deltaef1 is a transcriptional repressor of e-cadherin and regulates epithelial plasticity in breast...
TRANSCRIPT

DeltaEF1 is a transcriptional repressor of E-cadherin and regulates
epithelial plasticity in breast cancer cells
Andreas Eger*,1, Kirsten Aigner1, Stefan Sonderegger1, Brigitta Dampier2, Susanne Oehler2,Martin Schreiber2, Geert Berx3, Amparo Cano4, Hartmut Beug5 and Roland Foisner1,*
1Max F. Perutz Laboratories, University Departments at the Vienna Biocenter, Department of Medical Biochemistry, MedicalUniversity Vienna, Vienna, Austria; 2Division of Senology, Department of Obstetrics and Gynecology, Medical University Vienna;Austria; 3Department for Molecular Biomedical Research, VIB-Ghent University, Belgium; 4Instituto de Investigaciones Biomedicas‘Alberto Sols’ CSIC-UAM, Department of Biochemistry, UAM, Madrid, Spain; 5Research Institute of Molecular Pathology, Vienna;Austria
Downregulation of E-cadherin is a crucial event forepithelial to mesenchymal transition (EMT) in embryonicdevelopment and cancer progression. Using the EpFosERmammary tumour model we show that during EMT,upregulation of the transcriptional regulator deltaEF1coincided with transcriptional repression of E-cadherin.Ectopic expression of deltaEF1 in epithelial cells wassufficient to downregulate E-cadherin and to induce EMT.Analysis of E-cadherin promoter activity and chromatinimmunoprecipitation identified deltaEF1 as direct tran-scriptional repressor of E-cadherin. In human cancer cells,transcript levels of deltaEF1 correlated directly with theextent of E-cadherin repression and loss of the epithelialphenotype. The protein was enriched in nuclei of humancancer cells and physically associated with the E-cadherinpromoter. RNA interference-mediated downregulationof deltaEF1 in cancer cells was sufficient to derepressE-cadherin expression and restore cell to cell adhesion,suggesting that deltaEF1 is a key player in late stagecarcinogenesis.Oncogene (2005) 24, 2375–2385. doi:10.1038/sj.onc.1208429Published online 17 January 2005
Keywords: epithelial–mesenchymal transition; celladhesion; breast cancer; invasion; metastasis
Introduction
The epithelial cell–cell adhesion protein E-cadherin is apotent suppressor of tumour cell invasion and metas-tasis (Cavallaro et al., 2002; Conacci-Sorrell et al., 2002;Wheelock and Johnson, 2003). Tumour cells lose oracquire invasive properties when E-cadherin-mediatedadhesion is increased or inhibited, respectively (Behrens
et al., 1989; Frixen et al., 1991; Vleminckx et al., 1991).In line with these findings ectopic expression of E-cadherin in a transgenic mouse model prevented tumourcell invasion and metastasis (Perl et al., 1998). Althoughmutations of the E-cadherin gene have been found inspecific subtypes of cancers, the incidence of suchmutations in carcinogenesis is low (Risinger et al.,1994; Berx et al., 1995; Guilford et al., 1998). Loss ofE-cadherin in cancer cells frequently involves transcrip-tional repression (Strathdee, 2002; Thiery, 2002). Thisincludes epigenetic modifications, such as CpG islandhypermethylation of the E-cadherin regulatory promo-ter (Graff et al., 1995; Grady et al., 2000), as well asrepression by specific transcription factors, includingSnail (Batlle et al., 2000; Cano et al., 2000; Guaita et al.,2002; Peinado et al., 2004b), Slug (Hajra et al., 2002;Bolos et al., 2003), Smad-interacting protein 1 (SIP1,ZEB2) (Comijn et al., 2001) and the helix–loop–helixfactor E12/E47 (Perez-Moreno et al., 2001; Bolos et al.,2003).
Downregulation of E-cadherin is often accompaniedby a conversion of epithelial cells to migratory,fibroblastoid cells, a process collectively referred to asepithelial–mesenchymal transition (EMT). In nonpatho-logical conditions, EMT occurs during well-definedstages of embryonic development, tissue repair andreorganization (Hay, 1995). In the past years, however,EMT has been increasingly recognized as a key event inthe progression of tumour cells to dedifferentiated andinvasive phenotypes (Birchmeier et al., 1996; Thiery,2002; Grunert et al., 2003; Gotzmann et al., 2004).Various signalling proteins, such as receptor tyrosinekinases, small GTPases, MAP kinases, integrin-linkedkinase (ILK), PI3-kinase, TGF-b and c-Fos, as well asmatrix-metalloproteinases and extracellular matrix com-ponents have been implicated in the regulation of EMTand tumour progression, but crosstalks between thesepathways, their downstream targets, and their causalrelations to different steps of tumour developmentremain largely elusive (Birchmeier et al., 1996; Thiery,2002; Grunert et al., 2003; Gotzmann et al., 2004).Many of the signalling pathways interfere with thefunction and/or expression of E-cadherin, but in most
Received 3 September 2004; revised 3 December 2004; accepted 3December 2004; published online 17 January 2005
*Correspondence: A Eger and R Foisner, Department of MedicalBiochemistry, Medical University Vienna, Dr Bohrgasse 9, ViennaBiocenter, Vienna, Austria;E-mail: [email protected] or [email protected]
Oncogene (2005) 24, 2375–2385& 2005 Nature Publishing Group All rights reserved 0950-9232/05 $30.00
www.nature.com/onc

cases downstream effectors directly controlling E-cadherin function and expression are still unknown.Snail is the only protein that has recently beenimplicated in TGFb-, and ILK- dependent downregula-tion of E-cadherin (Tan et al., 2001; Peinado et al.,2003).
Several previously reported properties make thezinc-finger- and homeobox-containing transcriptionalregulator delta-crystallin enhancer-binding factor 1(deltaEF1, also known as ZEB1), a close homolog ofSIP1 (van Grunsven et al., 2001), a potential candidatefor a regulator of E-cadherin expression. First, expres-sion of deltaEF1 during development and in the adult isrestricted to E-cadherin-negative tissues of mesodermalorigin and of neural crest derivatives (Funahashi et al.,1993). Second, deltaEF1 bound to a short fragment ofthe E-cadherin promoter in vitro (Grooteclaes andFrisch, 2000). Third, deltaEF1 caused repression ofluciferase reporter constructs driven by the E-cadherinproximal regulatory promoter region (Grooteclaes andFrisch, 2000). However, despite these findings a directevidence for deltaEF1 being a bona fide E-cadherinrepressor in vivo is still lacking and, more importantly,its role in epithelial physiology and differentiation iscompletely elusive.
In this paper, we show that deltaEF1 is upregulatedduring EMT in a cell culture tumour model and thatectopic expression of deltaEF1 in epithelial cells issufficient to downregulate endogenous E-cadherin andto induce EMT on its own. Most importantly, we showthat deltaEF1 directly represses E-cadherin in humantumour cells and that knockdown of deltaEF1 expres-sion in breast cancer cells by RNA interference (RNAi)is sufficient to induce expression of E-cadherin.
Results
Expression of deltaEF1 is induced during EMT
EpFosER mouse mammary epithelial cells represent aninducible tumour model system that has been usedsuccessfully to unravel molecular mechanisms governingEMT (Eger et al., 2000; Stockinger et al., 2001; Egeret al., 2004). The cells constitutively express a fusionprotein of c-Fos and the hormone-binding domain ofthe oestrogen receptor. While these cells exhibit a fullypolarized epithelial phenotype in the absence of thehormone, they undergo EMT in a highly synchronousmanner upon oestradiol-dependent activation of thefusion protein (Eger et al., 2000). Molecular eventsunderlying FosER-induced EMT include loss of apico-basolateral polarity and growth in multilayers (2 daysafter FosER activation), downregulation of E-cadherin(3–5 days) and the formation of undifferentiatedfibroblastoid cells expressing a variety of mesenchymalmarker proteins (10 days). In addition, activation of theFosER fusion protein in vitro strongly enhanced thetumourigenic and metastatic potential of the cells inSCID mice (our unpublished data). In order to identifyFosER target genes that interfere with E-cadherin
expression and epithelial polarity, we have performeda comprehensive analysis of the genetic programme ofEpFosER cells at morphologically defined stages ofEMT. Polysome-bound mRNA representing activelytranslated RNA (Mikulits et al., 2000) was prepared 12,24, 48, 96 and 240 h after FosER activation and used forAffymetrix GeneChips analyses. Among the differen-tially regulated genes, we identified the transcriptionalregulator deltaEF1, whose RNA level was elevatedfourfold after 12 h of FosER activity and increased upto 12-fold after 96 h (Figure 1a). Oestradiol treatment ofthe parental EpH4 mammary epithelial cells for 48 h wasnot sufficient to induce deltaEF1 expression, indicatingthat upregulation of deltaEF1 is dependent on theactivity of the FosER fusion protein (Figure 1a).
A direct comparison of total E-cadherin and deltaEF1RNA and protein by RT–PCR and immunoblotting,respectively, revealed that upregulation of deltaEF1expression coincided with the downregulation ofE-cadherin mRNA and protein levels during EMT(Figure 1b and c). In contrast to deltaEF1, the expres-sion of Snail and SIP1, two previously describedtranscriptional repressors of E-cadherin, remainedlargely unaltered within the first 4 days of EMT(Figure 1b). Transcript levels of these genes wereelevated only upon long-term FosER activation(Figure 1b, 240 h oestradiol treatment).
Next, we investigated whether deltaEF1 is capable ofinterfering with E-cadherin promoter activity usingluciferase reporter constructs that contained the �319/þ 56 or the �178/þ 92 fragment of the murineE-cadherin proximal regulatory promoter. Transientexpression of deltaEF1 in differentiated epithelialEpFosER cells resulted in strong downregulation ofthe activities of both luciferase reporter genes(Figure 1d). Thus, deltaEF1 is a prime candidate fordownregulating E-cadherin expression during FosER-induced EMT.
Figure 1 Expression of deltaEF1 during EMT. (a) Polysome-bound mRNA was harvested from untreated and 12, 24, 48, 96 and240 h oestradiol-treated EpFosER mouse mammary epithelial cells.In addition, RNA was prepared from parental EpH4 cells treatedwith estradiol for 48 h. Polysome-bound RNA was used forAffymetrix GeneChips analyses. Experiments were performed intriplicates and bars represent standard deviation. (b) Total mRNAwas prepared from epithelial cells in the absence of estradiol (�E2)and from estradiol treated cultures (þE2, 6–240h). RT–PCR wasperformed with primers specific for E-cadherin, deltaEF1, Snailand SIP1. The expression of Actin was analysed in the samesamples as a control for the amount of cDNA. (c) Equivalentamounts of total cell lysates of untreated polarized EpFosER cells(�E2) and cells treated with estradiol for 24 h (24 h) to 240 h (240 h)were analysed by immunoblotting using antibodies to deltaEF1and E-cadherin. (d) Epithelial EpFosER cells were transientlytransfected with the E-cadherin reporter plasmids pGL3-Ecad(�319 to þ 56, wild-type sequence) or pGl2-E-cad (�178/þ 92,wild-type sequence) together with the deltaEF1 expression vector(dEF1) or empty plasmid (Control). The level of luciferase activitywas normalized to the expression of cotransfected b-galactosidaseand fold repression relative to the control vector is shown. Barsrepresent standard deviation
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2376
Oncogene
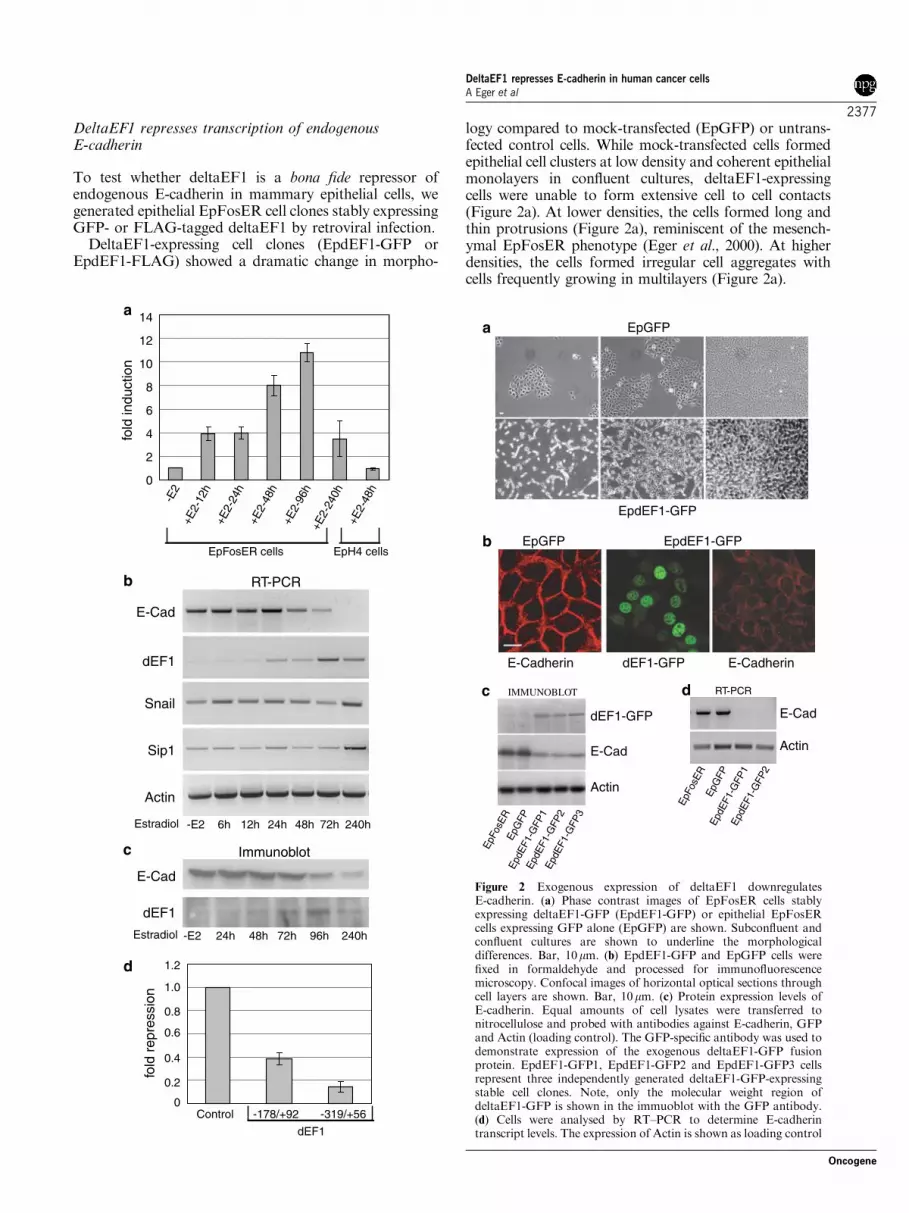
DeltaEF1 represses transcription of endogenousE-cadherin
To test whether deltaEF1 is a bona fide repressor ofendogenous E-cadherin in mammary epithelial cells, wegenerated epithelial EpFosER cell clones stably expressingGFP- or FLAG-tagged deltaEF1 by retroviral infection.
DeltaEF1-expressing cell clones (EpdEF1-GFP orEpdEF1-FLAG) showed a dramatic change in morpho-
logy compared to mock-transfected (EpGFP) or untrans-fected control cells. While mock-transfected cells formedepithelial cell clusters at low density and coherent epithelialmonolayers in confluent cultures, deltaEF1-expressingcells were unable to form extensive cell to cell contacts(Figure 2a). At lower densities, the cells formed long andthin protrusions (Figure 2a), reminiscent of the mesench-ymal EpFosER phenotype (Eger et al., 2000). At higherdensities, the cells formed irregular cell aggregates withcells frequently growing in multilayers (Figure 2a).
fold
indu
ctio
n
2
4
6
8
10
12
14
0
+E2-
48h
+E2-
12h
-E2
+E2-
48h
+E2-
96h
+E2-
240h
+E2-
24h
EpFosER cells
E-Cad
Snail
Estradiol 6h 12h 24h 48h 72h
Actin
dEF1
-E2
Sip1
EpH4 cells
240h
Control
fold
rep
ress
ion
0
0.2
0.4
0.6
0.8
1.0
1.2
dEF1
-319/+56-178/+92
RT-PCR
Immunoblot
E-Cad
dEF1
24h 48h 72h-E2 240h96hEstradiol
a
b
c
d
E-Cadherin
EpdEF1-GFP
dEF1-GFPE-Cadherin
EpGFP
IMMUNOBLOT
dEF1-GFP
E-Cad
Actin
EpFo
sER
EpG
FPEp
dEF1
-GFP
1Ep
dEF1
-GFP
2
a
b
c
EpGFP
EpdEF1-GFP
d RT-PCR
E-Cad
Actin
EpFo
sER
EpG
FPEp
dEF1
-GFP
1Ep
dEF1
-GFP
2
EpdE
F1-G
FP3
Figure 2 Exogenous expression of deltaEF1 downregulatesE-cadherin. (a) Phase contrast images of EpFosER cells stablyexpressing deltaEF1-GFP (EpdEF1-GFP) or epithelial EpFosERcells expressing GFP alone (EpGFP) are shown. Subconfluent andconfluent cultures are shown to underline the morphologicaldifferences. Bar, 10mm. (b) EpdEF1-GFP and EpGFP cells werefixed in formaldehyde and processed for immunofluorescencemicroscopy. Confocal images of horizontal optical sections throughcell layers are shown. Bar, 10mm. (c) Protein expression levels ofE-cadherin. Equal amounts of cell lysates were transferred tonitrocellulose and probed with antibodies against E-cadherin, GFPand Actin (loading control). The GFP-specific antibody was used todemonstrate expression of the exogenous deltaEF1-GFP fusionprotein. EpdEF1-GFP1, EpdEF1-GFP2 and EpdEF1-GFP3 cellsrepresent three independently generated deltaEF1-GFP-expressingstable cell clones. Note, only the molecular weight region ofdeltaEF1-GFP is shown in the immuoblot with the GFP antibody.(d) Cells were analysed by RT–PCR to determine E-cadherintranscript levels. The expression of Actin is shown as loading control
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2377
Oncogene
We next analysed expression and intracellular locali-zation of E-cadherin. In mock-transfected cells, E-cadherin was detected exclusively at lateral cell–cellcontact sites (Figure 2b). However, deltaEF1-expressingcells did not exhibit a significant E-cadherin-specificstaining at the cell periphery (Figure 2b). A weakstaining of the protein could only be detected atperinuclear regions (Figure 2b). Immunoblot analysesof total cell lysates confirmed that E-cadherin proteinlevels were strongly downregulated in three independentclones of EpdEF1-GFP versus control cells (Figure 2c).As tested by RT–PCR analyses, E-cadherin mRNAlevels also were strongly reduced (Figure 2d), suggestingthat E-cadherin was repressed on the transcriptionallevel.
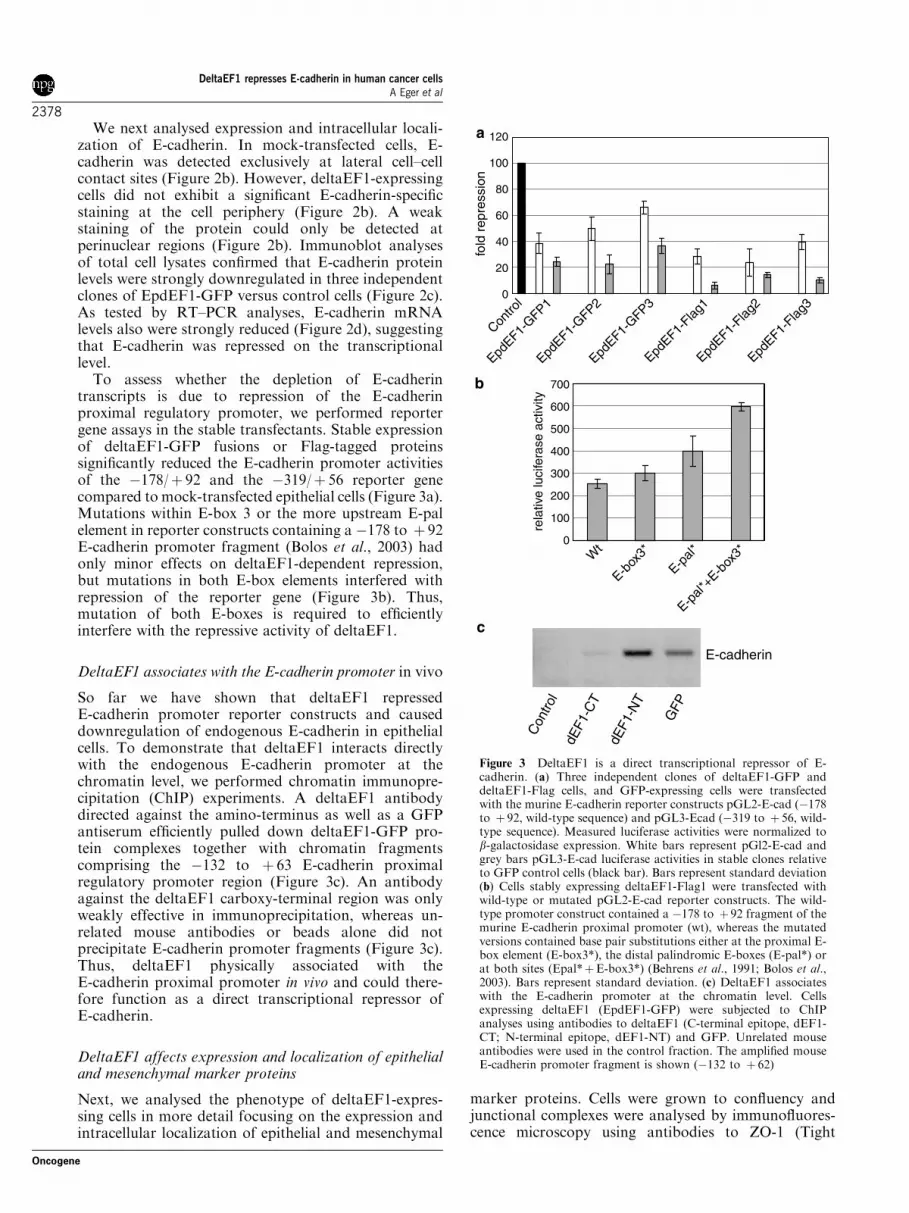
To assess whether the depletion of E-cadherintranscripts is due to repression of the E-cadherinproximal regulatory promoter, we performed reportergene assays in the stable transfectants. Stable expressionof deltaEF1-GFP fusions or Flag-tagged proteinssignificantly reduced the E-cadherin promoter activitiesof the �178/þ 92 and the �319/þ 56 reporter genecompared to mock-transfected epithelial cells (Figure 3a).Mutations within E-box 3 or the more upstream E-palelement in reporter constructs containing a �178 to þ 92E-cadherin promoter fragment (Bolos et al., 2003) hadonly minor effects on deltaEF1-dependent repression,but mutations in both E-box elements interfered withrepression of the reporter gene (Figure 3b). Thus,mutation of both E-boxes is required to efficientlyinterfere with the repressive activity of deltaEF1.
DeltaEF1 associates with the E-cadherin promoter in vivo
So far we have shown that deltaEF1 repressedE-cadherin promoter reporter constructs and causeddownregulation of endogenous E-cadherin in epithelialcells. To demonstrate that deltaEF1 interacts directlywith the endogenous E-cadherin promoter at thechromatin level, we performed chromatin immunopre-cipitation (ChIP) experiments. A deltaEF1 antibodydirected against the amino-terminus as well as a GFPantiserum efficiently pulled down deltaEF1-GFP pro-tein complexes together with chromatin fragmentscomprising the �132 to þ 63 E-cadherin proximalregulatory promoter region (Figure 3c). An antibodyagainst the deltaEF1 carboxy-terminal region was onlyweakly effective in immunoprecipitation, whereas un-related mouse antibodies or beads alone did notprecipitate E-cadherin promoter fragments (Figure 3c).Thus, deltaEF1 physically associated with theE-cadherin proximal promoter in vivo and could there-fore function as a direct transcriptional repressor ofE-cadherin.
DeltaEF1 affects expression and localization of epithelialand mesenchymal marker proteins
Next, we analysed the phenotype of deltaEF1-expres-sing cells in more detail focusing on the expression andintracellular localization of epithelial and mesenchymal
marker proteins. Cells were grown to confluency andjunctional complexes were analysed by immunofluores-cence microscopy using antibodies to ZO-1 (Tight
Contro
l0
20
40
60
80
100
120
fold
rep
ress
ion
rela
tive
luci
fera
se a
ctiv
ity
100
200
300
400
500
600
700
0
c
Con
trol
dEF1
-CT
dEF1
-NT
GFP
E-cadherin
EpdEF1-
GFP1
EpdEF1-
Flag1
EpdEF1-
GFP2
EpdEF1-
GFP3
EpdEF1-
Flag3
EpdEF1-
Flag2
Wt
E-box
3*
E-pal*
E-pal*
+E-b
ox3*
a
b
Figure 3 DeltaEF1 is a direct transcriptional repressor of E-cadherin. (a) Three independent clones of deltaEF1-GFP anddeltaEF1-Flag cells, and GFP-expressing cells were transfectedwith the murine E-cadherin reporter constructs pGL2-E-cad (�178to þ 92, wild-type sequence) and pGL3-Ecad (�319 to þ 56, wild-type sequence). Measured luciferase activities were normalized tob-galactosidase expression. White bars represent pGl2-E-cad andgrey bars pGL3-E-cad luciferase activities in stable clones relativeto GFP control cells (black bar). Bars represent standard deviation(b) Cells stably expressing deltaEF1-Flag1 were transfected withwild-type or mutated pGL2-E-cad reporter constructs. The wild-type promoter construct contained a �178 to þ 92 fragment of themurine E-cadherin proximal promoter (wt), whereas the mutatedversions contained base pair substitutions either at the proximal E-box element (E-box3*), the distal palindromic E-boxes (E-pal*) orat both sites (Epal*þE-box3*) (Behrens et al., 1991; Bolos et al.,2003). Bars represent standard deviation. (c) DeltaEF1 associateswith the E-cadherin promoter at the chromatin level. Cellsexpressing deltaEF1 (EpdEF1-GFP) were subjected to ChIPanalyses using antibodies to deltaEF1 (C-terminal epitope, dEF1-CT; N-terminal epitope, dEF1-NT) and GFP. Unrelated mouseantibodies were used in the control fraction. The amplified mouseE-cadherin promoter fragment is shown (�132 to þ 62)
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2378
Oncogene

junctions) and Desmoplakin (Desmosomes). In contrastto their peripheral localization at cell–cell contact sitesin GFP-expressing epithelial cells, both proteins werediffusely distributed throughout the cytoplasm indeltaEF1-expressing cells (Figure 4a). The overall ZO-1 protein level was significantly reduced, whereas theamount of Desmoplakin Iþ II was comparable to thatof mock-transfected epithelial cells (Figure 4a). Thus,deltaEF1-induced epithelial dedifferentiation includesboth delocalization and downregulation of epithelialproteins. In addition, ectopic expression of deltaEF1caused a strong induction of the mesenchymal markerprotein Vimentin (Figure 4b). Interestingly, N-cadherinprotein levels were also upregulated in the fibroblastoidcell cultures (Figure 4b). N-cadherin has previously beenshown to promote cell migration and tumour progres-sion and to accumulate in E-cadherin-negative tumourcells (Hazan et al., 2000; Tomita et al., 2000).Altogether, expression of deltaEF1 in epithelial cellswas sufficient to induce hallmarks of EMT, such asdownregulation of E-cadherin and ZO-1, disintegrationof cell–cell junctions and induction of mesenchymalmarker proteins.
DeltaEF1 represses E-cadherin in human breast cancercells
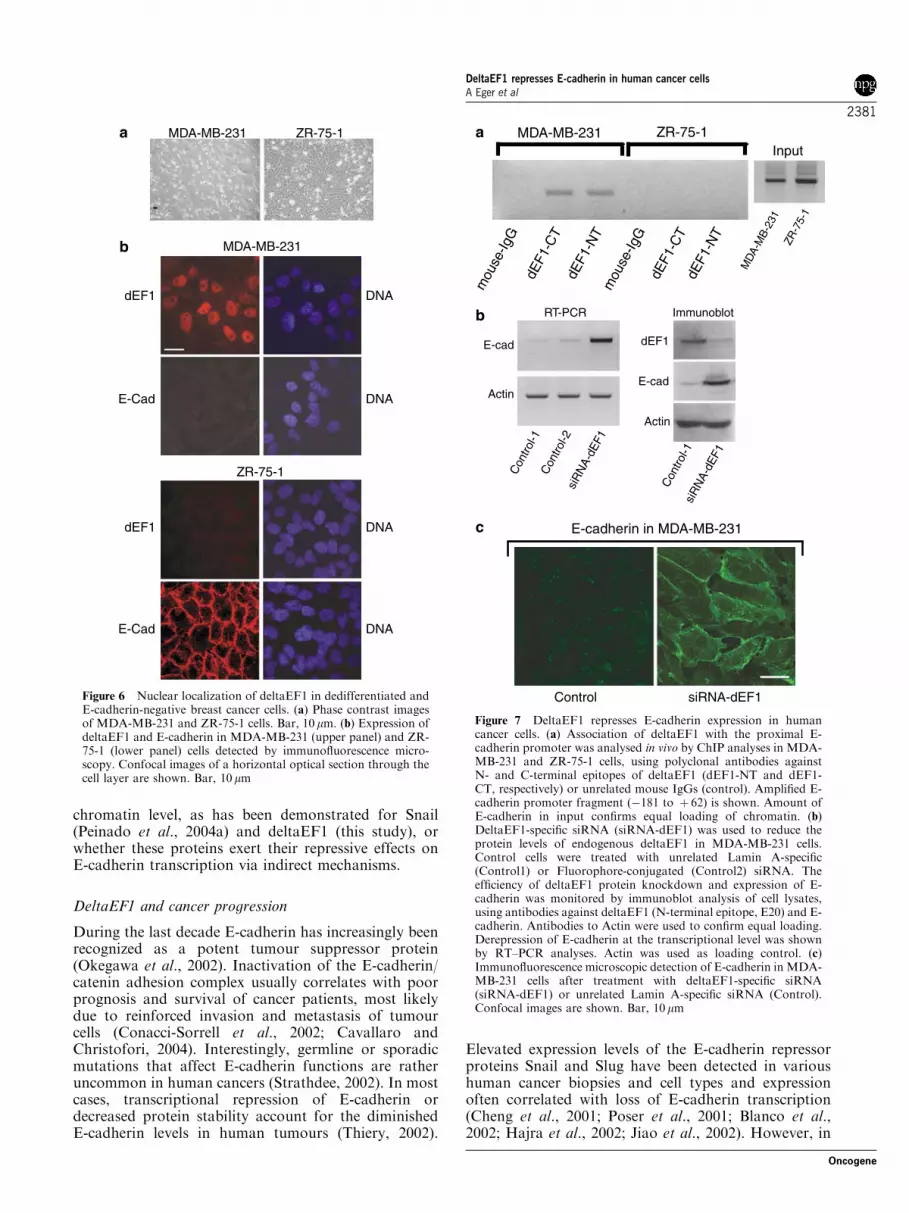
Loss of E-cadherin expression in human tumours isfrequently associated with poor prognosis and survival,most likely due to enhanced invasiveness and metastasis(Cavallaro and Christofori, 2004). To test whetherdeltaEF1 mediates transcriptional repression of E-cadherin in human tumour cells, we first performedreal-time PCR to examine the expression of deltaEF1and E-cadherin in a panel of 19 human cell lines derivedfrom normal breast (one), fibrocystic disease (two) orbreast cancer (16). The selected cells reflect manyfeatures of cancer cells in vivo and have been intensivelyused as tumour models in the last years (Lacroix andLeclercq, 2004). Interestingly, tumour cells that ex-pressed high levels of deltaEF1 exhibited very low orundetectable levels of E-cadherin transcripts (Figure 5).Conversely, all E-cadherin-positive cells had stronglyreduced deltaEF1 mRNA levels (Figure 5). Thus, thereexists a strong inverse correlation between expression ofdeltaEF1 and E-cadherin. Expression of Snail did notstrictly correlate with E-cadherin downregulation, sug-gesting that deltaEF1, rather than Snail is a keyregulator of E-cadherin expression in these humantumour cells. This is consistent with the high deltaEF1protein levels detected in the nuclei of E-cadherin-negative MDA-MB-231 (Figure 6b) or CAMA-1 cells(data not shown), whereas E-cadherin-positive cells,such as ZR-75-1, completely lacked nuclear deltaEF1(Figure 6b). Moreover, all cells with high deltaEF1levels exhibited a rather undifferentiated and spindle-shaped morphology (Figure 6a; compare also withLacroix and Leclercq, 2004). To support the model thatdeltaEF1 can serve as a direct transcriptional repressorof E-cadherin in these cancer cells, we performeddeltaEF1-specific ChIP experiments in MDA-MB-231cells, which lack E-cadherin, and ZR-75-1 cells, whichexpress high amounts of E-cadherin (Figure 5 and 6b).Unlike unrelated mouse antibodies, two deltaEF1-specific antibodies coprecipitated chromatin fragmentsof the human E-cadherin promoter in MDA-MB-231cells, whereas the same antibodies could not bring downthe promoter region in ZR-75-1 cells (Figure 7a). Thus,like ectopic deltaEF1 in murine cells (Figure 3c),endogenously expressed deltaEF1 in cancer cells directlyassociated with the E-cadherin promoter in vivo(Figure 7a).
These data suggest that deltaEF1 represents a majorregulator of E-cadherin transcription in human tumourcells. To test this model on a functional level, weemployed RNA interference (RNAi) to knock downendogenous deltaEF1 protein in cancer cells. Smallinterfering RNA (siRNA) targeting deltaEF1, or un-related control siRNA (Rhodamine-conjugated orLaminA-specific) were transfected into MDA-MB-231cells and expression of E-cadherin and deltaEF1 wasanalysed 3 days after transfection. Immunoblotting oftotal cell lysates revealed a significant decrease ofdeltaEF1 protein levels upon treatment with thedeltaEF1-specific siRNA (Figure 7b). In the same cells,
EpdEF1-GFPEpGFP
ZO-1
Dp
EpG
FPEp
dEF1
-GFP
1Ep
dEF1
-GFP
2
EpG
FPEp
dEF1
-GFP
1Ep
dEF1
-GFP
2
a
b
Vim
N-cad
EpdE
F1-G
FP3
EpdE
F1-G
FP3
ZO-1
Dp
Immunoblot
Immunoblot
Vim
N-cad
Figure 4 DeltaEF1 expression induces EMT. (a) Epithelial GFP-expressing (EpGFP) and deltaEF1-expressing (EpdEF1-GFP1)cells were processed for immunofluorescence using antibodies toZO-1 (Tight junctions) and Desmoplakin Iþ II (Desmosomes).Inserts show expression and nuclear localization of deltaEF1-GFP.Confocal images are shown. Bar, 10 mm. (b) Confocal images ofEpGFP and EpdEF1-GFP1 cells depicting the localization ofVimentin and N-cadherin are shown. Bar, 10mm. Immunoblots oftotal cell lysates in (a) and (b) show total levels of indicated proteins
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2379
Oncogene

expression of E-cadherin was highly induced, both onthe RNA as well as on the protein level (Figure 7b).E-cadherin accumulated at the plasma membrane andwas strongly enriched at sites of cell–cell contact(Figure 7c). The E-cadherin-expressing cells had oftenlost their fibroblastoid morphology and exhibited a well-spread, epithelial-like morphology. Thus, inhibition of asingle transcriptional regulator was sufficient to reacti-vate expression of a silenced E-cadherin gene in cancercells.
Discussion
DeltaEF1 – a novel member of the E-cadherin repressorfamily
DeltaEF1 (zfh1) and the related gene SIP1 (zfh2) werefirst isolated from a Drosophila cDNA expressionlibrary (Fortini et al., 1991). Both proteins contain twoC2H2 (Kruppel)-type zinc-finger clusters at their C- andN-termini as well as a central homeobox domain (vanGrunsven et al., 2001). Ectopic expression of SIP1 wasrecently found to reduce E-cadherin expression in canineepithelial cells (MDCK) (Comijn et al., 2001). AlsodeltaEF1 was shown to interact with the E-cadherinpromoter in vitro and to repress E-cadherin promoteractivity in reporter gene assays (Grooteclaes and Frisch,2000). However, the physiological significance of thesein vitro data remained completely elusive.
In this paper, we show that stable expression ofdeltaEF1 in mammary epithelial cells was sufficient tocause downregulation of endogenous E-cadherin mRNAand protein levels. Our findings strongly argue thatdeltaEF1 directly inhibits E-cadherin transcription, asdeltaEF1 physically associated with the E-cadherin
promoter at the chromatin level in vivo. In addition, wefound that deltaEF1 induced EMT in epithelial cellsincluding strong induction of Vimentin and N-cadherinand downregulation or relocalization of the epithelialproteins ZO-1 and Desmoplakin. Although E-cadherinsilencing is considered a hallmark of EMT, it is most likelynot sufficient to induce expression of N-cadherin orVimentin. Thus, deltaEF1 might regulate the expression ofvarious other proteins important for epithelial plasticity.
Two other zinc-finger proteins lacking homeoboxdomains, Snail and Slug, as well as the basic helix–loop–helix (bHLH) factor E47 have been reported to repressexpression of E-cadherin in different cell lines (Batlleet al., 2000; Cano et al., 2000; Perez-Moreno et al., 2001;Guaita et al., 2002; Hajra et al., 2002; Bolos et al., 2003).Repressive activity of Snail, Slug as well as SIP1 wasdependent on the integrity of E-box elements in theE-cadherin proximal promoter region (Batlle et al.,2000; Comijn et al., 2001; Hajra et al., 2002). Likewise,we could show that mutation of both E-boxes isrequired to significantly affect deltaEF1-mediated re-pression. At present it is unclear whether differentrepressor molecules can bind to adjacent E-box elementssimultaneously and repress transcription in a coopera-tive manner or whether the proteins function individu-ally in a cell-type and tissue-specific manner. In additionto proximal E-box elements, a complex interplay ofmultiple regulatory regions dispersed throughout largeparts of the E-cadherin locus has been identified recentlyin transgenic mice (Stemmler et al., 2003). Thus, thefunctional characterization of the different repressormolecules on the chromatin level in vivo represents amajor challenge of future research. In this context itremains to be demonstrated whether SIP1, Slug and E47can directly bind to the E-cadherin promoter at the
MD
A-M
B-46
8T-
47D
ZR-7
5-1
KPL-
1BT
-474
MD
A-M
B-45
3M
CF-
10F
HC
C11
43
MC
F-7
MC
F-10
AM
DA-
MB-
435S
CAL
-51
CAM
A-1
Hs-
578T
Hs-
578B
st
MD
A-M
B-23
1
AU56
5
HM
EC-1
84B5
Hcc
1937
0
0.5
1
1.5
2
2.5
3
rela
tive
tran
scrip
t lev
els
dEF1
Snail
E-cadherin
Figure 5 Expression of deltaEF1, Snail and E-cadherin in human breast cancer cell lines. Transcript levels of the three genes weredetermined in all cell-lines by real-time PCR. White bars denote relative E-cadherin, grey bars relative deltaEF1 and black bars relativeSnail transcript levels. HMEC-184B5 cells were established from normal mammary gland and MCF-10A and MCF-10F were derivedfrom mammary fibrocystic disease. All other cell lines were derived from breast cancer patients. Note that mRNA values of deltaEF1,Snail or E-cadherin are very low in some cell lines and corresponding bars not visible in this scale
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2380
Oncogene
chromatin level, as has been demonstrated for Snail(Peinado et al., 2004a) and deltaEF1 (this study), orwhether these proteins exert their repressive effects onE-cadherin transcription via indirect mechanisms.
DeltaEF1 and cancer progression
During the last decade E-cadherin has increasingly beenrecognized as a potent tumour suppressor protein(Okegawa et al., 2002). Inactivation of the E-cadherin/catenin adhesion complex usually correlates with poorprognosis and survival of cancer patients, most likelydue to reinforced invasion and metastasis of tumourcells (Conacci-Sorrell et al., 2002; Cavallaro andChristofori, 2004). Interestingly, germline or sporadicmutations that affect E-cadherin functions are ratheruncommon in human cancers (Strathdee, 2002). In mostcases, transcriptional repression of E-cadherin ordecreased protein stability account for the diminishedE-cadherin levels in human tumours (Thiery, 2002).
Elevated expression levels of the E-cadherin repressorproteins Snail and Slug have been detected in varioushuman cancer biopsies and cell types and expressionoften correlated with loss of E-cadherin transcription(Cheng et al., 2001; Poser et al., 2001; Blanco et al.,2002; Hajra et al., 2002; Jiao et al., 2002). However, in
MDA-MB-231
MDA-MB-231
dEF1 DNA
DNAE-Cad
a
b
ZR-75-1
dEF1 DNA
DNAE-Cad
ZR-75-1
Figure 6 Nuclear localization of deltaEF1 in dedifferentiated andE-cadherin-negative breast cancer cells. (a) Phase contrast imagesof MDA-MB-231 and ZR-75-1 cells. Bar, 10 mm. (b) Expression ofdeltaEF1 and E-cadherin in MDA-MB-231 (upper panel) and ZR-75-1 (lower panel) cells detected by immunofluorescence micro-scopy. Confocal images of a horizontal optical section through thecell layer are shown. Bar, 10 mm
mou
se-Ig
G
dEF1
-NT
dEF1
-CT
MDA-MB-231 ZR-75-1
dEF1
-NT
dEF1
-CT
a
b
Control siRNA-dEF1
E-cadherin in MDA-MB-231
mou
se-Ig
G
Input
MD
A-M
B-23
1ZR
-75-
1
Con
trol-1
siR
NA-
dEF1
dEF1
E-cad
Actin
siR
NA-
dEF1
Con
trol-1
Con
trol-2
E-cad
Actin
c
RT-PCR Immunoblot
Figure 7 DeltaEF1 represses E-cadherin expression in humancancer cells. (a) Association of deltaEF1 with the proximal E-cadherin promoter was analysed in vivo by ChIP analyses in MDA-MB-231 and ZR-75-1 cells, using polyclonal antibodies againstN- and C-terminal epitopes of deltaEF1 (dEF1-NT and dEF1-CT, respectively) or unrelated mouse IgGs (control). Amplified E-cadherin promoter fragment (�181 to þ 62) is shown. Amount ofE-cadherin in input confirms equal loading of chromatin. (b)DeltaEF1-specific siRNA (siRNA-dEF1) was used to reduce theprotein levels of endogenous deltaEF1 in MDA-MB-231 cells.Control cells were treated with unrelated Lamin A-specific(Control1) or Fluorophore-conjugated (Control2) siRNA. Theefficiency of deltaEF1 protein knockdown and expression of E-cadherin was monitored by immunoblot analysis of cell lysates,using antibodies against deltaEF1 (N-terminal epitope, E20) and E-cadherin. Antibodies to Actin were used to confirm equal loading.Derepression of E-cadherin at the transcriptional level was shownby RT–PCR analyses. Actin was used as loading control. (c)Immunofluorescence microscopic detection of E-cadherin in MDA-MB-231 cells after treatment with deltaEF1-specific siRNA(siRNA-dEF1) or unrelated Lamin A-specific siRNA (Control).Confocal images are shown. Bar, 10mm
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2381
Oncogene

human breast cancer cell lines, we were unable to find aclear correlation of Snail expression and the down-regulation of E-cadherin on the transcriptional level.Thus, expression of Snail is not always sufficient toblock E-cadherin transcription and may require coex-pression of other repressor molecules such as deltaEF1.Squamous cell carcinoma cells, for example, exhibitedelevated levels of both, deltaEF1 and Snail, which wereaccompanied by a strong reduction of E-cadherin proteinlevels and upregulation of mesenchymal marker proteins(Taki et al., 2003). Interestingly, ectopic expression ofSnail in various cell lines can result in induction ofdeltaEF1 expression (Guaita et al., 2002; Taki et al.,2003). Likewise, Snail was essential for proper expressionof deltaEF1 during mesoderm formation in Drosophilaembryonic development (Lai et al., 1993).
In this paper, we could show a strict inverse correlationof deltaEF1 and E-cadherin expression in a collection of19 human cell lines, most of them derived from breastcancer patients. Cells expressing elevated levels ofdeltaEF1 lacked E-cadherin and were unable to formepithelial cell sheets (for a detailed description of breastcancer cell lines used in this study, see Lacroix andLeclercq, 2004). Most importantly, a series of resultsshown here demonstrate a direct role of deltaEF1 inE-cadherin downregulation in these human tumour cells:First, in line with the RNA expression data, high proteinlevels of deltaEF1 were detected only in nuclei ofE-cadherin-negative cells. Second, deltaEF1 directly asso-ciated with the E-cadherin promoter at the chromatin levelin vivo. Third, downregulation of deltaEF1 by RNAi ledto derepression of the E-cadherin promoter on thetranscriptional level and accumulation of E-cadherin atthe plasma membrane. For this reason deltaEF1 mightenhance the malignant conversion of tumour cells to inva-sive and metastatic phenotypes. In line with our findings, arecent paper has also implied deltaEF1 in E-cadherinrepression in lung cancer cells (Ohira et al., 2003).
Thus, during cancer progression deltaEF1 couldfunction as a molecular switch allowing tumour cellsto change phenotypes rapidly and frequently. Thismight contribute to the cellular heterogeneity oftumours and explain the dynamics of E-cadherinexpression in different regions of solid tumours and atmetastatic lesions. DeltaEF1 and functionally related E-cadherin repressor molecules might regulate distinctstages of metastasis, such as initial dedifferentiation ofprimary tumour cells, the maintenance of the migratoryand/or undifferentiated phenotype, and the rate ofredifferentiation and settlement at distinct tissues andorgans. However, further studies in tumour samples andcell systems will be required for a better understandingof the diverse roles of these transcriptional repressors intumour formation, progression and metastasis.
Materials and methods
Cell culture
EpFosER and EpH4 cells were cultivated as describedpreviously (Reichmann et al., 1989; Eger et al., 2000).
EpdEF1-GFP, EpdEF1-Flag and EpGFP cells were cultivatedin DMEM and 10% FCS as described for the parental cells(Eger et al., 2000). Human cell lines were cultivated in mediarecommended by the American Type Culture Collection(ATCC).
DNA GeneChips
Polysome-bound poly(A)þ RNA was harvested as describedpreviously (Mikulits et al., 2000). Biotin-labelled cRNA forhybridization was prepared as described, using polysome-bound RNA fractions (Fambrough et al., 1999; Damm et al.,2001). Fragmented cRNA (10 mg) in 200 ml hybridizationsolution were hybridized onto the Affymetrix GeneChipMu11k array set (sub A, B), representing over 11 000 knownmurine genes and ESTs. Images were scanned at 3 mmresolution using a Hewlett-Packard GeneArray Scanner, andanalysed using Affymetrix GeneChip software. Experimentswere performed in triplicates, using independently isolatedmRNA preparations derived from different cell harvests.
Construction of deltaEF1 expression plasmids
The GFP- or FLAG-tag was inserted into the EcoRI and SalIsites of the pBabe-puro (Morgenstern and Land, 1990)expression vector. To allow rapid generation of GFP andFLAG fusion proteins, we inserted the Cassette B of theGateway cloning system (Invitrogen, Carlsbad, USA) into theSnaBI site located upstream of GFP and FLAG. Full-lengthdeltaEF1 cDNA was amplified using pCS3mDeltaEF1FLvector (Funahashi et al., 1993) as a template (Primer: forward50-CACCATGGCGGATGGCCCCAGGTG-30; reverse 50-AGCTTCATTTGTCTTCTCTTCAG-30) and cloned into theGateway Entry Vector pENTR/D-TOPO. To create C-terminally tagged fusion proteins, deltaEF1 was shuttled fromthe pENTR/D-TOPO entry vector into the Gateway compa-tible destination vector pBabe-puro-GFP or -FLAG via theGateway LR-reaction according to the manufacturer’s instruc-tions.
Retroviral infection and generation of stable cell clones
The helper-free, ecotropic Phoenix packaging cell line wastransfected with the deltaEF1 retroviral expression constructsusing Lipofectamine 2000 (Invitrogen, Carlsbad, USA). Cellswere cultivated for 30 h at 321C and supernatants (supple-mented with 5 mg/ml Polybrene) were used to infect EpFosERmammary epithelial cells. Target cells were incubated for 48 hat 371C in the presence of retroviral particles and for anotherday in fresh medium. Puromycin (5 mg/ml) was applied to thecultures to select for deltaEF1-expressing cells. DeltaEF1-GFPtransfectants were further enriched by Fluorescence-ActivatedCell Sorting. Three independent deltaEF1-GFP as well asdeltaEF1-FLAG subclones were obtained by limited dilution.These cell clones were used for all experiments.
Reporter gene assays
The mouse �319/þ 56 E-cadherin promoter fragment wasamplified by PCR and directionally cloned into the SacI andBglII sites of the pGL3 reporter vector (pGL3-Ecad). Themouse �178/þ 92 E-cadherin promoter sequences in its wild-type and mutant E-box versions (Behrens et al., 1991) wereexcised by XbaI from the chloramphenicol acetyltransferase(CAT) reporter gene (Behrens et al., 1991; Bolos et al., 2003)and cloned into the NheI site of pGL2 reporter vector (pGL2-Ecad) (Bolos et al., 2003). pGL2 and pGL3 reporter vectorswere purchased from Invitrogen (Carlsbad, USA).
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2382
Oncogene

E-cadherin promoter activity was determined in EpGFP andthree EpdEF1-GFP-expressing cell clones. Cells were trans-fected with 2 mg pGL2-Ecad (wild-type) or 2mg pGL3-Ecadreporter constructs together with 2mg of the b-galactosidaseexpression construct pAD-CMV1 : b-Gal (Eger et al., 2000) tonormalize for transfection. DeltaEF1-Flag1 cells were trans-fected with 2mg of wild-type or mutated pGL2-Ecad reporterconstructs (Behrens et al., 1991; Bolos et al., 2003) and 2mg ofpAD-CMV1 : b-Gal.To test the repressive activity of transiently expressed
deltaEF1 in epithelial EpFosER cells, the deltaEF1 expressionvector pCS3mDeltaEF1FL was cotransfected with wild-typepGL3-Ecad or wild-type pGL2-Ecad. In all, 2mg ofpCS3mDeltaEF1FL (or empty control vector) and 1 mgof E-cadherin promoter reporter constructs and 1 mg ofpAD-CMV1 : b-Gal were used for the assays. All transfectionswere performed in six-well plates with Lipofectamin 2000(Invitrogen, Carlsbad, USA). Luciferase and b-galactosidaseassays were performed 48 h after transfections as describedpreviously (Eger et al., 2000).
RT–PCR
Cells were lysed in TRIzol reagent (Invitrogen, Carlsbad,USA) and total RNA was prepared according to themanufacturer’s instructions. Poly(A)þ RNA was preparedusing the mRNA Isolation Kit (Roche, Mannheim, Germany)and cDNA was synthesized using the AMV first strand cDNAsynthesis kit (Roche, Mannheim, Germany). cDNAs were usedfor PCR analysis using the puReTaq-Ready-To-Go PCRbeads (Amersham Pharmacia Biotech, Uppsala, Sweden) andoligonucleotide primers specific for E-cadherin: forward 50-GAGCCTGAGTCCTGCAGTCC-30, reverse 50-TGTATTGCTGCTTGGCCTCA-30; deltaEF1: forward 50-CCTAGATCAGGACTCAAGAC-30, reverse 50-CACAGAAGGC AAGTGCTATC-30; SIP1: forward 50-TCCAAATAGCTTCTCTTCCGAGG-30, reverse 50-ACCTGTGATTCATGTGCTGCGA-30;Snail: forward 50-ACCTTCCAGCAGCCCT ACGACC-30,reverse 50-GTGTGGCTTCGGATGTGCATC; Actin: forward50- ATCTGGCACCACACCTTCT AC-30, reverse 50-CAGCCAGGTCCAGACGCAGG-30
Real-time PCR
For RNA isolation, cells were seeded at a density of B20 000cells/cm2 and incubated for 48 h. Medium was changed andcells were incubated for another 48 h to reach a cell density ofaround 70%. Medium was removed and cells were immedi-ately lysed in Tri-Reagent (Sigma, St Louis, USA). Total RNAwas prepared following the manufacturer’s instructions andfurther purified using the RNeasy Mini Kit (Qiagen, Hilden,Germany). RNA quality was assessed using the Bioanalyzer2100 and the RNA 6000 nanoLabChip Kit (Agilent Techno-logies, Palo Alto, CA, USA). Only RNAs with a 28S : 18Sratio higher than 1.8 were used for reverse transcription.cDNA was prepared with cDNA High Capacity cDNAArchive Kit (Applied Biosystems, Foster City, CA, USA).For the real-time PCR, the TaqMans-system (AppliedBiosystems, Foster City, CA, USA) with assay-on-demandt(Applied Biosystems) was used following the manufacturer’sinstructions (Tokuhiro et al., 2003). Assay ID numbers:deltaEF1: Hs00611018_m1; Snail Hs00195591_m1, E-cadherinHs00170423_m1. The relative expression of deltaEF1, Snailand E-cadherin was normalized to the amount of b-actin in thesame cDNA using the standard curve method described by themanufacturer.
Antibodies
The following antibodies (Abs) were used: mouse monoclonalAbs against Desmoplakin I and II (Progen Biotechnik,Heidelberg, Germany), E-cadherin (Transduction Labora-tories, Lexington, UK) and GFP (Roche, Mannheim, Ger-many); rat monoclonal Ab against ZO-1 (Chemicon,Temecula, USA); goat polyclonal Abs against deltaEF1(ZEB-R17 and ZEB-E20) and N-cadherin (N19) (all SantaCruz Biotechnology Inc., Santa Cruz, CA, USA); rabbitpolyclonal Abs to N-cadherin (H-63), GFP (Clontech, PaloAlto, USA), Actin and Vimentin (both Sigma, St.Louis, USA);secondary Abs coupled to Alexa Fluor 488 (Molecular Probes,Inc., Eugene, OR, USA), Texas Red or Peroxidase (JacksonLaboratories, West-Grove, USA).
Immunofluorescence microscopy and immunoblotting
Cells were fixed in 2.5% formaldehyde (Merck, Inc., White-house Station, NJ, USA) or methanol : acetone (1 : 1) andprocessed for immunofluorescence microscopy as describedpreviously (Eger et al., 2000). Samples were mounted inMowiol and viewed on a ZEISS Axiovert 100M equipped withan LSM510 confocal microscope.For immunblotting, total cell lysates obtained from equal
amounts of cells were separated by SDS–PAGE. Electro-transfer of proteins onto nitrocellulose (0.2 mm, Schleicher andSchuell, Inc., Dassel, Germany) was carried out in 40mM
glycine, 48mM Tris using the BioRad Mini Trans-blot system.Immunological detection of proteins was performed with theSuper Signal ECL system (Pierce Chemical Company, Rock-ford, IL, USA).
CHIP assay
Cells were crosslinked with 1% formaldehyde (Merck, Inc.,Whitehouse Station, NJ, USA) and processed for CHIPanalyses using the CHIP Assay Kit from Upstate Biotechnlogy(Lake Placid, NY, USA). Samples were sonicated 10 times for15 s in a SonoplusGM70 (Bandelin, Berlin, Germany) at cycle90% and output 40%. Chromatin–antibody complexes wereeluted from the ProteinA or ProteinG Sepharose beads(Amersham Pharmacia Biotech, Uppsala, Sweden) by additionof freshly prepared 1% SDS, 0.1M NaHCO3 and 10mM
dithiothreitol. Crosslinking was reversed by 5M NaCl andincubation for 16 h at 651C. The DNA was extracted withphenol–chloroform, precipitated with ethanol and dissolved inwater. PCR reactions were performed with the followingprimer: Murine E-cadherin promoter: forward 50-AGACAGGGGTGGAGGAAGTT-30, reverse 50-GGGCAG GAGTCTAGCAGAAG-30. Human E-cadherin promoter: forward 50-AACTCCA GGCTAGAGGGTCA-30, reverse 50-GGGCTGGAGTCTGAACTGA-30.
RNA inhibition experiments
DeltaEF1-specific siRNA targets the sequence UGAUCAGCCUCAAUCUGCA of the human deltaEF1 mRNA. Del-taEF1-specific RNA oligonucleotides were purchased fromRZPD (Deutsches Ressourcenzentrum fur GenomforschungGmbH, Berlin, Germany) and control siRNA-Rhodamineand LaminA-specific siRNA were purchased from Quiagen(Qiagen, Hilden, Germany). RNA was transfected into MDA-MB-231 cells using Oligofectamine (Invitrogen, Carlsbad,USA) according to the manufacturers’ instructions. Cells wereprocessed for immunofluorescence and immunoblot analyses3 days after transfection.
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2383
Oncogene

AcknowledgementsWe thank Bernd Schuttengruber and Christian Seiser, MedicalUniversity Vienna, for their technical advice regarding theChIP analyses, Josef Gotzmann, Medical University Vienna,for help with RNAi experiments and Gabriele Stengl and PeterSteinlein, Institute of Molecular Pathology, Vienna, for FlowCytometry (FACS) experiments. In addition, we thank Wolf-
gang Mikulits, Cancer Research Institute Vienna, for his criticalcomments on the manuscript. This study was supported bygrants from the Austrian Science Research Fund (FWF) No.SFB 006 to RF (603) and HB (612), by funds from the‘Hochschuljubilaumsstifting’ of the city of Vienna to AE, and byfunds of the Austrian Ministry of Education, Science, and theArts (Austrian Genome Research Program GEN-AU) to MS.
References
Batlle E, Sancho E, Franci C, Dominguez D, Monfar M,Baulida J and Garcia De Herreros A. (2000). Nat. Cell. Biol.,2, 84–89.
Behrens J, Lowrick O, Klein-Hitpass L and Birchmeier W.(1991). Proc. Natl. Acad. Sci. USA, 88, 11495–11499.
Behrens J, Mareel MM, Van Roy FM and Birchmeier W.(1989). J. Cell Biol., 108, 2435–2447.
Berx G, Cleton-Jansen AM, Nollet F, De Leeuw WJ, Van deVijver M, Cornelisse C and van Roy F. (1995). EMBO J.,14, 6107–6115.
Birchmeier C, Birchmeier W and Brand-Saberi B. (1996). ActaAnat., 156, 217–226.
Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A,Palacios J and Nieto MA. (2002). Oncogene, 21, 3241–3246.
Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, EstellerM and Cano A. (2003). J. Cell Sci., 116, 499–511.
Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, BlancoMJ, del Barrio MG, Portillo F and Nieto MA. (2000). Nat.Cell. Biol., 2, 76–83.
Cavallaro U and Christofori G. (2004). Nat. Rev. Cancer, 4,118–132.
Cavallaro U, Schaffhauser B and Christofori G. (2002).Cancer Lett., 176, 123–128.
Cheng CW, Wu PE, Yu JC, Huang CS, Yue CT, Wu CW andShen CY. (2001). Oncogene, 20, 3814–3823.
Comijn J, Berx G, Vermassen P, Verschueren K, vanGrunsven L, Bruyneel E, Mareel M, Huylebroeck D andvan Roy F. (2001). Mol. Cell, 7, 1267–1278.
Conacci-Sorrell M, Zhurinsky J and Ben-Ze’ev A. (2002).J. Clin. Invest., 109, 987–991.
Damm K, Hemmann U, Garin-Chesa P, Hauel N, KauffmannI, Priepke H, Niestroj C, Daiber C, Enenkel B, Guilliard B,Lauritsch I, Muller E, Pascolo E, Sauter G, Pantic M,Martens UM, Wenz C, Lingner J, Kraut N, Rettig WJ andSchnapp A. (2001). EMBO J., 20, 6958–6968.
Eger A, Stockinger A, Park J, Langkopf E, Mikula M,Gotzmann J, Mikulits W, Beug H and Foisner R. (2004).Oncogene, 23, 2672–2680.
Eger A, Stockinger A, Schaffhauser B, Beug H and Foisner R.(2000). J. Cell Biol., 148, 173–188.
Fambrough D, McClure K, Kazlauskas A and Lander ES.(1999). Cell, 97, 727–741.
Fortini ME, Lai ZC and Rubin GM. (1991). Mech. Dev., 34,113–122.
Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, WardaA, Lochner D and Birchmeier W. (1991). J. Cell Biol., 113,173–185.
Funahashi J, Sekido R, Murai K, Kamachi Y and Kondoh H.(1993). Development, 119, 433–446.
Gotzmann J, Mikula M, Eger A, Schulte-Hermann R, FoisnerR, Beug H and Mikulits W. (2004). Mutat. Res., 566,9–20.
Grady WM, Willis J, Guilford PJ, Dunbier AK, Toro TT,Lynch H, Wiesner G, Ferguson K, Eng C, Park JG, Kim SJand Markowitz S. (2000). Nat. Genet., 26, 16–17.
Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, JarrardDF, Isaacs WB, Pitha PM, Davidson NE and Baylin SB.(1995). Cancer Res., 55, 5195–5199.
Grooteclaes ML and Frisch SM. (2000). Oncogene, 19,3823–3828.
Grunert S, Jechlinger M and Beug H. (2003). Nat. Rev. Mol.Cell Biol., 4, 657–665.
Guaita S, Puig I, Franci C, Garrido M, Dominguez D, BatlleE, Sancho E, Dedhar S, De Herreros AG and Baulida J.(2002). J. Biol. Chem., 277, 39209–39216.
Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N,Harawira P, Taite H, Scoular R, Miller A and Reeve AE.(1998). Nature, 392, 402–405.
Hajra KM, Chen DY and Fearon ER. (2002). Cancer Res., 62,1613–1618.
Hay ED. (1995). Acta Anat., 154, 8–20.Hazan RB, Phillips GR, Qiao RF, Norton L and AaronsonSA. (2000). J. Cell Biol., 148, 779–790.
Jiao W, Miyazaki K and Kitajima Y. (2002). Br. J. Cancer, 86,98–101.
Lacroix M and Leclercq G. (2004). Breast Cancer Res. Treat.,83, 249–289.
Lai ZC, Rushton E, Bate M and Rubin GM. (1993). Proc.Natl. Acad. Sci. USA, 90, 4122–4126.
Mikulits W, Pradet-Balade B, Habermann B, Beug H, Garcia-Sanz JA and Mullner EW. (2000). FASEB J., 14, 1641–1652.
Morgenstern JP and Land H. (1990). Nucleic Acids Res., 18,3587–3596.
Ohira T, Gemmill RM, Ferguson K, Kusy S, Roche J,Brambilla E, Zeng C, Baron A, Bemis L, Erickson P, WilderE, Rustgi A, Kitajewski J, Gabrielson E, Bremnes R,Franklin W and Drabkin HA. (2003). Proc. Natl. Acad. Sci.USA, 100, 10429–10434.
Okegawa T, Li Y, Pong RC and Hsieh JT. (2002). J. Urol.,167, 1836–1843.
Peinado H, Ballestar E, Esteller M and Cano A. (2004a). Mol.Cell. Biol., 24, 306–319.
Peinado H, Marin F, Cubillo E, Stark HJ, Fusenig N, NietoMA and Cano A. (2004b). J. Cell Sci., 117, 2827–2839.
Peinado H, Quintanilla M and Cano A. (2003). J. Biol. Chem.,278, 21113–21123.
Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G,Portillo F, Nieto MA and Cano A. (2001). J. Biol. Chem.,276, 27424–27431.
Perl AK, Wilgenbus P, Dahl U, Semb H and Christofori G.(1998). Nature, 392, 190–193.
Poser I, Dominguez D, de Herreros AG, Varnai A, BuettnerR and Bosserhoff AK. (2001). J. Biol. Chem., 276,
24661–24666.Reichmann E, Ball R, Groner B and Friis RR. (1989). J. Cell
Biol., 108, 1127–1138.Risinger JI, Berchuck A, Kohler MF and Boyd J. (1994). Nat.
Genet., 7, 98–102.Stemmler MP, Hecht A, Kinzel B and Kemler R. (2003). Dev.
Dyn., 227, 238–245.
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2384
Oncogene

Stockinger A, Eger A, Wolf J, Beug H and Foisner R. (2001).J. Cell Biol., 154, 1185–1196.
Strathdee G. (2002). Semin. Cancer Biol., 12, 373–379.Taki M, Kamata N, Yokoyama K, Fujimoto R, Tsutsumi Sand Nagayama M. (2003). Cancer Sci., 94, 593–597.
Tan C, Costello P, Sanghera J, Dominguez D, Baulida J,de Herreros AG and Dedhar S. (2001). Oncogene, 20,133–140.
Thiery JP. (2002). Nat. Rev. Cancer, 2, 442–454.Tokuhiro S, Yamada R, Chang X, Suzuki A, Kochi Y,Sawada T, Suzuki M, Nagasaki M, Ohtsuki M, Ono M,Furukawa H, Nagashima M, Yoshino S, Mabuchi A, Sekine
A, Saito S, Takahashi A, Tsunoda T, Nakamura Y andYamamoto K. (2003). Nat. Genet., 35, 341–348.
Tomita K, van Bokhoven A, van Leenders GJ, Ruijter ET,Jansen CF, Bussemakers MJ and Schalken JA. (2000).Cancer Res., 60, 3650–3654.
van Grunsven LA, Schellens A, Huylebroeck D andVerschueren K. (2001). J. Bone Joint Surg. Am., 83-A(Suppl. 1), S40–S47.
Vleminckx K, Vakaet Jr L, Mareel M, Fiers W and van Roy F.(1991). Cell, 66, 107–119.
Wheelock MJ and Johnson KR. (2003). Annu. Rev. Cell. Dev.Biol., 19, 207–235.
DeltaEF1 represses E-cadherin in human cancer cellsA Eger et al
2385
Oncogene