000) 196] implicated rat hepatic cytochrome P450 (CYP) 1A2 as a likely modulatorof SA toxicity. Indeed, the in vitro toxicity of SA in primary culture of rat hepatocytes and human hepatic cell line HepG2,demonstrated as lactate dehydrogenase leakage and metabolic capability (MTT assay), was diminished following inductionof CYP1A by 2,3,7,8-tetrachlorodibenzo-p-dioxin, 3-methylcholanthrene, and�-naphtoflavone. Using microsomes containingrecombinant CYP1A1 or CYP1A2 we show that SA causes non-competitive inhibition of the former and competitive inhibition ofthe latter as assessed by ethoxyresorufin de-ethylation (EROD). In human hepatic microsomes SA exhibits competitive inhibitionof EROD activity with apparentKi of 2�M, a value identical to that observed for CYP1A2 inhibition in recombinant system.Pre-incubation of SA with human liver microsomes resulted in time-dependent, but not dose-dependent decline in EROD activitysuggesting CYP1A2 inhibition is not mechanism based. SA also inhibits activity of NADPH:CYP reductase, an enzyme requiredfor CYP activity, with IC50 very similar to that observed for EROD inhibition. Tentative mechanism for CYP1A involvement indecreased in vitro SA toxicity is discussed.© 2004 Elsevier Ireland Ltd. All rights reserved.
Keywords:Sanguinarine; Cytochrome P450; Toxicity; Human hepatic microsomes; NADPH:CYP reductase
1. Introduction
Sanguinarine (SA), a benzo[c]phenanthridine alka-loid (Fig. 1), exists in solution under physiological pH
∗ Corresponding author. Tel.:+420-585-632-316;fax: +420-585-632-302.
E-mail address:[email protected] (M. Modriansky).1 This work was submitted as a partial fulfillment of require-
ments for the Ph.D. degree.
as either an iminium (structure I) or ‘pseudobase’ form(structure II). The former structure is susceptible tonucleophilic attack and can reversibly react with hy-droxyl anion thus forming the latter. The ‘pseudobase’is an electroneutral substance which is the form ex-pected to penetrate cells where it can act as is or dis-sociate back into the positively charged iminium formof SA. The alkaloid is considered toxic and the pri-mary cause of numerous outbreaks of human poison-ing known as epidemic dropsy syndrome (Singh et al.,
0378-4274/$ – see front matter © 2004 Elsevier Ireland Ltd. All rights reserved.doi:10.1016/j.toxlet.2004.03.005

376 J. Vrba et al. / Toxicology Letters 151 (2004) 375–387
N+
O
O
O
O
CH3
I
N
O
O
O
O
OH
CH3
II
Fig. 1. Structure of sanguinarine iminium (I) and ‘pseudobase’ (II) forms.
2000). Contamination of mustard oil and grains withArgemone mexicanaL. seeds, in which SA is present,appears to be the reason of the outbreaks. On the otherhand, SA has been described to possess anti-microbialand anti-inflammatory activity (reviewed in Walterováet al., 1995). Especially the anti-microbial activityhas led to the use of extracts containing SA, e.g.from the plant Sanguinaria canadensisL., in tooth-pastes and oral rinse products (Frankos et al., 1990;Kuftinec et al., 1990). SA was also suggested as ananti-neoplastic and effective against multiple drug re-sistance in cervical cells (Ding et al., 2002). A recentin vivo study in mice stemming from structural sim-ilarity of SA with polycyclic aromatic hydrocarbons(PAHs) demonstrated mitigated toxicity of SA follow-ing 3-methylcholanthrene (3-MC), a cytochrome P450(CYP) inducer, administration for 3 days (Williamset al., 2000). Moreover, addition of S9 hepatic micro-somal fraction from Aroclor-treated rats to culturedhuman cells from oral tissue lessened the cytotoxicityof SA (Babich et al., 1996).
Indeed, scattered throughout literature data existsupporting the role of CYPs in SA metabolism. Whilethe early studies simply show interaction of SA withCYPs (Peeples and Dalvi, 1982), the latest narrow thefield down to CYP1A family (Stiborová et al., 2002;Williams et al., 2000). Therefore, CYP1A enzymesare likely candidates in faster detoxification of SA.
Cytochromes P450, the most important group ofphase I biotransformation enzymes, are involved indetoxification and elimination of foreign compoundsfrom the body (Anzenbacher and Anzenbacherová,2001). Comprised of vast number of enzymes, thisgene superfamily of proteins is expressed mainly inthe liver but some CYP enzymes are found in extra-hepatic tissues as well. The hepatic enzymes constitutethe basis of bioactivation and inactivation of endoge-
nous compounds, e.g. steroid hormones, as well asmedicaments. They are, however, also responsible foractivation of xenobiotics into carcinogens (Ioannidesand Parke, 1990). Important players in carcinogenactivation, and for that matter quite often used inenvironmental risk assessment, are the CYP1 familyenzymes (Fent, 2001). Their expression is controlledby aryl hydrocarbon (Ah) receptor and is inducible bycertain PAHs (Schrenk, 1998). Established inducersinclude 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD),3-methylcholanthrene, and �-naphtoflavone (BNF)(Correia, 1995). While the negative side of CYP1Asis often highlighted, i.e. their role in carcinogen acti-vation, induced expression of these proteins can alsopromote faster degradation of some toxicants.
We designed this study to verify whether otherCYP1A inducers, TCDD and BNF, also modulateSA toxicity in vitro and also to test whether SA ismetabolized into a less toxic substance by humanhepatic microsomal CYP1A2. We found that inducertreatment of primary rat hepatocytes and HepG2cells indeed attenuates SA toxicity but the extent ofmodulation differed between the two cell types. Ki-netic studies in microsomes containing recombinantCYP1A1 or CYP1A2, as well as in human liver mi-crosomes, demonstrate that SA is a strong inhibitorbut is not metabolized by CYP1A enzymes. Tentativecomplex formed between SA and CYP1A may beresponsible for lower in vitro toxicity of the alkaloid.
2. Materials and methods
2.1. Chemicals
Microsomes containing recombinant CYP1A1or CYP1A2 each in the presence of recombinant

J. Vrba et al. / Toxicology Letters 151 (2004) 375–387 377
rabbit NADPH:CYP reductase, foetal calf serum,Williams’ medium E, Eagle’s Minimum EssentialMedium with non essential amino acids, l-glutamine,7-ethoxyresorufin, resorufin, NADPH, dithiothre-itol (DTT), l-cysteine, lipoic acid, reduced glu-tathione (GSH), 3-methylcholanthrene, Triton X-100,�-naphtoflavone, cytochrome c, and superoxide dis-mutase (SOD) were purchased from Sigma Chemicals(St. Louis, MO). 2,3,7,8-tetrachlorodibenzo-p-dioxinwas a kind gift from Dr. Miroslav Machala (VeterinaryResearch Institute, Brno, Czech Republic). All otherchemicals were of the highest grade commerciallyavailable.
2.2. Isolation of sanguinarine
The procedure described for isolation of sanguinar-ine and chelerythrine from Chelidonium majuswasapplied (Slavık and Slavıková, 1954). The QBA ex-tract from Macleya cordata(Papaveraceae), purchasedfrom Camas Technologies, Inc., Broomfield, USA, andcontaining sanguinarine and chelerythrine in the ratio3:1, (1.0 g) was dissolved in water (150 ml), made al-kaline with ammonia, extracted ten times with ether(20 ml portions), organic fractions were pooled, driedwith Na2CO3, evaporated and the evaporation residuedissolved in the mixture acetic acid–benzene (1:99)(10 ml) was applied onto an Al2O3 (activated by wash-ing with 5% HCl, drying and heating at 170 ◦C for 6 h)column (150 g) and eluted with a gradient of ethanol(1–6%) in the mixture acetic acid–benzene (1:99).Fractions containing sanguinarine were pooled, evap-orated, evaporation residues were dissolved in ben-zene and alkaloid chloride was precipitated by the ad-dition of several drops of hydrogen chloride in di-ethyl ether. After filtration we obtained 148 mg of san-guinarine chloride, m.p. 281–285 ◦C, purity by HPLC99.3%. Published m.p. for sanguinarine chloride is277–280 ◦C (Southon and Buckingham, 1989).
2.3. Cell culture and cell viability assay
Primary rat hepatocytes were isolated by two stepcollagenase perfusion according to a published pro-tocol (Moldeus et al., 1978). Following isolation, thecells were plated on collagen coated culture dishes us-ing cell density 2×105 cells/cm2. Williams’ medium Esupplemented with 2 mM l-glutamine, 10 �M strepto-
mycin, 100 U/ml penicillin, 350 nM insulin, and 1 �Mdexamethasone, enriched for plating with 5% foetalcalf serum (v/v) was used for plating. The medium wasexchanged for a serum-free one 3 h later and the cul-ture was allowed to stabilize for additional 24 h. Cul-tures were maintained at 37 ◦C in 5% CO2 (air:CO2,95:5) humidified incubator.
HepG2 cells (ECACC No. 85011430) were seededon 24-well dishes in density 2.5 × 104 cells/cm2 us-ing Eagle’s Minimum Essential Medium with non es-sential amino acids and further supplemented with1 mM pyruvate, 100 U/ml penicillin, 100 �g/ml strep-tomycin, 2 mM glutamine, and 10% foetal calf serum(v/v).
Both primary rat hepatocytes and HepG2 cells weretreated, following 24 h of stabilization, with TCDD(1 nM final) and BNF (25 �M final) for 72 h, and 3-MC(5 �M final) for 20 h. Culture medium was changedevery 24 h, regardless of treatment. Following inducertreatment the medium was exchanged for a serum-freeone in case of HepG2 cells and cells were treated for3 h with SA (1–100 �M final) or DMSO as vehicle co-ntrol. Lactate dehydrogenase (LDH) leakage into themedium and MTT assay were measured to assess cy-tosolic membrane damage and metabolic capability ofcells, respectively. Cells treated with 1.5% (v/v) TritonX-100 represented 100% LDH activity in the medium.Cells treated with DMSO only (vehicle control) rep-resented 100% metabolic activity in the MTT assay.
2.4. Human liver microsomes
Liver samples were obtained from multi-organdonors who met an accidental death. Tissue acquisi-tion protocol was in accordance with the requirementsissued by local ethical commission of the Czech Re-public. Small pieces of tissue were well homogenizedand microsomal fraction was prepared by differen-tial centrifugation according to a published protocol(Schenkman and Jansson, 1998). Microsomes werethen stored at −80 ◦C in small aliquots of 25 mgprotein/ml. Protein content was estimated using themethod of Lowry et al. (1951). Typical content oftotal CYP was 500 pmol/mg protein.
2.5. Differential spectrophotometry
Human liver microsomes (0.5 mg protein/ml) weresuspended in 2 ml of 50 mM potassium phosphate

378 J. Vrba et al. / Toxicology Letters 151 (2004) 375–387
buffer, pH 7.4. The suspension was divided in halfand pipetted into two quartz microcuvettes. Follow-ing a baseline recording 7-ethoxyresorufin (4 �Mfinal concentration) was added into the sample cu-vette and absorbance spectrum was recorded on Shi-madzu UV-1601 spectrophotometer in the range of360–460 nm. Sequential additions of SA (1–15 �Mfinal concentration) were made into the sample cu-vette and absorbance spectrum was recorded aftereach addition. To circumvent interference of theligands with CYP absorbance spectrum, additionalabsorbance spectra were also recorded in the absenceof microsomes using the same sequence of substanceadditions. The corresponding spectra obtained in thepresence or absence of microsomes were subtractedusing the spectrophotometer software. RecombinantCYP enzymes (10 pmol) were used in the microsomalsuspension, other additions and set up were the sameas for liver microsomes.
2.6. CYP1A activity (EROD) assay
Protocol of Chang and Waxman (1998) was uti-lized in its entirety for time course measurements toassess SA pre-incubation effect on the activity. Aque-ous stock solution of SA (1 mM) was prepared forall experiments involving CYP enzymes in order toavoid organic solvent effects on activity of these en-zymes. All pre-incubations of microsomes with SAin the presence or absence of other effectors, includ-ing lipoic acid, DTT, GSH, l-cysteine, and NADPH,were conducted in total volume of 200 �l for 2, 4, and6 min at 37 ◦C in a separate eppendorf, then addedto 1.8 ml of assay buffer, 0.1 M HEPES, pH 7.8, in athermostatted fluorescence cuvette. Sixty seconds afterthe start of measurement 7-ethoxyresorufin was addedto initiate the reaction. NADPH, if omitted from thepre-incubation mixture, was added to the assay cu-vette 5 s prior to 7-ethoxyresorufin addition. Inhibitionstudies utilized the same protocol only adjusted to fit96-well plate format where 0.5 pmol of recombinantCYP1A1 or CYP1A2, or 15 �g of total protein fromhuman liver microsomes were used. All other param-eters were the same except methanol which was usedto terminate the reaction after 5 min. LuminometerLS50B (Perkin-Elmer Corp., Norwalk, CT) equippedwith either thermostatted cuvette holder with continu-ous mixing (time course measurements) or well plate
reader accessory was the detection apparatus. Excita-tion wavelength was set to 530 nm and emission wave-length to 585 nm. Readings from known amounts ofauthentic resorufin metabolite were used to constructa calibration curve.
2.7. HPLC detection of possible SA metabolites
SA (10 �M final concentration) was incubatedwith human liver microsomes (300 �g total protein)or microsomes containing recombinant CYP1A1 or1A2 (10 pmol) in 0.1 M HEPES, pH 7.8, in the pres-ence of 2 mM NADPH for up to 150 min at 37 ◦Cunder constant shaking (final volume 0.5 ml). Toaccount for non-enzymatic interaction between SAand NADPH, they were incubated as above in theabsence of microsomes. Reaction was stopped byadding an equal volume of methanol and vortexed.Following centrifugation for 1 min at 12,000 × g,the samples were injected onto an HPLC column.Analysis was carried out on a Shimadzu Class VPHPLC system (Shimadzu Corp., Tokyo, Japan) us-ing 250/4 Purospher®STAR RP-18e column withguard column 4/4 containing the same sorbent(Merck, Darmstadt, Germany). The HPLC systemis equipped with SPD-M10Avp UV detector andRF10AXL fluorescence detector. Samples were elutedin isocratic mode (1 ml/min) with mobile phase con-sisting of 0.1 M phosphate/acetonitrile/triethylamine70/30/1 (v/v/v), pH was adjusted to 2.9 with 85%H3PO4. Elution was monitored at 285 nm (UV) and327 nm excitation–577 nm emission (fluorescence).Retention time for SA under these conditions is7.6 min.
2.8. NADPH:CYP reductase activity assay
Human liver microsomes (15 �g total protein/assay)were suspended in 0.3 M phosphate buffer, pH 7.5 con-taining cytochrome c and the reaction was initiatedby NADPH addition (200 �M final). Absorbance at550 nm, the absorption peak for reduced cytochromec, was monitored for 4 min at 30 s intervals on Shi-madzu UV1601 spectrophotometer equipped with athermostatted cuvette holder (Shimadzu GmbH, Vi-enna). The absorbance change per minute was used incalculating half-maximal concentration of SA (IC50)necessary for inhibition of NADPH:CYP reductase ac-

J. Vrba et al. / Toxicology Letters 151 (2004) 375–387 379
tivity. Activity detected in samples containing 10 U/mlof SOD ±SA were used as 100% inhibition becausethis concentration of SOD caused complete inhibitionof the reaction.
2.9. Statistical analyses
Data were analyzed by ANOVA, followed byposthoc Tukey test. Results are presented as mean ±S.D. A P value of <0.05 was considered to be statis-tically significant.
3. Results
3.1. Toxicity of SA in primary cultures of rathepatocytes and HepG2 cells treated with CYP1Ainducers
3-MC is one of popular and well-known CYP induc-ers used in studying these enzymes since the 1960’s.This fact and a distant similarity of SA to PAHs ledto the use of 3-MC in assessing possible role of CYPsin SA toxicity (Williams et al., 2000). In the con-text of affecting toxicity of other substances 3-MC,a toxic agent itself, may be involved in ways otherthan inducing CYP1A. Therefore, we decided to usetwo other CYP1A inducers, TCDD and BNF (Correia,1995), besides 3-MC in testing their effect on SAtoxicity in primary rat hepatocytes. Human hepatomaHepG2 cell line was included in the in vitro testingbecause of scarce availability of human liver for pri-mary hepatocyte cultures and because this cell linedisplays CYP1A1 induction and activity (Krusekopfet al., 1997).
Cell viability was assessed by two assays: LDHleakage into culture medium, which signifies in-tegrity of cellular membrane, and MTT conversioninto insoluble formazan stain, which demonstratesfunctional mitochondrial dehydrogenases, i.e. func-tional mitochondria. The two assays were used ascomplementary because loss of MTT dye conversionactivity cannot distinguish compromised cells fromdead cells and as such is not necessarily a sign of atoxic effect of substances under investigation. There-fore, only cell populations displaying an increasein LDH activity in culture medium and comparabledecrease in MTT dye conversion may be considered
intoxicated. In preliminary experiments we estab-lished the half-maximal concentration of SA (IC50)causing cell death to be 10 �M in both primary rathepatocytes and HepG2 cells. The two tests showthat toxicity of SA at 10 �M was significantly dimin-ished by prior inducer treatment in both cell typeswith the exception of BNF effect in HepG2. All threeinducers are about equally potent in diminishing SAtoxicity in primary rat hepatocytes as judged by bothtests (Fig. 2). The same is true, with BNF beingthe most potent, for inducer effect on SA toxicity inHepG2 cells when tested by LDH leakage (Fig. 2).In contrast, BNF appears to increase SA toxicity asassessed by MTT assay, whereas 3-MC and TCDDsignificantly decrease SA toxicity in HepG2 cells(Fig. 2). Inducer effect became insignificant whentested on SA concentrations of 50 �M and higher.Observed morphological changes in cells dependedon SA concentration (data not shown) and corre-sponded to those described by others (Ding et al.,2002).
When comparing the CYP1A induction capabilityof all three inducers by evaluating EROD activity inmicrosomes from treated HepG2 cells, the order ofpotency was TCDD > BNF > 3-MC (data not shown).Induction periods selected based on preliminary exper-iments were 72 h for TCDD and BNF, and only 20 hfor 3-MC. The shorter induction period for 3-MC wasselected due to the substance toxic effects on HepG2cells.
3.2. Inhibition of CYP1A activity by SA
Effect of all three inducers on SA in vitro toxic-ity confirmed the hypothesis that increased CYP1Aexpression is likely responsible. Fig. 3 shows type IIligand binding spectrum, characterized by broad peakat 425 nm and trough at 390 nm, in human liver mi-crosomes even in the presence of 7-ethoxyresorufin.Same behavior was observed for both recombinantCYP1A enzymes (data not shown). Moreover, in-teraction of SA with either CYP1A1 or CYP1A2can be demonstrated as inhibition of enzymaticactivity specific to these two enzymes. Indeed,our data show that SA inhibits 7-ethoxyresorufinde-ethylation catalyzed by both of the enzymes. Thisinhibition is non-competitive in case of CYP1A1(Fig. 4, panel A) and competitive in case of CYP1A2

380 J. Vrba et al. / Toxicology Letters 151 (2004) 375–387
Fig. 2. Effect of SA on cell membrane integrity and viability of primary rat hepatocytes or human hepatoma cell line HepG2. Cellstreated for 72 h with DMSO (vehicle control, UT), 25 �M BNF, and 1 nM TCDD or for 20 h with 5 �M 3-MC were incubated for3 h at 37 ◦C with 10 �M SA. (A) Lactate dehydrogenase leakage into the culture medium was estimated in primary rat hepatocytes(RAT) and HepG2 cells (HepG2). Cells treated with 1.5% (v/v) Triton X-100 served as 100% of LDH activity in the culture medium.(B) Conversion of MTT into water insoluble formazan dye was used as a marker for mitochondrial function for RAT and HepG2.Cells treated with DMSO only instead of SA served as 100% of control activity. Data are mean ± S.D. from triplicate measurements.Same results were obtained in two additional independent experiments. Statistically significant from appropriate control cells (UT)*P < 0.05.
(Fig. 4, panel B). When using human liver micro-somes, we found the inhibition to be competitive(Fig. 4, panel C). Moreover, the Ki calculated forSA inhibition is 2 �M and identical to both re-combinant CYP1A2 microsomal system and humanliver microsomes. This evidence suggests that SA
may, besides causing unspecific and perhaps irre-versible changes in the three-dimensional structureof CYP1A, be a substrate and be metabolized aspreviously hypothesized (Hakim et al., 1961). Be-cause CYP1A1 shows non-competitive type inhibi-tion by SA, implicating SA as an unlikely substrate

J. Vrba et al. / Toxicology Letters 151 (2004) 375–387 381
Fig. 3. Ligand binding spectra of SA in human liver microsomes. Differential absorbance spectra of 4 �M 7-ethoxyresorufin (lowertrace) and 10 �M SA in the presence of 4 �M 7-ethoxyresorufin (upper trace) are shown. Spectra were recorded on a dual beamspectrophotometer in the presence or absence of human liver microsomes (0.5 mg total protein/ml). Corresponding spectra were subtractedusing the spectrophotometer software.
of CYP1A1, and CYP1A2 is represented by humanliver microsomes, these were used in all subse-quent experiments relating to possible metabolismof SA.
3.3. Effect of pre-incubation time and NADPH on SAinhibition of CYP1A2 activity
To test the possibility of metabolism we initiallyprobed the effect of different pre-incubation times andthe presence of NADPH on SA inhibition. Identical ap-proach was used in evidencing the mechanism-basedinactivation of CYP1A2 by trans-resveratrol (Changet al., 2001). Our data show that the decrease inthe magnitude of EROD activity in human liver mi-crosomes is time- but not concentration-dependent(Fig. 5). We also tested whether the presence ofNADPH is required for SA inactivation of EROD ac-tivity. This is not so because the initial rate of ERODactivity, shown as two parallel fluorescence traces inFig. 6, was the same regardless of NADPH presencein the microsomal pre-incubation mixture prior to7-ethoxyresorufin addition. The difference in fluores-cence intensity between the two traces in Fig. 5, maybe associated with non-enzymatic reaction betweenSA and NADPH.
3.4. Detection of possible SA metabolite inmicrosomal fraction
While the above described experiments dealing withpre-incubation effect suggest that SA is not metabo-lized, the result is ambiguous. The simplest approachto verify metabolism of a substance is incubating itwith a full enzymatic system, in this case comprisingmicrosomes with access to sufficient NADPH as notto limit the reaction, for a longer period of time. Mi-crosomes may be of different source including thosewith recombinant enzymes; optimum incubation timemust be sought but 2 h is usually sufficient to detecta metabolite by HPLC. We were unable to detect apresence of any other substance besides SA following15, 30, 60, and 150 min incubation periods selectedbecause 5 min pre-incubation was sufficient for SAto inhibit CYP1A activity. Chromatograms in Fig. 7demonstrate no difference in elution pattern betweenSA incubated with NADPH alone or in the presence ofhuman liver microsomes even after 150 min of incu-bation. Peaks flanking the major SA peak, which is asingle sharp peak when the alkaloid is used alone, aremost likely due to non-enzymatic interaction of SAwith NADPH or impurities in the NADPH stock usedfor the assay because these peaks are absent from the
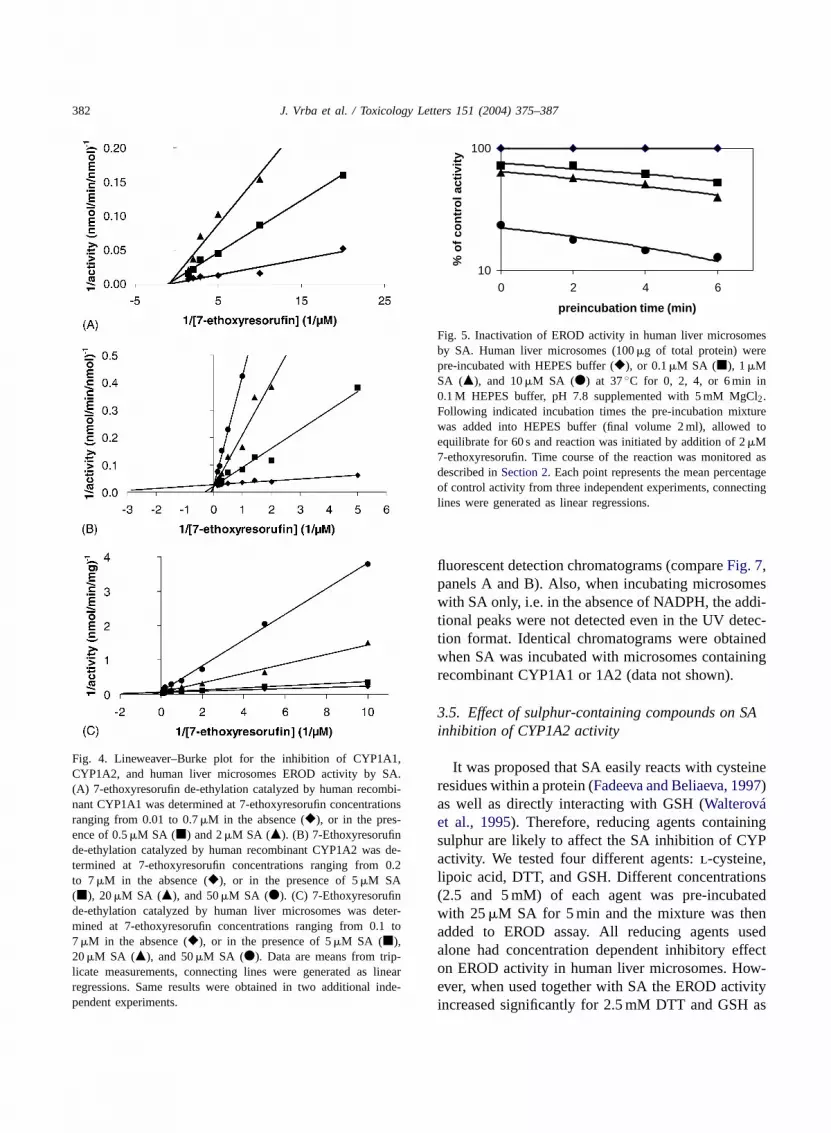
382 J. Vrba et al. / Toxicology Letters 151 (2004) 375–387
Fig. 4. Lineweaver–Burke plot for the inhibition of CYP1A1,CYP1A2, and human liver microsomes EROD activity by SA.(A) 7-ethoxyresorufin de-ethylation catalyzed by human recombi-nant CYP1A1 was determined at 7-ethoxyresorufin concentrationsranging from 0.01 to 0.7 �M in the absence (�), or in the pres-ence of 0.5 �M SA (�) and 2 �M SA (�). (B) 7-Ethoxyresorufinde-ethylation catalyzed by human recombinant CYP1A2 was de-termined at 7-ethoxyresorufin concentrations ranging from 0.2to 7 �M in the absence (�), or in the presence of 5 �M SA(�), 20 �M SA (�), and 50 �M SA (�). (C) 7-Ethoxyresorufinde-ethylation catalyzed by human liver microsomes was deter-mined at 7-ethoxyresorufin concentrations ranging from 0.1 to7 �M in the absence (�), or in the presence of 5 �M SA (�),20 �M SA (�), and 50 �M SA (�). Data are means from trip-licate measurements, connecting lines were generated as linearregressions. Same results were obtained in two additional inde-pendent experiments.
10
100
0 2 4 6
preincubation time (min)
% o
f co
ntr
ol a
ctiv
ity
Fig. 5. Inactivation of EROD activity in human liver microsomesby SA. Human liver microsomes (100 �g of total protein) werepre-incubated with HEPES buffer (�), or 0.1 �M SA (�), 1 �MSA (�), and 10 �M SA (�) at 37 ◦C for 0, 2, 4, or 6 min in0.1 M HEPES buffer, pH 7.8 supplemented with 5 mM MgCl2.Following indicated incubation times the pre-incubation mixturewas added into HEPES buffer (final volume 2 ml), allowed toequilibrate for 60 s and reaction was initiated by addition of 2 �M7-ethoxyresorufin. Time course of the reaction was monitored asdescribed in Section 2. Each point represents the mean percentageof control activity from three independent experiments, connectinglines were generated as linear regressions.
fluorescent detection chromatograms (compare Fig. 7,panels A and B). Also, when incubating microsomeswith SA only, i.e. in the absence of NADPH, the addi-tional peaks were not detected even in the UV detec-tion format. Identical chromatograms were obtainedwhen SA was incubated with microsomes containingrecombinant CYP1A1 or 1A2 (data not shown).
3.5. Effect of sulphur-containing compounds on SAinhibition of CYP1A2 activity
It was proposed that SA easily reacts with cysteineresidues within a protein (Fadeeva and Beliaeva, 1997)as well as directly interacting with GSH (Walterováet al., 1995). Therefore, reducing agents containingsulphur are likely to affect the SA inhibition of CYPactivity. We tested four different agents: l-cysteine,lipoic acid, DTT, and GSH. Different concentrations(2.5 and 5 mM) of each agent was pre-incubatedwith 25 �M SA for 5 min and the mixture was thenadded to EROD assay. All reducing agents usedalone had concentration dependent inhibitory effecton EROD activity in human liver microsomes. How-ever, when used together with SA the EROD activityincreased significantly for 2.5 mM DTT and GSH as

J. Vrba et al. / Toxicology Letters 151 (2004) 375–387 383
88
90
92
94
96
98
100
50 100 150 200 250 300time (s)
flu
ore
scen
ce (a
.u.)
+NADPH
-NADPH
Fig. 6. Effect of NADPH presence in the pre-incubation mixture. Actual fluorescence traces of the initial rate of 7-ethoxyresorufinde-ethylation are shown. Human liver microsomes (100 �g of total protein) were pre-incubated with 1 �M SA at 37 ◦C for 4 min in 0.1 MHEPES buffer, pH 7.8 supplemented with 5 mM MgCl2 in the presence (+NADPH) or absence (−NADPH) of the co-factor. Followingincubation the mixture was added into HEPES buffer (final volume 2 ml), allowed to equilibrate for 60 s and reaction was initiated byaddition of 2 �M 7-ethoxyresorufin. In case of NADPH absent in the pre-incubation, it was added 5 s prior to 7-ethoxyresorufin addition.Time course of the reaction was monitored as described in Section 2. Same result was obtained for pre-incubation times ranging from 0to 8 min.
compared to SA alone (Table 1). The data supportcrucial role of thiol groups within the active enzymecomplex as well as possible interaction of SA withthese.
Table 1Effect of sulphur-containing agents on SA inhibition of CYP1A2activity in human liver microsomes
Pre-incubation mixture EROD activity(pmol/min/mg protein)
None (control) 25 ± 2∗+SA 8 ± 1+2.5 mM DTT 20 ± 2∗+5 mM DTT 15 ± 2∗+2.5 mM GSH 22 ± 2∗+5 mM GSH 16 ± 1∗+SA + 2.5 mM DTT 16 ± 2∗+SA + 5 mM DTT 10 ± 1+SA + 2.5 mM GSH 16 ± 2∗+SA + 5 mM GSH 10 ± 1
l-Cysteine, DTT, GSH, or lipoic acid was added to thepre-incubation mixture of human liver microsomes and SA, andincubated at 37 ◦C for 5 min. After adding this mixture into athermostatted fluorescence cuvette to reach final volume of 2 ml,EROD activity was determined from the initial rate of fluores-cence increase as described in Section 2. Data are mean ± S.D.
from three independent experiments.∗ Significantly different from SA alone (P < 0.05).
3.6. Effect of SA on NADPH:CYP reductase
Perhaps often understood as a one-enzyme-gig,a reaction catalyzed by a CYP will generally pro-ceed with great difficulty, if at all, in the absenceof NADPH:CYP reductase and a steady supply ofNADPH. The NADPH binding site of NADPH:CYPreductase contains two important cysteine residues(Nisimoto and Shibata, 1981) which are an idealattack site for SA as a consequence of reduc-ing agent effects on SA presented here and else-where (Dršata et al., 1996; Fadeeva and Beliaeva,1997; Vespalec et al., 2003). Therefore, we testeddirectly the effect of SA on NADPH:CYP re-ductase activity assessed by cytochrome c reduc-tion.
SA inhibits NADPH:CYP reductase activity withIC50 of 21.8 �M (Fig. 8). This is close to the IC50of 16 �M SA obtained for EROD activity inhibitionin human liver microsomes. Other CYP isoenzymesmay therefore be affected as well. However, we foundthat CYP2C9 activity, diclofenac 4′-hydroxylation,was inhibited by SA with IC50 of 144 �M, i.e. almostten-fold higher than that for EROD inhibition (datanot shown).

384 J. Vrba et al. / Toxicology Letters 151 (2004) 375–387
Fig. 7. Representative HPLC chromatograms of SA metabolism. SA was incubated with excess NADPH in the presence (lower trace) orabsence (upper trace) of human liver microsomes for 150 min in 0.1 M HEPES buffer pH 7.8 at 37 ◦C. Following incubation the reactionwas stopped by addition of equal volume of methanol. Samples were centrifuged and loaded onto HPLC column. Elution conditions aredescribed in detail in Section 2. Panel A: UV detection at 285 nm; Panel B: fluorescent detection with ιex = 327 nm and ιem = 577 nm.

J. Vrba et al. / Toxicology Letters 151 (2004) 375–387 385
Fig. 8. Concentration-dependent inhibition of NADPH:CYP reduc-tase by SA. Activity of NADPH:CYP reductase in human livermicrosomes was monitored spectrophotometrically as reduction ofcytochrome c in the presence of NADPH and varying concen-trations of SA. Control samples also contained SOD (100 U/ml).Data are expressed as mean ± S.D. from three independent mea-surements. Activity detected in samples with only NADPH presentwere used as 0% inhibition, those containing SOD were used as100% inhibition.
4. Discussion
Knowledge of a substance fate within an organismis a prerequisite of its conscientious use as a medica-ment. While overuse is harmful with any substance,the borderline between positive and negative effectsis narrow for many. In such cases knowing the mech-anism of elimination is often of grave importance.Metabolism of SA in man has not been fully under-stood. Because of use of the alkaloid in medical prepa-rations and its discussed toxicity, metabolism of SAneeds to be elucidated.
Toxic effects of SA are ascribed to the alkaloid me-diated cell glutathione depletion accompanied by anincrease in lipid peroxidation (Das and Khanna, 1997).In this work we present evidence of a non-metabolicinteraction of SA with CYP1A enzymes and possi-bly NADPH:CYP reductase which may account forattenuated toxicity of the alkaloid. This is supportedby: (i) non-mechanism based inhibition of nativeCYP1A2; (ii) differing modes of inhibition of re-combinant CYP1A1 and CYP1A2 enzyme activity;(iii) decreased inhibition in the presence of reducingagents; and (iv) inhibition of NADPH:CYP reductaseactivity. Distinguishing between direct effect on CYPor NADPH:CYP reductase when monitoring CYP ac-
tivity is difficult. If the IC50 recorded for CYP activityand NADPH:CYP reductase were the same then SAwould act on the latter because NADPH:CYP reduc-tase is considered the limiting step in CYP-mediatedreactions. We observed marked differences in SA ef-fects on different CYP enzymes suggesting the alka-loid interacts with CYPs rather than the NADPH:CYPreductase.
Interaction of SA with CYP(s) is known from theliterature. It is therefore surprising that investiga-tions into the mechanism of the interaction have notbeen conducted to date. One of the reasons may bethe elusiveness of an SA metabolite. While found ingrazing animals (Hakim et al., 1961) and in urineor feces of rats after 72 h (Tandon et al., 1992),benzo[c]acridine was not detected in blood or urine ofhumans (Yi-Jong et al., 1990). Speculative pathwayfor SA metabolic clearance has been put forward andinvolves N-demethylation as the first step (Das andKhanna, 1997). However, no microsomal demethy-lase activity was detected in case of SA (Peeples andDalvi, 1982) suggesting routes other than CYP en-zymes for benzo[c]acridine formation. Still the pres-ence of a CYP-dependent metabolite was implicatedin SA genotoxicity study (Stiborová et al., 2002). Wefailed to detect substances other then SA resultingfrom enzyme-catalyzed reaction following incuba-tion of the alkaloid with human liver microsomes(Fig. 7). However, we observed a minute decreasein the amount of SA present in these samples whichmay be ascribed to loss of SA during deproteinizationprior to HPLC, i.e. SA to protein complexation (datanot shown). Indeed, SA forms non-covalent com-plex with albumins (Barták et al., 2003) and likelywith other proteins. Cysteine residues (Fadeeva andBeliaeva, 1997) or mercapto groups (Barták et al.,2003; Vespalec et al., 2003) are likely targets whichis supported by data presented here.
The idea that SA would attack the highly conservedproximal cysteine ligand of the heme present in CYPenzymes seems unlikely. Rather it would coordinateto ferric heme as suggested from ‘ type II’ bindingspectra (Peeples and Dalvi, 1982). Then dependingon the distal pocket environment, the presence of SAmay cause inhibition of a different type accountingfor the difference between inhibition of CYP1A1 andCYP1A2 (Fig. 4). Part of this phenomenon is un-doubtedly very different affinity of these two CYPs

386 J. Vrba et al. / Toxicology Letters 151 (2004) 375–387
towards 7-ethoxyresorufin: Km = 0.017 and 1.7 �Mfor CYP1A1 and CYP1A2, respectively. Followingthe same thought, variable Ki for different CYPsare to be expected based on their substrate cavitydimensions and primary sequence differences. For ex-ample CYP2E1, which can accommodate only smallmolecules, may not be inhibited by SA. This simpleidea of a fit versus no-fit of SA into a substrate cavitydoes not hold water for two reasons: (i) suitable bind-ing sites for SA, containing cysteine residues or not,located elsewhere on the protein may be attacked aswell causing conformational changes resulting in in-hibition; and (ii) inhibition of NADPH:CYP reductaseactivity by SA.
Considering that SA interacts with CYP1A en-zymes but is not metabolized by them, what can bethe reason for lower in vitro toxicity of the alka-loid? We postulate that SA binds to CYP enzymesas demonstrated by ‘ type II’ binding spectra, similar-ity of SA to PAHs may constitute higher affinity forCYP1As than for other CYP enzymes, e.g. CYP2C9.SA binds such that it bars electron transfer fromNADPH:CYP reductase to CYPs or other acceptorsas demonstrated by inhibition of EROD activity andinhibition of NADPH:CYP reductase activity. These‘diverted’ electrons likely cause damage to CYPsand/or NADPH:CYP reductase triggering their pro-teolysis. This is supported by the reported decreasein total CYP content (Ulrichová et al., 1996). Higherproteolytic activity associated with membrane span-ning proteins may activate other cellular mechanismsinvolved in membrane repair thereby preventing theSA-mediated membrane damage. The fate of SAwithin a cell following protease-mediated removalof its target protein is unclear but the alkaloid mayaccumulate in lysosomes and then be slowly excretedas shown in animal studies (Tandon et al., 1992). Wetherefore conclude that the basis for protective effectof CYP1A inducer treatment against SA toxicity isthe increased CYP1A expression.
Acknowledgements
This work was supported by the Ministry of Educa-tion of the Czech Republic grant no. MSM 151100003and by the Grant Agency of the Czech Republic grant303/01/P085.
References
Anzenbacher, P., Anzenbacherová, E., 2001. Cytochromes P450and metabolism of xenobiotics. Cell. Mol. Life Sci. 58, 737–747.
Babich, H., Zuckerbraun, H.L., Barber, I.B., Babich, S.B.,Borenfreund, E., 1996. Cytotoxicity of sanguinarine chlorideto cultured human cells from oral tissue. Pharmacol. Toxicol.78, 397–403.
Barták, P., Šimánek, V., Vlcková, M., Ulrichová, J., Vespalec,R., 2003. Interactions of sanguinarine and chelerythrine withmolecules containing mercapto group. J. Phys. Org. Chem. 16,803–810.
Chang, T.K.H., Chen, J., Lee, W.B.K., 2001. Differential inhibitionand inactivation of human CYP1 enzymes by trans-resveratrol:evidence for mechanism-based inactivation of CYP1A2. J.Pharm. Exp. Ther. 299, 874–882.
Chang, T.K.H., Waxman, D.J., 1998. Enzymatic analysis ofcDNA-expressed human CYP1A1, CYP1A2, and CYP1B1with 7-ethoxyresorufin as substrate. In: Phillips, I.R., Shephard,E.A. (Eds.), Methods in Molecular Biology. Cytochrome P450Protocols, vol. 107. Humana Press, Totowa, pp. 103–109.
Correia, M.A., 1995. Rat and human liver cytochromes P450.In: de Montellano, P.R.O. (Ed.), Cytochrome P450: Structure,Mechanism, and Biochemistry. Plenum Press, New York,pp. 607–630.
Das, M., Khanna, S.K., 1997. Clinicoepidemiological,toxicological, and safety evaluation studies on argemone oil.Crit. Rev. Toxicol. 27, 273–297.
Ding, Z., Tang, S.-C., Weerasinghe, P., Yang, X., Pater, A.,Liepins, A., 2002. The alkaloid sanguinarine is effective againstmultidrug resistance in human cervical cells via bimodal celldeath. Biochem. Pharmacol. 63, 1415–1421.
Dršata, J., Ulrichová, J., Walterová, D., 1996. Sanguinarineand chelerythrine as inhibitors of aromatic amino aciddecarboxylase. J. Enzyme Inhib. 10, 231–237.
Fadeeva, M.D., Beliaeva, T.N., 1997. Sanguinarine and ellipticinecytotoxic alkaloids isolated from well-known antitumor plants.Intracellular targets of their action. Tsitologia 37, 181–208.
Fent, K., 2001. Fish cell lines as versatile tools inecotoxicology: assessment of cytotoxicity, cytochrome P4501A induction potential and estrogenic activity of chemicals andenvironmental samples. Toxicol. In Vitro 15, 477–488.
Frankos, V.H., Brusick, D.J., Johnson, E.M., Maibach, H.I., Munro,I., Squire, R.A., Weil, C.S., 1990. Safety of sanguinaria extractas used in commercial toothpaste and oral rinse product. J.Can. Dent. Assoc. 56 (Suppl. 1), 41–47.
Hakim, S.A.E., Mijovic, V., Walker, J., 1961. Experimentaltransmission of sanguinarine in milk: detection of a metabolicproduct. Nature 189, 201–204.
Ioannides, C., Parke, D.V., 1990. The cytochrome P450 I genefamily of microsomal hemoproteins and their role in themetabolic activation of chemicals. Drug Metab. Rev. 22, 1–85.
Krusekopf, S., Kleeberg, U., Hildebrandt, A.G., Ruckpaul, K.,1997. Effects of benzimidazole derivatives on cytochrome P4501A1 expression in a human hepatoma cell line. Xenobiotica27, 1–9.

J. Vrba et al. / Toxicology Letters 151 (2004) 375–387 387
Kuftinec, M.M., Mueller-Joseph, L.J., Kopczyk, R.A., 1990.Sanguinaria toothpaste and oral rinse regimen clinical efficacyin short- and long-term trials. J. Can. Dent. Assoc. 56 (Suppl.),31–33.
Lowry, O.H., Rosebrough, N.J., Farr, A.L., Randall, R.J., 1951.Protein measurement with the Folin phenol reagent. J. Biol.Chem. 193, 265–275.
Moldeus, P., Hogberg, J., Orrenius, S., 1978. Isolation and use ofliver cells. Methods Enzymol. 52, 60–71.
Nisimoto, Y., Shibata, Y., 1981. Location of functional –SHgroups in NADPH cytochrome P450 reductase from rabbitliver microsomes. Biochim. Biophys. Acta 662, 291–299.
Peeples, A., Dalvi, R.R., 1982. Toxic alkaloids and their interactionwith microsomal cytochrome P-450 in vitro. J. Appl. Toxicol.2, 300–302.
Schenkman, J.B., Jansson I., 1998. Isolation and purificationof constitutive forms of microsomal cytochrome P450. In:Phillips, I.R., Shephard, E.A. (Eds.), Methods in MolecularBiology, Cytochrome P450 Protocols, vol. 107. Humana Press,Totowa, pp. 55–67.
Schrenk, D., 1998. Impact of dioxin-type induction ofdrug-metabolizing enzymes on the metabolism of endo- andxenobiotics. Biochem. Pharmacol. 55, 1155–1162.
Singh, N.P., Anuradha, S., Dhanwal, D.K., Singh, K., Prakash, A.,Madan, K., Agarwal, S.K., 2000. Epidemic dropsy—a clinicalstudy of the Delhi outbreak. J. Assoc. Physicians India 48,877–880.
Slavık, J., Slavıková, L., 1954. Alkaloidy rostlin makovitých(Papaveraceae) II. Delenı chelerythrinu a sanguinarinu a nálezdvou nových alkaloidu ve vlaštovicnıku (Chelidonium majusL.). Chem. Listy 48, 1382–1386.
Southon, I.W., Buckingham, J., 1989. Dictionary of Alkaloids.Chapman and Hall, London, p. 940.
Stiborová, M., Šimánek, V., Frei, E., Hobza, P., Ulrichová,J., 2002. DNA adduct formation from quaternary benzo-[c]phenanthridine alkaloids sanguinarine and chelerythrineas revealed by the 32P-postlabeling technique. Chem.-Biol.Interact. 140, 231–242.
Tandon, S., Das, M., Khanna, S.K., 1992. Biometabolic eliminationand organ retention profile of argemone alkaloid, sanguinarine,in rats and guinea pigs. Drug Metab. Dispos. 21, 194–197.
Ulrichová, J., Walterová, D., Vavrecková, C., Kamarád, V.,Šimánek, V., 1996. Cytotoxicity of benzo[c]phenanthridiniumalkaloids in isolated rat hepatocytes. Phytother. Res. 10, 220–223.
Vespalec, R., Barták, P., Šimánek, V., Vlcková, M., 2003.Electrophoretic investigation of interactions of sanguinarineand chelerythrine with molecules containing mercapto group.J. Chromatogr. B 797, 357–366.
Walterová, D., Ulrichová, J., Válka, I., Vicar, J., Vavrecková, C.,Táborská, E., Harkrader, R.J., Meyer, D.L., Cerná, H., Šimánek,V., 1995. Benzo[c]phenanthridine alkaloids sanguinarine andchelerythrine: biological activities and dental care applications.Acta Univ. Palacki. Olomuc. Fac. Med. 139, 7–16.
Williams, M.K., Dalvi, S., Dalvi, R.R., 2000. Influence of3-methylcholanthrene pretreatment on sanguinarine toxicity inmice. Vet. Hum. Toxicol. 42, 196–198.
Yi-Jong, L., McPherson, B.P., Smith, N.L., Taylor, C.D., Rizvi,P.Y., De Salva, S.J., 1990. The absence of benzo[c]acridineformation in in-vitro and in-vivo experiments with sanguinarineand Sanguinaria extract. FASEB J 4, A754, Abstract2827.