development and rheological investigation of novel alginate/n-succinylchitosan hydrogels
TRANSCRIPT

Development and Rheological Investigation of NovelAlginate/N-Succinylchitosan Hydrogels
M. R. NOBILE,1 V. PIROZZI,1 E. SOMMA,1 G. GOMEZ D’AYALA,2 P. LAURIENZO2
1Department of Chemical and Food Engineering, University of Salerno,Via Ponte Don Melillo 84084 Fisciano, Salerno, Italy
2Institute of Polymer Chemistry and Technology (ICTP) - CNR, Via Campi Flegrei 34, 80078 Pozzuoli, NA, Italy
Received 30 November 2007; revised 13 March 2008; accepted 17 March 2008
DOI: 10.1002/polb.21450
Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: In the present article alginate hydrogels and novel hydrogels based on
blends of alginate/N-succinylchitosan have been realized in water solution at neutral
conditions. The gels have been obtained by crosslinking via the internal setting
method using calcium carbonate (CaCO3) as calcium ions source. A rheological inves-
tigation of both the plain alginate and the alginate/N-succinylchitosan blend hydro-
gels has been performed by means of oscillatory dynamic measurements. The effect of
the inclusion of different amounts of CaCO3 on the critical deformation (cc) character-
izing the limit of the linear viscoelastic regime has been studied for the plain alginate
gels. The frequency response in small amplitude oscillatory experiments of the plain
alginate gels has been investigated in terms of the storage (G0) and loss (G00) modulus
behavior. The dynamic data have been interpreted in terms of the Friedrich and Hey-
mann model. The inclusion of the N-succinylchitosan, in the range 10–50% w/w, had
no effect on the cc values. On the contrary, when the 10% w/w of the N-succinylchito-
san is added to the plain alginate gels, a significant increase in the storage modulus
values is recorded for all the systems analyzed. The gelation kinetics has been inves-
tigated and the results indicate that the kinetics process can be accelerated increas-
ing the percentage of Ca12 ions and/or including the N-succinylchitosan in the plain
alginate systems. Finally, the morphological analysis of scaffolds obtained from the
hydrogels through freeze-drying revealed an interconnected porous structure. VVC 2008
Wiley Periodicals, Inc. J Polym Sci Part B: Polym Phys 46: 1167–1182, 2008
Keywords: alginate gels; biomaterials; gels; rheology
INTRODUCTION
Hydrogels are a class of materials of growing in-
terest in view of their numerous biomedical
applications. They are applied as space-filling
agents, as drug delivery vehicles and as three-
dimensional structures that organize cells to
direct the formation of a damaged tissue.1–3
Naturally derived polysaccharide-based hydro-
gels are appealing scaffold materials because
they are either components of or structurally
similar to the natural extracellular matrix of
many tissues, can often be processed under rela-
tively mild conditions and are able to absorb body
fluid for transfer of cell nutrients and metabolites
through the material. Such hydrogels play an
important role also in the field of drug release
and cosmetics, particularly for their high bio-
compatibility and low cost. Among them, algi-
nate-based hydrogels are ones of the commonly
investigated.4–8
Correspondence to: M. R. Nobile (E-mail: [email protected])
Journal of Polymer Science: Part B: Polymer Physics, Vol. 46, 1167–1182 (2008)
VVC 2008 Wiley Periodicals, Inc.
1167

Briefly, alginate is a safe biocompatible poly-
saccharide obtained from marine algae. It is a
linear polysaccharide copolymer of (1-4)-linked b-
mannuronic acid (M) and a-guluronic acid (G)
monomers. Within the alginate polymer, the M
and G monomers are sequentially assembled in
either repeating (MM or GG) or alternating (MG)
blocks. Alginate hydrogels are formed when diva-
lent cations, such as Ca21, cooperatively interact
with blocks of G monomers to form ionic bridges
between different polymer chains.9 Hydrogel sys-
tems based on alginate are also intended for
injectable scaffolds,10 that means that they can
be easily injected to the wanted sites and solidi-
fied in situ. Alginate crosslinking via calcium
ions is viewed as a mild process involving non-
toxic components and pH, osmolarity, and tem-
perature suitable for preserving mammalian
cells survive. On the other hand, it is important
to note that ionically crosslinked alginates show
low mechanical stability.11,12 To overcome single
polymer-based hydrogel drawbacks as weak me-
chanical properties, in the recent years heteroge-
neous hydrogels derived from polymer blends,
block copolymers, or interpenetrating polymer
networks, have been widely investigated. Blend-
ing is a simple method to combine the ad-
vantages of different polymers. The resulting
polymer blends may show synergistic proper-
ties,13,14 and are expected to have improved me-
chanical characteristics.
Because of its intrinsic antibacterial activity
and biocompatibility,15 chitosan is a good part-
ner for alginate in hydrogel development.16,17
Chitosan is a bioresorbable natural polysac-
charide, obtained by partial de-acetylation of
chitin.18–20 Structurally, chitosan is a linear
polysaccharide consisting of b(1-4) linked D-glu-
cosamine residues with a variable number of
randomly located N-acetylglucosamine groups.
Chitosan-based hydrogels can be formed by
change in pH value, ionic21 or covalent cross-
linking.22 A combination of chitosan with polyol
anionic salts has been reported to give tempera-
ture-controlled pH-dependent chitosan solu-
tions.23 Nevertheless, the acidic solubility and
gelation methods employed so far will surely
limit the application of chitosan as biopolymer
for in situ forming gels. Chemical derivatization
of chitosan, based on the reactivity of the pri-
mary amino groups, is a reported method to
induce solubility in neutral aqueous solutions,24
but the possibility to crosslink chitosan via ionic
interactions or by further reactions with chemi-
cal crosslinking agents such as glutaraldehyde
or genipin is strongly reduced upon modifica-
tion. The realization of blends with the easily
crosslinkable alginate may also represent a way
to overcome this problem.
In a previous work25 the preparation and
characterization of a novel composite based on
calcium sulfate (CaSO4) embedded in alginate or
in a blend of alginate with a water soluble chito-
san derivative (N-succinylchitosan, sCh) has
been reported. It has been found that the algi-
nate/sCh-based materials showed a considerable
improvement in Young modulus and yield
strength values with respect to the plain algi-
nate-based material, with a maximum corre-
sponding to a given composition of the alginate/
sCh blend. Such results have been tentatively
attributed to a synergistic effect of the N-succi-
nylchitosan in chelating calcium ions during the
alginate gelation process, which occurs as a con-
sequence of the slow calcium ions release from
CaSO4.
On the basis of this result, in the present ar-
ticle a novel hydrogel based on alginate and N-
succinylchitosan has been prepared in water so-
lution at neutral conditions. The gels have been
obtained by crosslinking via the internal setting
method,26,27 using calcium carbonate (CaCO3) as
calcium ions source. This technique allows a
controlled gelation of alginate through a slow
release of calcium ions at neutral pH, thus lead-
ing to the formation of a very regular gel
network. Uniform cell distribution and well-
controlled material properties are, indeed, neces-
sary in tissue engineering applications.28,29 The
plain alginate gels obtained by the CaCO3-GDL
(D-glucone-d-lactone) system are themselves
characterized by satisfactory mechanical pro-
perties for tissue engineering applications.28
Therefore, aim of this article is to show that the
inclusion of the N-succinylchitosan can lead to a
novel biocompatible hydrogel characterized by
good mechanical properties. At this regard, it
will be shown that a rheological study can be
successfully used to investigate the physical
characteristics of both the alginate gels and the
novel hydrogels, as also reported in the litera-
ture for different gel systems.30–47 Indeed, the
viscoelastic properties, the gelation kinetics, as
well as indications of the gels structure, will be
determined by the oscillatory dynamic study of
the gels. First, the effect of the calcium content
on the dynamic mechanical behavior of the plain
alginate will be presented. It is noteworthy that
1168 NOBILE ET AL.
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

there are no articles in the literature that inves-
tigate the effect of the inclusion of different
amounts of Ca21 ions both on the limit of the
linear viscoelastic regime as well as on the elas-
tic and viscous response of alginate gels on
the frequency. Then, alginate/N-succinylchitosan
blends of different compositions will be analyzed
to evidence the influence of the chitosan succi-
nate on the plain alginate properties, with the
goal to obtain a novel hydrogel with higher or
equivalent mechanical properties and structural
uniformity to satisfy the needs of different bio-
medical applications.
Furthermore, scaffolds obtained from the
hydrogels through freeze-drying have been
physically characterized by means of scanning
electron microscopy.
EXPERIMENTAL
Materials
Medium molecular weight chitosan (Ch) [75–
85% deacetylation degree; viscosity ¼ 200–800
cP; Mw ¼ 1 3 106 Da], succinic anhydride (SA),
calcium carbonate (CaCO3) and D-glucone-d-lac-
tone (GDL) were all purchased from Fluka-
Aldrich and used as received. Pyridine (Fluka)
was distilled under reduced pressure prior to
use. Medium molecular weight sodium alginate
(Alg) was supplied by Seaweed Company
(Shangai, China). The main chemical and physi-
cal characteristics of the alginate are reported
in Table 1. All the solvents were of analytical
grade.
N-Succynilchitosan Preparation Methodology
The succinylation reaction was carried out
according to a previously reported method.24
Briefly, a 2% chitosan solution (w/v) in aqueous
HCl (0.37%) was prepared and a 12.5% (w/v) an-
hydride succinic solution in pyridine was drop-
wise added at room temperature under vigorous
stirring. The reaction pH was maintained at 7.0
by simultaneous addition of a NaOH solution.
After 40 min of reaction, the modified polymer
was precipitated in a large excess of methanol,
washed with acetone and desiccated. To remove
the unreacted SA, the obtained product was dis-
solved in water and dialysed in a NaOH solution
for 48 h using a dialysis membrane bag with a
molecular weight cut-off of 10 kDa. (yield ¼95%)
FTIR: 1720 (C¼¼O acid stretching), 1640
(C¼¼O amide stretching), 1560 (N��H amide
bending) cm�1.1H NMR: d 4,6 (s, 1H), 3.7 (m, 6H), 2,5 (d,
4H).
Hydrogel Preparation Methodology
Alginate and alginate/N-succinylchitosan hydro-
gels were realized by the internal setting
method.27,28 To 10 mL of a 2% w/w polymeric so-
lution at different Alg/sCh ratios (100/0; 90/10,
80/20, 70/30, 60/40) a given amount of CaCO3
was added and the resulting mixture was left
under stirring for 30 min. Then a 9% D-glucone-
d-lactone (GDL) solution was added and the sys-
tem was kept for 45 s under vigorous stirring.
The concentrations of calcium carbonate used
were 0.1, 0.2, 0.5, and 0.7% w/w respect to poly-
meric total fraction. GDL was always used in
stoichiometric ratios respect to CaCO3 to obtain
a neutral gel.
The obtained solution was poured into a Petri
dish with diameter of 8 cm and left at room tem-
perature for 48 h to allow the complete alginate
crosslinking. The viscoelastic behavior of the al-
ginate and alginate/N-succinylchitosan hydrogels
was, then, studied in strain and frequency sweep
rheological tests, as described in details in the
‘‘Rheological measurements’’ section. Instead,
the obtained solution was quickly put on the rhe-
ometer plate to study the alginate and alginate/
N-succinylchitosan gelation kinetics.
It is noteworthy that the amount of Ca21 ions
theoretically necessary to saturate all the car-
boxylic groups in any alginate molecule corre-
sponds to the inclusion of the 0.2% w/w of
CaCO3.
As an example, to obtain 13 g of alginate
gel at 0.2% of CaCO3 concentration, 0.26 g of
Table 1. Main Physical (Weight Average Molecular
Weight, Mw; Number Average Molecular Weight, Mn;
P.I., Polydispersity Index) and Chemical (Percentage
of Overall Guluronic, FG, and Mannuronic, FM, Acidic
Residues; Number–Average of Guluronic Units in GG
Acidic Residues, NG) Characteristics of the Alginate
Used in This Work
Mw (Da) Mn (Da) P.I.
FG
(%)
FM
(%) NG
1.0838�106 1.9791�105 5.5 62 38 7.9 6 10%
RHEOLOGICAL INVESTIGATION OF NOVEL ALGINATE HYDROGELS 1169
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

alginate are dissolved in 11.3 g of demineralised
water under stirring for 1 h at room tempera-
ture; then, 0.03 g of CaCO3 are dispersed in the
solution by stirring at room temperature for
30 min over. Then 1.2 mL of a 9% GDL solution
is added and the mixture is kept under vigorous
stirring for 45 s.
Spectroscopic Measurements
FTIR characterization was carried out by using
a Perkin–Elmer spectrometer, model Paragon
500 (average of 20 scans at resolution of 4
cm�1). The dry materials were powdered,
ground with exhaustively dried KBr powder,
and the discs were prepared by compression
under vacuum.1H NMR spectra were recorded with a Bruker
avance DPX300 apparatus operating at 300
MHz. The sample was prepared by dissolving
15 mg of polymer in 0.75 mL of deuterated sol-
vents. Particularly, Ch was dissolved in a 20%
CD3COOD/D2O solution, while D2O was used
for sCh.
Rheological Measurements
The rheological investigation was performed on
the alginate and alginate/N-succinylchitosan
hydrogels with a strain controlled rotational
rheometer, ARES TA, equipped with a parallel
plate configuration (gap ¼ 1.5 mm, R ¼ 12.5
mm). Dynamic oscillatory strain sweep experi-
ments were performed on the hydrogels to deter-
mine the limit of the linear viscoelastic region.
Consequently, frequency sweep tests were per-
formed in the linear regime for all the samples.
The test temperature in all the experiments was
25 8C and the samples were tested under a con-
tinuous nitrogen purge.
The isothermal gelation kinetics was studied
at the temperature of 25 8C in quiescent condi-
tions by means of a rotational stress rheometer,
SR5000, Rheometric Inc., in a parallel plate con-
figuration (gap ¼ 1.5 mm, R ¼ 20 mm) at the
constant oscillation frequency of 1 rad/s under
nitrogen atmosphere. The formation of the gel
was analyzed by following the time evolution of
the storage (G0) and the loss (G00) moduli.
Scanning Electron Microscopy
The morphology of materials was investigated
by Scanning Electron Microscopy (SEM). The
hydrogels were prepared as described in Mor-
phological Analysis (left for 48 h at room tem-
perature). Then they were frozen in liquid nitro-
gen and lyophilized. The dried hydrogels were
fractured in liquid nitrogen and the surface of
fracture was observed. Samples were mounted
on a stub and coated with an Au/Pd alloy. Micro-
graphs were obtained using a scanning electron
microscope Philips XL 20.
RESULTS AND DISCUSSION
Synthesis of N-Succinylchitosan
To obtain a water-soluble polymer, the chitosan
was modified by reacting with succinic anhy-
dride. Further details of the synthesis and char-
acterization of the obtained N-succinylchitosan
polymer have been published elsewhere.25 The
succinylation reaction consists of a condensation
between polysaccharide amino groups and car-
bonylic groups of the anhydride, with conse-
quent formation of an amidic bond (Scheme 1).
The resulting product is a water-soluble poly-
mer. FTIR and 1H NMR analyses, performed on
both plain and modified chitosan, confirmed the
occurrence of succinylation.
In fact, the infrared spectrum of modified chi-
tosan (not shown), compared with that of chito-
san, shows the appearance of new absorption
peaks at 1720 cm�1 and 1640–1560 cm�1, which
correspond respectively, to carboxylic acid (C¼¼O
stretching of ��COOH) and amidic groups
(C¼¼O stretching and N��H bending). Moreover,
absorption increase of the peaks at 2930 and
1075 cm�1 (due to ��CH2��CH2�� of succinic
moieties) and broadening of the band at 3400
cm�1 (due to ��OH stretching) were also
detected.
The 1H NMR spectrum of the modified poly-
mer shows a new signal at 2.5 ppm correspond-
ing to the ��CH2�� moieties of succinic acid
(H-7, H-8). Furthermore, the shifting of the
peak relative to H-2 proton from 3 ppm in the
Ch spectrum to higher values in the sCh spec-
trum (where it overlaps the multiplet at 3.7
ppm) is in line with the formation of the
amidic bond. The degree of polymer succinyla-
tion has been estimated through the ratio
between peak areas of the grafted succinic
protons ��CH2�� CH2�� (2,5 ppm) and the
polysaccharide protons at 3,7 ppm, and turned
out to be around 90%. The high degree of
1170 NOBILE ET AL.
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

succinylation obtained is responsible for the
good water solubility of sCh.
Hydrogel Preparation
Alginate hydrogels have been widely investi-
gated in view of their numerous applications in
biomedical, cosmetic, and food fields. No exam-
ples exist in literature of hydrogels based on al-
ginate and chemically modified chitosan. In this
work, a novel alginate/N-succinylchitosan-based
hydrogel was prepared and investigated, and
compared with plain alginate gel.
The alginate and alginate/N-succinylchitosan
hydrogels were prepared by the internal setting
method.27,28 By this way it is possible to obtain
hydrogels with an uniform concentration of algi-
nate. In general, this method uses an inactive
form of the crosslinking ion, either bound by a
sequestering agent such as phosphate, citrate or
EDTA, or as an insoluble salt, for example
CaSO4 and CaCO3. In association with the diva-
lent ion, usually a solution of a slowly hydrolyz-
ing lactone, like D-glucono-d-lactone, is then
added to the mixture of alginate and crosslinker.
Because of the acidic pH generated by GDL, the
calcium ions are gradually released and cap-
tured by guluronic residues of alginate. In this
work, the CaCO3-GDL system was used to real-
ize the hydrogels. The slow release of calcium
ions allowed obtaining homogenous and trans-
parent hydrogels; the use of stoichiometric
CO2�3 /GDL ratio allowed for neutral pH. An
accurate premixing of alginate solution with
CaCO3 before the addition of GDL is necessary
to obtain a homogeneous dispersion of the
CaCO3 inside the alginate solution, to create a
regular network while the Ca21 ions are pro-
gressively released after the addition of GDL,
according to the following Scheme 2.
Rheological Analysis
The Alginate Hydrogels
The Strain Sweep Behavior. Alginate hydrogels
with a polymer concentration of 2% w/w and dif-
ferent amounts of Ca21 ions were investigated
under oscillatory strain sweep tests to define the
critical deformation (cc) characterizing the limit
of the linear viscoelastic regime as a function of
the calcium carbonate concentration. The values
of (cc) for gels are not often reported in litera-
ture, indeed only recently articles have ap-
peared on this subject.30–33,42,43,46,47 In particu-
lar, there are no articles that investigate the
effect of the inclusion of different amounts of
Ca21 ions on the limit of the linear viscoelastic
regime of alginate gels.
The existence and the extent of a linear visco-
elastic range for the alginate hydrogels studied
in this work has been first investigated at the
constant frequency of 0.5 rad/s. In Figure 1(a,b)
Scheme 2. Schematic representation of alginate gel-
ification process in presence of CaCO3/GDL system.29
Scheme 1. Succinylation reaction of chitosan.
RHEOLOGICAL INVESTIGATION OF NOVEL ALGINATE HYDROGELS 1171
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb
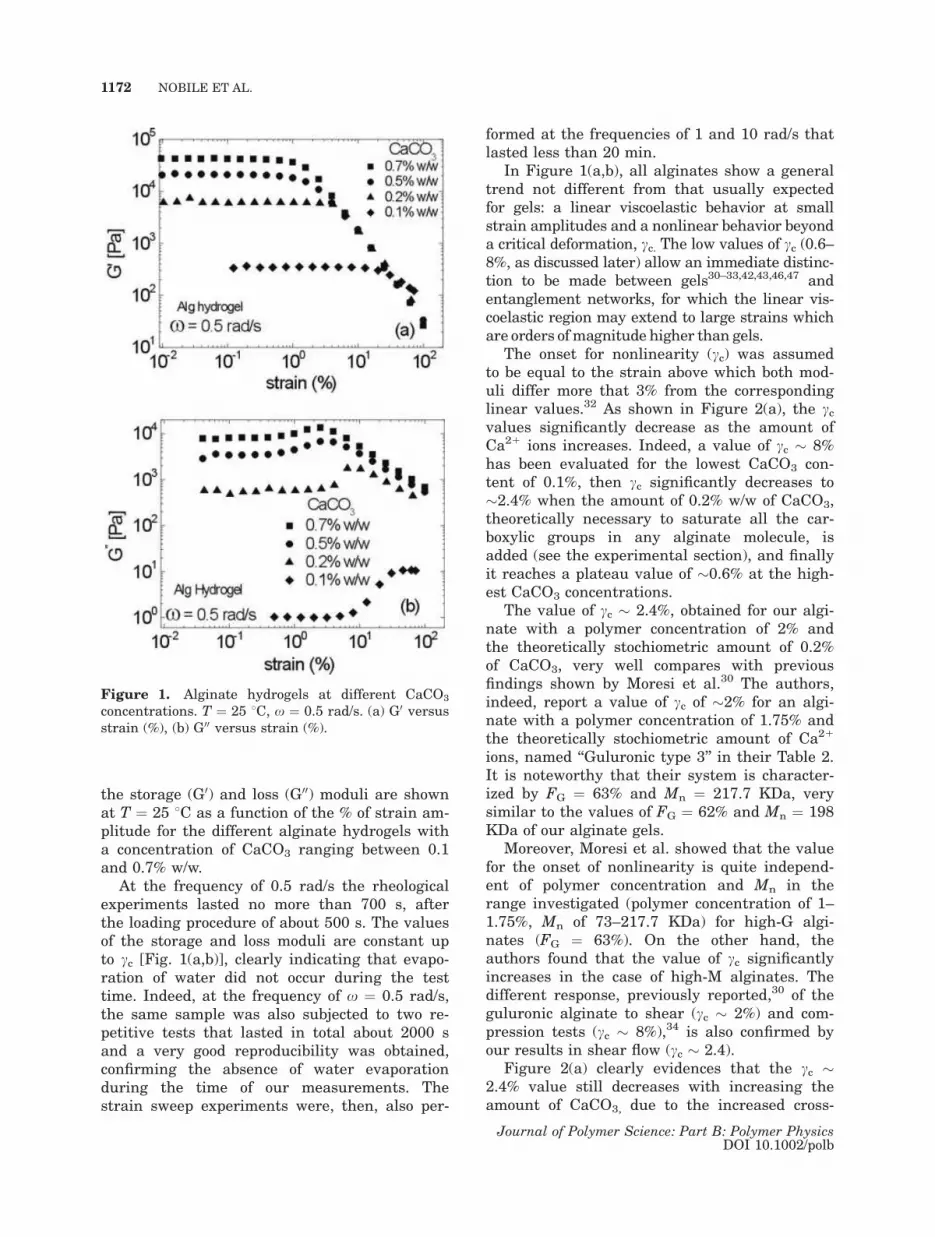
the storage (G0) and loss (G00) moduli are shown
at T ¼ 25 8C as a function of the % of strain am-
plitude for the different alginate hydrogels with
a concentration of CaCO3 ranging between 0.1
and 0.7% w/w.
At the frequency of 0.5 rad/s the rheological
experiments lasted no more than 700 s, after
the loading procedure of about 500 s. The values
of the storage and loss moduli are constant up
to cc [Fig. 1(a,b)], clearly indicating that evapo-
ration of water did not occur during the test
time. Indeed, at the frequency of x ¼ 0.5 rad/s,
the same sample was also subjected to two re-
petitive tests that lasted in total about 2000 s
and a very good reproducibility was obtained,
confirming the absence of water evaporation
during the time of our measurements. The
strain sweep experiments were, then, also per-
formed at the frequencies of 1 and 10 rad/s that
lasted less than 20 min.
In Figure 1(a,b), all alginates show a general
trend not different from that usually expected
for gels: a linear viscoelastic behavior at small
strain amplitudes and a nonlinear behavior beyond
a critical deformation, cc. The low values of cc (0.6–
8%, as discussed later) allow an immediate distinc-
tion to be made between gels30–33,42,43,46,47 and
entanglement networks, for which the linear vis-
coelastic region may extend to large strains which
are orders ofmagnitude higher than gels.
The onset for nonlinearity (cc) was assumed
to be equal to the strain above which both mod-
uli differ more that 3% from the corresponding
linear values.32 As shown in Figure 2(a), the ccvalues significantly decrease as the amount of
Ca21 ions increases. Indeed, a value of cc � 8%
has been evaluated for the lowest CaCO3 con-
tent of 0.1%, then cc significantly decreases to
�2.4% when the amount of 0.2% w/w of CaCO3,
theoretically necessary to saturate all the car-
boxylic groups in any alginate molecule, is
added (see the experimental section), and finally
it reaches a plateau value of �0.6% at the high-
est CaCO3 concentrations.
The value of cc � 2.4%, obtained for our algi-
nate with a polymer concentration of 2% and
the theoretically stochiometric amount of 0.2%
of CaCO3, very well compares with previous
findings shown by Moresi et al.30 The authors,
indeed, report a value of cc of �2% for an algi-
nate with a polymer concentration of 1.75% and
the theoretically stochiometric amount of Ca21
ions, named ‘‘Guluronic type 3’’ in their Table 2.
It is noteworthy that their system is character-
ized by FG ¼ 63% and Mn ¼ 217.7 KDa, very
similar to the values of FG ¼ 62% and Mn ¼ 198
KDa of our alginate gels.
Moreover, Moresi et al. showed that the value
for the onset of nonlinearity is quite independ-
ent of polymer concentration and Mn in the
range investigated (polymer concentration of 1–
1.75%, Mn of 73–217.7 KDa) for high-G algi-
nates (FG ¼ 63%). On the other hand, the
authors found that the value of cc significantly
increases in the case of high-M alginates. The
different response, previously reported,30 of the
guluronic alginate to shear (cc � 2%) and com-
pression tests (cc � 8%),34 is also confirmed by
our results in shear flow (cc � 2.4).
Figure 2(a) clearly evidences that the cc �2.4% value still decreases with increasing the
amount of CaCO3, due to the increased cross-
Figure 1. Alginate hydrogels at different CaCO3
concentrations. T ¼ 25 8C, x ¼ 0.5 rad/s. (a) G0 versus
strain (%), (b) G00 versus strain (%).
1172 NOBILE ET AL.
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

linking network density, and it reaches the cc �0.6% plateau value at the concentration of 0.5%
CaCO3 w/w. As known from literature,29 indeed,
a stoichiometric amount of calcium ions is not
sufficient to saturate all the carboxylate groups
of alginate, because of linkage conformations of
the guluronate residues in the chain, therefore
an excess of calcium is necessary to reach the
plateau value. Increasing the calcium ion
amount from 0.5 to 0.7% does not increase the
number of crosslinkings, since all the accessible
carboxylate groups have been saturated, conse-
quently the value of cc � 0.6% remains constant.
The G0 and G00 moduli, instead, still increase
going from 0.5 to 0.7% [Fig. 1(a,b)], because the
increase in modulus is generally caused not only
by a higher number of junction zones, but also
by a higher strength of these junction points in
gels, formed by ions of high binding strength.
In Figure 1(a,b), G0 and G00 data in the non-
linear region are also shown. Beyond the linear
viscoelastic limit, cc, the storage modulus, G0,
decreases with increasing the strain amplitude,
Figure 1(a). Looking at the Figure 1(b) a pecu-
liar behavior in the G00 profile is observed
beyond cc: in the nonlinear region, the loss mod-
uli data first increase, reaching a maximum,
and then decrease with increasing strains. A
similar trend in G0 and G00 has been reported for
different alginate30,31,43 and scleroglucan gels32
and it is a peculiar feature of structured sys-
tems. At this regard, it is noteworthy that differ-
ent papers have been recently published that
show how large amplitude oscillatory measure-
ments (LAOS) can be a very important tool to
study and classify complex fluids as biological
gel molecules, polyelectrolytes, surfactants, sus-
pensions.48–52 In particular, the article by Hyun
et al.48 classifies the types of large amplitude os-
cillatory shear (LAOS) behavior into four types:
Type I, strain thinning (G0 and G00 decreasing);
Type II strain hardening (G0 and G00 increasing);
Type III, weak strain overshoot (G0 decreasing,
G00 increasing followed by decreasing); Type IV,
strong strain overshoot (G0 and G00 increasing
followed by decreasing). Based on this classifica-
tion we, therefore, define our G0 and G00 results
as Type III.
The results of G00, reported in Figure 1(b), are
summarized in Figure 2(b,c). In particular, the
ratio between G00 maximum and the linear visco-
elastic loss modulus values (G00max/G
000) de-
creases as the amount of CaCO3 increases [as
shown in Figure 2(b)], reaching a plateau value
Figure 2. Alginate hydrogels x ¼ 0.5 rad/s. T ¼25 8C. (a) Linear viscoelastic limit (cc %) versus
CaCO3 concentration, (b) G00max/G
000 versus CaCO3
concentration, (c) cmax (%) values versus CaCO3 con-
centration.
RHEOLOGICAL INVESTIGATION OF NOVEL ALGINATE HYDROGELS 1173
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

at the concentration of 0.5% CaCO3 w/w. In Fig-
ure 2(c) it is evident that the strain value (cmax)
corresponding to G00max dramatically decreases
from �62% (for the lowest CaCO3 content) to a
value of �6.5% at 0.2% of CaCO3, finally reach-
ing a plateau value of cmax � 2.5% at the high-
est CaCO3 amounts investigated.
These results confirm that the viscoelastic
properties G0 and G00 are very sensitive to the
gel structure, with a dramatic variation in ccand cmax recorded in correspondence of the Ca21
ions amount theoretically necessary to saturate
all the carboxylic groups in any alginate mole-
cule. Our results also showed that plateau val-
ues in cc and cmax were obtained at CaCO3
amount of 0.5% w/w.
The onset for nonlinearity (cc) has been found
to be independent of frequency for tests per-
formed in the range 0.5–10 rad/s, as shown in
Figure 3(a,b) where the values of G0/G00 versus
strain amplitude is reported for the samples
with CaCO3 0.5 and 0.7% w/w, respectively. The
frequency sweep tests, discussed in next section,
are performed in the linear viscoelastic regime
with strain values lower than cc. The choice of
the frequency and strain range used in this
work has also ensured that no slipping of the
gel disks from the plates occurred during the os-
cillatory shear tests.
The Frequency Behavior. In this section the fre-
quency response of the alginate gels in the range
10�1 to 102 rad/s is analyzed and discussed. All
the frequency sweep tests were performed in the
linear viscoelastic regime at the constant strain of
0.25%, sensibly lower than cc. The measurements
lasted about 800 s after the loading procedure of
about 500 s. The test time is, then, comparable
with the test time of the strain sweep experiments
and therefore, as discussed previously, we can
assume that evaporation of the water did not
occur during our measurements.
Following Almdal et al.39 a gel is defined as a
soft solid or solid-like material which consists of
two or more components one of which is a sub-
stantial quantity of a liquid and shows an approx-
imately flat mechanical spectrum in small ampli-
tude oscillatory experiments. In Figure 4 the
G0(x), G00(x), and g*(x) data for our alginate gel
with CaCO3 amount of 0.2% w/w show profiles
characterized by two straight lines nearly parallel
to each other with a slight increase with fre-
quency. Moreover G0 is about one order of magni-
tude greater than G00. This behavior is shown by
all the alginate gels (Fig. 5) that, therefore, well
correspond to the definition of gels as given by
Figure 3. G0/G00 versus strain (%), at different fre-
quencies. T ¼ 25 8C. (a) Alginate hydrogel with
CaCO3 ¼ 0.5% w/w, (b) Alginate hydrogel with CaCO3
¼ 0.7% w/w.
Figure 4. G0, G00 and g* versus frequency, for the al-
ginate hydrogel with CaCO3 ¼ 0.2% w/w. T ¼ 25 8C.
1174 NOBILE ET AL.
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

Almdal et al.39 Indeed, the investigated alginate
gels can be classified as strong gels33 that under
small deformation conditions manifest the typical
behavior of viscoelastic solids. The results
reported in Figure 4 very well compare with pre-
vious findings by Segreen et al.35 for a Ca21-algi-
nate gel (1.4% w/w) and by Moresi et al. for a se-
ries of alginates gels.30,31
The effect of different amounts of Ca21 ions
on the G0(x) and G00(x) values has been studied
and the results are shown in Figure 5(a,b),
respectively. In all the cases, the mechanical
spectra are characterized by a quite flat depend-
ence of G0(x) and G00(x) on the frequency with
G0 about one order of magnitude greater than
G00. As expected, the storage and the loss moduli
both increase as the CaCO3 content is increased,
in agreement with previous findings by Kuo and
Ma28 that showed how compressive modulus
and strength increased with calcium content. In
particular, a strong increase in G0 is recorded at
the CaCO3 0.2% w/w concentration theoretically
necessary to saturate all the carboxylic groups
in any alginate molecule, confirming the results
discussed in the strain sweep section.
In the literature it has been proven that the
generalized Maxwell model33 can be successfully
applied to reconstruct the linear viscoelastic re-
gime of alginate gels submitted to compressive
tests.34 Nevertheless, Moresi et al.30 showed
that the evolution of G0 and G00 in small ampli-
tude oscillatory experiments of alginate gels
could be better described by means of the model
proposed by Friedrich and Heymann,36 that can
predict the linear viscoelastic behavior of gels
not only at the sol–gel transition, but also below
and above the gel point.
The proposed Friedrich and Heymann model
is an extension of the constitutive equation
introduced by Chambon and Winter37 to
describe only the gel point. In particular, the
extended model is also able to describe a start-
ing viscosity function for states below the gel
point and the existence of an equilibrium modu-
lus in the state above the gel point.
It is based on an extended relaxation function
depending on four parameters, namely
GðtÞ ¼ G1;a þS�a
Cð1� aÞt�a expð�t=kaÞ ð1Þ
where S�a is the strength, a is the order of the
relaxation function, G?,a and ka are the equilib-
rium modulus and the mean relaxation time per-
taining to a, respectively. In their article, Frie-
drich and Heymann36 clearly showed that the
extended relaxation function is able to describe
the evolution of G0 and G00 in linear viscoelasticity
during crosslinking reactions for all the reaction
stages in the entire frequency range.
In the linear regime, the knowledge of G(t)
allows the calculation of all the others visco-
elastic functions.53 In particular, the storage
and loss moduli can be computed as follows:
Figure 5. Alginate hydrogels at different CaCO3
concentrations. T ¼ 25 8C. (a) G0 versus frequency, (b)
G00 versus frequency.
G0ðxÞ ¼ G1;a þ x
Z
1
0
GðtÞ �G1;a
� �
cosðxtÞdt ð2aÞ
G00ðxÞ¼G1;aþx
Z
1
0
GðtÞ�G1;a
� �
sinðxtÞdt ð2bÞ
RHEOLOGICAL INVESTIGATION OF NOVEL ALGINATE HYDROGELS 1175
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

According to Friedrich and Heymann,36 eqs 1,
2a, and 2b turn out in
G0ðxÞ ¼ G1;a þ
ffiffiffi
2
p
r
S�aka
�aðxkaÞ
3sinbð1� aÞ arctanðxkaÞc
1þ ðxkaÞ2
h i1�a2
ð3aÞ
G00ðxÞ ¼
ffiffiffi
2
p
r
S�aka
�aðxkaÞcosbð1� aÞ arctanðxkaÞc
1þ ðxkaÞ2
h i1�a2
ð3bÞ
At moderate and high frequencies eqs 3a and
b give the same slope for G0 and G00 (G0 / xa,
G00 / xa), while at low frequencies they may
describe either liquid (G0 / x2, G00 / x) or solid
(G0 / G1;a, G00 / x) behavior.
It is noteworthy that Friedrich and Hey-
mann36 proved that, near the gel point or in the
high frequency range after the sol-gel transition
(xka � 1), the model reduces to:
G0ðxÞ ¼ G1;a þ
ffiffiffi
2
p
r
S�a xa cos
ap
2ð4aÞ
G00ðxÞ ¼
ffiffiffi
2
p
r
S�a xa sin
ap
2ð4bÞ
These equations are, then, independent of the
relaxation time ka. Moreover, the authors
pointed out that the ratio:
tan d ¼ G00=G0 ¼ tanap
2ð5Þ
is exact at the gel point, when the equilibrium
modulus is equal to zero (Chambon and Winter
model) and holds in the gel state at moderately
or high frequencies, when the equilibrium mod-
ulus can be neglected. Therefore, the article by
Friedrich and Heymann showed that eqs 4a and
b can be used in the gel state at high frequen-
cies when G?,a can be neglected.
In the literature, the evolution of the storage
and loss moduli, G0 and G00, in small amplitude os-
cillatory experiments of alginate gels in the high
frequency range has been successfully described
within the frame of the Friedrich and Heymann
model by means of eqs 4 and 5.30–31,43,47
Figure 5(a,b) show that the storage and loss
moduli of our alginate gels follow a power law
and remain parallel to each other in the high
frequency range investigated. Indeed, both mod-
uli are quite flat and behave as xa. Their ratio
is nearly constant and compares well with eq 5
within the experimental error. Following the lit-
erature suggestions we have, then, used eqs 4
and 5 to fit our dynamic moduli data. The agree-
ment is rather good, as it can be seen in Figure
6 for the CaCO3 0.2% w/w alginate gel. The best
values of the parameters S�a and a for the three
samples are reported in Table 2. The results
show a significant increase of the strength. S�a
with increasing the CaCO3 content from the
0.2% w/w concentration (corresponding to the
Ca21 ions amount theoretically necessary to sat-
urate all the carboxylic groups in any alginate
molecule) to the 0.5 and 0.7% w/w concentration.
The values of the order of the relaxation func-
tion a are in the range 0.05–0.1 confirming the
strong gel state of our samples. They well com-
pare with the a data reported in the literature
for alginate gels.30
Based on these results and on the strain
sweep behavior, the alginate with the 0.5% has
been chosen as the reference system to investi-
gate the influence of the chitosan succinate on
the plain alginate dynamic mechanical proper-
Figure 6. G0, G00 and tand versus frequency for the
alginate hydrogel with CaCO3 ¼ 0.2% w/w. T ¼ 25 8C.
Fit with the Friedrich and Heymann model (line).
Table 2. Strength (S�a) and Values of the Order of
the Relaxation Function (a) for Alginate Hydrogels at
Different CaCO3 Concentrations
CaCO3 Content (w/w %) a Sa* (Pa sa)
0.2 0.055 7,000
0.5 0.085 25,000
0.7 0.101 56,000
1176 NOBILE ET AL.
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

ties. This system, indeed, corresponds to a gel
with the Ca21 ions amount that reached the pla-
teau values in cc and cmax for the plain alginates
[see Fig. 2(a,c)] and shows a significant increase
in the strength values. For comparison the sys-
tems with 0.2 and 0.7% w/w CaCO3 content
have also been studied and the results are
shown in the following sections.
The Alginate/N-Succinylchitosan Hydrogels
With the intent to obtain a novel hydrogel with
improved biocompatibility, the hydrogels based
on the blends of alginate and N-succinylchitosan
with a total polymer concentration of 2% w/w
and different amounts of N-succinylchitosan
were examined under oscillatory tests. Indeed, it
was verified whether the dynamic mechanical
properties of the plain alginate would depend on
the amount of N-succinylchitosan.
The Strain Sweep Behavior. The critical defor-
mation (cc), characterizing the limit of the linear
viscoelastic regime, has been determined for the
different alginate/N-succinylchitosan blends.
The results, in terms of G0 and G00 versus % of
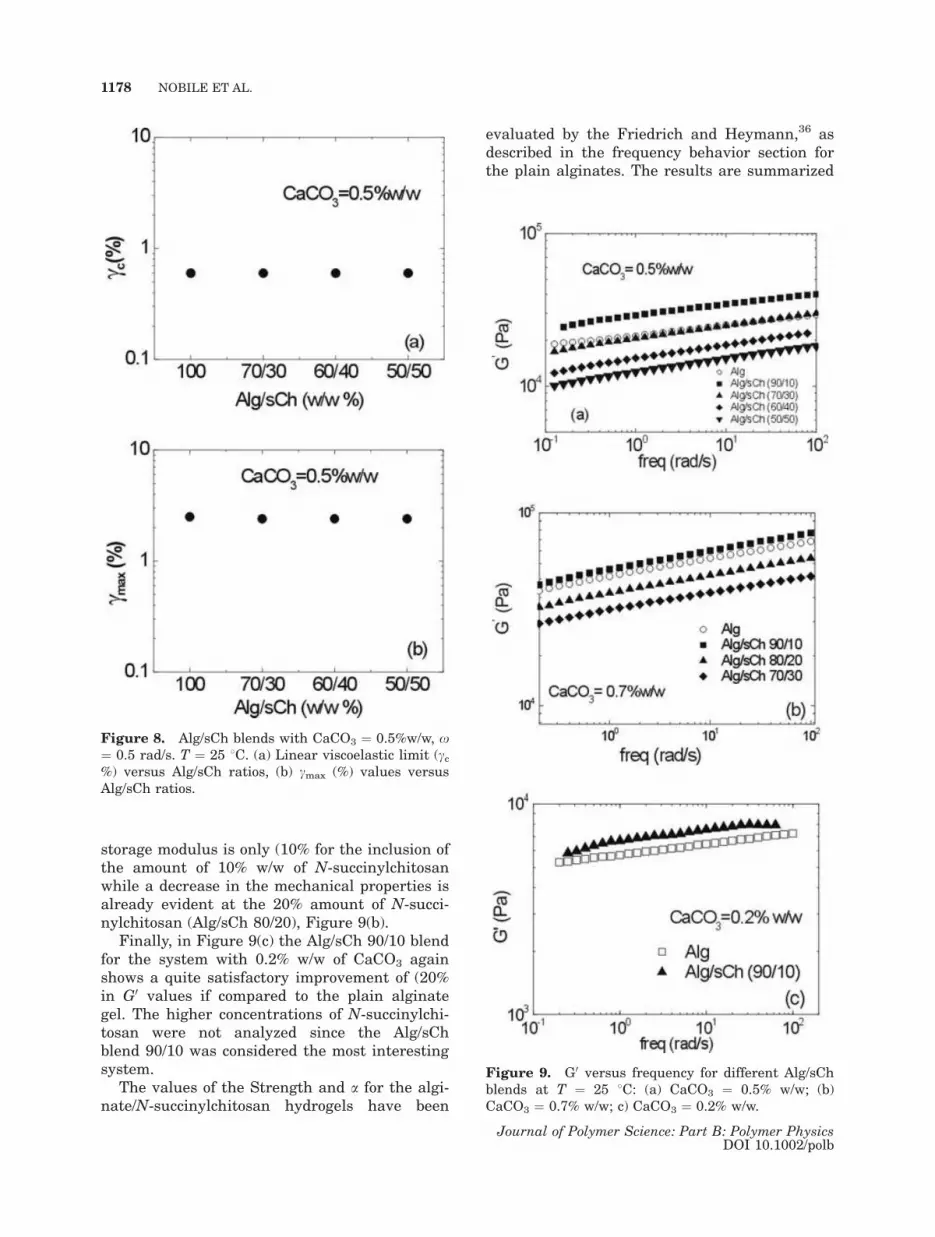
strain amplitude, are reported in Figure 7(a,b)
for the CaCO3 0.5% w/w system. As clearly
shown in the figures, the cc and cmax are equal
to the values previously reported for the plain
alginate gel with 0.5% w/w of CaCO3 [respec-
tively, of �0.6 and 2.5%, Fig. 2(a,c)], independ-
ently of the amount of N-succinylchitosan, in
the range analyzed. To better evidence this
behavior, the normalized G0/G00 and G00/G00
0 are
reported in the inset of Figure 7(a,b), while the
results in terms of cc and cmax as a function of
the amount of N-succinylchitosan are summar-
ized in Figure 8(a,b). Similar results have been
also obtained for the blends at CaCO3 concentra-
tions of 0.7 and 0.2% w/w.
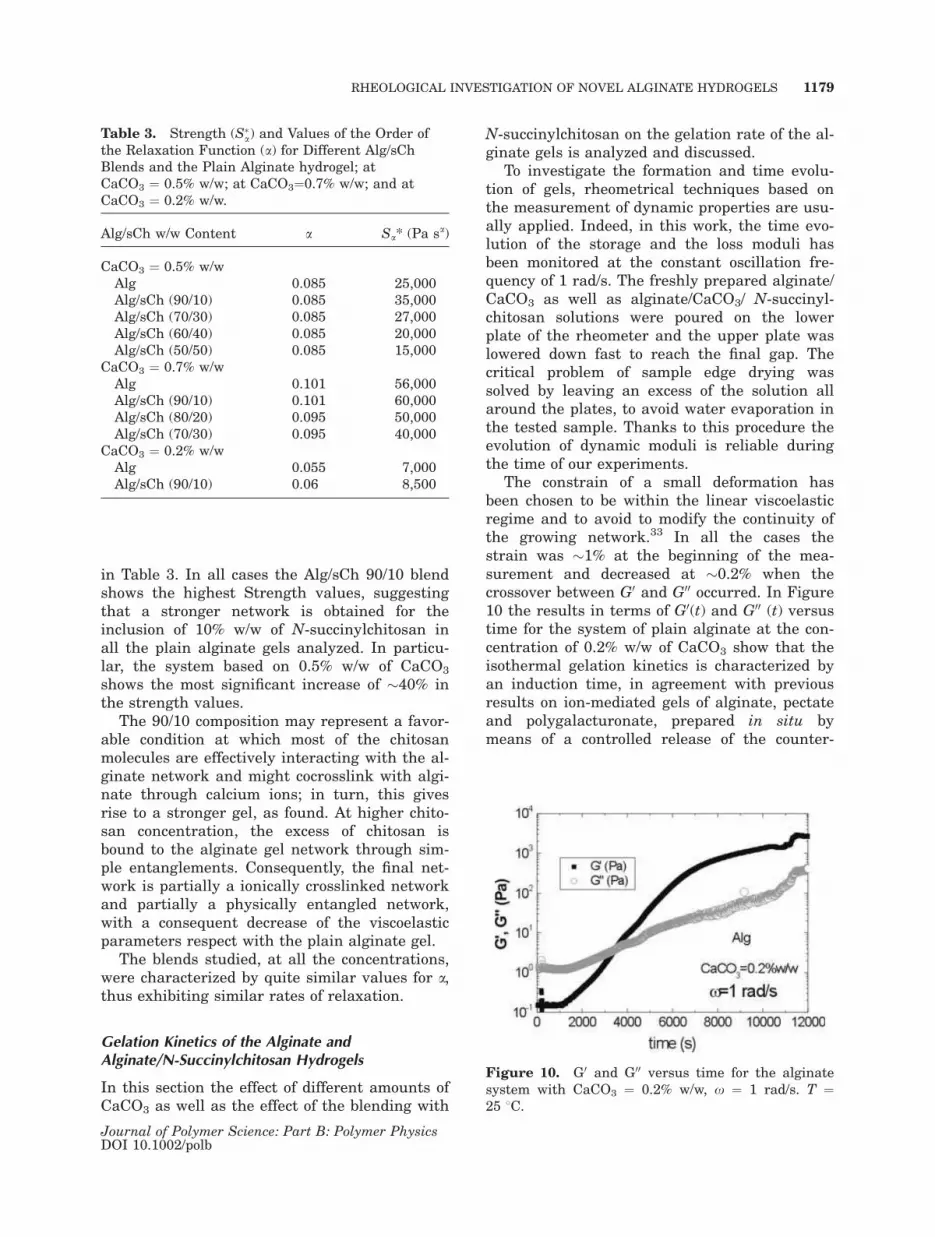
The Frequency Behavior. The effect of the inclu-
sion of the N-succinylchitosan on the frequency
behavior of the plain alginates gels has been,
then, studied in the range 10�1 to 102 rad/s. In
Figure 9(a–c) the results in terms of G0(x) for
the systems with 0.5, 0.7, and 0.2% w/w of
CaCO3, respectively, are reported. In all cases
the mechanical spectra are slightly dependent
on the frequency, with G0 about one order of
magnitude higher then G00 (not reported in the
figures). The hydrogels based on the alginate
and N-succinylchitosan blends can be, therefore,
classified as strong gels, analogously to the plain
alginate gels.
As reported in Figure 9(a), the alginate gel
with 0.5% w/w of CaCO3 shows significantly
increased values of the storage modulus ((40%)
when the amount of 10% w/w of N-succinylchito-
san (Alg/sCh 90/10) is blended to the plain algi-
nate, in all the frequency range tested. On the
other hand, G0 values for the Alg/sCh 70/30
blend are very similar to those for the plain algi-
nate gels, while a further increase in the N-suc-
cinylchitosan, determines a reduction in G0 val-
ues, as indicated for the Alg/sCh 60/40 and 50/
50 blends.
This behavior has been quite confirmed in the
case of the Alg/sCh system with 0.7% w/w of
CaCO3. Nonetheless, the improvement of the
Figure 7. Alg/sCh blends with CaCO3 ¼ 0.5%w/w, x
¼ 0.5 rad/s. T ¼ 25 8C. a) G0 versus strain(%), inset:
G0/G00 versus strain (%), b) G00 versus strain (%),
inset: G00/G000 versus strain (%).
RHEOLOGICAL INVESTIGATION OF NOVEL ALGINATE HYDROGELS 1177
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb
storage modulus is only (10% for the inclusion of
the amount of 10% w/w of N-succinylchitosan
while a decrease in the mechanical properties is
already evident at the 20% amount of N-succi-
nylchitosan (Alg/sCh 80/20), Figure 9(b).
Finally, in Figure 9(c) the Alg/sCh 90/10 blend
for the system with 0.2% w/w of CaCO3 again
shows a quite satisfactory improvement of (20%
in G0 values if compared to the plain alginate
gel. The higher concentrations of N-succinylchi-
tosan were not analyzed since the Alg/sCh
blend 90/10 was considered the most interesting
system.
The values of the Strength and a for the algi-
nate/N-succinylchitosan hydrogels have been
evaluated by the Friedrich and Heymann,36 as
described in the frequency behavior section for
the plain alginates. The results are summarized
Figure 8. Alg/sCh blends with CaCO3 ¼ 0.5%w/w, x
¼ 0.5 rad/s. T ¼ 25 8C. (a) Linear viscoelastic limit (cc%) versus Alg/sCh ratios, (b) cmax (%) values versus
Alg/sCh ratios.
Figure 9. G0 versus frequency for different Alg/sCh
blends at T ¼ 25 8C: (a) CaCO3 ¼ 0.5% w/w; (b)
CaCO3 ¼ 0.7% w/w; c) CaCO3 ¼ 0.2% w/w.
1178 NOBILE ET AL.
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb
in Table 3. In all cases the Alg/sCh 90/10 blend
shows the highest Strength values, suggesting
that a stronger network is obtained for the
inclusion of 10% w/w of N-succinylchitosan in
all the plain alginate gels analyzed. In particu-
lar, the system based on 0.5% w/w of CaCO3
shows the most significant increase of �40% in
the strength values.
The 90/10 composition may represent a favor-
able condition at which most of the chitosan
molecules are effectively interacting with the al-
ginate network and might cocrosslink with algi-
nate through calcium ions; in turn, this gives
rise to a stronger gel, as found. At higher chito-
san concentration, the excess of chitosan is
bound to the alginate gel network through sim-
ple entanglements. Consequently, the final net-
work is partially a ionically crosslinked network
and partially a physically entangled network,
with a consequent decrease of the viscoelastic
parameters respect with the plain alginate gel.
The blends studied, at all the concentrations,
were characterized by quite similar values for a,
thus exhibiting similar rates of relaxation.
Gelation Kinetics of the Alginate andAlginate/N-Succinylchitosan Hydrogels
In this section the effect of different amounts of
CaCO3 as well as the effect of the blending with
N-succinylchitosan on the gelation rate of the al-
ginate gels is analyzed and discussed.
To investigate the formation and time evolu-
tion of gels, rheometrical techniques based on
the measurement of dynamic properties are usu-
ally applied. Indeed, in this work, the time evo-
lution of the storage and the loss moduli has
been monitored at the constant oscillation fre-
quency of 1 rad/s. The freshly prepared alginate/
CaCO3 as well as alginate/CaCO3/ N-succinyl-
chitosan solutions were poured on the lower
plate of the rheometer and the upper plate was
lowered down fast to reach the final gap. The
critical problem of sample edge drying was
solved by leaving an excess of the solution all
around the plates, to avoid water evaporation in
the tested sample. Thanks to this procedure the
evolution of dynamic moduli is reliable during
the time of our experiments.
The constrain of a small deformation has
been chosen to be within the linear viscoelastic
regime and to avoid to modify the continuity of
the growing network.33 In all the cases the
strain was �1% at the beginning of the mea-
surement and decreased at �0.2% when the
crossover between G0 and G00 occurred. In Figure
10 the results in terms of G0(t) and G00 (t) versus
time for the system of plain alginate at the con-
centration of 0.2% w/w of CaCO3 show that the
isothermal gelation kinetics is characterized by
an induction time, in agreement with previous
results on ion-mediated gels of alginate, pectate
and polygalacturonate, prepared in situ by
means of a controlled release of the counter-
Table 3. Strength (S�a) and Values of the Order of
the Relaxation Function (a) for Different Alg/sCh
Blends and the Plain Alginate hydrogel; at
CaCO3 ¼ 0.5% w/w; at CaCO3¼0.7% w/w; and at
CaCO3 ¼ 0.2% w/w.
Alg/sCh w/w Content a Sa* (Pa sa)
CaCO3 ¼ 0.5% w/w
Alg 0.085 25,000
Alg/sCh (90/10) 0.085 35,000
Alg/sCh (70/30) 0.085 27,000
Alg/sCh (60/40) 0.085 20,000
Alg/sCh (50/50) 0.085 15,000
CaCO3 ¼ 0.7% w/w
Alg 0.101 56,000
Alg/sCh (90/10) 0.101 60,000
Alg/sCh (80/20) 0.095 50,000
Alg/sCh (70/30) 0.095 40,000
CaCO3 ¼ 0.2% w/w
Alg 0.055 7,000
Alg/sCh (90/10) 0.06 8,500
Figure 10. G0 and G00 versus time for the alginate
system with CaCO3 ¼ 0.2% w/w, x ¼ 1 rad/s. T ¼25 8C.
RHEOLOGICAL INVESTIGATION OF NOVEL ALGINATE HYDROGELS 1179
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

ions.38 In particular, as reported in Table 4, the
value of �1800 s for the induction time of the
plain alginate system has been evaluated at
0.2% w/w CaCO3. The sol–gel transition occurs
at the incipient prevalence of the elastic compo-
nent over the viscous one, observed at �3470 s,
corresponding to the G0-G00 crossover time.
The system with a concentration of 0.7% w/w
CaCO3 has also been studied and the corre-
sponding G0 versus time values are compared
with G0 values for the 0.2% w/w system in Fig-
ure 11. The induction time of �450 s and a G0-
G00 crossover time of �620 s are recorded for the
gel with 0.7% w/w CaCO3 (see Table 4). The
results, then, clearly indicate that the kinetics
process can be significantly increased if the per-
centage of the Ca21 ions is increased.
Finally the effect of the inclusion of the N-
succinylchitosan is analyzed for the 0.2% w/w
CaCO3 system. In Figure 12 it is evident that
the inclusion of the 10% w/w of the N-succinyl-
chitosan accelerates the gelation kinetics with
respect to the case of the plain alginate gel. The
induction time of �1450 s and the G0-G00 cross-
over time of �2550 s are measured for the Alg/
Ch (90/10) blend, as reported in Table 4. This
indicates once more that the sCh has a coopera-
tive effect in chelating calcium ions, at least for
the 10% wt amount here investigated.
Morphological Analysis
According to what already reported in litera-
ture,28 alginate hydrogels obtained by the
CaCO3/GDL system are uniform, transparent
and three-dimensionally well defined. Hydrogels
based on alginate/sCh blends retain these char-
acteristics.
One of the alginate most promising feature is
its ability to be processed into porous structures
for use in tissue regeneration. Porous structures
can be obtained by freezing and lyophilizing al-
ginate gels. Hydrogels crosslinked with 0.5%
CaCO3 have so been frozen and lyophilized and
then analyzed through scanning electron micros-
copy to investigate on their morphology. SEM
micrographs of scaffolds of plain alginate, Alg/
sCh 90/10 and Alg/Ch 60/40 are reported as
examples in Figure 13(a–c). The structures
shown are typical of scaffolds obtained by the
freeze-drying technique. It is evident that all
the samples show an open, interconnected pore
structure, with regular and adequate pores
dimension (100–200 lm). The addition of sCh to
Table 4. Onset Time (tonset) and G0-G00 Crossover
Time (tcrossover) for the Plain Alginate System and the
Blend (90/10) Alg/sCh with CaCO3 ¼ 0.2% w/w, and
the Plain Alginate System with CaCO3 ¼ 0.7% w/w
Sample tonset (s) tcross over (s)
Alg CaCO3 ¼ 0.7% w/w 450 620
Alg CaCO3 ¼ 0.2% w/w 1,800 3,470
Alg/sCh (90/10)
CaCO3 ¼ 0.2% w/w
1,450 2,550
Figure 11. G0 versus time. Comparison between the
alginate systems with CaCO3 ¼ 0.7% w/w and 0.2%
w/w, x ¼ 1 rad/s. T ¼ 25 8C.
Figure 12. G0 versus time. Comparison between the
plain alginate system and the Alg/sCh (90/10) blend,
CaCO3 ¼ 0.2% w/w, x ¼ 1 rad/s. T ¼ 25 8C
1180 NOBILE ET AL.
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

plain alginate does not seem to significantly
influence the cell size; a detailed insight into the
cell wall morphology at higher magnification did
not evidence any phase separation between the
polymers.
CONCLUSIONS
In this work, a novel alginate/N-succinylchito-
san-based hydrogel was prepared by cross-
linking via the internal setting method using
calcium carbonate (CaCO3) as calcium ions source.
It was shown that a rheological study can be
successfully used to investigate the physical
characteristics of both the alginate gels and the
novel hydrogels. Indeed, the viscoelastic proper-
ties G0 and G00 are very sensitive to the gel
structure, with a dramatic variation in cc and
cmax recorded in correspondence of the Ca21 ions
amount theoretically necessary to saturate all
the carboxylic groups in any alginate molecule.
Our results also showed that an excess of cal-
cium was necessary to reach the plateau value
of cc � 0.6%, since the stoichiometric amount of
calcium ions is not sufficient to saturate all the
carboxylate groups of alginate because of link-
age conformations of the guluronate residues in
the chain.
It was shown that blending is a simple
method to combine the advantages of the two
different polymers. The 90/10 alginate/N-succi-
nylchitosan composition may represent a favor-
able condition at which most of the chitosan
molecules are effectively interacting with the al-
ginate network and might cocrosslink with algi-
nate through calcium ions; in turn, this gives
rise to a strong gel with higher storage modulus
values.
The significant acceleration of the gelation ki-
netic observed for the Alg/sCh 90/10 with
respect to the case of the plain alginate gel was
attributed to a synergistic effect of the N-succi-
nylchitosan in chelating calcium ions during the
alginate gelation process. This result is quite
interesting in view of possible applications as
in situ gelling systems.
Finally, porous structures were obtained by
freezing and lyophilizing alginate/N-succinylchi-
tosan gels. SEM micrographs showed that all
the samples retain the pores dimension and
interconnection characteristic of plain alginate,
thus they are potentially useful as scaffolds for
use in tissue engineering.
The authors are very grateful to M. Malinconico for
the many helpful discussions. They thank G. Narciso
from ICTP for technical support in SEM analysis, and
S. Zambardino (NMR Service of Istituto di Chimica
Biomolecolare (ICB) of CNR, Pozzuoli, Italy) for tech-
nical support.
Figure 13. SEM micrographs of the freeze dired
hydrogels with CaCO3 ¼ 0.5% w/w: (a) plain alginate
hydrogel; (b) Alg/sCh (90/10) hydrogel; (c) Alg/sCh
(60/40) hydrogel.
RHEOLOGICAL INVESTIGATION OF NOVEL ALGINATE HYDROGELS 1181
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb

REFERENCES AND NOTES
1. Drury, J. L.; Mooney, D. J. Biomaterials 2003, 24,
4337–4351.
2. Peppas, N. A. Hydrogels in Medicine and Phar-
macy; CRC Press: Boca Raton, Florida, 1987.
3. Peppas, N. A.; Bures, P.; Leobandung, W.; Ichi-
kawa, H. Eur J Pharm Biopharm 2000, 50, 27–
46.
4. Holte, O.; Onsøyen, E.; Myrvold, R.; Karlsen, J.
Eur J Pharm Sci 2003, 403–407.
5. Tonnesen, H. H.; Karlsen, J Drug Dev Ind Pharm
2002, 28, 621–630.
6. Tapia, C.; Escobar, Z.; Costa, E.; Sapag-Hagar, J.;
Valenzuela, F.; Basualto, C.; Gai, M. N.; Yazdani-
Pedram, M. Eur J Pharm Biopharm 2004, 57, 65–
75.
7. Martinsen, A.; Skjak-Braek, G.; Smidsrod, O. Bio-
technol Bioeng 1989, 33, 79–89.
8. Rowley, J. A.; Madlambayan, G.; Mooney, D. J.
Biomaterials 1999, 20, 45–53.
9. Draget, K. I.; Ostgaar, K.; Smidsrod, O. Carbo-
hydr Polym 1991, 14, 159–178.
10. Balakrishnan, B.; Jayakrishnan, A. Biomaterials
2005, 26, 3941–3951.
11. Wang, X.; Spencer, H. G. Polymer 1998, 39, 2759–
2764.
12. LeRoux, M. A.; Guilak, F.; Setton, L. A. J Biomed
Mater Res 1999, 47, 46–53.
13. Bae, Y. H.; Kim, S. W. Adv Drug Deliv Rev 1993,
11, 109–135.
14. Liu, J.; Lin, S.; Li, L.; Liu, E. Int J Pharm 2005,
298, 117–125.
15. Hu, S. G.; Jou, C. H.; Yang, M. C. Biomaterials
2003, 24, 2685–2693.
16. Gaserod, O.; Sannes, A.; Skjak-Braek, G. Bioma-
terials 1999, 20, 773–783.
17. El-Kamel, A.; Sokar, M.; Naggar, V.; Al Gamal, S.
AAPS PharmSci 2002, 4, 1–7.
18. Chandy, T.; Sharma, C. P. Biomaterials 1993, 14,
939–944.
19. Hirano, S.; Tsuchida, H.; Nagao, N. Biomaterials
1989, 10, 574–576.
20. Lee, K. Y.; Shik Ha, W.; Ho Park, W. Biomaterials
1995, 16, 1211–1216.
21. Ko, J. A.; Park, H. J.; Hwang, S. J.; Park, J. B.;
Lee, J. S. Int J Pharm 2002, 249, 165–174.
22. Adekogbe, I.; Ghanem, A. Biomaterials 2005, 26,
7241–7250.
23. Chenite, A.; Chaput, C.; Wang, D.; Combes, C.;
Buschmann, M.; Hoemann, C. D.; Leroux, J. C.;
Atkinson, B. L. Biomaterials 2000, 21, 2155–2161.
24. Aiedeh, K.; Mutasem, O. T. Arch Pharm Pharm
Med Chem 1999, 332, 103–107.
25. Gomez d’Ayala, G.; De Rosa, A.; Laurienzo, P.;
Malinconico, M. J Biomed Mater Res A 2007, 81,
811–820.
26. Rassis, D. K.; Saguy, I. S.; Nussinovitch, A. Food
Hydrocolloids 2002, 16, 139–151.
27. Onsøyen, E. In Alginates. Thickening and Gelling
Agents for Food; Imeson, A., Ed.; Blackie Aca-
demic & Professional: London, 1992, pp 1–24.
28. Kuo, C. K.; Ma, P. X. Biomaterials 2001, 22, 511–
521.
29. Smidsrod, O.; Draget, K. I. Carbohydr Eur 1996,
14, 6–13.
30. Moresi, M.; Mancini, M.; Bruno, M.; Rancini, R. J
Texture Studies 2001, 32, 375–396.
31. Moresi, M.; Bruno, M.; Parente, E. J Food Eng
2004, 64, 179–186.
32. Bais, D.; Trevisan, A.; Lapasin, R.; Partal, P.; Galle-
gos, C. J Colloid Interface Sci 2005, 290, 546–556.
33. Lapasin, R.; Pricl, S. Rheology of Industrial Poly-
saccharides: Theory and Applications; Blackie
Academic & Professional: Glasgow, 1995.
34. Mancini, M.; Moresi, M.; Rancini, R. J Food Eng
1999, 39, 369–378.
35. Segeren, A. J. M.; Boskamp, J. V; Van Den Tem-
pel, M. J Chem Soc, Faraday Discussions 1974,
57, 255–262.
36. Friedrich, C.; Heymann, L. J Rheol 1988, 32,
235–241.
37. Chambon, F.; Winter, H. H. J Rheol 1987, 31,
683–697.
38. Lapasin, R.; Pricl, S.; Paoletti, S.; Zanetti, F. J
Appl Polym Sci 1990, 41, 1395–1410.
39. Almdal, K.; Dyre, J.; Hvidt, S.; Kramer, O. Polym
Gels Networks 1993, 1, 5–18.
40. Scanlan, J. C.; Winter, H. H. Macromolecules
1991, 24, 47–54.
41. Winter, H. H.; Mours, M.; In Advances in Polymer
Science. Springer: Berlin, Germany, 1997; pp 165–
234.
42. Gabriele, D.; de Cindio, B; D’Antona, P Rheolog-
ica Acta 2001, 40, 120–127.
43. Moresi, M.; Bruno, M. J of Food Engineering
2007, 82, 298–309.
44. Liu, X. X.; Qian, L. Y.; Shu, T.; Tong, Z. Polymer
2003, 44, 407–412.
45. Lu, L.; Liu, X. X.; Tong, Z.; Gao, Q. X. J Phys
Chem B 2006, 110, 25013–25020.
46. Coppetti, G.; Grassi, M.; Lapasin, R.; Pricl, S.
Glycoconj J 1997, 14, 951–961.
47. Rosalina, I.; Bhattacharya, M. Carbohydr Polym
2002, 48, 191–202.
48. Hyun, K.; Kim, S. H.; Ahn, K. H.; Lee, S. J. J
Non-Newtonian Fluid Mech 2002, 107, 51–65.
49. Shah, J. V.; Janmey, P. A. Rheol Acta 1997, 26,
262–268.
50. Pouzot, M.; Nicolai, T.; Benyahia L. Durand, D.
J Colloid Interface Sci 2006, 293, 376–383.
51. Rodd, A. B.; Dunstan, D. E.; Ross-Murphy, S. B.;
Boger, D. V. Rheol Acta 2001, 40, 23–29.
52. Bossard, F.; Moan, M.; Aubry, T. J Rheol 2007,
51, 1253–1270.
53. Ferry, J. D. Viscoelastic Properties of Polymers,
Wiley; New York, 1980.
1182 NOBILE ET AL.
Journal of Polymer Science: Part B: Polymer PhysicsDOI 10.1002/polb