integration techniques for modern bioinformatics workflows
TRANSCRIPT

University of Groningen
Integration techniques for modern bioinformatics workflowsKanterakis, Alexandros
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2018
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Kanterakis, A. (2018). Integration techniques for modern bioinformatics workflows. University of Groningen.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
The publication may also be distributed here under the terms of Article 25fa of the Dutch Copyright Act, indicated by the “Taverne” license.More information can be found on the University of Groningen website: https://www.rug.nl/library/open-access/self-archiving-pure/taverne-amendment.
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 14-07-2022

2 Creating transparent and reproduciblepipelines: Best practices for tools, dataand workflow management systems
Alexandros Kanterakis1, George Potamias2, Morris A. Swertz1
1Genomics Coordination Center, Department of Genetics, University Medical CenterGroningen, University of Groningen, Groningen, The Netherlands.2Institute of Computer Science, Foundation for Research and Technology Hellas(FORTH), Heraklion, Greece.
Submitted as a chapter to Human Genome Informatics: Translating Genes Into HealthEditors: Darrol Baker, Christophe Lambert and George Patrinos
Publication August 2018Publisher: Elsevier
https://www.elsevier.com/books/human-genome-informatics/baker/978-0-12-809414-3
25

AbstractToday, the practice of properly sharing the source code, analysis pipelines and protocolsof published studies has become commonplace in bioinformatics. Additionally, there is aplethora of technically mature Workflow Management Systems (WMS) that offer simpleand user-friendly environments where users can submit tools and build transparent,shareable and reproducible pipelines. Arguably, the adoption of open science policiesand the availability of efficient WMSs constitutes major progress towards battling thereplication crisis, advancing research dissemination and creating new collaborations.Yet, today we still see that it is very difficult to include a large range of tools in ascientific pipeline, whereas on the other side, certain technical and design choices ofmodern WMSs discourage users from doing just this. Here we present three sets ofeasily applicable “best practices” targeting (i) bioinformatics tool developers, (ii) datacurators and (iii) WMS engineers, respectively. These practices aim to make it easier toadd tools to a pipeline, to make it easier to directly process data, and to make WMSswidely hospitable for any external tool or pipeline. We also show how following theseguidelines can directly benefit the research community.
2.1 IntroductionToday, publishing the source code, data and other implementation details of a researchproject serves two basic purposes. The first is to allow the community to scrutinize,validate and confirm the correctness and soundness of a research methodology. Thesecond is to allow the community to properly utilize the methodology in novel data, orto adjust it to test new research hypotheses. This is a natural process that pertainsto practically every new invention or tool and can be reduced down to the over-simplistic sequence: Create a tool, test the tool, use the tool. Yet it is surprising thatin bioinformatics this natural sequence was not standard practice until the 1990s whenspecific groups and initiatives like BOSC [63] started advocating its use. Fortunately,today we can be assured that this process has become common practice, although thereare still grounds for improvement. Many researchers state that ideally, the ‘materialsand methods’ part of a scientific report should be an object that encapsulates thecomplete methodology, is directly available and executable, and should accompany(rather than supplement) any biologic investigation [69]. This highlights the need foranother set of tools that automate the sharing, replication and validation of results, andthe conclusions from published studies. Additionally, there is a need to include externaltools and data easily [58] and to be able to generate more complex or integrated
26

research pipelines. This new family of tools is referred to as Workflow ManagementSystems (WMS) or Data Workflow Systems [79].The tight integration of open source policies and WMSs is a highly anticipated
milestone that promises to advance many aspects of bioinformatics. In section 2.6we present some of these aspects, with fighting the replication crisis and advancinghealthcare through clinical genetics being the most prominent. One critical question iswhere do we stand now on our way to making this milestone a reality. Estimating thenumber of tools that are part of reproducible analysis pipelines is not a trivial task.Galaxy [55] has a publicly available repository of available tools called ‘toolshed’ 1 whichlists 3,356 different tools. myExperiment [56] is a social web-site where researcherscan share Research Objects such as scientific workflows; it contains approximately2,700 workflows. For comparison bio.tools [74], a community-based effort to list anddocument bioinformatics resources, lists 6,842 items2. Bioinformatics.ca [16] curatesa list of 1,548 tools and 641 databases, OMICtools [68] contains approximately 5,000tools and databases, and the Nucleic Acids Research journal curates a list of 1,700databases [53]. Finally, it is estimated there are approximately 100 different WMSs 3
with very diverse design principles.Despite the plethora of available repositories, tools, databases and WMSs, the task
of combining multiple tools in a research pipeline is still considered a cumbersomeprocedure that requires above-average IT skills [86]. This task becomes even moredifficult when the aim is to combine multiple existing pipelines, use more than one WMS,or to submit the analysis to a highly customized, High Performance Computing (HPC)environment. Since the progress and innovation in bioinformatics lies to a great extentin the correct combination of existing solutions, we should expect significant progresson the automation of pipeline building in the future[111]. In the meantime, today’sdevelopers of bioinformatics tools and services can follow certain software developmentguidelines that will help future researchers tremendously in building complex pipelineswith these tools. Additionally, data providers and curators can follow clear directionsin order to improve the availability, re-usability and semantic description of theseresources. Finally, WMS engineers should follow certain guidelines that can augmentthe inclusiveness, expressiveness and user-friendliness of these environments. Here wepresent a set of easy-to-follow guidelines for each of these groups.
1Galaxy Tool Shed: https://toolshed.g2.bx.psu.edu/2https://bio.tools/stats3Awesome Pipeline: https://github.com/pditommaso/awesome-pipeline
27

2.2 Existing workflow environmentThorough reviews on existing workflow environments in bioinformatics can be foundin many relevant studies [86], [79], [130]. Nevertheless, it is worthwhile to take a brieflook at some of the most well-known environments and frequently used techniques.The most prominent workflow environment and perhaps the only success story in
the area - with more than a decade of continuous development and use - is Galaxy [55].Galaxy has managed to build a lively community that includes biologists and IT-experts.It also acts as an analysis frontend in many research institutes. Other features that havecontributed significantly to Galaxy’s success are: (1) capability to build independentrepositories of workflows and tools and allow the inclusion of these from one installationto another, (2) having a very basic and simple wrapping mechanism for ambiguoustools, (3) native support for a large set of High Performance Environments (HPC)like (TORQUE, cloud, grid) [82], and (4) offering a simple, interactive web-basedenvironment for graph-like workflow creation. Despite Galaxy’s success, it still lacksmany of the qualitative criteria that we will present later. Perhaps most importantis the final criterion that discusses enabling users to collaborate in analyses, share,discuss and rate results, exchange ideas, and co-author scientific reports.The second most well-known environment is perhaps TAVERNA [150]. TAVERNA is
built around the integration of many web services and has limited adoption in the areaof genetics. The reason for this is that it is a more complex framework, it is not a webenvironment and forces users to adhere to specific standards (i.e. BioMoby, WSDL).We believe that the reasons why TAVERNA is lagging GALAXY, despite having aprofessional development team and extensive funding, should be studied more deeplyin order to generate valuable lessons for future reference.In this thesis we use a new workflow environment, MOLGENIS-compute [21], [19],
[20]. This environment offers tool integration and workflow description that is evensimpler than GALAXY and it is built around MOLGENIS, another successful toolused in the field [131]. Moreover it comes with “batteries included” tools and workflowsfor genotype imputation and RNA-sequencing analysis.Other environments with promising features include Omics Pipe [50] and EDGE
[89] for next generation sequencing data, and Chipster [77] for microarray data. Allthese environments claim to be decentralized and community-based although theirwide adoption by the community still needs to be proven.Besides integrated environments, it is worth noting some workflow solutions at the
level of programming languages for example, Python packages like Luigi and bcbio-
28

nextgen [61], and Java packages like bpipe [118]. Other solutions are Snakemake4,Nextflow5 and BigDataScript [29], which are novel programming languages dedicatedsolely to scientific pipelines6. Describing a workflow in these packages gives someresearchers the ability to execute their analyses easily in a plethora of environments,but unfortunately the packages are targeted at skilled IT users.Finally, existing workflows and languages for workflow description and exchange are
YAWL [140], Common Workflow Language7 and Workflow Description Language8 .The support of one or more workflow languages by an environment is essential andcomprises one of the most important qualitative criteria.
2.3 What software should be part of a scientific workflow?Even in the early phases of development of a bioinformatics tool, one should take intoconsideration that it will become part of a yet-to-be-designed workflow at some point.Since the needs of that workflow are unknown at the moment, all the design principlesused for the tool should focus on making it easy to configure, adapt and extend. Thisis on top of other quality criteria that bioinformatics methods should possess [149],[87]. After combining tens of different tools together for the scope of this thesis, wecan present a checklist of qualitative criteria for modern bioinformatics tools to covercoding, quality checks, project organization and documentation.
• On coding:– Is the code readable or else written in an extrovert way, assumming that
there is a community ready to read, correct and extend it?– Can people, unfamiliar with the project’s codebase get a good grasp of the
architecture and module organization of the project?– Is the code written according to the idiom used by the majority of program-
mers of the chosen language (i.e. camel case for Java or underscores forPython?)
– Is the code style (i.e. formatting, indentation, comment style) consistentthroughout the project?
4Similar to ‘make’ tool, with a targeted repository of workflows https://bitbucket.org/snakemake/snakemake/wiki/Home
5http://www.nextflow.io/6For more: https://www.biostars.org/p/91301/7https://github.com/common-workflow-language/common-workflow-language8https://github.com/broadinstitute/wdl
29

• On quality checks:– Do you provide test cases and unit tests that cover the entirety of the code
base?– When you correct bugs, do you make the previously failing input a new test
case?– Do you use assertions?– Does the test data come from “a real world problem”?– Does your tool generate logs that could easily trace errors on: input param-
eters, input data, user’s actions or implementation?
• On project organization– Is there a build tool that automates compilation, setup and installation?– Does the build tool check for necessary dependent libraries and software?– Have you tested the build tool in some basic commonly used environments?– Is the code hosted in an open repository? (i.e. github, bitbucket, gitlab)– Do you make incremental changes, with sufficient commit messages?– Do you “re-invent the wheel”? Or else, is any part of the project already
implemented in a mature and easily embedded way that you don’t use?
• On Documentation– Do you describe the tool sufficiently well?– Is some part of the text more targeted to novice users?– Do you provide tutorials or step-by-step guides?– Do you document interfaces, basic logic and project structure?– Do you document installation, setup and configuration?– Do you document memory, CPU, disk space or bandwidth requirements?– Do you provide execution time for average use?– Is the documentation self-generated? (i.e. Sphinx)– Do you provide means of providing feedback or contacting the main devel-
oper(s)?
30

• Having a user interface is always a nice choice, but does it always supportcommand line execution? Command line tools are far more easily adapted topipelines.
• Recommendations for command line tools in bioinformatics include [125]:alwayshave help screens, use stdout, stdin and stderr when applicable, check for as manyerrors as possible and raise exceptions when they occur, validate parameters, anddo not hard code paths.
• Create a script (e.g. with BASH) or a program that takes care of the installationof the tool in a number of known operating systems.
• Adopt open and widely used standards and formats for input and output data.
• If the tool will most likely be at the end of an analysis pipeline and will create alarge list of findings (e.g. thousands of variants) that require human inspection,consider generating an online database view. Excellent choices for this are MOL-GENIS [131] and BioMart. These systems are easily installed and populated withdata whilst they allow other researchers to explore the data with intuitive userinterfaces.
• Finally choose light web interfaces rather than local GUIs. Web interfaces alloweasy automatic interaction in contrast to local GUIs. Each tool can include amodular web interface that in turn can be a component of a larger web-site. Webframeworks like Django offer very simple solutions for this. An example of anintegrated environment in Django is given by Cobb [30]. Tools that include webinterfaces are Mutalyzer [148] and MutationInfo9.
Before checking these lists a novice bioinformatician might wonder what minimum ITknowledge is required in the area of bioinformatics. Apart from basic abstract notionsin computer programming, new comers should get experience in BASH scripting, theLinux operating system and modern interpreted languages like Python [110]. Theyshould also get accustomed to working with basic software development techniques[100] and tools that promote openness and reproducibility [73], [90] like GIT [114].These guidelines are not only targeted to novice but also to experienced users. The bio-
star project was formed to counteract the complexity, bad quality, poor documentationand organization of bioinformatics software, which was discouraging potential newcontributors. The purpose of this project was to create “low barrier entry” environments
9https://github.com/kantale/MutationInfo, http://www.epga.gr/MutationInfo/
31

[14] that enforce stability and scale-up community creation. Moreover, even if someof these guidelines are violated, publishing the code is still a very good idea [8]. It isimportant to note here that at one point the community was calling for early openpublication of code, regardless of the quality, while at the same time part of thecommunity was being judgmental and rejecting this step [35]. So having guidelines forqualitative software shouldn’t mean that the community does not support users whodo not follow them mainly due to inexperience.Another question is whether there should be guidelines for good practices on scientific
software usage (apart from creating new software). Nevertheless, since software isactually an experimental tool in a scientific experiment, the abuse of the tool mightactually be a good idea! For this reason the only guideline that is crucial whenusing scientific software regards tracking results and data provenance. This guidelineurges researchers to be extremely cautious and responsible when monitoring, trackingand managing scientific data and logs that are generated from scientific software. Inparticular, software logs are no difference to wet-lab notebook logs and should alwaysbe kept (and archived) for further validation and confirmation10. Here we argue thatthis responsibility is so essential to the scientific ethic that it should not be delegatedlightheartedly to automatic workflow systems without careful human inspection.
2.4 Preparing data for automatic workflow analysisScientific analysis workflows are autonomous research objects in the same fashion asindependent tools and data. This means that workflows need to be decoupled fromspecific data and should be able to analyze data from sources beyond the reach ofthe bioinformatician author. Being able to locate open, self-described and directlyaccessible data is one of the most sensitive areas of life sciences research. There aretwo reasons for this. The first covers law, ethical and privacy concerns regarding theindividuals participating in a study. The second is the reluctance of researchers torelease data that only they have the benefit of accessing, thus giving them an advantagein scientific exploitations. This line of thought has placed scientific data managementas a secondary concern, often given low priority in the scope of large projects andconsortia. It is interesting in this regard to take a look at the conservative views of themedical community on open data policies [91]. These views consider the latest trendsfor openness as a side-effect of the technology from which they should be protectedrather than as a revolutionary chance for progress. Instead of choosing sides in thiscontroversy, we argue that technology itself can be used to resolve the issue. This is10http://www.nature.com/gim/journal/v18/n2/full/gim2015163a.html
32

feasible by making data repositories capable of providing research data whilst protectingprivate sensitive information and ensuring proper citation.Today it is evident that the only way to derive clinically applicable results from the
analysis of genetic data is through the collective analysis of diverse datasets that areopen not only for analysis, but also for quality control and validation scrutiny [96]. Forthis reason we believe that significant progress has to be made in order to establishthe right legal frameworks that will empower political and decision-making boardsto create mechanisms that enforce and control the adaptation of open data policies.Even then, the most difficult change remains the paradigm shift that is needed: fromthe deep-rooted philosophy of researchers who treat data as personal or institutionalproperty towards more open policies [5]. Nevertheless, we are optimistic - some changeshave already started to take place due to the open data movement. Here, we present achecklist for open data management guidelines within a research project [134].
• Make long-term data storage plans early on. Most research data will remainvaluable even decades after publication.
• Release the consent forms, or other legal documents and policies under whichthe data were collected and released.
• Make sure that the data are discoverable. Direct links should not be more distantthan 2 or 3 clicks away from relevant search in a search engine.
• Consider submitting the data to an open repository, e.g. Gene Expression Omnibusor the European Genome-phenome Archive (EGA).
• Provide meta-data. Try to adopt open and widely used ontologies and formats,and make the datasets self-explanatory. Make sure all data are described uniformlyaccording to the meta-data and make the meta-data specifications available.
• Provide the “full story” of the data. Experimental protocols, equipment, pre-processing steps, data quality checks and so on.
• Provide links to software that has been used for pre-processing and links to toolsthat are directly applicable for downstream analysis.
• Make the data citable and suggest a proper citation for researchers use. Also showthe discoveries already made using the data and suggest further experiments.
• Provide a manager/supervisor’s email and contact information for potentialfeedback from the community.
33

2.5 Quality criteria for modern workflow environmentsAlthough the notion of a scientific workflow is a simple concept with a historic presencein the bioinformatics community there are many factors that affect their overall qualitythat are still being overlooked today. In this section we present some of these factors.
2.5.1 Being able to embed and to be embeddedAny workflow should have the ability to embed other workflows as components regardlessof their in-between unfamiliarity. Modern workflow environments tend to suffer fromthe “lock-in” syndrome. Namely, they demand their users invest considerable time andresources to wrap an external component with the required meta-data, libraries andconfiguration files into a workflow. Workflows should be agnostic regarding the possiblecomponents supported and should provide efficient mechanisms to wrap and includethem with minimum effort.Similarly, a workflow environment should not assume that it will be the primary
tool with which the researcher make the complete analysis. This behavior is selfishand reveals a desire to dominate a market rather than to contribute to a community.That being said, workflow systems should offer researchers the ability to export theircomplete analysis in formats that are easily digested by other systems. Examplesinclude simple BASH scripts with meta-data described in well-known and easily parsedformats like XML, JSON or YAML. Another option is to directly export meta-types,scripts and analysis code in serialized objects like PICKLE and CAMEL11 that can beeasily loaded as a whole from other tools.
2.5.2 Support ontologiesWorkflow systems tend to focus more on the analysis part of the tool and neglect thesemantic part. The semantic enrichment of a workflow can be achieved by adhering andconforming to the correct ontologies. Analysis pipelines that focus on high throughputgenomics, like Next Generation Sequencing (NGS), or proteomics have indeed a limitedneed for semantic integration mainly because a large part of this research landscape isuncharted so far. Nevertheless, when a pipeline approaches findings closer to the clinicallevel, the semantic enrichment is necessary. At this level, the plethora of findings, thevariety of research pipelines, and sometimes the discrepancies between conclusions,can create a bewildering terrain. Therefore, a semantic integration through ontologiescan provide common ground for direct comparison of findings and methods. Thus
11http://eev.ee/blog/2015/10/15/dont-use-pickle-use-camel/
34

ontologies, for example for biobanks [109], gene function [34], genetic variation [22] andphenotypes [117], can be extremely helpful. Of course, ontologies are not the panaceato all these problems since they have their own issues to be considered [94].
2.5.3 Support virtualizationThe computing requirements of a bioinformatics workflow are often unknown to theworkflow author or user but include the processing power, memory consumption andtime required. Additionally the underlying computing environment has its own require-ments (e.g. the operating system, installed libraries and preinstalled packages). Since aworkflow environment has its own requirements and dependencies, it is cumbersomefor even skilled and well IT-trained bioinformaticians to set it up and configure. Conse-quently, a considerable amount of valuable research time is spent in configuring andsetting up a pipeline or a workflow environment. Additionally, lack of documentation,IT inexperience and time pressure lead to misconfigured environments that in turnleads to waste of resources and may even produce erroneous results12. A solution forthis can be virtualization. Virtualization is the “bundling” of all required software,operating system, libraries and packages into a unique object (usually a file) that iscalled an “image” or “container”. This image can be executed in almost all knownoperating systems with the help of specific software, making this technique a very niceapproach for the “be embeddable” feature discussed above. Any workflow environmentthat can be virtualized is automatically easily embeddable in any other system by justincluding the created image.Some nice examples include the I2B2 (Informatics for Integrating Biology and the
Bedside) consortium13 which offers the complete framework in a VMare image [139].Another is the transMART software that is offered in a VWware or VirtualBox container[4]. Docker is also an open-source project that offers virtualization, as well as an openrepository where users can browse, download and execute a vast collection of communitygenerated containers. Docker borrows concepts from the GIT version control system.Namely, users can download a container, apply changes and “commit” the changes to anew container. The potential value of Docker in science has already been discussed [12],[41];for example BioShaDock is a Docker repository of tools that can be seamlesslyexecuted in Galaxy [104]. Other bioinformatics initiatives that are based on Docker
12Paper retracted due to software incompatibility: http://www.nature.com/news/error-found-in-study-of-first-ancient-african-genome-1.19258
13https://www.i2b2.org/
35

are the CAMI (Critical Assessment of Metagenomic Interpretation14), nucleotid.es15
and bioboxes.org. All these initiatives offer a testbed for comparison and evaluation ofexisting techniques mainly for NGS tasks.
2.5.4 Offer easy access to commonly used datasetsOver the last years, an increasing number of large research projects have generated largevolumes of qualitative data that are part of almost all bioinformatics analysis pipelines.So far although locating and collecting the data is straightforward, their volume andtheir dispersed descriptions makes their inclusion a difficult task. Modern workflowenvironments should offer automatic methods to collect and use them and examples ofrelevant datasets are ExAC [32] with 60.000 exomes, 1000 Genomes Project [1], GoNL[135], ENCODE project [33], METABRIC dataset [37] and data released from largeconsortia like GIANT16. Another impediment is that different consortia release datawith different consent forms, for example in the European Genome-phenome Archive(EGA) [84] each provider has a different access policy. Filling and submitting theseforms is another task that can be automated (with information provided by users).Another essential feature should be the automatic generation of artificial data wheneverthis is requested for testing and simulation purposes [105].
2.5.5 Support and standardize data visualizationA feature that is partially supported by existing workflow environments is the inherentsupport for data visualization. Over the course of genetics research, certain visualizationmethods have become standard and are easily interpreted and widely understood by thecommunity. Workflow environments should not only support the creation and inclusionof these visualization techniques, but also suggest them whenever a pipeline is beingwritten.For example in Genome-Wide Analysis Studies (GWAS), Manhattan plots for detect-
ing significance, Q-Q plots for detecting inflation and Principal Component Analysisplots for detecting population stratification or the inclusion of visualizations from toolsthat have become standard in certain analysis pipelines, like LocusView and Haploviewplots for GWAS. The environment should also support visualization for research datalike haplotypes [75], [9], reads from sequencing [71], NGS data [126], [136] and biologicalnetworks [103].
14http://cami-challenge.org/15Very interesting presentation on virtualization: http://nucleotid.es/blog/why-use-containers/16https://www.broadinstitute.org/collaboration/giant/index.php/Main_Page
36

As an implementation note, in the last few years we have seen a huge improvement inthe ability of Internet browsers to visualize large and complex data. This was possiblemainly due to the wide adoption and standardization of JavaScript. Today JavaScriptcan be used as a standalone genome browser (for example JBrowse [128] althoughUCSC also allows this functionality [115]) and genome viewer (e.g. pileup.js [142]).JavaScript has allowed the inclusion of interactive plots like those from RShiny17 andthe creation of aesthetically and informative novel forms of visualization with librarieslike D318. Today there are hundreds of very useful, context -pecific minor online toolsin the area of bioinformatics. The superior advantage of JavaScript is that it is alreadyinstalled (and used daily) in almost all Internet browsers by default. We thereforestrongly advocate workflow environments that support JavaScript data visualization.
2.5.6 Enable “batteries included” Workflow EnvironmentsA workflow environment, no matter how advanced or rich-in-features, is of no use tothe bioinformatics community if it does not offer a basic collection of widely adoptedand ready-to-use tools and workflows. The inclusion of these, even in early releases,will offer a valuable testing framework and will quickly attract users to become acontributing community. These tools and workflows can be split into the followingcategories:Tools:
• Tools that are essential and can be used for a variety of purposes such as plink[113] and GATK [39].
• Tools for basic text file manipulation (e.g. column extraction), format validation,conversion and quality control.
• Tools for basic analysis, e.g. for variant detection, sequence alignment, variantcalling, Principal Component Analysis, eQTL analysis, phasing, imputation,association analysis and meta-analysis.
• Wrappers for online tools. Interestingly, a large part of modern analysis demandsthe use of tools that require interaction through a web browser. These toolseither do not offer a local offline version or operate on large datasets that it isinconvenient to store locally. A modern workflow should include these tools viaspecial methods that emulate and automate browser interaction. Examples of
17http://shiny.rstudio.com/18https://d3js.org/
37

these tools are: for annotation (e.g. Avia [144], ANNOVAR [146], GEMINI [108]),function analysis of variation (e.g. Polyphen-2 [2], SIFT, CONSURF [24] ) andgene-set analysis (e.g. WebGestalt [145]).
Workflows:
• Existing workflows, e.g. for high throughput sequencing [102], RNA-sequencingdata [38], [138], identification of de novo mutations [119], and genotype imputation[78].
• Protocols for downstream analysis, e.g. for functional variation [85], finding ofnovel regulatory elements [120], investigation of somatic mutations for cancerresearch [28] and delivering clinically significant findings [152].
• Meta-workflows for comparison and benchmarking [7].
The environment should also provide basic indications of their performance andsystem requirements.
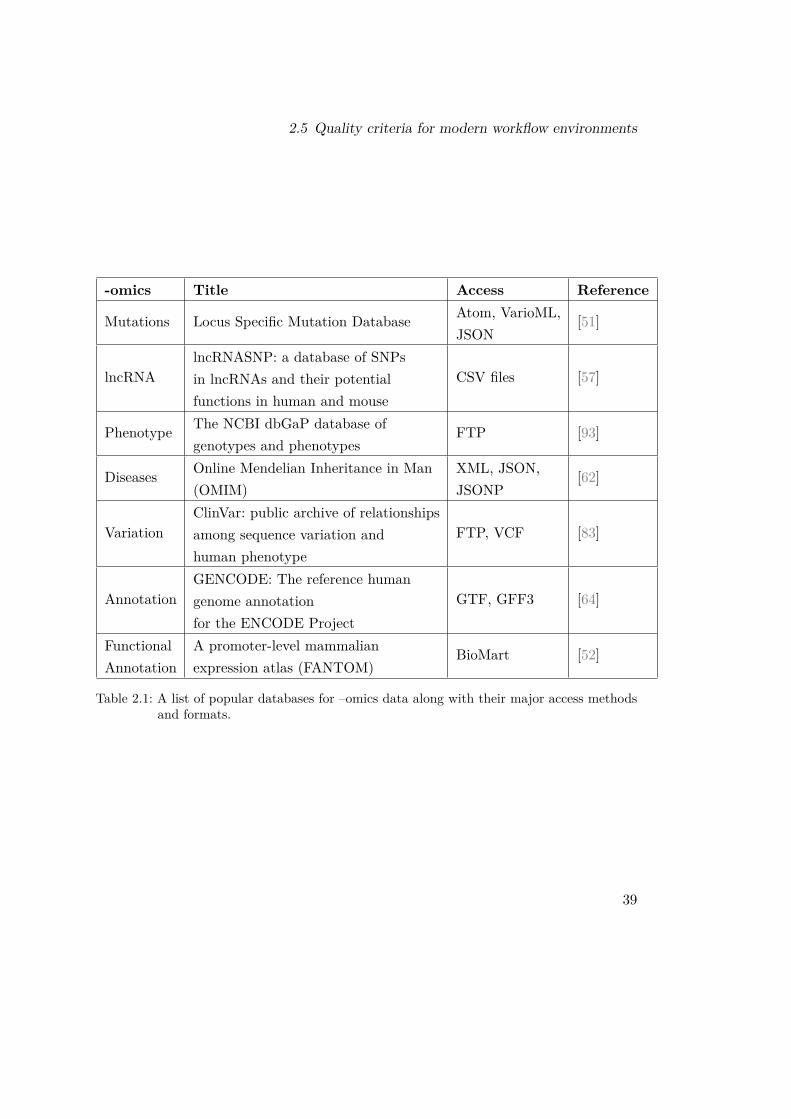
2.5.7 Facilitate data integration, both for import and exportToday, despite years of developing of formats, ontologies and standards in bioinformatics,there is still uncertainty over the correct or “right” standard when it comes to tool orservice interoperation [133]. To demonstrate this, we present a list of 7 commonly usedbioinformatics databases for various –omics datasets in Table 2.1. Each database isqualitative, built and maintained by professionals, offer modern user interfaces andhas been a standard source for information for numerous research workflows. Yet eachdatabase offers a different standard as a primary method for automated data exchange.Some are using raw data in a specific format, some have enabled API access, and somedelegate the access to secondary tools (i.e. BioMart). As long as the genetics fieldgenerates data with varying rates and complexity, we should not expect this situationto change in the near future. For this reason, a workflow environment that needs toinclude these databases must offer methods for accessing these sources tailored to thespecifics of each database. This underlines the need for “write once, use many” tools.A user who generates a tool that automates the access to a database and includes itin a workflow environment should also make it public for other users too, while theenvironment should allow and promote this policy after some basic quality checks ofcourse. The conclusion is that an environment that promotes the inclusion of manydiverse tools, workflows and databases will also promote the creation of tools thatautomate data access.
38
2.5 Quality criteria for modern workflow environments
-omics Title Access Reference
Mutations Locus Specific Mutation Database Atom, VarioML,JSON
[51]
lncRNAlncRNASNP: a database of SNPsin lncRNAs and their potentialfunctions in human and mouse
CSV files [57]
Phenotype The NCBI dbGaP database ofgenotypes and phenotypes
FTP [93]
Diseases Online Mendelian Inheritance in Man(OMIM)
XML, JSON,JSONP
[62]
VariationClinVar: public archive of relationshipsamong sequence variation andhuman phenotype
FTP, VCF [83]
AnnotationGENCODE: The reference humangenome annotationfor the ENCODE Project
GTF, GFF3 [64]
FunctionalAnnotation
A promoter-level mammalianexpression atlas (FANTOM)
BioMart [52]
Table 2.1: A list of popular databases for –omics data along with their major access methodsand formats.
39

2.5.8 Offer gateways for High Performance Computing EnvironmentsThe computational resources for a large part of modern workflows make their executionprohibitive in a local commodity computer. Fortunately, we have experienced anexplosion of HPC options in the area of –omics data [11]. These options can be splitinto two categories: the first refers to software abstractions that allow parallel executionin an HPC environment, and the second refers to existing HPC options.Regarding the first category the options are:
• Map-reduce systems [155], e.g. GATK [99].
• Specific Map-reduce implementations e.g. Hadoop, for example for NGS [107]data.
• SPARK [147].
• Multithread or multi-processing techniques, tailored for High Performance Work-stations, e.g. WiggleTools [153].
The second category includes the available HPC options:
• Computer clusters and computational grids, or else job-oriented approaches [21].
• Cloud, e.g. example for whole genome analysis [154].
• Single High Performance Workstations.
• Workstations with optimized hardware for specific tasks, e.g. GPUs19.
A workflow environment should be compatible with tools that belong to the firstcategory and offer execution options that belong to the second. This integration isperhaps the most difficult challenge for a workflow environment because each HPCenvironment has a completely different architecture and access policies. Access policiesinclude free-for-academic-use through nation-wide cluster infrastructure like Surf for theNetherlands and EDET for Greece, and paid for corporate providers like Amazon EC2or other bioinformatics tailored cloud solutions like sense20 and arvados21. Existingenvironments that offer execution for a plethora of HPC options are Yabi [72] foranalysis and BioPlanet [70] for benchmarking and comparison.
19http://richardcaseyhpc.com/parallel2/20https://sense.io/21https://arvados.org/
40

Another major consideration is the data policy. Submitting data for analysis in anexternal environment often violates strict (and sometimes very old) institutional datamanagement policies. For this reason some researchers suggest that for sensitive datawe should “bring computation to the data” [54] and not the opposite [47].
2.5.9 Engage users in collaborative experimentation and scientificauthoring
Existing workflow environments focus on automating the analysis part of the scientificprocess. We anticipate that the new generation of environments will also include thesecond and maybe more vital part of this process: active and fruitful engagement in acreative discourse that will enable the collaborative co-authoring of complete scientificreports, reaching perhaps the level of published papers. For these purposes, workflowenvironments should enable users build to communities, invite other users and easilyshare workflows and results. The system should include techniques to allow usersto comment, discuss and rate not only workflows, but results, ideas and techniques.It should also let users include rich text formatting descriptions of all parts of theanalysis and automatically generate publishable reports. Finally it should also giveproper credits to each user in the respective parts of the analysis to which they havecontributed [10]. By offering these features workflow environments will become thecenter of scientific exploration rather than just a useful analysis tool.This emerging trend -qualitative authoring pooled effort of an engaged community- is
called crowdsourcing. It has been used successfully to accomplish tasks that otherwisewould have required enormous amounts of work by a small group of researchersfor example the monitoring of infectious diseases [98], investigating novel drug-druginteractions [112] and creating valuable resources for personal genomics (e.g. OpenSNP[59] and SNPedia [23]). Of course crowdsourcing techniques can introduce a certainbias in the analysis [65] and they should be done with extreme caution especially inthe area of personal genomics [36]. This means the workflow environment should beable to apply moderation and detect cases where bias arises from a polarized user’scontribution.The most prominent tools for online interactive analysis are Jupyter22 and R Mark-
down23. The first mainly targets the Python community and the second the R commu-nity. In these environments the complete analysis can be easily shared and reproduced.Additionally, collaborative online scientific authoring is possible with services like
22http://jupyter.org/23http://rmarkdown.rstudio.com/
41

Overleaf24 and the direct hosting of generated reports can take place in a GIT enabledrepository for impartial analysis or in arXiv25 for close-to-publication reports. Hencean ideal workflow environment could integrate these techniques and enrich them withuser feedback via comments and ratings. Although this scenario sounds futuristic, ithas long been envisioned by the open science movement.
2.6 Benefits from integrated workflow analysis inBioinformatics
In this section we present the foreseeable short and long-term benefits for the bioinfor-matics community by following the criteria presented for workflow environments.
2.6.1 Enable meta-studies, combine datasets and increase statisticalpower.
One obvious benefit is to be able to perform integrated analysis and uncover previouslyunknown correlations or even causal mechanisms [92]. For example existing databasesalready include a huge amount if research data from GWAS that enable the meta-analysis and re-assessment of their findings [88], [76] and index them according todiseases and traits [18]. The improved statistical power can locate correlations for verydiverse and environmentally affected phenotypes such as Body Mass Index [129] or eveneducational attainment [116] although the interpretation of socially sensitive findingsshould always be very cautious.
2.6.2 Include methods and data from other research disciplinesAutomated analysis methods can generate the necessary abstraction layer, which willattract specialists from distant research areas with a minimum knowledge of genetics.For example, bioinformatics research can benefit greatly from “knowledge import” fromareas like machine learning, data mining, medical informatics, medical sensors, artificialintelligence and supercomputing. The Encode project had a special “machine learning”track that generated very interesting findings [33]. Existing studies can also be enrichedwith data from social networks [31].
24https://www.overleaf.com/25http://arxiv.org/
42

2.6.3 Fight the reproducibility crisisThe sparseness of data and analysis workflows from high-profile published studiesis a contributing factor to what has been characterized as the reproducibility crisisin bioinformatics. Namely, researchers are failing to reproduce results from manyprominent papers and some times-groundbreaking findings26. The consequences of thiscan be really harsh, since many retractions occur that stigmatize the community ingeneral [6], which in turn affect the overall financial viability of the field. One reportstated, “..mistakes such as failure to reproduce the data, were followed by a 50–73%drop in NIH funding for related studies”27.Moreover, irreproducible science is of practically no use for the community nor for
society and constitutes a waste of valuable funding [25]. Many researchers are blamingthe publication process (which favors positive findings and original work rather thatnegative findings or confirmation of results) and are demanding either the adoptionof open peer review policies [60], [13] or even the redesign of the complete publishingindustry [45]. Yet, a more simple and applicable remedy for this crisis is the wideadoption (and maybe enforcement) of open workflow environments that contain thecomplete analyses of published papers.
2.6.4 Spread of open source policies in genetics and privacy protectionToday a significant part of NGS data analysis for research purposes is taking placewith closed-source commercial or even academic software. The presence of this softwarein research workflows has been characterized as an impediment to science [143]. Thereis also an increase in commercial services that offer genetic data storage (e.g. Google28)and analysis29. Other worrisome trends are gene patents [97] and possible malevolentexploitation of easily de-anonymized traits and genomes that reside in public databases[3]. These trends raise ethical concerns and they also contribute to the reproducibilitycrises. Workflows that enable user collaboration can also monitor and guide thedevelopment of novel software from an initial draft to a mature and tested phase. Thisdevelopment can only happen of course in the open source domain. In the same waythat GIT promoted open source policies by “socializing” software development, webelieve that collaborative workflow environments can promote similar open sourcepolicies in genetics.26For a review of reports see: http://simplystatistics.org/2013/12/16/
a-summary-of-the-evidence-that-most-published-research-is-false/27http://blogs.nature.com/news/2012/11/retractions-stigmatize-scientific-fields-study-finds.html28https://cloud.google.com/genomics/29For example: https://insilicodb.com
43

2.6.5 Help clinical genetics researchThe clinical genetics field is tightly related to the availability of highly documented,well-tested and directly executable workflows. Even today there are genetic analysisworkflows that deliver clinically applicable results in a few days or even hours. Forexample, complete whole genome sequencing pipeline for neonatal diagnosis [121] [26]or pathogen detection in intensive care units [106], where speed is a of paramount im-portance. There are also clinical genetics workflows for the early diagnosis of Mendeliandisorders [151], while the automation of genetic analysis benefit the early diagnosis ofcomplex non-Mendelian rare [15] and common diseases like Alzheimer [43] and cansimplify or replace complex procedures like amniocentesis for the prenatal diagnosis ofTrisomy 21 [27]. Moreover cancer diagnosis and treatment has been revolutionized withthe introduction of NGS data [101] and guidelines for clinical applications are alreadyemerging [42], [127].Workflows in clinical genetics do not have to be “disease specific”. Although still in
its infancy, the idea of the universal inclusion of genetics in modern health care systems(called personalized or precision medicine) has been envisioned [66]. Probably the mostprominent public figure in this area30, Eric Topol, has emphasized the importance ofDNA and RNA level data on individualized medicine throughout the lifespan of anindividual [137]. This will allow the delivery of personalized drugs [49] and therapies[124] and will remedy current trends that threaten the survival of modern health caresystems31. The introduction of genetic screening as a routine test in healthcare, willguide medical practitioners (MPs) to take more informed decisions. Also, MPs willhave to adjust phenotyping in order to fully exploit the potentials of genetic screening[67]. Therefore we expect that clinical genetics have the potential to fundamentallychange the discipline of medicine.These visionary and futuristic ideas of course entail many challenges and considera-
tions [81]. So far, the most important are [132] the conflicting models [46], the lack ofstandards, and the uncertainty regarding the safety, effectiveness [40] and cost [44] ofthe clinical genetics guidelines generated. This is despite the initial promising resultsfrom GWAS [95] and much effort to create guidelines and mechanisms for reportingClinically Significantly Findings (CFS) to cohort or health care participants [17], [80].To tackle this, we suggest that modern workflow environments enabling multi-level
30Although there might be more prominent figures! http://www.nytimes.com/2015/01/25/us/obama-to-request-research-funding-for-treatments-tailored-to-patients-dna.html
31“An astonishing 86% of all full-time employees in the U.S. are now either overweight or suf-fer from a chronic (but often preventable) disease”, “the average cost of a family health insur-ance premium will surpass average household income by 2033”: http://www.forbes.com/sites/forbesleadershipforum/2012/06/01/its-time-to-bet-on-genomics/
44

data integration are the key to a solution. Adopting open data policies will allowlarge population level studies and meta-studies that will resolve existing discrepancies.A necessary prerequisite for this is also the inclusion of data from electronic HealthRecords [122] and BioBanks [141] (one successful initiative is the Dutch Lifelines cohort[123]). As long as findings from genetics research remains institutionally isolated anddisconnected from medical records the realization of clinical genetics and personalizedmedicine will remain only as vision.
2.7 DiscussionToday we have an abundance of high quality data that describe nearly all stages ofgenetic regulation and transcription for various cell types, diseases and populations.DNA sequences, RNA sequences and expression, protein sequence and structure, smallmolecule metabolite structure are all routinely measured in unprecedented rates [48].Moreover, the prospects in the area of data collection seem very bright as the throughputrates are increasing, the quality metrics are improving and the prices of obtainingsuch data are dropping. This leads us to the conclusion that the intermediate stepthat is missing between enormous data-collections and mechanistic or causal modelsis the data analysis. The recommended way to proceed should be via data-drivenapproaches. This in turn leads to a new question: How can we bring together diverseanalysis methods in order to qualitatively process heterogeneous big data in a constantlychanging technological landscape? This challenge requires the collective effort of thebioinformatics community and the effort can only flourish if development is focused onbuilding re-usable components rather than isolated solutions.
AcknowledgmentsFunding: The research leading to these results has received funding from the UbboEmmius Fund to AK and BBMRI-NL, a research infrastructure financed by theNetherlands Organization for Scientific Research (NWO project 184.021.007), to MS.
45


Bibliography
[1] Goncalo R Abecasis, Adam Auton, Lisa D Brooks, Mark A DePristo, Richard MDurbin, Robert E Handsaker, Hyun Min Kang, Gabor T Marth, and Gil AMcVean. An integrated map of genetic variation from 1,092 human genomes.Nature, 491(7422):56–65, 2012. ISSN 1476-4687. doi: 10.1038/nature11632. URLhttp://dx.doi.org/10.1038/nature11632.
[2] Ivan A Adzhubei, Steffen Schmidt, Leonid Peshkin, Vasily E Ramensky, AnnaGerasimova, Peer Bork, Alexey S Kondrashov, and Shamil R Sunyaev. A methodand server for predicting damaging missense mutations. Nature methods, 7(4):248–9, apr 2010. ISSN 1548-7105. doi: 10.1038/nmeth0410-248.
[3] Misha Angrist. Open window: when easily identifiable genomes and traitsare in the public domain. PloS one, 9(3):e92060, jan 2014. ISSN 1932-6203.doi: 10.1371/journal.pone.0092060. URL http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0092060.
[4] Brian D Athey, Michael Braxenthaler, Magali Haas, and Yike Guo. tranSMART:An Open Source and Community-Driven Informatics and Data Sharing Platformfor Clinical and Translational Research. AMIA Joint Summits on TranslationalScience Proceedings AMIA Summit on Translational Science, 2013:6–8, jan 2013.ISSN 2153-4063. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3814495tool=pmcentrezrendertype=abstract.
[5] Myles Axton. No second thoughts about data access. Nat Genet, 43(5):389, 2011.
[6] Pierre Azoulay, Jeffrey L Furman, Joshua L Krieger, and Fiona E Murray.Retractions. Working Paper 18499, National Bureau of Economic Research, oct2012. URL http://www.nber.org/papers/w18499.
[7] Orli G. Bahcall. Genomics: Benchmarking genome analysis pipelines. NatureReviews Genetics, 16(4):194–194, mar 2015. ISSN 1471-0056. doi: 10.1038/nrg3930.URL http://dx.doi.org/10.1038/nrg3930.
47

[8] Nick Barnes. Publish your computer code: it is good enough. Nature, 467(7317):753–753, 2010.
[9] J C Barrett, B Fry, J Maller, and M J Daly. Haploview: analysis andvisualization of LD and haplotype maps. Bioinformatics (Oxford, Eng-land), 21(2):263–5, jan 2005. ISSN 1367-4803. doi: 10.1093/bioinformatics/bth457. URL http://bioinformatics.oxfordjournals.org/content/21/2/263.full?keytype=refijkey=54SiAdNbKzbNg.
[10] Sean Bechhofer, Iain Buchan, David De Roure, Paolo Missier, John Ainsworth,Jiten Bhagat, Philip Couch, Don Cruickshank, Mark Delderfield, Ian Dunlop,Matthew Gamble, Danius Michaelides, Stuart Owen, David Newman, ShoaibSufi, and Carole Goble. Why linked data is not enough for scientists. FutureGeneration Computer Systems, 29(2):599–611, feb 2013. ISSN 0167739X. doi:10.1016/j.future.2011.08.004. URL http://www.sciencedirect.com/science/article/pii/S0167739X11001439.
[11] Bonnie Berger, Jian Peng, and Mona Singh. Computational solutions for omicsdata. Nature reviews. Genetics, 14(5):333–46, may 2013. ISSN 1471-0064. doi:10.1038/nrg3433. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3966295tool=pmcentrezrendertype=abstract.
[12] Carl Boettiger. An introduction to Docker for reproducible research. ACMSIGOPS Operating Systems Review, 49(1):71–79, jan 2015. ISSN 01635980.doi: 10.1145/2723872.2723882. URL http://dl.acm.org/citation.cfm?id=2723872.2723882.
[13] John Bohannon. Who’s afraid of peer review? Science (New York, N.Y.), 342(6154):60–5, oct 2013. ISSN 1095-9203. doi: 10.1126/science.342.6154.60. URLhttp://science.sciencemag.org/content/342/6154/60.abstract.
[14] Raoul J P Bonnal, Jan Aerts, George Githinji, Naohisa Goto, and MacLeanet al. Biogem: an effective tool-based approach for scaling up open sourcesoftware development in bioinformatics. Bioinformatics (Oxford, England), 28(7):1035–7, apr 2012. ISSN 1367-4811. doi: 10.1093/bioinformatics/bts080. URLhttp://bioinformatics.oxfordjournals.org/content/28/7/1035.full.
[15] Kym M Boycott, Megan R Vanstone, Dennis E Bulman, and Alex E MacKenzie.Rare-disease genetics in the era of next-generation sequencing: discovery totranslation. Nature reviews. Genetics, 14(10):681–91, oct 2013. ISSN 1471-0064.doi: 10.1038/nrg3555. URL http://dx.doi.org/10.1038/nrg3555.
48

[16] Michelle D Brazas, Joseph T Yamada, and BF Ouellette. Providing web serversand training in bioinformatics: 2010 update on the bioinformatics links directory.Nucleic acids research, 38(suppl_2):W3–W6, 2010.
[17] Catherine A Brownstein, Alan H Beggs, Nils Homer, and Merriman et al. Aninternational effort towards developing standards for best practices in analy-sis, interpretation and reporting of clinical genome sequencing results in theCLARITY Challenge. Genome biology, 15(3):R53, jan 2014. ISSN 1474-760X.doi: 10.1186/gb-2014-15-3-r53. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4073084tool=pmcentrezrendertype=abstract.
[18] Brendan Bulik-Sullivan, Hilary K Finucane, and Anttila et al. An atlas of geneticcorrelations across human diseases and traits. Nature genetics, 47(11):1236–41,sep 2015. ISSN 1546-1718. doi: 10.1038/ng.3406. URL http://www.ncbi.nlm.nih.gov/pubmed/26414676.
[19] H. V. Byelas, M. Dijkstra, and M. A. Swertz. Introducing data provenance anderror handling for NGS workflows within the molgenis computational frame-work. In Proceedings of the International Conference on Bioinformatics Models,Methods and Algorithms, pages 42–50. SciTePress - Science and and TechnologyPublications, jan 2012. ISBN 978-989-8425-90-4. doi: 10.5220/0003738900420050.
[20] Heorhiy Byelas, Alexandros Kanterakis, and Morris Swertz. Towards a MOLGE-NIS Based Computational Framework. In 2011 19th International EuromicroConference on Parallel, Distributed and Network-Based Processing, pages 331–338. IEEE, feb 2011. ISBN 978-1-4244-9682-2. doi: 10.1109/PDP.2011.53. URLhttp://ieeexplore.ieee.org/articleDetails.jsp?arnumber=5739032.
[21] Heorhiy Byelas, Martijn Dijkstra, Pieter BT Neerincx, Freerk Van Dijk, Alexan-dros Kanterakis, Patrick Deelen, and Morris A Swertz. Scaling bio-analyses fromcomputational clusters to grids. In IWSG, volume 993, 2013.
[22] Myles Byrne, Ivo Fac Fokkema, Owen Lancaster, and Tomasz et al. Adamu-siak. VarioML framework for comprehensive variation data representation andexchange. BMC bioinformatics, 13(1):254, jan 2012. ISSN 1471-2105. doi:10.1186/1471-2105-13-254. URL http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-13-254.
[23] Michael Cariaso and Greg Lennon. SNPedia: a wiki supporting personal genomeannotation, interpretation and analysis. Nucleic acids research, 40(Database
49

issue):D1308–12, jan 2012. ISSN 1362-4962. doi: 10.1093/nar/gkr798. URLhttp://nar.oxfordjournals.org/content/40/D1/D1308.short.
[24] Gershon Celniker, Guy Nimrod, Haim Ashkenazy, Fabian Glaser, Eric Martz,Itay Mayrose, Tal Pupko, and Nir Ben-Tal. ConSurf: Using Evolutionary Data toRaise Testable Hypotheses about Protein Function. Israel Journal of Chemistry,53(3-4):199–206, apr 2013. ISSN 00212148. doi: 10.1002/ijch.201200096. URLhttp://doi.wiley.com/10.1002/ijch.201200096.
[25] Iain Chalmers and Paul Glasziou. Avoidable waste in the production and reportingof research evidence. Lancet (London, England), 374(9683):86–9, jul 2009. ISSN1474-547X. doi: 10.1016/S0140-6736(09)60329-9. URL http://www.thelancet.com/article/S0140673609603299/fulltext.
[26] Colby Chiang, Ryan M Layer, Gregory G Faust, Michael R Lindberg, David BRose, Erik P Garrison, Gabor T Marth, Aaron R Quinlan, and Ira M Hall.SpeedSeq: ultra-fast personal genome analysis and interpretation. Nature Methods,12(10):966–8, aug 2015. ISSN 1548-7091. doi: 10.1038/nmeth.3505. URL http://www.ncbi.nlm.nih.gov/pubmed/26258291.
[27] Rossa W K Chiu, Ranjit Akolekar, Yama W L Zheng, and et al. Leung. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasmaDNA sequencing: large scale validity study. BMJ (Clinical research ed.), 342(jan11_1):c7401, jan 2011. ISSN 1756-1833. doi: 10.1136/bmj.c7401. URLhttp://www.bmj.com/content/342/bmj.c7401.long.
[28] Saket Kumar Choudhary and Santosh B Noronha. Galdrive: Pipeline for com-parative identification of driver mutations using the galaxy framework. bioRxiv,page 010538, 2014.
[29] Pablo Cingolani, Rob Sladek, and Mathieu Blanchette. BigDataScript:a scripting language for data pipelines. Bioinformatics (Oxford, Eng-land), 31(1):10–6, jan 2015. ISSN 1367-4811. doi: 10.1093/bioinformatics/btu595. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4271142tool=pmcentrezrendertype=abstract.
[30] Marea Cobb. NGSdb: An NGS Data Management and Analysis Platform forComparative Genomics. PhD thesis, University of Washington, 2015.
50

[31] Enrico Coiera. Social networks, social media, and social diseases. BMJ (Clinicalresearch ed.), 346(may22_16):f3007, jan 2013. ISSN 1756-1833. doi: 10.1136/bmj.f3007. URL http://www.bmj.com/content/346/bmj.f3007.abstract.
[32] Exome Aggregation Consortium. Analysis of protein-coding genetic variationin 60,706 humans. Nature, 536(7616):285–291, aug 2016. ISSN 0028-0836. doi:10.1038/nature19057. URL http://www.nature.com/articles/nature19057.
[33] The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNAElements) Project. Science (New York, N.Y.), 306(5696):636–40, oct 2004. ISSN1095-9203. doi: 10.1126/science.1105136. URL http://science.sciencemag.org/content/306/5696/636.abstract.
[34] The Gene Ontology Consortium. Gene Ontology Consortium: going forward.Nucleic Acids Research, 43(D1):D1049–D1056, 2015. doi: 10.1093/nar/gku1179.URL http://nar.oxfordjournals.org/content/43/D1/D1049.abstract.
[35] Manuel Corpas, Segun Fatumo, and Reinhard Schneider. How not to be abioinformatician. Source code for biology and medicine, 7(1):3, jan 2012. ISSN1751-0473. doi: 10.1186/1751-0473-7-3.
[36] Manuel Corpas, Willy Valdivia-Granda, Nazareth Torres, and Greshakeet al. Crowdsourced direct-to-consumer genomic analysis of a family quar-tet. BMC genomics, 16(1):910, jan 2015. ISSN 1471-2164. doi: 10.1186/s12864-015-1973-7. URL http://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-015-1973-7.
[37] Christina Curtis, Sohrab P Shah, Suet-Feung Chin, and Turashvili et al. Thegenomic and transcriptomic architecture of 2,000 breast tumours reveals novelsubgroups. Nature, 486(7403):346–52, jun 2012. ISSN 1476-4687. doi: 10.1038/nature10983. URL http://dx.doi.org/10.1038/nature10983.
[38] Patrick Deelen, Daria V Zhernakova, Mark de Haan, Marijke van der Si-jde, Marc Jan Bonder, Juha Karjalainen, K Joeri van der Velde, Kristin MAbbott, Jingyuan Fu, Cisca Wijmenga, Richard J Sinke, Morris A Swertz,and Lude Franke. Calling genotypes from public RNA-sequencing dataenables identification of genetic variants that affect gene-expression lev-els. Genome medicine, 7(1):30, jan 2015. ISSN 1756-994X. doi: 10.1186/s13073-015-0152-4. URL http://genomemedicine.biomedcentral.com/articles/10.1186/s13073-015-0152-4.
51

[39] Mark A DePristo, Eric Banks, Ryan Poplin, and Garimella et al. A framework forvariation discovery and genotyping using next-generation DNA sequencing data.Nature genetics, 43(5):491–8, may 2011. ISSN 1546-1718. doi: 10.1038/ng.806.URL http://dx.doi.org/10.1038/ng.806.
[40] Frederick E Dewey, Megan E Grove, Cuiping Pan, and Goldstein et al. Clin-ical interpretation and implications of whole-genome sequencing. JAMA,311(10):1035–45, mar 2014. ISSN 1538-3598. doi: 10.1001/jama.2014.1717. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4119063tool=pmcentrezrendertype=abstract.
[41] Paolo Di Tommaso, Emilio Palumbo, Maria Chatzou, Pablo Prieto, Michael L.Heuer, and Cedric Notredame. The impact of Docker containers on theperformance of genomic pipelines. PeerJ, 3:e1273, sep 2015. ISSN 2167-8359. doi: 10.7717/peerj.1273. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4586803tool=pmcentrezrendertype=abstract.
[42] Li Ding, Michael C. Wendl, Joshua F. McMichael, and Benjamin J. Raphael.Expanding the computational toolbox for mining cancer genomes. Nature ReviewsGenetics, 15(8):556–570, jul 2014. ISSN 1471-0056. doi: 10.1038/nrg3767. URLhttp://dx.doi.org/10.1038/nrg3767.
[43] James D Doecke, Simon M Laws, Noel G Faux, and Wilson et al. Blood-basedprotein biomarkers for diagnosis of Alzheimer disease. Archives of neurology, 69(10):1318–25, oct 2012. ISSN 1538-3687. doi: 10.1001/archneurol.2012.1282. URLhttp://archneur.jamanetwork.com/article.aspx?articleid=1217314.
[44] Michael P Douglas, Uri Ladabaum, Mark J Pletcher, Deborah A Marshall, andKathryn A Phillips. Economic evidence on identifying clinically actionablefindings with whole-genome sequencing: a scoping review. Genetics in medicine :official journal of the American College of Medical Genetics, 18(2):111–116, may2015. ISSN 1530-0366. doi: 10.1038/gim.2015.69. URL http://dx.doi.org/10.1038/gim.2015.69.
[45] Editorial. The future of publishing: A new page. Nature, 495(7442):425, mar2013. ISSN 1476-4687. doi: 10.1038/495425a. URL http://www.nature.com/news/the-future-of-publishing-a-new-page-1.12665.
[46] Editorial. Standards for clinical use of genetic variants. Nature genetics, 46(2):93, feb 2014. ISSN 1546-1718. doi: 10.1038/ng.2893. URL http://dx.doi.org/10.1038/ng.2893.
52

[47] Editorial. Peer review in the cloud. Nature Genetics, 48(3):223–223, mar2016. ISSN 1061-4036. doi: 10.1038/ng.3524. URL http://www.nature.com/articles/ng.3524.
[48] Michael Eisenstein. Big data: The power of petabytes. Nature, 527(7576):S2–S4,nov 2015. ISSN 0028-0836. doi: 10.1038/527S2a. URL http://dx.doi.org/10.1038/527S2a.
[49] Edward D Esplin, Ling Oei, and Michael P Snyder. Personalized sequencingand the future of medicine: discovery, diagnosis and defeat of disease. Phar-macogenomics, 15(14):1771–1790, nov 2014. ISSN 1744-8042. doi: 10.2217/pgs.14.117. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4336568tool=pmcentrezrendertype=abstract.
[50] Kathleen M Fisch, Tobias Meißner, Louis Gioia, Jean-Christophe Ducom, Tris-tan M Carland, Salvatore Loguercio, and Andrew I Su. Omics Pipe: a community-based framework for reproducible multi-omics data analysis. Bioinformatics(Oxford, England), 31(11):1724–8, jun 2015. ISSN 1367-4811. doi: 10.1093/bioinformatics/btv061. URL http://bioinformatics.oxfordjournals.org/content/31/11/1724.full.
[51] Ivo F A C Fokkema, Peter E M Taschner, Gerard C P Schaafsma, J Celli, JeroenF J Laros, and Johan T den Dunnen. LOVD v.2.0: the next generation in genevariant databases. Human mutation, 32(5):557–63, may 2011. ISSN 1098-1004. doi:10.1002/humu.21438. URL http://www.ncbi.nlm.nih.gov/pubmed/21520333.
[52] Alistair R R Forrest, Hideya Kawaji, Michael Rehli, and Baillie et al. Apromoter-level mammalian expression atlas. Nature, 507(7493):462–70, mar2014. ISSN 1476-4687. doi: 10.1038/nature13182. URL http://dx.doi.org/10.1038/nature13182.
[53] Michael Y. Galperin, Xosé M. Fernández-Suárez, and Daniel J. Rigden. The24th annual nucleic acids research database issue: a look back and upcomingchanges. Nucleic Acids Research, 45(D1):D1, 2017. doi: 10.1093/nar/gkw1188.URL http://dx.doi.org/10.1093/nar/gkw1188.
[54] Amadou Gaye, Yannick Marcon, Julia Isaeva, Philippe LaFlamme, and Turneret al. DataSHIELD: taking the analysis to the data, not the data to the anal-ysis. International journal of epidemiology, 43(6):1929–44, dec 2014. ISSN1464-3685. doi: 10.1093/ije/dyu188. URL http://ije.oxfordjournals.org/content/early/2014/09/26/ije.dyu188.short?rss=1.
53

[55] Belinda Giardine, Cathy Riemer, Ross C Hardison, Richard Burhans, and El-nitski et al. Galaxy: a platform for interactive large-scale genome analysis.Genome research, 15(10):1451–5, oct 2005. ISSN 1088-9051. doi: 10.1101/gr.4086505. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1240089tool=pmcentrezrendertype=abstract.
[56] Carole A Goble, Jiten Bhagat, Sergejs Aleksejevs, Don Cruickshank, DaniusMichaelides, David Newman, Mark Borkum, Sean Bechhofer, Marco Roos, andPeter et al. Li. myexperiment: a repository and social network for the sharing ofbioinformatics workflows. Nucleic acids research, 38(suppl 2):W677–W682, 2010.
[57] Jing Gong, Wei Liu, Jiayou Zhang, Xiaoping Miao, and An-Yuan Guo. lncR-NASNP: a database of SNPs in lncRNAs and their potential functions in humanand mouse. Nucleic acids research, 43(Database issue):D181–6, jan 2015. ISSN1362-4962. doi: 10.1093/nar/gku1000. URL http://nar.oxfordjournals.org/content/43/D1/D181.
[58] Alyssa Goodman, Alberto Pepe, Alexander W. Blocker, Christine L. Borgman,Kyle Cranmer, Merce Crosas, Rosanne Di Stefano, Yolanda Gil, Paul Groth,Margaret Hedstrom, David W. Hogg, Vinay Kashyap, Ashish Mahabal, AnetaSiemiginowska, and Aleksandra Slavkovic. Ten simple rules for the care andfeeding of scientific data. PLOS Computational Biology, 10(4):1–5, 04 2014. doi:10.1371/journal.pcbi.1003542. URL https://doi.org/10.1371/journal.pcbi.1003542.
[59] Bastian Greshake, Philipp E Bayer, Helge Rausch, and Julia Reda. openSNP–acrowdsourced web resource for personal genomics. PloS one, 9(3):e89204, jan 2014.ISSN 1932-6203. doi: 10.1371/journal.pone.0089204. URL http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0089204.
[60] Trish Groves. Is open peer review the fairest system? Yes. BMJ (Clin-ical research ed.), 341(nov16_2):c6424, jan 2010. ISSN 1756-1833. doi:10.1136/bmj.c6424. URL http://www.bmj.com/content/341/bmj.c6424?sid=60af7fc1-eb55-40af-b372-3db2a60593cc.
[61] Roman Valls Guimera. bcbio-nextgen: Automated, distributed next-gen se-quencing pipeline. EMBnet.journal, 17(B):30, feb 2012. ISSN 2226-6089.doi: 10.14806/ej.17.B.286. URL http://journal.embnet.org/index.php/embnetjournal/article/view/286.
54

[62] Ada Hamosh, Alan F Scott, Joanna S Amberger, Carol A Bocchini, and Victor AMcKusick. Online Mendelian Inheritance in Man (OMIM), a knowledgebaseof human genes and genetic disorders. Nucleic acids research, 33(Databaseissue):D514–7, jan 2005. ISSN 1362-4962. doi: 10.1093/nar/gki033. URL http://www.ncbi.nlm.nih.gov/pubmed/15608251.
[63] Nomi L. Harris, Peter J. A. Cock, Hilmar Lapp, Brad Chapman, Rob Davey,Christopher Fields, Karsten Hokamp, and Monica Munoz-Torres. The 2015bioinformatics open source conference (bosc 2015). PLOS Computational Biology,12(2):1–6, 02 2016. doi: 10.1371/journal.pcbi.1004691. URL https://doi.org/10.1371/journal.pcbi.1004691.
[64] Jennifer Harrow, Adam Frankish, Jose M Gonzalez, Electra Tapanari, andDiekhans et al. GENCODE: the reference human genome annotation for TheENCODE Project. Genome research, 22(9):1760–74, sep 2012. ISSN 1549-5469. doi: 10.1101/gr.135350.111. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3431492tool=pmcentrezrendertype=abstract.
[65] Robert T Hasty, Ryan C Garbalosa, Vincenzo A Barbato, and Valdes et al.Wikipedia vs peer-reviewed medical literature for information about the 10 mostcostly medical conditions. The Journal of the American Osteopathic Association,114(5):368–73, may 2014. ISSN 1945-1997. doi: 10.7556/jaoa.2014.035. URLhttp://www.ncbi.nlm.nih.gov/pubmed/24778001.
[66] Erika Check Hayden. Sequencing set to alter clinical landscape. Nature, 482(7385):288, feb 2012. ISSN 1476-4687. doi: 10.1038/482288a. URL http://www.nature.com/news/sequencing-set-to-alter-clinical-landscape-1.10032.
[67] Raoul C M Hennekam and Leslie G Biesecker. Next-generation se-quencing demands next-generation phenotyping. Human mutation,33(5):884–6, may 2012. ISSN 1098-1004. doi: 10.1002/humu.22048.URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3327792tool=pmcentrezrendertype=abstract.
[68] Vincent J Henry, Anita E Bandrowski, Anne-Sophie Pepin, Bruno J Gonzalez,and Arnaud Desfeux. Omictools: an informative directory for multi-omic dataanalysis. Database, 2014:bau069, 2014.
[69] Kristina Hettne, Stian Soiland-Reyes, Graham Klyne, Khalid Belhajjame,Matthew Gamble, Sean Bechhofer, Marco Roos, and Oscar Corcho. Workflow
55

forever: Semantic web semantic models and tools for preserving and digitally pub-lishing computational experiments. In Proceedings of the 4th International Work-shop on Semantic Web Applications and Tools for the Life Sciences, SWAT4LS’11, pages 36–37, New York, NY, USA, 2012. ACM. ISBN 978-1-4503-1076-5.doi: 10.1145/2166896.2166909. URL http://doi.acm.org/10.1145/2166896.2166909.
[70] Gareth Highnam, Jason J. Wang, Dean Kusler, Justin Zook, Vinaya Vijayan,Nir Leibovich, and David Mittelman. An analytical framework for optimizingvariant discovery from personal genomes. Nature Communications, 6:6275, feb2015. ISSN 2041-1723. doi: 10.1038/ncomms7275. URL http://www.nature.com/ncomms/2015/150225/ncomms7275/full/ncomms7275.html.
[71] Rolf Hilker, Kai Bernd Stadermann, Daniel Doppmeier, Jörn Kalinowski, JensStoye, Jasmin Straube, Jörn Winnebald, and Alexander Goesmann. ReadXplorer–visualization and analysis of mapped sequences. Bioinformatics (Oxford, Eng-land), 30(16):2247–54, aug 2014. ISSN 1367-4811. doi: 10.1093/bioinformatics/btu205. URL http://bioinformatics.oxfordjournals.org/content/30/16/2247.short?rss=1.
[72] Adam A Hunter, Andrew B Macgregor, Tamas O Szabo, Crispin A Welling-ton, and Matthew I Bellgard. Yabi: An online research environmentfor grid, high performance and cloud computing. Source code for bi-ology and medicine, 7(1):1, jan 2012. ISSN 1751-0473. doi: 10.1186/1751-0473-7-1. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3298538tool=pmcentrezrendertype=abstract.
[73] Darrel C Ince, Leslie Hatton, and John Graham-Cumming. The case for opencomputer programs. Nature, 482(7386):485–8, feb 2012. ISSN 1476-4687. URLhttp://dx.doi.org/10.1038/nature10836.
[74] Jon Ison, Kristoffer Rapacki, Hervé Ménager, Matúš Kalaš, Emil Rydza, PiotrChmura, Christian Anthon, Niall Beard, Karel Berka, and Dan et al. Bolser.Tools and data services registry: a community effort to document bioinformaticsresources. Nucleic acids research, 44(D1):D38–D47, 2016.
[75] Günter Jäger, Alexander Peltzer, and Kay Nieselt. inPHAP: inter-active visualization of genotype and phased haplotype data. BMCbioinformatics, 15(1):200, jan 2014. ISSN 1471-2105. doi: 10.1186/
56

1471-2105-15-200. URL http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-15-200.
[76] Andrew D Johnson and Christopher J O’Donnell. An open access databaseof genome-wide association results. BMC medical genetics, 10(1):6, jan 2009.ISSN 1471-2350. doi: 10.1186/1471-2350-10-6. URL http://bmcmedgenet.biomedcentral.com/articles/10.1186/1471-2350-10-6.
[77] M Aleksi Kallio, Jarno T Tuimala, Taavi Hupponen, Petri Klemelä, Massi-miliano Gentile, Ilari Scheinin, Mikko Koski, Janne Käki, and Eija I Kor-pelainen. Chipster: user-friendly analysis software for microarray and otherhigh-throughput data. BMC genomics, 12(1):507, jan 2011. ISSN 1471-2164. doi:10.1186/1471-2164-12-507. URL http://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-12-507.
[78] Alexandros Kanterakis, Joël Kuiper, George Potamias, and Morris A. Swertz.PyPedia: using the wiki paradigm as crowd sourcing environment for bioin-formatics protocols. Source Code for Biology and Medicine, 10(1):14, nov2015. ISSN 1751-0473. doi: 10.1186/s13029-015-0042-6. URL http://scfbm.biomedcentral.com/articles/10.1186/s13029-015-0042-6.
[79] Md Karim, Audrey Michel, Achille Zappa, Pavel Baranov, Ratnesh Sahay, andDietrich et al. Rebholz-Schuhmann. Improving data workflow systems withcloud services and use of open data for bioinformatics research. Briefings inBioinformatics, 2017.
[80] Jane Kaye, Matthew Hurles, Heather Griffin, Jasote Grewal, and Bobrow et al.Managing clinically significant findings in research: the UK10K example. Eu-ropean journal of human genetics : EJHG, 22(9):1100–4, sep 2014. ISSN 1476-5438. doi: 10.1038/ejhg.2013.290. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4026295tool=pmcentrezrendertype=abstract.
[81] Muin J Khoury, Marta Gwinn, M Scott Bowen, and W David Dotson. Beyondbase pairs to bedside: a population perspective on how genomics can improvehealth. American journal of public health, 102(1):34–7, jan 2012. ISSN 1541-0048.doi: 10.2105/AJPH.2011.300299. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3490552tool=pmcentrezrendertype=abstract.
[82] Michael T. Krieger, Oscar Torreno, Oswaldo Trelles, and Dieter Kranzlmüller.Building an open source cloud environment with auto-scaling resources for
57

executing bioinformatics and biomedical workflows. Future Generation ComputerSystems, feb 2016. ISSN 0167739X. doi: 10.1016/j.future.2016.02.008. URL http://www.sciencedirect.com/science/article/pii/S0167739X16300218.
[83] Melissa J Landrum, Jennifer M Lee, George R Riley, Wonhee Jang, Wendy SRubinstein, Deanna M Church, and Donna R Maglott. ClinVar: public archiveof relationships among sequence variation and human phenotype. Nucleic acidsresearch, 42(Database issue):D980–5, jan 2014. ISSN 1362-4962. doi: 10.1093/nar/gkt1113. URL http://nar.oxfordjournals.org/content/early/2013/11/14/nar.gkt1113.short.
[84] Ilkka Lappalainen, Jeff Almeida-King, Vasudev Kumanduri, Alexander Senf, andSpaldinget al. The European Genome-phenome Archive of human data consentedfor biomedical research. Nature Genetics, 47(7):692–695, jun 2015. ISSN 1061-4036. doi: 10.1038/ng.3312. URL http://dx.doi.org/10.1038/ng.3312.
[85] Tuuli Lappalainen, Michael Sammeth, Marc R Friedländer, Peter AC‘t Hoen,Jean Monlong, Manuel A Rivas, Mar Gonzàlez-Porta, Natalja Kurbatova, ThassoGriebel, and Pedro G et al. Ferreira. Transcriptome and genome sequencinguncovers functional variation in humans. Nature, 501(7468):506–511, 2013.
[86] Jeremy Leipzig. A review of bioinformatic pipeline frameworks. Briefingsin Bioinformatics, page bbw020, mar 2016. ISSN 1467-5463. doi: 10.1093/bib/bbw020. URL http://bib.oxfordjournals.org/content/early/2016/03/23/bib.bbw020.long.
[87] Felipe da Veiga Leprevost, Valmir C Barbosa, Eduardo L Francisco, YassetPerez-Riverol, and Paulo C Carvalho. On best practices in the development ofbioinformatics software. Frontiers in genetics, 5:199, jan 2014. ISSN 1664-8021.doi: 10.3389/fgene.2014.00199. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4078907tool=pmcentrezrendertype=abstract.
[88] Richard Leslie, Christopher J O’Donnell, and Andrew D Johnson. GRASP:analysis of genotype-phenotype results from 1390 genome-wide association studiesand corresponding open access database. Bioinformatics (Oxford, England), 30(12):i185–94, jun 2014. ISSN 1367-4811. doi: 10.1093/bioinformatics/btu273. URLhttp://bioinformatics.oxfordjournals.org/content/30/12/i185.short.
[89] Po-E Li, Chien-Chi Lo, Joseph J. Anderson, Karen W. Davenport, Kimberly A.Bishop-Lilly, Yan Xu, Sanaa Ahmed, Shihai Feng, Vishwesh P. Mokashi, and
58

Patrick S.G. Chain. Enabling the democratization of the genomics revolution witha fully integrated web-based bioinformatics platform. Nucleic Acids Research, 45(1):67–80, jan 2017. ISSN 0305-1048. doi: 10.1093/nar/gkw1027. URL https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkw1027.
[90] Zhifa Liu and Stan Pounds. An R package that automatically col-lects and archives details for reproducible computing. BMC bioin-formatics, 15(1):138, jan 2014. ISSN 1471-2105. doi: 10.1186/1471-2105-15-138. URL http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-15-138.
[91] Dan L Longo and Jeffrey M Drazen. Data Sharing. New England Journalof Medicine, 374(3):276–277, 2016. doi: 10.1056/NEJMe1516564. URL http://dx.doi.org/10.1056/NEJMe1516564.
[92] D G MacArthur, T A Manolio, D P Dimmock, H L Rehm, J Shendure, andet al. Abecasis. Guidelines for investigating causality of sequence variants inhuman disease. Nature, 508(7497):469–76, apr 2014. ISSN 1476-4687. doi:10.1038/nature13127. URL http://dx.doi.org/10.1038/nature13127.
[93] Matthew D Mailman, Michael Feolo, Yumi Jin, Masato Kimura, and Trykaet al. The NCBI dbGaP database of genotypes and phenotypes. Na-ture genetics, 39(10):1181–6, oct 2007. ISSN 1546-1718. doi: 10.1038/ng1007-1181. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2031016tool=pmcentrezrendertype=abstract.
[94] James Malone, Robert Stevens, Simon Jupp, Tom Hancocks, Helen Parkinson,and Cath Brooksbank. Ten Simple Rules for Selecting a Bio-ontology. PLOSComputational Biology, 12(2):e1004743, feb 2016. ISSN 1553-7358. doi: 10.1371/journal.pcbi.1004743. URL http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1004743.
[95] Teri A Manolio. Bringing genome-wide association findings into clinical use.Nature reviews. Genetics, 14(8):549–58, aug 2013. ISSN 1471-0064. doi: 10.1038/nrg3523. URL http://dx.doi.org/10.1038/nrg3523.
[96] Arjun K. Manrai, John P. A. Ioannidis, and Isaac S. Kohane. Clinical Genomics,From Pathogenicity Claims to Quantitative Risk Estimates. JAMA, feb 2016.ISSN 0098-7484. doi: 10.1001/jama.2016.1519. URL http://jama.jamanetwork.com/article.aspx?articleid=2498853#jvp160020r5.
59

[97] G Marchant. Genomics, ethics, and intellectual property. KRATTIGER, A.;MAHONEY, RT; NELSEN, L.; et al). Intellectual Property Management inHealth and Agricultural Innovation: a Handbook of best practices, pages 29–38,2007.
[98] David J McIver and John S Brownstein. Wikipedia usage estimates prevalenceof influenza-like illness in the United States in near real-time. PLoS computa-tional biology, 10(4):e1003581, apr 2014. ISSN 1553-7358. doi: 10.1371/journal.pcbi.1003581. URL http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003581.
[99] Aaron McKenna, Matthew Hanna, Eric Banks, Andrey Sivachenko, and Cibulskiset al. The Genome Analysis Toolkit: a MapReduce framework for analyzingnext-generation DNA sequencing data. Genome research, 20(9):1297–303, sep2010. ISSN 1549-5469. doi: 10.1101/gr.107524.110. URL http://genome.cshlp.org/content/20/9/1297.
[100] Zeeya Merali. Computational science: ...Error. Nature, 467(7317):775–7, oct 2010.ISSN 1476-4687. doi: 10.1038/467775a. URL http://www.nature.com/news/2010/101013/full/467775a.html.
[101] Matthew Meyerson, Stacey Gabriel, and Gad Getz. Advances in understandingcancer genomes through second-generation sequencing. Nature reviews. Genetics,11(10):685–96, oct 2010. ISSN 1471-0064. doi: 10.1038/nrg2841. URL http://dx.doi.org/10.1038/nrg2841.
[102] Takahiro Mimori, Naoki Nariai, Kaname Kojima, Mamoru Takahashi, AkiraOno, Yukuto Sato, Yumi Yamaguchi-Kabata, and Masao Nagasaki. iSVP: anintegrated structural variant calling pipeline from high-throughput sequencingdata. BMC systems biology, 7 Suppl 6:S8, jan 2013. ISSN 1752-0509. doi:10.1186/1752-0509-7-S6-S8.
[103] Koyel Mitra, Anne-Ruxandra Carvunis, Sanath Kumar Ramesh, and Trey Ideker.Integrative approaches for finding modular structure in biological networks.Nature reviews. Genetics, 14(10):719–32, oct 2013. ISSN 1471-0064. doi: 10.1038/nrg3552. URL http://dx.doi.org/10.1038/nrg3552.
[104] François Moreews, Olivier Sallou, Yvan Le Bras, Grosjean Marie, Cyril Monjeaud,Thomas A Darde, Olivier Collin, and Christophe Blanchet. A curated Domaincentric shared Docker registry linked to the Galaxy toolshed, jul 2015. URLhttps://hal.archives-ouvertes.fr/hal-01160514/.
60

[105] John C Mu, Marghoob Mohiyuddin, Jian Li, Narges Bani Asadi, Mark B Gerstein,Alexej Abyzov, Wing H Wong, and Hugo Y K Lam. VarSim: a high-fidelity simula-tion and validation framework for high-throughput genome sequencing with cancerapplications. Bioinformatics (Oxford, England), 31(9):1469–71, may 2015. ISSN1367-4811. doi: 10.1093/bioinformatics/btu828. URL http://bioinformatics.oxfordjournals.org/content/early/2014/12/17/bioinformatics.btu828.
[106] Samia N Naccache, Scot Federman, Narayanan Veeraraghavan, and Zaharia et al.A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identificationfrom next-generation sequencing of clinical samples. Genome research, 24(7):1180–92, jul 2014. ISSN 1549-5469. doi: 10.1101/gr.171934.113. URL http://genome.cshlp.org/content/early/2014/05/16/gr.171934.113.
[107] Matti Niemenmaa, Aleksi Kallio, André Schumacher, Petri Klemelä, Eija Kor-pelainen, and Keijo Heljanko. Hadoop-BAM: directly manipulating next gen-eration sequencing data in the cloud. Bioinformatics (Oxford, England), 28(6):876–7, mar 2012. ISSN 1367-4811. doi: 10.1093/bioinformatics/bts054. URLhttp://bioinformatics.oxfordjournals.org/content/28/6/876.
[108] Umadevi Paila, Brad A Chapman, Rory Kirchner, and Aaron R Quinlan.GEMINI: integrative exploration of genetic variation and genome annota-tions. PLoS computational biology, 9(7):e1003153, jan 2013. ISSN 1553-7358. doi: 10.1371/journal.pcbi.1003153. URL http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003153.
[109] Chao Pang, Dennis Hendriksen, Martijn Dijkstra, K Joeri van der Velde,Joel Kuiper, Hans L Hillege, and Morris A Swertz. BiobankConnect: soft-ware to rapidly connect data elements for pooled analysis across biobanksusing ontological and lexical indexing. Journal of the American Medical In-formatics Association : JAMIA, 22(1):65–75, jan 2015. ISSN 1527-974X. doi:10.1136/amiajnl-2013-002577.
[110] Jeffrey M Perkel. Programming: Pick up Python. Nature, 518(7537):125–6, feb2015. ISSN 1476-4687. doi: 10.1038/518125a. URL http://www.nature.com/news/programming-pick-up-python-1.16833.
[111] Jeffrey M Perkel. How bioinformatics tools are bringing genetic analysis to themasses. Nature News, 543(7643):137, 2017.
[112] Alexander Pfundner, Tobias Schönberg, John Horn, Richard D Boyce, andMatthias Samwald. Utilizing the wikidata system to improve the quality of
61

medical content in wikipedia in diverse languages: a pilot study. Journal of medicalInternet research, 17(5):e110, jan 2015. ISSN 1438-8871. doi: 10.2196/jmir.4163.URL http://www.jmir.org/2015/5/e110/.
[113] Shaun Purcell, Benjamin Neale, Kathe Todd-Brown, Lori Thomas, Manuel A RFerreira, David Bender, Julian Maller, Pamela Sklar, Paul I W de Bakker, Mark JDaly, and Pak C Sham. PLINK: a tool set for whole-genome association andpopulation-based linkage analyses. American journal of human genetics, 81(3):559–75, sep 2007. ISSN 0002-9297. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1950838tool=pmcentrezrendertype=abstract.
[114] Karthik Ram. Git can facilitate greater reproducibility and increased transparencyin science. Source code for biology and medicine, 8(1):7, jan 2013. ISSN 1751-0473.doi: 10.1186/1751-0473-8-7. URL http://www.scfbm.org/content/8/1/7.
[115] Brian J Raney, Timothy R Dreszer, Galt P Barber, Hiram Clawson, and Fujitaet al. Track data hubs enable visualization of user-defined genome-wide anno-tations on the UCSC Genome Browser. Bioinformatics (Oxford, England), 30(7):1003–5, apr 2014. ISSN 1367-4811. doi: 10.1093/bioinformatics/btt637. URLhttp://bioinformatics.oxfordjournals.org/content/30/7/1003.short.
[116] Cornelius A Rietveld, Sarah E Medland, Jaime Derringer, and Yang et al. GWASof 126,559 individuals identifies genetic variants associated with educationalattainment. Science (New York, N.Y.), 340(6139):1467–71, jun 2013. ISSN 1095-9203. doi: 10.1126/science.1235488. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3751588tool=pmcentrezrendertype=abstract.
[117] P N Robinson and S Mundlos. The human phenotype ontology. Clinical genetics,77(6):525–34, jun 2010. ISSN 1399-0004. doi: 10.1111/j.1399-0004.2010.01436.x.URL http://www.ncbi.nlm.nih.gov/pubmed/20412080.
[118] Simon P Sadedin, Bernard Pope, and Alicia Oshlack. Bpipe: a tool for runningand managing bioinformatics pipelines. Bioinformatics (Oxford, England), 28(11):1525–6, jun 2012. ISSN 1367-4811. doi: 10.1093/bioinformatics/bts167. URLhttp://www.ncbi.nlm.nih.gov/pubmed/22500002.
[119] Kaitlin E Samocha, Elise B Robinson, Stephan J Sanders, Christine Stevens,Aniko Sabo, and et al. McGrath. A framework for the interpretation of de novomutation in human disease. Nature genetics, 46(9):944–50, sep 2014. ISSN1546-1718. doi: 10.1038/ng.3050. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4222185tool=pmcentrezrendertype=abstract.
62

[120] David C Samuels, Leng Han, Jiang Li, Sheng Quanghu, Travis A Clark, Yu Shyr,and Yan Guo. Finding the lost treasures in exome sequencing data. Trendsin genetics : TIG, 29(10):593–9, oct 2013. ISSN 0168-9525. doi: 10.1016/j.tig.2013.07.006. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3926691tool=pmcentrezrendertype=abstract.
[121] C. J. Saunders, N. A. Miller, S. E. Soden, D. L. Dinwiddie, and Noll et al.Rapid Whole-Genome Sequencing for Genetic Disease Diagnosis in NeonatalIntensive Care Units. Science Translational Medicine, 4(154):154ra135–154ra135,oct 2012. ISSN 1946-6234. doi: 10.1126/scitranslmed.3004041. URL http://stm.sciencemag.org/content/4/154/154ra135.long.
[122] Maren T Scheuner, Han de Vries, Benjamin Kim, Robin C Meili, Sarah HOlmstead, and Stephanie Teleki. Are electronic health records readyfor genomic medicine? Genetics in medicine : Journal of the Ameri-can College of Medical Genetics, 11(7):510–7, jul 2009. ISSN 1530-0366.doi: 10.1097/GIM.0b013e3181a53331. URL http://dx.doi.org/10.1097/GIM.0b013e3181a53331.
[123] S. Scholtens, N. Smidt, M. A. Swertz, S. J. Bakker, A. Dotinga, J. M. Vonk, F. vanDijk, S. K. van Zon, C. Wijmenga, B. H. Wolffenbuttel, and R. P. Stolk. CohortProfile: LifeLines, a three-generation cohort study and biobank. InternationalJournal of Epidemiology, pages dyu229–, dec 2014. ISSN 0300-5771. doi: 10.1093/ije/dyu229. URL http://ije.oxfordjournals.org/content/early/2014/12/13/ije.dyu229.abstract?sid=4d7f00a7-ce80-4c6d-9b3e-3232bc06419f.
[124] Nicholas J. Schork. Personalized medicine: Time for one-person trials. Nature, 520(7549):609–611, apr 2015. ISSN 0028-0836. doi: 10.1038/520609a. URL http://www.nature.com/news/personalized-medicine-time-for-one-person-trials-1.17411.
[125] Torsten Seemann. Ten recommendations for creating usable bioinformaticscommand line software. GigaScience, 2(1):15, jan 2013. ISSN 2047-217X.doi: 10.1186/2047-217X-2-15. URL http://gigascience.biomedcentral.com/articles/10.1186/2047-217X-2-15.
[126] Helen Shen. Interactive notebooks: Sharing the code. Nature, 515(7525):151–2,nov 2014. ISSN 1476-4687. doi: 10.1038/515151a. URL http://www.nature.com/news/interactive-notebooks-sharing-the-code-1.16261.
63

[127] Derek Shyr and Qi Liu. Next generation sequencing in cancer research and clinicalapplication. Biological procedures online, 15(1):4, jan 2013. ISSN 1480-9222.doi: 10.1186/1480-9222-15-4. URL http://biologicalproceduresonline.biomedcentral.com/articles/10.1186/1480-9222-15-4.
[128] Mitchell E Skinner, Andrew V Uzilov, Lincoln D Stein, Christopher J Mungall,and Ian H Holmes. JBrowse: a next-generation genome browser. Genomeresearch, 19(9):1630–8, sep 2009. ISSN 1549-5469. doi: 10.1101/gr.094607.109. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2752129tool=pmcentrezrendertype=abstract.
[129] Elizabeth K Speliotes, Cristen J Willer, Sonja I Berndt, and Monda et al.Association analyses of 249,796 individuals reveal 18 new loci associated withbody mass index. Nature Genetics, 42(11):937–948, oct 2010. ISSN 1061-4036. doi:10.1038/ng.686. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3014648tool=pmcentrezrendertype=abstract.
[130] Ola Spjuth, Maria Krestyaninova, Janna Hastings, Huei-Yi Shen, and Heikkinenet al. Harmonising and linking biomedical and clinical data across disparatedata archives to enable integrative cross-biobank research. European journalof human genetics : EJHG, 24(4):521–528, aug 2015. ISSN 1476-5438. doi:10.1038/ejhg.2015.165. URL http://dx.doi.org/10.1038/ejhg.2015.165.
[131] Morris A Swertz, Martijn Dijkstra, Tomasz Adamusiak, Joeri K van der Velde,Alexandros Kanterakis, and Roos et al. The MOLGENIS toolkit: rapid proto-typing of biosoftware at the push of a button. BMC bioinformatics, 11 Suppl 1(Suppl 12):S12, jan 2010. ISSN 1471-2105. doi: 10.1186/1471-2105-11-S12-S12.URL http://www.biomedcentral.com/1471-2105/11/S12/S12.
[132] Jenny C Taylor, Hilary C Martin, Stefano Lise, and Broxholme et al. Factorsinfluencing success of clinical genome sequencing across a broad spectrum ofdisorders. Nature genetics, 47(7):717–26, jul 2015. ISSN 1546-1718. doi: 10.1038/ng.3304.
[133] Jessica D Tenenbaum, Susanna-Assunta Sansone, and Melissa Haendel. A seaof standards for omics data: sink or swim? Journal of the American MedicalInformatics Association : JAMIA, 21(2):200–3, jan 2014. ISSN 1527-974X.doi: 10.1136/amiajnl-2013-002066. URL http://jamia.oxfordjournals.org/content/21/2/200.abstract.
64

[134] Carol Tenopir, Suzie Allard, Kimberly Douglass, Arsev Umur Aydinoglu, Lei Wu,Eleanor Read, Maribeth Manoff, and Mike Frame. Data sharing by scientists:practices and perceptions. PloS one, 6(6):e21101, jan 2011. ISSN 1932-6203.doi: 10.1371/journal.pone.0021101. URL http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0021101.
[135] The Genome of the Netherlands Consortium. Whole-genome sequence variation,population structure and demographic history of the Dutch population. Naturegenetics, 46(8):818–825, jun 2014. ISSN 1546-1718. doi: 10.1038/ng.3021. URLhttp://dx.doi.org/10.1038/ng.3021.
[136] Helga Thorvaldsdóttir, James T Robinson, and Jill P Mesirov. IntegrativeGenomics Viewer (IGV): high-performance genomics data visualization andexploration. Briefings in bioinformatics, 14(2):178–92, mar 2013. ISSN 1477-4054.doi: 10.1093/bib/bbs017. URL http://bib.oxfordjournals.org/content/14/2/178.full?keytype=ref%2520ijkey=qTgjFwbRBAzRZWC.
[137] Eric J Topol. Individualized medicine from prewomb to tomb. Cell, 157(1):241–53, mar 2014. ISSN 1097-4172. doi: 10.1016/j.cell.2014.02.012. URL http://www.sciencedirect.com/science/article/pii/S0092867414002049.
[138] W. Torres-Garcia, S. Zheng, A. Sivachenko, R. Vegesna, Q. Wang, R. Yao,M. F. Berger, J. N. Weinstein, G. Getz, and R. G. W. Verhaak. PRADA:pipeline for RNA sequencing data analysis. Bioinformatics, 30(15):2224–2226,apr 2014. ISSN 1367-4803. doi: 10.1093/bioinformatics/btu169. URL http://bioinformatics.oxfordjournals.org/content/30/15/2224.full.
[139] Özlem Uzuner, Brett R South, Shuying Shen, and Scott L DuVall. 2010i2b2/VA challenge on concepts, assertions, and relations in clinical text. Jour-nal of the American Medical Informatics Association : JAMIA, 18(5):552–6, jan 2011. ISSN 1527-974X. doi: 10.1136/amiajnl-2011-000203. URLhttp://jamia.oxfordjournals.org/content/18/5/552.abstract.
[140] W.M.P. van der Aalst and A.H.M. ter Hofstede. YAWL: yet another workflowlanguage. Information Systems, 30(4):245–275, 2005. ISSN 03064379. doi:10.1016/j.is.2004.02.002. URL http://dl.acm.org/citation.cfm?id=1187495.1187496.
[141] Gert-Jan B van Ommen, Outi Törnwall, Christian Bréchot, and Dagher et al.BBMRI-ERIC as a resource for pharmaceutical and life science industries: the
65

development of biobank-based Expert Centres. European journal of humangenetics : EJHG, 23(7):893–900, nov 2014. ISSN 1476-5438. doi: 10.1038/ejhg.2014.235. URL http://dx.doi.org/10.1038/ejhg.2014.235.
[142] Dan Vanderkam, B. Arman Aksoy, Isaac Hodes, Jaclyn Perrone, and Jeff Hammer-bacher. pileup.js: a JavaScript library for interactive and in-browser visualizationof genomic data. Bioinformatics, 32(15):2378–2379, aug 2016. ISSN 1367-4803. doi: 10.1093/bioinformatics/btw167. URL https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btw167.
[143] Mauno Vihinen. No more hidden solutions in bioinformatics. Nature, 521(7552):261–261, may 2015. ISSN 0028-0836. doi: 10.1038/521261a. URL http://www.nature.com/news/no-more-hidden-solutions-in-bioinformatics-1.17587.
[144] Hue Vuong, Anney Che, Sarangan Ravichandran, Brian T Luke, Jack RCollins, and Uma S Mudunuri. AVIA v2.0: annotation, visualization and im-pact analysis of genomic variants and genes. Bioinformatics (Oxford, Eng-land), 31(16):2748–50, aug 2015. ISSN 1367-4811. doi: 10.1093/bioinformatics/btv200. URL http://bioinformatics.oxfordjournals.org/content/early/2015/04/22/bioinformatics.btv200.long.
[145] Jing Wang, Dexter Duncan, Zhiao Shi, and Bing Zhang. WEB-based GEne SeTAnaLysis Toolkit (WebGestalt): update 2013. Nucleic acids research, 41(WebServer issue):W77–83, jul 2013. ISSN 1362-4962. doi: 10.1093/nar/gkt439. URLhttp://nar.oxfordjournals.org/content/41/W1/W77.full.
[146] Kai Wang, Mingyao Li, and Hakon Hakonarson. ANNOVAR: functional an-notation of genetic variants from high-throughput sequencing data. Nucleicacids research, 38(16):e164, sep 2010. ISSN 1362-4962. doi: 10.1093/nar/gkq603. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2938201tool=pmcentrezrendertype=abstract.
[147] Marek S Wiewiórka, Antonio Messina, Alicja Pacholewska, Sergio Maffioletti,Piotr Gawrysiak, and Michał J Okoniewski. SparkSeq: fast, scalable andcloud-ready tool for the interactive genomic data analysis with nucleotideprecision. Bioinformatics (Oxford, England), 30(18):2652–3, sep 2014. ISSN1367-4811. doi: 10.1093/bioinformatics/btu343. URL http://bioinformatics.oxfordjournals.org/content/early/2014/05/19/bioinformatics.btu343.
66

[148] Martin Wildeman, Ernest van Ophuizen, Johan T den Dunnen, and Peter E MTaschner. Improving sequence variant descriptions in mutation databases andliterature using the Mutalyzer sequence variation nomenclature checker. Humanmutation, 29(1):6–13, jan 2008. ISSN 1098-1004. doi: 10.1002/humu.20654. URLhttp://www.ncbi.nlm.nih.gov/pubmed/18000842.
[149] Greg Wilson, DA Aruliah, C Titus Brown, Neil P Chue Hong, Matt Davis,Richard T Guy, Steven HD Haddock, Kathryn D Huff, Ian M Mitchell, and MarkD et al. Plumbley. Best practices for scientific computing. PLoS Biol, 12(1):e1001745, 2014.
[150] Katherine Wolstencroft, Robert Haines, Donal Fellows, and Williams et al.The Taverna workflow suite: designing and executing workflows of Web Ser-vices on the desktop, web or in the cloud. Nucleic acids research, 41(Web Server issue):W557–61, jul 2013. ISSN 1362-4962. doi: 10.1093/nar/gkt328. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3692062tool=pmcentrezrendertype=abstract.
[151] Yaping Yang, Donna M Muzny, Jeffrey G Reid, Matthew N Bainbridge, AleciaWillis, Patricia A Ward, Alicia Braxton, Joke Beuten, Fan Xia, and Zhiyv et al.Niu. Clinical whole-exome sequencing for the diagnosis of mendelian disorders.New England Journal of Medicine, 369(16):1502–1511, 2013.
[152] Tomasz Zemojtel, Sebastian Köhler, Luisa Mackenroth, and Jäger et al.Effective diagnosis of genetic disease by computational phenotype analy-sis of the disease-associated genome. Science translational medicine, 6(252):252ra123, sep 2014. ISSN 1946-6242. doi: 10.1126/scitranslmed.3009262. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4512639tool=pmcentrezrendertype=abstract.
[153] Daniel R Zerbino, Nathan Johnson, Thomas Juettemann, Steven P Wilder,and Paul Flicek. WiggleTools: parallel processing of large collections ofgenome-wide datasets for visualization and statistical analysis. Bioinfor-matics (Oxford, England), 30(7):1008–9, apr 2014. ISSN 1367-4811. doi:10.1093/bioinformatics/btt737. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3967112tool=pmcentrezrendertype=abstract.
[154] Shanrong Zhao, Kurt Prenger, Lance Smith, Thomas Messina, Hong-tao Fan, Edward Jaeger, and Susan Stephens. Rainbow: a tool for
67

large-scale whole-genome sequencing data analysis using cloud comput-ing. BMC genomics, 14:425, jan 2013. ISSN 1471-2164. doi: 10.1186/1471-2164-14-425. URL http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3698007tool=pmcentrezrendertype=abstract.
[155] Quan Zou, Xu-Bin Li, Wen-Rui Jiang, Zi-Yu Lin, Gui-Lin Li, and Ke Chen. Surveyof MapReduce frame operation in bioinformatics. Briefings in bioinformatics,15(4):637–47, jul 2014. ISSN 1477-4054. doi: 10.1093/bib/bbs088. URL http://bib.oxfordjournals.org/content/15/4/637.short?rss=1.
68