idevelopmentall pathology - ncbi
TRANSCRIPT

TeachingMonograph
IDevelopmentallPathologyRobert P. Bolande, MD
Professor of Pathology and PediatricsMcGill UniversityDirector of PathologyMontreal Children's HospitalMontreal, QuebecCanada

Copyright 0 1979, Universities Associated for Research and Education in Pathology, Inc.All rights reserved
The American Journal of PathologyOfficial publication of The American Association of Pathologists
Published by The American Association of Pathologists,9650 Rockvllle Pike, Bethesda, Maryland

DEVELOPMENTAL PATHOLOGY
NOMa Mo @RPhWn.ik PrOCSSS 627(1)The Embryo 627(1)The Fetus and Newborn 629(3)
Recton to Iniwy 630(4)Susceptibility of Developing Cells 630(4)Anoxia, Hypoperfusion, and Ischemic Injury 631(5)Drugs 634(8)Infection 635(9)The Inflammatory Response 637(11)
Acute Inflammation 637(11 )Chronic Inflammation and Repair 638(12)
Tata n 639(13)Congenital Defects as Reaction to Injurv 639(13)Incidence and Morbidity 640(14)Types of Anomalies and Their Pathologic Effects 641(15)Etiology 642(16)
Environmental teratogenesis 643(17)Human teratogens 644(18)Genetic factors 647(21)
Inbon Abnnaltis of Cd FUncto 650(24)
NopLsia 652(26)Definitions and Descriptions 653(27)Hamartomas 653(27)Teratomas 653(27)Embryomas 657(31)Leukemia 669(43)Lymphomas 669(43)Reticuloendotheliosis (Histiocytosis X) 670(44)The Fibromatoses of Infancy 670(44)
Benignity of Neonatal and Infantile Tumors 671(45)
Rdationships of Noas and Teatogens 672(46)Origin of Tumors in Dysplastic or Anomalous Tissue 672(46)Malignant Transformation in Hamartomas 674(48)Increased Expectancy of Neoplasms in Specific Teratologic Conditions 674(48)
Aniridia-Wilms' Tumor 674(48)Hemihypertrophy 675(49)Omphalocele-Macroglossia Syndrome (Beckwith-Wiedemann Syndrome) 676(50)Basal Cell Nevus Syndrome 676(50)Genitourinary Tract Malformations and Wilms' Tumor 676(50)Poland Syndrome and Leukemia 677(51)Nephroblastomatosis, Nodular Renal Blastema, and Related Anomalies 677(51)Saccrococcygeal Teratoma and Anomalies 677(51)
Cytogenetic Abnormalities and Chromosomal Breakage Syndromes 678(52)Pathogenetic Mechanisms 678(52)

Foreword to Teaching Monographs
This teaching monograph is being published by The American Journal ofPathology for Universities Associated for Research and Education in Pathologyas a service to medical students and their teachers of pathology. This venturerepresents a joint effort to make such teaching material available to a wideaudience. Separately bound copies of this Teaching Monograph can be pur-chased from Universities Associated for Research and Education in Pathology,Inc., 9650 Rockville Pike, Bethesda, MD 20014. The charge is $2.50 per copy fororders of up to ten and $1.50 per copy for orders of ten or more (prepaid).
The Editorial Board
John R. Carter, MD, Case Western Reserve University School of MedicineFrancis E. Cuppage, MD, University of Kansas Medical CenterJoe W. Grisham, MD, The University of North Carolina School of MedicineRobert B. Jennings, MD, Duke University Medical SchoolWerner H. Kirsten, MD, The University of ChicagoVincent R. Marchesi, MD, Yale University School of MedicineGoetz W. Richter, MD, The University of Rochester School of MedicineDante G. Scarpelli, MD, Northwestern University Medical SchoolRobert E. Stowell, MD, University of California, DavisBenjamin F. Trump, MD, University of Maryland
Series Editor: Dante G. Scarpelli, MD

Developmental PathologyRobert P. Bolande, MD
Pathologic reactions in very early life are quite different from those inmaturity. In general, the younger the organism, the greater its reactivedeviations from the adult. The peculiarities of each developmental stageare major determinants of host responsiveness to noxious influences. Weshall try to crystallize some of the basic features of these deviations,providing a basis for understanding the special way in which these patho-logic reactions cause disease in infants and children or predispose to moreprofound morbidity in later life. It is becoming clear that many diseases ofolder individuals have their origins in very early life. This plus the rapidprogress taking place in intrauterine diagnosis will soon exact a knowledgeof these pathologic concepts from all physicians.
Normal MorhonePIcProcessesDevelopment begins exuberantly with a fertilized ovum. This gener-
ative cell, bearing the genomic dicta of millions of years of evolution,quickly gives rise to a crude mass of rapidly dividing embryonal cells.These cells grow and diversify into highly specialized organs and tissues.Development proceeds at disparate rates throughout the body. The devel-opmental "''lan" begins to diminish in the fetus and progressively wanesthereafter. Aging also begins at the moment of conception and increasesin importance through the life cycle, ultimately overshadowing develop-ment, so that the end of development may be obscured. We shall focusour attention on the embryo, fetus, and young infant.
The Embyo
Intrauterine development occurs through a series of spatially organizedand temporally controlled cellular activities performed by a primordialmass of proliferating embryonal cells. These activities are mitosis, cyto-differentiation, morphogenetic movements, cell and tissue contact inter-actions (induction), and mass cell necroses. Implicit in these events is theability of the embryonal cells to sense their positional orientation so thatcephalo-caudal, dorso-ventral, and proximal-distal relationships arequickly established.
Cytodifferentiation is any change in the morphology or chemistry ofembryonal cells rendering them more specialized than their antecedents.0002-9440/79/0308-0627$01 .00 627OUAREP (1)

628 BOLANDE American Journal(2) of Pathology
It is the means by which the body becomes diversified into various organsand tissues. The process is dependent on the sequential turning-on orturning-off of specific gene activities which define the enzyme activity of acell and hence its ultimate biologic nature. Cytodifferentiation is evi-denced by an increasing complexity of cell structure. The mitochondriabecome prominent and ribosomes attach to an increasing endoplasmicreticulum. Special organelles and structures appear: cilia, the Golgi com-plex, myofibrils in muscle cells, secretory granules in exocrine or endo-crine cells, and collagen fibers in the intercellular ground substance.'The coordinated and directed migrations of individual cells or masses of
cells, known as morphogenetic movements, are exceedingly critical.2 Masscell migrations may involve entire primitive germ layers, eg, the ectodermin neurulation or the entoderm in primitive gut formation. Such mass cellmigrations characterize early embryogenesis and are known as morpho-genetic crises. The rapid movement and rearrangement of cells result ingreat structural instability and vulnerability. Injury sustained duringthese brief crises may have profound pathologic effects on morphogenesis.Later, many cells migrate to new locations in the embryo. Cells maymigrate either singly or in small groups. Epithelial cells tend to adhereand migrate as flat sheets.The most dramatic example of widespread migration of cells is the
neural crest cells, which begin to leave the dorsum of the closing neuraltube at approximately 4 weeks' gestation and eventually colonize virtuallyevery major area of the body.3 Derivatives of these neural crest cells giverise to the entire anatomic nervous system, the chromaffin system, thenonchromaffin paraganglia, many neuroendocrine cells of the APUD sys-tem of Pearse,4 Schwann cells of the nervous system, the leptomeninges,the skin melanoblasts, odontoblasts, and even certain connective andsupportive tissues of the face, jaw, and neck. So extensive are its contribu-tions that many investigators feel that the neural crest might merit dis-tinction as a "fourth" germ layer.
Intimately involved in cell movements are cell-to-cell contact andinductive reactions.5 When migrating cells come in contact, aggregation,repulsion, or migratory arrest may ensue. However, two apposed cellmasses may glide past each other with seeming indifference. Such cellularcontacts may result in inductive or morphogenetic tissue interactions.Induction occurs when two or more tissues become associated and analteration of the developmental course of the interactants ensues. Aggre-gation of like cells is the first step in organogenesis. Interactions of unlikecells are of importance in the induction and promotion of epithelial organdevelopment, particularly in organs formed by repeated branchings of

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 629March 1979 (3)
invaginated epithelial surfaces, eg, lungs, pancreas, liver, and salivaryglands.2 The orderly progression and orientation of this branched organmorphogenesis requires contact with a basement membrane and sub-jacent mesenchymal tissue constituents. These probably give structuralsupport and orientation as well as provide biochemical inductive effects.
Orderly morphogenesis depends as well on the disappearance or in-volution of certain transient structures at the appropriate time, eg, thepronephros and mesonephros, the m-ullerian duct in males, the wolffianduct in females, and portions of the aortic arch complex. This involutiontakes place by mass necrobiosis of cells, probably initiated by lysosomalenzyme activation.1The cellular and molecular mechanisms responsible for most of these
embryogenetic phenomena are poorly understood. Active cell movementsdepend, to a large extent, on the presence of contractile proteins in theircytoplasm.2 Microfilaments 40 to 70 X in diameter, representative of actinand myosin, have been identified.2 Cytoplasmic contractions produced bythese micofilaments may initiate folding, invaginations, or evaginations ofepithelial sheets. Cellular protrusions or elongations also result from theaction of microfilaments. Structural distortions of cells are supported by asemirigid cvtoskeleton formed by larger microtubules (250A in diameter).Many morphogenetic interactions, particularly the cell contact reac-
tions, must largely depend on the peculiarities of cell surface structures,ie, the glycocalyx and cytoplasmic membranes. Some embryonal cellshave specific receptor proteins on their surfaces for certain hormones.Inductive reactions and cytodifferentiation are probably initiated by thetransfer of these hormones from the receptor sites to the nucleus, wherethey in turn interact with DNA to initiate a change in protein biosyn-thesis, driving the tissue toward its ultimate structural and biochemicalstate of specialization.1 Despite recent progress, the mechanisms of induc-tion and cvtodifferentiation remain challenging mysteries.
The Fetus and Newbom
There follows a period of organogenesis characterized by gross mod-eling and further differentiation. The process is completed by 8 weeks'gestation, at which time the embryo has developed into a fetus, closelyapproximating the general morphology of an adult. Differentiation andgrowth continue but in a less spectacular fashion. The cessation of growthand stabilization of cell functions occur at different times for differentorgans, in many instances long after birth.
In some respects, birth is a developmental milestone of scant impor-tance. This is true for the developing kidney and nervous system. On the

630 BOLANDE American Journal(4) of Pathology
other hand, birth initiates profound changes in the lung and cardiovascu-lar system as a consequence of the adaptations required for respiration.Emergence of the fetus from the sterile confines of the uterus into theoutside world is an important step in initiating immunogenesis largelythrough microbial colonization of the skin and intestine. With the abruptparturient withdrawal of the hormonal, nutritional, and metabolic supportof the placenta, other profound effects are initiated in the endocrinesystem and gastrointestinal tract.
Although the histogenetic characteristics of the various organs andorgan systems have been well documented in embryology textbooks, thestructural, biochemical and physiologic descriptions of their completedevelopmental cycle are incomplete and uneven. Some organs have beenwell studied. Books can and have been written on the lung alone, describ-ing its evolution from an embryonic, glandular organ into a maturerespiratory organ at 18 years of age. Other works have dealt with someaspects of organogenesis in the fetal period alone.6 The importance ofcollating and integrating this ever-expanding body of knowledge into amore thorough understanding of developmental pathology is obvious. Acomplete review of the current status of this material is beyond the scopeof this monograph.
Reaction to InjurySusceptibility of Developing Cells
Injury to developing cells and tissues may be manifested by cell death,malformation, growth retardation, and, ultimately, inflammation. Thenature and timing of the injury will influence which reaction or combina-tion of reactions will be prevalent. Before differentiation begins, the earlyembryo is relatively resistant to most noxious stimuli unless the dosage isvery high. After organogenesis begins, injury to specific organs and tissuesis produced with relative ease. The enhanced susceptibility of most tissuesduring this period often results in death of the embryo. Growth arrest orretardation may be expected if an injury were to result in mass cellnecrosis. Teratogenic and inflammatory sequelae of such injuries will beconsidered later.
Injured embryonal cells display degeneration and necrosis, similar toadult cells. A complete analysis of the developmentally changing ten-dency to undergo necrosis is hampered by the limited number of agentswhose dose and pathogenicity can be precisely controlled. Foremost ofthese is irradiation. Here we see that young cells are more sensitive thanolder ones. The most sensitive cells are the primitive parenchymatous cells

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 631March 1979 (5)
of the various organs, ie, the organoblast. For example, a neuroblast of thedeveloping nervous system can be killed by 25 to 40 rad, while a matureneuron can tolerate 10,000 rad. The period of highest vulnerability toirradiation is between 2 and 8 weeks' gestation.' Before or after this time,overt manifestations of cell injury decrease.
-xia,H S-i-r and IschswckIwy
Because of its great nutritional requirements, developing tissue is sensi-tive to metabolic deprivations. Yet from mid gestation to the early neo-natal period, the fetus shows remarkable tenacity in withstanding anoxia.Newborn infants have been known to tolerate 30 minutes of total anoxiawith no untoward effects. This phenomenon has not been adequatelyexplained. It may be due to the ability of young tissue to anaerobicallyglycolyse faster, yet anoxia has not been shown to evoke increased anaero-bic glycolysis. The hardiness of fetal tissue in the face of anoxia may insome way be related to large stores of glycogen in the brain, heart, andliver near term; dissipation of these glycogen reserves is associated withdeath in anoxic fetuses. Of greater importance is the existence of a systemof vascular reflexes which, under anoxic conditions, is capable of redistrib-uting the circulating blood in the fetus and newbom. The lung, intestine,liver, and kidneys are not critical organs for fetal survival in utero; theirfunctions are provided by the placenta. Under adverse conditions, theperfusion of these organs may be curtailed by several mechanisms toasste perfusion of more vital organs. Shunts bypass their capillary net-works, eg, ductus venosus in the liver, the ductus arteriosus, and the lung.In addition, the small arterioles and precapillaries, particularly of the lungand gut, have relatively thick muscular walls and tiny lumens. Theirdistensibility is limited and they present high resistance to vascular flow.Normal postnatal development is followed by involution of the fetalmedia and increasing lumen: wall ratio (Figure 1). Anoxia increases thetonic state of these vessels, whereas increasing oxygenation of the bloodcauses these vessels to relax and dilate, allowing increased perfusion of thetissues. By sacrificing perfusion of gut, lungs, and liver, a life-sustaininglevel of perfusion of the developing brain, heart, and vital placenta ismaintained. Postnatal anoxia, particularly in prematurity, may elicit areversion to these fetal vascular patterns, usually with profound patho-logic effects, particularly on the lungs.
Anoxia, when severe and persistent, gives rise to metabolic acidosis,hypoglycemia, and shock. This metabolic state may be produced bysevere matemal bleeding or shock in the immediate prenatal period, butmore often it is caused by premature birth, wherein the infant's lungs
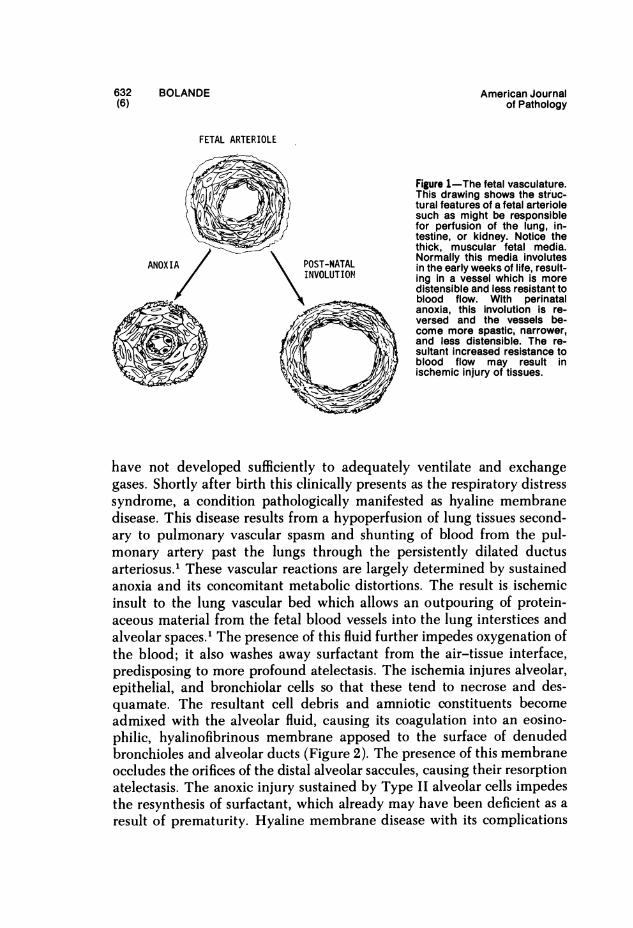
632 BOLANDE American Journal(6) of Pathology
FETAL ARTERIOLE
Figure 1-The fetal vasculature.This drawing shows the struc-tural features of a fetal arteriolesuch as might be responsiblefor perfusion of the lung, in-testine, or kidney. Notice thethick, muscular fetal media.
ANOXIA POST-NATAL ~~~~Normally this media involutesANOXIA \ POST-NATAL in the early weeks of life, result-INVOLUTION ing in a vessel which is more
distensible and less resistant toblood flow. With perinatal
-: anoxia, this involution is re-versed and the vessels be-come more spastic, narrower,
\,tS 8 and less distensible. The re-sultant increased resistance toblood flow may result in
i ischemic injury of tissues.
have not developed sufficiently to adequately ventilate and exchangegases. Shortly after birth this clinically presents as the respiratory distresssyndrome, a condition pathologically manifested as hyaline membranedisease. This disease results from a hypoperfusion of lung tissues second-ary to pulmonary vascular spasm and shunting of blood from the pul-monary artery past the lungs through the persistently dilated ductusarteriosus.1 These vascular reactions are largely determined by sustainedanoxia and its concomitant metabolic distortions. The result is ischemicinsult to the lung vascular bed which allows an outpouring of protein-aceous material from the fetal blood vessels into the lung interstices andalveolar spaces.1 The presence of this fluid further impedes oxygenation ofthe blood; it also washes away surfactant from the air-tissue interface,predisposing to more profound atelectasis. The ischemia injures alveolar,epithelial, and bronchiolar cells so that these tend to necrose and des-quamate. The resultant cell debris and amniotic constituents becomeadmixed with the alveolar fluid, causing its coagulation into an eosino-philic, hyalinofibrinous membrane apposed to the surface of denudedbronchioles and alveolar ducts (Figure 2). The presence of this membraneoccludes the orifices of the distal alveolar saccules, causing their resorptionatelectasis. The anoxic injury sustained by Type II alveolar cells impedesthe resynthesis of surfactant, which already may have been deficient as aresult of prematurity. Hyaline membrane disease with its complications
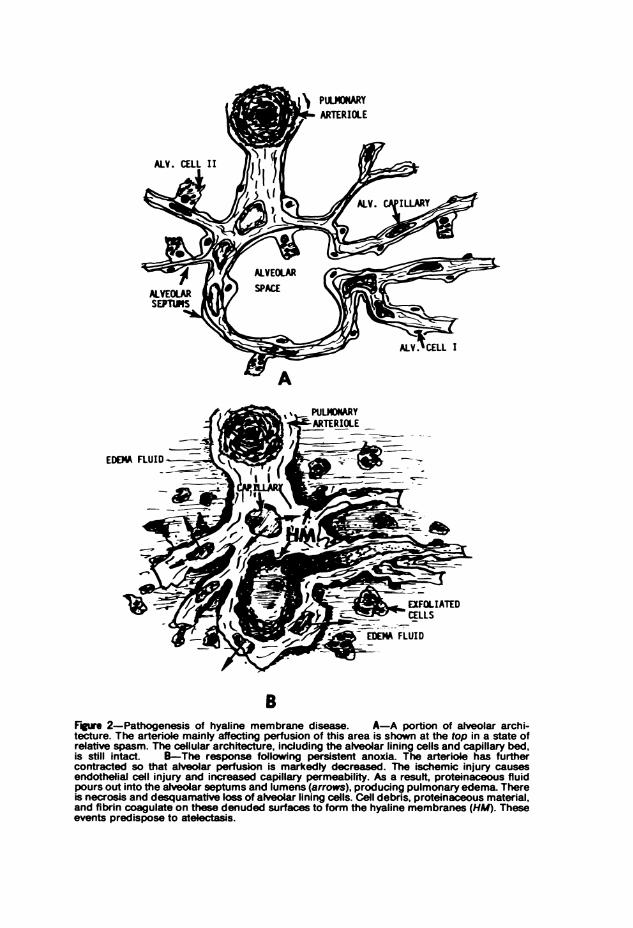
EDEMA FLUID
BFgr 2-Pathogenesis of hyaline membrane disease. A-A portion of alveolar archi-tecture. The arteriole mainty affecting perfusion of this area is shown at the top in a state ofrelative spasm. The cellular architecture, including the alveolar lining cells and capillary bed,is still intact. B-The response following persistent anoxia. The arteriole has furthercontracted so that alveolar perfusion is markedly decreased. The ischemic injury causesendothelial cell injury and increased capillary permeability. As a resuit, proteinaceous fluidpours out into the alveolar septums and lumens (arrows), producing pulmonary edema. Thereis necrosis and desquamative loss of alveolar lining cells. Cell debris, proteinaceous material,and fibrin coagulate on these denuded surfaces to form the hyaline membranes (HM). Theseevents predispose to atelectasis.

634 BOLANDE American Journal(8) of Pathology
and sequelae in the lung and other organs is one of the most importantdiseases of the perinatal period.7The ischemic injury of the lungs leading to hyaline membrane disease
may be visited on other organs as well. An ischemic enteropathy charac-terized by hemorrhagic necrosis of segments of the gastrointestinal tract,mainly in the ileocecal region may give rise to perforation and fatalperitonitis. This is often accompanied by the development of gas cysts inthe intestinal wall (pneumotosis cystoides intestinalis).8 The condition isoften referred to as "neonatal enterocolitis," although "ischemic enter-opathy" is a more accurate term. Ischemic injury of the bowel may, infact, be initiated in the fetus. Healing of this ischemic segment results infibrous obliteration of the intestinal lumen or congenital intestinal atresia.The perfusion of the brain is not limited by any special devices. Many
of the peculiarities of fetal circulation seem designed to maintain a highdegree of perfusion of this rapidly developing organ; even so, the brainmay be injured by anoxia. The respiratory distress syndrome may fatallyterminate with massive acute intraventricular hemorrhage. This bloodemanates from subependymal periventricular hemorrhages which thenrupture into the ventricles. The most proximate event is probably is-chemic injury to subependymal blood vessel walls, most likely followed byincreased brain perfusion. The latter ironically occurs as therapy improvesthe infant's vascular and metabolic status.1',
Perinatal anoxia has far greater effects on periventricular white matterthan on the cortical gray matter, where the greatest impact of anoxicdamage is found in older individuals. For example, infants surviving anepisode of anoxia caused by prolonged apnea or cardiac arrest develop aunique pattern of periventricular leukomalacia. This results from lique-faction necrosis of the sensitive periventricular white matter.9
Drugs
Unusual susceptibility to cell injury in the fetus and newborn may beencountered as a result of immaturity or latency of enzyme systems. Toxicsubstances of endogenous or exogenous origin may accumulate because ofthe organism's inefficiency in detoxifying or metabolizing them. Oneexample is the inability of the newborn to deal effectively with bilirubin, asubstance which is cytotoxic by virtue of its inhibition of oxidative phos-phorylation. It is detoxified mainly in the liver by conjugation withglucoronic acid, which renders it soluble and excretable in the bile. Theenzymes involved in this reaction comprise the bilirubin-glucuronyltransferase system, which does not become active until after the first fewdays of life. In premature infants, deficiencies of this enzyme system may

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 635March 1979 (9)
persist for longer periods. Thus, an unconjugated hyperbilirubinemia mayoccur in the presence of hemolysis or from liver enzyme immaturity alone.Unconjugated bilirubin can thus have severe pathologic effects, particu-larly on the brain. Here the lesion is known as kemicterus characterizedby a bright yellowish discoloration of brain tissue. It is the result of bilestaining and degeneration of groups of neurons in the basal ganglia,inferior olives, the cerebellum, hypothalamus, and floor of the fourthventricle. Survival with this type of brain injury results in mental retarda-tion.
Certain drugs may also have unusual toxic effects on the prematureinfant due to liver immaturity. Glucuronide formation is also involved inthe detoxification of the sulfonamides, morphine, and chloramphenicol.Immaturity of detoxifying enzyme systems may prolong the action of thedrug and exaggerate its toxic side effects.
Infwtiwi
Embryonal and fetal cells are susceptible to infectious agents, particu-larly viruses. These cells can be killed by lesser amounts of virus than arenecessary to kill mature cells. Newborn animals can be infected with smalldoses of virus by various routes of inoculation; older animals requirelarger doses, usually administered by a specific route. There is a tendencyfor virus infections to be widely disseminated in the fetus and newborn.This may be due to the inadequacy of interferon production by immaturecells.Unique portals of entry in the fetus and neonate provide easy access for
microorganisms into susceptible tissue. A number of bacteria, viruses, andparasites are capable of crossing from the maternal circulation into thefetal circulation through the placenta. When this occurs, fetal or neonatalsepticemia is the primal manifestation of transplacental infection. Themost important transplacental infections are syphilis, toxoplasmosis, cyto-megalovirus infection, and rubella. Infected amniotic fluid due to pre-mature rupture of the fetal membranes may be aspirated into the fetallungs, giving rise to neonatal pneumonia usually followed by septicemia.This is known as the amniotic infection syndrome.
Immediately before, during, or after birth, bacterial infection is usuallydue to gram-negative organisms of low virulence and invasiveness inadults, eg, Escherichia coli, Aerobacter aerogenes, Alcaligenes faecalis,and Proteus vulgaris. Infections with streptococci and staphylococci, al-though of lesser importance, persist as a hazard in the perinatal period.
After birth, the skin barrier is already disrupted by the umbilicalwound. Infection may begin here and penetrate into the body through the

636 BOLANDE American Journal(10) of Pathology
remnants of the umbilical stump and its vessels. The neonatal epidermis,particularly in prematures, is extremely thin and may be easily colonized,injured, and penetrated by certain microorganisms, even Nhen intact.During parturition, exposure of the fetal surfaces to maternal cervicova-ginal tissues infected with herpes simplex virus, for example, gives rise togeneralized, often fatal, disease.Newborn and young infants seem more prone than do older individuals
to the development of thrombotic microangiopathy and disseminatedintravascular coagulation in the presence of gram-negative endotoxemia.1At the same time, the young show a greater resistance to the lethal andpyrogenic actions of endotoxin. Thus, the disseminated intravascular co-agulation (DIC) syndromes are often encountered in septic infants.Whatever the portal of entry, infection is poorly contained and local-
ized by fetal or neonatal tissues. There is thus a tendency toward septi-cemia. The rapidity and ease with which septicemia follows local infectionis one of the characteristics of early life. This is due to a multiplicity offactors: a) the high water content and fiber paucity of the connectivetissues, facilitating diffusion of organisms or their toxic products, b) theinadequacy of the inflammatory response (see below), and c) the phago-cytic indolence of leukocytes in the newborn.
Phagocytic inertia may reflect the unstimulated condition of fixedtissue and circulating macrophages in their pristine intrauterine state.More important, there is a reduced opsonizing capacity as well as reducedantibody-mediated bacteriolysis. The placenta is selectively permeable toIgG antibodies derived from the mother, so that these are usually presentin adequate titers in the fetus and newborn. The opsonic deficiency is dueto low serum complement level in early life. C' activity in fetal serum isdependent on that which is synthesized by fetal tissues. Some componentsof C' activity appear before 20 weeks' gestation, and although C' increaseswith gestational age, it is only half the adult concentration at term. Adultlevels are attained at 6 to 12 months after birth. 10-12
After the first months of life, the hazard of severe generalized infectionlessens. The dissipation of maternally derived IgG, however, leaves in-fants susceptible to agents with which they have had no previous contactsuch as 3-hemolytic streptococcus, meningococcus, Haemophilus in-fluenzae, and a host of viruses which can give rise to serious respiratoryand gastrointestinal diseases. Later childhood is characterized by decreas-ing susceptibility to pneumonia, infectious diarrheas, croup, and bron-chiolitis. At school age, exposure to a large population of children in-creases the likelihood of contracting streptococcal infections and viralexanthemata.

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 637March 1979 (1 1)
The lnflanmiaao,y Response
Acute Inflammation
Inflammation in the classic sense appears late in phylogenesis.1 Lowerforms of life compensate for its absence by maintaining excellent regener-ative capacities. Serofibrinous exudate is seen only in the higher mam-mals. It is scarcely present in poikilothermal species. The exudation ofgranulocytes is found only in hematothermal animals; poikilothermalanimals do not possess these cells. The latter respond by exudation only ofplasma cells and lymphocytes. Histiocytic reactions and granulomas, in-cluding the formation of giant cells, are even more primitive inflamma-tory reactions. In higher forms of life such as birds and mammals, histio-cytic accumulations are prominent only after granulocytic exudationsubsides. In phylogenetically more primitive life forms, histiocytes appearprogressively earlier in inflammation, constituting the major primary cel-lular reactants to injury in bony and cartilaginous fishes. Angiofibroblasticproliferation is the most primitive reaction and is the main reactivecapability of invertebrates.
It appears that the inflammatory reaction in early life undergoes devel-opmental changes closely paralleling the phylogenetic progression.' Instudies involving a variety of injuries induced in fetal rats, monkeys, andrabbits, certain patterns emerge.1315 In early life, exudation of fluid andneutrophils is meagre and mononuclear cells are preponderant. In thelatter phases of gestation, the exudation of fluid and neutrophils increases,approximating that of the adult. In the human, these exudative reactionsbegin to appear at approximately 6 months of gestation and are fairlywell-developed at birth. There is a relative vascular unresponsiveness toinjury before the onset of exudative reactions. This vascular inertia maybe the result of tissue mast cell deficiencies at this stage of develop-ment.1'16 Sporadic mast cells appear in the human fetus between 3 and 4months of gestation. By 5 months, they are present in large numbers,coinciding with an increase in cell-bound histamine, probably within mastcells. It is after this time the adult-type exudative reactions start to appear.It is therefore likely that the presence of histamine and other vasoactivesubstances is necessary for the vasoactive component of the inflammatoryreactions. In addition, the tiny size and the sparseness of capillaries inyoung fetal tissue physically limits the endothelial surface area availablefor exudation.The early feebleness of neutrophilic exudative reactions depends on
additional factors. The predominant circulating leukocytes in the first halfof pregnancy are lymphocytes and monocytes. By 4 months of gestation,
638 BOLANDE American Journal(12) of Pathology
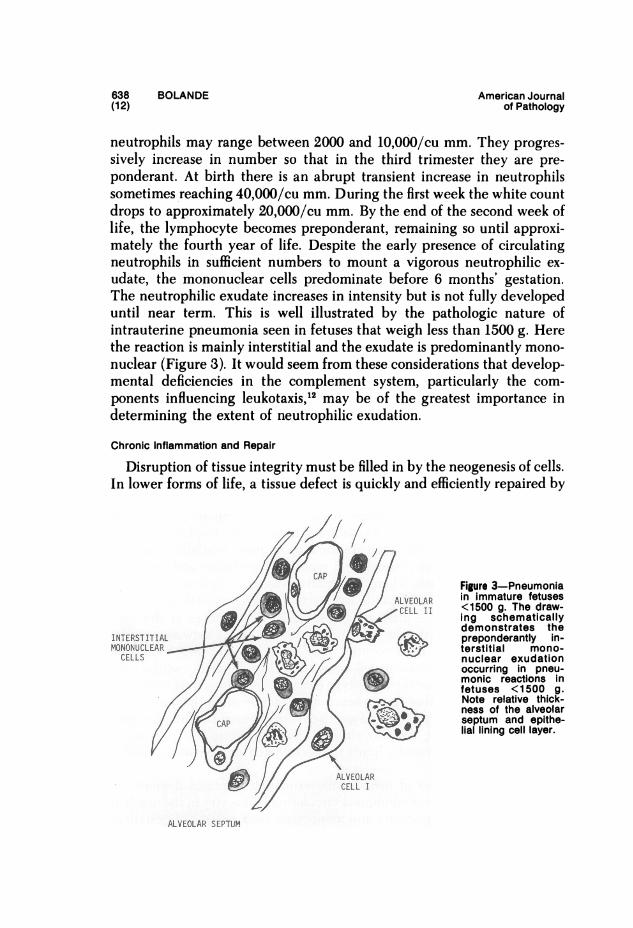
neutrophils may range between 2000 and 10,000/cu mm. They progres-sively increase in number so that in the third trimester they are pre-ponderant. At birth there is an abrupt transient increase in neutrophilssometimes reaching 40,000/cu mm. During the first week the white countdrops to approximately 20,000/cu mm. By the end of the second week oflife, the lymphocyte becomes preponderant, retnaining so until approxi-mately the fourth year of life. Despite the early presence of circulatingneutrophils in sufficient numbers to mount a vigorous neutrophilic ex-udate, the mononuclear cells predominate before 6 months' gestation.The neutrophilic exudate increases in intensity but is not fully developeduntil near term. This is well illustrated by the pathologic nature ofintrauterine pneumonia seen in fetuses that weigh less than 1500 g. Herethe reaction is mainly interstitial and the exudate is predominantly mono-nuclear (Figure 3). It would seem from these considerations that develop-mental deficiencies in the complement system, particularly the com-ponents influencing leukotaxis,12 may be of the greatest importance indetermining the extent of neutrophilic exudation.
Chronic Inflammation and Repair
Disruption of tissue integrity must be filled in by the neogenesis of cells.In lower forms of life, a tissue defect is quickly and efficiently repaired by
/ * / LELA nimaue eueFigure 3-Pneumonia
/X u / /2 tV -R ~~~~inimmature fetusescALVOAu <500 g. The draw-CELL 11 _hing schematicallyU_yX/_ ,,< X~ demonstrates theINTERSTITIAL - preponderantly in-MONOCLEAR terstitial mono-
CELLS nuclear exudationoccurring in pneu-monic reactions infetuses <1500 g.Note relative thick-
{f^a/ I/J/ - > . ' ness of the alveolar///& CAP Ni )j94 w aseptum and epithe-CAP li~~~~~~~~~~~~allining cell layer.
ALVEOLARCELL I
ALVEOLAR SEPTUM

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 639March 1979 (13)
regeneration, without the intervention of inflammatory or exudative reac-tions. This is exemplified by limb regeneration in amphibia followingamputation. Healing is virtually perfect, showing no stigmata of remoteinjury. As exudative reactions become phylogenetically more prominent,regenerative capabilities seem to subside A similar pattem is seen in thedeveloping mammalian conceptus. The result of early gestational cellinjury is not inflammation, but malformation.
Studies of the healing of experimental wounds in the fetus reveal aslowing of fibroblastic and epithelial regenerative reactions in the wake ofthe indolent exudative reactions described earlier. There is a poor granu-lation tissue development, no scab formation, and failure of epithelializa-tion.5l It has been suggested that the amniotic fluid itself may haverepressive effects on the healing of surface wounds.
In humans, stigmata of chronic inflammation such as fibrosis, gran-uloma formation, and smoldering exudative reactions may become appar-ent after the fifth month of gestation. When the fetus is the seat of achronic transplacental infection such as syphilis, rubella, or toxoplasmosis,there is a precocious development of the lymphoimmune system and largenumbers of plasma cells may appear in exudative foci after the sixthmonth of gestation. Normally, plasma cells are not prominent until severalmonths after birth. In such infected infants, elevated levels of IgM areusually present in the serum, which can readily be detected In umbilicalcord blood.A feature of chronic fetal inflammation is the predilection for dystro-
phic calcification and ferruginization.1 The fetal retention of calcium,iron, and phosphate predisposes to this type of reaction.' Selye showedthat tissues experimentally sensitized with vitamin D, or parathormone,respond to local acute injury with precipitious calcification, a processtermed "calciphylaxis."17 Administration of vitamin D to gestating ratsproduces calciphylaxis in their offspring. Young rats are much moresensitive to calciphylactic sensitizers than are older animals. Calciphylac-tic responses are suggested in humans by the calcification seen in meco-nium peritonitis, infantile arterial calcification, idiopathic infantile hyper-calcemia, cytomegalovirus, toxoplasmosis, and herpes infections of thebrain.
TeratenCil Defecs as Recto to Inly
Early cell injury, causing derangements or failures of programmedcellular phenomena at critical periods of embryogenesis may seriously

640 BOLANDE American Journal(14) of Pathology
distort the grand design of normal development. Either cell death ordegeneration, mitotic arrest or delay, or impedance of gene-enzymeactivation, whether environmentally produced or genetically determined,may seriously derange growth, differentiation, morphogenetic move-ments, and cell-to-cell contact and inductive interactions. This pathologicprocess is known as "teratogenesis," and its outcome is a structurallymalformed and/or functionally deficient offspring.
Genetically normal cells may be injured by noxious environmentalinfluences, whether indigenous to the fetal microenvironment or trans-mitted transplacentally from the mother. Such influences are known as"teratogens." Cell failures may also be genetically determined throughthe encoding of subversive information in the genome. A genetic defectmay, in addition, predispose a given cell to an environmental teratogen.The ultimate result of these teratogenic reactions is abnormality of
external bodily form or of the structure of one or more organs and tissues;these are known as "congenital anomalies or malformations." Con-stellations of anomalies are often found involving the same organs andstructures. When these constellations are repeatedly observed, they aredesignated "teratologic syndromes, often bearing an eponymic designa-tion derived from the individuals responsible for their recognition, eg,Ellis-van Creveld syndrome, Beckwith-Wiedemann syndrome, andDown's syndrome. The number and variety of congenital malformationsand teratologic syndromes are legion. We recommend Warkany's mon-umental work 18 on this subject to the interested reader.
Incidence and Morbidity 18,19
There is great variability in the incidence of congenital malformationsreported in newborn infants, depending on the nature of these studies.Ethnic, socioeconomic, and geographic factors are of importance. Al-though birth certificate records in the United States show an incidencebetween 1 and 2%, the actual figures are probably higher. The incidenceof malformations is higher in premature infants and in infants of low birthweight than in normal, term infants. Higher incidence figures are alsofound in stillborns, in spontaneously aborted fetuses, and in multiplebirths. Incidence figures found in the neonatal period are generally muchlower than those recorded in later life, since many malformations are notreadily detectable in infants. Thus, by the end of infancy the incidenceapproximates 10%, a more accurate figure. It has been estimated thatapproximately 21,000 deaths per year in the United States are directlyattributable to malformations. The importance of malformations in clini-cal pediatrics is indicated by the fact that 30 to 40% of admissions to

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 641March 1979 (15)
pediatric hospitals are due to disease arising from congenital malforma-tions.
Toe of A i Ther Pa Effects
In broad terms, congenital anomalies may be summarized as failures ofspecific morphogenetic processes '20:
1. Failure to form eg, renal agenesis, anencephaly2. Failure to form properly, eg, malformed external ear, hypoplastic
aorta, urethral stenosis, hypospadia3. Failure to involute, eg, double aortic arch4. Failure to fuse, eg, cleft lip and palate, spina bifida5. Failure to open eg, imperforate anus6. Failure to unite, eg, double uterus7. Failure to divide, eg, syndactyly8. Failure to divide properly, eg, polydactyly, tetralogy of Fallot,
transposition of the great vessels9. Failure to differentiate properly, eg, achondroplasia, congenital
and infantile neoplasms, hamartomas and hamartoses, heritabledisorders of metabolism
10. Failure to locate properly, eg, tissue heterotopias11. Failure to attach properly, eg, cecum mobile12. Failure to migrate, eg, Hirschsprung's disease (aganglionic mega-
colon)
Many congenital malformations, by virtue of their location, size, andconfiguration, may cause profound pathophysiologic effects, sometimesincompatible with prenatal life. Anencephaly or complete transposition ofthe great vessels are examples. Others predispose to life-threatening com-plications which develop at various times after birth. Intestinal obstruc-tion may result from Hirschsprung's disease or the progressive cysticenlargement of an intestinal duplication. Gastrointestinal bleeding mayoccur from the ulceration of heterotopic gastric mucosa within a Meckel'sdiverticulum. Certain congenital heart diseases predispose to bacterialendocarditis. Adenocystoid malformations of the lung or intralobar se-questrations predispose to recurrent lung infections. Congenital bladderneck or ureteral obstructions cause urinary retention which predisposes tourinary tract infections. The list of examples is great. In many instances,prompt diagnosis and corrective surgery may eliminate or help com-pensate for the presence of these defects to the extent that survival ispossible.

642 BOLANDE American Journal(16) of Pathology
The definition of teratogenesis as a failure of specific morphogeneticprocesses and programming may be too narrow. It is apparent that manycongenital malformations are initiated in the fetus after morphogenesis isrelatively complete; they often represent the inflammatory sequelae offetal injury. Examples of such late fetopathic effects are intestinal atresiaand transplacental infections (see below). In some instances, the injurymay be initiated at or shortly after birth, and because of the promptpostnatal expression of the disease process, it is presumed to have beeninitiated prenatally. This is true of "congenital" biliary atresia and neo-natal herpes simplex infections.
Etiology
Malformations are attributable to three factors: environmental terato-genic agents (teratogens), heredofamilial or genetic factors, and chromo-somal abnormalities. The majority (65 to 70%) of anomalies are of unde-termined origin (Table 1), if malformations due to multifactorialinheritance are left in the group of unknown etiology. When these areincluded among heredofamilial disorders, no cause can be recognized for
Table 1-Causes of Developmental Defects in Humans
Cause Incidence (%)
Known genetic transmission 20Chromosomal aberration 3-5Environmental causes
Radiation <1TherapeuticNuclear
Infections 2-3Rubella virusCytomegalovirusHerpesvirus hominisToxoplasma gondiiSyphilis
Maternal metabolic Imbalance 1-2Endemic cretinismDiabetesPhenylketonuriaVirilizing tumors
Drugs and environmental chemicals 4-6Androgenic hormoneFolic antagonistsThalidomideOrganic mercurySome hypoglycemics (?)Some anticonvulsants (?)
Combinations and interactions 7Unknown 65-70

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 643March 1979 (17)
approximately 30% of malformations. Each of these areas will be dis-cussed."'-"
Environmental Teratogenesis
Malformations can be produced in animals 21 by administering a widevariety of agents to the gestating female: a) physical agents: radiations,hypothermia, hypoxia, excess CO2, and mechanical trauma, b) matemalinfection, c) sex and adrenocortical hormones and hypoglycemic agents,d) vitamin deficiencies: vitamin A, riboflavin, niacin, pantothenic acid,folic acid, and vitamin E, e) antimetabolites, f) alkylating agents, g)antibiotics, and h) miscellaneous substances such as salicylates, azo dyes,and thalidomide.From experiments with such agents, Wilson 21 has crystallized most of
the important principles concerning the mode of action of such terato-genic agents. The deformities produced by a teratogenic insult depend onthe mode of action of the agent, its dosage, and the age of the embryo atthe time of administration. The genotype of the embryo is important inthat it determines the inherent susceptibility of an embryo at a given timein development.The susceptibility to teratogenic agents varies greatly during the course
of gestation. During the early stages of cleavage and blastulation, theembryo is relatively impervious to even high doses of teratogenic agents.The mechanism of this is unknown. Shortly thereafter, with the onset ofgastrulation and the establishment of the primitive germ layers, suscepti-bility reaches a maximum. Gastrulation is characterized by rapid physicalmovements of cells producing the characteristic rearrangement of tissues,on which many important inductions depend. Injury to the embryo atsuch a time may inhibit the movement of cells and restrict any inductiveactions required of them. By the time of gastrulation, localized areas ofthe embryo have differentiated specific organ-forming potentialities. Te-ratogenic action at this stage usually results in severe and widespreadanomalies.
Most organs have a period of particular susceptibility to a given terato-genic agent. Different anomalies are thus produced if a single given agentis applied at different times of gestation. It seems obvious that the maxi-mal teratogenic effect occurs when vital metabolic needs of a tissue at agiven stage in its development are rendered inadequate through theaction of the teratogenic agent. Susceptibility to teratogenesis decreases asdifferentiation and organogenesis proceed. When organogenesis is com-plete, the fetus responds to teratogenic insult by growth retardation orinflammation. It is too late to produce malformations (Figure 4).
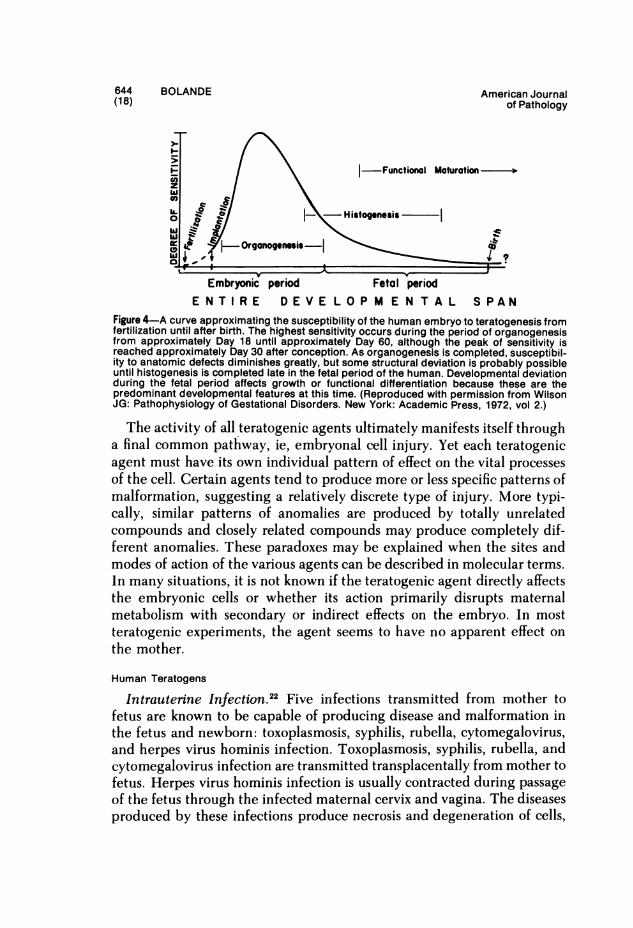
644 BOLANDE American Journal(18) of Pathology
I-/ \ Functional Maturation
U. i{,, -Histogenesiscj -QY+OgOrganog_nesis .
Embryonic period Fetal periodE N T I R E D E V E L O P M E N T A L S PAN
Figure 4-A curve approximating the susceptibility of the human embryo to teratogenesis fromfertilization until after birth. The highest sensitivity occurs during the period of organogenesisfrom approximately Day 18 until approximately Day 60, although the peak of sensitivity isreached approximately Day 30 after conception. As organogenesis is completed, susceptibil-ity to anatomic defects diminishes greatly, but some structural deviation is probably possibleuntil histogenesis is completed late in the fetal period of the human. Developmental deviationduring the fetal period affects growth or functional differentiation because these are thepredominant developmental features at this time. (Reproduced with permission from WilsonJG: Pathophysiology of Gestational Disorders. New York: Academic Press, 1972, vol 2.)
The activity of all teratogenic agents ultimately manifests itself througha final common pathway, ie, embryonal cell injury. Yet each teratogenicagent must have its own individual pattern of effect on the vital processesof the cell. Certain agents tend to produce more or less specific patterns ofmalformation, suggesting a relatively discrete type of injury. More typi-cally, similar patterns of anomalies are produced by totally unrelatedcompounds and closely related compounds may produce completely dif-ferent anomalies. These paradoxes may be explained when the sites andmodes of action of the various agents can be described in molecular terms.In many situations, it is not known if the teratogenic agent directly affectsthe embryonic cells or whether its action primarily disrupts maternalmetabolism with secondary or indirect effects on the embryo. In mostteratogenic experiments, the agent seems to have no apparent effect onthe mother.
Human Teratogens
Intrauterine Infection.22 Five infections transmitted from mother tofetus are known to be capable of producing disease and malformation inthe fetus and newborn: toxoplasmosis, syphilis, rubella, cytomegalovirus,and herpes virus hominis infection. Toxoplasmosis, syphilis, rubella, andcytomegalovirus infection are transmitted transplacentally from mother tofetus. Herpes virus hominis infection is usually contracted during passageof the fetus through the infected maternal cervix and vagina. The diseasesproduced by these infections produce necrosis and degeneration of cells,

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 645March 1979 (19)
sometimes with profound inflammation. These processes account for theprofound developmental defects and morbidity in these infants. Theresultant defects are not congenital malformations in the strictest sense ofthe word, ie, there is not altered programming or failure of normalembryonic development. Stigmata of inflammatory and necrobiotic reac-tions are characteristically present.The classic example of an infectious teratogen in humans is rubella. It
has been known since 1940 that maternal rubella infection in the firsttrimester of pregnancy leads to a complex of congenital malformations orfetal death in 20 to 40% of cases. The complex of congenital malforma-tions includes congenital heart defects, congenital cataracts, micro-cephaly, and deafness. Prospective studies have shown that rubella con-tracted in the first month of gestation results in 50% anomalous infants; inthe second month, 22%; and during the third to fifth months, 6 to 8%.After this time, the teratogenic effects of rubella infections are negligible.Heart and eye defects are more common when rubella infection occurs inthe first 2 months; hearing defects are most common when infectionoccurs in the second and third months. The virus crosses the placenta toinfect the fetus or embryo. It has been shown that infection of the embryowith rubella begins 10 to 12 days after the onset of maternal infection.The portal of entry appears to be the chorionic epithelium, with sub-sequent involvement of chorionic vasculature, the endothelium of theembryonic vascular bed, and the endocardium. Myocardial necrosis isfrequent in the left atrium, and massive necrosis of many other tissues isapparent. The viral infection persists throughout gestation and continuespostnatally, giving rise to a wide spectrum of inflammatory, degenerative,and proliferative lesions of the cardiovascular system, lungs, liver, bones,and teeth."
Irradiation.24 The sensitivity of embryonal cells to x-rays has beendiscussed earlier. The teratogenic effects depend on the dose, dose rate,and stage of gestation when exposed. Following a 2-week post-implantation period in which embryonic death occurs only after highdosages, teratogenicity becomes important until approximately 5 weeks'gestation. This sensitive period, as with other teratogens, occurs duringearly organogenesis. Heavy irradiation of the gestational matemal pelvisfor the treatment of cervical cancer results in a high incidence of infantswith microcephaly, blindness, spina bifida, cleft palate, skull defects,micromelia, and other deformities. The teratogenic potentialities of lowerdoses of radiation such as those resulting from diagnostic x-rays, radio-isotope therapy, or radioactive fallout from the atmosphere are poorlydefined. Exposure of pregnant women to the radioactivity following the

646 BOLANDE American Journal(20) of Pathology
atomic bomb explosions at Hiroshima and Nagasaki was associated withan increased incidence of microcephaly and mental and physical growthretardation. This was most prominent in offspring of women who were inthe 7th to 15th week of gestation and nearest the hypocenter of thebomb. It is likely that growth retardation overshadows teratogenesis in allperiods of early development. The most consistently affected structure isthe developing brain.Drugs and Chemical Agents.25'26 A number of therapeutic agents taken
by mothers in the first trimester have been clearly indicted as teratogenicor evoke a high index of suspicion (Tables 1 and 2). In 1961, a relationshipbetween the maternal ingestion of a new tranquilizing sedative, thalido-mide, and the birth of many deformed infants was established 20 Theinfants were born with stunted and misshapen limbs (phocomelia). Inaddition, anomalies of the ears, intestinal tract, and heart were present.Withdrawal of the drug from the market was associated with a diminish-ing incidence of phocomelia. The critical period for the ingestion of thedrug appeared to be between the 37th and 50th days of gestation. Dieth-ylstilbestrol has developed great notoriety since it was found that its useto prevent abortion in pregnant women gave rise to vaginal malforma-tions in their exposed offspring, sometimes followed by the developmentof clear cell adenocarcinoma of the vagina at adolescence.A large body of circumstantial evidence will continue to build, in-
criminating various drugs and agents in the production of congenitalanomalies in humans. Suspect are female sex hormones, tranquilizers,salicylates, certain antibiotics, antituberculous drugs, antimalarials, cer-tain anesthetic agents, anticonvulsants, and lithium carbonates.5 Epi-demiologic and experimental evidence will determine the guilt of thevarious agents. There will always remain a large group of anomalous
Table 2-Drugs Suspected of Embryotoxicity in Humans
Agent Types of defects
Maternal alcoholism Growth retardation, CNS, eye, jointAnticonvulsants Facial, digital, cardiac, mental
DiphenylhydantoinBarbiturates?Trimethadione
Neurotropic-anorectics Cardiac and variousAmphetamines
Oral anticoagulants Nasal, optic, mental, skeletalCoumarin (warfarin)
Alkylating agents Intrauterine death, few defectsOral hypoglycemics No definable syndrome
TolbutamideOther sulfonylureas

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 647March 1979 (21)
infants in whom maternal exposure to exogenous teratogenic agents can-not be detected. Subtle environmental influences must continually besought in these cases.Endogenous Conditions and Defciency States. In animals, it is impres-
sive how easily teratogenesis can be induced by metabolic deficiency inthe mother. This is striking in the case of maternal hypoxia, vitamindeficiency, or generalized malnutrition. Yet the evidence that such factorsplay a significant role in humans is not convincing."8
It seems reasonable to speculate on the role of the placenta in thepathogenesis of congenital malformation, particularly after it achievesmajor importance in regulating the internal milieu of the embryo. Abnor-malities of placentation might result in the deficiency states for theembryo or failure to bar toxic substances from the embryonal circulation.Implantation in extrauterine sites results in fetuses with a high incidenceof congenital anomalies.1 A higher incidence of malformation is noted inpregnancies complicated by antepartal bleeding, which is frequently asymptom of abnormal placentation. Toward the end of the third week ofgestation, the placenta and embryonic vascular systems have developed tothe extent that physiologic and even cellular interchange between the twocirculations can take place. It is possible that abnormalities in this rela-tionship during critical periods of development might have serious terato-genic sequelae.
Genetic Factors
Mendelian. Approximately 20% of congenital malformations are trans-mitted from generation to generation as are normal mendelian traits.Other common and familiar isolated malformations are due to multi-factorial inheritance. A tiny proportion of isolated major malformationshave been attributed to single mutant genes.19 Multiple congenital defectsare also recognized in children who have structural or numerical chromo-somal abnormalities. Chromosomal aberrations account for approximately5% of all developmental anomalies (Table 1).A long list of autosomal dominantly inherited, structural, congenital
malformations or teratologic syndromes can be cited. All of them areenumerated in McKusick's catalogue of human anomalies inherited asmendelian traits." Some represent only minor deviations from a normalsituation, eg, syndactyly or polydactyly. Others are major, life-threateningdisorders involving many organs, eg, Marfan's syndrome, von Reck-linghausen's disease. If the parents of a patient with a classic autosomaldominant disorder are normal and the dominant trait appears to besporadic, the disorder may be due to new mutation. Numerous kin-

648 BOLANDE American Journal(22) of Pathology
dreds are known in which such sporadic patients started pedigreestypical of dominant inheritance.'8Many congenital malformations are attributed to autosomal recessive
inheritance.28'29 An autosomal recessive trait is clinically manifested onlyin the homozygote. The parents of affected children are usually free of thephenotypic expression but are heterozygous for the abnormal gene. At themolecular level, the mutant gene typically results in an abnormal ordeficient enzyme. Since recessive genes are rare in the population, theaffected child often appears to be a sporadic case, because it is unlikelythat the mutant gene will become homozygous in the parental relatives,unless there is inbreeding.
Several congenital malformations can be listed under the heading ofsex-linked inheritance.28'29 In X-linked recessive inheritance, only malestend to be affected with the disease. Females are carriers of the trait andtransmit it to one half of their daughters, who are carriers like theirmothers, and to one half of their sons, who manifest the disease, sincethere is no corresponding normal gene in their Y chromosome. If a fatheris affected, all his daughters carry the abnormal recessive gene althoughthey are phenotypically normal; all his sons who received their X chromo-some from their genotypically normal mother are normal. Some forms ofhydrocephaly, imperforate anus, congenital cataract, and testicular femi-nization syndrome may serve as examples of congenital malformation,which is inherited as a sex-linked recessive trait. Sex-linked dominanttraits can be transmitted in kindreds from one generation to another likeautosomal dominant traits, but there is no male-to-male transmission. Theaffected heterozygous female will transmit the gene to one half of her sonsand daughters; the affected males will transmit their abnormal X chromo-some to all their daughters. Incontinentia pigmenti and hypophospha-temic ricket are clinical examples of this situation.
Multifactorial Inheritance. The term "multifactorial inheritance" refersto the process in which the malformation is due to the additive effect ofseveral minor abnormal genes and environmental factors. Many familial,relatively common congenital malformations fit the multifactorial model.These anomalies include cleft lip and palate, anencephaly, menin-gomyelocele, encephalocele, club foot, hypospadias, congenital hip dis-location, pyloric stenosis, and several cardiac malformations. The rate ofeach of these defects in general population is between 1:500 and 1:2000.19The recurrence rate for these defects among relatives in involved familiesis markedly increased but does not follow the pattern of mendelian in-heritance for a single mutant gene.Chromosomal Abnormalities.1"20'30 Chromosomal abnormalities are

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 649March 1979 (23)
typically associated with multiple developmental defects. It is estimatedthat approximately 0.5 to 1% of infants are born with recognizablechromosomal aberrations. Sex chromosome abnormalities account for ap-proximately 0.2% of these. Among liveborn babies with recognizablemalformations, approximately 4 to 5% have a detectable chromosomalaberration. Many embryos with chromosomal numerical or structuralaberrations do not reach term because the pregnancy is terminated byspontaneous abortion. The evidence is approximately 50 times higherthan in livebom infants, which indicates natural elimination of a consid-erable number of abnormal pregnancies. In studies published from differ-ent centers, approximately one half of the cytogenetically abnormal abor-tuses have one extra chromosome, resulting in trisomy. One quarter aremonosomic for the X chromosome, and nearly a quarter are triploid.Chromosomal aberrations are identified most commonly in abortions oc-curring during the first trimester. Among induced abortuses the incidenceof chromosomal aberrations is only approximately 6 to 7%.
It is still not clear what causes chromosome abnormalities. Some associ-ate increased matemal age with the errors in oogenesis leading to meioticnondisjunction; others blame over-ripeness of ova due to intrafollicularretention caused by hormonal maternal imbalance. Other observationspoint to environmental causes of nondisjunction such as ionizing radia-tion, streptococcal infection, viral infection, or chemical agents. It is notreally understood how a chromosome abnormality-leads to congenitaldefects. It was suggested that chromosomal aberration syndromes are notcaused by too many or too few classic structural genes but by disturbancein the function of regulatory DNA sites.Chromosome abnormalities associated with multiple congenital defects
can be subdivided into numerical and structural aberrations. Changes inchromosomal number are either aneuploid or polyploid. The cells may behypodiploid (usually 45) or hyperdiploid (usually 47 to 49). The loss of onechromosome leads to monosomy for that particular chromosome: an aneu-ploidic state with 45 chromosomes. Loss of one autosome or sex chromo-some is generally not compatible with survival; embryos missing achromosome usually die. More frequent is an absence of one sex chromo-some (Tumer syndrome 45,XO), although it is estimated that 95% ofembryos lacking a sex chromosome also die in utero.The presence of a specific, single extra chromosome is called "trisomy.''
Although causing profound anomalies of growth and development, thetrisomies are more frequently compatible with survival. They have beendetected in chromosomes 21, 18, 13, and 9 and in the sex chromosomes.The trisomies cause definite patterns of malformations, yet, for each

650 BOLANDE American Journal(24) of Pathology
trisomy, a spectrum of malformations occurs, with a significant degree ofclinical variability sometimes resulting in great dissimilarities of signs andsymptoms. Children with apparently identical trisomies may widely differclinically and morphologically. Apart from full trisomies when the wholeextra chromosome is present, many partial trisomies with a duplication ofonly a part of any autosome have been described. The cells containmultiples of the euploid number 23. The most common type of polyploidyin human embryos is triploidy (69 chromosomes). This can result from thesecond polar body failing to separate from the ovum or by an ovum beingfertilized by two sperms almost simultaneously. Although some suchfetuses have been born alive, they all died within a few days.
Aberrations in chromosomal structure may result from chromosomebreaks or be inherited from a parent who is a carrier of a balancedchromosomal translocation. A single chromosome break is a frequentresponse to environmental effects such as irradiation. In a single chromo-some, this is unlikely to lead to serious genetic consequences, since it willeither proceed to normal reunion or produce an unstable cell line whicheventually will be eliminated. However, if more than one break consis-tently occurs in a single chromosome, more serious consequences mayfollow such as chromosomal deletions, ring chromosome formation, in-versions, insertions, and reciprocal translocations. These may producedistinct teratologic syndromes.
There are individuals bearing two or more cell lines of different karyo-type. In the majority of cases, such persons are mosaics whose differingcell lines have arisen after fertilization by nondisjunction or anaphase lagduring mitosis or early cell divisions. Autosomes as well as sex chromo-somes may be involved in mosaicism that can lead to a great variety ofphenotypes: from normal to some mental and physical imperfection.Chimeras have two or more cell lines, like mosaics, but these cell lines arenot all indigenous to the individual. Such alien cells can, for example,originate from intrauterine maternal-fetal or twin-to-twin transfusionsthrough the placenta. Such maternal-fetal transfusions of immuno-competent lymphocytes may initiate a graft-versus-host reaction. This is aseverely debilitating and often fatal disease characterized by a peculiarskin rash, intractable diarrhea, and an immunodeficiency state associatedwith marked thymic destruction. This disease closely resembles some ofthe heritable immunodeficiency diseases and may be involved in theirpathogenesis.31
Inborn Abnormalities of Cell FunctionIn a broad sense, teratogenesis may be manifested as a derangement of
cellular function alone, without apparent structural malformation at birth.

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 651March 1979 (25)
The pathophysiologic expression of such derangements may not occuruntil long after birth. The conditions in question are usually hereditary.Such inborn cell dysfunction may ultimately evolve into a multiplicity ofdisease states involving every field of medicine.1'32
In general, these diseases may be broadly viewed and classified asfollows:
1. Synthesis of abnormal intracellular material, eg, the hemoglobin-opathies
2. Deficient cell synthesis and secretion, eg, immunoglobulins in im-munodeficiency disease, adrenal dyshormonogenesis in congenitaladrenal hyperplasia, a1-anti-trypsin deficiency
3. Synthesis of abnormal extracellular products, eg, exocrine secretionsin cystic fibrosis
4. Metabolic toxemias due to overproduction of circulating histotoxicmetabolites which affect a multiplicity of organs and tissues, eg,galactosemia, phenylketonuria, tyrosinemia, hereditary fructose in-tolerance, maple syrup disease, oxalosis, cystinosis
5. Storage diseases produced by the widespread overaccumulation ofmetabolic products in parenchymal and/or reticuloendothelial cells,eg, Gaucher's disease, Niemann-Pick disease, the gangliosidoses,the mucopolysaccharidoses, the glycogenoses
6. Heritable disorders of skeletogenesis and connective tissues, eg,chondrodysplasia, osteogenesis imperfecta, Marfan's syndrome
7. Neuromuscular derangements and abiotrophies, eg, muscular dys-trophy, hereditary ataxias, amyotonia congenita, certain dysmelina-tive leukodystrophies
8. Renal tubular defects, eg, de Toni-Fanconi syndrome, vitamin-D-resistant rickets, renal tubular acidosis
Over 60 of these diseases are the result of specific, genetically deter-mined enzyme deficiency. These enzymes may be degradative, synthetic,or lysosomal. They are responsible for the metabolic storage diseases andheritable metabolic toxemias and are characteristically transmitted asautosomal recessive traits. Prenatal identification of affected homozygoteshas become possible through the study of the enzymatic activity ofcultured amnion cells.
Typical of a heritable metabolic toxemia is galactosemia, a rare diseasetransmitted as an autosomal recessive trait, which occurs in approximately1 in 18,000 infants. The genetic defect is expressed as a congenital absenceof the enzyme galactose-l-phosphate uridyl transferase. This enzyme isrequired to utilize the galactose of milk, serving to convert galactose-l-

652 BOLANDE American Journal(26) of Pathology
phosphate to glucose-l-phosphate. Milk-feeding affected infants causesthe buildup of galactose-l-phosphate and galactitol; these substancesaccount for the toxemic state, which results in nutritional failure, hypogly-cemia, hepatomegaly, cataracts, and mental retardation. The most strik-ing pathologic changes are found in the liver, which at first shows fattymetamorphosis followed by cirrhosis. In time, the brain may show evi-dence of gliosis and neuronal degeneration.
Illustrative of a storage disease is Pompe's disease, the generalized formof the glycogenoses (Type II). There is intracellular accumulation ofchemically normal glycogen in virtually all tissues. The major clinicalfeatures are due to involvement of skeletal and cardiac muscle. Here theglycogen deposition results in muscular hypotonia and cardiac decompen-sation. The glycogen storage is due to the inborn deficiency of a lysosomalenzyme involved in glycogen breakdown, ie, a-(1 -l 4) glucosidase (acidmaltase). In many tissues studied ultrastructurally, the glycogen accumu-lations appear sequestered within huge lysosomes, reflective of the bio-chemical nature of the disease.
NeoplasiaCertain peculiar neoplasms of early life distinguish them in many
respects from those occurring in later life. Common adult cancers appearto arise by a regressive mutation of cells within mature tissue, generally incells retaining an ability to multiply and regenerate in adulthood. Someadult cancers may arise in developmentally anomalous tissue, or theirappearance is enhanced or predetermined by other inborn defects. Incontrast, the most common solid tumors of childhood are manifested invery early life, sometimes at birth, and are characterized by uniquecellular features indicating an origin in abnormal embryogenesis. Whenmalignant, they are rapidly progressive and highly lethal.
Neoplasms have become an extremely important cause of mortality inchildhood. The rank order of incidence rates of the most common neo-plasms of infancy and childhood in the United States is as follows 33:leukemia and lymphoma, 38.2/106 children/year; brain tumors, 23.9;Wilms' tumor, 7.8; neuroblastoma, 7.6; bone cancer, 4.8; rhabdomyosar-coma, 3.9; retinoblastoma, 3.0. These incidence rates vary widelythroughout the world, with the exception of Wilms' tumor, which seemsto occur at the same fixed rate in all areas studied.The susceptibility of the young host to the malignant process has been
demonstrated repeatedly by an enhanced growth of transplanted tumorsin young animals. Moreover, oncogenic viruses and chemical carcinogensmore readily induce tumors in the young host than in mature ones.

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 653March 1979 (27)
Paradoxically, regression and cytodifferentiation occur most often in hu-man tumors of early life."-"
Defir and Desc s
The important tumors to be considered here are hamartomas andhamartoses, teratomas, malignant embryomas, lymphomas and leuke-mias, reticuloendothelioses, and fibromatoses.
Hamartomas "
"Hamartoma" is a term used to describe a tumor-like mass of tissue,apparent at or near the time of birth, that is composed of an excess of moreor less normal tissue indigenous to its site of origin. Its capacity for growthis limited, paralleling that of the host, and its biologic behavior is benign.The distinction from malformation is often difficult. Some may be inter-mediate between malformation and true neoplasm.A hamartoma may be unifocal or multifocal. Multifocal hamartomas
are referred to as "hamartoses." Hamartomas may exhibit a multiplicityof forms at different sites within a given individual; in this case, they arereferred to as "pleiotropic hamartoses." The important hamartomas andhamartoses are listed in Table 3. Many of these are genetically deter-mined.
Teratomas
A teratoma is a tumor forned of a multiplicity of tissues derived frommore than one primitive germ layer.' Component tissues are often alien tothe site of development and tend to be arranged in a haphazard andconfused fashion. Besides the diverse and heterotopic constituents ofteratomatous tissue, there is also asynchronous maturation of its variousparts. Cells of embryonal, fetal, or adult character may be jumbledtogether. Frequently, much of the tissue is of glial and neuroid type,including ganglion cells, ependyma, and choroid plexus. Epithelium maybe differentiated into acinar, cystic, and ductlike structures. Abortiveattempts at organogenesis are apparent. Teratomatous tissue may besolid, multicystic, or arranged about a single large dermoid cyst. Increasein size of a teratoma may result from distention of cystic spaces with theaccumulated products of lining cells rather than true cell division. It mayalso result from the proliferation of fetal or embryonal constituents. Thisform of growth may be transient and rapid, only to abate with cyto-differentiation of the active tissues at some later stage of development. Insome instances, teratomas may become the seat of unbridled malignantgrowth, in which malignant embryonal tissue all but replaces the organoid

Table 3-Hamartomas and Hamartoses
Tissue of Neoplasticorigin Pathologic examples Genetics transformation
Congenital hemangiomas andvascular nevi of skin
Lymphangiomas andcystic hygromas
Milroy disease
Multiple glomanglomas
Autosomaldominant
Autosomaldominant
AngiomatosesSkin and VisceraHereditary mucocutaneous Autosomaltelangiectasia (Rendu-Osler) dominant
Facial and intracranial ?(Sturge-Weber)
Brain and retina Autosomal(von Hippel-Lindau) dominant
Congenital fibromatosis Autosomal
Familial cervical lipo-dystrophy (symmetriclipomatosis)
Multiple exostosis
Enchondromatosis(OIlier disease)
Fibrous dysplasia ofbone
Congenital melanotic neviLinear nevus sebaceous
(Jadassohn)Basal cell nevus syndrome
Multiple familialpolyposis
Peutz-Jeghersyndrome
Tuberous sclerosis
Maffucci syndrome
Gardner syndrome
Cowden syndrome
von Recklinghausen'sneurofibromatosis
Multiple mucosalneuroma syndrome
Hypernephroma,pheochromocytoma
dominant?Autosomal
dominant
Autosomal Chondrosarcoma,dominant rare
Chondrosarcoma,rare
Autosomal Osteogenicdominant? sarcoma, rare
Melanoma, rareBrain tumor
Autosomaldominant
Autosomaldominant
Autosomaldominant
Autosomaldominant
Autosomaldominant
Autosomaldominant
Autosomaldominant
Autosomaldominant
Sipple syndromeNeurocutaneous
melanosis
Basal cellcarcinomas
MedulloblastomaCarcinoma of
colon, commonCarcinoma, rare
Brain glioma
Chondrosarcomasof bone, angiosar-comas of skin, rare
Carcinoma of colon,fibrosarcomas ofbone or soft tissue
Carcinoma ofbreast, thyroid,bowel
Neurogenic sarcoma,phenochromocytoma,leukemia*
Medullarycarcinoma ofthyroid,pheochromocytoma
Same as aboveNeurogenic sarcomaMelanoma
* An unusual predilection for patients with von Recklinghausen's syndrome to developleukemia has been established.
Vascular
Connectivetissue
Skeletal
Skin
Intestine
Pleiotrophichamartoses(multiplesites oforigin)
Neurocris-topathichamartoses

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 655March 1979 (29)
patterns of the original tumor. Typically, teratomas develop in the testis,ovary, retroperitoneum, anterior mediastinum, and sacrococcygeal re-gions. Less common sites include the base of the skull, pineal region,brain, neck, and nasopharynx.
Gonadal Teratomas and Their Pathogenesis."-" Teratomas of the adultovary are usually benign dermoid cysts; testicular teratomas of adultmales are usually malignant. Conversely, prepubertal testicular teratomasare more likely to be benign and cystic; ovarian teratomas in young girlstend to be malignant. Malignant transformation in childhood is oftencarcinomatous, with morphologic features similar if not identical to thoseof yolk sac carcinoma or the endodermal sinus tumor of Teilum. The fea-tures of yolk sac tumors in childhood and their possible relationship toteratoma will be discussed later.
It is generally agreed that gonadal teratomas arise from totipotent germcells. It follows that testicular teratomas might be cytogenetically eitherXX or XY and that ovarian teratomas might be only XX. Recent studieshave suggested that malignant testicular teratomas may be only XX, incontrast to earlier studies indicating an equal number of chromatin-posi-tive and chromatin-negative tumors. The situation conceming ovarianteratomas is still vague.The explanation for the developmental peculiarities and sexually diver-
gent behavior of gonadal teratomas remains unclear. Investigators havelooked to the developmental differences in the ovary and testis for possibleclues. Germ cells in the ovary begin meiotic activity at 12 to 13 weeks'gestation. By mid gestation, most oocytes have entered meiotic prophase.After reaching the diplotene stage, further maturation is arrested; allviable oocytes begin a resting or dictyate stage by the time of birth.Meiotic activity recommences with ovulation at puberty. By contrast,meiotic activity does not occur in the testis before puberty and the onsetof spermatogenesis. A temporal relationship is seen to exist between peakperiods of meiotic activity and malignant transformation. In the ovary,malignancy occurs 5 to 15 years after the fetal period of meiosis; in thetestis, the same time elapses after the onset of spermatogenesis. Recentcytogenetic studies of benign ovarian teratomas show that these cells arediploid and homozygous for the centromeric region, suggesting that theyoriginate in an oocyte blocked in meiosis, possibly activated by par-thenogenesis. It has been theorized that testicular teratomas arise byfusion of haploid cells. If one accepts the germ cell origin of teratomas, itremains unclear whether malignancy is inherent in timing of develop-mental events or whether an additional mutagenic event occurs when thegerm cell is actively dividing and highly vulnerable to oncogenic influ-ences. The hormonal environment of the host may influence the course.

656 BOLANDE American Journal(30) of Pathology
For example, the menstrual cycle periodically provides a milieu receptiveto gestation which may in turn be conducive to cytodifferentiation in thepostpubertal ovary.
Sacrococcygeal Teratomas. Sacrococcygeal teratoma is the most com-mon teratoma in infancy, occurring once in every 20,000 to 40,000 livebirths. It is four times more frequent in females. This tumor is characteris-tically present at birth, bulging out the skin of the lower back, buttocks, orperitoneum.' Internally it may project into the pelvis or retroperitoneum.Prior to age 4 months, the tumor usually appears cytodifferentiated andquiescent, and local excision is usually curative. After 4 months of age,malignant transformation occurs in over 70% of patients and is associatedwith a high degree of lethality due to widespread metastases. Malignanttransformation usually is of the yolk sac carcinoma pattern."37
It is easy to accept the germ cell origin of gonadal teratomas since germcells are normally present in these structures. The origin of extragonadalteratomas is more problematic. Some believe that such teratomas arisefrom primordial germ cells entrapped in various sites during their migra-tion to the embryonic gonad."8 Others have proposed that they originatefrom undifferentiated pluripotent cells of the primitive streak that haveescaped the influence of embryonic organizers." The sacrococcygealprominence of these tumors in children might reflect the concentration ofthese cells at the primitive knot near the coccyx and the proximity of thegerm layers at this point.40A third possibility is that these tumors arederived from primitive yolk sac cells (see below).
Yolk Sac Tumors (Endodermal Sinus Tumor of Teilum). Yolk sac (endo-dermal sinus) tumors preferentially occur in the same sites as teratomas,ie, gonads, sacrococcygeal region, retroperitoneum. Yolk sac carcinomasaccount for one half to one third of all primary prepubertal testiculartumors. 42 These tumors are exceedingly malignant, although the infan-tile forms, particularly in patients younger than 1 year of age, may be lessaggressive. These tumors present a characteristic histologic pattern con-sisting of papilliform endodermal sinus structures (Schiller-Duval bodies),microcysts, and aggregates of embryonal clear cells (Figure 5). Globules ofintracellular or extracellular PAS (+) material are present, representativeof a-fetoprotein. This tumor index substance may be increased in thebloodstream as a result of its secretion by tumor cells. Teilum showed thatthese tumors, no matter where they occurred, reproduced the morpho-genetic features of yolk sac endoderm and allantoic mesoderm in the rat'splacenta. He coined the term "endodermal sinus tumor," which at onestroke dismissed the gaggle of diagnostic terminology associated withthese tumors and tied them all to a single embryopathic origin. The
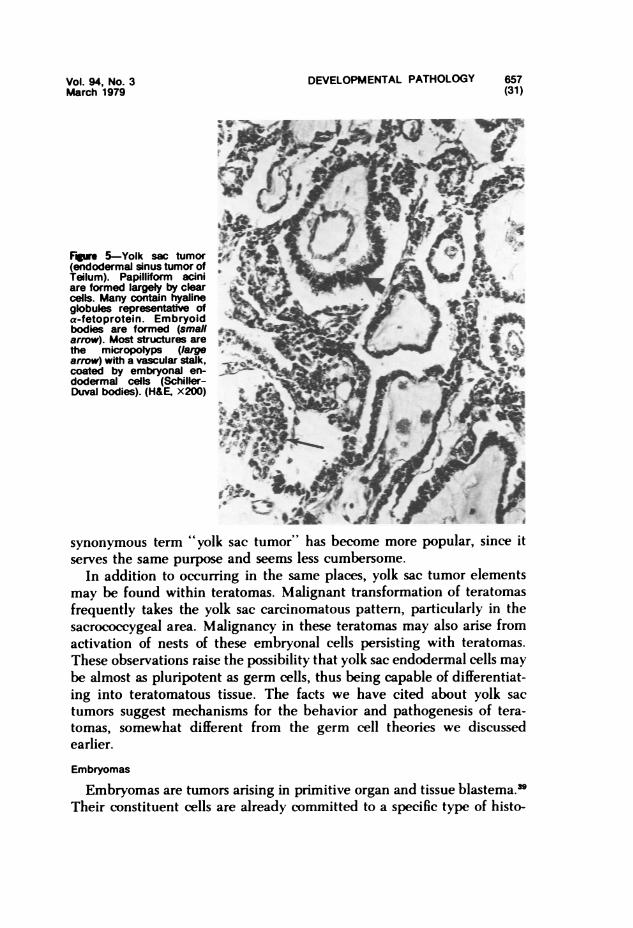
Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 657March 1979 (31)
Fe"e 5-Yolk sac tumor(endodermal sinus tumor of bTeilum). Papilliform aciniare formed largely by clcells. Many contain hyaline -globules representatfie ofa-fetoprotein. Embryoidbodies are formed (small-arrow). Most structures are _the micropoJyp (larg9e ~e^ zarrow) with a vascular stalk, ILcoated by embryonal en-dodermal cells (Schiller-Duval bodies). (H&E, X200)
synonymous term 'yolk sac tumor" has become more popular, since itserves the same purpose and seems less cumbersome.
In addition to occurring in the same places, yolk sac tumor elementsmay be found within teratomas. Malignant transformation of teratomasfrequently takes the yolk sac carcinomatous pattern, particularly in thesacrococcygeal area. Malignancy in these teratomas may also arise fromactivation of nests of these embryonal cells persisting with teratomas.These observations raise the possibility that yolk sac endodermal cells maybe almost as pluripotent as germ cells, thus being capable of differentiat-ing into teratomatous tissue. The facts we have cited about yolk sactumors suggest mechanisms for the behavior and pathogenesis of tera-tomas, somewhat different from the germ cell theories we discussedearlier.Embryomas
Embryomas are tumors arising in primitive organ and tissue blastema."Their constituent cells are already committed to a specific type of histo-

658 BOLANDE American Journal(32) of Pathology
genesis. As such, these tumors occur in specific organs and tissues. Theyare composed of organoblasts which retain many of the features of a moreprimitive stage of development of the involved tissue. It seems clear thatthese cells have failed to differentiate. The basis for this maturation arrestand its malignant sequelae are unknown.The tumors in this group are among the most common solid malignant
neoplasms of infancy and childhood. They include the Wilms' tumorcomplex, neuroblastoma, retinoblastoma, hepatoblastoma, medulloblas-toma, embryonal sarcomas, and rhabdomyosarcomas. We shall deal withthe Wilms' tumor group and neuroblastoma in greatest detail. Histologicillustrations of the other tumors discussed can be found elsewhere. 1,43
Wilms' Tumor. Wilms' tumor is a malignant embryoma of the kidney,derived from metanephric blastema. It is composed of an admixture ofmesoblastic stroma and primitive nephronoblastic epithelium arranged insheets and nests containing prominent foci of poorly formed or dysplastictubules and glomeruli (Figure 6A). It accounts for approximately 6% of allpediatric malignancies in patients younger than 15 years. Wilms' tumor isdiscovered most often in patients between 3 and 4 years of age. At thisage, it is extremely malignant and accounts for almost 20% of malignanttumors. Cure rates in tumors treated during the 1950s by prompt nephrec-tomy and postoperative irradiation were in the order of 30 to 40%. Theaddition of chemotherapy to the regimen has resulted in cure rates well inexcess of 50%. Cure rates in Wilms' tumor discovered in patients underage 1 year have always been much better, generally over 80%.
Wilms' tumors are bilateral in 5 to 10% of patients. The average age ofpatients with bilateral Wilms' tumor is 15 months, which is much youngerthan the peak age of incidence of unilateral Wilms' tumor.43 BilateralWilms' tumors may develop simultaneously or sequentially. Sequentialbilateral tumors, in which involvement of the contralateral kidney be-comes apparent some time after the treatment of an ostensibly unilateraltumor, are much less common than simultaneous bilateral tumors.
Infantile Congeners of Wilms' Tumor. Primary renal tumors of infantsand children are generally diagnosed as Wilms' tumor. They are thuspresumed to be possessed of an implacably malignant potentiality and,therefore, must be vigorously treated. In reality, this group of tumors isnot monolithic, showing considerable variability in clinical behavior andmorphologic characteristics, particularly when detected at birth or withinthe first few months of life. At this point of development, renal neoplasia isexpressed in several clinical and pathologic forms, all of which are signifi-cantly less aggressive than conventional Wilms' tumor, if not completelybenign. The cryptic presence of these entities in most series of Wilms'

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 659March 1979 (33)
tumors has contributed to the enhanced survival statistics generally re-corded in patients less than 1 year old. Careful reviews of the literaturebased on an appreciation of these lesions fail to reveal evidence of a trulylife-threatening, metastasizing Wilms' tumor of conventional morphologyoccurring within the first few months of life (congenital Wilms' tumor). Itwould be foolhardy to assert that such a lesion does not exist. It can onlybe said that it is exceedingly rare and not well described at this time."
Three more or less distinct clinical-pathologic entities are distinguish-able from conventional Wilms' tumor in the first months of life (Table 4).The foremost of these is the congenital mesoblastic nephroma of infancyor mesoblastic nephroma, sometimes referred to as "fetal renal mesenchy-mal hamartoma" or "leiomyomatous hamartoma."''" The tumor is typi-cally detected at birth or shortly thereafter by its huge size. It is composedpredominantly of a fibrous or mesenchymal stroma in which bizarre anddysplastic tubules are focally and irregularly scattered. The tumor isessentially benign and curable by nephrectomy alone, although a fewsuch tumors showing aggressive behavior have been encountered. Thistumor is the most common form of congenital renal neoplasia. It accountsfor approximately 5 to 10% of all pediatric renal neoplasms. It had beentypically misdiagnosed as "congenital Wilms' tumor" (Figure 6B).
Less appreciated is a group of well-differentiated monomorphic epithe-lial nephroblastomas." These tumors are essentially monomorphic, com-posed of well-differentiated, nephronoblastic epithelium forming discrete,closely apposed tubules, microcysts, or papillary forms. These are alsopredominantly benign. These tumors are considered to be cyto-differentiated kindred of conventional Wilms' tumor, evolving from meta-nephric blastema during fetal life in which the neoplastic process isinitiated earlier in development than in conventional Wilms' tumors.
In recent years, still another important type of congenital renal diseasehas been recognized, referred to as the 'nodular renal blastema/nephro-blastomatosis complex." This class of lesions is generally viewed as inter-mediate between malformation and true neoplasia.'7 Nodular renal blas-tema is characterized by the presence of discrete subcapsular clusters of
Table 4-4nfantile Congeners of Wilms' Tumor
Mesoblastic nephroma of infancyMonomorphic epithial nephroblastomata
Well-differentiated tubular adenomasMultikoculated cystic nephromaPolycystic nephroblastoma
The nodular renal blastema-nephroblastomatosis complex
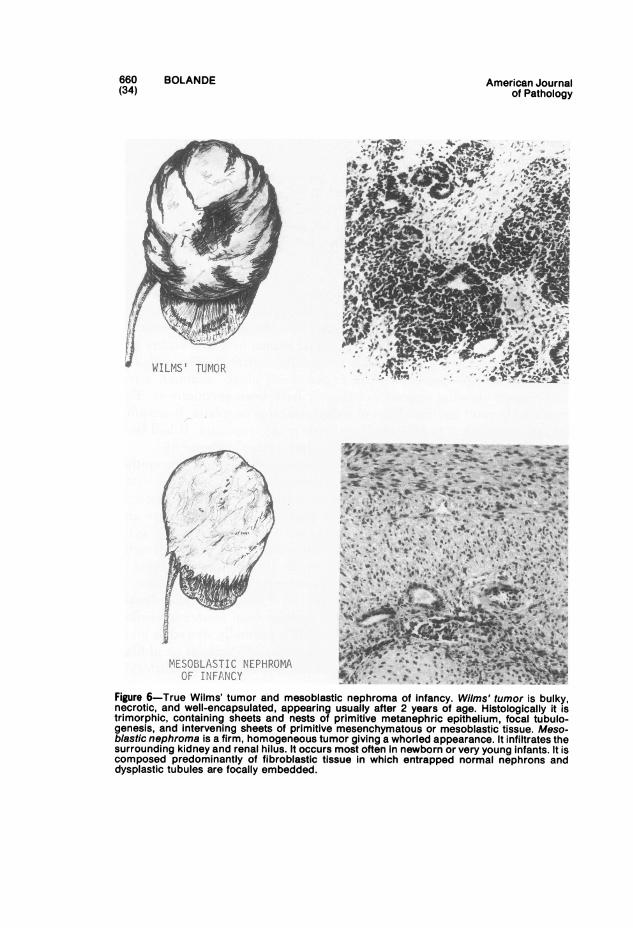
BOLANDE
WILMS' TUMOR
American Journalof Pathology
MESOBLASTIC NEPHROMAOF INFANCY
Figure 6-True Wilms' tumor and mesoblastic nephroma of infancy. Wilms' tumor is bulky,necrotic, and well-encapsulated, appearing usually after 2 years of age. Histologically it istrimorphic, containing sheets and nests of primitive metanephric epithelium, focal tubulo-genesis, and intervening sheets of primitive mesenchymatous or mesoblastic tissue. Meso-blastic nephroma is a firm, homogeneous tumor giving a whorled appearance. It infiltrates thesurrounding kidney and renal hilus. It occurs most often in newborn or very young infants. It iscomposed predominantly of fibroblastic tissue in which entrapped normal nephrons anddysplastic tubules are focally embedded.
660(34)
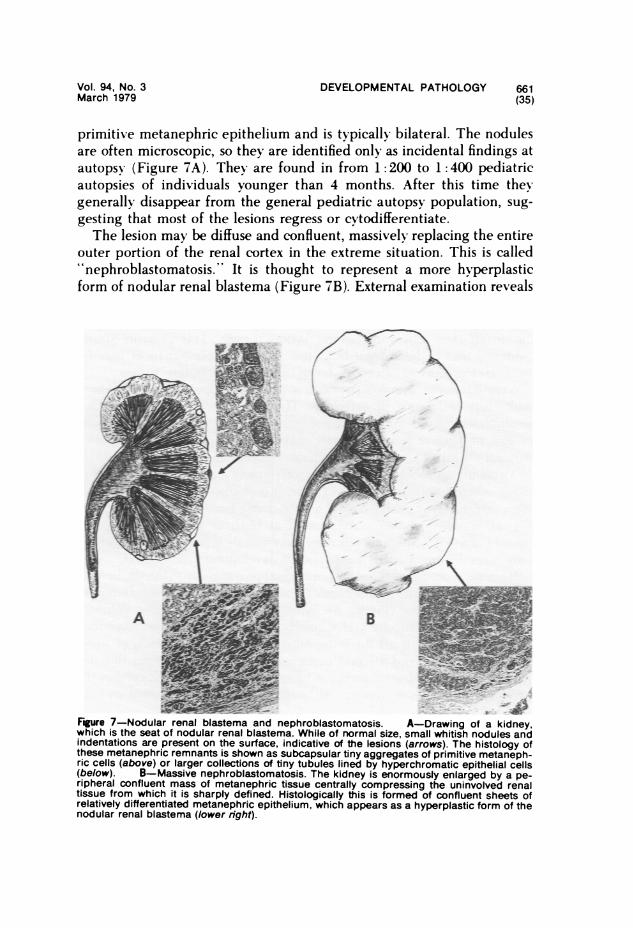
Vol. 94, No. 3March 1979
DEVELOPMENTAL PATHOLOGY 661(35)
primitive metanephric epithelium and is typically bilateral. The nodulesare often microscopic, so they are identified only as incidental findings atautopsy (Figure 7A). They are found in from 1: 200 to 1: 400 pediatricautopsies of individuals younger than 4 months. After this time theygenerally disappear from the general pediatric autopsy population, sug-gesting that most of the lesions regress or cytodifferentiate.The lesion may be diffuse and confluent, massively replacing the entire
outer portion of the renal cortex in the extreme situation. This is called''nephroblastomatosis." It is thought to represent a more hyperplasticform of nodular renal blastema (Figure 7B). External examination reveals
A
Fgure 7-Nodular renal blastema and nephroblastomatosis. A-Drawing of a kidney,which is the seat of nodular renal blastema. While of normal size, small whitish nodules andindentations are present on the surface, indicative of the lesions (arrows). The histology ofthese metanephric remnants is shown as subcapsular tiny aggregates of primitive metaneph-ric cells (above) or larger collections of tiny tubules lined by hyperchromatic epithelial cells(below). B-Massive nephroblastomatosis. The kidney is enormously enlarged by a pe-ripheral confluent mass of metanephric tissue centrally compressing the uninvolved renaltissue from which it is sharply defined. Histologically this is formed of confluent sheets ofrelatively differentiated metanephric epithelium, which appears as a hyperplastic form of thenodular renal blastema (lower right).

662 BOLANDE American Journal(36) of Pathology
bilateral renal enlargement with a bizarre external configuration mim-icking exaggerated fetal lobulations. A number of lesions fitting thisdescription have been reported as bilateral Wilms' tumor. Prolongedsurvival has been observed following treatment.48The ultimate fate and progression of nephroblastomatosis is unclear
and controversial, in particular the relationship to the development offrank Wilms' tumor. A number of investigators regard these lesions asnephroblastoma "in situ," implying that a certain proportion of thesecongenital lesions may give rise to Wilms' tumor later in life. The situa-tion is somewhat analogous to neuroblastoma "in situ." Bove et al 47found residua of nodular renal blastema in 25% of kidneys removed in thetreatment of patients with unilateral Wilms' tumor. Such foci were pres-ent in all their cases of bilateral Wilms' tumor. This presents strongevidence that nodular renal blastema and nephroblastomatosis may beimportant precursors of certain forms of malignant Wilms' tumor.
Neuroblastoma. Neuroblastoma is a clinically important and commonmalignant embryoma of early life. The tumor is formed of primitiveneuroblasts derived from the neural crest. It arises in the adrenal medullain 40% of cases or from some part of the abdominal, thoracic, pelvic, orcervical chains of autonomic ganglia in the remainder.
Although neuroblastoma may be an extremely malignant tumor, partic-ularly when it appears after age 1 year, it is characterized by higher curerates under 1 year (Table 5) and a remarkable incidence of spontaneousregression when clinically manifested at birth or within the first fewmonths of life.34'35 Spontaneous regression heavily influenced the excellentcure rates documented in this period of development.
Regresson occurs in three fashions: disappearance by cytolysis, hemor-rhagic necrosis leading to fibrocalcific residua, and cytodifferentiation toganglioneurofibroma (Figure 8). Nowhere is such regressive behaviordisplayed as dramatically and regularly as in congenital neuroblastoma.This phenomenon is all the more outstanding in the presence of wide-spread cutaneous and visceral metastases, which characterizes this dis-ease. Regressive behavior occurs in congenital neuroblastoma even in thepresence of osseous metastases, although skeletal involvement is an omi-
Table 5-Cure Rate and Age in Neuroblastoma
Age of onset Cure rate (%)
Neonatal 62-70Before 1 year 352nd year 19After 2nd year 5
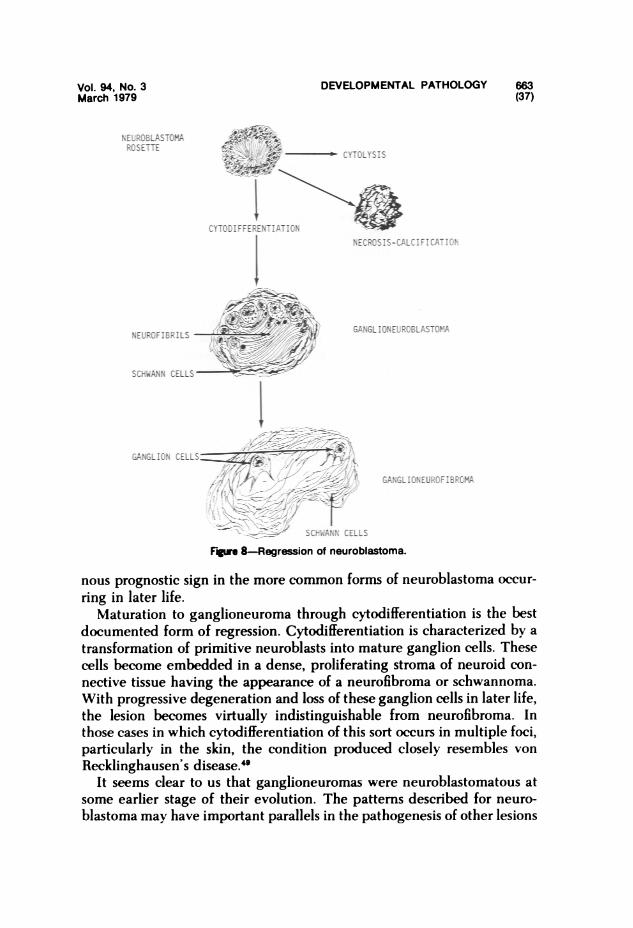
DEVELOPMENTAL PATHOLOGYVol. 94, No. 3March 1979
IICI1EUlIF F E E N TIIA T I Ul
CTI.LTSIS
Wl;
sim CB IL
IGANGLION
6MIllEWIfDI
Fgu 8-Regression of neuroblastoma.
nous prognostic sign in the more common forms of neuroblastoma occur-
ring in later life.Maturation to ganglioneuroma through cytodifferentiation is the best
documented form of regression. Cytodifferentiation is characterized by a
transformation of primitive neuroblasts into mature ganglion cells. Thesecells become embedded in a dense, proliferating stroma of neuroid con-
nective tissue having the appearance of a neurofibroma or schwannoma.With progressive degeneration and loss of these ganglion cells in later life,the lesion becomes virtually indistinguishable from neurofibroma. Inthose cases in which cytodifferentiation of this sort occurs in multiple foci,particularly in the skin, the condition produced closely resembles von
Recklinghausen's disease.4'It seems clear to us that ganglioneuromas were neuroblastomatous at
some earlier stage of their evolution. The patterns described for neuro-
blastoma may have important parallels in the pathogenesis of other lesions
663(37)

664 BOLANDE American Journal(38) of Pathology
of obscure origin. It is conceivable that cytodifferentiated or regressedresidua of certain embryonal or blastomatous tumors of early life arepathogeneically responsible for gliomas, chondrogenic hamartomas of thelung, medullary fibromas of the kidney, calcified adrenals, or some retinalgranulomas.
Neuroblastoma in situ. This condition merits serious consideration inany discussion of neuroblastoma.50 Neuroblastoma in situ refers to tinynodular aggregates of primitive neuroblasts in the central portion of theadrenal glands incidentally found in perinatal autopsies (Figure 9). De-pending on the intensity of search and the minimal size of the lesionacceptable to the investigator, neuroblastoma in situ has been estimatedto occur from 1 in 10 to 1 in 500 pediatric autopsies.51
It is generally agreed that the nodules are no longer identifiable afterage 3 months: they have either regressed or cytodifferentiated by this timeinto patterns indistinguishable from the normal constituents of the adre-nal medulla. A recent study suggests that such neuroblastic aggregates arealmost universally present in fetuses of 10 to 30 weeks' gestation but gen-erally regress following 20 weeks' gestation.51Neuroblastoma in situ, particularly over 2000 A, probably represents a
precursive or latent form of overt neuroblastoma. Its prevalence at birthcompared with the much lower incidence of clinically overt neuroblas-toma suggests a sizeable frequency of regression of incipient neuroblasto-mas. This pathogenetic relationship should be appreciated when attempt-ing to delineate heredofamilial patterns in neuroblastoma, as well as itsrelationship to other birth defects and neoplasms.
Neuroblastoma as a Neurocristopathy. The neurocristopathies havebeen defined as a group of dysgenetic, hamartomatous, and neoplastic
A D U L~~~AUT CORTEX
~~~~ ~~~FETAL CORTEX
NEUROBL STOMA "in situ" (>2 nwm)Figure 9-Neonatal adrenal containing a neuroblastoma "in situ" in the typical location. Atbirth, 80% of the cortex is composed of fetal reticularis cells (the fetal cortex), which begins toinvolute following birth. The definitive or adult cortex occupies the periphery of the cortex. Asthe fetal cortex involutes, the adult cortex expands into its typical pattern. In the first months oflife, a definitive medulla is not visible. The aggregations of primitive neuroblasts are thusreadily identified.

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 665March 1979 (39)
conditions sharing an origin in the maldevelopment of the neural crest orits derivatives 3 (Table 6). Neuroblastoma is a prominent and clear-cutmember of this cohort of diseases. The most primitive neuroblastomasconsist of cells identical to migratory neural crest cells in the embryo.Even in this primitive form, histochemical and ultrastructural evidenceof the neurosecretory character of these cells is present. Catecholaminegranules are present in their cytoplasm, demonstrable as dense-core gran-ules by electron microscopy or by their formalin-induced fluorescence. Ascvtodifferentiation of the tumor cells increases, the granules become moreprominent. The structural and histochemical features show that the cellsbelong to the APUD system of neuroendocrine cells as defined by Pearse,4indicating that they may be capable of secreting polypeptide hormonesin addition to biogenic amines.
Thus, a large proportion of neuroblastomas produce catecholamines,including norepinephrine, dopamine, metanephrine, normetanephrine,and vanilmandelic acid (VMA). These are secreted in the bloodstream andexcreated in the urine. VMA appears in approximately 85 to 90% ofneuroblastoma patients. Despite this secretory activity, less than 10% ofpatients develop hypertension. Opsomyoclonia, the jerky eye movementsreflective of severe cerebellar ataxia, may rarely develop, possibly fromthe action of neurosecretory products on the brain. Diarrhea is a morecommon symptom, and in some instances is intractable and so severe as tocause severe protein loss and malabsorption. This can occur with a rela-tivelv small tumor. It seems likely that these intestinal effects may be due
Table 6-The Neurocristopathies
SimpleDysgenetic
Hirschsprung's disease (aganglionic megacolon)AlbinismMandibulofacial dysostosisOtocephaly
NeoplasticNeuroblastomaPheochromocytomaMedullary carcinoma of thyroidMelanotic progonomaNonchromaffin paragangliomaCarcinoid tumors?Oat cell tumors?
Complex Neurocristopathic Syndromevon Recklinghausen's diseaseSipple's syndrome (MEN Type II)Multiple mucosal neuroma syndrome (MEN Type lIb)Neurocutaneous melanosisFamilial neuroblastoma and Hirschsprung's disease

666 BOLANDE American Journal(40) of Pathology
to action of other polypeptide hormones produced by these tumors. Casesof inappropriate ACTH production by neuroblastoma resulting in Cush-ing's syndrome have been described. Carcinoembryonic antigen is alsoproduced.The resemblance of the differentiated residua of neuroblastomatous
tissue to neurofibromatosis has been indicated. The coexistence of neuro-blastoma and neurofibromatosis has been described." Neuroblastomasoccurring with pheochromocytoma and non-chromaffin paragangliomaand with Hirschsprung's disease have also been described. The con-currences of these conditions are representative of neural crest mal-development and are beautifully illustrative of the neurocristopathy con-cept.
Medulloblastoma.'52 Medulloblastoma is the most common tumor ofthe brain found in young children. It arises in the region of the anteriormedullary velum, occupying a midline position within the cerebellum andthe lumen of the fourth ventricle. Because of its location, obstruction ofthe aqueduct of Sylvius and/or the fourth ventricle readily develops,giving rise to increased intracranial pressure. Medulloblasts constitutingthis tumor are generally small, round, and hyperchromatic. In moredifferentiated areas, the cells appear elongated, pyriform, or asteroid.Such areas suggest that the medulloblastomas are formed predominantlyof neuroglial precursors. Areas of rosette formation and rhabdomyoblasticdifferentiation may also be present.
Evidence of spontaneous maturation of medulloblastoma is not avail-able. Since the better differentiated areas of medulloblastomas may ap-proach the appearance of astrocytomas and oligodendrogliomas, Willis 3suggests that some medulloblastomas might mature into relatively lessaggressive gliomas.
Most medulloblastomas are malignant in their cellular behavior andclinical effects. Spread of the tumor occurs through the cerebrospinal fluidto involve the leptomeninges of the brain and spinal cord. Metastasesoutside the central nervous system may rarely occur. Peritoneal seedingmay occur when a ventriculoperitoneal shunt has been established torelieve the increased intracranial pressure.
Rhabdomyosarcoma.52'58 Rhabdomyosarcomas account for approxi-mately to 4 to 8% of malignant tumors of childhood, being less commonthan neuroblastoma or Wilms' tumor. Over one third of rhabdomyosar-comas occur in the head and neck, particularly in the orbit, paranasalsinuses, and middle ear. Approximately one quarter of cases occur in ex-tremities and the trunk, particularly in the skeletal muscles of the ex-tremities, retroperitoneum, buttocks, groin. or perianal region. Some 20%occur in the vaginal, vulval, vesical, and urethral submucosa in females

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 667March 1979 (41)
and in the prostatic, bladder, and urethral submucosa of males. Lesscommon sites are the extrahepatic bile ducts and nasopharynx. It isapparent that skeletal muscle may not normally be present at some ofthese sites of origin, indicating that these tumors may arise from a more orless pluripotent mesenchyme.
Four histologic types exist: embryonal, botryoid, alveolar (Riopellepattern), and pleomorphic (adult type). The embryonal type is composedof primitive undifferentiated cells. The botryoid form often occurs inhollow viscera and is characterized by the formation of polypoid grape-like clusters of tumor protruding into visceral lumina. This pattem is oftenencountered in the genitourinary tract, middle ear, or nasopharynx. Thetumor cells are primitive and embedded in myxoid matrix. The alveolartype is formed of clumps of embryonal cells separated by fibrous septums.Necroses of the centers of these cell masses give rise to an epithelial oralveolated pattern. In these latter three tumors, cytoplasm is scanty andcross-striations are only rarely found. However, myofibrillary material canbe readily found on electron microscopy. The pleomorphic or adult typeis composed of spindle cells with strap-like cytoplasm in which cross-striations can be more readily identified.
In general, rhabdomyosarcomas are highly invasive and lethal by virtueof invasion or hematogenous metastasis. The embryonal and botryoidtypes tend to be less aggressive. The prognosis tends to be better in younginfants.52
Retinoblastoma. 52 Retinoblastoma is an embryoma formed of the pre-cursors of rod and cone cells of the retina. These tumors arise in theposterior portions of the inner and outer nuclear layers of the retina; theymay be multifocal and bilateral. The disease occurs in approximately 1 in18,000 live-born children in the United States. Mortality is associated withdirect extension into the cranial cavity to involve the brain and lepto-meninges. Hematogenous metastases to bone, lymph nodes, liver, spleen,and kidney may occur. The mortality of this tumor has been so sharplyreduced by prompt enucleation of the affected globe(s) coupled withirradiation and chemotherapy that many affected individuals have lived toprocreate. The tumor, of neuroectodermal origin, shares many similaritieswith neuroblastoma in histologic appearance and behavior. Spontaneousregression may occur in retinoblastoma as in neuroblastoma and appearsto be heralded by hemorrhagic necrosis of the tumor, leading to fibroticand calcified residua. The frequency of spontaneous regression is notaccurately known but may be surprisingly high. It would also be inter-esting to learn if a retinoblastoma in situ existed, with biologic propertiesanalogous to neuroblastoma in situ.
Retinoblastoma is most notable among the embryomas of early life for

668 BOLANDE American Journal(42) of Pathology
its striking hereditary pattern, best shown in offspring of parents cured ofthe disease, particularly when the parent had bilateral tumors. In suchbilateral retinoblastomas, offspring are affected in a manner consistentwith the inheritance of a dominant gene. Yet offspring of parents withunilateral disease may also develop bilateral tumors and vice versa. Analy-sis of data indicates that 60% of retinoblastomas are nonhereditary andunilateral, 15% are unilateral and hereditary, and 25% are hereditary andbilateral. In the hereditary form, the tumors tend to appear a year earlierthan in the nonhereditary. It must be emphasized that the majority ofretinoblastomas are sporadic and without risk of tumor to the offspring,although some cases may be associated with a variety of malformations.Retinoblastoma kinships have been described in which children cured ofthe ocular lesions subsequently had osteogenic sarcoma of the longbones.36'51 The basis for this second cancer predilection is unknown.
Hepatoblastoma (Embryonal Carcinoma of the Liver)." 52 The term"hepatoblastoma" is used to designate embryonal malignancies of thehepatic parenchyma. The tumors in this group are of two types: a) theembryonal carcinoma composed solely or preponderantly of immatureliver cord cells and b) the mixed tumors containing, in addition, osteoidtissue, cartilage, bone, muscle, or sarcoma-like fibrous tissue. The imma-ture liver cells can be anaplastic or they may be arranged in a sinusoidalpattern clearly mimicking liver architecture. Within the sinusoids of suchtumors, islets of hematopoietic cells are found, despite the fact thathematopoiesis has ceased in the surrounding liver. This suggests that theembryonal hepatoblasts stimulate and support hematopoiesis. Differenti-ation of hepatoblasts into glycogen-containing cells approximating ma-ture hepatocytes can be seen. In some cases, atypical biliary ductules mayalso be formed in the tumor.
These tumors are usually found in children under 2 years of age andsometimes in newborn infants. Hepatoblastoma must be distinguishedfrom the adult-type of hepatocarcinoma because the prognosis is muchbetter in the former. The cells of the hepatoblastoma tend to be smallerand more closely backed than in hepatoma. Also, hepatoblastoma usuallyshows some foci of dysplastic or immature mesenchymal stroma, whileadult-type hepatoma does not.Some hepatoblastomas are extremely malignant, metastasizing early to
the liver itself, the lungs, and the peritoneum. Hepatoblastoma mayoccasionally be associated with the development of precocious puberty,due to ectopic gonadotrophin production. They also secrete a-fetoprotein.

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 669March 1979 (43)
Leukemia 15.4
The most common form of leukemia in childhood is the acute lympho-blastic form accounting for approximately 85% of cases. Its peak inci-dence occurs in patients between 3 and 5 years of age. The rest are acute:myelogenous, myelomonocytic leukemia, or erythroblastic leukemia.Chronic granulocytic leukemia rarely occurs in patients under 16 years ofage, and chronic lymphatic leukemia is virtually nonexistent in childhood.
In infants, acute myelogenous leukemia prevails. Leukemia may attimes be manifested at birth or shortly thereafter and is generally referredto as "congenital leukemia." It is typically of the acute myelogenous type.There is a 10- to 20-fold increase in the incidence of childhood leukemiain patients with Down's syndrome (mongolism); this is even more pro-nounced in this congenital type. Congenital leukemia is clinically mani-fested as purpura, skin infiltrates, and hepatosplenomegaly. There isusually marked leukocytosis, anemia, and thrombocytopenia. Myeloblastsand promyelocytes are present in the peripheral blood. The bone marrowis hyperplastic, consisting mainly of immature myeloid cells. The diseaseis easily confused with leukemoid reactions attending intrauterine or neo-natal infections, disseminated histiocytosis, and graft-vs-host reactions.The confusion with leukemoid reactions is furthered by the tendency forspontaneous and sometimes permanent remissions to occur in the first 4months of life. This phenomenon is striking in patients with Down'ssyndrome but has been observed in non-mongol infants as well.3 Ex-acerbation of the leukemic process later in the first year of life typicallyresults in rapid progression and death.
Pathologically, there is generalized infiltration of organs and tissueswith primitive myelopoietic elements. Particular caution must be used inbasing the diagnosis on infiltration of lymphoid tissues and liver, sincethese are often sites of extramedullary hematopoiesis, which might persistin a severe leukemoid reaction. Infiltration of organs, eg, kidney, skin, notnormally the site of extramedullary hematopoiesis in fetal life, is of greatdiagnostic significance.Lymphomas -
Malignant lymphomas account for approximately 10% of all solid tu-mors in childhood. The non-Hodgkin's group of lymphomas includelymphocytic, histiocytic, and stem-cell types and Burkitt lymphoma. Theprecise classification is still being formulated. These can present as a massin many locations, and there is marked variation in the site incidence in

670 BOLANDE American Journal(44) of Pathology
various series. The peak age is in mid-childhood. The development oflymphoma in childhood is often followed by the appearance of leukemia;the incidence varies widely with the site of presentation and histologictype. The incidence of this complication may be as high as 70% inchildren with undifferentiated lymphoma originating in lymph nodes.
Hodgkin's disease is rarely seen in patients younger than 2 years of age.Its incidence progressively rises after this time, reaching a peak in thethird decade. The pathology, progression, and therapy of the disease donot differ significantly from those in the adult. The staging laparotomy hasrecently fallen into disrepute in many pediatric centers since the markedlyenhanced risk of post-splenectomy sepsis in such children appears tooutweigh the positive aspects of this approach.55Reticuloendothelioses (Histiocytosis X)
It has been 35 years since the unitarian hypothesis was put forwarduniting Letterer-Siwe's disease, Hand-Schuller-Christian's disease, andeosinophilic granuloma as variations on the theme of reticuloendotheliosisor Histiocytosis X.1 The basic disease process involves the proliferation ofhistiocytes whose cytoplasm contains unique rods and filaments, ie, theLangerhans granules. Their proliferation results in the infiltration and/ornodular aggregation in many tissue and organs, including bone. The dis-ease occurs mainly in infants and young children, although adult caseshave been described.A simplified clinicopathologic approach to these disorders has recently
been proposed, suggesting that essentially two forms occur.56 In the first,there is a diffuse visceral infiltration of the proliferating histiocytes, whichoccurs in infants and is generally rapidly progressive and lethal; thisincludes cases previously called Letterer-Siwe's disease. In the secondform, there is the tendency for the histiocytes to form nodular gran-ulomatous masses, containing foam cells, multinucleated giant cells, andeosinophils. Often they involve bone. These lesions, while often destruc-tive and progressive, tend to regress and are rarely lethal. Hand-Schiuller-Christian's disease and eosinophilic granuloma fall in this category.Whether this simplistic approach is valid for all cases may be questioned.
The Fibromatoses of Infancy 57
The term "fibromatosis" refers to a host of lesions characterized by theproliferation of fibroblasts and/or fibromyoblasts. These have been vari-ously classified as to their location, histologic features, and age of onset.No matter how histologically aggressive these lesions appear, metastaticor true fibrosarcomatous behavior does not occur. Many are initiated inearly life. Their cellular features and behavior are similar.

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 671March 1979 (45)
We should like to call attention to the congenital generalized type.Congenital generalized fibromatosis is characterized by the presence ofmultiple discrete lesions throughout the body of newborn infants. Theseare presumed to represent multifocal origin. With the exception of thecentral nervous system, every organ and tissue seems susceptible to thedevelopment of the fibromatosis. The subcutis, gut, lung, and bone have aparticularly high incidence of involvement. In those with major visceralinvolvement, the prognosis is poor, particularly when the lung is involved.Despite the lethality of the visceral form, the majority of patients survive.More than half of the surviving patients have displayed spontaneousregression of the lesions. Singular congenital lesions of this sort may alsooccur and are usually embedded in the subcutaneous tissues of the upperextremities. This lesion is sometimes referred to as the "fibrous ham-artoma of infancy." Although a period of rapid growth may be presentearly on, local excision is usually curative.
Benignity of Neonatl ad Infane Twmors
It appears that certain tumors manifested at birth or shortly thereafterdisplay surprisingly benign behavior, sometimes despite malignant cellu-lar features, whereas a tumor of identical morphologic features would behighly lethal in later fife (Table 7). The realization of these facts abouttumors in early life has led to the postulation that an oncogenic "period ofgrace" exists, beginning in utero and extending through the first monthsof postnatal life.35 During this time, the host is resistant to the fullexpression or progression of malignant disease. Neoplasms seem repressedduring this period, tending toward benignity through arrested growth,regression, or cytodifferentiation. The mechanism(s) responsible for thisare unknown. It is conceivable that there is an immunologic basis for this,particularly where cytolysis and necrobiosis of cells are prominent. Cyto-
Table 7-Tumors of Infancy Showing Benign or Regressive Tendencies
Infantile congeners of Wilms' tumorNeuroblastoma
Neuroblastomas under 1 year of ageCongenital neuroblastoma (Type IV-S)Neuroblastoma in situ
Sacrococcygeal teratoma before 4 months of ageYolk sac carcinoma of the testis before 2 years of ageCongenital leukemiaCongenital and infantile fibromatosesInfantile embryonal rhabdomyosarcomasAdrenocortical carcinoma with hemihypertrophy before 1 year of ageRetinoblastoma?

672 BOLANDE American Journal(46) of Pathology
differentiation, on the other hand, may be biochemically induced by acertain hormonal milieu of the host at this time of life. The elucidation ofthese mechanisms is challenging and would have enormous implicationsin the understanding and, possibly, treatment of neoplasms in general.Although obviously not true for all neoplasms of very early life, the
oncogenic "grace period" concept probably has enough validity to bepractically applicable in clinical pediatric oncology. It should incline onetoward a certain prognostic optimism when confronted with a bizarreinfantile tumor whose natural behavior is uncertain. It should lead to anattenuation of the therapeutic intensity with which such a patient ismanaged. Chemotherapy, particularly in young infants, often results inbone marrow suppression, the consequences of which may be lethalsepticemia. Irradiation along the spinal axis may give rise to growthretardation and scoliosis. The mutagenicity of radiation and certainchemotherapeutic agents enhances the risk of developing a second canceror leukemia later in life.
Relationships of Neoplasia and TeratogenesisIn recent years, numerous and varied relationships have been shown to
exist between congenital malformations and neoplasms.36,56158'59 The ex-tent and complexity of these relationships is only beginning to be appreci-ated as more and more associations are documented. The kinship ofteratogenesis and oncogenesis appears to be of fundamental importancein human developmental pathobiology. Its appreciation significantly addsto the understanding of the neoplastic process in general. We shall exam-ine the existing body of data concerning these relationships and try toshow how in some instances they may be held accountable for the patho-genesis of certain neoplastic disorders.
Origin of Neoplasms in Anomalous or Dysplastic Tissue
An important factor predisposing to cancer in later life is the presenceof developmentally anomalous tissue. Thus cancer may develop in hetero-topias (fetal rests and choristomas), developmental vestiges, hamartomas,and dysgenetic gonads. Some oncologists have suggested that most tu-mors of early life arise in embryonal rests.36
Aberrant sexual development favors the development of gonadal neo-plasms in later life (Table 8). The undescended testes of male pseudo-hermaphrodites share the susceptibility to seminoma seen in cryptorchidnormal males.36 Of tumors occurring in undescended testes of malepseudohermaphrodites, 60% are seminomas. By contrast, Sertoli cell tu-mors are more common in the dysgenetic testes of the testicular femini-
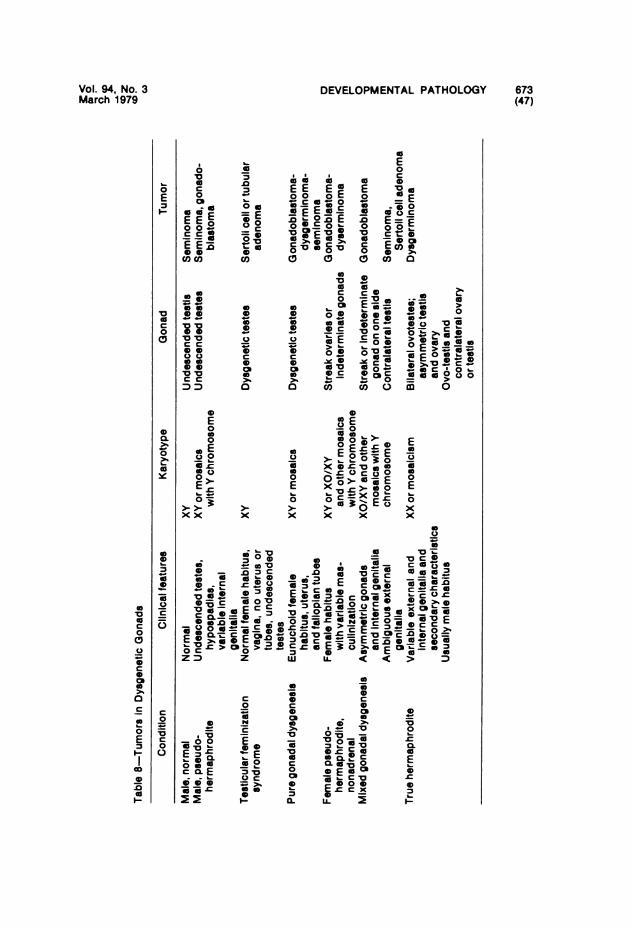
Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 673March 1979 (47)
0
~0 0 0oh 0 fl 0 .S-o E E4E E E sEo 00 oBE o
=E 0o. 0 = E0; 0 0
-0CI)0 a 0. CI-0cm
0 0e 0 Cl O 0c
0 0 0 0.0 -
V0 0 0 0 wo
0 0Z CI CO C.)a
0C 0E=ao -o
0 ~~~~~~~~050~~~~~~~
~ E E.r~ 0 0C)0
0 0~~E~~~~~E 0 E0N- ,>- 0 O
0 kV 0 E0 m E * >- 0
0 3: 0 0 0 0 'o000 j -* 0 x
0 =~~~~~.0:
o0 4D~0 'D ..: E ID~
& ~~~~~~E 0-a -a0 la 0 ,
CL O ~~~~~0- 0 0 '
0 ~~~~~~~ C ~ ~ ~ ~E0 E- EUs0. &-*0 =1 )cr
0 jC 00 2'
0 4I- I- 0. LL I-4

674 BOLANDE American Journal(48) of Pathology
zation syndrome,36 occurring in approximately 20% of these patients.A remarkable form of neoplasm occurring in dysgenetic gonads is the
gonadoblastoma or gonocytoma.36f51 These tumors most often arise in agonad of indeterminate nature in a phenotypic female. Generally theseindividuals are XY, although XO/XY mosaicism may be present. The Ychromosome is required for the development of a gonadoblastoma.
Malignant Transformation in Hamartomas
Hamartomas are sometimes the seat of malignant transformation. Thisusually occurs later in life. It is difficult to determine the actual increase insusceptibility to cancer over nonhamartomatous tissue, and in some ham-artomas the incidence of cancer is very high. Approximately 30% ofpatients with von Recklinghausen's disease may develop sarcomatoustransformation of neurofibroma after age 50.,36 Virtually all patients withmultiple familial polyposis of the colon eventually develop carcinoma ofthe colon.The salient features of malignant transformation in hamartomas and
hamartoses are documented in Table 3. Malignancy seems to be docu-mented most frequently in genetically determined hamartoses rather thanin the sporadic isolated lesions. The basis for this is unknown.
Increased Expectancy of Neoplasms in Specific Teratologic Conditions
Teratologic disorders predispose to neoplasms. These are summarizedin Table 9. It is important to emphasize specific instances.36
Aniridia-Wilms' Tumor
Aniridia is a dominantly inherited condition affecting no more than1:50,000 of the general population. Approximately 30% of cases aresporadic and presumed to represent new germinal mutations. The pres-ence of this congenital anomaly somehow renders the affected child proneto the development of Wilms' tumor. Aniridia is present in 1 of 80 cases ofWilms' tumor. Patients with sporadic aniridia seem more at risk, becauseapproximately one third of these develop Wilms' tumor. Additional butless common features of the aniridia-Wilms' tumor syndrome are micro-cephaly, physical and mental retardation, anomalies of the genitourinarytract, and recurved aural pinnae; a variety of developing Wilms' tumor ishighest when sporadic aniridia is accompanied by genitourinary malfor-mations and mental retardation.

DEVELOPMENTAL PATHOLOGY 675(49)
Table 9-Specific Teratologic Disorders Associated With Neoplasm
Anomaly Tu mor
Dysmorphic syndromesAniridiaHemihypertrophy
Beckwith syndrome
Genitourinary tract malformation andpseudohermaphroditism
Basal cell nevus syndrome
Poland syndromeChromosomal abnormalities
Mongolism (trisomy 21)
13q- syndromeChromosomal breakage syndromes
Fanconi anemia
Bloom syndrome
Ataxia telangiectasia
Wilms' tumorWilms' tumorHepatoblastomaAdrenocortical carcinomaAdrenocortical carcinomaWilms' tumor and nephroblastomatosisWilms' tumor and nephroblastomatosis
Basal cell carcinomaMedulloblastomaRhabdomyosarcomaLeukemia
LeukemiaRetinoblastomaRetinoblastoma
LeukemiaSquamous cell carcinomaHepatoma (androgen-induced)LeukemiaGastrointestinal carcinomaLymphomaLeukemiaOthers
Hemihypertrophy
Congenital hemihypertrophy is a condition characterized by gross asyrn-
metry of the body. Although detectable at birth, it usually becomes more
apparent with increasing age. Many cases are unrecognized by virtue oftheir subtlety. It seems to occur more often on the right side. Thesechildren are at unusually high risk for the development of one of threetypes of malignancy: Wilms' tumor, hepatoblastoma, and adrenocorticalcarcinoma.59 Although the majority of the tumors develop on the hyper-trophied side of the body, 30% develop contralaterally. It has been shownthat an excessive incidence of pigmented nevi and vascular hamartomasalso occur with hemihypertrophy.
Sixteen cases of adrenocortical carcinoma in patients with hemihyper-trophy have been described. These cases are clinically striking becausethey often present as an endocrinopathy, ie, Cushing's syndrome or
precocious puberty. Those patients who have their tumors diagnosed andremoved under 1 year of age seem to have a better prognosis than thosediagnosed and treated later in life. One of the surviving patients sub-
Vol. 94, No. 3March 1979

676 BOLANDE American Journal(50) of Pathology
sequently developed a fatal Wilms' tumor. These observations underlinethe necessity of the early diagnosis of hemihypertrophy and close surveil-lance of affected individuals.
Omphalocele-Macroglossia Syndrome (Beckwith-Wiedemann Syndrome)
The Beckwith-Wiedemann syndrome is a complex constellation ofanomalies, including oophalocele, fetal gigantism, macroglossia, cyto-megaly of the fetal adrenal cortex, hyperplasia of gonadal interstitial cells,renal medullary dysplasia, and hyperplastic visceromegaly in several otherorgans, including the kidneys and pancreas. The pancreas exhibits islethyperplasia. In some cases, neonatal hypoglycemia may prove fatal. Inapproximately 1 in 7 cases, postnatal development is associated with thedevelopment of hemihypertrophy. As in isolated hemihypertrophy, Beck-with's syndrome has been found to be excessively associated with adreno-cortical carcinomas and Wilms' tumor and nephroblastomatosis.60 Malig-nant neoplasm occurs in approximately 8% of cases. When hemihyper-trophy is present, tumors develop in 1 in 5 cases; when hemihypertrophyis absent, neoplasm occurs in only 1 in 18 cases. The cytomegaly of thefetal adrenal cortex has been viewed as a precursor of the adrenocorticalcarcinoma.
Basal Cell Nevus Syndrome
The principal features of the basal nevus or nevoid basal cell carcinomasyndrome consist of multiple basal cell hamartomas of the skin, multipleepithelial-lined jaw cysts, and multiple skeletal anomalies, including sco-liosis, spina bifida, bifid ribs, fused and hemivertebrae, and dolicho-cephaly with prominent supraorbital ridges and frontal eminences, abroad nasal root, and briding of the sella turcica. There may be agenesisof the corpus callosum and low intelligence. Ocular abnormalities may in-clude congenital cataracts, coloboma, and glaucoma. In addition, theremay be dyskeratosis of the palms and soles. The skin lesions tend totransform into frank basal cell carcinomas before 15 years of age. Inaddition, approximately 20% of children with the basal cell nevus syn-drome develop medulloblastoma at an early age. Lipomas and fibromasmay also be present. The basal cell nevus syndrome is inherited as anautosomal dominant trait with a high degree of penetrance.
Genitourinary Tract Malformations and Wilms' Tumor
There is an excess concurrence of Wilms' tumor with genitourinarytract malformations. Approximately 6% of patients with Wilms' tumorexhibit upper urinary tract anomalies, including horseshoe kidneys, ecto-

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 677March 1979 (51)
pic or solitary kidneys, hvpoplastic kidney, and duplication of the upperurinarv tract, ie, double kidneys, pelves, and ureters.
Wilms' tumor has been described in association with male pseudo-hermaphroditism manifested by cryptorchidism and hypospadias. Baraketet al 61 suggested that, in addition to Wilms' tumor, an unusual con-currence of congenital "nephron disorders" may occur in this teratologic-neoplastic complex. Bv nephron disorders, they refer to nephropathiesmanifested as the congenital nephrotic syndrome and infantile glomerulo-nephritis. Many of these disorders may represent a form of cvstic ne-phropathy resulting from the cytodifferentiation and regression of nodularrenal blastema or nephroblastomatosis. They point out that various com-binations of pseudohermaphroditism, congenital nephron disorders, andWilms' tumor have been reported. Thev suggest that these concurrencesare linked by common teratogenic factors affecting renal embry-ogenesis.
Poland Syndrome and Leukemia
The two main components of the Poland syndrome are svrm-brachydactyly and a pectoralis muscle defect. The muscle defect is aunilateral aplasia of the sternal portion of the pectoralis major muscle.The symbrachvdactJlv is ipsilateral and is characterized by syndactylyand short digits of the hand. At least 6 patients with acute leukemia havebeen described with this teratologic disorder.-"
Nephroblastomatosis, Nodular Renal Blastema, and Related Anomalies
There is a distinct association of these lesions with teratologic disorders.Bilateral nephroblastomatosis has been described in association with tri-somv 18, splenic agenesis and malformation of the liver, the Beckwith-Wiedemann syndrome, Klippel-Trenaunay syndrome, and familial fetalgigantism.47","The nephroblastomatosis complex, as pointed out earlier, must be
distinguished from Wilms' tumor, particularly bilateral Wilms' tumor. Itis our contention that nephroblastomatosis, or Wilms' tumor arising in it,shows the most striking teratologic associations and hereditary patterns.
Sacrococcygeal Teratoma and Anomalies
Approximately 105 of patients with sacrococcygeal teratomas haveassociated congenital anomalies. A strong familial history of twinning waspresent in 15% of the cases. The defects observed included imperforateanus, rectovaginal fistula, ectopia vesicae with epispadias, tracheo-esopha-geal fistulae, talipes equinovarus, hydrocephalus, and duplications ofhindgut and genitourinary tract. These anomalies occurred in 6 of 63

678 BOLANDE American Journal(52) of Pathology
sacrococcygeal teratomas. Some of the major malformations seen here arebest attributed to local growth disturbances secondary to the presence ofthe teratoma during intrauterine development.62
Cytogenetic Abnormalities and Chromosomal Breakage Syndromes
Specific chromosomal abnormalities are directly related to teratologicsyndromes, and in selected instances they are associated with an increasedincidence of neoplasms. Typical of this is mongolism, in which acutelymphoblastic leukemia occurs 10 to 20 times more often than in thegeneral pediatric population. In addition, chromosome fragility andbreakage without specific karyotype abnormality occur in a group ofheredofamilial disorders known as the chromosomal breakage syn-dromes.63 These syndromes, which are recessively inherited, are alsoassociated with a high incidence of neoplasms (Table 9).
Pathogenetic Mechanisms
It is clear that links between oncogenesis and teratogenesis are numer-ous and varied. An explanation for all these relationships is not available,yet certain speculations are possible. Rapidly dividing cells are a prerequi-site for teratogenesis and oncogenesis. During mitosis, the cell is mostvulnerable to structural changes in DNA, to disruption of transcription toRNA, and to subsequent translation into protein-enzyme synthesis. Theresting cell is more capable of a certain degree of repair and restoration ofthis system than is the dividing cell. Mitotic activity is more universallypresent throughout the body in intrauterine life than at any later stage inthe life cycle.Many agents known to be carcinogenic postnatally are teratogenic to
the fetus or embryo.58 Some such as irradiation, estrogens, and the alkyl-nitrosoureas give both effects when administered prenatally.36'64 Studies ofJapanese survivors of atomic bomb explosions have shown that thoseindividuals heavily exposed to radioactive fallout in utero have an in-creased incidence of microcephaly and mental retardation, but this popu-lation has not developed an excess of neoplasms. The population exposedduring childhood have developed an increased incidence of leukemia andother cancers.36 It seems clear from experimental studies with these agentsthat the timing of the initiating event(s) may be critical in determiningthe form of the outcome. The degree of cytodifferentiation and themetabolic or immunologic state of the organism may determine whetherthe effect is teratogenic and/or oncogenic. Teratogenesis and oncogenesismay represent developmentally different reactions to a special type ofinjury, with teratogenesis being the more primitive reaction. The effect of

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 679March 1979 (53)
an inciting agent in early gestation would be teratogenic, with a gradualshifting toward combined oncogenic-teratogenic forms in later gestationand, ultimately, pure oncogenic expression following late gestational orposnatal exposure. The coexistence of tumors and anomalies might beexplained on this basis.On the other hand, a primary teratologic event in the fetus may in some
fashion predispose the organism to a secondary oncogenic influence inlater life. This might explain the neoplastic transformation occurring inhamartomas and hamartoses, developmental vestiges, heterotopias, anddysgenetic tissues. It is also possible that these structures might harbor alatent oncogene which may have been expressed as an anomaly in intra-uterine life.
Environmental insults in later life, eg, trauma, irradiation, infection,hormones, drugs, and other chemical agents might cause a de-repressionor activation of this cryptic genome and result in the production of cancer.For example, the heightened hormonal stimulation accompanying pu-berty might be largely responsible for the development of tumors indysgenetic gonads. These anomalous tissues might respond abnormally toincreasing levels or cyclic fluctations in gonadotropins in adolescence. Ineffect, these hormones could exert a carcinogenic or cocarcinogenic influ-ence. Similar mechanisms may be involved in the development of theclear cell adenocarcinoma of the adolescent vagina transplacentally in-duced by diethylstilbestrol taken by their mothers during their earlygestation to prevent abortion." The transplacental effects of this agent onthe fetus leaves vestiges of anomalous sex duct differentiation such asvaginal adenosis. These anomalous vestiges respond in approximately 1 of4000 exposed girls by malignant transformation.
In those teratologic conditions associated with chromosomal instability,fragility, or imbalance, the unusual tendency to develop cancer is paral-leled by a predisposition of cell cultures from these patients to be trans-formed by oncogenic virus. Certain cytogenetic derangements rendercells unusually sensitive to malignant transformation by many carcino-genic agents. When this is associated with an immune-deficiency state, asis typical of many of the conditions in this group, a failure in the host'ssurveillance system against cancer might be anticipated. This would allowfor the establishment and growth of a clone of cancer cells, culminating ina frank neoplasm.The two-hit hypothesis of oncogenesis as forwarded by Knudson " has
special relevance in these considerations. The theory, originating fromstatistical analysis, embodies within a different semantic framework manyof the aforementioned concepts. It assumes that carcinogenesis is related

680 BOLANDE American Journal(54) of Pathology
to discrete mutational events occurring at a random and constant averagerate. For a tumor to develop, two such mutational events must occursequentially: the first may be prezygotic or postzygotic, and the second isalways postzygotic. When the first mutation is prezygotic, thus affectingall somatic cells, the tumor is hereditary, usually bilateral or multiple, andof very early appearance in life. When the first mutation is postzygotic,affecting only certain somatic cells, the tumor is nonhereditary and sin-gular. Knudson has gathered impressive statistical evidence in support ofthis hypothesis, particularly in comparing sporadic and familial embry-omas of early life, ie, retinoblastoma, Wilms' tumor, and possibly neuro-blastoma. It is not difficult to integrate this theory into the explanation ofthe relationships between teratogenesis and oncogenesis. It has relevanceif the first hit were expressed as a heritable anomaly, constellation ofanomalies, or hamartoid lesion and if the subsequent appearance of ma-lignant tumor were expressive of the second hit. The increased incidenceof tumor in these conditions depends on a heightened susceptibility ofthe defective genome to the oncogenic transformation of the second hit.The theory may be further applicable in nonhereditary combinations ofanomaly and tumor as well. Here one must assume a postzygotic eventoccurring in utero, which is represented as a birth defect or dysmorphicsyndrome. A population of cells must then be susceptible to malignanttransformation by a second hit, either within the anomalous tissue orextrinsic to it.
References1. Bolande RP: Cellular Aspects of Developmental Pathology. Philadelphia, Lea &
Febiger, 19672. Yamada KM: Cell morphogenetic movements. Handbook of Teratology. Vol 2:
Mechanisms and Pathogenesis. Edited by JG Wilson, FC Fraser. New York, PlenumPublishing Corp., pp 199-230
3. Bolande RP: The neurocristopathies: A unifying concept of disease arising inneural crest maldevelopment. Hum Pathol 5:409429, 1974
4. Pearse AG: The APUD cell concept and its implications in pathology. Pathol Annu9:2741, 1974
5. Saxen L: Abnormal cellular and tissue interactions.2 pp 171-1976. Roberts DF, Thomson AM: The Biology of Human Fetal Growth. London, Taylor
& Francis, 19767. Rosan RC, Lauweryns JM, Brand MM: Recent advances in the pathologic aspects
of neonatal respiratory distress. Pathol Annu 8:407452, 19738. DeSa DJ: The spectrum of ischemic bowel disease in the newborn. Perspect Pediatr
Pathol 3:273-309, 19769. Norman M: Perinatal brain damage. Perspect Pediatr Pathol 4:41-92, 1978
10. Ryan GB, Majno G: Acute inflammation: A review. Am J Pathol 86:185-276, 197711. Lynch MJ: Mechanisms and defects of the phagocytic systems of defense against
infection. Perspect Pediatr Pathol 1:33-115, 1973

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 681March 1979 (55)
12. Adinolfi M: Human complement: Onset and site of synthesis during fetal life. Am JDis Child 131:1015-1023, 1977
13. Dixon JB: Inflammation in the foetal and neonatal rat: The local reactions to skinbums. J Pathol Bacteriol 80:73-82, 1960
14. Schwartz LW, Osburn BI: An ontogenic study of the acute inflammatory reactionin the fetal rhesus monkey. I. Cellular response to bacterial and nonbacterial irri-tants. Lab Invest 31:441-453, 1974
15. Somasundaram K, Prathap K: Intra-uterine healing of skin wounds in rabbit foe-tuses. J Pathol 100:81-86, 1970
16. Dixon JB: Histamine, 5-hydroxytryptamine, and serum globulins in the foetal andneonatal rat. J Physiol 147:144-152, 1959
17. Selye H: Calciphylaxis. Chicago, The University of Chicago Press, 196218. Warkany J: Congenital Malformations. Chicago, Year Book Medical Publishers,
Inc., 197119. Holmes LB: Congenital malformations. N Engl J Med 295:204-207, 197620. Bolande RP, Kalousek DK: Teratogenesis and the pathogenesis of congenital mal-
formations. The Essentials of Human Reproductive Biology: A Basis for ClinicalPractice. Edited by F Naftolin, D Hamilton. Cambridge, Mass, M. I. T. Press, 1978
21. Wilson JG: Current status of teratology: General principles and mechanisms de-rived from animals studies. Handbook of Teratology. Vol 1: General Principles andEtiology. Edited by JG Wilson, FC Fraser. New York, Plenum Publishing Corp.,1977, pp 47-76
22. Kurent JE, Sever JL: Infectious disease.2' pp 2256023. Esterly JR, Oppenheimer EH: Intrauterine rubella infection. Perspect Pediatr
Pathol 1:313-8, 197324. Brent RL: Radiations and other physical agents.21 pp 153-22425. Wilson JG: Embryotoxicity of drugs in man.2' pp 3095626. Wilson JG: Environmental chemicals.2' pp 357-38627. McKusick VA: Mendelian inheritance in man. Catalogs of Autosomal Dominant,
Autosomal Recessive, and X-Linked Phenotypes, Fourth edition. Baltimore, JohnsHopkins University Press, 1975
28. Nora JJ, Fraser FC: Medical Genetics: Principles and Practice. Philadelphia, Lea &Febiger, 1974
29. Smith DW: Recognizable Patterns of Human Malformation: Genetic, Embryologicand Clinical Aspects. (Major Problems in Clinical Pediatrics, Vol 7.) Philadelphia,W. B. Saunders Co., 1970
30. Hamerton IL: Human Cytogenetics, Vol 2. New York, Academic Press, Inc., 197131. Seemayer TA, Bolande RP: The graft-vs-host reaction: A pathogenetic mechanism
of experimental and human disease. Perspect Pediatr Pathol (In press)32. Stanbury JB, Wyngaarden JB, Frederickson DS: The Metabolic Basis of Inherited
Disease, Third edition. New York, McGraw-Hill Book Co., 197233. Young JL Jr, Miller RW: Incidence of malignant tumors in U. S. children. J Pediatr
86:254-258, 197534. Everson TC, Cole WH: The Spontaneous Regression of Cancer. Philadelphia,
W. B. Saunders Co., 196635. Bolande RP: The benignity of neonatal tumors and the concept of cancer repres-
sion in early life. Am J Dis Child 122:12-14, 197136. Bolande RP: Neoplasia of early life and its relationships to teratogenesis. Perspect
Pediatr Pathol 3:145-183, 197637. Riley PA, Sutton PM: Why are ovarian teratomas benign whilst teratomas of the
testis are malignant? Lancet 1:1360-1362, 197538. Erickson RP, Gondos B: Altemative explanations of the differing behaviour of
ovarian and testicular teratomas. Lancet 1:407-409, 1976

682 BOLANDE American Journal(56) of Pathology
39. Willis RA: The embryonic tumors and teratomas. The Borderland of Embryologyand Pathology, Second edition. London, Butterworths, 1962
40. Gross RA, Clatworthy HW Jr, Meeker IA Jr: Sacrococcygeal teratomas in infantsand children: A report of 40 cases. Surg Obstet Gynecol 92:341-354, 1951
41. Gonzalez-Crussi F, Roth LM: The human yolk sac and yolk sac carcinoma: Anultrastructural study. Hum Pathol 7:675-691, 1976
42. Gonzalez-Crussi F: The human yolk sac (endodermal sinus) tumors. Perspect Pe-diatr Pathol (In press)
43. Kissane JM, Smith MG: Pathology of Infancy and Childhood, Second edition. St.Louis, C. V. Mosby, 1975
44. Bolande RP: Congenital and infantile neoplasia of the kidney. Lancet 2:1497-1499,1974
45. Bolande RP: Congenital mesoblastic nephroma of infancy. Perspect Pediatr Pathol1:227-250, 1973
46. Chatten J: Epithelial differentiation in Wilms' tumor: A clinicopathologic ap-praisal. Perspect Pediatr Pathol 3:225-254, 1976
47. Bove KE, McAdams AJ: The nephroblastomatosis complex and its relationship toWilms' tumor: A clinicopathological treatise. Perspect Pediatr Pathol 3:185-223,1976
48. de Chadarevian J-P, Fletcher BD, Chatten J, Rabinovitch HH: Massive infantilenephroblastomatosis: A clinical, radiological, and pathological analysis of four cases.Cancer 39:2294-2305, 1977
49. Bolande RP, Towler WF: A possible relationship of neuroblastoma to von Reck-linghausen's disease. Cancer 26:162-175, 1970
50. Beckwith JB, Perrin EV: In situ neuroblastomas: A contribution to the naturalhistory of neural crest tumors. Am J Pathol 43:1089-1104, 1963
51. Bolande RP: Childhood tumors and their relationship to birth defects. Genetics ofHuman Cancer. Edited by JJ Mulvihill, RW Miller, JF Fraumeni. New York, RavenPress, 1977, pp 43-75
52. Jones PG, Campbell PE: Tumors of Infancy and Childhood. Oxford, BlackwellScientific Publications, Ltd., 1976
53. Maurer HM, Moon T, Donaldson M, Fernandes C, Gehan EA, Hammmond D, HaysDM, Lawrence W, Newton W, Ragab A, Raney B, Soule EH, Sutow WW, Tefft M:The Intergroup Rhabdomyosarcoma Study: A preliminary report. Cancer 40:2015-2026,1977
54. Nathan DG, Oski FA: Hematology of Infancy and Childhood. Philadelphia, W. B.Saunderc Co., 1974
55. Singer DB: Postsplenectomy sepsis. Perspect Pediatr Pathol 1:285-312. 197356. Newton WA, Hamoudi AB: Histiocytosis: A histologic classification with clinical
correlation. Perspect Pediatr Pathol 1:251-283, 197357. Rosenberg H, Steinback WD, Spjut HJ: The fibromatoses of infancy and child-
hood. Perspect Pediatr Pathol 4:269-348, 197858. DiPaolo JA, Kotin P: Teratogenesis-oncogenesis: A study of possible relationships.
Arch Pathol 81:3-23, 196659. Miller RW: Relation between cancer and congenital defects in man. N Engl J Med
275:87-93, 196660. Sotelo-Avila C, Gooch WM: Neoplasms associated with the Beckwith-Wiedemann
syndrome. Perspect Pediatr Pathol 3:255-272, 197661. Barakat AY, Papadopoulou AL, Chandra RS, Hollerman CE, Calcagno PL: Pseu-
dohermaphroditism, nephron disorder and Wilms' tumor: A unifying concept. Pedi-atrics 54:366-369, 1974
62. Fraumeni JF Jr, Li FP, Dalager N: Teratomas in children: Epiaemoiogic features. JNatl Cancer Inst 51:1425-1430, 1973

Vol. 94, No. 3 DEVELOPMENTAL PATHOLOGY 683March 1979 (57)
63. Hecht F, McCaw BK: Chromosomal instabilitv svndromes.5 pp 105-12464. Rice JM: An ov,er-iew of transplacental chemical carcinogenesis. Teratolog- 8:113-
125, 197365. Herbst AL, Poskanzer DC, Robboy SJ, Friedlander L, Scully RE: Prenatal exposure
to stilbesterol: A prospective comparison of exposed female offspring with unexposedcontrols. N Engl J Med 292:334-339, 19735
66. Knudson AG Jr: Mutation and human cancer. Adv Cancer Res 17:317-352, 1973
AcknowledgnentsI am deeply grateful to Ms. Jean Pr&novost and Ms. Wendy Greenspoon for the typing of the
manuscript and to Ms. Jennifer French for the photographic reproductions.

684 BOLANDE American Journal(58) of Pathology
[End of Article]