intracellular calcium channels and their modulators
TRANSCRIPT

Author p
roof
Review
2003 © Ashley Publications Ltd ISSN 1354-3776 1
Ashley Publicationswww.ashley-pub.com
1. Introduction
2. Comparison of the ryanodine
receptor and the inositol-1,4,5-
triphosphate-receptor
3. Ryanodine receptor
4. Inositol-1,4,5-triphosphate-
receptor
5. Patent review of intracellular
Ca2+ channels
6. Expert opinion
Monthly Focus: Cardiovascular, Renal, Endocrine & Metabolic
Intracellular calcium channels and their modulatorsAndrei A KochegarovDepartment of Neurology, University of California Los Angeles, 5524B GONDA Center, 695 Charles E Young dr. S., UCLA, Los Angeles, CA 90095, USA
The ryanodine receptor (RyR) is an essential element in excitation–contrac-tion coupling. Ca2+ release from the sarcoplasmic reticulum (SR) is requiredfor skeletal and heart muscle contraction. The inositol-1,4,5-triphosphate-receptor (IP3R) plays an important function in the signal transduction ofmany hormones, cytokines, growth factors and antigens, and in fertilisation.Modulators of intracellular calcium channels are used for the treatment ofmalignant hyperthermia associated with abnormal Ca2+ transport and may beapplied in the treatment of cardiovascular and neurodegenerative disorders.
Keywords: inositol 1,4,5-triphosphate (IP3), intracellular calcium channel (ICC), ryanodine, ryanodine receptor (RyR), inositol 1,4,5-triphosphate receptor (IP3R)
Expert Opin. Ther. Patents (2003) 13(6):627-662
1. Introduction
Ca2+ influx and its release into the cytosol from intracellular stores stimulates acto-myosin contraction in muscle cells. Upon relaxation, Ca2+ ions are pumped out ofthe cytosol by plasma membrane Ca2+ ATPases (PMCAs) and pumped into intracel-lular compartments by sarcoplasmic/endoplasmic Ca2+ ATPases (SERCAs), whereCa2+ is stored and released upon necessity. The German physiologist, Lewis V Heil-brunn, believed that calcium ions played a ubiquitous role in cell signalling. In1947, in an experiment carried out with Dr F Wiercinski, it was demonstrated thatthe injection of Ca2+ ions into muscle produced muscle contraction [1]. He proposedthat Ca2+ influx, in response to an action potential, produces muscle contraction.However, the eminent English physiologist and founder of muscle physiology,AV Hill, estimated the rate of Ca2+ diffusion in muscle cytosol. According to Hill'sestimation the diffusion of a putative activator (i.e., Ca2+) from the surface to theinterior of a muscle fibre is greater than the rate of skeletal muscle contraction,which happens within a few milliseconds. Indeed, if Ca2+ diffused from the extracel-lular fluid, one would expect, in such a case, that the thinnest muscles would be thefastest muscles. This was known not to be the case and the discrepancy was termedHill’s paradox. Hill went on to predict that the activator of muscle should comefrom an internal source. The role of Ca2+ in muscle contraction was evident butowing to Hill’s authority in muscle physiology this discovery was buried in oblivionuntil the 1960s. Striated muscles are known to be penetrated by transverse tubules(TT) that are localised in the vicinity of the contractile filaments, a fact that did notrecieve attention by physiologists at the time. In actual fact, TT do not accumulateand release Ca2+ but rather deliver a depolarisation signal from the membraneperiphery to the vicinity of the actomyosin machinery where TTs are coupled withthe net of the sarcoplasmic reticulum (SR). In 1902, Emilio Veratti described anelaborate filigree of tubules surrounding each sarcomere (the contractile unit ofmuscle) in striated skeletal muscle tissue. Veratti could not ascribe any function tothis network and his discovery was forgotten until 1955 when Stanley Bennett andKeith Porter rediscovered Veratti’s system of tubules with the high power resolutionof an electron microscope. The Veratti system consists of two separate but

Author p
roof
Intracellular calcium channels and their modulators
2 Expert Opin. Ther. Patents (2003) 13(6)
functionally bound systems: the SR, a network of cisterns andtubules surrounding each sarcomere, and TT, a web of deepinvaginations of the cell membrane. TT of skeletal muscleform direct contacts with terminal cisterns of the SR, theresulting junctions being termed triada. In the 1960s, Dr LeePeachey showed that TT are a key element in excitation–con-traction (EC)-coupling by functioning to deliver the actionpotential into the depth of the muscle fibre [2]. In 1961, theGerman physiologist Hasselbach et al., and later in 1963 bythe Japanese physiologist Ebashi et al., provided experimentalproof for the active transport of Ca2+ into sarcoplasmic micro-somes [3,4]. In the 1960s this discovery put forward the ideathat Ca2+ release from the SR is essential for muscle contrac-tion [5-8]. It was shown that the electric stimulation of striatedmuscle produced Ca2+ release from the SR. At the same time,Katz and Ebashi showed that the interaction of Ca2+ with theactomyosin and troponin complex controls muscle contrac-tion [9,10]. The idea that Ca2+-release in muscles was responsi-ble for their contraction only became broadly accepted in1970 – 1980 and in that time the first mention and descrip-tion of an intracellular Ca2+-channel (ICC) in SR appeared inthe literature [11,12]. ICCs were ascribed by Franzini–Arm-strong to the junctions of SR with TT [13], where specialisedspanning proteins termed ‘feet’ (also junctional feet, pillars,bridges or spanning proteins) have been identifed.
Two types of intracellular calcium channel were described:the ryanodine receptor (RyR), abundant in muscle, and theinositol-1,3-4-triphosphate receptor (IP3R), located in brain.The heterogeneity of the endoplasmic reticulum (ER) withrespect to calcium channels was first recognised by Volpeet al., who proposed that in non-muscle cells, ER bearingIP3R represents discrete organelles with different immuno-chemical markers [14,15]. This calcium pool was termed calcio-some. To date, this compartment is considered to be aspecialised subcompartment of the ER as opposed to a dis-crete organelle. In some cell types, IP3R and RyR pools sharethe same compartments of ER whereas in others cell typesthey may be disconnected.
In 1957, Porter and Palade proposed that the ER in non-muscle tissues is the functional equivalent to SR in muscles.Almost all intracellular organelles, such as the ER/SR, Golgi,nuclear envelope (which is believed to be a continuum of theER), lysosomes and mitochondria have developed mechanismsto accumulate and release Ca2+. In mitochondria, however, themechanism of Ca2+ transport is quite different; influx of Ca2+
ions is through unidentified mitochondrial channels and isdriven by an electrical potential, and Ca2+ ions are pumped outby the Na+/Ca2+-exchanger. Ca2+ accumulation in intracellularorganelles plays an important role in the regulation of proteinsynthesis, transcription, protein sorting and protease activity inlysosomes and activity of oxidative phosphorylation in mito-chondria. Accordingly, recent estimations of the concentrationof free Ca2+ in the ER varies from the low micromolar range[16] up to the millimolar range [17,18]. It is thought that Ca2+
binds and co-precipitates with phosphate and oxalate in ER to
form insoluble deposits [19]. Co-transport of negativelycharged phosphate and oxalate facilitates Ca2+ transport byCa2+-ATPases, which prevent positive overcharging of the ER[20]. Recent findings suggest that phosphate enters throughspecialised phosphate ion channels in SR [21]. The lumen ofthe SR contains a low-affinity, high-capacity Ca2+ binding pro-tein, calsequestrin, which also increases the Ca2+-bufferingcapacity of the SR. Total (bound and free) calcium level in cel-lular compartments such as SR/ER most likely ranges from afew to 10 mM concentrations.
Many non-excitable cells display oscillations in cytosolicCa2+ levels resulting from the periodic release of Ca2+ fromintracellular reservoirs. Frequencies are varied, but most oscil-lations have periods ranging from a few seconds in animal cells to1 – 2 min in Physarum polycephalum. The most classicalexample of Ca2+ oscillations is in the Xenopus leavis oocyte,whereby they are triggered by contact of sperm with oocyte.Some stimuli, such as hormones, cytokines and antigens bindspecific receptors on the cell membrane and via second mes-sengers, generally IP3 and cADP-ribose (cADPR), activateCa2+ oscillations.
It was speculated that Ca2+-oscillations might encode spe-cial information. For example, the frequency and amplitudeof the calcium transients are crucial for proper egg activationand embryonic development. Until now, little was under-stood as to how these signals were deciphered. Ca2+-oscilla-tions in the fertilised oocyte may govern the pattern ofcellular split. In the slime mould, Physarum polycephalum,oscillations of [Ca2+]i control oscillations of contractile activ-ity, providing a shuttle streaming of protoplasm and in turn,culminating in the motion of this huge multinuclear cell. InPhysarum, Ca2+ oscillations persist continuously and theircontinuance does not depend on extracellular stimuli. TheRussian scientist, Dr Sophia Beylina, predicted thatPhysarum would become an interesting model for the studyof calcium and contractile oscillations. Proposals have beenmade suggesting that oscillations depend on the biphasiccharacteristic of RyR and IP3R. When [Ca2+]i is low thesechannels are closed but a small leakage persists that increases[Ca2+]i. Control of this leakage is important to the period ofoscillations since it determines the time at which [Ca2+]ibecomes high enough to activate Ca2+-release channels. Thispremise led to the idea for the existence of hypotheticalCa2+-channels that function to regulate Ca2+ leakage andthat play a pacemaker role in Ca2+ oscillations.
2. Comparison of the ryanodine receptor and the inositol-1,4,5-triphosphate-receptor
RyR and IP3R are tetramers consisting of four subunits. IP3Rhas 6 transmembrane domains (TM) in each subunit (a pore-loop exists between the 5th and 6th TM) and thereforebelongs to the 6 TM, P-superfamily as characterised by theprototype Shaker K+-channel and transient receptor potential(TRP), but does not share appreciable sequence similarity.

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 3
Estimation of the number of TM domains in RyR varies from4 to 12 [22,23]. Based on sequence analysis, a 6 TM modelresembling the IP3R topology was proposed (Figure 1) [24].The most recent data, based on recombinant protein (RyR/epidermal growth factor receptor (EGFR)) technique to markparticular regions of RyR, supported the existence of 8 TM: 6reliable TM and 2 tentative TM in the N-terminal region ofthe molecule [25]. The protein fragment corresponding to thetwo tentative TM do not appear to be critical for Ca2+ releasechannel function. Furthermore, deletion of the amino acidscorresponding to the hairpin loop (4274 – 4535) did not dis-rupt channel function, as measured in terms of [3H]-ryanod-ine binding and caffeine-induced Ca2+ release, thereforeimplying that this component does not form either the core ofthe channel conduction pore or a vital regulatory domain [25].The study confirmed the existence of a selectivity filterbetween M8 and M10 (between M5 and M6 in Figure 1),which itself is not a real transmembrane helix but a regionimmersed into the membrane. Therefore, the structure andtopology of RyR resembles, by way of its general features, thestructure of 6 TM cationic channels. RyR and IP3R containthe similar stretch of amino acids, GIGD and GVGD, in theP-loop that may be homologous to the sequence in K+ andvoltage-gated Ca2+-channels proposed to function as a selec-tivity filter [26]. Two Asp residues in this sequence were sug-gested to form Ca2+ binding sites.
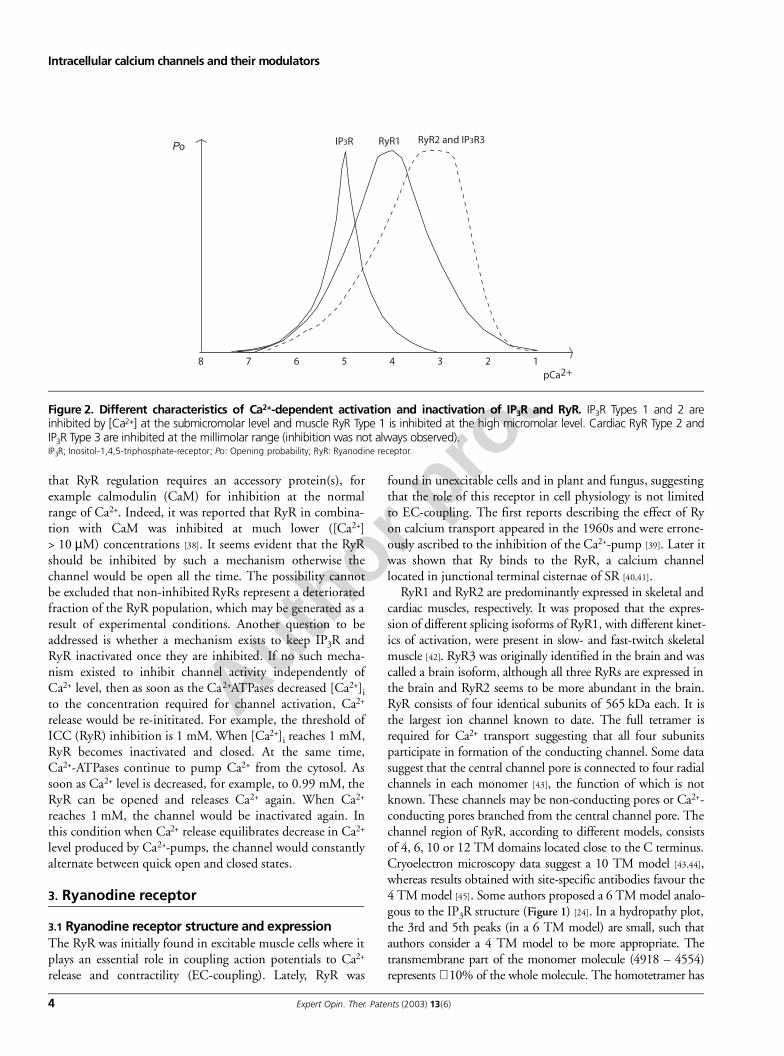
It was speculated that some of the similar sequences in RyRand IP3R confer the ability to interact with the same com-pounds, however, such compounds do not have the samephysiological effect. For example, caffeine has an activatingeffect on RyR but elicits a weak inhibitory effect on IP3R [27];the IP3R inhibitor, heparin, is a weak activator of RyR [28-30];
ryanodine (Ry), a modulator of RyR slightly activates IP3R[29]. Both receptors have a biphasic mode of regulation bycytoplasmic [Ca2+], so-called a bell-shaped dependencewhereby an increase in [Ca2+] activates RyR and IP3R, butupon reaching the threshold of activation further increases in[Ca2+] inhibit receptor function (Figure 2). The term‘Ca2+-induced Ca2+ release’ (CICR) was introduced by Fabi-ato in 1970s [31-33]. He found that a slight Ca2+ pulse pro-duced a larger Ca2+ response. The phenomenon of CICR isnot limited to muscle cells. In experiments with the slimemould Physarum polycephalum, CICR can be elicited by anystimuli producing [Ca2+]i elevation [34,35]. Today, it is probablethat only a few scientists remember that chlorotetracycline(CTC) can be used as a Ca2+ indicator. Fabiato (in the 1970s)used CTC to monitor Ca2+ release from intracellular Ca2+
stores because at that time penetrable Ca2+ indicators, such asfura-2 AM, were not yet available; their invention by Tsienwas not until the 1980s. CTC is still used however, in thestudy of Ca2+ transport in plants and fungus where ether dyesdo not penetrate the cell membrane, or have failed to accumu-late inside cells.
The IP3R is inhibited by [Ca2+] at the submicromolarlevel, whereas RyR is inhibited by [Ca2+] in the hundredmicromolar [36] to millimolar range (Figure 2) [37]. Someauthors were unable to demonstrate Ca2+-dependent inhibi-tion in slow-twitch muscles at all, or inhibition was observedat very high (millimolar) Ca2+ concentrations [37]. It seemsunlikely that such high [Ca2+]i may be in the normal physio-logical range but it may reflect concentrations present inabnormal conditions. One idea is that during Ca2+ releasethere is a local ‘cloud’ of millimolar [Ca2+] in the vicinity ofRyR, which is enough to inhibit RyR. An alternative idea is
FKBP
1 2 3 4 5 6
Imp
CaM CaM CaM
pAb129 4967
1000
1076
20002463
ATP2618
3609 2997 2876 CaM2774
4000
CaMK2808
3015 MR1 2618
MR2
Figure 1. RyR subunit according to the 6 TM model. This schematic shows the FKBP and Imp binding sites, the pAb129-bindingsite, which specifically recognises cardiac RyR2, MR1 and MR2. MR1 spans residues 2618 – 3015 and includes an ATP-binding site,three potential CaM-binding sites between residues 2774 – 2806, 2876 – 2897 and 2997–3015 and a CaMK phosphorylation site. CaM: Calmodulin; CaMK: CaM-dependent protein kinase; FKBP: FK-506-binding protein; Imp: Imperatoxin; MR: Modulator region; RyR: Ryanodine receptor;TM:Transmembrane domain.
Author p
roof
Intracellular calcium channels and their modulators
4 Expert Opin. Ther. Patents (2003) 13(6)
that RyR regulation requires an accessory protein(s), forexample calmodulin (CaM) for inhibition at the normalrange of Ca2+. Indeed, it was reported that RyR in combina-tion with CaM was inhibited at much lower ([Ca2+]> 10 µM) concentrations [38]. It seems evident that the RyRshould be inhibited by such a mechanism otherwise thechannel would be open all the time. The possibility cannotbe excluded that non-inhibited RyRs represent a deterioratedfraction of the RyR population, which may be generated as aresult of experimental conditions. Another question to beaddressed is whether a mechanism exists to keep IP3R andRyR inactivated once they are inhibited. If no such mecha-nism existed to inhibit channel activity independently ofCa2+ level, then as soon as the Ca2+ATPases decreased [Ca2+]ito the concentration required for channel activation, Ca2+
release would be re-inititated. For example, the threshold ofICC (RyR) inhibition is 1 mM. When [Ca2+]i reaches 1 mM,RyR becomes inactivated and closed. At the same time,Ca2+-ATPases continue to pump Ca2+ from the cytosol. Assoon as Ca2+ level is decreased, for example, to 0.99 mM, theRyR can be opened and releases Ca2+ again. When Ca2+
reaches 1 mM, the channel would be inactivated again. Inthis condition when Ca2+ release equilibrates decrease in Ca2+
level produced by Ca2+-pumps, the channel would constantlyalternate between quick open and closed states.
3. Ryanodine receptor
3.1 Ryanodine receptor structure and expressionThe RyR was initially found in excitable muscle cells where itplays an essential role in coupling action potentials to Ca2+
release and contractility (EC-coupling). Lately, RyR was
found in unexcitable cells and in plant and fungus, suggestingthat the role of this receptor in cell physiology is not limitedto EC-coupling. The first reports describing the effect of Ryon calcium transport appeared in the 1960s and were errone-ously ascribed to the inhibition of the Ca2+-pump [39]. Later itwas shown that Ry binds to the RyR, a calcium channellocated in junctional terminal cisternae of SR [40,41].
RyR1 and RyR2 are predominantly expressed in skeletal andcardiac muscles, respectively. It was proposed that the expres-sion of different splicing isoforms of RyR1, with different kinet-ics of activation, were present in slow- and fast-twitch skeletalmuscle [42]. RyR3 was originally identified in the brain and wascalled a brain isoform, although all three RyRs are expressed inthe brain and RyR2 seems to be more abundant in the brain.RyR consists of four identical subunits of 565 kDa each. It isthe largest ion channel known to date. The full tetramer isrequired for Ca2+ transport suggesting that all four subunitsparticipate in formation of the conducting channel. Some datasuggest that the central channel pore is connected to four radialchannels in each monomer [43], the function of which is notknown. These channels may be non-conducting pores or Ca2+-conducting pores branched from the central channel pore. Thechannel region of RyR, according to different models, consistsof 4, 6, 10 or 12 TM domains located close to the C terminus.Cryoelectron microscopy data suggest a 10 TM model [43,44],whereas results obtained with site-specific antibodies favour the4 TM model [45]. Some authors proposed a 6 TM model analo-gous to the IP3R structure (Figure 1) [24]. In a hydropathy plot,the 3rd and 5th peaks (in a 6 TM model) are small, such thatauthors consider a 4 TM model to be more appropriate. Thetransmembrane part of the monomer molecule (4918 – 4554)represents ∼ 10% of the whole molecule. The homotetramer has
8 7 6 5 4 3 2 1
Po IP3R RyR1 RyR2 and IP3R3
pCa2+
Figure 2. Different characteristics of Ca2+-dependent activation and inactivation of IP3R and RyR. IP3R Types 1 and 2 areinhibited by [Ca2+] at the submicromolar level and muscle RyR Type 1 is inhibited at the high micromolar level. Cardiac RyR Type 2 andIP3R Type 3 are inhibited at the millimolar range (inhibition was not always observed).IP3R; Inositol-1,4,5-triphosphate-receptor; Po: Opening probability; RyR: Ryanodine receptor.

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 5
a four-leaf clover shape and a size of 220 – 240 Å [43,46,47]. In thereconstruction of the RyR, ten independently-folded domainswere identified in the cytoplasmic part of each RyR monomer(Figure 3 [48]). Open-closed transition is accompanied by thedisposition of cytoplasmic domains 6, 9 and 10. The sites ofinteraction with FKBP12, calmodulin, imperatoxin(ImTxA) and dihydropyridine receptor (DHPR) were iden-tified. The cytoplasmic part of RyR is very large, whichaccounts for the regulation of its activity by numerous mod-ulatory factors, both small molecules and proteins.
In a medium of mixed cations, Ca2+ conductance ishigher than conductance of K+ and Na+ (I[Ca2+] > I[K+],[Na+], but in a monocation medium I[K+], [Na+] > I[Ca2+].It suggests that the filter selectivity of RyR may functionanalogously to the filter of the voltage-gated calcium chan-nel (VGCC) with multiple Ca2+-binding sites. The maxi-mum conductance is 80 pS and 172 pS for cardiac andmuscle channels, respectively. A low affinity of luminalCa2+-binding site (Kd ∼ 3 – 4 mM) suggests millimolarCa2+ levels in the ER/SR [49,50]. The central pore diameter is10 – 20 Å [43,47,51,52]. The opening of the channel is associ-ated with rotation of the TM region with respect to thecytoplasmic part of the channel tetramer [48]. In somereconstructions the central channel pore is plugged on thelumenal side of the RyR, perhaps representing a closed orinactivated state of the channel that may be related to theinactivation of the VGCC by the N-terminal part.
3.2 Excitation–contraction couplingThere are two mechanisms of EC-coupling. According tothe mechanical model in skeletal muscle, activation of RyRis mediated by conformational changes transmitted from theDHPR to RyR1 as a result of direct contact [53-55]. Accord-ing to this model, RyR1 is physically connected to theDHPR [56,57], although the participation of other protein(s)
is still under debate. In this model, DHPR functions as avoltage sensor rather than a Ca2+-channel (Figure 4).
In skeletal muscle, four DHPRs are clustered in atetramer on each RyR, so-called tetrads, which are organisedin an array on feet structures [58,59]. Data on the regions ofthe RyR that contact the DHPR are contradictive. In exper-iments with chimeric RyR2/RyR1 it was shown that transferof 1635 – 2636 residues from RyR1 to RyR2 confers amechanical type of EC-coupling [60]. Some experiments suggesta crucial role for the 1076 – 1112 ImTxA-binding region. Itwas proposed that ImTxA mimics the Glu666–Leu690 DHPRfragment which interacts with RyR1 [61]. Other experimentshighlight the importance of the variable D2 region (aminoacid residues 1303 – 1406 in RyR1) in EC-coupling, a regionhighly divergent between RyR1 and RyR2 [62]. The most signif-icant evidence of direct protein–protein interaction of DHPRand RyR1 is that the cytoplasmic fragment between TMrepeats II and III of the DHPR subunit α1 interacts with andactivates RyR1 [61,63-67]. Recently it was suggested that DHPRinteracts with the calmodulin (CaM) binding site (amino acids3609 – 3643) of RYR1 [68]. New findings suggest that DHPRinteraction with the CaM binding region of RyR1 in skeletalmuscle serves not only to activate Ca2+ release but also to turnoff RyR after its activation [69].
The second mechanism of EC-coupling, which takes placein cardiac muscle, is known as CICR [32-34]. According to thismodel, RyR does not make contact with the DHPR but ratherCa2+ influx via DHPR activates RyR and stimulates Ca2+
release. Although the relative importance of these two models issometimes discussed, there is firm evidence that in skeletal mus-cle Ca2+ influx is not necessary [53] and in the cardiac muscleCa2+ influx is absolutely indispensable for contraction [70,71].However, a significant number of skeletal muscle RyRs are notassociated with DHPRs [72] and it is discussed that CICR may,at least partly, contribute to skeletal muscle contraction [73,74].
65 4
10 9
38 7
FKBP12
CaM
TM
a10
6
4
9
32
pFKBP12
b CaM
3
10 9
8 7 3
ImTxA
c
5
Figure 3. Reconstruction of RyR. a) Lateral view. b) View from cytosol. c) View from SR/ER lumen. The numbers indicate distinctglobular domains. The small circles indicate regions of interaction with DHPR. The sites of interaction with CaM, FKBP and ImTxA areshown. Reproduced with permission from [340].CaM: Calmodulin; DHPR: Dihydropyridine receptor; ER: Endoplasmic reticulum; FKBP: FK-506-binding protein; ImTxA: Imperatoxin; SR: Sarcoplasmic reticulum; TM: Transmembrane domain.

Author p
roof
Intracellular calcium channels and their modulators
6 Expert Opin. Ther. Patents (2003) 13(6)
3.3 Interaction with proteinsCaM modulates RyR properties but its effects may bediverse. It may be a direct interaction with RyR and modula-tion via CaM-dependent protein kinase. The effect of CaMon skeletal RyR1 was demonstrated to be biphasic anddependent on Ca2+ level; CaM potentiates Ca2+ release at lowCa2+ concentrations (3 – 10 µM), while it showed an inhibi-tory effect at high Ca2+ concentrations (> 3 – 30 µM) [78-77].Cardiac RyR2 does not show activation by apoCaM but isinhibited by calcium-calmodulin (CaCaM) [78-80]. It was sug-gested that Ca2+ binding results in a CaM shift from 3625 – 3643toward 3614 – 3625 amino acids, regulating the functionalstate of RyR1 [81-83]. However, other authors suggest RyR1has a single CaM binding domain that is shared by apoCaMand CaCaM [84].
The FK-506-binding protein, FKBP12 (12 kDa), is closelyassociated with RyR1 (cardiac analogue FKBP12.6 is associ-ated with RyR2) [85,86]. It was proposed that FKBP stabilisesthe channel in both open and closed states reducing subcon-ductant current [87]. RyR forms a complex with junctional pro-teins triadin and junctin. It was shown that triadin and junctinanchor the luminal Ca2+-binding protein, calsequestrin, to theRyR enabling functional coupling between calsequestrin andRyR [88,89]. Most of the triadin and junctin molecules arelocated in the lumen of ER and their luminal segments bindthe Ca2+-calsequestrin complex [90]. Evidence has been pre-sented showing that calsequestrin activates RyR, whereas tria-din inhibits RyR [91]. It was proposed that luminal [Ca2+]controls RyR activity via calsequestrin binding and increasedluminal [Ca2+] facilitates Ca2+ release [92,93]. It was noticed thatcalsequestrin in the dephosphorylated state activated RyR [94].Some authors suggested that RyR forms a complex withcAMP-dependent protein kinase (PKA), PKA anchoring
protein and protein phosphatases PP1 and PP2A, which arevery important for channel regulation [95]. Interactions of RyRwith the cytoskeletal proteins ankyrin, annexin VI, sarcalu-menin and other proteins have also been reported.
Sorcin, a 22 kDa Ca2+-binding protein whose expression isupregulated during acquisition of multi-drug resistance,seems to be associated with cardiac RyR. The opening proba-bility (Po) of RyR was decreased significantly by sorcin bind-ing with an IC50 of 480 nM and this inhibition was shown tobe affected by PKA phosphorylation [96,97].
RyR clustering into a lattice structure has been demon-strated [98]. Cooperative interactions between receptors organ-ised into a cluster or array can increase their sensitivity toactivation and amplify the output of a Ca2+ signal by simulta-neous opening and closing [99]. Recently it was proposed thatthe KCa channel may be coupled to SR RyR [100].
3.4 Endogeneous modulators3.4.1 Ca2+, Mg2+
Ca2+ has a biphasic, general activating effect at physiologicalrange, whereas Mg2+ has an inhibitory effect. Some authors didnot find any evdience of Ca2+-dependent inhibition in slow-twitch [37] and in cardiac muscles [101]. Ca2+ inactivation occurswith an IC50 value of 0.7 – 3.7 mM for RyR1 but RyR2 showslittle, if any, inactivation. Inactivation of cardiac RyR2 occurredwith an IC50 of 15 mM [102], but others reported that RyR2shows little inactivation, even at [Ca2+] > 100 mM [103]. It wassuggested that RyR inhibition by high [Ca2+] was only a prop-erty of skeletal fast-twitch muscle. Some experiments demon-strated that the isolated cardiac isoform, RyR2, was notinhibited by Ca2+ at all, whereas in native vesicles from cardiacmuscles, Ca2+-induced inactivation was observed suggestingthat auxiliary protein(s) are necessary for RyR2 inactivation.
Cell membrane
DHPR DHPR
Depolarisation Depolarisation
Cytosol
SRSR
RyR1
RyR2
Ca2+
Ca2+
Ca2+
Ca2+Ca2+Ca2+Ca2+
Ca2+
a b
Figure 4. Two mechanisms of EC-coupling. a) In skeletal muscle, where DHPR contacts with RyR, depolarisation produces changes inDHPR conformation which are directly transferred to RyR; influx of Ca2+ is not necessary. b) In cardiac muscle DHPR works as a Ca2+
channel; action potentials induce opening of DHPR; influx of Ca2+ activates RyR. DHPR: Dihydropyridine receptor; EC-coupling: Excitation–contraction coupling; RyR: Ryanodine receptor; SR: Sarcoplasmic reticulum.
Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 7
Glu3987 in the mouse RyR2 is a high-affinity Ca2+ binding site[104]; a low-affinity site responsible for Ca2+-inactivation was notfound in RyR2. In RyR1, low-affinity Ca2+ binding sites weremapped to three adjacent regions lying between amino acids3726 and 5037 [105].
Mg2+ has an inhibitory effect, which is presumably condi-tioned by the interaction of Mg2+ with low-affinityCa2+-binding inhibitory sites on RyR. In skeletal muscle, theIC50 was ∼ 20 µM at 1 µM Ca2+ and 70 – 200 µM at10 µM Ca2+ [105,106]. As free [Mg2+]i is in the range of hun-dreds of micromoles, it is probable that at resting condi-tions, Mg2+ inhibits RyR but increasing [Ca2+] mayovercome this inhibitory effect.
3.4.2 Pyridine nucleotide: cADP-ribose and NAADP Traditionally, RyR was considered to be an effector of actionpotentials in excitable cells (neurons and muscles) but in the
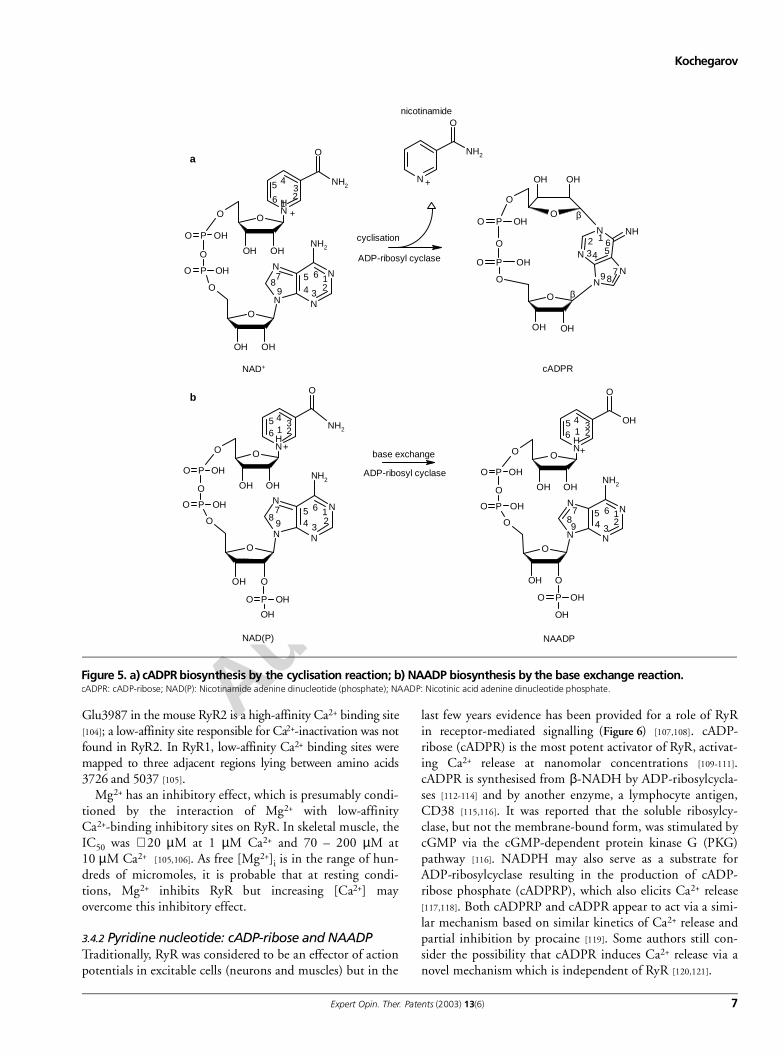
last few years evidence has been provided for a role of RyRin receptor-mediated signalling (Figure 6) [107,108]. cADP-ribose (cADPR) is the most potent activator of RyR, activat-ing Ca2+ release at nanomolar concentrations [109-111].cADPR is synthesised from β-NADH by ADP-ribosylcycla-ses [112-114] and by another enzyme, a lymphocyte antigen,CD38 [115,116]. It was reported that the soluble ribosylcy-clase, but not the membrane-bound form, was stimulated bycGMP via the cGMP-dependent protein kinase G (PKG)pathway [116]. NADPH may also serve as a substrate forADP-ribosylcyclase resulting in the production of cADP-ribose phosphate (cADPRP), which also elicits Ca2+ release[117,118]. Both cADPRP and cADPR appear to act via a simi-lar mechanism based on similar kinetics of Ca2+ release andpartial inhibition by procaine [119]. Some authors still con-sider the possibility that cADPR induces Ca2+ release via anovel mechanism which is independent of RyR [120,121].
Figure 5. a) cADPR biosynthesis by the cyclisation reaction; b) NAADP biosynthesis by the base exchange reaction.cADPR: cADP-ribose; NAD(P): Nicotinamide adenine dinucleotide (phosphate); NAADP: Nicotinic acid adenine dinucleotide phosphate.
OHOH
O
PO OH
O
PO OH
O
NH
NH2
O
O
OHOH
N
N
N
N
NH2
O
N
NH2
O
+
+
OH
OH OH
OH
NN
N
N
NH
O
O
O
PO OH
O
PO OH
O
NAD+ cADPR
cyclisation
ADP-ribosyl cyclase
nicotinamide
+
NAD(P)
65 4
321
87
95 6 1
234
1234 5
6
798
123
45
6
78
912
345 6
+
65 4
321
87
9
5 6 12
34
base exchange
ADP-ribosyl cyclase
NAADP
a
b
β
β
NH
NH2
O
O
PO OH
O
PO OH
O
OH OH
O
N
N
N
N
NH2
OH O
P OHO
OH
O
NH
OH
O
OHOH
O
PO OH
O
PO OH
O
O
OH
N
N
N
N
NH2
O
P OHO
OH
O
Author p
roof
Intracellular calcium channels and their modulators
8 Expert Opin. Ther. Patents (2003) 13(6)
Another Ca2+-releasing agent, NAADH, has been discov-ered originally as a minor contaminant of commercially avail-able NADH [122,123]. Ca2+-release induced by NADHtreatment occurs with characteristic delay caused by the enzy-matic conversion of NADH to active metabolites. In the pres-ence of nicotinic acid, ADP-ribosylcyclase and CD38 catalysethe base exchange reaction resulting in the synthesis ofNAADP from NADH and NADPH (Figure 5) [124]. It mayseem surprising that the same enzyme can catalyse suchdifferent reactions. The reaction requires millimolar levels ofreagents (IC50 = 0.2 mM for NADPH and 10 mM for nico-tinic acid) that exceeds physiological levels in the cell but itwas noticed that the reaction shifted towards NAADPH syn-thesis at acidic pH. It was hypothesised that the switchbetween these two reactions is regulated by a pH change [125].Both enzymes (soluble cyclase and CD38) can also producecGDP-ribose (cGDPR) from nicotinamide guanine dinucle-otide. CD38 in comparison to ADP-ribosylcyclase can cata-lyse the hydrolysis of cADPR to ADPR [126]. cADPR may alsobe hydrolysed spontaneously without enzyme activity and itshydrolysed metabolite inhibits RyR.
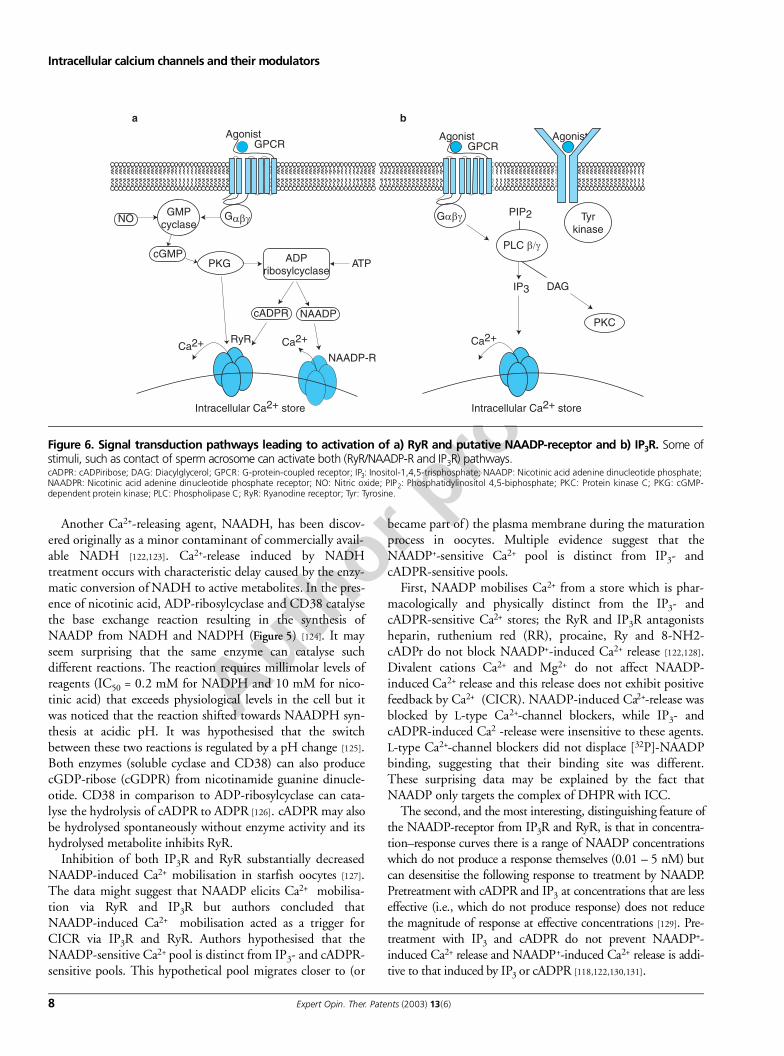
Inhibition of both IP3R and RyR substantially decreasedNAADP-induced Ca2+ mobilisation in starfish oocytes [127].The data might suggest that NAADP elicits Ca2+ mobilisa-tion via RyR and IP3R but authors concluded thatNAADP-induced Ca2+ mobilisation acted as a trigger forCICR via IP3R and RyR. Authors hypothesised that theNAADP-sensitive Ca2+ pool is distinct from IP3- and cADPR-sensitive pools. This hypothetical pool migrates closer to (or
became part of ) the plasma membrane during the maturationprocess in oocytes. Multiple evidence suggest that theNAADP+-sensitive Ca2+ pool is distinct from IP3- andcADPR-sensitive pools.
First, NAADP mobilises Ca2+ from a store which is phar-macologically and physically distinct from the IP3- andcADPR-sensitive Ca2+ stores; the RyR and IP3R antagonistsheparin, ruthenium red (RR), procaine, Ry and 8-NH2-cADPr do not block NAADP+-induced Ca2+ release [122,128].Divalent cations Ca2+ and Mg2+ do not affect NAADP-induced Ca2+ release and this release does not exhibit positivefeedback by Ca2+ (CICR). NAADP-induced Ca2+-release wasblocked by L-type Ca2+-channel blockers, while IP3- andcADPR-induced Ca2 -release were insensitive to these agents.L-type Ca2+-channel blockers did not displace [32P]-NAADPbinding, suggesting that their binding site was different.These surprising data may be explained by the fact thatNAADP only targets the complex of DHPR with ICC.
The second, and the most interesting, distinguishing feature ofthe NAADP-receptor from IP3R and RyR, is that in concentra-tion–response curves there is a range of NAADP concentrationswhich do not produce a response themselves (0.01 – 5 nM) butcan desensitise the following response to treatment by NAADP.Pretreatment with cADPR and IP3 at concentrations that are lesseffective (i.e., which do not produce response) does not reducethe magnitude of response at effective concentrations [129]. Pre-treatment with IP3 and cADPR do not prevent NAADP+-induced Ca2+ release and NAADP+-induced Ca2+ release is addi-tive to that induced by IP3 or cADPR [118,122,130,131].
Intracellular Ca2+ store Intracellular Ca2+ store
PIP2
IP3 DAG
PKC
Tyrkinase
AgonistGPCR
Gαβγ
Agonist AgonistGPCR
GαβγGMPcyclase
cGMPPKG ADP
ribosylcyclase
cADPR NAADP
Ca2+ Ca2+RyR
NAADP-R
NO
ATP
Ca2+
a b
PLC β/γ
Figure 6. Signal transduction pathways leading to activation of a) RyR and putative NAADP-receptor and b) IP3R. Some ofstimuli, such as contact of sperm acrosome can activate both (RyR/NAADP-R and IP3R) pathways.cADPR: cADPiribose; DAG: Diacylglycerol; GPCR: G-protein-coupled receptor; IP3: Inositol-1,4,5-trisphosphate; NAADP: Nicotinic acid adenine dinucleotide phosphate; NAADPR: Nicotinic acid adenine dinucleotide phosphate receptor; NO: Nitric oxide; PIP2: Phosphatidylinositol 4,5-biphosphate; PKC: Protein kinase C; PKG: cGMP-dependent protein kinase; PLC: Phospholipase C; RyR: Ryanodine receptor; Tyr: Tyrosine.

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 9
Third, agonists produce Ca2+ release from apparently differentpools: NAADP mobilises Ca2+ from a thapsigargin-insensitivepool whereas both cADPR and IP3 elicit Ca2+ release from tapsi-gargin-sensitive pool [132]. The location of the NAADP-sensitiveCa2+ pool is not known. It was proposed that this Ca2+ pool maybe close to the plasma membrane since, after NAADP injection,Ca2+ waves emanated from just beneath the plasma membrane[131]. In sea urchin eggs, NAADP mobilises Ca2+ from lysosome-related organelles in which active Ca2+ transport depends on aproton gradient maintained by a V-type proton pump but notfrom the ER [133]. Fourth, in sea urchin eggs, NAADP but notIP3 or cADPR induces long-term Ca2+ oscillations [134]. All thesedata in summary suggest that NAADP activates Ca2+-release via aNAADP-receptor distinctive from RyR and IP3R.
It was shown that stimulation of extracellular receptorssuch as bradykinin, angiotensin, adrenergic, cholecystokininand muscarinic receptors results in ribosylcyclase activationand cADPR synthesis [108,135]. It was proposed that theCa2+-response on receptor stimulation is mediated by recep-tor-coupled G proteins, which activate GMP-cylase increas-ing cGMP levels and activating PKG, which phosphorylatesand activates ADP-ribosylcyclase [107,135]. Locally releasedNO may trigger this pathway affecting GMP-cyclase. Addi-tionally, NO and PKG can directly modulate RyR. CyclicADP-ribose competes with ATP for the adenine binding siteon the cardiac RyR2 that suggests cADPR and ATP bind tothe same site [136]. A binding site for NAADP has not yetbeen identified in RyR.
3.4.3 Adenine nucleotidesAlmost all adenine nucleotides at millimolar levels activateRyR with an order of potency of ATP > cAMP > ADP > AMP,while non-adenine nucleotides, such as CTP, GTP, UTP andITP are ineffective [137-140]. ATP is the most potent activatoramong the adenines, probably because its concentration rep-resents energetic potential in the cell. There is a conservedATP-binding motif, GWGNFG, in 2618 – 2653 (for RyR2)[46,141]. RyR1 is more responsive to regulation by ATP,whereas cardiac RyR2 is more sensitive to Ca2+ levels reflect-ing the suitability of the cardiac isoform to CICR. It wasreported that at low calcium concentrations, RyR has twoATP-binding sites with half-activatory concentrations of 19and 350 µM, respectively [142]. These low affinity bindingsites may be responsible for the sometimes observed inhibi-tion of RyR by high levels of ATP (> 6 mM).
3.4.4 Phosphorylation by protein kinases RyR1 is phosphorylated by CaMK, PKA and PKG at Ser2843[143] and in RyR2 at Ser2809 [144]. Functional significance ofthis phosphorylation is still controversial; generally it isaccepted that phosphorylation results in RyR activation[145,146], however both channel inactivation [147,148] or noeffect have also been described.
Recently it was suggested that hyperphosphorylation ofRyR may be a contributing factor in heart failure [149,150].
Phosphorylation of proteins connected with RyR, such as tria-din and FKBP may also affect Ca2+ transport [151,152].
3.4.5 Fatty acid derivativesPalmitoyl carnitine and other acyl (chain length C > 14) carni-tine (IC50 = 10 – 15 µM) [153,154] and acyl-CoQ derivativeswith medium length C = 12 – 14 (IC50 = 6 µM) induced Ca2+
release in skeletal but not in cardiac muscles [154,155]. Activa-tion of Ca2+ release was slower than that produced by Ca2+ orATP and occurred with a lag time of 100 – 150 ms, which maysuggest that an enzymatic reaction was needed to convertpalmitoyl carnitine to an active releaser. Free fatty acids, suchas palmitic, stearic, arachidic, oleic and linoleic acids inducedCa2+-release [156,157]; however, in other studies these effectswere not reproduced [153] or fatty-acid-induced Ca2+ releasewas explained by inhibition of Ca2+-ATPase [158]. Arachidonicacid (AA; 50 µM) activated Ca2+ release in skeletal and cardiacmuscles [153] but AA-induced Ca2+ release was not inhibited byRR [159], posibly suggesting a target different from RyR. Morerecently it was reported that AA inhibited ryanodine binding,this may support the notion of targeting RyR but single-channelPo was not changed [160]. Sphingosine at low concentrationinhibited Ca2+-, caffeine- and doxorubicin-induced Ca2+ release[161,162] and also inhibited Ry binding in skeletal and cardiacmuscles, but at high concentration (30 – 50 µM), sphingosineand its derivative sphingosylphosphorylcholine activatedCa2+-release [161,163]. IP3R and RyR are differently modulatedby AA and its product leukotriene B4 (LTB4). The IP3R wasinhibited by AA (Ki = 27 nM), whereas the RyR was unaf-fected by this compound. In contrast, 100 nM LTB4 fullyactivated the RyR but did not influence the IP3R [164].
3.4.6 Endogenous polyaminesEndogenous polyamines, such as spermine (IC50 = 10 – 100 µM)and the less active intermediate compounds in spermine syn-thesis, spermidine and putrescine, inhibited in a voltage-dependent manner caffeine- and thymol-induced Ca2+
release [165,166]. It was suggested that polyamines either com-pete with Ca2+ ions in the channel pore or occlude the chan-nel pore. It was reported that endogenous polyamines alsoblock many types of ion channels and connexin (40)-formedchannels [167].
3.4.7 Reactive oxygen species and nitric oxideReactive oxygen intermediates and nitric oxide (NO) modu-late the contractile function of skeletal muscle fibres by cova-lent reversible or irreversible modifications of thiol groups.Oxidants produce disulfide bonds between subunits of thechannel tetramer and this may be responsible for alterations inchannel activity. On the whole, the channel can be modulatedby O2 tension (pO2), levels of glutathione and NO [168].There are 404 Cys residues in the RyR1 tetramer (101 persubunit) and under conditions comparable to resting muscle,∼ half of them are maintained in a reduced (free-thiol) state(i.e., ∼ 50 per subunit). Oxidation of 10 – 12 additional thiols

Author p
roof
Intracellular calcium channels and their modulators
10 Expert Opin. Ther. Patents (2003) 13(6)
(i.e., 63 oxi- and 38 free-thiol/subunit) had little effect onchannel activity [169]. Stronger oxidation (78 oxi- and 23 free-thiols) reversibly increased channel activity. More extensiveoxidation (13 free-thiols left/subunit) inactivated the channelirreversibly. This suggests three functional classes of RyR1 thi-ols, oxidation of which produces: no effect, reversible activa-tion and irreversible inactivation. Functionally inert thiols canprotect the channel from extensive oxidation, whereas activethiols may serve as sensors of redox potential produced byphysiological (e.g., glutathione, nitric oxide) and pathophysi-ologic (quinones, reactive oxygen species) conditions [169,170].
Effects of NO are controversial, they were described as acti-vating [171-174] in the prevalent cases but in some cases NOinhibited Ca2+-release [175,176]. This inhibition was mediatedby formation of S–S-bonds and was reversed by the S–Sreducer, 2-mercaptoethanol [176]. The effect of NO dependson its concentration and physiological conditions. It wasreported that initial release of NO activates RyRs. Strongerrelease of NO inhibits RyR activity and contraction but irre-versible activation was also reported [173,174].
Surprisingly, it was reported that NO, NO donors andN-ethylmaleimide, considered as an oxidant, may protect Cysresidues in the RyR against oxidation by other agents [82,177].Pretreatment with NO donors, at concentrations that have nodetectable effect on channel activity, block intersubunitcrosslinking and prevent activation of the channel by thedisulfide-inducing agent diamide. In contrast, higher concen-trations of NO donors produce intersubunit crosslinks andactivate the channel. Multiple effects of oxidants and NO canbe explained by different susceptibility of Cys residues: thiola-tion of high susceptible residues may result in one conforma-tion and thiolation by higher concentrations may involvemore residues rendering to different conformations. Higherconcentrations of oxidants and NO can produce oxidised andnitrosylated groups: -SO3H and -SNO that may have differ-ent effects [178]. The effect of NO may be further complicatedby its ability to modulate numerous pathways, such as cGMPand cADPR pathways that have been implicated in RyR regu-lation, so the resulting effect would depend on the particularconditions and on the cell type.
Cys3635 within the CaM-binding domain was proposed tobe responsible for skeletal muscle ryanodine receptor modula-tion by NO [179]. The cysteine is intercalated within a hydro-phobic CaM-binding domain. It was shown that CaM (bothApoCaM and Ca2+-CaM) prevents oxidation of this Cys, aswell as oxidation prevented CaM binding. Alkylation withN-ethylmaleimide or reaction with NO donors preferentiallyblocks apoCaM binding to RyR1, suggesting the existence ofa regulatory Cys within the ApoCaM binding site [82].
3.5 Pharmacological modulators3.5.1 Ryanodine and related compoundsRy is a natural compound isolated from the species of Ryania,slim trees and bushes growing in Central and South America.Extract from Ryania is a mixture of at least three biologically
active components: Ry (compound 1), 9,21- didehydroryano-dine and 18-hydroxyryanodine. The related compound, ryan-odol (compound 2), has been isolated from the wood of Perseaindica. Extract from Ryania species have been used by nativeAmericans to poison arrowheads. In 1948, Ry was isolated andcharacterised by Dr EF Rogers et al. in a publication based onplant insecticides [180]. Powder from Ryania wood was used asan insecticide before synthesis of DDT. Some authors supportthe use of ecologically safe natural insecticides such as Ry androtenone, decomposing with shorter half-life. The effect of Ryon contraction of cardiac and skeletal muscles (cardiac muscleis more sensitive) has been known since the 1960s [181]. Theeffect of Ry on Ca2+ release was first described in the mid 1960sand was erroneously ascribed to the interaction with the Ca2+
pump [39]. Ry was shown to bind a protein in the SR, whichwas identified as the RyR, an intracellular Ca2+ channel [182,183].In SR vesicles isolated from fast-twitch frog skeletal musclethere are two Ry binding sites with high (Kd ∼ 1 – 1 0nM) andlow (Kd = 1 – 10 µM) affinity. Ry binding to the high affinitybinding site (HABS) at nanomolar concentration results inRyR activation, which is characterised by increasing Po andopen state time, whereas binding to the low affinity bindingsite (LABS) at micromolar concentrations of Ry locks thechannel in a state of subnormal decreased conductance. Lowconcentrations of Ry (5 – 10 nM) activate a large conductancecalcium channel after a short delay (5 – 10 min) [184]. Bothhigh and low affinity Ry binding sites are located betweenArg4475 and the C terminus [185]. It was shown that Ry bindsto the tetrameric form, but not the momomeric form of RyRthat suggests the correct binding sites are formed by juxtaposi-tion of the monomers. Data obtained from [3H]-Ry bindingsuggest a 1:1 (Ry molecule per RyR tetramer) stoichiometryfor the high affinity binding site and either a 3:1 or 1:1 stoichi-ometry for the low affinity binding site. This discrepancyresulted in two different putative models describing Ry bind-ing to its receptor. The first model (the interconvertible sitemodel) suggests the existence of four initially identical HABSs
NH
O
O
OH
OH OH
OH
OH
OH
O
OH
OO
OH
OH OH
OH
OH
O
1 Ryanodine
2 Ryanodol
Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 11
in RyR. Binding of the first Ry molecule lowers the affinity ofthe three non-occupied Ry-binding sites. Binding of Ry mole-cules to the three other sites with low affinity results in channelclosure. One variation of this model proposes that Ry bindingto each site successively decreases the affinity of the unoccu-pied sites, producing four kinetics of Ry binding and fourchannel conductance states. The second model (the distinctsite model) proposes the existence of two distinct Ry-bindingsites, one with high affinity and another with low affinity.
Multiple concentration-dependent effects of Ry have beenreported [186]. Ry (5 – 40 nM, Kd1 = 0.7 nM) activates thechannel increasing Po (up to 300% of control) withoutchanging unitary conductance and significantly enhancingCa2+-induced Ca2+ release from SR vesicles. Ry, at concen-trations of 20 – 50 nM, induces occasional conductancefluctuations which correlate with Ry binding to a second site(Kd2 = 23 nM). Ry ≥ 50 nM stabilises the channel in a stateof subconductance which is not readily reversible. Higherconcentrations (≥ 70 µM) produce a unidirectional transi-tion from 0.5 to 0.25 conductance fluctuation, whereas con-centrations ≥ 200 µM cause almost complete closure of thechannel. These data suggest that Ry stabilises four discretestates of RyR and support the existence of multiple Ry effec-tor sites [186]. Binding and activation of RyR by nanomolarconcentrations of Ry shows functional use-dependence. Forexample, after Ry infusion in muscles, contracture devel-oped after stimulation by electrical impulses but not in non-stimulated muscles [187]. It may indicate that Ry only bindsto the high-affinity site on open RyR and does not bind toinactive RyR. [3H]-Ry binding can be used as an index ofRyR channel activation [188]. In acidic medium, Ry dehy-drates easily and gives anhydroryanodine, losing its typicalactivities on RyR [189].
The closely related compound, ryanodol, is a pentacyclicditerpene having 14 carbons and a single oxygen atom con-figured in four carbon rings, one ring containing the oxygenatom. The South-Asian tree, Cinnamomi cortex, containsmany diterpenoids, such as cinnzeylanol, cinnzeylanine andcinncassiol, having a carbon framework distantly related tothe ryanodol molecule. There are many chemical modifica-tions of Ry and ryanodol [189]. Ryanodol as distinguishedfrom Ry molecule has no pyrrole moiety and binds verte-brate RyR with 2000 times lower affinity (Kd = 7 nM for Ryand 12000 nM for ryanodol) [190,191]. These data suggestthat the pyrrole ring is the general requirement for highaffinity Ry binding. The carbon atoms 9 and 10 are protrud-ing from the binding site and form interactions leading tomodulation of RyR function. Ryanadol binds RyR withmuch less affinity and has a significantly lower toxic effecton vertebrates. However, surprisingly for certain insects rya-nadol and Ry have equivalent toxicity [192]. Ryanoid ester Abinds only HABS with a Kd value of 110 nM and onlycauses an increase in the Ca2+ release at concentrations ≤ 3mM [193]. Experiments with this compound could help toelucidate the model of Ry binding.
3.5.2 Local anaesthetics Analgesic effect of local anaesthetics (LA) is due to the block-age of Na+-channels, which are essential for the spreading ofaction potentials. Also, LAs bind and block RyR. The effect ofLAs on Ca2+ exchange and contraction has been known sincethe 1960s [194]. Procaine (3) and tetracaine (4) inhibited (inmillimolar range; EC50 = 1 – 2 mM) Ca2+-release from ERinduced by Ca2+, Ca2+ transporter ionomycin, inhibitor of SRCa2+-ATPase BHQ and quercetine (EC50 = 0.1 mM)[34,195-197]. Tetracaine applied to skeletal muscle was morepotent than procaine (IC50 = 0.1 – 0.6 mM and 4 mM,respectively). Other LAs, such as tertiary amines, etidocaine(5), bupivacaine (6), prilocaine (7), lidocaine (8) and mepi-vacaine, the quaternary amines QX 572 and QX 314 and theneutral anaesthetic benzocaine (9) displayed similar inhibitory
O
O
NH2
N • HCl
3 Procaine
O
O
N
NH
4 Tetracaine
8 Lidocaine
NH
O N
5 Etidocaine
NO
NH
6 Bupivacaine
NH
O NH
7 Prilocaine
N
NH
O
NH2
O
O
9 Benzocaine

Author p
roof
Intracellular calcium channels and their modulators
12 Expert Opin. Ther. Patents (2003) 13(6)
actions [198]. Some groups suggested that LAs stimulated Ca2+
release and muscle contraction, presumably by inhibition ofCa2+-ATPase [198,199]. The resulting effect on Ca2+ release maybe different; if LA blocking of ICC is more potent than Ca2+-pumps, Ca2+ would accumulate in the ER. If they block bothCa2+-ATPases and RyR to an equal degree, luminal Ca2+
would be trapped in the ER with some possible leakage. Ifthey block Ca2+-pumps but some ICC are still open, the Ca2+
efflux may exceed influx.Records of single-channel conductance showed two differ-
ent modes of LA action on skeletal muscle ICC [200,201]. Pro-caine and tetracaine decrease Po and reduced Ry binding.Lidocaine and quaternary amines, QX 314, and possiblyhigh concentrations of procaine produced slightly differenteffects, which is voltage-dependent and characterised byreduced channel conductance and increased Ry affinity. Thiseffect was explained by the interaction with low-affinity siteslocated close to the channel pore.
3.5.3 Volatile anaesthetics and halogenated hydrocarbonsSome volatile anaesthetics (VA) applied for general anaesthe-sia, mostly derivatives of halogenated hydrocarbons, such ashalothane (10), enflurane (11), isoflurane, halogenated phe-nols and chloroform (12), increase Ca2+-release via RyR fromthe ER [202]. During the last decade it was discovered thattheir anaesthetic effect is due to the interaction with GABAreceptors in the CNS and this effect is independent from theirinteraction with RyR. Ca2+-release was pH and[Ca2+]i-dependent with a maximum response at pH 7.1 and[Ca2+]i = 1 – 10 µM [203]. Concentrations of VA (∼ 0.75% forhalothane and 1.6% for enflurane) applied for general anaes-thesia are rich or exceed effective concentrations that affectRyR (0.002 – 3.8% (v/v) with an EC50 of 0.03 – 0.45% forgas concentration or 13 – 184 µM for aqueous phase concen-tration) [204-208]. The effect of VAs on the RyR has clinical rel-evance, such as a positive ionotropic effect on heart andmuscle contraction, blood pressure and on action potentialsin neurons. However, because VA may modulate ionic trans-port by Ca2+-ATPases, Na+/K+-ATPases, Na+/Ca2+-exchanger[209,210] and GABA receptors, their resulting effect may becomplicated. In cardiomyocytes, chloromethane, chloroethaneand chloroethylene derivatives attenuated the pick of electri-cally induced [Ca2+]i transients in a concentration-dependentand in a reversible manner (IC50 = 0.15 – 18.06 mM) [211].Halogenated phenol derivatives, such as chlorocresol(4-chloro-m-cresol), which is sometimes used for preservationof intravenously administered drugs, activated Ca2+ releaseand Ry binding (EC50 = 100 – 300 µM) [212,213]. Their effi-cacy was higher when it was added from the luminal side, sug-gesting intralumenal location of binding sites. The methylgroup located in the meta position is important for activationbecause other derivatives, such as 4-chloro-o-cresol andp-chlorophenol, are much less potent and the presence ofchloride is also critical since o-cresol was ineffective. The
other derivative, 4-chloro-m-ethylphenol induced Ca2+ releaseat a concentration of 100 – 500 µM. Several ortho-substi-tuted polychlorinated biphenyls increased Ry binding andCa2+ release. Non-halogenated 4-alkylphenol (ethylphenol tononylphenol) derivatives elicited Ca2+ release and their effica-cies were dependent on the length of the alkyl chain [214]. The4-alkylphenol concentration required to induce Ca2+ releasedecreases ∼ threefold for every methylene carbon increase inthe alkyl chain length, indicating that the Ca2+-releasingpotency of 4-alkylphenols is related to their ability to parti-tion into the membrane. Hexachlorocyclohexane (HCH),especially the δ isomer, which is 30-fold more potent than theγ isomer, stimulated Ca2+ release (EC50 = 22 µM) and inhib-ited Ry binding [215]. Dantrolene (13) effectively inhibits Ca2+
release induced by γ-HCH but only partially compared tothat induced by δ-HCH in the same preparation [216].
3.5.4 Malignant hyperthermia and central core diseaseAfter the introduction into practice in the mid 1950s of gen-eral anaesthetic drugs such as halothane, enflurane, isofluraneand the depolarising muscle relaxant, succinylcholine, it wasrevealed that some patients showed potentially fatal abnormaldrug reactions, characterised by muscle rigidity or uncon-trolled skeletal muscle contracture resulting in extensive mus-cle damage (rhabdomyolysis) and rapid increase in bodytemperature. For patients suspected of having malignanthyperthermia (MH), alternative methods of anaesthesia suchas ketamine, propofol and xenon can be applied. MH mayalso be triggered by extreme stress or exercise. MH is a rareautosomal dominant genetic disorder caused by mutations inthe gene encoding skeletal muscle RyR [217-219] and possibly inthe gene of the main subunit of DHPR [220,221] resulting in anabnormal increase in intracellular Ca2+. In RyR1, most of themutations resulting in MH are clustered within either anN-terminal domain (residues 35 – 614) or a central domain(residues 2163 – 2548). MH is characterised by an increasedmyoplasmic [Ca2+]i leading to muscle contracture rigidity andheat production [222]. [Ca2+]i elevation upregulates metabolicpathways such as glycogenolysis resulting in excess lactate pro-duction, metabolic acidosis and hyperactivation of the oxidative
FFF
Br Cl
10 Halothane
ClF
O F
F
F F
11 Enflurane
NH
NO
NO
O
N
O
O
(Na )
13 Dantrolene (1-[(5-(p-Nitrophenyl)furfurylidene)amino]hydantoin)
Cl Cl
Cl
12 Chloroform
Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 13
cycle with increased ATP depletion, high oxygen consump-tion and carbon dioxide production with hypoxaemia andhypercapnia. Long-term myoplasmic Ca2+ overload isthought to cause mitochondrial damage and subsequentdecreased metabolic activity. The crisis is accompanied bysubsequent creatine kinase elevation, hyperkalaemia poten-tially leading to tachycardia and ventricular fibrillation andmyoglobinuria with the possibility of renal failure. If apatient survives, normalisation of oedematous muscle andcreatine kinase (CK) levels occur within 10 – 15 days. With-out immediate treatment, up to 70% cases are lethal becauseof ventricular fibrillation, pulmonary oedema, intravasalcoagulopathy, cerebral hypoxic damage, cerebral oedema orrenal failure. During the crisis, treatment consists of earlyadministration of the RyR inhibitor, dantrolene. Earlyadministration of this drug has successfully reduced the mor-tality rate from 70% to 10%. It was shown that the dantro-lene binding site is located in a 590 – 609 amino acidsequence in RyR1 [223]. RyR1 and RyR3 isoforms were signif-icant, with a similar extent inhibited by dantrolene, whereasRyR2 was not inhibited [224]. Other myopathies, such as cen-tral core disease (CCD) characterised by hypotonia, proximalmuscle weakness and loss of muscle fibres may be also associ-ated with MH episodes [225] and can be as a result of abnor-mality in Ca2+ transport caused by mutations in RyR [226,227].
3.5.5 Methylxanthines (purine derivatives)Many plant methylxanthines are present in drugs and drinks.Coffee is the seed of a cherry from the tree genus Coffea andsome others such as the Kola Nut (Sterculiaceae cola) fromWest Africa, Yerba Mate (Ilex paraguariensis) plant from SouthAmerican tropics and Yaupon Holly or Cassina (Ilex vomitoria)from North America. Western Africans who mixed water withdried or fermented kola nuts made the first cola beverage. Cof-fee trees have been grown in Arabia near the Red Sea since the
7th century and were later introduced into coffee drinks as asource of caffeine. The Cocoa tree (Theobroma cacao) contain-ing theobromine and caffeine was found in Mexico and othercountries in Central and South America. Chocolate from thistree was the favourite beverage of the Aztec emperor Monte-zuma. According to the legend, tea, a beverage made from theleaves of Camellia sinensis, was discovered ∼ 5,000 years ago in2737 bc by a Chinese emperor when some tea leaves acciden-tally dropped into a pot of boiling water.
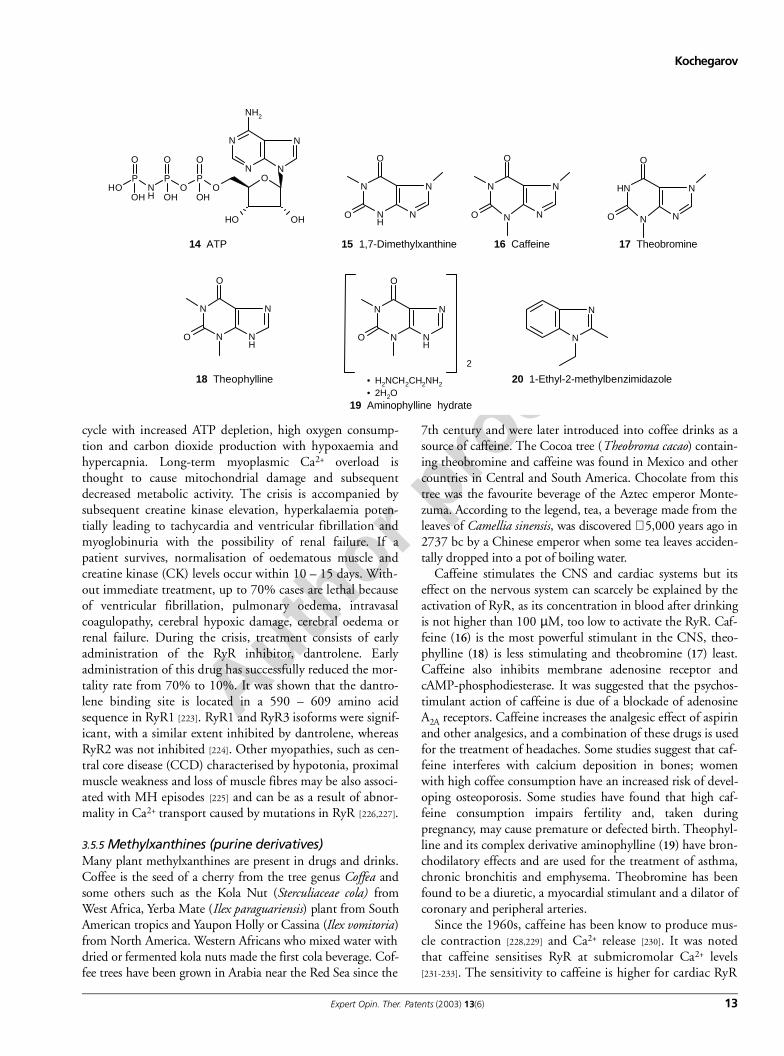
Caffeine stimulates the CNS and cardiac systems but itseffect on the nervous system can scarcely be explained by theactivation of RyR, as its concentration in blood after drinkingis not higher than 100 µM, too low to activate the RyR. Caf-feine (16) is the most powerful stimulant in the CNS, theo-phylline (18) is less stimulating and theobromine (17) least.Caffeine also inhibits membrane adenosine receptor andcAMP-phosphodiesterase. It was suggested that the psychos-timulant action of caffeine is due of a blockade of adenosineA2A receptors. Caffeine increases the analgesic effect of aspirinand other analgesics, and a combination of these drugs is usedfor the treatment of headaches. Some studies suggest that caf-feine interferes with calcium deposition in bones; womenwith high coffee consumption have an increased risk of devel-oping osteoporosis. Some studies have found that high caf-feine consumption impairs fertility and, taken duringpregnancy, may cause premature or defected birth. Theophyl-line and its complex derivative aminophylline (19) have bron-chodilatory effects and are used for the treatment of asthma,chronic bronchitis and emphysema. Theobromine has beenfound to be a diuretic, a myocardial stimulant and a dilator ofcoronary and peripheral arteries.
Since the 1960s, caffeine has been know to produce mus-cle contraction [228,229] and Ca2+ release [230]. It was notedthat caffeine sensitises RyR at submicromolar Ca2+ levels[231-233]. The sensitivity to caffeine is higher for cardiac RyR
O
N
NO
N
N
OHOH
NH2
PO
PNH
POH
OOO
OH OH OH
14 ATP
N
N
N
NH
O
O
15 1,7-Dimethylxanthine
N
N
N
N
O
O
16 Caffeine
N
N
NH
N
O
O
17 Theobromine
N
NH
N
N
O
O
18 Theophylline
N
NH
N
N
O
O
2
• H2NCH2CH2NH2
• 2H2O19 Aminophylline hydrate
N
N
20 1-Ethyl-2-methylbenzimidazole

Author p
roof
Intracellular calcium channels and their modulators
14 Expert Opin. Ther. Patents (2003) 13(6)
(EC50 = 0.2 – 0.5 mM) than for skeletal muscle isoforms.The effect of caffeine is similar to the effect of adeninenucleotides (ATP; 14) and their structure has some similaritythat may suggest caffeine and ATP have the same bindingsite. However, some evidence suggests that caffeine and ade-nine act on different, although partly overlapping, sites [234].In binding experiments, caffeine (1 – 30 mM) facilitated Rybinding [231]. Other methylxanthines, such as theophyllineand theobromine have a similar effect on RyR [235]. The fol-lowing order of potency of methylxanthines was reported[232]: 1,7-dimethylxanthine (15) > 3,7-dimethylxanthine(theobromine) > 1,3dimethylxanthine (theophylline) >1,3,7-trimethylxanthine (caffeine) and 3,9-dimethylxan-thine, whereas 1,9-dimethylxanthine and 1,3,9-trimethylx-anthine were only minimally effective, suggesting thatmethylation on the 9th position, and especially both the 9thand 3rd positions together, reduces their potency. 1-Ethyl-2-methylbenzimidazole (20) resembling methylxanthine struc-ture caused Ca2+ release from isolated triads and its effectiveconcentration was twice more than caffeine (5 and 2.5 mM,respectively) [202].
In experiments with Physarum polycephalum, treatment withcaffeine affected contractile activity; treatment with 10 – 15 mMresulted in an increase in the frequency of contractile oscillation,20 – 25 mM produced breach of regular period and 25 – 50 mMresulted in irreversible cessation of oscillations and fragmenta-tion of plasmodium into so-called caffeine spherules [236,237].This interesting phenomenon, caffeine spherulation, has not yetbeen explained. We can hypothesise that high caffeine levelsresults in cytosolic Ca2+ overload, which affects membrane-bound cytoskeleton resulting in membrane fragmentation. Sur-prisingly in experiments monitoring Ca2+ level in intracellularpools (using the fluorescent indicator CTC), treatment with caf-feine at a concentration of 10 – 25 mM resulted in the charac-teristic CICR only in rare cases, generally it produced only asmooth decrease in Ca2+ level [34]. This data suggests thepresence of two different RyR isoforms in Physarum: a char-acterised RyR isoform and an unusual new RyR isoform,which is only slightly, if at all, sensitive to caffeine and Rybut yet has the characteristic CICR and is inhibited by pro-caine. The new RyR isoform is located in a CTC-stainedCa2+ pool, which does not participate in contractile activity.
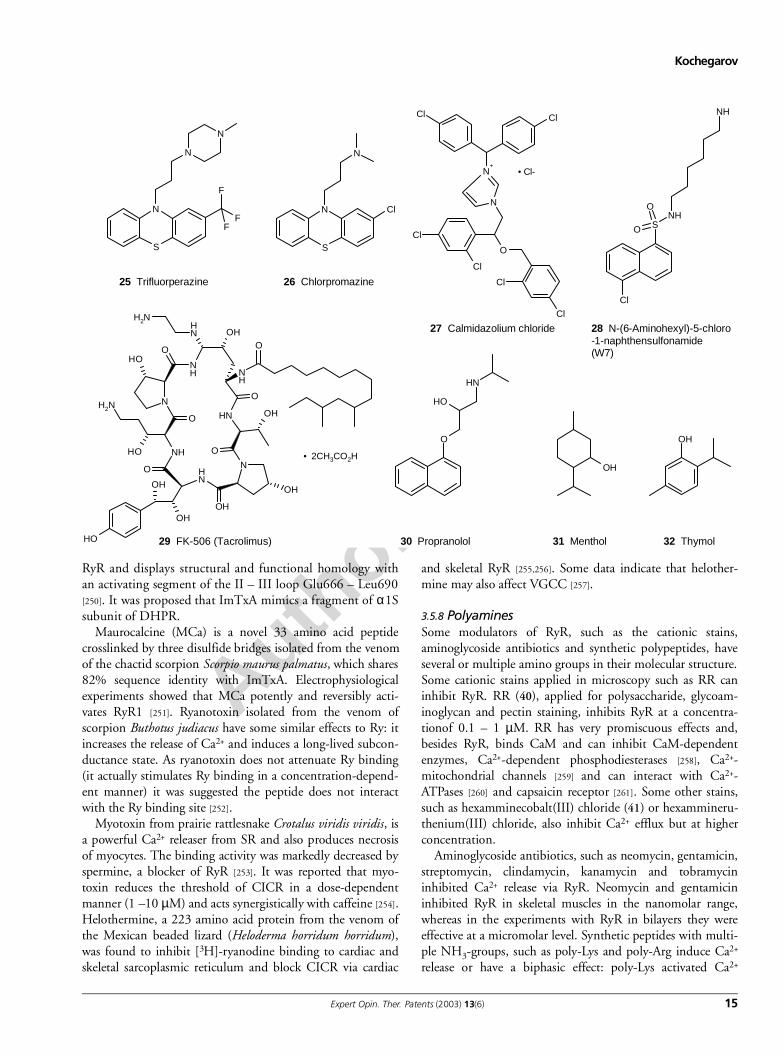
3.5.6 Carboline and carbazole derivativesMany α- and β-carboline derivatives elicited Ca2+-releasefrom SR and increased Ry binding. Carbolines halogenatedon C-5 and C-7 and methylated on N-9 were the most effec-tive. The most effective compounds are bromoeudistomin D(BED; 21) and its derivative 9-methyl-7-bromoeudistomin D(MBED; 22) [238]. Eudistomins are natural toxins isolatedfrom a Caribbean tunicate Eudistoma olivaceum [238-240]. TheEC50 effective concentrations of MBED and caffeine were∼ 1 µM and 1 mM, respectively, indicating that MBED is1000 times more potent than caffeine [241]. Procaine, RR andMg2+ inhibited MBED-induced Ca2+ release. The bell-shaped
profile of Ca2+ dependence for MBED is very similar to thatof caffeine. Since they share some structural similarity withcaffeine, it was proposed that they interact with the same site[241]. It was reported that in brain microsomes, caffeineand MBED bind to a common site, which was distinct fromthe ryanodine receptors [242] but the nature of this bindingwas not disclosed. 2-Hydroxycarbazole (23), a compoundstructurally related to MBED, has been proposed to activateboth Type 1 and Type 2 RyRs in skeletal and cardiac muscle,respectively [243]. The response to 2-hydroxycarbazole wascompetitively antagonised by caffeine. 2-hydroxycarbazoleelicits Ca2+ release from a thapsigargin-insensitive pool inboth muscle and non-muscle cell microsomes. Its effectiveconcentrations have been between 100 and 500 µM with anEC50 of 200 µM [244]. In contrast, the chemical analogueswith a carbazole skeleton and bromine at C-6, inhibit bothCa2+ and caffeine-induced Ca2+ release [245]. 4,6-Dibromo-3-hydroxycarbazole (24; 100 µM) abolished caffeine andCa2+-induced release and in comparison with RR and pro-caine, its effect was not suppressed by high [Ca2+]i.
3.5.7 Peptide toxinsSome proteins and peptides known as natural toxins interactand modulate channel properties of RyR. Their effect wasexplained in that they mimic some endogenous proteins inter-acting with RyR. As proteins can scarcely penetrate cell mem-branes it is not clear how they can be useful to a predator forthe intoxication of their victims. Two possibilities exist: i)peptide toxins can penetrate the cell membrane or ii) they canaffect other targets on the cell membrane, possibly a channelhomologous to RyR.
ImTxA, a 33-amino acid peptide isolated from an Africanscorpion, Pandinus imperator, venom, activated RyR(ED50 = 0.016 µg/ml) [246,247]. ImTxA selectively activatedRyR1 isoform [248] but recently evidence appeared that it canalso activate a cardiac RyR2 isoform [249]. A 138 amino acidcytoplasmic loop between repeats II and III of the α1 subu-nit DHPR (the II – III loop) was proposed to interact with aregion of the RyR to elicit Ca2+ release. Small segments (10– 20 amino acid residues) of the II – III loop retain thecapacity to activate Ca2+ release. ImTxA binds directly to the
NH
N
OH
Br
Br
21 Bromoeudistomin D
NN
OH
Br
Br
22 9-Methyl-7-bromoeudistomin D
NH
OH
BrBr
24 4,6-Dibromo-3-hydroxycarbazole
NH
OH
23 2-Hydroxycarbazole
Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 15
RyR and displays structural and functional homology withan activating segment of the II – III loop Glu666 – Leu690[250]. It was proposed that ImTxA mimics a fragment of α1Ssubunit of DHPR.
Maurocalcine (MCa) is a novel 33 amino acid peptidecrosslinked by three disulfide bridges isolated from the venomof the chactid scorpion Scorpio maurus palmatus, which shares82% sequence identity with ImTxA. Electrophysiologicalexperiments showed that MCa potently and reversibly acti-vates RyR1 [251]. Ryanotoxin isolated from the venom ofscorpion Buthotus judiacus have some similar effects to Ry: itincreases the release of Ca2+ and induces a long-lived subcon-ductance state. As ryanotoxin does not attenuate Ry binding(it actually stimulates Ry binding in a concentration-depend-ent manner) it was suggested the peptide does not interactwith the Ry binding site [252].
Myotoxin from prairie rattlesnake Crotalus viridis viridis, isa powerful Ca2+ releaser from SR and also produces necrosisof myocytes. The binding activity was markedly decreased byspermine, a blocker of RyR [253]. It was reported that myo-toxin reduces the threshold of CICR in a dose-dependentmanner (1 –10 µM) and acts synergistically with caffeine [254].Helothermine, a 223 amino acid protein from the venom ofthe Mexican beaded lizard (Heloderma horridum horridum),was found to inhibit [3H]-ryanodine binding to cardiac andskeletal sarcoplasmic reticulum and block CICR via cardiac
and skeletal RyR [255,256]. Some data indicate that helother-mine may also affect VGCC [257].
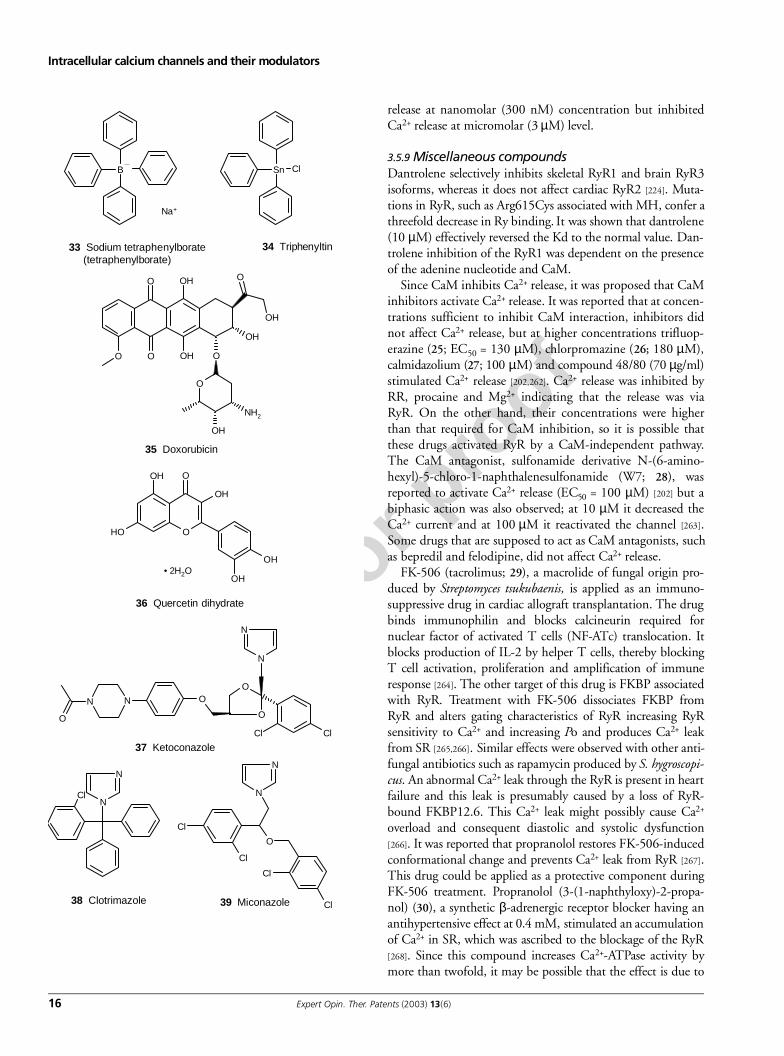
3.5.8 PolyaminesSome modulators of RyR, such as the cationic stains,aminoglycoside antibiotics and synthetic polypeptides, haveseveral or multiple amino groups in their molecular structure.Some cationic stains applied in microscopy such as RR caninhibit RyR. RR (40), applied for polysaccharide, glycoam-inoglycan and pectin staining, inhibits RyR at a concentra-tionof 0.1 – 1 µM. RR has very promiscuous effects and,besides RyR, binds CaM and can inhibit CaM-dependentenzymes, Ca2+-dependent phosphodiesterases [258], Ca2+-mitochondrial channels [259] and can interact with Ca2+-ATPases [260] and capsaicin receptor [261]. Some other stains,such as hexamminecobalt(III) chloride (41) or hexammineru-thenium(III) chloride, also inhibit Ca2+ efflux but at higherconcentration.
Aminoglycoside antibiotics, such as neomycin, gentamicin,streptomycin, clindamycin, kanamycin and tobramycininhibited Ca2+ release via RyR. Neomycin and gentamicininhibited RyR in skeletal muscles in the nanomolar range,whereas in the experiments with RyR in bilayers they wereeffective at a micromolar level. Synthetic peptides with multi-ple NH3-groups, such as poly-Lys and poly-Arg induce Ca2+
release or have a biphasic effect: poly-Lys activated Ca2+
S
NF
F
F
N
N
25 Trifluorperazine
S
N Cl
N
26 Chlorpromazine
N+
N
Cl
Cl
O
Cl
Cl
Cl Cl
• Cl-
27 Calmidazolium chloride
SNH
O
O
Cl
NH
28 N-(6-Aminohexyl)-5-chloro-1-naphthensulfonamide(W7)
OH
OH
N
N
NH
OH
O
NH
O
OHNH
NH
O
O
NH2
NH
O
OH
OH
OH
NH2
NH
O
OH
OH
• 2CH3CO2H
29 FK-506 (Tacrolimus)
O
OH
NH
30 Propranolol
OH
31 Menthol
OH
32 Thymol
Author p
roof
Intracellular calcium channels and their modulators
16 Expert Opin. Ther. Patents (2003) 13(6)
release at nanomolar (300 nM) concentration but inhibitedCa2+ release at micromolar (3 µM) level.
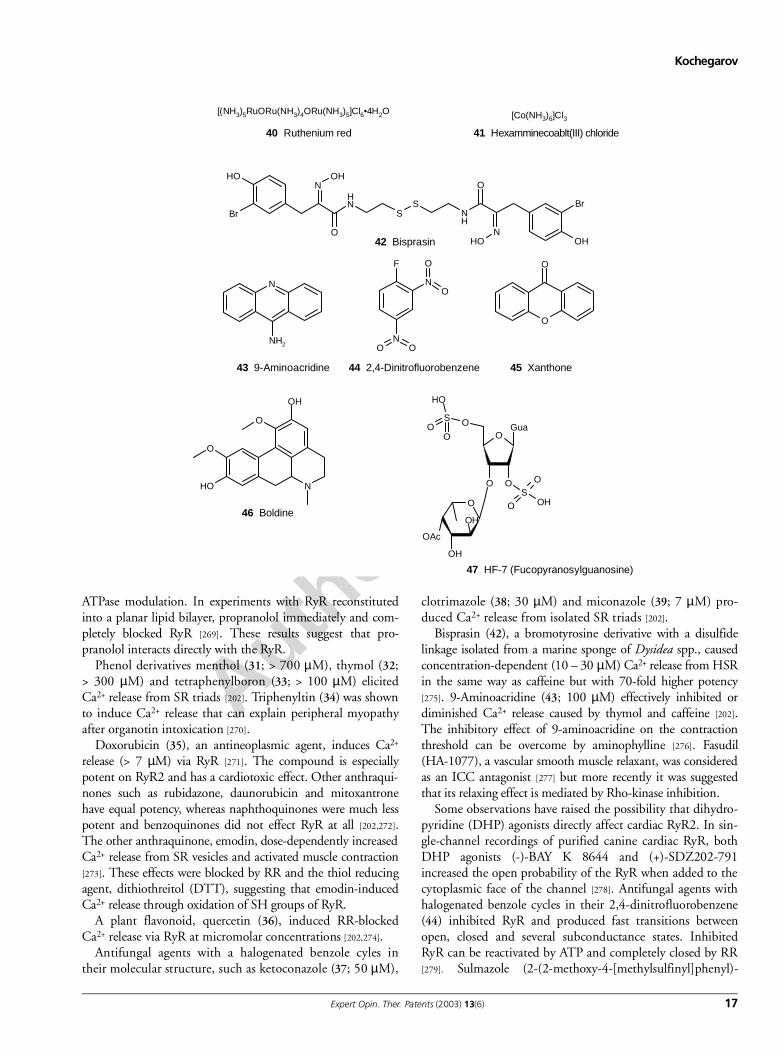
3.5.9 Miscellaneous compoundsDantrolene selectively inhibits skeletal RyR1 and brain RyR3isoforms, whereas it does not affect cardiac RyR2 [224]. Muta-tions in RyR, such as Arg615Cys associated with MH, confer athreefold decrease in Ry binding. It was shown that dantrolene(10 µM) effectively reversed the Kd to the normal value. Dan-trolene inhibition of the RyR1 was dependent on the presenceof the adenine nucleotide and CaM.
Since CaM inhibits Ca2+ release, it was proposed that CaMinhibitors activate Ca2+ release. It was reported that at concen-trations sufficient to inhibit CaM interaction, inhibitors didnot affect Ca2+ release, but at higher concentrations trifluop-erazine (25; EC50 = 130 µM), chlorpromazine (26; 180 µM),calmidazolium (27; 100 µM) and compound 48/80 (70 µg/ml)stimulated Ca2+ release [202,262]. Ca2+ release was inhibited byRR, procaine and Mg2+ indicating that the release was viaRyR. On the other hand, their concentrations were higherthan that required for CaM inhibition, so it is possible thatthese drugs activated RyR by a CaM-independent pathway.The CaM antagonist, sulfonamide derivative N-(6-amino-hexyl)-5-chloro-1-naphthalenesulfonamide (W7; 28), wasreported to activate Ca2+ release (EC50 = 100 µM) [202] but abiphasic action was also observed; at 10 µM it decreased theCa2+ current and at 100 µM it reactivated the channel [263].Some drugs that are supposed to act as CaM antagonists, suchas bepredil and felodipine, did not affect Ca2+ release.
FK-506 (tacrolimus; 29), a macrolide of fungal origin pro-duced by Streptomyces tsukubaenis, is applied as an immuno-suppressive drug in cardiac allograft transplantation. The drugbinds immunophilin and blocks calcineurin required fornuclear factor of activated T cells (NF-ATc) translocation. Itblocks production of IL-2 by helper T cells, thereby blockingT cell activation, proliferation and amplification of immuneresponse [264]. The other target of this drug is FKBP associatedwith RyR. Treatment with FK-506 dissociates FKBP fromRyR and alters gating characteristics of RyR increasing RyRsensitivity to Ca2+ and increasing Po and produces Ca2+ leakfrom SR [265,266]. Similar effects were observed with other anti-fungal antibiotics such as rapamycin produced by S. hygroscopi-cus. An abnormal Ca2+ leak through the RyR is present in heartfailure and this leak is presumably caused by a loss of RyR-bound FKBP12.6. This Ca2+ leak might possibly cause Ca2+
overload and consequent diastolic and systolic dysfunction[266]. It was reported that propranolol restores FK-506-inducedconformational change and prevents Ca2+ leak from RyR [267].This drug could be applied as a protective component duringFK-506 treatment. Propranolol (3-(1-naphthyloxy)-2-propa-nol) (30), a synthetic β-adrenergic receptor blocker having anantihypertensive effect at 0.4 mM, stimulated an accumulationof Ca2+ in SR, which was ascribed to the blockage of the RyR[268]. Since this compound increases Ca2+-ATPase activity bymore than twofold, it may be possible that the effect is due to
B
Na+
33 Sodium tetraphenylborate (tetraphenylborate)
Sn Cl
34 Triphenyltin
O
O OH
OH
O
O
OH
OH
O
O
OH
NH2
35 Doxorubicin
O
O
OH
OH
OH
OH
OH• 2H2O
36 Quercetin dihydrate
N N O
O
N
N
O
O
Cl Cl
37 Ketoconazole
N
NCl
38 Clotrimazole
N
N
Cl
Cl
O
Cl
Cl39 Miconazole
Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 17
ATPase modulation. In experiments with RyR reconstitutedinto a planar lipid bilayer, propranolol immediately and com-pletely blocked RyR [269]. These results suggest that pro-pranolol interacts directly with the RyR.
Phenol derivatives menthol (31; > 700 µM), thymol (32;> 300 µM) and tetraphenylboron (33; > 100 µM) elicitedCa2+ release from SR triads [202]. Triphenyltin (34) was shownto induce Ca2+ release that can explain peripheral myopathyafter organotin intoxication [270].
Doxorubicin (35), an antineoplasmic agent, induces Ca2+
release (> 7 µM) via RyR [271]. The compound is especiallypotent on RyR2 and has a cardiotoxic effect. Other anthraqui-nones such as rubidazone, daunorubicin and mitoxantronehave equal potency, whereas naphthoquinones were much lesspotent and benzoquinones did not effect RyR at all [202,272].The other anthraquinone, emodin, dose-dependently increasedCa2+ release from SR vesicles and activated muscle contraction[273]. These effects were blocked by RR and the thiol reducingagent, dithiothreitol (DTT), suggesting that emodin-inducedCa2+ release through oxidation of SH groups of RyR.
A plant flavonoid, quercetin (36), induced RR-blockedCa2+ release via RyR at micromolar concentrations [202,274].
Antifungal agents with a halogenated benzole cyles intheir molecular structure, such as ketoconazole (37; 50 µM),
clotrimazole (38; 30 µM) and miconazole (39; 7 µM) pro-duced Ca2+ release from isolated SR triads [202].
Bisprasin (42), a bromotyrosine derivative with a disulfidelinkage isolated from a marine sponge of Dysidea spp., causedconcentration-dependent (10 – 30 µM) Ca2+ release from HSRin the same way as caffeine but with 70-fold higher potency[275]. 9-Aminoacridine (43; 100 µM) effectively inhibited ordiminished Ca2+ release caused by thymol and caffeine [202].The inhibitory effect of 9-aminoacridine on the contractionthreshold can be overcome by aminophylline [276]. Fasudil(HA-1077), a vascular smooth muscle relaxant, was consideredas an ICC antagonist [277] but more recently it was suggestedthat its relaxing effect is mediated by Rho-kinase inhibition.
Some observations have raised the possibility that dihydro-pyridine (DHP) agonists directly affect cardiac RyR2. In sin-gle-channel recordings of purified canine cardiac RyR, bothDHP agonists (-)-BAY K 8644 and (+)-SDZ202-791increased the open probability of the RyR when added to thecytoplasmic face of the channel [278]. Antifungal agents withhalogenated benzole cycles in their 2,4-dinitrofluorobenzene(44) inhibited RyR and produced fast transitions betweenopen, closed and several subconductance states. InhibitedRyR can be reactivated by ATP and completely closed by RR[279]. Sulmazole (2-(2-methoxy-4-[methylsulfinyl]phenyl)-
[(NH3)5RuORu(NH3)4ORu(NH3)5]Cl6•4H2O
40 Ruthenium red
[Co(NH3)6]Cl3
41 Hexamminecoablt(III) chloride
OH
Br
NOH
NH
O
SS
NH
O
NOH
Br
OH42 Bisprasin
N
NH2
43 9-Aminoacridine
N
F
O
O
NO O
44 2,4-Dinitrofluorobenzene
O
O
45 Xanthone
N
OH
O
O
OH
46 Boldine
O
O
OAc
OH
O
OS
OS
O
O OH
Gua
OH
OH
OO
47 HF-7 (Fucopyranosylguanosine)

Author p
roof
Intracellular calcium channels and their modulators
18 Expert Opin. Ther. Patents (2003) 13(6)
1H-imidazo(4,5-b)pyridine) and its related enantiomers, (+)-and (-)-isomazole activate RyR with an EC50 of 445 µM [280].Sulmazole and isomazole, analogues, EMD 46512 and EMD41000, were 10 times more potent.
Xanthone (45) and norathyriol (1,3,6,7-tetrahydroxyxan-thone) can induce Ca2+ release from the SR of skeletal musclethrough a direct interaction with the Ca2+ release channel [281].Xanthone also activated the sarco(endo)plasmic reticulum Ca2+
ATPase (SERCA) pump. When tested in mouse diaphragm,norathyriol potentiated the muscle contraction followed bytwitch depression and contracture in either a Ca2+-containingor Ca2+-free media. Digitalis glycosides digoxin, digitoxin andactodigin activated Ca2+ release at nanomolar concentrationsvia cardiac RyR2 [282,283]. This effect may contribute to the ino-tropic activity of cardiac glycosides. Cardiac glycosides also acti-vated frog, but not mammalian, skeletal muscle RyR [284]. Abipyridine derivative, milrinone (1,6-dihydro-2-methyl-6-oxo-(3,4′-bipyridine)-5-carbonitrile), activates cardiac RyR2 at con-centrations of 100 µM – 2 mM. By contrast, another bipyrid-ine, 1,1′-diheptyl-4,4′-bipyridinium dibromide inhibited, theCa2+ release induced by 2 mM caffeine and polylysine [285].Milrinone is also a phosphodiesterase inhibitor. Another phos-phodiesterase inhibitor enoximone was without effect on singlechannel activity [286]. Boldine [(S)-2,9-dihydroxy-1,10-dimeth-oxyaporphine] (46), a major alkaloid in the leaves and bark ofboldo (Peumus boldus Mol.), has antioxidant and neuro/hepato-protective properties. The compound is a weak adrenoceptorand VGCC antagonist. It was shown that boldine induced Ca2+
release from SR, which was inhibited by RR at concentrationsof 300 µM and induced muscle contracture [287].
3.5.10 Sulfhydryl reagents and metal ionsReactive disulfides, metal ions, phthalocyanine dyes, H2O2and various anthraquinones, oxidise thiol (-SH) groups inRyR and produce Ca2+ release. Some metal ions, such as Hg2+
and Ag+ produce rapid Ca2+ release from the SR. Their effectcan be explained by either binding to sulfhydryl (SH) groupsof Cys residues in RyR or by catalysing the oxidation to adisulfide. Sulfhydryl reagents, such as N-ethylmaleimide (NEM,1 – 30 mM), thimerosal (200 – 400 µM), 5-5′-dithiobis-2-nitrobenzoic acid (> 300 µM) and N-succinimidyl 3-(2-pyri-dylthdithio) propionate (25 – 100 µM) activate Ca2+ release,which may be inhibited by thiol-reducing agents DTT andglutathione suggesting that it was mediated by sulfhydryl oxi-dation [202]. Ca2+ release induced by sulfhydryl reagents wasinhibited by RR and procaine indicating that Ca2+ wasreleased through RyR. Reactive disulfide compounds with apyridyl ring 2,2′dithiodipyridine and 4,4′ dithiodipyridineare very specific to free sulfhydryl (SH) groups. They induceCa2+ release at low concentration (> 2.5 µM). The benzo-phenanthrine alkaloid from Sanguinaria canadensis sanguinarineinduced Ca2+ release from loaded SR vesicles which wasblocked by RR and DTT, consistent with RyR as the site ofsanguinarine action [288]. Sanguinarine can also inhibit Ca2+-and Na+/K+-ATPases [289].
The sulfhydryl alkylating agent NEM has three distincteffects on Ca2+ release channel activity. First, channel activity isdecreased (phase 1); then with continued exposure the activityis dramatically increased (phase 2); and finally, the channel isagain inhibited (phase 3) [290]. In contrast, other authorsobserved activation, inhibition and then activation again [291].Both H2O2 and NO block the sometimes observed inhibitoryeffect of NEM. Low NEM concentrations (20 – 200 µM) elic-ited Ca2+ release from SR, which was fully reversed by reduc-ing agents or by washing. NEM (5 mM) decreased, increasedand then decreased the Po of the channel. The final reactionwas not reversed by reducing agents, as expected for an alkyla-tion reaction. A non-sulfhydryl reagent structurally related toNEM, N-ethylsuccinimide (0.1 – 0.5 mM), also elicited SRCa2+ release that was not reversed by DTT [291]. These datasuggest that at high concentrations, NEM may activate RyRswithout thiol alkylation.
The copper containing phthalocyanine dyes, alcian blue,copper phthalocyanine tetrasulfonic acid and Luxol fast blue,induce rapid calcium efflux from SR vesicles at micromolarconcentrations, which is RR-inhibited and reversible byreducing agents such as DDT [292]. The anthraquinones, dox-orubicin, mitoxantrone, daunorubicin and rubidazone areshown to be potent stimulators of Ca2+ release from skeletalmuscle SR vesicles and to trigger transient contractions inmuscle fibres [293]. Potent oxidising agents 5,5′-dithioni-trobenzoic acid and 5,5′-dithiobis-2-nitrobenzoic acid (syno-nyms: 2,2′-nitro-5,5′-dithiobenzoic acid, DTNB) producedan increase in Ca2+ release [202]. It was also reported that theireffect was insignificant and appeared only as a 2 – 3-foldincrease in the low rate of Ca2+ leak from the SR when allCa2+-uptake was blocked [294]. Related compounds, p-chlo-romercuribenzoic acid (pCMB) and p-chloromercuribenzenesulfonic acid also produced Ca2+ release [202].
Micromolar concentrations of some heavy metal ions, suchas Ag+ and Hg2+, activate Ca2+ release [295-298]. Some papersalso mentioned Cd2+ and Zn2+ as agents producing Ca2+
release [299] but others demonstrated that Cd2+ and Zn2+ wereineffective up to 200 µM. Low concentrations, 5 – 20 µM ofAg+ (AgNO), induced Ca2+ release and elicited muscle con-tracture [202]. It was concluded that contracture showed abiphasic mode: CICR-dependent and CICR-independent. Incaffeine solution, 0.5 µM Ag+ caused a long-lasting contrac-ture sometimes with two peaks and this contracture was inhib-ited by 2 mM procaine [300]. Pretreatment of fibres with Au3+
inhibited the Ag(+)-induced phasic contracture [300]. Themechanism of action of silver ions has not yet been clarified.One idea is that Ag+ may catalyse some reactions, such as oxi-dation of thiol groups. Another idea is that Ag+ indirectly acti-vates RyR by interaction with voltage-sensor and extracellularAu3+ at low concentrations modifies the interaction of Ag+
with voltage sensors in the TT [300]. Mercury (4 µM Hg2+),known to react with sulfhydryl groups, induces Ca2+ releasefrom SR [295,298]. Micromolar concentrations of Cu2+ and mer-captans such as cysteine, cysteamine and homocysteine trigger

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 19
large and rapid Ca2+ release from skeletal muscle SR vesicles[296]. It was suggested that Cu2+ catalyses H2O2 oxidation ofCys residues in RyR. The Ca2+ pump appears to be one of thetargets inhibited by heavy metals [295,301].
Organotin compound triethyltin induced Ca2+ release fromSR vesicles, and also inhibited SERCA pump and induced mus-cle contracture in mouse diaphragm [302]. Suramin is a polysul-fonated naphthylurea developed originally to treattrypanosomiasis and also as an anticancer agent. This compoundevokes Ca2+ release from skeletal muscle SR by activation of RyRand it can also interfere with the Ca2+-pump [303,304].
4. Inositol-1,4,5-triphosphate-receptor
The IP3R is a Ca2+-channel in the ER activated by IP3. Func-tional IP3R is a homo- or heterotetramer of ∼ 240 kDa glycopro-tein subunits. IP3R Type I (5IP3R-I) has been detected in heart, liver, kidney, ovary andPurkinje neurons of the cerebellum. IP3R-II is found predom-inantly in the brain. IP3R-III is expressed in pancreatic islets,kidney and the gastrointestinal tract. Rat IP3R-III is 2670amino acids in size, has 62 and 64% identity with IP3R-1 andIP3R-II, respectively [305]. IP3R has 6 TM, although a differ-ent model with 8 TM has also been predicted.
4.1 IP3-transduction pathwaysStimulation of a receptor in the cell membrane by its agonistresults in activation of phospholipase C (PLC). There are dif-ferent phospholipases: PLC, PLA and PLB recognising differ-ent sites in the phospholipid molecule. PLC producesdiacylglycerol (DAG) and IP3 by hydrolysing the membranelipid phosphatidylinositol 4,5-bisphosphate (PIP2) (Figure 7).DAG in combination with Ca2+ is necessary for activation ofprotein kinase C (PKC). Four isoforms of PLC weredescribed: PLC- α , β, γ and δ. PLC-α was later proven to be aproteolytic product of PLC-δ. These isoforms differ in theirability to hydrolyse PI, PIP or PIP2 and their dependence onCa2+. PLC-β is activated by the superfamily of G-protein-cou-pled receptors (GPCR) with 7 TM regions (Figure 6b) includ-ing: α adrenoreceptor, muscarinic acetylcholine-acivated M1,M3 and M5-receptors, purinergic ATP-activated P2y, P2u andP2t receptors, serotonin 5-HT receptor, histamine H1 recep-tor, T cell receptor (TCR), thrombocyte platelet activatingfactor (PAF) receptor, gonadotrophin-releasing hormone(GnRH) receptor, neutrophil N-formyl-methionyl-leucyl-phenylalanine (f-MLP) receptor, glucagon, vasopressin, oxito-cine, thrombin, thromboxane, bradykinin and tachykininreceptors etc., to name a few. Activating signal is transmittedby G protein types Gi and Gq. It is accepted that the Gα sub-unit participates in signal trunsduction but it was also shownthat the Gβγ dimer can interact with PLC-β [306].
Initially it was hypothesised that the IP3R may participate invision signalling in Drosophila photoreceptor neurons butrecent experiments demonstrated that Gq and PLC activation isrequired but IP3 production and Ca2+ release from intracellular
Ca2+ pools is not necessary [307]. Some data indicate that the IP3pathway may participate in bitter and sweet transduction.There are three predominant hypotheses about bitter and sweetreception: i) G protein/PLC/IP3 pathway, ii) G protein/cAMPpathway and iii) K+-channel as a receptor [308-310]. It is not clearwhether all these mechanisms co-exist in one bitter receptor cellor if only one of them is important for bitter transduction.
The second class of receptors activating PLC-γ includesgrowth factor receptors, including the epidermal growth fac-tor receptor (EGFR), fibroblast growth factor receptor(FGFR), platelet-derived growth factor receptor (PDGFR),ErbB2, and some antigen receptors, T and B cell receptors,IgE basophil (FcεRI) and IgG monocyte (FcγRII) receptors.After ligand binding the receptor is autophosphorylated andrecruits and phosphorylates PLC rendering it active.
4.2 IP3-receptor regulationActivity of IP3R is generally controlled by concentrations ofIP3 and [Ca2+]i. It was shown that when IP3 is absent, IP3R
Figure 7. Production of IP3 and DAG from membranephospholipid PIP2.DAG: Diacylglycerol; IP3: Inositol-1,4,5-triphosphate; PIP2: Phosphatidylinositol4,5-bisphosphate; PLC: Phospholipase C.
O-OPO
O-
O-
OH OH
OH
OPOO-
O-
P
O
O
O
PLC
PIP2 DAG
IP3
1
2
34 6
123
4
5
65
O
O
O-PO
O
O-OP
OO-
O-
OH OH
OH
OP
OO-
O-
O
O
O
O
OH
O
O

Author p
roof
Intracellular calcium channels and their modulators
20 Expert Opin. Ther. Patents (2003) 13(6)
has low ‘basal’ conductance. The other inositol derivative, tet-rakisphosphate (1,3,4,5IP4), inhibits IP3R [311]. The Po ofIP3R isoforms I and II have a bell-shaped dependence on[Ca2+]i, whereas the Po of the Type III isoform increasesmonotonically with increasing [Ca2+]i [312]. It was suggestedthis difference might have important functional conse-quences. Activation of Type III IP3R results in a single butglobal increase in [Ca2+]i that is more relevant for signal trans-duction, whereas the properties of types I and II IP3R are idealfor supporting Ca2+ oscillations. The cytoplasmic region ofIP3 has binding sites for regulatory molecules IP3, Ca2+, ATPand CaM. CaM plays a key role in the Ca2+-dependent inacti-vation of Type I IP3Rs [313]. Experiments with reconstitutedIP3R in non-cellular lipid bilayers revealed that in the absenceof CaM, Ca2+ acts positively on IP3R, but in the presence ofCaM, the high Ca2+ level inactivates the receptor [313]. CaMmight also affect IP3R via CaMKII [314]. There are two Serresidues, 1588 and 1755, potentially phosphorylated by PKA,PKG and CaMPK [315]. It was reported that Gi proteins coulddirectly associate and regulate IP3R [316].
Binding of adenine nucleotides increases Po of all three iso-forms but it was also reported that high levels of ATP(> 6 mM) may inhibit Type III IP3R. The binding specificityof Type 1 IP3R was ATP > ADP > AMP > GTP [317]. Somecompounds containing, or related to, adenine nucleotide, suchas CoA, adenophostin A, caffeine and cyclic ADP-ribose,which can modulate Ca2+ release via IP3R, were proposed toshare the adenine binding site. There are two splicing sites inthe IP3R gene: SI located within the IP3 binding site and SIIlocated between phosphorylation sites and near to theATP-binding site [315]. Inside SII there are three domains: A, Band C, which may be independently deleted. The existence ofspliced isoforms of rat brain IP3R have been reported: includ-ing SI or without SI and SIIABC, SIIAC, SIIA and full deletedSII (SII-) [315]. Splicing combinations might theoretically pro-duce ∼ 15 isoforms. Deletion of SI significantly decreases IP3binding. Deletion of SII located between Ser1588 andSer1755 may affect their susceptibility to phosphorylation andalso the binding of ATP to a nearby site (1773 – 1800) [315].Therefore, the differential expression of these slicing isoformsduring development and differenet tissue types may confer dif-ferent properties of IP3R and hence different responses.
PKA was shown to phosphorylate and enhance activation ofIP3R by IP3. Ser1589 and Ser1755 were determined as PKAsequence consensus in the neuronal Type 1 IP3R. IP3R2 andIP3R3 lack these consensus PKA phosphorylation sites, althoughthey may contain other putative PKA phosphorylation sites. Itwas demonstrated that PKA is able to phosphorylate types IIand III, albeit with lower efficiency (0.65 mol Pi Type I and0.04 and 0.14 mol Pi types II and III, respectively) [318]. It isgenerally suggested that phosphorylation of IP3R results inenhancement of IP3 potency but this effect is not very signifi-cant, ∼ 20% enhancement of IP3 potency and ∼ 10% the maxi-mal effect. Sometimes the opposite effect is observed. It washypothesised that in cells which express both types I and II, IP3R
may be heterotetrameric, which is more affected by PKA. PKGwas also shown to phosphorylate the same sites albeit with dif-ferent efficiency in the Type I IP3R [319]. PKC phosphorylatesnuclear IP3R and activate Ca2+ release [320]. Inhibition of IP3Rby AA was demonstrated with Ki 27 nM [321].
4.3 Modulators of IP3RThe pharmacology of IP3R is less developed than for RyR. Todate, there is no known reliable membrane-permeable inhibi-tor/activator of IP3R. Heparin is the most common inhibitorof IP3R for research purposes. Heparin is used as a blood anti-coagulant, which forms a complex with antithrombin III andinhibits several blood coagulation factors. Heparin is a hetero-geneous mixture of sulfated polysaccharide with the differentsugars (2-deoxy-2-sulfamino-α-D-glucose 6-sulfate >α-L-iduronic acid > 2-acetamido-2-deoxy-α-D-glucose >β-D-glucuronic acid) joined by glycosidic linkages. Heparincannot penetrate the cell membrane, therefore restricting itsapplication as an IP3R inhibitor.
Adenophostins A and B isolated from fungus Penicilliumbrevicompactum structurally resemble IP3. Adenophostin A,an agonist of IP3R, is the most potent, being ten times morepotent than IP3 [322]. Acyclophostin is a ribose-modified ana-logue of adenophostin [323]. They are not metabolised byIP3-converting enzymes and do not bind putative IP4-recep-tors. These compounds cannot penetrate the cell membrane.
Xestospongin (Xe) A, C, D, araguspongine B and demethyl-xestospongin B, a group of macrocyclic bis-1-oxaquinolizi-dines isolated from the Australian sponge Xestospongia sp., weresuggested to be membrane-permeable IP3R inhibitors [324-326].However, these compounds inhibited Ca2+ release in permea-bilised cells but not in cells with an intact plasma membrane[327]. Moreover, there have been contradictive data regardingthe effect of XeC. It has been reported that XeC induced Ca2+
release from the ER in neurons and this effect was similar tothat triggered by the Ca2+-ATPase inhibitor, thapsigargin[328-330]. Authors concluded that XeC is a potent inhibitor ofSR/ER Ca2+-ATPase and it cannot be regarded as a specificinhibitor of IP3Rs. 2-Aminoethoxydiphenyl borate (2-APB)was proposed as a membrane-permeable IP3R inhibitor. How-ever, it was reported that 2-APB and XeC also inhibits capaci-tative Ca2+ entry independently of the function of IP3R[331-334]. In Drosophila, 2-APB reversibly inhibited photorecep-tors activation acting on at least two processes thought tomediate the visual cascade. 2-APB inhibited both light andIP3-induced Ca2+ release, consistent with its role as an inhibi-tor of the IP3R. In addition, 2-APB reversibly inhibited theactivation of depolarising current flow through the plasmamembrane caused injection of calcium ions into the photore-ceptor and voltage-activated K+ current during depolarisation[335]. The most recent paper reported that ‘in experiments withpermeabilised cells’ 2-APB could inhibit IP3-induced release ofstored Ca2+ but could also release Ca2+ and the prevalence ofthese two effects varied between different cell types and did notcorrelate with the expression of a particular receptor type [336].

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 21
Recently, a new neuroactive compound, HF-7 (fucopyranosyl-guanosine; 47), was isolated from the venom of the funnel webspider, Hololena curta. HF-7 is an anionic nucleoside, whichaffects the intracellular concentration of Ca2+ in nerve cells.The exact target of HF-7 is not known, it may be an intracellu-lar Ca2+ channel such as IP3R [337].
5. Patent review of intracellular Ca2+ channels
Novel dioxobenzothiazole derivatives were patented as RyRmodulators for altering the concentration of free calcium inorder to treat diseases including congestive heart failure,migraine headache, hypertension, premature abortion, Par-kinson’s disease and Alzheimer’s disease (Table 1) [401].Although the specified compound did not exhibit a signifi-cant ability to close muscle RyR in a functional passive cal-cium efflux assay, it was active in a Ry binding affinity assay(IC50 = 275 nM). Some dioxobenzothiazoles can inhibit themitochondrial respiratory chain and are thus toxic.
Recently a high affinity polypeptide, G224, which has acomparable binding activity to IP3 was discovered [402]. It wasfound that this polypeptide acted as an IP3 sponge and had asignificant inhibitory effect on IP3-induced Ca2+ releasedepending on its concentration. It was suggested that G224might be used as a therapeutic agent for diseases associatedwith upregulations of IP3 and Ca2+ pathways. When pharma-cists deal with peptides or any membrane non-permeablecompounds, the first question to be addressed is how todeliver the compound inside cells. The drug can be used fortherapeutic application only if there is a technique to deliverthe drug to inside the cell.
It was thought that NAADP activated RyR but now itappears to target a different Ca2+-releasing channel, namely thehypothetical NAADP-activated intracellular Ca2+ channel,although in the literature there is no evidence about isolation ofthis channel, or else, this channel may represent an uncharacter-ised isoform (probably, a new splicing isoform) of RyR. Micro-injection of NAADP to Jurkat T cells selectively stimulates Ca2+
release at low concentrations (10 – 100 nM) but it self-inacti-vates release at higher concentrations (> 10 µM). The NAADP-mediated release was not affected by cADPR analogues but itwas partially reduced by an antagonist of the IP3-mediated sys-tem [403]. These results suggest that NAADP-induced Ca2+
release was not RyR-mediated; and IP3R-mediated Ca2+ releasemay be triggered by CICR mechanism. However, a techniquefor intracellular delivery of this compound should be developedbecause microinjection is not suitable for medical application.
Some synthetic cADPR analogues were patented, whichantagonise cADPR-mediated calcium release via RyR. Theywere claimed for treatment of autoimmune disorders includingthyroiditis, multiple sclerosis, uveitis, Addison’s disease, rheuma-toid arthritis and lupus erythematosus [404]. It is very importantthat pharmaceutical drugs designed for intracellular targets canpenetrate cell membranes or else they become useless or requireconjugation with special chemical carriers to deliver them into a
cell. Data regarding the ease of penetration of these compoundsacross the cell membrane is important and should be reportedwithin the patent literature. cADPR is considered not to be apermeant compound. In a recent patent, preincubation ofT cells (Jurkat cells) with the specified compound (1 µM) didnot affect calcium signalling [404]. If this compound cannot pen-etrate the cell membrane it is not surprising that there was noresponse. However, at higher concentrations this compoundinhibited long-lasting calcium entry but did not inhibit thapsi-gargin-induced calcium release. It is not clear if the compoundcan penetrate the cell membrane at higher concentrations orwhether it can affect calcium transport by a different pathway. Itis well known that Ca2+ channels of the TRP family mediatelong-lasting current. It could be speculated that cADPR deriva-tives may directly or indirectly affect TRP calcium channels butno evidence of such a mechanism exists in the literature. Thespecified compound dose-dependently (1 – 100 µM) inhibitedT cell proliferation and was shown to inhibit joint swelling in anantigen-induced arthritis model in mice.
Maurocalcine is a toxin isolated from the scorpion venomsof the Tunisian chactidae and Scorpio maurus palmatus. Addi-tion of the toxin to the cytoplasmic face of planar lipid bilayerscontaining RyR1 was found to modulate channel conductance[405]. The effects were blocked by the addition of RR and didnot appear to be mediated by the Ry site. The specified com-pound produced no effect on the luminal side of the lipidbilayers, indicating a cytoplasmic site of action. Synthetic andrecombinant maurocalcine and its bioactive analogues werepatented as immunosuppressant medicaments and in compo-sitions for treating disorders associated with calcium channeldysfunction [405]. It is an interesting finding, but first, it is nec-essary to figure out if this toxin can penetrate into the cell.
Myasthenia gravis (MG) is an autoimmune disease which ischaracterised by the production of abnormal antibodies whichtarget and disrupt pathways of electro-mechanical coupling(EMC) between nerves and muscles. MG patients have anti-bodies against muscle RyR and acetylcholine receptor whichmediate EC-coupling. Production of abnormal antibodiescorrelates with thymomas; tumours of the thymus where lym-phocytes maturate and differentiate. The antibodies againstRyR1 recognise a region near the N terminus, which seems tobe important for RyR regulation [338]. The antibodies causeallosteric inhibition of RyR function and Ca2+ release fromSR. Recently, new compounds were patented that inhibit thebinding of antibodies to the human RyR [406]. The use offusion proteins for the detection of antibodies specific forRyR in patient blood is also claimed.
Upregulation of cytosolic Ca2+ levels may be responsible forneuronal injury and neurodegenerative disorders. A novelmethod for treating neuronal reperfusion injury in patientswith ischaemic neuropathy, stroke and excitotoxic retinaldamage in glaucoma has been patented [407]. The methodcomprises the administration of the RyR antagonist, dantro-lene, to decrease cytosolic Ca2+ levels. Dantrolene has beenused for the treatment of malignant hyperthermia for many

Author p
roof
Intracellular calcium channels and their modulators
22 Expert Opin. Ther. Patents (2003) 13(6)
years and may be useful for the treatment of some neurode-generative disorders and stroke.
Recently, new methods of treating Huntington’s diseasewith multiple receptor antagonists and agonists, such as Ry,IP3, dopamine and retinoic acid receptors, a Ca2+-ATPaseenhancer, a calcium binding protein, a cAMP regulatingagent, a raft/caveolin structure normalising agent, nervegrowth factor (NGF), an agonist of the opioid or cannabinoidsignalling pathways and an anti-inflammatory agent were pat-ented [408]. It was reported that CD38-like enzymes that cata-lyse the synthesis of cADPR is upregulated by retinoids,retinoic acid, TNF and IL-1β. The only specified compoundin the patent is 9-cis-retinoic acid, a retinoic acid receptoragonist, which stimulates cADPR synthesis. This finding isquite opposite to previous data that suggests that an increasein [Ca2+]i contributes to neuronal injury. From this viewpointit seems unlikely that the curative effect of retinoic acid ismediated by regulation of intracellular [Ca2+].
Obesity is mostly a result of disorders of metabolic path-ways, when synthesis and accumulation of fatty acids exceedstheir degradation. Drugs increasing energy consumptionwould be promising therapeutic agents for the treatment ofobesity. Recently, compounds that induce Ca2+ releasethrough muscle type RyR1 were claimed for the treatment ofobesity [409]. The effect of RyR1 agonists on energy expendi-ture in rats, hamsters, mice and guinea-pigs was determined.The patented drug is a macrocyclic bromotyrosine derivativeoriginally isolated from the sponge Ianthella basta. Bastadinsact as selective agonists or modulators of skeletal muscleRyR1. The most potent activators were bastadins 5 and 7, andthe effect of bastadins were shown to be additive with that ofcaffeine and ATP and inhibited by the immunosuppresant,FK-506, suggesting that bastadins interact with FKBP12, asopposed to a direct interaction with RyR.
However, care must be taken with activators of RyR as some ofthese compounds, for example, general anaesthetics, activatingRyR and provoking Ca2+ release can result in drug-induced disor-ders like MH. More promising agents would specifically targetonly adipocytes, cells accumulating fatty acids. Interestingly,some small mammals living in cold environments or using hiber-nation to survive during the winter season developed a specialmechanism to enhance heat production. This mechanisminvolves the uncoupling protein (UCP) in mitochondria ofbrown adipose tissue. UCP-1 is exclusively expressed in brownadipocytes where it uncouples respiration from ATP synthesis,dissipating the proton gradient as heat. In adult humans,although UCP-1 can be detected there is a lack of brown adiposetissue. Understanding the mechanism which regulates the expres-sion of human UCP-1 will greatly facilitate the identification ofdrugs able to modulate energy expenditure in adult humans [339].
Xestospongins are a new group of macrocyclic compoundsisolated from the sponge Xestospongia sp. It was reported thatbis-1-oxaquinolizidine derivative, xestospongin C, could inhibitIP3R [410]. It is not yet clear if xestospongins are specific inhibi-tors of IP3R and if they can penetrate the cell membrane. Bis-1-
oxaquinolizidine derivatives, phospholipase C inhibitors andG protein inhibitors were patented by Contrimmune Biotech,Inc. for treating HIV and other viral infections, inflammatorydisease, psoriasis, pain, uncontrolled growth disorders such ascancer, cardiac arrhythmia and hypertension [410].
Some patents contain incredible information, for example, arecent patent claims that the vasodilating effect of NO donors isdue to the activation of the L-type calcium channel and the RyRby nitrosylating the thiol moieties of the proteins [412]. Actually,vasodilator drugs such as calcium channel blockers and β-block-ers decrease [Ca2+]i levels producing relaxation of smooth mus-cles in the blood vessels resulting in a decrease in blood pressure,improving cardiac output. NO activates cGMP-cyclase, andincreases in cGMP levels result in PKG activation, which phos-phorylates and inhibits VGCC. The eventual effect is a decreasein cytosolic Ca2+ levels and actomyosin relaxation. Direct effectsof S-nitrosylation and oxidation produced by NO on VGCC arenot yet fully understood with both channel activation and inhi-bition possible and dependent on NO concentration and partic-ular cell type. The activating effects of NO on Ca2+ channels,which are sometimes observed in experiments, cannot result invasodilatation. It would be interesting to test if NO can increase[Ca2+]i specifically in the heart whilst at the same time produc-ing a decrease in [Ca2+]i in vascular muscles.
Recently, new ether and heteroaromatic derivatives were pat-ented as activators of intracellular Ca2+ channels with neuronalactivity [412]. However, it is not disclosed as to what kind ofICC they can affect. It is scarcely possible that they can affectboth RyR and IP3R because no known up-to-date compoundcan activate both of these receptor types. In the ‘keywords’, IP3ligand is mentioned, which makes it possible that the patenteddrug is an IP3R ligand. Ether derivatives in combination withneurotrophic factors were claimed as useful for preventing neu-rodegeneration in the treatment of neurological, mental andcognitive disorders, cardiovascular disease, muscle disease, neu-ropathy, myasthenia gravis and spinal cord disorders [412].
Heteroaromatic compounds and their salts are claimed tobe useful for the treatment and prevention of neurodegenera-tion and neurological disease, such as Parkinson’s and Alzhe-imer’s diseases, neuralgia, myasthenia gravis, musculardystrophy, multiple sclerosis and stroke [413]. What kind ofcalcium channel the specified compound targets is not dis-closed but the specified compound is a pyridine derivative,which suggests the specified compound can modulate RyR.
The compounds were claimed to be useful for stimulatingneuronal regeneration and neurite growth in nerve cells andfor the treatment of nerve injury, peripheral neuropathies,brain and spinal cord injury, ischaemia and trauma. The com-pounds can also be optionally applied in combination with aneuronal growth factor such as NGF. The drugs are stated toincrease cytoplasmic calcium concentration via a calciumrelease channel such as the RyR or IP3R in the ER of the nervecell. It is not clear how increased [Ca2+]i can stimulate nerveregeneration. It is generally considered that increased [Ca2+]i isnot desirable for neurons.
Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 23
Table 1. New patent on the modulators of intracellular calcium channels.
Assignee Patent title (and compounds)
Chemical structure(if available)
Utility Ref.
Advanced Res. & Tech. Inst., Inc. et al.
Modulators of RyR comprising 2-(aryl)-4,7-dioxobenzothiazoles and analogues (WO9932115 )
Congestive heart failure, migraine headache, hypertension, premature abortion, Parkinson`s disease and Alzheimer`s disease
[401]
Riken W-S et al.
High affinity IP3-binding polypeptide (EP-0992587-A)
Disorders of calcium metabolism
[402]
Univ. of Bath cADPR analogues for modulating T cell activity(WO0037089)
Treating immune disorders including thyroiditis, multiple sclerosis, uveitis, Addison`s disease, rheumatoid arthritis and lupus erythematosus
[404]
Cellpep SA Maurocalcine, analogues thereof and their therapeutical uses (WO0164724)
Immunosuppressant medicaments and for treating disorders associated with calcium channel dysfunction
[405]
Unifob Stiftelsen Universitetsforskning Bergen
Detection of ryanodine receptor antibodies (WO0075658)
Recombinant fusion proteins Detection and treatment of myasthenia gravis
[406]
Univ. of South Florida
Method and composition for treatment of ischeamic neuronal reperfusion injury (dantrolene) (WO0141756)
For neuronal reperfusion injury associated with ischeamic neuropathy, including ischeamic optic retinopathy, ischeamic retinopathy, stroke, reperfusion injury after tPA treatment or carotid endoarterectomy, seizures and excitotoxic retinal damage of the glaucoma
[407]
cADPR: Cyclic ADP ribose; IP3: Inositol-1,3-4-triphosphate receptor; NO: Nitric oxide; RyR: Ryandodine receptor; tPA:Tissue plasminogen activator.
S
N
O
O
N
N
NH
O
N
O
OH
H
OH
Br
PO
OHO
POH
O
O
OHOH
O
NH
N
O
O
NO
N
O
O
Author p
roof
Intracellular calcium channels and their modulators
24 Expert Opin. Ther. Patents (2003) 13(6)
Table 1. New patents on the modulators of intracellular calcium channels (continued).
Assignee Patent title (and compounds)
Chemical structure(if available)
Utility Ref.
Fred Hutchinson Cancer Res. Center et al.
Methods for treatment of human Huntington’s disease and methods of screening for active agents (9-cis-retinoic acid)(WO0134633)
Treatment of Huntington’s disease
[408]
Novo Nordisk AS et al.
Use of compounds for the treatment of obesity (Bastadin 5) (WO0222122)
Treatment of obesity [409]
Univ. of Bath NAADP analogues for modulating T cell activity(WO0211736 )
Treatment of autoimmune diseases, rheumatoid arthritis, lupus erythematosus, graft rejection and immune disorders, or for modulating immune responses
[403]
Johns Hopkins Univ.
Treatment of advanced decompensated heart failure with NO donors(WO9955317 )
Decompensated heart failure
[411]
cADPR: Cyclic ADP ribose; IP3: Inositol-1,3-4-triphosphate receptor; NO: Nitric oxide; RyR: Ryandodine receptor; tPA:Tissue plasminogen activator.
OHO
O
NH
O
O
OH
Br
NOH O
N
Br
Br
OH
BrBr
NH
OH
N
N
N
NH2
ON
O
OH O
H
P
OH
OHO
P
O
OH
O
Br
PO
O
O
O
OHOH
N+
O
OH
H
C N+
NO
N
O
NH

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 25
6. Expert opinion
The IP3R plays an important function in the transduction ofsignals from cell peripheries. Extracellular stimuli, such ashormones, cytokines, growth factors, antigens, glucose, NOand sperm activate multiple receptors on the cell membraneand induce different pathways resulting in Ca2+ release fromintracellular stores. This release may be represented as a sin-gle Ca2+ burst, Ca2+ spikes or as continuous Ca2+ waves andoscillations, the significance of these are not yet understood.The RyR has been recognised for many years as a key ele-ment in EC-coupling. In recent years, considerable data hasdemonstrated an important role for RyR in signal transduc-tion via the cADP-ribose pathway, as well as hypotheticalNAADP-receptor via NAADP. RyR was also reported torelease Ca2+ from the nuclear envelope. It is not clear if, in
this case, the nuclear envelope is just an additional volumefor Ca2+ storage or if Ca2+ sinalling has special controllingfunctions for RNA/DNA processing and gene expression.Recently RyR, or a receptor closely identical to RyR, wasshown to serve as a sensor of extracellular Ca2+ in osteocytes[340,341]. Surprisingly, there are only a couple or a few knowndisorders associated with dysfunction of ICC: malignanthyperthermia associated with genetic mutation in RyR andmyasthenia gravis, autoimmune disease resulting fromabnormal antibodies against own RyR. Some data suggestthat presenilin mutation PS1-δE9 associated with familialAlzheimer’s disease results in enhanced PLC activity andincreased [Ca2+]i [342].
From all of the modulators of ICC, only dantrolene is usedtherapeutically. There are multiple compounds which modu-late RyR, whereas we know only a few compounds which can
Table 1. New patents on the modulators of intracellular calcium channels (continued).
Assignee Patent title (and compounds)
Chemical structure(if available)
Utility Ref.
Contrimmune Biotech., Inc.
Therapeutic uses for IP3R-mediated calcium channel modulators (Xestospongin C)(WO0238140)
Treatment of HIV and other viral infections, inflammatory disease, psoriasis, pain, uncontrolled growth disorders such as cancer, cardiac arrhythmia and hypertension
[410]
Schering AG et al.
Ether derivatives (WO0046193)
Treatment of neurological, mental and cognitive disorders, cardiovascular disease, muscle disease, neuropathy, myasthenia gravis and spinal cord disorders
[412]
Schering AG et al.
Heteroaromatic compounds with neuronal activity(WO0046222)
Peripheral neuropathies, stroke, brain and spinal cord injury, Parkinson`s and Alzheimer`s diseases, neuralgia, myasthenia gravis, muscular dystrophy, multiple sclerosis, ischeamia and trauma
[413]
cADPR: Cyclic ADP ribose; IP3: Inositol-1,3-4-triphosphate receptor; NO: Nitric oxide; RyR: Ryandodine receptor; tPA:Tissue plasminogen activator.
N O
O
N
H
H
H
HH H
N
O
O
N
O
N
N
O N
O
O
N

Author p
roof
Intracellular calcium channels and their modulators
26 Expert Opin. Ther. Patents (2003) 13(6)
modulate IP3R. Development of the pharmacology of intrac-ellular Ca2+ channels has great potential. Modulators of ICCmay be applied for the treatment of cardiovascular and neuro-degenerative disorders, such as Parkinson’s and Alzheimer’s dis-eases, brain and spinal cord injury, Huntington’s disease,autoimmune diseases, inflammatory disease and viral infec-tions. Ischaemic conditions are characterised by low ATP levels
and Ca2+ overload. There are multiple pathways of Ca2+ influxin cytosol including VGCC, Na+/Ca2+ exchanger and Ca2+
release from intracellular stores that makes it difficult to blockall these pathways. Theoretically, multi-drug combinationtherapy for treatment of stroke and ischaemia including block-ers of VGCC, Na+/Ca2+-exchanger and intracellular CCblocker dantrolene may be developed.
BibliographyPapers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
1. HEILBRUNN LV, WIERCINSKI F J: The action of various cations on muscle protoplasm. J. Cellular Comp. Physiol. (1947) 29:15-32.
2. PEACHEY LD: Transverse tubules in excitation–contraction coupling. Fed. Proc. (1965) 24(5):1124-1134.
3. HASSELBACH W, MAKINOSE M: Die calcium pumpe der ‘erschlaffungsgrana ‘des muskles und ihre abhangigkeit von der ATP-spaltung. Biochem. Z (1961) 333:518-528.
4. EBASHI S, LIPMANN F: Adenosine-triphosphate-linked concentration of calcium ions in a particular fraction of rabbit muscle. J. Cell Biol. (1962) 14:389-400.
5. LEE KS: Effect of electrical stimulation on uptake and release of calcium by the endoplasmic reticulum. Nature (1965) 207(992):85-86.
6. LEE KS, LADINSKY H, CHOI SJ, KASUYA Y: Studies on the in vitro interaction of electrical stimulation and Ca2+
movement in sarcoplasmic reticulum. J. Gen. Physiol. (1966) 49(4):689-715.
7. WINEGRAD S: Role of intracellular calcium movements in excitation–contraction coupling in skeletal muscle. Fed. Proc. (1965) 24(5):1146-1152.
8. JOBSIS FF, O’CONNOR MJ: Calcium release and reabsorption in the sartorius muscle of the toad. Biochem. Biophys. Res. Commun. (1966) 25(2):246-252.
9. KATZ AM, REPKE DI: Control of myocardial contraction: the sensitivity of cardiac actomyosin to calcium ion. Science (1966) 152(726):1242-1243.
10. EBASHI S, EBASHI F, KODAMA A: Troponin as the Ca2+ -receptive protein in the contractile system. J. Biochem. (Tokyo) (1967) 62(1):137-138.
11. DESMEDT JE, HAINAUT K: Excitation–contraction coupling in single muscle fibers and the calcium channel in sarcoplasmic reticulum. Ann. NY Acad. Sci. (1978) 307:433-435.
12. YAMAMOTO N, KASAI M: Mechanism and function of the Ca2+-gated cation channel in sarcoplasmic reticulum vesicles. J. Biochem. (Tokyo) (1982) 92(2):485-496.
13. FRANZINI-ARMSTRONG C: Studies of the triad. I. Structure of the junction in frog twitch fibers. J. Cell Biol. (1970) 47:488-499.
14. VOLPE P, KRAUSE KH, HASHIMOTO S, ZORZATO F et al.: ‘Calciosome’, a cytoplasmic organelle: the inositol 1,4,5-trisphosphate-sensitive Ca2+ store of non-muscle cells? Proc. Nat. Acad. Sci. USA (1988) 85(4):1091-1095.
15. POZZAN T, VOLPE P, ZORZATO F, RAVIN M et al.: The Ins(1,4,5)P3 –sensitive Ca2+ store of non-muscle cells: endoplasmic reticulum or calciosomes? J. Exp. Biol. (1988) 139:181-193.
16. KENDALL JM, RMER RL, CAMPBELL AK: Targeting aequorin to the endoplasmic reticulum of living cells. Biochem. Biophys. Res. Comm. (1992) 189(2):1008-1016.
17. MELDOLESI J, POZZAN T: The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem. Sci. (1998) 23(1):10-14.
18. BRINI M, CARAFOLI E: Calcium signalling: a historical account, recent developments and future perspectives. Cell Mol. Life Sci. (2000) 57(3):354-370.
19. DE MEIS L, HASSELBACH W, MACHADO RD: Characterization of calcium oxalate and calcium phosphate deposits in sarcoplasmic reticulum vesicles. J. Cell Biol. (1974) 62(2):505-509.
20. MENG XJ, TIMMER RT, GUNN RB, ABERCROMBIE RF: Separate entry pathways for phosphate and oxalate in rat brain microsomes. Am. J. Physiol. Cell Physiol. (2000) 278(6):C1183-C1190.
21. LAVER DR, LENZ GK, DULHUNTY AF: Phosphate ion channels in sarcoplasmic reticulum of rabbit skeletal muscle. J. Physiol. (2001) 535(Pt 3):715-728.
22. TAKESHIMA H, NISHIMURA S, MATSUMOTO T et al.: Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature (1989) 339(6224):439-445.
23. ZORZATO F, FUJII J, OTSU K, PHILLIPS M: Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. (1990) 265(4):2244-2256.
24. TUNWELL RE, WICKENDEN C, BERTRAND BM et al.: The human cardiac muscle ryanodine receptor-calcium release channel: identification, primary structure and topological analysis. Biochem. J. (1996) 318(Pt 2):477-487.
25. DU GG, SANDHU B, KHANNA VK, GUO XH, MACLENNAN DH: Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1). Proc. Natl. Acad. Sci. USA (2002) 99(26):16725-16730.
26. BOEHNING D, MAK D-OD, FOSKETT JK, JOSEPH SK: Molecular determinants of ion permeation and selectivity in inositol 1,4,5-trisphosphate receptor Ca2+ channels. J. Biol. Chem. (2001) 276:13509-13512.
27. BEZPROZVANNY I, BEZPROZVANNAYA S, EHRLICH BE: Caffeine-induced inhibition of inositol(1,4,5)-trisphosphate-gated calcium channels from cerebellum. Mol. Biol. Cell (1994) 5(1):97-103.
28. BEZPROZVANNY IB, ONDRIAS K, KAFTAN E et al.: Activation of the calcium release channel (ryanodine receptor) by heparin and other polyanions is calcium dependent. Mol. Biol. Cell (1993) 4(3):347-352.
29. EHRLICH BE, KAFTAN E, BEZPROZVANNAYA S, BEZPROZVANNY I: The pharmacology of intracellular Ca2+-release channels. Trends Pharmacol. Sci. (1994) 15(5):145-149.
30. TAYLOR CW, TRAYNOR D: Calcium and inositol trisphosphate receptors. J. Membr. Biol. (1995) 145(2):109-118.
31. FABIATO A, FABIATO F: Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J. Physiol. (1975) 249(3):469-495.

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 27
32. FABIATO A, FABIATO F: Use of chlorotetracycline fluorescence to demonstrate Ca2+-induced release of Ca2+ from the sarcoplasmic reticulum of skinned cardiac cells. Nature (1979) 281(5727):146-148.
33. FABIATO A: Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. (1983) 245(1):C1-C14.
34. KOCHEGAROV AA, BEYLINA SI, MATVEEVA NB, ZINCHENKO VP: Effects of Ca2+ -ATPase inhibitors, ionomycin, and pharmacological modulators of ryanodine receptor on calcium release from intracellular pools and on oscillatory contractile behavior in Physarum polycephalum. Biochemistry (Russian) (2000) 65(6):779-789.
35. KOCHEGAROV AA, BEYLINA SI, MATVEEVA NB, ZINCHENKO VP: Ionomycin and 2,5-di(tertbutyl)-1,4,-enzohydroquinone elicit Ca2+-induced Ca2+
release from intracellular pools in Physarum polycephalum. Comp. Biochem. Physiol. (2001) 128:279-288.
36. ANTARAMIÁN A, BUTANDA-OCHOA A, VÁZQUEZ-MARTÍNEZ O et al.: Functional expression of recombinant Type 1 ryanodine receptor in insect cells. Cell Calcium (2001) 30(1):9-17.
37. BASTIDE B, MOUNIER Y: Single-channel properties of the sarcoplasmic reticulum calcium-release channel in slow- and fast-twitch muscles of Rhesus monkeys. Pflugers Arch.IV. Eur. J. Physiol. (1998) 436(3):485-488.
38. TRIPATHY A, XU L, MANN G, MEISSNER G: CaM activation and inhibition of skeletal muscle Ca2+ release channel (ryanodine receptor). Biophys. J. (1995) 69:106-119.
39. FAIRHURST AS, JENDEN DJ: The distribution of a ryanodine-sensitive calcium pump in skeletal muscle fractions. J. Cell. Physiol. (1966) 67(2):233-238.
40. PESSAH IN, WATERHOUSE AL, CASIDA JE: The calcium-ryanodine receptor complex of skeletal and cardiac muscle. Biochem. Biophys. Res. Comm. (1985) 128:449-456.
41. CAMPBELL KP, KNUDSON CM, IMAGAWA T, LEUNG AT, SUTKO JL: Identification and characterization of the high affinity [3H]-ryanodine receptor of the junctional sarcoplasmic reticulum Ca2+
release channel. J. Biol.Chem. (1987)
42. MORRISSETTE J, XU L, NELSON A, MEISSNER G, BLOCK BA: Characterization of RyR1-slow, a ryanodine receptor specific to slow-twitch skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. (2000) 279(5):R1889-R1898.
43. WAGENKNECHT T, RADERMACHER M: Three-dimensional architecture of the skeletal muscle ryanodine receptor. FEBS Lett. (1995) 369:43-46.
44. SERYSHEVA II, ORLOVA EV, CHIU W, SHERMAN MB: Electron cryomicroscopy and angular reconstitution used to visualize the skeletal muscle calcium release channel. Nat. Struct. Biol. (1995) 2:18-24.
45. GRUNWALD R. MEISSNER G: Lumenal sites and C terminus accessibility of the skeletal muscle calcium release channel (ryanodine receptor). J. Biol. Chem. (1995) 270:11338-11347.
46. LAI FA, ERICKSON HP, ROUSSEAU E, LIU QY, MEISSNER G: Purification and reconstitution of the calcium release channel from skeletal muscle. Nature (London) (1988) 331:315-319.
47. ORLOVA EV, SERYSHEVA II, VAN HEEL M, HAMILTON SL: Two structural configurations of the skeletal muscle calcium release channel. Nature Struct. Biol. (1996) 3(6):547-552.
48. WAGENKNECHT T, SAMSO M: Three-dimensional reconstruction of ryanodine receptors. Front. Biosci. (2002) 7:d1464-dl474.
49. SMITH JS, IMAGAWA T, MA J, FILL M et al.: Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J. Gen. Physiol. (1988) 92:1-26.
50. LINDSAY AR, WILLIAMS AJ: Functional characterization of the ryanodine receptor purified from sheep cardiac muscle sarcoplasmic reticulum. Biochim. Biophys. Acta (1991) 1064:89-102.
51. MARKS AR, FLEISCHER S, TEMPST P: Surface topography analysis of the ryanodine receptor/junctional channel complex based on proteolysis sensitivity mapping. J. Biol. Chem. (1990) 265(22):13143-13149.
52. WILLIAMS AJ, WEST DJ, SITSAPESAN R: Light at the end of the Ca(2+)-release channel tunnel: structures and mechanisms involved in ion translocation in ryanodine receptor channels. Quarterly Reviews of Biophysics (2001) 34(1):61-104.
53. ARMSTRONG CM, BEZANILLA FM, HOROWICZ P: Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,Ν-tetracetic acid. Biochim. Biophys. Acta (1972) 267:605-608.
54. RIOS E, MA J, GONZALEZ A: The mechanical hypothesis of excitation–contraction (EC) coupling in skeletal muscle. J. Muscle Res. Cell Motil. (1991) 12:127-135.
55. MEISSNER G, LU X. Dihydropyridine receptor-ryanodine receptor interactions in skeletal muscle excitation–contraction coupling. Biosci. Rep. (1995) 15(5):399-408.
56. BLOCK BA, IMAGAWA T, CAMPBELL KP, FRANZINI-ARMSTRONG C: Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell. Biol. (1988) 107:2587-2600.
57. MARTY I, ROBERT M, VILLAZ M, DE JONGH KS et al.: Biochemical evidence for a complex involving dihydropyridine receptor and ryanodine receptor in triad junctions of skeletal muscle. Proc. Natl. Acad. Sci. USA (1994) 91:2270-2274.
58. FRANZINI-ARMSTRONG C, PROTASI F, RAMESH V: Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Annal. NY Acad. Sci. (1998) 853:20-30.
59. PROTASI F, FRANZINI-ARMSTRONG C, ALLEN PD: Role of ryanodine receptors in the assembly of calcium release units in skeletal muscle. J. Cell Biol. (1998) 140(4):831-842.
60. NAKAI J, SEKIGUCHI N, RANDO TA, ALLEN PD, BEAM KG: Two regions of the ryanodine receptor involved in coupling with L-type Ca2+ channels. J. Biol. Chem. (1998) 273(22):13403-13406.
61. GURROLA GB, ARÉVALO C, SREEKUMAR R et al.: Activation of ryanodine receptors by imperatoxin A and a peptide segment of the II – III loop of the dihydropyridine receptor. J. Biol. Chem. (1999) 274(12):7879-7886.
62. YAMAZAWA T, TAKESHIMA H, SHIMUTA M, IINO M: A region of the ryanodine receptor critical for excitation–contraction coupling in skeletal muscle. J. Biol. Chem. (1997) 272(13):8161-8164.
63. LU X, XU L, MEISSNER G: Activation of the skeletal muscle calcium release channel by a cytoplasmic loop of the dihydropyridine receptor. J. Biol. Chem. (1994) 269(9):6511-6516.
64. LU X, XU L, MEISSNER G: Phosphorylation of dihydropyridine receptor II – III loop peptide regulates skeletal muscle calcium release channel function. Evidence for an essential role of the β−OH group of Ser687. J. Biol. Chem. (1995) 270(31):18459-18464.

Author p
roof
Intracellular calcium channels and their modulators
28 Expert Opin. Ther. Patents (2003) 13(6)
65. EL-HAYEK R, ANTONIU B, WANG J, HAMILTON SL, IKEMOTO N: Identification of calcium release-triggering and blocking regions of the II – III loop of the skeletal muscle dihydropyridine receptor. J. Biol. Chem. (1995) 270(38):22116-22118.
66. LAMB GD, EL-HAYEK R, IKEMOTO N, STEPHENSON DG: Effects of dihydropyridine receptor II – III loop peptides on Ca2+ release in skinned skeletal muscle fibers. Am. J. Cell Physiol. (2000) 279(4):C891-C905.
67. STANGE M, TRIPATHY A, MEISSNER G: Two domains in dihydropyridine receptor activate the skeletal muscle Ca2+ release channel. Biophys. J. (2001) 81(3):1419-1429.
68. SENCER S, PAPINENI RVL, HALLING DB, PATE P et al.: Coupling of RyR1 and L-type calcium channels via CaM binding domains. J. Biol. Chem. (2001) 276:38237-38241.
69. PAPINENI RV, O’CONNELL KM, ZHANG H, DIRKSEN RT, HAMILTON SL: Suramin interacts with the calmodulin binding site on the ryanodine receptor, RyR1. J. Biol. Chem. (2002) 277(51):4916-49174.
70. NÄBAUER M, CALLEWAERT G, CLEEMANN L, MORAD M: Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science (1989) 244:800-803.
71. CALLEWAERT G: Excitation–contraction coupling in mammalian cardiac cells. Cardiovasc. Res. (1992) 26:923-932.
72. FRANZINI-ARMSTRONG C, JORGENSEN AO: Structure and development of EC-coupling units in skeletal muscle. Ann. Rev. Physiol. (1994) 56:509-534.
73. ANDERSON K, MEISSNER G: T-tubule depolarization-induced SR Ca2+ release is controlled by dihydropyridine receptor- and Ca2+-dependent mechanisms in cell homogenates from rabbit skeletal muscle. J. Gen. Physiol. (1995) 105:363-383.
74. KLEIN MG, CHENG H, SANTANA LF, JIANG YH et al.: Two mechanisms of quantized calcium release in skeletal muscle. Nature (1996) 379:455-458.
75. IKEMOTO T, IINO M, ENDO M: Enhancing effect of CaM on Ca2+-induced Ca2+ release in the sarcoplasmic reticulum of rabbit skeletal muscle fibres. J. Physiol. (1995) 487(Pt3):573-582.
76. IKEMOTO T, TAKESHIMA H, IINO M, ENDO M: Effect of CaM on Ca2+-induced Ca2+ release of skeletal muscle from mutant mice expressing either ryanodine receptor Type 1 or Type 3. Pflugers Arch. (1998) 437(1):43-48.
77. BURATTI R, PRESTIPINO G, MENEGAZZI P, TREVES S, ZORZATO F: Calcium dependent activation of skeletal muscle Ca2+ release channel (ryanodine receptor) by CaM. Biochem. Biophys. Res. Commun. (1995) 213(3):1082-1090.
78. BALSHAW DM, XU L, YAMAGUCHI N, PASEK DA, MEISSNER G: CaM binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. (2001) 276(23):20144-20153.
79. MEISSNER G, HENDERSON JS: Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg 2+, adenine nucleotide, and CaM. J. Biol. Chem. (1987) 262(7):3065-3073.
80. FRUEN BR, BARDY JM, BYREM TM et al.: Differential Ca2+ sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am. J. Physiol. (2000) 279:C724-C733.
81. RODNEY GG, MOORE CP, WILLIAMS BY, ZHANG JZ et al.: Calcium binding to CaM leads to an N-terminal shift in its binding site on the ryanodine receptor. J. Biol. Chem. (2001) 276(3):2069-2074.
82. HAMILTON SL, REID MB: RyR1 modulation by oxidation and CaM. Antioxid. Redox Signal (2000) 2(1):41-45.
83. SAMSO M, WAGENKNECHT T: ApoCaM and Ca2+-CaM bind to neighboring locations on the ryanodine receptor. J. Biol. Chem. (2002) 277(2):1349-1353.
84. YAMAGUCHI N, XIN C, MEISSNER G: Identification of ApoCaM and Ca2+-CaM regulatory domain in skeletal muscle Ca2+
release channel, ryanodine receptor. J. Biol. Chem. (2001) 276:22579-22585.
85. CARMODY M, MACKRILL JJ, SORRENTINO V, O’NEILL C: FKBP12 associates tightly with the skeletal muscle Type 1 ryanodine receptor, but not with other intracellular calcium release channels. FEBS Lett. (2001) 505(1):97-102.
86. MACKRILL JJ, O’DRISCOLL S, LAI FA, MCCARTHY TV: Analysis of Type 1 ryanodine receptor-12 kDa FK506-binding protein interaction. Biochem. Biophys. Res. Commun. (2001) 285(1):52-57.
87. GABURJAKOVA M, GABURJAKOVA J, REIKEN S, HUANG F et al.: FKBP12 binding modulates ryanodine receptor channel gating. J. Biol. Chem. (2001) 276(20):16931-16935.
88. GUO W, CAMPBELL KP: Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J. Biol. Chem. (1995) 270(16):9027-9030.
89. MURRAY BE, OHLENDIECK K: Complex formation between calsequestrin and the ryanodine receptor in fast- and slow-twitch rabbit skeletal muscle. FEBS Lett. (1998) 429(3):317-322.
90. CASWELL AH, BRANDT NR, BRUNSCHWIG JP, PURKERSON S: Localization and partial characterization of the oligomeric disulfide-linked molecular weight 95,000 protein (triadin) which binds the ryanodine and dihydropyridine receptors in skeletal muscle triadic vesicles. Biochem. (1991) 30(30):7507-7513.
91. OHKURA M, FURUKAWA K, FUJIMORI H, KURUMA A: Dual regulation of the skeletal muscle ryanodine receptor by triadin and calsequestrin. Biochem. (1998) 37(37):12987-12993.
92. HIDALGO C, DONOSO P: Luminal calcium regulation of calcium release from sarcoplasmic reticulum. Biosci. Rep. (1995) 15(5):387-397.
93. SITSAPESAN R, WILLIAMS AJ: Regulation of current flow through ryanodine receptors by luminal Ca2+. J. Membr. Biol. (1997) 159(3):179-185.
94. SZEGEDI C, SÁRKÖZI S, HERZOG A, JÓNA I, VARSÁNYI M: Calsequestrin: more than ‘only’ a luminal Ca2+ buffer inside the sarcoplasmic reticulum. Biochem. J. (1999) 337(Pt 1):19-22.
95. MARKS AR: Ryanodine receptors/calcium release channels in heart failure and sudden cardiac death. J. Mol. Cell. Cardiol. (2001) 33(4):615-624.
96. FISHMAN GI: Association of sorcin with the cardiac ryanodine receptor. J. Biol. Chem. (1995) 270:26411-26418.
97. LOKUTA AJ, MEYERS MB, SANDER PR, FISHMAN GI, VALDIVIA H: Modulation of cardiac ryanodine receptors by sorcin. J. Biol. Chem. (1997) 272:25333-25338.
98. YIN CC, LAI FA: Intrinsic lattice formation by the ryanodine receptor calcium-release channel. Nature Cell Biol. (2000) 2(9):669-671.

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 29
99. MARX SO, GABURJAKOVA J, GABURJAKOVA M, HENRIKSON C et al.: Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ. Res. (2001) 88:1151-1158.
100. WEN L, ZHANG L, LONGO LD: Cerebral artery KATP- and KCa-channel activity and contractility: changes with development. Am. Physiol. Soc. Abst. (2000) 7:0341R.
101. CHU A, FILL M, STEFANI E, ENTMAN ML: Cytoplasmic Ca2+ does not inhibit the cardiac muscle sarcoplasmic reticulum ryanodine receptor Ca2+ channel, although Ca2+-induced Ca2+ inactivation of Ca2+ release is observed in native vesicles. J. Membr. Biol. (1993) 135:49-59.
102. LAVER DR, RODEN LD, AHERN GP, EAGER KR et al.: Cytoplasmic Ca2+ inhibits the ryanodine receptor from cardiac muscle. J. Membr. Biol. (1995) 147:7-22.
103. DU GG, MACLENNAN DH: Ca2+
inactivation sites are located in the COOH-terminal quarter of recombinant rabbit skeletal muscle Ca2+ release channels (ryanodine receptors). J. Biol. Chem. (1999) 274:26120-26126.
104. LI P, CHEN SR: Molecular basis of Ca2+
activation of the mouse cardiac Ca2+ release channel (ryanodine receptor). J. Gen. Physiol. (2001) 118(1):33-44.
105. MEISSNER G, DARLING E, EVELETH J: Kinetics of rapid Ca2+ release by sarcoplasmic reticulum: effects of Ca2+ , Mg2+, and adenine nucleotides. Biochem. (1986) 25:236-244.
106. MOUTIN MJ, DUPONT Y: Rapid filtration studies of Ca2+ -induced Ca2+ release from skeletal sarcoplasmic reticulum. J. Biol. Chem. (1988) 263:4228-4235.
107. HIGASHIDA H, YOKOYAMA S, HASHII M, TAKETO M et al.: Muscarinic receptor-mediated dual regulation of ADP-ribosyl cyclase in NG108-15 neuronal cell membranes. J. Biol. Chem. (1997) 272:31272-31277.
108. CANCELA JM: Specific Ca2+ signaling evoked by cholecystokinin and acetylcholine: the roles of NAADP, cADPR, and IP3. Ann. Rev. Physiol. (2001) 63:99-117.
109. BERRIDGE MJ: Cell Signaling: a tale of two messengers. Nature (1993) 365(6445):388-389.
110. GUSE AH: Cyclic ADP-ribose. J. Mol. Med. (2000) 78(1):26-35
111. LI PL, TANG WX, VALDIVIA HH, ZOU AP, CAMPBELL WB: cADP-ribose activates reconstituted ryanodine receptors from coronary arterial smooth muscle. Am. J. Physiol. Heart Circ. Physiol. (2001) 280(1):H208-H215.
112. RUSINKO N, LEE HC: Widespread occurrence in animal tissues of an enzyme catalyzing the conversion of NAD+ into a cyclic metabolite with intracellular Ca2+-mobilizing activity J. Biol. Chem. (1989) 264:11725-11731.
113. ZIEGLER M: New functions of a long-known molecule: emerging roles of NAD in cellular signaling. Eur. J. Biochem. (2000) 267:1550-1564.
114. LEE HC: Physiological functions of cyclic ADP-ribose and NADPH as calcium messengers. Ann. Rev. Pharmacol. Toxicol. (2001) 41:317-345.
115. LEE HC, GRAEFF RM, WALSETH TF: ADP-ribosyl cyclase and CD38. Multi-functional enzymes in Ca2+ signaling. Adv. Exp. Med. Biol. (1997) 419:411-419.
116. GRAEFF RM, FRANCO L, DE FLORA, LEE HC: Cyclic GMP-dependent and -independent effects on the synthesis of the calcium messengers cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate. J. Biol. Chem. (1998) 273:118-125.
117. ZHANG FJ, GU QM, JING PC, SIH CJ: Enzymatic cyclization of nicotinamide adenine dinuleotide phosphate (NADP). Bioorg. Med. Chem. Lett. (1995) 5:2267-2272.
118. CHINI EN, DOUSA TP: Enzymatic synthesis and degradation of nicotinate adenine dinucleotide phosphate (NAADP), a Ca2+ -releasing agonist, in rat tissues. Biochem. Biophys. Res. Commun. (1995) 209:167-174.
119. VU CQ, LU PJ, CHEN CS, JACOBSON MK: 2-Phospho-cyclic ADP-ribose, a calcium-mobilizing agent derived from NADP. J. Biol. Chem. (1996) 271:4747-4754.
120. LAHOURATATE P, GUIBERT J, FAIVRE JF: cADP-ribose releases Ca2+ from cardiac sarcoplasmic reticulum independently of ryanodine receptor. Am. J. Physiol. (1997) 273(3 Pt 2):H1082-H1089.
121. COPELLO JA, QI Y, JEYAKUMAR LH, OGUNBUNMI E, FLEISCHER S: Lack of effect of cADP-ribose and NAADP on the activity of skeletal muscle and heart ryanodine receptors. Cell Calcium (2001) 30(4):269-284.
122. LEE HC, AARHUS R: A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J. Biol. Chem. (1995) 270:2152-2157.
123. CLAPPER DL, WALSETH TF, DARGIE PJ, LEE HC: Pyridine nucleotide metabolites stimulate calcium release from sea urchin microsomes desensitized to inositol trisphosphate. J. Biol. Chem. (1987) 262:9561-9568.
124. AARHUS R, DICKEY DM, GRAEFF RM, GEE KR et al.: Activation and inactivation of Ca2+ release by NADP+. J. Biol. Chem. (1996) 271:8513-8516.
125. CHINI EN, LIANG M, DOUSA TP: Differential effect of pH upon cyclic-ADP-ribose and nicotinate-adenine dinucleotidephosphate-induced Ca2+ release systems. Biochem. J. (1998) 335:499-504.
126. HOWARD M, GRIMALDI JC, BAZAN JF, LUND FE et al.: Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science (1993) 262:1056-1059.
127. SANTELLA L, KYOZUKA K, GENAZZANI AA, DE RISO L, CARAFOLI E: Nicotinic acid adenine dinucleotide phosphate-induced Ca2+ release. Interactions among distinct Ca2+ mobilizing mechanisms in starfish oocytes. J. Biol. Chem. (2000) 275:8301-8306.
128. NAVAZIO L, BEWELL MA, SIDDIQUA A, DICKINSON GD et al.: Calcium release from the endoplasmic reticulum of higher plants elicited by the NADP metabolite nicotinic acid adenine dinucleotide phosphate. Proc. Natl. Acad. Sci. USA (2000) 97(15):8693-8698.
129. GENAZZANI AA, GALIONE AA: Ca2+
release mechanism gated by the novel pyridine nucleotide, NAADP. Trends. Pharmacol. Sci. (9997) 18(4):108-110.
130. LEE HC, AARHUS R: Functional visualization of the separate but interacting calcium stores sensitive to NAADP and cyclic ADP-ribose. J. Cell Sci. (2000) 113:4413-4420.
131. GENAZZANI AA, EMPSON RM, GALIONE A: Unique inactivation properties of NAADP-sensitive Ca2+ release. J. Biol. Chem. (1996) 271:11599-11602.
132. GENAZZANI AA, GALIONE A: Nicotinic acid-adenine dinucleotide phosphate mobilizes Ca2+ from a thapsigargin-insensitive pool. Biochem. J. (1996) 315:721-725.
133. CHURCHILL GC, OKADA Y, THOMAS JM, GENAZZANI AA et al.: NAADP mobilizes Ca2+ from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell (2002) 111(5):703-708.
134. CHURCHILL GC, GALIONE A: NAADP induces Ca2+ oscillations via a two-pool mechanism by priming IP3 – and cADPR-sensitive Ca2+ stores. EMBO J. (2001) 20:2666-2671.

Author p
roof
Intracellular calcium channels and their modulators
30 Expert Opin. Ther. Patents (2003) 13(6)
135. HIGASHIDA H, YOKOYAMA S, HOSHI N, HASHII M et al.: Signal transduction from bradykinin, angiotensin, adrenergic and muscarinic receptors to effector enzymes, including ADP-ribosyl cyclase. Biol. Chem. (2001) 382(1):23-30.
136. SITSAPESAN R, MCGARRY SJ, WILLIAMS AJ: Cyclic ADP-ribose competes with ATP for the adenine nucleotide binding site on the cardiac ryanodine receptor Ca2+-release channel. Circ. Res. (1994) 75:596-600.
137. MORII H, TONOMURA Y: The gating behavior of a channel for Ca2+ -induced Ca2+
release in fragmented sarcoplasmic reticulum. J. Biochem. (1983) 93:1271-1285.
138. MEISSNER G: Adenine nucleotide stimulation of Ca2+ -induced Ca2+ release in sarcoplasmic reticulum. J. Biol. Chem. (1984) 259:2365-2374.
139. MEISSNER G, HENDERSON JS: Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg 2+ , adenine nucleotide, and CaM. J. Biol. Chem. (1987) 262:3065-3073.
140. CHAN WM, WELCH W, SITSAPESAN R: Structural factors that determine the ability of adenosine and related compounds to activate the cardiac ryanodine receptor. Br. J. Pharmacol. (2000) 130(7):1618-1626.
141. OTSU K, WILLARD HF, KHANNA VK, ZORZATO F et al.: Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J. Biol. Chem. (1990) 265(23):13472-13483.
142. JÓNA I, SZEGEDI C, SÁRKÖZI S et al.: Altered inhibition of the rat skeletal ryanodine receptor/calcium release channel by magnesium in the presence of ATP. Pflugers Arch.IV. Eur. J. Physiol. (2001) 441(6):729-738.
143. SUKO J, MAURER-FOGY I, PLANK B et al.: Phosphorylation of serine 2843 in ryanodine receptor-calcium release channel of skeletal muscle by cAMP-, cGMP- and CaM-dependent protein kinase. Biochim. Biophys. Acta (1993) 1175:193-206.
144. WITCHER DR, STRIFLER BA, JONES LR: Cardiac-specific phosphorylation site for multifunctional Ca2+
/CaM-dependent protein kinase is conserved in the brain ryanodine receptor. J. Biol. Chem. (1992) 267(7):4963-4967.
145. HERRMANN-FRANK A, VARSANYI M: Enhancement of Ca2+ release channel activity by phosphorylation of the skeletal muscle ryanodine receptor. FEBS Lett. (1993) 332:237-242.
146. GECHTMAN Z, ORR I, SHOSHAN-BARMATZ V: Involvement of protein phosphorylation in activation of Ca2+ efflux from sarcoplasmic reticulum. Biochem. J. (1991) 276:97-102.
147. WANG J, BEST PM: Inactivation of the sarcoplasmic reticulum channel by protein kinase. Nature (1992) 359:739-741.
148. WU Y, COLBRAN RJ, ANDERSON ME: CaM kinase is a molecular switch for cardiac excitation–contraction coupling. Proc. Natl. Acad. Sci. USA (2001) 98(5):2877-2881.
149. MARX SO, REIKEN S, HISAMATSU Y et al.: PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell (2000) 101(4):365-376.
150. ANTOS CL, FREY N, MARX SO, REIKEN S et al.: Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase A. Circ. Res. (2001) 89(11):997-1004.
151. COLPO P, NORI A, SACCHETTO R, DAMIANI E, MARGRETH A: Phosphorylation of the triadin cytoplasmic domain by CaM protein kinase in rabbit fast-twitch muscle sarcoplasmic reticulum. Mol. Cell. Biochem. (2001) 223(1-2):139-145.
152. MARKS AR, REIKEN S, MARX SO: Progression of heart failure: is protein kinase a hyperphosphorylation of the ryanodine receptor a contributing factor? Circulation (2002) 105(3):272-275.
153. EL-HAYEK R, VALDIVIA C, VALDIVIA HH, HOGAN K, CORONADO R: Activation of the Ca2+
release channel of skeletal muscle sarcoplasmic reticulum by palmitoyl carnitine. Biophys. J. (1993) 65:779-789.
154. DUMONTEIL E, BARRÉ H. MEISSNER G: Effects of palmitoyl carnitine and related metabolites on the avian Ca2+-ATPase and Ca2+
release channel. J. Physiol. (1994) 479:29-39.
155. FULCERI R, NORI A, GAMBERUCCI A, VOLPE P et al.: Fatty acyl-CoA esters induce calcium release from terminal cisternae of skeletal muscle. Cell Calcium (1994) 15:109-116.
156. CHEAH AM: Effect of long chain unsaturated fatty acids on the calcium transport of sarcoplasmic reticulum. Biochim. Biophys. Acta (1981) 648:113-119.
157. MESSINEO FC, RATHIER M, FAVREAU C, WATRAS J, TAKENAKA H: Mechanisms of fatty acid effects on sarcoplasmic reticulum: the effects of palmitic and oleic acids on sarcoplasmic reticulum function: a model for fatty acid membrane interactions. J. Biol. Chem. (1984) 259:1336-1343.
158. CARDOSO CM, DE MEIS L: Modulation by fatty acids of Ca2+ fluxes in sarcoplasmic-reticulum vesicles. Biochem. J. (1993) 296:49-52.
159. DETTBARN CA, PALADE P: Arachidonic acid-induced Ca2+ release from isolated sarcoplasmic reticulum. Biochem. Pharmacol. (1993) 45:1301-1309.
160. UEHARA A, YASUKOCHI M IMANAGA I: Modulation of ryanodine binding to the cardiac Ca2+ release channel by arachidonic acid. J. Mol. Cell. Cardiol. (1996) 28:43-51.
161. SABBADINI RA, BETTO R, TERESI A, FACHECHI-CASSANO G, SALVIATI G: The effects of sphingosine on sarcoplasmic reticulum membrane calcium release. J. Biol. Chem. (1992) 267:15475-15484.
162. MCDONOUGH PM, YASUI K, BETTO R, SALVIATI G et al.: Control of cardiac Ca2+ levels: inhibitory actions of sphingosine on Ca2+ transients and L-type Ca2+ channel conductance. Circ. Res. (1994) 75:981-989.
163. DETTBARN CA, BETTO R, SALVIATI G, SABBADINI RA, PALADE P: Involvement of ryanodine receptors in sphingosylphosphoryl choline-induced calcium release from brain microsomes. Brain Res. (1995) 669:79-85.
164. STRIGGOW F, EHRLICH BE: Regulation of intracellular calcium release channel function by arachidonic acid and leukotriene B4. Biochem. Biophys. Res .Commun. (1997) 237(2):413-418.
165. PALADE P: Drug-induced Ca2+ release from isolated sarcoplasmic reticulum: III block of Ca2+-induced Ca2+ release by organic polyamines. J. Biol. Chem. (1987) 262:6149-6154.
166. UEHARA A, FILL M, VELEZ P, YASUKOCHI M, IMANAGA I: Rectification of rabbit cardiac ryanodine receptor current by endogenous polyamines. Biophys. J. (1996) 71:769-777.
167. MUSA H, VEENSTRA D: Voltage-dependent blockade of connexin 40 gap junctions by spermine. Biophys. J. (2003) 84(1):205-219.
168. EU JP, SUN J, XU L, STAMLER JS, MEISSNER G: The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell (2000) 102(4):499-509.
169. SUN J, XU L, EU JP, STAMLE JS, MEISSNER G: Classes of thiols that influence the activity of the skeletal muscle calcium release channel. J. Biol. Chem. (2001) 276(19):15625-15630.

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 31
170. PESSAH IN, FENG W: Functional role of hyperreactive sulfhydryl moieties within the ryanodine receptor complex. Antioxid. Redox Signal (2000) 2(1):17-25.
171. EU JP, XU L, STAMLER JS, MEISSNER G: Regulation of ryanodine receptors by reactive nitrogen species. Biochem. Pharmacol. (1999) 57(10):1079-1084.
172. EAGER KR, DULHUNTY AF: Activation of the cardiac ryanodine receptor by sulfhydryl oxidation is modified by Mg 2+ and ATP. J. Membr. Biol. (1998) 163(1):9-18.
173. HART JD, DULHUNTY AF: Nitric oxide activates or inhibits skeletal muscle ryanodine receptors depending on its concentration, membrane potential and ligand binding. J. Membr. Biol. (2000) 173:227-236.
174. STOYANOVSKY D, MURPHY T, ANNO PR, KIM YM, SALAMA G: Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium (1997) 21:19-29.
175. ZAHRADNIKOVA A, MINAROVIC I, VENEMA RC, MESZAROS LG: Inactivation of the cardiac ryanodine receptor calcium release channel by nitric oxide. Cell Calcium (1997) 22:447-453.
176. MESZAROS LG, MINAROVIC I, ZAHRADNIKOVA A: Inhibition of the skeletal muscle ryanodine receptor calcium release channel by nitric oxide. FEBS Lett. (1996) 380:49-52.
177. AGHDASI B, ZHANG JZ, WU Y, REID MB, HAMILTON SL: Multiple classes of sulfhydryls modulate the skeletal muscle Ca2+ release channel. J. Biol. Chem. (1997) 272:3739-3749.
178. XU L, EU JP, MEISSNER G, STAMLER JS: Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science (1998) 279:234-237.
179. SUN J, XIN C EU JP, STAMLER JS, MEISSNER G: Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc. Natl. Acad. Sci.USA (2001) 98(20):11158-11162.
180. ROGERS EF et al.: Plant Insecticides. I. Ryanodine, a new alkaloid from Ryania speciosa Vahl. J. Am. Chem. Soc. (1948) 70:3086.
181. JENDEN DJ, FAIRHURST AS: The pharmacology of ryanodine. Pharmacol. Rev. (1969) 21:1-25.
182. FLEISCHER S, OGUNBUNMI EM, DIXON MC, FLEER EAM: Localization of Ca2+ release channels with ryanodine in junctional terminal cisternae of sarcoplasmic reticulum of fast skeletal muscle. Proc. Natl. Acad. Sci. USA (1985) 82(21):7256-7259.
183. MEISSNER G: Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J. Biol. Chem. (1986) 261:6300-6306.
184. BULL R, MARENGO J, SUAREZ-ISLA B et al.: Activation of calcium channels in sarcoplasmic reticulum from frog muscle by nanomolar concentrations of ryanodine. Biophys. J. (1989) 56:749-756.
185. CALLAWAY C, SERYSHEV A, WANG JP, SLAVIK KJ et al.: Localization of the high and low affinity [3H]-ryanodine binding sites on the skeletal muscle Ca2+ release channel. J. Biol. Chem. (1994) 269:15876-15884.
186. BUCK E, ZIMANYI I, ABRAMSON JJ, PESSAH IN: Ryanodine stabilizes multiple conformational states of the skeletal muscle calcium release channel. J. Biol. Chem. (1992) 267:23560-23567.
187. PROCITA L: The action of ryanodine on mammalian skeletal muscle in situ. J. Pharmacol. Exp. Ther. (1956) 117:363-373.
188. MEISSNER G, EL-HASHEM A: Ryanodine as functional probe of the skeletal muscle sarcoplasmic reticulum Ca2+ release channel. Mol. Cell. Biochem. (1992) 114:119-123.
189. SUTKO JL, AIREY JA, WELCH W, RUEST L: The pharmacology of ryanodine and related compounds. Pharmacol. Rev.(1997) 49:53-98.
• The most comprehensive review on the pharmacology of ryanodine.
190. BIDASEE KR, BESCH HR: Structure–function relationships among ryanodine derivatives. Pyridyl ryanodine definitively separates activation potency from high affinity. J. Biol. Chem. (1998) 273(20):12176-12186.
191. JEFFERIES PR, BLUMENKOPF TA, GENGO PJ et al.: Ryanodine action at calcium release channels: importance of hydroxyl substituents. J. Med. Chem. (1996) 39:2331-2338.
192. USHERWOOD PN, VAIS H: Towards the development of ryanoid insecticides with low mammalian toxicity. Toxicol. Lett. (1995) 82-83:247-254.
193. HUMERICKHOUSE RA, BESCH HR, GERZON K, RUEST L: Differential activating and deactivating effects of natural ryanodine congeners on the calcium release channel of sarcoplasmic reticulum: evidence for separation of effects at functionally distinct sites. Mol. Pharmacol. (1993) 44(2):412-421.
194. FEINSTEIN MB: Inhibition of contraction and calcium exchange-ability in rat uterus by local anesthetics. J. Pharmacol. Exp. Ther. (1966) 152(3):516-524.
195. CHAMBERLAIN BK, VOLPE P, FLEISCHER S: Inhibition of calcium-induced calcium release from purified cardiac sarcoplasmic reticulum vesicles. J. Biol. Chem. (1984) 259:7547-7553.
196. XU L, JONES RV, MEISSNER G: Effects of local anesthetics on single channel behavior of skeletal muscle calcium release channel. J. Gen. Physiol. (1993) 101:207-233.
197. ANTONIU B, KIM DH, MORII M, IKEMOTO N: Inhibitors of Ca2+ release from the isolated sarcoplasmic reticulum: Ica channel blockers. Biochim. Biophys. Acta (1985) 816:9-17.
198. VOLPE P, PALADE P, COSTELLO B, MITCHELL RD, FLEISCHER S: Spontaneous calcium release from sarcoplasmic reticulum: effect of local anesthetics. J. Biol. Chem. (1983) 258:12434-12442.
199. HERBETTE L, MESSINEO FC, KATZ AM: The interaction of drugs with the sarcoplasmic reticulum. Ann. Rev. Pharmacol. Toxicol. (1982) 22:413-434.
200. XU L, JONES RV, MEISSNER G: Effects of local anesthetics on single channel behavior of skeletal muscle calcium release channel. J. Gen. Physiol. (1993) 101:207-233.
201. ZAHRADNIKOVA A, PALADE P: Procaine effects on single sarcoplasmic reticulum Ca2+
release channels. Biophys. J. (1993) 64:991-1003.
202. PALADE P: Drug-induced Ca2+ release from isolated sarcoplasmic reticulum: II releases involving a Ca2+ -induced Ca2+ release channel. J. Biol. Chem. (1987) 262:6142-6148.
203. BELTRAN M, BULL R, DONOSO P, HIDALGO C: Ca2+ - and pH-dependent halothane stimulation of Ca2+ release in sarcoplasmic reticulum from frog muscle. Am. J. Physiol. (1996) 271:C540-C546.
204. BEELER TJ, GABLE K: Effect of halothane on Ca2+-induced Ca2+ release from sarcoplasmic reticulum vesicles isolated from rat skeletal muscle. Biochim. Biophys. Acta (1985) 821:142-152.
205. OHNISHI ST: Effects of halothane, caffeine, dantrolene and tetracaine on the calcium permeability of skeletal muscle sarcoplasmic reticulum of malignant hyperthermic pigs. Biochim. Biophys. Acta (1987) 897:261-268.
206. NELSON TE, SWEO T: Ca2+ uptake and Ca2+ release by skeletal muscle sarcoplasmic reticulum: different sensitivity to inhalational anesthetics. Anesthesiology (1988) 69:571-577.

Author p
roof
Intracellular calcium channels and their modulators
32 Expert Opin. Ther. Patents (2003) 13(6)
207. HERLAND JS, JULIAN FJ, STEPHENSON DG: Halothane increases Ca2+ efflux via Ca2+ channels of sarcoplasmic reticulum in chemically skinned rat myocardium. J. Physiol. (1990) 426:1-18.
208. FRAZER MJ, LYNCH C: Halothane and isoflurane effects on Ca2+ fluxes of isolated myocardial sarcoplasmic reticulum. Anesthesiology (1992) 77:316-323.
209. KOMAI H, RUSY BF: Effect of thiopental on Ca2+ release from sarcoplasmic reticulum in intact myocardium. Anesthesiology (1994) 81:946-952.
210. WHEELER DM, KATZ A, RICE RT, HANSFORD RG: Volatile anesthetics effects on sarcoplasmic reticulum Ca content and sarcolemmal Ca flux in isolated rat cardiac cell suspensions. Anesthesiology (1994) 80:372-382.
211. HOFFMANN P, HEINROTH K, RICHARDS D, PLEWS P, TORAASON M: Depression of calcium dynamics in cardiac myocytes: a common mechanism of halogenated hydrocarbon anesthetics and solvents. J. Mol. Cell. Cardiol. (1994) 26:579-589.
212. ZORZATO F, SCUTARI E, TEGAZZIN V, CLEMENTI E, TREVES S.: Chlorocresol: an activator of ryanodine receptor-mediated Ca2+
release. Mol. Pharmacol. (1993) 44:1192-1201.
213. HERRMANN-FRANK A, RICHTER M, SARKOZI S, MOHR U, LEHMANN-HORN F: 4-Chloro-m-cresol, a potent and specific activator of the skeletal muscle ryanodine receptor. Biochim. Biophys. Acta (1996) 1289(1):31-40.
214. BEELER TJ, GABLE K: Activation of Ca2+
release from sarcoplasmic reticulum vesicles by 4-alkylphenols. Arch. Biochem. Biophys. (1993) 301:216-220.
215. PESSAH IN, MOHR FC, SCHIEDT M, JOY RM: Stereoselective modulation of ryanodine-sensitive calcium channels by the isomer of hexachlorocyclohexane (-HCH). J. Pharmacol. Exp. Ther. (1992) 262:661-669.
216. ROSA R, SANFELIU C, RODRIGUEZ-FARRE E, FRANDSEN A, SCHOUSBOE A, SUNOL C: Properties of ryanodine receptors in cultured cerebellar granule neurons: effects of hexachlorocyclohexane isomers and calcium. J. Neurosci. Res. (1997) 47(1):27-33.
217. MCCARTHY TV, HEALY JMS, LEHANE M et al.: Localization of the malignant hyperthermia susceptibility locus to human chromosome 19q11.2-13.2. Nature (1990) 343:562-563.
218. GILLARD EF, OTSU K, FUJII J et al.: Polymorhisms and deduced amino acid substitutions in the coding sequence of the ryanodine receptor (RYR1) gene and individuals with malignant hyperthermia. Genomics (1992) 13:1247-1254.
219. QUANE KA, KEATING KE, HEALY JMS et al.: Mutation screening of the RYR1 gene in malignant hyperthermia: detection of a novel Tyr to Ser mutation in a pedigree with associated central cores. Genomics (1994) 23:236-239.
220. MONNIER N, PROCACCIO V, STIEGLITZ P, LUNARDI J: Malignant-hyperthermia susceptibility is associated with a mutation of the 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am. J. Hum. Genet. (1997) 60:1316-1325.
221. JURKAT-ROTT K, McCARTHY T, LEHMAN-HORN F: Genetics and pathogenesis of malignant hyperthermia. Muscle Nerve (2000) 23(1):4-17
222. HERBETTE L, MESSINEO FC, KATZ AM.: The interaction of drugs with the sarcoplasmic reticulum. Ann. Rev. Pharmacol. Toxicol. (1982) 22:413-434.
223. PAUL-PLETZER K, YAMAMOTO T, BHAT MB et al.: Identification of the dantrolene binding sequence on the skeletal muscle ryanodine receptor. J. Biol. Chem. (2002) 277(38):34918-34923.
224. ZHAO F, LI P, CHEN SR, LOUIS CF, FRUEN BR: Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J. Biol. Chem. (2001) 276(17):13810-13816.
225. SHUAIB A, PAASUKE RT, BROWNELL KW: Central core disease. Clinical features in 13 patients. Medicine (1987) 66:389-396.
226. LYNCH PJ, TONG J, LEHANE M et al.: A mutation in the transmembrane/lumenal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc. Natl. Acad. Sci. USA (1999) 277(38):34918-34923.
227. QUANE KA, HEALY JMS, KEATING KE et al.: Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat. Genet. (1993) 5:51-55.
228. GUTMANN E, SANDOW A: Caffeine-induced contracture and potentiation of contraction in normal and denervated rat muscle. Life Sci. (1965) 4(11):1149-1156.
229. ZETT L, KUCHLER G: Investigations on the action mechanism of caffeine-, KCl- and acid-induced contractions. Acta. Biol. Med. Ger. (1965) 14(6):687-699.
230. CARAFOLI E, PATRIARCA P, ROSSI CS: A comparative study of the role of mitochondria and the sarcoplasmic reticulum in the uptake and release of Ca2+ by the rat diaphragm. J. Cell. Physiol. (1969) 74(1):17-30.
231. HERNANDEZ-CRUZ A, DIAZ-MUNOZ M, GOMEZ-CHAVARIN M et al.: Properties of the ryanodine-sensitive release channel that underlie caffeine-induced Ca2+ mobilization from intracellular stores in mammalian sympathetic neurons. Eur. J. Neurosci. (1995) 7:1684-1699.
232. ROUSSEAU E, LADINE J, LIU QY, MEISSNER G: Activation of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum by caffeine and related compounds. Arch. Biochem. Biophys. (1988) 267:75-86.
233. ROUSSEAU E, MEISSNER G: Single cardiac sarcoplasmic reticulum Ca2+ release channel: activation by caffeine. Am. J. Physiol. (1989) 256:H328-H333.
234. MCGARRY SJ, WILLIAMS AJ: Adenosine discriminates between the caffeine and adenine nucleotide sites on the sheep cardiac sarcoplasmic reticulum calcium-release channel. J. Membr. Biol. (1994) 137:169-177.
235. SEIFERT J, CASIDA JE: Ca2+-dependent ryanodine binding site: soluble preparation from rabbit cardiac sarcoplasmic reticulum. Biochim. Biophys. Acta (1986) 861:399-405.
236. WOHLFARTH-BOTTERMANN KE: Oscillatory contraction activity in Physarum. J. Exp. Biol. (1979) 81:15-32.
237. MATTHEWS LM Jr: Ca2+ regulation in caffeine-derived microplasmodia of Physarum polycephalum. J. Cell Biol. (1977) 72(2):502-505.
238. TAKAHASHI Y, FURUKAWA K, ISHIBASHI M et al.: Structure–activity relationship of bromoeudistomin D, a powerful Ca2+ releaser in skeletal muscle sarcoplasmic reticulum. Eur. J. Pharmacol. (1995) 288(3):285-293.
239. OHIZUMI Y, MATSUNAGA K, NAKATANI K, KOBAYASHI J’I: Potent stimulation of myofilament force and ATPase activity of skeletal muscle by eudistomin M, a novel Ca2+-sensitizing agent from a Caribbean tunicate. J. Pharmacol. Exp. Ther. (1998) 285:695-699.

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 33
240. OHIZUMI Y: Application of physiologically active substances isolated from natural resources to pharmacological studies. Jpn. J. Pharmacol. (1997) 73(4):263-289.
241. SEINO A, KOBAYASHI M, KOBAYASHI J et al.: 9-Methyl-7-bromoeudistomin D, a powerful radio-labelable Ca2+ releaser having caffeine-like properties, acts on Ca2+-induced Ca2+ release channels of sarcoplasmic reticulum. J. Pharmacol. Exp. Ther. (1991) 256:861-867.
242. YOSHIKAWA K, FURUKAWA KI, YAMAMOTO M, MOMOSE K, OHIZUMI Y: [3H]-9-Methyl-7-bromoeudistomin D, a caffeine-like powerful releaser, binds to caffeine-binding sites distinct from the ryanodine receptors in brain microsomes. FEBS Lett. (1995) 373:250-254.
243. THOMAS JM, CHURCHILL GC, PATEL S, GALIONE A. Distinct pharmacology of 2-hydroxycarbazole-induced Ca2+ release in the sea urchin egg. J. Pharmacol. Exp. Ther. (2001) 298(2):644-650.
244. TOVEY SC, LONGLAND CL, MEZNA M, MICHELANGELI F: 2-Hydroxycarbazole induces Ca2+ release from sarcoplasmic reticulum by activating the ryanodine receptor. Eur. J. Pharmacol. (1998) 354(2-3):245-251.
245. TAKAHASHI Y, FURUKAWA KI, KOZUTSUMI D, ISHIBASHI M, KOBAYASHI J, OHIZUMI Y: 4,6-Dibromo- 3-hydroxycarbazole (an analogue of caffeine-like Ca2+ releaser), a novel type of inhibitor of Ca2+ -induced Ca2+
release in skeletal muscle sarcoplasmic reticulum. Br. J. Pharmacol. (1995) 114:941-948.
246. SHTIFMAN A, WARD CW, WANG J, VALDIVIA HH, SCHNEIDER MF: Effects of imperatoxin A on local sarcoplasmic reticulum Ca(2+) release in frog skeletal muscle. Biophys. J. (2000) 79(2):814-827.
247. SAMSO M, TRUJILLO R, GURROLA GB, VALDIVIA HH, WAGENKNECHT T: Three-dimensional location of the imperatoxin A binding site on the ryanodine receptor. J. Cell Biol. (1999) 146(2):493-499.
248. EL-HAYEK R, LOKUTA AJ, AREVALO C, VALDIVIA HH: Peptide probe of ryanodine receptor function. Imperatoxin A, a peptide from the venom of the scorpion Pandinus imperator, selectively activates skeletal-type ryanodine receptor isoforms. J. Biol. Chem. (1995) 270(48):28696-286704.
249. RIPATHY A, RESCH W, XU L, VALDIVIA HH, MEISSNER G: Imperatoxin A induces subconductance states in Ca2+ release channels (ryanodine receptors) of cardiac and skeletal muscle. J. Gen. Physiol. (1998) 111(5):679-690.
250. GURROLA GB, AREVALO C, SREEKUMAR R, LOKUTA AJ, WALKER JW, VALDIVIA HH: Activation of ryanodine receptors by imperatoxin A and a peptide segment of the II – III loop of the dihydropyridine receptor. J. Biol. Chem. (1999) 274(12):7879-7886.
251. FAJLOUN Z, KHARRAT R, CHEN L et al.: Chemical synthesis and characterization of maurocalcine, a scorpion toxin that activates Ca2+ release channel/ ryanodine receptors. FEBS Lett. (2000) 469(2-3):179-185.
252. MORRISSETTE J, BEURG M, SUKHAREVA M, CORONADO R: Purification and characterization of ryanotoxin, a peptide with actions similar to those of ryanodine. Biophys. J. (1996) 71(2):707-721.
253. OHKURA M, FURUKAWA K, OIKAWA K, OHIZUMI Y: The properties of specific binding site of 125 I-radioiodinated myotoxin a, a novel Ca2+ releasing agent, in skeletal muscle sarcoplasmic reticulum. J. Pharmacol. Exp. Ther. (1995) 273(2):934-939.
254. YUDKOWSKY ML, BEECH J, FLETCHER JE: Myotoxin a reduces the threshold for calcium-induced calcium release in skeletal muscle. Toxicon (1994) 32(3):273-278.
255. MORRISSETTE J, KRATZSCHMAR J, HAENDLER B et al.: Primary structure and properties of helothermine, a peptide toxin that blocks ryanodine receptors. Toxicon. (1990) 28(3):299-309.
256. MOCHCA-MORALES J, MARTIN BM, POSSANI LD: Isolation and characterization of helothermine, a novel toxin from Heloderma horridum horridum (Mexican beaded lizard) venom. Toxicon. (1990) 28(3):299-309.
257. NOBILE M, NOCETI F, PRESTIPINO G, POSSANI LD: Helothermine, a lizard venom toxin, inhibits calcium current in cerebellar granules. Exp. Brain Res. (1996) 110(1):15-20.
258. MASUOKA H, ITO M, NAKANO T, NAKA M, TANAKA T: effects of ruthenium red on activation of Ca2+-dependent cyclic nucleotide phosphodiesterase. Biochem. Biophys. Res. Comm. (1990) 169(1):315-322.
259. LUTHRA R, OLSON MS: The inhibition of calcium uptake and release by rat liver mitochondria by ruthenium red. FEBS Lett. (1977) 81(1):142-146.
260. CORBALAN-GARCIA S, TERUEL JA, GOMEZ-FERNANDEZ JC: Characterization of ruthenium red-binding sites of the Ca2+-ATPase from sarcoplasmic reticulum and their interaction with Ca2+-binding sites. Biochem. J. (1992) 287:767-774.
261. AMANN R, DONNERER J, LEMBECK F: Capsaicin-induced stimulation of polymodal nociceptors is antagonised by ruthenium red independently of extracellular calcium. Neuroscience (1989) 32(1):255-259.
262. BINDOLI A, AND FLEISCHER S: Induced Ca2+ release in skeletal muscle sarcoplasmic reticulum by sulfhydryl reagents and chlorpromazine. Arch. Biochem. Biophys. (1983) 221:458-466.
263. SMITH JS, ROUSSEAU E, MEISSNER G: CaM modulation of single sarcoplasmic reticulum Ca2+-release channels from cardiac and skeletal muscle. Circ. Res. (1989) 64:352-359.
264. PLOSKER GL, FOSTER RH: Tacrolimus: a further update of its pharmacology and therapeutic use in the management of organ transplantation. Drugs (2000) 59(2):323-389.
265. SU Z, SUGISHITA K, LI F, RITTER M, BARRY WH: Effects of FK506 on [Ca2+ ]I
differ in mouse and rabbit ventricular myocytes. J. Pharmacol. Exp. Ther. (2003) 304(1):334-341.
266. YANO M, ONO K, OHKUSA T et al.: Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation (2000) 102(17):2131-2136.
267. DOI M, YANO M, KOBAYASHI S et al.: Propranolol prevents the development of heart failure by restoring FKBP12.6-mediated stabilization of ryanodine receptor. Circulation (2002) 105(11):1374-1379.
268. SHOSHAN-BARMATZ V: ATP-dependent interaction of propranolol and local anesthetic with sarcoplasmic reticulum: stimulation of Ca2+ efflux. Biochem. J. (1988) 256:733-739.
269. ZCHUT S, FENG W, SHOSHAN-BARMATZ V: Ryanodine receptor/calcium release channel conformations as reflected in the different effects of propranolol on its ryanodine binding and channel activity. Biochem. J. (1996) 315:377-383.

Author p
roof
Intracellular calcium channels and their modulators
34 Expert Opin. Ther. Patents (2003) 13(6)
270. KANG JJ, CHEN IL, CHENG YW: Induction of calcium release from isolated sarcoplasmic reticulum by triphenyltin. J. Biochem. (1997) 122(1):173-177.
271. SAEKI K, OBI I, OGIKU N, SHI GEKAWA M, I MAGAWA T, MATSUMOTO T: Doxorubicin directly binds to the cardiac-type ryanodine receptor. Life Sci. (2002) 70(20):2377-2389.
272. ABRAMSON JJ, BUCK E, SALAMA G, CASIDA JE, PESSAH IN: Mechanism of anthraquinone-induced calcium release from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. (1988) 263(35):18750-18758.
273. CHENG YW, KANG JJ: Emodin-induced muscle contraction of mouse diaphragm and the involvement of Ca2+ influx and Ca2+
release from sarcoplasmic reticulum. Br. J. Pharmacol. (1998) 123(5):815-820.
274. LEE EH, MEISSNER G, KIM DO H: Effects of quercetin on single Ca2+ release channel behavior of skeletal muscle. Biophys. J. (2002) 82(3):1266-1277.
275. SUZUKI A, MATSUNAGA K, SHIN H et al.: A novel Ca2+ releaser with caffeine-like properties from a marine sponge, Dysidea spp., acts on Ca2+induced Ca2+ release channels of skeletal muscle sarcoplasmic reticulum. J. Pharmacol. Exp. Ther. (2000) 292(2):725-730.
276. LOSAVIO AS, KOTSIAS BA: The effect of aminophylline on the contraction threshold of rat diaphragm fibers and its modification by 9-aminoacridine. Life Sci. (1996) 58(13):1031-1037.
277. KONDOH Y, MIZUSAWA S, MURAKAMI M, NAKAMICHI H, NAGATA K: Fasudil (HA1077), an intracellular calcium antagonist, improves neurological deficits and tissue potassium loss in focal cerebral ischemia in gerbils. Neurol. Res. (1997) 19(2):211-215.
278. SAGAWA T, NISHIO M, SAGAWA K et al.: Activation of purified cardiac ryanodine receptors by dihydropyridine agonists. Am. J. Physiol. Heart Circ. Physiol. (2001) 280(3):H1201-H1207.
279. HADAD N, FENG W, SHOSHAN-BARMATZ V: Modification of ryanodine receptor/Ca2+ release channel with dinitrofluorobenzene. Biochem. J. (1999) 342(Pt 1):239-248.
280. MCGARRY SJ, WILLIAMS AJ: Activation of the sheep cardiac sarcoplasmic reticulum Ca2+-release channel by analogues of sulmazole. Br. J. Pharmacol. (1994) 111(4):1212-1220.
281. KANG JJ, CHENG YW, KO FN, KUO ML, LIN CN, TENG CM: Induction of calcium release from sarcoplasmic reticulum of skeletal muscle by xanthone and norathyriol. Br. J. Pharmacol. (1996) 118(7):1736-1742.
282. SAGAWA T, SAGAWA K, KELLY JE, TSUSHIMA RG, WASSERSTROM JA: Activation of cardiac ryanodine receptors by cardiac glycosides. Am. J. Physiol. Heart Circ. Physiol. (2002) 282(3):H1118-H1126.
283. MCGARRY SJ, SCHEUFLER E, WILLIAMS AJ: Effect of R56865 on cardiac sarcoplasmic reticulum function and its role as an antagonist of digoxin at the sarcoplasmic reticulum calcium release channel. Br. J. Pharmacol. (1995) 114(1):231-237.
284. SARKOZI S, SZENTESI P, JONA I, CSERNOCH L: Effects of cardiac glycosides on excitation–contraction coupling in frog skeletal muscle fibres. J. Physiol. (1996) 495(Pt 3):611-626.
285. KANG JJ, HSU KS, LIN-SHIAU SY: Effects of bipyridylium compounds on calcium release from triadic vesicles isolated from rabbit skeletal muscle. Br. J. Pharmacol. (1994) 112(4):1216-1222.
286. HOLMBERG SR, WILLIAMS AJ: Phosphodiesterase inhibitors and the cardiac sarcoplasmic reticulum calcium release channel: differential effects of milrinone and enoximone. Cardiovasc. Res. (1991) 25(7):537-545.
287. KANG JJ, CHENG YW: Effects of boldine on mouse diaphragm and sarcoplasmic reticulum vesicles isolated from skeletal muscle. Planta Med. (1998) 64(1):18-21.
288. HU CM, CHENG HW, CHENG YW, KANG JJ: Induction of skeletal muscle contracture and calcium release from isolated sarcoplasmic reticulum vesicles by sanguinarine. Br. J. Pharmacol. (2000) 130:299-306.
289. FADDEEVA MD, BELIAEVA TN: Inhibition of the activity of membrane-bound Ca2+-ATPase in the sarcoplasmic reticulum fragments of rabbit skeletal muscles by the alkaloid sanguinarine. Tsitologiia (1988) 30(6):685-690.
290. AGHDASI B, REID MB, HAMILTON SL: Nitric oxide protects the skeletal muscle Ca2+
release channel from oxidation induced activation. J. Biol. Chem. (1997) 272:25462-25467.
291. MENSHIKOVA EV, CHEONGE, SALAMA G: Low N-ethylmaleimide concentrations activate ryanodine receptors by a reversible interaction, not an alkylation of critical thiols. J. Biol. Chem. (2000) 275:36775-36780.
292. ABRAMSON JJ, CRONIN JR, SALAMA G: Oxidation induced by phthalocyanine dyes causes rapid calcium release from sarcoplasmic reticulum vesicles. Arch. Biochem. Biophys. (1988) 263(2):245-255.
293. ABRAMSON JJ, BUCK E, SALAMA G, CASIDA JE, PESSAH IN: Mechanism of anthraquinone-induced calcium release from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. (1988) 263(35):18750-18758.
294. POSTERINO GS, LAMB GD: Effects of reducing agents and oxidants on excitation-contraction coupling in skeletal muscle fibres of rat and toad. J. Physiol. (1996 ) 496(Pt 3):809-825.
295. BRUNDER DG, DETTBARN C, PALADE P: Heavy metal-induced Ca2+
release from sarcoplasmic reticulum. J. Biol. Chem. (1988) 263:18785-18792.
296. TRIMM JL, SALAMA G, ABRAMSON JJ: Sulfhydryl oxidation induces rapid calcium release from sarcoplasmic reticulum vesicles. J. Biol. Chem. (1986) 261:16092-16098.
297. SALAMA G, ABRAMSON JJ: Silver ions trigger Ca2+ release by acting at the apparent physiological release site in sarcoplasmic reticulum. J. Biol. Chem. (1984) 259:13363-13369.
298. ABRAMSON JJ, TRIMM JL, WEDEN L, SALAMA G: Heavy metals induce rapid calcium release from sarcoplasmic reticulum vesicles isolated from skeletal muscle. Proc. Natl. Acad. Sci. USA (1983) 80:1526-1530.
299. ZUCCHI R, RONCA-TESTONI S: The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol. Rev. (1997) 49(1):1-51.
• The most comprehensive review on the pharmacology of the ryanodine receptor.
300. NIHONYANAGI K, OBA T: Gold ion inhibits silver ion induced contracture and activates ryanodine receptors in skeletal muscle. Eur. J. Pharmacol. (1996)311(2-3):271-276.
301. TUPLING R, GREEN H: Silver ions induce Ca2+ release from the SR in vitro by acting on the Ca2+ release channel and the Ca2+ pump. J. Appl. Physiol. (2002) 92:1603-1610.

Author p
roof
Kochegarov
Expert Opin. Ther. Patents (2003) 13(6) 35
302. KANG JJ, LIU SH, CHEN IL, CHENG YW, LIN-SHIAU SY: Comparative studies on the induction of muscle contracture in mouse diaphragm and Ca2+ release from sarcoplasmic reticulum vesicles by organotin compounds. Pharmacol. Toxicol. (1998) 82(1):23-27.
303. EMMICK JT, KWON S, BIDASEE KR, BESCH KT, BESCH HR Jr: Dual effect of suramin on calcium fluxes across sarcoplasmic reticulum vesicle membranes. J. Pharmacol. Exp. Ther. (1994) 269(2):717-724.
304. SUKO J, HELLMANN G, DROBNY H: Short- and long-term functional alterations of the skeletal muscle calcium release channel (ryanodine receptor) by suramin: apparent dissociation of single channel current recording and [3H]-ryanodine binding. Mol. Pharmacol. (2001) 59(3):543-556.
305. BLONDEL O, TAKEDA J, JANSSEN H, SEINO S, BELL GI: Sequence and functional characterization of a third inositol trisphosphate receptor subtype, IP3
R-3, expressed in pancreatic islets, kidney, gastrointestinal tract, and other tissues. J. Biol. Chem. (1993) 268(15):11356-11363.
306. DIETRICH A, BRAZIL D, JENSEN ON et al.: Isoprenylation of the G protein γ subunit is both necessary and sufficient for βγ dimer-mediated stimulation of phospholipase C. Biochemistry (1996) 35(48):15174-15182.
307. HARDIE RC, RAGHU P: Activation of heterologously expressed Drosophila TRPL channels: Ca2+ is not required and InsP3 is not sufficient. Cell Calcium (1998) 24(3):153-163.
308. KOGANEZAWA M, SHIMADA I: Inositol 1,4,5-trisphosphate transduction cascade in taste reception of the fleshfly, Boettcherisca peregrina. J. Neurobiol. (2002) 51(1):66-83.
309. WONG GT, GANNON KS, MARGOLSKEE RF: Transduction of bitter and sweet taste by gustducin. Nature (1996) 381:796-800.
310. SPIELMAN AI, NAGAI H, SUNAVALA G et al.: Rapid kinetics of second messenger production in bitter taste. Am. J. Physiol. (1996) 270:C926-C931.
311. MAYRLEITNER M, SCHAFER R, FLEISCHER S: IP3 receptor purified from liver plasma membrane is an (1,4,5)IP3
activated and (1,3,4,5)IP4 inhibited calcium permeable ion channel. Cell Calcium (1995) 17(2):141-153.
312. HAGAR RE, BURGSTAHLER AD, NATHANSON MH, EHRLICH BE: Type III InsP3 receptor channel stays open in the presence of increased calcium. Nature (1998) 396(6706):81-84.
313. MICHIKAWA T, HIROTA J, KAWANO S et al.: CaM mediates calcium-dependent inactivation of the cerebellar type 1 inositol 1,4,5-trisphosphate receptor. Neuron (1999) 23:1-10.
314. MATIFAT F, HAGUE F, BRÛLÉ G, COLLIN T: Regulation of InsP3-mediated Ca2+ release by CaMKII in Xenopus oocytes. Pflugers Arch.iv. Eur. J. Physiol. (2001) 441(6):796-801.
315. NAKAGAWA T, OKANO H, FURUICHI T, ARUGA J, MIKOSHIBA K: The subtypes of the mouse inositol 1,4,5-trisphosphate receptor are expressed in a tissue-specific and developmentally specific manner. Proc. Natl. Acad. Sci. USA (1991) 88:6244-6248.
316. NIKASHIN AB, NEYLON KB, BOBIK A, TKACHUK VA: Regulation of inositol-1,4,5- triphosphate receptor by Gi-protein. Ross Fiziol. Zh. Im. I M Sechenova (1999) 85(8):1011-1021.
317. MAES K, MISSIAEN L, PARYS JB et al.: Adenine-nucleotide binding sites on the inositol 1,4,5-trisphosphate receptor bind caffeine, but not adenophostin A or cyclic ADP-ribose. Cell Calcium (1999) 25(2):143-152.
318. WOJCIKIEWICZ RJ, LUO SG: Phosphorylation of inositol 1,4,5-trisphosphate receptors by cAMP-dependent protein kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. J. Biol. Chem. (1998) 273(10):5670-5677.
319. HAUG LS, JENSEN V, HVALBY O, WALAAS SI, OSTVOLD AC: Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic nucleotide-dependent kinases in vitro and in rat cerebellar slices in situ. J. Biol. Chem. (1999) 274(11):7467-7473.
320. MATTER N, RITZ MF, FREYERMUTH S, ROGUE P, MALVIYA AN: Stimulation of nuclear protein kinase C leads to phosphorylation of nuclear inositol 1,4,5-trisphosphate receptor and accelerated calcium release by inositol 1,4,5- trisphosphate from isolated rat liver nuclei. J. Biol. Chem. (1993) 268(1):732-736.
321. STRIGGOW F, EHRLICH BE: Regulation of intracellular calcium release channel function by arachidonic acid and leukotriene B4. Biochem. Biophys. Res. Commun. (1997) 237(2):413-418.
322. MUTO A, MIKOSHIBA K: Activation of inositol 1,4,5-trisphosphate receptors induces transient changes in cell shape of fertilized Xenopus eggs. Cell Motil. Cytoskeleton (1998) 39(3):201-208.
323. BEECROFT MD, MARCHANT JS, RILEY AM: Acyclophostin: a ribose-modified analog of adenophostin A with high affinity for inositol 1,4,5-trisphosphate receptors and pH-dependent efficacy. Mol. Pharmacol. (1999) 55:109-117.
324. GAFNI J, MUNSCH JA, LAM TH et al.: Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron (1997) 19(3):723-733.
325. MIYAMOTO S, IZUMI M, HORI M, KOBAYASHI M, OZAKI H, KARAKI H: Xestospongin C, a selective and membrane-permeable inhibitor of IP 3 receptor, attenuates the positive inotropic effect of alpha-adrenergic stimulation in guinea-pig papillary muscle. Br. J. Pharmacol. (2000) 130(3):650-654.
326. SCHALOSKE R, SCHLATTERER C, MALCHOW DA: Xestospongin C-sensitive Ca2+ store is required for cAMP-induced Ca2+
influx and cAMP oscillations in Dictyostelium. J. Biol. Chem. (2000) 275(12):8404-8408.
327. OKA T, SATO K, HORI M, OZAKI H, KARAKI H: Xestospongin C, a novel blocker of IP3 receptor, attenuates the increase in cytosolic calcium level and degranulation that is induced by antigen in RBL-2H3 mast cells. Br. J. Pharmacol. (2002) 135(8):1959-1966.
328. CASTONGUAY A, ROBITAILLE R: Xestospongin C is a potent inhibitor of SERCA at a vertebrate synapse. Cell Calcium (2002) 32(1):39-47.
329. SOLOVYOVA N, FERNYHOUGH P, GLAZNER G, VERKHRATSKY A: Xestospongin C empties the ER calcium store but does not inhibit InsP3 –induced Ca2+ release in cultured dorsal root ganglia neurones. Cell Calcium (2002) 32(1):49-52.
330. DE SMET P, PARYS JB, CALLEWAERT G et al.: Xestospongin C is an equally potent inhibitor of the inositol 1,4,5-trisphosphate receptor and the endoplasmic-reticulum Ca2+ pumps. Cell Calcium (1999) 26(1-2):9-13.
331. IWASAKI H, MORI Y, HARA Y, UCHIDA K, ZHOU H, MIKOSHIBA K: 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Receptors Channels (2001) 7(6):429-439.

Author p
roof
Intracellular calcium channels and their modulators
36 Expert Opin. Ther. Patents (2003) 13(6)
332. BOOTMAN MD, COLLINS TJ, MACKENZIE L, RODERICK HL, BERRIDGE MJ, PEPPIATT CM: 2-Aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+
release. FASEB J. (2002) 16(10):1145-1150.
333. BISHARA NB, MURPHY TV, HILL MA: Capacitative Ca2+ entry in vascular endothelial cells is mediated via pathways sensitive to 2 aminoethoxydiphenyl borate and xestospongin C. Br. J. Pharmacol. (2002) 135(1):119-128.
334. MA HT, VENKATACHALAM K, PARYS JB, GILL DL: Modification of store-operated channel coupling and inositol trisphosphate receptor function by 2-aminoethoxydiphenyl borate in DT40 lymphocytes. J. Biol. Chem. (2002) 277(9):6915-6922.
335. WANG Y, DESHPANDE M, PAYNE R: 2-Aminoethoxydiphenyl borate inhibits phototransduction and blocks voltage-gated potassium channels in Limulus ventral photoreceptors. Cell Calcium (2002) 32(4):209-216.
336. SOULSBY M. DWOJCIKIEWICZ RJH: 2-Aminoethoxydiphenyl borate inhibits inositol 1,4,5-trisphosphate receptor function, ubiquitination and downregulation, but acts with variable characteristics in different cell types. Cell Calcium (2002) 32(4):175-181.
337. MCCORMICK J, LI Y, MCCORMICK K et al.: Structure and total synthesis of HF-7, a neuroactive glyconucleoside disulfate from the funnel-web spider Hololena curta. J. Am. Chem. Soc. (1999) 121(24):5661-5665.
338. SKEIE GO, MYGLAND A, TREVES S, GILHUS NE, AARLI JA, ZORZATO F: Ryanodine receptor antibodies in myasthenia gravis: epitope mapping and effect on calcium release in vitro. Muscle Nerve (2003) 27(1):81-89.
339. DEL MAR GONZALEZ-BARROSO M, RICQUIER D, CASSARD-DOULCIER AM: The human uncoupling protein-1 gene (UCP1): present status and perspectives in obesity research. Obes. Rev. (2000) 1(2):61-72.
340. MOONGA BS, LI S, IQBAL J et al.: Ca2+
influx through the osteoclastic plasma membrane ryanodine receptor. Am. J. Physiol. Renal Physiol. (2002) 282(5):F921-F932.
341. ZAIDI M, MOONGA BS, ADEBANJO OA: Novel mechanisms of calcium handling by the osteoclast: a review-hypothesis. Proc. Assoc. Am. Physicians (1999) 111(4):319-327.
342. CEDAZO-MINGUEZ A, POPESCU BO, ANKARCRONA M, NISHIMURA T, COWBURN RF: The presenilin 1 δ E9 mutation gives enhanced basal phospholipase C activity and a resultant increase in intracellular calcium concentrations. J. Biol. Chem. (2002) 277(39):36646-36655.
Patents
343. ADVANCED RES. & TECH. INST., INC. ET AL.: WO9932115 (1999).
344. RIKEN W-S ET AL.: EP-0992587-A (1999).
345. UNIV. OF BATH: WO0211736 (2000).
346. UNIV. OF BATH: WO0037089 (2000).
347. CELLPEP SA: WO0164724 (2001).
348. UNIFOB STIFTELSEN UNIVERSITETSFORSKNING BERGEN: WO0075658 (2000).
349. UNIV. OF SOUTH FLORIDA: WO0141756 (2001).
350. FRED HUTCHINSON CANCER RES. CENTER ET AL.: WO0134633 (2001).
351. NOVO NORDISK AS ET AL.: WO0222122 (2002).
352. CONTRIMMUNE BIOTECH., INC.: WO0238140 (2002).
353. JOHNS HOPKINS UNIV.: WO9955317 (1999).
354. SCHERING AG ET AL.: WO0046193 (2000).
355. SCHERING AG ET AL.: WO0046222 (2000).
AffiliationAndrei A KochegarovDepartment of Neurology, University of California Los Angeles, 5524B GONDA Center, 695 Charles E. Young dr. S., UCLA, Los Angeles, CA 90095, USATel: +1 310 825 4046;E-mail: [email protected]