cyclooxygenases, thromboxane, and atherosclerosis plaque destabilization by cyclooxygenase2...
TRANSCRIPT

Cyclooxygenases, Thromboxane, and AtherosclerosisPlaque Destabilization by Cyclooxygenase-2 Inhibition Combined With
Thromboxane Receptor Antagonism
Karine M. Egan, PhD; Miao Wang, PhD; Margaret B. Lucitt, BSc; Alicia M. Zukas, AB;Ellen Puré, PhD; John A. Lawson, MS; Garret A. FitzGerald, MD
Background—Antagonism or deletion of the receptor (the TP) for the cyclooxygenase (COX) product thromboxane(Tx)A2, retards atherogenesis in apolipoprotein E knockout (ApoE KO) mice. Although inhibition or deletion of COX-1retards atherogenesis in ApoE and LDL receptor (LDLR) KOs, the role of COX-2 in atherogenesis remainscontroversial. Other products of COX-2, such as prostaglandin (PG) I2 and PGE2, may both promote inflammation andrestrain the effects of TxA2. Thus, combination with a TP antagonist might reveal an antiinflammatory effect of a COX-2inhibitor in this disease. We addressed this issue and the role of TxA2 in the promotion and regression of diffuse,established atherosclerosis in Apobec-1/LDLR double KOs (DKOs).
Methods and Results—TP antagonism with S18886, but not combined inhibition of COX-1 and COX-2 with indomethacinor selective inhibition of COX-2 with Merck Frosst (MF) tricyclic, retards significantly atherogenesis in DKOs.Although indomethacin depressed urinary excretion of major metabolites of both TxA2, 2,3-dinor TxB2 (Tx-M), andPGI2, 2,3-dinor 6-keto PGF1� (PGI-M), only PGI-M was depressed by the COX-2 inhibitor. None of the treatmentsmodified significantly the increase in lipid peroxidation during atherogenesis, reflected by urinary 8,12-iso-iPF2�-VI.Combination with the COX-2 inhibitor failed to augment the impact of TP antagonism alone on lesion area. Rather,analysis of plaque morphology reflected changes consistent with destabilization of the lesion coincident with augmentedformation of TxA2. Despite a marked effect on disease progression, TP antagonism failed to induce regression ofestablished atherosclerotic disease in this model.
Conclusions—TP antagonism is more effective than combined inhibition of COX-1 and COX-2 in retarding atherogenesisin Apobec-1/LDLR DKO mice, which perhaps reflects activation of the receptor by multiple ligands during diseaseinitiation and early progression. Despite early intervention, selective inhibition of COX-2, alone or in combination witha TP antagonist, failed to modify disease progression but may undermine plaque stability when combined with theantagonist. TP antagonism failed to induce regression of established atherosclerotic disease. TP ligands, includingCOX-1 (but not COX-2)–derived TxA2, promote initiation and early progression of atherogenesis in Apobec-1/LDLRDKOs but appear unimportant in the maintenance of established disease. (Circulation. 2005;111:334-342.)
Key Words: thromboxane � atherosclerosis � lesion � prostaglandins � inflammation
Atherosclerosis bears many of the hallmarks of an inflam-matory disease1; however, there is scant evidence of a
causal role of inflammation in disease progression in humans.Prostaglandins (PGs), particularly PGE2
2 and PGI2,3 areestablished mediators of inflammation, and inhibition of theirbiosynthetic enzymes, the cyclooxygenases (COXs), by non-steroidal antiinflammatory drugs (NSAIDs) affords relief ofsymptoms in the inflammatory arthritides. Expression ofCOX-1 and COX-2 is augmented in inflamed synovia4 andalso in endothelium, vascular smooth muscle cells, andmacrophages in human atherosclerotic lesions,5,6 which raisesthe prospect that COX inhibitors might retard plaque progres-
sion. Recently, a placebo-controlled trial of rofecoxib re-vealed a 2-fold increase of myocardial infarction and stroke7
that led to withdrawal of the drug from the market. Thisprobably reflects a mechanism whereby depression of COX-2–derived prostacyclin (PGI2) removes a constraint on plate-let COX-1–derived thromboxane (Tx) A2 and other agoniststhat elevate blood pressure, promote atherogenesis, and aug-ment the thrombotic response to plaque rupture.7 Indeed,overview analysis of the experience with a structurallydistinct inhibitor, valdecoxib, reveals a 3-fold increase inmyocardial infarction and stroke,8 and a placebo-controlledclinical trial of a third, celecoxib, has been prematurely
Received June 29, 2004; revision received October 28, 2004; accepted November 3, 2004.From the Institute for Translational Medicine and Therapeutics (K.M.E., M.W., M.B.L., J.A.L., G.A.F.), University of Pennsylvania, Philadelphia, Pa,
The Wistar Institute and The Ludwig Institute for Cancer Research (A.M.Z., E.P.), Philadelphia, Pa.Correspondence to Garret A. FitzGerald, MD, 153 Johnson Pavilion, University of Pennsylvania, Philadelphia, PA 19104. E-mail
[email protected]© 2005 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/01.CIR.0000153386.95356.78
334
Molecular Cardiology
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from

terminated because of an excess of cardiovascular events onthe coxib. Despite these observations, a clinical trial designedto detect a cardioprotective effect of combining a thirdCOX-2 inhibitor (celecoxib) has been announced. In this trial,celecoxib will be combined with low-dose aspirin to suppressCOX-1–derived TxA2 in patients with osteoarthritis who areat high cardiovascular risk. It is assumed this strategy willreveal a beneficial effect of the COX-2 inhibitor.
COX-2–derived PGs mediate inflammation, a hallmark ofatherosclerosis. Additionally, descriptive analysis of plaquemorphology in case-control studies have implicated COX-2–derived PGE2 in the activation of matrix metalloproteinasesand consequent destabilization of atherosclerotic plaques inhumans.9,10 However, large-scale clinical trials of traditionalNSAIDs have not been performed in cardiovascular disease.
Cardioprotection from aspirin may reflect inhibition ofplatelet COX-1–dependent TxA2 formation.11 Indeed, indi-rect comparisons across clinical trials fail to detect anydifference in the degree of cardioprotection between lowdoses, which favor inhibition of COX-1 (75 to 150 mg/d), andhigher (�325 mg/d) antiinflammatory doses, which inhibitboth COX-1 and COX-2.12 A direct, controlled comparison ofthe effects of high- and low-dose aspirin on plaque burden orcardiovascular outcomes has not been performed.
These observations have prompted interest in mice asmore tractable models of atherosclerosis in which therelative importance of the 2 COX enzymes and theirproducts might be defined. Early evidence was obtainedfor the importance of COX-1. Thus, combined inhibitionof COX-1 and COX-2, but not of COX-2 alone, retardedatherogenesis in LDL receptor (LDLR) knockouts (KOs)despite a similar depression (�60%) of the cytokines,soluble intercellular adhesion molecule and monocytechemotactic protein-1, by the 2 regimens.13 Similarly, selec-tive inhibition and genetic deletion of COX-1 both retardatherogenesis in apolipoprotein (Apo) E KOs.14,15 Given theevidence for platelet activation in human atherosclerosis16
and in mutant mice,13,17 these data seem congruent withevidence that antagonism18 or deletion19 of the TP, the TxA2
receptor, retards atherogenesis in ApoE KOs.TxA2 is also a product of monocyte and macrophage
COX-2.20 Furthermore, products of lipid peroxidation, suchas the isoprostanes, which increase during atherogenesis,21,22
may activate the TP in a COX-independent manner.23 Thus, itis possible that selective antagonism of the TP might moreeffectively retard atherogenesis than COX-1 inhibition alone.Finally, it is unknown whether activation of the TP is relevantonly to lesion initiation and early development or whether TPantagonism can induce regression of established atheroscle-rotic disease.
The role of COX-2 in atherogenesis is much more contro-versial.24 COX-2 inhibitors have been shown variously toretard,25 accelerate,26 fail to accelerate,13 or leave unal-tered14,27,28 atherogenesis in LDLR and ApoE KO mice. Thecontradictory nature of these results has been attributed to thetiming of intervention, differences in the mouse models, anddifferences in the pharmacological probes used.24 The COX-2products PGE2 and PGI2 may also act as restraints on thecardiovascular effects of TxA2
19,29,30 to offset their proinflam-
matory action. Thus, despite the cardiovascular hazard fromselective COX-2 inhibition alone,7,8 TP antagonism or sup-pression of TxA2 by low-dose aspirin might theoreticallyreveal an antiinflammatory action of COX-2 inhibition,which might further augment its impact on atherogenesis.
We have addressed these outstanding issues in the Apobec-1/LDLR DKO. This model of atherosclerosis more faithfullyreplicates the human disease than either the LDLR KO or theApoE KO in several respects, including a multifocal origin ofdisease, predominance of the elevated cholesterol in LDL,and a pronounced gender dependence of the phenotype.31 Wehave compared the impact of TP antagonism with coinciden-tal inhibition of both COXs and selective inhibition of COX-2in the initiation and early progression of disease. Further-more, we have determined whether combination with a TPantagonist might reveal a beneficial effect of COX-2 inhibi-tion attributable to suppression of inflammation. Finally, weaddressed the possibility that TP antagonism might induceregression of established atherosclerotic disease.
MethodsAnimalsThe DKO mice were a gift from Drs Powell-Braxton and Bunting atGenentech Inc, South San Francisco, Calif. All animals were housedaccording to guidelines of the Institutional Animal Care and UsageCommittee (IACUC) of the University of Pennsylvania. All proce-dures were considered and approved by the IACUC. Animals werehoused in a controlled barrier environment that met universitystandards and US federal and statutory regulations. This includesclimate, air-exchange and filtration, photo-light period, and cagingspecifications.
Study DesignAlthough the DKOs spontaneously develop atherosclerotic lesionson a regular chow diet, the “Western” high-fat diet (0.2% cholester-ol, 21% saturated fat; formula TD 88137, Harlan Teklad) was chosenfor the initial study. Extensive atherosclerotic lesions develop inmice fed this diet for 92 days.31 Male mice were randomized to eachtreatment regimen at 6 weeks and were euthanized at 19 weeks ofage. Mice in this first study were randomized to receive the high-fatdiet in combination with (1) S18886 (3-((6R)-6-{[(4-chlorophenyl)sulfonyl]amino}-2-methyl-5,6,7,8 tetrahydro-1-naphthalenyl) propanoic acid, sodium salt), a nonprostanoid, highlyselective TP antagonist,32,33 in their drinking water (at 5 [n�8] or 10[n�8] mg/kg) or (2) indomethacin at 6 mg/L in their drinking water[n�10]) or diet alone (control group [n�13]) for 92 days. S18886was kindly provided by Dr Stefano Corda at Servier Laboratories,Technologie Servier, Orléans, France, and was administered in thedrinking water. The dose of drug was calculated on the basis of theaverage consumption of water (5 mL/d) and the body weight, asdetermined weekly. Solutions were prepared every 7 to 10 days.Indomethacin (Sigma) was added to the drinking water at a concen-tration of 6 mg/L. Solutions were prepared every 3 days. The dose ofdrug was calculated on the basis of the average consumption of water(5 mL/d) and the body weight, determined weekly. This corre-sponded to 10 to 20 ng/d. MF tricyclic [3-(3,4-difluorophenyl)-4-(4-(methylsulfonyl)phenyl)-2-(5H)-furanone], a selective COX-2 inhib-itor,26,27 was kindly provided by Dr Robert Gould of Merck ResearchLaboratories, West Pont, Pa. It was incorporated into a 1% choles-terol chow diet. The drug loading in the chow was 0.0075% wt/wtMF tricyclic, which is equivalent to a daily dose of 15 mg/kg if oneassumes a mouse weight of 25 g and that each mouse consumes 5 gof feed per day. Animals were randomized to either 1% cholesterolchow (ie, vehicle: n�11 males and n�14 females) or 1% cholester-ol/COX-2 inhibitor chow (n�12 males and n�11 females) adlibitum when they were 6 weeks of age.
Egan et al Cyclooxygenases, Thromboxane, and Atherosclerosis 335
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from

Male mice used for the second study, designed to assess theimpact of COX-2 inhibition on TP antagonism in atherosclerosis,were randomized to (1) the TP antagonist alone (1% cholesterolchow and 5 mg/kg S18886 in their drinking water [n�14]) or (2) thecombination regimen (1% cholesterol/COX-2 inhibitor [15 mg/kg]chow and 5 mg/kg S18886 in their drinking water [n�10]) at 6weeks of age. Again, they were euthanized for analysis at 19 weeksof age.
The DKOs develop atherosclerotic lesions spontaneously on aregular chow diet. The third study was designed to assess the abilityof TP antagonism to induce regression of established atheroscleroticdisease. Male DKOs progressed to 16 months of age on a regularchow diet and were then randomized to (1) euthanasia and immediateharvest of their aortas as a baseline control (n�9), (2) initiation oftreatment with the TP antagonist (n�7) S18886 (5 mg/kg/d) for afurther 16 weeks, or (3) no treatment until they were killed (n�8)and vascular harvesting was performed after 16 weeks (to control forlesion progression without intervention).
Lesion Analysis by the En Face MethodThe en face morphometric method quantifies the extent of athero-sclerosis in the entire murine aorta. Significant correlation betweenthe extent of lesions in the entire aorta calculated in this manner andlesion area measured at the aortic origin was demonstrated byTangirala and colleagues34 in both LDLR KO and ApoE KO mice.
Formalin-fixed aortas (from the heart to the iliac bifurcation) werecleaned of adventitial fat, opened longitudinally, and stained withSudan IV (Sigma). Images were photographed and digitized with theImage Pro analysis system (Phase 3 Imaging Systems). The total areaof the aorta and the atherosclerotic plaque area were measured. Thepercent lesion value was calculated as a ratio of the total aorta areato the lesion area for each aorta.
Histological Examination of Lesion MorphologySections (8 �m) of OCT embedded tissue were acetone fixed, andendogenous peroxidase activity was quenched with 0.3% hydrogenperoxide. The sections were then blocked with 20 �g/mL goat IgG(Jackson ImmunoResearch Laboratories Inc) followed by incubationwith 10 �g/mL rat anti-mouse CD16/CD32 to block Fc receptors(Pharmingen, USA). Serial sections were subjected to immunostain-ing with 5 �g/mL rabbit anti-laminin (Sigma), 10 �g/mL FITC-conjugated mouse anti-�-smooth muscle actin (SMA; clone 1A4,Sigma) followed by biotinylated goat anti-rabbit antibody (VectorLaboratories Inc), and biotinylated goat anti-FITC secondary anti-body (Vector Laboratories), respectively. This was followed byVectastain ABC avidin-biotin amplification (Vector Laboratories).The slides were then incubated with VIP (Vector Laboratories) andcounterstained with hematoxylin (Fisher Scientific). Isotype-matched controls were stained in parallel and in all cases showed nosignificant reactivity (data not shown).
Analysis of Cholesterol, Eicosanoids,and IsoeicosanoidsSerum cholesterol and urinary eicosanoids were measured as de-scribed previously.34,35 The urinary 2,3-dinor-TxB2 metabolite wasquantified by a stable isotope dilution reverse-phase (C18) HPLCcoupled tandem mass spectrometry assay, whereas 2,3-dinor-6-ketoPGF1� was measured with gas chromatography/mass spectrometry.A similar approach was taken to the quantification of 8,12-iso-iPF2�-VI, an abundant F2 isoprostane.36,37
Statistical AnalysisData are expressed as mean�SEM. Comparisons of multiple groupswere performed by ANOVA and a Dunn’s post-ANOVA multiplecomparison test when the ANOVA was significant. When only 2mean values were compared, the Student t test was used. Differenceswere considered statistically significant at P�0.05.
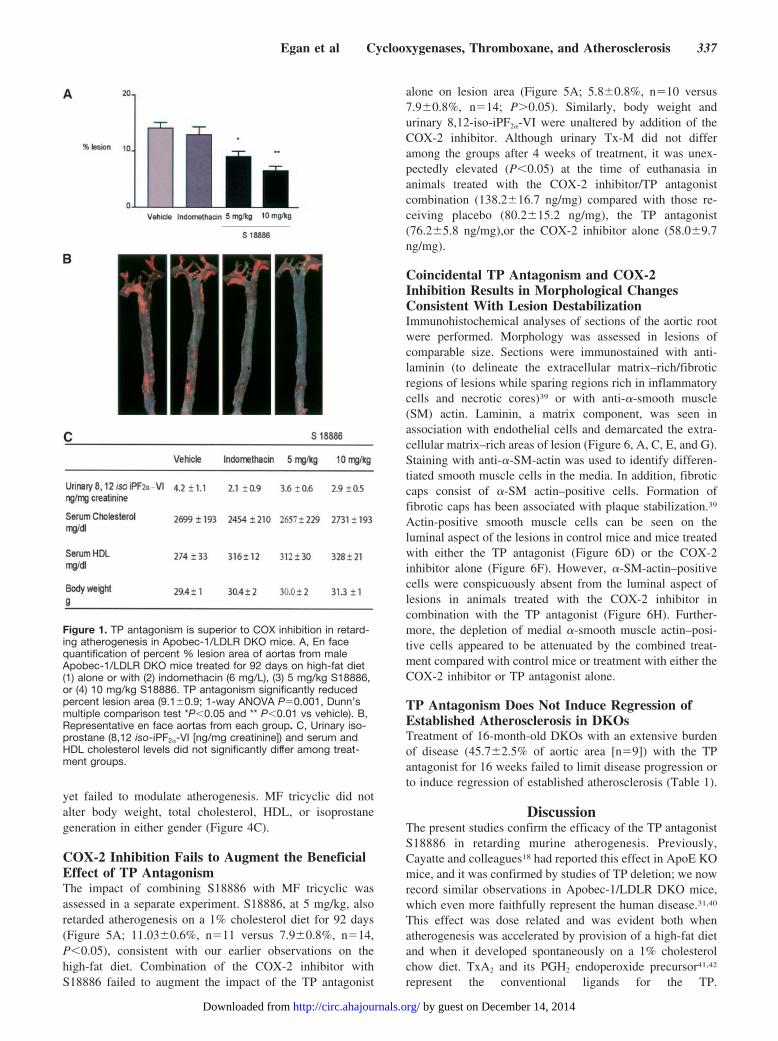
ResultsTP Antagonism, but Not Combined Inhibition ofCOX-1 and COX-2, Retards Atherogenesis inDKOs Fed a High-Fat DietAtherosclerotic lesion area was quantified in aortas harvestedfrom the animals after 92 days (13 weeks) of high-fat feedingin the absence (diet alone) or presence of indomethacin orS18886 to assess the impact of COX inhibition and TPantagonism on atherogenesis. ANOVA of the aortic lesionareas revealed significant (P�0.001) differences. The aver-age lesion area in control animals (diet alone) was14.1�1.0%. Treatment with S18886 at 5 (9.1�0.9%) and 10(6.6�0.8%) mg/kg significantly reduced lesion area in adose-dependent fashion (Figure 1A). Lesion area tended todecline in mice treated with indomethacin (12.9%�1.4), butthis difference did not attain statistical significance. Repre-sentative en face preparations from each group are presentedin Figure 1B. There were no significant differences in serumcholesterol, HDL cholesterol, urinary 8,12-iso-iPF2�-VI(ANOVA P�0.33) or body weight between groups at thetime of euthanasia (Figure 1C).
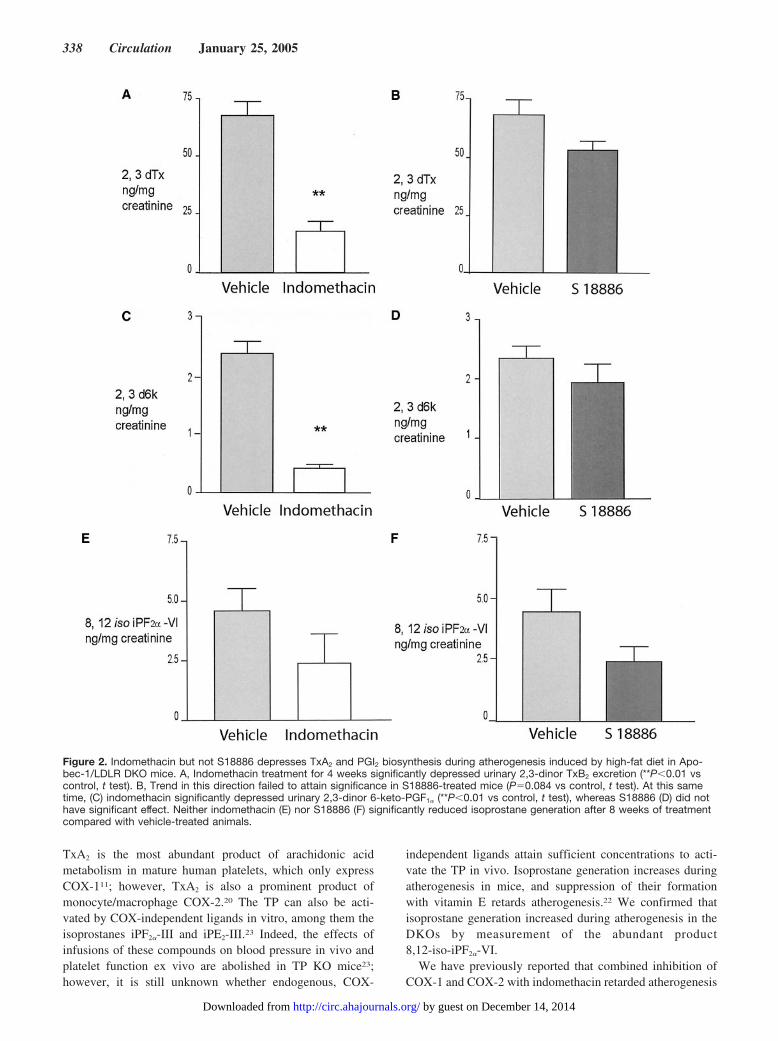
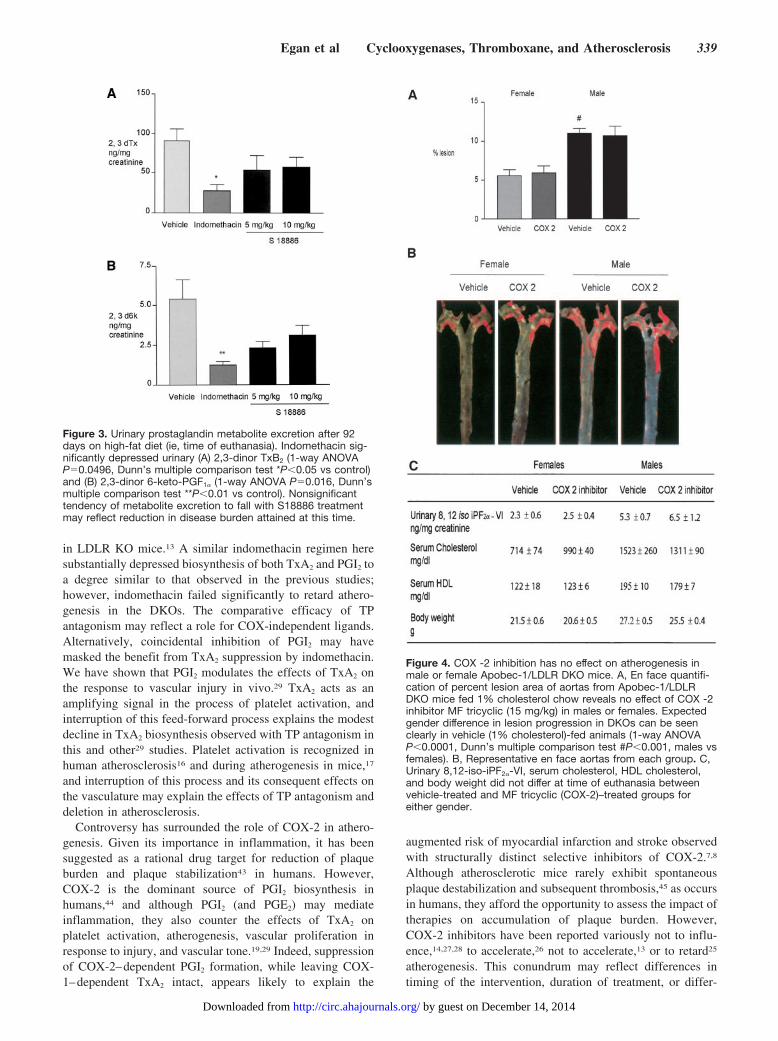
Biosynthesis of TxA2, as reflected by urinary Tx-M,increased during atherogenesis in the DKOs, as we previouslydescribed in ApoE and LDLR KO mice.35 Indomethacinreduced Tx-M significantly by roughly 70% from 68.1�6.4to 18.0�2.5 ng/mg (P�0.01) after 4 weeks’ feeding (Figure2A), and this effect was maintained at 8 weeks (data notshown). Although Tx-M tended to decline with the antago-nist, this difference (Figure 2B) failed to attain statisticalsignificance (P�0.08). Indomethacin alone maintained asignificant suppression of Tx-M excretion at 92 days oftreatment, immediately before animals were euthanized (Fig-ure 3A). A tendency to a reduction of Tx-M with S18886probably reflects an interruption of secondary formation ofTxA2 by activated platelets or the impact of the treatment onlesion burden and secondary platelet activation.29,38
Urinary PGI-M also increased during atherogenesis, as hadbeen observed in the other mouse models.35 Similarly, indo-methacin suppressed PGI-M excretion from 2.4�0.2 to0.61�0.2 ng/mg (P�0.01) at 4 weeks of feeding (Figure 2C),an effect retained at 8 weeks (data not shown). Animalstreated with S18886 failed to reduce PGI-M significantly(Figure 2F); however, as with Tx-M, a trend in that direction,which still failed to attain significance, was also evident attime of sacrifice (Figure 3B). Excretion of 8,12-iso-iPF2�-VIalso increased with progression of atherosclerosis. Neitherindomethacin nor S18886 significantly depressed isoprostanegeneration, although it tended to decline with both therapies.
Inhibition of COX-2 Fails to ModulateSpontaneous Atherogenesis in DKOsAs expected, male mice developed atherosclerosis signifi-cantly faster than females (Figure 4A), attaining 11.03�0.6%coverage of the aorta of males versus 5.57�0.8% in females(Figure 4B) in 1% cholesterol-fed mice at 19 weeks of age(P�0.001). Selective inhibition of COX-2 significantly de-pressed PGI-M excretion from 4.3�0.4 to 2.6�0.5 ng/mg(P�0.05) but not Tx-M (80.2�15.2 versus 58.0�9.7 ng/mg),
336 Circulation January 25, 2005
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from
yet failed to modulate atherogenesis. MF tricyclic did notalter body weight, total cholesterol, HDL, or isoprostanegeneration in either gender (Figure 4C).
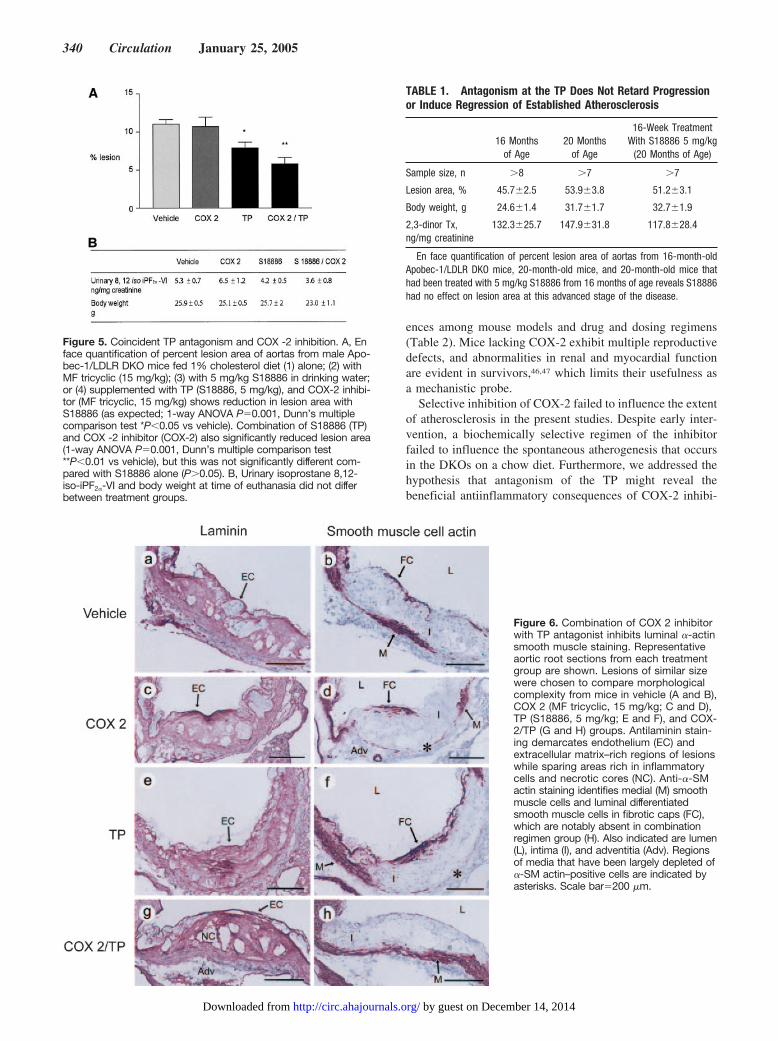
COX-2 Inhibition Fails to Augment the BeneficialEffect of TP AntagonismThe impact of combining S18886 with MF tricyclic wasassessed in a separate experiment. S18886, at 5 mg/kg, alsoretarded atherogenesis on a 1% cholesterol diet for 92 days(Figure 5A; 11.03�0.6%, n�11 versus 7.9�0.8%, n�14,P�0.05), consistent with our earlier observations on thehigh-fat diet. Combination of the COX-2 inhibitor withS18886 failed to augment the impact of the TP antagonist
alone on lesion area (Figure 5A; 5.8�0.8%, n�10 versus7.9�0.8%, n�14; P�0.05). Similarly, body weight andurinary 8,12-iso-iPF2�-VI were unaltered by addition of theCOX-2 inhibitor. Although urinary Tx-M did not differamong the groups after 4 weeks of treatment, it was unex-pectedly elevated (P�0.05) at the time of euthanasia inanimals treated with the COX-2 inhibitor/TP antagonistcombination (138.2�16.7 ng/mg) compared with those re-ceiving placebo (80.2�15.2 ng/mg), the TP antagonist(76.2�5.8 ng/mg),or the COX-2 inhibitor alone (58.0�9.7ng/mg).
Coincidental TP Antagonism and COX-2Inhibition Results in Morphological ChangesConsistent With Lesion DestabilizationImmunohistochemical analyses of sections of the aortic rootwere performed. Morphology was assessed in lesions ofcomparable size. Sections were immunostained with anti-laminin (to delineate the extracellular matrix–rich/fibroticregions of lesions while sparing regions rich in inflammatorycells and necrotic cores)39 or with anti-�-smooth muscle(SM) actin. Laminin, a matrix component, was seen inassociation with endothelial cells and demarcated the extra-cellular matrix–rich areas of lesion (Figure 6, A, C, E, and G).Staining with anti-�-SM-actin was used to identify differen-tiated smooth muscle cells in the media. In addition, fibroticcaps consist of �-SM actin–positive cells. Formation offibrotic caps has been associated with plaque stabilization.39
Actin-positive smooth muscle cells can be seen on theluminal aspect of the lesions in control mice and mice treatedwith either the TP antagonist (Figure 6D) or the COX-2inhibitor alone (Figure 6F). However, �-SM-actin–positivecells were conspicuously absent from the luminal aspect oflesions in animals treated with the COX-2 inhibitor incombination with the TP antagonist (Figure 6H). Further-more, the depletion of medial �-smooth muscle actin–posi-tive cells appeared to be attenuated by the combined treat-ment compared with control mice or treatment with either theCOX-2 inhibitor or TP antagonist alone.
TP Antagonism Does Not Induce Regression ofEstablished Atherosclerosis in DKOsTreatment of 16-month-old DKOs with an extensive burdenof disease (45.7�2.5% of aortic area [n�9]) with the TPantagonist for 16 weeks failed to limit disease progression orto induce regression of established atherosclerosis (Table 1).
DiscussionThe present studies confirm the efficacy of the TP antagonistS18886 in retarding murine atherogenesis. Previously,Cayatte and colleagues18 had reported this effect in ApoE KOmice, and it was confirmed by studies of TP deletion; we nowrecord similar observations in Apobec-1/LDLR DKO mice,which even more faithfully represent the human disease.31,40
This effect was dose related and was evident both whenatherogenesis was accelerated by provision of a high-fat dietand when it developed spontaneously on a 1% cholesterolchow diet. TxA2 and its PGH2 endoperoxide precursor41,42
represent the conventional ligands for the TP.
Figure 1. TP antagonism is superior to COX inhibition in retard-ing atherogenesis in Apobec-1/LDLR DKO mice. A, En facequantification of percent % lesion area of aortas from maleApobec-1/LDLR DKO mice treated for 92 days on high-fat diet(1) alone or with (2) indomethacin (6 mg/L), (3) 5 mg/kg S18886,or (4) 10 mg/kg S18886. TP antagonism significantly reducedpercent lesion area (9.1�0.9; 1-way ANOVA P�0.001, Dunn’smultiple comparison test *P�0.05 and ** P�0.01 vs vehicle). B,Representative en face aortas from each group. C, Urinary iso-prostane (8,12 iso-iPF2�-VI [ng/mg creatinine]) and serum andHDL cholesterol levels did not significantly differ among treat-ment groups.
Egan et al Cyclooxygenases, Thromboxane, and Atherosclerosis 337
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from
TxA2 is the most abundant product of arachidonic acidmetabolism in mature human platelets, which only expressCOX-111; however, TxA2 is also a prominent product ofmonocyte/macrophage COX-2.20 The TP can also be acti-vated by COX-independent ligands in vitro, among them theisoprostanes iPF2�-III and iPE2-III.23 Indeed, the effects ofinfusions of these compounds on blood pressure in vivo andplatelet function ex vivo are abolished in TP KO mice23;however, it is still unknown whether endogenous, COX-
independent ligands attain sufficient concentrations to acti-vate the TP in vivo. Isoprostane generation increases duringatherogenesis in mice, and suppression of their formationwith vitamin E retards atherogenesis.22 We confirmed thatisoprostane generation increased during atherogenesis in theDKOs by measurement of the abundant product8,12-iso-iPF2�-VI.
We have previously reported that combined inhibition ofCOX-1 and COX-2 with indomethacin retarded atherogenesis
Figure 2. Indomethacin but not S18886 depresses TxA2 and PGI2 biosynthesis during atherogenesis induced by high-fat diet in Apo-bec-1/LDLR DKO mice. A, Indomethacin treatment for 4 weeks significantly depressed urinary 2,3-dinor TxB2 excretion (**P�0.01 vscontrol, t test). B, Trend in this direction failed to attain significance in S18886-treated mice (P�0.084 vs control, t test). At this sametime, (C) indomethacin significantly depressed urinary 2,3-dinor 6-keto-PGF1� (**P�0.01 vs control, t test), whereas S18886 (D) did nothave significant effect. Neither indomethacin (E) nor S18886 (F) significantly reduced isoprostane generation after 8 weeks of treatmentcompared with vehicle-treated animals.
338 Circulation January 25, 2005
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from
in LDLR KO mice.13 A similar indomethacin regimen heresubstantially depressed biosynthesis of both TxA2 and PGI2 toa degree similar to that observed in the previous studies;however, indomethacin failed significantly to retard athero-genesis in the DKOs. The comparative efficacy of TPantagonism may reflect a role for COX-independent ligands.Alternatively, coincidental inhibition of PGI2 may havemasked the benefit from TxA2 suppression by indomethacin.We have shown that PGI2 modulates the effects of TxA2 onthe response to vascular injury in vivo.29 TxA2 acts as anamplifying signal in the process of platelet activation, andinterruption of this feed-forward process explains the modestdecline in TxA2 biosynthesis observed with TP antagonism inthis and other29 studies. Platelet activation is recognized inhuman atherosclerosis16 and during atherogenesis in mice,17
and interruption of this process and its consequent effects onthe vasculature may explain the effects of TP antagonism anddeletion in atherosclerosis.
Controversy has surrounded the role of COX-2 in athero-genesis. Given its importance in inflammation, it has beensuggested as a rational drug target for reduction of plaqueburden and plaque stabilization43 in humans. However,COX-2 is the dominant source of PGI2 biosynthesis inhumans,44 and although PGI2 (and PGE2) may mediateinflammation, they also counter the effects of TxA2 onplatelet activation, atherogenesis, vascular proliferation inresponse to injury, and vascular tone.19,29 Indeed, suppressionof COX-2–dependent PGI2 formation, while leaving COX-1–dependent TxA2 intact, appears likely to explain the
augmented risk of myocardial infarction and stroke observedwith structurally distinct selective inhibitors of COX-2.7,8
Although atherosclerotic mice rarely exhibit spontaneousplaque destabilization and subsequent thrombosis,45 as occursin humans, they afford the opportunity to assess the impact oftherapies on accumulation of plaque burden. However,COX-2 inhibitors have been reported variously not to influ-ence,14,27,28 to accelerate,26 not to accelerate,13 or to retard25
atherogenesis. This conundrum may reflect differences intiming of the intervention, duration of treatment, or differ-
Figure 3. Urinary prostaglandin metabolite excretion after 92days on high-fat diet (ie, time of euthanasia). Indomethacin sig-nificantly depressed urinary (A) 2,3-dinor TxB2 (1-way ANOVAP�0.0496, Dunn’s multiple comparison test *P�0.05 vs control)and (B) 2,3-dinor 6-keto-PGF1� (1-way ANOVA P�0.016, Dunn’smultiple comparison test **P�0.01 vs control). Nonsignificanttendency of metabolite excretion to fall with S18886 treatmentmay reflect reduction in disease burden attained at this time.
Figure 4. COX -2 inhibition has no effect on atherogenesis inmale or female Apobec-1/LDLR DKO mice. A, En face quantifi-cation of percent lesion area of aortas from Apobec-1/LDLRDKO mice fed 1% cholesterol chow reveals no effect of COX -2inhibitor MF tricyclic (15 mg/kg) in males or females. Expectedgender difference in lesion progression in DKOs can be seenclearly in vehicle (1% cholesterol)-fed animals (1-way ANOVAP�0.0001, Dunn’s multiple comparison test #P�0.001, males vsfemales). B, Representative en face aortas from each group. C,Urinary 8,12-iso-iPF2�-VI, serum cholesterol, HDL cholesterol,and body weight did not differ at time of euthanasia betweenvehicle-treated and MF tricyclic (COX-2)–treated groups foreither gender.
Egan et al Cyclooxygenases, Thromboxane, and Atherosclerosis 339
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from
ences among mouse models and drug and dosing regimens(Table 2). Mice lacking COX-2 exhibit multiple reproductivedefects, and abnormalities in renal and myocardial functionare evident in survivors,46,47 which limits their usefulness asa mechanistic probe.
Selective inhibition of COX-2 failed to influence the extentof atherosclerosis in the present studies. Despite early inter-vention, a biochemically selective regimen of the inhibitorfailed to influence the spontaneous atherogenesis that occursin the DKOs on a chow diet. Furthermore, we addressed thehypothesis that antagonism of the TP might reveal thebeneficial antiinflammatory consequences of COX-2 inhibi-
Figure 5. Coincident TP antagonism and COX -2 inhibition. A, Enface quantification of percent lesion area of aortas from male Apo-bec-1/LDLR DKO mice fed 1% cholesterol diet (1) alone; (2) withMF tricyclic (15 mg/kg); (3) with 5 mg/kg S18886 in drinking water;or (4) supplemented with TP (S18886, 5 mg/kg), and COX-2 inhibi-tor (MF tricyclic, 15 mg/kg) shows reduction in lesion area withS18886 (as expected; 1-way ANOVA P�0.001, Dunn’s multiplecomparison test *P�0.05 vs vehicle). Combination of S18886 (TP)and COX -2 inhibitor (COX-2) also significantly reduced lesion area(1-way ANOVA P�0.001, Dunn’s multiple comparison test**P�0.01 vs vehicle), but this was not significantly different com-pared with S18886 alone (P�0.05). B, Urinary isoprostane 8,12-iso-iPF2�-VI and body weight at time of euthanasia did not differbetween treatment groups.
Figure 6. Combination of COX 2 inhibitorwith TP antagonist inhibits luminal �-actinsmooth muscle staining. Representativeaortic root sections from each treatmentgroup are shown. Lesions of similar sizewere chosen to compare morphologicalcomplexity from mice in vehicle (A and B),COX 2 (MF tricyclic, 15 mg/kg; C and D),TP (S18886, 5 mg/kg; E and F), and COX-2/TP (G and H) groups. Antilaminin stain-ing demarcates endothelium (EC) andextracellular matrix–rich regions of lesionswhile sparing areas rich in inflammatorycells and necrotic cores (NC). Anti-�-SMactin staining identifies medial (M) smoothmuscle cells and luminal differentiatedsmooth muscle cells in fibrotic caps (FC),which are notably absent in combinationregimen group (H). Also indicated are lumen(L), intima (I), and adventitia (Adv). Regionsof media that have been largely depleted of�-SM actin–positive cells are indicated byasterisks. Scale bar�200 �m.
TABLE 1. Antagonism at the TP Does Not Retard Progressionor Induce Regression of Established Atherosclerosis
16 Monthsof Age
20 Monthsof Age
16-Week TreatmentWith S18886 5 mg/kg
(20 Months of Age)
Sample size, n �8 �7 �7
Lesion area, % 45.7�2.5 53.9�3.8 51.2�3.1
Body weight, g 24.6�1.4 31.7�1.7 32.7�1.9
2,3-dinor Tx,ng/mg creatinine
132.3�25.7 147.9�31.8 117.8�28.4
En face quantification of percent lesion area of aortas from 16-month-oldApobec-1/LDLR DKO mice, 20-month-old mice, and 20-month-old mice thathad been treated with 5 mg/kg S18886 from 16 months of age reveals S18886had no effect on lesion area at this advanced stage of the disease.
340 Circulation January 25, 2005
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from

tion. This is rendered all the more topical given the recentannouncement48 of a clinical trial designed to identify acardioprotective effect of the COX-2 inhibitor celecoxib inosteoarthritis patients with high risk of cardiovascular dis-ease. These patients will be taking low-dose aspirin tosuppress platelet COX-derived TxA2, a strategy that is sim-ulated by combination of the COX-2 inhibitor with the TPantagonist in the present studies.
TP antagonism delayed spontaneous atherogenesis in theDKOs, as it had when lesion formation was accelerated by ahigh-fat diet; however, addition of the COX-2 inhibitor failed toaugment the impact of TP antagonism on atherogenesis. Instead,it had 2 unexpected effects. First, biosynthesis of TxA2 wasaugmented, which perhaps reflects removal of a constraint onplatelet activation by suppression of PGI2.29 Second, althoughplaque burden was not increased compared with treatment withthe antagonist alone, lesion morphology was notably altered.Lesions contained more necrotic cores (Figure 6G), and al-though little if any luminal �-SM-actin staining (Figure 6H) wasevident to suggest that “cap” formation was inhibited, thereduction in medial �-SM actin–positive cells appeared to beless severe. Fibrotic caps can presumably be formed by themigration of actin-positive smooth muscle cells from the media.Alternatively, cells in the fibrotic cap may be derived from thedifferentiation of intimal cells that lack actin expression or fromthe redifferentiation of smooth muscle cells initially derivedfrom the media that downregulate expression of actin in associ-ation with their migration into the neointima. In either case, thelack of fibrotic caps suggests that the combined treatment withCOX-2 inhibitor and TP antagonist results in plaque destabili-zation. In view of these results, it will be of interest further toinvestigate the role of COX-2 and the TP in regulating smoothmuscle cell migration and phenotypic differentiation.
In the final study, we assessed the impact of TP antagonismon established atherosclerosis. Prolonged treatment of micewith extensive atherosclerosis that comprised �50% of aorticarea for up to 16 weeks failed to delay progression of diseaseor to induce regression of atherosclerosis.
In summary, we have provided further evidence that activa-tion of the TP promotes initiation and early development ofatherosclerotic lesions in mice. This may involve receptorligation both by conventional ligands, such as platelet COX-1–derived TxA2, and COX-independent ligands, such as the iso-prostanes. Indeed, this may occur in an integrated fashion,because TP ligation may activate NADPH oxidase and conse-quent free radical generation.49 Experience in this model sug-gests that clinical use of TP antagonists would be expected to beuseful in the earlier stages of disease, rather than in reversing
accumulated plaque burden in patients with diffuse, establishedatherosclerosis. Even early intervention with a selective inhibitorof COX-2 failed to modulate the modest, spontaneous athero-genesis that develops on a chow diet in this model or to augmentthe beneficial impact of a TP antagonist. Indeed, because TPantagonism may mimic the effect of low-dose aspirin, thesestudies suggest that coincidental treatment with a COX-2 inhib-itor may undermine the benefit of aspirin by predisposingatherosclerotic plaques to destabilization with subsequentvasoocclusive thrombosis. Given the clinical experience withrofeocoxib7 and valdecoxib,8 the impact of low-dose aspirin ongastroprotection in patients taking COX-2 inhibitors,50 and ourpresent findings on plaque morphology, the wisdom of conduct-ing trials to seek a cardiovascular benefit from selective inhibi-tors of COX-2 in atherothrombotic disease appears questionable.
AcknowledgmentsThese studies were supported by grants from the National Institutesof Health (HL545000, HL62250, and HL70121) and a TobaccoFormula Grant, Servier Laboratories, and by Merck Research Lab-oratories. Dr FitzGerald is the Robinette Professor of CardiovascularMedicine and the Elmer Bobst Professor of Pharmacology.
References1. Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med. 1999;
340:115–126.2. Hinson RM, Williams JA, Shacter E. Elevated interleukin 6 is induced by
prostaglandin E2 in a murine model of inflammation: possible role ofcyclooxygenase-2. Proc Natl Acad Sci U S A. 1996;93:4885–4890.
3. Murata T, Ushikubi F, Matsuoka T, Hirata M, Yamasaki A, Sugimoto Y,Ichikawa A, Aze Y, Tanaka T, Yoshida N, Ueno A, Oh-ishi S, NarumiyaS. Altered pain perception and inflammatory response in mice lackingprostacyclin receptor. Nature. 1997;388:678–682.
4. Crofford LJ, Wilder RL, Ristimaki AP, Sano H, Remmers EF, Epps HR,Hla T. Cyclooxygenase-1 and -2 expression in rheumatoid synovialtissues: effects of interleukin-1 beta, phorbol ester, and corticosteroids.J Clin Invest. 1994;93:1095–1101.
5. Schonbeck U, Sukhova GK, Graber P, Coulter S, Libby P. Augmentedexpression of cyclooxygenase-2 in human atherosclerotic lesions. Am JPathol. 1999;155:1281–1291.
6. Baker CS, Hall RJ, Evans TJ, Pomerance A, Maclouf J, Creminon C,Yacoub MH, Polak JM. Cyclooxygenase-2 is widely expressed in ath-erosclerotic lesions affecting native and transplanted human coronaryarteries and colocalizes with inducible nitric oxide synthase and nitroty-rosine particularly in macrophages. Arterioscler Thromb Vasc Biol. 1999;19:646–655.
7. Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709–1711.
8. Furburg CD, Psaty BM, FitzGerald GA. Parecoxib, valdecoxib, andcardiovascular risk. Circulation. 2005;111:249.
9. Shankavaram UT, Lai WC, Netzel-Arnett S, Mangan PR, Ardans JA,Caterina N, Stetler-Stevenson WG, Birkedal-Hansen H, Wahl LM.Monocyte membrane type 1-matrix metalloproteinase: prostaglandin-dependent regulation and role in metalloproteinase-2 activation. J BiolChem. 2001;276:19027–19032.
TABLE 2. Divergent Effects of COX-2 Inhibitors in Mouse Atherosclerosis
Mouse Model COX-2 Inhibitor Initiation Duration, wk Diet Phase of Disease Effect on Lesion Area Reference
ApoE KO MF tricyclic at 8 weeks of age 3 Regular chow Very early Increase 26
LDLR KO Rofecoxib at 10 weeks of age 6 High fat Early Decrease 25
LDLR KO Nimesulide at 8 weeks of age 18 High fat Intermediate No increase 13
ApoE KO SC-236 at 7 weeks of age 8 1% Cholesterol Intermediate-advanced No effect 14
ApoE KO MF tricyclic at 24 weeks of age 16 Regular chow Advanced No effect 27
ApoE KO Celecoxib at 26 weeks of age 15 Regular chow Advanced No effect 28
Egan et al Cyclooxygenases, Thromboxane, and Atherosclerosis 341
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from

10. Cipollone F, Fazia M, Iezzi A, Zucchelli M, Pini B, De Cesare D,Ucchino S, Spigonardo F, Bajocchi G, Bei R, Muraro R, Artese L,Piattelli A, Chiarelli F, Cuccurullo F, Mezzetti A. Suppression of thefunctionally coupled cyclooxygenase-2/prostaglandin E synthase as abasis of simvastatin-dependent plaque stabilization in humans. Circu-lation. 2003;107:1479–1485.
11. FitzGerald GA. Mechanisms of platelet activation: thromboxane A2 as anamplifying signal for other agonists. Am J Cardiol. 1991;68:11B–15B.
12. Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis ofrandomised trials of antiplatelet therapy for prevention of death, myo-cardial infarction, and stroke in high risk patients [published correctionappears in BMJ. 2002;324:141]. BMJ. 2002;324:71–86.
13. Pratico D, Tillmann C, Zhang ZB, Li H, FitzGerald GA. Acceleration ofatherogenesis by COX-1-dependent prostanoid formation in low densitylipoprotein receptor knockout mice. Proc Natl Acad Sci U S A. 2001;98:3358–3363.
14. Belton OA, Duffy A, Toomey S, Fitzgerald DJ. Cyclooxygenase isoformsand platelet vessel wall interactions in the apolipoprotein E knockoutmouse model of atherosclerosis. Circulation. 2003;108:3017–3023.
15. McClelland S, Toomey S, Hahren B, Fitzgerald DJ, Belton OA. Cyclo-oxygenase-1 gene deletion inhibits atherosclerosis in the ApoE�/� mousemodel. Arterioscler Thromb Vasc Biol. 2004;24:e73. Abstract..
16. FitzGerald GA, Smith B, Pedersen AK, Brash AR. Increased prostacyclinbiosynthesis in patients with severe atherosclerosis and platelet activation.N Engl J Med. 1984;310:1065–1068.
17. Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, BergmeierW, Richter T, Lorenz M, Konrad I, Nieswandt B, Gawaz M. A criticalrole of platelet adhesion in the initiation of atherosclerotic lesion for-mation. J Exp Med. 2002;196:887–896.
18. Cayatte AJ, Du Y, Oliver-Krasinski J, Lavielle G, Verbeuren TJ, CohenRA. The thromboxane receptor antagonist S18886 but not aspirin inhibitsatherogenesis in apo E-deficient mice: evidence that eicosanoids otherthan thromboxane contribute to atherosclerosis. Arterioscler ThrombVasc Biol. 2000;20:1724–1728.
19. Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T,Arai H, Oida H, Yurugi-Kobayashi T, Yamashita JK, Katagiri H, MajimaM, Yokode M, Kita T, Narumiya S. Roles of thromboxane A(2) andprostacyclin in the development of atherosclerosis in apoE-deficient mice.J Clin Invest. 2004;114:784–794.
20. Fu JY, Masferrer JL, Seibert K, Raz A, Needleman P. The induction andsuppression of prostaglandin H2 synthase (cyclooxygenase) in humanmonocytes. J Biol Chem. 1990;265:16737–16740.
21. Pratico D, Iuliano L, Mauriello A, Spagnoli L, Lawson JA, Rokach J, MacloufJ, Violi F, FitzGerald GA. Localization of distinct F2-isoprostanes in humanatherosclerotic lesions. J Clin Invest. 1997;100:2028–2034.
22. Pratico D, Tangirala RK, Rader DJ, Rokach J, FitzGerald GA. Vitamin Esuppresses isoprostane generation in vivo and reduces atherosclerosis inApoE-deficient mice. Nat Med. 1998;4:1189–1192.
23. Audoly LP, Rocca B, Fabre JE, Koller BH, Thomas D, Loeb AL,Coffman TM, FitzGerald GA. Cardiovascular responses to the iso-prostanes iPF(2alpha)-III and iPE(2)-III are mediated via thethromboxane A(2) receptor in vivo. Circulation. 2000;101:2833–2840.
24. FitzGerald GA. COX-2 and beyond: approaches to prostaglandin inhi-bition in human disease. Nat Rev Drug Discov. 2003;2:879–890.
25. Burleigh ME, Babaev VR, Oates JA, Harris RC, Gautam S, Riendeau D,Marnett LJ, Morrow JD, Fazio S, Linton MF. Cyclooxygenase-2promotes early atherosclerotic lesion formation in LDL receptor-deficientmice. Circulation. 2002;105:1816–1823.
26. Rott D, Zhu J, Burnett MS, Zhou YF, Zalles-Ganley A, Ogunmakinwa J, EpsteinSE. Effects of MF-tricyclic, a selective cyclooxygenase-2 inhibitor, on athero-sclerosis progression and susceptibility to cytomegalovirus replication inapolipoprotein-E knockout mice. J Am Coll Cardiol. 2003;41:1812–1819.
27. Olesen M, Kwong E, Meztli A, Kontny F, Seljeflot I, Arnesen H, LyngdorfL, Falk E. No effect of cyclooxygenase inhibition on plaque size in athero-sclerosis-prone mice. Scand Cardiovasc J. 2002;36:362–367.
28. Bea F, Blessing E, Bennett BJ, Kuo CC, Campbell LA, Kreuzer J,Rosenfeld ME. Chronic inhibition of cyclooxygenase-2 does not alterplaque composition in a mouse model of advanced unstable atheroscle-rosis. Cardiovasc Res. 2003;60:198–204.
29. Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T,Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascularresponse to thromboxane A2. Science. 2002;296:539–541.
30. Fabre JE, Nguyen M, Athirakul K, Coggins K, McNeish JD, Austin S,Parise LK, FitzGerald GA, Coffman TM, Koller BH. Activation of the
murine EP3 receptor for PGE2 inhibits cAMP production and promotesplatelet aggregation. J Clin Invest. 2001;107:603–610.
31. Powell-Braxton L, Veniant M, Latvala RD, Hirano KI, Won WB, Ross J,Dybdal N, Zlot CH, Young SG, Davidson NO. A mouse model of humanfamilial hypercholesterolemia: markedly elevated low density lipoproteincholesterol levels and severe atherosclerosis on a low-fat chow diet. NatMed. 1998;4:934–938.
32. Simonet S, Descombes JJ, Vallez MO, Dubuffet T, Lavielle G, VerbeurenTJ. S 18886, a new thromboxane (TP)-receptor antagonist is the activeisomer of S 18204 in all species, except in the guinea-pig. Adv Exp MedBiol. 1997;433:173–176.
33. Cimetiere B, Dubuffet T, Muller O, Descombes JJ, Simonet S, Laubie M,Verbeuren TJ, Lavielle G. Synthesis and biological evaluation of newtetrahydronaphthalene derivatives as thromboxane receptor antagonists.Bioorg Med Chem Lett. 1998;8:1375–1380.
34. Tangirala RK, Rubin EM, Palinski W. Quantitation of atherosclerosis in murinemodels: correlation between lesions in the aortic origin and in the entire aorta, anddifferences in the extent of lesions between sexes in LDL receptor-deficient andapolipoprotein E-deficient mice. J Lipid Res. 1995;36:2320–2328.
35. Pratico D, Cyrus T, Li H, FitzGerald GA. Endogenous biosynthesis ofthromboxane and prostacyclin in 2 distinct murine models of atheroscle-rosis. Blood. 2000;96:3823–3826.
36. Lawson JA, Li H, Rokach J, Adiyaman M, Hwang SW, Khanapure SP,FitzGerald GA. Identification of two major F2 isoprostanes, 8,12-iso- and5-epi-8,12-iso-isoprostane F2alpha-VI, in human urine. J Biol Chem.1998;273:29295–29301.
37. Zhang Z, Vezza R, Plappert T, McNamara P, Lawson JA, Austin S,Pratico D, Sutton MS, FitzGerald GA. COX-2-dependent cardiac failurein Gh/tTG transgenic mice. Circ Res. 2003;92:1153–1161.
38. Fitzgerald DJ, Doran J, Jackson E, FitzGerald GA. Coronary vascularocclusion mediated via thromboxane A2-prostaglandin endoperoxidereceptor activation in vivo. J Clin Invest. 1986;77:496–502.
39. Cuff CA, Kothapalli D, Azonobi I, Chun S, Zhang Y, Belkin R, Yeh C,Secreto A, Assoian RK, Rader DJ, Pure E. The adhesion receptor CD44promotes atherosclerosis by mediating inflammatory cell recruitment andvascular cell activation. J Clin Invest. 2001;108:1031–1040.
40. Rader DJ, FitzGerald GA. State of the art: atherosclerosis in a limitededition. Nat Med. 1998;4:899–900.
41. Hamberg M, Svensson J, Wakabayashi T, Samuelsson B. Isolation andstructure of two prostaglandin endoperoxides that cause platelet aggre-gation. Proc Natl Acad Sci U S A. 1974;71:345–349.
42. Hornby EJ, Skidmore IF. Evidence that prostaglandin endoperoxides caninduce platelet aggregation in the absence of thromboxane A2 production.Biochem Pharmacol. 1982;31:1158–1160.
43. Cipollone F, Rocca B, Patrono C. Cyclooxygenase-2 expression andinhibition in atherothrombosis. Arterioscler Thromb Vasc Biol. 2004;24:246–255.
44. McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA,FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase(COX)-2: the human pharmacology of a selective inhibitor of COX-2.Proc Natl Acad Sci U S A. 1999;96:272–277.
45. Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein E–deficientmouse: a decade of progress. Arterioscler Thromb Vasc Biol. 2004;24:1006–1014.
46. Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N,Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, Smithies O.Prostaglandin synthase 2 gene disruption causes severe renal pathology inthe mouse. Cell. 1995;83:473–482.
47. Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB,Contel NR, Eng VM, Collins RJ, Czerniak PM, Gorry SA, Trzaskos JM.Renal abnormalities and an altered inflammatory response in micelacking cyclooxygenase II. Nature. 1995;378:406–409.
48. Pollack A. A new trial of Celebrex, and questions on its timing. New YorkTimes. October 19, 2004;sect C:1.
49. Muzaffar S, Shukla N, Lobo C, Angelini GD, Jeremy JY. Iloprost inhibitssuperoxide formation and gp91phox expression induced by thethromboxane A2 analogue U46619, 8-isoprostane F2alpha, prostaglandinF2alpha, cytokines and endotoxin in the pig pulmonary artery. Br JPharmacol. 2004;141:488–496.
50. Schnitzer TJ, Burmester GR, Mysler E, Hochberg MC, Doherty M,Ehrsam E, Gitton X, Krammer G, Mellein B, Matchaba P, Gimona A,Hawkey CJ. Comparison of lumiracoxib with naproxen and ibuprofen inthe Therapeutic Arthritis Research and Gastrointestinal Event Trial(TARGET), reduction in ulcer complications: randomised controlled trial.Lancet. 2004;364:665–674.
342 Circulation January 25, 2005
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from

Because of an oversight, the name of Susanne Fries, MD, was excluded from the author list of thepaper by Egan et al, “Cyclooxygenases, Thromboxane, and Atherosclerosis: Plaque Destabiliza-tion by Cyclooxygenase-2 Inhibition Combined With Thromboxane Receptor Antagonism,” thatappeared in the January 25, 2005, issue of Circulation (Circulation. 2005;111:334–342).
DOI: 10.1161/CIRCULATIONAHA.105.5755
2412
Correction

and Garret A. FitzGeraldKarine M. Egan, Miao Wang, Margaret B. Lucitt, Alicia M. Zukas, Ellen Puré, John A. Lawson
Cyclooxygenase-2 Inhibition Combined With Thromboxane Receptor AntagonismCyclooxygenases, Thromboxane, and Atherosclerosis: Plaque Destabilization by
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2005 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/01.CIR.0000153386.95356.78
2005;111:334-342; originally published online January 17, 2005;Circulation.
http://circ.ahajournals.org/content/111/3/334World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/111/18/2412.full.pdfAn erratum has been published regarding this article. Please see the attached page for:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on December 14, 2014http://circ.ahajournals.org/Downloaded from