increased expression of visfatin in macrophages of human unstable carotid and coronary...
TRANSCRIPT

Increased Expression of Visfatin in Macrophages of HumanUnstable Carotid and Coronary AtherosclerosisPossible Role in Inflammation and Plaque Destabilization
Tuva B. Dahl, MSc; Arne Yndestad, MSc, PhD; Mona Skjelland, MD; Erik Øie, MD, PhD;Arve Dahl, MD, PhD; Annika Michelsen, MSc; Jan K. Damås, MD, PhD; Siv H. Tunheim, MSc;
Thor Ueland, PhD; Camilla Smith, MD; Bjørn Bendz, MD, PhD; Serena Tonstad MD, PhD;Lars Gullestad, MD, PhD; Stig S. Frøland, MD, PhD; Kirsten Krohg-Sørensen, MD, PhD;
David Russell, FRCPE; Pål Aukrust, MD, PhD; Bente Halvorsen, MSc, PhD
Background—Although the participation of inflammation in atherogenesis is widely recognized, the identification of thedifferent components has not been clarified. In particular, the role of inflammation in plaque destabilization is not fullyunderstood.
Methods and Results—Our main findings were as follows: (1) In a microarray experiment, we identified visfatin, one ofthe most recently identified adipokines, as a gene that was markedly enhanced in carotid plaques from symptomaticcompared with plaques from asymptomatic individuals. This finding was confirmed when carotid plaques from 7patients with asymptomatic and 14 patients with symptomatic lesions were examined with real-time reversetranscription polymerase chain reaction. (2) Immunohistochemistry showed that visfatin was localized in areas that wererich in lipid-loaded macrophages. (3) The relationship between visfatin and unstable lesions was also found in patientswith coronary artery disease, demonstrating a strong visfatin immunostaining in lipid-rich regions within the materialobtained at the site of plaque rupture in patients with acute myocardial infarction. (4) Both oxidized low-densitylipoprotein and tumor necrosis factor-� increased visfatin expression in THP-1 monocytes, with a particularly enhancingeffect when these stimuli were combined. (5) Visfatin increased matrix metalloproteinase-9 activity in THP-1monocytes and tumor necrosis factor-� and interleukin-8 levels in peripheral blood mononuclear cells. Both of theseeffects were abolished when insulin receptor signaling was blocked.
Conclusions—Our findings suggest that visfatin should be regarded as an inflammatory mediator, localized to foam cellmacrophages within unstable atherosclerotic lesions, that potentially plays a role in plaque destabilization. (Circulation.2007;115:972-980.)
Key Words: atherosclerosis � inflammation � leukocytes � plaque � coronary disease
Atherosclerosis is a progressive disease in which lipids,extracellular matrix, and activated vascular smooth muscle
cells accumulate in the arterial wall, resulting in growth of anatherosclerotic plaque.1 Recent research has shown that inflam-mation plays a key role in this process. Hence, immune cellsdominate early atherosclerotic plaques, their effector moleculesaccelerate progression of the lesions, and activation of inflam-mation can elicit various acute ischemic events such as acutecoronary syndromes, transient ischemic attacks, and stroke.2
However, although the participation of inflammatory mediators
in the atherosclerotic process has become widely recognized, theidentification of the different components, as well as theirrelative importance, is unclear. In particular, the way in whichinflammation may promote the transition from an asymptomaticfibroatheromatous plaque to a vulnerable and symptomaticlesion is not fully understood.
Clinical Perspective p 980The analysis of expressed genes in adipose tissue has
revealed that adipocytes produce and secrete a variety of
Received September 20, 2006; accepted January 5, 2007.From the Research Institute for Internal Medicine (T.B.D., A.Y., A.M., J.K.D., T.U., C.S., S.S.F., P.A., B.H.), Department of Neurology (M.S., A.D.,
D.R.), Section of Endocrinology (T.U.), Department of Cardiology (E.Ø., B.B., L.G.), Center for Occupational and Environmental Medicine (S.H.T.),Section of Clinical Immunology and Infectious Diseases (S.S.F., P.A.), Department of Thoracic and Cardiovascular Surgery (K.K.-S.), and Institute forSurgical Research (E.Ø.), Rikshospitalet-Radiumhospitalet Medical Center and University of Oslo; and Department of Preventive Cardiology, UllevålUniversity Hospital and University of Oslo (S.T.), Oslo, Norway.
The online-only Data Supplement, consisting of a table, is available with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.106.665893/DC1.
Correspondence to Bente Halvorsen, MSc, PhD, Research Institute for Internal Medicine, Rikshospitalet-Radiumhospitalet Medical Center, Universityof Oslo, N-0027 Oslo, Norway. E-mail [email protected]
© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.106.665893
972 by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from
bioactive substances, named adipokines, including growthfactors and cytokines. Several studies have suggested a rolefor these mediators in atherogenesis not only through regu-lation of lipid and glucose metabolism but also by modulatingthe inflammatory arm of atherosclerosis.3 Adipokines havealso been found to be expressed within atheroscleroticplaques, suggesting local and endocrine effects of thesemediators on atherosclerotic lesions.4,5 However, althoughseveral studies suggest a role for adipokines in atherogenesis,their role in plaque destabilization has not been clarified.
One of the most recently identified adipokines is visfatin,6
originally identified as pre–B-cell colony-enhancing factor-1.7 Visfatin appears to be preferentially produced by thevisceral adipose tissue and seems to have insulin mimeticactions.6 Interestingly, visfatin has also been shown to beproduced by immune cells (eg, neutrophils and macro-phages),7,8 and animal studies have suggested a role for thisadipokine in lung inflammation during septicemia.9 However,at present the pathophysiological role of visfatin in humansremains largely unknown.
In a screening experiment, we used high-density oligonu-cleotide microarrays to identify genes that may be differentlyregulated in atherosclerotic carotid plaques from asymptom-atic and symptomatic patients. This experiment identifiedvisfatin as a gene that was markedly enhanced in symptom-atic lesions. Herein we further examined the possible patho-genic role of this adipokine in plaque destabilization byseveral approaches including both clinical and experimentalstudies, particularly focusing on its role in inflammation andmatrix degradation.
MethodsPatientsCarotid plaques from 21 consecutive endarterectomy patients wereclassified into 2 groups depending on whether or not the patients hadexperienced ipsilateral stroke, transient ischemic attack, or amaurosisfugax in the 6 months before surgery. Plaques were characterized assymptomatic (n�14) or asymptomatic (n�7) according to thepresence or absence of cerebrovascular symptoms, respectively(Table 1). The carotid stenoses were diagnosed and classified byprecerebral color Duplex ultrasound10 and computed tomographicangiography11 according to consensus criteria. The asymptomaticcarotid stenoses were detected during clinical examinations ofpatients with coronary artery disease (CAD), peripheral arterydisease, or stroke/transient ischemic attack �6 months previously. Inother experiments, blood samples were collected from patients withstable (n�8) and unstable angina (n�8) (Table 2). Those withunstable disease had experienced ischemic chest pain at rest withinthe preceding 48 hours (ie, Braunwald class IIIB) but with noevidence of myocardial necrosis by enzymatic criteria. TransientST-T segment depression and/or T-wave inversion was present in allcases. All patients with stable angina had stable effort angina of �6months’ duration and a positive exercise test. Coronary angiographywas performed by standard techniques within 1 to 2 days afteradmission, and the diagnosis of CAD was confirmed by at least1-vessel disease, defined as �50% narrowing of luminal diameter, inall patients. Patients with concomitant inflammatory diseases andliver or kidney disease were excluded from the study. The protocolswere approved by the regional ethics committee. Signed informedconsent for participation in the study was obtained from allindividuals.
Carotid Endarterectomy SpecimensAtherosclerotic carotid plaques were retrieved from patients duringcarotid endarterectomy. Plaques that were used for protein and RNAextraction were rapidly frozen in liquid nitrogen. For Western blots,the tissue powders from the plaques were homogenized in ice-cold
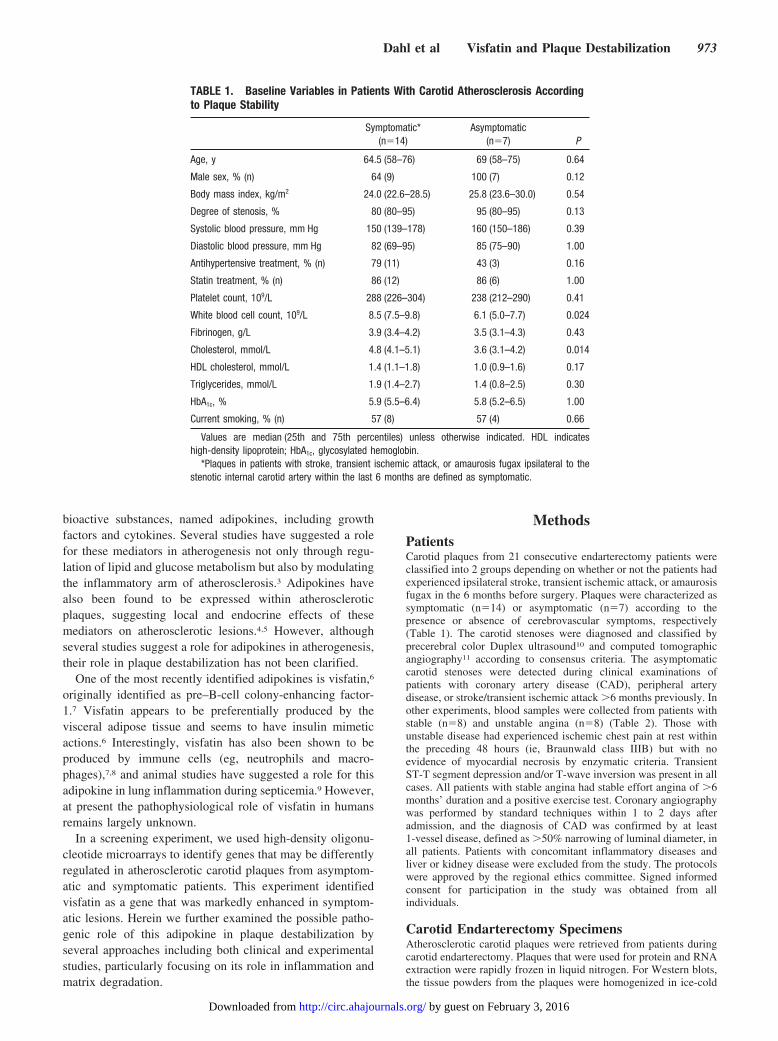
TABLE 1. Baseline Variables in Patients With Carotid Atherosclerosis Accordingto Plaque Stability
Symptomatic*(n�14)
Asymptomatic(n�7) P
Age, y 64.5 (58–76) 69 (58–75) 0.64
Male sex, % (n) 64 (9) 100 (7) 0.12
Body mass index, kg/m2 24.0 (22.6–28.5) 25.8 (23.6–30.0) 0.54
Degree of stenosis, % 80 (80–95) 95 (80–95) 0.13
Systolic blood pressure, mm Hg 150 (139–178) 160 (150–186) 0.39
Diastolic blood pressure, mm Hg 82 (69–95) 85 (75–90) 1.00
Antihypertensive treatment, % (n) 79 (11) 43 (3) 0.16
Statin treatment, % (n) 86 (12) 86 (6) 1.00
Platelet count, 109/L 288 (226–304) 238 (212–290) 0.41
White blood cell count, 109/L 8.5 (7.5–9.8) 6.1 (5.0–7.7) 0.024
Fibrinogen, g/L 3.9 (3.4–4.2) 3.5 (3.1–4.3) 0.43
Cholesterol, mmol/L 4.8 (4.1–5.1) 3.6 (3.1–4.2) 0.014
HDL cholesterol, mmol/L 1.4 (1.1–1.8) 1.0 (0.9–1.6) 0.17
Triglycerides, mmol/L 1.9 (1.4–2.7) 1.4 (0.8–2.5) 0.30
HbA1c, % 5.9 (5.5–6.4) 5.8 (5.2–6.5) 1.00
Current smoking, % (n) 57 (8) 57 (4) 0.66
Values are median (25th and 75th percentiles) unless otherwise indicated. HDL indicateshigh-density lipoprotein; HbA1c, glycosylated hemoglobin.
*Plaques in patients with stroke, transient ischemic attack, or amaurosis fugax ipsilateral to thestenotic internal carotid artery within the last 6 months are defined as symptomatic.
Dahl et al Visfatin and Plaque Destabilization 973
by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from

lysis buffer (PBS containing protease inhibitor cocktail [GIBCO,Paisley, UK] with 1% Triton X-100 and 0.1% Tween 20) at a ratioof 0.1 mL per 10 mg wet wt tissue by a metal blade homogenizer.Extracts were incubated on ice for 15 minutes and centrifuged at12 000g (15 minutes at 4°C). The supernatants were retained, andprotein concentrations in the samples were measured with thebicinchoninic acid method (Pierce, Cheshire, UK).
Tissue Sampling of Plaque Material DuringPercutaneous Coronary InterventionIn 4 patients with acute ST-elevation myocardial infarction under-going primary percutaneous coronary intervention (PCI), thrombusand plaque material at the site of the occlusion were aspiratedimmediately after the lesion was crossed with the guidewire. Amonorail aspiration catheter (Pronto, Vascular Solutions, Minneap-olis, Minn) was advanced over the wire, and a 20-mL air-filledsyringe was used to aspirate during advancement of the catheterthrough the occluded segment. The aspiration catheter was removed,and solid material was separated from liquid blood by means of asieve (pore filter size, 40 �m; Pronto). The solid material was fixedin 4% paraformaldehyde and embedded in paraffin.
Blood Sampling ProtocolPeripheral venous blood was drawn into pyrogen-free EDTA tubesthat were immediately immersed in melting ice and centrifuged at2500g for 20 minutes within 20 minutes to obtain platelet-poorplasma. All samples were stored at �80°C and thawed only once.
High-Density Oligonucleotide MicroarraysTotal RNA was isolated from frozen carotid tissue with the use ofMagNA Pure Kit III (Roche Applied Science, Indianapolis, Ind),quantified spectrophotometrically, and stored at �80°C. The HumanGenome U133A 2.0 Array encoding 14 500 genes was purchasedfrom Affymetrix (Santa Clara, Calif), and hybridization was per-formed according to the manufacturer’s 2-cycle target labelingprotocol. Briefly, cDNA was prepared from 100 ng total RNA, andcRNA was obtained from in vitro transcription of the cDNA. Thenthe cycle was repeated, making cDNA from 600 ng cRNA. There-after, biotin-labeled cRNA was generated from in vitro transcriptionof cDNA and fragmented before hybridization to the array. For dataanalyses, GeneChip Operation Software (1.3) and ArrayAssist (3.4)were used. Microarray suite calculation of the signal intensities was
performed, and genes with very low intensity were not included infurther analyses. Hypothesis testing with Benjamini-Hochberg cor-rection was performed between the 2 groups, resulting in nosignificant expression of genes/probes (P�0.05), possibly because ofthe low number of samples in each group and the high biologicalvariation between individuals. Therefore, a list of genes with aprobability value �0.05, without multiple correction, was obtained.To eliminate the effects of the calculation method chosen, we alsoused the same approach after robust multichip analysis calculation.To further strengthen our results, we compared each sample in onegroup with every sample in the other group (microarray suitecalculation). Lists of genes were made after these filtering criteria:genes with A (absent) or M (marginal) in both samples were
Figure 1. Increased visfatin expression in atherosclerotic carotidplaques from symptomatic patients. A, Gene expression of vis-fatin in 7 patients with asymptomatic and 14 patients withsymptomatic carotid plaques. mRNA levels were quantified withthe use of real-time reverse transcription polymerase chainreaction, and data are presented relative to the expression of18S ribosomal RNA. Horizontal lines represent median values.B, Representative Western blots of asymptomatic (As) andsymptomatic (S) plaques. C, Visfatin protein levels (Westernblot) from randomly selected plaques in the symptomatic group(n�10) and the asymptomatic group (n�6). In the Western blotassay, the primary antibody was directed against another visfa-tin epitope than the epitope that was used in immunohisto-chemistry (Figures 2A and 3B), ensuring the validity of positivedetection of visfatin in the plaques. The membranes werereprobed with 2 different housekeeping gene products (anti-MCM3 and anti–�-tubulin) to ensure equal loading on the gels.Western results are calculated as the visfatin/�-tubulin ratio, anddata are given as median and 25th to 75th percentiles. Similarresults were obtained with the use of the visfatin/MCM3 ratio.
TABLE 2. Baseline Variables in Patients With CAD
Stable AnginaPectoris* (n�8)
Unstable AnginaPectoris† (n�8)
Age, y 59 (52–70) 62 (55–74)
Male sex, % (n) 100 (8) 63 (5)
Body mass index, kg/m2 26 (25–28) 26 (25–28)
Systolic blood pressure, mm Hg 145 (131–159) 145 (120–148)
Diastolic blood pressure, mm Hg 80 (77–84) 80 (60–86)
Antihypertensive treatment, % (n) 88 (7) 100 (8)
Statin treatment, % (n) 100 (8) 100 (8)
Platelet count, 109/L 270 (250–284) 228 (205–269)
White blood cell count, 109/L 9.0 (7.5–9.5) 8.7 (5.8–9.6)
Cholesterol, mmol/L 4.1 (3.8–4.5) 5.1 (4.4–5.5)
HDL cholesterol, mmol/L 1.2 (1.2–1.2) 1.1 (1.1–1.2)
Triglycerides, mmol/L 1.7 (0.9–2.7) 1.8 (1.6–1.8)
Current smoking, % (n) 25 (2) 37 (3)
Values are median (25th and 75th percentiles) unless otherwise indicated.HDL indicates high-density lipoprotein.
*Patients used in the PCI study.†Patients used in the PBMC study.
974 Circulation February 27, 2007
by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from

removed, genes with NC (no change) were removed, and genes witha signal log ratio �2-fold were also removed.
Real-Time Quantitative Reverse TranscriptionPolymerase Chain ReactionTotal RNA was isolated from frozen THP-1 monocytes andcarotid tissue by the MagNA Pure LC instrument (Roche AppliedScience), with the use of MagNA Pure LC RNA isolation Kit IIand III, respectively, and stored in RNA storage solution (Am-bion, Houston, Tex) at �80°C until further analysis. Primers formatrix metalloproteinase (MMP)-9 (forward primer: 5�-GCTCACCTTCACTCGCGTGTA-3� and reverse primer: 5�-TCCGTGCTCCGCGACA-3�), visfatin (forward primer: 5�-CTTCTGGTAACTTAGATGGTCTGGAA-3� and reverseprimer: 5�-GCTCCTATGCCAGCAGTCTCTT-3�), 18S (forwardprimer: 5�-CGGCTACCACATCCAAGGAA-3� and reverseprimer: 5�-GCTGGAATTACCGCGGCT-3�), and �-actin (for-ward primer: 5�-AAGCACCAGGGCGTGAT-3� and reverseprimer: 5�-TCGTCCCAGTTGGTGACGA-3�) were designedwith the use of Primer Express software version 1 · 5 (AppliedBiosystems, Foster City, Calif). Quantification of mRNA wasperformed with the use of the ABI Prism 7000 (Applied Biosys-tems). SyBr Green assay was performed with the qPCR MasterMix for SYBR Green I (Eurogentec, Seraing, Belgium). Geneexpression of the housekeeping gene �-actin or 18S was used fornormalization.
ImmunohistochemistryParaformaldehyde-fixed sections of material obtained during PCIand acetone-fixed sections of asymptomatic and symptomatic carotidplaques were stained with the use of purified polyclonal rabbitanti-human visfatin IgG (Phoenix Pharmaceuticals, Belmont, Calif)and mouse anti-human monocytes/macrophages (calprotectin) IgG(MCA874G, Serotec Ltd, Oxford, UK). The primary antibodies werefollowed by biotinylated anti-rabbit or anti-mouse IgG (VectorLaboratories, Burlingame, Calif). The immunoreactivities were fur-ther amplified with avidin-biotin-peroxidase complexes (VectastainElite kit, Vector Laboratories). Diaminobenzidine was used as thechromogen in a commercial metal-enhanced system (Pierce Chem-ical, Rockford, Ill). The sections were counterstained with hematox-ylin. Omission of the primary antibody served as a negative control.
Western BlottingWestern blotting was performed as previously described,12 separat-ing equal amounts of protein from each sample by SDS-PAGE(10%) before transferring it onto polyvinyl difluoride membranes(NEN; Life Science, Boston, Mass). The membranes were incubatedwith rabbit antibody against visfatin (Phoenix Pharmaceuticals),stripped, and reprobed with mouse anti–�-tubulin (Sigma) and rabbitanti-MCM3 (DNA polymerase-� holoenzyme-associated protein P1;Abcam, Cambridge, UK) to ensure equal loading, followed byincubation with species-specific horseradish peroxidase–coupledsecondary antibodies (Cell Signaling, Beverly, Mass). The immunecomplex was visualized with the use of the Supersignal West PicoWestern blot detection system (Pierce) and exposure to HyperfilmECL (Amersham Biosciences, Buckinghamshire, UK) and detectedwith the use of the Kodak 440 CF imaging station (Boston, Mass).The software Total Laboratory v.1 · 10 (Phoretix, Newcastle, UK)was used for quantification.
Cell Culture ExperimentsThe human monocytic cell line THP-1 (American Type CultureCollection, Rockville, Md) was cultured in RPMI-1640 (Sigma, StLouis, Mo) with 10% fetal calf serum ([Sigma]), penicillin-streptomycin, and 2 mmol/L L-glutamine (Sigma) in 6-well trays(106 cells/mL; Costar, Cambridge, Mass). Before the experimentalstudies, the cells were washed once in RPMI-1640 and furtherincubated in serum-free medium (RPMI 1640 with 2 mmol/LL-glutamine supplemented without fetal calf serum) with and withoutdifferent concentrations of recombinant human (rh) visfatin (PhoenixPharmaceuticals, Calif), oxidized low-density lipoprotein (oxLDL)(20 �g/mL), tumor necrosis factor-� (TNF-�) (5 ng/mL; R&DSystems, Minneapolis, Minn), or a combination thereof. In a separateset of experiments, peripheral blood mononuclear cells (PBMCs)were obtained from heparinized blood by Isopaque-Ficoll (Lym-phoprep; Nycomed, Oslo, Norway) gradient centrifugation andincubated in flat-bottomed 96-well trays (2�106/mL; Costar) inmedium alone (RPMI 1640 with 2 mmol/L L-glutamine supple-mented with 5% fetal calf serum [Sigma]) or stimulated withrh-visfatin as described above. LDL was isolated from humanendotoxin-free heparin plasma and oxidatively modified by Cu2�
ions (10 �mol/L).13 In some experiments, hydroxy-2-naphthalenylmethylphosphonic acid tris-acetoxy-methyl ester(HNMPA-[AM]3, hereafter named HNMPA; 100 �mol/L; BiomolResearch Laboratories, Plymouth Meeting, Pa), a tyrosine kinase
Figure 2. Strong visfatin immunoreactivityin atherosclerotic carotid plaques fromsymptomatic patients. A, Visfatin immuno-reactivity in an atherosclerotic carotidplaque removed by endarterectomy from apatient with symptomatic disease. Visfatinimmunoreactivity was localized to parts ofthe lipid-rich core of the lesion with numer-ous CD68-positive macrophages (B). Inpatients with asymptomatic disease (C),both the atherosclerotic plaque and thelipid-rich visfatin-positive core were sub-stantially smaller than in lesions frompatients with symptomatic disease. Magni-fication �100.
Dahl et al Visfatin and Plaque Destabilization 975
by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from

inhibitor that blocks insulin receptor signaling, and polymyxin B (10�g/mL, Sigma) were added to cell cultures 30 minutes beforerh-visfatin stimulation. Control cells were given vehicle (mediumwith solvent used for the test substances). In all experiments, cellpellets and cell-free supernatants were stored at indicated time pointsat �80°C until analysis. Endotoxin levels in all media, buffers, andstimulants were �70 pg/mL (limulus amoebocyte lysate test).
ZymographyGelatinase activity was detected in THP-1 supernatant after incuba-tion for 24 hours.14 Human pro-MMP-9 and active-MMP-9 (Onco-gene, Cambridge, Mass) were run in a separate lane to ensureidentification of MMP activity. Duplicate gel was run and developedin buffer containing 200 �mol/L EDTA to ensure metal-dependentproteolysis. Gels were scanned by a Kodak 440 CF imaging station(Vector), and the software Total Laboratory (Phoretix) was used forquantification.
Enzyme ImmunoassayPlasma levels of visfatin were measured by enzyme immunoassay(Phoenix Pharmaceuticals). Levels of TNF-� and interleukin (IL)-8in PBMC supernatants were measured by DuoSet (R&D Systems).The sensitivity values for visfatin, TNF-�, and IL-8 enzyme immu-noassays were 2.8 ng/mL, 15 pg/mL, and 15 pg/mL, receptively. Theintra-assay and interassay coefficients of variation were �10% forall enzyme immunoassays.
Statistical AnalysesData are presented as median and interquartile range. We werelooking for large differences (ie, �2-fold increase or �50% de-crease), and we did not perform any sample size calculation.However, on the basis of previous experiments in our laboratory, asample size of 7 to 15 individuals in each group will be sufficient todetect large and biologically relevant differences in the gene expres-sion of inflammatory genes. The Mann-Whitney U test was used forcomparison of 2 independent groups, and the Wilcoxon signed ranktest was used in the paired situation. The Kruskal-Wallis test(Figures 4 and 5), Friedman test (Figure 3A), and 2-way ANOVA(Figure 6A and 6B) were used a priori where appropriate; the resultsof these tests are stated in the figure legends. The Fisher exact testwas used for comparison of proportions. Probability values (2-sided)were considered significant when �0.05. All authors have read and
agree to the manuscript as written. All authors had full access to andtake full responsibility for the integrity of the data.
ResultsExpression of Visfatin in SymptomaticCarotid PlaquesTo screen for genes that were regulated differently in symp-tomatic compared with asymptomatic carotid plaques, weused oligonucleotide microarrays encoding 14 500 humangenes to analyze the gene expression profiles in specimensfrom atherosclerotic carotid plaques isolated from 4 patientswith symptomatic and 4 patients with asymptomatic lesions.The patients were selected randomly from the total studypopulation. The analysis identified 136 genes to be regulateddifferently in symptomatic compared with asymptomaticplaques (�2-fold increase or �50% decrease; Table I in theonline-only Data Supplement), and visfatin was identified asone of these genes, being markedly upregulated in all symp-tomatic patients (2.1- to 3.2-fold increase). This increasedexpression of visfatin in symptomatic plaques was confirmedby real-time reverse transcription polymerase chain reactionwhen plaques from the total study population were analyzed(Figure 1A) and on the protein levels as assessed by Westernblot assays when carotid plaques from 6 asymptomatic and 10symptomatic patients were analyzed (Figure 1B and 1C).
Cellular Localization of Visfatin Protein inAtherosclerotic Carotid PlaquesTo determine the cellular localization of visfatin, immuno-histochemical analysis was performed on carotid plaquesfrom 2 patients with symptomatic disease and 2 patients withasymptomatic disease (Figure 2A). Staining of serial sectionsof these atherosclerotic lesions with anti-visfatin IgG showedstrong immunostaining in plaques from symptomatic patientsthat were localized to the lipid-rich core of the plaque withnumerous CD68-positive macrophages (Figure 2B). CD68-
Figure 3. Increased visfatin levels duringplaque rupture in patients with coronaryartery disease. A, Plasma levels of visfa-tin in 8 patients with stable angina before(baseline) and at different time pointsafter PCI, representing a mechanicallyinduced plaque rupture. Friedman test;P�0.011, *P�0.012 vs baseline. B, Im-munostaining and localization of visfatinin material removed from the site ofplaque rupture in patients withST-elevation myocardial infarction under-going PCI. Visfatin immunoreactivity wasseen in lipid-rich regions with strongCD68 immunostaining and in CD68-positive macrophages surrounding theseregions (C). Plaque material stained withomission of the primary antibody servedas control and demonstrated no immu-nostaining of any of the cellular elements(D). Magnification �200.
976 Circulation February 27, 2007
by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from
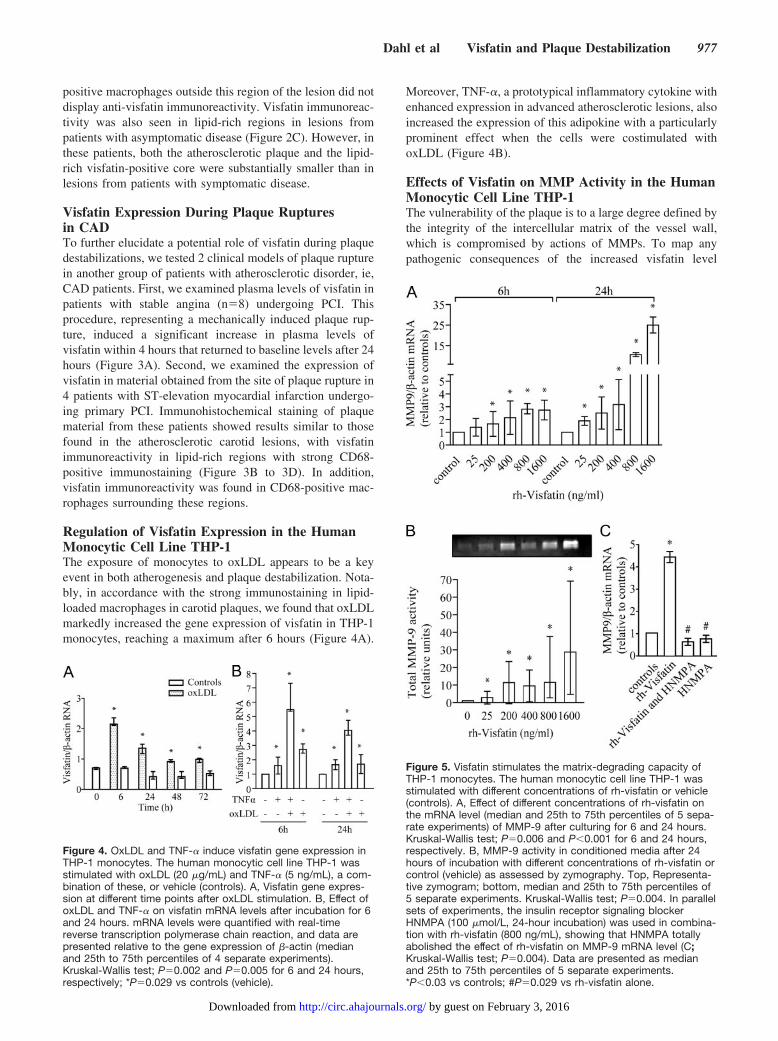
positive macrophages outside this region of the lesion did notdisplay anti-visfatin immunoreactivity. Visfatin immunoreac-tivity was also seen in lipid-rich regions in lesions frompatients with asymptomatic disease (Figure 2C). However, inthese patients, both the atherosclerotic plaque and the lipid-rich visfatin-positive core were substantially smaller than inlesions from patients with symptomatic disease.
Visfatin Expression During Plaque Rupturesin CADTo further elucidate a potential role of visfatin during plaquedestabilizations, we tested 2 clinical models of plaque rupturein another group of patients with atherosclerotic disorder, ie,CAD patients. First, we examined plasma levels of visfatin inpatients with stable angina (n�8) undergoing PCI. Thisprocedure, representing a mechanically induced plaque rup-ture, induced a significant increase in plasma levels ofvisfatin within 4 hours that returned to baseline levels after 24hours (Figure 3A). Second, we examined the expression ofvisfatin in material obtained from the site of plaque rupture in4 patients with ST-elevation myocardial infarction undergo-ing primary PCI. Immunohistochemical staining of plaquematerial from these patients showed results similar to thosefound in the atherosclerotic carotid lesions, with visfatinimmunoreactivity in lipid-rich regions with strong CD68-positive immunostaining (Figure 3B to 3D). In addition,visfatin immunoreactivity was found in CD68-positive mac-rophages surrounding these regions.
Regulation of Visfatin Expression in the HumanMonocytic Cell Line THP-1The exposure of monocytes to oxLDL appears to be a keyevent in both atherogenesis and plaque destabilization. Nota-bly, in accordance with the strong immunostaining in lipid-loaded macrophages in carotid plaques, we found that oxLDLmarkedly increased the gene expression of visfatin in THP-1monocytes, reaching a maximum after 6 hours (Figure 4A).
Moreover, TNF-�, a prototypical inflammatory cytokine withenhanced expression in advanced atherosclerotic lesions, alsoincreased the expression of this adipokine with a particularlyprominent effect when the cells were costimulated withoxLDL (Figure 4B).
Effects of Visfatin on MMP Activity in the HumanMonocytic Cell Line THP-1The vulnerability of the plaque is to a large degree defined bythe integrity of the intercellular matrix of the vessel wall,which is compromised by actions of MMPs. To map anypathogenic consequences of the increased visfatin level
Figure 4. OxLDL and TNF-� induce visfatin gene expression inTHP-1 monocytes. The human monocytic cell line THP-1 wasstimulated with oxLDL (20 �g/mL) and TNF-� (5 ng/mL), a com-bination of these, or vehicle (controls). A, Visfatin gene expres-sion at different time points after oxLDL stimulation. B, Effect ofoxLDL and TNF-� on visfatin mRNA levels after incubation for 6and 24 hours. mRNA levels were quantified with real-timereverse transcription polymerase chain reaction, and data arepresented relative to the gene expression of �-actin (medianand 25th to 75th percentiles of 4 separate experiments).Kruskal-Wallis test; P�0.002 and P�0.005 for 6 and 24 hours,respectively; *P�0.029 vs controls (vehicle).
Figure 5. Visfatin stimulates the matrix-degrading capacity ofTHP-1 monocytes. The human monocytic cell line THP-1 wasstimulated with different concentrations of rh-visfatin or vehicle(controls). A, Effect of different concentrations of rh-visfatin onthe mRNA level (median and 25th to 75th percentiles of 5 sepa-rate experiments) of MMP-9 after culturing for 6 and 24 hours.Kruskal-Wallis test; P�0.006 and P�0.001 for 6 and 24 hours,respectively. B, MMP-9 activity in conditioned media after 24hours of incubation with different concentrations of rh-visfatin orcontrol (vehicle) as assessed by zymography. Top, Representa-tive zymogram; bottom, median and 25th to 75th percentiles of5 separate experiments. Kruskal-Wallis test; P�0.004. In parallelsets of experiments, the insulin receptor signaling blockerHNMPA (100 �mol/L, 24-hour incubation) was used in combina-tion with rh-visfatin (800 ng/mL), showing that HNMPA totallyabolished the effect of rh-visfatin on MMP-9 mRNA level (C;Kruskal-Wallis test; P�0.004). Data are presented as medianand 25th to 75th percentiles of 5 separate experiments.*P�0.03 vs controls; #P�0.029 vs rh-visfatin alone.
Dahl et al Visfatin and Plaque Destabilization 977
by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from

within the unstable lesions, we examined the ability ofrh-visfatin to induce MMP-9 expression in THP-1 cells. Asshown in Figure 5A, rh-visfatin markedly increased MMP-9mRNA levels (�14-fold increase) in a dose-dependent man-ner, with the most marked effects after culturing for 24 hours.Importantly, such a visfatin-mediated increase was also seenwhen MMP-9 activity was analyzed by zymography in cellsupernatants (Figure 5B). The endotoxin level in the rh-visfatin preparation was �70 pg/mL in all experiments.Adding polymyxin B (10 �g/mL), a cationic polypeptide thatinhibits lipopolysaccharide activity, to the cell culture 30minutes before rh-visfatin activation had no effect on theMMP-9 levels (data not shown). This suggests strongly thatthe demonstrated effects of rh-visfatin were not related toendotoxin contamination of the culture medium or the rh-visfatin preparation. Visfatin has been reported to be bound toand activate the insulin receptor,6 and HNMPA, an inhibitorof insulin receptor signaling, totally abolished the visfatin-mediated increase in MMP-9 expression (Figure 5C).
Effect of Visfatin on PBMCs From UnstableAngina Patients and Healthy ControlsImmune-mediated plaque destabilization involves expressionof inflammatory cytokines. To examine the relevance of ourfindings on visfatin-mediated effects in THP-1 cells to humanatherosclerotic disorders, we examined the ability of rh-visfatin to stimulate IL-8 and TNF-� release in PBMCs from8 patients with unstable angina and 8 healthy controls withoutany CAD. As shown in Figure 6A and 6B, rh-visfatin (800ng/mL) markedly increased the IL-8 (�10-fold) and TNF-�(�80-fold) levels in PBMC supernatants in a dose-dependentmanner after culturing for 24 hours. As in the case of IL-8, aparticularly enhancing effect was seen in cells from patientswith unstable angina. Again, and comparable to the effect inTHP-1 cells, although HNMPA had no effect on its own, thisinsulin receptor signaling blocker totally abolished thevisfatin-mediated increase in IL-8 and TNF-� (Figure 6C and6D).
DiscussionVisfatin has been reported previously to be produced inadipose tissue, bone marrow, skeletal muscle, and liver.15 Inthis report, to the best of our knowledge, we demonstrate forthe first time that visfatin is strongly expressed withinlipid-loaded macrophages in atherosclerotic lesions, withincreased expression particularly in plaques from symptom-atic patients. On the functional level, visfatin was found to bea potent inducer of MMP-9 and inflammatory cytokines inTHP-1 monocytes and PBMCs, respectively. Visfatin haspreviously been indirectly linked to atherogenesis through itseffect on glucose homeostasis.6 Our findings in the presentstudy suggest that this adipokine could be related to athero-genesis and plaque destabilization in a more direct way.Moreover, visceral white adipose tissue seems to be a majorproducer of visfatin.15 Some reports also suggest a positivecorrelation between plasma levels of visfatin and visceraladiposity.6 On the basis of our findings in the present study,showing potent inflammatory effects of visfatin, it is temptingto hypothesize that this adipokine also could contribute to theinflammatory state and increased risk for cardiovascularevents characterizing patients with high abdominalfat/obesity.16
It was reported recently that macrophages from visceralwhite adipose tissue expressed higher levels of visfatin thandid mature adipocytes.17 In the present study we extend thisfinding by showing that visfatin is strongly expressed withinsymptomatic atherosclerotic carotid plaques and is localizedto areas with lipid-loaded macrophages. The relationshipbetween visfatin and unstable lesions was further supportedby our findings of strong visfatin immunostaining in lipid-rich regions within plaque material obtained at the site ofplaque rupture in patients with ST-elevation myocardialinfarction undergoing PCI as well as by the demonstration ofa marked increase in plasma visfatin levels during PCI instable angina patients, representing a mechanically inducedplaque rupture.
Figure 6. Visfatin increases the inflam-matory potential in PBMCs. PBMCs fromunstable angina patients (n�8) andhealthy controls (n�8) were cultured for24 hours with or without different con-centrations of rh-visfatin before IL-8 wasanalyzed (A) (2-way ANOVA; dose:P�0.001; group [patients andcontrols]�dose: P�0.031) and TNF-� (B)(2-way ANOVA; dose: P�0.001; group[patients and controls]�dose: P�0.862)levels in the conditioned media byenzyme immunoassays. In a parallel setof experiments, in PBMCs from 5 healthycontrols, the insulin receptor signalingblocker HNMPA (100 �mol/L, 24-hourincubation) was used in combination withrh-visfatin (800 ng/mL), showing thatHNMPA abolished the effect of rh-visfatinon IL-8 (C) and TNF-� (D). Data aremedian and 25th to 75th percentiles.*P�0.03, **P�0.006, ***P�0.001 vsunstimulated cells; †P�0.047 vs unstableangina patients; #P�0.029 vs rh-visfatinalone. ND indicates not detectable.
978 Circulation February 27, 2007
by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from

Previously, stimuli such as dexamethasone and peroxi-some proliferator-activated receptor agonists have beenfound to increase visfatin expression in adipocytes.18,19 Inthe present study we found that oxLDL is a potent stimulusfor visfatin expression in THP-1 monocytes, which is inagreement with localization of visfatin in regions withfoam cell macrophages. Although TNF-� seems to de-crease visfatin expression in adipocytes,6,20 this and otherinflammatory cytokines have been reported to increasevisfatin expression in neutrophils,9 suggesting that visfatinis regulated differently in different cell types. In thepresent study we found that a TNF-�–mediated upregula-tion of visfatin is also operating in THP-1 monocytes, witha particularly strong induction when TNF-� is acting inconcert with oxLDL. An unstable atherosclerotic lesion ischaracterized by increased levels of oxLDL and inflam-matory cytokines like TNF-�. On the basis of our findingsin THP-1 monocytes, such a milieu could be a potentstimulus for visfatin expression, potentially explaining itsenhanced expression in unstable atherosclerotic lesions.
Several reports have been published on the metaboliceffects on visfatin, including its insulin-mimicking ac-tions.6 Thus, studies addressing the molecular mechanismshave revealed that visfatin activates the intracellular sig-naling cascade for insulin, but, interestingly, visfatinactivates the insulin receptor in a manner distinct from thatof insulin.6 However, the effects of visfatin are notrestricted to the modification of glucose metabolism. Someprevious studies have suggested a role for this adipokine ininflammation and immune responses. In fact, visfatin wasoriginally isolated from a cDNA library derived fromactivated peripheral blood lymphocytes as a factor thatsynergizes with IL-7 to promote the differentiation ofB-cell precursors.7 More recently, visfatin has been re-ported to promote the survival of neutrophils duringexperimental inflammation and clinical sepsis,9 to stimu-late the expression of inflammatory cytokines in epithelialcells,21 and to be involved in the thrombin-induced endo-thelial cell dysfunction during various forms of acute lunginjury.8 In the present study we show that visfatin is apotent inducer of MMP-9 activity in THP-1 monocytes andthat it promotes a marked release of inflammatory cyto-kines (ie, IL-8 and TNF-�) in PBMCs, with a particularlyenhancing effect on IL-8 in cells from unstable anginapatients. As previously reported for the metabolic effectsof visfatin, these inflammatory actions were abolished byblocking insulin receptor signaling. MMP and inflamma-tory cytokines and their mutual interactions play an im-portant role in atherogenesis and plaque destabilization.The marked induction of these mediators by visfatin incells with relevance to atherosclerotic lesions suggests thatvisfatin, showing enhanced expression in symptomaticplaques, could be an important mediator in these pro-cesses. Moreover, the combined ability of visfatin toincrease TNF-� as well as to respond with increasedexpression on TNF-� stimulation suggests that the inter-action between TNF-� and visfatin could represent apathogenic loop operating in foam cell macrophages
within unstable atherosclerotic lesions, further promotingplaque destabilization.
Several reports have been published on the metaboliceffects of visfatin in particular in relation to its possible rolein diabetes and metabolic syndrome. Although relatively fewpatients were examined, our findings in the present studysuggest that visfatin also should be regarded as an inflamma-tory mediator, localized to foam cell macrophages withinunstable atherosclerotic lesions, that potentially plays a rolein plaque destabilization.
AcknowledgmentsThe authors thank Ellen Lund Sagen for excellent technicalassistance.
Sources of FundingThis work was supported by grants from the Norwegian Council ofCardiovascular Research, Research Council of Norway, Universityof Oslo, Medinnova Foundation, Eckbos Legater, Helse Sør,Rikshospitalet-Radiumhospitalet Medical Center, and the NorwegianFoundation for Health and Rehabilitation.
DisclosuresNone.
References1. Glass CK, Witztum JL. Atherosclerosis: the road ahead. Cell. 2001;104:
503–516.2. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease.
N Engl J Med. 2005;352:1685–1695.3. Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular
disease. Circ Res. 2005;96:939–949.4. Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regu-
latory pathways. Physiol Rev. 2006;86:515–581.5. Wu ZH, Zhao SP. Adipocyte: a potential target for the treatment of
atherosclerosis. Med Hypotheses. 2006;67:82–86.6. Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto
K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E,Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M,Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I.Visfatin: a protein secreted by visceral fat that mimics the effects ofinsulin. Science. 2005;307:426–430.
7. Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning andcharacterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–1437.
8. McGlothlin JR, Gao L, Lavoie T, Simon BA, Easley RB, Ma SF, RumalaBB, Garcia JG, Ye SQ. Molecular cloning and characterization of caninepre-B-cell colony-enhancing factor. Biochem Genet. 2005;43:127–141.
9. Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD, Marshall JC. Pre-Bcell colony-enhancing factor inhibits neutrophil apoptosis in experimentalinflammation and clinical sepsis. J Clin Invest. 2004;113:1318–1327.
10. Grant EG, Benson CB, Moneta GL, Alexandrov AV, Baker JD, Bluth EI,Carroll BA, Eliasziw M, Gocke J, Hertzberg BS, Katanick S, NeedlemanL, Pellerito J, Polak JF, Rholl KS, Wooster DL, Zierler RE. Carotid arterystenosis: gray-scale and Doppler US diagnosis: Society of Radiologists inUltrasound Consensus Conference. Radiology. 2003;229:340–346.
11. Anderson GB, Ashforth R, Steinke DE, Ferdinandy R, Findlay JM. CTangiography for the detection and characterization of carotid artery bifur-cation disease. Stroke. 2000;31:2168–2174.
12. Scholz H, Aukrust P, Damas JK, Tonstad S, Sagen EL, Kolset SO, HallC, Yndestad A, Halvorsen B. 8-Isoprostane increases scavenger receptorA and matrix metalloproteinase activity in THP-1 macrophages, resultingin long-lived foam cells. Eur J Clin Invest. 2004;34:451–458.
13. Halvorsen B, Aas UK, Kulseth MA, Drevon CA, Christiansen EN, KolsetSO. Proteoglycans in macrophages: characterization and possible role inthe cellular uptake of lipoproteins. Biochem J. 1998;331:743–752.
14. Staff AC, Ranheim T, Henriksen T, Halvorsen B. 8-Iso-prostaglandinf(2alpha) reduces trophoblast invasion and matrix metalloproteinaseactivity. Hypertension. 2000;35:1307–1313.
Dahl et al Visfatin and Plaque Destabilization 979
by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from

15. Sethi JK, Vidal-Puig A. Visfatin: the missing link between intra-abdominal obesity and diabetes? Trends Mol Med. 2005;11:344–347.
16. Wu JT, Wu LL. Linking inflammation and atherogenesis: soluble markersidentified for the detection of risk factors and for early risk assessment.Clin Chim Acta. 2006;366(1–2):74–80.
17. Curat CA, Wegner V, Sengenes C, Miranville A, Tonus C, Busse R,Bouloumie A. Macrophages in human visceral adipose tissue: increasedaccumulation in obesity and a source of resistin and visfatin. Diabe-tologia. 2006;49:744–747.
18. Choi KC, Ryu OH, Lee KW, Kim HY, Seo JA, Kim SG, Kim NH, ChoiDS, Baik SH, Choi KM. Effect of PPAR-alpha and -gamma agonist on
the expression of visfatin, adiponectin, and TNF-alpha in visceral fat ofOLETF rats. Biochem Biophys Res Commun. 2005;336:747–753.
19. Kralisch S, Klein J, Lossner U, Bluher M, Paschke R, Stumvoll M,Fasshauer M. Hormonal regulation of the novel adipocytokine visfatin in3T3-L1 adipocytes. J Endocrinol. 2005;185:R1–R8.
20. Stephens JM, Vidal-Puig AJ. An update on visfatin/pre-B cell colony-enhancing factor, an ubiquitously expressed, illusive cytokine that isregulated in obesity. Curr Opin Lipidol. 2006;17:128–131.
21. Ognjanovic S, Bryant-Greenwood GD. Pre-B-cell colony-enhancingfactor, a novel cytokine of human fetal membranes. Am J Obstet Gynecol.2002;187:1051–1058.
CLINICAL PERSPECTIVEThe analysis of expressed genes in adipose tissue has revealed that adipocytes produce and secrete a variety of bioactivesubstances, named adipokines, including growth factors and cytokines. Several studies have suggested a role for thesemediators in atherogenesis not only through regulation of lipid and glucose metabolism but also by modulating theinflammatory arm of atherosclerosis. Visfatin, one of the most recently identified adipokines, has previously been reportedto be produced in adipose tissue, bone marrow, skeletal muscle, and liver. Herein we demonstrate that visfatin is stronglyexpressed within lipid-loaded macrophages in carotid atherosclerotic lesions, with particularly increased expression incarotid plaques from symptomatic patients. On the functional level, visfatin was found to be a potent inducer of matrixdegradation and inflammation in monocytes and mononuclear cells from peripheral blood, respectively. Several reportshave been published on the metabolic effects of visfatin, in particular in relation to its possible role in diabetes andmetabolic syndrome. Our findings in the present study suggest that visfatin also should be regarded as an inflammatorymediator, localized to foam cell macrophages within unstable atherosclerotic lesions, potentially playing a role in plaquedestabilization. Our findings further underscore the link between lipid accumulation and inflammation in atherogenesis.
980 Circulation February 27, 2007
by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from

HalvorsenGullestad, Stig S. Frøland, Kirsten Krohg-Sørensen, David Russell, Pål Aukrust and BenteDamås, Siv H. Tunheim, Thor Ueland, Camilla Smith, Bjørn Bendz, Serena Tonstad, Lars
Tuva B. Dahl, Arne Yndestad, Mona Skjelland, Erik Øie, Arve Dahl, Annika Michelsen, Jan K.Coronary Atherosclerosis: Possible Role in Inflammation and Plaque Destabilization
Increased Expression of Visfatin in Macrophages of Human Unstable Carotid and
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2007 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/CIRCULATIONAHA.106.665893
2007;115:972-980; originally published online February 5, 2007;Circulation.
http://circ.ahajournals.org/content/115/8/972World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2007/02/05/CIRCULATIONAHA.106.665893.DC1.htmlData Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on February 3, 2016http://circ.ahajournals.org/Downloaded from